Ch. 31 Hematologic Problems ANEMIA Definition and Classification ANEMIA IS NOT A DISEASE; it is a manifestation of a pathologic process Definition of Anemia 1) deficiency in the number of erythrocytes (RBCs) 2) The quantity of hemoglobin 3) The volume of packed RBCs (hematocrit) Anemia is identified by o thorough history, o physical examination, o Labs of CBC, reticulocyte count, and peripheral blood smear. Various types of anemia 1) Morphologic classification (cellular characteristic) Based on descriptive, objective laboratory information about erythrocyte size and color. 2) Etiologic classification (underlying cause) Related to clinical conditions causing the anemia, such as blood loss, decreased erythrocyte production, or increased erythrocyte destruction.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Ch. 31 Hematologic Problems
ANEMIA
Definition and Classification ANEMIA IS NOT A DISEASE; it is a manifestation of a pathologic process
Definition of Anemia1) deficiency in the number of erythrocytes (RBCs)2) The quantity of hemoglobin3) The volume of packed RBCs (hematocrit)
Anemia is identified by o thorough history, o physical examination, o Labs of CBC, reticulocyte count, and peripheral blood smear.
Various types of anemia1) Morphologic classification (cellular characteristic)
Based on descriptive, objective laboratory information about erythrocyte size and color.2) Etiologic classification (underlying cause)
Related to clinical conditions causing the anemia, such as blood loss, decreased erythrocyte production, or increased erythrocyte destruction.
Although the morphologic system is the most accurate means of classifying anemias, it is easier to discuss patient care by focusing on the etiology of the anemia.
1) Impaired or decreased erythrocyte production
a. Decreased hemoglobin synthesis
iron deficiency Thalassemias (decreased globin synthesis) – high
incidence among African Americans Sideroblastic anemia
b. Defective DNA synthesis
Cobalamin (vit B12) deficiency Folic acid deficiency
c. Decreased number of erythrocyte precursors
Aplastic anemia Anemia of myeloproliferative diseases (ex; leukemia)
and myelodysplasia Chronic diseases or disorders Decreased erythropoietin in the kidneys
d. Chemotherapy2) Blood Loss a. acute trauma
blood vessel ruptureb. chronic gastritis
menstrual flow hemorrhoids
3) Increased destruction of erythrocytes.
a. Intrinsic Abnormal hemoglobin (Hb S-Sickle cell anemia) – high incidence among African Americans
Enzyme deficiency (G6PD) Membrane abnormalities (paroxysmal nocturnal
hemoglobinuria, hereditary spherocytosis)b. Extrinsic Physical trauma (prosthetic heart valves,
extracorporeal circulation, cardiopulmonary bypass) Antibodies (isoimmune and autoimmune) Infectious agents, medications, and toxins (malaria)

Clinical Manifestations Caused by body’s response to tissue hypoxia Specific manifestations can vary depending on the
1. Rate at which it has evolved2. The presence of coexisting disease3. Severity of the anemia – which is often determined by Hemoglobin (Hb) levels
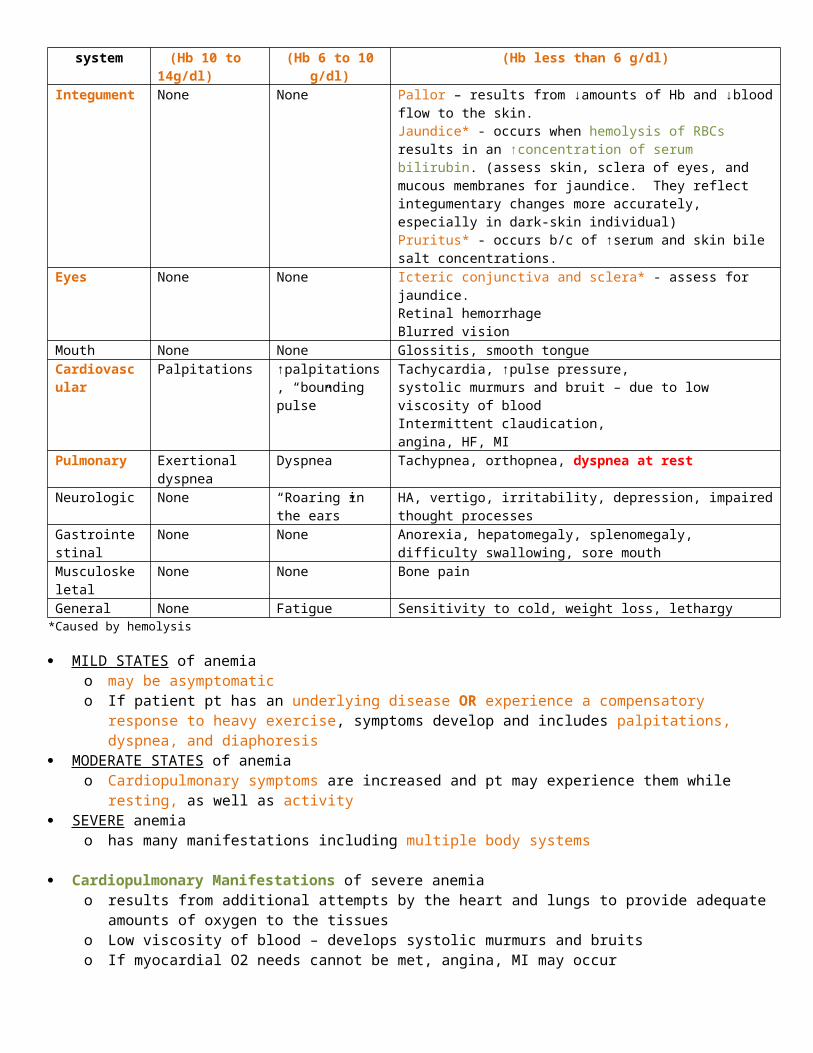
Clinical manifestations of anemia (table 31-3)Body system Mild
(Hb 10 to 14g/dl)Moderate
(Hb 6 to 10 g/dl)Severe
(Hb less than 6 g/dl)Integument None None Pallor – results from ↓amounts of Hb and ↓blood flow to the skin.
Jaundice* - occurs when hemolysis of RBCs results in an ↑concentration of serum bilirubin. (assess skin, sclera of eyes, and mucous membranes for jaundice. They reflect integumentary changes more accurately, especially in dark-skin individual)Pruritus* - occurs b/c of ↑serum and skin bile salt concentrations.
Eyes None None Icteric conjunctiva and sclera* - assess for jaundice.Retinal hemorrhageBlurred vision
Mouth None None Glossitis, smooth tongueCardiovascular Palpitations ↑palpitations,
“bounding pulse”Tachycardia, ↑pulse pressure, systolic murmurs and bruit – due to low viscosity of blood Intermittent claudication, angina, HF, MI
Pulmonary Exertional dyspnea Dyspnea Tachypnea, orthopnea, dyspnea at restNeurologic None “Roaring in the
ears”HA, vertigo, irritability, depression, impaired thought processes
Gastrointestinal None None Anorexia, hepatomegaly, splenomegaly, difficulty swallowing, sore mouth
Musculoskeletal None None Bone painGeneral None Fatigue Sensitivity to cold, weight loss, lethargy
*Caused by hemolysis
MILD STATES of anemia o may be asymptomatico If patient pt has an underlying disease OR experience a compensatory response to heavy exercise, symptoms
develop and includes palpitations, dyspnea, and diaphoresis MODERATE STATES of anemia
o Cardiopulmonary symptoms are increased and pt may experience them while resting, as well as activity SEVERE anemia
o has many manifestations including multiple body systems
Cardiopulmonary Manifestations of severe anemia o results from additional attempts by the heart and lungs to provide adequate amounts of oxygen to the tissueso Low viscosity of blood – develops systolic murmurs and bruitso If myocardial O2 needs cannot be met, angina, MI may occur o HF, cardiomegaly, pulmonary and systemic congestion, ascities, and peripheral edema may develop if heart is
overworked for an extended period of time
Nursing Management for AnemiaNursing assessment
Subjective Objective
o General – lethargy, apathy, general lymphadenopathy, fever
o Integumentary – pale skin and mucous membranes; blue, pale white, or icteric sclera; cheilitis’ poor skin turgor; brittle, spoon-shaped fingernails; jaundice; petechiae; ecchymoses; nose or gingival bleeding; poor healing; dry, brittle, thinning hair
o Respiratory – tachypneao Cardiovascular – tachycardia, systolic murmur, dysrhythmias; postural hypotension, widened pulse
pressure, buirts (especially carotid); intermittent claudication, ankle edemao Gastrointestinal – hepatosplenomegaly; glossitis; beefy, red tongue; stomatitis; abdominal distention;
anorexico Neurologic – HA, roaring in the ears, confusion, impaired judgment, irritability, ataxia, unsteady gait,
paralysis, loss of vibration senseo Possible findings - ↓RBCs, ↓ Hb, ↓Hct, ↑or↓ reticulocytes, MCV, serum iron, ferritin, folate, or cobalamin
(vit B12); heme (Guaiac)-positive stools; ↓serum erythropoietin level, ↑or↓ LDH, bilirubin, transferrin
Nursing Diagnosis Activity intolerance related to weakness and imbalance between oxygen supply/demand as evidenced by
increased pulse and blood pressure in response to activity and patient report of weakness Altered nutrition: less than body requirements related to inadequate nutritional intake and anorexia as evidenced
by weight loss, low serum albumin, decreased iron levels, vitamin deficiencies Ineffective management of therapeutic regimen related to lack of knowledge about appropriate nutrition and
medication regimen as evidenced by questioning about lifestyle adjustments, diet, and medications.
Planning1) Assume normal activities of daily living2) Maintain adequate nutrition3) Develop no complications related to anemia
Nursing Implementation GOAL – correcting the cause of the anemia Numerous causes of anemia necessitate different nursing interventions specific to the needs of the patient. Acute interventions
o Blood or blood product transfusiono Drug therapy (ex; erythropoietin, vitamin supplements)o Volume replacemento Oxygen therapy to stabilize the patient
Ongoing interventionso Dietary and lifestyle changes (described with specific types of anemia) can reverse some anemias.
Ongoing assessment of patient’s knowledge regarding adequate nutritional intake and compliance with safety precautions to prevent falls and injury and drug therapies.
Gerontologic Considerations of Anemia Changes in RBC mass Decreased production of androgens
o Decline of Hb (about 1 g/dl) between ages 70 and 88 years in healthy meno Minimal decline of Hb (about 0.2 g/dl) between ages 70 and 88 years in healthy women
Cobalamin (vit 12) deficiency due to pernicious anemia, insufficient dietary intake, malabsorption Multiple comorbid conditions in older adults Signs and symptoms include pallor, confusion, ataxia, fatigue, worsening angina, and HF May go unrecognized in older adults b/c manifestations of anemia may be mistaken as normal aging changes or
overlooked b/c another health problem.

Anemia Caused by Decreased Erythrocyte Production (↓ erythropoiesis) Equilibrium between RBC production (erythropoiesis) and RBC destruction/loss Normal life span of RBC is 120 days (around 4 months) Three alterations in erythropoiesis
1) Decreased Hb synthesis may lead to iron-deficiency anemia, thalassemia, and sideroblastic anemia2) Defective DNA synthesis in RBCs (ex; cobalamin (vit 12) deficiency, folic acid deficiency) may lead to
megaloblastic anemias3) Diminished availability of erythrocyte precursors may result in aplastic anemia and anemia of chronic disease.
Iron-Deficiency Anemia
One of the most common chronic hematologic disorders Found up to 30% of world’s population In US, occurs about 5 to 10% of people >45years Most susceptivle to iron-deficiency anemia
a. Very young b. Those on poor dietsc. Women in their reproductive years.
1 mg iron lost daily through feces, sweat, and urine in adult male and 1.5 mg/day in normal menstruating women
Median total iron loss with pregnancy is about 500 mg, or almost 2mg/day over 280 days of gestation.
Etiology 1) Dietary irons may be inadequate for those who have higher iron needs such as menstruating or pregnant
women.2) Malabsorption of iron may occur after GI surgery (removal or bypass of duodenum) and malabsorption
syndromes (disease of duodenum in which the absorption surface is altered or destroyed) – iron absorption occurs in the duodenum
3) Blood loss – major cause of iron deficiency in adults. 2 mL of whole blood contain 1 mg of iron. Major sources of chronic blood loss are from GI and GU systems GI blood loss
o often not apparent (not easily seen). o Loss of 50 to 75 ml of blood from upper GI tract –stools appear black (melena) – black color results
from iron in RBCo Common cause of BI bleed – peptic ulcer, gastritis, esophagitis, diverticuli, hemorrhoids, and
neoplasia GU blood loss–
o Average monthly menstrual blood loss is about 45 ml : loss about 22 mg of irono Common cause
menstrual bleeding post menopauseal bleeding
4) pregnancy - due to diversion of iron to the fetus for erythropoiesis, blood loss at delivery, and lactation5) chronic renal failure and dialysis treatment may induce iron-deficiency anemia b/c of the blood lost in
dialysis equipment 6) frequent blood sampling
Manifestations In early course – pt. may be asymptomatic When chronic – any general manifestations of anemia may develop Specific clinical symptoms of iron deficiency anemia – caused by lack of iron in the tissues
o Pallor – most common findings

o Glossitis (tongue inflammation) – 2nd most commono Cheilitis (lips inflammation)o Patient may report HA, paresthesias, burning sensation of the tongue
Diagnostic Studies Specific diagnostic studies to determine the cause of iron deficiency anemia
o Stool guaiac test - test for unseen blood in stoolo Endoscopy and colonoscopy – to detect GI bleedingo Bone marrow biopsy – if other tests are inconclusive
LABsHb/Hct MCV Reticulocytes Serum Iron TIBC Transferrin Ferritin Bilirubin
Iron deficiency ↓ ↓ Normal or slight ↓ or ↑
↓ ↑ N or ↓ ↓ N or ↓
Collaborative Care GOAL – treat the underlying disease that is causing reduced intake (ex; malnutrition, alcoholism) or absorption of
iron. TREATMENT – efforts are directed toward replacing iron.
o TEACH which foods are good sources of iron (Nutrition)o If nutrition is already adequate use oral or parenteral iron supplementso From acute blood loss transfusion of packed RBCs
Drug Therapyo Oral iron should be used whenever possible. (inexpensive and convenient)o Consideration in iron administration (5 factors)
1. NO EC or SR capsules. -b/c iron is absorbed best from duodenum and proximal jejunum. EC or SR release farther down in the GI tract.(counter productive and expensive)
2. Daily dose of 150 to 200 mg of iron. 3. Iron should be taken about an hour before meals, when duodenal mucosa is most acidic.
-b/c iron is best absorbed as ferrous sulfate Fe2+ in an acidic environment. Avoid binding the iron with food. Take iron with vit C or orange juice enhances absorption. Due to GI side effects, may need to ingest iron with meals.
4. Liquid forms should be diluted and ingested through a straw. -b/c undiluted liquid iron may stain patient’s teeth.
5. Adjust dose and type of iron supplement if side effects develop Ex) many individuals who need supplemental iron cannot tolerate ferrous sulfate b/c of the
effects of the sulfate base ferrous gluconate may be an acceptable substitute. TEACH – stools may turn black b/c GI excretes iron GI side effects
Heartburn, Constipation (common) –patient should be started on stool softeners and laxatives when
started on iron Diarrhea
o Parenteral use of iron may be necessary (IV or IM) For malabsorption, intolerance of oral iron, a need for iron beyond oral limits, or poor pt. compliance in
taking oral preparations of iron. Iron-dextran complex (INFeD) contains 50 mg/ml of elemental iron in 2 mL – longest use historically. Alternatives : Sodium ferrous gluconate and iron sucrose– may provide less risk of life-threatening
anaphylaxis and delayed serum sickness TEST DOSE of parenteral iron often done to assess potential allergic reaction IM
May stain skin – so use separate needles for withdrawing the solution and for injecting medication

Leave approx. 0.5 ml of air in synringe to completely clear iron from syringe Give deep IM in upper outer quadrant of buttocks Use 2 to 3 inch, 19 to 20 gauge needle No more than 2 ml of iron given in single injection Z-track technique to prevent leackage to subq No massage after injection
IV Iron dextran should not be mixed with other meds or added to parenteral nutrition solutions Give undiluted Rate should be no more than 1 ml/min IV line should be flushed with normal saline.
Nursing Management for Iron-Deficiency Anemia Recognize groups of individuals who are at risk - premenopausal women, pregnant women, persons from low
socioeconomic backgrounds, older adults, and individuals experiencing blood loss. TEACHING – diet (foods high in iron) Supplemental iron important for pregnant women Discuss patient needs for diagnostic studies to identify cause Hb and RBC counts are reassessed to evaluate response to therapy Compliance with dietary and drug therapy is emphasized Patient needs to continue to take iron therapy for 2 to 3 months after Hb levels returns to normal to replenish
body’s iron stores MONITOR potential liver problems related to iron storage for those who require lifelong iron supplementation.
Thalassemia
Etiology Thalassemia is a group of diseases that have an autosomal recessive genetic basis involving inadequate
production of normal hemoglobin. (abnormal Hb synthesis) Hemolysis also occurs in thalassemia But insufficient production of normal Hb is the predominant problem. Thalassemia is due to an absent or reduced globulin protein
α-thalassemia – absent or reduced α-globin chains β-thalassemia – absent or reduced β-globin chains
commonly found in members of ethnic groups whose origins are near the Mediterranean Sea and equatorial or near-equatorial regions of Asia, the Middle East, and Africa
person with thalassemia may have a heterozygous or homozygous form of dz. thalasemia minor (or thalassemic trait) –person who is heterozygous has one thalassemic gene and one
normal gene thalassemia major – a homozygous person has two thalassemic genes, causes severe condition
Manifestations Thalassemia major
o life threatening disease in which growth (both physical and mental) is retarded. o Pale and displays other general symptoms of anemia.o Symptoms develop in childhood by 2 years of age o Pronounced splenomegaly and hepatomegalyo Jaundice – from RBC hemolysiso Blood cell production is stimulated bone marrow becomes packed with immature erythroid precursors that
die further erythopoiesis stimulated Chronic bone marrow hyperplasia and expansion of the marrow space causes thickening of the cranium and maxillary cavity.

Thalassemia minoro Frequently asymptomatico Mild to moderate anemia o Patient has microcytosis (small cells) and hypochromia (pale cells)
DiagnosticsLabs
Hb/Hct MCV Reticulocytes Serum Iron TIBC Transferrin Ferritin BilirubinThalassemia major
↓ N or↓ ↑ ↑ ↓ ↓ N or ↑ ↑
Collaborative Care No specific drug or diet therapies Thalassemia minor
o Requires no treatment b/c body adapts to the reduction of normal Hb Thalassemia major Managed with blood transfusion or exchange transfusions in conjunction with IV deferoxamine (Desferal)
o deferoxamine(Desferal) is a chelating agent that binds to iron to reduce the iron overloading (hemochromatosis) that occurs with chronic transfusion therapy.
S/E: visual blurring, hearing loss, tinnitus, and knock-kneeso Keep the Hb level at approximately 10 g/dl – low enough to maintain pt’s own erythropoiesis without
enlarging the spleen. o Zinc supplementation may be needed – it is reduced with chelation therapyo Ascorbic acid supplementation during chelation therapy – increases urine excretion of iron (should not
be taken..otherwise,,as it increases the absorption of dietary iron)o NO iron supplementso May be treated by splenectomy since RBCs are sequestered in the enlarged spleen. o Hep C is present in majority of pts older than 25 years b/c of receiving blood transfusion before
screening for hep C virus. Hep C may result in cirrhosis and hepatocellular carcinoma
o Cardiac complications from iron overload are reported to cause 71% of the deaths.o Pulmonary dz and HTN also contribute to early deatho MONITOR hepatic, cardiac, pulmonary organ function o Endocrinopathies (hypogonadotrophic hypogonadism) and thrombosis may also be complications
Megaloblastic Anemias
Group of disorders caused by impaired DNA synthesis Megaloblasts – abnormal and large RBCs (macrocytic) – macrocytic RBCs are easily destroyed due to fragile cell
membranes C auses
cobalamin (vit B12) deficiency - common Folic acid deficiency - common Suppression of DNA synthesis by drugs Inborn errors of cobalamin and folic acid metabolism Erythroleukemia – malignant blood disorder characterized by a proliferation of erythropoietic cells in bone
marrowCobalamin (Vitamin B12) Deficiency Parietal cells of the gastric mucosa normally secretes a protein called intrinsic factor (IF). IF is required for
cobalamin (extrinsic factor) absorption in the distal ilium. If IF is not secreted, cobalamin will not be absorbed. C auses of cobalamin deficiency

Dietary deficiency Deficiency of gastric intrinsic factor
o Pernicious anemia – most common cause A disease in which the gastric mucosa is not secreting IF because of antibodies being disrected
against the gastric parietal cells and/or IF itself Insidious onset that begins in middle age or later (usually after age 40) with 60 years being the
most common age at diagnosis Occurs frequently in persons of Northern European ancestry (particularly Scandinavians) and
African Americans(disease begins early with higher frequence in women and often severe)o Gastrectomy
Intestinal malabsorption Increased requirement Chronic alcoholism
Etiology1. Cobalamin deficiency results from loss of IF-secreting gastric mucosal cells or impaired absorption of
cobalamin in the distal ileumo GI surgery such as gastrectomyo Pts who have had a small bowel resection involving the ileumo Patients with Crohn’s disease, ileitis, diverticuli of small intestine, chronic atrophic gastritis
2. Long-term users of H2-histamine receptor blockers3. Pernicious anemia is caused by absence of IF from either gastric mucosal atrophy or autoimmune destruction of
parietal cells – results in decrease of hydrochloric acid secretion by the stomach (acidic environment is required for IF secretion)
Clinical Manifestations General symptoms (table 31-3) GI manifestations – sore tongue, anorexia, nausea, vomiting, and abdominal pain Typical Neuromuscular manifestations – weakness, paresthesias of feet and hands, reduced vibratory and
position senses, ataxia, muscle weakness, impaired thought processes (confusion, dementia) Insicious onset – may take several months for manifestations to develop
Diagnostics Labs
Hb/Hct MCV Reticulocytes Serum Iron
TIBC Transferrin Ferritin Bilirubin Serum folate
Cobalamin Hycrochloric acid
Cobalamin deficiency
↓ ↑ N or ↓ N or ↑ N Slight ↑ ↑ N or slight ↑
normal low decreased
RBCs appear large (macrocytic) and have abnormal shapes Serum cobalamin levels are reduced If serum folate levels are normal and cobalamin levels are lowmegaloblastic anemia is due to a cobalamin
deficiency Patients with pernicious anemia are potential for gastric cancer – gastroscopy and biopsy of gastric mucosa may
be done Schilling test – assessing parietal cell function
o Administer radioactive cobalamin measure amount of cobalamin excreted in the urineo Who CANNOT absorb cobalamin excretes only a SMALL amount of this radioactive form.
Follow a parenteral administration of IFo Absorption of cobalamin when IF is added is diagnostic of pernicious anemia
Serum methylmalonic acid (MMA) and serum homocysteine can be used if other tests are not definite.

Collaborative Care Increasing dietary cobalamin does not correct the anemia – b/c not able to absorb cobalamin if IF is lacking or
imapred absorption in the ileum (but still…good nutrition is required) TREATMENT of CHOICE - Parenteral (cyanocobalamin or hydroxocobalamin) or intranasal (Nascobal)
administration Without administration die in 1 to 3 years Dosage and frequency may vary – typical treatment schedule consists of 1000mg of cobalamin IM daily for 2
weeks weekly until hematocrit is normalmonthly FOR LIFE High dose oral and sublingual cobalamin available Anemia can be REVERSED if supplemental cobalamin is used. May be irreversible if one has had long-standing
neuromuscular complications
Folic Acid Deficiency Also causes megaloblastic anemia (large RBCs with abnormal shapes) Folic acid is required for DNA synthesis leading to RBC formation Causes are
1. Poor nutrition -lack of leafy green vegetables, liver, citrus fruits, yeast, dried beans, nuts, and grains2. Malabsorption syndroms- small bowel disorders3. Drugs that impede the absorption and use of folic acid(ex; methotrexate), antiseizure drugs (ex;
Phenobarbital, diphenylhydantoin (Dilantin), and others (ex; trimethoprim, sulfasalazine)4. Alcohol abuse and anorexia5. Hemodialysis patients - folic acid is lost during dialysis
Manifestations Similar with cobalamin deficiency Develops insidiously GI disturbances include dyspepsia and a smooth, beefy red tongue ABSENCE OF NEUROLOGIC PROBLEMS is an important diagnostic finding – differentiates folic acid deficiency from
cobalamin deficiency. Labs
Hb/Hct MCV Reticulocytes Serum Iron
TIBC Transferrin Ferritin Bilirubin Serum folate
Cobalamin Hycrochloric acid
Folic acid deficiency
↓ ↑ N or ↓ N or ↑
N Slight ↑ ↑ N or slight ↑
low normal positive
*Serum folate (normal is 3 to 25 mg/ml)Collaborative Care Treatment – replacement therapy Usual dose is 1mg /day by mouth In malabsorption states, up to 5 mg/day may be required Duration depends on reason for deficiency Encourage to eat foods containing large amounts of folic acid
Nursing Management for Megaloblastic Anemia ATTENTION of signs and symptoms of possible megaloblastic anemias Early detection may REVERSE symptoms, (although dz development cannot be prevented) – so those with a positive
family history of pernicious anemia should be evaluated (b/c familial predisposition) NCP 31-1 Ensure that injuries are not sustained b/c diminished sensatiosn to heat and pain resulting from neurologic
impairment – cobalamin deficiency. Protect from falls, burns, and trauma. If heat therapy required assess skin at frequent intervals to detect redness
Ongoing care – compliance with treatment Careful follow-up to assess for neurologic difficulties

POTENTIAL FOR CANCER may be increased (ex; pts with atrophic gastritis-related pernicious anemia gastric carcinoma, alcohol increasesoral and esophageal cancer)
Anemia of Chronic Disease
Etiology Chronic inflammatory, autoimmune, infectious, or malignant disease can lead to anemia of chronic disease Associated with an underproduction of RBCs and mild shortening of RBC survivial. RBCs are normocytic, normochromic, and hypoproliferative Mild anemia, Can be more severe if primarily immune driven. Released cytokines causes an increased uptake and retention of
iron within macrophages diversion of iron from circulation into storage siteslimitation of iron availability for erythroid progenitor cells, and iron restricted erythropoiesis
Renal disease – o Primary factor causing anemia is decreased erythropoietin
Any condition that causes ↑RBC destruction (autoimmune hemolysis) + failure to augment erythropoiesis = anemia Myelosuppression and ↓erythropoiesis caused by disease, medications (chemotherapy), or radiation will
contribute to anemia Human immunodeficiency virus(HIV) and its treatment, hepatitis, malaria, and bleeding episodes Hypopituitary and hypothyroid states - leads to reduced tissue metabolism tissue oxygen needs are
diminished ↓production of erythropoietin by the kidneys. Adrenalectomy or Addison’s disease results in anemia due to adrenal dysfunction
LabsHb/Hct MCV Reticulocytes Serum
IronTIBC Transferrin Ferritin Bilirubin Serum
folateCobalamin
Chronic disease
↓ N or↓ N or ↓ N or ↓
↓ N or ↓ N or↑ N normal normal
Collaborative Care Must FIRST BE RECOGNIZED and DIFFERENTIATED from other anemias Best TREATMENT – correction of underlying disorder If severe – blood transfusion may be indicated, but are not recommended for long term Anemia related to renal disease or anemia related to cancer therapies – use Erythropoietin therapy (Epogen,
Procrit). Darbepoetin (Aranesp) – newer form of erythropoietin, longer duration of action than erythropoietin IV iron should ONLY be administered IF NECESSARY to improve erythropoietin therapy.
Aplastic Anemia
Disease in which patient has peripheral blood pancytopenia (↓ of all blood cell types – RBCs, WBCs, and platelets) and hypocellular bone marrow.
Etiology Low incidence; 4:1million Two major classification
1) Congenital origin – caused by chromosomal alterations. Approx. 30% of the aplastic anemias appear in childhood are inherited
o Fanconi syndromeo Congenital dyskeratosiso Amegakaryocytico Thrombocytopeniao Schwachman-Diamond Syndrome

2) Acquired aplastic anemia –o results from exposure to ionizing radiationo chemical agents (ex; benzene, insecticides, arsenic, alcohol)o viral and bacterial infections (ex; hepatitis, parvovirus, biliary tuberculosis),o Prescribed medications (ex; alkylating agents, antiseizure agents, antimetabolites, antimicrobials,
gold)o Pregnancyo Idiopathic
Manifestations Present abruptly(over days) or insidiously (over weeks to months) Vary from mild to severe Symptoms caused by suppression of any or all bone marrow elements Fatigue and dyspnea, cardiovascular, and cerebral responses, may be seen Nutropenia patients (low neutrophil count) – susceptible to infection, febrile T hrombocytopenia – predisposition to bleeding (ex; petechiae, ecchymosis, epistaxis)
Diagnostic Studies Labs
Hb/Hct MCV Reticulocytes Serum Iron
TIBC Transferrin Ferritin Bilirubin WBC platelet Bleeding time
Aplastic anemia
↓ N or slight ↑
↓ N or ↑ N or ↑
N N N low low prolong
Hb, WBC, platelet values are often decreased due to affected all marrow elements. Other RBC indices are generally normal.(normocytic, normochromic anemia)
Reticulocyte count is low Prolong bleeding time ↑serum iron and ↑total iron-binding capacity(TIBC) - initial signs of erythropoiesis suppression Bone marrow biopsy, aspiration, and pathologic examination may be done Because the marrow is hypocellular with increased yellow marrow (fat content), findings are very important .
Collaborative Care medical management
o hematopoietic stem cell transplant (HSCT)o immunosuppressive therapy with antithymocyte globulin (ATG) – horse serum that contains polyclonal
antibodies against human T cells. Can cause anaphylaxis and serum sickness. RATIONALE – aplastic anemia is an immune-mediated disease
o cyclosporine or high dose cyclophosphamide (Cytoxan) TREATMENT OF CHOICE is an HSCT
o For those who are <45 yrs who do not respond to immunosuppressive therapyo who have a human leukocyte antigen (HLA) – matched donor o Best results – younger pts, who did not have previous blood transfusions b/c prior transfusions increase
risk of graft rejection TREATMENT OF CHOICE is an Immunosuppression with ATG or cyclosporine or high-dose cyclophosphamide
o For older adult without an HLA-matched donorNursing Management for Aplastic Anemia Based on identifying and removing the causative agent and providing supportive care until pancytopenia REVERSES See nursing interventions for
a) anemia NCP 31-1b) thrombocytopenia NCP 31-2 c) neutropenia NCP 31-3
nursing actions DIRECTED at PREVENTING COMPLICATIONS from INFECTIONS and HEMORRHAGE

Anemia Caused by Blood Loss
Acute Blood Loss
Result of sudden hemorrhage Causes
o Traumao Complications of surgeryo Conditions or disease that disrupt vascular integrity
2 clinical concerns1) Hypovolemic shock – sudden reduction in total blood volume2) Body maintaining its blood volume by slowly increasing plasma volume. This occurs when acute loss is more
gradual. Although circulating fluid volume is preserved, # of RBCs available to carry oxygen is significantly diminished
Manifestations Manifestations are caused by body’s attempts to maintain adequate blood volume and meet oxygen requirements.
Understand clinical signs and symptoms of patient are more important than the lab values.o Ex) adult with peptic ulcer bleeding has 750 ml hematemesis (15% of normal total blood vlume) within
past 30 minutes , may have postural hypotension. BUT have normal Hb and Hct values. Be alert to patient’s expression of pain. Internal hemorrhage may cause pain due to tissue distention, organ
displacement, and nerve compression. Localized pain or referred pain.o Ex) retroperitoneal bleeding- patient may not have abdominal pain. But may have numbness and pain in
lower extremity secondary to compression of lateral cutaneous nerve, located in the first to third lumbar vertebrae
Major complications - shock
Diagnostic studies-When acute blood loss, values may seem NORMAL OR HIGH FOR 2 TO 3 DAYS because plasma volume has not yet had a chance to increase. Therefore, loss of RBCs is not showed in the lab.
-However, ONCE PLASMA IS REPLACED by endogenous and exogenous means, RBC mass is less concentrated. lab will reflect the blood loss by showing ↓RBC, ↓Hb, ↓Hct.
Hb/Hct MCV Reticulocytes Serum Iron
TIBC Transferrin Ferritin Bilirubin RBC
Aplastic anemia
↓ N or ↓ N or ↑ N N N N N low
*The blood for this lab results is drawn 2 to 3 days after acute blood loss.
Collaborative Care1) Replacing blood volume to prevent shock2) Identify the source of hemorrhage and stop the blood loss
IV fluids used in emergencies o Dextran, hetastarch, albumin, and/or crystalloid electrolyte solutions such as Lactated Ringers.
Once replaced the volume, CORRECT RBC LOSS!! Blood transfusions (packed RBCs) may be needed b/c it takes 2 to 5 days for the body to make RBCs. If bleeding is related to platelet or clotting disorder, replace those that are deficient.
May need IRON supplement (iron affects the marrow production of erythrocytes). If anemia still exists after the acute blood, give oral or parenteral iron b/c dietary iron is not adequate.

Nursing Management for Acute Blood Loss Postoperative Patients – MONITOR blood loss from drainage tubes and dressings and implement appropriate
actions NCP for pt with anemia resulting from acute blood loss most likely blood transfusion
There should be NO NEED for LONG TERM TREATMENT for this for acute blood loss anemia.
Chronic Blood Loss
Usually related to depletion of iron stores usually considered as iron-deficiency anemia. (ex; bleeding ulcer, hemorrhoids, menstrual and postmenopausal blood loss)
LabsHb/Hct MCV Reticulocytes Serum
IronTIBC Transferrin Ferritin Bilirubin
Aplastic anemia
↓ N or ↓ N or ↓ N or ↓ ↓ N or ↓ N or ↑ N
MANAGEMENT Identify source! STOP THE BLEEDING Supplemental IRON may be required. NCP 31-1
Anemia Caused by Increased Erythrocyte Destruction
Hemolytic Anemia 3rd major cause of anemia Caused by destruction of hemolysis of RBCs at a rate that exceeds production
Causes of hemolysis I ntrinsic hemolytic anemias (heredity) – results from RBC defects caused by
o Sickle cells (abnormal Hb)o Enzyme deficiencies that alter glycolysis (glucose-6 phosphate dehydrogenase [G6PD] deficiency)o RBC membrane abnormalities
Extrinsic hemolytic anemias (acquired) – RBCs are normal but damaged by external factors such aso Trapping of cells within the sinuses of the liver or spleeno Antibody-mediated destructiono Toxinso Mechanical injury (ex; prosthetic heart valves)
Two sites of hemolysis Intravascular hemolysis – occurs within the circulation Extravascular hemolysis – takes place in the macrophages of the spleen (primary site of destruction of old RBCs),
Liver, and bone marrow
Manifests general symptoms of anemia and clinical manifestations specific to the type of anemia JAUNDICE – ↑bilirubin levels due to ↑destruction of RBCs Splenomegaly, hepatomegaly due to hyperactivity MAJOR FOCUS OF TREATMENT – MAINTAIN RENAL FUNCTION (when RBCs are hemolyzed, Hb molecule is
released and filtered by kidneys. Hb molecules may accumulate and obstruct renal tubles lead to Acute tubular necrosis

Sickle Cell Anemia
Group of inherited, autosomal recessive disorders Mutation in β–globin gene located on chromosome 11 Presence of an abnormal form of hemoglobin, Hemoglobin S (Hb S) in the erythrocyte
Hemoglobin S – involves substitution of VALINE for glutamic acid on the β-globin chain of Hb. Hb S causes erythrocyte to stiffen and elongate taking on a sickle shape in response to low oxygen levels.
Identified during infancy or early childhood. Incurable Fatal by middle age from renal and pulmonary failure
Predominant in African Americans (400:1)Also affects people of Mediterranean, Caribbean, South and Central American, Arabian, or East Indian ancestry.
Etiology and PathophysiologyTypes of sickle cell disease
1) Sickle cell anemia – homozygous for Hb S (Hb SS)a. Inherits Hb S from both parentb. Most severe of SCD syndromes
2) Sickle cell thalassemia a. Inherits Hb S from one parent and another abnormal Hemoglobin (thalassemia) from other parent.b. Less common and less severe
3) Sickle cell Hb C dza. Person inhereits Hb S from one parent and another abnormal Hemoglobin (Hemoglobin C) from other
parent. b. Less common and less severe
4) Sickle cell trait – heterozygous for hemoglobin S (Hb AS)a. Person has inherited Hb S from one parent and normal Hb (Hb A) from other parentb. Very mild to asymptomatic condition
Sickling Episodes Major pathology is the sickling of RBCs.
-Most commonly triggered by low oxygen tension in the blood (hypoxia or deoxygenation of RBCs) and can be caused by viral or bacterial infection(most common factor), high altitude, emotional or physical stress, surgery, blood loss, dehydration, increased hydrogen ion concentration (acidosis), increased plasma osmolality, decreased plasma volume and low body temperature. Can also occur without an obvious cause. -Hemostasis promotes more local hypoxia, deoxygenation of more erythrocytes, and more sickling hemolyzed by the spleen, leading to anemia
Sickled RBCs become rigid, elongated, drescent shape. Cannot easily pass through capillaries, small vessels, and can cause vascular occlusion, leading to acute or chronic tissue injury
Sickling of cells is reversible with reoxygenation, BUT it EVENTUALLY becomes irreversible b/c recurrent sickling causes cell membrane damage.
CLINICAL HALLMARKS – vasoocclusive phenomena and hemolysis
S ickle cell crisis – severe, painful, acute exacerbation of RBC sickling causing a vasoocclusive crisis. -Blood flow impaired, vasospasm occurs, further restricting blood flow

-Severe capillary hypoxia causes changes in membrane permeability, leading to plasma loss, hemoconcentration, development of thrombi, further circulatory stagnation, tissue ischemia, infarction, and necrosis, and SHOCK (life threatening consequence of sickle cell crisis)-Can begin suddenly and persist for days to weeks
Sickling episodes are unpredictable with frequency, extent, and severity-Largely dependent on the percentabe of Hb S present (Hb SS is most severe form due to high percentage of Hb S)
Manifestations Effects vary from person to person Many people are in good health the majority of the time. But they may have chronic health problems and pain b/c
of organ tissue hypoxia and damage (ex; kidneys and/or liver) May be anemic but asymptomatic except during sickling episodes. Manifestations –
o PAIN – primary symptom, especially during sickle cell crisis b/c of ischemia of tissue. Can affect any area of body or several sites simultaneously (back, chest, extremities, abdomen are most common)
o Grayish cast skin o Pallor of mucous membranes – most sickle cell anemic pts have dark skin. So examine mucous
membranes for pallor.o Fatigueo Decreased exercise toleranceo Jaundice (common)o Prone to cholelithiasis (gallstones)o ½ of episodes accompany fever, swelling, tenderness, tachypnea, HTN, nausea, vomiting
Complications Spleen, lungs, kidneys and brain most often affected due to high oxygen needs
-INFECTION (major cause of morbidity and portality) due to failure of the SPLEEN to phagocytize foreign substances as it becomes infracted and dysfunctional (2 to 4 yrs of age)
o P neumonia (most common), aplastic crisis, hemolytic crisis, and gallstones- A cute chest syndrome – acute pulmonary complications that include pneumonia, tissue infarction, and fat embolism.
o Characterized by fever, chest pain, cough, pulmonary infiltrates, and dyspnea-Pulmonary infarctions may cause pulmonary HTN, MI, HF, and cor pulmonale.-Cardiomegaly leading to HF- A utosplenectomy – spleen becomes small because of repeated scarring-Retinal vessel obstruction hemorrhage, scarring, retinal detachment, and blindness-renal failure-Stroke – thrombosis and infarction of cerebral blood vessels-Osteoporosis and osteosclerosis after infarction-Chronic leg ulcers prevalent around ankles due to hypoxia- P riapism (persistent penile erection) if penile veins become occluded
Diagnostic Studies Peripheral blood smear – reveal sickled cells and abnormal reticulocytes. Sickling test – uses RBCs (in vitro) and exposes them to a deoxygenation agent Electrophoresis of hemoglobin readily identifies the abnormal Hb DNA testing - expensive Skeletal x-rays – bone and joint deformities and flattening MRI – diagnose a stroke caused by blocked cerebral vessels from sickled cells Doppler – assess DVT Chest x-ray – diagnose infection or organ malfunction

Labs – findings of hemolysis (jaundice, elevated serum bilirubin levels)Hb/Hct MCV Reticulocytes Serum
IronTIBC Transferrin Ferritin Bilirubin
Sickle cell ↓ N ↑ N or ↑ N or ↓ N N ↑
Nursing and Collaborative Management for Sickle cell DiseaseNO SPECIFIC TREATMENT for the diseaseCare is directed toward
1) Alleviating symptoms from complications of the disease2) Minimize end-organ damage
A cute chest syndrome – 1) treatment broad-spectrum antibiotics, 2) O2 therapy, 3) fluid therapy, 4) folic acid supplementation5) NO IRON THERAPY.
S ickle cell crisis – 1) May require hospitalization2) Oxygen – treat hypoxia and control sickling3) Rest – to reduce metabolic requirements4) Fluids and electrolytes – to reduce blood viscosity and maintain renal function5) Transfusion therapy when aplastic crisis occurs – may require chelation therapy to reduce transfusion –
produced iron overload (like thalassemia major)
6) PAIN – undertreatment is major problem due to lack of knowledge of health care professionalsDuring acute crisis – continous large doses of opioid analgesics(not PRN) + PCA pumpDrugs of Choice – Morphine and hycromorphoneContraindicated drugs – Meperidine (Demerol) - can cause seizure due to toxic accumulation.AFTER DISCHARGE, patients will often CONTINUE ON ORAL OPIOID ANALGESICS
7) Multimodal and multidisciplinary approach is often needed – NSAIDs, antineuropathic pain meds (TCA, antiseizure meds), local anesthetics, nerve blocks, transelectrodermal nerve stimulator (TENS), and acupuncture.
*DRUGSHydroxyurea (Hydrea): increases the production of hemoglobin F (fetal hemoglobin). Given in low dose to minimize the risk of drug-induced cancer and leukemia. If ↑Hb F, then, ↑ Hb concentration, ↓ sickle cells, and ↓ in hemolysis.
*Surgery Hemtaopoietic stem cell transplantation (HSCT) – only treatment that can CURE pts with SCD but the use is limited.
*TEACH patient and family about the disease and the reasons for supportive care. Teach to avoid crisis: reduce the chance of developing hypoxia (avoiding high altitudes). Maintain adequate fluid intake Treat infections promptly: Seek medical attention quickly when having problems with the upper respiratory tract
infections. Educate pain control: minor pain without infection or without symptoms warranting medical attention can
sometimes be managed at home Occupational therapists and physiotherapisis : help achieve optimum physical functioning and independence. psychologist may help cope with anxiety and depression Immunizations – pneumovax, Haemophilus influenza, influenza, and hepatitis

Chronic leg ulcers – bed rest, antibiotics, warm saline soaks, mechanical or enzyme debridement, grafting if necessary
Priapism – pain meds and nifedipine (Procardia)-CCB AFTER DISCHARGE, patients will often CONTINUE ON ORAL OPIOID ANALGESICS.
Acquired Hemolytic Anemia
Acquired Hemolytic Anemia is a destruction of RBCs from an extrinsic cause. PATIENT’S RBCs ARE NORMAL. The destruction is caused by an extrinsic factor.
Extrinsic cause of hemolysis has 3 categories1) Physical factor
Results from the exertion of extreme force on the cellso Traumatic events include hemodialysis, extracorporeal circulation used in cardiopulmonary
bypass, and prosthetic heart valveso Force needed to push blood through abnormal vessels (burned or affected by angiopathic
disease such as DM)
2) Immune reactions (antibodies destroy RBCs by mechanism in antigen-antibody reactions)a. Isoimmune Reactions occur when antibodies develop against antigens. Recipient’s antibodies hemolyze
donor cells when blood transfusionb. Autoimmune reactions result when individuals develop antibodies against their own RBCs.
i. May be idiopathicii. No prior hemolytic history as a result of the immunoglobulin IgG covering the RBCs
iii. Or secondary to other autoimmune disease (SLE, erythromatosus), leukemia, lymphoma, or medications (penicillin, indomethacin [Indocin], phenylbutazone [Butazolidine], phenacetin, quinidine, quinine, and methyldopa [Aldomet])
3) Infectious agents and toxinsa. Infectious agents foster hemolysis in four ways
i. By invading the RBC and destroying its contents (ex; parasites such as in malaria)ii. By releasing hemolytic substances (ex; Clostridium perfringens)
iii. By generating an antigen-antibody reactioniv. By contributing to splenomegaly as a means of increasing removal of damaged RBCs from the circulation
b. Toxins may cause RBC hemolysisChemicals such as oxidative drugs, arsenic, lead, copper, and snake venoms
LabsHb/Hct MCV Reticulocytes Serum
IronTIBC Transferrin Ferritin Bilirubin
Hemolytic anemia
↓ N or ↑ ↑ N or ↑ N or ↓ N N or ↑ ↑
Treatment and management General supportive care until causative agent can be eliminated or at least rendered less injurious to RBCs NURSE! Get ready to institute appropriate emergent therapy b/c HEMOLYTIC CRYSIS is a potential consequence. May need corticosteroids and blood products or removing the spleen.
Hemochromatosis

Hemochromatosis is an autosomal recessive disease. The characteristics of Increased intestinal iron abosorption causes INCREASED TISSUE IRON DEPOSITION
Etiology1) Primarily genetic
a. Most common genetic disorder among whites (3:5 per 1000 whites of European ancestry)b. Normal total body iron is 2 to 6g. hemochromatosis patients accumulate iron at a rate of 0.5 to
1.0g/year = may exceed total iron concentrations of 50gc. Symptoms usually develop between 40 to 60 years of age
2) Secondary to diseases such as thalassemia and sideroblastic anemia3) Liver disease4) Multiple blood transfusions
Manifestations Early symptoms – nonspecific , includes fatigue, arthralgia, impotence, abdominal pain, and weight loss Later symptoms – liver enlargement and eventually cirrhosis b/c of excess iron accumulations
Then, other organs become affected, resulting in DM, skin pigment changes (bronzing), cardiac changes(cardiomegaly), arthritis, and testicular atrophy.
Diagnostic Studies Physical examinations – enlarged liver and spleen and pigment changes in skin Labs - ↑serum iron, ↑ TIBC, ↑ serum ferritin
Hb/Hct MCV Reticulocytes
Serum Iron
TIBC Transferrin Ferritin Bilirubin
Hemochromatosis ↑ ↑ ↑
Molecular testing for KNOWN GENETIC MUTATIONS used to CONFIRM diagnosis. If not definitive Liver biopsy –quantify amount of iron and establish the diagnosis
Collaborative Care GOAL – remove excess iron from body and minimize any symptoms the patient may have
-REMOVE 500 ml of blood each week for 2 to 3 years until iron stored in the body are depleted. Less frequent afterwards. -Organ involvement management-Dietary modifications – avoid vit C, Iron supplements, uncooked seafood, and iron-rich foods-Most common cause of death are cirrhosis, liver failure, hepatic carcinoma, and cardiac failure.
Polycythemia
Polycythemia is the production and presence of INCREASED numbers of RBCs. Numbers are so great that the blood circulation is impaired due to INCREASED BLOOD VISCOSITY (hyperviscosity) and VOLUME (Hypervolemia).
Etiology and PathophysiologyTwo types of polycythemia
1. Primary polycythemia (or polycythemia vera)-Chronic myeloproliferative disorder -Caused by a chromosomal mutation in a single pluripotent stem cell-Leading to increased production of RBCs, WBCs, and platelets. patient has ↑blood viscosity and blood volume and congestion of organs and tissues with blood. Pts. have hypercoagulopathies.-Insidiously develops, and chronic, vacillating course.-Median age diagnosis is 60 years, slight male predominance

-Some suggested that the disease associates with environmental exposure, but no strong evidence.
2. Secondary polycythemiaa. H ypoxia driven – hypoxia stimulates erythropoietin(EPO) production in kidneys stimulates
erythrocyte production-May be due to high altitude, pulmonary disease, cardiovascular disease, alveolar hypoventilation, defective O2 transport, or tissue hypoxia.-Once Hb is stabilized at a higher level, EPO levels may return to normal.-Physiologic response in which body tries to compensate for a problem, rather than a pathologic response
b. H ypoxia independent -EPO is produced by malignant of benigh tumor tissues. EPO levels often remain elevated.
ManifestationsPolycythemia vera First symptoms are circulatory manifestations because of the HTN caused by hypervolemia and hyperviscosity.
Symptoms include HA, vertigo, dizziness, tinnitus, visual disturbances, generalized pruritus (exacerbated by hot bath. Related to histamine release from an increased number of basophils), paresthesias and erythromelalgia (painful burning and redness of hands and feet), angina, HF, intermittent claudication, and thrombophlebitis (may be complicated by embolization. MOST common SERIOUS acute complications is STROKE SECONDARY TO THROMBOSIS
These manifestations are caused by blood vessel distention, impaired blood flow, circulatory stasis, thrombosis, and tissue hypoxia caused by hypervolemia and hyperviscosity.
Hemorrhagic phenomena may result in petechiae, ecchymoses, epistaxis or GI bleeding due to vessel rupture from overdistention or inadequate platelet function. Hemorrhage can be acute and catastrophic
Hepatomegaly and splenomegaly – patient may complain of satiety and fullness Pain from peptic ulcer – due to increased gastric secretions or liver and spleen engorgement
Plethora (ruddy complexion) – congestion causing distention of the blood vessels
Hyperurecemia – uric acid is one of the products of cell destruction. If RBC destruction increases, uric acid production also increases. May cause GOUT
Diagnostic StudiesPolycythemia vera
1) Elevated HB and RBC count with microcytosis2) Low to normal EPO level (secondary polycythemia will have high level)3) Elevated WBC with basophilia4) Elevated platelets (thrombocytosis) and platelet dysfunction5) Elevated leukocyte alkaline phosphatase, uric acid, and cobalamin levels6) Elevated histamine levels
Bone marrow exam in polycythemia – shows hypercellularity of RBCs, WBCs, and plateletsSplenomegaly found in 90% with polycythemia vera but not secondary polycythemia.
Hb/Hct RBC WBC platelets Uric acid
Leukocyte alkaline phosphatase
histamine cobalamin EPO
Polycythemia vera
↑ ↑ ↑ with basophilia
↑Thrombocytosis
And platelet dysfunction
↑ ↑ ↑ ↑ Low to normal
*secondary polycythemia will have high levels of EPO

Collaborative CareTREATMENT is directoward
1) Reducing blood volume and viscosity and 2) Bone marrow activity
Phlebotomy - MAINSTAY of TREATMENT Surgical opening of a vein to withdraw bloodTo reduce hematocrit and keep it less than 45% to 48%. 300 to 500ml of blood removed starting from the time of diagnosis to every other day UNTIL HCT REDUCE TO NORMAL LEVELS. Repeated phlebotomies iron deficient, although asymptomatic, AVOID IRON SUPPLEMENT
Hydration therapy – to reduce blood viscosity
To inhibit bone marrow activity, myelosuppressive agents such as busulfam (Myleran), hydroxyurea (Hydrea), melphalan (Alkeran), and radioactive phosphorus may be given.
Nursing Management for Polycythemia Vera Polycythemia vera is not preventable. Requires ongoing evaluation
o Acute exacerbations of polycythemia vera – -Nurse may either assist with or perform the phlebotomy – depending on hospital policies.
Phlebotomy may be needed q 2 to 3 months, reducing blood volume by about 500ml each time. Evaluate for development of complications
-Hydration therapy – evaluate fluid intake and output to avoid fluid overload and underhydration-If myelosuppressive agents are used – administer drug as ordered, observe patient, and teach patient about side effects.-Nutritional status in collaboration with dietitian may be necessary – offset the inadequate food intake that results from GI symptoms of fullness, pain, and dyspepsia-Activities – to decrease thrombus formation –initiate active or passive leg exercise and ambulation when possible-Myelofibrosis and leukemia may develop (although low incidence) – may be caused by chemotherapeutic drugs used to treat the disease or secondary to a disorder in the stem cells that progresses to erythroleukemia
Secondary polycythemia - Maintaining adequate oxygenation may prevent problems b/c it is generated by any source of hypoxiaTEACH – stop smoking, avoid high altitudes, control chronic pulmonary disease
Thrombosis (ex; stroke) is the major cause of morbidity and mortality

Lymphomas Lymphomas are malignant neoplasms (cancer) Originates in the bone marrow and lymphatic structures resulting in the proliferation (rapid reproduction) of
lymphocytes. Two major types of lymphoma – Hodgkin’s lymphoma and non-Hodgkin’s lymphoma (NHL)
Hodgkin’s Lymphoma
Hodgkin’s lymphoma (=Hodgkin’s disease) Malignant condition Characteristics : proliferation of Reed-Sternberg cells which are giant, abnormal multinucleated cells located in the
lymph nodes. Bimodal age-specific incidence (occurs most frequently from 15 to 35 yrs of age and above 50 yrs old) Prevalent in men
Etiology Cause remains unknown Several key factors play a role:
o Infection with Epstein-Barr virus (EBV)o Genetic predispositiono Exposure to occupational toxinso Human immunodeficiency virus(HIV) infected patients have higher incidence
Pathophysiology In Hodgkin’s lymphoma, hyperplasia (excessive proliferation or reproduction) of monocytes and macrophages
destroys the normal structure of lymph nodes.
MAIN diagnostics -presence of Reed-Sternberg cells in the lymph node biopsy specimens
Disease arise in a single location (mostly lymph nodes) and then spreads along adjacent lymphatics. Cervical lymph nodes are first to be affected in most patients. If recurrent disease, more diffuse (spread, scattered), not necessarily contiguous. eventually infiltrates other organs, especially LUNGS, SPLEEN, AND LIVER-If above the diaphragm, it remains confined to lymph nodes for period of time. -If below diaphragm, frequently spreads to extralymphoid sites such as liver.
Clinical Manifestations Onset of symptoms is insidious
Initial development is lymphadenopathy (enlargement of nodes): separate nodes that remain movable and nontender.o Most common location is cervical, axilary, or inguinal lymphadenopathy o Second most common is mediastinal node mass – may have cough, dyspnea, stridor, and dysphagia
General Manifestations: Painless unless they exert pressure on adjacent nerves OR alcohol induced pain (cause is unknown), weight loss, fatige, weakness, fever, chills, tachycardia, night sweats, generalized pruritus without skin lesions

o Those who have B symptoms has worse prognosis. B symptoms include: fever, night sweats, and weight loss o H epatomegaly and splenomegaly, and anemia are seen in more advanced disease.
Other physical signs depends on disease location:o intrathoracic involvement – superior vena cava syndromeo Enlarged retroperitoneal nodes – palpable abdominal masses or interfere with renal functiono Liver involvement – jaundiceo Extradural involvement – paraplegia due to spinal cord compressiono Bone involvement – bone pain
Diagnostic Studies and Staging Studiesa) Peripheral blood analysis:
o shows microcytic hypochromic anemia, neutrophilic leukocytosis (15,000 to 28,000/µl), lymphopenia, and ↑platelet count, hypoferremia (excessive iron uptake by liver and spleen), leukopenia, thrombocytopenia, superimposed hypersplenism(splenomegaly with cytopenias), elevated leukocyte alkaline phosphate(from liver and bone involvement), hypercalcemia (bone involvement), hypoalbuminemia (liver involvement)
b) Excisional lymph node biopsy: o examine for presence of Reed-Sternberg cells and to identify the subtype (most common is nodular
sclerosing)c) Bone marrow biopsy:
o important aspect of stagingo Reed-Sternberg cells may also be found in the bone marrow
d) Radiologic evaluation: Define all sites and determine the clinical stage of the disease. o CT or MRI – intial staging toolso PET (positron emission tomography with or without CT scans - used to assess the response to therapy and
to differentiate residual tumor from fibrotic masses after treatment. These scans show mediastinal lymphadenopathy, renal displacement caused by retroperitoneal node enlargement, abdominal lymph node enlargement, and liver, spleen, bone, and brain infiltration.
Nursing and Collaborative Management Use diagnostic studies to stage the disease. Final staging is based on extent of the disease(clinical stage), as well as
the presence of B symptoms. Nomenclature used in staging involves
o Classification of A or B- A = no constitutional symptoms- B = fever, drenching sweats & 10% wt loss
o depending on whether symptoms are present when disease is foundo Roman numeral (I to IV) that reflects location and extent of the diseaseo Elevated sedimentation rateo Age ≥50 yrso Presence of a large mediastinal mass and low serum albumin, hemoglobin, and lymphocyte counts may
move an early stage (I or II) to an unfavorable prognosis, needs more aggressive therapy. Tx- begins w accurate classification & staging C hemotherapy
o ABVD: Adriamycin, Vinblastine, Bleomycin , Dacarbazine. – STANDARD -Those in early stage will receive 2 to 4 cycles of chemotherapy. -Those with early-stage but unfavorable prognostic features (ex; presence of B symptoms) or intermediate-stage disease will receive 4 to 6 cycles of chemotherapy-Advanced stage: more aggressive treatment using 6 to 8 cycles
o MOPP (alternating with ABVD): Mechlorethamine, Oncovin, Procarbazine, and Prednisone. o Stanford V and BEACOPP and ICE : more aggressive regimens.
Radiation therapy

-Depends on sites of disease and presence of resistant disease after chemotherapy.
TREATMENT OF CHOICE for advanced, refractory or relapsed Hodgkin’s lymphoma (stages IIIB and IV) : Intensive chemotherapy with or without autologous or allogeneic HSCT “AND”Hematopoietic growth factors(*HSCT allow pts to receive higher, potentially curative doses of chemotherapy while reducing life-threatening leukopenia)
The development of secondary malignancies such as acute myelogenous leukemia, non-Hodgkin’s lymphoma, and solid tumors are serious consequences of the treatment for Hodgkin’s lymphoma. Endocrine, cardiac, and pulmonary dysfunction may develop due to long-term toxicities from treatment.
Nursing Care -Based on managing problems related to the disease (pain due to tumor), pancytopenia, and other side effects of therapy. Supporting patient is important. Physical, psychosocial, and spiritual consequences of patient’s disease must be addressed. -TEACH! Fertility issues soon after diagnosis b/c this disease is frequently seen in adolescents and young adults.
Staging: HD is divided into stages according to the microscopic appearance of involved LN, the extent & severity & prognosis
Stage I -Involvement of a single lymph node or a single extranodal site (spleen, thymus)
Stage II -Involvement of 2 or more lympn node on same side of diaphragm OR localized involvement of an extranodal site and one or more lympn node regions of the same side of the diaphragm. The # of anatomic sites are indicated by a subscript (e.g. IIз )
Stage III -Involvement of Lymph nodes on both sides of the diaphragm. May include a single extranodal site, the spleen, or both. -Now subdivided into lymphatic involvement
-Stage III1: upper abdomen in the spleen (splenic, celiac, and portal nodes) -Stage III2: lower abdominal nodes in the periaortic, mesenteric, and iliac regions
Stage IV -Diffuse or disseminated disease of one or more extralymphatic organs or tissues with or without associated lymph node involvement. -Extranodal site is identified as H, hepatic; L, Lung; P, pleura; M, marrow; D, dermal; O, osseous
Non-Hodgkin’s Lymphoma
Most commonly occurring hematologic cancer, fifth leading cause of cancer death. Affects all ages Heterogenous group of malignant neoplasms of primarily B-cell or T-cell origin which B-cell lymphomas constitute
about 90%. Variety of clinical presentations; may be slow or rapid developing disease
Etiology and Pathophysiology Cause is unknown Risk factors include:
o More common in those who have immunosuppressive medications (ex; to prevent rejection following an organ transplant or treat autoimmune disorders)
o Those who received chemotherapy or radiation therapyo Occupational exposure to carcinogens

No hallmark feature in NHL. However, all NHLs involve lymphocytes arrested in various stages of development.Examples:
o lymphoblastic lymphoma and lymphoblastic leukemia(majority of dz within bone marrow: result from malignant proliferation of small naïve B lymphocytes.
o Diffuse large B-cell lymphoma (most common in adults): neoplasm that originates in the lymph nodeso Burkitt’s lymphoma: highly aggressive, thought to originate from B-cell blast cells in the lymph nodes.o Follicular lymphoma: most common indolent(inactive) or low-grade lymphoma, malignant group of B cells
that occupy center of the lymph node as they approach the end of maturation.
Clinical Manifestations Can originate outside the lymph nodes Unpredictable spreading Painless lymph node enlargement Majority have already disseminated disease at the time of diagnosis. Therefore, other symptoms will be present
depending the spread. (hepatomegaly with liver involvement, neurologic symptoms with CNS disease) Other manifestations: airway obstruction, hyperuricemia, renal failure from tumor lysis syndrome, pericardial
tamponade, and GI complaints. High-grade lymphomas may have lymphadeopathy and B symptoms such as fever, night sweats, and weight l oss. Usually normal peripheral blood but some lymphomas manifest in “leukemic” phase.
Diagnostic and Staging Similar to Hodgkin’s lymphoma but more studies may be done due to extranodal sites MRI: to rule out CNS or bone marrow infiltration Barium enema or CT: to visualize suspected GI involvement Lymph node biopsy: establishes the cell type and pattern
Establishing the precise histologic subtype is extremely important – use lymph node biopsy the establish cell types and pattern.
NHL is classified based on morphologic, genetic, immunophenotypic, and clinical features.
International Working Formulation (IWF) & Nursing and Collaborative Management for Non-Hodgkin’s Lymphoma
Low-grade (Indolent) lymphomas*Difficult to effectively treat.
Small lymphocytic, plasmacytoidFollicular, predominantly small cleaved cellFollicular, mixed small cleaved and large cell
- Median overall survival of 9 yrs- Relapse several times- Cure is very unlikely- Observation until dz progression for asymptomatic patients with low-volume tumors and normal blood counts.- External beam irradiation for local, limited disease. - Therapy indicated when local symptoms from progressive, bulky, or painful disease or compromise of normal organ function- OPTIONS: - *Rituximab q weekly and then every 2 months as maintenance therapyOnce disease is symptomatic, rituximab with chemotherapy such as cyclophosphamide (Cytoxan) with or without prednisone or even the CHOP regimen may be used.- Complete remissions are uncommon- Majority responds with improvement in adenopathy(swelling in lymph nodes) and symptoms.
Intermediate-grade (Aggressive)
Follicular, predominantly large cellDiffuse, small cleaved cell

lymphomas Diffuse, mixed, small and large cellDiffuse, large cell; cleaved cellPeripheral T cell
High-grade (Very aggressive) Lymphomas*more aggressive the lymphoma, the more responsive to treatment and curing.
Large cell immunoblastic LymphoblasticSmall noncleaved cell; Burkitt’s
- Combination chemotherapy (high dose), aggressive with Rituximab and with localized radiation if needed-Most common standard chemotherapeutic regimen is CHOP-R-RICE, EPOCH, CHAP, ESHAP -Dose dense(intensified) treatment with CHOP-14 q 2 weeks OR q 3 to 4 weeks with hematopoietic growth factor -High-dose chemotherapy with autologous HSCT – better outcome
Some subtypes may be treated differently than general standards b/c NHL represents a large variety of neoplasmsCutaneous T-cell lymphoma
- Topical corticosteroids or topical chemotherapy for limited-stage disease
More diffuse disease - Phototherapy, α-interferon, oral bexarotene (Targretin), a retinoid, or denileukin diftitox (Ontak), which is a novel fusion protein consisting of interleukin-2 and diphtheria toxin
Other therapiesPrimary used for patients with indolent lymphomas, particularly those with chemotherapy-refractory disease.
- Monoclonal antibodies ibritumomab tiuxetan (Zevalin) and tositumomab (Bexxar). – these antibodies targets the CD20 antigen which is on the surface of mature B cells and B-cell tumors. These antibodies are linked to radioactive isotope so it allows for the delivery of radiation directly to the malignant cells. - SIDE EFFECTS: Pantocytopenia - Minimize rhe risk of radiation exposure to staff and others
Factors considered with NHLs: advanced disease, number of extranodal sites, older than 60 yrs of age, high serum lactate dehydrogenase, and performance status.
For making therapeutic decisions and assessing prognosis: immunologic, cytogenetic, and molecular studies are useful
Hodgkin’s lymphoma has better prognosis than NHLs
Nursing and Collaborative Management for Non-Hodgkin’s Lymphoma
*Rituximab –- used to treat NHL-Genetically engineered monoclonal antibody against the CD20 antigen on the surface of normal and malignant B lymphocytes. Once bound to cell, causes lysis and cell death.
Nursing Care Similar to Hodgkin’s lymphoma Manage problems related to the disease (ex; pain due to tumor, spinal cord compression, tumor lysis syndrome),
pancytopenia, and other side effects of therapy. Important for the nurse to have an understanding of the subtype and extent of the disease b/c NHL are more
extensive and involves specific organs (ex; CNS, spleen, liver, GI tract, bone marrow)Hannah’s bonus question #1
Q: patient with NHL involving the colon shows symptoms such as abdominal guarding and enlarged and tympanic abdomen. As a nurse, what do you think is happening to the patient ?
A: could indicate bowel perforation and considered a medical emergency. Patient with Burkiitt’s NHL starting chemotherapy – high risk for tumor lysis syndrome, have frequent lab drawn and
monitored, strict intake and output Care for radiation therapy: skin and safety issues

Psychosocial considerations are important Fertility issues Long term evaluation is importantMultiple myeloma
Multiple myeloma, or plasma cell myeloma, is a condition in which neoplastic plasma cells infiltrate the bone marrow and destroy bone.
More common in men, African Americans, usually develops after 40 years of age Lives approx 2 years after diagnosis IF untreated. Myeloma cells tend to collect in the bone marrow and in the hard, outer part of bones. Sometimes they collect in
only one bone and form a single mass, or tumor, called a plasmacytoma. In most cases, however, the myeloma cells collect in many bones, often forming many tumors and causing other problems. When this happens, the disease is called multiple myeloma.
Etiology Cause unknown Genetic factors, viral infection, exposure to radiation, organic chemicals (ex;benzene), herbicides, and insecticides
may play a role of developing multiple myeloma.
Pathophysiology Neoplastic plasma cells (activated B cells) infiltrate bone marrow, produce abnormal and excessive amounts of
immunoglobulin (myeloma protein or M protein)(usually IgG –most common, IgA,IgD, or IgE) and cytokines (interleukins ; IL-4, IL-5, and IL-6) – plays an important role in bone destruction.
Normal plasma cells are reduced-Result in end-organ effects of myeloma, destroying the bone, and bone marrow, nodes, lymph, liver, spleen, kidneys, and heart muscles are invaded
Bence Jones proteins (free light-chain proteins) from the myeloma cell can be detected in urine in some patients.
Manifestations Multiple myeloma develops slowly and insidiously. The patient often does not manifest symptoms until the disease
is advanced. SKELETAL PAIN (major symptom) – pain in the pelvis, spine, and ribs is common and it’s triggered by movement. Diffuse osteoporosis develops, osteolytic lesions are seen in the skull, vertebrae, and ribs. Vertebral destruction, possible paraplegia, and fractures due to loss of bone integrity Hypercalcemia; renal, GI, or neurologic manifestations such as polyuria, anorexia, confusion, and seizures, coma and
cardiac problems High protein levels(myeloma protein); renal tubular obstruction, interstitial nephritis, renal failure Anemia, thrombocytopenia, neutropenia, granulocytopenia
Diagnostic Studies Monoclonal (M) an antibody protein can be found in blood and urine Pancytopenia, hypercalcemia, presence of Bence Jones protein in the urine, and elevated serum creatinine x-rays – show bone erosions, generalized thinning of bones, fractures, vertebrae, ribs, pelvis, and bones of thigh and
upper arm. Bone marrow analysis – shows increased numbers of plasma cells TWO MARKERS to measure prognosis β 2-microglobulin and albumin (higher levels of β2-microglobulin and lower levels of albuminpoorer prognosis)

Collaborative Care GOAL - Manage both the disease and its symptom, control pain, prevent fractures.
Multiple myeloma is seldom cured, but treatment can relieve symptoms, produce remission, and prolong life.Current treatment options – watchful waiting (for early multiple myeloma), corticosteroids, chemotherapy, biologic therapy, and hematopoietic stem cell transplantation (HSCT)
Ambulation, adequate hydration – treats hypercalcemia, dehydration, and potential renal damage
Analgesics, orthopedic support, localized radiation therapy help reduce skeletal painB iphosphonates such as pamidronate (Aredia), zoledronic acid (Zometa), and etidronate (Didronel) – give monthly by IV infusion. inhibits bone breakdown and used from skeletal pain and hypercalcemia
Vertebroplasty to support degenerative vertebrae - surgery
CHEMOTHERAPY - first treatment recommended for multiple myeloma – to reduce number of plasma cellsAlkylating drugs includes melphalan (Alkeran), cyclophosphamide (Cytoxan), vincristine (Oncovin), and doxorubicin (Adriamycin). Corticosteroids (prednisone, dexamethasone[Decadron]) may be added – they show antitumor effect in some patients.
COMMON REGIMEN is VAD – vincristine , doxorubicin (Adriamycin ), and dexamethason ---give once a month over several months before an autologous HSCT. After autologous HSCT, high-dose chemotherapy such as melphalan evolved as the standard of care
Other meds to keep myeloma under controlBortezomib (Velcade) – new, proteasome inhibitor.
Proteasomes are present in all cells and regulate cell growth. (Normal cells recover from proteasome inhibition, but Cancer cells die when inhibited).Indicated for patients whos disease relapsed after two prior treatments and who have demonstrated resistance to their last treatment.
Thalidomide (Thalomid)Immune-modulating and antiangiogenic drug – slow the growth of plasma cells and reduce their numbersGive alone or with corticosteroids or chemotherapy drugsContraindicated in pregnant womenAssociates in DVT, neuropathies, and excess somnolence
α-interferon therapy following chemotherapy - altersthe receptors for IL-6 (growth factor for myeloma cells)
TREATMENT FOR COMPLICATIONS Allopurinol (Zyloprim) – reduce hyperuricemiaIV furosemide (Lasix) – promote renal excretion of calciumCalcitonin and pamidronate –treats hypercalcemia and decrease risk of fractures and reduce bone pain
Nursing management for Multiple Myeloma
Maintain adequate hydration:3-4L/d for output of 1.5-2L/d. MONITOR electrolyte and fluid balance for renal dysfunction
Assist with ambulation: weight bearing to reabsorb calcium Adm. Corticosteroids to help excrete calcium; allopurinol (antigout-xanthine oxidase inhibitors)for hyperuremia Pain management with analgesics (NSAIDs, acetaminophen, or acetaminophen/opioid combination) better than
opioid alone in diminishing bone pain, braces, local radiation Prompt treatment of Infection nurse must be careful when moving and ambulating the patient – due to potential for pathologic fractures.

Peripheral neuropathy is common – prevent falls. Psycosocial: support adaptation to chronic nature- symptoms remit and exacerbate, ultimately disease becomes
resistant to treatment See anemia (NCP 31-1), thrombocytonemia (NCP 31-2), neutropenia (NCP 31-3)
Disorders of the Spleen
Located in the upper left quadrant next to kidney Functions can be classified as
– Hematopoietic: Able to produce RBCs during fetal development– Filtration
Remove old and damaged RBCs from circulation Removes hemoglobin from RBCs and returns iron component to the bone marrow for reuse Filters out bacteria, especially encapsulated organisms
Normal spleen contains 20 to 40 ml of blood. Do not usually serve as reservoir for blood volume and erythrocytes.
Spleen can be affected by many illness may cause splenomegaly (enlarged spleen). Degree of enlargement varies with dz. Ex) Chronic myelogenous leukemia, hairy cell leukemia, and thalassemia major causes massive splenic enlargement. Heart failure and SLE causes mild splenic enlargement.
H ypersplenism – occurrence of splenomegaly and peripheral cytopenias (anemia, leukemia, and thrombocytopenia)
Manifestations When spleen enlarges, normal filtering and sequestering capacity increases. reduction in number of circulating
blood cells Howell –Jolly bodies or pitted or pocked erythrocytes – unusual findings in the peripheral smear Slight to moderate enlargement : usually asymptomatic, may be found during routine examination of abdomen. Massive enlargement : some tolerate, but complain of abdominal discomfort and early satiety.
Techniques to assess size of spleen (diagnostic tests) Physical examination, Tc-sulfer colloid liver-spleen scan, CT scan, MRI, and ultrasound scan
Treatment of splenomegaly Laparoscopy or open laparotomy and splenectomy( has dramatic effect in increasing peripheral RBC, WBC, platelet)
Splenectomy may be done due to splenomegaly, splenic rupture (from trauma, during other surgical procedures, disease such as mononucleosis, malaria, and lymphoid, neoplasms)
Nursing Management Pain management : may require analgesics May impair diaphragmatic excursion : evaluate lung expansion care in moving, turning and positioning prevent life-threatening complications if hypersplenism post op splenectomy : special observation for hemorrhage which can lead to shock, fever, and abdominal
distention. Immunologic deficiencies may develop. (↓IgM levels, normal IgG and IgA). May have lifelong risk for infection (especially pneumococcus) – TEACH for pneumovax immunization
Blood Component Therapy

Blood component therapy is frequently used in managing hematologic diseases. However, blood component therapy only temporarily supports the patient until the underlying problem is resolved.
Use only if necessary Avoid developing a complacent attitude about this common but potentially dangerous therapy Make sure PHYSICIAN discuss risks, benefits, and alternatives with the patient and that this is documented. Blood Transfusion has a broader meaning today. Not just whole blood (used rarely for massive hemorrhage) but
also specific components of blood such as platelets, packed red blood cells (PRBCs), or plasma
Administration Procedure If using small guage needles : split unit (the blood bank issues half of the unit at a time) or dilute with NS to ensure
that it does not run over the MAXIMUM TIME OF 4 HOURS due to bacterial growth. Blood unrefrigerated for 4 hoursreturn to blood bank.
o 23-guage needle: safe, but usually impedes flow, cause hemolysis particularly when using pressured infusion device. Smaller needles can be used for platelets, albumin, and clotting factor replacement.
o 19-guage needle preferred running into a free-flowing line. o 16 to 18 guage needle : for rapid transfusions
“Y-type” tubing with a microaggregate filter (filters out particulate) with one Y for the isotonic saline solution and other Y for the blood product.
DO NOT USE Dextrose Solutions or Lactated Ringers : they induce RBC hemolysisNo other additives (inducing medications) should be given via same tubing as blood UNLESS tubing is cleared with saline solution
When the blood or blood components have been obtained from the blood bank, POSITIVE IDENTIFICATION OF THE DONOR BLOOD AND RECIPIENT MUST BE MADE. Improper product-to-patient identification causes 90% of hemolytic transfusion reactions.
The blood bank is responsible for typing and crossmatching the donor’s blood with recipient’s blood : result of testing should be noted on the product bag or tag.
Nursing Management Make sure pt agrees and understands the procedure, and signs and symptoms to report. Take V/S for baseline: If ANY abnormals, report to MD to clarify when to administer blood. Administer as SOON AS it is brought to patient: DO NOT refrigerate in nursing unit. IF NOT USED WITHIN 30
MINUTES, RETURN TO BLOOD BANK!! DURING FIRST 15 MINUTES or 50ML OF INFUSION- STAY WITH THE PT b/c untoward reactions most likely to occur
at this time. Infusion rate should be no more than 2ml/min during thise period. PRBCs should not be infused quickly UNLESS EMERGENCY b/c may cause chills due to cold product. IF rapid
replacement is necessary, blood-warming device may be used. Fresh frozen plasma and platelets, may be infused over 15 to 30 minutes.
After 15 minutes – retake V/S. rate of infusion is governed by clinical condition of the patient AND the product being infused. If not danger of fluid overload, most patients can tolerate of 1 unit of PRBCs over 2 hours.
Blood Transfusion Reactions A blood transfusion reaction is an adverse reaction to blood transfusion therapy that can range in severity from
mild symptoms to a life-threatening condition. Blood transfusion reactions can be classified as acute or delayed.If acute transfusion raction occurs …
1) Stop the transfusion2) Maintain a patent IV line with saline solution3) Notify blood bank and MD immediately4) Recheck identifying tags and numbers5) Monitor vital signs and urine output6) Treat symptoms per physician order

7) Save the blood bag and tubing and send them to blood bank for examination8) Complete transfusion reaction reports9) Collect required blood and urine specimens at intervals stipulated by hospital policy to evaluate for hemolysis10) Document on transfusion reaction form and patient chart.
Blood bank and Labs are responsible for identifying the type of reaction
Acute Transfusion Reactions
Reaction Cause Manifestatiosn Management PreventionAcute Hemolytic
- infusion of ABO-incompatible components or components containing 10 mL or more of RBCs.
- Antibodies in recipient’s plasma attach to antigens on transfused red blood cells, causing agglutination of cells(blocks blood flow) and RBC destruction
- Mislabeling specimens
- Adm. To wrong pt.
- Chills, fever, low back pain, flushing, tachycardia, dyspnea, tachypnea, hypotension, vascular collapse, hemoglobinuria, acute jaundice, dark urine, bleeding, acute renal failure, shock, cardiac arrest, death.
- Acute : first 15 min of transfusion
- Delayed: 2 to 14 days after transfusion
- Treat shock and DIC if present
- Draw blood samples for serologic testing slowly to avoid hemolysis from procedures. Send urine specimen to lab
- Maintain BP with IV colloid solutions. Give diuretics as prescribed to maintain output.
- Insert urinary catheter or measure voided amounts to monitor urine output
- Dialysis may be required if renal failure occurs
- Do not transfuse additional RBC containing components until blood bank has provided newly crossmatched units
Meticulously verify and document pt’s identification from sample collection to component infusion.
Febrile (nonhemolytic)Reactions
MOST COMMON
- Sensitization to donor WBCs (leukocyte incompatibility-most common) platelets, or plasma proteins.
- Sudden chills and fever >1C rise, HA, flushing, anxiety, vomiting, muscle pain
- Give antipyretics as prescribed-AVOID ASPIRIN in thrombocytopenic patients
- DO NOT RESTART TRANSFUSION unless physician orders
consider leukocyte-poor blood products (filtered, washed, or frozen) for patients with a history of two or more such reactions.
Mild allergic reactions
- Sensitivity to foreign plasma proteins
- More common in individuals with Hx of allergies
- Flushing, itching, uticaria(hives)
- Give antihistamine, corticosteroid as directed
- If symptoms are mild and transient, transfusion may be restarted slowly
- Do not restart transfusion if FEVER or pulmonary symptoms develop
-Treat prophylactically with antihistamines-Consider washed RBCs and platelets.
Anaphylactic and severe allergic reactions
- Sensitivity to donor plasma proteins
- Infusion of IgA proteins to IgA-deficient recipient who has developed IgA antibody
- Anxiety, urticaria, dyspnea, wheezing, progressint to dyanosis, bronchospasm, hypotension, shock, and possible cardiac arrest.
- Initiate CPR, if indicated- Have EPINEPHRINE ready
for injection- Do NOT RESTART
TRANSFUSION
-Transfuse extensively washed RBC products, from which all plasma has been removed.-Use blood from IgA-deficient donor-Use autologous components
Circulatory overload
- Fluid administered faster than the
- Cough, dyspnea, pulmonary
- Place pt upright with feet in dependent position
-Adjust transfusion volume and flow rate

circulation can accommodate
- Individual w/cardiac or renal insufficiency are at risk
- Elderly at risk
congestion, HA, HTN, tachycardia, JVD
- Complain of SOB and adventitious breath sounds
- Adm. Prescribed diuretics, oxygen, morphine
- Phlebotomy may be indicated
based on pt. size and clinical status-Have blood bank divide unti into smaller aliquots for better spacing of fluid input
Sepsis Transfusion of bacterially infected blood
- Rapid onset of chills, high fever, vomiting, diarrhea, marked hypotension, or shock
- Obtain culture of pt’s blood and send bag with remaining blood and tubing to blood bank for further study
- Treat septicemia as directed – antibiotics, IV fluids, vasopressors
-collect, process, store, and transfuse blood products according to blood banking standards and infuse within 4 hr of starting time.
Transfusion related acute lung injury (TRALI)
- Reaction btwn transfused antileukocyte antibodies and recipient’s leukocytes, causing pulmonary inflammation and capillary leak
- Fever, hypotension, tachypnea, dyspnea, decreased O2 sat, frothy sputum
- Non-cardiogenic pulmonary edema (acute lung injury)
- Send bag with remaining blood and tubing to blood bank for further study; draw blood for arterial blood gases and HLA or antileukocyte antibodies; obtain chest x-ray
- Provide oxygen and administer corticosteroids (diuretics of no value?)
- Initiate CPR if needed and provide ventilator and blood pressure support if needed
Provide leukocyte reduced products. Identify donors who are implicated in TRALI reactions and DO NOT ALLOW THEM TO DONATE
- - -
Massive Blood Transfusion Reaction An acute complication of transfusing large volumes of blood products is termed massive blood transfusion
reaction. Massive blood transfusion reactions can occur when replacement of RBCs or blood exceeds the total blood volume within 24 hours.
Delayed Transfusion Reactions
Delayed transfusion reactions include delayed hemolytic reactions, infections, iron overload, and graft-versus-host disease.
Infectious agents transmitted by blood transfusion include hepatitis B and C viruses, HIV, human herpesvirus type 6, Epstein-Barr virus, human T cell leukemia, cytomegalovirus, and malaria.
AutoTransfusion Autotranfusion, or autologous transfusion, consists of removing whole blood from a person and transfusing that
blood back into the same person. The problems of incompatibility, allergic reactions, and transmission of disease can be avoided.
Related Documents











