Chemical Etiology of Nucleic Acids: Aminopropyl Nucleic Acids (APNAs) by Ding Zhou, Matheus Froeyen, Jozef Rozenski, Arthur Van Aerschot, and Piet Herdewijn* Laboratory of Medicinal Chemistry, Rega Institute for Medical Research, Katholieke Universiteit Leuven, Minderbroedersstraat 10, B-3000 Leuven (phone: þ 32-16-337387; fax: þ 32-16-337340; e-mail: [email protected]) Dedicated to Leslie Orgel on the occasion of his 80th birthday Aminopropyl nucleic acids (APNAs) are constitutionally simple nucleic acid alternatives with one stereogenic center per nucleotide, and with the potential to hybridize with RNA and to exert catalytic functions. We have developed a protecting group strategy to synthesize APNAs, although in a not very efficient way. Isolation and purification of APNAs proved to be difficult. Their structures might be more suited to function as potential catalytic polymers than as information systems that may evolve into RNA. Introduction. – It could be expected that an evolutionary progenitor of RNA would be constitutionally a simpler polymer. This means that it could be assembled from simple prebiotic organic material (such as phosphate, ammonia, formaldehyde, glycolaldehyde), and that it possesses no or few stereogenic centers. A further prerequisite is that it could store and transfer information (this could be done by using nucleobases or their predecessors). As this potential progenitor has prepared the appearance of RNA, it might be advantageous that it is formed as a constituent of a kinetically labile library with high catalytic potential [1]. In the best-case scenario, it could also contribute to homochirality of nucleic acids, as chirality plays a role in the kinetic and thermodynamic stability of a polymer (selection of homochirality may be based on the elimination of the kinetically most labile congeners and/or of the least (or most) soluble competitor, and/or of the polymers least prone to thermodynamic selection by base pairing). Recently, we published on a new type of acyclic nucleic acids (3’-aminopropyl nucleic acids, 3’-APNAs) [2] which was designed based on the same principles as Eschenmoser)s 3’-NH-TNA [3] and reducing the number of C-atoms to only one stereogenic centre. In principle, such a nucleic acid could have been assembled in nature from phosphorylated glycolaldehyde (C 2 ), formaldehyde (C 1 ), NH 3 , and a nucleobase (the disadvantage, though, is that the nucleobase is alkylated in (APNAs), and no anomeric center is present). Incorporation of one, two, or three aminopropyl nucleotides in DNA leads to a large destabilization of a DNA duplex [2]. Fully modified 3’-aminopropyl nucleic acids (3’-APNA) could not be obtained due to competing side reactions during oligonucleotide synthesis such as intramolecular attack of the nucleophilic N-atom on the electrophilic P-atom [2]. However, APNAs are simple nucleic acid alternatives, with only one stereogenic center per nucleotide, CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 740 # 2007 Verlag Helvetica Chimica Acta AG, Zɒrich

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Chemical Etiology of Nucleic Acids: Aminopropyl Nucleic Acids(APNAs)
by Ding Zhou, Matheus Froeyen, Jozef Rozenski, Arthur Van Aerschot, and Piet Herdewijn*
Laboratory of Medicinal Chemistry, Rega Institute for Medical Research, Katholieke UniversiteitLeuven, Minderbroedersstraat 10, B-3000 Leuven
(phone: þ32-16-337387; fax: þ32-16-337340; e-mail: [email protected])
Dedicated to Leslie Orgel on the occasion of his 80th birthday
Aminopropyl nucleic acids (APNAs) are constitutionally simple nucleic acid alternatives with onestereogenic center per nucleotide, and with the potential to hybridize with RNA and to exert catalyticfunctions. We have developed a protecting group strategy to synthesize APNAs, although in a not veryefficient way. Isolation and purification of APNAs proved to be difficult. Their structures might be moresuited to function as potential catalytic polymers than as information systems that may evolve into RNA.
Introduction. – It could be expected that an evolutionary progenitor of RNAwouldbe constitutionally a simpler polymer. This means that it could be assembled fromsimple prebiotic organic material (such as phosphate, ammonia, formaldehyde,glycolaldehyde), and that it possesses no or few stereogenic centers. A furtherprerequisite is that it could store and transfer information (this could be done by usingnucleobases or their predecessors). As this potential progenitor has prepared theappearance of RNA, it might be advantageous that it is formed as a constituent of akinetically labile library with high catalytic potential [1]. In the best-case scenario, itcould also contribute to homochirality of nucleic acids, as chirality plays a role in thekinetic and thermodynamic stability of a polymer (selection of homochirality may bebased on the elimination of the kinetically most labile congeners and/or of the least (ormost) soluble competitor, and/or of the polymers least prone to thermodynamicselection by base pairing).
Recently, we published on a new type of acyclic nucleic acids (3’-aminopropylnucleic acids, 3’-APNAs) [2] which was designed based on the same principles asEschenmoser=s 3’-NH-TNA [3] and reducing the number of C-atoms to only onestereogenic centre. In principle, such a nucleic acid could have been assembled innature from phosphorylated glycolaldehyde (C2), formaldehyde (C1), NH3, and anucleobase (the disadvantage, though, is that the nucleobase is alkylated in ?APNAs=,and no anomeric center is present). Incorporation of one, two, or three aminopropylnucleotides in DNA leads to a large destabilization of a DNA duplex [2].
Fully modified 3’-aminopropyl nucleic acids (3’-APNA) could not be obtained dueto competing side reactions during oligonucleotide synthesis such as intramolecularattack of the nucleophilic N-atom on the electrophilic P-atom [2]. However, APNAsare simple nucleic acid alternatives, with only one stereogenic center per nucleotide,
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)740
G 2007 Verlag Helvetica Chimica Acta AG, ZJrich

with the potential to hybridize with RNA (dependent on chirality and constitution),with high catalytic potential due to their flexibility and the presence of severalnucleophilic centers, and, most probably, kinetically labile. It could be a potentialpredecessor of RNA as a genetic molecule and, as a catalyst, for a primitive form of life.Therefore, we continued our effort to synthesize the fully modified APNAs, a potentialattainable goal in function of the recently described synthesis of the glycol nucleic acids(GNA), which showed successful self-hybridization [4]. As it is difficult to predict theoptimal constitution (2’-amino- or 3’-aminopropyl nucleic acids) and decide uponchirality ((R)- or (S)-monomers) in function of chemical stability and hybridizationproperties, we envisioned the synthesis of all possible isomeric nucleic acids with theadenine base as well as with the thymine base (Fig. 1).
To obtain the fully modified APNAs and to avoid intramolecular attack of thepotential amino group on PIII, which would cause side reactions during synthesis [2], thenucleophilicity of the N-atom of the respective building blocks has to be reduced duringthe oligonucleotide assembly. This would probably be attained by using a protectinggroup like the Boc ((tert-butoxy)carbonyl) group for the amino group of aminopropylnucleosides to obtain a carbamate. However, the protecting group should be easilycleaved by mild acidic conditions used in oligonucleotide assembly (3% Cl3CCOOHacid, 2 min). From the literature reported, there are several protecting groupcandidates such as t-Bumeoc (¼1-{[3,5-di-(tert-butyl)phenyl]-1-methylethoxy}carbon-yl) [5], Ddz (¼ [2-(3,5-dimethoxyphenyl)-1-methylethoxy]carbonyl), and Bpoc (¼ [2-(1,1’-biphenyl-4-yl)-1-methylethoxy]carbonyl), which could match this requirement,and which were already used before in peptide synthesis (Fig. 2) [6]. However, the Ddzgroup was shown to be somewhat too stable. A prolonged contact with acid (at least16 min) [6] was required to accomplish complete deprotection, which resulted in somedegree of depurination of the acid-sensitive DNA chain. Therefore, we considered thehighly acid-labile amino protecting groups already developed for peptide chemistry: t-Bumeoc and Bpoc. The t-Bumeoc group proved to be more difficult to synthesize.Therefore, the focus was shifted to the Bpoc group that can easily be prepared fromcommercially available reagents [6].
Fig. 1. Constitution and configuration of members of two APNA families (aminopropyl nucleic acids)
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 741

Two classes of Bpoc protected and phosphitylated 3’-aminopropyl- and 2’-amino-propyl nucleoside building blocks (3’- and 2’-APNps) were synthesized and used for thepreparation of the corresponding modified oligonucleotides (Fig. 3).
Results and Discussion. – The nucleoside derivatives required for our study, namely,3’-aminopropyl and 2’-aminopropyl nucleosides (3’- or 2’-APNs) with either an adenineor thymine base moiety were prepared from enantiomerically pure (S)-1,2-O-isopropylidene-glycerol (being less expensive than the (R)-isomer).
Fig. 3. Aminopropyl nucleoside building blocks used for oligonucleotide synthesis
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)742
Fig. 2. Amino-protecting groups cleaved by mild acid and considered for APNA synthesis

Synthesis of Protected (R)- and (S)-3’-Acyclic Nucleosides (3’-APNs). Thesynthesis of nucleosides 3a, 3b, 5a and 5b starting from the commercially available(S)-1,2-O-isopropylideneglycerol (1; R¼OH) is outlined in Scheme 1. The synthesis of2a, 2b, 4a, and 4b was published recently [2] [7]. Condensation of 2a, 2b, 4a, and 4b withBpoc-OC6H4(p-CO2Me) using Triton B catalyst (benzyl(trimethyl)ammonium hydrox-ide soln. 40 wt-% in MeOH) afforded 3a, 3b, 5a, and 5b, respectively, of which theTBDMS (¼ (t-Bu)Me2Si) protecting group was slowly removed during the reaction.All the desired (R)- and (S)-isomers were isolated and characterized by opticalrotation, mass spectrometry, and NMR spectroscopy.
Synthesis of Protected (R)- and (S)-2’-Aminopropyl Nucleosides (2’-APNs). Thesynthesis of 12a and 12b starting from 6a and 6b, respectivley, [2] [7] is outlined inScheme 2. Mesylation of the free 2’-OH group of 6a or 6b, followed by substitution ofthe MsO group by azide ions affords azido derivatives 8a or 8b, respectively, in one potwithout isolation of the mesylated intermediates 7a and 7b. Due to neighboring groupparticipation reaction of the thymine base, introduction of the N3 group proceeded withretention of configuration (which is not the case for the adenine congener). The tritylmoiety of 8a or 8b was cleaved in the presence of 2% TsOH in a solvent mixtureCH2Cl2/MeOH 2 :1 (v/v) affording 9a and 9b respectively, which were silylated with (t-Bu)Me2SiCl (TBDMSCl). Catalytic hydrogenation of the N3 group, followed byprotection of the amino group by Bpoc, furnished the nucleosides (R)-12a and (S)-12b.
The synthesis of the isomeric nucleoside 18a (Scheme 3) also started from thecommercially available 1. Silylation of the primary OH group (3-OH) of compound 13[8] was carried out with TBDMSCl. Subsequent selective cleavage of the MMTr groupin the presence of 1% TsOH in a solvent mixture CH2Cl2/MeOH 2 :1 (v/v) gave 15.Reaction of compound 15 under Mitsunobu condition [9] with N3-benzoylthyminefurnished the protected nucleoside, which was debenzoylated to provide 10b (identicalwith the compound described in Scheme 2). However, the introduction of the adeninebase could not be carried out under Mitsunobu conditions due to the difficulties with
Scheme 1. Preparation of the (R)-Configured Protected Nucleosides 3a and 3b, and (S)-IsomericNucleosides 5a and 5b, Starting from (S)-Isopropylidene-glycerol 1
a) Bpoc-OC6H4(p-CO2Me) (Bpoc¼ {[2-(1,1’-biphenyl-4-yl)prop-2-yl]oxy}carbonyl), Triton B, dioxane,DMF, 558 ; T: thymin-1-yl; ABz: N6-benzoyladenin-9-yl.
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 743

the separation of the obtained N(7)- and N(9)-isomers. Alternatively, the primary OHfunction of 15 was tosylated to give 16a, followed by reaction with the sodium salt ofadenine, which was prepared in situ by treatment of adenine with NaH. Subsequentprotection of the exocyclic amine with benzoyl chloride (BzCl) in pyridine was carriedout to give 16c. Catalytic hydrogenation of the azide derivatives gave the free aminoderivatives 17a and 11b. Condensation of 17a and 11b with Bpoc-OC6H4(p-CO2Me)with Triton B as a catalyst afforded the nucleosides 18a and 12b, respectively. Theidentical [a]20
D values for 12b obtained via Schemes 2 and 3 further established theretention of configuration when the N3 group is introduced after the introduction of thethymine base moiety (Scheme 2). The stereochemical purity of the compounds can bedemonstrated by the identical but opposite [a]20
D values for compound 12a and 18a.
Synthesis of Oligonucleotides with 3’- or 2’-Phosphoramidate Linkages. Phosphity-lation was carried out on the Bpoc protected derivatives, 3a, 3b, 5a, 5b, 12a, 12b, and 18ain CH2Cl2 using freshly distilled EtN(i-Pr)2 and 2-cyanoethyl N,N-diisopropylchlor-ophosphoramidite under Ar to afford 3’-amino analogues 19a ((R), B¼A), 19b ((R),
Scheme 2. Preparation of the (R)- and (S)-Configured Protected Nucleosides 12a and 12b, respectively,Starting from (S)-Isopropylidene-glycerol 1
a) MsCl, pyridine (not isolated). b) NaN3, DMF, 1008. c) 2% TsOH in CH2Cl2 and MeOH (9a : 54% overtwo steps, 9b : 47% over two steps). d) (t-Bu)Me2SiCl (TBDMSCl), 1H-imidazole, DMF. e) H2, THF,
Pd/C. f) Bpoc-OC6H4(p-CO2Me), Triton B, dioxane, DMF, 558. A: adenin-9-yl; T: thymin-1-yl.
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)744

B¼T), 20a ((S), B¼A), 20b ((S), B¼T), and 2’-amino analogues 21a ((R), B¼A),22b ((S), B¼T), 22a ((S), B¼A) in high yield (Scheme 4).
The assembly on a DNA synthesizer of oligonucleotide strands (3’- and 2’-APNA)containing exclusively phosphoramidate linkages was accomplished according to amodified version of the phosphoramidite method [10] [11]. Coupling steps involve
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 745
Scheme 3. Preparation of the (S)-Configured Protected Nucleosides 18a and 12b Starting from 1
a) TBDMSCl, 1H-imidazole, DMF. b) 1% TsOH in CH2Cl2 and MeOH. c) TsCl, pyridine (not isolated).d) A, 80% NaH, 808, DMF, 1 h; 1008, 15 h. e) BzCl, pyridine; sat. NH3 in MeOH, 08. f) TBz, Diisopropylazodicarboxylate (DIAD), Ph3P, THF; sat. NH3 in MeOH, 08. g) H2, THF, Pd/C. h) Bpoc-OC6H4(p-
CO2Me), Triton B, dioxane, DMF, 558 (18a, 46%; 12b, 46%). A: adenin-9-yl; T: thymin-1-yl.
Scheme 4. Conversion of the Target Nucleosides to the Corresponding Phosphoramidites (Fig. 3 showsthe detailed structures)
a) EtN(i-Pr)2, 2-cyanoethyl N,N-diisopropylchlorophosphoramidite in CH2Cl2. a series: adenine; bseries: thymine; (R)-series: 19, 21; (S)-series: 20, 22.

exchange reaction of the iPr2N-group of the approaching phosphoramidite (i.e., 19a,19b, 20a, 20b, 21a, 22a, 22b). In accordance with equilibrium theory for an amine-exchange reaction, this ?couple-oxidize-couple-cap-oxidize= approach was moreefficient than a single coupling, even when using twice the equivalents of monomerfor the single coupling. After double coupling, unreacted amino groups were cappedwith Ac2O, after which the second oxidation of the internucleotide phosphoramiditediester linkage into the phosphoramidate group was carried out with aqueous I2.Subsequent deprotection of the Bpoc group of the newly added residue with 3%Cl3CCOOH soln. in CH2Cl2, enabled additional chain elongation steps to be repeatedfor the construction of the desired oligomeric phosphoramidates. All modifiedoligonucleotides were assembled on a universal support comprising a propane-1,3-diol linker. This enables assembly starting with any modified building block via anormal phosphodiester linkage. The detachment of the oligomers from the solidsupport with concomitant deprotection of the phosphate and nucleobase protectinggroups was achieved by treatment with a mixture of MeNH2 (40% in H2O) andconcentrated aqueous NH3 1 :1 (308). The fully deprotected oligonucleotide phos-phoramidate analogues were purified by ion exchange chromatography and analyzedby mass spectrometry to establish the correct incorporation of the acyclic building blocks.
Synthesis of Oligonucleotides with Incorporation of 3’-Aminopropyl Thymidine19b. Test Reactions. To evaluate the incorporation conditions, a single, triple, andfivefold consecutive incorporation was carried out with the Bpoc protected thymidineanalogue 19b using a single coupling with 19 equiv. of monomer for 3 min in eachsynthetic cycle. This was followed by the standard capping (extended to 1 min) andoxidation protocol with 0.02m I2 in THF/pyridine/H2O. Incorporation within a regularthymidine homopolymer allowed evaluation of the overall coupling efficiencyaccording to trityl yield. The Bpoc protecting group itself does not allow immediatevisual inspection of the coupling reaction. Hence, with fully modified sequences, thereaction products, after complete assembly and deprotection of the oligonucleotide, areanalyzed by ion-exchange chromatography. The overall trityl yield spanning the gapwith the modified part for the sequences 23 and 25, dropped from 95 over 80 to 64% forthe oligomer with five phosphoramidate linkages. The ion-exchange profiles at pH 12clearly show the incomplete coupling at the different phosphoramidate steps (Figs. 4and 5).
In addition, one starts noticing a complex diffuse pattern for the full lengtholigomer 25 with five phosphoramidate linkages, which we could not directly explain(Fig. 5). Correct masses were found for the isolated oligomers (Table 1). Also the full-length oligonucleotide gradually diminishes in yield. However, for sequence 25 themass spectrum for the isolated oligomer corresponding to the top last part of theanalytical profile included substantial amounts of the n�1, n�2 and even n�3detections (masses found following deconvolution: 2806.5, 2545.5, and 2284.4).
We also noticed n�1 and n�2 detections in the mass spectrum for 24. Never-theless, as seen in Fig. 4, sequence 24 displayed a relatively sharp peak for the full-length oligomer. This is remarkable in view of the fact that thymidine homopolymers,even 13-nucleotides-long, can easily be separated from failure sequences even on apreparative scale using a salt gradient at pH 12. (Analytical profile of a deliberatemixture of a 13-mer homopolymer with failure sequences; Fig. 6).
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)746
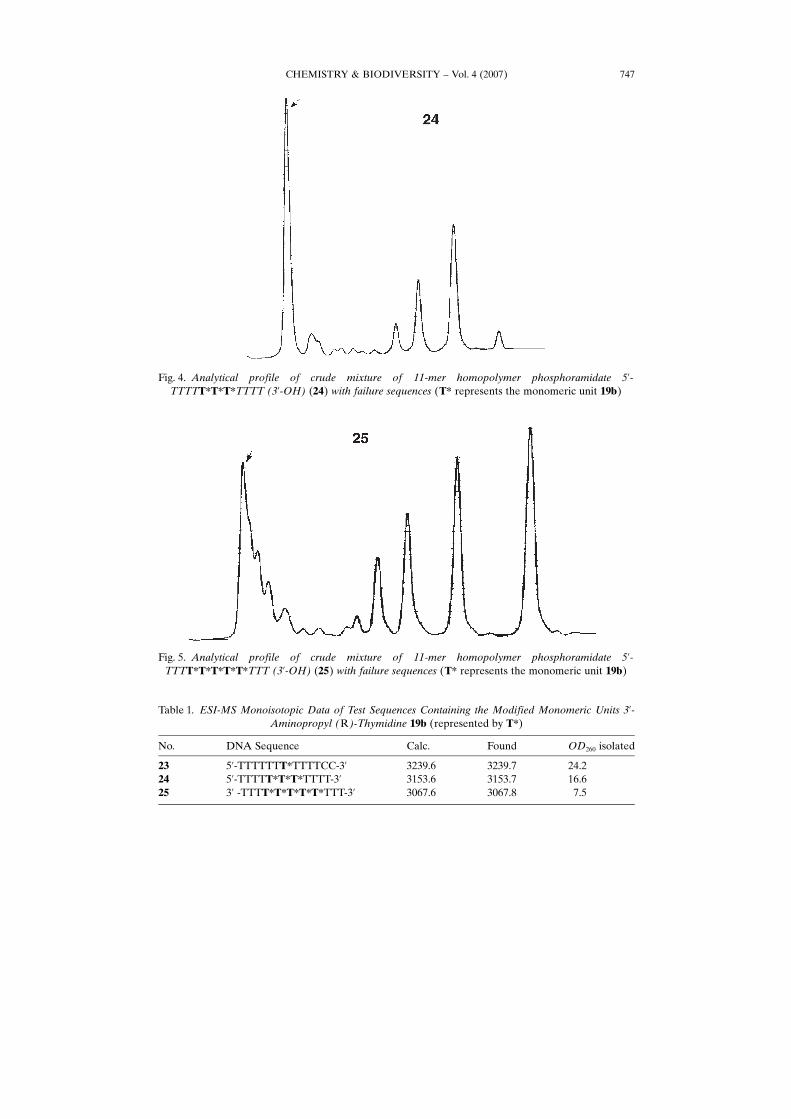
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 747
Fig. 5. Analytical profile of crude mixture of 11-mer homopolymer phosphoramidate 5’-TTTT*T*T*T*T*TTT (3’-OH) (25) with failure sequences (T* represents the monomeric unit 19b)
Fig. 4. Analytical profile of crude mixture of 11-mer homopolymer phosphoramidate 5’-TTTTT*T*T*TTTT (3’-OH) (24) with failure sequences (T* represents the monomeric unit 19b)
Table 1. ESI-MS Monoisotopic Data of Test Sequences Containing the Modified Monomeric Units 3’-Aminopropyl (R)-Thymidine 19b (represented by T*)
No. DNA Sequence Calc. Found OD260 isolated
23 5’-TTTTTTT*TTTTCC-3’ 3239.6 3239.7 24.224 5’-TTTTT*T*T*TTTT-3’ 3153.6 3153.7 16.625 3’ -TTTT*T*T*T*T*TTT-3’ 3067.6 3067.8 7.5

Synthesis of Fully Modified Oligonucleotides with 3’- or 2’-PhosphoramidateLinkages. Considering the decreasing yield of the full-length oligonucleotide uponincorporation of multiple modifications, and, in accordance with literature forribonucleotide phosphoramidates [12], we then focused on a double coupling protocol.Hereto, coupling was allowed to proceed twice in the presence of 11 equiv. of monomer(0.1m) for 3 min, with an intermediate oxidation and washing step (a total of 22 equiv.consumed). The second coupling was followed first by a capping and then by anoxidation step. In view of the obtained quantities of phosphoramidates and the vastexcess needed per coupling, the synthesis of only homopolymers could be envisionedfor the different modified building blocks. On the other hand, the sequences needed tobe sufficiently long to hopefully allow stable Watson–Crick base pairing (A–T) as wellas to allow analysis of the pairing potential to DNA and RNA complements. We,therefore, opted for 13-mers, which, for some building blocks, was the synthetic limit.Table 2 gives an overview of the different planned and obtained sequences.
Although oligonucleotide phosphoramidate synthesis is well-established – albeit inlower yield compared to regular oligonucleotide synthesis – a first general conclusion isthe very cumbersome synthesis for the different acyclic phosphoramidate congeners.This can be easily seen from the HPLC profiles shown in Figs. 7–9.
In general, synthesis of the polymers comprising 2’-aminopropyl analogues provedslightly better (i.e., 31, and 32 in Fig. 8). The phosphitylation is an amine-exchangereaction, and, apparently, this equilibrium is shifted more towards the intermediate
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)748
Fig. 6. Analytical profile of deliberate mixture of 13-mer homopolymer of thymidine with failure 12-merand 11-mer sequences

phosphoramidite (chain elongation, prior to oxidation) with the sterically morehindered 2’-amino groups. Separation and isolation of the different products anddeletion sequences, in general, is straightforward on an ion exchange column up to thechain lengths synthesized here.
Overall, the mirror-image oligonucleotides displayed the same HPLC profile. Theadenine polymers, 27 and 32, also perform better than the thymine-containing ones, 29and 31 (hereto, compare sequence 27 (same profile for 26, not shown) to 29 (or 28 ; notshown)). For the latter 29, no substantial amounts of the desired sequence could be
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 749
Table 2. Fully Modified Envisioned 13-mer Sequencesa) [3]
No. Modification Fraction MS OD260 isolated
Calc.b) Found
26 3’NH2(R)A (19a) 26a 3628.9 (13-mer) NDc) 0.626b 3358.8 (12-mer) 4631.7 0.4
27 3’NH2(S)A (20a) 27a 3628.9 (13-mer) 3629.2 0.627b 3358.8 (12-mer) NDc) 0.3
28 3’NH2(R)T (19b) 28a 2989.7 (11-mer) 3012.0 (þ Na), 4672.4,2989.5 (608)
0.2
28c 2467.5 (9-mer) 2467.9 0.428f 1684.4 (6-mer) 1684.6 1.3
29 3’NH2(S)T (20b) 29a 2989.7 (11-mer) 3011.9 (þ Na) 0.229c 2467.5 (9-mer) 2467.8 0.529f 1684.4 (6-mer) 1684.6 1.5
30 2’NH2(S)T (22b) 30a 3511.7 (13-mer) 3251.1 (12-mer) 3.231 2’NH2(S)T (22b’)d) 31a 3511.7 (13-mer) NAe) (258)
3511.6 (608)1.60.9
31b 2989.7 (11-mer) 3012.0 (þ Na) 0.931d 2467.5 (9-mer) 2467.9 1.331g 1684.4 (6-mer) 1684.6 0.15
32 2’NH2(S)A (22a) 32z (nþ1) 3628.9 (13-mer) 3629.3 (13-mer),3359.3 (12-mer)
2.3
32a 3628.9 (13-mer) 3629.3 (13-mer),3359.2 (12-mer)
1.3
32b 3359.2 (12-mer) 3359.2 (12-mer),3629.3 (13-mer)
1.6
33 2’NH2(R)A (21a) 33z 3586.9 (13-mer) 3587.7 (13-mer),3317.6 (12-mer),3047.5 (11-mer)
1.1
33a 3586.9 (no capping) 13-merþ12-merþ11-mer 1.333b 3316.8 (no capping) 13-merþ12-merþ11-mer 5.133c 3046.7 (no capping) 13-merþ12-merþ11-mer 2.6
a) All sequences were assembled on a solid support containing a propane-1,3-diol linker. The 3’-tail thusgenerated (propane-1,3-diol phosphate) does not interfere with physico-chemical studies. b) Allcalculated masses correspond to the terminal N-acetylated oligonucleotides. c) No clean signal couldbe obtained. d) The (S)-phosphoramidites with a thymine base obtained according to Schemes 2 and 3were both investigated in the oligomerization reaction (series 30 and 31). e) Not assignable: multiplemolecular ions found.

isolated in view of the declining yield. On the contrary, the 2’-aminopropyl congeners31 (and 30 ; not shown) did yield the expected 13-mers. Likewise, the yields with 32 and33 improved further. However, the 2’-aminopropyl oligonucleotides all do show a muchmore complicated profile, with either more than the expected intermediate peaks forthe thymine analogues 30 and 31, or broad diffuse peaks for the adenine polymers 32and 33 (33 shown in Fig. 9). In addition, a so far not explained interaction seems to beat play for 32 and 33, where a late-eluting peak is noticed in the HPLC profile. Re-analysis of the peaks isolated at 40 and 50% of the salt gradient, respectively, both 32aand 32z yield a mixture of both peaks again, as shown in Fig. 8.
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)750
Fig. 7. Analytical profiles of crude mixture of 13-mer homopolymers 27 (from 3’NH2(S)A (20a)) and 29(from 3’NH2(S)T(20b)) with failure sequences
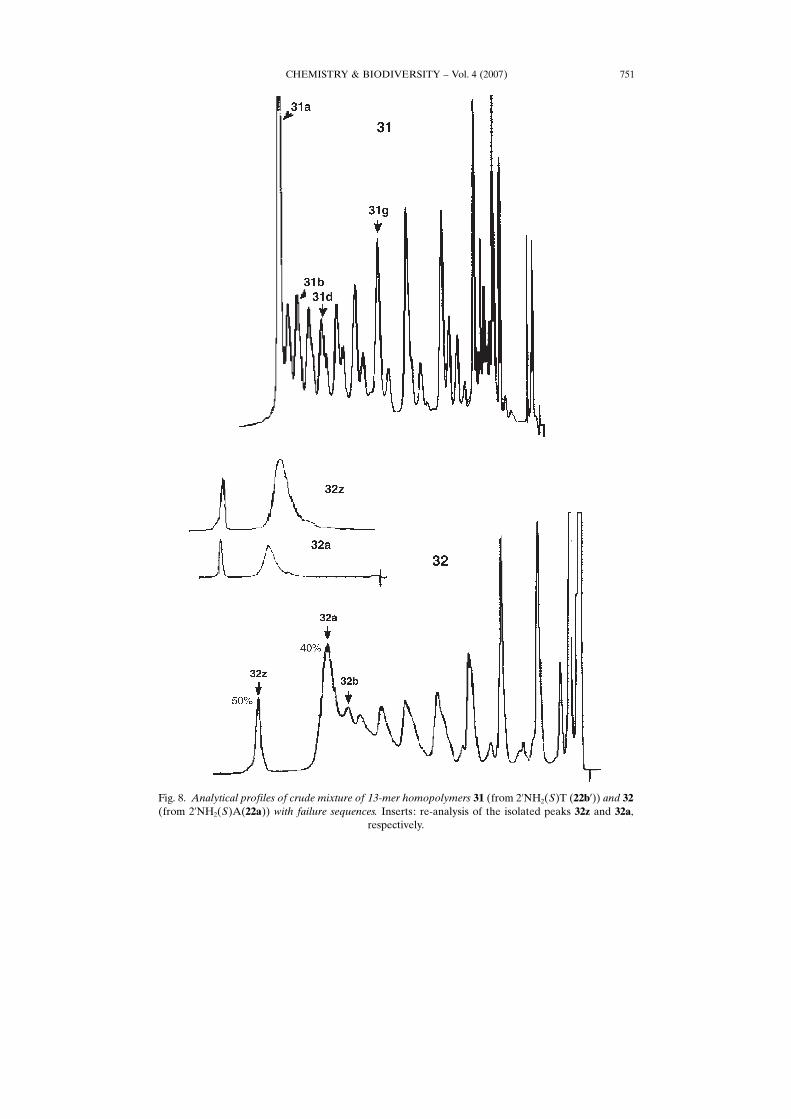
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 751
Fig. 8. Analytical profiles of crude mixture of 13-mer homopolymers 31 (from 2’NH2(S)T (22b’)) and 32(from 2’NH2(S)A(22a)) with failure sequences. Inserts: re-analysis of the isolated peaks 32z and 32a,
respectively.
Mass-spectrometric analysis of all products isolated proved very complicated aswell, where, in many cases, several salt adducts were detected (Naþ , Kþ as well asmultiple adducts, Table 2). Although systematically a cap-LC is coupled to the massspectrometer, which should greatly reduce the amount of salt adducts, in some cases noproducts could be detected without having any salt adducts. In other cases, the analysiswas improved running the LC at 608 (28a and 31a in Table 2). However, for someproducts we still could not provide an interpretable signal, or otherwise unidentifiedhigh-molecular-weight compounds (including a series of salt adducts) were detected.
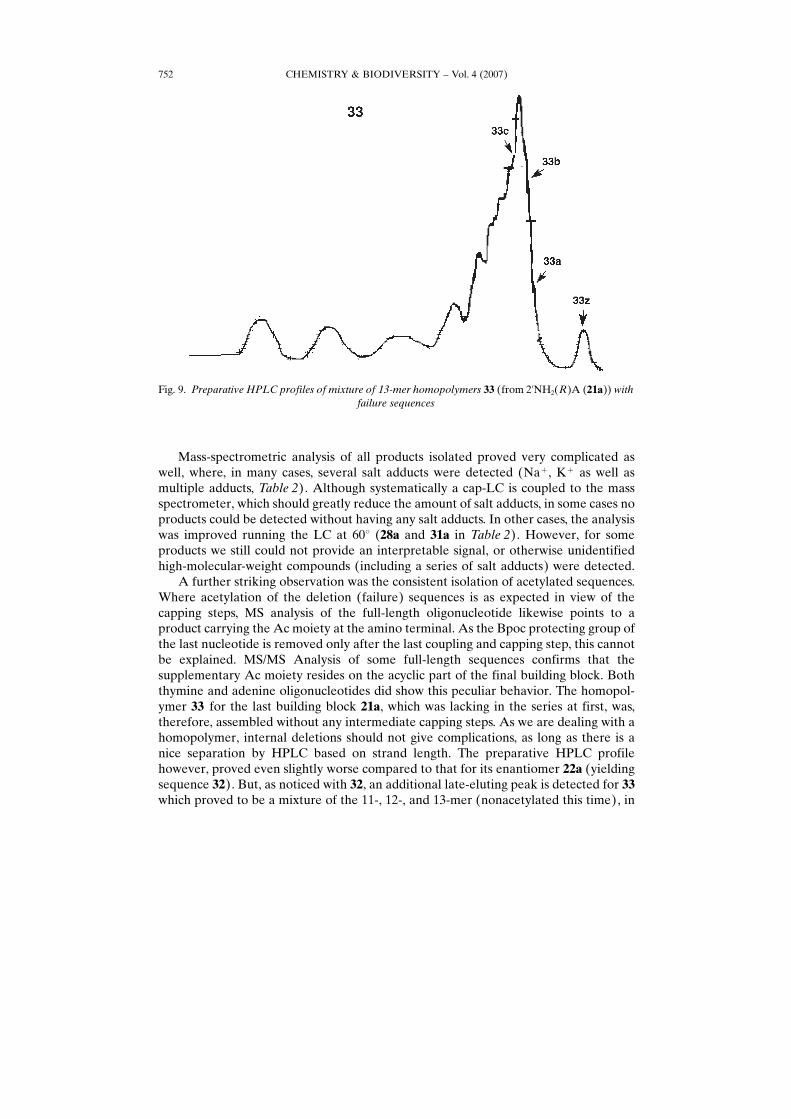
A further striking observation was the consistent isolation of acetylated sequences.Where acetylation of the deletion (failure) sequences is as expected in view of thecapping steps, MS analysis of the full-length oligonucleotide likewise points to aproduct carrying the Ac moiety at the amino terminal. As the Bpoc protecting group ofthe last nucleotide is removed only after the last coupling and capping step, this cannotbe explained. MS/MS Analysis of some full-length sequences confirms that thesupplementary Ac moiety resides on the acyclic part of the final building block. Boththymine and adenine oligonucleotides did show this peculiar behavior. The homopol-ymer 33 for the last building block 21a, which was lacking in the series at first, was,therefore, assembled without any intermediate capping steps. As we are dealing with ahomopolymer, internal deletions should not give complications, as long as there is anice separation by HPLC based on strand length. The preparative HPLC profilehowever, proved even slightly worse compared to that for its enantiomer 22a (yieldingsequence 32). But, as noticed with 32, an additional late-eluting peak is detected for 33which proved to be a mixture of the 11-, 12-, and 13-mer (nonacetylated this time), in
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)752
Fig. 9. Preparative HPLC profiles of mixture of 13-mer homopolymers 33 (from 2’NH2(R)A (21a)) withfailure sequences

about the same ratio as for the last eluting regular peak (Fig. 9). Again, we cannotexplain this peculiar behavior, but it hints at formation of complexes (as is the detectionof the correct mass of 28 and 30 only at 608).
However, it is clear now that the capping step is not the cause of the irregular HPLCprofiles. MS/MS Experiments at different voltages could not detect rearrangedfragments (i.e., phosphoramidates to phosphodiesters for 2’-APNA). The P�N linkageproved to be stable and only PO�
3 could be detected. In case of rearrangement, amixture of posphodiesters would have generated H2PO�
4 fragments, which were notdetected either in our products. We, therefore, have no explanation for the unusualHPLC profiles of the 2’-aminopropyl polymers with broad or multiple peaks.
Overall, it can be concluded that the synthetic strategy for these acyclicphosphoramidate oligonucleotides is still not optimal.
Pairing Properties of the Acyclic Phosphoramidates. Incorporation of a singlemodification into oligonucleotides caused a substantial drop in affinity [2]. Incorpo-ration of several dimers, resulting in acyclic phosphoramidate linkages alternating withregular deoxyribose phosphodiesters, had an additive effect [2]. The homothymidinepolymers synthesized here, which contained a single, triple, or five-fold modification,allowed to study the effect of consecutive acyclic building blocks. The affinity ofsequences 23–25 for DNA as well as RNA was studied by thermal denaturation(Table 3). At physiological conditions (0.1m NaCl), only sequence 23 with a singlemodification displayed a measurable Tm, but only at high salt conditions (1m NaCl) anice transition could be detected. Even at these high salt conditions, the multiplymodified sequences 24 and 25 did not display a detectable interaction with either aDNA or RNA complement. As a control sequence, we did have a (dT)10 available. It isclear, however, that consecutive incorporations of acyclic phosphoramidates have adeleterious effect on the pairing potential of these oligonucleotides.
The minute quantities available of the fully modified sequences were used to studyself-pairing and base pairing between the different acyclic oligomers, as well asinteraction with DNA and RNA complements (except for (R)-2’-APNA with athymine base which was not available). Unfortunately, no interactions whatsoevercould be detected by thermal denaturation experiments. One of the questions thatcould be asked is whether APNAs are able to form A- or B-type helixes withoutgenerating steric clashes due to the presence of the NH groups. Therefore, a static
Table 3. Thermal Stability for Incorporation of One, Three, and Five Monomeric Units 19b (T*) intoDNAOligomers
Oligonucleotide Target (dA)13
0.1m NaCl(dA)13
1m NaCl(rA)13
1m NaClTm [8] Tm [8] Tm [8]
5’-TTTTTT*TTTTT-3’ (23) 12.5 27.2 28.55’-TTTTT*T*T*TTTT-3’ (24) <10 <10 <105’-TTTT*T*T*T*T*TTT-3’ (25) –a) –a) –a)5’-TTTTTTTTTTT-3’ (dT)10 25.5 39.0 35.0
a) No interaction detectable.
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 753

model of (S)-2’-APNA in complex with RNA was built. This model was constructedstarting from the X-ray structure of a DNA·TNA hybrid [13]. The isolated TNAresidues (thymine base) were fit on a poly T ·A duplex (in the A-type conformation) inorder to obtain a model of an (S)-GNA·RNA duplex (by removing part of the sugarmoieties of the TNA residues) [14]. The 2’-O-atoms were then exchanged for NHgroups [15]. Introduction of the NH group does not give steric clashes in the model(Fig. 10), although the NH atoms are situated in a rather hydrophobic environment.The (S)-2’-APNA is sterically possible. Although, given the potential for the formationof intramolecular H-bonds involving the NH groups which are now located in ahydrophobic environment, it is very unlikely that 2’-APNAwill be pre-organized in thishelix structure, and a more dynamic model is needed to analyze the potentialalternative conformations.
Fig. 10. Picture of the (S)-2’-APNA backbone which might occur in a 2’-APNA·RNA duplex showing nosteric clash for the NH atom. This structure is highly unlikely given the absence of H-bonds.
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)754

Conclusions. – In conclusion, two series of aminopropyl nucleosides 3’-APNA and2’-APNA were obtained with a Bpoc protecting group. With this new acid labileprotecting group, the nucleophilicity of the N-atom of the respective building blockshas been reduced during the oligonucleotide assembly. However, because the amine-exchange coupling step in phosphoramidate synthesis is an equilibrium reaction, thesynthesis of fully modified oligomers was not highly efficient. The adenine polymersand polymers comprising 2’-aminopropyl analogues performed slightly better, whichcan also be observed by the HPLC purification profiles and mass spectrometry analysis.
The study of the effect of consecutive acyclic building blocks in homothymidinepolymers showed that their incorporation decreased the stability of the duplex withcomplementary DNA. Unfortunately, no interactions (as measured by hypochromicitystudies) could be detected by thermal denaturation experiments for self-pairing,homochiral pairing between APNAs, or cross-pairing with DNA or RNA. (It should benoted that we do not have information for (R)-2’-APNA with thymine base.)
Interesting observations from this research project are: a) the efficiency of thesynthesis of the aminopropyl nucleic acids is as well base- as backbone-dependent; b) itis very difficult to isolate a single oligomer by HPLC which might be due to self-aggregation or rearrangement; c) side reactions easily occur during the oligomerizationreaction, which, itself, is a non-efficient process using phosphoramidite chemistry.These results are in contrast to the ease of synthesis and isolation of the oxygencongener, GNA [3]. Apparently, the introduction of the N-atom converts GNA into apotential constituent of a reaction mixture with mainly catalytic power [1]. Given theirsimple constitution, the prebiotic synthetic contingency of APNAs is reasonable.However, their structure might not be very well suited to function as informationsystem with the ability to communicate with RNA or to evolve into RNA. It is notunreasonable to suggest that APNAs may occur in conformations which do not belongto the typical DNA helical structure due to the formation of intramolecular H-bonds. Incontrast, oligomers such as APNAs might be potential catalytic polymers fitting in ametabolism-first scenario (where prebiotic nucleic acids might have played a role ascatalyst and evolved in a later stage to a genetic system). However, to further provesuch a hypothesis, new synthetic methods need to be developed to increase theefficiency of the coupling reaction and to obtain longer sequences with mixed bases toanalyze structure, hybridization properties, and catalytic properties of APNA in itsdifferent regio- and stereoisomeric forms.
Experimental Part
General. For all reactions, anal. grade solvents were used. Column chromatography (CC): ICN silicagel 60-200 60A. TLC: Merck silica gel 60 F254 aluminium sheets; visualization under UV or by sprayingwith H2SO4/anisaldehyde. Optical rotations: Optical Activity AA-10 polarimeter. 1H-NMR Spectra:Varian Unity 500-MHz spectrometer; TMS as internal standard; d in ppm, J in Hz. 13C-NMR Spectra:Varian Gemini 200-MHz apparatus; in (D6)DMSO (39.6 ppm) or CDCl3 (76.9 ppm), using the solventpeaks as reference; d in ppm. HR-MS: Quadrupole time-of-flight mass spectrometer (Q-Tof-2,Micromass, UK-Manchester) equipped with a standard electrospray-ionization (ESI) interface; sampleswere infused in i-PrOH/H2O 1 :1 at 3 ml/min; in m/z.
1-([1,1’-Biphenyl]-4-yl)-1-methylethyl (R)/(S)-3-[6-(Benzoylamino)-9H-purin-9-yl]-2-hydroxypro-pylcarbamate (3a/5a). To a soln. of 2a/4a (2a, 0.94 g, 2.19 mmol; 4a, 0.81 g, 1.90 mmol) in 10 ml of dry
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 755

dioxane was injected Triton B (4 wt-% benzyl(trimethyl)ammonium hydroxide soln. in MeOH; 2a,0.97 ml, 2.19 mmol; 4a, 0.84 ml, 1.90 mmol). The soln. was concentrated under vacuum (water bath�408). Then, the residue was dissolved in 5 ml of DMFand then co-evaporated with 15 ml of dry dioxanetwice. The residue was dissolved in 3 ml of DMFand 15 ml of dioxane. Then, the Bpoc reagent (2a, 1.11 g,2.85 mmol; 4a, 0.96 g, 2.47 mmol) was added, and the mixture was stirred at 558 for 3 h. The mixture wasquenched with 25 ml of sat. NaHCO3 soln. and then extracted with CH2Cl2 (2�20 ml). The org. layer wasdried (Na2SO4) and concentrated, and the residue was purified by CC (CH2Cl2/MeOH/Et3N 98 :1.5 : 0.5).
Yields: 3a : 0.53 g (44%); 5a : 0.50 g (48%). [a]20D (3a)¼ �2.4 (c¼1.1, DMF); [a]20
D (5a)¼ þ2.2 (c¼1.0, DMF). Rf (CH2Cl2 :MeOH 95 :5) 0.56. 1H-NMR ((D6)DMSO): 1.74 (s, 2 Me); 3.02 (m, CH2(3’); 3.89(m, H�C(2’)); 4.02–4.10 (m, Ha�C(1’)); 4.33–4.38 (m, Hb�C(1’)); 5.31–5.33 (d, J ¼ 3.4; OH); 7.30–7.36 (m, 2 H of Bpoc); 7.40–7.48 (m, 4 H of Bpoc); 7.52–7.66 (m, H�C(3,4,5) of Bz, 3 H of Bpoc); 8.04–8.06 (d, J¼5.0, H�C(2,6) of Bz); 8.36 (s, H�C(8)); 8.73 (s, H�C(2)); 11.14 (s, NH). 13C-NMR((D6)DMSO): 29.2 (Me of Bpoc); 29.3 (Me of Bpoc); 44.4 (C(3’)); 47.4 (C(1’)); 68.2 (C(2’)); 79.7 (Me2Cof Bpoc); 125.0 (C(5)); 125.4, 126.5 126.7, 127.4 (C of Bpoc); 128.6 (H�C(3,5) of Bz); 129.1 (H�C(2,6)of Bz); 132.5 (H�C(4) of Bz); 140.1 (C(1) of Bz); 138.5, 140.1, 145.6 (C of Bpoc); 146.4 (C(8)); 150.1(C(4)); 151.4 (C(2)); 152.8 (C(6)); 155.3 (C¼O). HR-ESI-MS: 551.2419 ([MþH]þ , C31H31N6Oþ
4 ; calc.551.2407).
1-([1,1’-Biphenyl]-4-yl)-1-methylethyl (R)/(S)-2-Hydroxy-3-(5-methyl-2,4-dioxo-3,4-dihydro-1-pyri-midin-1(2H)-yl)propylcarbamate (3b/5b). To a soln. of 2b/4b (2b, 620 mg, 1.98 mmol; 4b, 530 mg,1.70 mmol) in 10 ml of dry dioxane was injectedTriton B (40 wt-% benzyltrimethylammonium hydroxidesoln. in MeOH; 2b, 0.87 ml, 1.98 mmol; 4b, 0.75 ml, 1.70 mmol). The soln. was concentrated in vacuo(water bath �408). Then, the residue was dissolved in 5 ml of DMF and then co-evapotated with 15 mldry dioxane twice. The residue was dissolved in 3 ml of DMF and 15 ml of dioxane. Then, the Bpocreagent (2b, 1.0 g, 2.57 mmol; 4b, 860 mg, 2.21 mmol) was added, and the mixture was stirred at 558 for3 h. The reaction was quenched with 25 ml of sat. NaHCO3, and then the mixture was extracted withCH2Cl2 (2�20 ml). The org. layer was dried (Na2SO4) and concentrated, and the residue was purified byCC (CH2Cl2/MeOH/Et3N 98 :1.5 : 0.5).
Yields: 3b : 450 mg (52%); 5b : 370 mg (50%). [a]20D (3b)¼ �13.8 (c¼0.97, DMF); [a]20
D (5b)¼þ14.1 (c¼0.5, DMF). Rf (CH2Cl2/MeOH 95 :5) 0.55. 1H-NMR (CDCl3): 1.79 (s, Me of T); 1.81 (s, 2 Meof Bpoc); 3.44–3.47 (m, CH2(3’)); 3.49–3.51 (dd, J(1’a,2’)¼7.0, Jgem¼14.0, Ha�C(1’)); 3.83–3.87 (dd,Hb�C(1’)); 4.02 (m, H�C(2’)); 4.24 (d, OH); 5.58 (q, J¼5.6, NH�C¼O, NH�C¼O); 7.02 (s, 1 H of T);7.32–7.34 (m, 2 H of Bpoc); 7.39–7.46 (m, 4 H of Bpoc); 7.54–7.57 (m, H�C(3,4,5) of Bz, 1 H of Bpoc);9.66 (br. s, NH of T). 13C-NMR (CDCl3): 12.1 (Me of T); 29.0 (Me of Bpoc); 43.8 (C(3’)); 51.9 (C(1’));69.4 (C(2’)); 81.2 (CMe2 of Bpoc); 109.8 (C(5)); 124.7, 127.0, 127.5, 128.7, 139.7, 140.6 (C of Bpoc); 142.4(C(6)); 145.3 (C of Bpoc); 151.7 (C(2)); 156.5 (C¼O); 164.7 (C(4)). HR-ESI-MS: 438.2018 ([MþH]þ ,C24H28N3Oþ
5 ; calc. 438.2029).N-[9-((R)-2-Azido-3-{[(tert-butyl)dimethylsilyl]oxy}propyl)-9H-purin-6-yl]benzamide (10a). Com-
pound 8a (3.88 g, 6.36 mmol, in Scheme 2) was treated with 2% TsOH in CH2Cl2 and MeOH (2 :1, 50 ml)for 3 h, the reaction was quenched with 30 ml of sat. NaHCO3 aq. soln., and then the mixture wasextracted with CH2Cl2 (3�20 ml). The org. layer was dried (Na2SO4) concentrated, and co-evaporatedwith toluene twice. The residue was dissolved in 25 ml of DMF. Then, 1H-imidazole (1.08 g, 15.9 mmol)and TBDMSCl (1.92 g, 12.7 mmol) were added, and the mixture was stirred overnight. The mixture wasconcentrated in vacuo, and the residue was dissolved in 30 ml of CH2Cl2. The soln. was washed with H2O(3�20 ml). The org. layer was dried (Na2SO4) and concentrated, and the residue was purified by CC(CH2Cl2). 2.1 g (73%) of 10a. 1H-NMR (CDCl3): 0.08 (s, 2 Me); 0.90 (s, 3 Me); 3.68–3.74 (dd, J(3’a,2’)¼5.7, Jgem¼10.0, Ha�C(3’)); 3.85–3.90 (dd, J(3’b,2’)¼3.0, Jgem¼12.4, Hb�C(3’)); 3.95 (m, H�C(2’));4.16–4.23 (dd, J(1’a,2’)¼8.1, Jgem¼14.0, Ha�C(1’)); 4.42–4.48 (dd, J(1’b,2’)¼3.8, Jgem¼14.0, Hb�C(1’));7.44–7.58 (m, H�C(3,4,5) of Bz); 8.00–8.04 (d, J¼7.5, H�C(2,6) of Bz); 8.07 (s, H�C(8)); 8.73 (s,H�C(2)); 9.29 (s, NH). 13C-NMR (CDCl3): �5.7, �5.6 (Me2Si); 18.1 (SiC); 25.7 (Me3C); 43.9 (C(1’));61.4 (C(3’)); 63.5 (C(2’)); 122.8 (C(5)); 127.8, 128.7, 132.6, 133.6 (C of Bz); 143.5 (C(8)); 149.5 (C(4));152.1 (C(2)); 152.6 (C(6)); 164.7 (C¼O). HR-ESI-MS: 453.2199 ([MþH]þ , C21H29N8O2Siþ ; calc.453.2183).
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)756

N-[9-((R)-2-Amino-3-{[(tert-butyl)dimethylsilyl]oxy}propyl)-9H-purin-6-yl]benzamide (11a). Asoln. of 10a (2.0 g, 4.42 mmol) in 15 ml of THF was treated with 10% Pd/C and placed under H2
atmosphere. The mixture was stirred for 3 h and filtered throughCelite. The solid was rinsed with MeOH,and the combined filtrates were evaporated under reduced pressure. The residual oil was purified by CC(CH2Cl2/MeOH 92 :8): 1.50 g (80%) of 11a. 1H-NMR (CDCl3): 0.08 (s, 2 Me); 0.93 (s, 3 Me); 1.76 (br. s,NH2); 3.33 (m, H�C(2’)); 3.57–3.58 (d, J¼4.4, CH2(1’)); 4.15–4.22 (dd, J(3’a,2’)¼7.4, Jgem¼14.0,Ha�C(3’)); 4.37–4.44 (dd, J(3’b,2’)¼4.6, Jgem¼14.0, Hb�C(3’)); 7.48–7.61 (m, H�C(3,4,5) of Bz); 8.01–8.04 (d, J¼7.5, H�C(2,6) of Bz); 8.11 (s, H�C(8)); 8.77 (s, H�C(2)); 9.29 (s, NH). 13C-NMR (CDCl3):�5.5 (Me2Si); 18.2 (SiC); 25.8 (Me3C); 47.5 (C(1’)); 52.3 (C(2’)); 65.2 (C(3’)); 122.9 (C(5)); 127.9, 128.7,132.6, 133.7 (C of Bz); 143.9 (C(8)); 149.4 (C(4)); 152.4 (C(2)); 152.6 (C(6)); 164.7 (C¼O). HR-ESI-MS:427.2279 ([MþH]þ , C21H31N6O2Siþ ; calc. 427.2278).
1-[1,1’-Biphenyl]-4-yl-1-methylethyl (R)-2-[6-(Benzoylamino)-9H-purin-9-yl]-1-(hydroxymethyl)-ethylcarbamate (12a). To a soln. of 11a (680 mg, 1.58 mmol) in 10 ml of dry dioxane was injected TritonB (40 wt-% in MeOH, 0.70 ml, 2.58 mmol). The soln. was concentrated in vacuo (water bath �408).Then, the residue was dissolved in 5 ml of DMF, and 15 ml of dry dioxane was added twice, and themixture was evaporated to remove possible traces of H2O. To the remaining DMF soln. were added 15 mlof dioxane and the Bpoc reagent (807 mg, 2.07 mmol), and the mixture was stirred at 558 for 3 h. Thereaction was quenched with 25 ml of sat. NaHCO3 and then the mixture was extracted with CH2Cl2 (3�20 ml). The org. layer was dried (Na2SO4) and concentrated, and the residue was purified by CC(CH2Cl2/MeOH/Et3N 97 :2.5 :0.5): 0.40 g (46%) of 12a. Rf (CH2Cl2/MeOH 95 :5) 0.46. [a]20
D ¼ þ8.2 (c¼0.47, DMF). 1H-NMR ((D6)DMSO): 1.53, 1.62 (s, 2 Me); 3.46 (m, CH2(1’)); 3.90 (m, H�C(2’)); 4.18–4.26 (m, Ha�C(3’)); 4.48–4.53 (m, Hb�C(3’)); 5.06 (q, J ¼ 3.4, OH); 7.20–7.22 (m, 2 H of Bpoc); 7.30–7.40 (m, 4 H of Bpoc); 7.51–7.67 (m, H�C(3,4,5) of Bz, 3 H of Bpoc); 8.04–8.06 (d, J¼7.5, H�C(2,6) ofBz); 8.36 (s, H�C(8)); 8.73 (s, H�C(2)); 11.18 (s, NH). 13C-NMR ((D6)DMSO): 28.9 (Me of Bpoc); 30.0(Me of Bpoc); 44.9 (C(1’)); 52.6 (C(2’)); 62.0 (C(3’)); 80.0 (OCMe2 of Bpoc); 124.9 (C(5)); 125.6, 126.8127.0, 127.7 (C of Bpoc); 128.9 (H�C(3,5) of Bz); 129.2 (H�C(2,6) of Bz); 132.8 (H�C(4) of Bz); 140.4(C(1) of Bz); 134.2, 138.7, 145.2 (C of Bpoc); 146.4 (C(8)); 150.5 (C(4)); 151.6 (C(2)); 153.2 (C(6)); 154.8(C¼O). HR-ESI-MS: 551.2419 ([MþH]þ , C31H31N6Oþ
4 ; calc. 551.2407).1-{(S)-2-Azido-3-[(4-methoxyphenyl)(diphenyl)methoxy]propyl}-5-methylpyrimidine-2,4(1H,3H)-
dione (8b). To a soln. of 6b (5.38 g, 11.4 mmol) in 30 ml of dry pyridine was added MsCl (1.43 g,12.5 mmol). The mixture was stirred for 2 h and the reaction was quenched by addition of sat. aq.NaHCO3 (30 ml), and then the mixture was extracted with CH2Cl2 (2�30 ml). The org. layer was dried(Na2SO4) and evaporated, and the residue was co-evaporated twice with toluene. The residue wasdissolved in 20 ml of DMF. Then, NaN3 (1.11 g, 17.1 mmol) was added, and the mixture was stirred at 1008overnight. The mixture was concentrated, and the residue was dissolved in CH2Cl2 and washed with sat.aq. NaHCO3 (2�30 ml). The org. layer was dried (Na2SO4) and concentrated in vacuo. The residue waspurified by CC (CH2Cl2): 3.81 g (67%) of 8b. Rf (CH2Cl2/MeOH 99 :1) 0.25. 1H-NMR (CDCl3): 1.87 (s,Me of T); 3.15–3.22 (dd, J(3’a,2’)¼5.6, Jgem¼10.0, Ha�C(3’)); 3.34–3.39 (dd, J(3’b,2’)¼3.4, Jgem¼10.0,Hb�C(3’)); 3.41–3.47 (dd, J(1’a,2’)¼4.2, Jgem¼9.3, Ha�C(1’)); 3.79 (s, Me); 3.94 (m, H�C(2’)); 3.90–4.01 (dd, J(1’b,2’)¼4.4, Jgem¼9.3, Hb�C(1’)); 6.83–6.84 (d, 2 H of MTr); 6.95–6.96 (d, J ¼ 2.4, H�C(6));7.20–7.47 (m, 12 H of MTr); 9.38 (br. s, NH of T). 13C-NMR (CDCl3): 12.2 (Me); 49.3 (C(3’)); 55.3 (Me);60.2 (C(2’)); 63.4 (C(1’)); 87.1 (C of MTr); 110.5 (C(5)); 113.3 (C of MTr); 127.2, 128.0, 128.2, 130.3, 134.8,141.2, 158.8 (C of MTr); 143.7 (C(6)); 151.0 (C(2)); 164.6 (C(4)). HR-ESI-MS: 520.1962 ([MþNa]þ ,C28H27N5NaOþ
4 ; calc. 520.1960).(S)-2,3-Dihydro-2-{[(4-methoxyphenyl)(diphenyl)methoxy]methyl}-6-methyl-7H-[1,3]oxazolo[3,2-
a]pyrimidin-7-one (8a’). Yield: 3.81 g (67%). Rf (CH2Cl2/MeOH 98 :2) 0.33. 1H-NMR (CDCl3): 2.01 (s,Me of T); 3.17–3.21 (dd, J(3’a,2’)¼3.0, Jgem¼10.9, Ha�C(3’)); 3.65–3.70 (dd, J(3’b,2’)¼3.3, Jgem¼10.9,Hb�C(3’)); 3.80 (s, Me of MMTr); 3.97–4.02 (dd, J(1’a,2’)¼5.4, Jgem¼9.3, Ha�C(1’)); 4.18–4.24 (t, J¼9.3, Hb�C(1’)); 5.00 (m, H�C(2’)); 6.82–6.85 (d, J¼8.8, 2 H of MTr); 7.12 (s, H�C(6)); 7.22–7.37 (m,12 H of MTr). 13C-NMR (CDCl3): 14.0 (Me); 48.3 (C(3’)); 55.2 (Me); 63.4 (C(1’)); 77.2 (C(2’)); 86.8 (C ofMTr); 113.3 (C of MTr); 118.6 (C(5)); 127.2, 128.0, 128.1, 130.2, 134.4, 143.3, 158.8 (C of MTr); 143.6(C(6)); 151.0 (C(2)); 160.6 (C(4)). HR-ESI-MS: 455.1964 ([MþNa]þ , C28H27N2Oþ
4 ; calc. 455.1971).
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 757

1-((S)-2-Azido-3-{[(tert-butyl)dimethylsilyl]oxy}propyl)-5-methylpyrimidine-2,4(1H,3H)-dione(10b). Synthesis of 10b was accomplished as described for 10a starting from 8b (3.38 g, 6.80 mmol): 1.5 g(65%) of 10b. 1H-NMR (CDCl3): 0.10 (s, 2 Me); 0.92 (s, 3 Me); 1.93 (s, Me of T); 3.45–3.52 (dd,J(3’a,2’)¼8.2, Jgem¼13.8, Ha�C(3’)); 3.68–3.73 (dd, J(1’b,2’)¼4.5, Jgem¼9.8, Hb�C(1’)); 3.80–3.89 (m,H�C(2’), Ha�C(1’)); 3.97–4.03 (dd, J(3’b,2’)¼3.9, Jgem¼13.8, Hb�C(3’)); 7.05 (s, H�C(6) of T); 9.57 (s,HN of T). 13C-NMR (CDCl3): �5.7, �5.6 (Me2Si); 12.2 (Me of T); 18.1 (SiC); 25.7 (Me3C); 48.8(C(1’)); 61.0 (C(3’)); 63.5 (C(2’)); 110.5 (C(5)); 141.2 (C(6)); 151.0 (C(2)); 164.6 (C(4)). HR-ESI-MS:340.1801 ([MþH]þ , C14H26N5O3Siþ ; calc. 340.1805).
1-((S)-2-Amino-3-{[(tert-butyl)dimethylsilyl]oxy}propyl)-5-methylpyrimidine-2,4(1H,3H)-dione(11b). The protected derivative 11b was obtained as described for 11a starting from 10b (1.09 g,3.19 mmol): 800 mg (80%) of 11b. 1H-NMR (CDCl3): 0.07 (s, 2 Me); 0.90 (s, 3 Me); 1.89 (s, Me of T);3.20 (m, H�C(2’)); 3.56–3.58 (d, J¼4.4, CH2(1’); 3.58–3.67 (dd, J(3’a,2’)¼5.2, Jgem¼13.6, Ha�C(3’));3.70–3.82 (dd, J(3’b,2’)¼5.6, Jgem¼13.6, Hb�C(3’)); 7.09 (s, H�C(6) of T). 13C-NMR (CDCl3): �5.7, �5.6 (Me2Si); 12.1 (Me of T); 18.1 (SiC); 25.7 (Me3C); 51.5 (C(1’)); 51.7 (C(3’)); 65.1 (C(2’)); 110.0(C(5)); 141.7 (C(6)); 151.5 (C(2)); 164.6 (C(4)). HR-ESI-MS: 314.1897 ([MþH]þ , C14H28N3O3Siþ ; calc.314.1900).
1-([1,1’-Biphenyl]-4-yl)-1-methylethyl (S)-2-Hydroxy-1-[(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl]ethylcarbamate (12b). Synthesis of 12b was accomplished as described for 12a startingfrom compound 11b (620 mg, 1.98 mmol): 0.53 g (44%) of 12b. [a]20
D ¼ �13.7 (c¼0.8, DMF). 1H-NMR(CDCl3): 1.57, 1.67 (s, 2 Me of Bpoc); 1.79 (s, Me of T); 3.33 (m, CH2(3’); 3.73–3.74 (m, H�C(2’)); 3.94–4.00 (dd, J¼3.3, Jgem¼13.6, CH2(1’)); 4.88 (q, J¼5.4, OH); 6.92–6.95 (d, J¼9.3, 1 H of T); 7.32–7.40 (m,2 H of Bpoc); 7.43–7.47 (m, 3 H of Bpoc); 7.52–7.55 (d, J¼7.8, 2 H of Bpoc); 7.63–7.65 (d, J¼7.5, 2 H ofBpoc); 11.20 (br. s, NH of T). 13C-NMR (CDCl3): 12.6 (Me of T); 28.8, 30.2 (Me of Bpoc); 49.7 (C(1’));51.3 (C(3’)); 62.2 (C(2’)); 79.9 (C of Bpoc); 108.0 (C(5)); 124.9, 126.7, 127.1, 127.7, 129.3, 138.7, 140.4 (C ofBpoc); 142.8 (C(6)); 146.6 (C of Bpoc); 151.3 (C(2)); 155.0 (C¼O); 164.9 (C(4)). HR-ESI-MS: 438.2023([MþH]þ , C24H28N3Oþ
5 ; calc. 438.2029).({(S)-2-Azido-3-[(4-methoxyphenyl)(diphenyl)methoxy]propyl}oxy)(tert-butyl)dimethylsilane (14).
Compound 13 (8.79 g, 22.6 mmol) was dissolved in DMF (20 ml) under cooling with an ice-bath, and 1H-imidazole (3.84 g, 56.5 mmol) was added, followed by TBDMSCl (5.40 g, 34.2 mmol). Stirring at r.t. wascontinued overnight. The reaction was quenched with 50 ml of sat. NaHCO3, and then the mixture wasextracted with hexane (2�40 ml). The org. layer was dried (Na2SO4) and concentrated, the residue waspurified by CC (CH2Cl2/hexane 1 :1): 10.8 g (95%) of 14. 1H-NMR (CDCl3): 0.08, 0.09 (s, Me2Si); 0.90 (s,3 Me); 3.21–3.35 (m, CH2OSi); 3.56–3.68 (m, CHN3); 3.74–3.77 (d, J¼5.6, CH2OC); 3.83 (s, MeO);6.87–6.91 (d, J¼7.0, 2 H of MMTr); 7.28–7.42 (m, 8 H of MMTr); 7.51–7.54 (d, J¼7.0, 2 H of MMTR).13C-NMR (CDCl3): �5.7 (Me2Si); 18.0 (SiC); 25.6 (Me3C); 55.1 (MeO); 63.0 (COSi); 63.1 (CN3); 63.3(COMMTr); 86.7 (C of MMTr); 119.0 (C(5)); 141.7 (C(8)); 149.9 (C(2)); 152.6 (C(6)); 156.2 (C(4)). HR-ESI-MS: 526.2524 ([MþNa]þ , C29H37N3NaO3Siþ ; calc. 526.2502).
(S)-2-Azido-3-{[(tert-butyl)dimethylsilyl]oxy}propan-1-ol (15). Compound 14 (9.64 g, 19.2 mmol)was treated with 1% TsOH (60 ml) in an ice-bath for 1 h. The mixture was worked up by addition of sat.aq. NaHCO3 soln. and extracted with CH2Cl2 (2�30 ml). After drying the org. layer (Na2SO4) andconcentrating, the residue was purified by CC (CH2Cl2): 3.40 g (77%) of 15. 1H-NMR ((D6)DMSO):�0.11 (s, Me2Si); 0.92 (s, 3 Me); 3.54–3.69 (m, CH2(3’)); 3.71–3.77 (m, H�C(2’)); 3.80–3.81 (m,CH2(1’)). 13C-NMR ((D6)DMSO): �5.6 (Me2Si); 18.2 (SiC); 25.6 (Me3C); 62.7 (C(1’)); 63.8 (C(3’));64.0 (C(2’)). HR-ESI-MS: 232.1475 ([MþH]þ , C9H22N3O2Siþ ; calc. 232.1481).
(S)-2-Azido-3-{[(tert-butyl)dimethylsilyl]oxy}propyl 4-Methylbenzenesulfonate (16a). To a soln. of15 (1.75 g, 7.54 mmol) in 20 ml of dry pyridine was added TsCl (1.54 g, 9.05 mmol). The mixture wasstirred for 4 h, the reaction was quenched by addition of sat. aq. NaHCO3 soln., and the mixture wasextracted with CH2Cl2 (2�30 ml). The org. layer was dried (Na2SO4) and concentrated, and the residuewas purified by CC (CH2Cl2/hexane 2 :1): 2.75 g (95%) of 16. 1H-NMR ((D6)DMSO): 0.04, 0.05 (s,Me2Si); 0.86 (s, 3 Me); 2.45 (s, MeO); 3.60 (m, H�C(2’)); 3.69–3.71 (d, J¼5.4, CH2(3’)); 3.99–4.05 (dd,J(3’a,2’)¼7.2, Jgem¼9.8, Ha�C(3’)); 4.12–4.16 (dd, J(3’b,2’)¼3.9, Jgem¼9.8, Hb�C(3’)); 7.35–7.37 (d, J¼7.5, 2 arom. H); 7.79–7.82 (d, J¼7.2, 2 arom. H). 13C-NMR ((D6)DMSO): �5.6 (Me2Si); 18.1 (SiC); 21.6
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)758

(MeO); 25.6 (Me3C); 60.8 (C(1’)); 62.4 (C(2’)); 68.2 (C(3’)); 128.0 130.0, 132.5, 145.2 (arom. C). HR-ESI-MS: 386.1575 ([MþH]þ , C16H28N3O4SSiþ ; calc. 386.1570).
N-[9-((S)-2-Azido-3-{[(tert-butyl)dimethylsilyl]oxy}propyl)-9H-purin-6-yl]benzamide (16c) . Amixture of adenine (1.38 g, 10.2 mmol) and NaH (80% dispersion in oil, 0.30 g, 10.2 mmol) wasdissolved in DMF (40 ml) and stirred for 90 min at 808. Compound 16a (2.38 g, 6.80 mmol) in DMF(2 ml) was added dropwise, and the mixture was stirred at 1008 overnight. Following filtration throughCelite, the solvent was evaporated. The residue was dissolved in CH2Cl2 (30 ml) and washed with H2O(2�20 ml). The aq. layer was extracted with CH2Cl2 (30 ml). The combined org. layer was dried(Na2SO4) and concentrated to yield the crude 16b. Compound 16b was dissolved in pyridine (20 ml) andcooled to 08, and BzCl (1.81 g, 12.9 mmol) was added. Stirring at 08 was continued for 1 h after which themixture was allowed to warm to r.t. overnight. After quenching the mixture with MeOH, the volatileswere removed. The residue was cooled and treated with sat. NH3/MeOH (20 ml) for 30 min. Afterevaporation of the solvent, the remaining residue was dissolved in CH2Cl2 (20 ml) and washed with sat.aq. NaHCO3 soln. (2�10 ml). The org. layer was dried (Na2SO4) and concentrated, and the residue waspurified by CC (CH2Cl2) to afford 1.60 g (52%) of 16c. 1H-NMR (CDCl3): 0.09 (s, 2 Me); 0.90 (s, 3 Me);3.69–3.74 (dd, J(3’a,2’)¼6.0, Jgem¼9.8, Ha�C(3’)); 3.86–3.95 (m, Hb�C(3’), H�C(2’)); 4.16–4.24 (dd,J(1’a,2’)¼8.0, Jgem¼14.2, Ha�C(1’)); 4.43–4.49 (dd, J(1’b,2’)¼3.6, Jgem¼14.0, Hb�C(1’)); 7.44–7.59 (m,H�C(3,4,5) of Bz); 8.01–8.05 (d, J¼7.9, H�C(2,6) of Bz); 8.07 (s, H�C(8)); 8.75 (s, H�C(2)); 9.25 (s,NH). 13C-NMR (CDCl3): �5.6, �5.6 (Me2Si); 18.2 (SiC); 25.7 (Me3C); 44.0 (C(1’)); 61.5 (C(3’)); 63.5(C(2’)); 122.8 (C(5)); 127.8, 128.7, 132.7, 133.6 (C of Bz); 143.5 (C(8)); 149.5 (C(4)); 152.1 (C(2)); 152.6(C(6)); 164.8 (C¼O). HR-ESI-MS: 453.2154 ([MþH]þ , C21H29N8O2Siþ ; calc. 453.2183).
1-((S)-2-Azido-3-{[(tert-butyl)dimethylsilyl]oxy}propyl)-5-methylpyrimidine-2,4(1H,3H)-dione(10b). Compound 15 (1.35 g, 5.85 mmol), N3-benzoylthymine (1.90 g, 6.49 mmol), and Ph3P (3.07 g,11.7 mmol) were dissolved in dry THF (20 ml) and cooled to 08. DIAD (2.36 g, 5.20 mmol) in THF (5 ml)was added dropwise over 1/2 h. The mixture was allowed to warm to r.t., and stirring was continued for 2more h. After concentrating the mixture, the residue was dissolved in CH2Cl2 (25 ml) and washed withH2O (2�20 ml). The org. layer was dried (Na2SO4), and the solvent was evaporated. The residual oil wastreated with NH3/MeOH for 3 h at r.t. After removal of the volatiles, the residue was purified by CC(CH2Cl2/MeOH 99 :1): 1.33 g (67%) of 10b. 1H-NMR (CDCl3): 0.12 (s, 2 Me); 0.93 (s, 3 Me); 1.94 (s, Meof T); 3.46–3.53 (dd, J(3’a,2’)¼8.3, Jgem¼13.9, Ha�C(3’)); 3.69–3.74 (dd, J(1’b,2’)¼5.0, Jgem¼10,Hb�C(1’)); 3.81–3.91 (m, H�C(2’), Ha�(1’)); 3.98–4.04 (dd, J(3’b,2’)¼4.0, Jgem¼13.9, Hb�C(3’)); 7.06(s, H�C(6) of T); 9.40 (s, HN of T). 13C-NMR (CDCl3): �5.7, �5.6 (Me2Si); 12.3 (Me of T); 18.2 (SiC);25.7 (Me3C); 48.8 (C(1’)); 61.1 (C(3’)); 63.5 (C(2’)); 110.5 (C(5)); 141.2 (C(6)); 151.0 (C(2)); 164.3(C(4)). HR-ESI-MS: 340.1808 ([MþH]þ , C14H26N5O3Siþ ; calc. for 340.1805).
N-[9-((S)-2-Amino-3-{[(tert-butyl)dimethylsilyl]oxy}propyl)-9H-purin-6-yl]benzamide (17a). Asoln. of 16a (1.46 g, 3.23 mmol) in 15 ml of THF was treated with 10% Pd/C and placed under a H2
atmosphere. The mixture was stirred for 3 h and filtered. The solid was rinsed with MeOH, and thecombined filtrates were evaporated under reduced pressure. The residual oil was purified by CC (CH2Cl2/MeOH 92 :8): 1.10 g (80%) of 17a. 1H-NMR (CDCl3): 0.09 (s, 2 Me); 0.94 (s, 3 Me); 1.78 (br. s, NH2);3.34–3.60 (m, H�C(2’)); 3.58–3.60 (d, J¼4.7, CH2(1’)); 4.16–4.23 (dd, J(3’a,2’)¼7.8, Jgem¼13.9,Ha�C(3’)); 4.39–4.45 (dd, J(3’b,2’)¼5.0, Jgem¼13.9, Hb�C(3’)); 7.50–7.61 (m, H�C(3,4,5) of Bz); 8.03–8.06 (d, J ¼ 7.3, H�C(2,6) of Bz); 8.12 (s, H�C(8)); 8.80 (s, H�C(2)); 9.17 (s, NH). 13C-NMR (CDCl3):�5.4 (Me2Si); 18.3 (SiC); 25.9 (Me3C); 47.5 (C(1’)); 52.4(C(2’)); 65.2 (C(3’)); 122.9 (C(5)); 127.9, 128.8,132.7, 133.7 (C of Bz); 143.9 (C(8)); 149.4 (C(4)); 152.4 (C(2)); 152.6 (C(6)); 164.7 (C¼O). HR-ESI-MS:427.2283 ([MþH]þ , C21H31N6O2Siþ ; calc. 427.2278).
1-[1,1’-Biphenyl]-4-yl-1-methylethyl (1S)-2-[6-(Benzoylamino)-9H-purin-9-yl]-1-(hydroxymethyl)-ethylcarbamate (18a). Synthesis of 18a was accomplished as described for 12a starting from 17a(1.06 g, 1.58 mmol). Yield: 0.40 g (46%). Rf (CH2Cl2/MeOH 95 :5) 0.46. [a]20
D ¼ �8.9 (c¼0.24, DMF).1H-NMR ((D6)DMSO): 1.53, 1.62 (s, 2 Me); 3.47 (m, CH2(1’)); 3.89 (m, H�C(2’)); 4.18–4.26 (m,Ha�C(3’)); 4.48–4.54 (m, Hb�C(3’)); 5.06 (q, J ¼ 3.5, OH); 7.20–7.22 (m, 2 H of Bpoc); 7.29–7.40 (m,4 H of Bpoc); 7.51–7.68 (m, H�C(3,4,5) of Bz, 3 H of Bpoc); 8.04–8.06 (d, J¼7.5, H�C(2,6) of Bz); 8.36(s, H�C(8)); 8.73 (s, H�C(2)); 11.16 (s, NH). 13C-NMR ((D6)DMSO): 29.0 (Me of Bpoc); 30.0 (Me ofBpoc); 44.9 (C(1’)); 52.5 (C(2’)); 62.1 (C(3’)); 80.0 (O�CMe2 of Bpoc); 125.0 (C(5)); 125.6, 126.9, 127.1,
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 759

127.8 (C of Bpoc); 128.9 (H�C(3,5) of Bz); 129.3 (H�C(2,6) of Bz); 132.8 (H�C(4) of Bz); 140.4 (C(1)of Bz); 134.3, 138.7, 145.2 (C of Bpoc); 146.5 (C(8)); 150.5 (C(4)); 151.6 (C(2)); 153.1 (C(6)); 154.9(C¼O). HR-ESI-MS: 551.2391 ([MþH]þ , C31H31N6Oþ
4 ; calc. 551.2407).1-((S)-2-Amino-3-{[(tert-butyl)dimethylsilyl]oxy}propyl)-5-methylpyrimidine-2,4(1H,3H)-dione
(11b). Synthesis of 11b was accomplished as described for 17a starting from 10b (1.23 g, 3.63 mmol).Yield: 910 mg (80%). 1H-NMR ((D6)DMSO): 0.08 (s, 2 Me); 0.92 (s, 3 Me); 1.91 (s, Me of T); 3.19 (m,H�C(2’)); 3.57–3.58 (d, J¼3.6, CH2(1’)); 3.58–3.65 (dd, J(3’a,2’)¼4.8, Jgem¼13.8, Ha�C(3’)); 3.69–3.81 (dd, J(3’b,2’)¼5.4, Jgem¼13.8, Hb�C(3’)); 7.09 (s, H�C(6) of T). 13C-NMR (CDCl3): �5.5, �5.5(Me2Si); 12.2 (Me of T); 18.2 (SiC); 25.8 (Me3C); 51.6 (C(1’)); 51.8 (C(3’)); 65.2 (C(2’)); 109.9 (C(5));141.6 (C(6)); 151.1 (C(2)); 164.1 (C(4)). HR-ESI-MS: 314.1912 ([MþH]þ , C14H28N3O3Siþ ; calc.314.1900).
1-([1,1’-Biphenyl]-4-yl)-1-methylethyl (S)-2-Hydroxy-1-[(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl]ethylcarbamate (12b). Synthesis of 12b was accomplished as described for 12a startingfrom 11b (620 mg, 1.98 mmol). Yield: 0.55 g (46%). [a]20
D ¼ �14.1 (c¼0.8, DMF). 1H-NMR (CDCl3):1.58, 1.68 (s, 2 Me of Bpoc); 1.80 (s, Me of T); 3.33 (m, CH2(3’)); 3.74 (m, H�C(2’)); 3.95–4.00 (dd, J¼3.5, Jgem¼13.6, CH2(1’)); 4.88 (q, J¼5.4, OH); 6.93–6.96 (d, J¼9.1, 1 H of T); 7.29–7.37 (m, 2 H ofBpoc); 7.43–7.48 (m, 3 H of Bpoc); 7.53–7.56 (d, J¼8.4, 2 H of Bpoc); 7.63–7.66 (d, J¼7.5, 2 H of Bpoc);11.21 (br. s, NH of T). 13C-NMR (CDCl3): 12.5 (Me of T); 28.7, 30.2 (Me of Bpoc); 49.8 (C(1’)); 51.3(C(3’)); 62.2 (C(2’)); 79.8 (C of Bpoc); 107.9 (C(5)); 124.9, 126.6, 127.0, 127.9, 129.3, 138.7, 140.3 (C ofBpoc); 142.7 (C(6)); 146.5 (C of Bpoc); 151.3 (C(2)); 155.0 (C¼O); 164.8 (C(4)). HR-ESI-MS: 460.1835([MþNa]þ , C24H27N3NaOþ
5 ; calc. 460.1848).General Procedure for Phosphoramidite Synthesis (19a (or 19b)–22a (or 22b)). Phosphitylation was
carried out on the derivatives 3a, 3b, 5a, 5b, 12a, 12b, and 18a, resp., in CH2Cl2 using freshly distilledEtN(i-Pr)2 and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite at 08 under Ar. The mixture wasstirred at 08 for 90 min after which completeness of the reaction was indicated by TLC. H2O (3 ml) wasadded, the soln. was stirred for another 10 min, and partitioned between CH2Cl2 (50 ml) and aq. NaHCO3
(30 ml). The org. layer was washed with brine (3�30 ml), and the aq. phases were back-extracted withCH2Cl2 (30 ml). After evaporation of the solvent, the resulting oil was purified by FC. Dissolution inCH2Cl2 (3 ml) and precipitation from cold hexane (160 ml, �608) was carried out twice to afford thedesired product as a white powder. The obtained product was dried in vacuo and stored under N2 at �208(22b and 22b’ are identical compounds from which the starting material was obtained by two differentroutes i.e., Schemes 2 and 3).
19a : 759 mg (94%). 31P-NMR: 149.28, 150.77. HR-ESI-MS: 749.3328 ([M�H]� , C40H46N8O5P� ;calc. 749.3329).
19b : 528 mg (75%). 31P-NMR: 149.34, 150.67. HR-ESI-MS: 636.2953 ([M�H]� , C33H43N5O6P� ;calc. 636.2951).
20a : 842 mg (92%). 31P-NMR: 149.30, 150.77. HR-ESI-MS: 749.3326 ([M�H]� , C40H46N8O5P� ;calc. 749.3329).
20b : 333 mg (64%). 31P-NMR: 149.37, 150.68. HR-ESI-MS: 636.2953 ([M�H]� , C33H43N5O6P� ;calc. 636.2951).
21a : 286 mg (63%). 31P-NMR: 150.66. HR-ESI-MS: 749.3317 ([M�H]� , C40H46N8O5P� ; calc.749.3329).
22a : 290 mg (78%). 31P-NMR: 149.39. HR-ESI-MS: 749.3346 ([M�H]� , C40H46N8O5P� ; calc.749.3329).
22b : 479 mg (77%). 31P-NMR: 149.35, 150.41. HR-ESI-MS: 636.2960 ([M�H]� , C33H43N5O6P� ;calc. 636.2951).
22b’: 235 mg (45%). 31P-NMR: 149.32, 150.38. HR-ESI-MS: 636.2943 ([M�H]� , C33H43N5O6P� ;calc. 636.2951).
Synthesis of Oligonucleotides. Oligonucleotide assembly was performed on an ExpediteTM DNAsynthesizer (Applied Biosystems) by using the phosphoramidite approach. The standard DNA assemblyprotocol was adjusted to double coupling and oxidation. The coupling time was prolonged to 3 min. Theoligomers were deprotected and cleaved from the solid support by treatment with MeNH2 (40% in H2O)/conc. aq. NH3 1 :1 (308). After gel filtration on a NAP-10S column (Sephadex G25-DNA grade;
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007)760

Pharmacia) with H2O as eluent, the crude was analyzed on a Mono-QS HR 5/5 anion exchange column,after which purification was achieved on a Mono-QS HR 10/10 column (Pharmacia) with the followinggradient system (A: 10 mm NaOH, pH 12.0, 0.1m NaCl; B: 10 mm NaOH, pH 12.0, 0.9m NaCl).
The low-pressure liquid chromatography system consisted of a Merck-Hitachi L 6200 A intelligentpump, a Mono-QS-HR 10/10 column (Pharmacia), a Uvicord SII 2138 UV detector (Pharmacia-LKB),and a recorder. The product-containing fraction was desalted on a NAP-10S column and lyophilized.
Oligonucleotides were characterized, and their purity was checked by HPLC/MS on a cap.chromatograph (CapLC, Waters, Milford, MA). Columns of 150 mm�0.3 mm length (LCPackings, SanFrancisco, CA) were used. Oligonucleotides were eluted with a triethylammonium/1,1,1,3,3,3-hexa-fluoropropan-2-ol/MeCN solvent system. Flow rate was 5 ml/min. Electrospray mass spectra wereacquired on an orthogonal acceleration / time-of-flight mass spectrometer (Q-Tof-2, Micromass,Manchester, UK) in negative ion mode. Scan time used was 2 s. The combined spectra from achromatographic peak were deconvoluted using the MaxEnt algorithm of the software (Masslynx 3.4,Micromass, UK-Manchester). Theoretical oligonucleotide masses were calculated using the monoiso-topic element masses.
Melting Temperatures. Analysis of melting temp. of complementary duplexes was carried out asdescribed in [2].
The authors thank the K. U. Leuven for financial support (GOA) and C. Biernaux for editorial help.
REFERENCES
[1] K. Schçning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy, A. Eschenmoser, Science 2000, 290,1347.
[2] D. Zhou, I. M. Lagoja, J. Rozenski, R. Busson, A. Van Aerschot, P. Herdewijn, ChemBioChem 2005,6, 2298.
[3] X. Wu, S. Guntha, M. Ferencic, R. Krishnamurthy, A. Eschenmoser, Org. Lett. 2002, 4, 1283.[4] L. Zhang, A. Peritz, E. Meggers, J. Am. Chem. Soc. 2005, 127, 4174.[5] W. Voelter, J. Muller, Liebigs Ann. Chem. 1983, 248.[6] S. Zaramella, E. Yeheskiely, R. Strçmberg, J. Am. Chem. Soc. 2004, 126, 14029.[7] D. Zhou, I. M. Lagoja, A. Van Aerschot, P. Herdewijn, Collect. Czech. Chem. Soc. 2006, 71, 15.[8] P. Sieber, B. Iselin, Helv. Chim. Acta 1968, 51, 614.[9] J. Wang, P. Herdewijn, J. Org. Chem. 1999, 64, 7820.
[10] S. Gryaznov, J. K. Chen, J. Am. Chem. Soc. 1994, 116, 3143.[11] R. G. Schultz, S. M. Gryaznov, Nucleic Acids Res. 1996, 24, 2966.[12] D. A. Flosser, R. A. Olofson, Tetrahedron Lett. 2002, 43, 4275.[13] P. S. Pallan, C. J. Wilds, Z. Wawrzak, R. Krishnamurthy, A. Eschenmoser, M. Egli, Angew. Chem.,
Int. Ed. 2003, 42, 5893.[14] L. Zhang, A. Peritz, E. Meggers, J. Am. Chem. Soc. 2006, 127, 4174.[15] V. Tereshko, S. Gryaznov, M. Egli, J. Am. Chem. Soc. 1998, 120, 269.
Received July 20, 2006
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 761
Related Documents











