Claudin-1 Involved in Neonatal Ichthyosis Sclerosing Cholangitis Syndrome Regulates Hepatic Paracellular Permeability Brigitte Grosse, 1 Doris Cassio, 1 Nadya Yousef, 1,2 Ce ´line Bernardo, 3 Emmanuel Jacquemin, 1,4 and Emmanuel Gonzales 1,4 Neonatal ichthyosis and sclerosing cholangitis (NISCH) syndrome is a liver disease caused by mutations of CLDN1 encoding Claudin-1, a tight-junction (TJ) protein. In this syn- drome, it is speculated that cholestasis is caused by Claudin-1 absence, leading to increased paracellular permeability and liver injuries secondary to paracellular bile regurgitation. We studied the role of claudin-1 in hepatic paracellular permeability. A NISCH liver and polar- ized rat cell lines forming TJs, the hepatocellular Can 10 and the cholangiocellular normal rat choloangiocyte (NRC), were used. In contrast to NRC, Can 10 does not express claudin- 1. Can 10 cells were transfected with a plasmid encoding Claudin-1, and stable Claudin-1- expressing clones were isolated. Claudin-1 expression was silenced by transfection with short interfering RNA in Can 10 clones and with short hairpin RNA in NRC. Claudin-1 expres- sion was evaluated by quantitative reverse-transcriptase polymerase chain reaction, immuno- blotting, and immunolocalization. Paracellular permeability was assessed by fluorescein isothiocyanate-dextran passage in both lines and by transepithelial resistance measurements in NRC. In the NISCH liver, Claudin-1 was not detected in hepatocytes or cholangiocytes. In Claudin-1 expressing Can 10 clones, Claudin-1 was localized at the TJ and paracellular permeability was decreased, compared to parental Can 10 cells, this decrease correlating with claudin-1 levels. Silencing of Claudin-1 in Can 10 clones increased paracellular perme- ability to a level similar to that of parental cells. Similarly, we observed an increase of para- cellular permeability in NRC cells silenced for claudin-1 expression. Conclusion: Defect in claudin-1 expression increases paracellular permeability in polarized hepatic cell lines, sup- porting the hypothesis that paracellular bile leakage through deficient TJs is involved in liver pathology observed in NISCH syndrome. (HEPATOLOGY 2012;55:1249-1259) N eonatal ichthyosis and sclerosing cholangitis (NISCH) syndrome (MIM 607626) is a rare autosomal recessive disease caused by muta- tions in CLDN1 encoding Claudin-1, an integral membrane protein of tight junctions (TJs). 1,2 To date, 12 patients have been reported as presenting with neo- natal-onset sclerosing cholangitis, ichthyosis, hypotri- chosis, and dental anomalies. 2-5 Two deletions result- ing in premature stop codon have been identified. 2,3 In skin and fibroblasts of NISCH patients, Claudin-1 was not detected by immunoblotting or immunostain- ing. 2,3,6 In the 2 patients reported on, so far, in whom liver claudin-1 expression was studied, expression in cholangiocytes was not studied. Claudin-1 was not detected by immunoblotting using a whole liver extract in 1 patient or by immunofluorescence in he- patocytes in the other patient. 2 TJs are junctional complexes that contribute to the formation of polarized epithelial barriers by controlling the extent and selectivity of permeability along the Abbreviations: BC, bile canaliculi; BSEP, bile salt export pump; DMEM, Dulbecco’s modified Eagle’s medium; FITC, fluorescein isothiocyanate; HCV, hepatitis C virus; JAMs, junction adhesion molecules; MDR3, multidrug resistance 3 P-glycoprotein; MEM, minimal essential medium; mRNA, messenger RNA; NISCH, neonatal ichthyosis and sclerosing cholangitis; NRC, normal rat cholangiocyte; qRT-PCR, quantitative reverse-transcriptase polymerase chain reaction; shRNA, short hairpin RNA; siRNA, short interfering RNA; TER, transepithelial resistance; TJ, tight junction; ZO, zona occludens. From the 1 INSERM UMR-S757, Orsay, University Paris-Sud 11, Paris, France; and 2 Pediatric Intensive Care Unit, 3 Pathology Unit, and 4 Pediatric Hepatology Unit and National Reference Center for Biliary Atresia, CHU Biceˆtre, Assistance Publique–Hoˆpitaux de Paris, University Paris-Sud 11, Paris, France. Received July 31, 2011; accepted October 5, 2011. N.Y. was supported by the Groupe Francophone d’He´patologie, Gastroente´rologie et Nutrition Pe´diatriques (GFHGNP), France. Address reprint requests to: Emmanuel Gonzales, M.D., Ph.D., Service d’He´patologie Pe´diatrique, Hoˆpital Biceˆtre, 78 rue du Ge´ne´ral Leclerc, Le Kremlin-Biceˆtre, 94275 Paris, France. E-mail: [email protected]; fax: þ33 1 45 21 28 16. 1249

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Claudin-1 Involved in Neonatal Ichthyosis SclerosingCholangitis Syndrome Regulates Hepatic
Paracellular PermeabilityBrigitte Grosse,1 Doris Cassio,1 Nadya Yousef,1,2 Celine Bernardo,3 Emmanuel Jacquemin,1,4
and Emmanuel Gonzales1,4
Neonatal ichthyosis and sclerosing cholangitis (NISCH) syndrome is a liver disease causedby mutations of CLDN1 encoding Claudin-1, a tight-junction (TJ) protein. In this syn-drome, it is speculated that cholestasis is caused by Claudin-1 absence, leading to increasedparacellular permeability and liver injuries secondary to paracellular bile regurgitation. Westudied the role of claudin-1 in hepatic paracellular permeability. A NISCH liver and polar-ized rat cell lines forming TJs, the hepatocellular Can 10 and the cholangiocellular normalrat choloangiocyte (NRC), were used. In contrast to NRC, Can 10 does not express claudin-1. Can 10 cells were transfected with a plasmid encoding Claudin-1, and stable Claudin-1-expressing clones were isolated. Claudin-1 expression was silenced by transfection with shortinterfering RNA in Can 10 clones and with short hairpin RNA in NRC. Claudin-1 expres-sion was evaluated by quantitative reverse-transcriptase polymerase chain reaction, immuno-blotting, and immunolocalization. Paracellular permeability was assessed by fluoresceinisothiocyanate-dextran passage in both lines and by transepithelial resistance measurementsin NRC. In the NISCH liver, Claudin-1 was not detected in hepatocytes or cholangiocytes.In Claudin-1 expressing Can 10 clones, Claudin-1 was localized at the TJ and paracellularpermeability was decreased, compared to parental Can 10 cells, this decrease correlatingwith claudin-1 levels. Silencing of Claudin-1 in Can 10 clones increased paracellular perme-ability to a level similar to that of parental cells. Similarly, we observed an increase of para-cellular permeability in NRC cells silenced for claudin-1 expression. Conclusion: Defect inclaudin-1 expression increases paracellular permeability in polarized hepatic cell lines, sup-porting the hypothesis that paracellular bile leakage through deficient TJs is involved in liverpathology observed in NISCH syndrome. (HEPATOLOGY 2012;55:1249-1259)
Neonatal ichthyosis and sclerosing cholangitis(NISCH) syndrome (MIM 607626) is a rareautosomal recessive disease caused by muta-
tions in CLDN1 encoding Claudin-1, an integralmembrane protein of tight junctions (TJs).1,2 To date,12 patients have been reported as presenting with neo-natal-onset sclerosing cholangitis, ichthyosis, hypotri-chosis, and dental anomalies.2-5 Two deletions result-ing in premature stop codon have been identified.2,3
In skin and fibroblasts of NISCH patients, Claudin-1
was not detected by immunoblotting or immunostain-ing.2,3,6 In the 2 patients reported on, so far, in whomliver claudin-1 expression was studied, expression incholangiocytes was not studied. Claudin-1 was notdetected by immunoblotting using a whole liverextract in 1 patient or by immunofluorescence in he-patocytes in the other patient.2
TJs are junctional complexes that contribute to theformation of polarized epithelial barriers by controllingthe extent and selectivity of permeability along the
Abbreviations: BC, bile canaliculi; BSEP, bile salt export pump; DMEM, Dulbecco’s modified Eagle’s medium; FITC, fluorescein isothiocyanate; HCV, hepatitisC virus; JAMs, junction adhesion molecules; MDR3, multidrug resistance 3 P-glycoprotein; MEM, minimal essential medium; mRNA, messenger RNA; NISCH,neonatal ichthyosis and sclerosing cholangitis; NRC, normal rat cholangiocyte; qRT-PCR, quantitative reverse-transcriptase polymerase chain reaction; shRNA, shorthairpin RNA; siRNA, short interfering RNA; TER, transepithelial resistance; TJ, tight junction; ZO, zona occludens.From the 1INSERM UMR-S757, Orsay, University Paris-Sud 11, Paris, France; and 2Pediatric Intensive Care Unit, 3Pathology Unit, and 4Pediatric Hepatology
Unit and National Reference Center for Biliary Atresia, CHU Bicetre, Assistance Publique–Hopitaux de Paris, University Paris-Sud 11, Paris, France.Received July 31, 2011; accepted October 5, 2011.N.Y. was supported by the Groupe Francophone d’Hepatologie, Gastroenterologie et Nutrition Pediatriques (GFHGNP), France. Address reprint requests to:
Emmanuel Gonzales, M.D., Ph.D., Service d’Hepatologie Pediatrique, Hopital Bicetre, 78 rue du General Leclerc, Le Kremlin-Bicetre, 94275 Paris, France.E-mail: [email protected]; fax: þ33 1 45 21 28 16.
1249

paracellular pathway (i.e., the gate function) and byforming an apical/basolateral intramembrane diffusionbarrier to membrane proteins in the outer leaflet ofthe plasma membrane (i.e., the fence function).7,8 Thestructure of TJs is complex, involving integral mem-brane proteins (e.g., occludin, claudins, and junctionadhesion molecules; JAMs) linked to a dense cytoplas-mic network of scaffolding and adaptor proteins, sig-naling components, and actin-binding cytoskeletallinkers. In various diseases, including familial hyper-cholanemia, causative mutations in genes encoding TJproteins have been identified.9 Moreover, claudindefects have been reported in skin, kidney, inner ear,and eye genetic diseases, revealing the crucial roleplayed by TJs and claudins in epithelia.10 In the liver,TJs separate bile from plasma. They play a critical rolein establishing the biliary canalicular pole, maintainingcanalicular localization of specific proteins, sealing bilecanaliculi (BC), and protecting liver cells from toxicbile components, in particular bile acids.11 Secondarychanges in the expression pattern of TJ proteins andtheir transcripts have been described in different ani-mal models of cholestasis.12-14 In various human cho-lestatic diseases, secondary alterations of TJs have beenreported, both in hepatocytes and in cholangiocytes,suggesting that paracellular permeability plays a role inthe mechanism of cholestasis.15-19
In patients with NISCH syndrome, it is speculatedthat increased paracellular permeability caused by pri-mary Claudin-1 defect leads to bile leakage, resulting inbile duct and hepatocellular injury. Unfortunately, thebasis for the sclerosing cholangitis in NISCH patientscould not be studied in Cldn1-knockout mice becauseof severe impairment of epidermal barrier function andsubsequent dehydration, which results in early lethal-ity.10,20 Claudin-1-related modifications of paracellularpermeability have been reported in Madin-Darby caninekidney cells,21,22 but, so far, comparable studies in livercell lines have not been carried out. Herein, we studiedthe effect of claudin-1 expression on paracellular perme-ability in rat hepatocellular (Can 10) and biliary (i.e.,normal rat cholangiocyte; NRC) polarized cell lines.Our results show that the level of Claudin-1 expressioncorrelates with paracellular permeability in both lines,supporting the pathophysiological mechanism of bileleakage caused by a primary Claudin-1 defect in theliver disease observed in NISCH syndrome.
Materials and Methods
Chemicals. Fetal calf serum, Dulbecco’s modifiedEagle’s medium (DMEM) F12 medium, some supple-ments for NRC culture (minimal essential medium[MEM] non-amino-acid, lipid concentrate, vitamin so-lution, and L-glutamine), Opti-MEM, and Geneticinwere from Gibco (Grand Island, NY). Puromycin wasfrom Ozyme (Paris, France). Type I collagen was fromBD Biosciences (Franklin Lakes, NJ). Coon’s modifiedF12 medium, some supplements for NRC culture(e.g., spite medium, trypsin inhibitor soybean, bovinepituitary extract, dexamethasone, and triodothyronine),TRI Reagent, 4-kDa-fluorescein isothiocyanate(FITC)-dextran, and antibody directed against actinwere from Sigma-Aldrich (St. Louis, MO). Antibodiesdirected against human or mouse claudin-1, -2, -3, -4,and -5, occludin, and JAM-A were from Zymed(South San Francisco, CA). Antibodies against E-cad-herin and zona occludens (ZO)-2 were from Transduc-tion Laboratories (Lexington, KY), and anti-ZO-1 wasfrom Dr. Bruce Stevenson (Salk Institute, La Jolla,CA).23 FITC-dextran (3-kDa) and secondary antibod-ies conjugated to Alexa Fluor and horseradish peroxi-dase were from Molecular Probes (Eugene, OR).Cell Lines and Cell Culture. Can 10 cells were cul-
tured as previously described.24 The NRC cell line,kindly provided by Dr. Nicholas LaRusso (MayoClinic, Rochester, MN), was cultured on type I colla-gen (except for inserts), as previously described.25 Cellswere cultured on plastic dishes (RNA and proteinextraction), on glass coverslips (immunostaining andparacellular permeability measurement for Can 10),and on inserts (transepithelial resistance [TER] andfluorescent dextran passage measurements of NRC).Generation of Claudin-1 Expressing Can 10
Clones (Can 10-CLDN-1). pCMV6-A, encodinghuman Claudin-1 (OriGene Technologies, Inc., Rock-ville, MD), was transfected into Can 10 cells, usingthe Microporator (MP-100; Labtech International,East Sussex, UK), according to the manufacturer’sinstructions. Clonal selection of stably transfected cellswas obtained with 400 lg/mL of Geneticin, added 2days after transfection. Stable Geneticin-resistant Can10-CLDN-1 clones were obtained at a frequency of8 � 10�5; their chromosome content, determined aspreviously described,24 was similar to that of parental
CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24761Potential conflict of interest: Nothing to report.Additonal Supporting Information may be found in the online version of this article.
1250 GROSSE ET AL. HEPATOLOGY, April 2012

Can 10 cells (mean number, 50; range, 46-52). Clau-din-1 expression was examined by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR), im-munoblotting, and immunolocalization in cells platedat 2-4 � 103/cm2 and cultured for 7-8 days.Silencing of Claudin-1.Short interfering RNA. Can 10-CLDN-1 clones were
transfected with short interfering RNA (siRNA) (5’-GAUCCAGUGCAAAGUCUU-3’) duplex targetingclaudin-1 and with scrambled siRNA (Eurogentec, SanDiego, CA). Cells were plated at 2.5 � 104/cm2 andimmediately transfected using Lipofectamine RNAi-MAX (Invitrogen, Carlsbad, CA), according to themanufacturer’s instructions. Final concentrations were40 nM for siRNA and 2 lL/mL for LipofectamineRNAiMAX. Cells were studied 2-3 days after siRNAtransfection.Short hairpin RNA. NRC cells were transfected 24
hours after plating (2 � 104/cm2) with a pool of threemouse claudin-1 short hairpin RNA (shRNA)-lentivi-ral vector plamids (Santa Cruz Biotechnology, SantaCruz, CA); among them, two were identical insequence to rat claudin-1 and one was not. NRC cellswere also transfected with scrambled shRNA (SantaCruz). Transfection was performed using FuGENEHD reagent (Roche, Mannheim, Germany), accordingto the manufacturer’s instructions (transfectionreagent:DNA ¼ 5:2). Puromycin (10 lg/mL) wasadded 48 hours after transfection; two independentstably transfected populations (claudin-1 shRNA pop-A and pop-B) and one scrambled shRNA populationwere isolated after 1-2 months in puromycin contain-ing medium.The silencing of claudin-1 expression was studied in
cells plated at high density (3 � 104/cm2 for Can10-CLDN-1 clones and 4-5 � 105/cm2 for NRC)after 2-3 days (Can 10-CLDN1 clones) and 3-4 days(NRC) of culture.Real-Time qRT-PCR. Total RNA was isolated using
TRI Reagent and was converted to complementaryDNA (cDNA) using SuperScript II Reverse Transcrip-tase (Invitrogen). Real-time PCR was performed usingiQ SYBR Green SuperMix (Bio-Rad, Hercules, CA),according to the manufacturer’s instructions. Relativeexpression of claudin-1 was quantified using glyceral-dehyde 3-phosphate dehydrogenase and bactin ashousekeeping genes. Primers are detailed in SupportingTable 1.Immunoblotting. Protein lysates were processed as
previously described.26 Primary antibody dilutionswere as follows: ZO-1, undiluted; JAM-A, 1/100; clau-din-1, -2, -3, and -5, 1/125; ZO-2, 1/200; claudin-4,
1/250; occludin, 1/500; and actin, 1/1000. ImageJ1.37v software was used for densitometric analysis.Immunolocalization.Cells. Cells were fixed in ethanol (4�C, 30 minutes),
then in acetone (room temperature, 1 minute). There-after, indirect immunofluorescence staining was per-formed as previously described.26 Antibodies againstclaudin-1, E-cadherin, and ZO-1 were used at dilu-tions of 1/100, 1/200, and undiluted, respectively.Appropriate Alexa Fluor–conjugated secondary anti-bodies were used at a dilution of 1/500. Cells wereobserved using a Zeiss Axioskop fluorescence micro-scope (Carl Zeiss, Inc., Gottingen, Germany). Confo-cal analysis was performed using a Zeiss LSM 510confocal microscope equipped with a �63 objective,and xy sections were taken in 0.3-lm steps.Liver sections. Immunostaining was performed as
previously described.27 Liver samples of a NISCHpatient (patient II-1, family 2, reference 2) withan homozygous CLDN1 truncating mutation werestudied.2 A healthy human liver served as a control.Anti-claudin-1 antibody was used, at a dilution of1/100. Anti-MDR3 (multidrug resistance P-glyco-protein) (Sigma-Aldrich) and anti-BSEP (bile saltexport pump) antibodies were used as previouslydescribed.27
Epithelial Permeability Assay in Can 10Cells. Paracellular permeability of Can 10 cells wasassessed by measuring paracellular diffusion into theBC of 3- and 4-kDa FITC-dextrans, as previouslydescribed.28 Cells were plated on glass coverslips at4 � 103/cm2, and paracellular permeability was exam-ined after 7-8 days. For cells transfected with siRNA, ahigher plating density (3 � 104cells/cm2) was used,and cells were examined after 2-3 days. Cells wereincubated with 2 mg/mL of FITC-dextran for 10minutes at 37�C, washed twice with cold, serum-freemedium, and observed within 5-10 minutes aftermounting. Fluorescent BC were counted and expressedas a percentage of total number of BC observed byphase contrast. Three coverslips were examined fromeach culture (four fields per coverslip and at least 200BC were observed).TER and Paracellular Permeability of NRC
Cells. Paracellular permeability of wild-type NRC,NRC transfected with scrambled shRNA, and the twopopulations transfected with claudin-1 shRNA wasassessed by measuring the TER and transepithelial fluxof 4-kDa FITC-dextran. Experiments were performed onconfluent monolayers obtained by culturing on inserts(0.4 lM pore size, BD Falcon; BD Biosciences), for 3-4days, with cells plated at 3-4 � 105/cm2. TER was
HEPATOLOGY, Vol. 55, No. 4, 2012 GROSSE ET AL. 1251

measured once a day using a Millicell-ERS volt-ohmmeter (Millipore, Billerica, MA). TER was expressed asohm.cm2 after background subtraction. On the lastday, 4-kDa FITC-dextran (2 mg/mL of serum-free cul-ture medium) was added to the upper chamber. Sam-ples were collected from the lower compartment at 30-minute intervals for 2 hours. Fluorescence intensitywas measured using a Wallac fluorometer (VICTOR3;PerkinElmer, Walthamn, MA). FITC-dextran flux wasdetermined by linear regression after correction forbackground and expressed as ng/h.cm2, using a stand-ard curve.Statistical Analysis. Data are expressed as means 6
standard deviation. Statistical analyses were performedusing the Student’s t test, with a P value <0.05 beingconsidered statistically significant.
Results
We first studied Claudin-1 expression in healthyhuman liver and in a liver of a NISCH patient. Inhealthy human liver, Claudin-1 was detected in hepa-tocytes (Fig. 1A) and cholangiocytes (Fig. 1B). InNISCH patient liver, we confirmed the absence ofClaudin-1 in hepatocytes (Fig. 1C), we showed the
absence of claudin-1 in cholangiocytes (Fig. 1D), andwe observed a normal canalicular localization of BSEP(Fig. 1E) and MDR3 (Fig. 1F). To test the role ofclaudin-1 in paracellular permeability in the liver, weused two polarized rat cell lines: the hepatocellularCan 10 and the biliary NRC. Both form TJs. How-ever, in contrast to NRC,29 Can 10 cells do notexpress claudin-1, although they express and properlytarget a large panel of TJ proteins.24,30 Therefore, weexperimentally manipulated claudin-1 levels in thesecells and subsequently determined their paracellularpermeability.Generation and Characterization of Stable Can
10 Clones Expressing Claudin-1. Can 10 is derivedfrom the rat hepatoma Fao line after a transient cul-ture in spheroids.24 It exhibits the typical hepatocytepolarity with functional long BC mimicking in vivoBC. It has been shown to be an appropriate model tostudy canalicular vectorial transport.24,30-32
The pCMV6-CLDN-1 plasmid was successfullytransfected into Can 10 cells. We isolated a total of 23stable Geneticin-resistant Can 10-CLDN-1 clones.Among those, nine clones expressed Claudin-1 andfour were selected for further analysis based on theirdifferent expression levels of claudin-1. High levels ofclaudin-1 messenger RNA (mRNA) and protein werefound in clone 10 and lower levels in clones 1, 21,and 23 (Fig. 2A,B). One negative clone, clone 6, wasused as a control. As expected, Claudin-1 mRNAand protein were not detected in negative clone 6and parental Can 10 cells (Fig. 2A,B). All clonesexpressed other TJ proteins, claudin-2, -3, -4, and -5,JAM-A, occludin, ZO-1, and ZO-2 (Fig. 2B). Mini-mal changes were observed in the expression of theseTJ proteins, except for claudin-4, which was decreasedin claudin-1-expressing Can 10-CLDN-1 clones. Todetermine the localization of Claudin-1 and homoge-neity of cell populations, Can 10-CLDN-1 cloneswere analyzed by immunofluorescence, using ZO-1 asa TJ control marker. In all four expressing clones,Claudin-1 was present in most cells and colocalizedwith ZO-1 at the TJ, as illustrated for clones 10 and21 (Fig. 2C).Paracellular Permeability Is Decreased in Can 10
Clones Expressing Claudin-1. Paracellular permeabil-ity was assessed by measuring the passage of FITC-dextrans into the BC. All Can 10-CLDN-1 clonesexpressing Claudin-1 showed reduced paracellular pas-sage of 3- (Fig. 3A) and 4-kDa (Fig. 3B), FITC-dex-trans, when compared to negative clone 6 and parentalCan 10 cells. Moreover, the decrease in permeabilitycorrelates with Claudin-1 expression level (Fig. 3C)
Fig. 1. Claudin-1 expression in healthy human liver and in a NISCHpatient liver. Liver sections from healthy human liver (A and B) or aNISCH patient (C-F) were immunostained with anti-claudin-1 (A-D),anti-BSEP (E), and anti-MDR3 (F) antibodies. In healthy human liver,Claudin-1 was concentrated at the TJ, sealing canaliculi, in hepatocyte(A, arrows), and at the upper part of the lateral plasma membrane incholangiocyte (B, arrowhead). In a NISCH patient liver, Claudin-1 wasnot detected in hepatocytes (C) or cholangiocytes (D), whereas BSEP(E) and MDR3 (F) were appropriately expressed at the canalicularmembrane. Bar, 30 lm.
1252 GROSSE ET AL. HEPATOLOGY, April 2012

(for Claudin-1 expressing clones, r2 ¼ 0.992 and0.953 for 3- and 4-kDa dextrans, respectively).Claudin-1 Silencing by siRNA Increases Paracellu-
lar Permeability to the Level of Parental Can 10cells. To further demonstrate that Claudin-1 expres-sion is directly implicated in the decrease of paracellu-lar permeability, we used siRNA to silence Claudin-1in three Can 10-CLDN-1 clones expressing various lev-els of Claudin-1. As assessed by qRT-PCR (data notshown) and immunoblotting (Fig. 4A), treatment withsiRNA targeting claudin-1 resulted in a 70%-90%decrease of Claudin-1 expression, when compared toscrambled siRNA. This decrease in Claudin-1 expres-sion was also observed by immunofluorescence (Fig.4B). In the three clones tested, 4-kDa FITC-dextranpassage increased in Claudin-1-silenced clones, reachingthe level observed in parental Can 10 cells (Fig. 4C).Claudin-1 Silencing by shRNA Increases Paracel-
lular Permeability of NRC Cells. NRC is a cell line
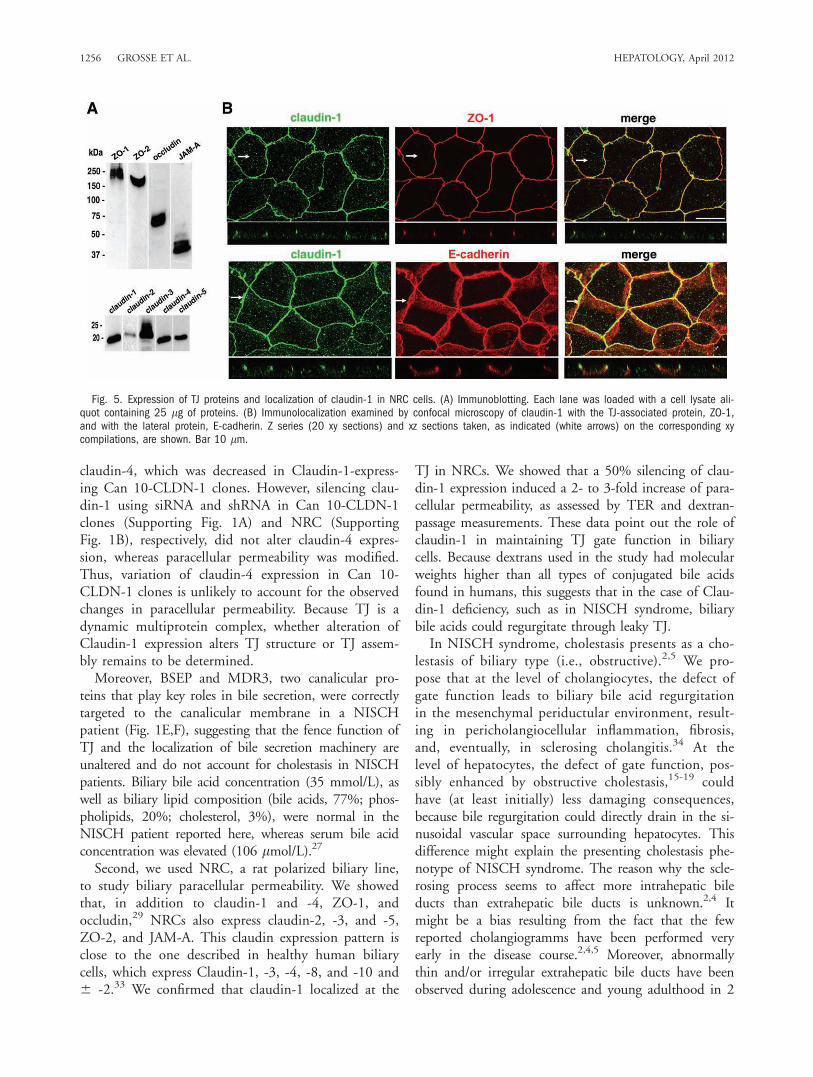
derived from a primary culture of normal rat cholan-giocytes. It exhibits the typical simple polarity of chol-angiocytes and has been shown to be an appropriatemodel to study the TJ barrier function of the bile ductepithelium.25,29 Expression of TJ proteins in NRCcells was only partially characterized.29 Using immuno-blotting, we determined that in addition to claudin-1and -4, ZO-1, and occludin,29 NRC cells also expressclaudin-2, -3, and -5, ZO-2, and JAM-A (Fig. 5A).Using confocal microscopy, we studied the localizationof claudin-1 in NRC cells (Fig. 5B). As expected, clau-din-1 was properly targeted and colocalized with ZO-1at the upper part of the lateral plasma membrane (i.e.,the TJ level), whereas E-cadherin was present through-out the lateral plasma membrane.To evaluate the role of claudin-1 in NRC paracellu-
lar permeability, cells were transfected with claudin-1shRNA. Two independent stably transfected popula-tions, claudin-1 shRNA pop-A and pop-B, were
Fig. 2. Can 10-CLDN1 clones express various levels of Claudin-1 at the TJ. (A) Expression levels of Claudin-1 mRNA were determined in fiveCan 10-CLDN1 clones (including negative clone 6) and in Can 10 parental cells, using qRT-PCR. Claudin-1 mRNA level is expressed in arbitraryunits, with a level of 1 being attributed to clone 1, the lowest producer of claudin-1. (B) Immunoblotting. Each lane was loaded with 25 lg ofproteins. Claudin-1, -2, -3, -4, and -5, JAM-A, occludin, ZO-1, and ZO-2 were detected. Actin was used as a loading control. (C) Immunolocaliza-tion of Claudin-1 and ZO-1 in three Can 10-CLDN1 clones (including negative clone 6) and in Can 10 parental cells. The corresponding phase-contrast image is shown on the left. Some BC, well visible in phase contrast, are indicated by arrows. Bar, 10 lm.
HEPATOLOGY, Vol. 55, No. 4, 2012 GROSSE ET AL. 1253

isolated and characterized (Fig. 6). In both popula-tions, we obtained a 50% decrease of claudin-1mRNA (data not shown) and claudin-1 protein (Fig.6A), when compared to cells transfected withscrambled shRNA and wild-type NRC cells. Usingimmunolocalization, we found that in contrast to wild-type cells and cells transfected with scrambled shRNA,claudin-1 is not homogeneously expressed in claudin-1shRNA pop-A and pop-B. In both populations, negativefields for claudin-1 were observed (Fig. 6B). Paracellularpermeability was examined by measuring TER (Fig.6C,D) and FITC-dextran passage (Fig. 6E,F). In wild-type NRC and in NRC transfected with scrambledshRNA, TER increased with cell growth from days 2 to4, whereas this increase was not observed in NRC trans-fected with shRNA targeting claudin-1 (Fig. 6C). Atconfluency, the TER was reduced in both cell popula-tions transfected with claudin-1 shRNA (200-220ohm.cm2), compared to the TER in cells transfectedwith scrambled shRNA or wild-type cells (550-570ohm.cm2) (Fig. 6D).Consistent with the decrease of TER, 4-kDa FITC-
dextran passage was altered in claudin-1 shRNA pop-Aand pop-B (Fig. 6E), resulting in a 2- to 3-foldincrease in paracellular flux of 4-kDa FITC-dextran inclaudin-1 shRNA pop-A and pop-B (Fig. 6F).
Discussion
NISCH syndrome is caused by mutations inCLDN1 encoding claudin-1, an integral TJ protein. Inhumans and rats, claudin-1 is expressed by hepatocytesthroughout the whole liver lobule and by cholangio-cytes.11,33 NISCH patients show radiological andhistological features of sclerosing cholangitis, whereasabsence of Claudin-1 has not been proven in cholan-giocytes.2,5 It is believed that absence of Claudin-1 inthe liver alters TJ integrity, which is crucial in prevent-ing paracellular passage of bile components, and thatthe ensuing leakage and paracellular regurgitation oftoxic bile components (i.e., bile acids) leads to bileduct injury and cholestasis in NISCH patients. Here,we showed in a NISCH patient, that claudin-1 wasnot detected in hepatocytes or cholangiocytes. Weshowed, in hepatocellular and biliary cell lines, the twomain liver cell types affected in NISCH syndrome,that a decrease in the expression level of claudin-1leads to an increase in paracellular permeability,whereas an increase in claudin-1 expression leads toparacellular permeability decrease. These data reinforcethe primary pathogenic role of mutated Claudin-1 inNISCH syndrome.
Fig. 3. Claudin-1 expression decreases paracellular permeability ofCan 10-CLDN-1 clones. (A and B) Paracellular permeability was assessedby measuring paracellular diffusion into the BC of 3- and 4-kDa FITC-dextrans. Values are means from three to five independent cell cultures.**P < 0.01; ***P < 0.001, as compared with negative clone 6 andparental Can 10 cells. (C) Relation between the paracellular permeabilityand the level of Claudin-1, calculated by densitometric analysis of immu-noblots. Claudin-1 level is expressed in arbitrary units, with a level of 1being attributed to clone 1, the lowest producer of Claudin-1.
1254 GROSSE ET AL. HEPATOLOGY, April 2012

First, we used the Can 10 line as a hepatocellularpolarized model to study hepatocellular paracellularpermeability.24,30-32 Can 10 cells express claudin-2, -3,-4, and -5, occludin, JAM-A, E-cadherin, and ZO-1and -2 proteins.30 With the exception of the absenceof claudin-1 and expression of claudin-4, the Can10 TJ protein repertoire closely resembles that ofhuman hepatocytes.11 In stable Can 10 clones express-ing human Claudin-1, paracellular permeability was
decreased. This decrease was proportional to the levelof Claudin-1. Silencing Claudin-1 with siRNArestored paracellular permeability to the level observedin Claudin-1 nonexpressing cells. Collectively, thesefindings show that Claudin-1 expression regulates para-cellular permeability in Can 10 cells and point out therole of Claudin-1 in maintaining TJ gate function in ahepatocellular cell type. Our expression studies ofother TJ proteins showed minimal changes, except for
Fig. 4. Silencing of Claudin-1 in Can 10-CLDN1 clones increases paracellular permeability to the level of parental Can 10 cells. Cells trans-fected by lipofectamine with no siRNA, scrambled siRNA, and Claudin-1 siRNA were examined 2-3 days after transfection. (A) Immunoblotting.Each lane was loaded with 25 lg of proteins. Claudin-1 was codetected with actin. (B) Immunolocalization of Claudin-1 and ZO-1 with the cor-responding phase-contrast image shown on the left. Some BC, well visible in phase contrast, are indicated by arrows. Bar, 10 lm. (C) Paracellu-lar permeability (4-kDa FITC-dextrans). Values are means from three independent cell cultures of each clone. ***P < 0.001, as compared withno siRNA and scrambled siRNA-transfected cells.
HEPATOLOGY, Vol. 55, No. 4, 2012 GROSSE ET AL. 1255
claudin-4, which was decreased in Claudin-1-express-ing Can 10-CLDN-1 clones. However, silencing clau-din-1 using siRNA and shRNA in Can 10-CLDN-1clones (Supporting Fig. 1A) and NRC (SupportingFig. 1B), respectively, did not alter claudin-4 expres-sion, whereas paracellular permeability was modified.Thus, variation of claudin-4 expression in Can 10-CLDN-1 clones is unlikely to account for the observedchanges in paracellular permeability. Because TJ is adynamic multiprotein complex, whether alteration ofClaudin-1 expression alters TJ structure or TJ assem-bly remains to be determined.Moreover, BSEP and MDR3, two canalicular pro-
teins that play key roles in bile secretion, were correctlytargeted to the canalicular membrane in a NISCHpatient (Fig. 1E,F), suggesting that the fence function ofTJ and the localization of bile secretion machinery areunaltered and do not account for cholestasis in NISCHpatients. Biliary bile acid concentration (35 mmol/L), aswell as biliary lipid composition (bile acids, 77%; phos-pholipids, 20%; cholesterol, 3%), were normal in theNISCH patient reported here, whereas serum bile acidconcentration was elevated (106 lmol/L).27
Second, we used NRC, a rat polarized biliary line,to study biliary paracellular permeability. We showedthat, in addition to claudin-1 and -4, ZO-1, andoccludin,29 NRCs also express claudin-2, -3, and -5,ZO-2, and JAM-A. This claudin expression pattern isclose to the one described in healthy human biliarycells, which express Claudin-1, -3, -4, -8, and -10 and6 -2.33 We confirmed that claudin-1 localized at the
TJ in NRCs. We showed that a 50% silencing of clau-din-1 expression induced a 2- to 3-fold increase of para-cellular permeability, as assessed by TER and dextran-passage measurements. These data point out the role ofclaudin-1 in maintaining TJ gate function in biliarycells. Because dextrans used in the study had molecularweights higher than all types of conjugated bile acidsfound in humans, this suggests that in the case of Clau-din-1 deficiency, such as in NISCH syndrome, biliarybile acids could regurgitate through leaky TJ.In NISCH syndrome, cholestasis presents as a cho-
lestasis of biliary type (i.e., obstructive).2,5 We pro-pose that at the level of cholangiocytes, the defect ofgate function leads to biliary bile acid regurgitationin the mesenchymal periductular environment, result-ing in pericholangiocellular inflammation, fibrosis,and, eventually, in sclerosing cholangitis.34 At thelevel of hepatocytes, the defect of gate function, pos-sibly enhanced by obstructive cholestasis,15-19 couldhave (at least initially) less damaging consequences,because bile regurgitation could directly drain in the si-nusoidal vascular space surrounding hepatocytes. Thisdifference might explain the presenting cholestasis phe-notype of NISCH syndrome. The reason why the scle-rosing process seems to affect more intrahepatic bileducts than extrahepatic bile ducts is unknown.2,4 Itmight be a bias resulting from the fact that the fewreported cholangiogramms have been performed veryearly in the disease course.2,4,5 Moreover, abnormallythin and/or irregular extrahepatic bile ducts have beenobserved during adolescence and young adulthood in 2
Fig. 5. Expression of TJ proteins and localization of claudin-1 in NRC cells. (A) Immunoblotting. Each lane was loaded with a cell lysate ali-quot containing 25 lg of proteins. (B) Immunolocalization examined by confocal microscopy of claudin-1 with the TJ-associated protein, ZO-1,and with the lateral protein, E-cadherin. Z series (20 xy sections) and xz sections taken, as indicated (white arrows) on the corresponding xycompilations, are shown. Bar 10 lm.
1256 GROSSE ET AL. HEPATOLOGY, April 2012

of the 4 originally reported patients with NISCH syn-drome2 (unpublished results, E.J.). This observation isin accord with the expression of claudin-1 in extrahe-patic bile ducts.33
It was found that discrete residues within the firstextracellular loop of claudin-1 are critical for hepatitisC virus (HCV) entry.35 Monoclonal antibodies target-ing claudin-1 have been shown to prevent HCV entry
Fig. 6. Silencing of claudin-1 increased paracellular permeability in NRC cells. NRC cells were transfected with scrambled or claudin-1 shRNA.Two populations, claudin-1 shRNA pop-A and pop-B, were isolated. (A) Immunoblotting. The upper panel shows a representative immunoblot.The lower panel illustrates densitometric analysis of scanned immunoblots obtained from at least three different cultures of each type of cells.*P < 0.05, as compared to scrambled shRNA-transfected cells and wild-type NRC. (B) Immunolocalization of claudin-1 and ZO-1. In each case,the corresponding phase-contrast image is shown on the left. Bar, 10 lm. (C and D) TER. (C) A representative experiment is presented (cellsplated at 3-4 � 105/insert; confluent cell monolayers obtained 3-4 days after seeding). (D) Compilation of TER values obtained at confluencyfrom three different cultures of each type of cells. ***P < 0.001, as compared to scrambled shRNA-transfected cells and wild-type NRC. (Eand F) Dextran flux measurements. The passage of 4-kDa FITC-dextran from the apical to the basal side of confluent cell monolayers was fol-lowed during 2 hours. (E) A representative experiment is shown. (F) Mean values of dextran flux obtained from three different confluent culturesof each type of cells are presented. *P < 0.05; **P < 0.01, as compared to scrambled shRNA-transfected cells and wild-type NRC. The sameinserts were used for TER and dextran flux measurements.
HEPATOLOGY, Vol. 55, No. 4, 2012 GROSSE ET AL. 1257

in human primary hepatocytes and the HepG2line.36,37 TJ was not altered by such treatment in thepoorly polarized HepG2 line.37 However, our resultsobtained in highly polarized hepatocellular and stableclaudin-1-expressing Can 10 cells indicate that inhibi-tion of claudin-1 expression alters paracellular perme-ability. Consequently, the effect of anti-claudin-1antibody therapy on TJ functional integrity shouldbe studied in highly polarized hepatocellular cellsexpressing claudin-1, before translating such findingsto human studies.Herein, we showed that claudin-1 expression level
correlates with paracellular permeability, both in hepa-tocellular and biliary cell lines. Our results support apathophysiological model for the liver disease observedin NISCH syndrome, in which a bile leakage throughClaudin-1-deficient TJs of hepatocellular and biliarycells might result in direct hepatocellular and/or biliaryinjuries and in cholestasis. Such a pathophysiologicalmodel fits well with the sclerosing cholangitis featuresobserved in NISCH syndrome. More generally, ourresults support a primary role for paracellular perme-ability modifications induced by abnormal TJ proteinexpression in the pathophysiology of cholestatic liverdiseases, especially sclerosing cholangitis. Nevertheless,because TJ proteins may be involved in cellular mech-anisms other than regulation of paracellular permeabil-ity, such as cell proliferation and adhesion, we cannotexclude that other mechanisms might contribute to thepathogenesis of NISCH syndrome.8 We suggest thatthe cell systems we have generated represent usefultools for studying hepatic paracellular permeability.
Acknowledgments: The authors thank Dr. A. Hub-bard (Johns Hopkins University, Baltimore, MD) forfruitful discussion and editing advice, Dr V. Nicolas(IFR 141, Plateforme d’Imagerie Cellulaire, Chatenay-Malabry, France) for help with confocal analysis,Dr B. Stieger (Division of Clinical Pharmacology, Uni-versity Hospital, Zurich, Switzerland) for providinganti-BSEP antibody, S. Prigent (INSERM UMR-S757,University Paris-Sud 11, Orsay, France) for technicalsupport, and M.J. Redon and Dr. C. Guettier (Pathol-ogy Unit, CHU Bicetre, Assistance Publique–Hopitauxde Paris, University Paris-Sud 11, Paris, France) fortechnical help with liver immunostaining.
References1. Baala L, Hadj-Rabia S, Hamel-Teillac D, Hadchouel M, Prost C, Leal
SM, et al. Homozygosity mapping of a locus for a novel syndromic ich-thyosis to chromosome 3q27-q28. J Invest Dermatol 2002;119:70-76.
2. Hadj-Rabia S, Baala L, Vabres P, Hamel-Teillac D, Jacquemin E, FabreM, et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis
associated with ichthyosis: a tight junction disease. Gastroenterology2004;127:1386-1390. Erratum in: Gastroenterology 2005;128:524.
3. Feldmeyer L, Huber M, Fellmann F, Beckmann JS, Frenk E, Hohl D.Confirmation of the origin of NISCH syndrome. Hum Mutat 2006;27:408-410.
4. Nagtzaam IF, van Geel M, Driessen A, Steijlen PM, van Steensel MA.Bile duct paucity is part of the neonatal ichthyosis-sclerosing cholangitisphenotype. Br J Dermatol 2010;163:205-207.
5. Paganelli M, Stephenne X, Gilis A, Jacquemin E, Henrion Caude A,Girard M, et al. NISCH syndrome: extremely variable liver diseaseseverity from claudin-1 deficiency. J Pediatr Gastroenterol Nutr 2011;53:350-354.
6. Zimmerli SC, Kerl K, Hadj-Rabia S, Hohl D, Hauser C. Human epi-dermal Langerhans cells express the tight junction protein claudin-1and are present in human genetic claudin-1 deficiency (NISCH syn-drome). Exp Dermatol 2008;17:20-23.
7. Cereijido M, Contreras RG, Shoshani L, Flores-Benitez D, Larre I.Tight junction and polarity interaction in the transporting epithelialphenotype. Biochim Biophys Acta 2008;1778:770-793.
8. Balda MS, Matter K. Tight junctions at a glance. J Cell Sci 2008;121:3677-3682.
9. Anderson JM, Van Itallie CM. Physiology and function of the tightjunction. Cold Spring Harb Perspect Biol 2009;1:a002584.
10. Elkouby-Naor L, Ben-Yosef T. Functions of claudin tight junction pro-teins and their complex interactions in various physiological systems.Int Rev Cell Mol Biol 2010;279:1-32.
11. Kojima T, Sawada N, Yamaguchi H, Fort AG, Spray DC. Gap andtight junctions in liver: composition, regulation and function. In: AriasIM, ed., Alter HJ, Boyer JL, Cohen DE, Fausto N, Shafritz DA, Wolk-off AW, assoc. eds. The Liver: Biology and Pathobiology, 5th ed. WestSussex, UK: Wiley-Blackwell; 2010:201-220.
12. De Vos R, Desmet VJ. Morphologic changes of the junctional complexof the hepatocytes in rat liver after bile duct ligation. Br J Exp Pathol1978;59:220-227.
13. Elias E, Hruban Z, Wade JB, Boyer JL. Phalloidin-induced cholestasis:a microfilament-mediated change in junctional complex permeability.Proc Natl Acad Sci U S A 1980;77:2229-2233.
14. Rahner C, Stieger B, Landmann L. Structure-function correlation oftight junctional impairment after intrahepatic and extrahepatic cholesta-sis in rat liver. Gastroenterology 1996;110:1564-1578.
15. Robenek H, Herwig J, Themann H. The morphologic characteristics ofintercellular junctions between normal human liver cells and cells frompatients with extrahepatic cholestasis. Am J Pathol 1980;100:93-114.
16. Boyer JL. Tight junctions in normal and cholestatic liver: does the par-acellular pathway have functional significance? HEPATOLOGY 1983;3:614-617.
17. Anderson JM. Leaky junctions and cholestasis: a tight correlation. Gas-troenterology 1996;110:1662-1665.
18. Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis.N Engl J Med 1998;339:1217-1227.
19. Sakisaka S, Kawaguchi T, Taniguchi E, Hanada S, Sasatomi K, Koga H,et al. Alterations in tight junctions differ between primary biliary cirrho-sis and primary sclerosing cholangitis. HEPATOLOGY 2001;33:1460-1468.
20. Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, et al.Claudin-based tight junctions are crucial for the mammalian epidermalbarrier: a lesson from claudin-1-deficient mice. J Cell Biol 2002;156:1099-1111.
21. Inai T, Kobayashi J, Shibata Y. Claudin-1 contributes to the epithelialbarrier function in MDCK cells. Eur J Cell Biol 1999;78:849-855.
22. McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, SkareIB, et al. Inducible expression of claudin-1-myc but not occludin-VSV-G results in aberrant tight junction strand formation in MDCK cells.J Cell Sci 2000;113:3387-3398.
23. Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identifi-cation of ZO-1: a high molecular weight polypeptide associated withthe tight junction (zonula occludens) in a variety of epithelia. J CellBiol 1986;103:755-766.
1258 GROSSE ET AL. HEPATOLOGY, April 2012

24. Peng X, Grosse B, Le Tiec B, Nicolas V, Delagebeaudeuf C, Bedda T,et al. How to induce non-polarized cells of hepatic origin to expresstypical hepatocyte polarity: generation of new highly polarized cellmodels with developed and functional bile canaliculi. Cell Tissue Res2006;323:233-243.
25. Vroman B, LaRusso NF. Development and characterization of polarizedprimary cultures of rat intrahepatic bile duct epithelial cells. Lab Invest1996;74:303-313.
26. Bender V, Buschlen S, Cassio D. Expression and localization of hepato-cyte domain-specific plasma membrane proteins in hepatoma x fibro-blast hybrids and in hepatoma dedifferentiated variants. J Cell Sci1998;111:3437-3450.
27. Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E,Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children withnormal gamma-glutamyl transferase progressive familial intrahepaticcholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2and natural history. HEPATOLOGY 2010;51:1645-1655.
28. Ihrke G, Neufeld EB, Meads T, Shanks MR, Cassio D, Laurent M,et al. WIF-B cells: an in vitro model for studies of hepatocyte polarity.J Cell Biol 1993;123:1761-1775.
29. Sheth P, Delos Santos N, Seth A, LaRusso NF, Rao RK. Lipopolysac-charide disrupts tight junctions in cholangiocyte monolayers by a c-Src-,TLR4-, and LBP-dependent mechanism. Am J Physiol Gastrointest LiverPhysiol 2007;293:G308-G318.
30. Decaens C, Durand M, Grosse B, Cassio D. Which in vitro modelscould be best used to study hepatocyte polarity? Biol Cell 2008;100:387-398.
31. Cassio D, Macias RI, Grosse B, Marin JJ, Monte MJ. Expression,localization, and inducibility by bile acids of hepatobiliary transportersin the new polarized rat hepatic cell lines, Can 3-1 and Can 10. CellTissue Res 2007;330:447-460.
32. Hernandez S, Tsuchiya Y, Garcıa-Ruiz JP, Lalioti V, Nielsen S, CassioD, et al. ATP7B copper-regulated traffic and association with the tightjunctions: copper excretion into the bile. Gastroenterology 2008;134:1215-1223.
33. Nemeth Z, Szasz AM, Tatrai P, Nemeth J, Gyorffy H, Somoracz A,et al. Claudin-1, -2, -3, -4, -7, -8, and -10 protein expression in biliarytract cancers. J Histochem Cytochem 2009;57:113-121.
34. Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H,et al. Regurgitation of bile acids from leaky bile ducts causes sclerosingcholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004;127:261-274.
35. Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B,et al. Claudin-1 is a hepatitis C virus co-receptor required for alate step in entry. Nature 2007;446:801-805. Erratum in: Nature 2007;446:1.
36. Krieger SE, Zeisel MB, Davis C, Thumann C, Harris HJ, SchnoberEK, et al. Inhibition of hepatitis C virus infection by anti-claudin-1antibodies is mediated by neutralization of E2-CD81-claudin-1 associa-tions. HEPATOLOGY 2010;51:1144-1157.
37. Fofana I, Krieger SE, Grunert F, Glauben S, Xiao F, Fafi-Kremer S,et al. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virusinfection of primary human hepatocytes. Gastroenterology 2010;139:953-964.
HEPATOLOGY, Vol. 55, No. 4, 2012 GROSSE ET AL. 1259
Related Documents











