Circadian rhythm disruption in a mouse model of Rett syndrome circadian disruption in RTT ☆ Quan Li a,b,c,1 , Dawn H. Loh a,1 , Takashi Kudo a , Danny Truong a , Matthew Derakhshesh a , Zoë MacDowell Kaswan a , Cristina A. Ghiani a , Rosemarie Tsoa b , Yin Cheng b , Yi E. Sun b, ⁎, Christopher S. Colwell a, ⁎ a Laboratory of Circadian and Sleep Medicine, Department of Psychiatry, University of California, Los Angeles, 760 Westwood Plaza, Los Angeles 90095-1759, USA b Department of Psychiatry, University of California, Los Angeles, Box 957332, 635 S. Charles E. Young Dr, Los Angeles, CA 90095-7332, USA c Molecular, Cellular and Integrative Physiology Program, University of California, Los Angeles, Los Angeles, CA 90095, USA abstract article info Article history: Received 18 October 2014 Revised 8 March 2015 Accepted 9 March 2015 Available online 14 March 2015 Keywords: Circadian rhythms MeCP2 Rett syndrome (RTT) Sleep Suprachiasmatic nucleus (SCN) VIP Disturbances in the sleep/wake cycle are prevalent in patients with Rett syndrome (RTT). We sought to determine whether the circadian system is disrupted in a RTT model, Mecp2 -/y mice. We found that MeCP2 mutants showed decreased strength and precision of daily rhythms of activity coupled with extremely fragmented sleep. The central circadian clock (suprachiasmatic nucleus) exhibited significant reduction in the number of neurons expressing vasoactive intestinal peptide (VIP) as well as compromised spontaneous neural activity. The molecular clockwork was disrupted both centrally in the SCN and in peripheral organs, indicating a general disorganization of the circadian system. Disruption of the molecular clockwork was observed in fibroblasts of RTT patients. Finally, MeCP2 mutant mice were vulnerable to circadian disruption as chronic jet lag accelerated mortality. Our finds suggest an integral role of MeCP2 in the circadian timing system and provides a possible mechanistic explanation for the sleep/wake distrubances observed in RTT patients. The work raises the possibility that RTT patients may benefit from a temporally structured environment. © 2015 Elsevier Inc. All rights reserved. Rett syndrome (RTT) is an X-linked progressive genetic disorder characterized by regression of learned hand and language abilities, coupled with gait abnormalities, stereotypic hand movements and microcephaly (Hagberg et al., 1983; Neul et al., 2010). Sleep disruption is common in RTT patients (Glaze et al., 1987; Nomura, 2005; Young et al., 2007) with nighttime autonomic dysfunction also being reported later in the disease progression (Rohdin et al., 2007; Weese-Mayer et al., 2008). At least some of these sleep symptoms are disruptions in timing with RTT patients reporting difficulty sleeping at night and staying awake during the day, raising the possibility of an underlying circadian deficit. The majority of RTT cases are caused by mutations in the gene encoding methyl-CpG-binding protein 2, Mecp2 (Amir et al., 1999), which encodes a transcriptional regulator that binds to methylated CpG sites (Nan et al., 1997) and both activates and represses transcrip- tion (Chahrour et al., 2008). The Mecp2 gene is highly expressed in the master pacemaker of the circadian timing system (suprachiasmatic nucleus, SCN; Dragich et al., 2007) and is phosphorylated upon photic stimulation (Zhou et al., 2006). Importantly, MeCP2 binds and transcriptionally activates the circadian clock genes, Per1 and Per2, indicating a direct link between this chromatin regulator and core molecular clockwork (Alvarez-Saavedra et al., 2011). Therefore, we became interested in exploring possible circadian dysfunction in a mouse model of RTT as a strategy to understanding the sleep deficits in RTT patients. Due to the depth of background information, we utilized a mouse model of RTT with the loss of the methyl-CpG-binding domain (MBD) of the MeCP2 protein through the deletion of exon 3 of the Mecp2 gene (Chen et al., 2001). Methods Animals All experimental protocols used in this study were approved by the UCLA Animal Research Committee. UCLA Division of Laboratory animal recommendations for animal use and welfare, as well as National Institutes of Health guidelines were followed. Experiments for behavior and physiology assays used 7–8 week-old Mecp2 1lox mutant mice with a deletion of exon 3 of the Mecp2 gene (strain B6.Cg-Mecp2 tm1.1Jae /Mmucd; Chen et al., 2001) on a C57BL/6J back- ground (N7 generation speed congenic) henceforth referred to as Neurobiology of Disease 77 (2015) 155–164 ☆ The authors declare no competing financial interests. ⁎ Corresponding authors. E-mail addresses: [email protected] (Y.E. Sun), [email protected] (C.S. Colwell). 1 Both authors contributed equally to the work. Available online on ScienceDirect (www.sciencedirect.com). http://dx.doi.org/10.1016/j.nbd.2015.03.009 0969-9961/© 2015 Elsevier Inc. All rights reserved. Contents lists available at ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Neurobiology of Disease 77 (2015) 155–164
Contents lists available at ScienceDirect
Neurobiology of Disease
j ourna l homepage: www.e lsev ie r .com/ locate /ynbd i
Circadian rhythm disruption in a mouse model of Rett syndromecircadian disruption in RTT☆
Quan Li a,b,c,1, Dawn H. Loh a,1, Takashi Kudo a, Danny Truong a, Matthew Derakhshesh a,Zoë MacDowell Kaswan a, Cristina A. Ghiani a, Rosemarie Tsoa b, Yin Cheng b,Yi E. Sun b,⁎, Christopher S. Colwell a,⁎a Laboratory of Circadian and Sleep Medicine, Department of Psychiatry, University of California, Los Angeles, 760 Westwood Plaza, Los Angeles 90095-1759, USAb Department of Psychiatry, University of California, Los Angeles, Box 957332, 635 S. Charles E. Young Dr, Los Angeles, CA 90095-7332, USAc Molecular, Cellular and Integrative Physiology Program, University of California, Los Angeles, Los Angeles, CA 90095, USA
☆ The authors declare no competing financial interests.⁎ Corresponding authors.
E-mail addresses: [email protected] (Y.E. Sun), c(C.S. Colwell).
1 Both authors contributed equally to the work.Available online on ScienceDirect (www.sciencedir
http://dx.doi.org/10.1016/j.nbd.2015.03.0090969-9961/© 2015 Elsevier Inc. All rights reserved.
a b s t r a c t
a r t i c l e i n f oArticle history:Received 18 October 2014Revised 8 March 2015Accepted 9 March 2015Available online 14 March 2015
Keywords:Circadian rhythmsMeCP2Rett syndrome (RTT)SleepSuprachiasmatic nucleus (SCN)VIP
Disturbances in the sleep/wake cycle are prevalent in patients with Rett syndrome (RTT). We sought todetermine whether the circadian system is disrupted in a RTT model, Mecp2−/y mice. We found that MeCP2mutants showed decreased strength and precision of daily rhythms of activity coupled with extremelyfragmented sleep. The central circadian clock (suprachiasmatic nucleus) exhibited significant reduction in thenumber of neurons expressing vasoactive intestinal peptide (VIP) as well as compromised spontaneous neuralactivity. The molecular clockwork was disrupted both centrally in the SCN and in peripheral organs, indicatinga general disorganization of the circadian system. Disruption of the molecular clockwork was observed infibroblasts of RTT patients. Finally, MeCP2 mutant mice were vulnerable to circadian disruption as chronic jetlag acceleratedmortality. Our finds suggest an integral role ofMeCP2 in the circadian timing system and providesa possiblemechanistic explanation for the sleep/wake distrubances observed in RTT patients. Thework raises thepossibility that RTT patients may benefit from a temporally structured environment.
© 2015 Elsevier Inc. All rights reserved.
Rett syndrome (RTT) is an X-linked progressive genetic disordercharacterized by regression of learned hand and language abilities,coupled with gait abnormalities, stereotypic hand movements andmicrocephaly (Hagberg et al., 1983; Neul et al., 2010). Sleep disruptionis common in RTT patients (Glaze et al., 1987; Nomura, 2005; Younget al., 2007) with nighttime autonomic dysfunction also being reportedlater in the disease progression (Rohdin et al., 2007;Weese-Mayer et al.,2008). At least some of these sleep symptoms are disruptions in timingwith RTT patients reporting difficulty sleeping at night and stayingawake during the day, raising the possibility of an underlying circadiandeficit.
The majority of RTT cases are caused by mutations in the geneencoding methyl-CpG-binding protein 2, Mecp2 (Amir et al., 1999),which encodes a transcriptional regulator that binds to methylatedCpG sites (Nan et al., 1997) and both activates and represses transcrip-tion (Chahrour et al., 2008). The Mecp2 gene is highly expressed in themaster pacemaker of the circadian timing system (suprachiasmatic
ect.com).
nucleus, SCN; Dragich et al., 2007) and is phosphorylated upon photicstimulation (Zhou et al., 2006). Importantly, MeCP2 binds andtranscriptionally activates the circadian clock genes, Per1 and Per2,indicating a direct link between this chromatin regulator and coremolecular clockwork (Alvarez-Saavedra et al., 2011). Therefore, webecame interested in exploring possible circadian dysfunction in amouse model of RTT as a strategy to understanding the sleep deficitsin RTTpatients. Due to thedepth of background information,we utilizeda mouse model of RTT with the loss of the methyl-CpG-binding domain(MBD) of the MeCP2 protein through the deletion of exon 3 of theMecp2 gene (Chen et al., 2001).
Methods
Animals
All experimental protocols used in this study were approvedby the UCLA Animal Research Committee. UCLA Division of Laboratoryanimal recommendations for animal use and welfare, as well asNational Institutes of Health guidelines were followed. Experimentsfor behavior and physiology assays used 7–8 week-old Mecp21lox
mutant mice with a deletion of exon 3 of the Mecp2 gene (strainB6.Cg-Mecp2tm1.1Jae/Mmucd; Chen et al., 2001) on a C57BL/6J back-ground (N7 generation speed congenic) henceforth referred to as

156 Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164
Mecp2−/y mutants or KO. To determine the impact of the Mecp2−/y
mutation on circadian gene expression, we crossed Mecp2−/+ femalemutants to male mice homozygous for the luciferase reporter insertedas an in-frame fusion to the endogenous Period2 gene (mPer2Luc/Luc,backcrossed to C57BL/6 for a minimum of 12 generations; Yoo et al.,2004). Hemizygous Mecp2−/y;mPer2Luc/+ mice were compared toMecp2+/+;mPer2Luc/+ littermate males (WT) for the explanted tissuebioluminescence assays.
Immobility-defined sleep assay
Methods used were similar to those described previously (Loh et al.,2013).MaleMecp2−/ymutantmice andWT littermates (7–8weeks old)were housed in 12:12 LD conditions. A side-on view of each cage wasobtained and video was captured using surveillance cameras with visi-ble light filters (Gadspot Inc., City of Industry, CA) connected to thevideo-capture card (Adlink Technology Inc., Irvine, CA). The AnyMazesoftware (Stoelting Co., Wood Dale, IL) was used to track the animals.Immobility detectionwas set to 95% of the area of the animal immobile,as previously determined by correlation with simultaneous EEG/EMGrecordings (Pack et al., 2007; Fisher et al., 2012). We recorded over 3consecutive days, and used an average of days 2 and 3 for furtheranalysis. Immobility-defined sleep was determined in 1 min bins andtotal sleep was determined by summing immobility durations. A sleepbout was defined as a duration of continuous immobility (maximumgap: 1 min; threshold: 40 counts/min), and the number of bouts andthe average duration of bouts were determined for both day and nightusing ClockLab software (Actimetrics, Wilmette, IL). Sleep latency wasdetermined by visually examining individual records of sleep behaviorand measuring the time relative to lights-on at which each mouseachieved sleep onset (half-max of daily sleep).
Wheel running activity assay
Methods used were similar to those described previously (Kudoet al., 2011; Loh et al., 2013). Male Mecp2−/y mutant mice and WTlittermates were housed in cages containing running wheels(Mini Mitter Co., Bend, OR) from 6 to 7 weeks of age and their wheel-running activity recorded as revolutions (rev) per 3 min intervals.Animals were exposed to a 12:12 h light–dark cycle (LD; light intensity300 lx) for 10 days, and then released to 24 h of constant darkness (DD)to assess their free-running activity pattern for 10 days. Locomotoractivity rhythms were analyzed by periodogram analysis combinedwith the χ2 test with P = 0.05 significance levels (El Temps, Barcelona,Spain) on the raw data. The periodogram refers to behavioral rhythmamplitude as “power” (%V) or periodicities in the time series correctedfor amount of activity and normalized to the percentage of variancederived from peak significance (P = 0.05). Fragmentation wascalculated using the number of activity bouts (maximum gap: 21 min;threshold 3 counts/min) per day using ClockLab software. Precisionwas calculated by determining the difference between the daily onsetof activity and lights off under LD conditions, and the differencebetween the daily onset of activity from the best-fit line through thedaily activity onsets under DD conditions. All handling of mice in DDwas carried out with the aid of night vision goggles (FJW Industries,Palatine, IL). Negative masking experiments were conducted using 1 hof 50 lx full spectrum white light at zeitgeber time (ZT) 14 (2 h afterlights off) and activity levels (rev/h) during the light pulse werecompared to the previous day's activity to determine the effect of photicsuppression of activity.
Histology and immunohistochemical (IHC) of the SCN
Control and mutant mice were anesthetized with isoflurane andtranscardially perfused with saline solution (NaCl 0.15 M) followed byphosphates buffered (PB, 0.1M, pH 7.4) containing 4% paraformaldehyde
(PFA) in 0.15 M phosphate buffer (PB) pH 7.4 at 4 °C. The brains weredissected out, post-fixed in 4% PFA at room temperature for 2 h andthen cryopreserved in 30% sucrose in PB. Coronal sections (20–30 μm)were cut on a cryostat (Leica, Buffalo Grove, IL), collected sequentiallyinto 24-well dishes and then processed for IHC or Nissl staining usingcresyl violet.
Nissl staining was performed as previously reported (Colwell et al.,1996). Stained sections (20 μm) were used to estimate the volume,the height and width of the SCN. Images were acquired on a ZeissAxioskop with an Axiocam using the AxioVision software (Zeiss, Pleas-anton, CA, USA). Measurements (in μm) were performed using theAxioVision software. Because the borders of the Nissl-defined SCNare somewhat arbitrary, measurements were taken by two observersblind to the animal genotype. For each animal, the three measurementsweremade in consecutive slices of the central SCN.Measurements fromthe 2most central sections, the 2 sections anterior and 2 posterior werethen averaged. Data are shown as the average ± SEM of 6 consecutivesections from 8 MeCP2 KO and 7 littermate wild-type (C57) mice.
IHC was performed as previously reported (Loh et al., 2013) withminor modifications. Briefly, brain sections (30 μm) were incubated inblocking serum overnight at 4 °C (3% bovine serum albumin, 0.1% tritonX-100 and 0.025% sodium azide in PB), and then incubated withprimary antibodies against VIP (rabbit polyclonal 1:2000; ImmunoStar;Hudson, WI, USA) for 24 h at 4 °C with gentle shaking. Three washes(15 min. each) were carried out in blocking solution, then thecorresponding biotinylated secondary antibodies were incubated for90 min at room temperature (1:500; Vector Laboratories, Burlingame,CA, USA). Slices were then incubated with the avidin-peroxidase kitfollowing the manufacturer's protocol (ABC kit; Vector). Peroxidaseactivity was revealed by using 3,3′-diaminobenzidine as a chromogenand intensified with nickel chloride according to the protocol providedby the supplier (Vector). The primary antibody was omitted in controlsections. Slices were mounted on gelatin-coated slides and coverslipped with Depex mounting medium (Fisher Scientific). Specificimmunostaining was detected as a brown precipitate.
All stereological analysis was performed by a single investigatorusing an AxioImager M2 ApoTome microscope (Zeiss, Pleasanton,CA, USA) equipped with a motorized stage controlled by theStereoInvestigator software (MicroBrightField Biosciences, Williston,VT, USA). The SCN was identified by location and cell density, and thearea of interest was defined as the ventral SCN. Due to the low numberof VIP+ neurons and the small area of interest, the stereologicalparameters were designed to cover the entire area of interest and allVIP+ neurons were counted directly. The area of interest was outlinedat 10× magnification and cells were counted at 40× magnificationunder Köhler illumination.
Patch-clamp electrophysiology of SCN neurons
Methods used were similar to those described previously (Kudoet al., 2011, 2013). Male Mecp2−/y mutant mice and WT littermates(6–7 weeks old) entrained on a 12:12 LD or DL cycle were anesthetizedin an isoflurane chamber at ZT 2.5 and ZT 12.5 for ZT 4–6 and ZT 14–16recordings respectively. Anesthetized mice were decapitated, brainswere extracted and incubated in ice-cold slice solution for 5 min.Coronal slices (300 μm) of mid-SCN were collected in slice solution(in mM: 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 125 NaCl, 3 KCl, 5MgCl2, 1 CaCl2) using a vibratome, and then incubated for 30 min at30 °C followed by 1 h at room temperature in artificial cerebrospinalfluid (ACSF, in mM; 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 125 NaCl, 3KCl, 2 MgCl2, 2 CaCl2) while continually being aerated with 95% O2/5%CO2. Slices were placed in a recording chamber (PH-1, WarnerInstruments, Hamden, CT) attached to the stage of a fixed stage uprightDICmicroscope (OLYMPUS, Tokyo, Japan), and superfused continuously(2 ml/min) with room temperature ACSF aerated with 95% O2/5% CO2.Whole cell patch clamp SCN recordings were made using electrode

157Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164
micropipettes (7–9 MΩ) pulled from glass capillaries and recordingelectrodes were filled with standard internal solution (in mM):112.5 K-gluconate, 1 EGTA; 10 Hepes; 5 MgATP; 1 GTP; 0.1 leupeptin;10 phosphocreatine; 4 NaCl; 17.5 KCl; 0.5 CaCl2; and 1 MgCl2. Internalsolution pH was adjusted to 7.25–7.3 and osmolarity adjusted to 290–300 mOsm. Recordings were obtained with the AXOPATCH 200Bamplifier (Molecular Devices, Sunnyvale, CA) and monitored on-linewith pCLAMP (Ver 10, Molecular Devices). SFR were recordedwith pCLAMP for 1 min using current-clamp in the whole cell patchconfiguration.
Bioluminescence assay of PER2-driven luciferase activity in ex vivo explants
Methods used for real-time monitoring of bioluminescence ofPER2::LUC ex vivo tissue explants were similar to those describedpreviously (Nakamura et al., 2011; Loh et al., 2011). Male Mecp2−/y
mutant mice and WT littermates (6–7 weeks old) were sacrificedbetween ZT 10 and ZT 11 from mice housed in a 12:12 LD cycle, andthe SCN, heart, lung, liver, and adrenal explantswere collected. Explantswere transferred onto Millicell membranes (0.4 μm, PICMORG50,Millipore, Bedford, MA) resting on 1.2 ml of recording media thatcontained freshly added 0.1 mM luciferin (sodium salt monohydrate,Biosynth, Staad, Switzerland), and the 35 mm dishes were sealedusing autoclaved vacuum grease (Dow Corning, Midland, MI). Tissueexplants were immediately inserted in the Lumicycle photometer(Actimetrics, Wilmette, IL), and bioluminescence was monitored at37 °C for 6–7 consecutive uninterrupted days. Raw bioluminescencevalues were normalized by subtracting a running average of 24 h ofbioluminescence, and next performing a 2-hour smoothing average.Period, amplitude, rate of damping, fit to regression line (R2), and firstcalculated peak for phase relationships were calculated as previouslydescribed (Loh et al., 2011).
Cell culture
Primary MEFs were prepared from E13.5 embryos following a stan-dard protocol described by Takahashi and Yamanaka (2006).Mecp2−/y
MEFs and WT Mecp2+/y MEFs were dissected and collected from indi-vidual embryo isolated from E13.5-day-pregnant female Mecp2−/+
(heterozygous) mice on the C57BL/6 background. Following dissection,genotyping was performed to confirmMEF genotype and onlyMecp2−/
y and Mecp2+/y MEFs from the same littermate embryos were used forsubsequent qRT-PCR experiments. Homogenous population werecultured and expanded with Dulbecco's modified eagle medium(DMEM, Life Technologies), supplemented with glucose (4.5 g/l),10% Fetal Bovine Serum (FBS), 2 mM L-glutamine, and 1× penicillin/streptomycin. Human Foreskin Fibroblasts (HFFs) from RTT individualsand control human subject were obtained from the American TypeCulture Collection or Coriell Cell Repository and maintained accordingto the recommended protocol. Three types of HFFs were used for qRT-PCR: GM17880 (RTT T158M), GM11273 (RTT R106W) and a normalsubject line (WT HFF). HFFs were cultured and expanded with 10%FBS MEFs medium. Prior to measuring circadian rhythms in geneexpression, cell cultures were synchronized using 0.1 μM dexametha-sone (D2915, Sigma-Aldrich). Cells were incubated with 0.1 μM dexa-methasone for 2 h at 37 °C after cells reached confluence for 3 days.After 2 h, medium was changed to 10% normal MEFs medium attime = 0. Then at each indicated time, cells were washed with coldPBS and collected for qRT-PCR.
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
Total RNA was extracted from MEFs or HFFs using the Trizol(Invitrogen, Carlsbad, CA) protocol provided by the manufacturer.cDNA was obtained by reverse transcription (RT) with Superscript IIISuperMix Kit (Invitrogen) following manufacturer's protocol and non-
RT controls were performed simultaneously to monitor genomic DNAcontamination. Quantitative real time PCRs were performed withSYBR Green (Fast SYBR Green Master Mix, Life Technologies) usingthe StepOnePlus system (Applied Biosystems, Life Technology, FosterCity, CA). Oligonucleotide primer sequences were designed to crossintron–exon boundaries and validated for specificity and efficiency.Forward and reverse primers with corresponding transcripts wereused: mPer2 (100 bp): Forward: 5′-GGGCATTACCTCCGAGTATA-3′Reverse: 5′-GGCCACTTGGTTAGAGATGTA-3′; mGapdh (200 bp):Forward: 5′-AGAGAGGGAGGAGGGGAAATG; Reverse: 5′-AACAGGGAGGAGCAGAGAGCAC; hPER2 (125 bp) Forward: 5′-TGGATGAAAGGGCGGTCCCT-3′; Reverse: 5′-ACTGCAGGATCTTTTTGTGGA-3′; hGAPDH(200 bp) Forward: 5′-CCGCATCTTCTTTTGCGT-3′; Reverse: 5′-TAAAAGCAGCCCTGGTGACC-3′. Reactions were performed with the equivalentof 20 ng of starting total RNA in a 20-μl reaction volume comprising ofthe 1× SYBR Green Master Mix and 0.5 μM each of the forward andreverse primers. Melting curves were calculated at the end of eachreaction to check PCR product specificity. The relative levels of clockgene transcripts were determined using the 2−ΔΔCt method withGapdh/GAPDH as the normalizing reference gene. Studies confirmedthat the levels of the housekeeping gene transcript Gapdh did notfluctuate within all indicated time points.
Chromatin immunoprecipitation (ChIP)
MEFs plated on 10 cm2 plate at each time pointwere cross-linked byadding 270 μl of 37% formaldehyde per 10ml at a final concentration of1% (vol/vol). Cells were cross-linked for 10min at room temperature ona slowly rotating stage. MEFs were collected by centrifugation at 1500 gfor 10 min at 4 °C and pellets were obtained and washed with cold PBStwice. Nuclei of cross-linked pellets were isolated by stepwise hypoton-ic process with protease inhibitor cocktail (Roche). Nuclei pelletswere collected by centrifugation and suspended in 0.3 ml lysis buffer.The re-suspended nuclei were sonicated using Bioruptor (Diagenode,Denville, NJ) for 30 cycles (30 s ON/30 s OFF) at high power, resultingin DNA fragments of 400–600 base pairs (bp) in length. 100 μg ofsheared chromatin were then added to antibody pre-coupledDynabeads (Dynabeads® M-280 Sheep Anti-Mouse IgG or DynabeadsM-280 Sheep anti-Rabbit IgG, Invitrogen) for overnight immunoprecip-itation. After reverse cross-linking, the eluted samples were purifiedwith DNA Clean & Concentrator™-5 kit (Zymo, Irvine, CA). PurifiedDNA fragmentswere subjected to real timeqPCRs to analyze the proteinto DNA interaction within Per2 promoter regions. Results were oftenpresented as “percent input” values which were calculated by quantify-ing the abundance of DNA fragments of interest added to the ChIPreaction with respect to the abundance of total input DNA fragmentsfound in the final immunoprecipitate. Primers sequences were alreadyvalidated and listed as below: mPer2 (CpG binding site, 178 bp):Forward: 5′-ACGTCGTCGCAGGTGATAG-3′ Reverse: 5′-CGAGTAGGCTCGTCCACTTC-3′ mPer2 (Cre1 binding site, 183 bp): Forward: 5′-CCAGGTGGATGAGCTGTGTA-3′ Reverse: 5′-AGCACCTCTGGTTCCTCTGA-3′mPer2 (Cre2 binding site, 162 bp): Forward: 5′-GAACCTCTGAGGGAACCACA-3′ Reverse: 5′-GTGCTCCCCATGTCTTGAGT-3′.
Circadian destabilization by chronic jet lag
Mecp2−/y mutants at 7 weeks of age were housed under one of twolighting conditions. The stable circadian environment consisted of a12:12 LD cycle that matched the lighting cycle the mice had previouslybeen weaned from. For the destabilized group, weekly phase advanceswere applied from 8 weeks of age to advance the time of lights off by6 h tomimic chronic jet lag conditions. Previouswork has demonstratedthat this protocol does not alter the mortality of young adult C57 mice(Davidson et al., 2006). We confirmed this lack of effect on mortalitywith a small cohort of mice (n = 4) but did not repeat the previouslypublished study with a full cohort of mice. Food and water were made

158 Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164
easily accessible and available ad libitum. Mice were examined daily forhealth status andmice deemed to bemoribundwere euthanized wherepossible.
Statistical analysis
Statistical analysis was performed using Sigma Stat (ver. 12) orGraphPad (v. 4). Unpaired t-tests were applied to compare activity,sleep, cell counts, SFR, and bioluminescence parameters of WT andMecp2−/y mutants. One factor analysis of variance (ANOVA) wasapplied to determine the rhythmicity and peak/trough values of qRT-PCR results for the generation of the fold-change histograms, with twoway ANOVA used to determine effect of time and genotype on Per2and PER2 transcript expression. Acceleration of disease progression bycircadian instability was analyzed using the Kaplan–Meier survivalanalysis test. Statistical significance was defined by P b 0.05 in allanalyses.
Results
Sleep is delayed and fragmented in the Mecp2−/y mutants
We examined behaviorally-defined sleep in male hemizygous nullmice (Mecp2−/y) to determine if the mutation alters the amount ortemporal pattern of sleep under LD conditions (Fig. 1). We found thatMecp2−/y mutants slept for the same amount of time per 24 h day aswild-type (WT) littermates (Fig. 1b), but exhibited more sleep boutsand of shorter duration. Similar to RTT patients,Mecp2−/y hemizygotesexhibit increased sleep latency (42.2 ± 18.6 min after lights on)
Fig. 1. The loss of MeCP2 altered the timing but not the total amount of sleep. Videorecording was used to measure sleep as defined by time spent immobile, in combinationwith automated mouse tracking analysis software. (a) Hourly running averages ofimmobility-defined sleep in WT (n = 8) andMecp2−/y mutants (n = 7) are plotted. Themutants exhibit difficulty falling asleep (P b 0.05) and wake later relative to the time oflights off (P b 0.05). (b) The total amount of immobility-defined sleep is not significantlydifferent between WT and Mecp2−/y mutants. However, sleep in Mecp2−/y mutants issignificantly more fragmented as determined by the number of bouts and the averageduration of the bouts. *: indicates P b 0.05 as determined by post hoc t-tests.
compared to WT littermates (−4.0 ± 8.2 min after lights on, P =0.015).Mecp2−/y mutants also wake later (71.0 ± 17.3 min after lightsoff) than WT littermates (−0.1 ± 13.6 min after lights off, P = 0.001).We conclude that while Mecp2−/y mutants sleep for the same amountof time as WT, the quality of sleep is compromised due to the highlyfragmented nature of their sleeping pattern. Furthermore, the increasedsleep latency and inertia exhibited by the mutants is in keeping withaltered temporal patterning due to deficits in the circadian timingsystem.
Diurnal and circadian rhythms of locomotor activity are severely disruptedin the Mecp2−/y mutants
We examined the impact of the MeCP2 mutation on Mecp2−/y
mutants using a behavioralwheel running assay.Wecompared locomo-tor activity rhythms of Mecp2−/y mutants (n = 8) to WT littermatemales (n = 8) between 6 and 8 weeks of age in LD and constantdarkness (DD) (Table 1). The diurnal and circadian rhythms of activitywere dramatically altered in Mecp2−/y mutants (Fig. 2), as evidencedby the significantly reduced strength (power) of locomotor activity.Mecp2−/y mutants exhibited reduced levels of activity under both LDand DD conditions and activity rhythms were significantly morefragmented and of lower precision than age-matched WT littermates.Under DD, 4 out of 8 Mecp2−/y mutants were arrhythmic and therewas a small but significant increase in the free-running period of theremaining weakly rhythmic mutants (Table 1). Strikingly, our resultsindicate that Mecp2−/y mice have difficulty synchronizing to the LDcycle (Fig. 2) as 6 out of 8 mutants continue to exhibit wheel runningbehavior in the presence of light. Masking experiments conductedunder LD confirmed a significant reduction in masking behavior. Forexample, WT mice exhibited significant negative masking behaviorevident by suppression of running-wheel activity during the lightperiod (486 ± 113 rev/h, n = 8) compared to their baseline activity(2052 ± 165 rev/h, P b 0.001). In contrast, Mecp2−/y mice did notshow significant masking when exposed to 1-h light pulse (light:331 ± 103 rev/h; baseline: 456 ± 125 rev/h, n = 7, P = 0.79). Thus,behaviorally the MeCP2 mutants show significant deficits in both thephotic regulation and the generation of circadian rhythms.
Table 1Parameters of wheel running activity in male WT andMecp2−/y mice.
WT Mecp2−/y
LDLD activity (rev/h) 547.1 ± 113.7 361.1 ± 46.0⁎,⁎⁎
Alpha (min) 690.8 ± 52.5 361.1 ± 46.0⁎⁎
LD precision (min) −17.1 ± 2.8 −143.6 ± 60.3⁎⁎
LD fragmentation, bouts/day 5.9 ± 0.5 9.9 ± 0.6⁎⁎
Power (%V) 49.8 ± 5.2 29.2 ± 3.3⁎⁎
DDLD to DD phase angle (min) −32.9 ± 10.0 naArrhythmic 0/8 4/8Tau (h) 23.5 ± 0.1 24.4 ± 0.3⁎
DD activity (rev/h) 956.1 ± 129.5 86.7 ± 22.0⁎⁎
Alpha (min) 502.1 ± 20.2 257.3 ± 32.9⁎⁎
DD precision (min) −8.0 ± 0.7 −126.3 ± 42.9⁎⁎
DD fragmentation (bouts/day) 5.3 ± 0.5 9.9 ± 1.1⁎
DD power (%V) 60.3 ± 3.9 19.0 ± 1.2⁎⁎
Phase shift to light at CT 16 81.6 ± 11.2 na
Values are reported as mean± S.E.M. n=8 forWT and n=8 forMecp2−/y mice. LD, lightdark cycle; DD, constant darkness; rev, wheel revolutions, and %V, normalized Qp.Significant differences between WT and Mecp2−/y mice are measured by t-test orMann–Whitney rank sum t-test in the event of failed normality or equal variance tests.Tau in DD of the mutants was measured from 4 rhythmic animals. The activity recordswere too poor to measure the LD to DD phase angle or the light induced phase shift andare indicated with “na”.⁎ P b 0.01.⁎⁎ P b 0.001.
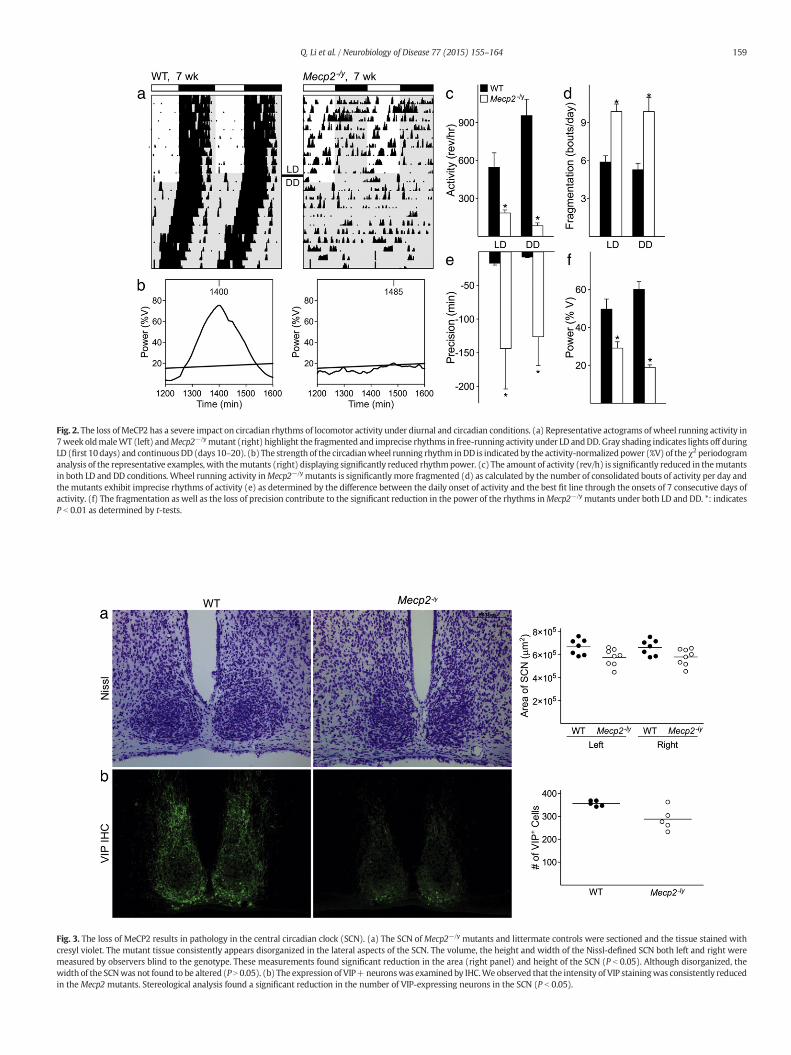
Fig. 2. The loss of MeCP2 has a severe impact on circadian rhythms of locomotor activity under diurnal and circadian conditions. (a) Representative actograms ofwheel running activity in7week oldmaleWT (left) andMecp2−/ymutant (right) highlight the fragmented and imprecise rhythms in free-running activity under LD andDD. Gray shading indicates lights off duringLD (first 10days) and continuousDD (days 10–20). (b) The strength of the circadianwheel running rhythm inDD is indicated by the activity-normalizedpower (%V) of the χ2 periodogramanalysis of the representative examples, with themutants (right) displaying significantly reduced rhythmpower. (c) The amount of activity (rev/h) is significantly reduced in themutantsin both LD and DD conditions. Wheel running activity inMecp2−/y mutants is significantly more fragmented (d) as calculated by the number of consolidated bouts of activity per day andthe mutants exhibit imprecise rhythms of activity (e) as determined by the difference between the daily onset of activity and the best fit line through the onsets of 7 consecutive days ofactivity. (f) The fragmentation as well as the loss of precision contribute to the significant reduction in the power of the rhythms inMecp2−/y mutants under both LD and DD. *: indicatesP b 0.01 as determined by t-tests.
Fig. 3. The loss of MeCP2 results in pathology in the central circadian clock (SCN). (a) The SCN ofMecp2−/y mutants and littermate controls were sectioned and the tissue stained withcresyl violet. The mutant tissue consistently appears disorganized in the lateral aspects of the SCN. The volume, the height and width of the Nissl-defined SCN both left and right weremeasured by observers blind to the genotype. These measurements found significant reduction in the area (right panel) and height of the SCN (P b 0.05). Although disorganized, thewidth of the SCNwas not found to be altered (P N 0.05). (b) The expression of VIP+neuronswas examined by IHC.We observed that the intensity of VIP stainingwas consistently reducedin theMecp2 mutants. Stereological analysis found a significant reduction in the number of VIP-expressing neurons in the SCN (P b 0.05).
159Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164

160 Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164
Structural abnormalities in the suprachiasmatic nucleus of the Mecp2−/y
mutants
It is well established that the suprachiasmatic nucleus (SCN) inthe hypothalamus is the brain region responsible for controllingcircadian rhythms and the temporal patterning of sleep. To examinethe possibility that the loss of MeCP2 caused an abnormal organizationof the SCN, Nissl-stained sections were examined. The mutant SCNconsistently (8/8) exhibited a dense cytoarchitecture which appeareddisorganized especially in the lateral aspects of the structure (Fig. 3a).To explore this further, we measured the volume, the height andwidth of the Nissl-defined SCN. These measurements indicated thatarea was significantly reduced by about 14% in both the left and rightmutant SCN (Fig. 3a; P b 0.05) compared to WT. The height of theventral–dorsal axis was also significantly decreased by about 11%(left and right mutant SCN 996 μm ± 17, P = 0.0002 and 1019 μm ±29, P = 0.0143 respectively, versus 1113 μm ± 15 and 1113 μm ±12 left, right WT SCN; P b 0.05), while the reduction in the width(9–10%) of the mutant SCNwas not statistically significant. The neuro-peptide VIP is known to be important for the synchronization of the SCNcell population so we examined the expression of the neuropeptide VIPusing IHC. We found that the intensity of VIP staining was consistentlyreduced in the Mecp2 mutants (Fig. 3b). Stereological analysis found asignificant reduction in the number of VIP-expressing neurons in theSCN (WT: 301 ± 31, n = 4; KO: 188 ± 37, n = 4; P b 0.05). This dataindicates that the loss of MeCP2 causes pathology in the central clock.
Neural activity in the central clock is compromised in the Mecp2−/y
mutants
We examined the spontaneous firing rate (SFR) of the dorsalSCN neurons in Mecp2−/y mutants using whole-cell patch clampelectrophysiology. Each of these cells was determined to be within thedorsal region of the SCN by directly visualizing the location of the cellwith infrared DIC video microscopy. We found a significant decreasein the daytime and nighttime SFR (Figs. 4a, b). There was still a day/night difference in firing rate in the Mecp2−/y mutant SCN, suggestingthat the molecular clockwork that underlies SCN rhythms is stillfunctional. The resting membrane potential (WT: 40 ± 1; KO: 42 ±2 mV), conductance (WT: 9.88 ± 1.03; KO: 7.77 ± 0.89 μS) or inputresistance (WT: 2.87± 1.29; KO: 3.45± 1.25 GΩ) did not vary betweenthe genotypes in the day. The SFR is a directmeasure of the output of theSCN and our data indicate that the Mecp2−/y mutants show greatlyreduced spontaneous neural activity. This data indicates that MeCP2 isessential for the normal physiological function of the central clock.
Fig. 4. TheMeCP2 protein is critical formaintaining spontaneous firing rate (SFR) of SCN neuronSCN. Two factor ANOVA revealed significant differences (P b 0.05) between genotypes and day/genotype and # for day/night differences.
Molecular clockwork is disrupted in the Mecp2−/y mutants
To examine the impact of the loss of MeCP2 on the molecularfeedback loop, the PERIOD2::Luciferase fusion protein reporter knock-in transgene (PER2::LUC; Yoo et al., 2004) was crossed into theMecp2−/y mutant line. Explants of PER2::LUC SCN from Mecp2−/y
hemizygotes exhibited lower amplitude rhythms of PER2-drivenrhythms in bioluminescence compared to WT SCN explants (Figs. 5a,c) without a change in the period (Table 2). We examined the timingof the first peak of PER2::LUC ex vivo rhythms of other central andperipheral oscillators that may be relevant to RTT patient symptoms,including the heart, lung, liver, and adrenals (Fig. 5b). We foundsignificantly altered phase of the PER2::LUC rhythm in Mecp2−/y
explants using a two way ANOVA (P = 0.03 effect of genotype), withpost hoc pairwise comparisons revealing significantly advanced phasein adrenal explants. Due to variability, the phases of the other peripheraloscillators were not significantly different from WT tissue. Theamplitude of oscillations in the peripheral organs of Mecp2−/y explantswere generally reducedwith the strongest impact on the lung (Table 2).This bioluminescence data indicates that while the rhythms inPER2::LUC-driven luciferase activity continue in the SCN and peripheralorgans of the MeCP2 mutant mice; the amplitude of the SCN andsynchrony of the network of oscillators is reduced in the mutants.
Mutations in MeCP2 alter the molecular oscillator in mouse and humanfibroblasts
To further examine the impact ofMeCP2 deficiency on themolecularoscillator, we took advantage of prior work showing that intrinsicproperties of the circadian oscillator can be measured from synchro-nized fibroblast cell populations (Balsalobre et al., 1998; Welsh et al.,2004). In 6 separate experiments, mouse embryonic fibroblasts(MEFs) were synchronized with dexamethasone and then sampledevery 6 h for a total of 48 h. Per2 expression was measured by qRT-PCR and confirmed to be rhythmic in WT MEFs with peaks 24 h post-synchronization (one way ANOVA on ranks; P b 0.05; Fig. 6a). MEFsfrom Mecp2−/y mutant embryos were still rhythmic but had loweramplitude compared to WT. The peak to trough ratio was reduced forPer2 (WT: 12.2 ± 0.6; KO: 7.1 ± 0.9; t-test, P b 0.001). Next, weperformed ChIP-qPCRs with MEFs to examine the impact of MeCP2mutation in regulation of chromatin states in promoter region of Per2gene. MEFs were synchronized with dexamethasone and chromatincollected at 12 and 24 h post-synchronization. Two antibodiesH3K4me3 (transcriptional activator) and K3K27me3 (transcriptionalrepressor) were used for immunoprecipitation followed by qPCRs. The
s. (a, b) The loss ofMeCP2 results in lower daytime and nighttime SFR inMecp2−/ymutantnight SFR, and significant differences determined by post hoc analysis are indicated by * for

Fig. 5. Altered phase and amplitude of PER2::LUC rhythms in some but not all tissue explants of Mecp2−/y mutants. (a) The loss of MeCP2 affects the molecular oscillator in the SCN asmeasured by baseline-subtracted bioluminescence rhythms driven by the PER2::LUC reporterMecp2−/y mutants exhibit significantly lower amplitude of SCN PER2::LUC rhythms(t-test P b 0.05, *). (b) Two factor ANOVA comparisons revealed a significant effect of genotype (P = 0.001) and tissue (P b 0.001) on the phase of the first peak in cultured explants ofPER2::LUC reporter tissues, with a significant interaction of genotype and tissue (P=0.034). Post hoc Bonferroni t-tests revealed a significant impact of the mutation on the phase of theliver (P=0.004) but not of the other tissues examined. The trend towards an advanced phase of the non-SCN oscillatorsmay be in part negated by the aberrant daytime activity observedin some but not allMecp2−/ymutants. Therewas also a significant effect of genotype (P b 0.001) on amplitude of PER2::LUC rhythms,with post hoc analysis revealing significantly reducedamplitudes of SCN and lung rhythms (both P b 0.001) (b).
161Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164
WTMEFs exhibited a significant (one way ANOVA, P b 0.05) increase inH3K4me3 enrichment at Per2 promoter regions (3.30 ± 1.55) asmeasured 24 h after synchronization whereas the mutant MEFs didnot show any H3K4me3 enrichment (0.13 ± 0.07). The K3K27me3binding of the Per2 promoter did not vary between the genotypes atthis same time point. Finally, Per2 expression was measured by qRT-PCR in fibroblasts from two RTT patients (T158M, 7 experiments andR106W, 4 experiments; Fig. 6b). Again, PER2 expression was found tobe rhythmic (one way ANOVA on ranks; P b 0.05). We did not see adecrease in amplitude in either patient cell line but did see a changein phase of peak expression in R106W cells. Therefore, while thehuman lines show more variability, our measurements of gene expres-sion rhythms indicated abnormalities in the molecular clockwork ofboth mouse and human fibroblasts.
Destabilization of the circadian system accelerates fatality of Mecp2−/y
mutants
The circadian disruption exhibited by the Mecp2−/y mouse modelsuggests an unstable circadian system, which may render the animalsvulnerable to further environmental instability. Previous work hasshown that weekly phase advances of the LD cycle (mimicking jet lag)were very disruptive to the circadian systemand even increasedmortal-ity in aged, but not young, adult mice (Davidson et al., 2006). Wesubjected Mecp2−/y mutants to circadian instability by chronic jet lagand observed an acceleration of disease progression as measured bypost-weaning fatality (Fig. 7). We saw an increase in mortality in thetreated group, which started within the first week and continuedthroughout the experiment. Our results demonstrate that the fatalitydue to the genetic loss of MeCP2 can be accelerated by environmentalperturbation.
Discussion
Several experimental models of RTT have been created (Chen et al.,2001; Guy et al., 2001; Shahbazian et al., 2002; Pelka et al., 2006) thatrecapitulate the key deficits observed in RTT. Our study uses themouse model of RTT with the loss of function of the MeCP2 proteinthrough the deletion of exon 3 of the Mecp2 gene (Chen et al., 2001;Stearns et al., 2007). We focused on the hemizygous null male in thisstudy as the mutation results in the uniform loss of MeCP2 in all cells.The femaleMecp2+/– mutants are likely to show phenotypic variabilitydue to X chromosome inactivation effects. We found delayed sleeponset, increased sleep fragmentation, and increased sleep inertia thatrecapitulate sleep problems described in RTT patients, suggesting thatthis model is suitable for examining the impact of MeCP2 mutation onsleep and circadian rhythms. This is not a conditional transgenicmodel so the observed deficits could a secondary consequence ofneuro-developmental abnormalities.
Video-based immobility-defined behavior was performed to assessthe sleep cycle in Mecp2−/y mice. Such video measurements of sleepand wake have been validated and are highly correlated to simulta-neously EEG-defined sleep (Pack et al., 2007; Fisher et al., 2012) withthe caveat that the depth of sleep or the relative progression from onesleep stage to another (NREM vs. REM) cannot be determined. MeCP2mutant mice exhibited increased sleep fragmentation characterized byan increase in the number of sleep bouts and a decrease in the durationof each sleep bout (Fig. 1). These data clearly indicate that althoughMecp2−/y mice demonstrated the same amount of sleep time comparedtoWT during both day and night, the quality of sleepwas compromiseddue to increased fragmentation. The fragmentation in sleep observed inMecp2−/y mice parallels what we observed with the locomotor activitymeasurements. The mutant mice showed difficulty initiating sleep
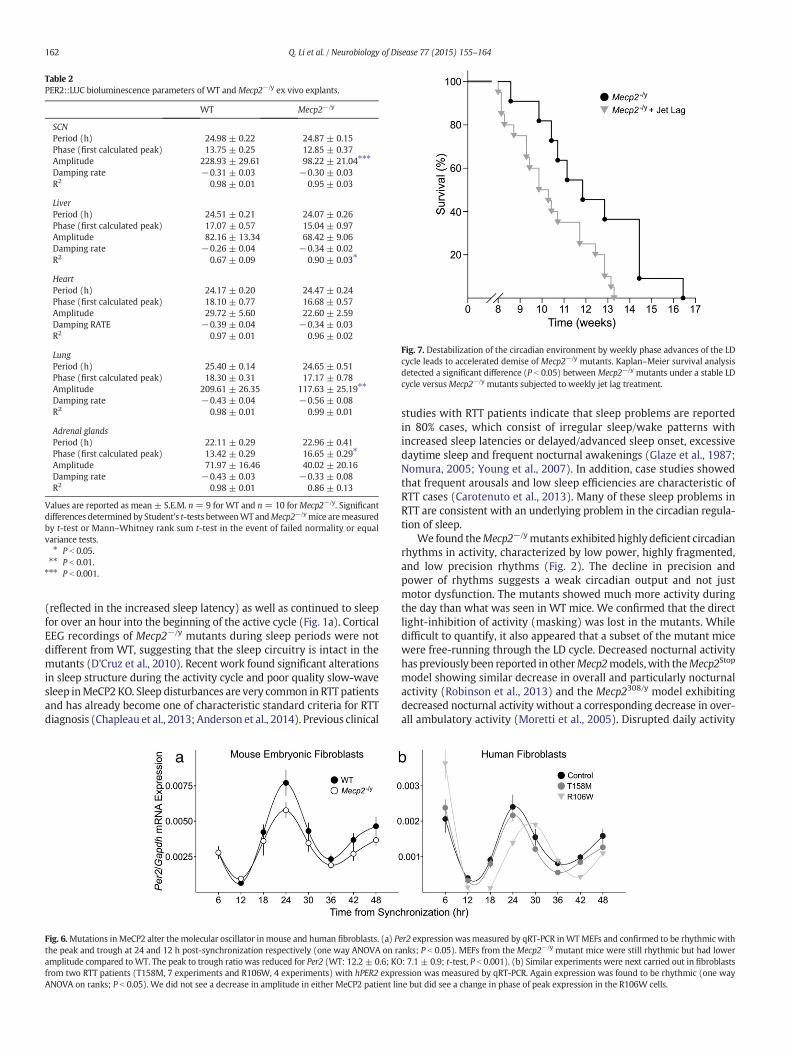
Table 2PER2::LUC bioluminescence parameters of WT andMecp2−/y ex vivo explants.
WT Mecp2−/y
SCNPeriod (h) 24.98 ± 0.22 24.87 ± 0.15Phase (first calculated peak) 13.75 ± 0.25 12.85 ± 0.37Amplitude 228.93 ± 29.61 98.22 ± 21.04⁎⁎⁎
Damping rate −0.31 ± 0.03 −0.30 ± 0.03R2 0.98 ± 0.01 0.95 ± 0.03
LiverPeriod (h) 24.51 ± 0.21 24.07 ± 0.26Phase (first calculated peak) 17.07 ± 0.57 15.04 ± 0.97Amplitude 82.16 ± 13.34 68.42 ± 9.06Damping rate −0.26 ± 0.04 −0.34 ± 0.02R2 0.67 ± 0.09 0.90 ± 0.03⁎
HeartPeriod (h) 24.17 ± 0.20 24.47 ± 0.24Phase (first calculated peak) 18.10 ± 0.77 16.68 ± 0.57Amplitude 29.72 ± 5.60 22.60 ± 2.59Damping RATE −0.39 ± 0.04 −0.34 ± 0.03R2 0.97 ± 0.01 0.96 ± 0.02
LungPeriod (h) 25.40 ± 0.14 24.65 ± 0.51Phase (first calculated peak) 18.30 ± 0.31 17.17 ± 0.78Amplitude 209.61 ± 26.35 117.63 ± 25.19⁎⁎
Damping rate −0.43 ± 0.04 −0.56 ± 0.08R2 0.98 ± 0.01 0.99 ± 0.01
Adrenal glandsPeriod (h) 22.11 ± 0.29 22.96 ± 0.41Phase (first calculated peak) 13.42 ± 0.29 16.65 ± 0.29⁎
Amplitude 71.97 ± 16.46 40.02 ± 20.16Damping rate −0.43 ± 0.03 −0.33 ± 0.08R2 0.98 ± 0.01 0.86 ± 0.13
Values are reported as mean ± S.E.M. n = 9 for WT and n= 10 forMecp2−/y. Significantdifferences determined by Student's t-tests betweenWT andMecp2−/ymice aremeasuredby t-test or Mann–Whitney rank sum t-test in the event of failed normality or equalvariance tests.⁎ P b 0.05.⁎⁎ P b 0.01.⁎⁎⁎ P b 0.001.
Fig. 7. Destabilization of the circadian environment by weekly phase advances of the LDcycle leads to accelerated demise of Mecp2−/y mutants. Kaplan–Meier survival analysisdetected a significant difference (P b 0.05) between Mecp2−/y mutants under a stable LDcycle versusMecp2−/y mutants subjected to weekly jet lag treatment.
162 Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164
(reflected in the increased sleep latency) as well as continued to sleepfor over an hour into the beginning of the active cycle (Fig. 1a). CorticalEEG recordings of Mecp2−/y mutants during sleep periods were notdifferent from WT, suggesting that the sleep circuitry is intact in themutants (D'Cruz et al., 2010). Recent work found significant alterationsin sleep structure during the activity cycle and poor quality slow-wavesleep inMeCP2 KO. Sleep disturbances are very common in RTT patientsand has already become one of characteristic standard criteria for RTTdiagnosis (Chapleau et al., 2013; Anderson et al., 2014). Previous clinical
Fig. 6.Mutations inMeCP2 alter the molecular oscillator inmouse and human fibroblasts. (a) Pthe peak and trough at 24 and 12 h post-synchronization respectively (one way ANOVA on ramplitude compared toWT. The peak to trough ratio was reduced for Per2 (WT: 12.2 ± 0.6; KOfrom two RTT patients (T158M, 7 experiments and R106W, 4 experiments) with hPER2 exprANOVA on ranks; P b 0.05). We did not see a decrease in amplitude in either MeCP2 patient li
studies with RTT patients indicate that sleep problems are reportedin 80% cases, which consist of irregular sleep/wake patterns withincreased sleep latencies or delayed/advanced sleep onset, excessivedaytime sleep and frequent nocturnal awakenings (Glaze et al., 1987;Nomura, 2005; Young et al., 2007). In addition, case studies showedthat frequent arousals and low sleep efficiencies are characteristic ofRTT cases (Carotenuto et al., 2013). Many of these sleep problems inRTT are consistent with an underlying problem in the circadian regula-tion of sleep.
We found theMecp2−/ymutants exhibited highly deficient circadianrhythms in activity, characterized by low power, highly fragmented,and low precision rhythms (Fig. 2). The decline in precision andpower of rhythms suggests a weak circadian output and not justmotor dysfunction. The mutants showed much more activity duringthe day than what was seen in WT mice. We confirmed that the directlight-inhibition of activity (masking) was lost in the mutants. Whiledifficult to quantify, it also appeared that a subset of the mutant micewere free-running through the LD cycle. Decreased nocturnal activityhas previously been reported in otherMecp2models,with theMecp2Stop
model showing similar decrease in overall and particularly nocturnalactivity (Robinson et al., 2013) and the Mecp2308/y model exhibitingdecreased nocturnal activity without a corresponding decrease in over-all ambulatory activity (Moretti et al., 2005). Disrupted daily activity
er2 expression wasmeasured by qRT-PCR inWTMEFs and confirmed to be rhythmic withanks; P b 0.05). MEFs from the Mecp2−/y mutant mice were still rhythmic but had lower: 7.1 ± 0.9; t-test, P b 0.001). (b) Similar experiments were next carried out in fibroblasts
ession was measured by qRT-PCR. Again expression was found to be rhythmic (one wayne but did see a change in phase of peak expression in the R106W cells.

163Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164
rhythms have been shown in adult female MeCP2-deficient mice(Wither et al., 2012). Taken together with our data, we conclude thatnormal MeCP2 expression is essential for normal temporal patterningof daily activity.
The deficits observed in circadian rhythms of activity are likely tooriginate from deficits within the SCN. There is compelling evidencethat a subset of neurons expressing the neuropeptide VIP play a criticalrole in the SCN circuit (Aton et al., 2005; Brown et al., 2007; Ciarleglioet al., 2009; Maywood et al., 2011; An et al., 2011; Kudo et al., 2013).Hence, the reduction in the number of VIP neurons (Fig. 3b) can providean explanation for a weakening of the light response as well as theoutput of the SCN circuit. The changes in the number of VIP expressingneurons and Nissl-defined SCN volume were seen at 2–3 months of ageso we believe that these changes are a result of an altered developmen-tal program rather than a degenerative process. MeCP2 mutant micealso exhibited a significant reduction in spontaneous electrical activityin SCN neurons in the daytime (Fig. 4a). High daytime SFR is a hallmarkfeature of SCN neurons (Fig. 4b) and is critical for the central clock tocommunicatewith the rest of the organs (Colwell, 2011). This reductionin daytime electrical activity is likely to underlie the disrupted behavior-al rhythms in thesemice. A reduction in daytimeelectrical activity in theSCNhas been found in agingmice (Nakamura et al., 2011; Farajnia et al.,2012) as well as in mouse models of neurodegenerative disease(Kudo et al., 2011; Loh et al., 2013). In each of these cases, the strengthof the behavioral rhythms was significantly reduced in parallel to thereduction in neural activity. Neurophysiologic consequences of MeCP2dysfunction varies as a function of the cell population. For example,studies demonstrated a significant reduction in SFR in somatosensorycortical pyramidal neurons in MeCP2 mutant mice (Dani et al., 2005;Chang et al., 2006). Other studies in the locus coeruleus (Taneja et al.,2009) and hippocampus (Zhang et al., 2008) have found evidencefor neural hyper-excitability in mutant compared to WT cells. In thecortex, MeCP2 dysfunction induces a shift of the homeostatic balancebetween excitation and inhibition (E/I) leading to enhanced inhibition(Dani et al., 2005; Wood et al., 2009). Reduced spontaneous activitythat we see in the SCN could arise from a change in intrinsic membranecurrents, a decrease in the E/I ratio, or as a result of the decrease in VIP.
Epigenetic mechanisms involving histone modifications haverecently emerged as important regulators of the central clock (Doiet al., 2006; Katada and Sassone-Corsi, 2010). The core mechanism re-sponsible for generating circadian oscillations is composed ofinteracting transcription and translation feedback loops driving rhyth-mic expression of key clock genes such as Per2. MeCP2 is a brain-abundant epigenetic factor that is highly expressed in the SCN(Dragich et al., 2007) and could be critical for the epigenetic regulationof the molecular clockwork there. As the first experiment testing forpotential deficits in the molecular clock system, rhythms in PER2-driven luciferase activity were examined within the SCN (Fig. 5a). Exvivo measurements with Mecp2−/y SCN explants revealed a significantdecline in the amplitude of PER2-driven bioluminescence activityrhythms. With the weakening of the SCN clock as measured by neuralactivity as well as the PER2 rhythms, we expected to see phasedispersion in the peripheral organs. We did see phase changes in theliver and adrenals but the heart and lung were just slightly shifted(Fig. 5b). The amplitude of the rhythms from peripheral organs wasmore consistently reduced (Fig. 5c) suggesting that while MeCP2 isnot essential for rhythms in circadian gene expression, its absence altersthe robustness of rhythms. This reduction was also seen in the Per2rhythm measured from MEFs (Fig. 6a). While not the focus of thepresent study, we did see an increased binding for both H3K4me3 inthe Per2 promoter that was lost in the mutants. This reduction of tran-scription activation in the mutant MEFs suggests that alterations inhistone methylation are likely part of the molecular explanationfor the observed changes in Per2 expression. The majority of RTTcases result from missense mutations within the MBD or C-terminaltruncations (Ghosh et al., 2008). Common MBD mutations include
T158M and R106W that cause profound effects on MeCP2 structure,stability and DNA-binding properties that correlate with the severityof RTT patient symptoms. To examine the impact of human MeCP2mutations in molecular clock gene expression, we measured the PER2expression rhythms from RTT patient's fibroblasts (T158M andR106W) and normal human subject fibroblasts (Fig. 6b). The impactvaried with the mutation with the T158M line showing normal PER2rhythms, while R106W fibroblasts showed a phase delay. This datasuggests that at least some of RTT patients may have direct alterationsin themolecular timingmechanisms responsible for circadian rhythms.
The altered circadian and sleep behavior that we have documentedin theMecp2−/y line is likely to have profound consequences on patienthealth. In recent years, a wide range of studies have demonstratedthat disruption of the circadian system leads to a cluster of symptomsincluding metabolic and cardiovascular disease, as well as cognitivedeficits (Hastings et al., 2003; Bechtold et al., 2010; Schroeder andColwell, 2013). Many of these same symptoms are seen in RTT patients.We therefore should consider circadian dysfunction is not just a symp-tom of RTT, but also as a key part of the disease mechanism. If thishypothesis is correct, circadian disruption may make the symptoms ofRTT worse. We tested this by exposing the mutant mice to a weeklyphase shift of the LD cycle and demonstrated increased mortality(Fig. 7). Previous work (Davidson et al., 2006) has already establishedthat this protocol does alter the mortality of young C57 mice. Theacceleration of mortality due to circadian destabilization raises severalcritical issues for the management of the disease. These patients maywell be vulnerable to the ill-effects of circadian disruption. There are anumber of strategies for improving circadian output (Schroeder andColwell, 2013) that can be tried in the animal models and ultimatelyin the RTT patients. Besides the possible effect on longevity, the disrup-tion of the sleep/wake cycle has a significantly negative effect on thepatients and family members who care for chronically ill patients.
Contributions
QL, DHL, YES and CSC designed the study. DHL, DT and MD carriedout the behavioral analyses. ZMK and CAG carried out the anatomicalanalysis. TK performed the electrophysiological analysis. DHL and QLperformed the bioluminescence experiments. QL, RT, and YC performedthe MEF and human fibroblast experiments. DHL, QL, and CSC draftedthe manuscript.
Acknowledgments
Wewould like to thankDr. A. Schroeder for her invaluable assistancewith the bioluminescence recordings, Prof. G. Block for helpfuldiscussions, Donna Crandall for help with the graphics, and Olivia NHitchcock and Jin H Choi for tracing Nissl-defined SCN.
References
Alvarez-Saavedra, M., Antoun, G., Yanagiya, A., Oliva-Hernandez, R., Cornejo-Palma, D.,Perez-Iratxeta, C., Sonenberg, N., Cheng, H.Y., 2011. miRNA-132 orchestrates chromatinremodeling and translational control of the circadian clock. Hum. Mol. Genet. 20,731–751.
Amir, R.E., et al., 1999. Rett syndrome is caused by mutations in X-linked MECP2,encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
An, S., Irwin, R.P., Allen, C.N., Tsai, C., Herzog, E.D., 2011. Vasoactive intestinal polypeptiderequires parallel changes in adenylate cyclase and phospholipase C to entraincircadian rhythms to a predictable phase. J. Neurophysiol. 105, 2289–2296.
Anderson, A., Wong, K., Jacoby, P., Downs, J., Leonard, H., 2014. Twenty years ofsurveillance in Rett syndrome: what does this tell us? Orphanet J. Rare Dis. 9, 87.
Aton, S.J., Colwell, C.S., Harmar, A.J., Waschek, J., Herzog, E.D., 2005. Vasoactive intestinalpolypeptide mediates circadian rhythmicity and synchrony in mammalian clockneurons. Nat. Neurosci. 8, 476–483.
Balsalobre, A., Damiola, F., Schibler, U., 1998. A serum shock induces circadian geneexpression in mammalian tissue culture cells. Cell 93, 929–937.
Bechtold, D.A., Gibbs, J.E., Loudon, A.S.I., 2010. Circadian dysfunction in disease. TrendsPharmacol. Sci. 31, 191–198.

164 Q. Li et al. / Neurobiology of Disease 77 (2015) 155–164
Brown, T.M., Colwell, C.S., Waschek, J.A., Piggins, H.D., 2007. Disrupted neuronal activityrhythms in the suprachiasmatic nuclei of vasoactive intestinal polypeptide-deficientmice. J. Neurophysiol. 97, 2553–2558.
Carotenuto, M., et al., 2013. Polysomnographic findings in Rett syndrome: a case–controlstudy. Sleep Breath. 17, 93–98.
Chahrour, M., Jung, S.Y., Shaw, C., Zhou, X., Wong, S.T., Qin, J., Zoghbi, H.Y., 2008. MeCP2, akey contributor to neurological disease, activates and represses transcription. Science320, 1224–1229.
Chang, Q., Khare, G., Dani, V., Nelson, S., Jaenisch, R., 2006. The disease progression ofMecp2mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348.
Chapleau, C.A., Lane, J., Larimore, J., Li, W., Pozzo-Miller, L., Percy, A.K., 2013. Recentprogress in Rett syndrome and MeCP2 dysfunction: assessment of potentialtreatment options. Future Neurol. 8 (1). http://dx.doi.org/10.2217/fnl.12.79.
Chen, R.Z., Akbarian, S., Tudor, M., Jaenisch, R., 2001. Deficiency of methyl-CpG bindingprotein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27,327–331.
Ciarleglio, C.M., Gamble, K.L., Axley, J.C., Strauss, B.R., Cohen, J.Y., Colwell, C.S., McMahon,D.G., 2009. Population encoding by circadian clock neurons organizes circadianbehavior. J. Neurosci. 29, 1670–1676.
Colwell, C.S., 2011. Linking neural activity and molecular oscillations in the SCN. Nat. Rev.Neurosci. 12, 553–569.
Colwell, C.S., Altemus, K.L., Cepeda, C., Levine, M.S., 1996. Regulation of N-methyl-D-aspartate-induced toxicity in the neostriatum: a role for metabotropic glutamatereceptors? Proc. Natl. Acad. Sci. U. S. A. 93 (3), 1200–1204.
Dani, V.S., Chang, Q., Maffei, A., Turrigiano, G.G., Jaenisch, R., Nelson, S.B., 2005. Reducedcortical activity due to a shift in the balance between excitation and inhibition in amouse model of Rett syndrome. Proc. Natl. Acad. Sci. U. S. A. 102, 12560–12565.
Davidson, A.J., Sellix, M.T., Daniel, J., Yamazaki, S., Menaker, M., Block, G.D., 2006. Chronicjet-lag increases mortality in aged mice. Curr. Biol. 16, R914–R916.
D'Cruz, J.A., Wu, C., Zahid, T., El-Hayek, Y., Zhang, L., Eubanks, J.H., 2010. Alterations ofcortical and hippocampal EEG activity in MeCP2-deficient mice. Neurobiol. Dis. 38,8–16.
Doi, M., Hirayama, J., Sassone-Corsi, P., 2006. Circadian regulator CLOCK is a histoneacetyltransferase. Cell 125, 497–508.
Dragich, J.M., Kim, Y.-H., Arnold, A.P., Schanen, N.C., 2007. Differential distribution of theMeCP2 splice variants in the postnatal mouse brain. J. Comp. Neurol. 501, 526–542.
Farajnia, S., Michel, S., Deboer, T., vanderLeest, H.T., Houben, T., Rohling, J.H.,Ramkisoensing, A., Yasenkov, R., Meijer, J.H., 2012. Evidence for neuronaldesynchrony in the aged suprachiasmatic nucleus clock. J. Neurosci. 32, 5891–5899.
Fisher, S.P., et al., 2012. Rapid assessment of sleep–wake behavior inmice. J. Biol. Rhythms27, 48–58.
Ghosh, R.P., Horowitz-Scherer, R.A., Nikitina, T., Gierasch, L.M., Woodcock, C.L., 2008. Rettsyndrome-causing mutations in human MeCP2 result in diverse structural changesthat impact folding and DNA interactions. J. Biol. Chem. 283, 20523–20534.
Glaze, D.G., Frost, J.D., Zoghbi, H.Y., Percy, A.K., 1987. Rett's syndrome: characterization ofrespiratory patterns and sleep. Ann. Neurol. 21, 377–382.
Guy, J., Hendrich, B., Holmes, M., Martin, J.E., Bird, A., 2001. Amouse Mecp2-null mutationcauses neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326.
Hagberg, B., Aicardi, J., Dias, K., Ramos, O., 1983. A progressive syndrome of autism,dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: reportof 35 cases. Ann. Neurol. 14, 471–479.
Hastings, M.H., Reddy, A.B., Maywood, E.S., 2003. A clockwork web: circadian timing inbrain and periphery, in health and disease. Nat. Rev. Neurosci. 4, 649–661.
Katada, S., Sassone-Corsi, P., 2010. The histone methyltransferase MLL1 permits theoscillation of circadian gene expression. Nat. Struct. Mol. Biol. 17 (12), 1414–1421.
Kudo, T., Schroeder, A., Loh, D.H., Kuljis, D., Jordan, M.C., Roos, K.P., Colwell, C.S., 2011.Dysfunctions in circadian behavior and physiology in mouse models of Huntington'sdisease. Exp. Neurol. 228, 80–90.
Kudo, T., Tahara, Y., Gamble, K.L., McMahon, D.G., Block, G.D., Colwell, C.S., 2013.Vasoactive intestinal peptide produces long-lasting changes in neural activity in thesuprachiasmatic nucleus. J. Neurophysiol. 110 (5), 1097–1106.
Loh, D.H.-W., et al., 2011. Effects of vasoactive intestinal peptide genotype on circadiangene expression in the suprachiasmatic nucleus and peripheral organs. J. Biol.Rhythms 26, 200–209.
Loh, D.H.-W., Kudo, T., Truong, D., Wu, Y., Colwell, C.S., 2013. The Q175 mouse model ofHuntington's disease shows gene dosage- and age-related decline in circadianrhythms of activity and sleep. PLoS ONE 8, e69993.
Maywood, E.S., Chesham, J.E., O'Brien, J.A., Hastings, M.H., 2011. A diversity of paracrinesignals sustains molecular circadian cycling in suprachiasmatic nucleus circuits.Proc. Natl. Acad. Sci. U. S. A. 108, 14306–14311.
Moretti, P., Bouwknecht, J.A., Teague, R., Paylor, R., Zoghbi, H.Y., 2005. Abnormalities ofsocial interactions and home-cage behavior in a mouse model of Rett syndrome.Hum. Mol. Genet. 14, 205–220.
Nakamura, T.J., et al., 2011. Age-related decline in circadian output. J. Neurosci. 31,10201–10205.
Nan, X., Campoy, F.J., Bird, A., 1997. MeCP2 is a transcriptional repressor with abundantbinding sites in genomic chromatin. Cell 88, 471–481.
Neul, J.L., Kaufmann, W.E., Glaze, D.G., Christodoulou, J., Clarke, A.J., Bahi-Buisson, N.,Leonard, H., Bailey, M.E., Schanen, N.C., Zappella, M., Renieri, A., Huppke, P., Percy,A.K., RettSearch Consortium, 2010. Rett syndrome: revised diagnostic criteria andnomenclature. Ann. Neurol. 68, 944–950.
Nomura, Y., 2005. Early behavior characteristics and sleep disturbance in Rett syndrome.Brain Dev. 27 (Suppl. 1), S35–S42.
Pack, A.I., et al., 2007. Novel method for high-throughput phenotyping of sleep in mice.Physiol. Genomics 28, 232–238.
Pelka, G.J., et al., 2006. Mecp2 deficiency is associated with learning and cognitive deficitsand altered gene activity in the hippocampal region of mice. Brain 129, 887–898.
Robinson, L., Plano, A., Cobb, S., Riedel, G., 2013. Long-term home cage activity scansreveal lowered exploratory behaviour in symptomatic female Rett mice. Behav.Brain Res. 250, 148–156.
Rohdin, M., et al., 2007. Disturbances in cardiorespiratory function during day and nightin Rett syndrome. Pediatr. Neurol. 37, 338–344.
Schroeder, A.M., Colwell, C.S., 2013. How to fix a broken clock. Trends Pharmacol. Sci. 34(11), 605–619.
Shahbazian, M., et al., 2002. Mice with truncated MeCP2 recapitulate many Rettsyndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254.
Stearns, N.A., et al., 2007. Behavioral and anatomical abnormalities in Mecp2 mutantmice: a model for Rett syndrome. NSC 146, 907–921.
Takahashi, K., Yamanaka, S., 2006. Induction of pluripotent stem cells from mouseembryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676.
Taneja, P., et al., 2009. Pathophysiology of locus ceruleus neurons in a mouse model ofRett syndrome. J. Neurosci. 29, 12187–12195.
Weese-Mayer, D.E., Lieske, S.P., Boothby, C.M., Kenny, A.S., Bennett, H.L., Ramirez, J.M.,2008. Autonomic dysregulation in young girls with Rett syndrome during nighttimein-home recordings. Pediatr. Pulmonol. 43, 1045–1060.
Welsh, D.K., Yoo, S.-H., Liu, A.C., Takahashi, J.S., Kay, S.A., 2004. Bioluminescence imagingof individual fibroblasts reveals persistent, independently phased circadian rhythmsof clock gene expression. Curr. Biol. 14, 2289–2295.
Wither, R.G., Colic, S., Wu, C., Bardakjian, B.L., Zhang, L., Eubanks, J.H., 2012. Daily rhythmicbehaviors and thermoregulatory patterns are disrupted in adult female MeCP2-deficient mice. PLoS ONE 7, e35396.
Wood, L., Gray, N.W., Zhou, Z., Greenberg, M.E., Shepherd, G.M.G., 2009. Synaptic circuitabnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interferencemodel of methyl-CpG-binding protein 2 deficiency. J. Neurosci. 29, 12440–12448.
Yoo, S.-H., et al., 2004. PERIOD2::LUCIFERASE real-time reporting of circadian dynamicsreveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl.Acad. Sci. U. S. A. 101, 5339–5346.
Young, D., Nagarajan, L., de Klerk, N., Jacoby, P., Ellaway, C., Leonard, H., 2007. Sleepproblems in Rett syndrome. Brain Dev. 29, 609–616.
Zhang, L., He, J., Jugloff, D.G.M., Eubanks, J.H., 2008. The MeCP2-null mouse hippocampusdisplays altered basal inhibitory rhythms and is prone to hyperexcitability.Hippocampus 18, 294–309.
Zhou, Z., et al., 2006. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52,255–269.
Related Documents











