1 Welcome! Mass Spectrometry meets Cheminformatics Tobias Kind and Julie Leary UC Davis Course 4: Mass Spectrometry Tools & Concepts Class website: CHE 241 - Spring 2008 - CRN 16583 Slides: http://fiehnlab.ucdavis.edu/staff/kind/Teaching/ PPT is hyperlinked – please change to Slide Show Mode

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
Welcome!
Mass Spectrometry meets CheminformaticsTobias Kind and Julie Leary
UC Davis
Course 4: Mass Spectrometry Tools & Concepts
Class website: CHE 241 - Spring 2008 - CRN 16583Slides: http://fiehnlab.ucdavis.edu/staff/kind/Teaching/PPT is hyperlinked – please change to Slide Show Mode

2
Atomic Mass
Hexachlorobenzene (C6Cl6)
average mass - 284.7804 u
integer mass - 282.0 u
monoisotopic mass - 281.81312 u
Correct unit is [u] – unified atomic mass unit or [Da] Dalton see SI units1 u = 1 Da = 1/12th of mass of carbon 12C = 1.66053886 x 10-27 kg
C6Cl6: C6 Cl6 p(gss, s/p:40) Chrg 0R: 1000 Res.Pwr...
282 284 286 288 290 292 294 296m/z
0
10
20
30
40
50
60
70
80
90
100
Rel
ativ
e A
bund
ance
283.81
285.81
281.81
287.80
289.80284.81
286.81
282.82
288.81 291.80 292.80 294.80 295.80
Always (always) check molecular masses obtained from databases or publications.For mass spectrometry the monoisotopic mass is used.
ClCl
ClCl
Cl
Cl
InChIKey:CKAPSXZOOQJIBF-UHFFFAOYAV

3
MWTWIN
Example: Molecular Weight Calculator (Matthew Monroe / PNNL)

4
Mass Accuracy
50-200 ppmLinear IonTrap
3 - 5 ppmTriple Quad
3 - 5 ppmQ-TOF
3 - 5 ppmTOF-MS
1 - 2 ppmMagnetic Sector
0.5 - 1 ppmOrbitrap
0.1 - 1 ppmFT-ICR-MS
Mass AccuracyType
(10 ppm in Ultra-Zoom)
Instruments must be calibrated to obtain high mass accuracy.In case of FT-ICR-MS mass calibration can be stable over weeks.Post- mass calibration can be performed if calibrant was run with samples.Mass of electron becomes important at around 500 Da.
6E1)m
m-m(ppmexp
calcexp+∗=
m(e-) = 0.00054858026 u = mass of electronm(1H) = 1.0078246 u = mass of proton

5
Resolving Power
High resolving power is helpful forseparation of species with almostsame mass (isobars).
High resolving power can not beused to distinguish betweenstructural isomers.
Example:C8H10N2O has 100,082,479 isomers.
RP = 1700
RP = 48,250
Example Solanine (CID=30185)

6
Isotopic Pattern Generators
Elements can be a) monoisotopic (F, Na, P, I)b) polyisotopic (H, C, N, O, S, Cl, Br)
Isotopic pattern generators generate the isotopic abundances for a given mass value.Calculation is very time-consuming and based on Fast Fourier algorithms.
866.00 868.00 870.00 872.00 874.00 876.00 878.00.0
20.0
40.0
60.0
80.0
100.0
Example: Molecular Weight Calculator (Matthew Monroe / PNNL)

7
Isotopic pattern generators
C6Cl6: C6 Cl6 p(gss, s/p:40) Chrg 0R: 1000 Res.Pwr...
282 284 286 288 290 292 294 296m/z
0
10
20
30
40
50
60
70
80
90
100
Rel
ativ
e A
bund
ance
283.81
285.81
281.81
287.80
289.80284.81
286.81
282.82
288.81 291.80 292.80 294.80 295.80
Example form Thermo Xcalibur with a very versatile isotopic pattern generator

8
Charge states
CID: 3081765MW = 1125.50082C50H72N13O15P
charge state 2
charge state 1

9
Different charge states and peak resolutions
562 563 564 565 566 567 568m/z
0
10
20
30
40
50
60
70
80
90
1000
10
20
30
40
50
60
70
80
90
100 562.75
563.25
563.76
564.26564.76 565.77 566.78 567.78
562.75
563.25
563.75
564.25 564.76 565.76 566.76 567.26
C 50 H72 N13 O 15 P: C 50 H72 N13 O 15 P 1p (gss, s /p:40) Chrg 2R: 2000 Res .Pwr . @FWHM
C 50 H72 N13 O 15 P: C 50 H72 N13 O 15 P 1p (gss, s /p:40) Chrg 2R: 200000 Res .Pwr . @FWHM
2000 Resolving PowerCharge state 2
200,000 Resolving PowerCharge state 2
1125 1130 1135m/z
0
10
20
30
40
50
60
70
80
90
1000
10
20
30
40
50
60
70
80
90
100 1125.50
1126.51
1127.52
1128.521130.54 1132.55 1134.55
1125.50
1126.50
1127.51
1128.51 1129.51 1131.52 1133.52 1135.53
C50 H72 N13 O 15 P: C50 H72 N13 O 15 P 1p (gss, s /p:40) Chrg 1R: 2000 Res .Pwr . @FWHM
C50 H72 N13 O 15 P: C50 H72 N13 O 15 P 1p (gss, s /p:40) Chrg 1R: 200000 Res .Pwr . @FWHM
2000 Resolving PowerCharge state 1
200,000 Resolving PowerCharge state 1
1125 1130 1135m/z
0
10
20
30
40
50
60
70
80
90
1000
10
20
30
40
50
60
70
80
90
100
1125 1130 1135m/z
0
10
20
30
40
50
60
70
80
90
1000
10
20
30
40
50
60
70
80
90
100 1125.50
1126.51
1127.52
1128.521130.54 1132.55 1134.55
1125.50
1126.50
1127.51
1128.51 1129.51 1131.52 1133.52 1135.53
C50 H72 N13 O 15 P: C50 H72 N13 O 15 P 1p (gss, s /p:40) Chrg 1R: 2000 Res .Pwr . @FWHM
C50 H72 N13 O 15 P: C50 H72 N13 O 15 P 1p (gss, s /p:40) Chrg 1R: 200000 Res .Pwr . @FWHM
2000 Resolving PowerCharge state 1
200,000 Resolving PowerCharge state 1
Example of Phosphorylated Angiotensin isotopic pattern without adduct [M+H]+ simulated by Thermo XCalibur
0.5 1.0

10
Charge state deconvolution
z = 2
z = 1
Monoisotopic peaks are shown on the mass spectrum, where z = 1
Example from Decon2Ls Tutorial (PNNL)A Software Tool for Deconvolution of High Resolution Mass Spectra
Peaks are detected and charge states are automatically calculated for whole datasets
Download Decon2LSPicture sources: PNNL/ Decon2Ls Tutorial

11
Adduct formation
http://fiehnlab.ucdavis.edu/staff/kind/Metabolomics/MS-Adduct-Calculator/
Ion name Ion mass Charge Mult Mass Result:1. Positive ion mode M+H M + 1.007276 1+ 1 1.007276 2.007276M+NH4 M + 18.033823 1+ 1 18.033823 19.033823M+Na M + 22.989218 1+ 1 22.989218 23.989218M+CH3OH+H M + 33.033489 1+ 1 33.033489 34.0334891. Negative ion mode M-H M - 1.007276 1- 1 -1.007276 -0.007276M+Na-2H M + 20.974666 1- 1 20.974666 21.974666M+Cl M + 34.969402 1- 1 34.969402 35.969402
Adduct formation is observed in ESI, CI and APCI ionization modes (and others).Adduct detection is problematic for small molecules, can be influenced by solvent selection.Adduct detection can be automated if two or more adducts are detected in mass spectrum.Switching polarity (+/-) can be used for confirmation of adduct.
Download the Mass Spectral Adduct Calculator

12
Molecular Formula Generators
Formula generators are used to create molecular formulae from accurate masses.Input requires 1) accurate isotopic mass (with or without adduct) and
2) error in ppm or mDa (milli Dalton)
Accurate mass
Mass errorExample MWTWIN

13
The molecular formula space of small moleculescalculated by the Seven Golden Rules
Source http://fiehnlab.ucdavis.edu/projects/Seven_Golden_Rules/Molecular-Formula-Space/
Each molecular formula can expand to billions of structural isomers.Molecular Formula ≠ Molecular Isomer

14
Frequency distribution of molecular formulas
Source: Kind and Fiehn BMC Bioinformatics 2006 7:234 doi:10.1186/1471-2105-7-234

15
Impact of mass accuracy on number of formulas

16
Mass accuracy and isotopic pattern
Example from: http://fiehnlab.ucdavis.edu/projects/Seven_Golden_Rules/Examples/Solanine/
Example:ESI-MS (+) of Solanine on a LTQResolving Power: 1700Mass Accuracy: 46 ppmIsotopic Abundance Error: ±1.46%
C45H73NO15MW = 867.49799
[M+H]+

17
Isotopic abundances as orthogonal filter
Source: Kind and Fiehn BMC Bioinformatics 2006 7:234 doi:10.1186/1471-2105-7-234

18
Molecular Isomer Generators
Isomer generators are used to create all possible structural isomers from a given molecular formula. Deterministic and stochastic (random) generators are in use.
Example: MOLGEN DEMO (Bayreuth)

19
The molecular isomer space is unknown
Accurate mass Formula Number Isomers in Beilstein DB
77.99531 CH2O4 6 0
78.04293 CH6N2O2 28 1
78.03169 C2H6O3 10 8
78.02180 C4H2N2 465 2
78.01056 C5H2O 151 2
78.04695 C6H6 217 29
150.04293 C7H6N2O2 100,082,479 153
150.09054 C7H10N4 66,583,863 105
150.03169 C8H6O3 6,717,404 90
150.07931 C8H10N2O 76,307,072 542
150.06808 C9H10O2 6,843,602 667
150.11569 C9H14N2 9,459,132 568
150.02180 C10H2N2 65,563,828 0
150.10446 C10H14O 1,548,361 1938
150.01056 C11H2O 9,414,509 0
150.14084 C11H18 84,051 762
150.04695 C12H6 34,030,905 12
Source: http://fiehnlab.ucdavis.edu/projects/Seven_Golden_Rules/Molecular-Isomer-Generator/Meringer M: Mathematische Modelle für die kombinatorische Chemie und die molekulare Strukturaufklärung.
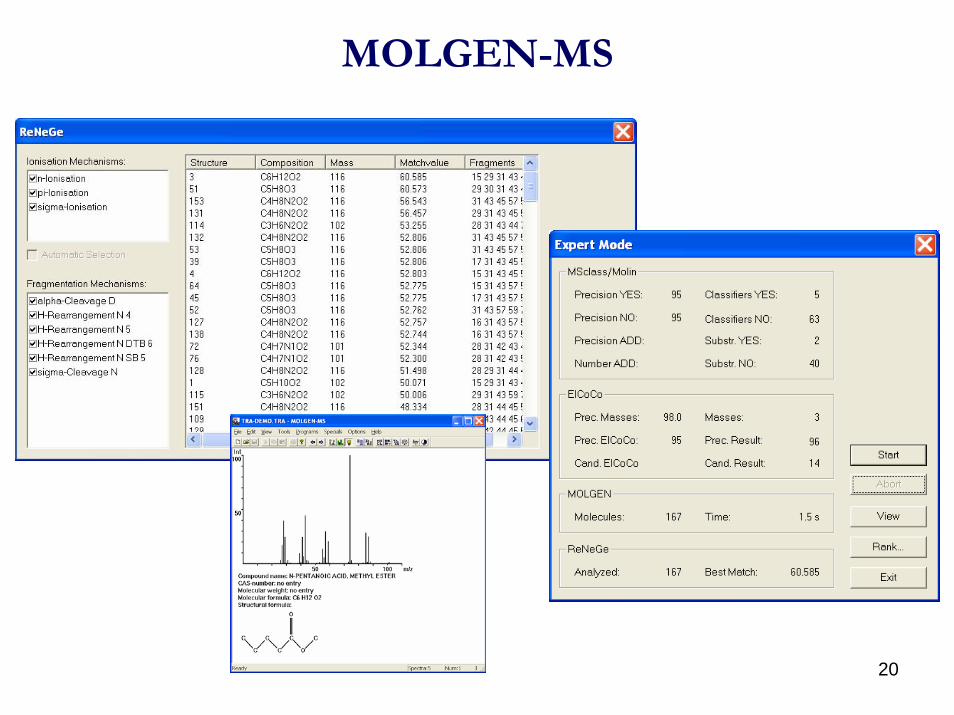
20
MOLGEN-MS

21
Substructure predictions
(mainlib) Coronene40 130 220 310
0
50
100
100
150
168 222
300
Use computer algorithms (machine learning) for automated interpretation of fragmentsand corresponding substructures. Algorithms creates present/absent list of substructures.
Example from NIST-MS search

22
Simulation of mass spectra
Why is simulation or prediction of mass spectra important?
Molecular isomers (structures) can be generated very fast from molecular formulasOnly certain mass spectra can be simulated (MS/MS of peptides, oligosaccharides, lipids)Problematic is abundance determinationProblematic are all complex rearrangement reactions (gas phase ion chemistry)Simulation of mass spectra from small molecules is new and important research
Experimentalspectra
Simulatedspectra
Perform comparison or matching

23
Barcode spectrum from Mass Frontier
Example: MassFrontier Helpfile

24
Peptide Sequence Fragmentation Modelling
Example: Taken from Molecular Weight Calculator (Matthew Monroe / PNNL)

25
Matching experimental vs. theoretical sequence
Example: RHPEYAVSVLLR

26
The Last Page - What is important to remember:
Important performance parameters are accurate mass, resolving power, scan speedand accurate isotopic abundances
Even high resolving power MS can not distinguish between structural isomers
Accurate Mass Molecular Formula Structural Isomers MS/MS
Mass spectra of only some substance classes can be simulated

27
Tasks (12 min):
(1)Use a Molecular Formula Finder Molecular Weight Calculator or any other tool (HR2 in 7 Golden Rules)
Calculate how many formulae (elements CHNO) can be calculated fora) m/z 500.5 at 100 ppm, 10 ppm, 1 pm b) m/z 500.0 at 100 ppm, 10 ppm, 1 pm
(2)Calculate the number of isomers for C12H12 using MOLGEN or CDK
(3)Generate the isotopic pattern for Chlorophyll a and Hexachlorobenzene.
(4) Download Decon2LS and perform example calculations from tutorial

28
Literature (35 min):
Debating resolution and mass accuracy (MP Balogh)
Enumerating molecules (Faulon)
Milestones in fourier transform ion cyclotron resonance mass spectrometry technique development

29
Links:
Used for research: (right click – open hyperlink)http://download.enovatia.com/images/promass/help/promassxcaliWebMain.htmlhttp://download.enovatia.com/images/promass/protein_MW_determination/bsa.html#1.36zoomhttp://www.bioc.aecom.yu.edu/labs/angellab/http://www.ads.tuwien.ac.at/publications/bib/pdf/mujezinovic_07.pdfhttp://mendel.imp.ac.at/mass-spectrometry/http://www.chm.bris.ac.uk/sillymolecules/sillymols.htmhttp://ncrr.pnl.gov/training/workshops/2007HUPO/LCMSBasedProteomicsDataProcessing.pdfhttp://www.iupac.org/publications/pac/2003/pdf/7506x0683.pdf"Sic transit gloria chimica”http://www.google.com/search?hl=en&q=%22Sic+transit+gloria+chimica&btnG=Search
Of general importance for this course:http://fiehnlab.ucdavis.edu/staff/kind/Metabolomics/Structure_Elucidation/
Related Documents











