Cell surface receptors activate p21-activated kinase 1 via multiple Ras and PI3-kinase-dependent pathways Raymond E. Menard a , Raymond R. Mattingly a,b, * a Department of Pharmacology, Wayne State University, 540 E. Canfield, Room 6326, Detroit, MI 48201, USA b Barbara Ann Karmanos Cancer Institute, 540 E. Canfield, Detroit, MI, USA Received 10 February 2003; received in revised form 15 April 2003; accepted 12 May 2003 Abstract p21-activated kinases (PAKs) were the first identified mammalian members of a growing family of Ste20-like serine–threonine protein kinases. In this study, we show that PAK1 can be stimulated by carbachol, lysophosphatidic acid (LPA), epidermal growth factor (EGF), and phorbol 12-myristate 13-acetate (PMA) by multiple independent and overlapping pathways. Dominant-negative Ras, Rac, and Cdc42 inhibited PAK1 activation by all of these agonists, while active Rac1 and Cdc42 were sufficient to maximally activate PAK1 in the absence of any treatment. Active Ras induced only a weak activation of PAK1 that could be potentiated by muscarinic receptor stimulation. Studies using inhibitors of the EGF receptor tyrosine kinase, phosphatidylinositol 3-kinase (PI3-kinase) and protein kinase C (PKC) revealed that all of the cell surface agonists could activate PAK1 through pathways independent of PKC, that EGF stimulated a PI3-kinase dependent pathway to stimulate PAK1, and that muscarinic receptor stimulation of PAK1 was predominantly mediated through this EGF-R-dependent mechanism. Activation of PAK1 by LPA was independent of PI3-kinase and the EGF receptor, but was inhibited by dominant-negative RhoA. These results identify multiple Ras-dependent pathways to activation of PAK1. D 2003 Elsevier Inc. All rights reserved. Keywords: p21-activated kinase; Protein kinase C; Muscarinic receptor; Epidermal growth factor receptor 1. Introduction Cell transformation requires modulation of signals from oncogenes to stimulate proliferation, actin cytoskeleton rearrangement, and cell survival. Mitogen-activated protein kinase (MAPK) cascades control the expression of genes that are important for the regulation of these cell functions. In mammalian cells, several parallel MAPK pathways have been extensively studied in recent years [1]. The ERK/ MAPK cascade is regulated by Ras GTPase in response to agonists of tyrosine kinase and G protein-coupled receptors (GPCRs). The MAPK p38 and Jun kinase (JNK) cascades are involved in the stress response of mammalian cells. The JNK pathway has been shown to be activated by the Rho family GTPases Cdc42 and Rac1 [2]. The members of the Rho subfamily of small GTPases are thought to be primarily involved in the organization of the actin cytoskeleton; Rac regulates lamellipodia and then ruffling behavior, Rho controls stress fiber formation, and Cdc42 has been shown to control the formation of filopodia [3]. Rac- and Rho-controlled pathways involved in the organization of the actin cytoskeleton are distinct from those involved in cell transformation [3,4]. In Swiss 3T3 fibroblasts, Cdc42, Rac, and Rho have been placed in a hierarchical cascade, where Cdc42 activates Rac, which in turn activates Rho; additionally, Ras has been found to activate Rac [4–7]. The Rho family GTPases thus link plasma membrane receptors to the assembly and organiza- tion of the actin cytoskeleton. In a variety of cell types, including fibroblasts, extracellular stimuli have been shown to activate the Rho GTPase cascade at different points [8]. Addition of LPA to quiescent fibroblasts induces the for- mation of actin stress fibers, while growth factors such as PDGF and insulin stimulate polymerization of actin at the 0898-6568/$ - see front matter D 2003 Elsevier Inc. All rights reserved. doi:10.1016/S0898-6568(03)00087-1 Abbreviations: PAK, p21-activated kinase; MBP, myelin basic protein; LPA, lysophosphatidic acid; EGF, epidermal growth factor; PMA, phorbol 12-myristate 13-acetate; GPCR, G protein coupled receptor; MAPK, mitogen-activated protein kinase; HA, haemagglutinin; PAGE, polyacryla- mide gel electrophoresis; PI3-kinase, phosphoinositide 3-kinase; GAP, GTPase-activating protein. * Corresponding author. Department of Pharmacology, Wayne State University, 540 E. Canfield, Room 6326, Detroit, MI 48201, USA. Tel.: +1- 313-577-6022; fax: +1-313-577-6739. E-mail address: [email protected] (R.R. Mattingly). www.elsevier.com/locate/cellsig Cellular Signalling 15 (2003) 1099 – 1109

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

www.elsevier.com/locate/cellsig
Cellular Signalling 15 (2003) 1099–1109
Cell surface receptors activate p21-activated kinase 1 via multiple Ras and
PI3-kinase-dependent pathways
Raymond E. Menarda, Raymond R. Mattinglya,b,*
aDepartment of Pharmacology, Wayne State University, 540 E. Canfield, Room 6326, Detroit, MI 48201, USAbBarbara Ann Karmanos Cancer Institute, 540 E. Canfield, Detroit, MI, USA
Received 10 February 2003; received in revised form 15 April 2003; accepted 12 May 2003
Abstract
p21-activated kinases (PAKs) were the first identified mammalian members of a growing family of Ste20-like serine– threonine protein
kinases. In this study, we show that PAK1 can be stimulated by carbachol, lysophosphatidic acid (LPA), epidermal growth factor (EGF), and
phorbol 12-myristate 13-acetate (PMA) by multiple independent and overlapping pathways. Dominant-negative Ras, Rac, and Cdc42
inhibited PAK1 activation by all of these agonists, while active Rac1 and Cdc42 were sufficient to maximally activate PAK1 in the absence of
any treatment. Active Ras induced only a weak activation of PAK1 that could be potentiated by muscarinic receptor stimulation. Studies
using inhibitors of the EGF receptor tyrosine kinase, phosphatidylinositol 3-kinase (PI3-kinase) and protein kinase C (PKC) revealed that all
of the cell surface agonists could activate PAK1 through pathways independent of PKC, that EGF stimulated a PI3-kinase dependent pathway
to stimulate PAK1, and that muscarinic receptor stimulation of PAK1 was predominantly mediated through this EGF-R-dependent
mechanism. Activation of PAK1 by LPA was independent of PI3-kinase and the EGF receptor, but was inhibited by dominant-negative
RhoA. These results identify multiple Ras-dependent pathways to activation of PAK1.
D 2003 Elsevier Inc. All rights reserved.
Keywords: p21-activated kinase; Protein kinase C; Muscarinic receptor; Epidermal growth factor receptor
1. Introduction
Cell transformation requires modulation of signals from
oncogenes to stimulate proliferation, actin cytoskeleton
rearrangement, and cell survival. Mitogen-activated protein
kinase (MAPK) cascades control the expression of genes
that are important for the regulation of these cell functions.
In mammalian cells, several parallel MAPK pathways have
been extensively studied in recent years [1]. The ERK/
MAPK cascade is regulated by Ras GTPase in response to
agonists of tyrosine kinase and G protein-coupled receptors
(GPCRs). The MAPK p38 and Jun kinase (JNK) cascades
0898-6568/$ - see front matter D 2003 Elsevier Inc. All rights reserved.
doi:10.1016/S0898-6568(03)00087-1
Abbreviations: PAK, p21-activated kinase; MBP, myelin basic protein;
LPA, lysophosphatidic acid; EGF, epidermal growth factor; PMA, phorbol
12-myristate 13-acetate; GPCR, G protein coupled receptor; MAPK,
mitogen-activated protein kinase; HA, haemagglutinin; PAGE, polyacryla-
mide gel electrophoresis; PI3-kinase, phosphoinositide 3-kinase; GAP,
GTPase-activating protein.
* Corresponding author. Department of Pharmacology, Wayne State
University, 540 E. Canfield, Room 6326, Detroit, MI 48201, USA. Tel.: +1-
313-577-6022; fax: +1-313-577-6739.
E-mail address: [email protected] (R.R. Mattingly).
are involved in the stress response of mammalian cells. The
JNK pathway has been shown to be activated by the Rho
family GTPases Cdc42 and Rac1 [2].
The members of the Rho subfamily of small GTPases are
thought to be primarily involved in the organization of the
actin cytoskeleton; Rac regulates lamellipodia and then
ruffling behavior, Rho controls stress fiber formation, and
Cdc42 has been shown to control the formation of filopodia
[3]. Rac- and Rho-controlled pathways involved in the
organization of the actin cytoskeleton are distinct from
those involved in cell transformation [3,4]. In Swiss 3T3
fibroblasts, Cdc42, Rac, and Rho have been placed in a
hierarchical cascade, where Cdc42 activates Rac, which in
turn activates Rho; additionally, Ras has been found to
activate Rac [4–7]. The Rho family GTPases thus link
plasma membrane receptors to the assembly and organiza-
tion of the actin cytoskeleton. In a variety of cell types,
including fibroblasts, extracellular stimuli have been shown
to activate the Rho GTPase cascade at different points [8].
Addition of LPA to quiescent fibroblasts induces the for-
mation of actin stress fibers, while growth factors such as
PDGF and insulin stimulate polymerization of actin at the

R.E. Menard, R.R. Mattingly / Cellular Signalling 15 (2003) 1099–11091100
plasma membrane of many cell types to induce lamellipodia
formation and surface ruffling in a PI3-kinase-dependent
manner [6]. Active Rac and Cdc42 regulate distinct down-
stream signaling pathways by interacting with specific
effector proteins, including a family of serine–threonine
protein kinases termed PAKs (p21-activated kinases).
PAKs were the first identified mammalian members of a
growing family of Ste20-like serine– threonine protein
kinases. PAKs were found in screens for binding targets of
Rac and Cdc42 GTPases in rat brain [9]. This provided some
of the first evidence that Cdc42 and Rac act like other
traditional GTP-binding proteins to stimulate a target/effec-
tor protein. Additional studies revealed three proteins in
neutrophil cytosol of molecular size 62, 65 and 68 kDa,
which interacted in a GTP-dependent manner with Rac and
Cdc42, but not Rho [10]. Peptide sequencing of these
proteins showed identity to rat brain PAK65. It is generally
accepted that in mammalian tissues there are at least three
main PAK isoforms; PAK1, a 68-kDa protein that is highly
expressed in brain, muscle, and spleen; PAK2, a 62-kDa
protein with ubiquitous tissue distribution; and PAK3, a 65-
kDa protein that is expressed in the brain, but with a cell type
distribution that is different from that of PAK1 [11]. Three
additional PAKs have since been identified. These PAKs,
termed 4–6, have been classified as Group II PAKs [12–15].
All PAK proteins identified to date share a similar N-
terminal motif stretching over 18 amino acids that mediates
the interaction with Rac or Cdc42 and is referred to as CRIB
(Cdc42/Rac interactive binding domain) [16,17]. There are
many proteins from a wide range of species that contain a
CRIB homology domain, including WASP. WASP, the
Wiskott–Aldrich Syndrome Protein, may link Cdc42 to
the actin cytoskeleton. WASP binds to Cdc42 in a GTP-
dependent manner and has been shown to associate with
Nck, an SH3 domain containing adaptor protein [18]. The
adaptor protein Nck has also been shown to recruit PAK1 to
cell membranes and this membrane localization is sufficient
for PAK1 activation [19]. Autophosphorylation of PAK
blocks binding of Nck and PIX to PAK1, which provides
a way of regulating PAK interactions [20,21].
The PAK proteins also contain a highly homologous C-
terminal kinase domain [22]. PAKs 1 and 2 play similar roles
in cytoskeletal rearrangements and cellular signaling [23].
PAK2 is also involved in apoptotic cell death, following its
proteolytic cleavage by DEVD-sensitive caspases [24,25].
The role of PAKs in Raf1 activation has come to the forefront
in recent years. PAK3 is capable of phosphorylating Raf-1 on
serine 338 in vitro and in vivo [26], in a process that requires
the interaction of PI3-kinase and Ras [27]. PAK1 has also
been shown to be involved in Raf1 regulation via PI3-kinase
[28] and the interaction between active PAK1 and Raf1 is
necessary for Raf1 phosphorylation [29]. Microtubule integ-
rity also has been shown to regulate PAK and Raf1 activation
in a Ras-independent manner [30].
In this study, we delineated multiple overlapping and
independent pathways through which PAK1 could be acti-
vated. We stimulated COS7 cells, SKNSH neuroblastoma
cells and NIH-3T3 fibroblasts with agonists for G protein
coupled receptor pathways, with epidermal growth factor
(EGF), and with a cell permeable activator of protein kinase
C, phorbol 12-myristate 13-acetate (PMA). Our results
support a model where multiple signaling pathways con-
verge on the activation of PAK1 through G protein and
tyrosine kinase-dependent mechanisms.
2. Materials and methods
2.1. Plasmids and antibodies
Antibodies to the N and C terminus (N20 and C19) of
PAK1 were purchased from Santa Cruz Biotechnology. The
PhosphoPAK1 (Thr 423)/PAK2 (Thr 402) antibody was
purchased from Cell Signalling Technology. Plasmids used
in this study include, pCDhM2 [31], pRK7mycPAK1 pre-
pared from pCMV6mycPAK1 plasmid (that was a kind gift
from Gary Bokoch) by subcloning a BamH1/EcoR1 frag-
ment into pRK7, and pKH3 with inserts for RacN17,
Cdc42N17, Ras N17, RhoAN19, RacV12, Cdc42L61,
RasV12, and RhoAV14. The pKH3 plasmid expresses
inserts with a triple HA epitope tag at the N-terminus as
described previously [8,32].
2.2. Cell culture and transfection assays
COS7 cells, NIH-3T3 fibroblasts, and SKNSH neuro-
blastoma cells were cultured as described previously [32–
34]. COS7 cells were transfected by calcium phosphate
precipitation with 1 Ag pRK7myc-PAK1 plasmid, 1 AgpCDhM2, and 3 Ag test or vector control plasmids. Cells
were grown for 48 h after transfection and for cells treated
with LPA, serum starved for 24 h prior to treatment.
2.3. Agonist treatment and immunoprecipitation for COS7
transfectants
The COS7 cells were treated with 100 AM carbachol, 4
AM LPA, 200 nM PMA or 100 nM EGF for time points
ranging from 0 to 60 min. The maximal time to activation
was 5 min for EGF, 15 min for LPA and PMA and 30 min
for carbachol. For inhibition studies, pretreatment with 200
nM Wortmannin, 200 nM AG1478, or 5 AM bisindolylma-
leimide was done for 30 min prior to agonist treatment. The
medium was then aspirated, the cells washed twice with
cold PBS, and lysed on ice as described [35], except that 0.5
mM DTT, 20 mM h-glycerophosphate, 20 Ag/ml aprotinin,
20 Ag/ml leupeptin, 20 Ag/ml pepstatin, 1 mM PMSF, and 1
AM okadaic acid were added to the lysis buffer just prior to
use. Material was scraped into microfuge tubes and centri-
fuged at maximum speed for 10 min. The supernatant was
pre-cleared with 20 Al of a 50% suspension of Protein A
Sepharose beads and then re-centrifuged.
R.E. Menard, R.R. Mattingly / Cellular Signalling 15 (2003) 1099–1109 1101
The cleared extracts were then split into two aliquots.
One 200-Al portion was used for TCA precipitation of total
protein. To the other 200-Al portion, 0.48 Ag 9e10 anti-
body was added for 1 h at 4 jC, followed by 0.24 Agrabbit anti-mouse antibody for 1 h and then 40 Al of a 50%suspension of Protein A Sepharose beads for an additional
hour. The beads were washed once in lysis buffer, two
times in RIPA buffer, pH 8.0, containing 300 mM NaCl,
1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris, 5
mM orthovanadate, 50 mM Na h-glycerophosphate, 50
mM NaF, 20 mM Na pyrophosphate, and 1 mM PMSF
and finally once in a kinase buffer containing 12.5 mM
HEPES, 12.5 mM Na h-glycerophosphate, 7.5 mM Mg
Cl2, 0.5 mM EGTA, 0.5 mM NaF, and 0.5 mM Na
Vanadate, pH 7.0. One half of the immunoprecipitated
sample was used for the kinase assay and one half was
used for Western blot analysis. The blots were probed with
9e10 to detect myc-tagged proteins and 12Ca5 to detect
HA-tagged proteins.
2.4. In vitro kinase assay
The in vitro kinase assay is a modification of a procedure
by Knaus et al. [35]. Forty microliters of kinase buffer
containing 4 ACi g-32P-ATP, 500 ng GTP, and 1 Ag RacV12,per sample was added to the immunoprecipitates. Where
indicated, 4 Ag Myelin Basic Protein (MBP) was used as a
substrate to detect PAK1 activity. After 20 min at 30 jC, thekinase reaction was stopped by the addition of sample
loading buffer and boiling of samples for 5 min. The results
were visualized using SDS-polyacrylamide electrophoresis
(PAGE), autoradiography, and phosphoimaging. Data
reported are meanF S.E.M. from three independent experi-
ments.
2.4.1. Agonist treatment and immunoprecipitation of
endogenous PAK1
The procedure is the same as described above expect that
NIH-3T3/hM1 fibroblasts or SKNSH neuroblastoma cells
were lysed with a buffer, pH 7.4 containing 25 mM HEPES,
0.3 M NaCl, 1.5 mM MgCl2, 1 mM Na Vanadate, 0.1%
Triton X-100, 0.5 mM DTT, 20 mM h-glycerophosphate, 20Ag/ml aprotinin, 20 Ag/ml leupeptin, 1 mM PMSF, and 1
AM okadaic acid, and then PAK1 was immunoprecipitated
using 0.6 Ag N-20 anti-PAK1 antibody.
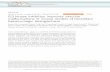
Fig. 1. Time course of PAK1 activation in COS7 cells treated with EGF,
carbachol, LPA, or PMA. COS7 cells were transfected with myc-tagged
PAK1, treated as shown and the myc-PAK immunoprecipitated and assayed
for activity against an MBP substrate. (A) Kinase assay of PAK1
phosphorylation following treatment with agonists. Maximal activation of
mycPAK1 activity was determined using phosphoimaging. The times to
maximal activation were 5 min for EGF, 15 min for LPA and PMA, and 30
min for carbachol. (B) Representative Western blot showing quantitative
recovery of mycPAK1 in the kinase assay protocol is independent of
treatment protocol. –Ve shows cells transfected with an empty vector
control. (C) Graph representing time course activation of mycPAK1.
MeanF S.E.M. from n= 5.
3. Results
3.1. Time course of PAK1 activation in COS7 cells
To investigate the pathways through which PAK1 can be
activated, COS7 cells were transiently transfected with a
myc-tagged PAK1 construct and subtype 2 human musca-
rinic receptors, and then stimulated with EGF (to activate
growth factor tyrosine kinase signaling), carbachol or LPA
(to stimulate G protein-coupled receptors), and PMA (to
activate PKC). Time course analysis showed that maximal
PAK1 activation with EGF was at 5 min, while LPA and
PMA took 15 min to reach peak activation, and carbachol
was the slowest to activate PAK1 taking 30 min (Fig. 1).
The peak increase in activity compared to untreated cells
was 4.8 fold for EGF, 4.5 fold for carbachol, and 3.1 fold for
both LPA and PMA. These results may indicate a hierarchy
to PAK1 activation in these cells. Once GPCRs are activat-
ed, a transactivation with the EGF receptor may take place
[36,37], and the Ghg subunits that are released may also
activate down stream effectors such as PI3-kinase [38–40]
to achieve maximal activation of PAK1, but with slower
kinetics.
3.2. Activation of PAK1 by EGF requires Ras, Rac and
Cdc42 activity
Mutations that occur outside of the GTPase effector
domain can determine the activity of Ras, Rac, Cdc42,
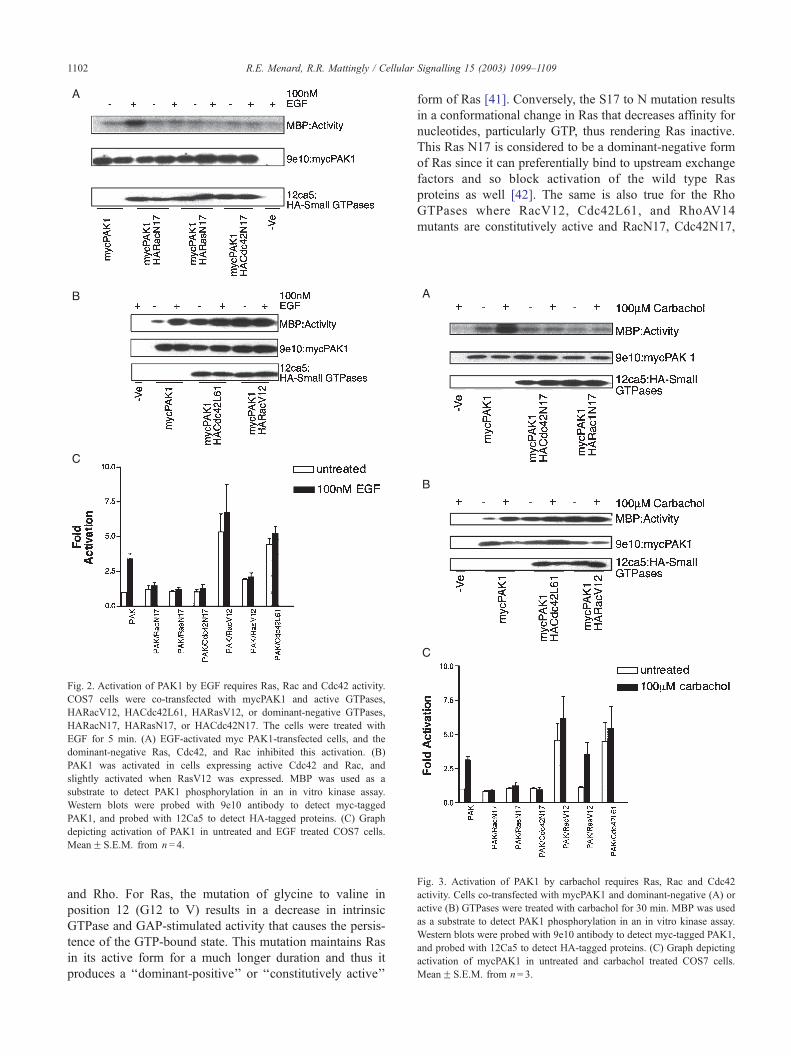
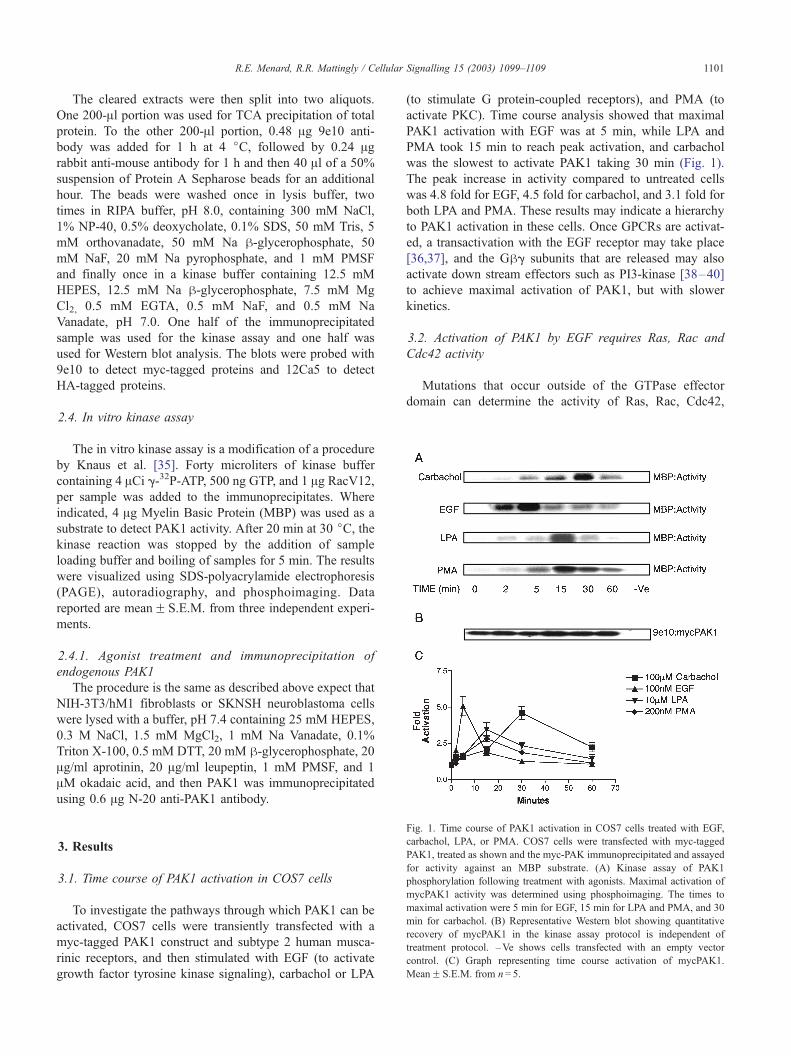
Fig. 2. Activation of PAK1 by EGF requires Ras, Rac and Cdc42 activity.
COS7 cells were co-transfected with mycPAK1 and active GTPases,
HARacV12, HACdc42L61, HARasV12, or dominant-negative GTPases,
HARacN17, HARasN17, or HACdc42N17. The cells were treated with
EGF for 5 min. (A) EGF-activated myc PAK1-transfected cells, and the
dominant-negative Ras, Cdc42, and Rac inhibited this activation. (B)
PAK1 was activated in cells expressing active Cdc42 and Rac, and
slightly activated when RasV12 was expressed. MBP was used as a
substrate to detect PAK1 phosphorylation in an in vitro kinase assay.
Western blots were probed with 9e10 antibody to detect myc-tagged
PAK1, and probed with 12Ca5 to detect HA-tagged proteins. (C) Graph
depicting activation of PAK1 in untreated and EGF treated COS7 cells.
MeanF S.E.M. from n= 4.
Fig. 3. Activation of PAK1 by carbachol requires Ras, Rac and Cdc42
activity. Cells co-transfected with mycPAK1 and dominant-negative (A) or
active (B) GTPases were treated with carbachol for 30 min. MBP was used
as a substrate to detect PAK1 phosphorylation in an in vitro kinase assay.
Western blots were probed with 9e10 antibody to detect myc-tagged PAK1,
and probed with 12Ca5 to detect HA-tagged proteins. (C) Graph depicting
activation of mycPAK1 in untreated and carbachol treated COS7 cells.
MeanF S.E.M. from n= 3.
R.E. Menard, R.R. Mattingly / Cellular Signalling 15 (2003) 1099–11091102
and Rho. For Ras, the mutation of glycine to valine in
position 12 (G12 to V) results in a decrease in intrinsic
GTPase and GAP-stimulated activity that causes the persis-
tence of the GTP-bound state. This mutation maintains Ras
in its active form for a much longer duration and thus it
produces a ‘‘dominant-positive’’ or ‘‘constitutively active’’
form of Ras [41]. Conversely, the S17 to N mutation results
in a conformational change in Ras that decreases affinity for
nucleotides, particularly GTP, thus rendering Ras inactive.
This Ras N17 is considered to be a dominant-negative form
of Ras since it can preferentially bind to upstream exchange
factors and so block activation of the wild type Ras
proteins as well [42]. The same is also true for the Rho
GTPases where RacV12, Cdc42L61, and RhoAV14
mutants are constitutively active and RacN17, Cdc42N17,
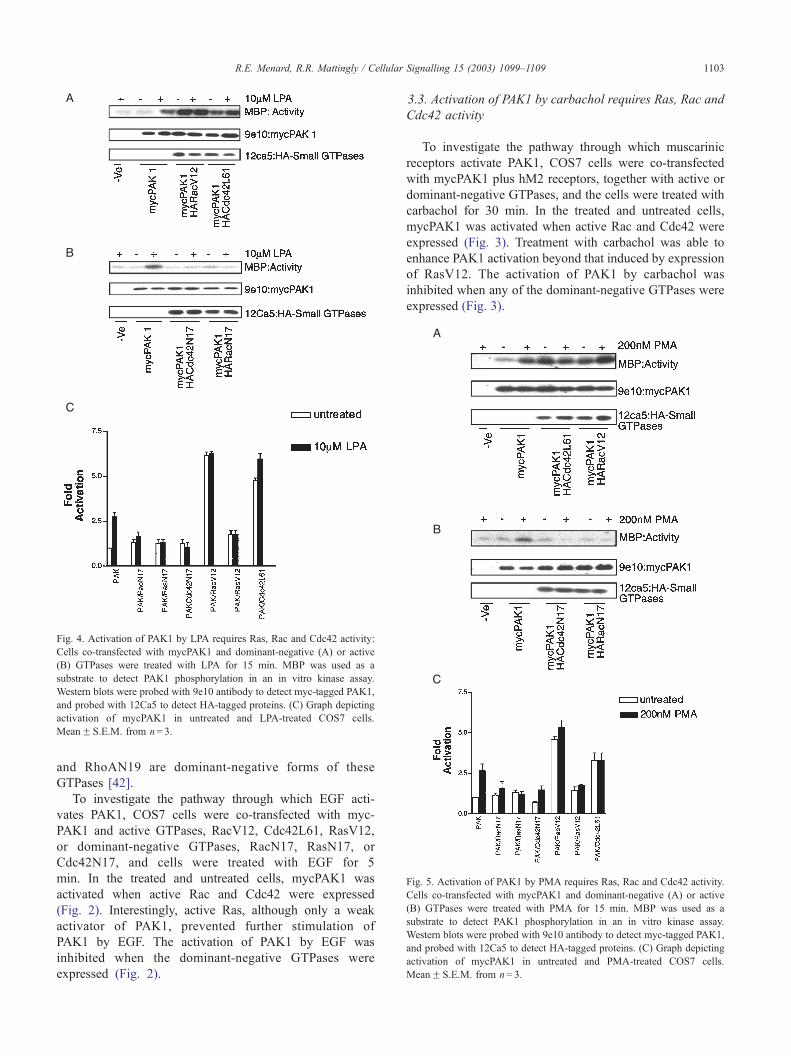
Fig. 4. Activation of PAK1 by LPA requires Ras, Rac and Cdc42 activity:
Cells co-transfected with mycPAK1 and dominant-negative (A) or active
(B) GTPases were treated with LPA for 15 min. MBP was used as a
substrate to detect PAK1 phosphorylation in an in vitro kinase assay.
Western blots were probed with 9e10 antibody to detect myc-tagged PAK1,
and probed with 12Ca5 to detect HA-tagged proteins. (C) Graph depicting
activation of mycPAK1 in untreated and LPA-treated COS7 cells.
MeanF S.E.M. from n= 3.
Fig. 5. Activation of PAK1 by PMA requires Ras, Rac and Cdc42 activity.
Cells co-transfected with mycPAK1 and dominant-negative (A) or active
(B) GTPases were treated with PMA for 15 min. MBP was used as a
substrate to detect PAK1 phosphorylation in an in vitro kinase assay.
Western blots were probed with 9e10 antibody to detect myc-tagged PAK1,
and probed with 12Ca5 to detect HA-tagged proteins. (C) Graph depicting
activation of mycPAK1 in untreated and PMA-treated COS7 cells.
MeanF S.E.M. from n= 3.
R.E. Menard, R.R. Mattingly / Cellular
and RhoAN19 are dominant-negative forms of these
GTPases [42].
To investigate the pathway through which EGF acti-
vates PAK1, COS7 cells were co-transfected with myc-
PAK1 and active GTPases, RacV12, Cdc42L61, RasV12,
or dominant-negative GTPases, RacN17, RasN17, or
Cdc42N17, and cells were treated with EGF for 5
min. In the treated and untreated cells, mycPAK1 was
activated when active Rac and Cdc42 were expressed
(Fig. 2). Interestingly, active Ras, although only a weak
activator of PAK1, prevented further stimulation of
PAK1 by EGF. The activation of PAK1 by EGF was
inhibited when the dominant-negative GTPases were
expressed (Fig. 2).
3.3. Activation of PAK1 by carbachol requires Ras, Rac and
Cdc42 activity
To investigate the pathway through which muscarinic
receptors activate PAK1, COS7 cells were co-transfected
with mycPAK1 plus hM2 receptors, together with active or
dominant-negative GTPases, and the cells were treated with
carbachol for 30 min. In the treated and untreated cells,
mycPAK1 was activated when active Rac and Cdc42 were
expressed (Fig. 3). Treatment with carbachol was able to
enhance PAK1 activation beyond that induced by expression
of RasV12. The activation of PAK1 by carbachol was
inhibited when any of the dominant-negative GTPases were
expressed (Fig. 3).
Signalling 15 (2003) 1099–1109 1103
llular Signalling 15 (2003) 1099–1109
3.4. Activation of PAK1 by LPA requires Ras, Rac and
Cdc42 activity
To investigate the pathway through which LPA activates
PAK1, COS7 cells were co-transfected with mycPAK1 and
active or dominant-negative GTPases, and the cells were
treated with LPA for 15 min. In the treated and untreated
cells, mycPAK1 was activated when active Rac and Cdc42
were expressed (Fig. 4). As with the other agonists, the
R.E. Menard, R.R. Mattingly / Ce1104
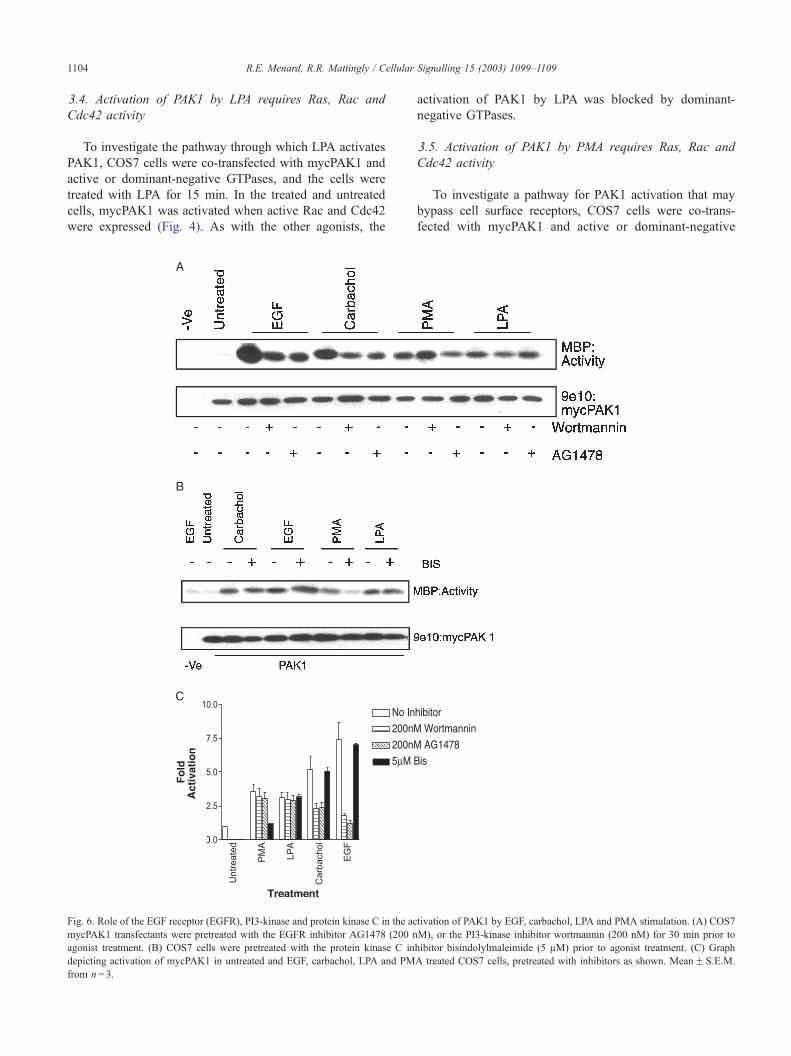
Fig. 6. Role of the EGF receptor (EGFR), PI3-kinase and protein kinase C in the ac
mycPAK1 transfectants were pretreated with the EGFR inhibitor AG1478 (200 n
agonist treatment. (B) COS7 cells were pretreated with the protein kinase C in
depicting activation of mycPAK1 in untreated and EGF, carbachol, LPA and PM
from n= 3.
activation of PAK1 by LPA was blocked by dominant-
negative GTPases.
3.5. Activation of PAK1 by PMA requires Ras, Rac and
Cdc42 activity
To investigate a pathway for PAK1 activation that may
bypass cell surface receptors, COS7 cells were co-trans-
fected with mycPAK1 and active or dominant-negative
tivation of PAK1 by EGF, carbachol, LPA and PMA stimulation. (A) COS7
M), or the PI3-kinase inhibitor wortmannin (200 nM) for 30 min prior to
hibitor bisindolylmaleimide (5 AM) prior to agonist treatment. (C) Graph
A treated COS7 cells, pretreated with inhibitors as shown. MeanF S.E.M.

Fig. 7. Detection of active PAK1 using an antibody that recognizes
phosphorylated threonine 423. COS7 cells were transfected with myc-
tagged PAK1 and treated as shown. Western blots were probed first with
9e10 to detect myc-tagged PAK1, then stripped and re-probed with a
PhosphoPAK1 (Thr 423)/PAK2 (Thr 402) antibody (Cell Signalling
Technology).
Fig. 8. LPA stimulation of COS7 cells may regulate PAK1 activation
through RhoA. COS7 cells were transfected with mycPAK1 alone or co-
transfected with HARhoAN19 or HARhoV14 and then treated with LPA.
(A) Data from a representative experiment. (B) Graph depicting activation
of PAK1 by LPA in COS7 cells. MeanF S.E.M. from n= 3.
R.E. Menard, R.R. Mattingly / Cellular Signalling 15 (2003) 1099–1109 1105
GTPases, and the cells were then treated with PMA for 15
min (Fig. 5). All of the active GTPases caused stimulation
of PAK1 that was insensitive to further activation by PMA
and all of the dominant-negative GTPases blocked PMA
stimulation of PAK1.
3.6. Role of the EGF receptor (EGFR), PI3-kinase and
protein kinase C in the activation of PAK1 by EGF,
carbachol, LPA and PMA stimulation
To further investigate the signaling pathways through
which PAK1 is activated, COS7mycPAK1 transfectants were
pretreated with the selective EGFR inhibitor AG1478 (200
nM), or the PI3-kinase inhibitor wortmannin (200 nM), or the
protein kinase C inhibitor bisindolylmaleimide (5 AM) for 30
min prior to agonist treatment. Pretreatment of cells with
AG1478 decreased PAK1 activation by EGF from 7.4 to 1.2
fold and by carbachol from 5.2 to 2.4 fold. Pretreatment with
wortmannin also inhibited the stimulatory effects of EGF,
from 7.4- to 1.8-fold increase, and carbachol from 5.2- to 2.3-
fold increase. These inhibitors did not significantly affect the
stimulation of PAK1 by LPA or PMA (Fig. 6). Pretreatment
with bisindolylmaleimide did not affect PAK1 activation by
EGF, carbachol, or LPA, however activation by PMA was
inhibited, as there was a decrease in fold activation from 3.6
to 1.2 (Fig. 6). These results indicate that PAK1 stimulation
by EGF and carbachol relies on EGFR and PI3-kinase
activation, and that LPA and PMA stimulate independent
pathways to PAK1 activation.
3.7. Detection of active PAK1 using an antibody which
recognizes phosphorylated threonine 423
In order to confirm the results of the PAK1 kinase
activity assays performed by MBP phosphorylation, West-
ern blots from experiments in which COS7 cells were
pretreated with the inhibitors for 30 min prior to agonist
treatment were stripped and re-probed with a PhosphoPAK1
(Thr 423)/PAK2 (Thr 402) antibody [43]. The results are
shown in Fig. 7. Activation of PAK1 by carbachol, EGF,
LPA, and PMA can be detected by this antibody. Further-
more, the results of the inhibition study depicted in Fig. 6
are confirmed by this antibody. This antibody may thus
prove to be a more time efficient method of PAK1 activation
than radioactive kinase assays.
3.8. LPA stimulation of COS7 cells may regulate PAK1
activation through RhoA
As none of the pharmacological inhibitors blocked LPA
stimulation of PAK1 activity, we concluded that another
R.E. Menard, R.R. Mattingly / Cellular1106
signal transduction pathway was involved. We therefore
investigated the possible role of RhoA as a potential effector
of LPA activation [7,8,44]. COS7 cells were transfected
with mycPAK1 alone or co-transfected with RhoAN19 or
RhoV14 and then treated with LPA. Cotransfection with
RhoAN19 inhibits PAK1 stimulation from 2.8- to 1.1-fold
activation in LPA-treated cells (Fig. 8). The mycPAK1 co-
transfectants containing RhoAV14, revealed increased
PAK1 activity of 2.3-fold activation in the presence or
absence of LPA (Fig. 8). These results indicate a critical
role for RhoA in PAK1 activation.
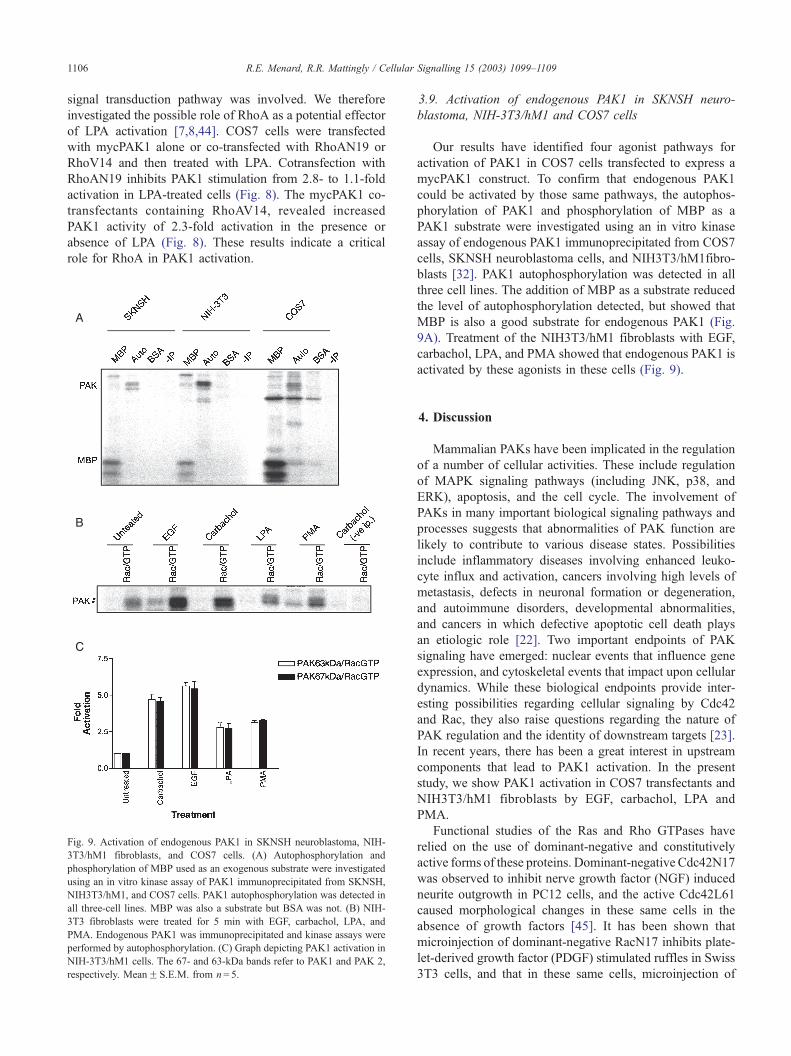
Fig. 9. Activation of endogenous PAK1 in SKNSH neuroblastoma, NIH-
3T3/hM1 fibroblasts, and COS7 cells. (A) Autophosphorylation and
phosphorylation of MBP used as an exogenous substrate were investigated
using an in vitro kinase assay of PAK1 immunoprecipitated from SKNSH,
NIH3T3/hM1, and COS7 cells. PAK1 autophosphorylation was detected in
all three-cell lines. MBP was also a substrate but BSA was not. (B) NIH-
3T3 fibroblasts were treated for 5 min with EGF, carbachol, LPA, and
PMA. Endogenous PAK1 was immunoprecipitated and kinase assays were
performed by autophosphorylation. (C) Graph depicting PAK1 activation in
NIH-3T3/hM1 cells. The 67- and 63-kDa bands refer to PAK1 and PAK 2,
respectively. MeanF S.E.M. from n= 5.
3.9. Activation of endogenous PAK1 in SKNSH neuro-
blastoma, NIH-3T3/hM1 and COS7 cells
Our results have identified four agonist pathways for
activation of PAK1 in COS7 cells transfected to express a
mycPAK1 construct. To confirm that endogenous PAK1
could be activated by those same pathways, the autophos-
phorylation of PAK1 and phosphorylation of MBP as a
PAK1 substrate were investigated using an in vitro kinase
assay of endogenous PAK1 immunoprecipitated from COS7
cells, SKNSH neuroblastoma cells, and NIH3T3/hM1fibro-
blasts [32]. PAK1 autophosphorylation was detected in all
three cell lines. The addition of MBP as a substrate reduced
the level of autophosphorylation detected, but showed that
MBP is also a good substrate for endogenous PAK1 (Fig.
9A). Treatment of the NIH3T3/hM1 fibroblasts with EGF,
carbachol, LPA, and PMA showed that endogenous PAK1 is
activated by these agonists in these cells (Fig. 9).
Signalling 15 (2003) 1099–1109
4. Discussion
Mammalian PAKs have been implicated in the regulation
of a number of cellular activities. These include regulation
of MAPK signaling pathways (including JNK, p38, and
ERK), apoptosis, and the cell cycle. The involvement of
PAKs in many important biological signaling pathways and
processes suggests that abnormalities of PAK function are
likely to contribute to various disease states. Possibilities
include inflammatory diseases involving enhanced leuko-
cyte influx and activation, cancers involving high levels of
metastasis, defects in neuronal formation or degeneration,
and autoimmune disorders, developmental abnormalities,
and cancers in which defective apoptotic cell death plays
an etiologic role [22]. Two important endpoints of PAK
signaling have emerged: nuclear events that influence gene
expression, and cytoskeletal events that impact upon cellular
dynamics. While these biological endpoints provide inter-
esting possibilities regarding cellular signaling by Cdc42
and Rac, they also raise questions regarding the nature of
PAK regulation and the identity of downstream targets [23].
In recent years, there has been a great interest in upstream
components that lead to PAK1 activation. In the present
study, we show PAK1 activation in COS7 transfectants and
NIH3T3/hM1 fibroblasts by EGF, carbachol, LPA and
PMA.
Functional studies of the Ras and Rho GTPases have
relied on the use of dominant-negative and constitutively
active forms of these proteins. Dominant-negative Cdc42N17
was observed to inhibit nerve growth factor (NGF) induced
neurite outgrowth in PC12 cells, and the active Cdc42L61
caused morphological changes in these same cells in the
absence of growth factors [45]. It has been shown that
microinjection of dominant-negative RacN17 inhibits plate-
let-derived growth factor (PDGF) stimulated ruffles in Swiss
3T3 cells, and that in these same cells, microinjection of

R.E. Menard, R.R. Mattingly / Cellular Signalling 15 (2003) 1099–1109 1107
active RacV12 is sufficient to induce membrane ruffles in the
absence of growth factors [5,6]. The use of dominant-nega-
tive Cdc42 and Rac has been utilized in studying anchorage
independent growth in Ras transformation [46], and also Rac
regulation of actin polymerization [47]. Active GTPases can
also effect cell transformation. Active RacV12 can cause
malignant transformation on its own [48], and active RasV12
can induce focus formation in NIH3T3 fibroblasts that can be
inhibited by co-expression of dominant-negative RhoA19
[49]. It has also been reported that dominant-negative forms
of Rac, Cdc42, and RhoA are able to inhibit Ras activation of
PAK in Rat-1 fibroblasts [50].
The activation of PAK1 by active Cdc42 and Rac1 (Figs.
2–5) has been reported by many groups [9,10,22,35,51,52],
and our results coincide with what others have found. In
contrast, the effects of active Ras on PAK1 are more
surprising. The low level of PAK1 activation by RasV12
in these cells, except when cells are treated with carbachol,
is intriguing. It has been reported that RasV12 activates
PAK1 in certain cell lines, such as RAT1and Swiss-3T3, but
not NIH3T3 fibroblasts [50,53]. It is possible that other
factors are required for the activation of PAK by RasV12
and that muscarinic receptor stimulation can supply these
signals in COS7 cells. Alternatively, there may be down-
regulation of endogenous receptors and the PKC pathway
by RasV12, but the transfected muscarinic receptors may
remain functional under these conditions. All agonist stim-
ulation of PAK1 could be inhibited by dominant-negative
forms of Ras, Rac, and Cdc42, indicating that EGF, carba-
Fig. 10. A potential scheme whereby multiple signaling pathways converge on
mechanisms. The results show that PAK1 is activated by four separate pathways
activate PAK1. Carbachol can transactivate EGFR and go through PI3-kinase to a
EGFR-independent pathways (as indicated by the dashed arrows). LPA activation
activation of PAK1. The activation by EGF goes through PI3-kinase and PMA act
chol, LPA, and PMA act through mechanisms in which
these small GTPases are required. Thus, there is a definite
involvement of Ras in PAK1 activation, but the exact
pathway through Ras to PAK1 remains unclear.
In recent years, evidence of cross-communication be-
tween EGFR and GPCRs has emerged. It has been observed
that GPCR-induced EGFR tyrosine phosphorylation is rap-
id, transient, and comparable with receptor activation by
low amounts of EGF [36,54–56]. The signal capacity of the
transactivated EGFR is not restricted to Ras–MAP kinase
pathway but rather functions as an important mediator for
Gq, Gi, and G12/13-coupled receptors to induce a diversity
of cellular responses such as activation of PI3-kinase,
modulation of ion channels, and cytoskeletal reorganization
[36,57]. The time course study we performed showed EGF
and carbachol activate PAK1 following 5 and 30 min of
treatment, respectively (Fig. 1). In addition, the carbachol-
dependent activation of PAK1 was blocked by the selective
EGFR inhibitor AG1478 (Fig. 6). The results of our study
clearly indicate that muscarinic receptors require EGFR
transactivation for the major part of their ability to activate
PAK1. It is likely that muscarinic receptors can also couple
to additional pathways for PAK1 activation, however, as
carbachol still stimulates PAK1 in the presence of active Ras
when EGF cannot, and the inhibition of carbachol-depen-
dent PAK1 activation by AG1478 is incomplete. It is
noteworthy that the pathway from endogenous G protein-
coupled receptors for LPA does not require the EGF
receptor [36,54].
the activation of PAK1 through G protein and tyrosine kinase-dependent
. The two G protein coupled receptor agonists act differently in how they
ctivate PAK1 via Rac/Cdc42. It can also apparently couple weakly through
of PAK1 goes through RhoA, which can possibly lead to Ras-dependent
ivates PAK1 via PKC. PKC may activate an upstream effector such as Ras.

R.E. Menard, R.R. Mattingly / Cellular Signalling 15 (2003) 1099–11091108
The role of PAKs in cytoskeletal re-organization led us to
investigate what other proteins may be involved in this
process. Phorbol esters, such as PMA, can induce actin
assembly in neutrophils and this cytoskeletal alteration is
due to PKC activation [58]. Various isotypes of PKC may
play different roles in cells. Both PKCa and PKCu can be
activated by diacylglycerol but PKCa can potentially activate
PKC~ leading to InB stimulation, and PKCu may activate
Ras leading to Erk2 activation [59,60]. The PMA activation
of PAK1 is inhibited by dominant-negative forms of Rac and
Cdc42, therefore it is likely that PKC interacts with a protein
upstream of PAK1. It is likely that PMA induces Ras
activation, as dominant-negative Ras can inhibit PMA stim-
ulation of PAK1. The inhibition of PMA-dependent PAK1
activation by bisindolylmaleimide shows that there is a role
for protein kinase C in PAK1 stimulation, but the lack of
effect of PKC inhibition on any of the cell surface receptor
agonists suggests that PKC is not a necessary component of
the activation of PAK1 by any of these pathways.
The lack of inhibition of LPA-dependent PAK1 activity
by inhibitors of PKC and PI3-kinase led us to investigate
other proteins that may be involved in PAK1 activation.
LPA receptors have been shown to act through G12,13 G-
proteins to stimulate RhoA and PAK1 [6,61,62]. The exact
mechanism by which RhoA induces PAK1 activation is
unclear. In a study of PAK1 and Rho GTPases in rat brain,
there was an association between Rac and PAK1, Cdc42 and
PAK1, but not between RhoA and PAK1 [63]. In our
experiments, RhoAN19 was able to block the activation of
PAK1 by LPA, and in addition RhoAV14 was able to
activate PAK1 (Fig. 8). How then could RhoA lead to
PAK activation? It has been reported that co-transfection
of active RhoA and active Raf can activate PAK in Rat1
fibroblasts [50]. In the COS7 cells in this study, active RhoA
was sufficient to induce PAK1 activation. The role of RhoA
in cell adhesion has been studied and it has been determined
that FAK and paxillin are phosphorylated in response to
RhoA and this may lead to Ras/Raf activation [64,65].
There is also evidence that cAMP-dependent protein kinase
(PKA) phosphorylates PAK1, paxillin and Nck [66], and it
is known PAK1 interacts with paxillin and Nck [19,21,67–
70]. It is thus quite possible that activation of PAK1 by
RhoA is indirect through other upstream effectors (Fig. 10).
Further investigation of the role of components upstream of
PAK1, such as Ghg and Akt, may yield a more complete
insight as to the exact mechanism of PAK1 activation.
Acknowledgements
We thank Dr. G. Bokoch for the gift of the plasmid
encoding PAK1, Dr. C. Neudauer for the subcloning of
PAK1 and A. Jovanovski for technical assistance.
This work was supported in part by RO1-CA81150
(to R.R.M.) and T32-CA09531 (to R.E.M.).
References
[1] Lopez-Ilasaca M. Biochem Pharmacol 1998;56(3):269–77.
[2] Brown JL, Stowers L, Baer M, Trejo J, Coughlin S, Chant J. Curr
Biol 1996;6(5):598–605.
[3] Symons M. Curr Opin Biotechnol 1995;6(6):668–74.
[4] Downward J. Nature 1992;359(6393):273–4.
[5] Ridley AJ, Hall A. Cell 1992;70(3):389–99.
[6] Nobes CD, Hawkins P, Stephens L, Hall A. J Cell Sci 1995;
108(Pt. 1):225–33.
[7] Mackay DJ, Hall A. J Biol Chem 1998;273(33):20685–8.
[8] Beqaj S, Jakkaraju S, Mattingly RR, Pan D, Schuger L. J Cell Biol
2002;156(5):893–903.
[9] Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. Nature
1994;367(6458):40–6.
[10] Martin GA, Bollag G, McCormick F, Abo A. EMBO J 1995;14(9):
1970–8.
[11] Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM,
Chernoff J. Curr Biol 1997;7(3):202–10.
[12] Abo A, Qu J, Cammarano MS, Dan C, Fritsch A, Baud V, et al.
EMBO J 1998;17(22):6527–40.
[13] Yang F, Li X, Sharma M, Zarnegar M, Lim B, Sun Z. J Biol Chem
2001;276(18):15345–53.
[14] Pandey A, Dan I, Kristiansen TZ, Watanabe NM, Voldby J, Kajikawa
E, et al. Oncogene 2002;21(24):3939–48.
[15] Jaffer ZM, Chernoff J. Int J Biochem Cell Biol 2002;34(7):713–7.
[16] Hoffman GR, Cerione RA. Cell 2000;102(4):403–6.
[17] Knaus UG, Wang Y, Reilly AM, Warnock D, Jackson JH. J Biol
Chem 1998;273(34):21512–8.
[18] Van Aelst L, D’Souza-Schorey C. Genes Dev 1997;11(18):2295–322.
[19] Lu W, Katz S, Gupta R, Mayer BJ. Curr Biol 1997;7(2):85–94.
[20] Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, et al. Mol
Cell 1998;1(2):183–92.
[21] Zhao ZS, Manser E, Lim L. Mol Cell Biol 2000;20(11):3906–17.
[22] Knaus UG, Bokoch GM. Int J Biochem Cell Biol 1998;30(8):
857–62.
[23] Bagrodia S, Cerione RA. Trends Cell Biol 1999;9(9):350–5.
[24] Rudel T, Bokoch GM. Science 1997;276(5318):1571–4.
[25] Rudel T, Zenke FT, Chuang TH, Bokoch GM. J Immunol 1998;
160(1):7–11.
[26] King AJ, Sun H, Diaz B, Barnard D, Miao W, Bagrodia S, et al.
Nature 1998;396(6707):180–3.
[27] Sun H, King AJ, Diaz HB, Marshall MS. Curr Biol 2000;10(5):
281–4.
[28] Chaudhary A, King WG, Mattaliano MD, Frost JA, Diaz B, Morrison
DK, et al. Curr Biol 2000;10(9):551–4.
[29] Zang M, Hayne C, Luo Z. J Biol Chem 2002;277(6):4395–405.
[30] Zang M, Waelde CA, Xiang X, Rana A, Wen R, Luo Z. J Biol Chem
2001;276(27):25157–65.
[31] Mattingly RR, Macara IG. Nature 1996;382(6588):268–72.
[32] Mattingly RR, Sorisky A, Brann MR, Macara IG. Mol Cell Biol
1994;14(12):7943–52.
[33] Mattingly RR, Milstein ML, Mirkin BL. Cell Signal 2001;13(7):
499–505.
[34] Mattingly RR, Gibbs RA, Menard RE, Reiners Jr JJ. J Pharmacol Exp
Ther 2002;303(1):74–81.
[35] Knaus UG, Morris S, Dong HJ, Chernoff J, Bokoch GM. Science
1995;269(5221):221–3.
[36] Leserer M, Gschwind A, Ullrich A. IUBMB Life 2000;49(5):405–9.
[37] Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C,
et al. Nature 1999;402(6764):884–8.
[38] Nakano T, Kontani K, Kurosu H, Katada T, Hoshi M, Chiba K. Dev
Biol 1999;209(1):200–9.
[39] Maier U, Babich A, Nurnberg B. J Biol Chem 1999;274(41):29311–7.
[40] Kurosu H, Maehama T, Okada T, Yamamoto T, Hoshino S, Fukui Y,
et al. J Biol Chem 1997;272(39):24252–6.

R.E. Menard, R.R. Mattingly / Cellular Signalling 15 (2003) 1099–1109 1109
[41] McCormick F. Curr Opin Genet Dev 1994;4(1):71–6.
[42] Lim L, Manser E, Leung T, Hall C. Eur J Biochem 1996;242(2):
171–85.
[43] Vadlamudi RK, Adam L, Wang RA, Mandal M, Nguyen D, Sahin A,
et al. J Biol Chem 2000;275(46):36238–44.
[44] Ridley AJ. Curr Biol 1996;6(10):1256–64.
[45] Daniels RH, Hall PS, Bokoch GM. EMBO J 1998;17(3):754–64.
[46] Qiu RG, Abo A, McCormick F, Symons M. Mol Cell Biol 1997;17(6):
3449–58.
[47] Joneson T, McDonough M, Bar-Sagi D, Van Aelst L. Science
1996;274(5291):1374–6.
[48] Qiu RG, Chen J, Kirn D, McCormick F, Symons M. Nature
1995;374(6521):457–9.
[49] Qiu RG, Chen J, McCormick F, Symons M. Proc Natl Acad Sci
U S A 1995;92(25):11781–5.
[50] Tang Y, Yu J, Field J. Mol Cell Biol 1999;19(3):1881–91.
[51] Manser E, Leung T, Lim L. Methods Mol Biol 1998;84:295–305.
[52] Manser E, Lim L. Prog Mol Subcell Biol 1999;22:115–33.
[53] Reeder MK, Serebriiskii IG, Golemis EA, Chernoff J. J Biol Chem
2001;276(44):40606–13.
[54] Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. EMBO J
1997;16(23):7032–44.
[55] Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T,
Utsunomiya H, et al. J Biol Chem 1998;273(15):8890–6.
[56] Keely SJ, Uribe JM, Barrett KE. J Biol Chem 1998;273(42):
27111–7.
[57] Gohla A, Offermanns S, Wilkie TM, Schultz G. J Biol Chem
1999;274(25):17901–7.
[58] Downey GP, Chan CK, Lea P, Takai A, Grinstein S. J Cell Biol
1992;116(3):695–706.
[59] Goekjian PG, Jirousek MR. Curr Med Chem 1999;6(9):877–903.
[60] Mellor H, Parker PJ. Biochem J 1998;332(Pt. 2):281–92.
[61] Nobes C, Hall A. Curr Opin Genet Dev 1994;4(1):77–81.
[62] Schmitz U, Thommes K, Beier I, Vetter H. Biochem Biophys Res
Commun 2002;291(3):687–91.
[63] Terashima T, Yasuda H, Terada M, Kogawa S, Maeda K, Haneda M,
et al. J Neurochem 2001;77(4):986–93.
[64] Nobes CD, Hall A. J Cell Biol 1999;144(6):1235–44.
[65] Clark EA, King WG, Brugge JS, Symons M, Hynes RO. J Cell Biol
1998;142(2):573–86.
[66] Howe AK, Juliano RL. Nat Cell Biol 2000;2(9):593–600.
[67] Brown MC, West KA, Turner CE. Mol Biol Cell 2002;13(5):
1550–65.
[68] Hashimoto S, Tsubouchi A, Mazaki Y, Sabe H. J Biol Chem
2001;276(8):6037–45.
[69] Bokoch GM, Wang Y, Bohl BP, Sells MA, Quilliam LA, Knaus UG.
J Biol Chem 1996;271(42):25746–9.
[70] Wunderlich L, Goher A, Farago A, Downward J, Buday L. Cell
Signal 1999;11(4):253–62.
Related Documents



![Diacylglycerol kinase ζ generates dipalmitoyl-phosphatidic ... · kinase C [6], and p21 activated protein kinase 1 [7,8].PAasan intracellular signaling lipid is generated by phosphorylation](https://static.cupdf.com/doc/110x72/5fe275ed0f93ac2b35696d07/diacylglycerol-kinase-generates-dipalmitoyl-phosphatidic-kinase-c-6-and.jpg)