Applied Catalysis A: General 185 (1999) 137–152 Characterization of γ -alumina and borated alumina catalysts Cristina Flego, Wallace O’Neil Parker Jr. * Department of Physical Chemistry, EniTecnologie SpA, 20097 San Donato, MI, Italy Received 4 February 1999; received in revised form 27 April 1999; accepted 28 April 1999 Abstract The acidity and surface structure of pure g-alumina and a borated alumina (AB), containing a low amount of boria (1.9 wt.%), are investigated by a variety of techniques. The coordination states of B, and the distribution of hydroxyls on the surface, are studied by 11 B MAS NMR and UV–Vis-NIR spectroscopies, respectively. Partially hydrated AB has trigonal boron (BO 3 ) on the surface, as found in B 2 O 3 (via 11 B quadrupolar parameters), and a small part (ca. 50 mmol/g of AB, or 10% mole) of the boron nuclei exhibit 11 B resonances narrowed by exchange with water (BO 4 ). Boria introduction creates new types of surface hydroxyl groups, giving rise to a B–OHNIR band at 1382nm. However, quantification via NIR bands reveals no significant change in the total number of hydroxyl groups. The structural types of borate surface species on dried AB, consistent with this finding, are presented. Pyrrole adsorption shows that AB contains no basic sites. The acidities (quantity, strength) of dehydrated samples are evaluated by IR spectroscopy and static volumetric adsorption using pyridine and ammonia as basic probes, respectively. In-situ 13 C NMR is also used to study the acid/base strength by monitoring the low energy model reactions (at 25 ◦ C) of 1-butene double-bond isomerization (DBI) and isobutene dimerization. All three methods concur that dried AB has greater acidity than g-alumina due to Lewis acid sites with greater strength. Volumetry and in-situ NMR find that only ca. 6% of the boron nuclei (34 mmol/g) on dried AB furnish (Lewis acid) chemisorption sites for butene. This corresponds closely to the number of sites in AB adsorbing water (forming BO 4 by 11 B NMR) and to those chemisorbing pyridine strongly (desorption above 400 ◦ C). UV–Vis-NIR spectra reveal that the Lewis sites of AB perturb the double bond of chemisorbed olefins (partial hydride transfer) and the surface hydroxyl groups physisorb olefins by H-bonding. ©1999 Elsevier Science B.V. All rights reserved. Keywords: Alumina; Boron oxide; Acidity; 11 B and 13 C NMR; UV–Vis-NIR; Adsorption (butene, pyridine, ammonia, pyrrole) 1. Introduction Aluminas have an important role in heterogeneous catalysis, both as catalysts and supports [1]. Alumina is an amphoteric oxide. Its acidity is increased by sur- face modification with supported oxides [2], mineral acids [3] and halogens atoms [4]. Borated alumina (AB) is an example of the first two types. AB is * Corresponding author. Tel.: +39-02-520-56319; fax: +39-02- 520-36347; e-mail: [email protected] usually prepared by impregnation of alumina (i.e. g-Al 2 O 3 ) with aqueous boric acid followed by cal- cination [5–8]. Other methods involve vapor decom- position with ethyl borate [9,10] and co-precipitation from an aluminum nitrate and boric acid solution using ammonium hydroxide [6,11]. AB is used for reactions such as selective ox- idation of ethane [12], Beckmann rearrangement [5,9,10], m-xylene isomerization [10,13], toluene dis- proportionation [10,13] and skeletal isomerization of n-butenes [8], where the maximal activities occur 0926-860X/99/$ – see front matter ©1999 Elsevier Science B.V. All rights reserved. PII:S0926-860X(99)00137-4

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Applied Catalysis A: General 185 (1999) 137–152
Characterization ofγ -alumina and borated alumina catalysts
Cristina Flego, Wallace O’Neil Parker Jr.∗Department of Physical Chemistry, EniTecnologie SpA, 20097 San Donato, MI, Italy
Received 4 February 1999; received in revised form 27 April 1999; accepted 28 April 1999
Abstract
The acidity and surface structure of pureg-alumina and a borated alumina (AB), containing a low amount of boria (1.9 wt.%),are investigated by a variety of techniques. The coordination states of B, and the distribution of hydroxyls on the surface, arestudied by11B MAS NMR and UV–Vis-NIR spectroscopies, respectively. Partially hydrated AB has trigonal boron (BO3) onthe surface, as found in B2O3 (via 11B quadrupolar parameters), and a small part (ca. 50mmol/g of AB, or 10% mole) of theboron nuclei exhibit11B resonances narrowed by exchange with water (BO4). Boria introduction creates new types of surfacehydroxyl groups, giving rise to a B–OHNIR band at 1382 nm. However, quantification via NIR bands reveals no significantchange in the total number of hydroxyl groups. The structural types of borate surface species on dried AB, consistent withthis finding, are presented. Pyrrole adsorption shows that AB contains no basic sites. The acidities (quantity, strength) ofdehydrated samples are evaluated by IR spectroscopy and static volumetric adsorption using pyridine and ammonia as basicprobes, respectively. In-situ13C NMR is also used to study the acid/base strength by monitoring the low energy model reactions(at 25◦C) of 1-butene double-bond isomerization (DBI) and isobutene dimerization. All three methods concur that dried ABhas greater acidity thang-alumina due to Lewis acid sites with greater strength. Volumetry and in-situ NMR find that onlyca. 6% of the boron nuclei (34mmol/g) on dried AB furnish (Lewis acid) chemisorption sites for butene. This correspondsclosely to the number of sites in AB adsorbing water (forming BO4 by 11B NMR) and to those chemisorbing pyridine strongly(desorption above 400◦C). UV–Vis-NIR spectra reveal that the Lewis sites of AB perturb the double bond of chemisorbedolefins (partial hydride transfer) and the surface hydroxyl groups physisorb olefins by H-bonding. ©1999 Elsevier ScienceB.V. All rights reserved.
Keywords:Alumina; Boron oxide; Acidity;11B and13C NMR; UV–Vis-NIR; Adsorption (butene, pyridine, ammonia, pyrrole)
1. Introduction
Aluminas have an important role in heterogeneouscatalysis, both as catalysts and supports [1]. Aluminais an amphoteric oxide. Its acidity is increased by sur-face modification with supported oxides [2], mineralacids [3] and halogens atoms [4]. Borated alumina(AB) is an example of the first two types. AB is
∗ Corresponding author. Tel.: +39-02-520-56319; fax: +39-02-520-36347; e-mail: [email protected]
usually prepared by impregnation of alumina (i.e.g-Al2O3) with aqueous boric acid followed by cal-cination [5–8]. Other methods involve vapor decom-position with ethyl borate [9,10] and co-precipitationfrom an aluminum nitrate and boric acid solutionusing ammonium hydroxide [6,11].
AB is used for reactions such as selective ox-idation of ethane [12], Beckmann rearrangement[5,9,10],m-xylene isomerization [10,13], toluene dis-proportionation [10,13] and skeletal isomerizationof n-butenes [8], where the maximal activities occur
0926-860X/99/$ – see front matter ©1999 Elsevier Science B.V. All rights reserved.PII: S0926-860X(99)00137-4

138 C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152
at boria loadings of 25, 24, 14, 15 and 10 wt.%, re-spectively. The surface acidity/basicity of aluminas,and modified aluminas, determine their activities andlifetimes in reactions since they have no internal porestructure. The need to relate surface structure/aciditywith catalytic activity has solicited physical–chemicalinvestigations.
Spectroscopic or adsorption/desorption studiesof AB have been made by various groups [5–18].The AB samples studied contained 2–40 wt.% ofboria. Acidities were evaluated by four differentmethods. (i) Temperature programmed desorption(TPD) of ammonia [5,6,9,11,15,16]. (ii) Compari-son of TPD profiles for 2,6-dimethylpyridine andpyridine [8]. (iii) IR spectroscopy of pyridine ad-sorption [14]. (iv) Titration withn-butylamine andHammett indicators [13]. Few works have ad-dressed acid site quantification [5,8,15,16], andthere is no detailed study of AB with low boriacontent.
Sato and co-workers [8] recently showed, by11BMAS (magic angle spinning) NMR, that at boria con-tents above 2 wt.% (or 100mg/m2) BO4 species areformed. Curtin et al. [5] characterized AB with 2%boria (200mg/m2) by surface area measurements, X-ray diffraction and TPD of ammonia. They found novariation in morphology compared to alumina, and ob-tained no information on the nature of the acid sites orif basic sites were present. We chose to study AB at aboria content (100mg/m2) sufficiently high to destroythe basicity of alumina yet low enough to provide the‘simplest’ acidity.
The aim of this work is to characterize and differen-tiateg-alumina and AB at low boria content. Multiplemethods of characterization will be used: (i) multin-uclear NMR and UV–Vis-NIR (Ultraviolet–Visible-Near InfraRed) studies to analyze the surface struc-tures, (ii) a combination of spectroscopic (IR, NMR)and volumetric adsorption studies to identify/quantifythe surface adsorption sites and (iii) adsorption mea-surements (volumetric and spectroscopic) with probemolecules (basic, acidic and small olefins) along within-situ NMR of model butene reactions to investigateacid–base properties. The combined use of these tech-niques (some applied for the first time) will permittheir relative potentials for describing these materialsto be verified. Adequate catalyst characterization isvery important, since a detailed knowledge of sur-
face properties is required to rationalize catalyticphenomena.
2. Experimental
2.1. Materials
Commercialg-Al2O3 was purchased from Akzo(A), Sudchemie and Condea. A was used without pu-rification to obtain AB by a one-step impregnationwith aqueous boric acid followed by calcination at500◦C. All the aluminas have ca. 200 m2/g of surfacearea and 0.45 ml/g of pore volume (from nitrogen ad-sorption, BET). Introduction of boria, 1.9 wt.%, to thesurface of A did not change these values. All reagents(gases from SIAD, liquids from Merck and Aldrich)were of reagent grade and purified by cryotreatmentsbefore use.
2.2. UV–Vis-NIR spectroscopy
UV–Vis-NIR spectra were registered (25◦C) overthe range 190–1500 nm using a Lambda 19 (Perkin-Elmer) spectrophotometer equipped with a reflectancesphere. The resolution was 1 nm, and the reflection wasmeasured in Kubelka-Munk (K-M) units. The pow-dered samples were lodged in a Suprasil quartz celland all thermal and adsorption treatments were per-formed in-situ. Before each treatment, the catalystswere evacuated at 350◦C for 8 h (10 Pa). Spectral de-convolution was performed by the Peak Solve (Galac-tic) program. Curve fit quality was measured by thecorrelation coefficient (R2), standard deviation (σ ) andthe Levenberg–Marquardt algorithm (χ2).
2.3. NMR spectroscopy
Spectra were collected on a Bruker CXP-300 orAMX-300 at 25◦C. All NMR experiments were madewith powders.11B (96 MHz) MAS NMR spectrawere collected using 4 mm zirconia rotors (spinningat 14 kHz), 10◦ rf pulses (ca. 0.7ms), 1 s cycle delayand with a Bruker probe having a low11B background(no boronitride components). B(OCH3)3 in chloro-form (18.1 ppm) was the chemical shift reference.Simulations of spectra to give anisotropic parameters

C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152 139
and deconvolutions were made with WIN-FIT andWIN-NMR, respectively.
In-situ 13C NMR (75 MHz) spectra were collectedon the catalysts (300 mg) contained in 8 mm non-spinning flame sealed glass tubes, with Rotaflo valvesfor dosing with gaseous reagents from a vacuum line,using 30◦ rf pulses (4ms), 2 s cycle delay, 300 scansand1H decoupling only during acquisition. These con-ditions provided quantitative spectra since the longestlongitudinal relaxation time (T1) was less than 2 s. Thecatalysts were dried in the tubes at 350◦C for 8 h un-der vacuum (10 Pa) prior to study (1H quantification)or adsorption, via cold trap transfer, of 1-13C-1-butene(0.15 mmoles olefin per gram of catalyst), 1-butene orisobutene (both 1.56 mmol/g).
1H (300 MHz) and13C (75 MHz) MAS NMR spec-tra were collected using 7 mm zirconia rotors spin-ning at 5 kHz.13C acquisition conditions (andT1s)were the same as for in-situ experiments except morescans were used (up to 40 000).1H spectra were col-lected using 25◦ rf pulses (3ms) and 10 s cycle delay.The longestT1swere found on dried samples (3–5 s).Chemical shifts for1H and 13C were referenced towater (4.8 ppm) and adamantane (38.5, 29.5 ppm)or ethylbenzene (–CH3 at 15.9 ppm), respectively.Quantification of protons was made by referring1HMAS NMR areas to those of a water capillary (3ml,0.17 mmoles) and a highly crystalline silicalite (typeMFI) with 0.86 mmoles of SiOH per gram (determinedby 29Si NMR). Spinning side bands were includedand the background signal was subtracted out. Theerror is estimated to be +/−10% of the value given.
2.4. FT-IR spectroscopy
Pyridine adsorption–desorption was monitored byFT-IR (Fourier Transform Infrared) spectroscopy aspreviously [19]. Self-supported wafers of 15–20 mg/cm2 thickness were evacuated in situ (500◦C, 1 h,10 Pa) in a pyrex cell with KBr windows, contactedwith probe at 200◦C and desorbed at increasing tem-perature (200–500◦C) in dynamic vacuum. Spectrawere registered (at 25◦C) with a Perkin-Elmer 2000FT-IR spectrometer at 1 cm−1 resolution. The errorin peak area determination was around 15%. Acidstrength distribution was determined as for ammonia(below).
2.5. Volumetry
Adsorption measurements were performed in apyrex volumetric apparatus equipped with pressureand vacuum detectors. The powders were evacuatedat 500◦C for 1 h at 10 Pa, and increasing amounts ofadsorbate were added until saturation. The amountadsorbed (mmol/g) was calculated by the general gaslaw knowing the expansion coefficient of the system.The error in this measurement is estimated to be ca.3%.
To determine the acid strength distribution, am-monia was adsorbed at 150◦C and desorptions weremade at increasing temperatures (150–450◦C) un-der dynamic vacuum. An adsorption isotherm wasalways made at 150◦C after each thermal treatmentto determine the amount desorbed. This provided anacid strength scale, with higher desorption temper-atures corresponding to stronger acid sites. Desorp-tion at 150◦C gave the number of very weak acidsites. The differences between the amounts desorbedat 150◦C/250◦C, 250◦C/350◦C and 350◦C/450◦Cyielded the number of weak, medium and medium-strong sites, respectively. Any NH3 remaining ad-sorbed at 450◦C corresponds to the number of strongacid sites.
Pyrrole adsorption was performed at 65◦C followedby desorption at 25◦C (under dynamic vacuum) andby a second isotherm at 65◦C, in order to evaluatethe chemi- and physi-sorbed species. The same volu-metric apparatus was used to measure the adsorptionof 1-butene and isobutene at 25◦C. After saturationthe sample was evacuated overnight at 25◦C andanother adsorption was performed to determine thephysisorbed amount.
3. Results
3.1. Structural characterization
3.1.1. NMR spectroscopyBoron in solids usually occurs in tetrahedral BO4
or trigonal BO3 configurations. The11B MAS NMRspectrum of solid boron oxide (B2O3), in Fig. 1,shows a quadrupolar ‘doublet’ pattern characteristicof trigonal boron (Tg-B). The anisotropic parameters
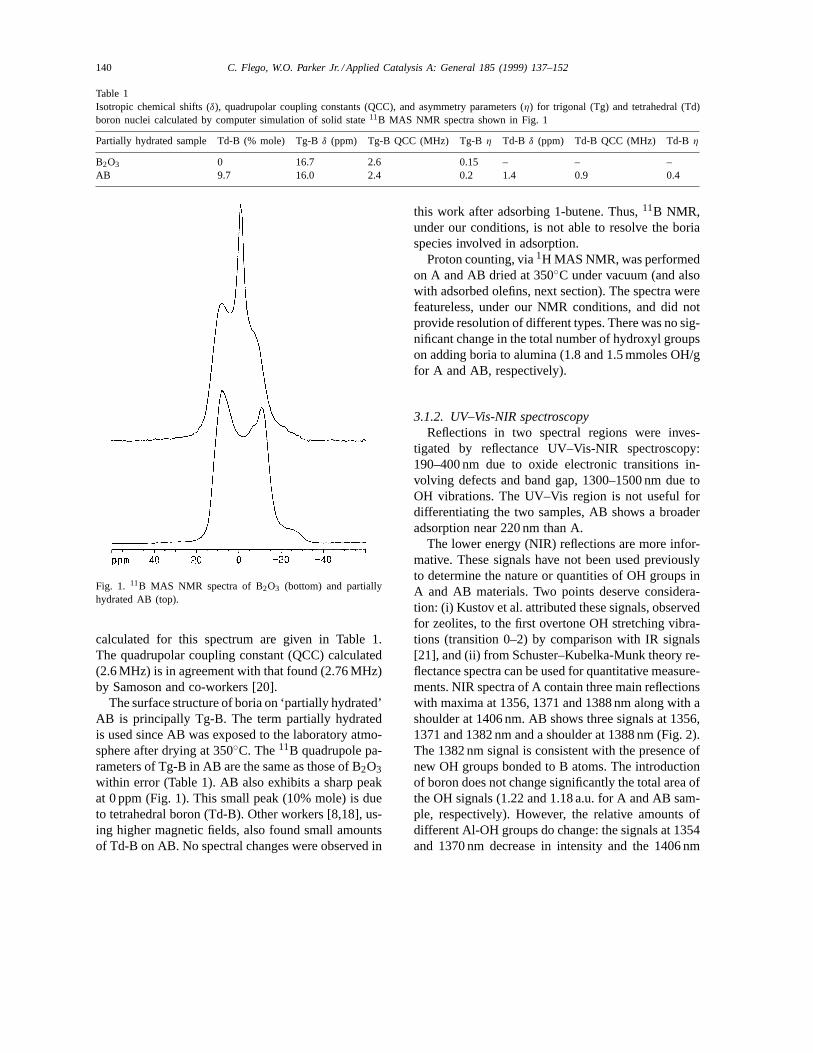
140 C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152
Table 1Isotropic chemical shifts (δ), quadrupolar coupling constants (QCC), and asymmetry parameters (η) for trigonal (Tg) and tetrahedral (Td)boron nuclei calculated by computer simulation of solid state11B MAS NMR spectra shown in Fig. 1
Partially hydrated sample Td-B (% mole) Tg-Bδ (ppm) Tg-B QCC (MHz) Tg-Bη Td-B δ (ppm) Td-B QCC (MHz) Td-Bη
B2O3 0 16.7 2.6 0.15 – – –AB 9.7 16.0 2.4 0.2 1.4 0.9 0.4
Fig. 1. 11B MAS NMR spectra of B2O3 (bottom) and partiallyhydrated AB (top).
calculated for this spectrum are given in Table 1.The quadrupolar coupling constant (QCC) calculated(2.6 MHz) is in agreement with that found (2.76 MHz)by Samoson and co-workers [20].
The surface structure of boria on ‘partially hydrated’AB is principally Tg-B. The term partially hydratedis used since AB was exposed to the laboratory atmo-sphere after drying at 350◦C. The11B quadrupole pa-rameters of Tg-B in AB are the same as those of B2O3within error (Table 1). AB also exhibits a sharp peakat 0 ppm (Fig. 1). This small peak (10% mole) is dueto tetrahedral boron (Td-B). Other workers [8,18], us-ing higher magnetic fields, also found small amountsof Td-B on AB. No spectral changes were observed in
this work after adsorbing 1-butene. Thus,11B NMR,under our conditions, is not able to resolve the boriaspecies involved in adsorption.
Proton counting, via1H MAS NMR, was performedon A and AB dried at 350◦C under vacuum (and alsowith adsorbed olefins, next section). The spectra werefeatureless, under our NMR conditions, and did notprovide resolution of different types. There was no sig-nificant change in the total number of hydroxyl groupson adding boria to alumina (1.8 and 1.5 mmoles OH/gfor A and AB, respectively).
3.1.2. UV–Vis-NIR spectroscopyReflections in two spectral regions were inves-
tigated by reflectance UV–Vis-NIR spectroscopy:190–400 nm due to oxide electronic transitions in-volving defects and band gap, 1300–1500 nm due toOH vibrations. The UV–Vis region is not useful fordifferentiating the two samples, AB shows a broaderadsorption near 220 nm than A.
The lower energy (NIR) reflections are more infor-mative. These signals have not been used previouslyto determine the nature or quantities of OH groups inA and AB materials. Two points deserve considera-tion: (i) Kustov et al. attributed these signals, observedfor zeolites, to the first overtone OH stretching vibra-tions (transition 0–2) by comparison with IR signals[21], and (ii) from Schuster–Kubelka-Munk theory re-flectance spectra can be used for quantitative measure-ments. NIR spectra of A contain three main reflectionswith maxima at 1356, 1371 and 1388 nm along with ashoulder at 1406 nm. AB shows three signals at 1356,1371 and 1382 nm and a shoulder at 1388 nm (Fig. 2).The 1382 nm signal is consistent with the presence ofnew OH groups bonded to B atoms. The introductionof boron does not change significantly the total area ofthe OH signals (1.22 and 1.18 a.u. for A and AB sam-ple, respectively). However, the relative amounts ofdifferent Al-OH groups do change: the signals at 1354and 1370 nm decrease in intensity and the 1406 nm
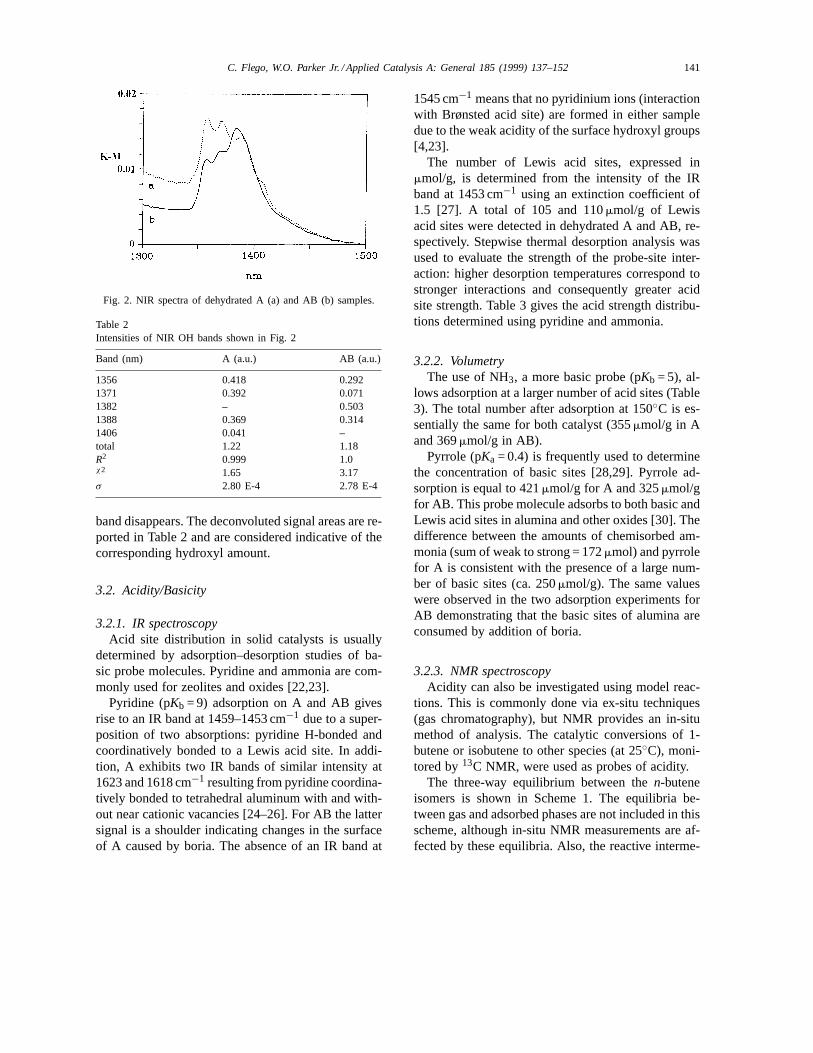
C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152 141
Fig. 2. NIR spectra of dehydrated A (a) and AB (b) samples.
Table 2Intensities of NIR OH bands shown in Fig. 2
Band (nm) A (a.u.) AB (a.u.)
1356 0.418 0.2921371 0.392 0.0711382 – 0.5031388 0.369 0.3141406 0.041 –total 1.22 1.18R2 0.999 1.0χ2 1.65 3.17σ 2.80 E-4 2.78 E-4
band disappears. The deconvoluted signal areas are re-ported in Table 2 and are considered indicative of thecorresponding hydroxyl amount.
3.2. Acidity/Basicity
3.2.1. IR spectroscopyAcid site distribution in solid catalysts is usually
determined by adsorption–desorption studies of ba-sic probe molecules. Pyridine and ammonia are com-monly used for zeolites and oxides [22,23].
Pyridine (pKb = 9) adsorption on A and AB givesrise to an IR band at 1459–1453 cm−1 due to a super-position of two absorptions: pyridine H-bonded andcoordinatively bonded to a Lewis acid site. In addi-tion, A exhibits two IR bands of similar intensity at1623 and 1618 cm−1 resulting from pyridine coordina-tively bonded to tetrahedral aluminum with and with-out near cationic vacancies [24–26]. For AB the lattersignal is a shoulder indicating changes in the surfaceof A caused by boria. The absence of an IR band at
1545 cm−1 means that no pyridinium ions (interactionwith Brønsted acid site) are formed in either sampledue to the weak acidity of the surface hydroxyl groups[4,23].
The number of Lewis acid sites, expressed inmmol/g, is determined from the intensity of the IRband at 1453 cm−1 using an extinction coefficient of1.5 [27]. A total of 105 and 110mmol/g of Lewisacid sites were detected in dehydrated A and AB, re-spectively. Stepwise thermal desorption analysis wasused to evaluate the strength of the probe-site inter-action: higher desorption temperatures correspond tostronger interactions and consequently greater acidsite strength. Table 3 gives the acid strength distribu-tions determined using pyridine and ammonia.
3.2.2. VolumetryThe use of NH3, a more basic probe (pKb = 5), al-
lows adsorption at a larger number of acid sites (Table3). The total number after adsorption at 150◦C is es-sentially the same for both catalyst (355mmol/g in Aand 369mmol/g in AB).
Pyrrole (pKa = 0.4) is frequently used to determinethe concentration of basic sites [28,29]. Pyrrole ad-sorption is equal to 421mmol/g for A and 325mmol/gfor AB. This probe molecule adsorbs to both basic andLewis acid sites in alumina and other oxides [30]. Thedifference between the amounts of chemisorbed am-monia (sum of weak to strong = 172mmol) and pyrrolefor A is consistent with the presence of a large num-ber of basic sites (ca. 250mmol/g). The same valueswere observed in the two adsorption experiments forAB demonstrating that the basic sites of alumina areconsumed by addition of boria.
3.2.3. NMR spectroscopyAcidity can also be investigated using model reac-
tions. This is commonly done via ex-situ techniques(gas chromatography), but NMR provides an in-situmethod of analysis. The catalytic conversions of 1-butene or isobutene to other species (at 25◦C), moni-tored by13C NMR, were used as probes of acidity.
The three-way equilibrium between then-buteneisomers is shown in Scheme 1. The equilibria be-tween gas and adsorbed phases are not included in thisscheme, although in-situ NMR measurements are af-fected by these equilibria. Also, the reactive interme-
142 C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152
Table 3Acid strength distribution by pyridine and ammonia adsorption–desorption
Basic probe molecule 1T desorption (◦C) Acid strength Lewis sites A (mmol/g) Lewis sites AB (mmol/g)
Pyridine 200–300 Weak 37 55300–400 Medium 41 22400–500 Medium-strong 16 10>500 Strong 11 23
NH3 150 Very weak 181 160150–250 Weak 111 129250–350 Medium 34 17350–450 Medium-strong 29 55>450 Strong 0 8
Scheme 1. Three-way gas phase equilibrium forn-butenes.
diates (in the H scrambling reactions) involved in theconversion of the isomers are not considered. It is thenature of these intermediates which gives informationon the type of active site operating. NMR did not de-tect any intermediates, they were inferred by indirectmeans (below in butene reactions).
At thermodynamic equilibrium (25◦C) in the gasphase 2-butene predominates: 1-butene (3%),cis(21%) andtrans (76%) 2-butene [31]. The reactionrates toward thermodynamic equilibrium can be usedto study acidity. This is conveniently done by adsorb-ing 1-13C-1-butene and collecting in-situ13C NMRspectra [32–35] as a function of time. The overallrate of double-bond isomerization (DBI) of 1-butene(k1 + k2) provides a relative measure of acid/basestrength. The stereoselectivity of DBI (defined as thecis/trans ratio of the 2-butene initially formed) isexpressed as (C/T)0 = k1/k2. It gives information onthe type of acidity (reactive intermediate). For exam-ple, alkyl carbenium ions forming on Brønsted acidsites should give no stereoselectivity (C/T)0 = 1, sincethere is free rotation about the C2–C3 bond. On thecontrary, Lewis acid sites and basic sites can causelarge stereoselectivity effects. Reaction profiles forn-butenes isomerization have been used to classify cata-lysts [36] and to characterizeg-aluminas [31,37–39].
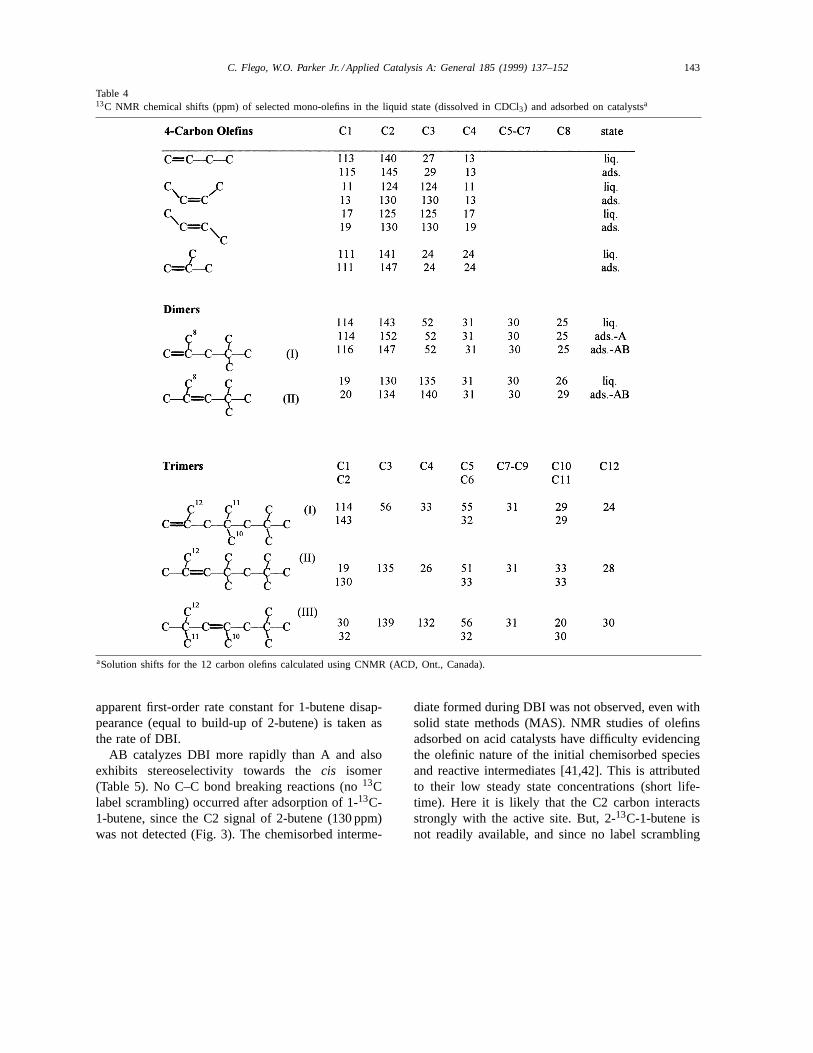
In-situ 13C NMR spectra (e.g. for AB, Fig. 3)
Fig. 3. In-situ13C NMR spectra, as a function of time at 25◦C, afteradsorption of 1-13C-1-butene on AB. Chemical shifts indicatedabove selected peaks.
clearly show the conversion (at 25◦C) of 1-13C-1-butene into 1-13C-2-butene even without performingMAS on the sample, because the butene moleculesdetected are very mobile (liquid-like). The C1 carbonsignal (115 ppm) of 1-butene is gradually replacedby the C1 signals oftrans (19 ppm) andcis (13 ppm)2-butene. Olefin signals are assigned in Table 4. The
C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152 143
Table 413C NMR chemical shifts (ppm) of selected mono-olefins in the liquid state (dissolved in CDCl3) and adsorbed on catalystsa
aSolution shifts for the 12 carbon olefins calculated using CNMR (ACD, Ont., Canada).
apparent first-order rate constant for 1-butene disap-pearance (equal to build-up of 2-butene) is taken asthe rate of DBI.
AB catalyzes DBI more rapidly than A and alsoexhibits stereoselectivity towards thecis isomer(Table 5). No C–C bond breaking reactions (no13Clabel scrambling) occurred after adsorption of 1-13C-1-butene, since the C2 signal of 2-butene (130 ppm)was not detected (Fig. 3). The chemisorbed interme-
diate formed during DBI was not observed, even withsolid state methods (MAS). NMR studies of olefinsadsorbed on acid catalysts have difficulty evidencingthe olefinic nature of the initial chemisorbed speciesand reactive intermediates [41,42]. This is attributedto their low steady state concentrations (short life-time). Here it is likely that the C2 carbon interactsstrongly with the active site. But, 2-13C-1-butene isnot readily available, and since no label scrambling
144 C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152
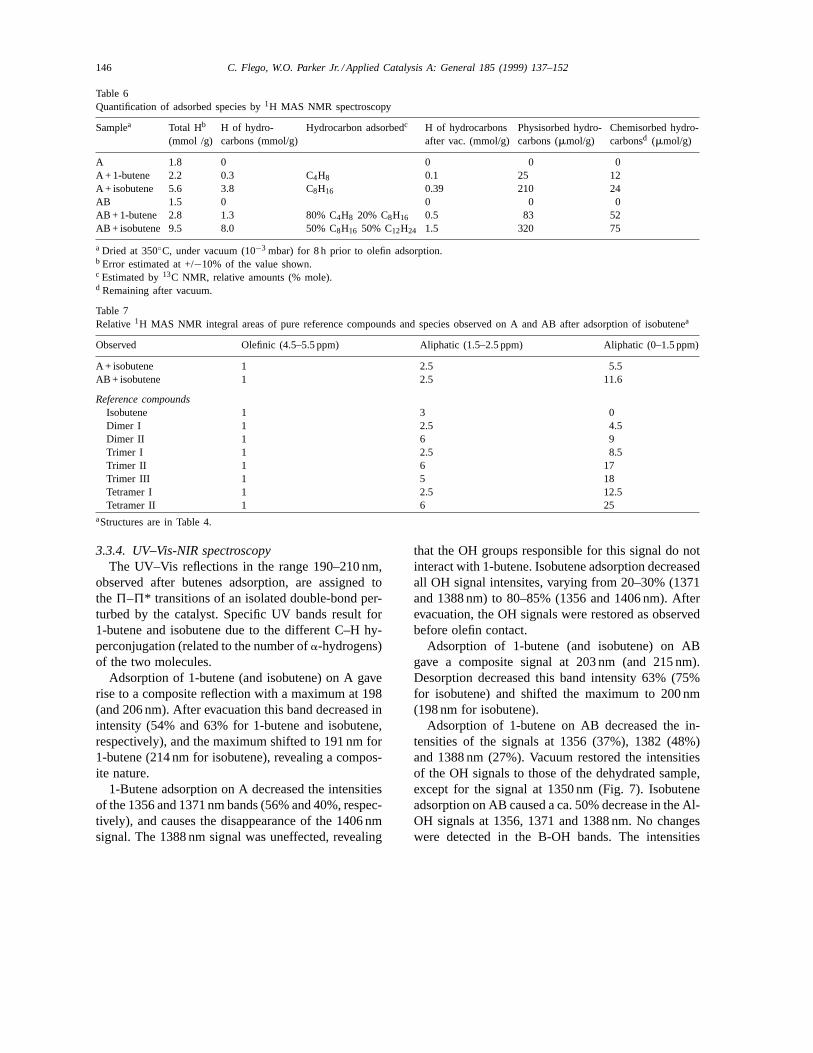
Table 5Characterization ofg-aluminas and AB using butene model reactions (at 25◦C). 13C NMR studies of this work made using 0.15 and1.56 mmole doses of 1-13C-1-butene and isobutene per gram of dried catalyst, respectively
Apparent DBI rate (h−1) Stereo-selectivity (C/T)0 Apparent isobutene dimerization rate (h−1)
g-Al2O3(lab. syn.) 0.08a 2–6b –g-Al2O3(Cyanamid) 0.09c
g-Al2O3(Sudchemie) 0.30 2.5 0.02–0.1g-Al2O3(Condea) 0.27 2.3 0.02–0.1g-Al2O3 (Akzo)A 0.05 0.9 <0.02AB 0.95 2.5 >3.0
a From [39], alumina pretreated at 310◦C.b From [38].c From [40], ex-situ GC measurement.
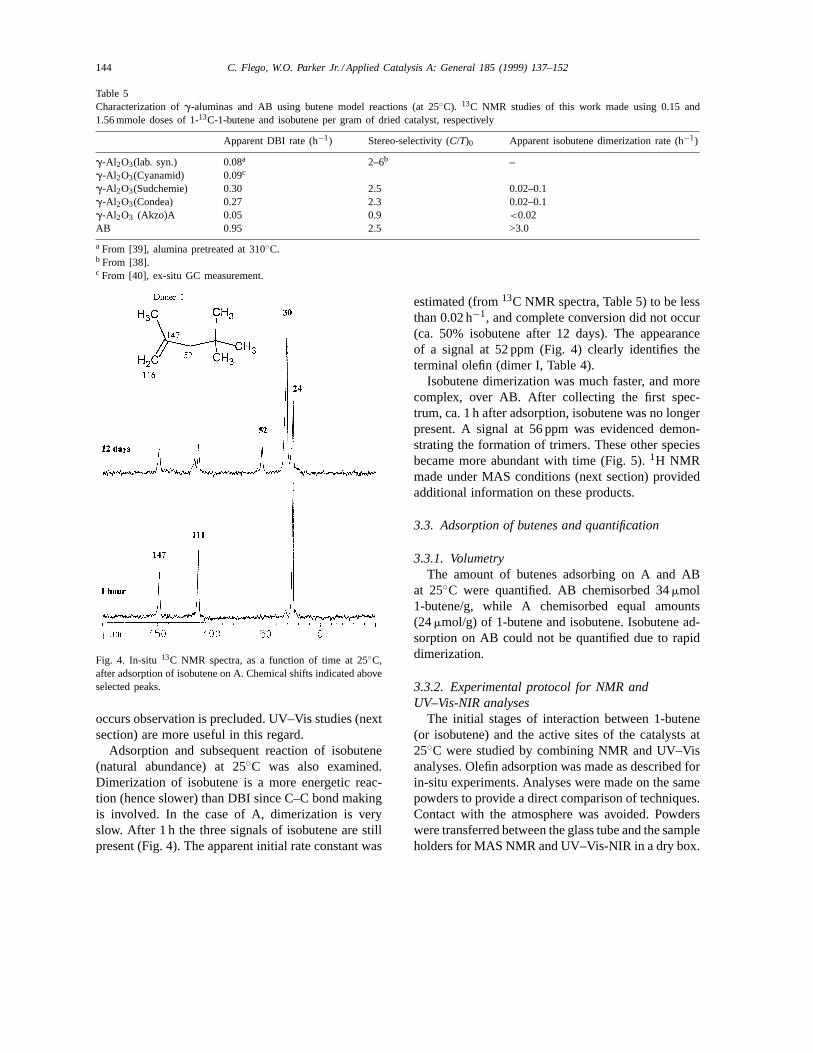
Fig. 4. In-situ 13C NMR spectra, as a function of time at 25◦C,after adsorption of isobutene on A. Chemical shifts indicated aboveselected peaks.
occurs observation is precluded. UV–Vis studies (nextsection) are more useful in this regard.
Adsorption and subsequent reaction of isobutene(natural abundance) at 25◦C was also examined.Dimerization of isobutene is a more energetic reac-tion (hence slower) than DBI since C–C bond makingis involved. In the case of A, dimerization is veryslow. After 1 h the three signals of isobutene are stillpresent (Fig. 4). The apparent initial rate constant was
estimated (from13C NMR spectra, Table 5) to be lessthan 0.02 h−1, and complete conversion did not occur(ca. 50% isobutene after 12 days). The appearanceof a signal at 52 ppm (Fig. 4) clearly identifies theterminal olefin (dimer I, Table 4).
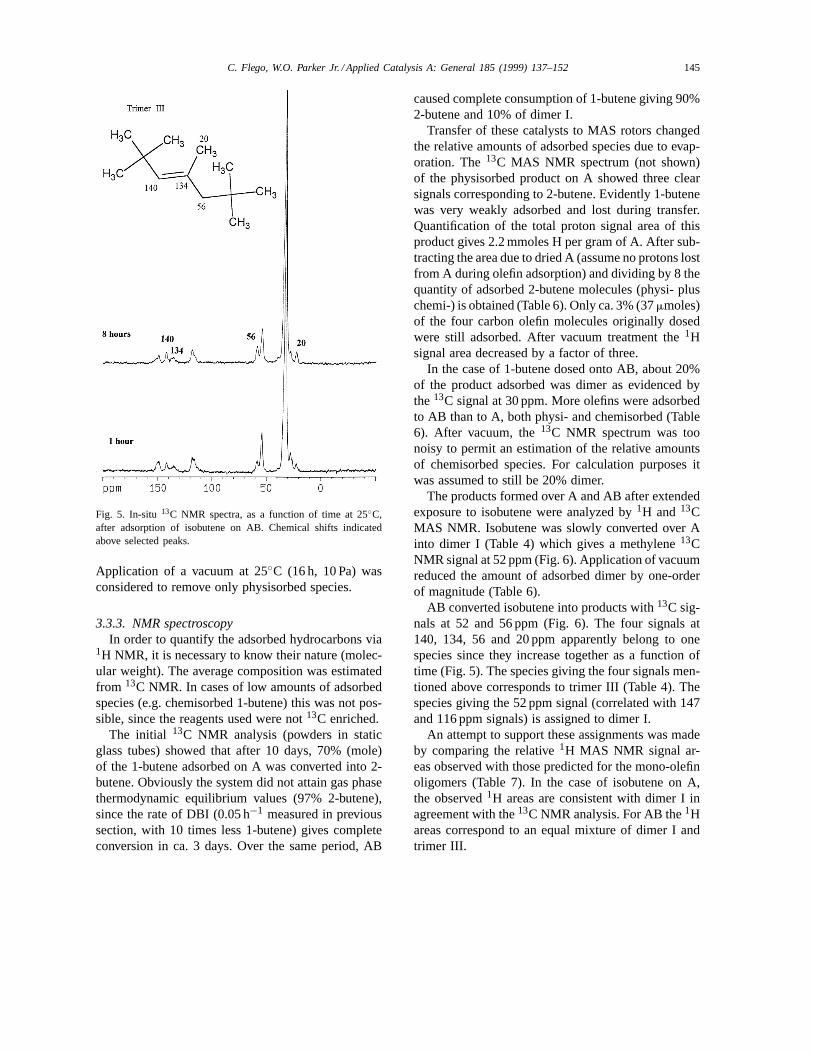
Isobutene dimerization was much faster, and morecomplex, over AB. After collecting the first spec-trum, ca. 1 h after adsorption, isobutene was no longerpresent. A signal at 56 ppm was evidenced demon-strating the formation of trimers. These other speciesbecame more abundant with time (Fig. 5).1H NMRmade under MAS conditions (next section) providedadditional information on these products.
3.3. Adsorption of butenes and quantification
3.3.1. VolumetryThe amount of butenes adsorbing on A and AB
at 25◦C were quantified. AB chemisorbed 34mmol1-butene/g, while A chemisorbed equal amounts(24mmol/g) of 1-butene and isobutene. Isobutene ad-sorption on AB could not be quantified due to rapiddimerization.
3.3.2. Experimental protocol for NMR andUV–Vis-NIR analyses
The initial stages of interaction between 1-butene(or isobutene) and the active sites of the catalysts at25◦C were studied by combining NMR and UV–Visanalyses. Olefin adsorption was made as described forin-situ experiments. Analyses were made on the samepowders to provide a direct comparison of techniques.Contact with the atmosphere was avoided. Powderswere transferred between the glass tube and the sampleholders for MAS NMR and UV–Vis-NIR in a dry box.
C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152 145
Fig. 5. In-situ 13C NMR spectra, as a function of time at 25◦C,after adsorption of isobutene on AB. Chemical shifts indicatedabove selected peaks.
Application of a vacuum at 25◦C (16 h, 10 Pa) wasconsidered to remove only physisorbed species.
3.3.3. NMR spectroscopyIn order to quantify the adsorbed hydrocarbons via
1H NMR, it is necessary to know their nature (molec-ular weight). The average composition was estimatedfrom 13C NMR. In cases of low amounts of adsorbedspecies (e.g. chemisorbed 1-butene) this was not pos-sible, since the reagents used were not13C enriched.
The initial 13C NMR analysis (powders in staticglass tubes) showed that after 10 days, 70% (mole)of the 1-butene adsorbed on A was converted into 2-butene. Obviously the system did not attain gas phasethermodynamic equilibrium values (97% 2-butene),since the rate of DBI (0.05 h−1 measured in previoussection, with 10 times less 1-butene) gives completeconversion in ca. 3 days. Over the same period, AB
caused complete consumption of 1-butene giving 90%2-butene and 10% of dimer I.
Transfer of these catalysts to MAS rotors changedthe relative amounts of adsorbed species due to evap-oration. The13C MAS NMR spectrum (not shown)of the physisorbed product on A showed three clearsignals corresponding to 2-butene. Evidently 1-butenewas very weakly adsorbed and lost during transfer.Quantification of the total proton signal area of thisproduct gives 2.2 mmoles H per gram of A. After sub-tracting the area due to dried A (assume no protons lostfrom A during olefin adsorption) and dividing by 8 thequantity of adsorbed 2-butene molecules (physi- pluschemi-) is obtained (Table 6). Only ca. 3% (37mmoles)of the four carbon olefin molecules originally dosedwere still adsorbed. After vacuum treatment the1Hsignal area decreased by a factor of three.
In the case of 1-butene dosed onto AB, about 20%of the product adsorbed was dimer as evidenced bythe13C signal at 30 ppm. More olefins were adsorbedto AB than to A, both physi- and chemisorbed (Table6). After vacuum, the13C NMR spectrum was toonoisy to permit an estimation of the relative amountsof chemisorbed species. For calculation purposes itwas assumed to still be 20% dimer.
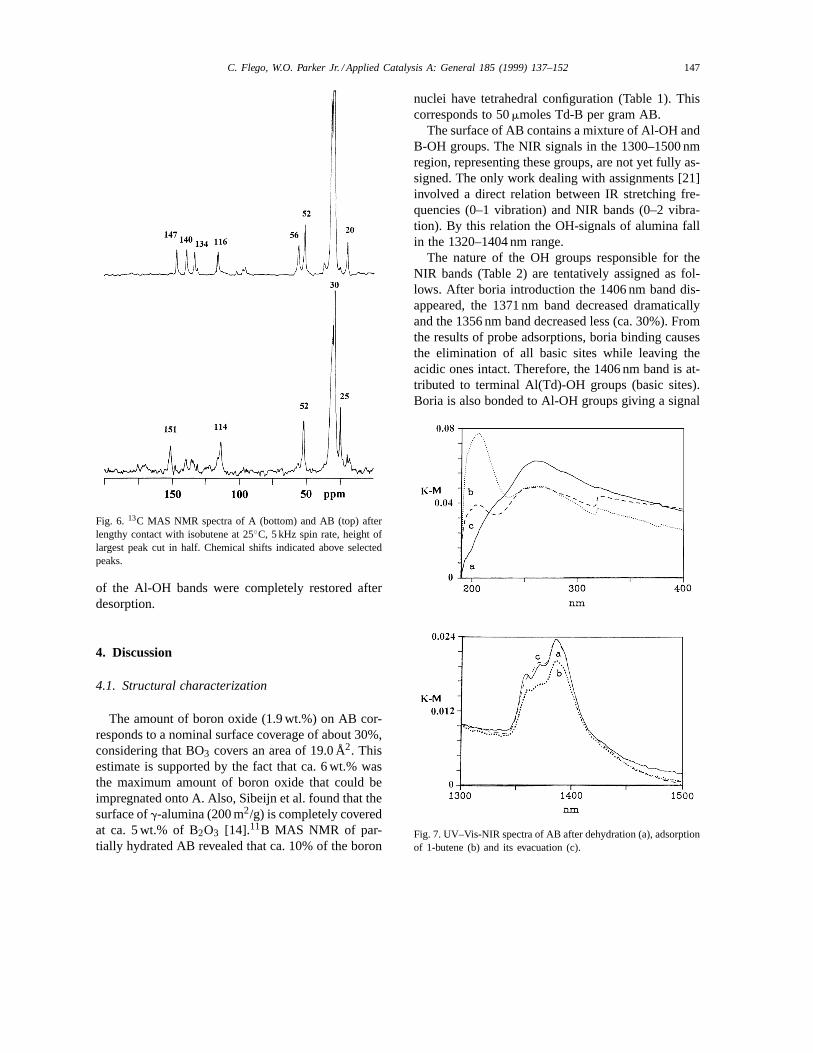
The products formed over A and AB after extendedexposure to isobutene were analyzed by1H and 13CMAS NMR. Isobutene was slowly converted over Ainto dimer I (Table 4) which gives a methylene13CNMR signal at 52 ppm (Fig. 6). Application of vacuumreduced the amount of adsorbed dimer by one-orderof magnitude (Table 6).
AB converted isobutene into products with13C sig-nals at 52 and 56 ppm (Fig. 6). The four signals at140, 134, 56 and 20 ppm apparently belong to onespecies since they increase together as a function oftime (Fig. 5). The species giving the four signals men-tioned above corresponds to trimer III (Table 4). Thespecies giving the 52 ppm signal (correlated with 147and 116 ppm signals) is assigned to dimer I.
An attempt to support these assignments was madeby comparing the relative1H MAS NMR signal ar-eas observed with those predicted for the mono-olefinoligomers (Table 7). In the case of isobutene on A,the observed1H areas are consistent with dimer I inagreement with the13C NMR analysis. For AB the1Hareas correspond to an equal mixture of dimer I andtrimer III.
146 C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152
Table 6Quantification of adsorbed species by1H MAS NMR spectroscopy
Samplea Total Hb H of hydro- Hydrocarbon adsorbedc H of hydrocarbons Physisorbed hydro- Chemisorbed hydro-(mmol /g) carbons (mmol/g) after vac. (mmol/g) carbons (mmol/g) carbonsd (mmol/g)
A 1.8 0 0 0 0A + 1-butene 2.2 0.3 C4H8 0.1 25 12A + isobutene 5.6 3.8 C8H16 0.39 210 24AB 1.5 0 0 0 0AB + 1-butene 2.8 1.3 80% C4H8 20% C8H16 0.5 83 52AB + isobutene 9.5 8.0 50% C8H16 50% C12H24 1.5 320 75
a Dried at 350◦C, under vacuum (10−3 mbar) for 8 h prior to olefin adsorption.b Error estimated at +/−10% of the value shown.c Estimated by13C NMR, relative amounts (% mole).d Remaining after vacuum.
Table 7Relative1H MAS NMR integral areas of pure reference compounds and species observed on A and AB after adsorption of isobutenea
Observed Olefinic (4.5–5.5 ppm) Aliphatic (1.5–2.5 ppm) Aliphatic (0–1.5 ppm)
A + isobutene 1 2.5 5.5AB + isobutene 1 2.5 11.6
Reference compoundsIsobutene 1 3 0Dimer I 1 2.5 4.5Dimer II 1 6 9Trimer I 1 2.5 8.5Trimer II 1 6 17Trimer III 1 5 18Tetramer I 1 2.5 12.5Tetramer II 1 6 25
aStructures are in Table 4.
3.3.4. UV–Vis-NIR spectroscopyThe UV–Vis reflections in the range 190–210 nm,
observed after butenes adsorption, are assigned tothe5–5* transitions of an isolated double-bond per-turbed by the catalyst. Specific UV bands result for1-butene and isobutene due to the different C–H hy-perconjugation (related to the number ofa-hydrogens)of the two molecules.
Adsorption of 1-butene (and isobutene) on A gaverise to a composite reflection with a maximum at 198(and 206 nm). After evacuation this band decreased inintensity (54% and 63% for 1-butene and isobutene,respectively), and the maximum shifted to 191 nm for1-butene (214 nm for isobutene), revealing a compos-ite nature.
1-Butene adsorption on A decreased the intensitiesof the 1356 and 1371 nm bands (56% and 40%, respec-tively), and causes the disappearance of the 1406 nmsignal. The 1388 nm signal was uneffected, revealing
that the OH groups responsible for this signal do notinteract with 1-butene. Isobutene adsorption decreasedall OH signal intensites, varying from 20–30% (1371and 1388 nm) to 80–85% (1356 and 1406 nm). Afterevacuation, the OH signals were restored as observedbefore olefin contact.
Adsorption of 1-butene (and isobutene) on ABgave a composite signal at 203 nm (and 215 nm).Desorption decreased this band intensity 63% (75%for isobutene) and shifted the maximum to 200 nm(198 nm for isobutene).
Adsorption of 1-butene on AB decreased the in-tensities of the signals at 1356 (37%), 1382 (48%)and 1388 nm (27%). Vacuum restored the intensitiesof the OH signals to those of the dehydrated sample,except for the signal at 1350 nm (Fig. 7). Isobuteneadsorption on AB caused a ca. 50% decrease in the Al-OH signals at 1356, 1371 and 1388 nm. No changeswere detected in the B-OH bands. The intensities
C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152 147
Fig. 6. 13C MAS NMR spectra of A (bottom) and AB (top) afterlengthy contact with isobutene at 25◦C, 5 kHz spin rate, height oflargest peak cut in half. Chemical shifts indicated above selectedpeaks.
of the Al-OH bands were completely restored afterdesorption.
4. Discussion
4.1. Structural characterization
The amount of boron oxide (1.9 wt.%) on AB cor-responds to a nominal surface coverage of about 30%,considering that BO3 covers an area of 19.0 Å2. Thisestimate is supported by the fact that ca. 6 wt.% wasthe maximum amount of boron oxide that could beimpregnated onto A. Also, Sibeijn et al. found that thesurface ofg-alumina (200 m2/g) is completely coveredat ca. 5 wt.% of B2O3 [14].11B MAS NMR of par-tially hydrated AB revealed that ca. 10% of the boron
nuclei have tetrahedral configuration (Table 1). Thiscorresponds to 50mmoles Td-B per gram AB.
The surface of AB contains a mixture of Al-OH andB-OH groups. The NIR signals in the 1300–1500 nmregion, representing these groups, are not yet fully as-signed. The only work dealing with assignments [21]involved a direct relation between IR stretching fre-quencies (0–1 vibration) and NIR bands (0–2 vibra-tion). By this relation the OH-signals of alumina fallin the 1320–1404 nm range.
The nature of the OH groups responsible for theNIR bands (Table 2) are tentatively assigned as fol-lows. After boria introduction the 1406 nm band dis-appeared, the 1371 nm band decreased dramaticallyand the 1356 nm band decreased less (ca. 30%). Fromthe results of probe adsorptions, boria binding causesthe elimination of all basic sites while leaving theacidic ones intact. Therefore, the 1406 nm band is at-tributed to terminal Al(Td)-OH groups (basic sites).Boria is also bonded to Al-OH groups giving a signal
Fig. 7. UV–Vis-NIR spectra of AB after dehydration (a), adsorptionof 1-butene (b) and its evacuation (c).

148 C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152
Fig. 8. Schematic structures of borate species on an alumina surface.
at 1371 nm and, in part, 1356 nm. A 1382 nm band iscreated on binding boria indicating it is due to B-OHgroups. The other OH signals are assigned to Al-OHgroups with different local environments.
The various trigonal (BO3) and tetrahedral (BO4)forms for the surface bound borate species are shownin Fig. 8. The comprehensive results presented herepermit an objective choice of the most probable sur-face structures present on AB. Dried AB will be con-sidered, since the more detailed spectroscopic analysis(UV–Vis-NIR) necessarily involves this form. There-fore, all the tetrahedral structures (1–3) can be elimi-nated, since they exist only in the hydrated form. Thefinding (NMR and NIR) that the total number of hy-droxyl groups remains essentially constant after bond-ing borate species is an important constraint. Only verysmall pieces of 7 (e.g. a dimer) satisfy this constraint,as do equal amounts of structures 4 and 5. Sibeijn et al.
[14] suggested structure 7 based on IR studies of ABwith higher amounts of boria.
4.2. Acidity/ Basicity
Pyrrole adsorption reveals a rather large number ofbasic sites on A (ca. 0.25 mmol/g) which are elimi-nated by boria binding, consistent with the findings ofothers [15]. The boron nuclei on AB (0.54 mmol/g)outnumber the basic sites on A. Thus, in additionto basic sites, boron nuclei are also attached to(non-basic) alumina hydroxyl groups (i.e. 1356 and1371 nm reflections) and located in cationic vacanciesnear Al(Td) sites.
Pyridine adsorption revealed both H-bond and co-ordinative interactions with A and AB. No Brønstedacidity was evidenced for dried A or AB using pyri-dine, in agreement with the findings of others [4,23].

C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152 149
The presence of hydroxyl groups do not assureBrønsted acid sites, rather these OH groups interactweakly with olefins, probably by H-bonding (seebelow).
Greater amounts of Lewis acid sites were measuredas the basicity of the probe molecule increased: 1-butene< pyridine< ammonia. AB and A have essen-tially the same number of total acid sites by pyridineadsorption (ca. 110mmol/g Lewis sites, Table 3) andby ammonia adsorption (ca. 360mmol/g). Thus, thegreater acidity of AB, as evidenced by DBI and 1-butene chemisorption, is due to greater acid strength.The higher acid strength of AB is consistent with ionictheory. Lewis acidity of anhydrous oxides increaseswith ionic potential (8.298 and 5.989 eV for B and Al,respectively).
Only the strongest acid sites of AB are able tochemisorb butene molecules. The number of 1-butenemolecules chemisorbed at 25◦C (ca. 34mmol/g, deter-mined by NMR and volumetric analyses) is compa-rable to the number of pyridine molecules desorbingabove 400◦C and about half the ammonia moleculesdesorbing above 350◦C (Table 3).
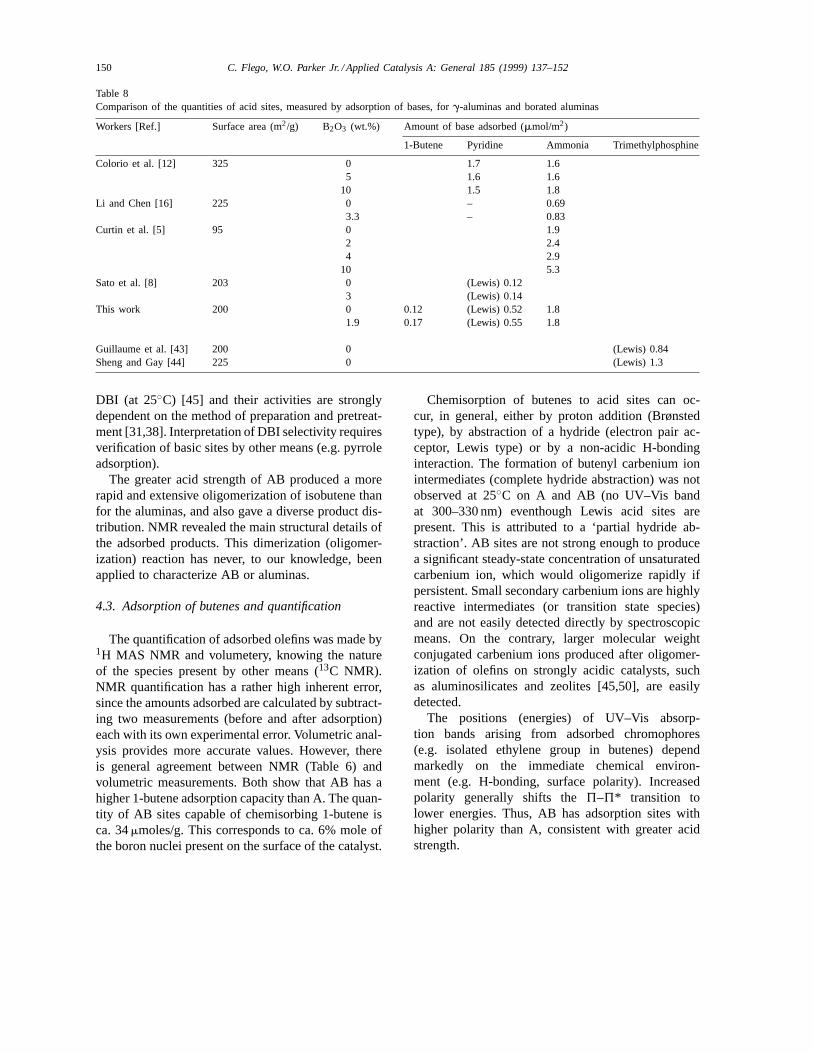
Volumetry has been used by other workers to quan-tify acid adsorption sites ong-alumina and AB. A listof the most recent works is shown in Table 8. Pyri-dine and ammonia were used as bases. Olefins areemployed here for the first time.
Volumetric measurements using the same basesshould give similar values for the total number ofacid sites (per surface area) in pureg-aluminas. Thereis good agreement that ca. 1.8mmol/m2 of acid sitesin alumina adsorb ammonia (Table 8). Li and Chen[16] reported 0.69mmol/m2 of ammonia desorbedabove 120◦C, which excludes the very weak acidsites. Subtraction of the very weak acid sites (ca.160mmol/g, Table 3) from our value gives a similarresult (0.90mmol/m2).
We find that approximately one-third of the totalammonia adsorption sites on A are able to adsorbpyridine (Table 8). This amount (0.52mmol/m2), de-termined via IR spectroscopy, is much less than thatreported by Colorio et al. (1.7mmol/m2) using vol-umetry [12]. This is because IR measured only Lewisacid sites while volumetry measured all chemisorbed(Lewis plus H-bonded) pyridine. Also, the above au-thors have suspect data, since a larger amount of theweaker base (pyridine) was adsorbed. Sato et al. also
found much less pyridine chemisorbed to Lewis sitesthan we did [8]. This is attributed to their using thedifference between the amount of desorbed pyridineand desorbed 2,6-dimethylpyridine. The latter base isstronger than pyridine and not completely selective forBrønsted sites.
Acid sites on pure alumina have also been quantifiedby other workers using trimethylphosphine and31PNMR (Table 8). The values are intermediate betweenthose for ammonia and pyridine, consistent with itsintermediate basicity (pKb = ca. 6).
According to Li and Chen and Curtin et al., the ad-dition of small amounts of boria (3.3% and 2%, re-spectively), to alumina increases slightly the numberof acid sites adsorbing ammonia (20% and 30%, re-spectively). However, the results of this work and thedata of Sato et al. (Table 8) agree that no real change inthe number of acid sites occurs. The claims of strongBrønsted acidity on AB by other workers [8,10] con-cerned catalysts with high boria loadings.
The rate of DBI demonstrates that AB is more acidicthan A (Table 5). The stereoselectivity of DBI (to-wardscis-2-butene), observed for bothg-Al2O3 andAB, can be explained by the formation of an allylicintermediate. There is general agreement that DBI onaluminas involves allylic intermediates and Lewis acidsites. Two different intermediates have been proposed,an allylic cation (butenyl carbenium ion) [4,45] and anallylic carbanion formed at a Lewis acid (Al3+) andbase (O2−) pair site [4,46–48]. As shown above, ABhas no basic sites capable of forming carbanion inter-mediates and since there are no Bronsted acid sitesonly Lewis acidity is driving DBI.
Allylic intermediates retain their stereochemistriesuntil they react because there is a large barrier to ro-tation about the C2–C3 bond. Thecis isomer is pro-duced faster since the product determining step is thechemisorption of 1-butene and the approach of thegauche 1-butene conformer (which becomes thecisbutenyl intermediate) to the surface is less hindered.However, if DBI activity is very low an apparent non-stereoselectivity may be observed, even with a Lewissite [45]. The low activity of the Akzo alumina (A)apparently causes its lack of stereoselectivity, eventhough Lewis acid sites (or basic sites) are operating.The other two commercial aluminas studied here havehigher basicities than A [49] causing higher DBI ratesand (C/T)0 (Table 5). Pure aluminas catalyze very slow
150 C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152
Table 8Comparison of the quantities of acid sites, measured by adsorption of bases, forg-aluminas and borated aluminas
Workers [Ref.] Surface area (m2/g) B2O3 (wt.%) Amount of base adsorbed (mmol/m2)
1-Butene Pyridine Ammonia Trimethylphosphine
Colorio et al. [12] 325 0 1.7 1.65 1.6 1.6
10 1.5 1.8Li and Chen [16] 225 0 – 0.69
3.3 – 0.83Curtin et al. [5] 95 0 1.9
2 2.44 2.9
10 5.3Sato et al. [8] 203 0 (Lewis) 0.12
3 (Lewis) 0.14This work 200 0 0.12 (Lewis) 0.52 1.8
1.9 0.17 (Lewis) 0.55 1.8
Guillaume et al. [43] 200 0 (Lewis) 0.84Sheng and Gay [44] 225 0 (Lewis) 1.3
DBI (at 25◦C) [45] and their activities are stronglydependent on the method of preparation and pretreat-ment [31,38]. Interpretation of DBI selectivity requiresverification of basic sites by other means (e.g. pyrroleadsorption).
The greater acid strength of AB produced a morerapid and extensive oligomerization of isobutene thanfor the aluminas, and also gave a diverse product dis-tribution. NMR revealed the main structural details ofthe adsorbed products. This dimerization (oligomer-ization) reaction has never, to our knowledge, beenapplied to characterize AB or aluminas.
4.3. Adsorption of butenes and quantification
The quantification of adsorbed olefins was made by1H MAS NMR and volumetery, knowing the natureof the species present by other means (13C NMR).NMR quantification has a rather high inherent error,since the amounts adsorbed are calculated by subtract-ing two measurements (before and after adsorption)each with its own experimental error. Volumetric anal-ysis provides more accurate values. However, thereis general agreement between NMR (Table 6) andvolumetric measurements. Both show that AB has ahigher 1-butene adsorption capacity than A. The quan-tity of AB sites capable of chemisorbing 1-butene isca. 34mmoles/g. This corresponds to ca. 6% mole ofthe boron nuclei present on the surface of the catalyst.
Chemisorption of butenes to acid sites can oc-cur, in general, either by proton addition (Brønstedtype), by abstraction of a hydride (electron pair ac-ceptor, Lewis type) or by a non-acidic H-bondinginteraction. The formation of butenyl carbenium ionintermediates (complete hydride abstraction) was notobserved at 25◦C on A and AB (no UV–Vis bandat 300–330 nm) eventhough Lewis acid sites arepresent. This is attributed to a ‘partial hydride ab-straction’. AB sites are not strong enough to producea significant steady-state concentration of unsaturatedcarbenium ion, which would oligomerize rapidly ifpersistent. Small secondary carbenium ions are highlyreactive intermediates (or transition state species)and are not easily detected directly by spectroscopicmeans. On the contrary, larger molecular weightconjugated carbenium ions produced after oligomer-ization of olefins on strongly acidic catalysts, suchas aluminosilicates and zeolites [45,50], are easilydetected.
The positions (energies) of UV–Vis absorp-tion bands arising from adsorbed chromophores(e.g. isolated ethylene group in butenes) dependmarkedly on the immediate chemical environ-ment (e.g. H-bonding, surface polarity). Increasedpolarity generally shifts the5–5* transition tolower energies. Thus, AB has adsorption sites withhigher polarity than A, consistent with greater acidstrength.

C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152 151
The adsorption of 1-butene and isobutene decreasesthe intensities of OH signals in the 1300–1500 nmrange. This is attributed to H-bonding betweenbutenes and surface OH groups. Adsorption is weak(physisorption) as confirmed by the restoration ofthe original OH band intensity after evacuation at25◦C. The band in the 190–215 nm range decreases,with evacuation, meaning that some olefin moleculesremain chemisorbed on Lewis acid sites.
The broad UV–Vis band near 205 nm observed foradsorbed olefins on AB contains absorptions for bothphysi- and chemisorbed species. Based on spectralchanges caused by evacuation, the 194 nm compositeband is assigned to physisorbed (H-bonded) 1-butene(207 nm for isobutene) while the low energy reflec-tions of the composite bands are due to chemisorbed(partially hydride abstracted) and reacted species (i.e.dimers, trimers).
5. Conclusions
The boron sites on dried AB have trigonal coordina-tion (Lewis acidity).11B MAS NMR shows that 10%mole of the boron nuclei in partially hydrated AB at-tain tetrahedral coordination (Td) after adsorbing wa-ter. This analysis could not distinguish the nuclei in-teracting with butene molecules.
NIR spectra evidenced changes in OH reflectionbands on binding a low amount of boria to A: strongintensity decrease for the 1371 nm band, disappear-ance of the 1406 nm band (basic Al-OH sites) and ap-pearance of a new band at 1382 nm. The total numberof hydroxyl groups remained constant, consistent withborate surface species on dried AB being composedof equal amounts of structures 4 and 5 (Fig. 8).
The acidity of both dried A and AB, responsible forchemisorption, is of Lewis type as found by pyridineadsorption and the stereoselectivity of DBI (allyl inter-mediate). IR and volumetric adsorption studies (withpyridine and ammonia, respectively), along with therates of DBI and isobutene oligomerization, revealedthat g-Al2O3 has weaker acidity than AB. The in-creased acidity of AB is not due to a larger number oftotal acid sites, but rather to increased acid strength.The isobutene reaction is more sensitive than DBI forclassifying the acidities.
NIR spectra revealed that approximately half ofthe AB hydroxyl groups interacted with physisorbedbutenes. The interactions were very weak, consistentwith hydrogen bonding. The positions of UV–Visbands of chemisorbed olefins indicate they are specieswith the hydride only ‘partially’ abstracted by theLewis acid sites on AB.
NMR and volumetry were used to quantify thesites adsorbing 1-butene molecules at 25◦C. Volume-try found 24 and 34mmol/g of sites for A and AB,respectively. These values correspond to the sites withmedium-strong plus strong acid strength as definedby pyridine desorption.1H MAS NMR of adsorbedolefins gives similar values but is less accurate thanvolumetry. 13C NMR (natural abundance) identifiedonly dimer I (terminal olefin) on A after isobutene ad-sorption. AB produced equimolar amounts of dimer Iand trimer III from isobutene.
Two considerations are noteworthy regarding thepotentials of the various techniques used. First, thecombination of NMR and volumetry allows quantifica-tion of probe molecules and the necessary in-situ veri-fication of any chemical transformations occuring dur-ing volumetry measurements. Second, UV–Vis-NIRspectroscopy is equally informative, if not more so,than IR for studying the coordination states of metalatoms and the structures of surface hydroxyls on thesematerials.
References
[1] H. Knozinger, P. Ratnasamy, Catal. Rev. Sci. Eng. 17 (1978)31.
[2] S. Meijers, L.H. Gielgens, V. Ponec, J. Catal. 156 (1995) 147.[3] E. Garbowski, J.P. Candy, M. Primet, J. Chem. Soc., Faraday
Trans. 79 (1983) 835.[4] Z.X. Cheng, V. Ponec, J. Catal. 148 (1994) 607.[5] T. Curtin, J.B. Mc Monagle, B.K. Hodnett, Appl. Catal. 93
(1992) 91.[6] C. Li, Y.W. Chen, Catal. Lett. 10 (1991) 297.[7] G. Colorio, A. Auroux, B. Bonnetot, J. Thermal Anal. 38
(1992) 2565.[8] S. Sato, M. Kuroki, T. Sodesawa, F. Nozaki, G.E. Maciel, J.
Mol. Catal. A 104 (1995) 171.[9] S. Sato, H. Hasebe, H. Sakurai, K. Urabe, Y. Izumi, Appl.
Catal. 29 (1987) 107.[10] H. Sakurai, S. Sato, K. Urabe, Y. Izumi, Chem. Lett. (1985)
1783.[11] W.J. Wang, Y.W. Chen, Catal. Lett. 10 (1991) 297.[12] G. Colorio, J.C. Vedrine, A. Auroux, B. Bonnetot, Appl.
Catal. 137 (1996) 55.

152 C. Flego, W.O. Parker Jr. / Applied Catalysis A: General 185 (1999) 137–152
[13] K.P. Peil, L.G. Galya, G. Marcelin, J. Catal. 115 (1989) 441.[14] M. Sibeijn, J.A.R. van Veen, A. Bliek, J.A. Moulijn, J. Catal.
145 (1994) 416.[15] G.C. Colorio, A. Auroux, B. Bonnetot, J. Thermal Anal. 40
(1993) 1267.[16] C. Li, Y.-.W. Chen, Catal. Lett. 19 (1993) 99.[17] A. Delmastro, G. Gozzelino, D. Mazza, M. Vallino, G. Busca,
V. Lorenzelli, J. Chem. Soc., Faraday Trans. 88 (1992) 2065.[18] F.M. Bautista, J.M. Campelo, A. Garcia, D. Luna, J.M.
Marinas, M.C. Moreno, A.A. Romero, J.A. Naulo, M.Macias, J. Catal. 173 (1998) 333.
[19] C. Flego, I. Kiricsi, C. Perego, G. Bellussi, Catal. Lett. 35(1995) 125.
[20] A. Samoson, E. Kundla, E. Lippmaa, J. Magn. Reson. 49(1982) 350.
[21] L.M. Kustov, V.Y. Borovkov, V.B. Kazansky, J. Catal. 72(1981) 149.
[22] J. Kijenski, A. Baiker, Catal. Today 5 (1989) 1.[23] L. Forni, Catal. Rev. 8 (1973) 65.[24] E.P. Parry, J. Catal. 2 (1963) 371 K.H. Bowne, F.R. Cannings,
R.C. Pitkethly, J. Phys. Chem. 74 (1970) 2197.[25] P. Nortier, P. Fourre, A.B. Mohammed Saad, O. Saur, J.C.
Lavalley, Appl. Catal. 61 (1990) 141.[26] G. Busca, V. Lorenzelli, G. Ramis, R.J. Willey, Langmuir 9
(1993) 1492.[27] J. Take, T. Yamaguchi, K. Miyamoto, H. Ohyama, M. Misono,
Stud. Surf. Sci. Catal. 28 (1986) 495.[28] D. Barthomeuf, J. Phys. Chem. 88 (1984) 42 P.O. Scokart,
P.G. Rouxhet, Bull. Soc. Chim. Belg. 90 (1980) 983.[29] M. Huang, S. Kaliaguine, M. Muscas, A. Auroux, J. Catal.
157 (1995) 266.[30] P.O. Scokart, P.G. Rouxhet, J.C.S. Faraday Trans. I 76 (1980)
1476.
[31] Y. Hong, F.R. Chen, J.J. Fripiat, Catal. Lett. 17 (1993) 187.[32] W.O. Parker, Jr., Stud. Surf. Sci. Catal. 94 (1995) 568; and
references therein.[33] D. Michel, W. Meiler, H. Pfeifer, H.J. Rauscher, H. Siegel,
J. Mol. Catal. 5 (1979) 263.[34] G. Engelhardt, D. Michel, High Resolution Solid-State NMR
of Silicates and Zeolites, Wiley, New York, 1987, p. 449.[35] J.B. Nagy, G. Engelhardt, D. Michel, Adv. Colloid Interface
Sci. 23 (1985) 67.[36] S. Tsuchiya, S. Kawasaki, M. Mikami, H. Imamura, Zeolites
7 (1987) 4.[37] W.O. Haag, H. Pines, J. Am. Chem. Soc. 82 (1960) 2488.[38] J.W. Hightower, W.K. Hall, J. Phys. Chem. 71 (1967) 1014.[39] J.F. Kriz, I.D. Gay, J. Phys. Chem. 80 (1976) 2951.[40] J.H. Lunsford, L.W. Zingery, M.P. Rosynek, J. Catal. 38
(1975) 179.[41] A.G. Stepanov, M.V. Luzgin, V.N. Romannikov, K.I.
Zamaraev, Catal. Lett. 24 (1994) 271.[42] J.F. Haw, B.R. Richardson, I.S. Oshiro, N.D. Lazo, J.A. Speed,
J. Am. Chem. Soc. 111 (1989) 2052.[43] D. Guillaume, S. Gautier, I. Despujol, F. Alario, P. Beccat,
Catal. Lett. 43 (1997) 213.[44] T.-.C. Sheng, I.D. Gay, J. Catal. 145 (1994) 10.[45] H.P. Leftin, Carbonium Ions, vol. 1, Interscience, New York,
1968, Chap. 10.[46] M. Trombetta, G. Busca, S.A. Rossini, V. Piccoli, U. Cornaro,
J. Catal. 168 (1997) 334.[47] M.P. Rosynek, F.L. Strey, J. Catal. 41 (1976) 312.[48] J.B. Peri, J. Phys. Chem. 69 (1965) 231.[49] M. Trombetta, G. Busca, unpublished results.[50] C. Flego, I. Kiricsi, W.O. Parker Jr., M.G. Clerici, Appl.
Catal. 124 (1995) 107.
Related Documents











