Recent Patents on Chemical Engineering, 2008, 1, 113-125 113 1874-4788/08 $100.00+.00 © 2008 Bentham Science Publishers Ltd. Catalysis in the Petroleum Naphtha Catalytic Reforming Process Ahmed K. Aboul-Gheit* and Salwa A.-W. Ghoneim Process Development Department, Egyptian Petroleum Research Institute, Nasr City, P.O. Box 9540, Cairo 11787, Egypt Received: November 14, 2007; Accepted: January 31, 2008; Revised: February 24, 2008 Abstract: Before the US. Patent (1949) by Haensel wherein platinum was used as the active metal component in the industrial catalytic reforming catalysts, molybdenum and /or chromium oxides were used but were suffering from very rapid deactivation by coke even when used under high pressures. Platinum containing catalysts enjoyed very high selectivity via producing high yields of high octane C 5 + reformates even at relatively much lower pressures. Improvements were then carried out; some concerned with modifying the metal component, and others concerning modifying the support as well as improving the processing schemes. So many patents were disclosing bimetallic or even polymetallic catalysts containing primarily platinum and/or rhenium or iridium, or their combinations, in addition to tin, zinc, germanium, bismuth, phosphorus, or chlorine, etc. These catalysts acquired advantageous activities, selectivities and long time-on stream with maintaining high yields of high octane motor gasoline. Keywords: Platinum, rhenium, iridium, Al 2 O 3 , aromatization, isomerization. INTRODUCTION In commercial reforming operations using such catalysts, one or a series of reactions constitute the heart of the reforming unit. Each reactor is generally provided with fixed bed, or beds, of the catalyst which receive down-flow feed, and each is provided with a preheater or interstage heater, because the reactions which take place are endothermic. During the operating cycle, a naphtha feed, with hydrogen, usually recycle hydrogen gas is concurrently passed through a reactor. The sequences of reforming reactions take place as a continuum throughout the series of staged reactors of the reforming unit. The product from the last reactor of the series is separated into a liquid fraction, and a vapor effluent. The former is recovered as a C 5 + liquid product; the latter is a gas rich in hydrogen, and usually contains small amounts of gaseous hydrocarbons, from which hydrogen is separated and recycled to the first reactor of the process to minimize coke production. The activity of the catalyst gradually declines during the time-on-stream of an operating cycle due to the build-up of coke, which is believed to result from the deposition of coke precursors such as anthracene, etc. and other condensed ring aromatic molecules on the catalyst, these polymerizing to form coke. During the operation, the process temperature is gradually raised to compensate for the activity loss caused by the coke deposition. Eventually, however, economics dictate the necessity of reactivating the catalyst. Conse- quently, in all processes, the feed must be cut out and the catalyst must necessarily be periodically regenerated by burning off the coke at controlled conditions. Regeneration and reactivation of catalysts apply two major methods in the multi-reactor units of catalytic refor- ming. Reactivation of the catalyst is completed in a sequence of steps wherein the agglomerated metal components are *Address correspondence to this author at the Process Development Department, Egyptian Petroleum Research Institute, Cairo 11787, Nasr City, P.O. Box 9540, Egypt; Fax: 22747433; Tel: 0020222608600; E-mail: [email protected] automatically redispersed. In the semi-regenerative process, the entire unit is operated by gradual and progressive increase of temperature to maintain the activity of the catalyst until finally the hydrocarbon is cut out and the entire unit is shut down for regeneration of the catalyst. In cyclic processes, the reactors are individually isolated or taken off oil and swung out of line by various manifolding arrange- ment, motor operated adjustment by valves and the like. The catalyst is regenerated to remove the coke deposits, and then reactivated while the other reactors of the series remain on oil. A swing reactor temporarily replaces a reactor which is removed from the series for regeneration and reactivation of the catalyst until it is put back in the series. An advantage of the cyclic operation is that higher on-oil operating severities can be employed. Since there is no necessity to shut down the unit for catalyst regeneration and reactivation. Environmentally, lead phase-down and lead phase-out are required and refiners are under pressure to improve operation efficiency by employing better reforming tech- nology. Higher C 5 + liquid yields of higher octane product are being demanded. A traditional approach is to modify existing reforming catalysts or find new catalysts to improve yield by suppressing metal and acid cracking reactions. Another approach has been to reduce the operating pressure of the unit, which though favors increased yield and aromatization, leads to premature catalyst deactivation due to an increased rate of coke deposition. Although to some extent coke can be overcome by high hydrogen recycle rates, the combination of low pressure and high hydrogen recycle rate is incompatible with existing equipment. Reduction in hydrogen recycle rate at low pressure, though desirable, can lead to catastrophic catalyst deactivation, especially when the unit is operated at ultra-low pressures where C 5 + liquid yield is optimized. This makes conventional operations represent a compromise in process conditions where increased yield potential is sacrificed to maintain unit operability. Catalysts of high activity and high stability at such harsh severity have to be designed.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Recent Patents on Chemical Engineering, 2008, 1, 113-125 113
1874-4788/08 $100.00+.00 © 2008 Bentham Science Publishers Ltd.
Catalysis in the Petroleum Naphtha Catalytic Reforming Process
Ahmed K. Aboul-Gheit* and Salwa A.-W. Ghoneim
Process Development Department, Egyptian Petroleum Research Institute, Nasr City, P.O. Box 9540, Cairo 11787,
Egypt
Received: November 14, 2007; Accepted: January 31, 2008; Revised: February 24, 2008
Abstract: Before the US. Patent (1949) by Haensel wherein platinum was used as the active metal component in the
industrial catalytic reforming catalysts, molybdenum and /or chromium oxides were used but were suffering from very
rapid deactivation by coke even when used under high pressures. Platinum containing catalysts enjoyed very high
selectivity via producing high yields of high octane C5+ reformates even at relatively much lower pressures. Improvements
were then carried out; some concerned with modifying the metal component, and others concerning modifying the support
as well as improving the processing schemes. So many patents were disclosing bimetallic or even polymetallic catalysts
containing primarily platinum and/or rhenium or iridium, or their combinations, in addition to tin, zinc, germanium,
bismuth, phosphorus, or chlorine, etc. These catalysts acquired advantageous activities, selectivities and long time-on
stream with maintaining high yields of high octane motor gasoline.
Keywords: Platinum, rhenium, iridium, Al2O3, aromatization, isomerization.
INTRODUCTION
In commercial reforming operations using such catalysts, one or a series of reactions constitute the heart of the reforming unit. Each reactor is generally provided with fixed bed, or beds, of the catalyst which receive down-flow feed, and each is provided with a preheater or interstage heater, because the reactions which take place are endothermic. During the operating cycle, a naphtha feed, with hydrogen, usually recycle hydrogen gas is concurrently passed through a reactor. The sequences of reforming reactions take place as a continuum throughout the series of staged reactors of the reforming unit. The product from the last reactor of the series is separated into a liquid fraction, and a vapor effluent. The former is recovered as a C5
+ liquid product; the latter is a
gas rich in hydrogen, and usually contains small amounts of gaseous hydrocarbons, from which hydrogen is separated and recycled to the first reactor of the process to minimize coke production.
The activity of the catalyst gradually declines during the time-on-stream of an operating cycle due to the build-up of coke, which is believed to result from the deposition of coke precursors such as anthracene, etc. and other condensed ring aromatic molecules on the catalyst, these polymerizing to form coke. During the operation, the process temperature is gradually raised to compensate for the activity loss caused by the coke deposition. Eventually, however, economics dictate the necessity of reactivating the catalyst. Conse-quently, in all processes, the feed must be cut out and the catalyst must necessarily be periodically regenerated by burning off the coke at controlled conditions.
Regeneration and reactivation of catalysts apply two major methods in the multi-reactor units of catalytic refor-ming. Reactivation of the catalyst is completed in a sequence of steps wherein the agglomerated metal components are
*Address correspondence to this author at the Process Development
Department, Egyptian Petroleum Research Institute, Cairo 11787, Nasr City,
P.O. Box 9540, Egypt; Fax: 22747433; Tel: 0020222608600; E-mail: [email protected]
automatically redispersed. In the semi-regenerative process, the entire unit is operated by gradual and progressive increase of temperature to maintain the activity of the catalyst until finally the hydrocarbon is cut out and the entire unit is shut down for regeneration of the catalyst. In cyclic processes, the reactors are individually isolated or taken off oil and swung out of line by various manifolding arrange-ment, motor operated adjustment by valves and the like. The catalyst is regenerated to remove the coke deposits, and then reactivated while the other reactors of the series remain on oil. A swing reactor temporarily replaces a reactor which is removed from the series for regeneration and reactivation of the catalyst until it is put back in the series. An advantage of the cyclic operation is that higher on-oil operating severities can be employed. Since there is no necessity to shut down the unit for catalyst regeneration and reactivation.
Environmentally, lead phase-down and lead phase-out are required and refiners are under pressure to improve operation efficiency by employing better reforming tech-nology. Higher C5
+ liquid yields of higher octane product are
being demanded. A traditional approach is to modify existing reforming catalysts or find new catalysts to improve yield by suppressing metal and acid cracking reactions. Another approach has been to reduce the operating pressure of the unit, which though favors increased yield and aromatization, leads to premature catalyst deactivation due to an increased rate of coke deposition. Although to some extent coke can be overcome by high hydrogen recycle rates, the combination of low pressure and high hydrogen recycle rate is incompatible with existing equipment. Reduction in hydrogen recycle rate at low pressure, though desirable, can lead to catastrophic catalyst deactivation, especially when the unit is operated at ultra-low pressures where C5
+ liquid
yield is optimized. This makes conventional operations represent a compromise in process conditions where increased yield potential is sacrificed to maintain unit operability. Catalysts of high activity and high stability at such harsh severity have to be designed.

114 Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 Aboul-Gheit and Ghoneim
The catalysts are constituted of composite particles which contain beside a carrier (support material), the platinum, rhenium and iridium metal components, and a halide component. The support frequently contains alumina, bento-nite, clay, diatomaceous earth, zeolite, silica, activated carbon, magnesia, zirconia, titania, etc. It is preferred that the support material acquires a surface area of more than 50 m
2g
1, preferably from 100-300 m
2g
-1, a bulk density of 0.3-
1.0 g. cm-3
, preferably 0.4-0.8 g. cm-3
, and average pore volume of 0.2-1.1 cm
3g
-1 , preferably 0.3-0.8 cm
3g
-1 and an
average pore diameter of 30-300 .
1. REFORMING CATALYSTS
The petroleum industry has been extremely active in attempting to convert straight-run naphtha and natural gasoline stocks of low octane number into high octane motor fuel so that it can be used in more efficient high compression engines. The modern catalytic reforming process is the most efficient outlet realizing this target. At first, this process used less active and less selective non-noble metal oxide catalysts. Although this process has come to commercial application, yet it didn’t provide an ultimate solution for production of high yields of high octane motor fuel from straight-run naphtha and natural gasoline feed stocks. These catalysts were principally composed of molybdenum or chromium or their mixtures on alumina, but their primary defect was the excessive coke deposition in the pores and on the surface of the catalysts and hence, their regeneration after short-time-on-stream periods was frequent, such that their overall life periods were very short.
1.1. Platinum Containing Catalysts
Haensel has achieved [1, 2] the most important discovery in the catalytic reforming science; he introduced platinum as the active metal component in catalytic reforming catalysts. Platinum has greatly suppressed carbon deposition and hence extended the periods between regenerations, beside its strong activity for dehydrogenation, dehydrocyclization and isomerization reactions. These patents were assigned to Universal Oil Products Company.
Practically, the full range naphtha fraction distilled from a crude oil can be separated into light and heavy naphthas. The former is subjected to a hydroisomerization process, whereas the latter is catalytically reformed. The former process is carried out at less severe operating conditions to minimize hydrocracking. Both upgraded (isomerized and reformed) naphthas are mixed to provide a high octane C5
+
gasoline pool and hydrogen. Catalytic reforming converts low octane n-paraffines and naphthenes to high octane aromatic-rich C5
+ liquid reformate and hydrogen.
Searching continues for improved reforming catalysts that offer high selectivity, i.e., high liquid C5
+ and H2 yields,
high activity, low coking rates and high stability. High selectivity should minimize the yield of undesirable C1-C4
gaseous products. Catalysts with acceptable selectivities have higher activity because they allow operation at lower temperatures while maintaining the same octane level and allow operation at the same temperature but at higher octane level. This will also allow for significant extension of the cycle length and reduce frequency of regeneration.
Steam treatment of H-mordenite (H-MOR) containing catalysts increased the hydroconversion activities of n-hexane, whereas this treatment decreased the activity of the platinum/NH4-MOR catalyst [3].
1.2. Promoted Platinum-Containing Catalysts
1.2.1. Platinum-Rhenium Catalysts
The original commercial catalyst employs platinum deposited on halogen- acidified - alumina support [1]. Kluksdhal [4] discovered the high efficiency of rhenium when combined with platinum at atomic ratios of rhenium to platinum between 0.2 and 2.0, then in 1971 he launched another patent [5] showing the importance of holding the atomic ratio of rhenium to platinum to less than 1.0. Higher ratios than 1.0 can be used but generally no further significant improvement is obtained. The patent also discloses that the naphtha feed should be essentially free of sulfur. At least contains less than 5 ppm, and still more preferably less than 1 ppm. In patent [4], Kluksdhal again indicates that the atomic ratio of rhenium to platinum has to be essentially not greater than 1.0. More preferably, the atom ratio is less than about 0.7. Rhenium and platinum have about the same atomic weight; the atomic ratio is essentially the same as the weight ratio.
An article entitled "New development in reforming by Pollitzer, Haensel and Hayes [6], shows that the yield of C5
+
liquid product reformate reaches a maximum when the Re constitutes 50% of the total catalytic metal (platinum/ rhenium =1) and that thereafter this yield declines as the relative platinum/rhenium wt ratio is either increased or decreased. The relationship holds true over a fairly wide range of platinum content, indicating that the modifying effect of rhenium is indeed exerted on the platinum.
Rhenium was found to greatly stabilize platinum during operation [7] and several authors [8-14] attempted to reveal the role of rhenium in the platinum- rhenium/ alumina catalysts. Wagstaff and Prins [8] suggested that the oxides of the two metals exist separately on the support but their reduction is initiated by some reduced platinum nuclei, probably through dissociating hydrogen. These authors proposed that Re2O7, in the hydrated form, becomes mobile and able to migrate to the platinum reduction centers where the two oxides reduce simultaneously and an alloy may be formed.
Isaacs and Petersen [15] supported these findings and proposed that a variety of hydration degrees of Re2O7 can exist that result in varying mobilities of this oxide. These mobilities depend on the drying temperature of the catalyst. The higher the temperature, the lower the degree of hydration and, hence, the lower the mobility of Re2O7. These authors [15] also indicated that interaction between platinum and rhenium takes place during reduction in all cases. A patent by Buss [16] discloses the inclusion of a minor amount (< 0.1%) of iridium in a catalyst containing up to 0.3% rhenium and 0.3% platinum. Whereas Gallagher and Yarrington [17] disclose a bimetallic reforming catalyst containing rhenium and platinum in atomic ratios between 2 and 5.

Catalytic Reforming of Naphtha Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 115
Platinum-Rhenium catalysts are of two types: those with equal amounts of Pt and Re, with typical weight percentages of Pt (0.3%)-Re (0.3%)-S (0.04%)/Al2O3-Cl (0.9%), and the so called skewed metal catalyst with more rhenium than platinum-typically 0.2% Pt and 0.4% Re [18].
Galperin et al. [19] have disclosed the use of a support, and e.g., alumina impregnated extrudate with EDTA and a tin compound. The active component is platinum group metal and rhenium (may be used). These catalysts give enhanced yield, activity and stability for conversion of naphtha into high octane gasoline and aromatics.
1.2.2. Platinum-Iridium Catalysts
The second metal that can be a peer competitor of rhenium in the industrial catalytic reforming catalysts is iridium. The platinum-iridium catalysts are particularly resis-tive to carbon deposition by virtue of the hydrogenolyzing activity of iridium metal. Additives may be also used during the impregnation step to improve the dispersion of the metal into the support [20].
A bimetallic catalyst (platinum-iridium/alumina(Cl)) of some industrial importance appeared in the early 1970's in a US patent by Buss [21], it can be prepared from chloro-platinic and chloroiridic acids, either by two successive impregnation or by coimpregnation. The behavior of the two metals in reducing or oxidizing atmospheres are so different that there is doubt about easily maintaining intimate contact between both metals once it has been formed. Foger and Jaeger [22], concluded that attaining a single phase platinum-iridium alloy is possible only if the concentration of platinum and iridium are nearly equal. After oxidation below 300ºC, Iridium alone is transferred to the oxide and above this temperature Iridium oxide crystals segregate. Above 550ºC, IrO2 is transported through the gas phase. This was in accordance with those obtained by Garten and Sinfelt [23]. Through coimpregnation of H2PtCl6 and H2IrCl6, if the exposure to air is maintained <375ºC, highly disperse bime-tallic clusters are obtained. Around 600ºC, large crystallites of iridium oxide are formed, and after reduction, the catalyst transforms to highly disperse platinum or platinum rich clusters and crystallites. This emphasizes the importance of applying a physical treatment subsequent to catalyst impregnation.
Several studies are concerned with the application of iridium and platinum - iridium catalysts in hydrocarbon hydroconversion reactions. Aboul-Gheit and Abdel-Hamid [24] explain how two precursors of platinum and iridium possessing equal rates of adsorption on -alumina support and equal platinum-rhenium ratios are homogeneously dispersed through adding a proper additive to prepare bimetallic catalysts. Two metal precursors possessing diffe-rent adsorption rates could not be homogeneously distributed in the support. Chloroplatinic (H2PtCl6) and chloroiridic (H2IrCl6) acids are two homogeneously dispersed precursors which are used for preparing highly active platinum- iridium/ alumina catalysts. Here, the platinum and iridium precursors can be successfully co-impregnated and homogeneously dispersed in a single impregnating solution. Activities may be also used during the impregnation step of catalyst preparation to improve the dispersion of the metal(s) into the
support. For instance, in preparing platinum-iridium / alumina catalysts, Aboul-Gheit and Abdel-Hamid [24] show how two precursors of platinum and iridium possessing equal rates of adsorption on - alumina support are homo-geneously dispersed through adding a proper additive to prepare the bimetallic catalyst, whereas two precursors possessing different adsorption rates (coefficients) could not be homogeneously dispersed in the support extrudate.
Huang et al. [25], concluded that incorporation of plati-num into iridium cluster retards the oxidative agglomeration of iridium. When bimetallic catalysts were oxidized at 320ºC, the majority of surface species were bimetallic platinum-iridium oxichlorides; no significant IrO2 agglome-rates were observed. A few patents claim improved results with addition of a 3
rd element to platinum-iridium couple
which is capable of promoting change of the metal support interaction.
A platinum-iridium-chromium catalyst is prepared by co-impregnation of alumina with H2PtCl6, H2IrCl6 and chromic acid. Before impregnation, the alumina beads were pretreated under CO2 flow [26]. In all cases of preparation an eggshell profile of the catalyst pellets appeared. Pretreatment of alumina with CO2 at room temperature promotes a desirable uniform metal distribution. A uniform distribution profile is normally obtained by competitive impregnation of H2PtCl6 and a higher HCl concentration. Such low platinum and high chloride concentration, make the exchange sites of alumina so numerous and the chloride legends of platinum well protected.
CO2 is not acidic enough to maintain low pH and does not protect the chloride environment of platinum. The lower acid sites number of the support and the formation of hydrolyzed platinum species having a lower affinity for the support lead to a uniform distribution profile of platinum. This positive effect of CO2 as also being used during the introduction of platinum and iridium on alumina modified by other elements introduced either before silicon, magnesium or after calcium, phosphorous, barium, selenium shaping [27].
1.2.3. Promotion by Non-Metals
Halides may be added to the reaction zone during refor-ming, such as by injecting HCl gas, carbon tetrachloride, or an alkyl halide in the naphtha feed and /or into recycle hydrogen gas stream entering the reaction zone of the reformer. The amount of water in the reaction zone should be adjusted to maintain a molar ratio of water to chloride that permits high Cl
- stability. HCl was a successful additive that
increases the metals dispersion as a function of increasing its concentration.
It is preferable that the platinum-rhenium catalysts be presulfided prior to reforming to avoid excessive hydro-cracking. The presulfiding is conducted via passing a gaseous stream containing H2S, alkylmercaptan or carbon disulfide on the catalyst until the catalyst contains 0.1-0.5 wt to wt of sulfur: rhenium ratio.
In reforming catalysts since at least 1959, phosphorous has been known to enhance aromatic yields. Haensel [2] has taught this in his US patent. In a patent by Alley [28] it is

116 Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 Aboul-Gheit and Ghoneim
disclosed that a catalyst containing chelating ions of a group VIII noble metal with phosphoric acid enhanced the isomeri-zation activity.
Antos [29-32] shows that addition of phosphorous to a noble metal reforming catalyst results in improved C5
+ yield,
and Wilhelm taught that the inclusion of bismuth in a platinum group reforming catalyst improves selectivity, activity, and stability characteristics [33-36]. Antos [37] again disclosed that platinum, bismuth, nickel and halogen components, are affection catalytic components in hydro-carbon conversion catalysts.
Wu and Drake [38, 39], have described a reforming cata-lyst of mixed composition or stage-loaded comprising a first catalyst containing platinum and rhenium on a porous carrier material and a second catalyst comprising a bismuth and silica components. However, no one has disclosed the benefits of including both bismuth and phosphorous in a noble metal naphtha reforming catalysts.
Tanev [40] disclosed a bismuth and phosphorous con-taining -alumina as catalyst supports in naphtha reforming catalysts comprising chlorine and optionally rhenium. This catalyst unexpectedly exhibited significantly lower coking rates and C5
+ yields with higher stability relative to catalysts
containing only either bismuth or phosphorous. The effective amount of bismuth and phosphorous are distributed in the support particles, 0.05-0.1 wt% of bismuth, 0.05-0.6 wt% of phosphorous and the support composition is 62% alumina powder plus 38% alumina sol (prepared using alumina, acetic acid and water).
Bogdan`s US patent [41] discloses a catalyst comprising a refractory inorganic oxide, platinum-group metal, group IVA metal, indium and Lanthanide-series metal. Signi-ficantly improved selectivity of the naphtha reforming reactions as well as of aromatics production are realized. Bogdan and Bricker (UOP, Des Plaines) have disclosed the use of refractory inorganic oxide, platinum group metal, uniform IVA and a surface- layer Lanthanide series metal (cerium) [42]. The catalyst is particularly selective for aromatics rich product. An organic chloride is contacted with a water free reformer catalyst reactor in an amount and for a time period that are effective to restore at least a portion of the activity of the reformer catalyst.
Bogdan et al. [43] have provided a reforming process, selective for paraffins dehydrocyclization to aromatics, using a large-pore molecular sieve catalyst containing a uniformly distributed platinum group metal component, and a tin component incorporated into the large-pore molecular sieve (L-Zeolite) by secondary synthesis. The use of this catalyst results in greater selectivity of conversion of paraffins to aromatics and improved catalyst stability.
A group of inventors participating in a US Patent [44] used a catalytic system comprising a metal of group VIII, a metal of group VI, a metal oxide as a carrier and suitable quantities of components selected from a zeolite of the FER type, phosphorous and a mixture thereof, in upgrading of hydrocarbons boiling in the naphtha range containing sulfur impurities, namely in hydrodesulphurization with contem-poraneous skeletal isomerization of olefins contained in this
naphtha, together with reaction of olefin hydrogenation, carried out in a single step.
Lin and Parsons [45] have contacted an organic chloride with the reformer catalyst in an amount and for a time period that are effective to restore at least a portion of the activity of the reformer catalyst. Also, Lin et al. [46] have prepared a catalyst containing a group VIII metal, or group VII B metal or tin, germanium, copper, selenium or combination of two or more metals or oxides thereof is activated by:
a) Reduction by a flow of a reducing gas.
b) Flowing a halogen-containing compound for a first time period (>1 min and < 60 min).
c) Discontinuing the flow of the halogen for a period > 1 min. In an activation zone the catalyst is activated via removing water.
Moreover, Lin [47], discloses that, the presence of an organic aluminum halide in the feed can be effective for inhibiting deactivation of the reformer catalyst.
Verduijn et al. [48], have provided a process for reforming petroleum hydrocarbon stocks and in particular, aromatizing, with a catalyst of zeolite KL impregnated with a metal promoter e.g., platinum, in which the zeolite crystals are hockeypuck shape. The process has a good yield and selectivity for the desired reformed products and the catalyst is stable, associated with a low rate of coke formation and has a long catalyst active life.
In Fig. (1) the C6-C10 feed reforms at low severity and product separation to C6-C7 saturates and C6-C10 aromatics. The saturates are then reformed at ultra-low pressure. The second route differ from the first in that the feed is primarily fractionated to C8-C10 cut which is subjected to conventional reforming and the C6-C7 cut which is reformed at ultra-low pressure.
Touvelle et al. [49], have disclosed a method for treating naphtha. The naphtha feed rich in naphthenes is contacted with a ring opening catalyst containing group VIII metal and conditions suitable for ring opening of naphthene rings to form a ring opened products. The ring open product can then be contacted with a catalytic cracking catalyst under effective cracking conditions to form an olefin product which is particularly high in ethylene and propylene content.
Ducreux and Jolimaitre [50], introduced a process com-bining hydroisomerization and separation using a zeolitic adsorbent with a mixed structure for the production of high octane number gasoline. Multibranched paraffins contained in C5-C8 cut are separated by at least one zeolite adsorbent with a mixed structure and principal channels with openings defined by a ring containing 10 oxygen atoms and secondary channels with openings by a ring of at least 12 oxygen atoms. The secondary channels only being accessible to the feed to be separated via the principal channels.
Gillespie and Cohn [51], have carried out a selective upgrading of paraffinic feedstock to obtain isoparaffin-rich product for blending into gasoline. They used catalysts containing a support composed of sulfated oxide or hydroxide of a group IVB metal, a component of at least one lanthanide element or yttrium component (preferably

Catalytic Reforming of Naphtha Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 117
ytterbium) and at least one platinum group metal component (preferably platinum).
Zhao and Wagner [52], have devised a process for catalytic reforming of a hydrocarbon feed stream containing H2O, CO2, CH4 and CO at levels such that the H2O/CH4 is less than 0.8 and the CO2/CH4 is greater than 0.5 and the feed stream further contains quantities of sulfur compounds up to about 20 ppm. The catalyst used in this process con-tains from ~0.5 wt% to about 25 wt% of a calcium com-pound additive, from about 2 wt% to about 30 wt% nickel, and from about 25 wt% to about 98 wt% of an aluminum compound carrier, wherein substantially all of the calcium is combined with the alumina. The reforming process can be utilized to produce syngas, especially low hydrogen to carbon monoxide ratio syngas for applications such as iron ore reduction.
Glover in an EP patent [53] improved a catalytic refor-ming process which enhanced the product yield structure by the combination of bifunctional catalytic reforming and zeolitic reforming catalysts in a sandwich configuration which shows a surprising improvement in aromatics yield. Contacting the feedstock sequentially with a catalyst system which comprises a first bifunctional catalyst containing platinum, a metal promoter, a refractory inorganic oxide and a halogen in the first catalytic zone. The zeolitic reforming catalyst comprising a nonacidic zeolite (L-zeolite) and a platinum-group metal in the zeolitic-reforming zone. A terminal bifunctional catalyst comprising platinum, a metal promoter, a refractory inorganic oxide and a halogen is also combined. The first and terminal bifuctional catalysts are the same, the metal promoter of the first and terminal catalysts are selected from group IVA metals; rhenium and indium.
The terminal catalyst zone comprises a moving-bed system with continuous catalyst regeneration.
In the following paragraph, we introduce a further knowledge to the reader about the catalytic activity. It is very important to clarify how a metal promoter for platinum behaves differently towards different reactions in the same catalytic process.
Aboul-Gheit and coworkers [54] found that in benzene hydrogenation, the activity of a catalyst containing 0.35wt% platinum promoted with 0.35wt% tungsten or 0.35wt% germanium supported on alumina in presence or absence of Cl are in the order:
Pt-W (Cl) > Pt-W > Pt (Cl) > Pt > Pt-Ge > Pt-Ge (Cl).
For n-heptane isomerization, the activity order of these catalysts is as follows:
Pt-Ge (Cl) > Pt (Cl) > Pt > Pt-Ge > Pt-W (Cl) > Pt-W
For n-heptane hydrocracking, the activities of those catalysts are in the order:
Pt-Ge (Cl) > Pt-Ge > Pt (Cl) > Pt-W (Cl) > Pt > Pt-W.
Evidently, germanium can be a successful promoter for platinum, particularly in presence of Cl
- ion in the isome-
rization and hydrocracking of n- heptane, but may act as an inhibitor for platinum in benzene hydrogenation. On the contrary, tungsten acts as a successful promoter for platinum in benzene hydrogenation, whereas germanium acts as an inhibitor in benzene hydrogenation, particularly when Cl
- is
included. In some patents, where improvement is directed towards promoting a target reaction, the inventors should
Fig. (1). Flow chart of the catalytic reforming process of C6-C10 feed.
C6 – C7
Saturates
C6 – C10
Blend Feed C6 – C10
Aromatics
C8 – C10
Cut
C6 – C10 Feed Blend C6-C7
Cut
Low severity reforming
Separation Ultra-low pressure reforming
Fractionation
Ultra-low pressure reforming
Conventional reforming
118 Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 Aboul-Gheit and Ghoneim
sufficiently take care of the negative effect accomplished on another reaction taking place in the same process.
Three -alumina supported catalysts containing 0.6wt% of the transition metals platinum, ruthenium, and palladium as well as combination of each of these metals with rhenium have been studied for hydrogenating the aromatics present in a desulfurised white spirit fraction. The catalyst containing 0.6wt% rhenium, which represents complete replacement of the transition metal, appears to be completely inactive for aromatics hydrogenation. The relative activities of the catalysts containing 0.6wt% of the metals platinum, ruthenium, and palladium at 100ºC are in the order 4.2: 2.6: 1, respectively, whereas for the 50/50 combinations of these metals with rhenium are of the order 4.1:1.9:1 respectively, indicating that the replacement of ruthenium with rhenium may lower hydrogenation activity of ruthenium by 30% [55,56].
However, the platinum-rhenium combination has been used in catalysts containing H-MOR. The incorporation of 0.35 wt% rhenium or thorium in catalyst containing 0.35wt% platinum has decreased n-heptane isomers in product from 57.0% to 18.5% and 17.0%, respectively, although these two metals have increased the effectiveness factor for this reaction [57]. Aboul-Gheit et al. [58] have studied the hydroconversion for n-heptane using the dealuminated H-MOR. Again platinum, rhenium and platinum-rhenium supported on Na-MOR catalysts show high effectiveness factor values which assumed that the rate-determining steps in n-heptane hydroconversion are chemical and not physical factors.
In coimpregnating ammonium para-tungstate with chloroplatinic or chloroiridic acid, a poor dispersion occurs
and a portion of the para-tungstate precipitates. To overcome their precipitation, the para-tungstate was dissolved in H2O2. Eventhough, the dispersion was still poor because the adsorption rates of the two precursors were not equal. Similarly, the authors prepared the bimetallic combination [59]; Platinum-germanium; platinum-molybdenum; plati-num-rhenium; platinum-ruthenium; platinum-tungsten; ruthenium-rhenium and palladium- rhenium, to be used for deep aromatics hydrogenation and n-paraffins isomerization (reforming reactions).
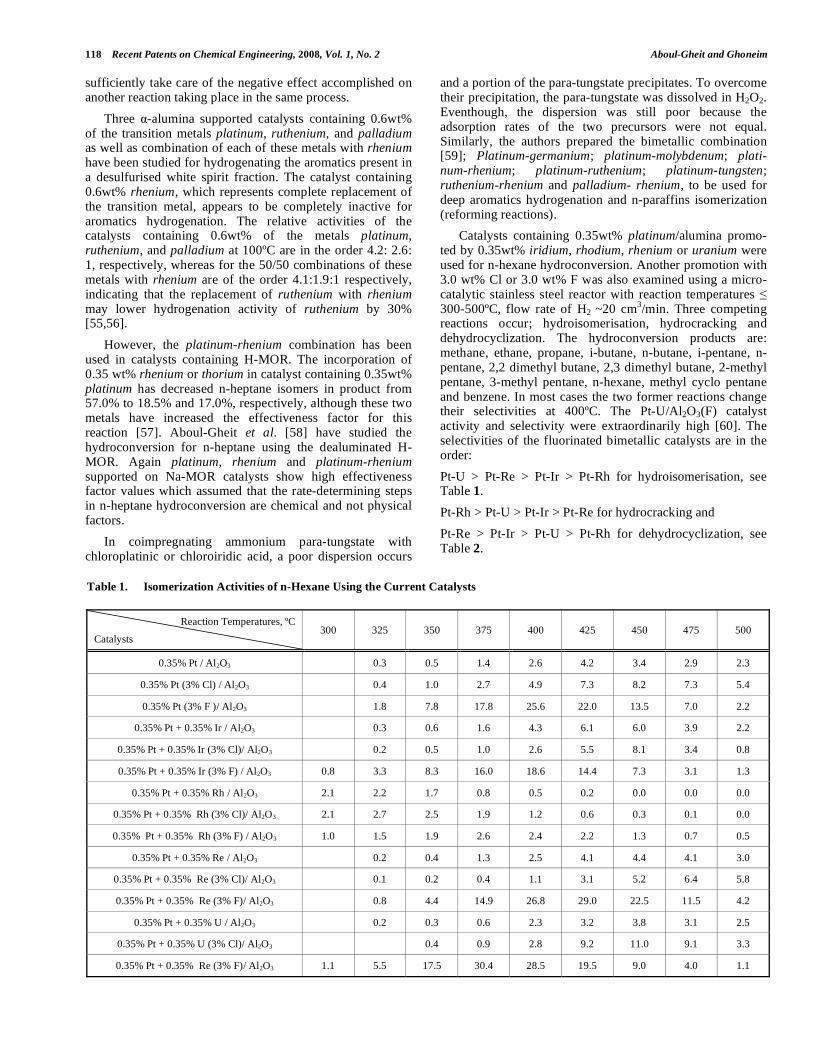
Catalysts containing 0.35wt% platinum/alumina promo-ted by 0.35wt% iridium, rhodium, rhenium or uranium were used for n-hexane hydroconversion. Another promotion with 3.0 wt% Cl or 3.0 wt% F was also examined using a micro-catalytic stainless steel reactor with reaction temperatures 300-500ºC, flow rate of H2 ~20 cm
3/min. Three competing
reactions occur; hydroisomerisation, hydrocracking and dehydrocyclization. The hydroconversion products are: methane, ethane, propane, i-butane, n-butane, i-pentane, n-pentane, 2,2 dimethyl butane, 2,3 dimethyl butane, 2-methyl pentane, 3-methyl pentane, n-hexane, methyl cyclo pentane and benzene. In most cases the two former reactions change their selectivities at 400ºC. The Pt-U/Al2O3(F) catalyst activity and selectivity were extraordinarily high [60]. The selectivities of the fluorinated bimetallic catalysts are in the order:
Pt-U > Pt-Re > Pt-Ir > Pt-Rh for hydroisomerisation, see Table 1.
Pt-Rh > Pt-U > Pt-Ir > Pt-Re for hydrocracking and
Pt-Re > Pt-Ir > Pt-U > Pt-Rh for dehydrocyclization, see Table 2.
Table 1. Isomerization Activities of n-Hexane Using the Current Catalysts
Reaction Temperatures, ºC
Catalysts 300 325 350 375 400 425 450 475 500
0.35% Pt / Al2O3 0.3 0.5 1.4 2.6 4.2 3.4 2.9 2.3
0.35% Pt (3% Cl) / Al2O3 0.4 1.0 2.7 4.9 7.3 8.2 7.3 5.4
0.35% Pt (3% F )/ Al2O3 1.8 7.8 17.8 25.6 22.0 13.5 7.0 2.2
0.35% Pt + 0.35% Ir / Al2O3 0.3 0.6 1.6 4.3 6.1 6.0 3.9 2.2
0.35% Pt + 0.35% Ir (3% Cl)/ Al2O3 0.2 0.5 1.0 2.6 5.5 8.1 3.4 0.8
0.35% Pt + 0.35% Ir (3% F) / Al2O3 0.8 3.3 8.3 16.0 18.6 14.4 7.3 3.1 1.3
0.35% Pt + 0.35% Rh / Al2O3 2.1 2.2 1.7 0.8 0.5 0.2 0.0 0.0 0.0
0.35% Pt + 0.35% Rh (3% Cl)/ Al2O3 2.1 2.7 2.5 1.9 1.2 0.6 0.3 0.1 0.0
0.35% Pt + 0.35% Rh (3% F) / Al2O3 1.0 1.5 1.9 2.6 2.4 2.2 1.3 0.7 0.5
0.35% Pt + 0.35% Re / Al2O3 0.2 0.4 1.3 2.5 4.1 4.4 4.1 3.0
0.35% Pt + 0.35% Re (3% Cl)/ Al2O3 0.1 0.2 0.4 1.1 3.1 5.2 6.4 5.8
0.35% Pt + 0.35% Re (3% F)/ Al2O3 0.8 4.4 14.9 26.8 29.0 22.5 11.5 4.2
0.35% Pt + 0.35% U / Al2O3 0.2 0.3 0.6 2.3 3.2 3.8 3.1 2.5
0.35% Pt + 0.35% U (3% Cl)/ Al2O3 0.4 0.9 2.8 9.2 11.0 9.1 3.3
0.35% Pt + 0.35% Re (3% F)/ Al2O3 1.1 5.5 17.5 30.4 28.5 19.5 9.0 4.0 1.1

Catalytic Reforming of Naphtha Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 119
Dehydrogenation of cyclohexane on catalysts containing platinum, rhodium, rhenium, uranium, platinum-iridium, platinum-rhodium, platinum-rhenium, and platinum-uranium on alumina were studied and the platinum-uranium catalyst was the most active. The rhodium/alumina catalyst was the most hydrogenolysing catalyst, producing propane; due to the higher percentage of d-bond character of rhodium. Introduction of uranium inhibits the activity of platinum. The effects of Cl
- and F
- were optimum at 3.0%, which may
be due to improving the spillover of hydrogen as well as to increase the dispersion of the metal in the support. The inclusion of Cl reduces the activity of platinum-iridium, whereas both Cl and F reduce the activity of the platinum-rhodium containing catalyst [61].
In Table 1, the monometallic Pt/alumina gives very low isomerization activity (maximum 4.2%), the activity was modestly enhanced by chlorine (maximum 8.2). A further increase of isomerization activity was obtained by fluorine treatment (maximum 25.6). Combining Ir with Pt gives maximum 6.1%, addition of chlorine attains a maximum of 8.1% whereas addition of fluorine gives maximum isomers of 18.6%. Again, combining Rh with Pt gives isomers of 2.2%, addition of chlorine to the catalyst slightly raise the activity to 2.7%, however, the addition of fluorine also does not give further activation. When Re was combined with Pt, the maximum activity is 4.4%, addition of chlorine gives a maximum of 6.4%, whereas, addition of fluorine enhances the isomerization activity to reach a maximum of 29%. Uranium combination with Pt gives a maximum of 3.8% isomers. Addition of chlorine enhances the activity to obtain
a maximum of 11%, whereas fluorination of this catalyst gives as high as 30%.
Table 2 indicates that, although the isomerization activi-ties shown in Table 1 increase to reach a maximum as a function of temperature, beyond which this activity dec-reases with the further increase of temperature. However, the dehydrocyclization activity on the contrary increases as a function of temperature. Iridium combination with platinum increases benzene production via chlorine or fluorine treatment from 10.0 to 12.6 and 18.6%, respectively, at 500ºC. Rhodium on the contrary, acts as an inhibitor in all cases, which fails to give measurable activation.
Re combined with Pt gives 7.1% at 500ºC; addition of chlorine does not give an increase, but addition of fluorine increases the activity to as high as 35.5% (the best treat-ment). On the other hand, combination of Pt with U gives 5% at 500ºC; addition of chlorine gives 16.2% and addition of fluorine gives 29%.
Benzene and toluene were hydrogenated to cyclohexane and methyl cyclohexane, respectively. To a catalyst containing 0.35wt% platinum/Al2O3:
(a) 0.35wt % of the metals iridium, rhodium, rhenium or uranium was incorporated, and (b) fluorination or chlorination was accomplished.
1- All catalysts gave good activities at 125-150°C.
2- Iridium or rhenium combination with platinum enhances the activity of platinum, whereas, uranium acts as an inhibitor.
Table 2. Dehydrocyclization Activities of n-Hexane Using the Current Catalysts
Reaction Temperatures, ºC
Catalysts 300 325 350 375 400 425 450 475 500
0.35% Pt / Al2O3 0.0 0.1 0.3 0.7 2.1 3.1 4.8 5.9
0.35% Pt (3% Cl) / Al2O3 0.0 0.2 0.8 2.2 5.0 7.9 11.8 17.1
0.35% Pt (3% F )/ Al2O3 0.0 0.2 0.8 3.4 11.0 18.3 20.4 24.2
0.35% Pt + 0.35% Ir / Al2O3 0.0 0.2 0.4 1.6 3.6 5.3 8.6 10.0
0.35% Pt + 0.35% Ir (3% Cl)/ Al2O3 0.0 0.0 0.2 0.5 1.8 5.7 8.7 12.6
0.35% Pt + 0.35% Ir (3% F) / Al2O3 0.0 0.2 0.4 2.2 5.7 10.4 14.7 16.2 18.6
0.35% Pt + 0.35%Rh / Al2O3 0.4 1.4 3.3 3.9 3.7 2.5 1.3 1.5 0.4
0.35% Pt +0.35% Rh (3% Cl)/ Al2O3 0.3 1.2 2.8 4.5 4.9 3.3 3.8 1.9 1.7
0.35% Pt +0.35% Rh (3% F) / Al2O3 0.2 0.8 1.6 2.7 3.0 3.4 3.6 3.8 3.7
0.35% Pt + 0.35%Re / Al2O3 0.0 0.0 0.4 0.9 2.5 3.5 4.8 7.1
0.35% Pt +0.35% Re (3% Cl)/ Al2O3 0.0 0.0 0.1 0.4 1.1 2.5 4.2 7.4
0.35% Pt + 0.35% Re (3% F)/ Al2O3 0.0 0.0 0.7 2.8 7.3 16.5 25.3 35.5
0.35% Pt + 0.35% U / Al2O3 0.0 0.1 0.2 0.7 1.4 2.8 3.7 5.0
0.35% Pt + 0.35% U (3% Cl)/ Al2O3 0.0 0.1 0.6 1.9 4.8 11.9 16.2
0.35% Pt + 0.35% Re (3% F)/ Al2O3 0.0 0.1 1.7 5.4 10.1 19.7 28.0 29.0

120 Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 Aboul-Gheit and Ghoneim
3- Halogenation promotes the activity.
4-Alkyl substituent enhances the aromatic ring hydrogenation [62].
Catalysts exhibiting larger values of effectiveness factor ( ) and hence lower Thiele modulus values (ø) were found to enhance production of 2,3-dimethylpentane on account of 3-methylhexane, whereas the reverse is true with respect to catalysts exhibiting lower values of and higher values of ø [63].
2. CATALYTIC REFORMING PROCESSING
In multiple reactor reformer systems with at least two reactors serially connected and each containing a reformer catalyst and simultaneously injected with the charge in which a chlorinating agent is sequentially introduced (in water free conditions) in an amount and for a period of time effective to inhibit deactivation of the reforming catalyst. During operation of a conventional catalytic reforming system, the activity of the catalyst declines gradually over time due to the following reactions:
1. Formation of coke within the pores, as well as on the surface of the catalyst
2. Agglomeration of the catalyst metal component, and
3. Loss of the halogen component of the catalyst
The deactivation of a catalyst exhibits the following:
1. Lower product octane number
2. Requirement of higher reaction temperature
3. Higher required reaction pressure
4. Decreased time intervals between regenerations (cycle time)
5. Increased requirement for hydrogen, and
6. Decreased selectivity
A previous knowledge is that the deactivation of a reformer catalyst can be inhibited via chlorinating agent addition during reforming. This chlorination of the reformer catalyst is thought to inhibit deactivation by:
1. Counteracting the formation of coke on the catalyst
2. Redispersing the metal component(s) of the catalyst in a more uniform manner, and
3. Replacing the halogen component which has been stripped from the catalyst during reforming
Lin et al. [46] indicated that the catalyst can be activated in an activation zone by
1. removing water from the activation zone
2. reducing iron oxide contained in the activation zone in presence of a reducing gas thereby forming reduced iron and water
3. removing water from the activation zone
4. reducing the metal oxide of the catalyst in presence of a reducing gas thereby forming reduced metal and water
5. removing water from the activation zone
6. during step 4 flowing a halogen-containing compound over the catalyst for contact with the catalyst for a first time period; and
7. following step 6 and during step 4, substantially discontinuing the flow of halogen-containing compound over the catalyst for a second time period.
In an EP patent, McInnes [64] provides a catalytic reforming process (Fig. 2) preformed in a reforming unit comprising a plurality of catalyst-containing reaction zone or reactors connected in series, wherein feed naphtha is passed into the first (or upstream) reactor of the series of reactors in the unit and product naphtha having a higher octane rating than the feed naphtha is recovered from the last (or downstream) reactor of the series of reactors in the unit, the process comprising, in sequence, the following steps:
1. passing naphtha into and through the series of reactors of the unit and progressively increasing the temperature of at least the last reactor of the series of reactors to maintain the octane rating of the product naphtha at least at a given value
2. substituting a spare reactor containing active reforming catalyst in place of the last reactor of the series when the temperature of at least the last reactor of the series attains specified maximum temperature
3. passing feed naphtha through the reactors of the unit and recovering a naphtha product having at least the given octane rating from the last reactor of the series
4. progressively raising the temperature of at least the last reactor of the series of reactors to maintain at least the given octane of the product naphtha;
5. optionally repeating steps 2, 3 and 4
6. interrupting the passage of naphtha into and through the reactors of the unit, and regenerating catalyst (by in situ and /or ex situ regeneration) in at least the last reactor of the series. This step may effected by in situ regeneration and reactivation of catalyst in the reactors of the unit and at least one spare reactor, the spare reactor (s) being connected to form part of the unit; and
7. optionally repeating steps 1 to 6
The unit may comprise at least three reactors connected in series.
The amount of the catalyst in the last reactor of the series in the unit may comprise from 50 to 90 wt% of the total catalyst in the unit. The first reactor of the series in the unit contain from 5 to 20 wt% of the total catalyst in the unit. Whereas, this amount in the reactors between the first and the last reactors of the series may be in a rage of 5 to 45 wt% of the total catalyst in the unit
In step 1, the temperature of other reactor (s) between the first and the last reactors may be progressively raised to maintain the octane rating of the naphtha product.
The conventional practice of chlorinating a reformer catalyst in the reactors of a multiple-reactor reformer system is to inject a chlorinating agent into the hydrocarbon feed charged to the 1
st reactor of the series. The chlorinating agent
is then carried out with the hydrocarbon feed to the reaction

Catalytic Reforming of Naphtha Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 121
zone of the 1st reformer reactor and subsequently to the reac-
tion zones of the downstream reactors where it is contacted with the reformer catalyst. The water concentration in the feed to the 1
st reactor of the multiple-reactor system has to be
maintained within a certain concentration range while adding the chlorinating agent. The water-chloride ratio within the reformer reaction zone should be kept at an appropriate level so as to maintain both catalytic activity and stability by suppressing excessive hydrocracking occurring during conventional chlorination. The water concentration in the reformer feed is also maintained at certain levels in order to aid in carrying the chlorinating agent through the series of reformer reactors so as to properly expose the catalysts contained in the downstream reactors to the chlorinating agent.
However, the conventional reforming method has the disadvantage of requiring the presence of water in the hydrocarbon feed charged to the multiple reactors reforming system which can cause accelerated coking and thus accelerated deactivation of the catalyst. A further disad-vantage of requiring the presence of water in the hydro-carbon feed is that water can strip the halogen component from the reformer catalyst causing decreased activity and decreased stability. A further still disadvantage is that the reformer catalyst contained in the downstream reactors of the multiple-reactor reformer system experiences an accelerated rate of deactivation compared to the reformer catalyst in the upstream reactors of the system, thus decreasing the time between which the entire system must be shut down for regeneration of the reformer catalyst, i.e., decreased cycle time.
Lin and Parsons have achieved an improved reforming process (Fig. 3). They disclosed in a US patent [65] that the stability of the reformer catalyst is increased and also problems associated with the use of water as a means for
aiding in conveyance of the chlorinating agent from the first reactor of the series of reactors to the downstream reactors of the series are achieved. The stability in all reactors is improved significantly as compared with other conventional reforming processes as follows: A substantially water-free reformer feed comprising a reformable hydrocarbon stock is charged to a reformer system comprising at least two reactors serially connected in fluid flow communication, with each reactor containing at least a volume of reforming catalyst and operating under reforming conditions. While the substantially water-free reformer feed is being charged to the multiple-reactor reformer system, a chlorinating agent is introduced, without simultaneously introducing water, immediately upstream from the inlets of all the reformer reactors in an amount and for a period of time that is effective to inhibit the deactivation of the reformer catalyst. The introduction of the chlorinating agent into all the reformer reactors must occur sequentially, with only one reactor at a time receiving an injection of the chlorinating agent. The chlorinating agent may be injected in pure form or with a carrier. The carrier should dissolve the chlorinating agent, which may not be water; it is preferably a hydrocarbon of the same composition as the reformable hydrocarbon of the dry reformer feed. The injection rate is a rate sufficient to provide a concentration of the chlorinating agent in the dry reformer feed of from more than ~ 0.05 ppm of the dry reformer feed to less than about 50 ppm.
Lin and Parsons disclosed in a WO patent [66] that the presence of water in the reformer reaction zone before, during, or after chloriding is necessary to counteract the excessive hydrocracking which is typically encountered when chloriding a reformer catalyst. They discovered that chloriding of a reformer catalyst in a substantially water-free reaction zone without adding water to the reaction zone before, during or after chlording was thought to cause catalyst deactivation.
Fig. (2). Schematic flow diagram for catalytic naphtha reforming.
55 54
17
60 57 58
41 64
42
65
64
62
44 27
43
55 54
61
28
34
23
26
59
17 20 24
21
19
45
51 52
30
31
29
35 63 14
33
47
11
50
16 15 13
12
22 25 40
32
18

122 Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 Aboul-Gheit and Ghoneim
Again, Lin and Parsons [45] added that perchloro-ethylene can be injected into the substantially water-free hydrocarbon feed of from 0.5 to 50 ppm. They claimed that the reformer catalyst comprises platinum-rhenium/ Al2O3, and chlorine. Furthermore, their US patent in 2003 [67] gave some variety of chlorine-containing additives, and termed them nonmetallic organic chlorine containing additives, for example hexachloroethane, carbon tetrachloride, 1-chloro-2-methylpropane, 2-chloro-2-methylpropane, tert-butylchlo-rides, propylene dichloride, perchloroethylene and mixtures of two or more thereof. The presently more preferred nonmetallic organic chloride is perchloroethylene (PCE).
Walsh et al. [68], have disclosed that a process comprises separating a naphtha feed into a fraction comprising C7
-
hydrocarbons and heavy C8+ fraction. The C8
+ fraction is
separated into a light fraction comprising C8 and/or C8-C9, which then is reformed to produce gaseous and/or a desired distribution of aromatics. Boger and Sorensen [69], have provided a system of processes and reactors for the catalytic reforming of naphtha. The reactors comprising monometallic catalysts having honeycomb-type structure that allow for improved efficiency, productivity and reaction selectivity over conventional catalytic reforming technology.
In a WO patent, Salmon [70] provides a process and apparatus for increasing the concentration of aromatic compounds during catalyst reforming reactions by taking advantage of thermodynamic and chemical equilibrium considerations while utilizing readily available reactors to create a process and new apparatus that is more efficient and less costly than other alternate technologies.
PERFORMANCE MONITORING OF THE CATA-LYSTS
Catalytic reforming units operate best when the process variables are adjusted to produce the maximum yield of the
desired products within the limits of acceptable catalyst cycle life. The primary concerns of a refiner are the yield and the quality of the reformate and the catalyst performance in general. The starting point is a unit material balance, which is seldom found to be 100%. If the material balance is in the range of 98 to 102% wt%, the yield data can be used after adjusting the charge or reformate volumes to bring the balance to 100%. To allow optimization of the process conditions for best performance, any loss or gain in material should be viewed carefully.
The performance of a catalytic reformer depends on a combination of controllable process variables. Reactor temperatures and pressures, space velocity, chloride-water balance, and the level of feed contaminants all influence catalyst activity, selectivity, and stability. When the catalyst is performing properly, the reformer may be operated to attain the maximum yield of the desired product. A close monitoring of the process parameters during reformer operation is required to understand the variations in the catalyst performance. The major parameters used to monitor reformer-catalyst performance are as follows:
1. WAIT/WABT (weighted average inlet/ bed temperature)
2. Delta temperatures across the reactors
3. Octane number of feed and reformate
4. Precursor conversion/ recovery
5. Aromatics production
6. C5+ reformate yield
7. C3 / C1 ratio
8. Recycle rate and H2/HC ratio
9. Hydrogen production and purity
Fig. (3). schematic representation of the inventive process.
20 54 40 16 30 46
10 66
68
14
72
12 24
70
26
28 18
22 32
36
34
44
74
48
52
50
62 58
60
64
42 56
76

Catalytic Reforming of Naphtha Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 123
The reactor inlet temperatures, WAIT and WABT, are invariably the most effective parameters which can provide information about the relative activity of the catalyst. Generally, higher inlet temperatures indicate lower catalyst activity. This means that the catalyst has been deactivated and the increase in temperature is required to maintain the desired level of activity. Hence, these temperatures are used to monitor the performance of catalyst.
However, reactor temperature differentials convey the most crucial information regarding the sequence of reactions taking place in individual reactors /beds. Reactor tempe-rature differentials are strongly dependent on feedstock composition and circulating gas rate, which can be modified as required. These temperature variables, along with recycle gas composition and specific gravity, provide instantaneous indication of any change in reformate yield. Reactor pressure differentials are used to monitor catalyst bed fouling by particulate material. Iron is the most common particulate material and not only causes bed plugging but also interferes with catalyst chloride retention and acidity.
An increase in feed rate requires that reactor temperatures be increased to maintain a specified level of catalyst activity. For example, a 10% increase in feed rate necessitates approximately 3 to 4ºC increase in temperature in order to maintain octane. Conversely, reduction in feed rate warrants reduction in WABT to maintain octane. Although the feedstock composition is not an easily controllable variable, any change in the feed quality will be reflected in the performance to the catalyst. To ascertain the feed quality, Universal Oil Products Inc. (UOP) developed an expression N + 2A (naphthenes plus two times aromatics; liquid vol.%) using correlations of ASTM distillation and API gravity with feed composition in terms of paraffins, naphthenes, and aromatics. This formula essentially gives a measure of the aromatic precursors available in the feed which should contribute to the production of aromatics. This expression
also enables the calculation of precursor conversion or recovery to judge the overall performance of the catalyst. Table 3 shows briefly the effect of changes in feed quality and the process variables on the catalyst performance.
The hydrogen to hydrocarbon ratio has a significant effect on catalyst deactivation. The cycle life varies directly with hydrogen to hydrocarbon ratio to approximately the 1.6 power. The purity of recycle hydrogen varies with separator pressure and temperature, octane, and feedstock charac-teristics. Lower purity of recycle hydrogen indicates a lower hydrogen to hydrocarbon ratio and greater deactivation of the catalyst. Yield of aromatics in the product increases with increase in temperature and as a result the reformate octane also increases. But reformate yield decreases with increasing temperature. Thus, increasing octane results in lower reformate yields. Hydrogen yield also increases as octane rises, as do the other light ends. Good catalyst performance results in high hydrogen and reformate yields.
Composition of recycle gas and stabilizer overhead liquids and gases are best used in conjunction with accurate flow rates to determine overall methane (C1), ethane (C2), propane (C3), and butane (C4) yields. The molar ratio of propane to methane (C3/C1) obtained from these overall yields is an indication of cracking activity and is frequently monitored. Ideally, the C3/C1 mole ratio during normal operation should be in the range 0.7-1.2. Performance outside this range indicates that the catalyst is out of acid-metal balance. During a normal operating cycle, metal activity is not an easily moderated variable; therefore, the acid activity is the primary alterable variable. Consequently, if the performance of the catalyst indicates acid-metal imbalance, optimization of water-chloride injection rates will be essential. However, if the modification of the water-chloride balance is inadequate to restore the activity, the temperature is the only parameter which can be adjusted to achieve the desired performance [71].
Table 3. Effect of Feed Properties and Process Variables on Catalyst Performance
Increase H2 purity H2
yield
C5+
yield Delta temperature
Reactor
temperature
Feed property
API - - - - +
Paraffins - - - - +
Naphthenes + + + + -
Aromatics - - + - 0
Initial boiling point - - - - -
Final boiling point 0(-) + + + +
Process variable
Separator pressure + 0 + 0 0
Separator temperature - 0 - 0 0
Reactor pressure - - - - -
Reformate RONC - + - + +
“+, increase; -, decrease; 0, no effect.

124 Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 Aboul-Gheit and Ghoneim
DEACTIVATION BY COKING
Although numerous studies of the deactivation by coking of industrial or model reforming catalysts have been carried out, interpretations of the results sometimes diverge. The reason is that coking reactions are very complex and their rate can be defined only by the total amounts of coke deposited. However, this total coke loading is due to combination of the formation of coke precursors, their polymerization (on the support or on the metal), and their destruction by metallic or acidic mechanisms.
The following conclusions are well established:
-Coke can be deposited on the metal and on the acidic support.
-The metallic surface, at the steady state of the reaction, is covered by an amount of coke that remains constant when coke continues to be accumulated on the acidic support.
-The higher the metallic dispersion, the lower the coverage by coke of the metallic phase at steady state.
The catalytic properties of the metal are a responsible factor in the coking of bifunctional catalysts:
1. Olefins are produced and can be polymerized on the acidic sites of the support.
2. Polymers produced on the support can be stabilized through dehydrogenation by a reverse spillover of hydrogen.
3. Precursors of coke can be destroyed at high temperature by the metal (and also on the support by spillover of hydrogen).
Coke deposition on the support is defined by the acid-base properties of the catalyst-coking precursor pair.
Location of coke (on the metal or on the support) and nature of coke (light or graphitic) are more important parameters for catalyst stability than coke content.
Modification of Pt by Re or Ir decreases the amount of deposited coke and induces preferential deposition of coke on the support.
Modifications of Pt by Group IV components (Sn, Pb, Ge) or sulfurization of the metallic function also improve the reforming catalyst stability. This improvement is linked to preferential deposition of coke on the support.
The very low selectivity of catalytic reforming for the coking reaction (only one atom of carbon out of 200,000 activated by a Pt/Al2IO3-Cl catalyst is transformed into nondesorbable coke), catalyst lifetime in the reforming process can still be enhanced by modification of the metallic phase. In fact, such improvements allow the use of reforming catalysts under the more severe reaction conditions needed for the production of lead-free gasoline [72].
In a EP patent, schorfheide [73] disclosed that, in a process wherein, a series of reforming zones, or reactors, each of which contains a bed, or beds of a sulfur sensitive polymetallic platinum-containing catalyst, the beds of catalyst are connected with a hydrocarbon or naphtha feed, and hydrogen, at reforming conditions to produce a hydro-carbon, or naphtha product of improved octane, the
improvement wherein, at start-up, sulfur is add to the tail reactor of the series, and excluded from the lead reactor. Increased hydrogen purity, aromatics, and C5
+ liquid yield
are obtained and there is less gas make.
Fersing and Nasclmento [74] enhanced a method for the isomerization of a hydrocarbon charge containing a substantial quantity of paraffin base hydrocarbons with 5 or 6 carbon atoms and a benzene content 2 wt%, in which the charge to be treated passes, in the presence of hydrogen, at a total pressure 10 bars and at an average temperature ranging between 100 and 200ºC, through at least one reactor containing a catalyst. An adjunctive fluid is introduced in the upstream section of the reaction zone; a fluid that at 40ºC, and under atmospheric pressure, is in a gaseous phase and has a density n-pentane density taken into account under the same conditions.
CURRENT & FUTURE DEVELOPMENTS
In the 50’s, the catalytic reforming process was direc-ted towards maximizing the production of aromatics (~50%); by the virtue of their high octane numbers. Nevertheless, not only aromatics have been restricted to 20% and also TEL was banded for its high toxicity. The outlet was to maximize isomerization as well as to convert 6-memberd ring naphthenes to 5-membered ones which acquire higher octane numbers. Other etheric components were then synthesized and mixed with the motor gasoline to > 15%, e.g., methyl tert. butyl ether (MTBE) which acquires octane numbers around 140. However, it was contradicted for its sweeping from its storage tanks to the underground water and condemned to be toxic, therefore its addition to motor gasoline was prevented. Another newly welcomed gasoline component is recently coming into use, i.e., ethanol, which is produced via fermentation of agricultural products such as wheat.
This shows that in spite of the changes occurring to substitute one gasoline component by another, still the catalytic reforming processes persist and still requiring research to produce more and more active catalysts.
REFERENCES
[1] Haensel, V.: US2479110A (1949).
[2] Haensel, V.: US2890167A (1959). [3] Aboul-Gheit AK, Ghoneim SA, Al-owais AA. Effect of
hydrothermal treatment and ammonium ion incorporation in platinum-mordenite catalysis for n-hexane hydroconversion. Appl
Catal: A-General 1998;170:277-283. [4] Kluksdahl, H.E.: US3415737A (1968).
[5] Kluksdahl, H.E.: US3558477A (1971). [6] Haensel V, Pollitzer EL, Hayes JC. New developments in
reforming Proceeding of the 8th World Petrol. Congress, Moscow, 1971; 4: 255-261.
[7] Jassens LW, Petersen EE. Fouling of a platinum-rhenium refor-ming catalyst using model reforming reactions. J Catal 1982; 76:
265-273. [8] Wagstaff N, Prins R. Alloy formation and metal oxide segregation
in Pt-Re/ Al2O3 catalysts as investigated by temperature-programmed reduction. J Catal 1979; 59: 434-445.
[9] Bolivar C, Charcosset H, Frety R, et al. Platinum-rhenium/alumina catalysts I- Investigation of reduction by hydrogen. J Catal 1975;
39: 249-259. [10] Bolivar C, Charcosset H, Frety R, et al. Platinum-rhenium/alumina
catalysts II- Study of the metallic phase after reduction. J Catal 1976; 45: 163-178.

Catalytic Reforming of Naphtha Recent Patents on Chemical Engineering, 2008, Vol. 1, No. 2 125
[11] McNicols BD. The reducibility of rhenium in Re on -Al2O3and Pt-
Re on -Al2O3 catalysts. J Catal 1977; 46: 438-440. [12] Peri JB. Infrared studies of Pt and Pt-Re reforming catalysts. J
Catal 1978; 52: 144-156. [13] Charcosset H, Frety R, Leclerg G, Mendes E, Primet M, Tournayan
L. The state of Re in the Pt-Re/ -Al2O3 catalysts. J Catal 1979; 56: 468-471.
[14] Biloen P, Helle JN, Verbeek H, Dautzenberg FM, Sachtler WMH. The role of rhenium and sulfur in platinum-based hydrocarbon-
conversion catalysts. J Catal 1980; 63: 112-118. [15] Isaacs BH, Petersen EE. The effect of drying temperature on the
temperature-programmed reduction profile of a platinum/rhenium/ alumina catalyst. J Catal 1982; 77: 43-52.
[16] Buss, W.C.: US3578583 (1971). [17] Gallagher, J.P., Yarrington, R. M.: US4356081 (1982).
[18] Parera JM, Figoll NS. Chemistry and processing of petroleum; in Catalytic naphtha reforming science and technology, Antos, G.J.,
Aitani, A.M. and Parera, J.M. Ed. 1995; 15. [19] Galperin, L. B., Modica, F. S., McBride, T. K.: US20056872300B1
(2005). [20] Aboul-Gheit AK. The role of additives in the impregnation of
platinum and ruthenium on alumina catalysts. J Chem Tech Biotechnol 1979; 29: 480-486.
[21] Buss, W.C.: US3554902 (1971). [22] Foger K, Jaeger H. The structure of Pt particles on -Al2O3 support.
J Catal 1981; 70: 53. [23] Garten RI, Sinfelt JH. Temperature-programmed reduction of
bimetallic Ir Fe/Al2O3 catalysts. J Catal 1980;62:127. [24] Aboul-Gheit AK, Hamid SM. Impregnation design for preparing
bimetallic catalysts. Proceeding of the Sixth International Symposium on The Scientific Bases for the Preparation of
Heterogeneous Catalysis “Studies in surface science and catalysis” G. Poncelet, J. Martines, B. Delmon, BA. Jacobs and B. Grange
(Eds.), Elsevier, Amsterdam, 1995; 91: 1131-1136. [25] HuangY J, Fung S C, Gates W E, McVicker GB. Pt---Ir/Al2O3
catalysts-The effect of Pt---Ir interaction on Ir agglomeration and catalytic performance. J Catal 1989; 118: 192.
[26] Kresge CT, Chester AW, Oleck SM. Control of metal radial profiles in alumina supports by carbon dioxide. Appl Catal 1992;
81: 215-226. [27] Antos, G.J.: US4312788 (1992).
[28] Alley, Jr.: US3706 815 (1972). [29] Antos, G.J., Chao, T. H.: US4367137 (1983).
[30] Antos, G.J., Chao, T.H.: US4416804 (1983). [31] Antos, G. J., Chao, T. H.: US4426279 (1984). [32] Antos, G. J., Chao, T.H.: US44463104 (1984). [33] Wilhelm, F. C.: US3798155 (1974).
[34] Wilhelm, F.C.: US3859 201 (1975). [35] Wilhelm, F C.: US3888763 (1975).
[36] Wilhelm, F.C.: US3900387 (1975). [37] Antos, G.J.: US19774036743 (1977).
[38] Wu, A. H, Drake, C. A.: US20006083 867 (2000). [39] Wu, A. H, Drake, C. A.: US20016172273B1 (2001).
[40] Tanev, P. T.: US20036667270B2 (2003). [41] Bogdan, P. L.: US20056884340B1 (2005).
[42] Bogdan, P. L., Bricker, M. L.: US20026419820B1 (2002). [43] Bogdan, P. L, Chen, Q., Moscoso, J. G., Bricker, J. C.:
US20026358400B1 (2002). [44] Zanibelli, L., Arrigonl, V., Albertos, F., Atanes, E., Cholley, T.,
Panarello, F.: US20046746598 (2004). [45] Lin, F.N., Parsons, J.S.: US20036558532B1 (2003).
[46] Lin, F.N., Parsons, J.S., Macahan, D.H., Limoges, B.H.:
US20036593264 (2003). [47] Lin, F.N.: US20026478952B1 (2002).
[48] Verduijin, J.P., McVicker, G.B., Ziemiak, J.J.: US20046740228B1 (2004).
[49] Touvelle, M.S., Kleln, D.P., Chen, T.-J., Martens, L.R., Ellis, E.S.: US20036 652737B2 (2003).
[50] Ducreux, O., Jolimaitre, E.: US20046809228B2 (2004). [51] Gillespie, R. D., Cohn, M. J.: US20056881873 B2 (2005).
[52] Zhao, S., Wagner, J.P.: US20066984371B2 (2006). [53] Glover, B.K.: EP20001038943 A1 (2000).
[54] Aboul-Gheit AK, Menoufy MF, Ebeid FM. Platinum-germanium and platinum- tungsten on alumina catalysts for hydroconversion
reactions. Appl Catal 1982; 4: 181-188. [55] Aboul-Gheit AK, Cosyns J. Rhenium, tungsten and molybdenum as
substitutes for platinum in aromatics hydrogenation catalysts. J Appl Chem Biotechnol 1976; 26: 536-540.
[56] Aboul-Gheit AK. The hydrogenation of aromatics on catalysts having their platinum, ruthenium and palladium replaced with
rhenium. J Appl Chem Biotechnol 1977; 27: 121-124. [57] Aboul-Gheit AK, Menoufy MF, El-morsi AK, Abdel-Hamid SM.
Hydroconversion and diffusion of n-heptane on mordenite catalysts. Zeolites 1987; 7: 353-359.
[58] Aboul-Gheit AK, Menoufy MF, El-morsi AK. Hydroconversion of n-heptane on catalysts containing platinum, rhenium and platinum-
rhenium on sodium mordenite. Appl Catal 1990; 61: 283-292. [59] Aboul-Gheit A K, Cosyns J. The life-time of platinum-tungsten
catalysts supported on alumina for the hydrogenation of aromatic hydrocarbons. Rev Inst Mex Petrol 1975; 7(3): 61-63.
[60] Aboul-Fotouh SM. Thesis, Faculty of Education. Ain Shams Univ 1997.
[61] Ali LI, Ali AA, Aboul-Fotouh SM, Aboul-Gheit AK. Dehydroge-nation of cyclohexane on catalysts containing noble metals and
their combinations with platinum on alumina support. Appl Catal: A- General 1999; 177: 99-110.
[62] Ali AA, Ali L, Aboul-Fotouh SM, Aboul-Gheit AK. Hydro-genation of aromatics on modified platinum-alumina catalysts.
Appl Catal: A-General 1998; 170: 285-296. [63] Aboul-Gheit AK, Menofy MF, El-Morsi AK, Abdel-Hamid SM.
Effect of alumina-deficiency and promotion with platinum and rhenium on the catalytic and diffusional behaviour of mordenite
catalysts. J Chem Tech Biotechnol 1987; 39: 37-43. [64] McInnes, B.J.: EP0567700A1 (1992).
[65] Lin, F.N., Parsons, J.S.: US20026458266B1 (2002). [66] Lin, F.N.., Parsons, J. S.: WO0303000827 A1 (2003)
[67] Lin, F.N., Parsons, J.S.: US20036610196B1 (2003). [68] Walsh, J.F., Go, A., McGregor, D.R., Rebeck, J.W., Sanchez, L.E.:
US20036602404B2 (2003). [69] Boger, T.R., Sorensen, C.M.: US20046773580B2 (2004).
[70] Salmon, E.J.: WO05010128A1 (2005). [71] Murthy KR, Sharma N, George, N. Structure and performance of
reforming catalyst. Antos GJ, Aitani AM, Parera JM Editors in Catalytic naphtha reforming sci and technol. Marcel Dekker, New
York 1995; 243. [72] Marecot P, Barbler J. Deactivation by coking; in Catalytic naphtha
reforming science and technology, Antos GJ, Aitani AM, Parera JM Editors in Catalytic naphtha reforming sci and technol. Marcel
Dekker, New York 1995; 305. [73] Schorfheide, J.J.: EP0256184 (1988).
[74] Fersing, M., Nasclmento, P.: US20026416657 B1 (2002).
Related Documents











