Biochem. J. (2012) 447, 427–436 (Printed in Great Britain) doi:10.1042/BJ20121103 427 Characterization of ML-IAP protein stability and physiological role in vivo Eugene VARFOLOMEEV*, Elham MORADI†, Jasmin N. DYNEK* 1 , Jiping ZHA‡ 2 , Anna V. FEDOROVA*, Kurt DESHAYES*, Wayne J. FAIRBROTHER*, Kim NEWTON§, Jennifer LE COUTER† and Domagoj VUCIC* 3 *Department of Early Discovery Biochemistry, Genentech Inc., South San Francisco, CA 94080, U.S.A., †Department of Molecular Biology, Genentech Inc., South San Francisco, CA 94080, U.S.A., ‡Department of Pathology, Genentech Inc., South San Francisco, CA 94080, U.S.A., and §Department of Physiological Chemistry, Genentech Inc., South San Francisco, CA 94080, U.S.A. ML-IAP [melanoma IAP (inhibitor of apoptosis)] is an anti- apoptotic protein that is expressed highly in melanomas where it contributes to resistance to apoptotic stimuli. The anti-apoptotic activity and elevated expression of IAP family proteins in many human cancers makes IAP proteins attractive targets for inhibition by cancer therapeutics. Small-molecule IAP antagonists that bind with high affinities to select BIR (baculovirus IAP repeat) domains have been shown to stimulate auto-ubiquitination and rapid proteasomal degradation of c-IAP1 (cellular IAP1) and c-IAP2 (cellular IAP2). In the present paper, we report ML- IAP proteasomal degradation in response to bivalent, but not monovalent, IAP antagonists. This degradation required ML-IAP ubiquitin ligase activity and was independent of c-IAP1 or c-IAP2. Although ML-IAP is best characterized in melanoma cells, we show that ML-IAP expression in normal mammalian tissues is restricted largely to the eye, being most abundant in ciliary body epithelium and retinal pigment epithelium. Surprisingly, given this pattern of expression, gene-targeted mice lacking ML-IAP exhibited normal intraocular pressure as well as normal retinal structure and function. The results of the present study indicate that ML-IAP is dispensable for both normal mouse development and ocular homoeostasis. Key words: ciliary body expression, E3 ubiquitin ligase, genetic ablation, inhibitor of apoptosis (IAP) antagonist, melanoma inhibitor of apoptosis (ML-IAP), retinal pigment epithelium expression. INTRODUCTION Apoptosis is a genetically regulated form of cell death that is mediated by death receptors belonging to the TNF (tumour necrosis factor) receptor family or through mitochondrial pathways [1]. Both pathways activate caspases, the cysteine- dependent aspartyl-specific proteases that execute demolition of the cell [2]. The IAP (inhibitor of apoptosis) proteins interact with a variety of inducers and effectors of apoptosis [3]. The ability to block cell death induced by many different pro-apoptotic stimuli make IAP proteins central regulators of cell fate. ML- IAP (melanoma IAP; also known as BIRC7, livin or KIAP) is expressed highly in melanomas and several other tumour types, where it contributes to resistance to death stimuli [4–9]. The related XIAP (X-linked IAP) binds and inhibits caspases 3, 7 and 9 directly, whereas ML-IAP limits amplification of the apoptotic signal by Smac (second mitochondrial-derived activator of caspase) [2,10–12]. Located in the mitochondria of healthy cells, Smac is released into the cytosol during apoptosis, where it binds and inhibits XIAP [6,13,14]. ML-IAP binds Smac with high affinity and sequesters Smac away from XIAP [15,16]. Certain IAP proteins are also E3 ubiquitin ligases that use their C-terminal RING domains to ubiquitinate themselves and several of their binding partners [17,18]. Small-molecule IAP antagonists (Smac mimetics) bind select BIR (baculovirus IAP repeat) domains of IAP proteins, thereby blocking IAP protein interactions with caspases and Smac [6]. When IAP antagonists bind the BIR3 domain of c-IAP1 (cellular IAP1) and c-IAP2 (cellular IAP2), they stimulate c-IAP auto-ubiquitination and proteasomal degradation [19–21]. IAP ubiquitin ligase activity is critical for IAP-mediated pro- survival signalling as well as IAP antagonist functions [17,18]. IAP RING domain dimerization is essential for IAP ubiquitin ligase activity, and antagonists appear to stimulate this E3 activity by promoting dimer formation [22,23]. Recent biochemical and structural studies have shown that, in the absence of IAP antagonists, BIR3 through RING domains of c-IAP1 form a compact monomeric structure that prevents RING dimerization [22,24]. Binding of IAP antagonists to the c-IAP1 BIR3 domain blocks critical BIR3–RING interactions, and conformational rearrangements allow RING dimerization and instigate E3 ligase activity [24]. In the present study we demonstrate that a bivalent IAP antagonist causes proteasomal degradation of ML-IAP, which is dependent on the BIR domain of ML-IAP but independent of c-IAP1, c-IAP2 or XIAP. Interestingly, monovalent IAP antagonists did not stimulate ML-IAP E3 ligase activity and promote its degradation. ML-IAP expression in normal mouse tissues appears restricted to the eye, but genetic ablation of ML-IAP did not disrupt general mouse development or any retinal structure and ocular function, suggesting that ML-IAP is not essential for eye homoeostasis in mammals. Abbreviations used: BIR, baculovirus inhibitor of apoptosis repeat; CBE, ciliary body epithelium; cIAP, cellular inhibitor of apoptosis; cSLO, confocal scanning laser ophthalmoscopy; ERG, electroretinography; FA, fluorescein angiography; IAP, inhibitor of apoptosis; IOP, intraocular pressure; IPTG, isopropyl β-D-thiogalactopyranoside; MEF, mouse embryonic fibroblast; ML-IAP, melanoma IAP; OP, oscillatory potential; PR, photoreceptor; RPE, retinal pigment epithelium; SD-OCT, spectral domain-optical coherence tomography; Smac, second mitochondrial-derived activator of caspase; XIAP, X-linked IAP; Z-VAD-FMK, benzyloxycarbonyl-Val-Ala-DL-Asp-fluoromethylketone. 1 Present address: Health Interactions, San Francisco, CA 94111, U.S.A. 2 Present address: Crown Bioscience Inc., 6 Beijing West Road, Jiangsu Province, 215400, People’s Republic of China 3 To whom correspondence should be addressed (email [email protected]). c ⃝ The Authors Journal compilation c ⃝ 2012 Biochemical Society Biochemical Journal www.biochemj.org

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Biochem. J. (2012) 447, 427–436 (Printed in Great Britain) doi:10.1042/BJ20121103 427
Characterization of ML-IAP protein stability and physiological role in vivoEugene VARFOLOMEEV*, Elham MORADI†, Jasmin N. DYNEK*1, Jiping ZHA‡2, Anna V. FEDOROVA*, Kurt DESHAYES*,Wayne J. FAIRBROTHER*, Kim NEWTON§, Jennifer LE COUTER† and Domagoj VUCIC*3
*Department of Early Discovery Biochemistry, Genentech Inc., South San Francisco, CA 94080, U.S.A., †Department of Molecular Biology, Genentech Inc., South San Francisco, CA94080, U.S.A., ‡Department of Pathology, Genentech Inc., South San Francisco, CA 94080, U.S.A., and §Department of Physiological Chemistry, Genentech Inc., South San Francisco,CA 94080, U.S.A.
ML-IAP [melanoma IAP (inhibitor of apoptosis)] is an anti-apoptotic protein that is expressed highly in melanomas where itcontributes to resistance to apoptotic stimuli. The anti-apoptoticactivity and elevated expression of IAP family proteins in manyhuman cancers makes IAP proteins attractive targets for inhibitionby cancer therapeutics. Small-molecule IAP antagonists thatbind with high affinities to select BIR (baculovirus IAP repeat)domains have been shown to stimulate auto-ubiquitinationand rapid proteasomal degradation of c-IAP1 (cellular IAP1) andc-IAP2 (cellular IAP2). In the present paper, we report ML-IAP proteasomal degradation in response to bivalent, but notmonovalent, IAP antagonists. This degradation required ML-IAPubiquitin ligase activity and was independent of c-IAP1 or c-IAP2.Although ML-IAP is best characterized in melanoma cells, we
show that ML-IAP expression in normal mammalian tissues isrestricted largely to the eye, being most abundant in ciliary bodyepithelium and retinal pigment epithelium. Surprisingly, giventhis pattern of expression, gene-targeted mice lacking ML-IAPexhibited normal intraocular pressure as well as normal retinalstructure and function. The results of the present study indicatethat ML-IAP is dispensable for both normal mouse developmentand ocular homoeostasis.
Key words: ciliary body expression, E3 ubiquitin ligase, geneticablation, inhibitor of apoptosis (IAP) antagonist, melanomainhibitor of apoptosis (ML-IAP), retinal pigment epitheliumexpression.
INTRODUCTION
Apoptosis is a genetically regulated form of cell death thatis mediated by death receptors belonging to the TNF (tumournecrosis factor) receptor family or through mitochondrialpathways [1]. Both pathways activate caspases, the cysteine-dependent aspartyl-specific proteases that execute demolition ofthe cell [2]. The IAP (inhibitor of apoptosis) proteins interactwith a variety of inducers and effectors of apoptosis [3]. Theability to block cell death induced by many different pro-apoptoticstimuli make IAP proteins central regulators of cell fate. ML-IAP (melanoma IAP; also known as BIRC7, livin or KIAP)is expressed highly in melanomas and several other tumourtypes, where it contributes to resistance to death stimuli [4–9].The related XIAP (X-linked IAP) binds and inhibits caspases3, 7 and 9 directly, whereas ML-IAP limits amplification of theapoptotic signal by Smac (second mitochondrial-derived activatorof caspase) [2,10–12]. Located in the mitochondria of healthycells, Smac is released into the cytosol during apoptosis, where itbinds and inhibits XIAP [6,13,14]. ML-IAP binds Smac with highaffinity and sequesters Smac away from XIAP [15,16]. CertainIAP proteins are also E3 ubiquitin ligases that use their C-terminalRING domains to ubiquitinate themselves and several of theirbinding partners [17,18].
Small-molecule IAP antagonists (Smac mimetics) bind selectBIR (baculovirus IAP repeat) domains of IAP proteins, thereby
blocking IAP protein interactions with caspases and Smac[6]. When IAP antagonists bind the BIR3 domain of c-IAP1(cellular IAP1) and c-IAP2 (cellular IAP2), they stimulatec-IAP auto-ubiquitination and proteasomal degradation [19–21].IAP ubiquitin ligase activity is critical for IAP-mediated pro-survival signalling as well as IAP antagonist functions [17,18].IAP RING domain dimerization is essential for IAP ubiquitinligase activity, and antagonists appear to stimulate this E3 activityby promoting dimer formation [22,23]. Recent biochemicaland structural studies have shown that, in the absence of IAPantagonists, BIR3 through RING domains of c-IAP1 form acompact monomeric structure that prevents RING dimerization[22,24]. Binding of IAP antagonists to the c-IAP1 BIR3 domainblocks critical BIR3–RING interactions, and conformationalrearrangements allow RING dimerization and instigate E3 ligaseactivity [24].
In the present study we demonstrate that a bivalent IAPantagonist causes proteasomal degradation of ML-IAP, which isdependent on the BIR domain of ML-IAP but independent ofc-IAP1, c-IAP2 or XIAP. Interestingly, monovalent IAPantagonists did not stimulate ML-IAP E3 ligase activity andpromote its degradation. ML-IAP expression in normal mousetissues appears restricted to the eye, but genetic ablation ofML-IAP did not disrupt general mouse development or any retinalstructure and ocular function, suggesting that ML-IAP is notessential for eye homoeostasis in mammals.
Abbreviations used: BIR, baculovirus inhibitor of apoptosis repeat; CBE, ciliary body epithelium; cIAP, cellular inhibitor of apoptosis; cSLO, confocalscanning laser ophthalmoscopy; ERG, electroretinography; FA, fluorescein angiography; IAP, inhibitor of apoptosis; IOP, intraocular pressure; IPTG,isopropyl β-D-thiogalactopyranoside; MEF, mouse embryonic fibroblast; ML-IAP, melanoma IAP; OP, oscillatory potential; PR, photoreceptor; RPE, retinalpigment epithelium; SD-OCT, spectral domain-optical coherence tomography; Smac, second mitochondrial-derived activator of caspase; XIAP, X-linkedIAP; Z-VAD-FMK, benzyloxycarbonyl-Val-Ala-DL-Asp-fluoromethylketone.
1 Present address: Health Interactions, San Francisco, CA 94111, U.S.A.2 Present address: Crown Bioscience Inc., 6 Beijing West Road, Jiangsu Province, 215400, People’s Republic of China3 To whom correspondence should be addressed (email [email protected]).
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society
Bio
chem
ical
Jou
rnal
w
ww
.bio
chem
j.org

428 E. Varfolomeev and others
EXPERIMENTAL
Cell lines, antibodies, transfections and reagents
MeWo, Colo 829, Malme-3M and SK-MEL28 human mela-noma cells were obtained from A.T.C.C. KMS18 multiplemyeloma cells were from JCRB (Japanese Collection of ResearchBioresources). c-IAP1- and c-IAP2-knockout, and matched wild-type MEFs (mouse embryonic fibroblasts) were provided byDr John Silke (Walter and Eliza Hall Institute of MedicalResearch, Melbourne, Australia) [20,25]. All cells were grownon 50:50 DMEM (Dulbeco’s modified Eagle’s medium)/FK12medium supplemented with 10% FBS (fetal bovine serum),10000 units/ml penicillin and 10000 µg/ml streptomycin.KMS18 cells were transfected by electroporation using AmaxaNucleofactor technology (Lonza). Plasmids expressing FLAG–c-IAP1, FLAG–c-IAP2, FLAG–c-IAP1 RING mutant (H588A),Myc–ML-IAP, Myc–ML-IAP !RING and Myc–ML-IAP BIRmutant (D138A) have been described previously [5,19,26].ML-IAP RING mutant (H251A) was generated by site-directed mutagenesis. The primary antibodies against c-IAP1were purchased from R&D Systems (affinity-purified goatantibody) or Alexis Biochemicals, anti-ubiquitin and anti-XIAP antibodies were from Cell Signaling Technology, anti-FLAG M2 antibody was from Sigma, anti-Myc antibody wasfrom Roche, anti-tubulin antibody was from ICN Biomedicals,and monoclonal and polyclonal antibodies against ML-IAPwere generated at Genentech [4]. MG132 and Z-VAD-FMK(benzyloxycarbonyl-Val-Ala-DL-Asp-fluoromethylketone) werepurchased from Calbiochem, and IAP antagonists MV1, BV6[19] and GDC-0152 [27] were from Genentech. Western blotanalyses were performed as described previously [15].
Recombinant protein production and ubiquitination assays
Full-length ML-IAP (residues 1–298) was subcloned into the pET-15b bacterial expression vector (Novagen) and the protein wasexpressed in Escherichia coli strain BL21-Gold (DE3) (AgilentTechnologies). Overnight cultures were diluted 1:100 and grownat 37 ◦C in Luria–Bertani medium with 50 mg/ml carbenicillin to aD600 of 0.8 with vigorous shaking (200 rev./min). IPTG (isopropylβ-D-thiogalactopyranoside) was added to a final concentrationof 0.5 mM to induce protein expression. After IPTG induction,expression was allowed to proceed overnight at 16 ◦C, withshaking at 200 rev./min. Cells were pelleted and resuspended in50 ml/l buffer A [50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM DTT(dithiothreitol) and Roche complete protease inhibitors]. Cellswere homogenized, microfluidized and centrifuged (27000 g for30 min at 4 ◦C). Protein was purified by passing through a Ni-NTA (Ni2 + -nitrilotriacetate) agarose column (Qiagen), followedby gel-filtration chromatography. Protein was then loaded onto a Superdex 75 gel-filtration column (Pharmacia) equilibratedwith buffer A (without protease inhibitors). Eluted fractions wereanalysed by SDS/PAGE and ESI (electrospray ionization)–TOF(time-of-flight) LC (liquid chromatography)-MS and stored at4 ◦C. Reconstituted ubiquitination assays with recombinant ML-IAP and E2 enzymes were performed as described previously[19,28]. Recombinant UbcH5a/b/c, UbcH6, UbcH7, E1 enzymeand ubiquitin were purchased from Boston Biochem.
SD-OCT (spectral domain-optical coherence tomography) and FA(fluorescein angiography)
All procedures involving animal experiments adhered to theARVO (Association for Research in Vision and Opthalmology)
guidelines for the use of animals in ophthalmic and visionresearch, using protocols approved and monitored by Genentech’sInstitutional Animal Care and Use Committee. Longitudinalassessments of SD-OCT and FA (Spectralis HRA + OCT,Heidelberg Engineering) were obtained from 1- to 6-month-old wild-type and homozygous mutant mice. This cohortcomprised wild-type males (n = 10) and females (n = 8), and ML-IAP− / − males (n = 8) and females (n = 10). An aged cohort, 20–25 months old, was also evaluated. For these experiments, micewere anaesthetized with an intraperitoneal injection of a solutionof ketamine (16 mg/kg) and xylazine (5 mg/kg) and maintainedon a heating pad at 37 ◦C. The pupils were dilated with a topicaldrop of 1 % tropicamide (Bausch & Lomb). Bilateral SD-OCTvolume scans of 19 slices with a distance between slices of200 µm were performed for at least three regions of each eyeunder study (superior nasal, superior temporal and central inferiorareas). Retinal thickness was obtained in all cases.
Angiography assessment was performed in a single eye foreach mouse as part of the SD-OCT session. Following anintraperitoneal injection of 0.1 ml of 2.5% fluorescein sodium(Alcon), images were obtained for early (30–180 s) and late (300–400 s) phases after injection.
IOP (intraocular pressure)
Non-invasive tonometry was measured in conscious 6-month-oldmice restrained in a plastic sleeve in the early afternoon hours(13:30–15:00 h). Bilateral assessments were performed using theTonoLab rebound tonometer with disposable probes (Icare). Anaverage of six tests were recorded per eye. Each group comprised16–22 eyes, wild-type male (n = 10 mice; 20 eyes) and female(n = 8 mice; 16 eyes) mice, and ML-IAP− / − male (n = 8 mice;16 eyes) and female (n = 10 mice; 20 eyes) mice.
ERG (electroretinography)
Prior to ERG assessment, mice were adapted to darknessovernight and subsequently the whole manipulation was per-formed in dim red light. For these studies, the groups consisted ofeyes from wild-type males (n = 10) and females (n = 8), and eyesfrom ML-IAP− / − male (n = 8) and female (n = 10) mice. Micewere anaesthetized with an intraperitoneal injection of a solutionof ketamine (16 mg/kg) and xylazine (5 mg/kg) and maintainedon a heating pad at 37 ◦C. The pupils were dilated as describedabove. A topical drop of 2.5% hypromellose (Goniovisc, SigmaPharmaceuticals) was instilled in each eye immediately beforeplacing the contact lens electrodes (The Electrode Store). Areference electrode was placed on the forehead, with a groundelectrode placed in the tail. Flash-induced ERG was recordedbilaterally using the Espion E2 system and Ganzfeld dome(Diagnosys). The electrical responses of the retina were recordedsimultaneously from both eyes. Retinal responses were recordedunder dark adaptation to white light flashes at five steps rangingfrom − 4 to 1.5 log(cd · s · m− 2), capturing rod-driven rod–conemixed responses as well as cone-dominated responses at thehighest intensities [28a]. For each light-intensity stimulus, ten 3-sconsecutive stimuli were averaged, with an interval between lightflashes in scotopic conditions of 10 s for dim flashes and up to 60 sfor the highest intensity flashes. OPs (oscillatory potentials) wereisolated using white flashes of 1.5 log(cd · s · m− 2) in a recordingfrequency range of 100–10 000 Hz. The amplitudes of the a-waveand b-wave, and peak-to-peak OPs were measured off-line.
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

Bivalent IAP antagonist promotes degradation of ML-IAP 429
Figure 1 IAP antagonist BV6 promotes degradation of ML-IAP
(A) Colo 829 or MeWo cells were treated with BV6 (2 µM) for the times indicated in the absence or presence of MG132 or Z-VAD-FMK (20 µM each). Cellular lysates were examined by Westernblotting with ML-IAP-, c-IAP1- or tubulin-specific antibodies. (B) ML-IAP degrades slower than c-IAP1 following BV6 treatment. MeWo cells were treated for the times indicated with BV6 and cellularlysates were examined as described in (A). (C) KMS18 cells were transiently transfected with the indicated ML-IAP constructs and 24 h later were treated with BV6 (2 µM) and MG132 (20 µM) for4 h. Cellular lysates were examined with anti-Myc (ML-IAP) and anti-actin antibodies.
RESULTS
The IAP antagonist BV6 promotes ML-IAP degradation
We, and others, have shown that IAP antagonists stimulate c-IAP1and c-IAP2 auto-ubiquitination and proteasomal degradation[19,20]. We examined the fate of ML-IAP following IAPantagonist treatment in several melanoma cell lines. Additionof the bivalent IAP antagonist BV6 [19] stimulated degradation ofML-IAP in MeWo and Colo 829 cells. Consistent with previousstudies [19], c-IAP1 was also degraded (Figure 1A). Theproteasome inhibitor MG132 inhibited degradation of ML-IAP,whereas the pan-caspase inhibitor Z-VAD-FMK had no effect,indicating ubiquitin–proteasome system involvement in ML-IAPdegradation (Figure 1A). We found that c-IAP1 was degraded
much faster than ML-IAP. For example, c-IAP1 was not detectedin cellular lysates after 10 min of BV6 treatment, whereascomplete ML-IAP degradation took more than 1 h (Figure 1B).
Next, we mutated the ML-IAP BIR or RING domains(Supplementary Figure S1 at http://www.BiochemJ.org/bj/447/bj4470427add.htm) and determined the effect of these mutationson ML-IAP stability in KMS18 cells. Wild-type ML-IAP wasdegraded fully after BV6 treatment, but mutation of the Zn2 + -co-ordinating residue His251 to alanine or deletion of the entireRING domain prevented ML-IAP degradation in response to BV6(Figure 1C). Similarly, mutation of an IAP antagonist-bindingresidue in the BIR domain (Asp138 to alanine) of ML-IAP alsoprohibited ML-IAP degradation (Figure 1C). Therefore the IAPantagonist BV6 stimulates ML-IAP degradation, and requires the
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

430 E. Varfolomeev and others
Figure 2 c-IAP1, c-IAP2 or XIAP are not required for degradation of ML-IAP
(A and B) Knockdown of other IAP proteins does not affect ML-IAP degradation. MeWo cells were transfected with c-IAP1-specific and c-IAP2-specific (A), or XIAP-specific (B), or control siRNA(short interfering RNA) oligonucleotides. At 48 h later, cells were treated with BV6 (2 µM) in the absence or presence of MG132 (20 µM) and cellular lysates were examined by Western blotting withML-IAP-, c-IAP1-, XIAP- or tubulin-specific antibodies. (C) Wild-type, c-IAP1-knockout (KO) or c-IAP2-KO MEFs were treated with BV6 for the times indicated in the absence or presence of MG132(20 µM). Cellular lysates were examined as described in (A). (D) KMS18 cells were transfected with Myc–ML-IAP and vector or FLAG–c-IAP1, FLAG–c-IAP1 RING mutant (H588A), FLAG–c-IAP2,or a combination of FLAG–c-IAP1 and FLAG–c-IAP2. At 24 h later cells were treated with BV6 for the times indicated in the absence or presence of MG132. Cellular lysates were examined withanti-Myc (ML-IAP), anti-FLAG (c-IAP1/2) or anti-tubulin antibodies.
RING domain and functional antagonist-binding site on the BIRdomain of ML-IAP.
Degradation of ML-IAP occurs independently of other IAP proteins
Next we investigated whether other IAP proteins are importantfor IAP antagonist-stimulated ML-IAP degradation using severaldifferent cellular models wherein IAP protein expression wasdown-regulated, ablated genetically or accomplished through
ectopic transfection. Simultaneous knockdown of c-IAP1 andc-IAP2, or knockdown of XIAP, did not affect BV6-inducedML-IAP degradation (Figures 2A and 2B). Consistent withthese data, BV6 caused degradation of ML-IAP expressedectopically in wild-type, c-IAP1-deficient or c-IAP2-deficientMEFs (Figure 2C). Ectopically expressed ML-IAP was alsodegraded after BV6 treatment of KMS18 multiple myelomacells, which lack Birc2 (c-IAP1 gene) and Birc3 (c-IAP2 gene)[29,30] (Figure 2D). Finally, co-expression of ML-IAP with
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society
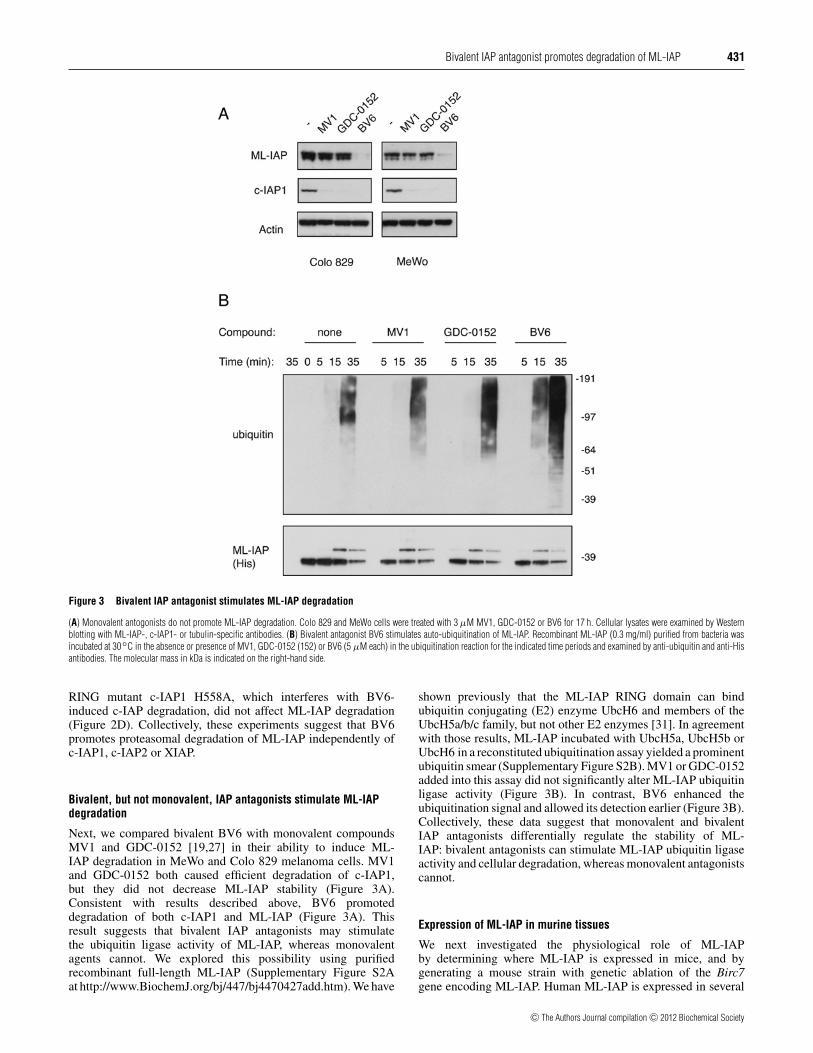
Bivalent IAP antagonist promotes degradation of ML-IAP 431
Figure 3 Bivalent IAP antagonist stimulates ML-IAP degradation
(A) Monovalent antogonists do not promote ML-IAP degradation. Colo 829 and MeWo cells were treated with 3 µM MV1, GDC-0152 or BV6 for 17 h. Cellular lysates were examined by Westernblotting with ML-IAP-, c-IAP1- or tubulin-specific antibodies. (B) Bivalent antagonist BV6 stimulates auto-ubiquitination of ML-IAP. Recombinant ML-IAP (0.3 mg/ml) purified from bacteria wasincubated at 30◦C in the absence or presence of MV1, GDC-0152 (152) or BV6 (5 µM each) in the ubiquitination reaction for the indicated time periods and examined by anti-ubiquitin and anti-Hisantibodies. The molecular mass in kDa is indicated on the right-hand side.
RING mutant c-IAP1 H558A, which interferes with BV6-induced c-IAP degradation, did not affect ML-IAP degradation(Figure 2D). Collectively, these experiments suggest that BV6promotes proteasomal degradation of ML-IAP independently ofc-IAP1, c-IAP2 or XIAP.
Bivalent, but not monovalent, IAP antagonists stimulate ML-IAPdegradation
Next, we compared bivalent BV6 with monovalent compoundsMV1 and GDC-0152 [19,27] in their ability to induce ML-IAP degradation in MeWo and Colo 829 melanoma cells. MV1and GDC-0152 both caused efficient degradation of c-IAP1,but they did not decrease ML-IAP stability (Figure 3A).Consistent with results described above, BV6 promoteddegradation of both c-IAP1 and ML-IAP (Figure 3A). Thisresult suggests that bivalent IAP antagonists may stimulatethe ubiquitin ligase activity of ML-IAP, whereas monovalentagents cannot. We explored this possibility using purifiedrecombinant full-length ML-IAP (Supplementary Figure S2Aat http://www.BiochemJ.org/bj/447/bj4470427add.htm). We have
shown previously that the ML-IAP RING domain can bindubiquitin conjugating (E2) enzyme UbcH6 and members of theUbcH5a/b/c family, but not other E2 enzymes [31]. In agreementwith those results, ML-IAP incubated with UbcH5a, UbcH5b orUbcH6 in a reconstituted ubiquitination assay yielded a prominentubiquitin smear (Supplementary Figure S2B). MV1 or GDC-0152added into this assay did not significantly alter ML-IAP ubiquitinligase activity (Figure 3B). In contrast, BV6 enhanced theubiquitination signal and allowed its detection earlier (Figure 3B).Collectively, these data suggest that monovalent and bivalentIAP antagonists differentially regulate the stability of ML-IAP: bivalent antagonists can stimulate ML-IAP ubiquitin ligaseactivity and cellular degradation, whereas monovalent antagonistscannot.
Expression of ML-IAP in murine tissues
We next investigated the physiological role of ML-IAPby determining where ML-IAP is expressed in mice, and bygenerating a mouse strain with genetic ablation of the Birc7gene encoding ML-IAP. Human ML-IAP is expressed in several
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

432 E. Varfolomeev and others
Figure 4 Analysis of BRIC7 mRNA expression in mouse, rat and human tissues
Quantitative real-time PCR analysis of BRIC7 mRNA abundance in RNA samples derived from mouse (A) and human (B) tissue samples, and from rat and mouse eyes (C). Values for the indicatedmRNA abundances were normalized to that of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (A and C) or RPL19 (B) mRNA as internal controls. BC, bone, calvia; BL, bone, long; BM, bonemarrow; LG, lacrimal gland; LN , lymph node; MG, mammary gland; PBL, peripheral blood lymphocyte; PP, Peyer’s patches; SC, spinal cord; SGM, salivary gland, mandibular; SGP, salivary gland,parotid; SV, seminal vesicle; VC, vena cava; VD, vas deferens; VJ, vein, jugular; VP, vein, portal; RPE + CC, RPE and choroid capillaries.
human cancers and, to a lesser degree, in a few normal tissues,such as adult kidney [6,32]. Unexpectedly, mouse Birc7 mRNAwas detected exclusively in eye samples (Figure 4A). When werevisited ML-IAP expression in normal human tissues, BIRC7expression was higher in eye tissue than in the kidney (Figure 4B).In both rat and mouse eyes Birc7 showed the highest expressionin the complex of RPE (retinal pigment epithelium) and choroidcapillaries (Figure 4C). BIRC2, BIRC3 and XIAP showed a muchbroader pattern of expression in the same samples and were notmore abundant in mouse or human eyes (Supplementary FiguresS3–S5 at http://www.BiochemJ.org/bj/447/bj4470427add.htm).
ML-IAP-deficient mice were generated in which the firstfour coding exons were replaced with a LacZ/Neo cassette(Supplementary Figure S3). Quantitative real-time PCR analysisand Western blotting with an anti-mouse ML-IAP antibodyconfirmed ML-IAP protein and mRNA expression in wild-type,but not ML-IAP− / − , RPE (Supplementary Figure S3). Expressionof Birc2, Birc3 or Xiap mRNA was not affected by ML-IAP
deficiency (Supplementary Figures S4A–S4C). Mice lacking ML-IAP did not exhibit any developmental abnormalities or immune-system-related disorders, and they aged comparably with theirwild-type littermates (results not shown), suggesting that ML-IAP is not critical for overall homoeostasis.
Normal retinal and vessel architecture in ML-IAP-deficient mice
SD-OCT, an increasingly important diagnostic tool inophthalmology, provides ultra-high-resolution analysis of retinalarchitecture in the live animal [33–35]. To assess whether thedeletion of ML-IAP alters the general morphological structure ofthe retina or contributes to any retinal degeneration over time, weperformed a series of SD-OCT and cSLO (confocal scanning laserophthalmoscopy) assessments on mutant and wild-type animals.The overall morphological structure in the ML-IAP− / − retinawas unaltered (Figure 5A) at first assessment relative to wild-type
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

Bivalent IAP antagonist promotes degradation of ML-IAP 433
Figure 5 Normal retinal and retinal vessel architecture in ML-IAP− / − eyes
B-scans (cross-sectional views) of the wild-type and mutant retinas obtained by SD-OCT. Representative images are shown for 1-month-old male mice (A) and 6-month-old mice (same eyes asshown in A at 1-month assessment) (B). Up to 25 months of age, in a distinct cohort of mice, SD-OCT imaging revealed a thinning outer nuclear layer (segment between OPL and ELM). Retinalpigment epithelial cell loss was also evidenced by increased SD-OCT laser penetration into the choroidal layer (arrowhead) and hypofluorescent regions (arrows) in the cSLO fundus images (C).Representative fundus fluorescein angiographic early- (results not shown) and late-phase images were obtained from 6-month-old mice (D). Paired images for the right eye are presented: fundusphoto (left-hand panel) and fundus fluorescein angiographic images (right-hand panel). No evidence of extravascular leakage of the dye was observed. The retinal vessel patterning and appearanceis indistinguishable between wild-type and ML-IAP− / − eyes. BM, Bruch’s membrane; ELM, external limiting membrane; CC, choriocapillaris; ILM, inner limiting membrane; KO, knock out; ONH,optic nerve head; OPL, outer plexiform layer; WT, wild-type.
littermates, and no further changes were noted over the first 6months of life (Figure 5B). At 6 months of age the average retinalthickness (mean +− S.D.) was 262 +− 6 µm for wild-type males,258 +− 7 µm for ML-IAP− / − males, 258 +− 7 µm for wild-typefemales and 257 +− 14 µm for ML-IAP− / − females. Assessmentsin a much older cohort (20–25 months) revealed some alterations(Figure 5C), including geographic atrophy, that were not uniqueto the mutant eyes, but rather appeared to be age-related andsimilarly manifested in an age-matched wild-type group ofmice.
Vascular structure and integrity were also examined over thesame time frame by using FA in addition to the acquired fundusimages. For these studies, we restricted our imaging to the right
eye. Early- (results not shown) and late-phase recordings revealeda normal vessel morphology and pattern (Figure 5D). As therewas no evidence of leakage from the retinal vasculature, weconclude that loss of ML-IAP had no effect on vessel stabilizationof the inner blood–retinal barrier. The outer barrier, comprisedof the single layer of RPE, also appeared to be intact as therewas no fluorescein signal derived from the underlying choroidcapillaries.
Normal IOP and retinal function in ML-IAP-deficient mice
ML-IAP is expressed in the CBE (ciliary body epithelium),a circumferential tissue in the front of the eye that performs
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

434 E. Varfolomeev and others
Figure 6 Normal retinal function in ML-IAP-deficient eyes
Representative scoptic flash ERG responses for the mid-intensity stimulus [1.0 log(cd · s · m2)] are shown for wild-type and ML-IAP− / − eyes at 1 month (A) and 6 months of age (B). ML-IAP− / −
eyes demonstrated normal responses. KO, knock out; WT, wild-type.
several functions including aqueous humour production. Eyesof examined animals appeared normal by gross inspection,indicating that IOP was not extremely abnormally low or high.We determined the IOP in wild-type and ML-IAP− / − male andfemale cohorts. Pressures were monitored through 20–24 weeksof age. IOP at weeks 22–24 for wild-type males was 15.3 +− 4mmHg (1 mmHg = 0.133 kPa) (mean +− S.D.), 16 +− 3 mmHgfor ML-IAP− / − males, 20 +− 6 mmHg for wild-type femalesand 18 +− 5 mmHg for ML-IAP− / − females. These pressuremeasurements were not significantly different between male orfemale ML-IAP− / − animals in relation to their respective wild-type littermates, nor were differences between genders significant.This is consistent with the normal outward appearance of the eyesand the health of the anterior and posterior segment.
To determine possible effects of ML-IAP deficiency on retinalfunction, a series of full-field scotopic ERG experiments wasperformed on ML-IAP− / − mice, and on their corresponding ML-IAP+ / + littermates in a longitudinal manner from 1 to 6 months ofdevelopment. At 1 month, the amplitude and timing of the scotopica- or b-waves did not show significant differences between wild-type and mutant mice (Figure 6A). To determine the contributionof third-order neurons to light-induced ERG, OPs were isolatedfrom the electrophysiological recordings. The OP recorded inresponse to high-intensity light stimuli under scotopic conditionsshowed no significant differences between genotypes (results notshown). At 6 months (Figure 6B) there was again no indication ofreduced retinal function. Throughout the assessment period, allelectrophysiological responses indicated that the retinal functionin the ML-IAP− / − mutants was not altered.
DISCUSSION
The ability of IAP proteins to block cell death mediated throughdeath receptor and mitochondrial pathways and to promote
survival signalling pathways, as well as their elevated expressionin many human cancers, makes these proteins attractive targets foranti-cancer therapies [36]. One of the major strategies for targetingIAP proteins involves Smac-mimicking IAP antagonists, whosepotential to fight human tumours is currently under investigationin several clinical trials [6]. Both monovalent and bivalent IAPantagonists induce rapid auto-ubiquitination and proteasomaldegradation of c-IAP1 and c-IAP2 [19,20]. IAP antagoniststrigger this proteolytic outcome by causing conformationalchanges in c-IAP proteins that allow RING dimerization andunlocking of their ubiquitin ligase activity [24]. However, it wasnot known whether IAP antagonists affect the stability of ML-IAP. In the present study we have shown that, similarly to cIAPs,IAP antagonists can promote proteasomal degradation of ML-IAP. Although c-IAP1 was reported to influence the stabilityof other IAP proteins, especially c-IAP2 and XIAP [37,38],down-regulation of any of these IAPs did not affect steady-state levels of ML-IAP or IAP antagonist-stimulated degradationof ML-IAP. Interestingly, only the bivalent antagonist BV6induced ML-IAP degradation, whereas monovalent agents did notaffect its stability. The explanation for this stark difference withcIAP degradation was provided by a reconstituted ubiquitinationreaction, where the bivalent antagonist, but not monovalentcompounds, stimulated ML-IAP E3 ligase activity. ML-IAPis a prototype of a simple IAP protein with a single BIR anda RING domain [5]. Lacking the UBA domain and CARD(caspase-recruitment domain) present in c-IAP proteins [6], ML-IAP is not expected to engage conformational states that couldpotentially regulate its E3 ligase activity. Thus bivalent antagonistmay bridge two ML-IAP molecules to boost its ubiquitin ligaseactivity.
Surprisingly, in normal tissues, ML-IAP is almost exclusivelyexpressed in eyes. For 12 years we have referred to thisprotein as ML-IAP and discussed its expression in a varietyof human cancers [6]. High expression of ML-IAP in the
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

Bivalent IAP antagonist promotes degradation of ML-IAP 435
eyes has eluded the scientific community, possibly because eyetissues are not included in most commercially available tissuecollections. Nevertheless, it is now clear that in humans androdents ML-IAP is principally expressed in ocular structures. Therestricted expression of ML-IAP in the RPE and CBE, both ofneuroectoderm origin, focused our phenotype characterization tothe structures and functions of the eye.
The RPE, a single polarized layer of cuboidal-columnarpigmented cells between the retina and the choroid, provides theouter blood–retinal barrier and functions at a very high metabolicrate. The apical RPE surface has an intimate interaction withthe light transducing PRs (photoreceptors) of the retina. Withmicrovilli and cylindrical cytoplasmic sheaths that enclose theends of the PRs, the RPE provides nourishment and pigmentsof the visual cycle to the rods and cones and also removes thecontinuously shed outer segments of these cells (reviewed in[39]). Imaging studies including SD-OCT, cSLO and FA, alongwith functional ERG indicated that this RPE/PR functional unitwas intact in the mutant eye.
The ciliary body is a double layer of cuboidal epitheliumcomposed of an inner transparent layer and outer pigmentedlayer [40]. Like the RPE, the pigmented CBE also performs abarrier function referred to as the blood aqueous barrier. Theprimary function of this triangular tissue located near the frontof the eye are 3-fold: (i) to secrete the aqueous humour thatnourishes the avascular lens and cornea, (ii) to maintain IOP forthe appropriate shape and optical properties of the globe and (iii)for accommodation/ to change the lens shape for near or distantvision (reviewed in [41]). The normal outward appearance, healthof the anterior and posterior segment and normal IOP of the ML-IAP− / − eye indicated overall normal development and functionof the ciliary body.
Genetic ablation of ML-IAP did not reveal any apparentdefect during development or at later stages of life. It ispossible that ML-IAP is more relevant in cancer cells that areconstantly on the verge of death, which requires that they maintainexpression of all anti-apoptotic weapons in their arsenal. Inaddition to ML-IAP, eyes of humans and rodents express otherIAP proteins, making ML-IAP a potentially redundant factorfor regular ocular functions. Future studies involving crossesof ML-IAP-deficient mice with mice lacking other apoptoticregulators might yet reveal a potential role for ML-IAP in eyehomoeostasis.
AUTHOR CONTRIBUTION
Eugene Varfolomeev performed ML-IAP expression studies; Jasmin Dynek conductedML-IAP protein stability studies; Kim Newton managed the ML-IAP-knockout colony;Anna Fedorova and Kurt Deshayes produced recombinant ML-IAP protein; Elham Moradiand Jennifer Le Couter performed ocular studies; Jiping Zha performed pathologicalexamination; and Domagoj Vucic conceived and supervised the study, and wrote the paperwith input from Eugene Varfolomeev, Wayne Fairbrother, Kim Newton, Jasmin Dynek,and Jennifer Le Couter. All authors provided intellectual input in study design and dataanalyses.
ACKNOWLEDGEMENTS
We thank Erin Dueber, Tanya Goncharov, Dorothy French and members of theEarly Discovery Biochemistry and Physiological Chemistry Departments for reagents,suggestions and comments.
FUNDING
All authors are employees of Genentech, Inc.
REFERENCES
1 Steller, H. (1995) Mechanisms and genes of cellular suicide. Science 267, 1445–14492 Salvesen, G. S. and Abrams, J. M. (2004) Caspase activation: stepping on the gas or
releasing the brakes? Lessons from humans and flies. Oncogene 23, 2774–27843 Salvesen, G. S. and Duckett, C. S. (2002) IAP proteins: blocking the road to death’s door.
Nat. Rev. Mol. Cell Biol. 3, 401–4104 Dynek, J. N., Chan, S. M., Liu, J., Zha, J., Fairbrother, W. J. and Vucic, D. (2008)
Microphthalmia-associated transcription factor is a critical transcriptional regulator ofmelanoma inhibitor of apoptosis in melanomas. Cancer Res. 68, 3124–3132
5 Vucic, D., Stennicke, H. R., Pisabarro, M. T., Salvesen, G. S. and Dixit, V. M. (2000)ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in humanmelanomas. Curr. Biol. 10, 1359–1366
6 Fulda, S. and Vucic, D. (2012) Targeting IAP proteins for therapeutic intervention incancer. Nat. Rev. Drug Discovery 11, 109–124
7 Kasof, G. M. and Gomes, B. C. (2001) Livin, a novel inhibitor of apoptosis protein familymember. J. Biol. Chem. 276, 3238–3246
8 Lin, J. H., Deng, G., Huang, Q. and Morser, J. (2000) KIAP, a novel member of theinhibitor of apoptosis protein family. Biochem. Biophys. Res. Commun. 279, 820–831
9 Crnkovic-Mertens, I., Hoppe-Seyler, F. and Butz, K. (2003) Induction of apoptosis intumor cells by siRNA-mediated silencing of the livin/ML-IAP/KIAP gene. Oncogene 22,8330–8336
10 Eckelman, B. P., Salvesen, G. S. and Scott, F. L. (2006) Human inhibitor of apoptosisproteins: why XIAP is the black sheep of the family. EMBO Rep. 7, 988–994
11 Varfolomeev, E. and Vucic, D. (2011) Inhibitor of apoptosis proteins: fascinating biologyleads to attractive tumor therapeutic targets. Future Oncol. 7, 633–648
12 Silke, J. and Brink, R. (2010) Regulation of TNFRSF and innate immune signallingcomplexes by TRAFs and cIAPs. Cell Death Differ. 17, 35–45
13 Du, C., Fang, M., Li, Y., Li, L. and Wang, X. (2000) Smac, a mitochondrial protein thatpromotes cytochrome c-dependent caspase activation by eliminating IAP inhibition.Cell 102, 33–42
14 Verhagen, A. M., Ekert, P. G., Pakusch, M., Silke, J., Connolly, L. M., Reid, G. E., Moritz,R. L., Simpson, R. J. and Vaux, D. L. (2000) Identification of DIABLO, a mammalianprotein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102,43–53
15 Vucic, D., Franklin, M. C., Wallweber, H. J., Das, K., Eckelman, B. P., Shin, H., Elliott,L. O., Kadkhodayan, S., Deshayes, K., Salvesen, G. S. and Fairbrother, W. J. (2005)Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor:implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem. J. 385,11–20
16 Duckett, C. S. (2005) IAP proteins: sticking it to Smac. Biochem. J. 385, e1–e217 Vucic, D., Dixit, V. M. and Wertz, I. E. (2011) Ubiquitylation in apoptosis: a
post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 12,439–452
18 Vaux, D. L. and Silke, J. (2005) IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol.6, 287–297
19 Varfolomeev, E., Blankenship, J. W., Wayson, S. M., Fedorova, A. V., Kayagaki, N., Garg,P., Zobel, K., Dynek, J. N., Elliott, L. O., Wallweber, H. J. et al. (2007) IAP antagonistsinduce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis.Cell 131, 669–681
20 Vince, J. E., Wong, W. W., Khan, N., Feltham, R., Chau, D., Ahmed, A. U., Benetatos,C. A., Chunduru, S. K., Condon, S. M., McKinlay, M. et al. (2007) IAP antagonists targetcIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693
21 Bertrand, M. J., Milutinovic, S., Dickson, K. M., Ho, W. C., Boudreault, A., Durkin, J.,Gillard, J. W., Jaquith, J. B., Morris, S. J. and Barker, P. A. (2008) cIAP1 and cIAP2facilitate cancer cell survival by functioning as E3 ligases that promote RIP1ubiquitination. Mol. Cell 30, 689–700
22 Feltham, R., Bettjeman, B., Budhidarmo, R., Mace, P. D., Shirley, S., Condon, S. M.,Chunduru, S. K., McKinlay, M. A., Vaux, D. L., Silke, J. and Day, C. L. (2011) Smacmimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domaindimerization. J. Biol. Chem. 286, 17015–17028
23 Mace, P. D., Linke, K., Feltham, R., Schumacher, F. R., Smith, C. A., Vaux, D. L., Silke, J.and Day, C. L. (2008) Structures of the cIAP2 RING domain reveal conformationalchanges associated with ubiquitin-conjugating enzyme (E2) recruitment. J. Biol. Chem.283, 31633–31640
24 Dueber, E. C., Schoeffler, A. J., Lingel, A., Elliott, J. M., Fedorova, A. V., Giannetti, A. M.,Zobel, K., Maurer, B., Varfolomeev, E., Wu, P. et al. (2011) Antagonists induce aconformational change in cIAP1 that promotes autoubiquitination. Science 334,376–380
25 Varfolomeev, E., Goncharov, T., Fedorova, A. V., Dynek, J. N., Zobel, K., Deshayes, K.,Fairbrother, W. J. and Vucic, D. (2008) c-IAP1 and c-IAP2 are critical mediators of tumornecrosis factor α (TNFα)-induced NF-κB activation. J. Biol. Chem. 283,24295–24299
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

436 E. Varfolomeev and others
26 Varfolomeev, E., Wayson, S. M., Dixit, V. M., Fairbrother, W. J. and Vucic, D.(2006) The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulatesNF-κB activation independently of TRAF1 AND TRAF2. J. Biol. Chem. 281,29022–29029
27 Flygare, J. A., Beresini, M., Budha, N., Chan, H., Chan, I. T., Cheeti, S., Cohen, F.,Deshayes, K., Doerner, K., Eckhardt, S. G. et al. (2012) Discovery of a potentsmall-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidatefor the treatment of cancer (GDC-0152). J. Med. Chem. 55, 4101–4113
28 Blankenship, J. W., Varfolomeev, E., Goncharov, T., Fedorova, A. V., Kirkpatrick, D. S.,Izrael-Tomasevic, A., Phu, L., Arnott, D., Aghajan, M., Zobel, K. et al. (2009) Ubiquitinbinding modulates IAP antagonist-stimulated proteasomal degradation of c-IAP1 andc-IAP2. Biochem. J. 417, 149–160
28a Tanimoto, N., Muehlfriedel, R. L., Fischer, M. D., Fahl, E., Humphries, P., Biel, M. andSeeliger, M. W. (2009) Vision tests in the mouse: functional phenotyping withelectroretinography. Front. Biosci. 14, 2730–2737
29 Annunziata, C. M., Davis, R. E., Demchenko, Y., Bellamy, W., Gabrea, A., Zhan, F., Lenz,G., Hanamura, I., Wright, G., Xiao, W. et al. (2007) Frequent engagement of the classicaland alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma.Cancer Cell 12, 115–130
30 Keats, J. J., Fonseca, R., Chesi, M., Schop, R., Baker, A., Chng, W. J., Van Wier, S.,Tiedemann, R., Shi, C. X., Sebag, M. et al. (2007) Promiscuous mutationsactivate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 12,131–144
31 Dynek, J. N., Goncharov, T., Dueber, E. C., Fedorova, A. V., Izrael-Tomasevic, A., Phu, L.,Helgason, E., Fairbrother, W. J., Deshayes, K., Kirkpatrick, D. S. and Vucic, D. (2010)c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling.EMBO J. 29, 4198–4209
32 Wagener, N., Crnkovic-Mertens, I., Vetter, C., Macher-Goppinger, S., Bedke, J., Grone,E. F., Zentgraf, H., Pritsch, M., Hoppe-Seyler, K., Buse, S. et al. (2007) Expression ofinhibitor of apoptosis protein Livin in renal cell carcinoma and non-tumorous adultkidney. Br. J. Cancer 97, 1271–1276
33 Hee, M. R., Izatt, J. A., Swanson, E. A., Huang, D., Schuman, J. S., Lin, C. P., Puliafito,C. A. and Fujimoto, J. G. (1995) Optical coherence tomography of the human retina.Arch. Ophthalmol. 113, 325–332
34 Huang, D., Swanson, E. A., Lin, C. P., Schuman, J. S., Stinson, W. G., Chang, W., Hee,M. R., Flotte, T., Gregory, K., Puliafito, C. A. et al. (1991) Optical coherence tomography.Science 254, 1178–1181
35 Puliafito, C. A., Hee, M. R., Lin, C. P., Reichel, E., Schuman, J. S., Duker, J. S., Izatt, J.A., Swanson, E. A. and Fujimoto, J. G. (1995) Imaging of macular diseases with opticalcoherence tomography. Ophthalmology 102, 217–229
36 Vucic, D. and Fairbrother, W. J. (2007) The inhibitor of apoptosis proteins as therapeutictargets in cancer. Clin. Cancer Res. 13, 5995–6000
37 Conte, D., Holcik, M., Lefebvre, C. A., Lacasse, E., Picketts, D. J., Wright, K. E. andKorneluk, R. G. (2006) Inhibitor of apoptosis protein cIAP2 is essential forlipopolysaccharide-induced macrophage survival. Mol. Cell. Biol. 26, 699–708
38 Silke, J., Kratina, T., Chu, D., Ekert, P. G., Day, C. L., Pakusch, M., Huang, D. C. and Vaux,D. L. (2005) Determination of cell survival by RING-mediated regulation of inhibitor ofapoptosis (IAP) protein abundance. Proc. Natl. Acad. Sci. U.S.A. 102, 16182–16187
39 Strauss, O. (2005) The retinal pigment epithelium in visual function. Physiol. Rev. 85,845–881
40 Beebe, D. C. (1986) Development of the ciliary body: a brief review. Trans. Ophthalmol.Soc. U.K. 105, 123–130
41 Goel, M., Picciani, R. G., Lee, R. K. and Bhattacharya, S. K. (2010) Aqueous humordynamics: a review. Open Ophthalmol. J. 4, 52–59
Received 11 July 2012/31 July 2012; accepted 2 August 2012Published as BJ Immediate Publication 2 August 2012, doi:10.1042/BJ20121103
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

Biochem. J. (2012) 447, 427–436 (Printed in Great Britain) doi:10.1042/BJ20121103
SUPPLEMENTARY ONLINE DATACharacterization of ML-IAP protein stability and physiological role in vivoEugene VARFOLOMEEV*, Elham MORADI†, Jasmin N. DYNEK*1, Jiping ZHA‡2, Anna V. FEDOROVA*, Kurt DESHAYES*,Wayne J. FAIRBROTHER*, Kim NEWTON§, Jennifer LE COUTER† and Domagoj VUCIC*3
*Department of Early Discovery Biochemistry, Genentech Inc., South San Francisco, CA 94080, U.S.A., †Department of Molecular Biology, Genentech Inc., South San Francisco, CA94080, U.S.A., ‡Department of Pathology, Genentech Inc., South San Francisco, CA 94080, U.S.A., and §Department of Physiological Chemistry, Genentech Inc., South San Francisco,CA 94080, U.S.A.
EXPERIMENTAL
Generation of Birc7-deficient mice
ML-IAP− / − mice are described in detail in Figure S3. Allexperiments involving animals were approved by the Genentechanimal care and use committee.
Analysis of gene expression
The abundance of mRNAs for human, mouse and rat BIRC7,BIRC2, BIRC3 and XIAP were determined as described previously[1,2]. For collection of eye tissues, eyes were enucleatedimmediately following killing, the anterior portion of the eye wasremoved and the retina was dissected free of any other tissue. Theremaining eye-cup was then flatten for removal of RPE + choroidcomplex. All tissues were snap-frozen before processing.Eye-derived mouse and rat mRNA samples were purified bythe mRNA Plus RNA Isolation kit (Qiagen). Mouse and humantissue-specific RNA samples were purchased from ZyagenLaboratories and BioChain Institute respectively. The followingsequences were used for detection, where F indicates the forwardprimer, R indicates the reverse primer and P indicates the probe:human BIRC7 (ML-IAP): (F) 5′-TTCCCCGACCACCGC, (R)5′-AAACAGGTACAGTTAAGCCATCCC and (P) 5′-FAM-AGGAGGCCCTTGCTTGGCGTG-TAMRA (FAM is 6-carboxyfluorescein and TAMRA is tetramethylrhodamine;human BIRC2 (cIAP1): (F) 5′-TGTTTGGTGAACTATATT-AGTATGTATGTGTACC, (R) 5′-GAAAGAACAACAAATCC-AGTAACTCCT and (P) 5′-FAM-AAGGGAGTAGTGTCAC-TGCTTGTTATGCATCATTT-TAMRA; human BIRC3(cIAP2): (F) 5′-CCGTCAAGTTCAAGCCAGTT, (R) 5′-TCT-CCTGGGCTGTCTGATGTG and (P) 5′-FAM-AAGGGAGTA-GTGTCACTGCTTGTTATGCATCATTT-TAMRA; humanXIAP: (F) 5′-ACCTTGTGATCGTGCCTGG, (R) 5′-TTCC-GGCCCAAAACAAAGA and (P) 5′-FAM-CAGAACAC-AGGCGACACTTTCCTAATTGC-TAMRA; mouse Birc7: (F)5′-CCAGACCTACACCCCTTTGC, (R) 5′-GGGTCTCTCTTC-TGGATCTGAGTAA and (P) 5′-FAM-CCACTCCCTCAGC-TCCTGCCCAT-TAMRA; mouse Birc2: (F) 5′-TTCAGGCT-TGTCATTGGAAG, (R) 5′-AGATGACCACACGGAATGAAand (P) 5′-FAM-CGTTCTTCTTGTAATCTCCGCAACTGC-TAMRA; mouse Birc3: (F) 5′-GCAGCAACCTCATTCAGAAA,(R) 5′-GCAATGTCATCTGTGGGAAG and (P) 5′-FAM-
CTTCGGGAAATTGACCCTGCG-TAMRA; mouse Xiap: (F)5′-CGGAGGATGAGTCAAGTCAA, (R) 5′-GTGACCAGAT-GTCCACAAGG and (P) 5′-FAM-TCCTCTTGTAGGCGC-CTTAGCTGC-TAMRA; rat Birc7: (F) 5′-CCAGGCC-TACACCCCCTTGC, (R) 5′-GAGTCTCTCTTCTGGATC-TGAGTAA and 5′-FAM-CCACTCCCTCAGCTCCCACCCAT-TAMRA; rat Birc2: (F) 5′-TTCAGGCTTGTCATTGGAAG,(R) 5′-AGATGACCACAAGGAATGAA and (P) 5′-FAM-CTTTCTTCTTGTAATCTCCGCAGCTGT-TAMRA; andrat Xiap: (F) 5′-CGCAGGATGAGTCAAGTCAG, (R)5′-GTGACCAGATGTCCACAAGG and (P) 5′-FAM-TCCTCTTGTAGGCGCCTTAGCTGC-TAMRA.
Figure S1 IAP antagonist BV6 promotes degradation of ML-IAP
(A) SK-MEL28 or Malme-3M cells were treated with BV6 (2 mM) for the times indicated. Cellularlysates were examined by Western blotting with ML-IAP-, c-IAP1- or tubulin-specific antibodies.(B) Schematic representation of ML-IAP indicating the location of the BIR domain mutation(D138A) and RING domain mutation (H251A). Hrs, hours.
1 Present address: Health Interactions, San Francisco, CA 94111, U.S.A.2 Present address: Crown Bioscience Inc., 6 Beijing West Road, Jiangsu Province, 215400, People’s Republic of China3 To whom correspondence should be addressed (email [email protected]).
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society
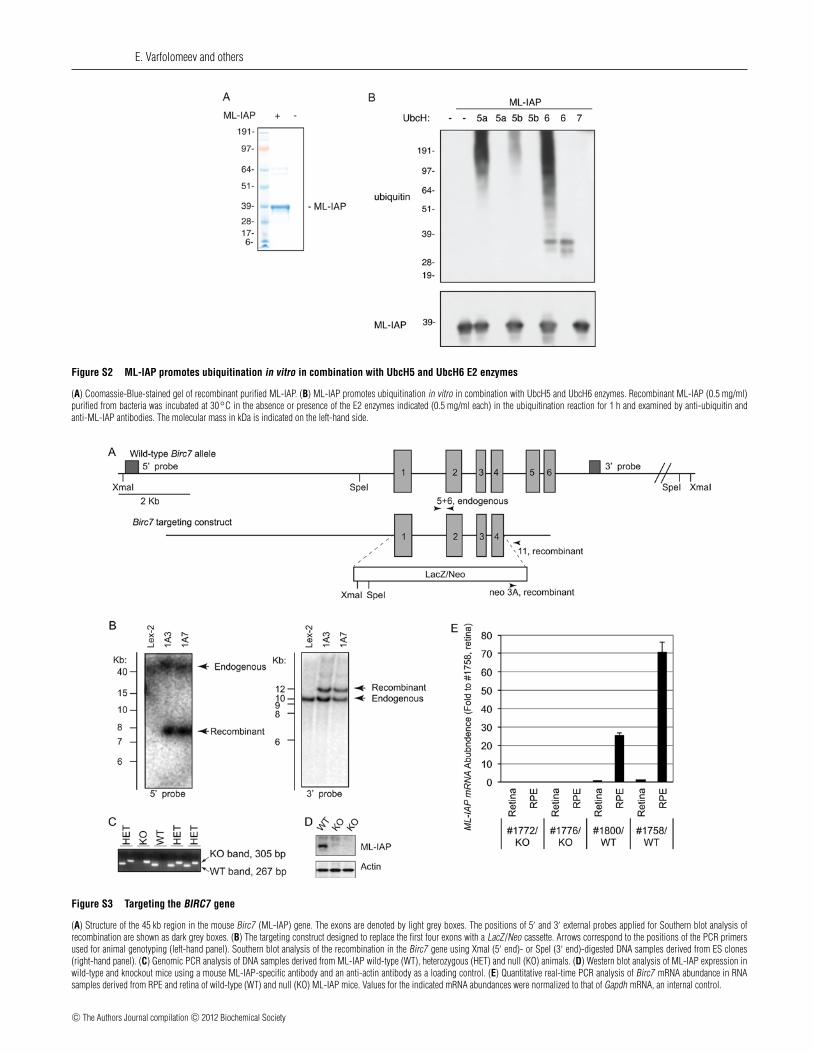
E. Varfolomeev and others
Figure S2 ML-IAP promotes ubiquitination in vitro in combination with UbcH5 and UbcH6 E2 enzymes
(A) Coomassie-Blue-stained gel of recombinant purified ML-IAP. (B) ML-IAP promotes ubiquitination in vitro in combination with UbcH5 and UbcH6 enzymes. Recombinant ML-IAP (0.5 mg/ml)purified from bacteria was incubated at 30◦C in the absence or presence of the E2 enzymes indicated (0.5 mg/ml each) in the ubiquitination reaction for 1 h and examined by anti-ubiquitin andanti-ML-IAP antibodies. The molecular mass in kDa is indicated on the left-hand side.
Figure S3 Targeting the BIRC7 gene
(A) Structure of the 45 kb region in the mouse Birc7 (ML-IAP) gene. The exons are denoted by light grey boxes. The positions of 5′ and 3′ external probes applied for Southern blot analysis ofrecombination are shown as dark grey boxes. (B) The targeting construct designed to replace the first four exons with a LacZ/Neo cassette. Arrows correspond to the positions of the PCR primersused for animal genotyping (left-hand panel). Southern blot analysis of the recombination in the Birc7 gene using XmaI (5′ end)- or SpeI (3′ end)-digested DNA samples derived from ES clones(right-hand panel). (C) Genomic PCR analysis of DNA samples derived from ML-IAP wild-type (WT), heterozygous (HET) and null (KO) animals. (D) Western blot analysis of ML-IAP expression inwild-type and knockout mice using a mouse ML-IAP-specific antibody and an anti-actin antibody as a loading control. (E) Quantitative real-time PCR analysis of Birc7 mRNA abundance in RNAsamples derived from RPE and retina of wild-type (WT) and null (KO) ML-IAP mice. Values for the indicated mRNA abundances were normalized to that of Gapdh mRNA, an internal control.
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

Bivalent IAP antagonist promotes degradation of ML-IAP
Figure S4 Expression of Birc2, Birc3 and Xiap mRNA in ML-IAP-null mice
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society

E. Varfolomeev and others
Quantitative real-time PCR analysis of Birc2, Birc3 and Xiap mRNA abundance in RNA samples derived from RPE and retina of wild-type (WT) and null (KO) ML-IAP mice. Values for the indicatedmRNA abundances were normalized to that of Gapdh mRNA, an internal control. (A) Analysis of BIRC2 mRNA expression in mouse, rat and human tissues. Quantitative real-time PCR analysis ofBIRC2 mRNA abundance in RNA samples derived from mouse (a) and human (b) tissue samples, and from rat and mouse eyes (c). Values for the indicated mRNA abundances were normalized tothat of GAPDH (a and c) or RPL19 (b) mRNA, internal controls. (B) Analysis of Birc3 mRNA expression in mouse tissues. Quantitative real-time PCR analysis of Birc3 mRNA abundance in RNAsamples derived from mouse tissue samples. Values for the indicated mRNA abundances were normalized to that of Gapdh, an internal control. (C) Analysis of XIAP mRNA expression in mouse, ratand human tissues. Quantitative real-time PCR analysis of XIAP mRNA abundance in RNA samples derived from mouse (a) and human (b) tissue samples, and from rat and mouse eyes (c). Valuesfor the indicated mRNA abundances were normalized to that of GAPDH (a and c) or RPL19 (b) mRNA internal controls. BC, bone, calvia; BL, bone, long; BM, bone marrow; LG, lacrimal gland; LN ,lymph node; MG, mammary gland; PBL, peripheral blood lymphocyte; PP, Peyer’s patches; SC, spinal cord; SGM, salivary gland, mandibular; SGP, salivary gland, parotid; SV, seminal vesicle; VC,vena cava; VD, vas deferens; VJ, vein, jugular; VP, vein, portal; RPE + CC, RPE and choroid capillaries.
REFERENCES
1 Varfolomeev, E., Blankenship, J. W., Wayson, S. M., Fedorova, A. V., Kayagaki, N., Garg, P.,Zobel, K., Dynek, J. N., Elliott, L. O., Wallweber, H. J. et al. (2007) IAP antagonists induceautoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131,669–681
2 Varfolomeev, E., Goncharov, T., Maecker, H., Zobel, K., Komuves, L. G., Deshayes, K. andVucic, D. (2012) Cellular inhibitors of apoptosis are global regulators of NF-κB and MAPKactivation by members of the TNF family of receptors. Sci. Signal. 5, ra22
Received 11 July 2012/31 July 2012; accepted 2 August 2012Published as BJ Immediate Publication 2 August 2012, doi:10.1042/BJ20121103
c⃝ The Authors Journal compilation c⃝ 2012 Biochemical Society
Related Documents











