THE JOURNAL 0 1984by The American Society of Biolosrcal Chemists, Inc. OF BIOLOGICAL CHEMIST~Y Val. 259. No. 21. Iseue of November 10, PP. 13413-13423 1984 Printed in C.S.A. Microsomal Enzymesof Cholesterol Biosynthesis from Lanosterol CHARACTERIZATION, SOLUBILIZATION, AND PARTIAL PURIFICATION OF NADPH-DEPENDENT A8*14-STEROID 14-REDUCTASE* (Received for publication, June 29,1984) Young-Ki PaikS, James M. Trzaskos, Ali Shafied, and James L. Gaylor From the Central Research and Development Department, E. I. du Pont de Nemours and Company, Glenolden Laboratory, Glenolden, Pennsylvania 19036 The membrane-bound enzyme of microsomes that catalyzes NADPH-dependent reduction of the 14-dou- ble bond of conjugated and A7~'"-stero1s has been studied both as collected in microsomes from broken cell preparations of rat liver and after solubilization. Optimal incubation conditions for assay of the mem- brane-bound enzyme have been determined, and prop- erties of the microsomal enzyme have been established with respect to cofactor requirements, kinetics, pH, addition of inhibitors, addition of glycerol phospha- tides, and sterol substrate specificity. The 14-reductase is readily solubilized with a mix- ture of octylglucoside and taurodeoxyeholic acid. The solubilized enzymehas been enriched by precipitation with polyethylene glycol and chromatography on DEAE-Sephacel and hydroxylapatite columns. The re- sulting partially purified enzyme has been obtained free of other microsomal enzymes of cholesterol bio- synthesis: 4-methyl sterol oxidase, A6-7-stero1 7-reduc- tase, A8~24-sterol 24-reductase, 3-ketosteroid reduc- tase, and steroid 8 + 7-ene isomerase, plus microsomal cytochrome P-450, cytochrome P-460 reductase, cy- tochrome ba reductase, and cytochrome b6. The par- tially purified enzyme is stimulated by addition of phospholipids. All of the properties exhibited by partially purified 14-reductase are consistent with the suggestion that the solubilized and enriched enzyme catalyzesthe mi- crosomal reduction of the 14-double bond of the sterol- conjugated dienes. However, presence of the enzyme does not prove that the sterol-conjugated dienes are obligatory precursors of cholesterol. A possible role of formation and metabolism of A'"'-diene sterols during rat liver microsomal synthesis of cholesterol from lanosterol' has been investigated in several laboratories * This is contribution 3295 from the Central Research and Devel- opment Department, E. I. du Pont de Nemours and Co. The costa of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "adver- tisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. 3 Present address, The Gladstone Foundation Laboratory, Univer- sity of California, San Francisco, CA 94140. 8 Present address, Medicinal Chemistry Department, College of Pharmacy, University of Wisconsin, Madison, WI 53713. The abbreviations and trivial names used are: lanosterol, 4,4,14a- trimethyl-5a-cholesta-8,24-dien-3p-ol; zymosterol, 5a-cholesta-8,24- dien-3p-ol; desmosterol, cholesta-5,24-dien-3f?-ol; 7-debydrocholes- terol, cholesta-5,7-dien-3,T-ol; AY-9944, tram-1,4-bis(2-chloroben- zylaminomethy1)cyclohexane dihydrochloride; triparanol, 4-chloro- (1-7). Veryfew properties of an enzyme capable of catalyzing reduction of the 14-double bond of the conjugated diene have been reported. To date, we know that enzymic reduction of the 14-doublebond of A7,14-diene and A'"4-diene sterols by rat liver microsomes occurs well under anaerobic conditions, re- quires NADPH, and is strongly inhibited by AY-9944 (5, 10, 17). During the course of study of 14a-demethylation of lanosterol, it has become evident that a A'*I4-diene sterol indeed accumulates as a result of C-32 demethylation (11,12); therefore, characterization, solubilization, and purification of the A14-reductase whichis responsible for metabolism of the accumulated 4,4-dimethyl~terol-A~-'~-diene to the A'-monoene have been carried out. Although some properties such as thestereochemical reac- tion mechanism of 14-reduction by the membrane-bound enzyme have been extensively studied, the physical and en- zymic properties plus purification of microsomal 14-reductase have not been established. In this study, wenow report the development of an assay and partial characterization of the membrane-bound enzyme with respect to kinetics, pH, effects of inhibitors, and dependence upon phospholipids and/or other membrane components. The enzyme was solubilized with a mixture of detergents and the 14-reductase was partially purified by polyethylene glycol precipitation plus chromatography on DEW-Sephacel and hydroxylapatite columns which resolves the solubilized enzyme from other rat liver microsomal enzymes of choles- terol biosynthesis. Properties of the solubilized and partially purified enzyme have been compared tothe microsomal- boundenzyme. Particular attention also has been paid to evidence that differentiates the NADPH-dependent 14-reduc- tase activity from the other NADPH-dependent reductases of sterol biosynthesis: 7-reductase, !&reductase, and 3-ketoste- roid reductase. Resolution of the solubilized enzymes is nec- essary to determine whether or not different microsomal enzymes catalyze each of these NADPH-dependent steps of cholesterol biosynthesis (13). Also, resolution and reconsti- tution is needed to demonstrate that NADPH-dependent cytochrome P-450 reductase, the most abundant NADPH- dependent enzyme of rat liver microsomes, does not partici- pate in the direct transfer of reducing equivalents to these steroids (13, 14). EXPERIMENTAL PROCEDURES Chemicals and Reagents taurodeoxycholate (Na+ salt), NADPH (tetrasodium salt), @-NADH The following chemicals and reagents were purchased from Sigma: cy-[4-[2-diethylamino)ethoxylphenyl]-cr-(4-methylphenyl)benze- methanol. HFLC, high-performance liquid chromatography; rrt, rel- ative retention time; PEG, polyethylene glycol. 13413

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

THE JOURNAL 0 1984by The American Society of Biolosrcal Chemists, Inc.
OF BIOLOGICAL CHEMIST~Y Val. 259. No. 21. Iseue of November 10, PP. 13413-13423 1984 Printed in C.S.A.
Microsomal Enzymes of Cholesterol Biosynthesis from Lanosterol CHARACTERIZATION, SOLUBILIZATION, AND PARTIAL PURIFICATION OF NADPH-DEPENDENT A8*14-STEROID 14-REDUCTASE*
(Received for publication, June 29,1984)
Young-Ki PaikS, James M. Trzaskos, Ali Shafied, and James L. Gaylor From the Central Research and Development Department, E. I. du Pont de Nemours and Company, Glenolden Laboratory, Glenolden, Pennsylvania 19036
The membrane-bound enzyme of microsomes that catalyzes NADPH-dependent reduction of the 14-dou- ble bond of conjugated and A7~'"-stero1s has been studied both as collected in microsomes from broken cell preparations of rat liver and after solubilization. Optimal incubation conditions for assay of the mem- brane-bound enzyme have been determined, and prop- erties of the microsomal enzyme have been established with respect to cofactor requirements, kinetics, pH, addition of inhibitors, addition of glycerol phospha- tides, and sterol substrate specificity.
The 14-reductase is readily solubilized with a mix- ture of octylglucoside and taurodeoxyeholic acid. The solubilized enzyme has been enriched by precipitation with polyethylene glycol and chromatography on DEAE-Sephacel and hydroxylapatite columns. The re- sulting partially purified enzyme has been obtained free of other microsomal enzymes of cholesterol bio- synthesis: 4-methyl sterol oxidase, A6-7-stero1 7-reduc- tase, A8~24-sterol 24-reductase, 3-ketosteroid reduc- tase, and steroid 8 + 7-ene isomerase, plus microsomal cytochrome P-450, cytochrome P-460 reductase, cy- tochrome ba reductase, and cytochrome b6. The par- tially purified enzyme is stimulated by addition of phospholipids.
All of the properties exhibited by partially purified 14-reductase are consistent with the suggestion that the solubilized and enriched enzyme catalyzes the mi- crosomal reduction of the 14-double bond of the sterol- conjugated dienes. However, presence of the enzyme does not prove that the sterol-conjugated dienes are obligatory precursors of cholesterol.
A possible role of formation and metabolism of A'"'-diene sterols during rat liver microsomal synthesis of cholesterol from lanosterol' has been investigated in several laboratories
* This is contribution 3295 from the Central Research and Devel- opment Department, E. I. du Pont de Nemours and Co. The costa of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "adver- tisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
3 Present address, The Gladstone Foundation Laboratory, Univer- sity of California, San Francisco, CA 94140.
8 Present address, Medicinal Chemistry Department, College of Pharmacy, University of Wisconsin, Madison, WI 53713.
The abbreviations and trivial names used are: lanosterol, 4,4,14a- trimethyl-5a-cholesta-8,24-dien-3p-ol; zymosterol, 5a-cholesta-8,24- dien-3p-ol; desmosterol, cholesta-5,24-dien-3f?-ol; 7-debydrocholes- terol, cholesta-5,7-dien-3,T-ol; AY-9944, tram-1,4-bis(2-chloroben- zylaminomethy1)cyclohexane dihydrochloride; triparanol, 4-chloro-
(1-7). Very few properties of an enzyme capable of catalyzing reduction of the 14-double bond of the conjugated diene have been reported. To date, we know that enzymic reduction of the 14-double bond of A7,14-diene and A'"4-diene sterols by rat liver microsomes occurs well under anaerobic conditions, re- quires NADPH, and is strongly inhibited by AY-9944 (5, 10, 17). During the course of study of 14a-demethylation of lanosterol, it has become evident that a A'*I4-diene sterol indeed accumulates as a result of C-32 demethylation (11,12); therefore, characterization, solubilization, and purification of the A14-reductase which is responsible for metabolism of the accumulated 4,4-dimethyl~terol-A~-'~-diene to the A'-monoene have been carried out.
Although some properties such as the stereochemical reac- tion mechanism of 14-reduction by the membrane-bound enzyme have been extensively studied, the physical and en- zymic properties plus purification of microsomal 14-reductase have not been established. In this study, we now report the development of an assay and partial characterization of the membrane-bound enzyme with respect to kinetics, pH, effects of inhibitors, and dependence upon phospholipids and/or other membrane components.
The enzyme was solubilized with a mixture of detergents and the 14-reductase was partially purified by polyethylene glycol precipitation plus chromatography on DEW-Sephacel and hydroxylapatite columns which resolves the solubilized enzyme from other rat liver microsomal enzymes of choles- terol biosynthesis. Properties of the solubilized and partially purified enzyme have been compared to the microsomal- bound enzyme. Particular attention also has been paid to evidence that differentiates the NADPH-dependent 14-reduc- tase activity from the other NADPH-dependent reductases of sterol biosynthesis: 7-reductase, !&reductase, and 3-ketoste- roid reductase. Resolution of the solubilized enzymes is nec- essary to determine whether or not different microsomal enzymes catalyze each of these NADPH-dependent steps of cholesterol biosynthesis (13). Also, resolution and reconsti- tution is needed to demonstrate that NADPH-dependent cytochrome P-450 reductase, the most abundant NADPH- dependent enzyme of rat liver microsomes, does not partici- pate in the direct transfer of reducing equivalents to these steroids (13, 14).
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
taurodeoxycholate (Na+ salt), NADPH (tetrasodium salt), @-NADH The following chemicals and reagents were purchased from Sigma:
cy-[4-[2-diethylamino)ethoxylphenyl]-cr-(4-methylphenyl)benze- methanol. HFLC, high-performance liquid chromatography; rrt, rel- ative retention time; PEG, polyethylene glycol.
13413

13414 Sterol 14-Reductase
(disodium salt), B-NAD+ (grade III), N-ethylmaleimide, glucose oxi- dase (Type V, from Aspergillus Niger), glutathione (reduced form), dithiothreitol, HgClZ, phospholipase A2 (from porcine pancrease), Tris' HCl, and nicotinamide. Octyl-j3-D-glucopyranoside was pur- chased from Calbiochem-Behring. AY-9944 was obtained from Ayerst Laboratory (New York, NY). Polyethylene glycol and Silica Gel G 60 H F z ~ were purchased from EM Science (Gibbstown, NJ). The chro- matographic gels, such as DEAE-Sephacel, Sephadex G-25, hydrox- ylapatite were purchased from Pharmacia Fine Chemicals Co. (Upp- sala, Sweden) and Bio-Rad. Egg L-a-lecithin was purchased from Avanti Polar Lipids (Birmingham, AL). All other chemicals were of the best grade available.
Preparation of Microsomes from Rat Liver Microsomes were prepared from male, Sprague-Dawley rats (150-
250 g) that were maintained on a cholestyramine-containing diet and a reversed light cycle as previously described (12), using 0.1 M potas- sium phosphate buffer, pH 7.4, including 1 mM reduced glutathione, 0.5 mM EDTA, and 20% (v/v) glycerol (Buffer A). Prior to prepara- tion of the homogenates, residual blood was removed from livers by exhaustive perfusion in situ with cold 0.25 M sucrose solution. Micro- somal pellets obtained by centrifugation were washed three times with fresh Buffer A, and the pellets from the final centrifugation could be stored at -80 "C for several weeks without loss of enzymic activity.
Preparation of Sterols HPLC was performed on an Ultrasphere-octyl column (4.6 mm X
25 cm) with a mobile phase of acetonitrilelmethanol/HzO (45:45:10). The temperature was maintained at 45 "C, and the flow rate was 1.00 ml/min. Solutions of sterols in ethanol (1-5 mg/ml) were injected (10-50 pl). Sterol mass was detected by UV absorbance at 214 nm. Relative retention times are reported with authentic cholesterol as standard
Cn&o
Found: 386.3545 Calculated 386.3549
High-resolution mass spectroscopy was performed on a VG"M 7070H mass spectrometer. Mass fragmentation showed 386 (96%, M), 371 (39%, "CHI), 368 (52%, M-HZO), 353 (45%, M-CHI-HZO), 273 (28%, M-CsHI,), 255 (37%, M-CaH17-H20), 231 (30%, M-C&I17- C3He), and 213 (53%, M-CEH~~-C&-H~O); direct probe analysis was performed on a Finnigan 4021 spectrometer with initial temperatures a t 40 'C for 0.5 min then ramp speed of 50 "C/min to a final temper- ature of 300 "C; scan range was 40-600 a.m.u., scan time was 2.0 s, multiplier voltage was 1300 V, 70 EV with methane as ionizing gas. NMR was performed on either a Varian EM 390 (90 MHz) or a Nicolet 360 (360 MHz) using CDCl, assolvent; cholesterol (360 MHz): 0.68 (s,3H, C-lS-CHd, 0.88 (dd, 6H, C-26-CHI), 3.52 (m, 1H, 3a-H), and 5.34 (d, lH, C-6-H). GLC was carried out on a packed column (2 mm X 6 foot) of OV-17, 3% (w/w) on Chromosorb W H P (100-120 mesh). Nitrogen was the flow gas maintained at 30 ml/min, the oven temperature was 260 "C, and the flame ionization detector was 300 "C. Authentic cholesterol exhibited a symmetrical peak (>99.5% purity); all relative retention times are reported against the cholesterol stand- ard. When reported, diffuse reflectance IR analysis was performed on a Nicolet 7199 FTIR. All melting points were uncorrected.
Synthesk, of 4,4-Dimethyl-5a-ch~lesta-8,14- and -7,14-dkn-3@-sterok
Cholesta-4,7-dien-3-one, which was used as starting material for most sterol syntheses, was prepared from recrystallized 7-dehydro- cholesterol in 80% yield as previously described (15). The isolated product displayed m.p. 85-87 "C and X,-238 nm (c = 15,800); m.p. (literature) (16) 86-88 "C; X, = 238 nm (e = 15,500). 4,4-Dimethyl-5a-cholesta-8,14-dien-3j3-ol and 4,4-dimethyl-5a-
cholesta-7,14-dien-3j3-01 were prepared by previously published pro- cedures (17, 18) with the following modifications. Cholesta-l,i'-dien- 3-one was first methylated according to the procedure described by Woodward et al. (19). Potassium t-butoxide (13.17 g) was dissolved in 200 ml of dry t-butanol with gentle heating. Cholesta-4,7-dien-3- one (15 g) was dissolved in 100 ml of t-butanol, and the resulting solution was added to the t-butoxide solution. Freshly distilled methyl iodide (14.6 ml) dissolved in 10 ml of t-butanol was immediately added to the above reaction mixture and heated under reflux and NZ
for 1 h. The mixture was cooled to room temperature, water was added, and the mixture was extracted with ether. The extract was washed sequentially with water and saturated NaCl, and then the ether extract was dried over sodium sulfate. Solvent was evaporated under reduced pressure, and the residue was crystallized from chlo- roform/methanol to give 4,4-dimethylcholesta-5,7-dien-3-one (12 g, 80% yield), m.p. 158-159 "C; [a], -20.5 f 0.8 "C (C, 1.00); X, = 282.5 nm (e = 9,635) and 273 nm (e = 9,020); m.p. (literature) (18)
nm (c = 9,900). 2 g of 4,4-dimethylcholesta-5,7-dien-3-one were dissolved in 125
ml of dry ether, and the resulting solution was added to a slurry of lithium aluminum hydride (0.95 g) in 25 ml of dry ether. The reaction mixture was heated under reflux for 1 h with stirring. After cooling to room temperature, crushed ice was carefully added followed by the addition of 150 ml of a saturated solution of ammonium chloride. The resulting mixture was shaken extensively and extracted with 20% methylene chloride in ether. The ether extract was successively washed with water, washed with saturated sodium chloride solution, and dried over sodium sulfate. Solvent was evaporated under reduced pressure, and the resulting residue was crystallized from chloroform/ methanol to give 4,4-dirnethylcholesta-5,7-dien-3~-01 (70% yield), m.p. 137.5-139 "C; [.]I, -155.6 f 0.8 "C (C, 1.0); X, = 282.5 nm (e = 9,970) and 237 nm (e = 9,764) m.p. (literature) (18) 139-141 "C; [a], -159 "C (C, 1.39); X, = 282.5 nm (e = 10,300 and 273 nm (t = 10,300)]. NMR (90 MHz): 0.60 (8, 3H, C-18-CH3), 0.97 (8, 3H, C-19- CHI), 3.40 (m, lH, C-3-aH), 5.55 (m, lH, C-7-H), 5.93 (d, lH, C-6- H).
150 mg of 4,4-dimethylcholesta-5,7-dien-3j3-01 was dissolved in a mixture of 95% ethanol (3 ml), benzene (0.4 ml), and 36% hydrochlo- ric acid (0.75 ml). The mixture was heated under reflux for 3 h and then allowed to cool to room temperature. Water was added, and the mixture was extracted with ether. The ether extract was successively washed with water, sodium biocarbonate solution, and a saturated solution of sodium chloride. The solution was dried over sodium sulfate. The solvent was evaporated, and the resulting residue was crystallized 3 times from methanol/ethyl acetate to give 4,4-dimethyl- 5a-cholesta-8,14-dien-3@-01(76% yield), m.p. 141-142 "c; [CY], -14.6 f 0.06 "C (C, 1.58); X, = 248 nm; m.p. (literature) (18) 144-145 "c; [a], -13 "C (C, 1.61); X,. = 248 nm]. High-resolution/mass-spectral analysis showed precise mass of 412.3727 (calculated for C&uO: 412.3705). Prominent fragmentation ions were 412 (loo%, M), 397
162-164 "c; [Cy], -19 'c (c, 1.33); 1, = 282.5 ( t = 10,100) and 273
(38%, "CHI), 394 (9%, M-HZO), 379 (42%, M-CHs-HzO), 351 (5%, M-HZO-CIH?), 299 (13%, M-CeH17), 281 (8%, M-HzO-CeHl7). NMR (360 MHz): 0.81 (s, 3H, C-18-CH3), 0.84 (8, 3H, c-lg-CH~), 3.25 (m, lH, C-3-aH), 5.36 (m, lH, C-15-H). 4,4-Dimethylcholesta-7,14-dien-3j3-ol was prepared by acid isomer-
ization of the AS*7-sterol acetate in pyridine as follows. 2 g of 4,4- dimethylcholesta-5,7-dien-3@-01 were dissolved in 100 ml of a mixture of acetic anhydride (50 ml) and pyridine (50 ml) and heated for 1 h under nitrogen. The reaction mixture was then cooled to room tem- perature and poured into water. Sterol was extracted with ether. The ether extract was washed successively with cold 2 N hydrochloric acid, saturated sodium bicarbonate solution, and water, then dried over sodium sulfate. The residue was crystallized from ethyl acetate/ methanol to give 4,4-dimethylcholest-5,7-dien-3~-01 acetate (85% yield), m.p. 143 "C; [a], -97.1 f 0.8 "C (C, 1.0); X, = 282.5 nm (e = 10,078) and 273 nm (6 = 9912) (m.p. (literature) (18) 136-137 "C; [a], - 106 "C (C, 1.08); X,, = 282.5 nm (e = 11,100) and 273 nm (e = 11,400)). High-resolution/mass-spectral analysis showed precise mass of 454.3835 (calculated for CI1Hs0O2: 454.3810). Prominent fragmentation ions were 454 (25%, M), 394 (18%, M-CH&OOH), 379 (loo%, M-CH~COOH-CHI), 351 (7%, M-CH~COOH-CIH~), 325 (43%, M-CH~COOH-CSH~). NMR (360 MHz): 0.60 (8 , 3H, C-18- CHI), 1.0 (8, 3H, C-19-CH3), 2.08 (8, 3H, CHI of acetoxy function), 4.62 (m, 1H, C-3-aH), 5.55 (m, lH, C-6-H), 5.90 (d, lH, C-7-H).
500 mg of 4,4-dimethylcholesta-5,7-dien-3@-01 acetate was dis- solved in 20 ml of chloroform, and the solution was cooled to -40 "C; the cold solution was treated with dry hydrochloric acid gas for 2 h. The reaction mixture was then cooled to -60 "C and neutralized with a solution of anhydrous ammonia in methanol; after neutralization the temperature was allowed to adjust slowly to 20 "C. The mixture was then successively washed with water and saturated sodium chlo- ride solution; the solvent was evaporated under reduced pressure. The residue was crystallized from chloroform/methanol (75% yield). For enzyme studies, product obtained by crystallization was further sub- jected to silver nitrate (12%) preparative TLC on Silica Gel GFw. Plates were developed in benzene petroleum ether (73) and the band

Sterol 14-Reductase 13415
with RF = 0.45 was eluted. After normal workup, the sterol acetate was crystallized again for final saponification with lithium aluminum hydride. 4,4-Dimethyl-5a-cholesta-7,14-dien-3~-0~acetate obtained by crys-
tallization had m.p. 112-114 "e [ a ] D -129.8 f 0.8 "c (c, 1.0); X- = 248 nm (e = 9,125) m.p. (literature) (18) 117-119 "c; [ah -136 "c (c, 1.65); X, = 243 nm (e = 9,900). NMR (360 MHz): 0.82 (s,3H, C-18- CH3), 0.96 (5, 3H, C-19-CH3), 2.06 (8, 3H, CH3 of acetoxy function), 4.51 (m, 1H, C-3-aH), 5.51 (m, lH, C-15-H), 5.81 (m, lH, C-7-H).
200 mg of 4,4-dimethyl-5~-cholesta-7,14-dien-3@-ol acetate was dissolved in 12.5 ml of anhydrous ether, and the solution was added to a slurry of lithium aluminum hydride (95 mg) in anhydrous ether (2.5 ml). The resulting suspension was gently heated with reflux for 1 h, and then the mixture was allowed to cool to roqm temperature. Crushed ice was carefully added followed by the addition of 15 ml of a saturated solution of ammonium chloride. The ammonium chloride mixture was extensively shaken and extracted with ether. The ether extract was washed with water and a saturated sodium chloride solution; the solution was then dried over sodium sulfate. The solvent was evaporated under reduced pressure, and the residue was subjected to TLC on Silica Gel GFm. Preparative TLC plates were developed in chloroform, and the band corresponding to the alcohol was col- lected; the sterol was crystallized from chloroform/methanol (75% yield), m.p. 115-116 "C; [ a ] D -150.3 f 0.8 "C (C, 1.0); X, = 245 (e = 8940). High-resolution/mass-spectral analysis showed a precise mass of 412.3689 (calculated for C,H,O 412.3705). Prominent ions in the high-mass region of the spectrum were 412 (loo%, M), 397 (30%, M-CHs), 379 (23%, M-CH~-HZO-C~HT), 299 (74%, "Ca17), 281 (35%, M-CaH17-HzO). NMR (360 MHz): 0.84 (9, 3H, C-lS-CHd, 0.98 (s, 3H, C-19-CH3), 3.25 (m, lH, C-3-aH), 5.51 (m, lH, C-15-H), 5.83 (m, lH, C-7-H).
Each sterol diene exhibited single peaks on HPLC and GLC: 4,4- dimethyl-5a-cholesta-8,14-dien-3j3-ol, rrt = 1.100 and 1.591; 4,443- methyl-5a-cholesta-7,14-dien-3~-ol, rrt = 1.097 and 1.565.
Modifications of previously described procedures (18,19) have been employed for the production of the 4,4-gern-dimethyl-substituted monoene sterols. 4,4-Dirnethyl-5a-cholesta-8,14-dien-3~-ol was first acetylated in a mixture of acetic anhydride and pyridine. The acetate was extracted with ether, and the extract was washed successively with water and a saturated sodium chloride solution. After removal of the solvent under reduced pressure, the residue was crystallized from acetone/methanol.
100 mg of 4,4-dimethyl-5a-cholesta-8,14-dien-3~-ol .acetate was dissolved in 20 ml of absolute ethanol, and the solution was added to 100 mg of freshly prepared activated Raney Nickel which had been previously hydrogenated for 1 h. Hydrogenation at atmospheric pres- sure was continued for 4 h. The reaction mixture was filtered, and the filtrate was evaporated to dryness under reduced pressure. The residue was subjected to TLC on 10% silver nitrate-Silica Gel G plates, and the band corresponding to 4,4-dimethyl-5a-cholest-8-en- 38-01 acetate was isolated and crystallized from ether/methanol (46 mg). This material softens a t 110 "C, starts melting at 115 "C with clearing at 120 "C (m.p. (literature) (19) 122-125 "C). The resulting product was saponified with lithium aluminum hydride, and after normal workup, the product was crystallized from chloroform/meth- anol. The crystallized product showed two peaks upon gas chroma- tography-mass spectroscopy analysis both with expected molecular ions of 414 a.m.u.
The sterols in the mixture were resolved by normal phase HPLC (Zorbax-Si, 10 mm X 25 cm, E. I. DuPont, Wilmington, DE) employ- ing hexane/tetrahydrofuran/propanol-2 (95:5:0.25) as the mobile phase. The isolated fractions corresponded to 4,4-dimethyl-5a-cho- lest-8(14)-en-3@-01 and 4,4-dimethy1-5a-cholest-8-en-3@-01. Charac- terization of the recovered A'-monoene showed m.p. 158-160 "C; m.p. (literature) (17, 18) 159-161 "C. High-resolution/mass-spectral anal- ysis indicated a precise mass of 414.3793 (calculated for CmHWO: 414.3862). Prominent ions in the high-mass region of the spectrum were 414 (40%, M), 399 (16%, M-CH3), 381 ( l l%, M-CH3-HzO), 301
19), and 3.25 (m, lH, C-3-aH).
of the method described by Adkins and Pavlic (20). 2 g of 44- 4,4-Dimethy1-5a-cholest-7-en-3j3-01 was synthesized by adaptation
dimethylcholesta-5,7-dien-3~-01 acetate were dissolved in 120 ml of absolute ethanol, and the solution was shaken with 2 g of freshly prepared Raney Nickel (activity W-4) a t 50 "C and 45 pounds of hydrogen pressure for 12 h. After removal of the catalyst, the solvent was evaporated and the recovered solid residue was crystallized from acetone/methanol to give 4,4-dimethyl-5a-cholest-7-en-3~-01 acetate,
(12%, M-CSH17). NMR (360 MHz): 0.62 (s,3H, C-lS), 0.86 (s,3H, C-
m.p. 138-140 "C; [ a ] D +16.5 f 0.8 "C (C, 1.0) m.p. (literature) (18)
182CH3), 2.06 (s,3H, CHs of acetoxy function); 4.57 (m, lH, C-3-aH) 5.22 (m, lH, C-7-H).
1 g of 4,4-dimethyl-5a-cholest-7-en-3j3-01 acetate was dissolved in 60 ml of anhydrous ether, and the solution was added to a slurry of lithium aluminum hydride (400 mg) in 15 ml of anhydrous ether. The suspension was heated under reflux for 1 h (nitrogen atmosphere), and after the usual workup, the resulting residue was crystallized from chloroform/methanol to give the desired alcohol (SO% yield), m.p. 140-142 "C; [ a ] D -2.4 f 0.84 "C (C, 1.0) m.p. (literature) (18) 145-147 "C; [a], + 5 "C (C, 1.09). High-resolution/mass-spectral anal- ysis showed a precise mass of 414.3843 (calculated for CzSHwO: 414.3862). Prominent ions in the high-mass region of the spectrum were: 414 (loo%, M), 399 (22%, M-CHs), 396 (9%, M-HzO), 381 (13%,
134-136 "C; [ a ] D +25 "C (C, 1.05)). NMR (360 MHz): 0.52 (8,3H, C-
M-CHa-HzO), 353 (3%, M-HzO-C3H7), 301 (9%, M-C&I17), 283 (56%, M-HZO-CeH17), 261 (15%, M- HzO-CloHla). NMR (360 MHz): 0.53 (8,
3H, C-18-CH3), 3.20 (m, lH, C-3-aH), 5.25 (m, lH, C-7-H). Both 4,4-dimethyl-5a-cholest-8-en-3@-01 and 4,4-dimethyl-5a-cho-
lest-7-en-38-01 exhibited single peaks on HPLC and GLC: rrt = 1.412 and 1.493 for 8-ene and 1.334 and 1.723 for 7-ene, respectively.
Synthesis of Other Sterol Substrates The methods for the preparation of 5a-cholesta-8,14-dien-38-01
and 5a-cholesta-7,14-dien-3B-o1 were those previously described by others (21, 22). These sterols were used as alternate substrates for 14-reductase. Each sterol was judged homogeneous on GLC; rrt = 1.084 and 1.062, respectively, for the 8,14- and 7,14-dien, respectively. Mass-spectral analysis, ultraviolet spectra, and NMR were consistent with literature values.
7-Dehydrocholesterol was purified as substrate for steroid 5,7-diene 7-reductase. High- and low-resolution/mass-spectral analysis and NMR data were as expected. HPLC and GLC relative retention times were 0.8109 and 1.167, respectively. For assays of both Au-sterol 24- reductase and A' + A7-sterol isomerase, zymosterol, 5a-cholesta- 8,24-dien-38-01, was isolated as described previously (23). In addition to the purification described in Ref. 23, an additional purification step on preparative TLC was carried out as described by Ditullio et al. (24). Expected physical constants were observed. Purity of >98% was demonstrated on GLC, rrt = 1.289. Pure standards of each enzymic reaction product, 5a-cholest-8-en-3~-o1(24-reductase), were prepared and analyzed for purity on GLC, rrt = 1.422 and 1.071, respectively. 4,4-Dimethyl-Sa-cholest-7-en-3-one was prepared by Jones oxida-
tion (25) for use as substrate for the NADPH-dependent 3-ketosteroid reductase of cholesterol biosynthesis (26). High-resolution/mass- spectral analysis showed precise mass of 412.3339 (calculated for CBHBO: 412.3342) and IR spectroscopy showed the characteristic peak at 1705 cm", thus agreeing with expected oxidation of the 38- hydroxyl group to a ketone.
For assay of 4-methyl sterol oxidase [30, 31-"CH3]4,4-dimethyl- 5a-cholest-7-en-3@-01 was prepared as described previously (27). The labeled sterol (1.05 X lo7 dpm/mg) was shown to be homogenous on GLC and HPLC. 3~-Hydroxy-4,4-dimethyl-5a-cholest-8(14)-en-l5-one was pre-
pared to determine if the 15-ketone might inhibit the 14-reductase (see below). The procedure was that previously described by Wood- ward et al. (28, 29). Ultraviolet spectra agreed with literature values and NMR spectra were as expected. High-resolution/mass-spectral analysis of the compound showed a precise mass of 428.3645 (calcu- lated for CmH,02: 428.3654). Prominent ions in the high-mass region of the spectrum were 428 (loo%, M+), 413 (16.8%, "CHI), 410 (23.7%, M-HzO), and 395 (55.6%, M-CH3-H20).
Preparation of Sterol Substrate Suspensions Sterols were suspended in the assay buffer solution with the aid of
the non-ionic detergent, Triton WR-1339, to a final ratio of 751 (detergentxterol, w/w). To prepare the suspensions, warm buffer was added to solutions of substrate and detergent in acetone. The acetone was evaporated. All suspensions were clear and stable upon storage at -80 "C.
Assay of A7*"-Sterol A"-Reductase 60 nmol of 4,4-dimethy1-5a-cholesta-7,14-dien-3j3-01 was added to
an assay mixture (total volume 1.0 ml) containing 2 mg of microsomal protein, 2 mM NADPH, and 25 mg of glucose plus 20 units of glucose oxidase with preincubations under nitrogen at 37 'C for 4 min unless

13416 Sterol 14-Reductase
otherwise specified to establish anaerobic conditions. All buffers (Buffer A) used for incubation had been equilibrated with nitrogen, and nitrogen was exchanged for air in all sealed reaction flasks prior to the start of incubations. Incubation of the complete mixture was carried out anaerobically in sealed flasks for 10 min at 37 "C unless otherwise indicated. Incubations were terminated by the addition of 1 ml of ethanolic KOH followed by heating under reflux for 10 min. Sterols were extracted four times with 4 ml of petroleum ether, and the solvent was evaporated to dryness under a nitrogen stream. The resulting residue was dissolved in 200-500 pl of n-hexane for quan- titation by GLC at high sensitive attenuation. Relative retention times ( t o cholesterol standard) were 1.565 (4,4-dimethyl-5a-cholesta- 7,14-dien-3@-01) and 1.723 (4,4-dimethyl-5a-cholest-7-en-3j3-ol), re- spectively. HPLC (reverse phase) was utilized to analyze other iso- meric sterol substrates in the substrate specificity study (Table 11) in addition to the GLC assay. The extents of enzymic activity were calculated from the relative amounts of diene substrate and monwne product in incubated samples that were compared to unincubated controls. Anaerobic conditions prevented other transformations of sterols in the incubation mixture. Throughout this study, the only chromatographically detectable endogeneous sterol of consequence in 2 mg of microsomal protein was cholesterol. Furthermore, there was only a trace of endogeneous cholesterol detected in the partially purified 14-reductase (see below).
Assay of AK*7-Steroid 7"Reductase The assay was the same as for the 14-reductase with 100 nmol of
7-dehydrocholesterol as substrate. The assay contained 1 mg of microsomal protein, 10 mM reduced glutathione, 10 mM nicotinamide, and 4 mM MgCl, in 1.0 ml of Buffer A. Incubation was carried out for 30 min under anaerobic conditions. The extent of reduction was calculated by the rate of disappearance of UV-detectable substrate on HPLC.
Assay of 3-Ketosteroid Reductase Previously established incubation conditions were used (26), with
GLC detection of enzymic reaction products. Relative retention times on GLC of 4,4-dimethyl-5a-cholest-7-en-3-one and 4,4-dimethyl-5a- cholest-7-en-38-01 uersus cholesterol were 1.584 and 1.723, respec- tively.
Assay of Sterol 24-Reductase 100 nmol of zymosterol, 5a-cholesta-8,24-dien-3j3-01 that was sus-
pended in buffer with the aid of Triton WR-1339, were added to a reaction mixture (total volume 1 ml) containing 3 mg of microsomal protein, 2 mM NADPH, 10 mM GSH, 10 mM nicotinamide, 4 mM MgCl,, and 10 p~ AY-9944 in 0.1 M Tris.HC1 buffer, pH 6.2 (at 37 "C), including 20% (v/v) glycerol. The reaction was allowed to proceed under anaerobic conditions for 30 min at 37 "C after temper- ature equilibration and enzymic removal of dissolved oxygen. Reac- tions were terminated by the addition of 1 ml of ethanolic KOH and workup was as described above. Quantitation was performed by GLC analysis in which 5a-cholest-8-en-3j3-01, the product, was easily sep- arated from zymosbrol. AY-9944 is well resolved from sterols in GLC separation (relative retention time to cholesterol standard was 2.0 at 260 "C).
Assay of Sterol 8-Isomerase and 4-Methyl Sterol Oxidase
scribed in Refs. 23 and 30. Gas-liquid chromatography was utilized Assays of these two enzymes were carried out as previously de-
to determine the amounts of isomerase enzyme product, 5a-cholesta- 7,24-dien-3@-01. Methyl sterol oxidase activity was coupled to the NAD+-dependent la-oic acid decarboxylase (27) for ease of assay in the absence of NADPH resulting in accumulation of 3-ketosteroid (also detected by GLC).
Other Assays Assays of A'-sterol 5-desaturase, cytochrome P-450, NADPH cy-
tochrome P-450 reductase, and NADH-cytochrome b, reductase were performed exactly as previously described (30, 31). Lowry assay (32) was used for all protein determinations employing bovine serum albumin as a standard. Sodium dodecyl sulfate (l%, w/v) was added to the alkaline carbonate solution for assay of protein in solutions containing detergents.
Solubilization and Purification of the NADPH-dependent Sterol 14-Reductase
Step I: Solubilization of the Membrane-bound Enzyme-All proce- dures were conducted at 4 "C mless otherwise stated. Washed micro- somes were suspended to a protein concentration of 20 mg/ml in Buffer A. 4% (w/v) solutions of octyl-j3-D-glucopyranoside (octylglu- coside) and sodium taurodeoxycholate were made fresh with cold Buffer A, and each detergent solution was added dropwise to the protein suspension such that the final detergent ratios, with respect to protein, were 1.51 (octylglucoside) and 0.51 (taurodeoxycholate). This procedure was conducted at ice temperature with gentle stirring which was continued for 30 min following the final addition of detergent solution. Additional buffer solution was added to adjust the final octylglucoside concentration to approximately 1.0% (w/v) before centrifugation. A clear supernatant fraction (Sl) was obtained by centrifugation at 105,000 X g for 1 h. This procedure generally resulted in >90% solubilization of all membrane-bound proteins and total recovery of 14-reductase activity in the supernatant fraction. For enzymic assay, a small aliquot of S1 was diluted with fresh Buffer A (concentration, 2-3 mg/ml) to reduce slightly adverse detergent ef- fects. (Details of development of this solubilization protocol that was first applied successfully to the steroid 8 + 7-ene isomerase will be reported later upon completion of the investigation of that enzyme of sterol biosynthesis.)
Step XI: Polyethylene Glycol 3000 Precipitation of Solubilized En- zyme-A fresh 40% (w/v) solution of polyethylene glycol 3000 (M, = 3000-3700) was prepared in Buffer A. The pH of the solution was adjusted to 7.4 with 5 M phosphoric acid. An appropriate volume of cold PEG solution was added dropwise to the S1 fraction (protein concentration adjusted to 6-8 mg/ml) and mixed well with gentle stirring in an ice bath. After the final addition, the protein suspension was stirred on ice for 2 h and then centrifuged to sediment the precipitated protein (35,000 X g for 15 rnin). In this study, PEG precipitates were isolated from three fractions: P-A, 0-8%; P-B, 8- 16%; P-C, 16-23%; the final 16-23% fraction was necessarily obtained by sedimenting at 105,000 X g for 1 h. The 8-16% (P-B) fraction was used for enrichment of the sterol 14-reductase. Enzymic activity was measured on a small aliquot of each PEG fraction which was SUS- pended in fresh Buffer A.-
SteD III: DEAE-SeDhacel Column Chromatomaohv-DEAE-Se- phacei resins were exhaustively washed with Bufier B il0 mM potas- sium phosphate, pH 7.9, containing 1 mM dithiothreitol, 0.5 mM EDTA, and 20% glycerol) in a large column until the conductivity and pH detected for the eluent was the same as the starting buffer. The washed gel was packed into a column (2.5 X 32 cm) and equili- brated with 1.5 liters of Buffer C (0.2% octylglucoside, 0.05% sodium taurodeoxycholate in Buffer B). Protein solution obtained by sus- pending PEG fractions (8-16% precipitate) in Buffer C was applied to the gel. The column was washed with 320 ml of equilibration buffer and 500 ml of Buffer D (0.1 M KC1 in Buffer C). The proteins tightly bound to the gel were eluted with a 600-ml linear salt gradient that was composed of equal volumes of Buffers D and E (0.1 M potassium phosphate, pH 7.0, including 0.6 M KC1 in Buffer C). The gradient was run at a flow rate of 0.5 ml/min, and 10-min fractions were collected; protein in the eluate was monitored at 280 nm. The enzymic activity was measured anaerobically on a dialyzed aliquot (against 200 X Buffer B, for 2 h) of each fraction.
Step IV: Hydroxylapatite Column Chromatography-Hydroxylapa- tite resin (Bio-Gel-HT, Bio-Rad) was suspended in Buffer F (5 mM potassium phosphate, pH 7.0, containing 1 mM dithiothreitol, 0.5 mM EDTA, and 20% glycerol) which was prepared with double-distilled, deionized water. The resins were packed into a small column (1 X 12 cm) with a final bed volume of 8 ml. The column was washed and equilibrated with 10 volumes of Buffer F and Buffer G (0.2% octyl- glucoside and 0.05% sodium taurodeoxycholate in Buffer F) in se- quence. The pooled, active fractions obtained from the DEAE-Sepha- cel column were concentrated on a Diaflo YM-10 membrane (Amicon Corp., Lexington, MA) and subsequently filtered through two pre- packed Sephadex G-25 columns (PD-10, Pharmacia Fine Chemicals Co., Uppsala, Sweden) which had been equilibrated with the same buffer to remove dissolved salts. The filtrate was dialyzed against 200 volumes of the equilibration buffer for 3 h. The resulting samples were applied to the gel and washed with 5 column volumes of equili- bration buffer containing 20 mM phosphate (Buffer H). The proteins were eluted with a 500-ml linear gradient of Buffers H and J (0.2 M phosphate in Buffer G) followed by 200 ml of a second gradient composed of Buffers J and K (0.4 M phosphate in Buffer G). The

Sterol 14-Reductase 13417
eluate was monitored at 280 nm and 4-ml fractions were collected. A small aliquot of each fraction (protein: 0.2 mg) was assayed for sterol 14-reductase activity following a 3-h dialysis against 200 vol- umes of Buffer B.
RESULTS AND DISCUSSION
Localization of the Sterol 14-Reductase-The enzyme that catalyzes reduction of the 14-double bond of the conjugated ~ i~* '~-s te ro l diene is bound to the endoplasmic reticulum and is isolated with the microsomal pellets of rat liver homoge- nates (8, 9). Based on activity present in liver homogenates, approximately 90% of 14-reductase was found in the micro- somal pellets when the enzymic activity was examined in the microsomal pellets, 105,000 x g supernatant ( S d , and mi- tochondria. No activity was detected in the Slm fraction. Based on this observation, attempts to characterize and purify the membrane-bound 14-reductase were made using only washed microsomal pellets that were sedimented at 105,000 x €!.
Properties of Microsomal-bound Sterol 14-Reductase-As shown in Table I, the microsomal membrane-bound enzyme that catalyzes reduction of the 14-double bond of 4,4-di- rnethyl-5a-cholesta-7,14-dien-3@-01 requires NADPH for 14- double bond reduction. Although the reaction occurs under oxygen, anaerobic conditions yielded maximal rates of con- version without any 02-dependent reactions occurring simul- taneously. NADH, under either aerobic or anaerobic condi- tions, only poorly supported reduction. All subsequent exper- iments were carried out with 2 mM NADPH under anaerobic conditions unless otherwise indicated.
The enzyme was also sensitive to sulfhydryl-binding agents such as N-ethylmaleimide and HgC12 as observed for other nonoxidative cholesterol synthetic enzymes such as the A24- reductase of desmosterol (33) and the As-isomerase of zy-
TABLE I Properties of microsomal membrane-bound sterol 14-reductase
The complete system contained 60 p M 4,4-dimethyl-5a-cholesta- 7,14-dien-3@-01, 2 mg of microsomal protein, 2 mM NADPH, 25 mg of glucose, 20 units of glucose oxidase (Type V) in 0.1 M potassium phosphate buffer, pH 7.4, including 20% glycerol, 0.5 mM EDTA, and 1 mM reduced glutathione (unless omitted as indicated). Incubations were conducted anaerobically at 37 "C for 10 min. Values represent the means and S.D. of six samples obtained from three separate experiments.
Incubation conditions
specific Relative activity reaction
rates nmol/min f mg
protein %
Complete system 0.85 f 0.04 100
Substitute NADH, NZ 0.069 -C 0.004 8.1 Substitute NADH, Oz 0.004 f 0.001 <0.5 +oze 0.61 f 0.058 72 +N-Ethylmaleimideb 0.40 f 0.047 47 N-Ethylmaleimide + GSH' 0.82 f 0.031 96
-NADPH (+Nz, fO2) 0 0
+HgC1zd 0.008 f 0.001 4 . 0 HgC12 + GSH' 0.76 f 0.02 90 In Tris . HCl buffer' 0.61 -C 0.100 72 Mg2" 0.80 f 0.017 94 Ca2+g 0.82 f 0.042 96.5 Glucose and glucose oxidase were omitted from the incubation
5 mM N-ethylmaleimide, and the buffer was free of GSH.
3 mM HgClZ.
mixture which was prepared in air-saturated phosphate buffer.
e 10 mM GSH was added before N-ethylmaleimide.
e 10 mM GSH was added before HgC12. '0.1 M Tris. HCl buffer (pH 7.4 at 37 "C) which contains the same
constituents as in phosphate buffer. Concentration of the metal ions tested was 4 mM.
O u 1 2 3 4 5 7.5 10 u 5102030405060 90 120
Protein (ma) Incubation Time (min)
FIG. 1. Effect of incubation length and amount of micro- somal protein. A, amount of microsomal protein was varied with a 10-min incubation; B, length of incubation was varied with 2 mg of protein. Assays were conducted anaerobically at 37 "C under condi- tions as described under "Experimental Procedures." The results are the averages of duplicate samples from two separate experiments.
mosterol (23). For both sulfhydryl-binding agents the extent of inactivation of enzyme was negligible in the presence of 10 mM glutathione. Replacement of the phosphate buffer with the Tris-HCl buffer (0.1 M, pH 7.4, at 37 "C) resulted in approximately a 27% decrease in catalytic activity which was not restored by the addition of potassium ion (KC1) at various concentrations. It was observed, however, that the other NADPH-dependent reductases, such as the 24-reductase of A8*24-diene (zymosterol), the 3-ketosteroid reductase, and the A7-reductase were not affected by the Tris. HC1 buffer treat- ment.2 Effects of additions of various metal ions were not extensively examined, but neither M p n o r Ca2+ (0.5-20 mM were tested) stimulated activity.
With incubation conditions as shown in Table I, the en- zymic velocity was directly proportional to the amount of protein added up to 2 mg (Fig. 1). Although addition of larger amounts of microsomal protein resulted in more extensive conversion, initial velocity conditions were not maintained (Fig. lA). Similarly, when 2 mg of microsomes were incubated, the rate was constant for almost 20 min (Fig. 1B). For the 14- double bond reductase, there was no significant difference between the microsomal activity which was obtained from animals that were fed either a regular diet or a diet that contained cholestyramine (cholestyramine is a nonabsorbable anion exchange resin that was obtained from Mead Johnson Co., Evansville, IN).
Somewhat in contrast to other microsomal enzymes of cholesterol biosynthesis (13, 23), the microsomal-bound 14- reductase appeared to have a relatively broad pH profile in the acidic region, whereas drastically decreased activity was observed with only moderate elevation of pH above 7.8 (Fig. 2). Even among other nonoxidative enzymes of cholesterol synthesis these results are in sharp contrast to that of 8- isomerase (23), which is also an anaerobic, microsomal en- zyme. In subsequent experiments, the pH was strongly buff- ered at 7.4.
With the conditions established for measurement of initial velocities, the apparent K,,, and Vmar of the membrane-bound 14-reductase were determined with 4,4-dimethyl-5a-cholesta- 7,14-dien-3@-01. KA and V- from the Lineweaver-Burk re- ciprocal plot of A14-reductase were 29.4 pM and 1.13 nmol/ min/mg of protein (Fig. 3). Substrate inhibition was observed at concentrations in excess of 100 p ~ . KA for NADPH was
Y.-K. Paik, J. M. Trzaskos, A. Shafiee, and J. L. Gaylor, unpub- lished observations.
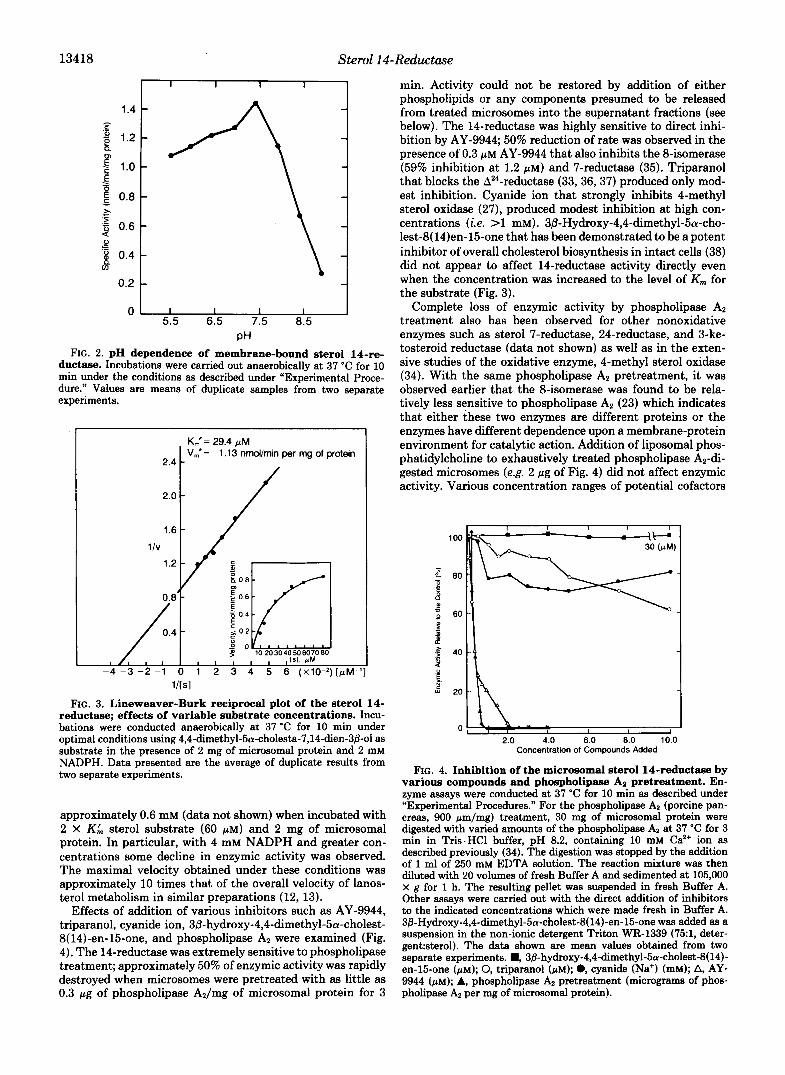
13418 Sterol 14-Reductase
0 1 I 1 I I I 5.5 6.5 7.5 8.5
PH FIG. 2. pH dependence of membrane-bound sterol 14-re-
ductase. Incubations were carried out anaerobically at 37 "C for 10 min under the conditions as described under "Experimental Proce- dure." Values are means of duplicate samples from two separate experiments.
2.4
2.0
1.6
l l v
1.2
0.8
, /A -4 -3 -2 -1
K,'= 29.4 pM 4:= 1 . 1 3 nmol/min per mg of protein
/ $ 0 6
2 0 4
0.2
B o >" 1020304050607080
1 2 3 4 5 6 ( x ~ O - ' ) [pM" IS I . CM
1lIsl
FIG. 3. Lineweaver-Burk reciprocal plot of the sterol 14- reductase; effects of variable substrate concentrations. Incu- bations were conducted anaerobically at 37 "C for 10 min under optimal conditions using 4,4-dimethyl-5a-cholesta-7,14-dien-3~-ol as substrate in the presence of 2 mg of microsomal protein and 2 mM NADPH. Data presented are the average of duplicate results from two separate experiments.
approximately 0.6 mM (data not shown) when incubated with 2 X KA sterol substrate (60 p ~ ) and 2 mg of microsomal protein. In particular, with 4 mM NADPH and greater con- centrations some decline in enzymic activity was observed. The maximal velocity obtained under these conditions was approximately 10 times that of the overall velocity of lanos- terol metabolism in similar preparations (12, 13).
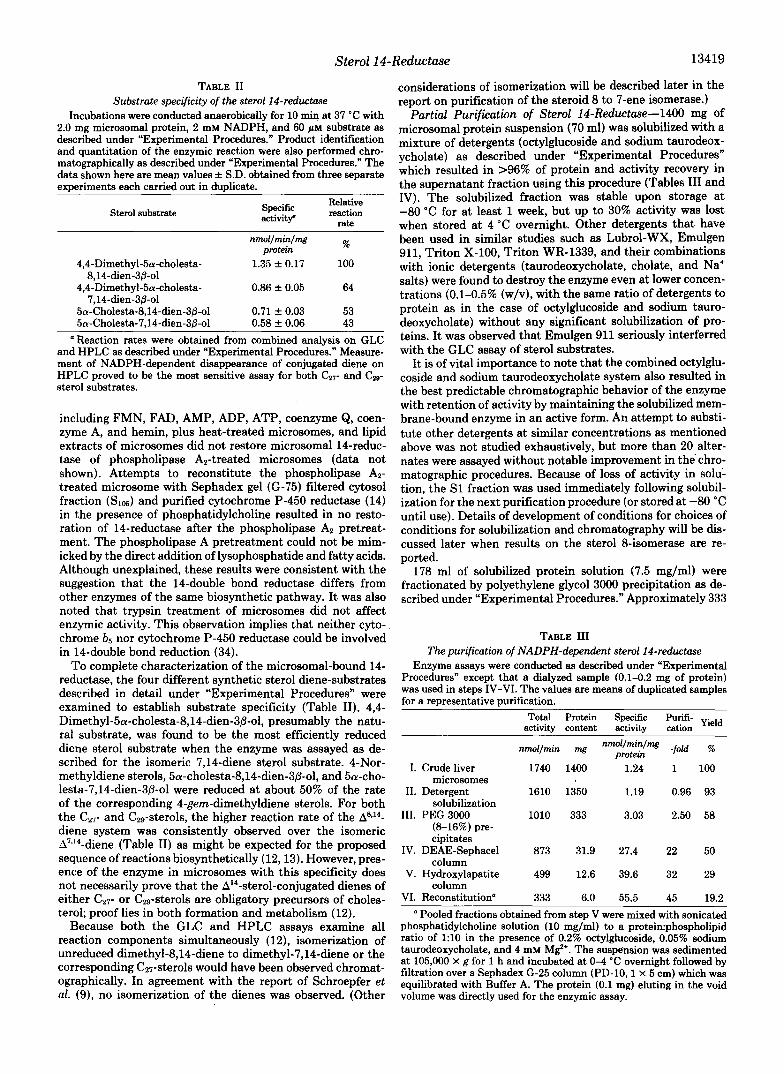
Effects of addition of various inhibitors such as AY-9944, triparanol, cyanide ion, 3~-hydroxy-4,4-dimethyl-5a-cholest- 8(14)-en-15-one, and phospholipase AP were examined (Fig. 4). The 14-reductase was extremely sensitive to phospholipase treatment; approximately 50% of enzymic activity was rapidly destroyed when microsomes were pretreated with as little as 0.3 pg of phospholipase AP/mg of microsomal protein for 3
min. Activity could not be restored by addition of either phospholipids or any components presumed to be released from treated microsomes into the supernatant fractions (see below). The 14-reductase was highly sensitive to direct inhi- bition by AY-9944; 50% reduction of rate was observed in the presence of 0.3 p~ AY-9944 that also inhibits the 8-isomerase (59% inhibition at 1.2 p ~ ) and 7-reductase (35). Triparanol that blocks the AZ4-reductase (33,36,37) produced only mod- est inhibition. Cyanide ion that strongly inhibits 4-methyl sterol oxidase (27), produced modest inhibition at high con- centrations ( i e . >1 mM). 3@-Hydroxy-4,4-dimethyl-5a-cho- lest-8(14)en-15-one that has been demonstrated to be a potent inhibitor of overall cholesterol biosynthesis in intact cells (38) did not appear to affect 14-reductase activity directly even when the concentration was increased to the level of K , for the substrate (Fig. 3).
Complete loss of enzymic activity by phospholipase Az treatment also has been observed for other nonoxidative enzymes such as sterol 7-reductase, 24-reductase, and 3-ke- tosteroid reductase (data not shown) as well as in the exten- sive studies of the oxidative enzyme, 4-methyl sterol oxidase (34). With the same phospholipase AP pretreatment, it was observed earlier that the 8-isomerase was found to be rela- tively less sensitive to phospholipase A, (23) which indicates that either these two enzymes are different proteins or the enzymes have different dependence upon a membrane-protein environment for catalytic action. Addition of liposomal phos- phatidylcholine to exhaustively treated phospholipase Az-di- gested microsomes (e.g. 2 pg of Fig. 4) did not affect enzymic activity. Various concentration ranges of potential cofactors
2.0 4.0 6.0 8.0 10.0 Concentration of Compounds Added
FIG. 4. Inhibition of the microsomal sterol 14-reductase by various compounds and phospholipase A2 pretreatment. En- zyme assays were conducted at 37 "C for 10 min as described under "Experimental Procedures." For the phospholipase A, (porcine pan- creas, 900 pm/mg) treatment, 30 mg of microsomal protein were digested with varied amounts of the phospholipase A, at 37 "C for 3 min in Tris.HC1 buffer, pH 8.2, containing 10 mM Ca2+ ion as described previously (34). The digestion was stopped by the addition of 1 ml of 250 mM EDTA solution. The reaction mixture was then diluted with 20 volumes of fresh Buffer A and sedimented at 105,000 X g for 1 h. The resulting pellet was suspended in fresh Buffer A. Other assays were carried out with the direct addition of inhibitors to the indicated concentrations which were made fresh in Buffer A. 3~-Hydroxy-4,4-dimethyl-5a-cholest-8(14)-en-15-one was added as a suspension in the non-ionic detergent Triton WR-1339 (75:1, deter- genksterol). The data shown are mean values obtained from two separate experiments. W, 3~-hydroxy-4,4-dimethyl-5a-cholest-8(14)- en-15-one ( p ~ ) ; 0, triparanol ( p ~ ) ; 0, cyanide (Na') (mM); A, AY- 9944 ( p ~ ) ; A, phospholipase AP pretreatment (micrograms of phos- pholipase A2 per mg of microsomal protein).
Sterol 14-Reductase 13419
TABLE I1 Substrate specificity of the sterol 14-reductase
Incubations were conducted anaerobically for 10 min at 37 “C with 2.0 mg microsomal protein, 2 mM NADPH, and 60 FM substrate as described under “Experimental Procedures.’’ Product identification and quantitation of the enzymic reaction were also performed chro- matographically as described under ”Experimental Procedures.” The data shown here are mean values & S.D. obtained from three separate experiments each carried out in duplicate.
Sterol substrate specific Relative
activity” reaction
rate
nmol/min/mg % protein
4,4-Dimethyl-5a-cholesta- 1.35 & 0.17 100
4,4-Dimethyl-5a-cholesta- 0.86 & 0.05 64
5a-Cholesta-8,14-dien-3j3-01 0.71 & 0.03 53 5a-Cholesta-7,14-dien-3j3-01 0.58 & 0.06 43
8,14-dien-3@-01
7,14-dien-3/3-01
a Reaction rates were obtained from combined analysis on GLC and HPLC as described under “Experimental Procedures.” Measure- ment of NADPH-dependent disappearance of conjugated diene on HPLC proved to be the most sensitive assay for both CZ7- and Cm- sterol substrates.
including FMN, FAD, AMP, ADP, ATP, coenzyme Q, coen- zyme A, and hemin, plus heat-treated microsomes, and lipid extracts of microsomes did not restore microsomal 14-reduc- tase of phospholipase A,-treated microsomes (data not shown). Attempts to reconstitute the phospholipase AZ- treated microsome with Sephadex gel (G-75) filtered cytosol fraction (SI,) and purified cytochrome P-450 reductase (14) in the presence of phosphatidylcholine resulted in no resto- ration of 14-reductase after the phospholipase A2 pretreat- ment. The phospholipase A pretreatment could not be mim- icked by the direct addition of lysophosphatide and fatty acids. Although unexplained, these results were consistent with the suggestion that the 14-double bond reductase differs from other enzymes of the same biosynthetic pathway. It was also noted that trypsin treatment of microsomes did not affect enzymic activity. This observation implies that neither cyto- chrome b5 nor cytochrome P-450 reductase could be involved in 14-double bond reduction (34).
To complete characterization of the microsomal-bound 14- reductase, the four different synthetic sterol diene-substrates described in detail under “Experimental Procedures” were examined to establish substrate specificity (Table 11). 4,4- Dimethyl-5a-cholesta-8,14-dien-3~-ol, presumably the natu- ral substrate, was found to be the most efficiently reduced diene sterol substrate when the enzyme was assayed as de- scribed for the isomeric 7,14-diene sterol substrate. 4-Nor- methyldiene sterols, 5a-cholesta-8,14-dien-3/3-01, and 5a-cho- lesta-7,14-dien-3/3-01 were reduced at about 50% of the rate of the corresponding 4-gem-dimethyldiene sterols. For both the C27- and Cz9-sterols, the higher reaction rate of the A’*’’- diene system was consistently observed over the isomeric A7*14-diene (Table 11) as might be expected for the proposed sequence of reactions biosynthetically (12,13). However, pres- ence of the enzyme in microsomes with this specificity does not necessarily prove that the A14-sterol-conjugated dienes of either c27- or Cm-sterols are obligatory precursors of choles- terol; proof lies in both formation and metabolism (12).
Because both the GLC and HPLC assays examine all reaction components simultaneously ( E ) , isomerization of unreduced dimethyl-8,14-diene to dimethyl-7,14-diene or the corresponding Cz7-sterols would have been observed chromat- ographically. In agreement with the report of Schroepfer et al. (9), no isomerization of the dienes was observed. (Other
considerations of isomerization will be described later in the report on purification of the steroid 8 to 7-ene isomerase.)
Partial Purification of Sterol 14-Reductase-1400 mg of microsomal protein suspension (70 ml) was solubilized with a mixture of detergents (octylglucoside and sodium taurodeox- ycholate) as described under “Experimental Procedures” which resulted in >96% of protein and activity recovery in the supernatant fraction using this procedure (Tables 111 and IV). The solubilized fraction was stable upon storage at -80 “C for at least 1 week, but up to 30% activity was lost when stored at 4 “C overnight. Other detergents that have been used in similar studies such as Lubrol-WX, Emulgen 911, Triton X-100, Triton WR-1339, and their combinations with ionic detergents (taurodeoxycholate, cholate, and Na+ salts) were found to destroy the enzyme even at lower concen- trations (0.1-0.5% (w/v), with the same ratio of detergents to protein as in the case of octylglucoside and sodium tauro- deoxycholate) without any significant solubilization of pro- teins. It was observed that Emulgen 911 seriously interferred with the GLC assay of sterol substrates.
It is of vital importance to note that the combined octylglu- coside and sodium taurodeoxycholate system also resulted in the best predictable chromatographic behavior of the enzyme with retention of activity by maintaining the solubilized mem- brane-bound enzyme in an active form. An attempt to substi- tute other detergents at similar concentrations as mentioned above was not studied exhaustively, but more than 20 alter- nates were assayed without notable improvement in the chro- matographic procedures. Because of loss of activity in solu- tion, the S1 fraction was used immediately following solubil- ization for the next purification procedure (or stored at -80 “C until use). Details of development of conditions for choices of conditions for solubilization and chromatography will be dis- cussed later when results on the sterol 8-isomerase are re- ported.
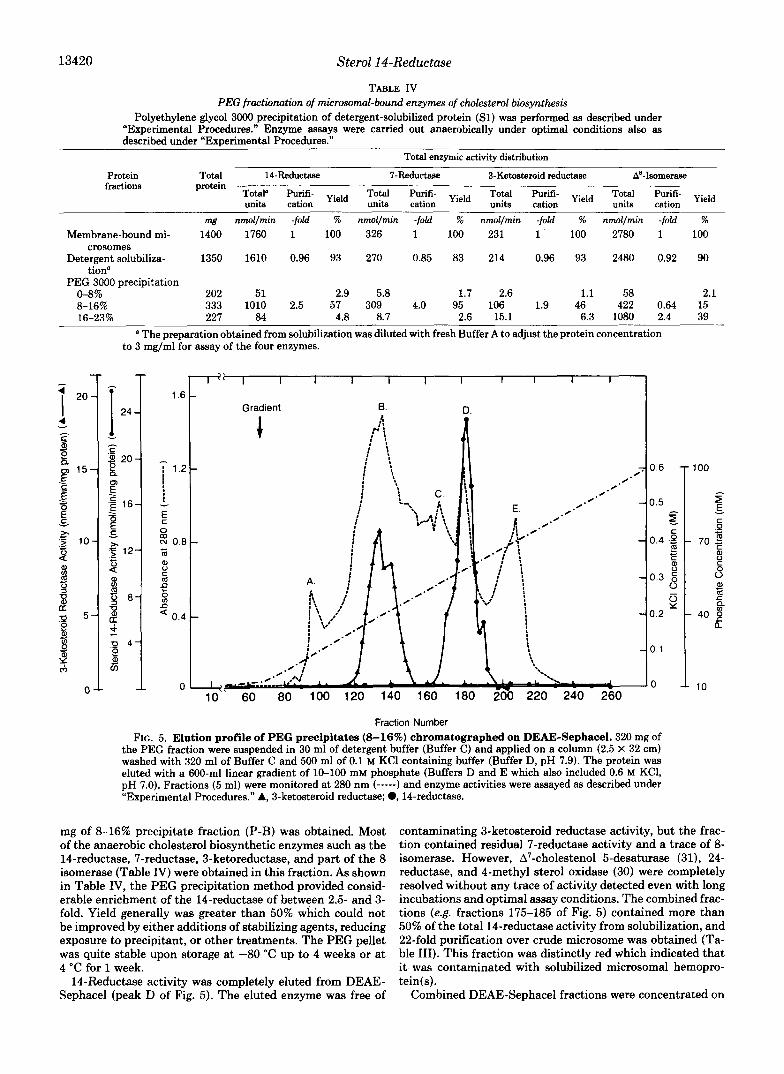
178 ml of solubilized protein solution (7.5 mg/ml) were fractionated by polyethylene glycol 3000 precipitation as de- scribed under “Experimental Procedures.” Approximately 333
TABLE 111 The purification of NADPH-&pendent sterol 14-reductase
Enzyme assays were conducted as described under “Experimental Procedures” except that a dialyzed sample (0.1-0.2 mg of protein) was used in steps IV-VI. The values are means of duplicated samples for a representative purification.
Total Protein Specific Purifi- activity content activity cation
nmollmin mg nmollminlng -fold %
I. Crude liver 1740 1400 1.24 1 100 microsomes
11. Detergent 1610 1350 1.19 0.96 93
111. PEG 3000 solubilization
1010 333 3.03 2.50 58 (8-16%) pre- cipitates
IV. DEAE-Sephacel 873 31.9 27.4 22 50
V. Hydroxylapatite column
499 12.6 39.6 32 29 column
VI. Reconstitution” 333 6.0 55.5 45 19.2 a Pooled fractions obtained from step V were mixed with sonicated
phosphatidylcholine solution (10 mg/ml) to a protein:phospholipid ratio of 1 : lO in the presence of 0.2% octylglucoside, 0.05% sodium taurodeoxycholate, and 4 mM M$+. The suspension was sedimented at 105,000 X g for 1 h and incubated at 0-4 “C overnight followed by filtration over a Sephadex G-25 column (PD-10,l x 5 cm) which was equilibrated with Buffer A. The protein (0.1 mg) eluting in the void volume was directly used for the enzymic assay.
13420 Sterol 14-Reductase
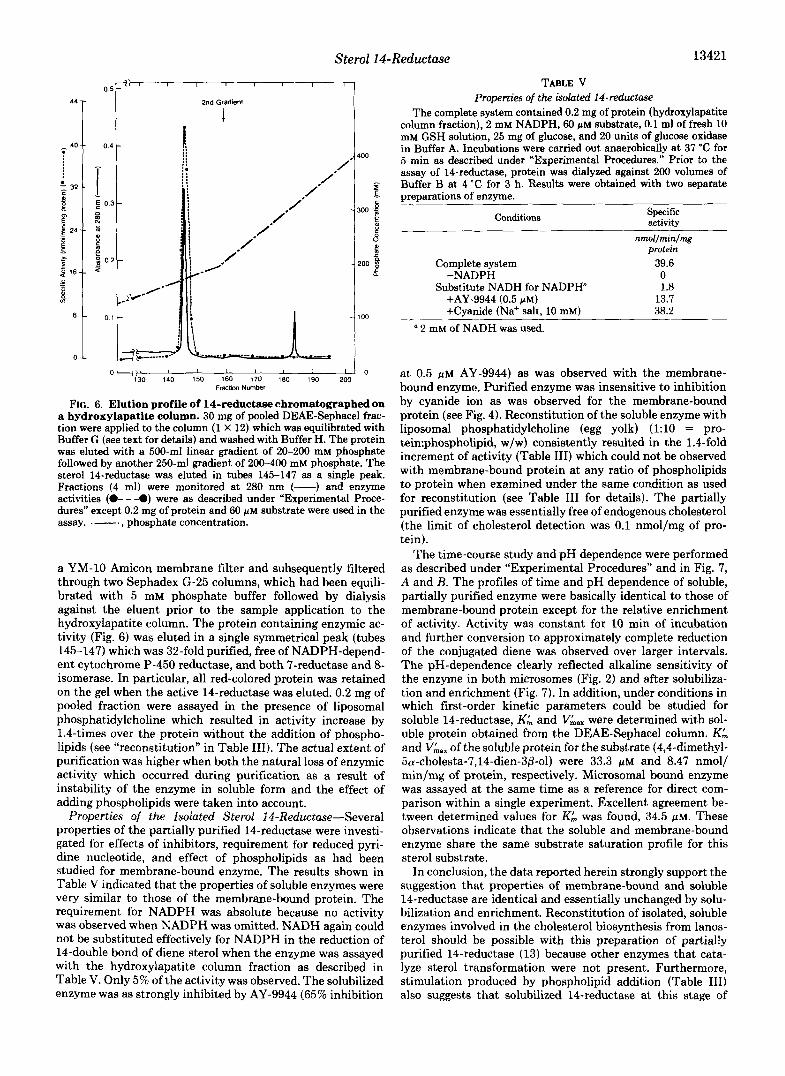
TABLE IV PEG fractionation of microsod-bound enzymes of cholesterol biosynthesis
Polyethylene glycol 3000 precipitation of detergent-solubilized protein (SI) was performed as described under ”Experimental Procedures.” Enzyme assays were carried out anaerobically under optimal conditions also as described under “Experimental Procedures.”
Total enzymic activity distribution
Protein Total Il-FkductaSe 7-R.eductaee fractions
3-Ketosteroid reductase A’-Isomerase protein
Totala Purifi- Yield Total Purifi- Yield Total Purifi- Yield Total Purifi- Yield units cation units cation units cation units cation
mg nmollmin -fold % nmollmin -fold 76 nmol/min -fold % nmollmin -fold % Membrane-bound mi- 1400 1760 1 100 326 1 100 231 1 100 2780 1 100
Detergent solubiliza- 1350 1610 0.96 93 270 0.85 83 214 0.96 93 2480 0.92 90
PEG 3000 precipitation
crosomes
tion”
0-8% 202 51 2.9 5.8 1.7 2.6 1.1 58 2.1 am 333 1010 2.5 57 309 4.0 95 106 1.9 46 422 0.64 15 16-2396 227 84 4.8 8.7 2.6 15.1 6.3 1080 2.4 39
a The oreoaration obtained from solubilization was diluted with fresh Buffer A to adiust the protein concentration to 3 mg/ml for assay of the four enzymes.
. .
1.6
- 1.2
v !
E % 0.8 0
c m a, 0 C m f! 9
a 0.4
T loo
Fraction Number FIG. 5. Elution profile of PEG precipitates (8-16%) chromatographed on DEAE-Sephacel. 320 mg of
the PEG fraction were suspended in 30 ml of detergent buffer (Buffer C) and applied on a column (2.5 X 32 cm) washed with 320 ml of Buffer C and 500 ml of 0.1 M KC1 containing buffer (Buffer D, pH 7.9). The protein was eluted with a 600-ml linear gradient of 10-100 mM phosphate (Buffers D and E which also included 0.6 M KCl, pH 7.0). Fractions (5 ml) were monitored at 280 nm (-----) and enzyme activities were assayed as described under “Experimental Procedures.” A, 3-ketosteroid reductase; 0,14-reductase.
mg of 8-16% precipitate fraction (P-B) was obtained. Most of the anaerobic cholesterol biosynthetic enzymes such as the 14-reductase, 7-reductase, 3-ketoreductase, and part of the 8 isomerase (Table IV) were obtained in this fraction. As shown in Table IV, the PEG precipitation method provided consid- erable enrichment of the 14-reductase of between 2.5- and 3- fold. Yield generally was greater than 50% which could not be improved by either additions of stabilizing agents, reducing exposure to precipitant, or other treatments. The PEG pellet was quite stable upon storage at -80 “C up to 4 weeks or at 4 “C for 1 week.
14-Reductase activity was completely eluted from DEAE- Sephacel (peak D of Fig. 5). The eluted enzyme was free of
contaminating 3-ketosteroid reductase activity, but the frac- tion contained residual 7-reductase activity and a trace of 8- isomerase. However, A7-cholestenol 5-desaturase (31), 24- reductase, and 4-methyl sterol oxidase (30) were completely resolved without any trace of activity detected even with long incubations and optimal assay conditions. The combined frac- tions (e.g. fractions 175-185 of Fig. 5) contained more than 50% of the total 14-reductase activity from solubilization, and 22-fold purification over crude microsome was obtained (Ta- ble 111). This fraction was distinctly red which indicated that it. was contaminated with solubilized microsomal hemopro- tein(s).
Combined DEAE-Sephacel fractions were concentrated on
Sterol 14-Reductase 13421
100
- I -
300 f
m r L
200 3 a
IO0
0
FIG. 6. Elution profile of 14-reductase chromatographed on a hydroxylapatite column. 30 mg of pooled DEAE-Sephacel frac- tion were applied to the column (1 X 12) which was equilibrated with Buffer G (see text for details) and washed with Buffer H. The protein was eluted with a 500-ml linear gradient of 20-200 mM phosphate followed by another 250-ml gradient of 200-400 mM phosphate. The sterol 14-reductase was eluted in tubes 145-147 as a single peak. Fractions (4 ml) were monitored at 280 nm (-) and enzyme activities ( 0 - - - 0 ) were as described under "Experimental Proce- dures" except 0.2 mg of protein and 60 p~ substrate were used in the assay. , phosphate concentration.
a YM-10 Amicon membrane filter and subsequently filtered through two Sephadex G-25 columns, which had been equili- brated with 5 mM phosphate buffer followed by dialysis against the eluent prior to the sample application to the hydroxylapatite column. The protein containing enzymic ac- tivity (Fig. 6) was eluted in a single symmetrical peak (tubes 145-147) which was 32-fold purified, free of NADPH-depend- ent cytochrome P-450 reductase, and both 7-reductase and 8- isomerase. In particular, all red-colored protein was retained on the gel when the active 14-reductase was eluted. 0.2 mg of pooled fraction were assayed in the presence of liposomal phosphatidylcholine which resulted in activity increase by 1.4-times over the protein without the addition of phospho- lipids (see "reconstitution" in Table 111). The actual extent of purification was higher when both the natural loss of enzymic activity which occurred during purification as a result of instability of the enzyme in soluble form and the effect of adding phospholipids were taken into account.
Properties of the Isolated Sterol 14-Reductase-Several properties of the partially purified 14-reductase were investi- gated for effects of inhibitors, requirement for reduced pyri- dine nucleotide, and effect of phospholipids as had been studied for membrane-bound enzyme. The results shown in Table V indicated that the properties of soluble enzymes were very similar to those of the membrane-bound protein. The requirement for NADPH was absolute because no activity was observed when NADPH was omitted. NADH again could not be substituted effectively for NADPH in the reduction of 14-double bond of diene sterol when the enzyme was assayed with the hydroxylapatite column fraction as described in Table V. Only 5% of the activity was observed. The solubilized enzyme was as strongly inhibited by AY-9944 (65% inhibition
TABLE V Properties of the isolated 14-reductase
The complete system contained 0.2 mg of protein (hydroxylapatite column fraction), 2 mM NADPH, 60 p~ substrate, 0.1 ml of fresh 10 mM GSH solution, 25 mg of glucose, and 20 units of glucose oxidase in Buffer A. Incubations were carried out anaerobically at 37 "C for 5 min as described under "Experimental Procedures." Prior to the assay of 14-reductase, protein was dialyzed against 200 volumes of Buffer B at 4 "C for 3 h. Results were obtained with two separate preparations of enzyme.
Conditions Specific activity
nmolfrnin/rng protein
Complete system 39.6
Substitute NADH for NADPH" 1.8
+Cyanide (Na+ salt, 10 mM) 38.2
-NADPH 0
+AY-9944 (0.5 pM) 13.7
a 2 mM of NADH was used.
at 0.5 WM AY-9944) as was observed with the membrane- bound enzyme. Purified enzyme was insensitive to inhibition by cyanide ion as was observed for the membrane-bound protein (see Fig. 4). Reconstitution of the soluble enzyme with liposomal phosphatidylcholine (egg yolk) ( 1 : l O = pro- tein:phospholipid, w / ~ ) consistently resulted in the 1.4-fold increment of activity (Table 111) which could not be observed with membrane-bound protein at any ratio of phospholipids to protein when examined under the same condition as used for reconstitution (see Table I11 for details). The partially purified enzyme was essentially free of endogenous cholesterol (the limit of cholesterol detection was 0.1 nmol/mg of pro- tein).
The time-course study and pH dependence were performed as described under "Experimental Procedures" and in Fig. 7, A and B. The profiles of time and pH dependence of soluble, partially purified enzyme were basically identical to those of membrane-bound protein except for the relative enrichment of activity. Activity was constant for 10 min of incubation and further conversion to approximately complete reduction of the conjugated diene was observed over larger intervals. The pH-dependence clearly reflected alkaline sensitivity of the enzyme in both microsomes (Fig. 2) and after solubiliza- tion and enrichment (Fig. 7). In addition, under conditions in which first-order kinetic parameters could be studied for soluble l.l-reductase, IC,, and V,!,,., were determined with sol- uble protein obtained from the DEAE-Sephacel column. K& and VLaX of the soluble protein for the substrate (4,4-dimethyl- 5a-cholesta-7,14-dien-3&01) were 33.3 PM and 8.47 nmol/ min/mg of protein, respectively. Microsomal bound enzyme was assayed at the same time as a reference for direct com- parison within a single experiment. Excellent agreement be- tween determined values for Kh was found, 34.5 PM. These observations indicate that the soluble and membrane-bound enzyme share the same substrate saturation profile for this sterol substrate.
In conclusion, the data reported herein strongly support the suggestion that properties of membrane-bound and soluble 14-reductase are identical and essentially unchanged by solu- bilization and enrichment. Reconstitution of isolated, soluble enzymes involved in the cholesterol biosynthesis from lanos- terol should be possible with this preparation of partially purified 14-reductase (13) because other enzymes that cata- lyze sterol transformation were not present. Furthermore, stimulation produced by phospholipid addition (Table 111) also suggests that solubilized 14-reductase at this stage of
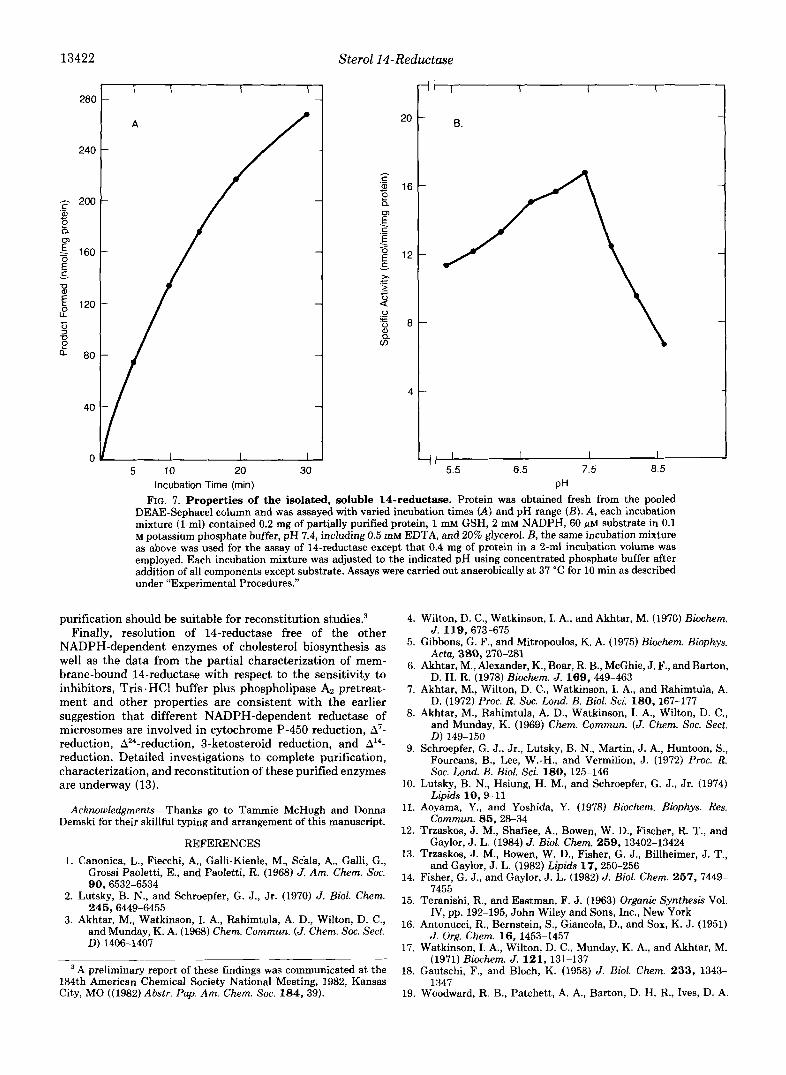
13422
280 lL 240
.- 200
a, 0 c
L
- 160
E
2 E 120
a 80 P
0
v
V aJ
c u 3 V
40
0 5 10 20
Incubation Time (min)
Sterol 14-Reductase
I I 1 I I \
0.
ii I I I I
5.5 6.5 7.5 8.5 PH
FIG. 7. Properties of the isolated, soluble 14-reductase. Protein was obtained fresh from the pooled DEAE-Sephacel column and was assayed with varied incubation times (A) and pH range (B). A, each incubation mixture (1 ml) contained 0.2 mg of partially purified protein, 1 mM GSH, 2 mM NADPH, 60 WM substrate in 0.1 M potassium phosphate buffer, pH 7.4, including 0.5 m M EDTA, and 20% glycerol. B, the same incubation mixture as above was used for the assay of 14-reductase except that 0.4 mg of protein in a 2-ml incubation volume was employed. Each incubation mixture was adjusted to the indicated pH using concentrated phosphate buffer after addition of all components except substrate. Assays were carried out anaerobically at 37 “C for 10 min as described under “Experimental Procedures.”
purification should be suitable for reconstitution studies? Finally, resolution of 14-reductase free of the other
NADPH-dependent enzymes of cholesterol biosynthesis as well as the data from the partial characterization of mem- brane-bound 14-reductase with respect to the sensitivity to inhibitors, Tris . HC1 buffer plus phospholipase Az pretreat- ment and other properties are consistent with the earlier suggestion that different NADPH-dependent reductase of microsomes are involved in cytochrome P-450 reduction, A7- reduction, AZ4-reduction, 3-ketosteroid reduction, and AI4-
reduction. Detailed investigations to complete purification, characterization, and reconstitution of these purified enzymes are underway (13).
Acknowledgments-Thanks go to Tammie McHugh and Donna Demski for their skillful typing and arrangement of this manuscript.
REFERENCES 1. Canonica, L., Fiecchi, A., Galli-Kienle, M., Scala, A., Galli, G.,
Grossi Paoletti, E., and Paoletti, R. (1968) J. Am. Chem. SOC. 90,6532-6534
2. Lutsky, B. N., and Schroepfer, G. J., Jr. (1970) J. Bwl. Chem.
3. Akhtar, M., Watkinson, I. A., Rahimtula, A. D., Wilton, D. C., and Munday, K. A. (1968) Chern. Cornrnun. (J. Chern. SOC. Sect.
245,6449-6455
D) 1406-1407
A preliminary report of these findings was communicated at the
City, MO ((1982) Abstr. Pap. Am. Chem. SOC. 184 , 39). 184th American Chemical Society National Meeting, 1982, Kansas
4. Wilton, D. C., Watkinson, I. A., and Akhtar, M. (1970) Biochern.
5. Gibbons, G. F., and Mitropoulos, K. A. (1975) Biochem. Biophys. Acta, 380,270-281
6. Akhtar, M., Alexander, K., Boar, R. B., McGhie, J. F., andBarton, D. H. R. (1978) Biochem. J. 169, 449-463
7. Akhtar, M., Wilton, D. C., Watkinson, I. A., and Rahimtula, A. D. (1972) Proc. R. SOC. L a n d . B. Bid. Sci. 180. 167-177
8. Akhtar, M., Rahimtula, A. D., Watkinson, I. A., Wilton, D. C., and Munday, K. (1969) Chem. Commun. (J . Chem. SOC. Sect.
9. Schroepfer, G. J., Jr., Lutsky, B. N., Martin, J. A., Huntoon, S., Fourcans, B., Lee, W.-H., and Vermilion, J. (1972) Proc. R. SOC. Lond. B. Biol. Sci. 180 , 125-146
10. Lutsky, B. N., Hsiung, H. M., and Schroepfer, G. J., Jr. (1974) Lipids 10,9-11
11. Aoyama, Y., and Yoshida, Y. (1978) Biochem. Biophys. Res. Comnun. 85,28-34
12. Trzaskos, J. M., Shafiee, A., Bowen, W. D., Fischer, R. T., and Gaylor, J. L. (1984) J. Biol. Chem. 259, 13402-13424
13. Trzaskos, J. M., Bowen, W. D., Fisher, G. J., Billheimer, J. T., and Gaylor, J. L. (1982) Lipids 1 7 , 250-256
14. Fisher, G. J., and Gaylor, J. L. (1982) J. Biol. Chem. 257, 7449- 7455
15. Teranishi, R., and Eastman, F. J. (1963) Organic Synthesis Vol. IV, pp. 192-195, John Wiley and Sons, Inc., New York
16. Antonucci, R., Bernstein, S., Giancola, D., and Sox, K. J. (1951) J. Org. Chem. 16, 1453-1457
17. Watkinson, I. A., Wilton, D. C., Munday, K. A., and Akhtar, M. (1971) Biochem. J. 121 , 131-137
18. Gautschi, F., and Bloch, K. (1958) J . Biol. Chem. 2 3 3 , 1343- 1347
19. Woodward, R. B., Patchett, A. A., Barton, D. H. R., Ives, D. A.
J. 119,673-675
D) 149-150

Sterol 14-Reductase 13423
J., and Kelley, R. B. (1957) J. Chem. SOC. (Lond.) I, 1131-1144 20. Adkins, H., and Pavlic, A. A. (1947) J. Am. Chem. SOC. 69,3039-
3041 21. Fieser, L. F., and Ourisson, G. (1953), J. Am. Chem. SOC. 75,
22. Knight, J. C. , Klein, P. D., Szczepanik, P. A. (1966) J. Bwl. Chern.
23. Yamaga, N., and Gaylor, J . L. (1978) J . Lipid Res. 19, 375-382 24. Ditullio, N. W., Jacobs, C. S., Jr., and Holmes, W. L. (1965) J.
25. Djerassi, C., Engle, R. R., and Bowers, A. (1956) J. Org. Chem.
26. Billheimer, J. T., Alcorn, M., and Gaylor, J. L. (1981) Arch.
27. Miller, W. L., and Gaylor J. L. (1970) J . Bwl. Chem. 245,5375-
28. Woodward, R. B., Patchett, A. A., Barton, D. H. R., Ives, D. A.
4404-4414
241,1502-1508
Chromatogr. 20,354-357
21,1547-1549
Biochem. Biophys. 2 11,430-438
5381
J., and Kelly, R. B. (1954) J . Am. Chem. SOC. 76, 2852-2853
29. Woodward, R. B., Patchett, A. A., Barton, D. H. R., Ives, D. A.
30. Fukushima, H., Grinstead, G. F., and Gaylor, J . L. (1981) J. Biol.
31. Grinstead, G. F., and Gaylor, J . L. (1982) J. Biol. Chem. 257,
32. Lowry, 0. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J.
33. Steinberg, D., and Avigan, J. (1969) Methods Enzymol. 15, 514-
34. Bechtold, M. M., Delwiche, C . V., Comai, K., and Gaylor, J. L.
35. Dempsey, M. (1969) Methods Enzymot. 15, 501-514 36. Avigan, J., Goodman, D. S., and Steinberg, D. (1963) J. Bwl.
37. Schroepfer, G. J., Jr. (1961) J. Biol. Chern. 236, 1668-1673 38. Parish, E. J., and Schroepfer, G. J., Jr. (1981) Fed. Proc. 40,
J., and Kelly, R. B. (1957) J. Chem. SOC. I, 1131-1144
Chem. 256,4822-4826
13937-13944
(1951) J. Biol. Chem. 193, 265-275
521
(1972) J. Biol. Chem. 247, 7650-7656
Chem. 238, 1283-1286
1681
Related Documents











