20104 Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 This journal is c the Owner Societies 2011 Cite this: Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 DSD-PBEP86: in search of the best double-hybrid DFT with spin-component scaled MP2 and dispersion correctionsw Sebastian Kozuch* a and Jan M. L. Martin* ab Received 11th August 2011, Accepted 27th September 2011 DOI: 10.1039/c1cp22592h Spin-component scaled double hybrids including dispersion correction were optimized for many exchange and correlation functionals. Even DSD-LDA performs surprisingly well. DSD-PBEP86 emerged as a very accurate and robust method, approaching the accuracy of composite ab initio methods at a fraction of their computational cost. Double hybrid (DH) DFT has shown to be a successful method for the accurate energy estimation of small and medium sized molecules. 1–6 DH-DFT in its more typical formulation mixes an exact exchange term (‘‘HF-like’’) with the DFT exchange functional as simple hybrids, but adds a perturbational correlation term (‘‘MP2-like’’) to the DFT correlation in the basis of the Kohn–Sham orbitals. 7 The first DH of this form was the B2-PLYP of Grimme, 3 with other examples being the general purpose B2GP-PLYP 2 and the long-range corrected oB97X-2. 5 Some additional flexibility can be provided by setting a different weight to the same-spin and opposite-spin MP2, in the spin-component-scaled MP2 method (SCS-MP2). 8 Same- spin MP2 reflects mostly long range correlation, while the opposite-spin is related to short range interactions. 1,8 It is possible (and advisable) to add a dispersion correction to DFT methods, as typically they do not account for long range interactions. 9 In the case of DH-DFT this is less critical, as the MP2 term does account for those interactions (at least partially); 2 nevertheless, the accuracy expected for DHs cannot be attainable for non-bonding interactions without a dispersion add-on. When the SCS distinction and a dispersion term are included, we obtain the DSD-DFT (Dispersion corrected, Spin-component scaled Double Hybrid). 1 The total exchange– correlation term is then expressed as (see eqn (1)): E XC =c X E X DFT + (1 c X )E X HF +c C E C DFT +c O E O MP2 +c S E S MP2 +s 6 E D (1) where c X is the amount of DFT exchange, c C that of DFT correlation, c O and c S of opposite and same-spin MP2, and s 6 of the D2 dispersion correction. In this form (eqn (1)) we have already presented the DSD-BLYP functional, which had a remarkable performance. 1,4,6,10 Herein we will seek for the best combination of exchange– correlation functional for the DSD-DFT method. The different functionals were selected from the ‘‘Popularity Poll of Density Functionals’’, 11 with some choices of our own added. The parametrization procedure is virtually the same as the one discussed in our previous DSD-BLYP paper. 1 (Further technical details are relegated to the electronic supporting information.) Six training sets were used to optimize the parameters of eqn (1): W4-08 (atomization energies), 12 DBH24 (reaction kinetics), 13 Pd (oxidative additions on a bare Pd atom), 14 Grubbs (olefin metathesis with a Ru catalyst), 15 Grimme’s ‘‘Mindless benchmark’’ (quasi-random main group reactions), 16 and S22 (for van der Waals forces and H-bonds). 17,18 These training sets cover thermochemistry and kinetics of main group and transition metals, plus long range interactions. Fig. 1 presents the overall error statistics over our training sets for the different functional combinations. Table 1 shows the parameters of selected combinations (detailed error statistics over all the subsets are reported in the ESIw). Perhaps the most stunning result to the present investigators is the surprisingly good performance of the DSD-SVWN5 (that is, DSD-LDA) functional, with an average error of just 1.83 kcal/mol. Even though all double hybrids are, strictly speaking, on the 5th rung of the ‘‘Jacob’s Ladder’’, 19 we can climb the underlying ‘‘sub-ladder’’ functional. For DSD-PBE (2nd sub-rung, a GGA) this is improved to 1.75 kcal/mol, while DSD-TPSS (3rd sub-rung, a meta-GGA) actually does worse at 2.09 kcal/mol. Essentially all of the improvement for DSD-PBE derives from a 25% drop in the RMSD for the ‘‘mindless benchmark’’. Returning to the DSD-LDA result, we note that replace- ment by a GGA of either the exchange part (DSD-BVWN5, DSD-PBEVWN5) or the correlation part (DSD-SLYP, DSD- SPBE, or DSD-SP86) leads to a very significant deterioration. Perhaps this is not surprising as the performance of LDA itself, such as it is, would be even worse were it not for an error compensation between underestimated exchange and excess correlation. a Department of Organic Chemistry, Weizmann Institute of Science, IL-76100 Rechovot, Israel. E-mail: [email protected] b Center for Advanced Scientific Computing and Modeling (CASCAM), Department of Chemistry, University of North Texas, Denton, TX 76203-5017, USA. E-mail: [email protected] w Electronic supplementary information (ESI) available: Complete statistical errors of the training and validation sets, detailed theoretical methods, plus guidelines to run DSD-PBEP86 with various software packages. See DOI: 10.1039/c1cp22592h PCCP Dynamic Article Links www.rsc.org/pccp COMMUNICATION Downloaded by University of North Texas on 16 November 2011 Published on 12 October 2011 on http://pubs.rsc.org | doi:10.1039/C1CP22592H View Online / Journal Homepage / Table of Contents for this issue

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

20104 Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 This journal is c the Owner Societies 2011
Cite this: Phys. Chem. Chem. Phys., 2011, 13, 20104–20107
DSD-PBEP86: in search of the best double-hybrid DFT with
spin-component scaled MP2 and dispersion correctionsw
Sebastian Kozuch*aand Jan M. L. Martin*
ab
Received 11th August 2011, Accepted 27th September 2011
DOI: 10.1039/c1cp22592h
Spin-component scaled double hybrids including dispersion
correction were optimized for many exchange and correlation
functionals. Even DSD-LDA performs surprisingly well.
DSD-PBEP86 emerged as a very accurate and robust method,
approaching the accuracy of composite ab initio methods at a
fraction of their computational cost.
Double hybrid (DH) DFT has shown to be a successful
method for the accurate energy estimation of small and
medium sized molecules.1–6 DH-DFT in its more typical
formulation mixes an exact exchange term (‘‘HF-like’’) with
the DFT exchange functional as simple hybrids, but adds a
perturbational correlation term (‘‘MP2-like’’) to the DFT
correlation in the basis of the Kohn–Sham orbitals.7 The first
DH of this form was the B2-PLYP of Grimme,3 with other
examples being the general purpose B2GP-PLYP2 and the
long-range corrected oB97X-2.5
Some additional flexibility can be provided by setting a
different weight to the same-spin and opposite-spin MP2, in
the spin-component-scaled MP2 method (SCS-MP2).8 Same-
spin MP2 reflects mostly long range correlation, while the
opposite-spin is related to short range interactions.1,8
It is possible (and advisable) to add a dispersion correction
to DFT methods, as typically they do not account for long
range interactions.9 In the case of DH-DFT this is less critical,
as the MP2 term does account for those interactions (at least
partially);2 nevertheless, the accuracy expected for DHs
cannot be attainable for non-bonding interactions without a
dispersion add-on.
When the SCS distinction and a dispersion term are
included, we obtain the DSD-DFT (Dispersion corrected,
Spin-component scaled Double Hybrid).1 The total exchange–
correlation term is then expressed as (see eqn (1)):
EXC = cXEXDFT + (1 � cX)EX
HF + cCECDFT
+ cOEOMP2 + cSES
MP2 + s6ED (1)
where cX is the amount of DFT exchange, cC that of DFT
correlation, cO and cS of opposite and same-spin MP2, and s6of the D2 dispersion correction. In this form (eqn (1)) we have
already presented the DSD-BLYP functional, which had a
remarkable performance.1,4,6,10
Herein we will seek for the best combination of exchange–
correlation functional for the DSD-DFT method. The
different functionals were selected from the ‘‘Popularity Poll of
Density Functionals’’,11 with some choices of our own added.
The parametrization procedure is virtually the same as the
one discussed in our previous DSD-BLYP paper.1 (Further
technical details are relegated to the electronic supporting
information.)
Six training sets were used to optimize the parameters of
eqn (1): W4-08 (atomization energies),12 DBH24 (reaction
kinetics),13 Pd (oxidative additions on a bare Pd atom),14
Grubbs (olefin metathesis with a Ru catalyst),15 Grimme’s
‘‘Mindless benchmark’’ (quasi-randommain group reactions),16
and S22 (for van der Waals forces and H-bonds).17,18 These
training sets cover thermochemistry and kinetics of main
group and transition metals, plus long range interactions.
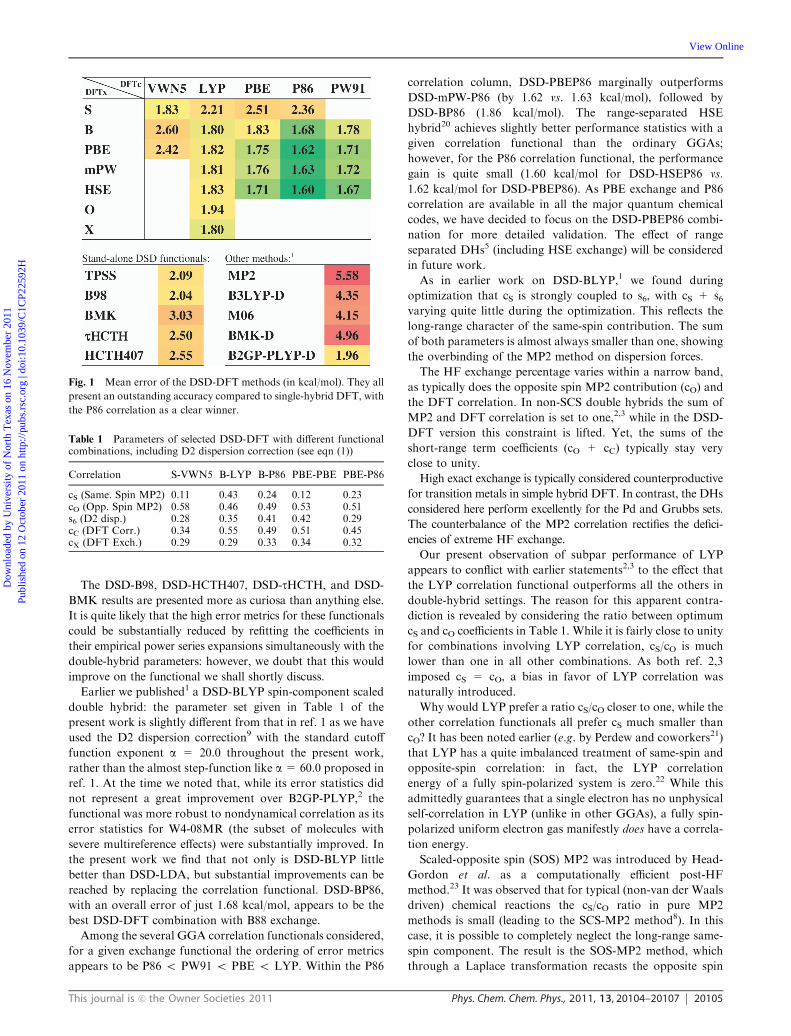
Fig. 1 presents the overall error statistics over our training
sets for the different functional combinations. Table 1 shows
the parameters of selected combinations (detailed error
statistics over all the subsets are reported in the ESIw).Perhaps the most stunning result to the present investigators
is the surprisingly good performance of the DSD-SVWN5
(that is, DSD-LDA) functional, with an average error of just
1.83 kcal/mol. Even though all double hybrids are, strictly
speaking, on the 5th rung of the ‘‘Jacob’s Ladder’’,19 we can
climb the underlying ‘‘sub-ladder’’ functional. For DSD-PBE
(2nd sub-rung, a GGA) this is improved to 1.75 kcal/mol,
while DSD-TPSS (3rd sub-rung, a meta-GGA) actually does
worse at 2.09 kcal/mol. Essentially all of the improvement for
DSD-PBE derives from a 25% drop in the RMSD for the
‘‘mindless benchmark’’.
Returning to the DSD-LDA result, we note that replace-
ment by a GGA of either the exchange part (DSD-BVWN5,
DSD-PBEVWN5) or the correlation part (DSD-SLYP, DSD-
SPBE, or DSD-SP86) leads to a very significant deterioration.
Perhaps this is not surprising as the performance of LDA
itself, such as it is, would be even worse were it not for an error
compensation between underestimated exchange and excess
correlation.
aDepartment of Organic Chemistry, Weizmann Institute of Science,IL-76100 Rechovot, Israel. E-mail: [email protected]
b Center for Advanced Scientific Computing and Modeling(CASCAM), Department of Chemistry, University of North Texas,Denton, TX 76203-5017, USA. E-mail: [email protected]
w Electronic supplementary information (ESI) available: Completestatistical errors of the training and validation sets, detailed theoreticalmethods, plus guidelines to run DSD-PBEP86 with various softwarepackages. See DOI: 10.1039/c1cp22592h
PCCP Dynamic Article Links
www.rsc.org/pccp COMMUNICATION
Dow
nloa
ded
by U
nive
rsity
of
Nor
th T
exas
on
16 N
ovem
ber
2011
Publ
ishe
d on
12
Oct
ober
201
1 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
2592
HView Online / Journal Homepage / Table of Contents for this issue
This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 20105
The DSD-B98, DSD-HCTH407, DSD-tHCTH, and DSD-
BMK results are presented more as curiosa than anything else.
It is quite likely that the high error metrics for these functionals
could be substantially reduced by refitting the coefficients in
their empirical power series expansions simultaneously with the
double-hybrid parameters: however, we doubt that this would
improve on the functional we shall shortly discuss.
Earlier we published1 a DSD-BLYP spin-component scaled
double hybrid: the parameter set given in Table 1 of the
present work is slightly different from that in ref. 1 as we have
used the D2 dispersion correction9 with the standard cutoff
function exponent a = 20.0 throughout the present work,
rather than the almost step-function like a= 60.0 proposed in
ref. 1. At the time we noted that, while its error statistics did
not represent a great improvement over B2GP-PLYP,2 the
functional was more robust to nondynamical correlation as its
error statistics for W4-08MR (the subset of molecules with
severe multireference effects) were substantially improved. In
the present work we find that not only is DSD-BLYP little
better than DSD-LDA, but substantial improvements can be
reached by replacing the correlation functional. DSD-BP86,
with an overall error of just 1.68 kcal/mol, appears to be the
best DSD-DFT combination with B88 exchange.
Among the several GGA correlation functionals considered,
for a given exchange functional the ordering of error metrics
appears to be P86 o PW91 o PBE o LYP. Within the P86
correlation column, DSD-PBEP86 marginally outperforms
DSD-mPW-P86 (by 1.62 vs. 1.63 kcal/mol), followed by
DSD-BP86 (1.86 kcal/mol). The range-separated HSE
hybrid20 achieves slightly better performance statistics with a
given correlation functional than the ordinary GGAs;
however, for the P86 correlation functional, the performance
gain is quite small (1.60 kcal/mol for DSD-HSEP86 vs.
1.62 kcal/mol for DSD-PBEP86). As PBE exchange and P86
correlation are available in all the major quantum chemical
codes, we have decided to focus on the DSD-PBEP86 combi-
nation for more detailed validation. The effect of range
separated DHs5 (including HSE exchange) will be considered
in future work.
As in earlier work on DSD-BLYP,1 we found during
optimization that cS is strongly coupled to s6, with cS + s6varying quite little during the optimization. This reflects the
long-range character of the same-spin contribution. The sum
of both parameters is almost always smaller than one, showing
the overbinding of the MP2 method on dispersion forces.
The HF exchange percentage varies within a narrow band,
as typically does the opposite spin MP2 contribution (cO) and
the DFT correlation. In non-SCS double hybrids the sum of
MP2 and DFT correlation is set to one,2,3 while in the DSD-
DFT version this constraint is lifted. Yet, the sums of the
short-range term coefficients (cO + cC) typically stay very
close to unity.
High exact exchange is typically considered counterproductive
for transition metals in simple hybrid DFT. In contrast, the DHs
considered here perform excellently for the Pd and Grubbs sets.
The counterbalance of the MP2 correlation rectifies the defici-
encies of extreme HF exchange.
Our present observation of subpar performance of LYP
appears to conflict with earlier statements2,3 to the effect that
the LYP correlation functional outperforms all the others in
double-hybrid settings. The reason for this apparent contra-
diction is revealed by considering the ratio between optimum
cS and cO coefficients in Table 1. While it is fairly close to unity
for combinations involving LYP correlation, cS/cO is much
lower than one in all other combinations. As both ref. 2,3
imposed cS = cO, a bias in favor of LYP correlation was
naturally introduced.
Why would LYP prefer a ratio cS/cO closer to one, while the
other correlation functionals all prefer cS much smaller than
cO? It has been noted earlier (e.g. by Perdew and coworkers21)
that LYP has a quite imbalanced treatment of same-spin and
opposite-spin correlation: in fact, the LYP correlation
energy of a fully spin-polarized system is zero.22 While this
admittedly guarantees that a single electron has no unphysical
self-correlation in LYP (unlike in other GGAs), a fully spin-
polarized uniform electron gas manifestly does have a correla-
tion energy.
Scaled-opposite spin (SOS) MP2 was introduced by Head-
Gordon et al. as a computationally efficient post-HF
method.23 It was observed that for typical (non-van der Waals
driven) chemical reactions the cS/cO ratio in pure MP2
methods is small (leading to the SCS-MP2 method8). In this
case, it is possible to completely neglect the long-range same-
spin component. The result is the SOS-MP2 method, which
through a Laplace transformation recasts the opposite spin
Fig. 1 Mean error of the DSD-DFT methods (in kcal/mol). They all
present an outstanding accuracy compared to single-hybrid DFT, with
the P86 correlation as a clear winner.
Table 1 Parameters of selected DSD-DFT with different functionalcombinations, including D2 dispersion correction (see eqn (1))
Correlation S-VWN5 B-LYP B-P86 PBE-PBE PBE-P86
cS (Same. Spin MP2) 0.11 0.43 0.24 0.12 0.23cO (Opp. Spin MP2) 0.58 0.46 0.49 0.53 0.51s6 (D2 disp.) 0.28 0.35 0.41 0.42 0.29cC (DFT Corr.) 0.34 0.55 0.49 0.51 0.45cX (DFT Exch.) 0.29 0.29 0.33 0.34 0.32
Dow
nloa
ded
by U
nive
rsity
of
Nor
th T
exas
on
16 N
ovem
ber
2011
Publ
ishe
d on
12
Oct
ober
201
1 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
2592
H
View Online

20106 Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 This journal is c the Owner Societies 2011
MP2 in a form with O(N4) computational cost scaling, rather
than the O(N5) of full MP2. This approach was already
applied on the B2OS3LYP and PWPB95 double-hybrids.6,24
For the sake of clarity, we will refer as ‘‘DOD-DFT’’ to
DSD-DFT parametrizations with constraint cS = 0. Not
surprisingly, we found a putative DOD-BLYP to have a rather
poor RMSD of 2.82 kcal/mol for the training set. However,
DOD-PBE seems a natural choice, as the optimal cS/cO ratio
is small to begin with. Its RMSD is only 1.82 kcal/mol,
comparable with some of the best double-hybrids (see complete
statistics and parameters in the ESIw).To improve the long-range behaviour of the selected DSD-
PBEP86, we substituted the D2 dispersion correction by the
D3BeckeJohnson version.25 The latter’s parameters were fitted
against the S130 set of weak interaction data employed by
Grimme and coworkers in the original D326 and D3BJ25
papers (it is also a subset of the GMTKN30 benchmark4).
Its 130 datapoints include the S22 set at equilibrium and two
stretched geometries (inter-monomer distances 1.5 and 2 times
equilibrium),27 n-alkane conformer energies28 (which are
particularly sensitive toward proper treatment of dispersion28),
alkane dimers from ethane to n-heptane, small peptide con-
formers, sugar conformers, cysteine conformers, and six rare
gas dimers. The latter were given a weight of 20 as in ref. 25,26.
As the published D3BJ parameters for other functionals25
have zero or insignificant a1 for double-hybrids, a1 was con-
strained to zero. In addition, as the reduction in RMSD from
allowing a nonzero s8 was found to be statistically insignificant
during the fitting, s8 was fixed at zero as well. The resulting
D3BJ parameters were s6 = 0.418 and a2 = 5.65 (a procedure4
based on asymptotic behavior of noble gas dimers yielded a
somewhat higher s6 = 0.50). As the superposition of the DFT
and the dispersion correction parts must be shaped according
to the method, we re-optimized the DH parameters, now with
D3BJ instead of D2. The final and recommended DSD-PBEP86
parameters with the D3BJ dispersion scheme are cS = 0.25,
cO = 0.53, cX = 0.30, cC = 0.43. For this functional, the mean
error for the training set is 1.51. (If D3BJ is not available, the
optimal D2 has s6 = 0.276).
Strangely, the original DSD-BLYP1 with D3BJ dispersion6
actually worsens in the training set compared to D2 (mean
error = 2.34 vs. 1.80). The reason for this deterioration lies in
the parameters for this functional being optimized for D2
dispersion, and not for D3. This shows that the dispersion
must be optimized jointly with the DFT part, taking into
account not only dispersion forces but also the short-range
interactions (as we did for the final DSD-PBEP86 parameters).
Several validation sets were studied to test the validity of the
method. Some of them were already considered in our previous
DSD-BLYP paper,1 such as the NHTBH38 (Non Hydrogen
Transfer Barrier Heights) and HTBH38 (Hydrogen Transfer
Barrier Heights),29 H-bonded dimers,30 van der Waals
complexes,31 pericyclic reactions,32,33 alkanes thermochemistry
and isomerization,34 and harmonic frequencies.1 Selected
results are presented in Table 2, while the rest is relegated to
the ESI.w However, it must be noted that all the DSD-DFT
methods have an outstanding performance compared to
lower rungs on Perdew’s ladder.19 To these validation sets,
we add here the anthracene dimerization35 and W4-11
(an expanded version of the W4-08 small molecule thermo-
chemistry benchmark).36
For the W4-11 set, restricting ourselves to the subset of 124
molecules not dominated by nondynamical correlation effects
leads to an RMSD of 1.9 kcal/mol, on par with the G4
composite ab initio method. The latter is still more resilient
for strongly multireference species, although much of the
DSD-PBEP86 RMSD there comes from a few pathological
systems such as trans-HOOO.
Improvements over the earlier DSD-BLYP and B2GP-
PLYP functionals are seen especially for the Houk pericyclic
reaction set and, to a lesser extent, for hydrogen-bonded
dimers. Unlike DSD-BLYP, DSD-PBEP86 does quite well
for alkane TAEs.
The [4 + 4] cycloadditive photodimerization of anthracene
to a D2h ‘‘sandwich’’ is a challenging test for DFT methods35
as it involves both covalent bonding between the central rings
and dispersion forces between the outer rings. (The mechanism
has been explored in detail by Bendikov and coworkers37).
At B3LYP-D/Def2-TZVP reference geometries, we obtain
11.15 kcal/mol at the CCSD(T)/cc-pVTZ(no f on H) level:
lower level results with this basis set are: MP2 20.59, SCS-MP2
13.13, and CCSD 9.85 kcal/mol. At the MP2/cc-pVQZ level,
we obtained 20.21 kcal/mol (SCS-MP2 12.55 kcal/mol): using
these latter two results (which stretched our hardware to the
limit) for estimated basis set expansion effects beyond
cc-pVTZ(no d on H), we obtain 10.77 and 10.57 kcal/mol,
respectively, leading us to propose a best estimate of
10.7� 1 kcal/mol where the error bar is somewhat conservative.
The earlier best estimate35 of 9 � 3 kcal/mol was obtained from
MP2/TZV(2d,2p) geometries at a combination of QCISD(T)/
cc-pVDZ and MP2/cc-pVQZ levels.
As noted earlier,35,38 most conventional DFT methods get
the sign wrong even with dispersion corrections. We do note
that M06-2X38,39 yields a semiquantitative answer of about
6 kcal/mol. At the B2GP-PLYP/TZVPP level, we obtain
–0.90 kcal/mol raw and +7.39 kcal/mol with a D2 dispersion
correction. DSD-BLYP yields 3.04 (raw) and 10.50 (D2 or
D3BJ) kcal/mol.
DSD-PBEP86 yields 6.32 (raw) 12.04 (D2), and 12.07
(D3BJ) kcal/mol. The dispersion-corrected results are in rather
pleasing agreement with the best estimate, but even the
raw answer is still semiquantitative. This is not the case for
Table 2 Selected RMSD errors for validation sets1 (in kcal/mol)
DSD-BLYPa
DSD-PBEP86b
B2GP-PLYPa,c B3LYPa,c M06c
NHTBH38 2.08 2.18 1.61 5.95 2.83HTBH38 1.14 1.12 1.33 5.78 2.41H-Bonds 0.45 0.38 0.47 0.95 0.42VdW 0.43 0.47 0.40 0.82 0.49Pericyclicd 3.15 2.05 2.65 2.56 2.08W4-11 non MR 2.2 1.9 1.9 4.2 3.9W4-11 all 2.6 2.5 3.0 5.0 4.4Alkanes:TAEs 3.48 1.07 1.00 0.61 3.67Isomeriz. 0.40 0.16 0.19 0.43 0.25
a With D2 dispersion correction; b With D3BJ dispersion correction.c As appeared in ref. 1; d Using W1 data as reference.
Dow
nloa
ded
by U
nive
rsity
of
Nor
th T
exas
on
16 N
ovem
ber
2011
Publ
ishe
d on
12
Oct
ober
201
1 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
2592
H
View Online

This journal is c the Owner Societies 2011 Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 20107
B2GP-PLYP (-0.90 raw, 7.39 with D2) or even DSD-BLYP
(3.04 raw, 10.50 with D2). SCS-CCSD40 overshoots
(15.69 kcal/mol) and SCS(MI)-MP241 severely undershoots
(4.69 kcal/mol) despite being ostensibly optimized for inter-
molecular interactions.
Performance of double hybrids for harmonic frequencies
was considered in ref. 1 for the HFREQ27 set of harmonic
frequencies of 27 small molecules. The optimum double hybrid
for frequencies was found to be 43% exchange and 18% MP2
(not useful parameters for thermochemistry nor, especially,
kinetics), with an RMS deviation of 18.6 cm�1. B2GP-PLYP
and DSD-BLYP yields much worse RMSD of 29.8 and
30.9 cm�1, respectively, compared to 33.9 for B3LYP and
44.0 cm�1 for MP2. Using scaling factors (given in parentheses),
this can be reduced to 17.7 (0.989) for B2GP-PLYP and
22.4 (0.990) for DSD-BLYP.
For DSD-PBEP86, even the unscaled frequencies have an
RMSD = 19.7 cm�1, which drops to 17.7 cm�1 when F2 is
removed as an outlier. A fitted scaling factor of 0.995 yields a
just marginally better RMSD = 16.6 cm�1. We wish to stress
that no parametrization whatsoever was made for this property:
therefore, this strongly suggests that DSD-PBEP86 is a more
‘universal’ functional than either B2GP-PLYP or DSD-BLYP.
For the sake of completeness, DSD-BP86 and DSD-mPWP86
yield unscaled RMSDs of 19.2 and 18.8 cm�1, respectively.
Conclusions
Following up on our earlier DSD-BLYP functional,1 we have
optimized spin-component scaled, dispersion-corrected9,25,26
double hybrids using many different exchange and correlation
functional forms. The resulting functionals have been validated
against a variety of thermodynamic and kinetic benchmarks.
Somewhat surprisingly, the quality of the results is only mildly
sensitive to the choice of the underlying DFT exchange and
correlation components, and even DSD-LDA yields respectable
performance. Simple, nonempirical GGAs appear to work best:
meta-GGAs offer no advantage. P86 correlation is a clear
winner, followed by PW91C and PBEC. The choice of the
exchange DFT part is less critical, and we selected the PBE
exchange for its accuracy and wide availability. Our earlier
assertion that LYP correlation is essential for double hybrids
is found to be an artifact of constraining the same-spin and
opposite-spin coefficients to be equal (although other non-LYP
based double hybrids were found to be efficient5,6). Our best
functional, DSD-PBEP86 (with D3BJ dispersion25), emerges as a
very accurate and robust quantum mechanical method, also
performing well for properties not included in its parametriza-
tion (such as harmonic frequencies). It approaches the accuracy
of ab initio composite methods at a fraction of their computa-
tional cost. Lastly, it can be run using unmodified versions of
several common quantum chemistry programs (see ESIw).
Notes and references
1 S. Kozuch, D. Gruzman and J. M. L. Martin, J. Phys. Chem. C,2010, 114, 20801–20808.
2 A. Karton, A. Tarnopolsky, J.-F. Lamere, G. C. Schatz andJ. M. L. Martin, J. Phys. Chem. A, 2008, 112, 12868–12886.
3 S. Grimme, J. Chem. Phys., 2006, 124, 034108.
4 L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13,6670–6688.
5 J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2009,131, 174105.
6 L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7,291–309.
7 A. Gorling and M. Levy, Phys. Rev. B: Condens. Matter, 1993,47, 13105.
8 S. Grimme, J. Chem. Phys., 2003, 118, 9095.9 S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473.10 T.-Y. Lai, C.-Y. Yang, H.-J. Lin, C.-Y. Yang and W.-P. Hu,
J. Chem. Phys., 2011, 134, 244110.11 M. Swart, F. M. Bickelhaupt and M. Duran, Popularity Poll
Density Functionals, 2010; http://www.marcelswart.eu/dft-poll/newsitem.pdf.
12 A. Karton, A. Tarnopolsky, J.-F. Lamere, G. C. Schatz andJ. M. L. Martin, J. Phys. Chem. A, 2008, 112, 12868–12886.
13 J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput.,2009, 5, 808–821.
14 M. M. Quintal, A. Karton, M. A. Iron, A. D. Boese and J. M. L.Martin, J. Phys. Chem. A, 2006, 110, 709–716.
15 Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5,324–333.
16 M. Korth and S. Grimme, J. Chem. Theory Comput., 2009, 5,993–1003.
17 P. Jurecka, J. Sponer, J. Cerny and P. Hobza, Phys. Chem. Chem.Phys., 2006, 8, 1985.
18 T. Takatani, E. G. Hohenstein, M. Malagoli, M. S. Marshall andC. D. Sherrill, J. Chem. Phys., 2010, 132, 144104.
19 J. P. Perdew and K. Schmidt, in AIP Conference Proceedings, 2001,vol. 577, pp. 1–20.
20 J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003,118, 8207.
21 J. P. Perdew and K. Burke, Int. J. Quantum Chem., 1996, 57,309–319.
22 For UEG follows trivially from eqn (2) in B. Miehlich, A. Savin,H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206;C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789.
23 Y. Jung, R. C. Lochan, A. D. Dutoi and M. Head-Gordon,J. Chem. Phys., 2004, 121, 9793.
24 T. Benighaus, R. A. DiStasio, R. C. Lochan, J.-D. Chai andM. Head-Gordon, J. Phys. Chem. A, 2008, 112, 2702–2712.
25 S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32,1456–1465.
26 S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys.,2010, 132, 154104.
27 L. Grafova, M. Pitonak, J. Rezac and P. Hobza, J. Chem. TheoryComput., 2010, 6, 2365–2376.
28 D. Gruzman, A. Karton and J. M. L. Martin, J. Phys. Chem. A,2009, 113, 11974–11983.
29 Y. Zhao, N. Gonzalez-Garcıa and D. G. Truhlar, J. Phys. Chem.A, 2005, 109, 2012–2018.
30 A. D. Boese, J. M. L. Martin and W. Klopper, J. Phys. Chem. A,2007, 111, 11122–11133.
31 Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2005, 1,415–432.
32 D. H. Ess and K. N. Houk, J. Phys. Chem. A, 2005, 109,9542–9553.
33 V. Guner, K. S. Khuong, A. G. Leach, P. S. Lee, M. D. Bartbergerand K. N. Houk, J. Phys. Chem. A, 2003, 107, 11445–11459.
34 A. Karton, D. Gruzman and J. M. L. Martin, J. Phys. Chem. A,2009, 113, 8434–8447.
35 S. Grimme, C. Diedrich and M. Korth, Angew. Chem., Int. Ed.,2006, 45, 625–629.
36 A. Karton, S. Daon and J. M. L. Martin, Chem. Phys. Lett., 2011,510, 165–178.
37 S. S. Zade, N. Zamoshchik, A. R. Reddy, G. Fridman-Marueli,D. Sheberla and M. Bendikov, J. Am. Chem. Soc., 2011, 133,10803–10816.
38 Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167.39 Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241.40 T. Takatani, E. G. Hohenstein and C. D. Sherrill, J. Chem. Phys.,
2008, 128, 124111.41 R. A. Distasio and M. Head-Gordon, Mol. Phys., 2007, 105,
1073–1083.
Dow
nloa
ded
by U
nive
rsity
of
Nor
th T
exas
on
16 N
ovem
ber
2011
Publ
ishe
d on
12
Oct
ober
201
1 on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
2592
H
View Online
Related Documents











