Am. J. Hum. Genet. 52:144-151, 1993 Cloning of Functional Alpha Propionyl CoA Carboxylase and Correction of Enzyme Deficiency in pccA Fibroblasts Jozsef Stankovics' and Fred D. Ledley Howard Hughes Medical Institute, Departments of Cell Biology and Pediatrics, Baylor College of Medicine, Houston Summary Propionyl CoA carboxylase (PPC) is a heteromeric enzyme composed of a subunits (PCCA) and P (PCCB) subunits. We describe cDNA clones expressing human PCCA and complementation of the genetic defect in pccA fibroblasts by DNA-mediated gene transfer. Two cDNA clones were constructed. The first corresponds to the previously reported, putatively full-length, open reading frame. The second encodes a chimera composed of the mitochondrial leader sequence of human methylmalonyl CoA mutase and the mature PCCA protein. Both clones reconstitute propionate flux to normal levels in fibroblasts from patients genetically deficient in PCCA (pccA). The maximal level of propionate flux approached, but never exceeded, the levels seen in control plates of normal cells. In contrast, the maximal level of PPC holoenzyme activity reached only 10%-20% that of normal controls, which corresponded roughly to the fraction of cells actually trans- formed with the recombinant gene. These data suggest that the level of PCCA expression in fibroblasts does not normally limit PCC holoenzyme activity or propionate flux. The fact that a small fraction of cells reconsti- tutes propionate flux to normal levels suggests that metabolic cooperation between cells is capable of increasing the metabolic capacity of recombinant enzyme in a subpopulation of cells. These factors may have important implications for the rational design of somatic gene therapy for PCCA deficiency. Introduction Metabolism of propionate in higher eukaryotes pro- ceeds by derivatization of this volatile fatty acid to propionyl CoA, followed by degradation via the en- zymes propionyl CoA carboxylase (PCC; E.C.6.4. 1.3), methylmalonyl CoA racemase, and methylmalo- nyl CoA mutase (MCM; E.C.5.4.99.2). PCC is a het- eromeric dodecamer composed of a subunits (PCCA) and i (PCCB) subunits. Genetic deficiency of PCC causes an inborn error of metabolism, termed "propi- onic acidemia." Two distinct genotypic forms of pro- pionic acidemia are classically distinguished by so- matic cell complementation: pccA (McKusick 23200), resulting from inherited defects in the a (PCCA) gene, Address for correspondence and reprints: Fred D. Ledley, M.D., Department of Cell Biology, Baylor College of Medicine, Houston, TX 77030. 1. Present address: University Medical School of PNcs, Depart- ment of Pediatrics, P~cs, Hungary. i 1993 by The American Society of Human Genetics. All rights reserved. 0002-9297/93/5201-0017$02.00 144 and pccBC (McKusick 23205), resulting from inher- ited defects in the t3 (PCCB) gene. PCCA and PCCB cDNA clones have been reported elsewhere (Kraus et al. 1986; Lamhonwah et al. 1986), and mutations in the PCCA gene have been identified which are associ- ated with pccA forms of propionic acidemia (Ohura et al. 1989). Both pccA deficiency and pccBC deficiency cause profound disturbances in intermediary metabolism. Infants can present with severe metabolic acidosis, hyperglycinemia, and hyperammonemia in the new- born period, which is often fatal. Survivors often have recurrent episodes of life-threatening acidemia and mental retardation, despite dietary and pharmacologi- cal management (Rosenberg and Fenton 1989). Our laboratory is interested in the molecular and biochemical processes which will be involved in so- matic gene therapy for organic acid disorders. In stud- ies directed at developing somatic gene therapy for MCM deficiency (methylmalonic acidemia; McKu- sick 251000), we have demonstrated the complemen- tation of MCM deficiency in fibroblasts from patients

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Am. J. Hum. Genet. 52:144-151, 1993
Cloning of Functional Alpha Propionyl CoA Carboxylase andCorrection of Enzyme Deficiency in pccA FibroblastsJozsef Stankovics' and Fred D. Ledley
Howard Hughes Medical Institute, Departments of Cell Biology and Pediatrics, Baylor College of Medicine, Houston
Summary
Propionyl CoA carboxylase (PPC) is a heteromeric enzyme composed of a subunits (PCCA) and P (PCCB)subunits. We describe cDNA clones expressing human PCCA and complementation of the genetic defectinpccA fibroblasts by DNA-mediated gene transfer. Two cDNA clones were constructed. The first correspondsto the previously reported, putatively full-length, open reading frame. The second encodes a chimeracomposed of the mitochondrial leader sequence of human methylmalonyl CoA mutase and the mature PCCAprotein. Both clones reconstitute propionate flux to normal levels in fibroblasts from patients geneticallydeficient in PCCA (pccA). The maximal level of propionate flux approached, but never exceeded, the levelsseen in control plates of normal cells. In contrast, the maximal level of PPC holoenzyme activity reachedonly 10%-20% that of normal controls, which corresponded roughly to the fraction of cells actually trans-formed with the recombinant gene. These data suggest that the level of PCCA expression in fibroblasts doesnot normally limit PCC holoenzyme activity or propionate flux. The fact that a small fraction of cells reconsti-tutes propionate flux to normal levels suggests that metabolic cooperation between cells is capable ofincreasing the metabolic capacity of recombinant enzyme in a subpopulation of cells. These factors may haveimportant implications for the rational design of somatic gene therapy for PCCA deficiency.
Introduction
Metabolism of propionate in higher eukaryotes pro-ceeds by derivatization of this volatile fatty acid topropionyl CoA, followed by degradation via the en-zymes propionyl CoA carboxylase (PCC; E.C.6.4.1.3), methylmalonyl CoA racemase, and methylmalo-nyl CoA mutase (MCM; E.C.5.4.99.2). PCC is a het-eromeric dodecamer composed of a subunits (PCCA)and i (PCCB) subunits. Genetic deficiency of PCCcauses an inborn error of metabolism, termed "propi-onic acidemia." Two distinct genotypic forms of pro-pionic acidemia are classically distinguished by so-matic cell complementation: pccA (McKusick 23200),resulting from inherited defects in the a (PCCA) gene,
Address for correspondence and reprints: Fred D. Ledley, M.D.,Department of Cell Biology, Baylor College of Medicine, Houston,TX 77030.
1. Present address: University Medical School of PNcs, Depart-ment of Pediatrics, P~cs, Hungary.i 1993 by The American Society of Human Genetics. All rights reserved.0002-9297/93/5201-0017$02.00
144
and pccBC (McKusick 23205), resulting from inher-ited defects in the t3 (PCCB) gene. PCCA and PCCBcDNA clones have been reported elsewhere (Kraus etal. 1986; Lamhonwah et al. 1986), and mutations inthe PCCA gene have been identified which are associ-ated with pccA forms of propionic acidemia (Ohuraet al. 1989).
Both pccA deficiency and pccBC deficiency causeprofound disturbances in intermediary metabolism.Infants can present with severe metabolic acidosis,hyperglycinemia, and hyperammonemia in the new-born period, which is often fatal. Survivors often haverecurrent episodes of life-threatening acidemia andmental retardation, despite dietary and pharmacologi-cal management (Rosenberg and Fenton 1989).Our laboratory is interested in the molecular and
biochemical processes which will be involved in so-matic gene therapy for organic acid disorders. In stud-ies directed at developing somatic gene therapy forMCM deficiency (methylmalonic acidemia; McKu-sick 251000), we have demonstrated the complemen-tation ofMCM deficiency in fibroblasts from patients

Complementation of pccA by Gene Transfer
with mut forms of methylmalonic acidemia, by DNA-mediated gene transfer (Wilkemeyer et al. 1991, andin press; Andrews et al., submitted), as well as perma-nent restoration ofMCM activity in fibroblasts trans-duced with a recombinant retroviral vector carryingthe humanMCM gene (Sawada and Ledley, in press).These studies have also addressed the metabolic conse-quences of overexpressing recombinant MCM, theconstraints on metabolic flux which will determine theoptimal somatic target for gene therapy, and the levelof recombinant gene expression required to obtain abiological effect (Wilkemeyer et al., in press).The present studies are directed at considering so-
matic gene therapy for propionic acidemia. We de-scribe the cloning of a functional PCCA cDNA clone,demonstrate that DNA gene transfer of this clone willcomplement the genetic defect in pccA fibroblasts, andprovide an initial assessment of the metabolic interac-tions which will be involved in constituting propionateflux for the purposes of somatic gene therapy.
Material and Methods
Amplification and Cloning of the Open ReadingFrame of PCCA
Oligonucleotides corresponding to the published se-quences of the proposed translation initiation and ter-mination codons ofhumanPCCA (GenBank accessionnumber X14608) were synthesized (Lamhonwah et al.1986). Additional oligonucleotides were synthesizedto internal sequences. Oligonucleotide sequences weremodified to include restriction-endonuclease sites, with-out altering the reported amino acid sequence (fig. 1).
Total RNA was reverse-transcribed using randomoligonucleotide primers, and this material was ampli-fied by PCR as described elsewhere (Innis et al. 1990).PCR was performed on phage constituting a humanliver cDNA library (Kwok et al. 1985), as describedelsewhere (Jansen and Ledley 1990). PCR conditionsincluded 30 cycles at 921C for 1 min, 61 0C for 1 min,and 740C for 1.5 min. PCR products were subclonedafter restriction-enzyme digestion into pGEM-3Zf(Promega). The complete open reading frame was re-constituted by three-part ligation (fig. 1, top).The human MCM cDNA sequence has been re-
ported elsewhere (Jansen et al. 1989). A chimericclone encoding the mitochondrial leader sequence ofhuman MCM (Jansen et al. 1989) and the maturePCCA protein was constructed (fig. 1, bottom). PCRwas performed to amplify sequences encoding the mi-tochondrial leader sequence from human MCM, and
PCC mRNA'IAAAAAA
5' SEGMENT#1 .0. +4- #2
P K3' SEGMENT
#3 -* -4- #4K S
K
-1
MCMUL4[fNA
0kilobases
+S
2
B D#5 04-#6
il l
Sm-O-#7
PCCA cl
.14?I, I,
DNA
I
.m:1.:1.,
+-
-1 0kilobases
I I~~~~~~~~~~~~~
S4-#4
I
2
3
3
Figure I Cloning of human PCCA and expression vectors.Top, Bases 39-970 of PCCA, amplified by PCR after reverse tran-scription of total liver RNA, and bases 971-2157, amplified froma human liver cDNA library. These segments were recombinedby three-part ligation and were cloned into the expression vectorpNAss-CMV. Bottom, Chimeric gene constructed from the mito-chondrial leader sequence of the humanMCM cDNA and from theregion coding the mature (processed) protein of PCCA. Segmentswere amplified from the respective cDNA clones by PCR and wererecombined by three-part ligation into the expression vector pNAss-CMV. P = PstI; K = KpnI; S = Sall; B = BamHI; D = Dral; Sm =
SmaI; and CMV = cytomegalovirus immediate early promoter.Oligonucleotide sequences are as follows: 1, GCGCTGCAGCT-GATGCTGAGC; 2, CAAGGAACTCCACGGTACCAGCAG; 3,ATATTCCTCTGCTGGTACCGTGG; 4, TATGTCGACTTCA-TTCCAGCTCCACGAGCA; 5, GTGTTTGGATCCTCCACCA-TGTTAAGAGCT; 6, CTGGTTTAAAAGTCGTTGCTGTATG-AGCCT; and 7, CTGAACCCGGGTTCAGTGGGATATGATCC.
a DraI (blunt) restriction site was inserted at the 3' endof these sequences. Similarly, sequences encoding themature PCCA protein were amplified by PCR, and aSmaI (blunt) restriction site was inserted at the 5' endof these sequences. These two fragments were recom-bined in pGEM-3Zf plasmid by three-part ligation(fig. 1, bottom). Three independent clones were se-
cmv
+
.I..
145
Up
.,. _.,IV VL

Stankovics and Ledley
quenced in both directions by using double-stranded,dideoxy sequencing (USB Sequenase version 2.0 kit).
Gene Transfer and Expression of PCCASequences constituting the open reading frame of
PCCA or the chimeric MCM/PCCA construct weresubcloned into the vector pNAss-CMV (MacGregorand Caskey 1989) by blunt-end ligation (fig. 1). Pri-mary pccA and pccBC fibroblasts were provided byDr. Wayne Fenton (Yale University). Mut and normalfibroblasts have been described elsewhere (Ledley etal. 1990). Expression-vector plasmids were intro-duced into primary fibroblasts by electroporation us-ing a BioRad GenepulserTm according to a methoddescribed elsewhere (Wilkemeyer et al. 1991). Elec-troporation was performed with 5 x 105 cells in 300gl, with 20 jg of DNA (unless varied as indicated),260 V, and 960 jF. Cells were plated at 1.5 x 105cells/ 12-mm round culture chamber (12-well plate).Control experiments with 3-galactosidase were per-formed periodically to ensure consistency of electro-poration conditions.
Incorporation of [14C]-propionate into trichloro-acetic acid-precipitable material was measured ac-cording to a method described elsewhere (Ledley et al.1990), 48 h after electroporation, during the transientphase of gene expression. PCC holoenzyme activitywas measured in protein extracts harvested 48 h afterelectroporation, by using the method of Gravel et al.(1977), which measures the fixation of [14C]-HCO3and incorporation into acid-precipitable material.Assays were performed on 25 jig of protein harvestedfrom trypsinized cells by three freeze-thaw cycles(- 700C and 20'C) in 0.05M Tris-HCl (pH 8), 0.025M KCl. Protein concentration was determined by Bio-Rad assay. For cocultivation, cells were plated on 12-
1064 1070 1080 1090 1100I I
REPORTED HUMAN PCCA SEQUENCEAA IleHisTr2ProGlXProSerProGlvLvs Thr VaILeufin
well (12-mm) plates at a density of 150,000 cells/well after overnight treatment with mitomycin-C at aconcentration of 5 ig/106 cells.
Results
Cloning and Sequencing the Open Reading Frameof Human PCCA
Sequences corresponding to the reported open read-ing frame ofPCCA were amplified by PCR, subcloned,and sequenced. The 5' end of the cDNA from base39 to base 970 was cloned from reverse-transcribedhuman liver RNA, and the 3' end of the cDNA frombase 971 to base 2157 was cloned from a human livercDNA library (Kwok et al. 1985). A substantial dis-crepancy was identified between the sequence of ourclones and that reported elsewhere (Lamhonwah et al.1986), involving nine missing or inserted nucleotidesbetween base 1066 and base 1141 (fig. 2). Thesechanges alter the predicted reading frame comprising26 amino acids between amino acid 339 and aminoacid 365. The reading frame predicted from our se-quence preserves apparent homology to the rat PCCAsequence reported by Browner at al. (1989) (fig. 2).
Reconstitution of Propionate Incorporation in PCCA-deficientCells with PCCA or MCMIPCCA cDNA Clones
Sequences corresponding to either the putative read-ing frame ofPCCA or the chimeric MCM-PCCA con-struct were recombined in an expression vector whichutilizes the CMV immediate early promoter (fig. 3)and were electroporated into PCCA-deficient cells aswell as into control cells including normal, pccBC, andmut fibroblasts. Incorporation of [14C]-propionateinto TCA-precipitable material is dependent on theactivity of both PCC and MCM, as evidenced by the
1110 1120 1130 1140| I
1150
GluHissLeu SerGlvThrAsnLvsLeullePheAlaPheAasnDNA ATTCACTGGCCTGGACCTAGTCCAGGAAA*GA*CCGTGTTGC*A*GGAGC*ACC*TCTCAGGCACAAACAAGCTGATATTCGCATTCAACGG...DNA ATT*ACTGGCCTGGACCTAGTCCAGGAAATGATCCGTGTTGCTAAGG*GCTACCCTCTCAGGCACAAACAAGCTGATATTCGCAT*CAACGG...AA Ile ThrGlyLeuAsDLeuValGlnGluMetIleArgValAlaLysGly TyrProLeuAraHisLvsGlnAlaAsIleAraIle Asn...REVISED HUMAN PCCA SEQUENCE
AA Ile ThrGlyLeuAspLeuValGlnGluMetIleLeuValAlaLysGly TyrProLeuArgLisLysGlnGluAspIleProIle SerDNA ATT*ACTGGCCTGGACTTAGTCCAAGAAATGATCCTGGTTGCTAAGG*GTTACCCACTCAGGCACAAGCAAGAGGATATTCCCAT*CAGTGG...RAT PCCA SEQUENCE
Figure 2 Revised sequence of human PCCA. Differences between the PCCA sequence published by Lamhonwah et al. (1986) andthe sequence determined in these studies are shown. The revised protein sequence (underlined) has a different reading frame and preservesidentity to the rat PCCA protein sequence reported by Browner et al. (1989) and revised by W. Fenton and L. E. Rosenberg (personalcommunication).
146
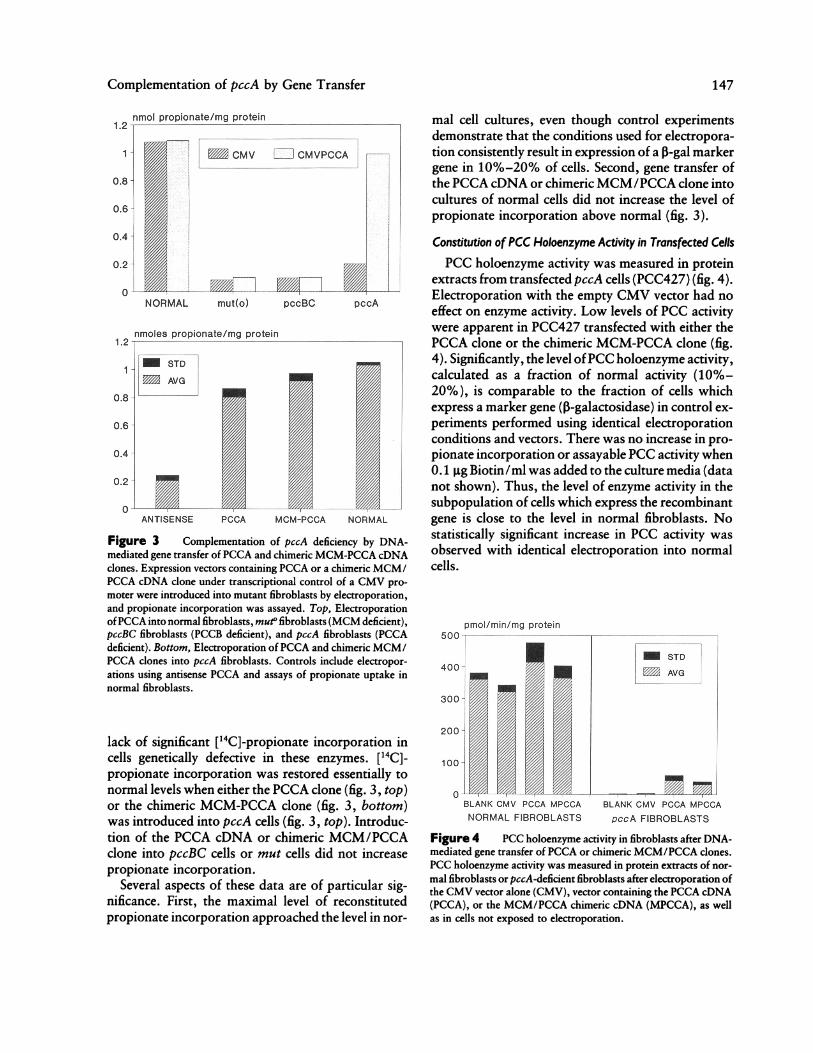
Complementation of pccA by Gene Transfer
nmol propionate/mg protein
ANTISENSE PCCA MCM-PCCA NORMAL
Figure 3 Complementation of pccA deficiency by DNA-mediated gene transfer ofPCCA and chimeric MCM-PCCA cDNAclones. Expression vectors containing PCCA or a chimeric MCM/PCCA cDNA clone under transcriptional control of a CMV pro-moter were introduced into mutant fibroblasts by electroporation,and propionate incorporation was assayed. Top, ElectroporationofPCCA into normal fibroblasts, mut fibroblasts (MCM deficient),pccBC fibroblasts (PCCB deficient), and pccA fibroblasts (PCCAdeficient). Bottom, Electroporation ofPCCA and chimeric MCM/PCCA clones into pccA fibroblasts. Controls include electropor-ations using antisense PCCA and assays of propionate uptake innormal fibroblasts.
lack of significant [14C]-propionate incorporation incells genetically defective in these enzymes. [14C]-propionate incorporation was restored essentially tonormal levels when either the PCCA clone (fig. 3, top)or the chimeric MCM-PCCA clone (fig. 3, bottom)was introduced into pccA cells (fig. 3, top). Introduc-tion of the PCCA cDNA or chimeric MCM/PCCAclone into pccBC cells or mut cells did not increasepropionate incorporation.
Several aspects of these data are of particular sig-nificance. First, the maximal level of reconstitutedpropionate incorporation approached the level in nor-
mal cell cultures, even though control experimentsdemonstrate that the conditions used for electropora-tion consistently result in expression of a 3-gal markergene in 10%-20% of cells. Second, gene transfer ofthe PCCA cDNA or chimericMCM/PCCA clone intocultures of normal cells did not increase the level ofpropionate incorporation above normal (fig. 3).
Constitution of PCC Holoenzyme Activity in Transfected CellsPCC holoenzyme activity was measured in protein
extracts from transfected pccA cells (PCC427) (fig. 4).Electroporation with the empty CMV vector had noeffect on enzyme activity. Low levels of PCC activitywere apparent in PCC427 transfected with either thePCCA clone or the chimeric MCM-PCCA clone (fig.4). Significantly, the level ofPCC holoenzyme activity,calculated as a fraction of normal activity (10%-20%), is comparable to the fraction of cells whichexpress a marker gene (13-galactosidase) in control ex-periments performed using identical electroporationconditions and vectors. There was no increase in pro-pionate incorporation or assayable PCC activity when0.1 gg Biotin/ml was added to the culture media (datanot shown). Thus, the level of enzyme activity in thesubpopulation of cells which express the recombinantgene is close to the level in normal fibroblasts. Nostatistically significant increase in PCC activity wasobserved with identical electroporation into normalcells.
pmol/min/mg protein
BLANK CMV PCCA MPCCA BLANK CMV PCCA MPCCANORMAL FIBROBLASTS pccA FIBROBLASTS
Figure 4 PCC holoenzyme activity in fibroblasts after DNA-mediated gene transfer ofPCCA or chimeric MCM/PCCA clones.PCC holoenzyme activity was measured in protein extracts of nor-mal fibroblasts or pccA-deficient fibroblasts after electroporation ofthe CMV vector alone (CMV), vector containing the PCCA cDNA(PCCA), or the MCM/PCCA chimeric cDNA (MPCCA), as wellas in cells not exposed to electroporation.
147

Stankovics and Ledley
Dynamics of Propionate Metabolism in Transfected Cells
To further investigate the level of PCC activity re-quired to reconstitute propionate flux, propionate in-corporation was measured in response to electropora-tion with different amounts of the vector DNA (fig. 5,top). In these experiments the conditions for electro-poration were identical in each reaction, with the totalamount of DNA maintained at 20 jg by diluting thePCCA vector with the empty CMV vector. Significantlevels of [14C]-propionate incorporation are observed
150
100
50
with < 1-2 gg of DNA. Moreover, propionate incor-poration was nonlinearly related to DNA concentra-tion, with levels approaching, but never exceeding,the level in normal fibroblasts.PCC holoenzyme was assayed in protein extracts
from cells electroporated under identical conditions.PCCA activity paralleled the level of propionate flux,approaching levels which were 20% of the level innormal cell cultures. As in previous experiments, thisfraction was similar to the fraction of cells trans-formed in control experiments with the 3-galactosi-dase marker gene. In control experiments (not shown),we have demonstrated that the level of expression ofanother gene (MCM) continues to increase propor-tionally to the amount of DNA added in a range of1-60 jg of DNA.
Metabolic Cooperation between Normal and PCCA Cells
To assess whether the disproportionately greaterreconstitution of propionate flux relative to holoen-zyme activity was an artifact of exposing cells to elec-troporation, [14C]-propionate incorporation was mea-sured in cocultures containing different dilutions ofnormal fibroblasts and pccA fibroblasts (fig. 6). Theseresults demonstrate that propionate incorporation inmixed cultures is disproportionately greater than thefraction of normal cells in the culture.
Discussion
Inherited deficiencies of PCC are associated withsignificant mortality and morbidity, despite conven-
800 5 10 15 20 NORMAL
PCCA EXPRESSION VECTOR (ug DNA)
Figure 5 Relationship between amount of DNA in electro-poration and complementation of propionate incorporation andPCC holoenzyme activity. Top, Propionate incorporation in two
pccA cell lines (PCCA1749 and PCCA427) electroporated with vary-
ing amounts of the PCCA expression vector. The total amount ofDNA was maintained at a constant level by addition of the emptyCMV vector. Maximal activity in both cell lines approached butdid not exceed the levels observed in normal control fibroblasts.Bottom, PCC holoenzyme activity in PCCA1749 cells electropor-ated under identical conditions. The increase of activity parallelsthat observed for propionate incorporation, but maximal levelsare only 20% that in normal controls. This fraction resembles thefraction of cells which express the 13-galactosidase marker gene in thesame CMV vector after electroporation using identical conditions.
60
40
20
% normal activity
v0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
FRACTION OF NORMAL CELLS1
Figure 6 Propionate incorporation in cocultures of normaland pccA fibroblasts. Propionate incorporation in cocultures con-
taining different ratios of normal and pccA fibroblast. The level ofpropionate incorporation was observed to be disproportionatelygreater than the fraction of normal cells.
A* COCULTURE
+ proportional
Inn._Z uu r
n
148

Complementation of pccA by Gene Transfer
tional therapy, and may be appropriate disorders forsomatic gene therapy. Both the PCCA subunit and thePCCB subunit of this multimeric enzyme have beencloned previously (Kraus et al. 1986; Lamhonwah etal. 1986), but these studies have not demonstratedenzymatic activity of the putative full-length cDNAclones. The present work describes (a) cloning andsequencing the putative open reading frame of the asubunit of the human PCC gene and (b) complementa-tion of the defect in PCC enzyme activity and propio-nate metabolism in pccA fibroblasts by DNA-medi-ated gene transfer. The ability to complement thedefect in pccA cells by gene transfer of these clonesconfirms the association of the pccA defect with thisgenetic locus (Hsia et al. 1971; Gravel et al. 1977;Wolfet al. 1978,1980; Lamhonwah et al. 1983; Lam-honwah and Gravel 1987) and suggests that in thefuture it may be feasible to consider somatic gene ther-apy for this deficiency state.One of the problems encountered in this work was
the uncertainty whether the published human PCCAsequence was complete and whether the reported read-ing frame was entirely correct. Significant differenceswere identified in the open reading frame of our clonesand in the sequences reported previously. Moreover,there is some uncertainty whether the reported humanPCCA sequence contains the complete mitochondrialleader sequence. The reported cDNA clone has anAUG and putative mitochondrial leader sequence up-stream from the determined amino-terminal end ofthemature protein. The rat mitochondrial leader se-quence (provided by Dr. Wayne Fenton) contains anadditional 16 amino acids 5' to the human AUG.When the putative 5' untranslated sequences from thehuman cDNA are translated, there is evident homol-ogy with the rat mitochondrial leader sequence (notshown), suggesting that there is evolutionary pressurefor preservation of these sequences. Primer extensiondata from our laboratory (data not shown) suggestthat there are >50 bases 5' to the putative AUG of thehuman open reading frame, although thus far we havenot been able to clone these sequences. Because of ourconcern about the completeness of the PCCA mito-chondrial leader sequence, we constructed clones cor-responding to both the reported human sequence anda chimera with sequences encoding the mitochondrialleader sequence from human MCM and the maturePCCA protein. Significantly, both the chimeric genecontaining the MCM leader sequence and the genecontaining the reported open reading frame of PCCAexpressed biological activity. Thus, there is sufficient
information in the described mitochondrial leader se-quence to direct assembly of a functional PCCA sub-unit even if this sequence is incomplete.The ability to complement the defect in pccA cells
by gene transfer may provide means for differentialdiagnosis of pccA deficiency vis-a-vis pccBC defi-ciency. This method for diagnosis would be importantfor predicting the susceptibility of individual patientsto gene-replacement therapy (Wilkemeyer et al. 1991).The reconstitution of propionate metabolism in
pccA cells after gene transfer exhibited several quanti-tatively important characteristics. The first observa-tion is that constituting low levels of assayable enzymeactivity restored propionate flux to essentially normallevels. This would provide an explanation for the clini-cal observation that mutations which leave little resid-ual activity may cause a less severe clinical phenotype(Wolf et al. 1979).The second observation is that the maximal level of
PCC holoenzyme activity never exceeds 10%-20%of normal cultures. This fraction correlates with theefficiency of electroporation-namely, the number ofcells which stain with X gal after control electropor-ation with identical constructs containing the s-ga-lactosidase reporter gene. In addition, no consistentincrease in holoenzyme activity was observed afterelectroporation of normal cells. In light ofthe fact thatPCC is a heteromeric protein, the failure to overex-press PCC holoenzyme activity could be explained bylimitations in the amount of the 13 subunit in eachcell under conditions used in these experiments. Thishypothesis differs from the predictions of Ohura et al.(1989), which suggest that the 0i subunit is normallysynthesized in excess and is rapidly degraded unlesscomplexed with a subunit. In the future, both hypoth-eses can be tested with expression vectors for PCCB.The third observation is that, with increasing
amounts ofDNA, the maximal level of [14C]-propion-ate incorporation in transfected cultures approached,but never exceeded, the levels in normal cell cultures.Again, this level of propionate flux was achieved withconditions which introduce the recombinant gene intoonly 10%-20% of cells. A disproportionately highlevel of propionate incorporation relative to the frac-tion of metabolically competent cells was also observedin cocultures of PCCA and normal cells, suggestingthat this was not simply an artifact of electroporation.A similar disproportionality between propionate
flux and the fraction of transfected cells has been ob-served after genetic complementation of MCM defi-ciency (Wilkemeyer et al., in press). These studies sug-
149

150 Stankovics and Ledley
gested that there was a rate-limiting reaction inpropionate flux, which could not be increased by over-expression ofMCM. These studies also suggested thatmetabolic cooperation between adjacent cells enabledthe products of the limiting reaction to move betweencells, thus increasing the flux through MCM in thesubpopulation of genetically reconstituted cells. Thepotential for metabolic cooperation was demon-strated by cocultivation of pcc and mut. These cocul-tures exhibit higher levels of propionate metabolismthan does either mutant cell line alone, suggesting thatthe products of the PCC reaction can move betweencells and serve as substrates for MCM (Wilkemeyeret al., in press). In typical experiments the level of[14C]-propionate incorporation, which is 1.8-2.5nmol propionate/mg protein in mut or pcc cells, willincrease to 4-5 nmol propionate/mg protein in cul-tures containing 1:2 or 2:1 mixtures of these two celllines. In contrast, when one cell line is grown on aTranswellTM membrane and the other is grown in thesame well below this membrane, there is no increasein propionate incorporation (Wilkemeyer et al., inpress).The saturation ofpropionate incorporation in pccA
cells transfected with PCCA provides more evidencefor the presence of a rate-limiting reaction in this path-way. We believe that the rate-limiting process involvesa step in the provision of the acyl-CoA substrate forPCC. However, since we were unable to overexpressPCC holoenzyme activity, the present experiments donot exclude the theoretical possibility that PCC itselfmay be the rate-limiting reaction. The fact that themetabolic flux through the subpopulation of normalor genetically reconstituted cells cocultivated withpccA cells is greater than normal suggests that meta-bolic cooperation can provide the limiting substratefor reactions in metabolically competent cells.
These data illustrate how the potential for somaticgene therapy raises novel questions concerning holo-enzyme assembly, the kinetics of metabolic flux, andthe metabolic interactions among cells. The challengeofunderstanding the consequences of genetic reconsti-tution may prove to be much like those facing meta-bolic engineering in simple organisms, where the pres-ence of accessory pathways and metabolic rigidityoften confound efforts to attain predictable ends (Bai-ley 1991; Stephanopoulos and Vallino 1991).
AcknowledgmentsWe thank Dr. Wayne Fenton for contributing the pccA
cell lines, Amira Elgawly for technical assistance, and
Tammy Reid for preparation of the manuscript. This workwas supported by National Institutes of Health grants HD-24186 and HD-24064. F.D.L. is an Assistant Investigatorof the Howard Hughes Medical Institute and has equityinterest in Vector Therapeutics, Inc.
ReferencesAndrews E, Jansen R, Crane AM, Cholin S, McDonnell M,
Ledley FD. Activity of a recombinant human methylma-lonyl CoA mutase expressed in primary fibroblasts andSaccharomyces cerevisiae (submitted)
Bailey JE (1991) Towards a science of metabolic engi-neering. Science 252:1668-1675
Browner MF, Taroni F, Sztul E, Rosenberg LE (1989) Se-quence analysis, biogenesis, and mitochondrial import ofthe a-subunit of rat liver propionyl CoA carboxylase. JBiol Chem 21:12685-12689
Gravel RA, Lam KF, Scully KJ, Hsia YE (1977) Geneticcomplementation of propionyl CoA carboxylase defi-ciency in cultured human fibroblasts. Am J Hum Genet29:378-388
Hsia YE, Scully KJ, Rosenberg LE (1971) Inherited propio-nyl CoA carboxylase deficiency in "ketotic hyperglyci-nemia." J Clin Invest 50:127-130
Innis M, Gelfand D, Sninsky J, White T (1990) PCR proto-cols. Academic Press, New York
Jansen R, Kalousek F, Fenton W, Rosenberg LE, Ledley FD(1989) Cloning of full-length methylmalonyl-CoA mutasefrom a cDNA library using the polymerase chain reaction.Genomics 4:198-205
Jansen R, Ledley FD (1990) Heterozygous mutations at themut locus in fibroblasts with mut° methylmalonic acide-mia identified by polymerase-chain-reaction cDNA clon-ing. Am J Hum Genet 47:808-814
Kraus JP, Williamson CL, Firgaira FA, Yang-Feng TL,Munke M, Francke U, Rosenberg LE (1986) Cloning andscreening with nanogram amounts of immunopurifiedmRNAs: cDNA cloning and chromosomal mapping ofcystathione 0-synthase and the 1B subunit of propionylCoA carboxylase. Proc Natl Acad Sci USA 83:2047-2051
Kwok SC, Ledley FD, Dilella AG, Robson KJH, Woo SL(1985) Nucleotide sequence of a full-length complemen-tary DNA done and amino acid sequence of human phe-nylalanine hydroxylase. Biochemistry 24:556-561
Lamhonwah AM, Barankiewicz TJ, Willard HF, MahuranDJ, Quan F, Gravel RA (1986) Isolation of cDNA clonescoding for the a and 13 chains of human propionyl CoAcarboxylase: chromosomal assignment and DNA poly-morphism associated with PCCA and PCCB genes. ProcNatl Acad Sci USA 83:4864-4868
Lamhonwah A-M, Gravel RA (1987) Propionicacidemia:absence of alpha-chain mRNA in fibroblasts from patientsof the pccA complementation group. Am J Hum Genet41:1124-1131
Lamhonwah AM, Lam KF, Tsui F, Robinson B, Saunders

Complementation of pccA by Gene Transfer 151
ME, Gravel RA (1983) Assignment of the a and 1i chainsof human propionyl CoA carboxylase to genetic comple-mentation groups. Am J Hum Genet 35:889-899
Ledley FD, Crane AM, Lumetta M (1990) Heterogeneousalleles and expression of methylmalonyl CoA mutase inmut methylmalonic acidemia. Am J Hum Genet 46:539-547
MacGregor G, Caskey CT (1989) Construction of plasmidsthat express E. coli 13 galactosidase in mammalian cells.Nucleic Acids Res 17:2365
Ohura T, KrausJP, Rosenberg LE (1989) Unequal synthesisand differential degradation of propionyl CoA carboxyl-ase in cells from normal and propionic acidemia patients.Am J Hum Genet 45:33-40
Rosenberg LE, Fenton WA (1989) Disorders of propionateand methylmalonate metabolism. In: Sciver CR, BeaudetAL, Sly WS, Valle D (eds) The metabolic basis of inheriteddisease, 6th ed, vol 1. McGraw-Hill, New York, pp 821-844
Sawada T. Ledley FD. Correction of methylmalonyl CoAmutase deficiency in mut0 fibroblasts and constitution ofgene expression in primary human hepatocytes by retrovi-ral mediated gene transfer. Somat Cel Mol Genet (in press)
Stephanopoulos G, Vallino JJ (1991) Network rigidity andmetabolic engineering in metabolite overproduction. Sci-ence 252:1675-1681
Wilkemeyer MF, Crane AM, Ledley FD (1991) Differentialdiagnosis ofmut and cbl methylmalonic aciduria byDNAmediated gene transfer in primary fibroblasts. J Clin Invest87:915-918
Wilkemeyer M, StankovicsJ, Foy T, Ledley FD. Propionatemetabolism in cultured human cells after overexpressionof recombinant methylmalonyl CoA mutase: implicationsfor somatic gene therapy. Somat Cell Mol Genet (in press)
Wolf B. Hsia YE, Rosenberg LE (1978) Biochemical differ-ences between mutant propionyl CoA carboxylase fromtwo complementation groups. Am J Hum Genet 30:455-464
Wolf B, Paulson EP, Hsia YE (1979) Asymptomaticpropio-nyl CoA carboxylase deficiency in a 13 year old girl. JPediatr 95:563-566
Wolf B, Willard HF, Rosenberg LE (1980) Kinetic analysisof genetic complementation in heterokaryons of propio-nyl CoA carboxylase-deficient human fibroblasts. Am JHum Genet 32:16-25
Related Documents











