Cerebellar Alterations and Gait Defects as Therapeutic Outcome Measures for Enzyme Replacement Therapy in α-Mannosidosis Markus Damme, PhD, Stijn Stroobants, Steven U. Walkley, DVM, PhD, Renate Lüllmann- Rauch, MD, Rudi D`Hooge, PhD, Jens Fogh, DVM, Paul Saftig, PhD, Torben Lübke, PhD, and Judith Blanz, PhD Department of Biochemistry 2 (MD), Georg-August University Göttingen; Department of Biochemistry 1 (TL), University of Bielefeld; Biochemical Institute (PS, JB), University of Kiel; Laboratory of Biological Psychology (SS, RH), University of Leuven; Sidney Weisner Laboratory of Genetic Neurological Disease, Department of Neuroscience, Rose F. Kennedy Center for Research in Mental Retardation and Human Development, Albert Einstein College of Medicine (SUW), Bronx, New York; Anatomisches Institut (RLM), Christian-Albrechts Universitity Kiel; and Zymenex A/S, Hillerød (JF) Abstract α-Mannosidosis is a rare lysosomal storage disease with accumulation of undegraded mannosyl- linked oligosaccharides in cells throughout the body, most notably in the CNS. This leads to a broad spectrum of neurological manifestations, including progressive intellectual impairment, disturbed motor functions and cerebellar atrophy. To develop therapeutic outcome measures for enzyme replacement therapy (ERT) that could be used for human patients, a gene knockout model of α-mannosidosis in mice was analyzed for CNS pathology and motor deficits. In the cerebellar molecular layer, α-mannosidosis mice display clusters of activated Bergman glia, infiltration of phagocytic macrophages and accumulation of free cholesterol and gangliosides (GM1), notably in regions lacking Purkinje cells. α-mannosidosis brain lysates also displayed increased expression of Lamp1 and hyperglycosylation of the cholesterol binding protein NPC2. Detailed assessment of motor function revealed age-dependent gait defects in the mice that resemble the disturbed motor function in human patients. Short-term ERT partially reversed the observed cerebellar pathology with fewer activated macrophages and astrocytes but unchanged levels of hyperglycosylated NPC2, gangliosides and cholesterol. The present study demonstrates cerebellar alterations in α- mannosidosis mice that relate to the motor deficits and pathological changes seen in human patients and can be used as therapeutic outcome measures. Keywords α-Mannosidase; α-Mannosidosis; Cerebellar atrophy; Enzyme replacement therapy; Gait defects; Knockout mouse model; Lysosomal storage disease Send correspondence and reprint requests to: Judith Blanz, PhD, Biochemical Institute, University of Kiel, Eduard-Buchner-Haus, Otto-Hahn-Platz 9, 24118 Kiel, Germany. [email protected]. Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. NIH Public Access Author Manuscript J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1. Published in final edited form as: J Neuropathol Exp Neurol. 2011 January ; 70(1): 83–94. doi:10.1097/NEN.0b013e31820428fa. NIH-PA Author Manuscript NIH-PA Author Manuscript NIH-PA Author Manuscript

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Cerebellar Alterations and Gait Defects as Therapeutic OutcomeMeasures for Enzyme Replacement Therapy in α-Mannosidosis
Markus Damme, PhD, Stijn Stroobants, Steven U. Walkley, DVM, PhD, Renate Lüllmann-Rauch, MD, Rudi D`Hooge, PhD, Jens Fogh, DVM, Paul Saftig, PhD, Torben Lübke, PhD,and Judith Blanz, PhDDepartment of Biochemistry 2 (MD), Georg-August University Göttingen; Department ofBiochemistry 1 (TL), University of Bielefeld; Biochemical Institute (PS, JB), University of Kiel;Laboratory of Biological Psychology (SS, RH), University of Leuven; Sidney Weisner Laboratoryof Genetic Neurological Disease, Department of Neuroscience, Rose F. Kennedy Center forResearch in Mental Retardation and Human Development, Albert Einstein College of Medicine(SUW), Bronx, New York; Anatomisches Institut (RLM), Christian-Albrechts Universitity Kiel; andZymenex A/S, Hillerød (JF)
Abstractα-Mannosidosis is a rare lysosomal storage disease with accumulation of undegraded mannosyl-linked oligosaccharides in cells throughout the body, most notably in the CNS. This leads to abroad spectrum of neurological manifestations, including progressive intellectual impairment,disturbed motor functions and cerebellar atrophy. To develop therapeutic outcome measures forenzyme replacement therapy (ERT) that could be used for human patients, a gene knockout modelof α-mannosidosis in mice was analyzed for CNS pathology and motor deficits. In the cerebellarmolecular layer, α-mannosidosis mice display clusters of activated Bergman glia, infiltration ofphagocytic macrophages and accumulation of free cholesterol and gangliosides (GM1), notably inregions lacking Purkinje cells. α-mannosidosis brain lysates also displayed increased expression ofLamp1 and hyperglycosylation of the cholesterol binding protein NPC2. Detailed assessment ofmotor function revealed age-dependent gait defects in the mice that resemble the disturbed motorfunction in human patients. Short-term ERT partially reversed the observed cerebellar pathologywith fewer activated macrophages and astrocytes but unchanged levels of hyperglycosylatedNPC2, gangliosides and cholesterol. The present study demonstrates cerebellar alterations in α-mannosidosis mice that relate to the motor deficits and pathological changes seen in humanpatients and can be used as therapeutic outcome measures.
Keywordsα-Mannosidase; α-Mannosidosis; Cerebellar atrophy; Enzyme replacement therapy; Gait defects;Knockout mouse model; Lysosomal storage disease
Send correspondence and reprint requests to: Judith Blanz, PhD, Biochemical Institute, University of Kiel, Eduard-Buchner-Haus,Otto-Hahn-Platz 9, 24118 Kiel, Germany. [email protected]'s Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to ourcustomers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review ofthe resulting proof before it is published in its final citable form. Please note that during the production process errors may bediscovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
NIH Public AccessAuthor ManuscriptJ Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
Published in final edited form as:J Neuropathol Exp Neurol. 2011 January ; 70(1): 83–94. doi:10.1097/NEN.0b013e31820428fa.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

INTRODUCTIONLysosomal storage disorders (LSDs) are a group of individually rare human genetic diseasescharacterized by defects in lysosomal hydrolysis of macromolecules (e.g. lipids andglycoproteins), leading to accumulation of undegraded material in the lysosome (1). α-Mannosidosis is a progressive LSD caused by deficiency of the lysosomal hydrolase α-mannosidase (LAMAN) (2,3), and accumulation of polymannose oligosaccharides in theendosomal/lysosomal system. Abnormalities in α-mannosidosis patients include intellectualand psychiatric disability and prominent motor defects (4). These motor defects are due tomultiple factors including orthopedic pathology, but also ataxia that results from cerebellaratrophy and demyelination (5,6). Patients with α-mannosidosis patients present a variety ofclinical symptoms. At present, 3 clinical types have been suggested: a mild and moderateform with late onset (>10 years) and slow disease progression (type I); development ofataxia at the age of 20 to 30 years (type II); and severe forms leading to an early death fromCNS involvement or myopathy (type III). Most patients have the moderate form (type II) ofthe disease (4).
Animal models of α-mannosidosis in guinea pigs, cats, and mice have been used to study theconsequences of α-mannosidase deficiency and to develop therapeutic approaches for thiscarbohydrate storage disorder. In particular, mice with targeted disruption of the LAMANgene Man2b1 (7) were shown to be a valid model for α-mannosidosis (8,9). Cerebellarpathology comparable to that in patients has been described in both guinea pigs and catswith this disease, but not yet in α-mannosidosis mice. Behavioral analyses revealedneurocognitive impairments in adult α-mannosidosis mice that mimic many aspects ofhuman α-mannosidosis (8–10). Detailed assessment of motor system dysfunction, includingsensitive methods of quantitative gait analysis, and cerebellar pathology in these mice hasnot been performed. At present, no therapeutic treatment other than bone marrowtransplantation is available for α-mannosidosis patients but preclinical enzyme replacementtherapy (ERT) in the mouse model has shown promise (10,11). Short-term, high-dose ERTsuccessfully decreased neuronal storage of sugars in the brain of α-mannosidosis mice andcorrelated with improved neuromotor abilities (10). Inasmuch as clearance of stored sugarswas prominent in the hippocampus and other brain regions but not in the cerebellum, it isunclear as to how ERT contributed to the observed improvements in motor function.
To investigate whether neuromotor deficits in α-mannosidosis mice relate to the cerebellarpathology that is comparable to that seen in humans, and that can be used as outcomemeasures in therapy studies, we performed detailed histological and biochemical analyses onthe cerebellum of α-mannosidosis mice. Sensitive methods for quantitative gait analyses areincluded to determine the functional consequences of the cerebellar findings. In addition, theeffects of a short-term, high-dose ERT regimen on the pathological alterations wereevaluated.
MATERIALS AND METHODSAnimals
α-Mannosidase knockout (KO) mice, (referred to here as “α-mannosidosis mice”) weregenerated by targeted disruption of the Man2b1 gene leading to a deficiency in LAMANactivity (7). KO mice and their wild-type (WT) littermates were bred on a C57Bl/6background and maintained under standard housing conditions. Genotyping was carried outby polymerase chain reaction, as described (7). Six- to 14-month-old α-mannosidosisanimals and age-matched WT animals were studied. All animal experiments were approvedby local authorities.
Damme et al. Page 2
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Primary Antibodies and ReagentsMonoclonal mouse anti-glial fibrillary acidic protein (GFAP) and Filipin were purchasedfrom Sigma-Aldrich (St. Louis, MO). Monoclonal mouse anti-Lamp1 (1D4B) and mouseanti-Lamp2 (Abl-93) were purchased from Developmental Studies Hybridoma Bank (IowaCity, Iowa). Antibodies to myelin basic protein (MBP) and calbindin were from Calbiochem(Darmstadt, Germany), to GM1 from Biomol (Hamburg, Germany), and to Actin fromSigma-Aldrich. Rat anti-CD68 and mouse anti-F4/80 were from Abd Serotec (Oxford, UK).Rabbit anti-NPC2 protein antiserum was a kind gift from Shutish Patel (12). Goat anti-mouse IgG, goat anti-rabbit IgG and Vectastain ABC kit were from Vector Laboratories(Burlingame, CA). Fluorophore conjugated secondary antibodies (Alexa Fluor 488 and 546)and DAPI were purchased from Molecular Probes (Eugene, OR). Appropriate negativecontrols omitting primary antibodies were included in immunohistochemistry (IHC)protocols to confirm specificity. Chemicals were purchased from Sigma-Aldrich.
Intravenous Injection of Recombinant Human LAMANRecombinant human LAMAN was purified from CHO cells as described (11). It wasinjected twice a week (4 ×) into the tail vein of adult mice in a dose of 500 U per kg bodyweight. Mock-injected KO and WT mice given the same volume of PBS served as controls.LAMAN activity was determined as previously described (10).
Tissue CollectionAnimals were deeply anesthetized and subsequently perfused with 0.1 M phosphate buffer(PB), pH 7.4 followed by 4 % paraformaldehyde in PB (for free floating sections), or 6 %glutaraldehyde supplemented with 1 % procaine (for electron microscopy). Brains wereremoved and cut sagittally into halves. For free-floating sections, brains were post-fixed in4% paraformaldehyde overnight at 4°C, and stored in 30% sucrose until sectioning. Samplesfor electron microscopy were processed as previously described (10).
Western Blot AnalysisFor Western blots, brains were removed after PB perfusion and homogenized in 9 volumes(v/w) of ice-cold lysis buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 1 %Triton X-100 with protease inhibitors). After incubation on ice for 30 minutes followed bysonification, homogenates were cleared by centrifugation at 13,000 rpm for 15 minutes at4°C. Equal amounts of protein were separated on 12.5 % or 15 % SDS-PAGE gels, blottedonto nitrocellulose membranes and incubated with the respective primary antibodies.Horseradish peroxidase-conjugated secondary antibodies were detected bychemiluminescence (SuperSignalWest, Pierce, Pittsburgh, PA). Actin was used as a loadingcontrol.
ImmunohistochemistryFor IHC, sagittally cut, frozen brain halves were sectioned on a Leica 9000s microtome into50-µm-thick free-floating slices. When staining for gangliosides and cholesterol, unfrozenbrain halves were cut sagittally into 35-µm-thick free-floating sections using a Leicavibratome. The sections were rinsed in 0.1 M PB and subsequently blocked with 4% normalgoat serum in 0.1 M PB with 0.2 % bovine serum albumin and 0.25% Triton X-100 forpermeabilization at room temperature (RT) for 2 hours. Primary antibodies were diluted inblocking solution and incubated overnight at 4°C with the sections at gentle agitation. Afterwashing 3× with washing buffer (0.1 M PB with 0.25 % TX-100), appropriate secondaryantibodies (fluorophore conjugated or biotinylated) diluted in washing buffer were incubatedwith the sections for 2 hours at RT. After additional 3 washes with washing buffer, thesections were coverslipped with MOWIOL/DABCO (fluorophore-labelled sections) or
Damme et al. Page 3
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

processed for 3, 3’diaminobenzidine (DAB)-visualization. DAB-stained sections weremounted, dehydrated and coverslipped with Permount™. Some sections were counterstainedwith Nissl stain. DAB-stained sections with the anti-GFAP-antibody were detected withHorseradish peroxidase-labelled secondary antibody omitting Biotin/Streptavidin-step.
HistochemistryFor filipin histochemistry of brain sections, mice were perfused with 4% paraformaldehydein 0.1 M PB and post-fixed in the same fixative overnight followed by storage in 0.1 M PBat 4°C. Filipin histochemistry was performed on 35-µm-thick vibratome sections. All stepswere performed at RT. Sections were washed 2 × 10 minutes in PBS and 2 × 10 minutes in0.2% Saponin/PBS. Slices were incubated in Filipin (0.05 mg/ml) for 20 minutes, washed 2× 10 minutes in 0.02% Saponin/PBS and 2 × 10 minutes in PBS. Slices were mounted inProlong antifade mounting solution (Invitrogen).
Isolation of Neutral OligosaccharidesNeutral oligosaccharides were extracted as described (11). In brief, brains werehomogenized in 9 volumes (v/w) HPLC-grade water. After sonification, proteins wereprecipitated by methanol and extracted by addition of chloroform/water. Supernatants weredesalted by incubation with mixed-bed ion-exchange resin (AG 501-X8, Bio-Rad, Hercules,CA) and soluble material was lyophilized.
Thin Layer Chromatography of Neutral OligosaccharidesLyophilized oligosaccharides were resuspended in HPLC-grade water and spotted ontoSilica gel thin layer chromatography plates (20 × 20 Silica gel F60, Merck, Darmstadt,Germany). Oligosaccharides were separated by n-butanol/acetic acid/water (100:50:50)development overnight followed by n-propanol/nitromethan/water (100:80:60) developmentfor 4 hours. After drying, plates were sprayed with 0.2% (w/v) orcinol-solution (20% H2SO4dissolved in water) and heated at 110°C until dark bands appeared.
Quantitative Gait Analysisα-Mannosidosis mice 6 and 19 months of age underwent treadmill gait analysis, as described(13). Following a 30-second habituation period, 4 trials of 60-second treadmill walking wereconducted, during which mice were filmed by a ventrally placed webcam. These trials wereexecuted at different combinations of speed and slope (16 cm/s and 0°; 16 cm/s and 10°; 22cm/s and 0°; 22 cm/s and 10°). Mice were encouraged to keep pace with the treadmill usingan electric grid placed at the end of the treadmill. The number of required electricalstimulations (errors) was registered during each trial. Several gait variables including base-width (distance between contralateral paws), front/hind distance (distance between front pawprint and subsequent hind paw print) and stride length (distance between 2 subsequent printsof the same paw) were extracted from these video data using an automated algorithm (14).Additionally, incongruity coefficients (ICs) for related variables were calculated as ameasure of disturbed gait. These coefficients are defined as the absolute value of thedifference between the z-scores of 2 related gait variables, for example:
With IC = incongruity coefficient; FB = front base (distance between front paws); HB =hind base (distance between hind paws); stdev = standard deviation. Dissimilar deviationsfrom the group mean result in a higher IC based on these variables and is a sign of ill-balanced or uncoordinated gait.
Damme et al. Page 4
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

RESULTSRegional Differences in Carbohydrate Storage in Brains of α-Mannosidosis Mice
Neutral oligosaccharides were extracted from different brain regions including cortex,cerebellum, brainstem and midbrain of α-mannosidosis mice and separated by thin layerchromatography (Fig. 1). Whereas mannosyl-linked oligosaccharides were present in allbrain regions, remarkably high levels of stored sugars were detected in the cerebellum of α-mannosidosis mice.
Storage of Gangliosides, Cholesterol and Autofluorescent Material in the Cerebellum of α-Mannosidosis Mice
Based on our thin layer chromatography analyses, the cerebellum is the site of highestprimary storage. To investigate whether primary lysosomal storage secondarily affects thedistribution of gangliosides as in many other LSDs (15), we performed IHC on brainsections of 12-month-old KO and WT mice using antibodies specific for GM1, GM2 andGM3 gangliosides. The secondary accumulation of gangliosides in the brain is a commonhallmark of LSDs (15). Among the tested gangliosides, only GM1 was elevated in the KObrains (Fig. 2B, D–F). In WT, GM1 was barely detectable (Fig. 2A, C), but pyramidalneurons of the hippocampus of KO mice displayed slightly increased levels of GM2 andGM3 (data not shown). Vesicular GM1 accumulation was apparent throughout the brain,including the cerebellum (Fig. 2A–D, F), cerebral cortex and midbrain (Fig. 2E). In thecerebellum, GM1 accumulated exclusively within the molecular layer where it was foundwithin the dendritic tree of Purkinje cells and in vesicular-like structures within thecytoplasm of glial and neuronal cells (Fig. 2D, F).
Because ganglioside storage correlates at least partially with cholesterol storage (16), weused the fluorescent dye filipin to stain free cholesterol in the KO and WT tissue (Fig. 2G–J). In KO (Fig. 2 H, J), but not in WT (Fig. 2 G, I) brains, cholesterol accumulatedspecifically in the cerebellar molecular layer in a pattern comparable to that of GM1.However, other brain regions with elevated GM1 levels lack stored cholesterol. IHC onbrain sections of 6-month-old mice revealed less prominent storage of both lipids, indicatingtheir progressive accumulation (not shown). Consistent with these observations, subcellularfractionation of whole brain homogenates showed a significantly decreased density oflysosomal compartments in the KO brains, suggesting an accumulation of metabolites withlow density like lipids within α-mannosidosis lysosomes (Supplemental Fig. 1).
AutofluorescenceUnstained vibratome brain sections of KO and WT brains at 12 months were used to studytheir autofluorescent properties following excitation of different wavelengths (UV light,350nm, 488 nm and 633 nm) (Fig. 3B, D). Autofluorescent material was detected inPurkinje cells within the molecular layer and the deep nuclei of the cerebellum in KO mice.Autofluorescence was also visible in Purkinje cells of WT mice, a finding consistent withthe reported accumulation of lipofuscin in aged Purkinje cells (Fig. 3B, upper panel) (17).Autofluorescence was detected in all excitation wavelengths with highest emission afterUV-excitation at 350 nm. Cholesterol was present within most of these autofluorescentstructures as indicated by co-staining with Filipin (Fig. 3B, lower panel).
Activation of Macrophages and AstrogliosisActivation of CNS resident macrophages and astrocytes is characteristic of LSDneuropathology. To investigate whether microglial activation also occurs in α-mannosidosis,the expression of CD68/macrosialin (18) (Fig. 3A) and F4/80 (Fig. 3D) was studied by IHCof WT and KO brains. The KO brains displayed numerous CD68-positive macrophages with
Damme et al. Page 5
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

plump, amoeboid shapes indicative of activation mainly in the cerebellar molecular layerwhereas WT brains showed ramified microglial cells with fine processes characteristic of anon-activated state. Activated microglia were often found in a cluster-like assembly andgradually increased in number from anterior to posterior lobes (Fig. 3A). The granular celllayer, white matter tracts and deep cerebellar nuclei of KO cerebellum were mostly devoidof activated microglia. Intracellular autofluorescence was visible in F4/80-positivemacrophages, suggesting phagocytic uptake of autofluorescent storage material (Fig. 3D).Electron micrographs of macrophages in the cerebellar molecular layer of KO micedisplayed cytoplasmic multilamellar and lipofuscin like material indicative of lipid storage(Fig. 3C).
Because microglial activation is often accompanied by reactive astrogliosis anddemyelination, we performed immunofluorescence and Western blot analyses of GFAP andMBP, which are specific for astrocytes and myelin, respectively. As in the cerebellum, WTand KO brains showed comparable MBP distribution (Fig. 4A, upper panel) and expressionlevels (Fig. 4C), suggesting normal myelination the KO mice. Myelin staining with Luxolfast blue also showed no differences in the overall appearance of cerebellar myelin betweenthe 2 genotypes (not shown). In contrast, GFAP was slightly upregulated in the KO mice(Fig. 4A, lower panel) that was confirmed by Western blot analysis (Fig. 4C), suggesting anactivation of astrocytes upon disruption of LAMAN activity. Upregulation was also evidentfor the lysosome-associated membrane proteins Lamp1 and Lamp2 (Fig. 4C).
Regional Restricted Astrogliosis and Partial Loss of Purkinje CellsTo study reactive astrocytosis in more detail, we performed GFAP IHC that revealed aregional restricted astrogliosis (arrowhead) of Bergmann glia within the molecular and thePurkinje cell layer of the cerebellum of KO mice (Fig. 4B). To determine whether theregionally restricted astrogliosis was associated with changes within the Purkinje cell layeras in other murine LSD models (19–21), sections of KO and WT mice were co-stained forcalbindin, (a protein dominantly expressed in the soma and dendritic tree of Purkinje cells[22]) and GFAP (Fig. 4D). The reactive astrogliosis occurred predominantly within regionsdevoid of Purkinje cells, as indicated by absence of their dendritic trees. Immunolabeling ofcalbindin revealed a comparable distribution between the Purkinje cell layers of KO and WTmice (Fig. 4E), but KO cerebellum displayed scattered regions lacking Purkinje soma anddendritic trees (Fig. 4F), indicating partial loss of this cell type in the KO mice. SurvivingPurkinje cells displayed spheroids that were close to Purkinje cell bodies (Supplemental Fig.2).
Quantitative Gait AnalysisAt the age of 6 months there was no difference between the WT and KO mice in any of thegait parameters (Table). However, at age 19 months KO mice showed an increasedincongruity coefficient (IC) for left and right stride lengths (p < 0.05). Left and right stridelengths were closely related in WT mice but there was much more incongruity in the KOmice. Scatter plot and respective regression lines illustrate this difference (Fig. 5). A similartrend occurred in the relationship between front strides and hind strides (Table). Theseresults indicate that aging KO mice show increasingly inharmonious gait dynamics anduncoordinated gait; they also displayed a higher number of errors in their treadmill walkingat both ages (data not shown), as described previously (10).
Enzyme Replacement TherapyShort-term, high-dose treatment with recombinant human LAMAN resulted in partialcorrection of the primary storage of oligosaccharides in different brain regions (10). Here,we investigated the effect of this ERT regimen on the cerebellar phenotype. Detailed
Damme et al. Page 6
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

examination of the cerebellum by thin layer chromatography did not reveal any effects onoligomannose storage after 4 injections with 500 U per kg body weight LAMAN, althoughhigh amounts of LAMAN activity were present in this brain region (Supplemental Fig. 3).No obvious alterations in the size or number of storage vacuoles were observed for thePurkinje cells (Supplemental Fig. 3B). To evaluate changes in microglia activation,astrogliosis and secondary storage of lipids in response to the ERT, IHC and Western blotswere performed.
Untreated KO mice displayed microglia of the amoeboid, phagocytic shape, whereas in thebrains of ERT-treated mice, CD68-positive microglia were reduced slightly in number andhad less of the amoeboid shape but with fine processes (Fig. 6A, first panel). Lamp1immunoreactivity was markedly reduced in all LAMAN-treated animals (Fig. 6B, upperpanel) and large Lamp1-positive inclusions in the molecular layer, probably located inmicroglial cells, disappeared almost completely (Fig. 6A, second panel). After ERT,hypertrophy of Bergmann glia with processes ranging deep into the molecular layer were atleast partially normalized to the WT level, as demonstrated by GFAP immunostaining (Fig.6A, third panel).
Finally, we evaluated the ERT effects on the secondary storage of lipids in the cerebellumand on NPC2, a soluble lysosomal protein mediating the export of free cholesterol fromlysosomes. NPC2 was recently shown to be hyperglycosylated in KO mice due to impairedprocessing of its glycan moiety by missing LAMAN activity (23). Filipin labelling of freecholesterol did not change after high dose ERT regimen (Fig. 6A, lowermost panel) and wasaccompanied by persistent storage of GM1 gangliosides (data not shown). Thehyperglycosylation of NPC2 also did not change in KO mice after ERT (Fig. 6B, lowermostpanel), suggesting insufficient amounts of active LAMAN for glycan trimming.
DISCUSSIONUnderstanding the neuropathological characteristics of α-mannosidosis will be important forfuture ERT trials in humans. The present study characterizes cerebellar pathology as a futureoutcome measure to assess therapeutic efficacy. Neuropathological changes occurredespecially in the molecular layer of the cerebellum, the location of highest oligosaccharideaccumulation in the brain. Progressive gait disturbances in aged KO mice indicate that gaitimpairments are prominent features in the mouse model, as well as in human patients.Pathological changes further included secondary accumulation of GM1 ganglioside, freecholesterol and lipophilic autofluorescent material that was partially engulfed by infiltratedCD68/ F4/80-positive macrophages. Regionally restricted gliosis of Bergman glia was foundin regions that were devoid of Purkinje cells, and surviving Purkinje cells showed dendriticswelling and axonal spheroids.
Pathogenesis of α-MannosidosisAlthough no detailed neuropathological reports are available for cases of human of α-mannosidosis, different animal models have been described to examine the pathogenesis ofthe disease (7,24–26). Previous studies focussed on the hippocampus (10) and did not revealsignificant activation of microglia and astroglia; however, the investigated cohort of micewas younger (>3 months) than those in the present study.
The clinical disease course described here is more subtle than that observed in cattle, catsand guinea pigs with prominent cerebellar atrophy and progressive motor dysfunctions(27,28). The disease in cats is most prominent and leads to premature death (29). Despite thelack of demyelination in the mouse model and different patterns of brain ganglioside andcholesterol accumulation, overall histopathological courses of the disease are similar in all
Damme et al. Page 7
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

animal models. The comparison of the clinical and histopathological course of the diseaseacross different animal models suggests that loss of Purkinje cells contributes to the severityof the disease. Accordingly, α-mannosidosis cats show the most rapid loss of this cell type(30), whereas later onset loss is obvious in guinea pigs (27), and there is only mild Purkinjecell death in mice. Thus, factors other than storage in the different species must be crucialfor the survival of affected cells.
A possible explanation for this obvious discrepancy could be the difference in lysosomaloligosaccharide degradation between cats and cattle on one hand, and rodents and humanson the other. In contrast to cattle and cats, rodents and humans express a lysosomalchitobiose and a second lysosomal α-mannosidase (31) that is specific for cleavage of theα-1,6-mannosidic core of glycoproteins (3,32). These differences in enzyme expression leadto considerably different primary storage products (33,34), and possibly to a differentpathology between species.
Lipid StorageGanglioside and cholesterol storage are prominent features in LSDs with neurologicalinvolvement. They have been described in LSDs with primary defects in lipid-degradation,gangliosidoses and Niemann-Pick type C, but also in LSDs unrelated to lipid-degradation,such as mucopolysaccharidoses and glycoproteinoses (15). Secondary storage ofgangliosides and cholesterol is well documented in the animal models of α-mannosidosis(27,35,36). Interestingly, α-mannosidosis cats, guinea pigs and mice show a different patternof accumulated gangliosides and cholesterol. In cats, GM2 storage predominates over GM3whereas guinea pigs store mainly GM3. In both animal models, GM1 storage has not beenreported and cholesterol accumulation appears after that of ganglioside storage (27). Incontrast, KO mouse brains display prominent storage of GM1 but not of GM2 and GM3.Cholesterol was only found to be accumulated in the molecular layer of the cerebellum,where it coincided with GM1 storage. These data indicate that in mice, cholesterol storage isnot secondary to that of GM2 and/or GM3 gangliosides as suggested for the cat and guineapig models, but rather depends on GM1. Further developmental analyses have to clarifywhether GM1 storage in the cerebellum is secondary to cholesterol or vice versa. Recentdata suggest that LAMAN is involved in the trimming of lysosomal glycoproteins includingthe cholesterol binding enzyme NPC2 (23) and in mouse tissues deficient for α-mannosidase, NPC2 is hyperglycosylated. Increased glycosylation of NPC2 slows thecholesterol transfer rate of the protein (37), suggesting that this effect could alter thecholesterol and ganglioside metabolism in LAMAN deficient tissues. So far, NPC2hyperglycosylation has only been shown in the mouse model of α-mannosidosis and furtherstudies have to clarify how this protein contributes to the disturbed lipid metabolism in theother animal models for α-mannosidosis.
Astrogliosis and InflammationCNS inflammation (i.e. activation of astrocytes and microglia) is a common feature ofneurodegenerative diseases including LSDs (38–41). Recent publications describe cerebellarspecific pathological alterations in patients and in different mouse models of neuronal ceroidlipofuscinosis (19–21), including localized astrogliosis of Bergmann glia. Patches ofhypertrophic Bergman glia were found in close proximity to degenerated Purkinje cells orbreaches within the Purkinje layer (19,20), alterations that resemble those found in thepresent study. These results support the idea that impaired function and/or degeneration ofPurkinje cells leads to a restricted astrogliosis of the functional closely linked Bergmannglia. However, we cannot completely rule out the possibility of deleterious effects of theactivated Bergmann glia on the surrounding Purkinje cells, leading to their impairedfunction. In this regard it should be noted that mouse models for Niemann-Pick C disease
Damme et al. Page 8
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

that are deficient for either NPC1 or NPC2, show a similar distribution of accumulatedcholesterol within the cerebellum although the NPC proteins are differentially expressed inBergmann glia and Purkinje cells (12), respectively, highlighting the crosstalk between bothcell types.
Microglia activation is well documented for several LSD mouse models and patients (40–43), but the mechanism of induction of this inflammatory response is unclear. In principle,self-activation of microglia due to the accumulation of primary or secondary storagematerial as well as exogenous stimuli of inflammation and/or phagocytosis is possible.Macrophages are highly active in the catabolism of mannose-containing glycoproteins and,therefore, are the most prominently affected cell type in α-mannosidosis mice, at least inperipheral tissues (7). The presence of undegraded, autofluorescent material within brainmicroglia argues for only partial clearance of phagocytosed lipophilic material due todefects in lysosomal breakdown. Therefore, it is likely that the prominent accumulation ofundegraded substrates within CNS resident macrophages triggers their self-activation.However, the focal distribution of highly activated microglia / phagocytes within themolecular layer of the KO cerebellum rather suggests an exogenous activation of microgliarather than self-activation. In mouse models of GM1 and GM2 gangliosidoses, CNSinflammation is prominent and the reduction of these stored gangliosides decreasedmicroglia activation in vivo (40), whereas mixed brain gangliosides and GM1 alone caninduce microglia ramification in vitro (44), indicating that the amount of stored gangliosidesin the brain is crucial for microglia activation possibly via toll-like receptors (45,46). Wetherefore speculate that in α-mannosidosis mice, GM1 storage is the main factor stimulatingmicroglia activation but our data suggest that both exogenous stimuli as well as endogenousstorage likely contribute to the inflammatory responses.
Gait AnalysisIn accordance with our previous observations, the youngest group of KO mice displayedmore walking errors on the treadmill device (10). Previously employed methods andmeasures failed to demonstrate neuromotor dysfunction in these mice (8,9). Here also, 6-month-old KO mice did not show alterations in any of the measured gait variables or in theirinterrelationships. This could mean that the observed treadmill errors might not be attributedto uncoordinated gait performance proper, but rather to deficits in gross ambulatory ability,tardiness, or procedural learning defects. Treadmill performance is typically impaired incerebellum-lesioned mice (47,48). The observed defect in the youngest mice could representa decrease in cerebellum-dependent gait adaptability, since treadmill walking mechanicssubstantially differ from normal over ground locomotion in rodents (49,50). Blanz et alreported amelioration of treadmill performance in LAMAN KO mice after high-dose ERT inthe absence of effects on Purkinje and granule cells, which might to indicate that reductionof cerebral storage, reinstates compensatory neural mechanisms (10).
Gait and coordinated movements obviously further deteriorated as the mice grew older andaged KO mice showed reduced congruence of stride lengths. Significant incongruityoccurred in contralateral stride lengths, whereas concomitant tendencies appeared in front-versus-hind stride lengths. Divergence of normally concurrent gait variables was alsoreported in another LSD murine model (51). Notably, mice with electrolytic lesions of thecerebellum show a similar gait profile (unpublished observations during validation of thetreadmill device), and α-mannosidosis guinea pigs show gait changes that might be theconsequence of cerebellar dysfunction and spinal cord pathology (52). The presentlydescribed progressive degeneration of Purkinje cells following metabolic perturbation andlysosomal storage may underlie the progressive decline of coordinated movements,neuromotor adaptability and gait in KO mice.
Damme et al. Page 9
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

ERTIt has previously been shown that short-term ERT with high doses of recombinant LAMANcan reduce storage and reverse the ataxic phenotype of α-mannosidosis mice (10). We usedthe same treatment regimen to investigate this therapeutic effect on the cerebellarneuropathology. Even though the cerebellum showed highest uptake of recombinantenzyme, storage reduction in the cerebellum was not obvious with storage vacuolespersisting in Purkinje cells. Despite the limited effect on cerebellar neurons, Lamp1immunoreactivity (most probably located to microglia) was markedly reduced and anti-CD68 and -GFAP staining revealed a partial effect on microglia and Bergmann gliaactivation, respectively. These data suggest an uptake of recombinant LAMAN either intoglial cells or a decrease in exogenously induced activation. Analyses of the glycan structuresof recombinant LAMAN revealed a low degree of phosphorylation coinciding with cellularLAMAN uptake that has been shown to be at least partially independent of mannose 6-phosphate (10). Because glia express several carbohydrate specific receptors, we speculatethat some of the recombinant enzyme was taken up preferentially by these cells leading to apartial decrease in Lamp1 expression and glial activation. The precise mechanisms ofLAMAN uptake into neural cells has to be investigated in detail and will be object of futureresearch. In contrast to the observed normalization of Lamp1 and the slight reduction of glialactivation, cholesterol and GM1 accumulation and NPC2 glycosylation did not respond toERT, suggesting that the short duration of the treatment may limit the potential effect ofERT on neuronal storage reduction. The effect of long-term ERT on neuronal storage couldnot be assessed since humoral immune responses following frequent injections of therecombinant enzyme precluded longer ERT treatment in the classical α-mannosidosis KOmouse model. To overcome these limitations, we are currently generating an immune-tolerant mouse model. Induction of immune-tolerance by transgenic approaches has beenshown to be a valuable tool for long-term enzyme treatment in other LSDs (53,54).
In summary, cerebellar alterations including loss of Purkinje cells, inflammation and motordeficits are common to all α-mannosidosis animal models and relate to the observedpathology in human patients. The observed cerebellar alterations in α-mannosidosis micemay underlie the progressive motor deficits that occur in these mice as they age. Short-termhigh-dose ERT effectively corrected some aspects of cerebellar pathology in α-mannosidosismice including macrophage activation, astrogliosis and Lamp1 upregulation, but only long-term studies will clarify whether ERT has the potential to ameliorate completely the CNSpathology. The observed cerebellar pathology and ataxia characterized herein may be usedin forthcoming ERT studies as possible therapeutic outcome measures.
Supplementary MaterialRefer to Web version on PubMed Central for supplementary material.
AcknowledgmentsWe thank Ellen Eckermann-Felkl and Inez Götting for excellent technical assistance and Nafeeza Ali, CristinDavidson and Matt Micsenyi for helping with filipin and ganglioside immunohistochemistry and their great supportduring the stay in the Walkley Lab. This publication does not necessarily represent the opinion of the EuropeanCommunity and the Community is not responsible for any use that might be made of the data appearing in thispublication.
This work was supported by the HUE-MAN consortium (European Commission FP VI contract LHSM-CT-2006-018692)), the EMBO association (ASTF 183.00-2008) and by the NIH (HD045561) (SUW).
Damme et al. Page 10
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

REFERENCES1. Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005; 128:413–431. [PubMed: 15686451]2. al Daher S, de Gasperi R, Daniel P, et al. The substrate-specificity of human lysosomal alpha-D-
mannosidase in relation to genetic alpha-mannosidosis. Biochem J. 1991; 277:743–751. [PubMed:1872811]
3. Aronson NN Jr, Kuranda MJ. Lysosomal degradation of Asn-linked glycoproteins. Faseb J. 1989;3:2615–2622. [PubMed: 2531691]
4. Malm D, Nilssen O. Alpha-mannosidosis. Orphanet J Rare Dis. 2008; 3:21. [PubMed: 18651971]5. Dietemann JL, Filippi de la Palavesa MM, Tranchant C, et al. MR findings in mannosidosis.
Neuroradiology. 1990; 32:485–487. [PubMed: 2287376]6. Niemann S, Beck M, Seidel G, et al. Neurology of adult alpha-mannosidosis. J Neurol Neurosurg
Psychiatry. 1996; 61:116–117. [PubMed: 8676143]7. Stinchi S, Lullmann-Rauch R, Hartmann D, et al. Targeted disruption of the lysosomal alpha-
mannosidase gene results in mice resembling a mild form of human alpha-mannosidosis. Hum MolGenet. 1999; 8:1365–1372. [PubMed: 10400983]
8. Caeyenberghs K, Balschun D, Roces DP, et al. Multivariate neurocognitive and emotional profile ofa mannosidosis murine model for therapy assessment. Neurobiol Dis. 2006; 23:422–432. [PubMed:16766199]
9. D'Hooge R, Lullmann-Rauch R, Beckers T, et al. Neurocognitive and psychotiform behavioralalterations and enhanced hippocampal long-term potentiation in transgenic mice displayingneuropathological features of human alpha-mannosidosis. J Neurosci. 2005; 25:6539–6549.[PubMed: 16014715]
10. Blanz J, Stroobants S, Lullmann-Rauch R, et al. Reversal of peripheral and central neural storageand ataxia after recombinant enzyme replacement therapy in alpha-mannosidosis mice. Hum MolGenet. 2008; 17:3437–3445. [PubMed: 18713755]
11. Roces DP, Lullmann-Rauch R, Peng J, et al. Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study. Hum Mol Genet. 2004; 13:1979–1988. [PubMed:15269179]
12. Ong WY, Sundaram RK, Huang E, et al. Neuronal localization and association of Niemann PickC2 protein (HE1/NPC2) with the postsynaptic density. Neuroscience. 2004; 128:561–570.[PubMed: 15381285]
13. Matzner U, Lullmann-Rauch R, Stroobants S, et al. Enzyme replacement improves ataxic gait andcentral nervous system histopathology in a mouse model of metachromatic leukodystrophy. MolTher. 2009; 17:600–606. [PubMed: 19174759]
14. Leroy T, Stroobants S, Aerts JM, et al. Automatic analysis of altered gait in arylsulphatase A-deficient mice in the open field. Behav Res Methods. 2009; 41:787–794. [PubMed: 19587193]
15. Walkley SU. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin CellDev Biol. 2004; 15:433–444. [PubMed: 15207833]
16. McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol,gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders.J Comp Neurol. 2004; 480:415–426. [PubMed: 15558784]
17. Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence influorescently labeled tissue. J Histochem Cytochem. 1999; 47:719–730. [PubMed: 10330448]
18. Holness CL, da Silva RP, Fawcett J, et al. Macrosialin, a mouse macrophage-restrictedglycoprotein, is a member of the lamp/lgp family. J Biol Chem. 1993; 268:9661–9666. [PubMed:8486654]
19. Chang M, Cooper JD, Sleat DE, et al. Intraventricular enzyme replacement improves diseasephenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. 2008;16:649–656. [PubMed: 18362923]
20. Macauley SL, Wozniak DF, Kielar C, et al. Cerebellar pathology and motor deficits in thepalmitoyl protein thioesterase 1-deficient mouse. Exp Neurol. 2009; 217:124–135. [PubMed:19416667]
Damme et al. Page 11
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

21. Weimer JM, Benedict JW, Getty AL, et al. Cerebellar defects in a mouse model of juvenileneuronal ceroid lipofuscinosis. Brain Res. 2009; 1266:93–107. [PubMed: 19230832]
22. Takahashi-Iwanaga H, Kondo H, Yamakuni T, et al. An immunohistochemical study on theontogeny of cells immunoreactive for spot 35 protein, a novel Purkinje cell-specific protein, in therat cerebellum. Brain Res. 1986; 394:225–231. [PubMed: 3533215]
23. Damme M, Morelle W, Schmidt B, et al. Impaired lysosomal trimming of N-linkedoligosaccharides leads to hyperglycosylation of native lysosomal proteins in mice with alpha-mannosidosis. Mol Cell Biol. 2010; 30:273–283. [PubMed: 19884343]
24. Crawley AC, Jones MZ, Bonning LE, et al. Alpha-mannosidosis in the guinea pig: a new animalmodel for lysosomal storage disorders. Pediatr Res. 1999; 46:501–509. [PubMed: 10541310]
25. Hocking JD, Jolly RD, Batt RD. Deficiency of alpha-mannosidase in Angus cattle. An inheritedlysosomal storage disease. Biochem J. 1972; 128:69–78. [PubMed: 4673577]
26. Jolly RD, Slack PM, Winter PJ, et al. Mannosidosis: patterns of storage and urinary excretion ofoligosaccharides in the bovine model. Aust J Exp Biol Med Sci. 1980; 58:421–428. [PubMed:7436888]
27. Crawley AC, Walkley SU. Developmental analysis of CNS pathology in the lysosomal storagedisease alpha-mannosidosis. J Neuropathol Exp Neurol. 2007; 66:687–697. [PubMed: 17882013]
28. Walkley SU, Baker HJ, Rattazzi MC, et al. Neuroaxonal dystrophy in neuronal storage disorders:evidence for major GABAergic neuron involvement. J Neurol Sci. 1991; 104:1–8. [PubMed:1919594]
29. Vite CH, McGowan JC, Braund KG, et al. Histopathology, electrodiagnostic testing, and magneticresonance imaging show significant peripheral and central nervous system myelin abnormalities inthe cat model of alpha-mannosidosis. J Neuropathol Exp Neurol. 2001; 60:817–828. [PubMed:11487056]
30. Walkley SU. Neurobiology and cellular pathogenesis of glycolipid storage diseases. Philos Trans RSoc Lond B Biol Sci. 2003; 358:893–904. [PubMed: 12803923]
31. Daniel PF, Evans JE, De Gasperi R, et al. A human lysosomal alpha(1----6)-mannosidase active onthe branched trimannosyl core of complex glycans. Glycobiology. 1992; 2:327–336. [PubMed:1421754]
32. Daniel PF, Winchester B, Warren CD. Mammalian alpha-mannosidases--multiple forms but acommon purpose? Glycobiology. 1994; 4:551–566. [PubMed: 7881169]
33. Abraham D, Blakemore WF, Jolly RD, et al. The catabolism of mammalian glycoproteins.Comparison of the storage products in bovine, feline and human mannosidosis. Biochem J. 1983;215:573–579. [PubMed: 6661184]
34. Michalski JC, Klein A. Glycoprotein lysosomal storage disorders: alpha- and beta-mannosidosis,fucosidosis and alpha-N-acetylgalactosaminidase deficiency. Biochim Biophys Acta. 1999;1455:69–84. [PubMed: 10571005]
35. Goodman LA, Livingston PO, Walkley SU. Ectopic dendrites occur only on cortical pyramidalcells containing elevated GM2 ganglioside in alpha-mannosidosis. Proc Natl Acad Sci U S A.1991; 88:11330–11334. [PubMed: 1763046]
36. Walkley SU, Siegel DA, Dobrenis K. GM2 ganglioside and pyramidal neuron dendritogenesis.Neurochem Res. 1995; 20:1287–1299. [PubMed: 8786814]
37. Cheruku SR, Xu Z, Dutia R, et al. Mechanism of cholesterol transfer from the Niemann-Pick typeC2 protein to model membranes supports a role in lysosomal cholesterol transport. J Biol Chem.2006; 281:31594–31604. [PubMed: 16606609]
38. Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronicneurodegeneration. Nat Rev Immunol. 2007; 7:161–167. [PubMed: 17220915]
39. Jeyakumar M, Dwek RA, Butters TD, et al. Storage solutions: treating lysosomal disorders of thebrain. Nat Rev Neurosci. 2005; 6:713–725. [PubMed: 16049428]
40. Jeyakumar M, Thomas R, Elliot-Smith E, et al. Central nervous system inflammation is a hallmarkof pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003; 126:974–987.[PubMed: 12615653]
Damme et al. Page 12
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

41. Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoffdisease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci U S A. 2000;97:10954–10959. [PubMed: 11005868]
42. Baudry M, Yao Y, Simmons D, et al. Postnatal development of inflammation in a murine model ofNiemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. ExpNeurol. 2003; 184:887–903. [PubMed: 14769381]
43. Ohmi K, Greenberg DS, Rajavel KS, et al. Activated microglia in cortex of mouse models ofmucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003; 100:1902–1907. [PubMed:12576554]
44. Park JY, Kim HY, Jou I, et al. GM1 induces p38 and microtubule dependent ramification of ratprimary microglia in vitro. Brain Res. 2008; 1244:13–23. [PubMed: 18930716]
45. Jou I, Lee JH, Park SY, et al. Gangliosides trigger inflammatory responses via TLR4 in brain glia.Am J Pathol. 2006; 168:1619–1630. [PubMed: 16651628]
46. Yoon HJ, Jeon SB, Suk K, et al. Contribution of TLR2 to the initiation of ganglioside-triggeredinflammatory signaling. Mol Cells. 2008; 25:99–104. [PubMed: 18319620]
47. Le Marec N, Caston J, Lalonde R. Impaired motor skills on static and mobile beams in lurchermutant mice. Exp Brain Res. 1997; 116:131–138. [PubMed: 9305822]
48. Le Marec N, Lalonde R. Treadmill performance of mice with cerebellar lesions: 2. Lurcher mutantmice. Neurobiol Learn Mem. 2000; 73:195–206. [PubMed: 10775492]
49. Herbin M, Hackert R, Gasc JP, et al. Gait parameters of treadmill versus overground locomotion inmouse. Behav Brain Res. 2007; 181:173–179. [PubMed: 17521749]
50. Pereira JE, Cabrita AM, Filipe VM, et al. A comparison analysis of hindlimb kinematics duringoverground and treadmill locomotion in rats. Behav Brain Res. 2006; 172:212–218. [PubMed:16777243]
51. Stroobants S, Leroy T, Eckhardt M, et al. Early signs of neurolipidosis-related behaviouralalterations in a murine model of metachromatic leukodystrophy. Behav Brain Res. 2008; 189:306–316. [PubMed: 18336930]
52. Robinson AJ, Crawley AC, Auclair D, et al. Behavioural characterisation of the alpha-mannosidosis guinea pig. Behav Brain Res. 2008; 186:176–184. [PubMed: 17889945]
53. Matzner U, Matthes F, Herbst E, et al. Induction of tolerance to human arylsulfatase A in a mousemodel of metachromatic leukodystrophy. Mol Med. 2007; 13:471–479. [PubMed: 17660863]
54. Sands MS, Vogler CA, Ohlemiller KK, et al. Biodistribution, kinetics, and efficacy of highlyphosphorylated and non-phosphorylated beta-glucuronidase in the murine model ofmucopolysaccharidosis VII. J Biol Chem. 2001; 276:43160–43165. [PubMed: 11562370]
Damme et al. Page 13
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 1.Thin layer chromatography of extracted oligosaccharides from whole brain of wild-type andα-mannosidosis (KO) mice and of different brain regions of 2 KO mice. The cerebellumshows the highest amount of storage material. Oligosaccharides with one N-acetylglucosamine and 2 – 9 mannose residues are labelled as M2 – M9. Arrows markorcinol-positive bands insensitive to lysosomal hydrolase α-mannosidase.
Damme et al. Page 14
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 2.(A–F) Vibratome sections of formalin-fixed brains from 11-month-old wild-type (WT; A,C) and α-mannosidosis (KO) (B, D–F) mice immunostained for GM1-gangliosides (brown)with Nissl counterstain (nuclei, blue). GM1-positive vesicles accumulate in the molecularlayer (ml) of the KO mouse cerebellum (B, D, F; black arrows), and in neurons within themidbrain (mb, E). No vesicular GM1 accumulation was found in the cerebellar granularlayer (gl) or white matter (wm). (G–J) Filipin-positive free cholesterol accumulates in theKO mouse brain specifically in the molecular layer of the cerebellum (H–J), but not in WTbrain (G, I). Scale bars: A–B, G–H, 100 µm; C–F, I–J, 50 µm.
Damme et al. Page 15
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 3.Macrophage/microglia in α-mannosidosis (KO) mice. (A, B, D) Vibratome sections offormalin fixed brains from 12-month-old wild-type and KO mice were analyzed foractivation of macrophages by either 3, 3’ diaminobenzidine (DAB) immunohistochemistry(A) or immunofluorescence (D) using anti-CD68 (A) or F4/80 (D) antibodies.Autofluorescence images (B, D) were taken with excitation wavelengths of 488 nm, 633 nmand UV light. In the KO brain anti-CD68 strongly labels activated microglia that are foundpredominantly in the cerebellar molecular layer (ml) where there is abundant storage ofautofluorescent material (B, upper panel) (pcl = Purkinje cell layer; gl = granular layer).Cholesterol-containing autofluorescence material demonstrated by Filipin staining (B, lowerpanel in red) is taken up by F4/80-positive macrophages (D, green). (C) Electronmicrographs from macrophages in the cerebellar molecular layer of a KO mouse displaycytoplasmic (left) multilamellar (*) and (right) lipofuscin like (❋) material. Scale bars: A(upper panel): 400 µm, (lower panel) 50 µm; B (upper panel): 500 µm, (lower panel) 15 µm;D: 50 µm.
Damme et al. Page 16
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
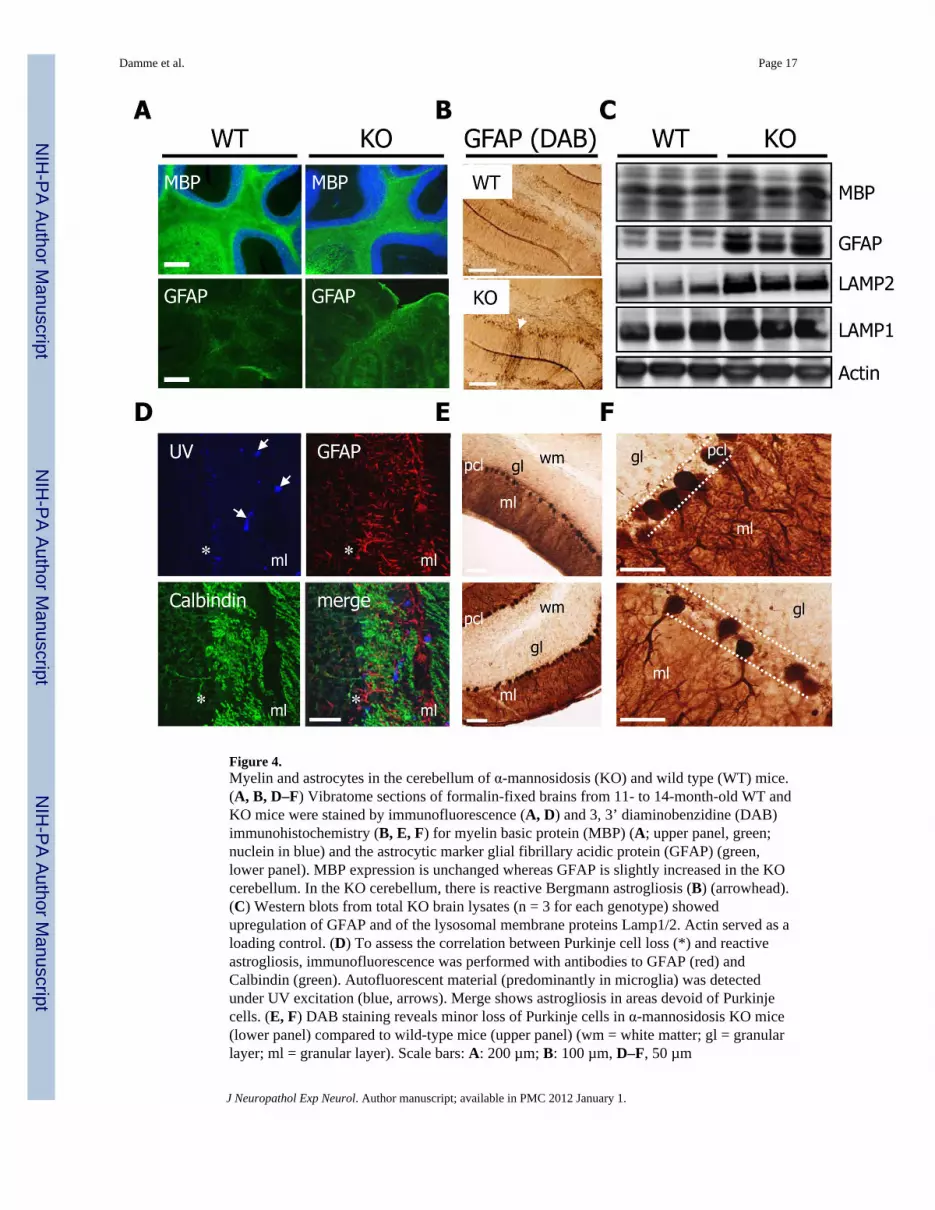
Figure 4.Myelin and astrocytes in the cerebellum of α-mannosidosis (KO) and wild type (WT) mice.(A, B, D–F) Vibratome sections of formalin-fixed brains from 11- to 14-month-old WT andKO mice were stained by immunofluorescence (A, D) and 3, 3’ diaminobenzidine (DAB)immunohistochemistry (B, E, F) for myelin basic protein (MBP) (A; upper panel, green;nuclein in blue) and the astrocytic marker glial fibrillary acidic protein (GFAP) (green,lower panel). MBP expression is unchanged whereas GFAP is slightly increased in the KOcerebellum. In the KO cerebellum, there is reactive Bergmann astrogliosis (B) (arrowhead).(C) Western blots from total KO brain lysates (n = 3 for each genotype) showedupregulation of GFAP and of the lysosomal membrane proteins Lamp1/2. Actin served as aloading control. (D) To assess the correlation between Purkinje cell loss (*) and reactiveastrogliosis, immunofluorescence was performed with antibodies to GFAP (red) andCalbindin (green). Autofluorescent material (predominantly in microglia) was detectedunder UV excitation (blue, arrows). Merge shows astrogliosis in areas devoid of Purkinjecells. (E, F) DAB staining reveals minor loss of Purkinje cells in α-mannosidosis KO mice(lower panel) compared to wild-type mice (upper panel) (wm = white matter; gl = granularlayer; ml = granular layer). Scale bars: A: 200 µm; B: 100 µm, D–F, 50 µm
Damme et al. Page 17
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
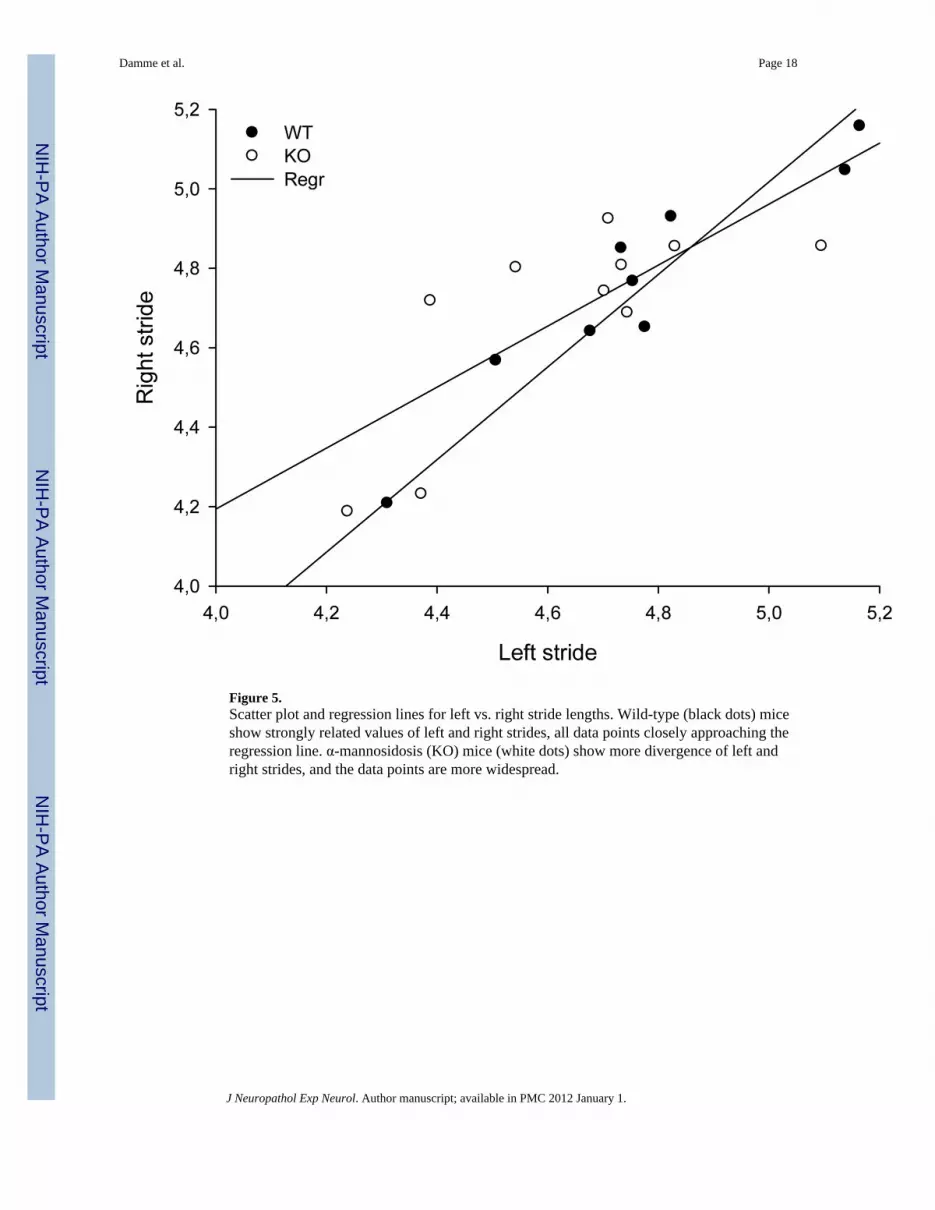
Figure 5.Scatter plot and regression lines for left vs. right stride lengths. Wild-type (black dots) miceshow strongly related values of left and right strides, all data points closely approaching theregression line. α-mannosidosis (KO) mice (white dots) show more divergence of left andright strides, and the data points are more widespread.
Damme et al. Page 18
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 6.Effects of enzyme replacement therapy (ERT) in α-mannosidase (KO) mice. (A) Vibratomesections of cerebella from wild-type (WT) (left column), KO (middle column) and ERT-treated KO mice (right column) were stained for CD68, Lamp1 and glial fibrillary acidicprotein (GFAP). For detection of cholesterol, sections were stained with Filipin andevaluated by fluorescence (lowermost panels). Lamp1 immunoreactivity and astrogliosis ofBergmann glia were nearly comparable to those in WT mice after ERT in KO mice; slighteffects of ERT were observed on microglia (CD68). No differences of cholesterolaccumulation were observed after ERT. (B) Western blots of whole brain lysates (WT, KOand ERT treated KO; n = 2 each genotype), showed almost complete normalization of
Damme et al. Page 19
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Lamp1-specific bands after ERT and only faint effects on the amount of the cholesterolbinding protein NPC2. No effects on the molecular weight differences between WT and KOmice were detected after ERT. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH)served as loading control. Scale bars: A: 200 µm; B: 100 µm.
Damme et al. Page 20
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Damme et al. Page 21
Table
Gait Parameter Values for Wild-Type and α-Mannosidosis (Knockout) Mice at ages 6 and 19 Months
6 months 19 months
WT KO WT KO
(n = 20) (n = 17) (n = 10) (n = 11)
FB (cm) 1.57 1.56 1.67 1.76
HB (cm) 2.56 2.60 2.73 2.76
LFHD (cm) 1.36 1.46 1.64 1.56
RFHD (cm) 1.36 1.45 1.63 1.31
LFS (cm) 4.87 4.65 4.69 4.30
RFS (cm) 4.77 4.68 4.71 4.63
LHS (cm) 4.85 4.83 4.70 4.67
RHS (cm) 4.96 4.76 4.61 4.79
IC (FB-HB) 0.88 0.77 0.55 0.80 (p = 0.16)
IC (LFHD-RFHD) 0.75 0.69 0.61 0.58
IC (LS-RS) 0.27 0.31 0.21 0.92* (p < 0.05)
IC (FS-HS) 0.21 0.22 0.28 0.90 (p = 0.06)
Gait variables including base-width (distance between contralateral paws), and stride length (distance between 2 subsequent prints of the samepaw) were extracted from video data using an automated algorithm (14).
Abbreviations: WT, wild type, KO, knockout, FB = Front Base (distance between front paws); HB = Hind Base (distance between hind paws;LFHD = left front/hind distance (distance between front paw print and subsequent hind paw print); RFHD = right front/hind distance; lfs = leftfront limb stride; rfs = right front limb stride; lhs = left hind limb stride; rhs = right hind limb stride; IC = incongruity coefficient; ls, left strides; rs,right strides; fs, front strides; hs, hind strides.
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 January 1.
Related Documents











