International Journal of Biological Macromolecules 74 (2015) 155–161 Contents lists available at ScienceDirect International Journal of Biological Macromolecules j ourna l h o mepa ge: www.elsevier.com/locate/ijbiomac Carboxymethyl cellulose/silica hybrids as templates for calcium phosphate biomimetic mineralization Ahmed Salama ∗ , Ragab E. Abou-Zeid, Mohamed El-Sakhawy, Ahmed El-Gendy Cellulose and Paper Department, National Research Center, El-Tahrir Street, Dokki, Cairo, Egypt a r t i c l e i n f o Article history: Received 21 October 2014 Received in revised form 20 November 2014 Accepted 22 November 2014 Available online 16 December 2014 Keywords: Hybrid Sol–gel Bioactivity Carboxymethyl cellulose Calcium phosphate a b s t r a c t Multiphase hybrid materials were synthesized using carboxymethyl cellulose (CMC) as bioactive poly- mer, silica gel as matrix assisted networks and calcium phosphate as inorganic mineral phase. These hybrids were investigated with infrared spectroscopy, X-ray diffraction, scanning electron microscopy, energy dispersive X-ray spectroscopy and transmission electron microscopy. Biomimetic crystal growth nucleated from the CMC/silica hybrids was suggested as amorphous calcium phosphate with an evidence that hydroxyapatite, the mineralized component of bone, may be formed at high CMC content. This study provides an efficient approach toward bone-like hybrids with potential bone healing applications. © 2014 Elsevier B.V. All rights reserved. 1. Introduction More than 2.2 million bone grafting process is carried out every year for regenerative bone surgery [1]. However, due to the limitations associated with these bone surgery, searching for new alternative biocompatible regenerative scaffolds for bone defects becomes a clinical challenge. Bone tissue engineering is emerging as a novel solution that can be used to promote near- natural materials for using in implantology and traumatology. Last decade, significant efforts have been devoted to develop bone graft substitutes from biomaterials that can augment or regener- ate injured bone [2–6]. The ideal biomaterials are required to be bioactive, biodegradable, have sufficient mechanical integrity for implantation in load bearing defects, and have a highly porous and interconnected architecture for bone and vascular ingrowth. Low-bioactive materials need firstly surface modifications by incorporation of functional groups to be mineralized by the biomimetic method. Some reports revealed that the introduction of compounds have bioactive functional groups such as proteins [7,8], charged polysaccharides [9,10] and amino acids [11] as ini- tiations for calcium ions nucleation can promote hydroxyapatite formation when immersed in a biological fluid containing ion con- centrations nearly equal to those of human blood plasma [12]. A ∗ Corresponding author. Tel.: +20 1008842629. E-mail address: ahmed [email protected] (A. Salama). recent study investigated the effects of anionic polysaccharides like alginate and phosphorylated alginate on the rate of hydroxyapatite growth and mineralization. The results of the study showed that alginate had no large effect on development of calcium phosphate crystals comparing to phosphorylated alginate which exhibited strong nonspecific binding to the crystals [3]. Carboxymethyl cel- lulose (CMC) has gained increasing interest in the recent years as a candidate material for bone tissue engineering due to its biocompatibility, biodegradability and anionic properties [13–17]. In most cases, CMC was mechanically mixed with pre-prepared hydroxyapatite for hybrid material formation. The additions of nanohydroxyapatite to carboxymethyl cellulose/chitosan film had improved the mechanical properties, swelling behavior and bioac- tivity of CMC/chitosan films [14]. However, few attempts have been made for studying bioactivity and biomematic mineralization of ionic polysaccharides like chitosan [18], and carboxymethyl cellu- lose [5] in simulated body fluids. Silica has numerous favorable properties such as biocompati- bility, adjustable surface area, and easy modification with a large number of functional molecules. In addition, the effect of silicon on bone formation has been studied early and the results revealed that silicon deficiency in animal trials results in abnormal bone forma- tion with reduced precipitation of bone apatite [19]. Also, there is evidence that the presence of polysilanol groups of the amorphous silica may promote the role of collagen as a template for intrafib- rillar apatite precipitation. The fragility and difficult processing in the form of 3D scaffold architecture of sol–gel derived bioactive http://dx.doi.org/10.1016/j.ijbiomac.2014.11.041 0141-8130/© 2014 Elsevier B.V. All rights reserved.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Cp
AC
a
ARR2AA
KHSBCC
1
etndendgabii
ibo[tfc
h0
International Journal of Biological Macromolecules 74 (2015) 155–161
Contents lists available at ScienceDirect
International Journal of Biological Macromolecules
j ourna l h o mepa ge: www.elsev ier .com/ locate / i jb iomac
arboxymethyl cellulose/silica hybrids as templates for calciumhosphate biomimetic mineralization
hmed Salama ∗, Ragab E. Abou-Zeid, Mohamed El-Sakhawy, Ahmed El-Gendyellulose and Paper Department, National Research Center, El-Tahrir Street, Dokki, Cairo, Egypt
r t i c l e i n f o
rticle history:eceived 21 October 2014eceived in revised form0 November 2014ccepted 22 November 2014vailable online 16 December 2014
a b s t r a c t
Multiphase hybrid materials were synthesized using carboxymethyl cellulose (CMC) as bioactive poly-mer, silica gel as matrix assisted networks and calcium phosphate as inorganic mineral phase. Thesehybrids were investigated with infrared spectroscopy, X-ray diffraction, scanning electron microscopy,energy dispersive X-ray spectroscopy and transmission electron microscopy. Biomimetic crystal growthnucleated from the CMC/silica hybrids was suggested as amorphous calcium phosphate with an evidencethat hydroxyapatite, the mineralized component of bone, may be formed at high CMC content. This study
eywords:ybridol–gelioactivityarboxymethyl cellulose
provides an efficient approach toward bone-like hybrids with potential bone healing applications.© 2014 Elsevier B.V. All rights reserved.
alcium phosphate
. Introduction
More than 2.2 million bone grafting process is carried outvery year for regenerative bone surgery [1]. However, due tohe limitations associated with these bone surgery, searching forew alternative biocompatible regenerative scaffolds for boneefects becomes a clinical challenge. Bone tissue engineering ismerging as a novel solution that can be used to promote near-atural materials for using in implantology and traumatology. Lastecade, significant efforts have been devoted to develop boneraft substitutes from biomaterials that can augment or regener-te injured bone [2–6]. The ideal biomaterials are required to beioactive, biodegradable, have sufficient mechanical integrity for
mplantation in load bearing defects, and have a highly porous andnterconnected architecture for bone and vascular ingrowth.
Low-bioactive materials need firstly surface modifications byncorporation of functional groups to be mineralized by theiomimetic method. Some reports revealed that the introductionf compounds have bioactive functional groups such as proteins7,8], charged polysaccharides [9,10] and amino acids [11] as ini-
iations for calcium ions nucleation can promote hydroxyapatiteormation when immersed in a biological fluid containing ion con-entrations nearly equal to those of human blood plasma [12]. A∗ Corresponding author. Tel.: +20 1008842629.E-mail address: ahmed [email protected] (A. Salama).
ttp://dx.doi.org/10.1016/j.ijbiomac.2014.11.041141-8130/© 2014 Elsevier B.V. All rights reserved.
recent study investigated the effects of anionic polysaccharides likealginate and phosphorylated alginate on the rate of hydroxyapatitegrowth and mineralization. The results of the study showed thatalginate had no large effect on development of calcium phosphatecrystals comparing to phosphorylated alginate which exhibitedstrong nonspecific binding to the crystals [3]. Carboxymethyl cel-lulose (CMC) has gained increasing interest in the recent yearsas a candidate material for bone tissue engineering due to itsbiocompatibility, biodegradability and anionic properties [13–17].In most cases, CMC was mechanically mixed with pre-preparedhydroxyapatite for hybrid material formation. The additions ofnanohydroxyapatite to carboxymethyl cellulose/chitosan film hadimproved the mechanical properties, swelling behavior and bioac-tivity of CMC/chitosan films [14]. However, few attempts have beenmade for studying bioactivity and biomematic mineralization ofionic polysaccharides like chitosan [18], and carboxymethyl cellu-lose [5] in simulated body fluids.
Silica has numerous favorable properties such as biocompati-bility, adjustable surface area, and easy modification with a largenumber of functional molecules. In addition, the effect of silicon onbone formation has been studied early and the results revealed thatsilicon deficiency in animal trials results in abnormal bone forma-tion with reduced precipitation of bone apatite [19]. Also, there is
evidence that the presence of polysilanol groups of the amorphoussilica may promote the role of collagen as a template for intrafib-rillar apatite precipitation. The fragility and difficult processing inthe form of 3D scaffold architecture of sol–gel derived bioactive
156 A. Salama et al. / International Journal of Biological Macromolecules 74 (2015) 155–161
Table 1Sample codes for the prepared CMC/silica hybrids.
Samples CMC%
Pure silica* 0CMC/silicaA 5CMC/silicaB 10CMC/silicaC 15CMC/silicaD 20
*
gapp[ctfcp[
oCtimaCgc
2
2
cCccpbw
2
CitTv3athawwv
Table 2Composition of double concentration simulated body fluid.
Phosphate part* [gm] Calcium part [gm]
Tris 1.2110 1.2110Na2SO4·10H2O 0.0644 –NaHCO3 0.1411 –K2HPO4 0.0696 –NaCl – 3.1980KCl – 0.0880MgCl2·6H2O – 0.1220
2.5.3. Compressive strength testHybrids (diameter: 20 mm; height/diameter ratio: 4/1)
Prepared with the same condition at 8 ml TEOS.
lasses restricted their application for bone regeneration. Severalttempts have been carried out to combine organic polymers likeolysaccharides with silica [20–26] to improve the mechanicalroperties [22,24], washing durability [27] and thermal stability28] of the produced hybrid. Porous bioactive glass microspheresontained chitosan as biomolecular template initiate the precipi-ation of apatite crystals on their surface when immersed in SBFor 24 h at 37 ◦C [22]. The hydrogen bonding between the polysac-harides and the inorganic mineral has a positive effect in avoidinghase separation and producing transparent free standing hybrids29].
The threshold task of this article was to study the developmentf a novel CMC-silica-calcium phosphate multiphase biocomposite.MC could be used as assistant reagent for silica gel forma-ion at low silica precursor concentration, and as a material formproving mechanical properties and a template for biomimatic
ineralization of bone like inorganic. Moreover, silica gel was useds a network matrix for preparing of bioactive water insolubleMC/silica hybrids. Additionally, the study aims to understand therowth of bioactive calcium phosphate crystals as a function of CMConcentration.
. Materials and methods
.1. Materials
Carboxymethyl cellulose sodium salt (>99.5%) with high vis-osity was purchased from Fluka Biochemika. The viscosity of 4%MC in water at 25 ◦C is 1000–1500 mPa s. Tetraethyl orthosili-ate (TEOS), (99.9%) was purchased from Sigma Aldrich. The otherhemicals, such as calcium chloride dihydrate, dibasic potassiumhosphate, Magnesium chloride hexahydrate and other simulatedody fluid chemicals were analytical grade and used as receivedithout further purification.
.2. CMC/silica hybrids preparation
Organic-inorganic hybrids with different weight ratios ofMC:TEOS (w:w) were synthesized via a sol–gel process. As shown
n Table 1, a given amount of CMC was dissolved in double dis-illed water at room temperature under continuous stirring for 24 h.hen, 4 ml TEOS and 4 ml acetic acid solution (acetic acid:water 2:1/v) were added to form a mixture of pH 4 with a total volume2 ml. The former was used as a silica precursor and the latter as
catalyst. Subsequently, the mixture was stirred at 60 ◦C for 2 ho ensure a complete hydrolysis of TEOS molecules. The resultingomogenous solution was cast in 15 ml Falcon tube in a water batht 40 ◦C for 48 h to form a homogenous gel. The unreacted residueas extracted from the hybrid materials via washing for three days
ith distilled water and subsequently dried at 40 ◦C for 24 h in aacuum oven.
CaCl2·2H2O – 0.1470
* Every part dissolves in 100 ml double distilled water.
2.3. Biomimetic mineralization of calcium phosphate on thehybrids
All solutions were prepared just before use. The in vitro bioac-tivity of the CMC/silica hybrid disk samples (10 mm × 10 mm) wascarried out by immersing in double concentration simulated bodyfluid (2 × SBF). This solution was prepared in two parts as shown inTable 2, to accelerate the hydroxyapatite formation [30].
CMC/silica hybrids in swellable form were incubated in 50 ml2 × SBF (25 ml from each part) in Falcon tubes for 5 days. Thesolution was renewed after centrifugation every 24 h and the pHwas checked on samples regularly. pH values were maintainedat constant physiological pH (7.4) over the entire course of themineralization to minimize problems associated with SBF prepara-tion and stabilization [31]. Finally, the samples were washed withdouble distilled water and dried at room temperature for furtheranalysis.
2.4. Cyclic water absorbance of CMC/silica hybrid
Water absorbance for the hybrid samples was done with dou-ble distilled water. Discs of the hybrid materials with a definedweight were immersed in water. The weights of the hydrated sam-ples were recorded over various time intervals after removal ofthe surface liquid using Whitman filter paper until reach equilib-rium. The percent of water uptake was calculated by the followingequation:
Water absorption% =[
Wt − W0
W0
]× 100
where W0 is the initial weight and Wt the final weight of the hybridsamples at time t.
The study was continued for 4 times to observe the stability ofthe hybrids toward absorption and drying cycles.
2.5. Characterization methodology
2.5.1. ATR–FTIRAttenuated total reflection–Fourier transform infrared spec-
troscopy (ATR–FTIR) was done on a Thermo Nicolet FT-IR Nexus470 with a diamond crystal. Spectra were recorded from 500 to4000 cm−1 with a resolution of 2 cm−1.
2.5.2. XRDX-ray diffraction (XRD) patterns were recorded with an
Empyrean Powder Diffractometer (Cu K�, 0.154 nm) between 3 and70◦ 2� with a step size of 0.01◦ s−1. Samples were mounted on asilicon support.
were tested using a LLOYD universal testing machine witha 500 N load cell by simultaneously determining force and
Biolog
casdAtcs
2
EEl
2
ww
F(
A. Salama et al. / International Journal of
orresponding length variation. The rate of strain was set bydjusting the cross-head speed to 2 mm/min until 50% compres-ion ultimately. Stress–strain relationships were determined inry state for at least three samples per composition and condition.pparent density of the scaffolds was calculated as the quotient of
he weight and the cylindrical volume. Compressive modulus wasalculated from the slope of a linear fit to the elastic range of thetress–strain curve.
.5.4. SEM and EDXScanning electron microscopy was done on a JEOL JXA-840A
lectron probe microanalyzer with tungsten filament (30 kV). ForDX experiments an Oxford INCAx-sight SN detector with a reso-ution of 128 eV at 5.9 keV was used.
.5.5. TEMTransmission electron microscope (TEM) images were taken
ith a JEOL JEM-2100 electron microscopy at 100k× magnification,ith an acceleration voltage of 120 kV.
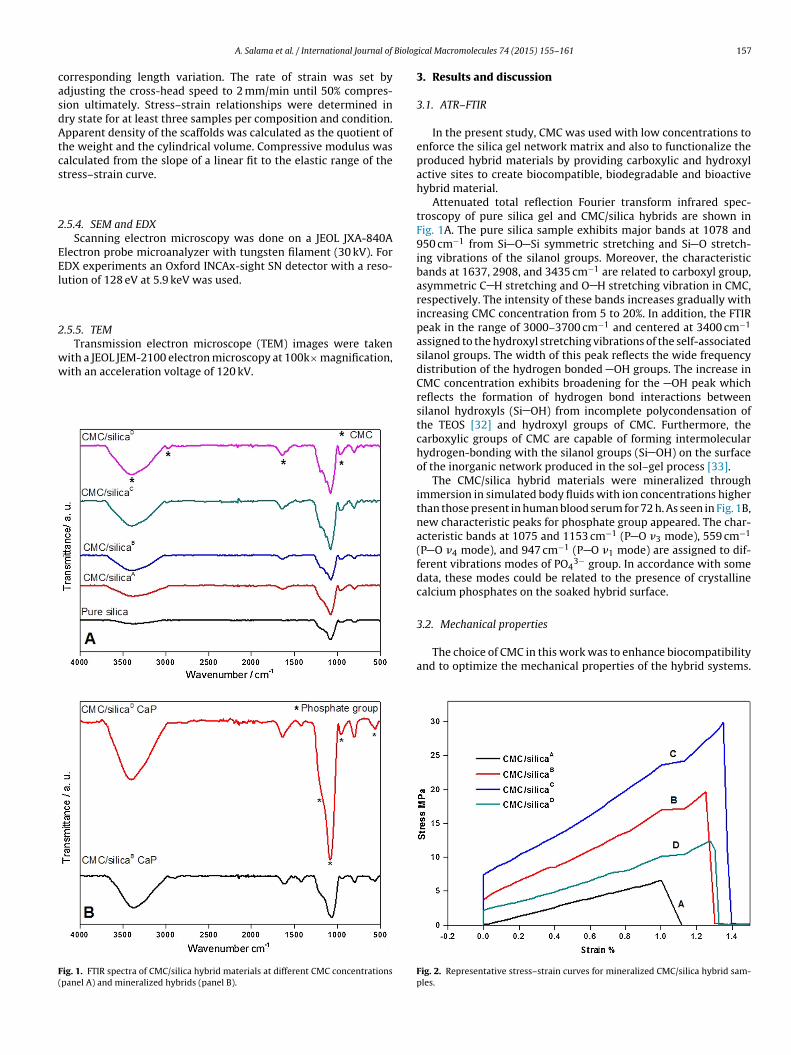
ig. 1. FTIR spectra of CMC/silica hybrid materials at different CMC concentrationspanel A) and mineralized hybrids (panel B).
ical Macromolecules 74 (2015) 155–161 157
3. Results and discussion
3.1. ATR–FTIR
In the present study, CMC was used with low concentrations toenforce the silica gel network matrix and also to functionalize theproduced hybrid materials by providing carboxylic and hydroxylactive sites to create biocompatible, biodegradable and bioactivehybrid material.
Attenuated total reflection Fourier transform infrared spec-troscopy of pure silica gel and CMC/silica hybrids are shown inFig. 1A. The pure silica sample exhibits major bands at 1078 and950 cm−1 from Si O Si symmetric stretching and Si O stretch-ing vibrations of the silanol groups. Moreover, the characteristicbands at 1637, 2908, and 3435 cm−1 are related to carboxyl group,asymmetric C H stretching and O H stretching vibration in CMC,respectively. The intensity of these bands increases gradually withincreasing CMC concentration from 5 to 20%. In addition, the FTIRpeak in the range of 3000–3700 cm−1 and centered at 3400 cm−1
assigned to the hydroxyl stretching vibrations of the self-associatedsilanol groups. The width of this peak reflects the wide frequencydistribution of the hydrogen bonded OH groups. The increase inCMC concentration exhibits broadening for the OH peak whichreflects the formation of hydrogen bond interactions betweensilanol hydroxyls (Si OH) from incomplete polycondensation ofthe TEOS [32] and hydroxyl groups of CMC. Furthermore, thecarboxylic groups of CMC are capable of forming intermolecularhydrogen-bonding with the silanol groups (Si OH) on the surfaceof the inorganic network produced in the sol–gel process [33].
The CMC/silica hybrid materials were mineralized throughimmersion in simulated body fluids with ion concentrations higherthan those present in human blood serum for 72 h. As seen in Fig. 1B,new characteristic peaks for phosphate group appeared. The char-acteristic bands at 1075 and 1153 cm−1 (P O �3 mode), 559 cm−1
(P O �4 mode), and 947 cm−1 (P O �1 mode) are assigned to dif-ferent vibrations modes of PO4
3− group. In accordance with somedata, these modes could be related to the presence of crystallinecalcium phosphates on the soaked hybrid surface.
3.2. Mechanical properties
The choice of CMC in this work was to enhance biocompatibilityand to optimize the mechanical properties of the hybrid systems.
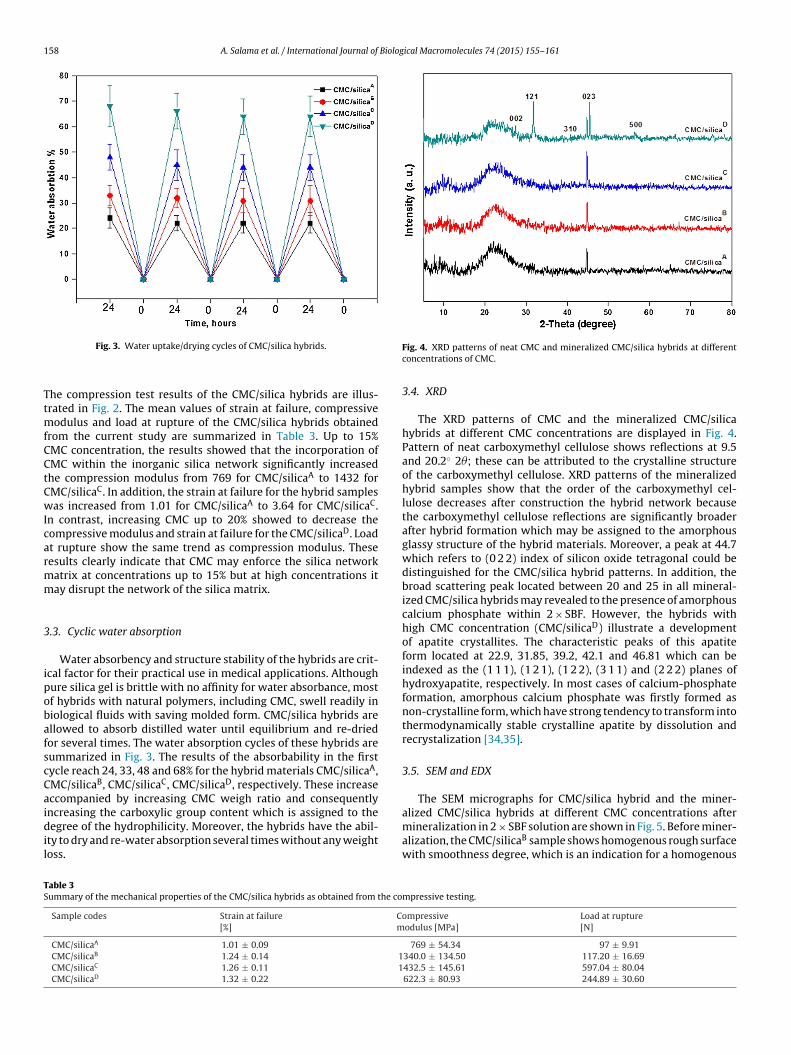
Fig. 2. Representative stress–strain curves for mineralized CMC/silica hybrid sam-ples.
158 A. Salama et al. / International Journal of Biological Macromolecules 74 (2015) 155–161
TtmfCCtCwIcarmm
3
ipobafscCaidil
alized CMC/silica hybrids at different CMC concentrations after
TS
Fig. 3. Water uptake/drying cycles of CMC/silica hybrids.
he compression test results of the CMC/silica hybrids are illus-rated in Fig. 2. The mean values of strain at failure, compressive
odulus and load at rupture of the CMC/silica hybrids obtainedrom the current study are summarized in Table 3. Up to 15%MC concentration, the results showed that the incorporation ofMC within the inorganic silica network significantly increasedhe compression modulus from 769 for CMC/silicaA to 1432 forMC/silicaC. In addition, the strain at failure for the hybrid samplesas increased from 1.01 for CMC/silicaA to 3.64 for CMC/silicaC.
n contrast, increasing CMC up to 20% showed to decrease theompressive modulus and strain at failure for the CMC/silicaD. Loadt rupture show the same trend as compression modulus. Theseesults clearly indicate that CMC may enforce the silica networkatrix at concentrations up to 15% but at high concentrations itay disrupt the network of the silica matrix.
.3. Cyclic water absorption
Water absorbency and structure stability of the hybrids are crit-cal factor for their practical use in medical applications. Althoughure silica gel is brittle with no affinity for water absorbance, mostf hybrids with natural polymers, including CMC, swell readily iniological fluids with saving molded form. CMC/silica hybrids arellowed to absorb distilled water until equilibrium and re-driedor several times. The water absorption cycles of these hybrids areummarized in Fig. 3. The results of the absorbability in the firstycle reach 24, 33, 48 and 68% for the hybrid materials CMC/silicaA,MC/silicaB, CMC/silicaC, CMC/silicaD, respectively. These increaseccompanied by increasing CMC weigh ratio and consequentlyncreasing the carboxylic group content which is assigned to the
egree of the hydrophilicity. Moreover, the hybrids have the abil-ty to dry and re-water absorption several times without any weightoss.
able 3ummary of the mechanical properties of the CMC/silica hybrids as obtained from the co
Sample codes Strain at failure[%]
Cm
CMC/silicaA 1.01 ± 0.09
CMC/silicaB 1.24 ± 0.14 1CMC/silicaC 1.26 ± 0.11 1CMC/silicaD 1.32 ± 0.22
Fig. 4. XRD patterns of neat CMC and mineralized CMC/silica hybrids at differentconcentrations of CMC.
3.4. XRD
The XRD patterns of CMC and the mineralized CMC/silicahybrids at different CMC concentrations are displayed in Fig. 4.Pattern of neat carboxymethyl cellulose shows reflections at 9.5and 20.2◦ 2�; these can be attributed to the crystalline structureof the carboxymethyl cellulose. XRD patterns of the mineralizedhybrid samples show that the order of the carboxymethyl cel-lulose decreases after construction the hybrid network becausethe carboxymethyl cellulose reflections are significantly broaderafter hybrid formation which may be assigned to the amorphousglassy structure of the hybrid materials. Moreover, a peak at 44.7which refers to (0 2 2) index of silicon oxide tetragonal could bedistinguished for the CMC/silica hybrid patterns. In addition, thebroad scattering peak located between 20 and 25 in all mineral-ized CMC/silica hybrids may revealed to the presence of amorphouscalcium phosphate within 2 × SBF. However, the hybrids withhigh CMC concentration (CMC/silicaD) illustrate a developmentof apatite crystallites. The characteristic peaks of this apatiteform located at 22.9, 31.85, 39.2, 42.1 and 46.81 which can beindexed as the (1 1 1), (1 2 1), (1 2 2), (3 1 1) and (2 2 2) planes ofhydroxyapatite, respectively. In most cases of calcium-phosphateformation, amorphous calcium phosphate was firstly formed asnon-crystalline form, which have strong tendency to transform intothermodynamically stable crystalline apatite by dissolution andrecrystalization [34,35].
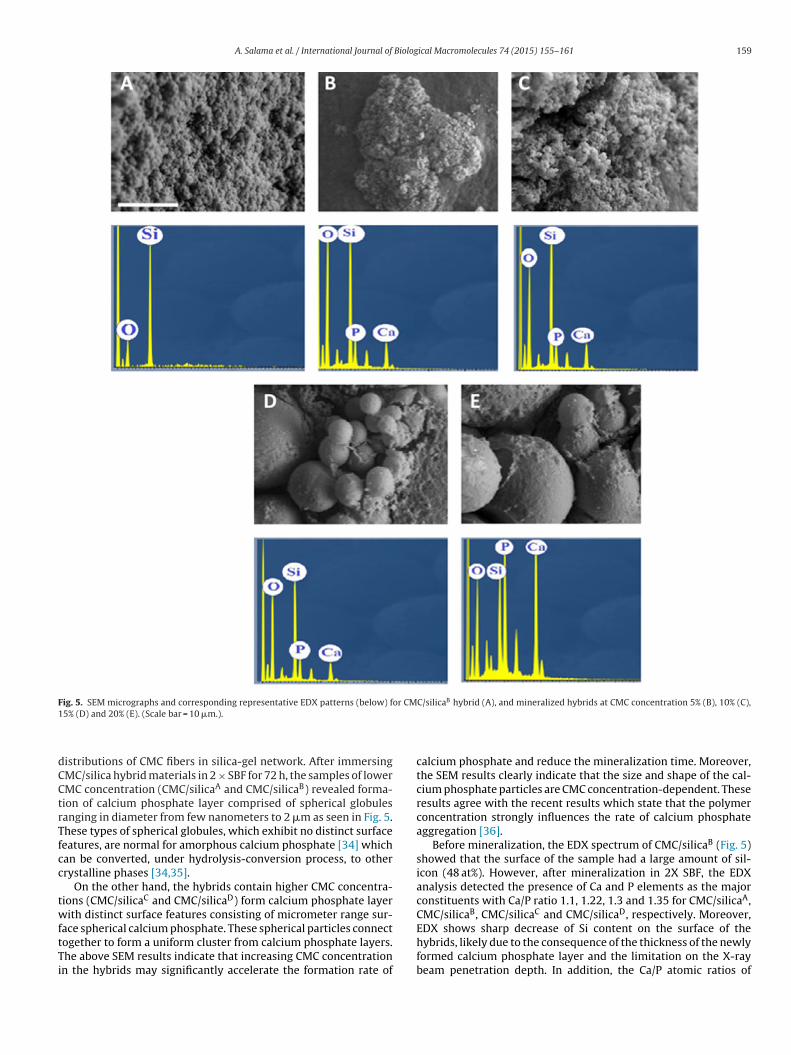
3.5. SEM and EDX
The SEM micrographs for CMC/silica hybrid and the miner-
mineralization in 2 × SBF solution are shown in Fig. 5. Before miner-alization, the CMC/silicaB sample shows homogenous rough surfacewith smoothness degree, which is an indication for a homogenous
mpressive testing.
ompressiveodulus [MPa]
Load at rupture[N]
769 ± 54.34 97 ± 9.91340.0 ± 134.50 117.20 ± 16.69432.5 ± 145.61 597.04 ± 80.04622.3 ± 80.93 244.89 ± 30.60
A. Salama et al. / International Journal of Biological Macromolecules 74 (2015) 155–161 159
Fig. 5. SEM micrographs and corresponding representative EDX patterns (below) for CMC/silicaB hybrid (A), and mineralized hybrids at CMC concentration 5% (B), 10% (C),1
dCCtrTfcc
twftTi
5% (D) and 20% (E). (Scale bar = 10 �m.).
istributions of CMC fibers in silica-gel network. After immersingMC/silica hybrid materials in 2 × SBF for 72 h, the samples of lowerMC concentration (CMC/silicaA and CMC/silicaB) revealed forma-ion of calcium phosphate layer comprised of spherical globulesanging in diameter from few nanometers to 2 �m as seen in Fig. 5.hese types of spherical globules, which exhibit no distinct surfaceeatures, are normal for amorphous calcium phosphate [34] whichan be converted, under hydrolysis-conversion process, to otherrystalline phases [34,35].
On the other hand, the hybrids contain higher CMC concentra-ions (CMC/silicaC and CMC/silicaD) form calcium phosphate layerith distinct surface features consisting of micrometer range sur-
ace spherical calcium phosphate. These spherical particles connect
ogether to form a uniform cluster from calcium phosphate layers.he above SEM results indicate that increasing CMC concentrationn the hybrids may significantly accelerate the formation rate ofcalcium phosphate and reduce the mineralization time. Moreover,the SEM results clearly indicate that the size and shape of the cal-cium phosphate particles are CMC concentration-dependent. Theseresults agree with the recent results which state that the polymerconcentration strongly influences the rate of calcium phosphateaggregation [36].
Before mineralization, the EDX spectrum of CMC/silicaB (Fig. 5)showed that the surface of the sample had a large amount of sil-icon (48 at%). However, after mineralization in 2X SBF, the EDXanalysis detected the presence of Ca and P elements as the majorconstituents with Ca/P ratio 1.1, 1.22, 1.3 and 1.35 for CMC/silicaA,CMC/silicaB, CMC/silicaC and CMC/silicaD, respectively. Moreover,EDX shows sharp decrease of Si content on the surface of the
hybrids, likely due to the consequence of the thickness of the newlyformed calcium phosphate layer and the limitation on the X-raybeam penetration depth. In addition, the Ca/P atomic ratios of
160 A. Salama et al. / International Journal of Biological Macromolecules 74 (2015) 155–161
Fig. 6. Representative TEM images of the mineralized CMC/silicaC hybrid.
ttpststtrw
he mineralized surfaces which range from 1.1 to 1.35 are belowhe stoichiometric value of 1.5 and 1.67 for amorphous calciumhosphate and hydroxyapatite, respectively. Deviation from thetoichiometic values are known in the literature and may be dueo cationic substitutions at the Ca2+ sites by Mg2+ or Na+ or anionicubstitution at PO4
3− sites by CO32− or HPO4
2− or a combination of
hese substitutions [34]. In conclusion, increasing the CMC concen-ration in the hybrid materials had a significant effect on the Ca/Patio of the calcium phosphate mineralized layer. The Ca/P ratioas increased gradually with increasing CMC concentration whichFig. 7. Illustration of the CMC/silica hybrids and biom
in so far exhibits the role of anionic carboxylate group in CMC toadsorb Ca2+ and hence initiate the nucleation process. The resultsfrom SEM and EDX studies clearly confirmed that the hybrid sur-faces have new surface enriched by Ca and P, which are the mainconstituents of hydroxyapatite.
3.6. TEM
To provide better qualitative understanding of the internalstructure and give more detailed information about biomimetic
imetic mineralization processes using 2 × SBF.

Biolog
gTsmpc
3h
watmphathlaAflpgimoabll2
4
Ctccmaaidfpadpm
[[
[[[
[
[[[
[[[[
[
[
[[
[[
[
[[[[[
A. Salama et al. / International Journal of
rowth of calcium phosphate onto CMC/silica hybrid materials,EM observation for the mineralized CMC/silicaC was recorded. Ashown in the TEM images in Fig. 6, calcium phosphate nano andicroparticles are tightly closed to CMC fibers which confirm the
revious suggestion that CMC may act as mineralization sites foralcium phosphate rather than silica.
.7. Growth mechanism of calcium phosphate on CMC/silicaybrid
The effect of polymer concentration, surfaces and moleculareight on the calcium phosphate crystal phases, particle sizes
nd crystal shapes were investigated [37]. This study showedhat CMC can be used as an effective template for biomimetic
ineralization to synthesize micro and nano-spherical calciumhosphate with rough surface. The construction of CMC/silicaybrids and the subsequent biomimetic mineralization mechanismre illustrated in Fig. 7. Calcium phosphate layer coating forma-ion from 2 × SBF onto the surface of the biocompatible CMC/silicaybrids was promoted by negatively charged functional carboxy-
ate groups [34]. These negatively charged functional groups acts nucleation sites which attract Ca2+ ions on the hybrid surface.fter that, calcium ions adsorb PO4
3− ions from simulated bodyuid solution which in turn starts to form a nucleus of calciumhosphate. These nuclei are formed on the hybrid surface androw into a dense calcium phosphate layer by additional precip-tation of calcium and phosphate ions from the SBF. Since the
ain factor for calcium phosphate precipitation is the densityf carboxylate ion groups, increasing the CMC concentration wasccompanied by increasing the bioactivity of the hybrids and alsoy increasing the Ca/P ratio. The XRD, FTIR, and EDX data estab-
ish that various shapes and composition of calcium phosphateayers are mineralized on CMC/silica hybrid after immersing in
× SBF.
. Conclusion
In vitro biomimetic calcium phosphate formation ability onMC/silica hybrids was investigated. It was shown that the bioac-ivity of sol–gel derived hybrids is mainly governed by the CMContent. After immersing in 2 × SBF, the hybrids with lower CMContent (5 and 10%) showed amorphous calcium phosphate for-ation. In contrast, the hybrids with higher CMC content (15
nd 20%) showed amorphous calcium phosphate and/or hydroxy-patite growing. The compressive modulus and strain at failurencreased with an increase in CMC up to 15% content thenecreased at 20% concentration. These results suggest that theormed Ca/P ratio and the morphology of the formed calcium phos-hate layers are governed by the active carboxylate sites which
re represented by CMC concentration. CMC can be incorporatedirectly with the biomimetic mineralization process of calciumhosphate, which is useful to prepare new categories of hybridaterials.[[
[
ical Macromolecules 74 (2015) 155–161 161
Acknowledgement
This work was supported by the National Research Centerin Cairo, Egypt. Project of cellulose and paper department (No.10130101).
References
[1] J.J. Li, E.S. Gil, R.S. Hayden, C. Li, S.I. Roohani-Esfahani, D.L. Kaplan, H. Zreiqat,Biomacromolecules 14 (2013) 2179–2188.
[2] Z. Sun, Q. An, Q. Zhao, Y. Shangguan, Q. Zheng, Cryst. Growth Des. 12 (2012)2382–2388.
[3] R.J. Coleman, K.S. Jack, S. Perrier, L. Grøndahl, Cryst. Growth Des. 13 (2013)4252–4259.
[4] Z. Li, H.R. Ramay, K.D. Hauch, D. Xiao, M. Zhang, Biomaterials 26 (2005)3919–3928.
[5] K. Rodríguez, S. Renneckar, P. Gatenholm, ACS Appl. Mater. Interfaces 3 (2011)681–689.
[6] A. Salama, M. Neumann, C. Günter, A. Taubert, Beilstein J. Nanotechnol. 5 (2014)1553–1568.
[7] C. Zhang, W. Zhang, H. Yao, H. Zhu, L. Mao, S. Yu, Cryst. Growth Des. 13 (2013)3505–3513.
[8] S. Prieto, A. Shkilnyy, C. Rumplasch, A. Ribeiro, F.J. Arias, J.C. Rodríguez-cabello,A. Taubert, Biomacromolecules 12 (2011) 1480–1486.
[9] G. Toskas, C. Cherif, R.-D. Hund, E. Laourine, B. Mahltig, A. Fahmi, C. Heinemann,T. Hanke, Carbohydr. Polym. 94 (2013) 713–722.
10] F. Khan, S.R. Ahmad, Macromol. Biosci. 13 (2013) 395–421.11] B. Palazzo, D. Walsh, M. Iafisco, E. Foresti, L. Bertinetti, G. Martra, C.L. Bianchi,
G. Cappelletti, N. Roveri, Acta Biomater. 5 (2009) 1241–1252.12] T. Kokubo, H. Takadama, Biomaterials 27 (2006) 2907–2915.13] A. Salama, M. El-Sakhawy, Carbohydr. Polym. 113 (2014) 500–506.14] J. Liuyun, L. Yubao, X. Chengdong, J. Mater. Sci. Mater. Med. 20 (2009)
1645–1652.15] L. Jiang, Y. Li, X. Wang, L. Zhang, J. Wen, M. Gong, Carbohydr. Polym. 74 (2008)
680–684.16] R. Barbucci, A. Magnani, M. Consumi, Macromolecules 33 (2000) 7475–7480.17] S. Schweizer, A. Taubert, Macromol. Biosci. 7 (2007) 1085–1099.18] E.J. Lee, D.S. Shin, H.E. Kim, H.W. Kim, Y.H. Koh, J.H. Jang, Biomaterials 30 (2009)
743–750.19] E.M. Carlisle, Science (80–) 178 (1972) 619–621.20] M.R. Gandhi, S. Meenakshi, Int. J. Biol. Macromol. 50 (2012) 650–657.21] S. Park, J. You, H. Park, S.J. Haam, W. Kim, Biomaterials 22 (2001) 323–330.22] B. Lei, K.H. Shin, Y.W. Moon, D.Y. Noh, Y.H. Koh, Y. Jin, H.E. Kim, J. Am. Ceram.
Soc. 95 (2012) 30–33.23] T. Uragami, T. Katayama, T. Miyata, H. Tamura, T. Shiraiwa, A. Higuchi,
Biomacromolecules 5 (2004) 1567–1574.24] K. Molvinger, F. Quignard, D. Brunel, M. Boissière, J. Devoisselle, Chem. Mater.
16 (2004) 3367–3372.25] L. Yu, J. Gong, C. Zeng, L. Zhang, Ind. Eng. Chem. Res. 51 (2012) 2299–2308.26] J.G. Varghese, R.S. Karuppannan, M.Y. Kariduraganavar, J. Chem. Eng. Data 55
(2010) 2084–2092.27] W. Huang, Y. Song, Y. Xing, J. Dai, Ind. Eng. Chem. Res. 49 (2010) 9135–9142.28] Y. Kim, S. Ha, S. Jeon, D.W. Yoo, S. Chun, B. Sohn, J. Lee, Langmuir 26 (2010)
7555–7560.29] N. Rangelova, L. Radev, S. Nenkova, I.M.M. Salvado, M.H.V. Fernandes, M. Her-
zog, Cent. Eur. J. Chem. 9 (2011) 112–118.30] C. Ohtsuki, T. Kokubo, T. Yamamuro, J. Non Cryst. Solids 143 (1992) 84–92.31] M. Bohner, J. Lemaitre, Biomaterials 30 (2009) 2175–2179.32] R. Li, a.E. Clark, L.L. Hench, J. Appl. Biomater. 2 (1991) 231–239.33] K. Nie, W. Pang, Y. Wang, F. Lu, Q. Zhu, Mater. Lett. 59 (2005) 1325–1328.34] D.O. Costa, B.A. Allo, R. Klassen, J.L. Hutter, S.J. Dixon, A.S. Rizkalla, Langmuir 28
(2012) 3871–3880.
35] F. Abbona, H.E.L. Madsen, R. Boistelle, J. Cryst. Growth 74 (1986) 581–590.36] J. Ye, D. Wang, D.N. Zeiger, W.C. Miles, S. Lin-Gibson, Biomacromolecules 14(2013) 3417–3422.37] A. Shkilnyy, A. Friedrich, B. Tiersch, S. Schöne, M. Fechner, J. Koetz, C.-W.
Schläpfer, A. Taubert, Langmuir 24 (2008) 2102–2109.
Related Documents











