University of Groningen Carbon-nitrogen bond formation via catalytic alcohol activation Yan, Tao IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2017 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Yan, T. (2017). Carbon-nitrogen bond formation via catalytic alcohol activation. University of Groningen. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 23-07-2022

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

University of Groningen
Carbon-nitrogen bond formation via catalytic alcohol activationYan, Tao
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2017
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Yan, T. (2017). Carbon-nitrogen bond formation via catalytic alcohol activation. University of Groningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 23-07-2022

Carbon-Nitrogen Bond Formation
via Catalytic Alcohol Activation
Tao Yan

The work described in this thesis was carried out at the Stratingh Institute for
Chemistry, University of Groningen, The Netherlands.
This work was financially supported by University of Groningen.
Cover design by Tao Yan.
Printed by Ipskamp Printing, Enschede, The Netherlands.
ISBN: 978-94-034-0047-1 (printed version)
ISBN: 978-94-034-0046-4 (digital version)
faculty of science and engeneering
stratingh institute for chemistry

Carbon-Nitrogen Bond Formation via Catalytic Alcohol Activation
PhD thesis
to obtain the degree of PhD at the University of Groningen on the authority of the
Rector Magnificus Prof. E. Sterken and in accordance with
the decision by the College of Deans.
This thesis will be defended in public on
Monday 18 September 2017 at 12.45 hours
by
Tao Yan
born on 27 June 1990 in Anhui, China

Supervisors
Prof. B. L. Feringa
Prof. K. Barta
Assessment Committee
Prof. J. G. de Vries
Prof. B. de Bruin
Prof. M. Beller

Contents
Chapter 1
Borrowing hydrogen meets metal-ligand bifunctional catalysis, an introduction to the thesis
1
Chapter 2
Iron catalyzed direct alkylation of amines with alcohols
21
Chapter 3
Benzylamines via iron catalyzed direct amination of benzyl alcohols
49
Chapter 4
Pyrroles via Iron-Catalyzed N-Heterocyclization from Unsaturated Diols and Primary Amines
75
Chapter 5
Direct N-alkylation of unprotected amino acids with alcohols
93
Chapter 6
Ruthenium catalyzed N-alkylation of amino acid esters with
121
Nederlandse samenvatting 141
English summary 143
Acknowledgements 145


1
Chapter 1
Borrowing hydrogen meets metal-ligand bifunctional catalysis,
an introduction to the thesis
1.1 Introduction
1.1.1 Catalysis: key to a sustainable future
1.1.2 Catalytic carbon-nitrogen bond formation: the focus of this thesis
1.2 Metal-ligand bifunctional catalysis
1.2.1 Background
1.2.2 The Shvo catalyst
1.2.3 The Knölker complex
1.2.4 Recent progress in bifunctional catalysis
1.3 Catalysis based on the borrowing hydrogen strategy
1.3.1 Alkylation of amines with alcohols through borrowing hydrogen
1.3.2 Challenges and recent discoveries
1.4 Conclusion
1.5 Outline of the thesis

Chapter 1
2
1.1 Introduction
1.1.1 Catalysis: the key to a sustainable future
Catalysis is the tool to tune the kinetics of a chemical transformation, that allows
desired reactions to be conducted selectively, under mild reaction conditions.[1]
Catalysis is one of the few areas with a direct influence on our daily life.[2] It
contributes directly and indirectly to 35% of the global GDP.[3]
Currently, a variety of chemical transformations still rely on the use of
stoichiometric reagents and low atom economy processes.[1,2,4] These include the
use of protecting groups or harsh reaction conditions, mainly due to high activation
energy of the desired transformations. During these processes, stoichiometric
amount of side-products are produced and frequently discarded. Also, the
increasing demand for a more environmental benign society and the changing
landscape of accessible chemical feedstocks and energy sources, indicate we are
facing a transition period of energy and chemical production.[5]
Catalysis, beyond accelerating chemical transformations, allows the use of
renewable carbon sources through converting bio-based molecules and CO2 to
more valuable chemicals, as well as accessing alternative energy such as
converting and storing solar energy in chemicals.[5,6] Consequently, the new
advances in catalysis will not only lead to considerable economic benefit, but more
importantly, are key to build a sustainable society.[7]
1.1.2 Catalytic carbon-nitrogen bond formation: the focus of this thesis
This thesis discusses an alternative methodology to construct carbon-nitrogen
bonds[8], using widely abundant alcohols as substrates instead of the traditional
alkyl halides or aldehydes, promoted by metal-ligand bifunctional catalytic systems.
As an introduction to this thesis, this chapter gives background to the field of
ligand-metal bifunctional catalysis which is dramatically changing the face of
chemistry, in particular redox chemistry.[9] The introduction will include catalysis
based on the borrowing hydrogen strategy which will be also extensively involved
in the following chapters. Further, literature background to the Shvo catalyst[10a]
and the Knölker complex[10b] which are important catalysts employed in this thesis
will also be discussed.
1.2 Metal-ligand bifunctional catalysis
1.2.1 Background
Transition metal catalyzed chemical transformations promote the efficient and
environmentally benign synthesis of molecular targets[11]. In conventional
transition metal based catalysis, the coordination and further transformation of the
substrate is performed at the metal center and the role of the ligand is to keep the
metal complex in solution as well as regulate the electronic and steric properties
of the transition metal complex[12]. However, there is a class of catalysts, in that
the coordination and chemical transformation occurs on both the metal center as
well as the ligand. An early example from the Noyori group described the

Introduction to the thesis
3
asymmetric hydrogenation of ketones using a ruthenium diphosphine-diamine
complex.[13] The diamine ligand acts as the proton donor that stabilizes the alkoxy
before forming an alcohol by reduction of a simple ketone (Scheme 1, intermediate
1). It was proposed that the substrate is in the second coordination sphere of the
catalyst complex, not directly coordinated to the metal center[14] (Scheme 1, A).
The original halogen contained BINAP-Ru(II) complexes were only found to be
active for hydrogenation of functionalized ketones with nitrogen, oxygen or
halogen atoms near the carbonyl group (Scheme 1, B)[15]. In the latter case, the
additional heteroatom is required to form a metallacycle (Scheme 1, intermediate
2) to stabilize the alkoxy ligand before protonation. The type of catalysis described
in Scheme 1A was coined as ‘metal-ligand bifunctional catalysis’ by Noyori in
2001.[9a,16]
Scheme 1: A Asymmetric hydrogenation of acetophenone with a ruthenium BINAP
diamine complex; B asymmetric hydrogenation of activated carbonyls with
ruthenium BINAP complex.
1.2.2 The Shvo catalyst
In 1984, it was reported by the Shvo group that the reactivity of transfer
hydrogenation reactions catalyzed by triruthenium dodecacarbonyl was
significantly improved by adding diphenylacetylene (Scheme 2, A).[17] Later on, it
was proven that a ruthenium complex bearing cyclopentadienone was formed and
played an essential role.[18] The structure of the complex was determined by X ray
spectroscopic analysis (Scheme 2, B) by the Shvo group.[10a] The initial synthetic
approach to obtain this complex took 2 steps (Scheme 2, C, a) during which
Ru3(CO)12 (3) and tetraphenylcyclopentadienone (4) were heated to reflux in
benzene, forming [Ph4(4-C4CO)]-Ru(CO)3 (Cat 2)[19]. Subsequently, Cat 2 was
refluxed in isopropanol to give Shvo’s catalyst (Cat 1)[10a]. An alternative synthetic
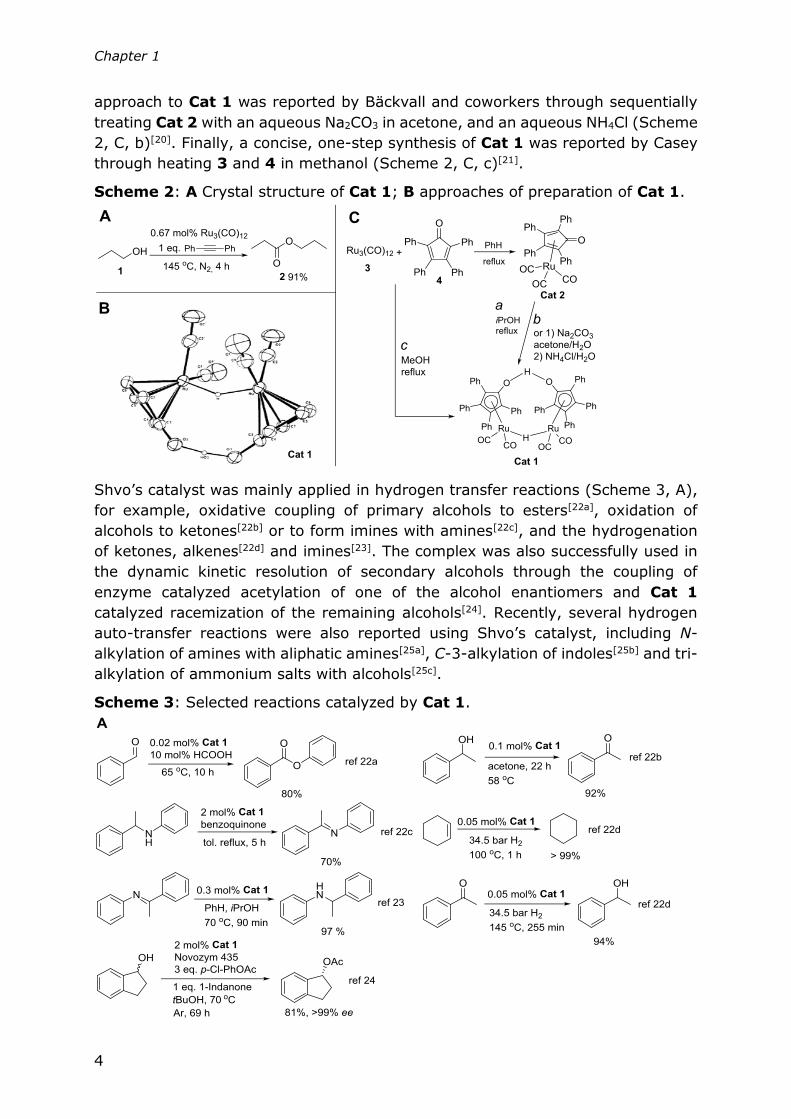
Chapter 1
4
approach to Cat 1 was reported by Bäckvall and coworkers through sequentially
treating Cat 2 with an aqueous Na2CO3 in acetone, and an aqueous NH4Cl (Scheme
2, C, b)[20]. Finally, a concise, one-step synthesis of Cat 1 was reported by Casey
through heating 3 and 4 in methanol (Scheme 2, C, c)[21].
Scheme 2: A Crystal structure of Cat 1; B approaches of preparation of Cat 1.
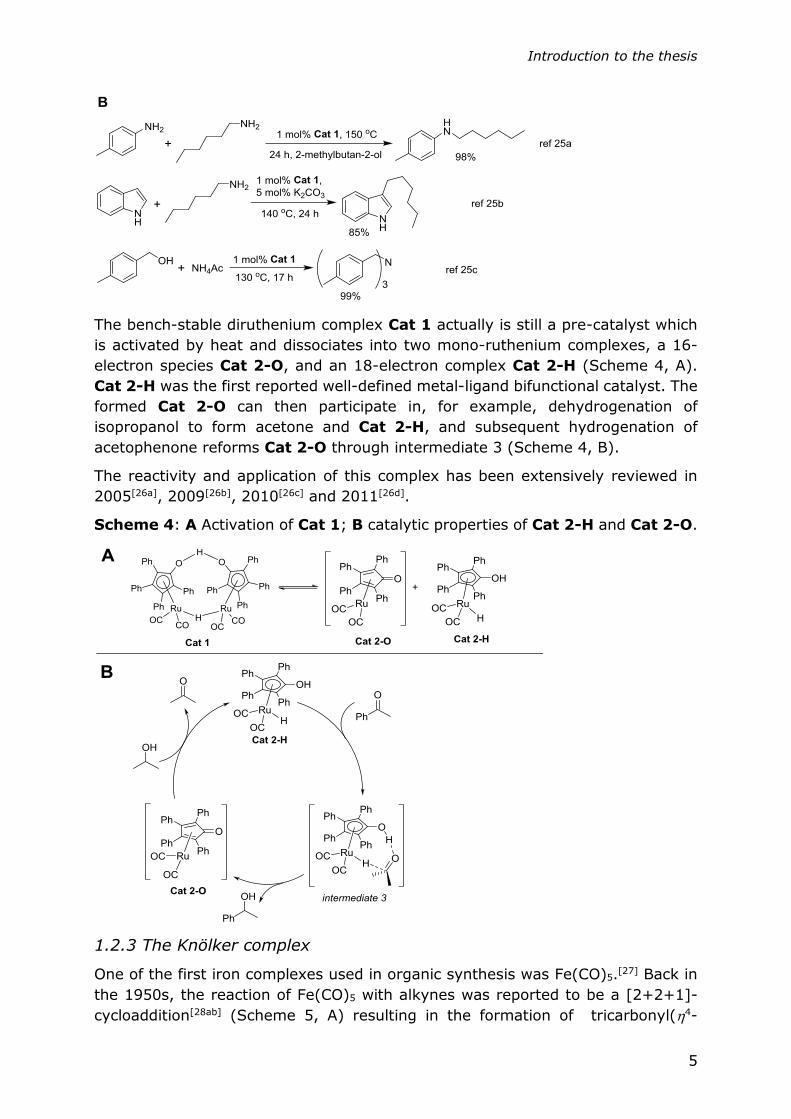
Shvo’s catalyst was mainly applied in hydrogen transfer reactions (Scheme 3, A),
for example, oxidative coupling of primary alcohols to esters[22a], oxidation of
alcohols to ketones[22b] or to form imines with amines[22c], and the hydrogenation
of ketones, alkenes[22d] and imines[23]. The complex was also successfully used in
the dynamic kinetic resolution of secondary alcohols through the coupling of
enzyme catalyzed acetylation of one of the alcohol enantiomers and Cat 1
catalyzed racemization of the remaining alcohols[24]. Recently, several hydrogen
auto-transfer reactions were also reported using Shvo’s catalyst, including N-
alkylation of amines with aliphatic amines[25a], C-3-alkylation of indoles[25b] and tri-
alkylation of ammonium salts with alcohols[25c].
Scheme 3: Selected reactions catalyzed by Cat 1.
Introduction to the thesis
5
The bench-stable diruthenium complex Cat 1 actually is still a pre-catalyst which
is activated by heat and dissociates into two mono-ruthenium complexes, a 16-
electron species Cat 2-O, and an 18-electron complex Cat 2-H (Scheme 4, A).
Cat 2-H was the first reported well-defined metal-ligand bifunctional catalyst. The
formed Cat 2-O can then participate in, for example, dehydrogenation of
isopropanol to form acetone and Cat 2-H, and subsequent hydrogenation of
acetophenone reforms Cat 2-O through intermediate 3 (Scheme 4, B).
The reactivity and application of this complex has been extensively reviewed in
2005[26a], 2009[26b], 2010[26c] and 2011[26d].
Scheme 4: A Activation of Cat 1; B catalytic properties of Cat 2-H and Cat 2-O.
1.2.3 The Knölker complex
One of the first iron complexes used in organic synthesis was Fe(CO)5.[27] Back in
the 1950s, the reaction of Fe(CO)5 with alkynes was reported to be a [2+2+1]-
cycloaddition[28ab] (Scheme 5, A) resulting in the formation of tricarbonyl(4-
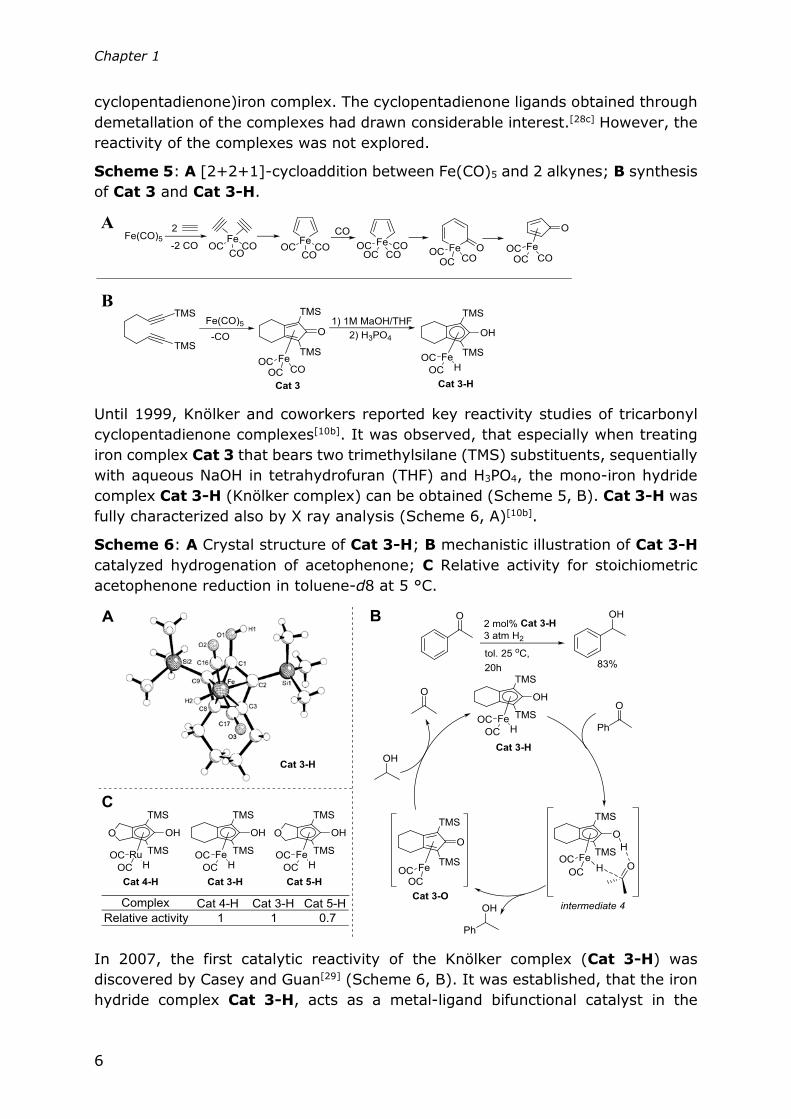
Chapter 1
6
cyclopentadienone)iron complex. The cyclopentadienone ligands obtained through
demetallation of the complexes had drawn considerable interest.[28c] However, the
reactivity of the complexes was not explored.
Scheme 5: A [2+2+1]-cycloaddition between Fe(CO)5 and 2 alkynes; B synthesis
of Cat 3 and Cat 3-H.
Until 1999, Knölker and coworkers reported key reactivity studies of tricarbonyl
cyclopentadienone complexes[10b]. It was observed, that especially when treating
iron complex Cat 3 that bears two trimethylsilane (TMS) substituents, sequentially
with aqueous NaOH in tetrahydrofuran (THF) and H3PO4, the mono-iron hydride
complex Cat 3-H (Knölker complex) can be obtained (Scheme 5, B). Cat 3-H was
fully characterized also by X ray analysis (Scheme 6, A)[10b].
Scheme 6: A Crystal structure of Cat 3-H; B mechanistic illustration of Cat 3-H
catalyzed hydrogenation of acetophenone; C Relative activity for stoichiometric
acetophenone reduction in toluene-d8 at 5 °C.
In 2007, the first catalytic reactivity of the Knölker complex (Cat 3-H) was
discovered by Casey and Guan[29] (Scheme 6, B). It was established, that the iron
hydride complex Cat 3-H, acts as a metal-ligand bifunctional catalyst in the
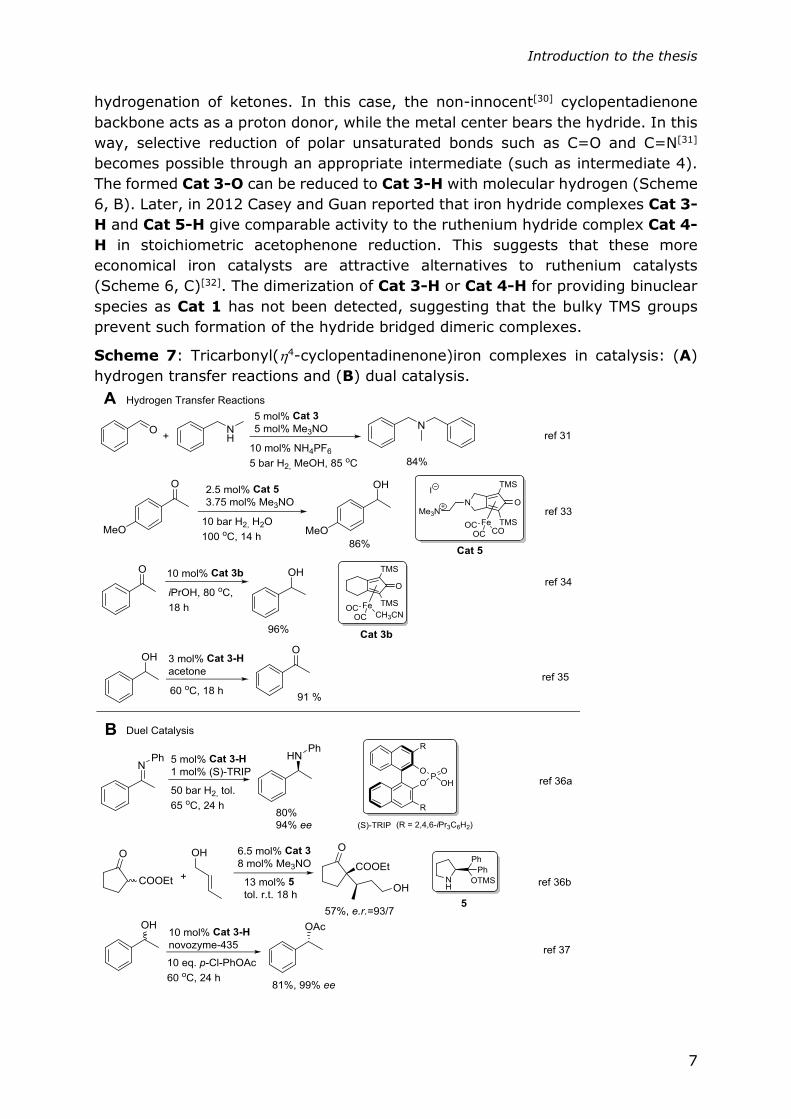
Introduction to the thesis
7
hydrogenation of ketones. In this case, the non-innocent[30] cyclopentadienone
backbone acts as a proton donor, while the metal center bears the hydride. In this
way, selective reduction of polar unsaturated bonds such as C=O and C=N[31]
becomes possible through an appropriate intermediate (such as intermediate 4).
The formed Cat 3-O can be reduced to Cat 3-H with molecular hydrogen (Scheme
6, B). Later, in 2012 Casey and Guan reported that iron hydride complexes Cat 3-
H and Cat 5-H give comparable activity to the ruthenium hydride complex Cat 4-
H in stoichiometric acetophenone reduction. This suggests that these more
economical iron catalysts are attractive alternatives to ruthenium catalysts
(Scheme 6, C)[32]. The dimerization of Cat 3-H or Cat 4-H for providing binuclear
species as Cat 1 has not been detected, suggesting that the bulky TMS groups
prevent such formation of the hydride bridged dimeric complexes.
Scheme 7: Tricarbonyl(4-cyclopentadinenone)iron complexes in catalysis: (A)
hydrogen transfer reactions and (B) dual catalysis.

Chapter 1
8
After Casey and Guan’s initial discovery on the catalytic behaviour of the Knölker
complex, several studies involving this complex have been reported. These
included reductive amination[31], hydrogenation of carbonyl compounds and imines
in water[33], transfer hydrogenation of carbonyl compounds and imines[34],
Oppenauer-type oxidation of alcohols[35] (Scheme 7, A). Furthermore, catalytic
systems involving dual catalysis were described, in which the Knölker complex
catalyzed hydrogen transfer reactions were coupled with organo-catalysis[36] or
enzyme promoted transformations[37] (Scheme 7, B). Moisture and air stable
complex Cat 3 is frequently used as a pre-catalyst. One CO ligand in Cat 3 can be
easily removed by Me3NO, generating Me3N, CO2 and active species Cat 3-O
(Scheme 6A).
Several analogues of the original complex have been reported through steric and
electronic modifications[38] of Cat 3 (Scheme 8). Modifications of the proton donor
site on the non-innocent ligand were reported by Nakazawa[39] and Guan[40], and
chirality was introduced to the metal complex by Wills[41], Berkessel[42] and
Gennari[43]. The field has been reviewed by Knölker[44], Guan[45], as well as
Quintard and Rodriguez[46] recently.
Scheme 8: Analogues of Knölker’s complex.
1.2.4 Recent progress in metal-ligand bifunctional catalysis
Since Noyori’s ruthenium diphosphine-diamine system was reported in 1995,
besides the development of the Shvo and Knölker complexes, considerable
progress has been established in this area[47,59]. For example, Morris and coworkers
reported well-defined iron complexes bearing PNNP ligands (such as Cat 8, shown
in Scheme 9) for the asymmetric transfer hydrogenation of carbonyl compounds
and imines. The reactions were completed within minutes in most cases, and the
ee reached 99% when imines were employed as the substrates[48]. Related to these
excellent results established by the Morris group, Bullock highlighted the potential
of iron-based catalysts to reach reactivity comparable to that obtained with noble
metal catalysts[49] (Scheme 9).

Introduction to the thesis
9
Scheme 9: Comparison of bifunctional catalyst systems developed by Noyori and
Morris.
In 2004, Milstein and coworkers reported a new type of ruthenium pincer complex,
that operates via the aromatization-rearomatization of a pyridine based
heteroaromatic ligand. This complex catalyzed the acceptorless dehydrogenation
of alcohols to ketones[50] and esters[51]. Subsequently the acceptorless
dehydrogenation and coupling between alcohols and amines to form amides was
reported[52] (Scheme 10, A). Comparing to classical metal-ligand bifunctional
catalysts that bear N-donors to activate H2 or alcohols and subsequently reducing
polar unsaturated bonds, the Milstein-type pincer complex has a C-donor (Scheme
10, B), and is able to activate a wider variety of bonds, including N-H bonds[53],
CO2[54], nitriles[55] and O2
[56].
Scheme 10: A Milstein pincer complex catalyzed acceptorless dehydrogenative
coupling for the synthesis of esters and amides; B activation of H2 by the Milstein
pincer complex Cat 9.
Recently, a number of new metal-ligand bifunctional catalysts have been
reported[57]. Selected examples are shown in Scheme 11, in which the structures
shown are after dihydrogen activation. In these complexes, the p- or π electron on
the ligand offers a proton acceptor site that is involved in the heterolytic splitting
of dihydrogen which results in the formation of the corresponding metal hydride.
Interestingly, Harman and Peters recently reported a nickel complex featuring a
borane moiety in the supporting ligand scafford (Scheme 11)[57e]. The property of
this complex is more comparable to a heterobimetallic complex (boron mimics a
second metal)[58] instead of a Noyori-type bifunctional complex. The catalytic
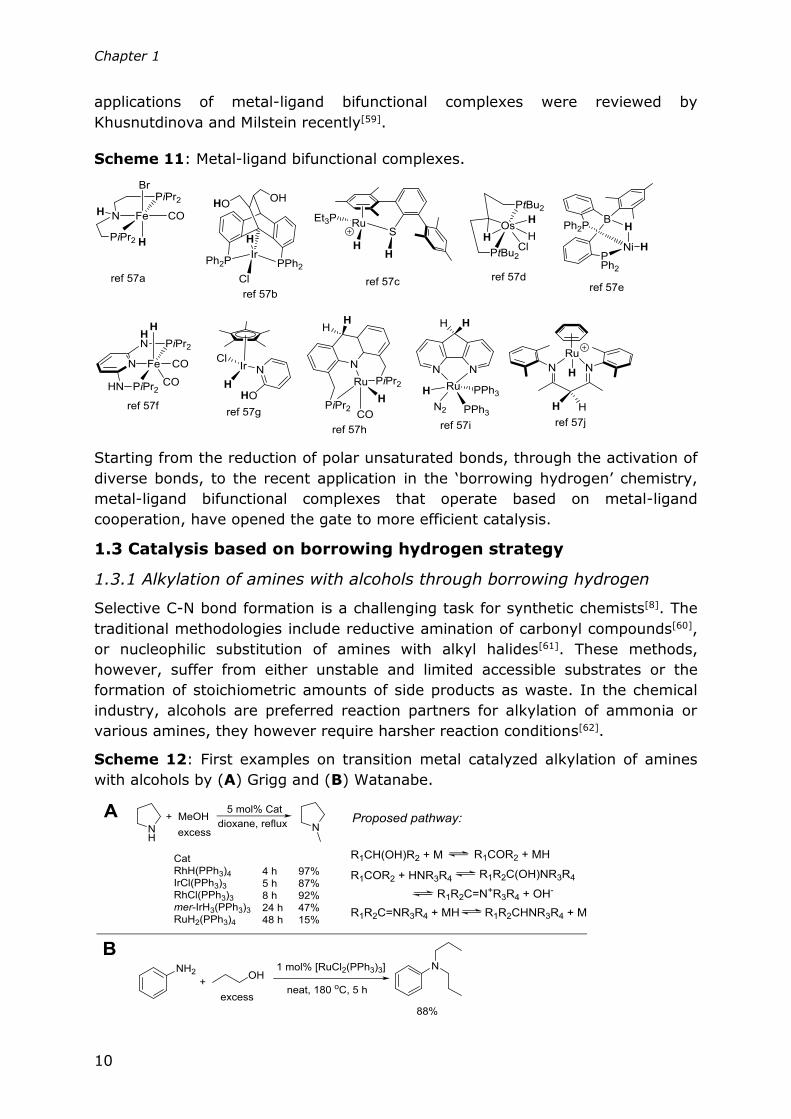
Chapter 1
10
applications of metal-ligand bifunctional complexes were reviewed by
Khusnutdinova and Milstein recently[59].
Scheme 11: Metal-ligand bifunctional complexes.
Starting from the reduction of polar unsaturated bonds, through the activation of
diverse bonds, to the recent application in the ‘borrowing hydrogen’ chemistry,
metal-ligand bifunctional complexes that operate based on metal-ligand
cooperation, have opened the gate to more efficient catalysis.
1.3 Catalysis based on borrowing hydrogen strategy
1.3.1 Alkylation of amines with alcohols through borrowing hydrogen
Selective C-N bond formation is a challenging task for synthetic chemists[8]. The
traditional methodologies include reductive amination of carbonyl compounds[60],
or nucleophilic substitution of amines with alkyl halides[61]. These methods,
however, suffer from either unstable and limited accessible substrates or the
formation of stoichiometric amounts of side products as waste. In the chemical
industry, alcohols are preferred reaction partners for alkylation of ammonia or
various amines, they however require harsher reaction conditions[62].
Scheme 12: First examples on transition metal catalyzed alkylation of amines
with alcohols by (A) Grigg and (B) Watanabe.

Introduction to the thesis
11
In 1981, Grigg and coworkers reported the first example of alkylation of amines
with alcohols under significantly milder conditions using transition metal
catalysts[63] (Scheme 12, A). They proposed that the reactivity of the alcohol was
improved by the formation of the corresponding carbonyl compound, which
subsequently underwent imine formation with the amine reaction partner.
Reduction of this imine intermediate resulted in the alkylated amine. At around the
same time, Watanabe and coworkers also reported the alkylation of anilines with
various alcohols catalyzed by a ruthenium complex[64] (Scheme 12, B).
Following these studies, during the past 3 decades, various catalytic systems have
been developed for direct alkylation of amines with alcohols, mostly using
ruthenium or iridium based catalysts[65] (Scheme 13). This field has been
extensively reviewed[66].
Scheme 13: Alkylation of amines with alcohols catalyzed by ruthenium or iridium
based catalytic systems.

Chapter 1
12
To characterize these types of reactions, Williams et al. coined the term ‘borrowing
hydrogen’[67] in 2004. During the catalytic cycle, an alcohol is dehydrogenated to
the corresponding carbonyl compound, which reacts with the amine to form an
imine (Scheme 14, A). The hydrogen delivered from the alcohol is temporarily
stored at the metal complex. The imine is reduced in situ to the alkylated amine
by the hydrogen stored on the metal complex. Key features are that the process
is hydrogen neutral, no other reagents are needed and the only stoichiometric by-
product is water. Variations of this reaction have also been reported, for instance,
C-C bond formation through alcohol activation[68,69] (Scheme 14, B) and alkane
metathesis through dehydrogenation of alkane to alkene, alkene metathesis and
hydrogenation of alkene to alkane[70] (Scheme 14, C).
Scheme 14: A Proposed mechanism of alkylation of amines with alcohols through
borrowing hydrogen; B C-C bond formation through alcohol activation; C alkane
metathesis through borrowing hydrogen.
Alkylation of amines with alcohols through borrowing hydrogen has been applied
in the pharmaceutical industry due to its significant economic benefit compared to
traditional methodologies of N-alkylations[66]. For example, Pfizer recently
developed a new pathway for synthetizing a GlyT1 inhibitor (9) (Scheme 15)[65h].
Comparing to conventional pathway, the key optimization was a direct amination
of alcohol 6 with amine 7 to provide the key intermediate 8 through the borrowing
hydrogen strategy. It is a redox-neutral process that avoids the use of
stoichiometric amount of oxidant and reductant.

Introduction to the thesis
13
Scheme 15: Conventional and Pfizer’s pathway for the synthesis of a GlyT1
Inhibitor.
1.3.2 Challenges and recent discoveries
The borrowing hydrogen strategy has been recognized as a key concept in catalysis
and sustainable chemistry, as it is a highly atom economic process[71]. Since the
first examples reported by the groups of Grigg[63] and Watanabe[64], and till recent
discoveries[65,66], most reactions were promoted by ruthenium or iridium based
catalytic systems. After three decades of discovery, the scientific community
realized that the main challenge was to developa non-precious metal based
catalyst for promoting this transformation[66d,72]. Iron with its high Earth-
abundance[73a], low cost[73b] and toxicity[73c], has been identified as an attractive
candidate.
In 2014, our group reported the first example of alkylation of amines with alcohols
with the well-defined bifunctional iron complex (Knölker complex)[74] (Scheme 16,
A). Subsequently, Wills and coworkers[75a] reported the same transformation with
an analogue complex Cat 16. Zhao and coworkers[75d] showed that with the
assistance of Lewis acids, the yields of alkylated amines could be significantly
improved when secondary alcohols were employed. The synthesis of allylic
amines[75c] and pyrroles[76], and -alkylation of ketones with alcohols[75d] were
further explored using the same catalytic system. In 2016, Kirchner and
coworkers[77] reported a PNP pincer type iron complex Cat 17 catalyzed alkylation
of amines with alcohols.
Besides iron, cobalt pincer complexes were successfully applied in the same
transformation by the groups of Kempe[78a], Zhang[78b] and Kirchner[78c] (Scheme
16). Also, novel catalytic systems based on manganese pincer complexes were
reported by the groups of Beller[79ab] and Sortais[79c] (Scheme 16, B). Several types
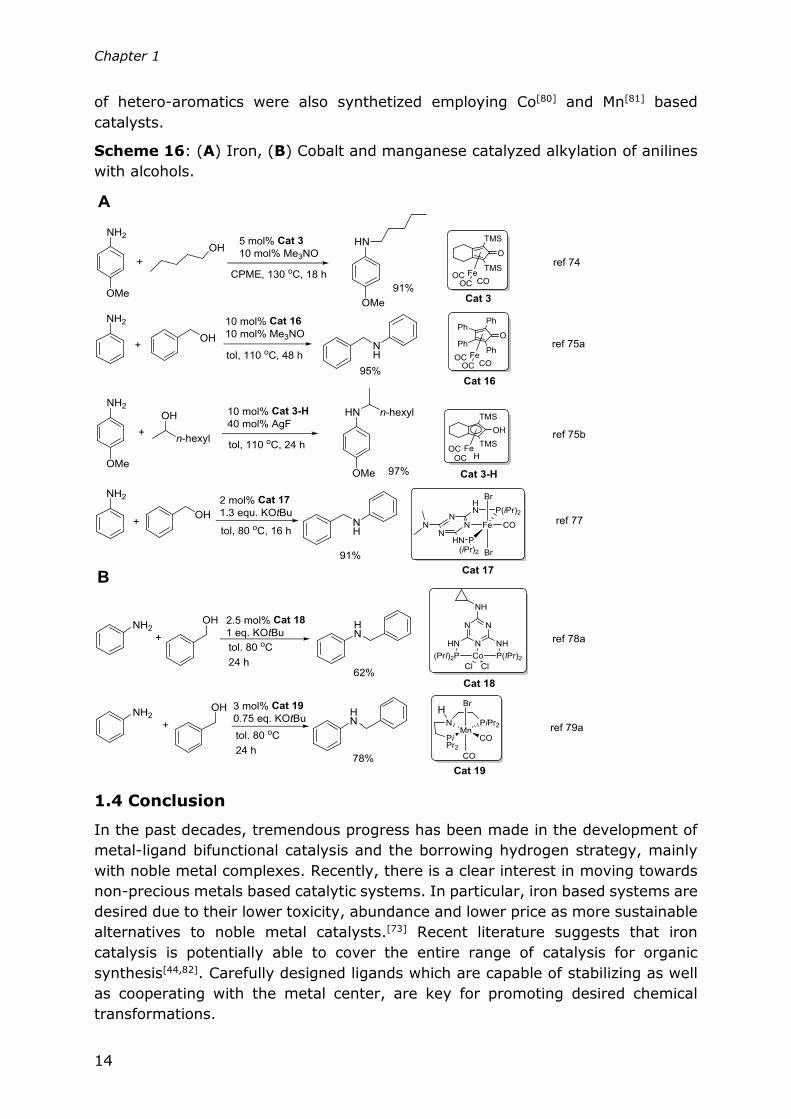
Chapter 1
14
of hetero-aromatics were also synthetized employing Co[80] and Mn[81] based
catalysts.
Scheme 16: (A) Iron, (B) Cobalt and manganese catalyzed alkylation of anilines
with alcohols.
1.4 Conclusion
In the past decades, tremendous progress has been made in the development of
metal-ligand bifunctional catalysis and the borrowing hydrogen strategy, mainly
with noble metal complexes. Recently, there is a clear interest in moving towards
non-precious metals based catalytic systems. In particular, iron based systems are
desired due to their lower toxicity, abundance and lower price as more sustainable
alternatives to noble metal catalysts.[73] Recent literature suggests that iron
catalysis is potentially able to cover the entire range of catalysis for organic
synthesis[44,82]. Carefully designed ligands which are capable of stabilizing as well
as cooperating with the metal center, are key for promoting desired chemical
transformations.

Introduction to the thesis
15
1.5 Outline of the thesis
This thesis describes the development of novel catalytic methods for the selective
alkylation of amines with alcohols through the borrowing hydrogen methodology,
using metal-ligand bifunctional complexes, in particular the Knölker complex and
the Shvo catalyst.
In Chapter 2, the discovery of the first well-defined iron complex catalyzed
alkylation of amines with alcohol is described. Chapter 3 describes the application
of the discovered method for transformations involving benzyl alcohols in order to
obtain benzylamines. In Chapter 4, iron catalyzed pyrrole synthesis is described
by N-heterocyclization of amines with unsaturated diols. Chapter 5 describes the
direct N-alkylation of unprotected amino acids with alcohols using the Knölker
complex and the Shvo catalyst with retention of stereochemistry. Chapter 6
describes the use of the Shvo catalyst in alkylation of amino acids esters with
alcohols, without racemization.

Chapter 1
16
References
[1] M. Beller, A. Renken, R. A. van Santen, Catalysis: From Principles to Applications,
Wiley-VCH, 2012.
[2] J. Hagen, Industrial Catalysis: A Practical Approach, 3rd Edition, John Wiley & Sons,
2015.
[3] J. Armor, North American Catalysis Society, report on “What is Catalysis or Catalysts,
So what?”, 2012, http://nacatsoc.org/above/what-is-catalysis/ (accessed 8th May
2017).
[4] M. B. Smith, J. March, March’s advanced organic chemistry: reactions, mechanisms,
and structure, 6th ed., John Wiley & Sons, Inc.: Hoboken, New Jersey, 2007.
[5] a) P. Lanzafame, G. Centi, S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562-7580;
b) S. Abate, P. Lanzafame, S. Perathoner, G. Centi, ChemSusChem, 2015, 8, 2854–
2866.
[6] a) S. Perathoner, S. Gross, E. J. M. Hensen, H. Wessel, H. Chraye, G. Centi,
ChemCatChem, 2017, 9, 904–909; b) M. Beller, G. Centi, L. Sun, ChemSusChem,
2017, 10, 6-13; c) P. J. Deuss, K. Barta, J. G. de Vries, Catal. Sci. Technol., 2014,
4, 1174-1196; d) K. Barta, P. C. Ford, Acc. Chem. Res., 2014, 47, 1503-1512.
[7] Catalysis—Key to a Sustainable Future Science and Technology Roadmap for
Catalysis in the Netherlands, Jan. 2015, http://www.niok.eu/en/wp-
content/files/catalysis-key-to-a-sustainable-future-web1.pdf (accessed 12th May
2017).
[8] a) J. Bariwalab, E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283-9303; b) Y.
Park, Y. Kim, S. Chang, Chem. Rev., 2017, 117, 9247–9301; c) J. F. Hartwig, Acc.
Chem. Res., 2008, 41, 1534−1544; d) J. F. Hartwig, Nature, 2008, 455, 314−322.
[9] Selected review for metal-ligand bifunctional catalysis, see: a) R. Noyori, M.
Yamakawa, S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944; b) R. Noyori, M.
Kitamura, T. Ohkuma, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 5356–5362; c) S. E.
Clapham, A. Hadzovic, R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237; d)
T. Ikariya, K. Murata, R. Noyori, Org. Biomol. Chem., 2006, 4, 393–406; e) D.
Milstein, Top. Catal., 2010, 53, 915–923.
[10] a) Y. Shvo, D. Czarkie, Y. Rahamim, J. Am. Chem. Soc., 1986, 108, 7400-7402; a)
H.-J. Knölker, E. Baum, H. Goesmann, R. Klauss, Angew. Chem. Int. Ed., 1999, 38,
2064-2066.
[11] J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University
Science Books, Sausalito, 2009.
[12] For a well-studied example on electronic and steric effect of phosphorus ligand on
homogenous catalysis, see: C. A. Tolman, Chem. Rev., 1977, 77, 313–348.
[13] T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc., 1995,
117, 2675-2616.
[14] Recent studies suggest that inner-sphere mechanism cannot be excluded in typical
outer-sphere mechanism systems, one recent paper see: P. A. Dub, B. L. Scott, J. C.
Gordon, J. Am. Chem. Soc., 2017, 139, 1245−1260.
[15] M. Kitamura, T. Ohkuma, S. Inoue, N. Sayo, H. Kumobayashi, S. Akutagawa, T. Ohta,
H. Takaya, R. Noyori, J. Am. Chem. Soc., 1988, 110, 629-631.
[16] For other relevant concepts, a) a review on bimetallic catalysis, see: E. K. van den
Beuken, B. L. Feringa, Tetrahedron, 1998, 54, 12985-13011; b) an example on
bifunctional enzyme, see: V. Fϋlöp, J. W. B. Moir, S. J. Ferguson, J. Hajdu, Cell, 1995,
81, 369-377; c) a review on multifunctional catalysis, see: G. J. Rowlandsp,
Tetrahedron, 2001, 57, 1865-1882.
[17] Y. Blum, D. Reshef, Y. Shvo, Tetrahedron Lett., 1981, 22, 1541-1544.
[18] Y. Blum, Y. Shvo, Isr. J. Chem., 1984, 24, 1984, 144-148.
[19] M. I. Bruce, J. R. Knight, J. Organometal. Chem., 1968, 12, 411-413.
[20] B. A. Persson, A. L. E. Larsson, M. Le Ray, J.-E. Bäckvall, J. Am. Chem. Soc., 1999,
121, 1645-1650.
[21] C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi, M. Kavana, J. Am. Chem. Soc.,
2001, 123, 1090-1100.

Introduction to the thesis
17
[22] a) N. Menashe, Y. Shvo, Organometallics, 1991, 10, 3885-3891; b) M. L. S. Almeida,
M. Beller, G.-Z. Wang, J.-E. Bäckvall, Chem. Eur. J., 1996, 2, 1533–1536; c) A. H.
Ell, J. S. Samec, C. Brasse, J.-E. Bäckvall, Chem. Commun., 2002, 10, 1144-1145;
d) Y. Blum, D. Czarkie, Y. Rahamim, Y. Shvo, Organometallics, 1985, 4, 1459–1461.
[23] J. S. Samec, J.-E. Bäckvall, Chem. Eur. J., 2002, 8, 2955-2961.
[24] A. L. E. Larsson, B. A. Persson, J.-E. Bäckvall, Angew. Chem. Int. Ed. Engl., 1997,
36, 1211-1212.
[25] a) D. Hollmann, S. Bähn, A. Tillack, M. Beller, Angew. Chem. Int. Ed., 2007, 46,
8291–8294; b) S. Imm, S. Bähn, A. Tillack, K. Mevius, L. Neubert, M. Beller, Chem.
Eur. J., 2010, 16, 2705–2709; c) C. Segarra, E. Mas-Marza, J. A. Mata, E. Peris, Adv.
Synth. Catal., 2011, 353, 2078–2084.
[26] a) R. Karvembu, R. Prabhakaran, K. Natarajan, Coord. Chem. Rev., 2005, 249, 911–
918; b) J. S. M. Samec, J.-E. Bäckvall, Hydroxytetraphenycyclo-
pentadienyl(tetraphenyl-2,4-cyclopentadien-1-one)-m-hydrotetracarbonyl-
diruthenium(II), In: Fuchs PL (ed) Encyclopedia of reagents for organic synthesis,
vol 7, 2nd ed., 2009, Wiley, New York, 5557–5564; c) B. L. Conley, M. K.
Pennington-Boggio, E. Boz, T. J. Williams, Chem. Rev., 2010, 110, 2294–2312; d)
M. C. Warner, C. P. Casey, J.-E. Bäckvall, Top. Organomet. Chem., 2011, 37, 85–
125.
[27] S. Samson, G. R. Stephenson, J. P. Stambuli, Pentacarbonyliron, e-EROS
Encyclopedia of Reagents for Organic Synthesis, 2008, DOI:
10.1002/047084289X.rp019.pub2.
[28] a) W. Reppe, H. Vetter, Justus Liebigs Ann. Chem., 1953, 582, 133-161; b) E. Weiss,
W. Hubel, J. Inorg. Nucl. Chem., 1959, 11, 42-55; c) G. N. Schrauzer, J. Am. Chem.
Soc., 1959, 81, 5307-5310.
[29] C. P. Casey, H. Guan, J. Am. Chem. Soc., 2007, 129, 5816-5817.
[30] For a review on non-innocent ligands, see: V. Lyaskovskyy, B. de Bruin, ACS Catal.,
2012, 2, 270–279.
[31] A. Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard, J.-L. Renaud, Angew.
Chem. Int. Ed., 2012, 51, 4976-4980.
[32] C. P. Casey, H. Guan, Organometallics, 2012, 31, 2631−2638.
[33] D. S. Merel, M. Elie, J.-F. Lohier, S. Gaillard, J.-L. Renaud, ChemCatChem, 2013, 5,
2939-2945.
[34] T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal., 2012, 354, 597-
601.
[35] M. G. Coleman, A. N. Brown, B. A. Bolton, H. Guan, Adv. Synth. Catal., 2010, 352,
967–970.
[36] a) S. Zhou, S. Fleischer, K. Junge, M. Beller, Angew. Chem. Int. Ed., 2011, 50, 5120–
5124; b) A. Quintard, T. Constantieux, J. Rodriguez, Angew. Chem. Int. Ed., 2013,
52, 12883–12887.
[37] O. El-Sepelgy, N. Alandini, M. Rueping, Angew. Chem. Int. Ed., 2016, 55, 13602 –
13605.
[38] a) S. Moulin, H. Dentel, A. Pagnoux-Ozherelyeva, S. Gaillard, A. Poater, L. Cavallo,
J.-F. Lohier, J.-L. Renaud, Chem. Eur. J., 2013, 19, 17881–17890; b) R. Hodgkinson,
A. Del Grosso, G. Clarkson, M. Wills, Dalton Trans., 2016, 45, 3992–4005; c) S.
Elangovan, S. Quintero-Duque, V. Dorcet, T. Roisnel, L. Norel, C. Darcel, J.-B. Sortais,
Organometallics, 2015, 34, 4521–4528; d) S. V. Facchini, J.-M. Neudörfl, L.
Pignataro, M. Cettolin, C. Gennari, A. Berkessel, U. Piarulli, ChemCatChem, DOI:
10.1002/cctc.201601591.
[39] M. Kamitani, Y. Nishiguchi, R. Tada, M. Itazaki, H. Nakazawa, Organometallics, 2014,
33, 1532–1535.
[40] A. Chakraborty, R. G. Kinney, J. A. Krause, H. Guan, ACS Catal., 2016, 6,
7855−7864.
[41] T. C. Johnson, G. J. Clarkson, M. Wills, Organometallics, 2011, 30, 1859–1868.
[42] A. Berkessel, S. Reichau, A. von der Hoh, N. Leconte, J.-M. Neudorfl, Organometallics,
2011, 30, 3880–3887.

Chapter 1
18
[43] P. Gajewski, M. Renom-Carrasco, S. V. Facchini, L. Pignataro, L. Lefort, J. G. de Vries,
R. Ferraccioli, A. Forni, U. Piarulli, C. Gennari, Eur. J. Org. Chem., 2015, 1887–1893.
[44] I. Bauer, H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387.
[45] A. Chakraborty, H. Guan, Iron, dicarbonylhydro[(1,2,3,3a,7a-η)-4,5,6,7-tetrahydro-
2-hydroxy-1,3-bis(trimethylsilyl)-1H-inden-1-yl], e-Encyclopedia of Reagents for
Organic Synthesis, 2014, DOI: 10.1002/047084289X.rn-01666.
[46] A. Quintard, J. Rodriguez, Angew. Chem. Int. Ed., 2014, 53, 4044–4055.
[47] a) R. Noyori, T. Ohkuma, Angew. Chem. Int. Ed., 2001, 40, 40-73; b) H.
Grützmacher, Angew. Chem. Int. Ed., 2008, 47, 1814–1818.
[48] W. Zuo, A. J. Lough, Y. F. Li, R. H. Morris, Science, 2013, 342, 1080-1083.
[49] R. M. Bullock, Science, 2013, 342, 1054-1055.
[50] J. Zhang, M. Gandelman, L. J. W. Shimon, H. Rozenberg, D. Milstein, Organometallics,
2004, 23, 4026-4033.
[51] J. Zhang, G. Leitus, Y. Ben-David, D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840-
10841.
[52] C. Gunanathan, Y. Ben-David, D. Milstein, Science, 2007, 317, 790-792.
[53] E. Khaskin, M. A. Iron, L. J.W. Shimon, J. Zhang, D. Milstein, J. Am. Chem. Soc.,
2010, 132, 8542–8543.
[54] M. Vogt, M. Gargir, M. A. Iron, Y. Diskin-Posner, Y. Ben-David, D. Milstein, Chem.
Eur. J., 2012, 18, 9194–9197.
[55] M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J.W. Shimon, Y. Ben-
David, D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018.
[56] M. Feller, E. Ben-Ari, Y. Diskin-Posner, R. Carmieli, L. Weiner, D. Milstein, J. Am.
Chem. Soc., 2015, 137, 4634–4637.
[57] a) S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C.
Holthausen, W. D. Jones, S. Schneider, ACS Catal., 2014, 4, 3994–4003; b) S. Musa,
I. Shaposhnikov, S. Cohen, D. Gelman, Angew. Chem. Int. Ed., 2011, 50, 3533–
3537; c) T. Stahl, K. Müther, Y. Ohki, K. Tatsumi, M. Oestreich, J. Am. Chem. Soc.,
2013, 135, 10978–10981; d) D. G. Gusev, A. J. Lough, Organometallics, 2002, 21,
2601–2603; e) W. H. Harman, J. C. Peters, J. Am. Chem. Soc., 2012, 134,
5080−5082; f) B. Bichler, C. Holzhacker, B. Stöger, M. Puchberger, L. F. Veiros, K.
Kirchner, Organometallics, 2013, 32, 4114–4121; g) A. M. Royer, T. B. Rauchfuss,
D. L. Gray, Organometallics, 2010, 29, 6763–6768; h) C. Gunanathan, B.
Gnanaprakasam, M. A. Iron, L. J. W.Shimon, D. Milstein, J. Am. Chem. Soc., 2010,
132, 14763–14765; i) E. Stepowska, H. Jiang, D. Song, Chem. Commun., 2010, 46,
556-558; j) A. D. Phillips, G. B. Laurenczy, R. Scopelliti, P. J. Dyson, Organometallics,
2007, 26, 1120-1122.
[58] J. P. Krogman, B. M. Foxman, C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582–
14585.
[59] J. R. Khusnutdinova, D. Milstein, Angew. Chem. Int. Ed., 2015, 54, 12236–12273.
[60] E. W. Baxter, A. B. Reitz, Reductive Aminations of Carbonyl Compounds with
Borohydride and Borane Reducing Agents, Organic Reactions, John Wiley and Sons,
Inc., DOI: 10.1002/0471264180.or059.01, ISBN 978-0-471-17655-8, 2004.
[61] M. B. Smith, J. March, March’s Advanced Organic Chemistry: Reactions, Mechanisms,
and Structure, 6th ed.; Wiley: Hoboken, NJ, 2007, Chapter 10.
[62] Methylamines can be produced at 350−500 °C and under 15−30 bar of pressure
using an aluminum-based heterogeneous catalyst from ammonia and methanol. See:
K. Weissermel, H.-J. Arpe, Industrial Organic Chemistry, 4th ed.; Wiley-VCH:
Weinheim, Germany, 2003, page 51.
[63] R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit, N. J. Tongpenyai, Chem. Soc. Chem.
Commun., 1981, 611−612.
[64] Y. Watanabe, Y. Tsuji, Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667−2670.
[65] a) M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A.
Watson, J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774; b) B.
Sundararaju, Z. Tang, M. Achard, G. V. M. Sharma, L. Toupet, C. Bruneau, Adv.
Synth. Catal., 2010, 352, 3141–3146; c) M. Zhang, S. Imm, S. Bahn, H. Neumann,
M. Beller, Angew. Chem. Int. Ed., 2011, 50, 11197–11201; d) B. Blank, S. Michlik,

Introduction to the thesis
19
R. Kempe, Chem. Eur. J., 2009, 15, 3790–3799; e) S. Imm, S. Bahn, M. Zhang, L.
Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas, M. Beller, Angew. Chem. Int.
Ed., 2011, 50, 7599–7603; f) R. Kawahara, K.-i. Fujita, R. Yamaguchi, J. Am. Chem.
Soc., 2010, 132, 15108–15111; g) R. Yamaguchi, S. Kawagoe, C. Asai, K.-i. Fujita,
Org. Lett., 2008, 10, 181–184; h) M. A. Berliner, S. P. A. Dubant, T. Makowski, K.
Ng, B. Sitter, C. Wager, Y. Zhang, Org. Process Res. Dev., 2011, 15, 1052–1062.
[66] a) M. H. S. A. Hamid, P. A. Slatford, J. M. J. Williams, Adv. Synth. Catal., 2007, 349,
1555−1575; b) T. D. Nixon, M. K. Whittlesey, J. M. J. Williams, Dalton Trans., 2009,
753−762; c) G. Guillena, D. J. Ramon, M. Yus, Chem. Rev., 2010, 110, 1611−1641;
d) S. Bahn, S. Imm, L. Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem,
2011, 3, 1853−1864; e) G. E. Dobereiner, R. H. Crabtree, Chem. Rev., 2010, 110,
681−703. f) C. Gunanathan, D. Milstein, Science, 2013, 341, 1229712; g) Q. Yang,
Q. Wang, Z. Yu, Chem. Soc. Rev., 2015, 44, 2305—2329.
[67] M. G. Edwards, R. F. R. Jazzar, B. M. Paine, D. J. Shermer, M. K. Whittlesey, J. M. J.
Williams, D. D. Edney, Chem. Commun., 2004, 90–91.
[68] M. G. Edwards, J. M. J. Williams, Angew. Chem. Int. Ed., 2002, 41, 4740–4743.
[69] R. Martínez, D. J. Ramón, M. Yus, Tetrahedron, 2006, 62, 8982–8987.
[70] A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski, M. Brookhart, Science,
2006, 312, 257–261.
[71] B. M. Trost, Science, 1991, 254, 1471–1477.
[72] A. J. A. Watson, J. M. J. Williams, Science, 2010, 329, 635−636.
[73] a) Rare Earth Elements—Critical Resources for High Technology, U.S. Geological
Survey, Fact Sheet 087-02; b) S. Enthaler, K. Junge, M. Beller, Angew. Chem. Int.
Ed., 2008, 47, 3317–3321; c) Guideline on the Specification Limits for Residues of
Metal Catalysts or Metal Reagents, EMEA/CHMP/ SWP/4446/2000, European
Medicines Agency, London, February 21, 2008.
[74] T. Yan, B. L. Feringa, K. Barta, Nat. Commun., 2014, 5, 5602.
[75] a) A. J. Rawlings, L. J. Diorazio, M. Wills, Org. Lett., 2015, 17, 1086-1089; b) H.-J.
Pan, T. W. Ng, Y. Zhao, Chem. Commun., 2015, 51, 11907-11910; c) B.
Emayavaramban, M. Roy, B. Sundararaju, Chem. Eur. J., 2016, 22, 3952–3955; d)
S. Elangovan, J.-B. Sortais, M. Beller, C. Darcel, Angew. Chem. Int. Ed., 2015, 54,
14483 –14486.
[76] T. Yan, K. Barta, ChemSusChem, 2016, 9, 2321–2325; b) B. Emayavaramban, M.
Sen, B. Sundararaju, Org. Lett., 2017, 19, 6–9.
[77] M. Mastalir, B. Stöger, E. Pittenauer, M. Puchberger, G. Allmaier, K. Kirchner, Adv.
Synth. Catal., 2016, 358, 3824–3831.
[78] a) S. Rosler, M. Ertl, T. Irrgang, R. Kempe, Angew. Chem. Int. Ed., 2015, 54, 15046–
15050; b) G. Zhang, Z. Yin, S. Zheng, Org. Lett., 2016, 18, 300−303; c) M. Mastalir,
G. Tomsu, E. Pittenauer, G. Allmaier, K. Kirchner, Org. Lett., 2016, 18, 3462−3465.
[79] a) S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel, M. Beller, Nat.
Commun., 2016, 7, 12641; b) J. Neumann, S. Elangovan, A. Spannenberg, K. Junge,
M. Beller, Chem. Eur. J., DOI: 10.1002/chem.201605218; c) A. Bruneau-Voisinea, D.
Wang, V. Dorcet, T. Roisnel, C. Darcel, J.-B. Sortais, J. Catal., 2017, 347, 57–62.
[80] a) P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David, D. Milstein, Angew. Chem. Int.
Ed., 2016, 55, 14373–14377.
[81] a) M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier, K. Kirchner, J. Am. Chem. Soc.,
2016, 138, 15543–15546; b) N. Deibl, R. Kempe, Angew. Chem. Int. Ed., 2017, 56,
1663–1666.
[82] A. Fϋrstner, ACS Cent. Sci., 2016, 2, 778−789.

Chapter 1
20

21
Chapter 2
Iron catalyzed direct alkylation of amines with alcohols
The selective conversion of carbon-oxygen bonds into carbon-nitrogen bonds to
form amines is one of the most important chemical transformations for the
production of bulk and fine chemicals and pharma intermediates. An attractive
atom economic way of carrying out such C-N bond formations is the direct N-
alkylation of simple amines with alcohols through the so-called borrowing
hydrogen strategy. Recently, transition metal complexes based on precious noble
metals have emerged as suitable catalysts for this transformation; however, the
crucial change towards highly selective methodologies, which use abundant,
inexpensive and environmentally friendly metals, in particular iron, has not yet
been accomplished. In this chapter, the homogeneous, iron-catalyzed, direct
alkylation of amines with alcohols is described. The scope of this new methodology
includes the selective monoalkylation of anilines and benzyl amines with a wide
range of alcohols as well as the use of diols in the formation of five-, six- and
seven- membered nitrogen heterocycles, which are privileged structures in
numerous pharmaceuticals.
Part of this chapter was published:
T. Yan, B. L. Feringa, K. Barta, Nat. Commun., 2014, 5, 5602.

Chapter 2
22
Introduction
Amines are among the most valuable classes of compounds in chemistry,
omnipresent in natural products, in particular alkaloids[2a], and widely used as
pharmaceuticals, agrochemicals, lubricants and surfactants[1,2,3]. Therefore the
development of efficient catalytic methodologies for C-N bond formation is a
paramount goal in organic chemistry.
The choice of an alcohol[4,5] as substrate for direct C-N bond formation is highly
desirable in order to produce secondary and tertiary amines and N-heterocyclic
compounds (Figure 1).
Figure 1: Catalytic, direct N-alkylation of amines with alcohols. a Conventional conversion
of alcohol into amine via installing a leaving group before nucleophilic substitution with an
amine donor or oxidizing alcohols to carbonyl compounds followed by reductive amination.
b The direct C–N bond formation via coupling of primary alcohols and amines forms
secondary amine products R1–NH–R2. R1–OH is a short- or long-chain aliphatic alcohol and
may contain aromatic functionality. R2–NH2 is an aromatic or aliphatic amine. c Using diols
of various chain lengths the products are 5- (n=1), 6- (n=2) or 7- (n=3) membered N-
heterocycles.
Alcohols are readily available through a variety of industrial processes and are
highly relevant starting materials in view of recent developments in the field of
renewables as they can be obtained via fermentation or catalytic conversion of
lignocellulosic biomass.[6,7] Conventional non-catalytic transformations of an amine
with an alcohol take place via installing a suitable leaving group instead of the
alcohol functionality followed by nucleophilic substitution, or oxidizing alcohols to
carbonyl compounds followed by reductive amination; these multistep pathways
suffer from low atom economy[8] or limited selectivity and the production of
stoichiometric amounts of waste (Figure 1a).[9]
A privileged catalytic methodology for the direct coupling of alcohols with amines
is based on the so-called borrowing hydrogen strategy (Figure 1b, 1c and Figure
2). During the catalytic cycle an alcohol is dehydrogenated to the corresponding
carbonyl compound, which reacts with the amine to form an imine. The imine is in

Alkylation of amines with alcohols
23
situ reduced to the alkylated amine and the metal complex facilitates the required
hydrogen shuttling. In fact the hydrogen delivered by the alcohol is temporarily
stored in the metal complex. Key features are that the process is hydrogen neutral,
no other reagents are needed and the only stoichiometric by-product is water.
A number of transition metal complexes have proven effective in this catalytic C-
N bond formation, in particular those based on ruthenium[10-12] and iridium[13].
These and related methodologies have been extensively reviewed[1,3,4,14-16].
Despite recent progress, the key challenge is the development of catalysts that
rely on the use of widely abundant, inexpensive metals[3,17]. Iron is considered to
be the ultimate, sustainable alternative for ruthenium[18], however, no unequivocal
reductive amination via a borrowing hydrogen mechanism has been reported[19].
Direct N-alkylation of amines with alcohols is limited to iron-halogenides under
rather harsh reaction conditions (160-200 °C), not proceeding via a hydrogen
autotransfer pathway[20].
This work shows that a well-defined homogenous Fe-based catalyst can be
successfully used in this atom-economic process, with a broad substrate scope.
These direct, Fe-catalyzed transformations are highly modular, and provide water,
as the only stoichiometric byproduct. The products are valuable secondary and
tertiary amines or heterocycles, which contain diverse moieties R1 (from the
alcohol substrate) and R2 (from the amine substrate) (Figure 1).
The presented direct waste-free alcohol to amine functional group interconversions
are important toward the development of sustainable iron based catalysis and will
enable the valorization of biomass-derived alcohols in environment-benign
reaction media.
Results and discussion
We reasoned that direct C-N bond formation with an iron catalyst is possible
provided by the catalytic complex which shows high activity both in alcohol
dehydrogenation (Figure 2a, Step 1) and imine hydrogenation (Figure 2a, Step 3).
The realization of this concept for direct alkylation of amines with alcohols using
cyclopentadieneone iron tricarbonyl complex Cat 3, the precurse to form Knölker’s
complex[21] Cat 3-H, is presented here (Figure 2). Iron cyclopentadienone complex
Cat 3 (Figure 2b), has been recently employed in catalysis including hydrogenation
of ketones[22], reductive amination[23], transfer hydrogenation of carbonyl
compounds and imines[24a], Oppenauer-type oxidation of alcohols[24b] as well as
cooperative dual catalysis[25]. Given its unique reactivity, we considered Cat 3 a
promising candidate for the development of the desired iron-catalyzed hydrogen
borrowing methodology. To achieve the direct amination of alcohols, the key
challenge is to match alcohol dehydrogenation and imine hydrogenation steps by
establishing conditions under which the formed Fe-H species (Cat 3-H) from the
initial alcohol dehydrogenation step is able to reduce the imine at a sufficient rate,
not requiring the use of dihydrogen.

Chapter 2
24
Catalytic N-alkylation with alcohols Preliminary experiments using 4-
methoxyaniline (1a) and a simple aliphatic alcohol, 1-pentanol (2a), with 5 mol%
pre-catalyst Cat 3 and 10 mol% Me3NO oxidant (to form active Cat 3-O) at 110 °C
in toluene showed the formation of 4-methoxy-n-pentylaniline (3aa), albeit with
only 30% selectivity at 48% substrate conversion (Table 1, entry 1). Although
promising, the initial results using common organic solvents indicated low
conversion of 1a or low selectivity towards 3aa and analysis of the reaction
mixtures identified insufficient imine reduction as the key problem. We reasoned
that most probably a weakly coordinating ethereal solvent is needed to stabilize
the key iron intermediates Cat 3-O[27]. In addition, imine formation might be
facilitated in solvents, which have limited miscibility with water. The major
breakthrough came when the green solvent cyclopentyl methyl ether (CPME), one
that uniquely combines such properties, was selected as reaction medium. CPME
recently emerged as a low-toxicity, sustainable solvent alternative for
tetrahydrofuran[28].
Figure 2 Individual reaction steps in the iron-catalyzed N-alkylation of amines using iron
cyclopentadienone complexes. a The overall transformation is the direct coupling of an
alcohol (R1–OH) with an amine (R2–NH2) to form the product amine (R1–NH–R2). The
sequence of reaction steps starts with the dehydrogenation of R1–OH to the corresponding
aldehyde with iron complex Cat 3-O (Step 1). Thereby one ‘hydrogen equivalent’ is
temporarily stored at the bifunctional iron complex Cat 3-O, which is converted to its
reduced, hydride form Cat 3-H. In Step 2, the carbonyl intermediate reacts with amine
R2–NH2 to form an imine intermediate and water. In Step 3, the hydrogen equivalent
“borrowed” from alcohol R1–OH are used in the reduction of the imine intermediate to
obtain the desired product R1–NH–R2. The reduction is accompanied by conversion of iron
hydride Cat 3-H to Cat 3-O, thereby a vacant coordination site is regenerated and the
catalytic cycle closed. b Reactivity of Fe cyclopentadienone complexes: Cat 3 is an air- and
moisture-stable iron tricarbonyl precatalyst. The active Cat 3-O complex is in situ
generated from Cat 3 by the addition of Me3NO to remove one CO. Cat 3-O is readily
converted to Cat 3-H by reaction with an alcohol.
Alkylation of amines with alcohols
25
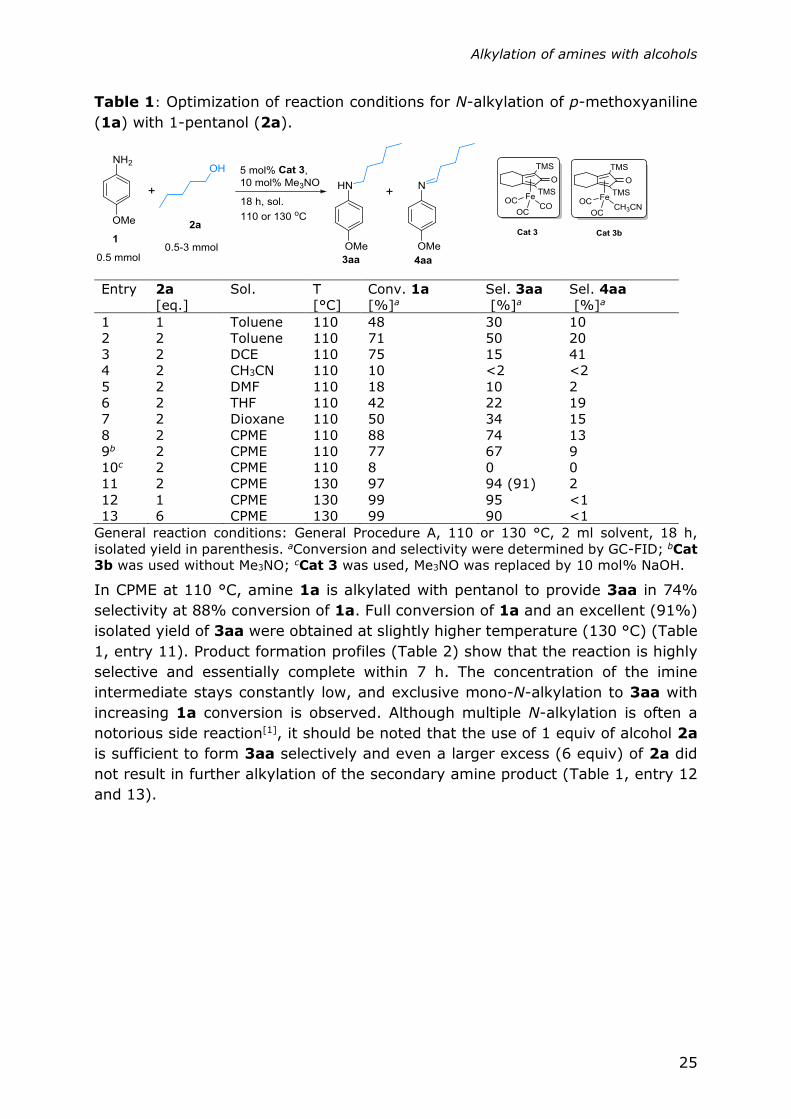
Table 1: Optimization of reaction conditions for N-alkylation of p-methoxyaniline
(1a) with 1-pentanol (2a).
Entry 2a
[eq.]
Sol. T
[°C]
Conv. 1a
[%]a
Sel. 3aa
[%]a
Sel. 4aa
[%]a
1 1 Toluene 110 48 30 10
2 2 Toluene 110 71 50 20
3 2 DCE 110 75 15 41
4 2 CH3CN 110 10 <2 <2
5 2 DMF 110 18 10 2
6 2 THF 110 42 22 19
7 2 Dioxane 110 50 34 15
8 2 CPME 110 88 74 13
9b 2 CPME 110 77 67 9
10c 2 CPME 110 8 0 0
11 2 CPME 130 97 94 (91) 2
12 1 CPME 130 99 95 <1
13 6 CPME 130 99 90 <1
General reaction conditions: General Procedure A, 110 or 130 °C, 2 ml solvent, 18 h,
isolated yield in parenthesis. aConversion and selectivity were determined by GC-FID; bCat
3b was used without Me3NO; cCat 3 was used, Me3NO was replaced by 10 mol% NaOH.
In CPME at 110 °C, amine 1a is alkylated with pentanol to provide 3aa in 74%
selectivity at 88% conversion of 1a. Full conversion of 1a and an excellent (91%)
isolated yield of 3aa were obtained at slightly higher temperature (130 °C) (Table
1, entry 11). Product formation profiles (Table 2) show that the reaction is highly
selective and essentially complete within 7 h. The concentration of the imine
intermediate stays constantly low, and exclusive mono-N-alkylation to 3aa with
increasing 1a conversion is observed. Although multiple N-alkylation is often a
notorious side reaction[1], it should be noted that the use of 1 equiv of alcohol 2a
is sufficient to form 3aa selectively and even a larger excess (6 equiv) of 2a did
not result in further alkylation of the secondary amine product (Table 1, entry 12
and 13).
Chapter 2
26
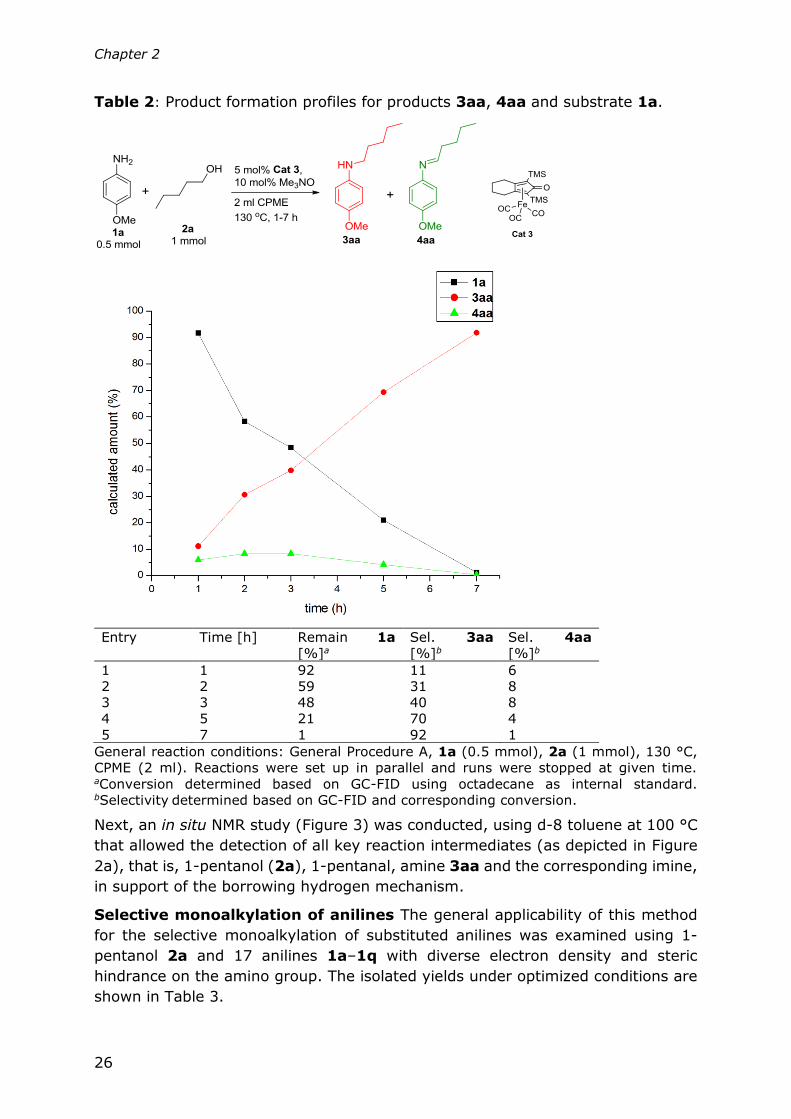
Table 2: Product formation profiles for products 3aa, 4aa and substrate 1a.
Entry Time [h] Remain 1a
[%]a
Sel. 3aa
[%]b
Sel. 4aa
[%]b
1 1 92 11 6
2 2 59 31 8
3 3 48 40 8
4 5 21 70 4
5 7 1 92 1
General reaction conditions: General Procedure A, 1a (0.5 mmol), 2a (1 mmol), 130 °C,
CPME (2 ml). Reactions were set up in parallel and runs were stopped at given time. aConversion determined based on GC-FID using octadecane as internal standard. bSelectivity determined based on GC-FID and corresponding conversion.
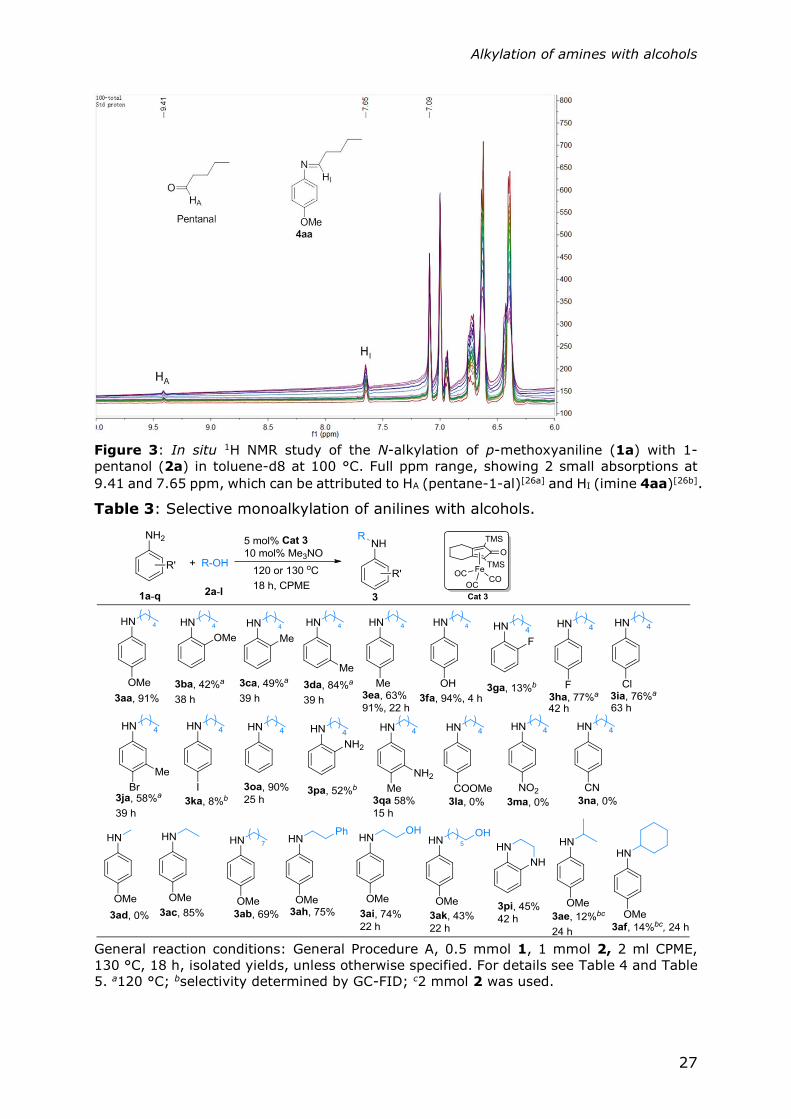
Next, an in situ NMR study (Figure 3) was conducted, using d-8 toluene at 100 °C
that allowed the detection of all key reaction intermediates (as depicted in Figure
2a), that is, 1-pentanol (2a), 1-pentanal, amine 3aa and the corresponding imine,
in support of the borrowing hydrogen mechanism.
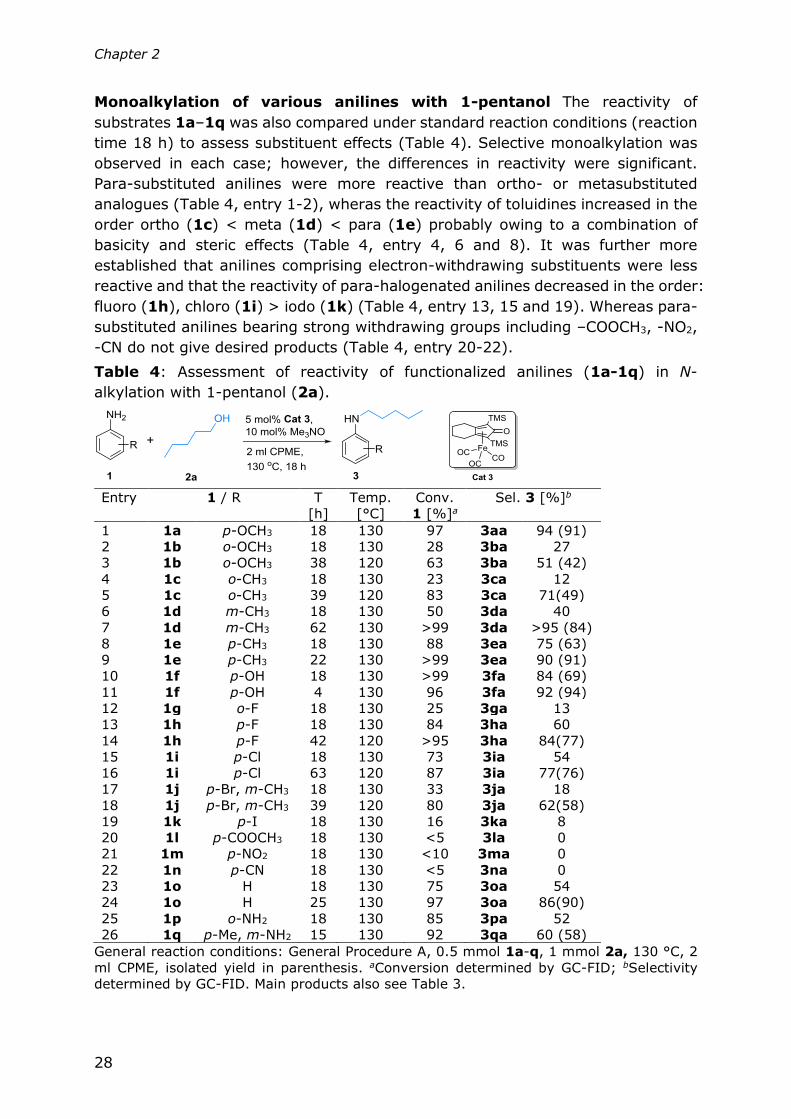
Selective monoalkylation of anilines The general applicability of this method
for the selective monoalkylation of substituted anilines was examined using 1-
pentanol 2a and 17 anilines 1a–1q with diverse electron density and steric
hindrance on the amino group. The isolated yields under optimized conditions are
shown in Table 3.
Alkylation of amines with alcohols
27
Figure 3: In situ 1H NMR study of the N-alkylation of p-methoxyaniline (1a) with 1-
pentanol (2a) in toluene-d8 at 100 °C. Full ppm range, showing 2 small absorptions at
9.41 and 7.65 ppm, which can be attributed to HA (pentane-1-al)[26a] and HI (imine 4aa)[26b].
Table 3: Selective monoalkylation of anilines with alcohols.
General reaction conditions: General Procedure A, 0.5 mmol 1, 1 mmol 2, 2 ml CPME,
130 °C, 18 h, isolated yields, unless otherwise specified. For details see Table 4 and Table
5. a120 °C; bselectivity determined by GC-FID; c2 mmol 2 was used.
Chapter 2
28
Monoalkylation of various anilines with 1-pentanol The reactivity of
substrates 1a–1q was also compared under standard reaction conditions (reaction
time 18 h) to assess substituent effects (Table 4). Selective monoalkylation was
observed in each case; however, the differences in reactivity were significant.
Para-substituted anilines were more reactive than ortho- or metasubstituted
analogues (Table 4, entry 1-2), wheras the reactivity of toluidines increased in the
order ortho (1c) < meta (1d) < para (1e) probably owing to a combination of
basicity and steric effects (Table 4, entry 4, 6 and 8). It was further more
established that anilines comprising electron-withdrawing substituents were less
reactive and that the reactivity of para-halogenated anilines decreased in the order:
fluoro (1h), chloro (1i) > iodo (1k) (Table 4, entry 13, 15 and 19). Whereas para-
substituted anilines bearing strong withdrawing groups including –COOCH3, -NO2,
-CN do not give desired products (Table 4, entry 20-22).
Table 4: Assessment of reactivity of functionalized anilines (1a-1q) in N-
alkylation with 1-pentanol (2a).
Entry 1 / R T
[h]
Temp.
[°C]
Conv.
1 [%]a
Sel. 3 [%]b
1 1a p-OCH3 18 130 97 3aa 94 (91)
2 1b o-OCH3 18 130 28 3ba 27
3 1b o-OCH3 38 120 63 3ba 51 (42)
4 1c o-CH3 18 130 23 3ca 12
5 1c o-CH3 39 120 83 3ca 71(49)
6 1d m-CH3 18 130 50 3da 40
7 1d m-CH3 62 130 >99 3da >95 (84)
8 1e p-CH3 18 130 88 3ea 75 (63)
9 1e p-CH3 22 130 >99 3ea 90 (91)
10 1f p-OH 18 130 >99 3fa 84 (69)
11 1f p-OH 4 130 96 3fa 92 (94)
12 1g o-F 18 130 25 3ga 13
13 1h p-F 18 130 84 3ha 60
14 1h p-F 42 120 >95 3ha 84(77)
15 1i p-Cl 18 130 73 3ia 54
16 1i p-Cl 63 120 87 3ia 77(76)
17 1j p-Br, m-CH3 18 130 33 3ja 18
18 1j p-Br, m-CH3 39 120 80 3ja 62(58)
19 1k p-I 18 130 16 3ka 8
20 1l p-COOCH3 18 130 <5 3la 0
21 1m p-NO2 18 130 <10 3ma 0
22 1n p-CN 18 130 <5 3na 0
23 1o H 18 130 75 3oa 54
24 1o H 25 130 97 3oa 86(90)
25 1p o-NH2 18 130 85 3pa 52
26 1q p-Me, m-NH2 15 130 92 3qa 60 (58)
General reaction conditions: General Procedure A, 0.5 mmol 1a-q, 1 mmol 2a, 130 °C, 2
ml CPME, isolated yield in parenthesis. aConversion determined by GC-FID; bSelectivity
determined by GC-FID. Main products also see Table 3.

Alkylation of amines with alcohols
29
Accordingly, the best result was obtained with p-hydroxyaniline, which was
converted to 3fa in excellent 94% isolated yield already after 4 h (Table 4, entry
11). This increased reactivity might be attributed to the enhanced nucleophilicity
of 1f or the presence of the relatively acidic phenol moiety, which likely catalyzes
the imine formation step. p-Methoxyaniline was converted to 3aa within 7 h (Table
2), and para-methylaniline (1e), being less reactive, gave 3ea in 91% isolated
yield after 22h (Table 4, entry 9). Unsubstituted aniline (1o) gave 3oa 90%
isolated yield after 25 h (Table 4, entry 24).
Other substrates required further optimization, which was initially conducted using
one of the least reactive substrates 1b. It was shown that prolonged reaction times
and the addition of molecular sieves (to accelerate the imine formation step) lead
to satisfactory results and products 3ba (Table 4, entry 3), as well as 3ca, 3da,
3ia and 3ja were isolated in moderate-to-high yields (42–84%) (Table 4, entry 5,
7, 16 and 18). Taking advantage of the distinct difference in reactivity between
substituted anilines, the selective monoalkylation of 1-methyl-2,4-diamino-
benzene (1q) resulted in preferential formation of 3qa in 58% isolated yield (Table
4, entry 26).
Table 5: N-alkylation of p-methoxyaniline (1a) with various alcohols (2b-2l).
Entry 2 Time [h] Conv.
1a [%]a
Sel. 3 [%]b Sel. 4 [%]b
1 2b n-Octanol 18 85 3ab 79 (69) 4ab 6
2 2c Ethanol 18 94 3ac 90 (85) 4ac 2
3 2d Methanol 18 8 3ad 0 4ad 0
4c 2e Propane-2-ol 24 42 3ae 12 4ae 24
5c 2f Cyclohexanol 24 50 3af 14 4af 32
6 2g Benzyl-alcohol 18 79 3ag 12 4ag 66
7 2h Phenylethanol 18 93 3ah 87 (75) 4ah 5
8 2i Ethane-1,2-diol 22 82 3ai 70 (74) 4ai 0
9 2k Hexane-1,6-diol 22 83 3ak 65 (43) 4ak 0
10d 2i Ethane-1,2-diol 24 80 3pi 40 4pi 0
11d 2i Ethane-1,2-diol 42 90 3pi 60 (45) 4pi 0
General reaction conditions: General Procedure A, 0.5 mmol 1a, 1 mmol 2a, 130 oC, 2 ml
CPME, isolated yield in parenthesis. aConversion determined by GC-FID; bSelectivity
determined by GC-FID; c2 mmol alcohol 2 was used; d3 mmol 2i was used and 2-
aminoaniline (1p) was used instead of 1a. Main products also see Table 3.
Monoalkylation of p-methoxyaniline with various alcohols Having
established the reactivity pattern of anilines, we found that the selective
monoalkylation of p-methoxyaniline (1a) proceeds with a variety of alcohols (2b-
2l) with excellent results (Table 5). For instance, octanol (2b), ethanol (2c) as
well as 2-phenyl-ethane-1-ol (2h) were readily converted to the corresponding
amines 3ab, 3ac and 3ah in 69–85% isolated yields (Table 5, entry 1, 2 and 7).

Chapter 2
30
Alkylations using methanol were not successful (Table 5, entry 3), probably
because the dehydrogenation of MeOH with employed catalytic system is
unfavored. Interestingly, selective mono-alkylation with ethane-1,2-diol (2i) and
hexane-1,6-diol (2k) delivered valuable amino-alcohols 3ai (74%) and 3ak (43%)
(Table 5, entry 8-9). Notably, 2,3-dihydro-quinoxaline (3pi) containing a
heterocyclic structure could also be constructed directly from inexpensive ethylene
glycol (Table 3; Table 5, entry 10-11).
N-alkylation of aliphatic amines with aliphatic alcohols It is important that
aliphatic amines could also be successfully used as reaction partners with various
alcohols (Figure 4). For instance, pentane-1-amine (5a) was alkylated with
benzylalcohol (2g), providing 6ag in 67% yield. The same monoalkylated amine
was obtained in 62% yield by a reverse route from benzylamine (7a) and 1-
pentanol (2a), showing the flexibility and versatility of the new catalytic
transformation. The reaction of piperidine (5b) with 2-phenylethylamine (2h)
afforded tertiary amine 6bh in 53% isolated yield. Interestingly, furfuryl-amine
(5c), which can be derived from lignocellulosic biomass through furfural, displayed
an interesting reactivity towards bis-N-alkylation. In the reaction of 5c with 3 equiv
of 2a, preferentially the corresponding tertiary amine (6caa) was formed.
Figure 4: N-Alkylation of various aliphatic amines with alcohols. a Modular synthesis with
aliphatic amines and alcohols. Pentyl-1-amine (5a) can be coupled with benzyl-alcohol (2g)
or benzyl amine (7a) is coupled with 1-pentanol (2a) to afford the same amine product. b
The methodology can be extended to secondary amines, piperidine (5b) is alkylated with
2-phenyl-1-ethanol (2h). c The reaction of furfuryl-amine (5c) with 1-pentanol (2a)
results in bis-N-alkylation product 6caa. aMolecular sieves were added.
Alkylation of amines with alcohols
31
N-alkylation of benzylamines N-alkylated benzyl amines are particularly
important targets, as these moieties are present in a variety of drug molecules[29].
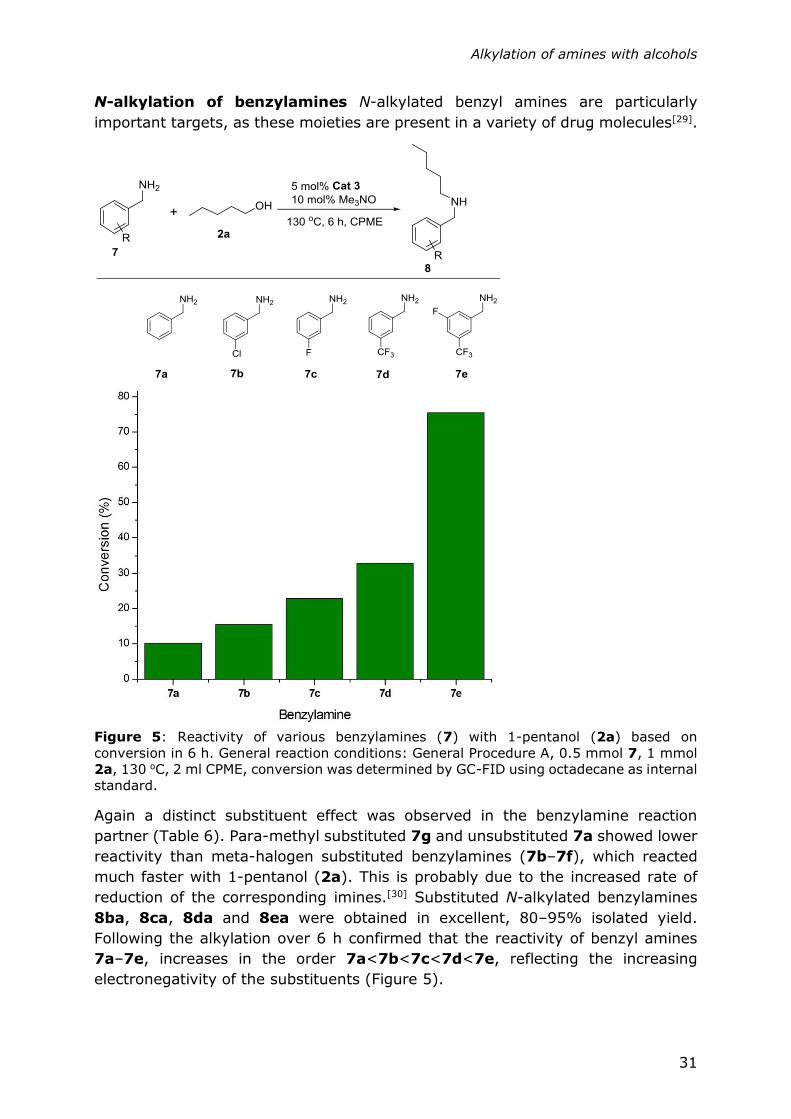
Figure 5: Reactivity of various benzylamines (7) with 1-pentanol (2a) based on
conversion in 6 h. General reaction conditions: General Procedure A, 0.5 mmol 7, 1 mmol
2a, 130 oC, 2 ml CPME, conversion was determined by GC-FID using octadecane as internal
standard.
Again a distinct substituent effect was observed in the benzylamine reaction
partner (Table 6). Para-methyl substituted 7g and unsubstituted 7a showed lower
reactivity than meta-halogen substituted benzylamines (7b–7f), which reacted
much faster with 1-pentanol (2a). This is probably due to the increased rate of
reduction of the corresponding imines.[30] Substituted N-alkylated benzylamines
8ba, 8ca, 8da and 8ea were obtained in excellent, 80–95% isolated yield.
Following the alkylation over 6 h confirmed that the reactivity of benzyl amines
7a–7e, increases in the order 7a<7b<7c<7d<7e, reflecting the increasing
electronegativity of the substituents (Figure 5).

Chapter 2
32
Table 6: N-Alkylation of benzylamines with 1-pentanol and diols.
General reaction conditions: General procedure A, 0.5 mmol 7, 1 mmol 2, 130 oC, 18-50
h, isolated yields after purification. aSelectivity determined by GC-FID.
Heterocycle formation with various benzylamines and diols Building on the
excellent reactivity of benzyl amines 7c–7e, we attempted the formation of
nitrogen-containing heterocycles of various sizes using diols of different chain
lengths (butane-1,4-diol 2m, pentane-1,5-diol 2l or hexane- 1,6-diol 2k) (Table
6). An additional advantage of the use of benzylamines is that the free N-
heterocycles can be readily obtained by common debenzylation procedures.
Despite the fact that a sequential catalytic N,N-dialkylation at the same nitrogen
has to occur involving each of the hydroxyl groups of the diol, we reasoned that
the first intermolecular alkylation is followed by an iminium ion-based alkylation,
likely facilitated by the intramolecular nature of the second alkylation step. To our
delight, pyrrolidine 9cm, containing a key five-membered heterocycle, was
obtained in 60% yield from 7c and butane-1,4-ol (2m). Six-membered piperidine
derivative 9cl was constructed using pentane-1,5 diol (2l) and 7c. As seven-
membered ring formation is generally challenging in organic synthesis, it is a
notable feature of our new Fe-based catalytic procedure that various azepane type
N-heterocyclic compounds were readily obtained using benzyl amines 7b–7e and
hexane-1,6-diol (2k). It is remarkable that in all these reactions, full conversion
and perfect product selectivity was observed. Among all derivatives, the chloro-
substituted 9bk was obtained with the highest isolated yield (85%). In contrast,

Alkylation of amines with alcohols
33
the reaction of unsubstituted benzylamine 7a with 2k afforded the monoalkylation
product, showing that the second cyclization step was much slower in this case.
These results highlight the value of this methodology in accessing biological
important nitrogen heterocycles, which contain -F and -CF3 substituents. These are
frequently encountered motives in pharmaceutically active compounds (Figure 6).
Synthesis of pharmaceutically relevant molecules To demonstrate the power
of the new C–N bond formation, we applied the Fe-catalyzed direct amine
alkylation as a key connection step in the synthesis of the N-arylpiperazine drug
Piribedil (12) (Figure 6), a dopamine antagonist used in the treatment of
Parkinson’s disease[11,31]. The application of iron-based homogeneous catalyst in
the preparation of pharmaceutically active molecules is highly desired, given the
limited allowed ppm levels of noble metal catalysts in the final products[29].
Commercially available 1-(2-pyrimidyl)-piperazine (10) and 4 equiv of piperonyl
alcohol (11) were used as the substrates, providing Piribedil in 54% isolated yield.
This direct, base free catalytic coupling illustrates several advantages for drug
syntheses: no auxiliary reagents are required, an iron catalyst and the green
solvent CPME are used, and water is the only waste product.
Figure 6: Direct synthesis of Piribedil 12.
Conclusion
We have developed a versatile Fe-based catalytic approach for the direct coupling
of amines and alcohols. Key elements are a single bifunctional Fe-catalyst for
alcohol dehydrogenation and imine hydrogenation, and the use of the green
solvent CPME. The variety of primary amines and alcohols, including those derived
from biomass, which can be coupled, provides an excellent basis for highly flexible
methodology to synthesize a broad array of secondary amines and nitrogen
heterocycles and holds promise for future application in diversity-oriented
synthesis strategies. The first upscaling experiments showed that this direct N-
alkylation can provide up to gram amounts of secondary amines. Mechanistic
investigations are needed to elucidate the role of steric and electronic parameters
in the rate-determining step, which can be both the imine formation as well as the
imine reduction. For example, p-hydroxy aniline was rapidly converted within 4 h,
whereas o-methoxy aniline was much less reactive. Similarly, benzyl-amines
display marked reactivity difference, depending on the nature and position of the
substituents at the aromatic ring. We foresee that control of the reactivity of well-
defined iron catalysts, as shown here successfully for the borrowing hydrogen
strategy in the direct alkylation of amines with alcohols, will enable the discovery
of a range of other new catalyst structures and sustainable transformations, using
abundant iron.

Chapter 2
34
Experimental section
General methods
Chromatography: Merck silica gel type 9385 230-400 mesh or Merck Al2O3 90
active neutral, TLC: Merck silica gel 60, 0.25 mm or Al2O3 60 F254 neutral.
Components were visualized by UV, Ninhydrin or I2 staining. Progress of the
reactions was determined by GC-MS (GC: HP 6890, MS: HP 5973) with an HP012
column (Agilent Technologies, Palo Alto, CA). Mass spectra were recorded on an
AEI-MS-902 mass spectrometer (EI+) or a LTQ Orbitrap XL (ESI+). Conversions
were determined by GC-FID (GC: HP 6890) with an HP-5 column (Agilent
Technologies, Palo Alto, CA). GC-MS and GC-FID analysis method: 60 °C 5 min,
180 °C 5 min (10 °C/min), 260 oC 5 min (10 °C/min). 1H- and 13C NMR spectra
were recorded on a Varian AMX400 (400 and 100.59 MHz, respectively) using
CDCl3 or CD2Cl2 as solvent. In situ 1H NMR spectra were recorded on a Varian Unity
Plus Varian-500 (500 MHz) using toluene-d8 as solvent. Chemical shift values are
reported in ppm with the solvent resonance as the internal standard (CDCl3: 7.26
for 1H, 77.00 for 13C; CD2Cl2: 5.32 for 1H, 53.84 for 13C; toluene-d8: 7.09 for 1H).
Data are reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet,
t = triplet, q = quartet, br = broad, m = multiplet), coupling constants (Hz), and
integration. All reactions were carried out under an Argon atmosphere using oven
dried glassware and using standard Schlenk techniques. THF and toluene were
collected from a MBRAUN solvent purification system (MB SPS-800). Dioxane
(99.5%, extra dry), dichloroethane (DCE, 99.8%, extra dry), N,N-
dimethylformamide (DMF, 99.8%, extra dry) and acetonitrile (CH3CN, 99.9%,
extra dry) were purchased from Acros without further purification. Aniline, 4-
methylbenzylamine, benzylamine, 3-trifluoromethylbenzylamine were purified by
distillation. 4-Methylaniline was purified by recrystallization. 2-Methoxyaniline was
purified by column chromatography. All other reagents were purchased from
Sigma or Acros in reagent or higher grade and were used without further
purification. Complex Cat 3, Cat 3b was synthetized according to literature
procedures[32].
Preparation of Cat 3 and Cat 3b
1,8-Bis(trimethylsilyl)-1,7-octadiyne To a solution of 1,7-octadiyne (2 mL,
1.56 g, 15 mmol) in THF (20 mL), a solution of n-BuLi (20.6 mL of a 1.6M in
hexane solution, 33 mmol, 2.2 equiv) was added dropwise over 20 min at -78 °C
under N2, in a 100ml Schlenk tube. After stiring for 10 min at -78 °C, and 1 h
under room temperature, trimethylsilyl chloride (8.4 ml, 66 mmol, 4.4 equiv) was
added slowly and the mixture was stirred at room temperature for 16 hours. The
cloudy, white mixture was quenched with saturated aqueous NH4Cl (5 mL) followed
by water until all of the white precipitate dissolved. The organic layer was removed

Alkylation of amines with alcohols
35
and the aqueous layer was extracted with pentane. The combined organic layers
were dried over anhydrous Na2SO4 and the solvent was evaporated under reduced
pressure to give a yellow oil. Purification of the crude product by Kugelrohr
distillation afforded 3.56 g (95% yield) of a light yellow oil/solid mixture (product
is a low-melting solid). 1H NMR (400 MHz, CDCl3, ppm): δ 2.23–2.28 (m, 4H),
1.60–1.64 (m, 4H), 0.15 (s, 18H). The physical data were identical in all respects
to those previously reported.[32]
Tricarbonyl(1,3-bis(trimethylsilyl)-4,5,6,7-tetrahydro-2H-inden-2-
one)iron (Cat 3) A solution of 1,8-bis(trimethylsilyl)-1,7-octadiyne (1.2 g, 4.8
mmol) and diiron nonacarbonyl (1.74 g, 4.8 mmol, 1 equiv) in toluene (60 mL) in
a 250 ml oven-dried Schlenk tube was heated to 120 °C and stirred for 24 hours
under argon. After cooling, the mixture was filtered over celite. Then the solution
was concentrated and filtered over silica to remove all the iron black. The solution
was evaporated under reduced pressure. The remaining solid residue was washed
with cold pentane and crystalized in heptane affording yellow a crystalline solid,
which is air and moisture stable (1.36g, 68% yield). 1H NMR (400 MHz, CDCl3): δ
2.50–2.62 (m, 4H), 1.77–1.87 (m, 4H), 0.26 (s, 18 H). 13C NMR (100 MHz, CDCl3):
δ 209.04, 181.22, 110.99, 71.74, 24.77, 22.41, –0.28. The physical data were
identical in all respects to those previously reported.[32]
Acetonitrile dicarbonyl(1,3-bis(trimethylsilyl)-4,5,6,7-tetrahydro-2H-
inden-2-one)iron (Cat 3b) An oven-dried 250 ml Schlenk tube was charged with
100 ml dry acetone and 2 ml dry CH3CN and degassed with N2 for 20 min. Then 1
g Cat 3 (2.38 mmol) was added under N2, stirring for 1 min until it fully solubilized.
216 mg Me3NO (1.2eq) was added under N2. A direct color change from yellow to
orange was observed in 5 s. The conversion of Cat 3 can be monitored by TLC
(pentane/ethyl acetate = 1/1, on silica gel, RfCat1a = 0.95, RfCat1b = 0.35). After 1
h, the solvent was removed under a vacuum; Cat 3b was purified through flash
chromatography, and obtained as brown solid (0.91g, 88% yield). 1H NMR (400
MHz, CDCl3): δ 2.05 – 2.48 (m, 4H), 2.21 (s, 3H), 1.38 – 1.73 (m, 4H), 0.22 (s,
18 H). 13C NMR (100 MHz, CDCl3): δ 212.80, 180.12, 126.00, 106.58, 69.91, 24.83,
22.31, 4.43, -0.12. The physical data were identical in all respects to those
previously reported.[32]
Cat 3 and Cat 3b are slightly light sensitive but air stable.
Representative procedures
General procedure A: An oven-dried 20 ml Schlenk tube, equipped with a stirring
bar, was charged with amine (0.500 mmol, 1 equiv), alcohol (given amount), iron
complex Cat 3 (0.025 mmol, 10.5 mg), Me3NO (0.050 mmol, 3.75 mg) and
cyclopentyl methyl ether (solvent, 2 ml). Solid materials were weighed into the
Schlenk tube under air, the Schlenk tube was subsequently connected to an argon
line and vacuum-argon exchange was performed three times. Liquid starting
materials and solvent were charged under an argon stream. The Schlenk tube was
capped and the mixture was rapidly stirred at room temperature for 1 min, then it
was placed in a pre-heated oil bath at the appropriate temperature and stirred for

Chapter 2
36
a given time. The reaction mixture was cooled down to room temperature and the
crude mixture was filtered through celite, eluted with ethyl-acetate, and
concentrated in vacuo. The residue was purified by flash column chromatography
to provide the pure amine product.
General procedure B (With molecular sieves): An oven-dried 20 ml Schlenk tube,
equipped with stirring bar, was charged with amine (0.5 mmol, 1 equiv), alcohol
(given amount), iron complex Cat 3 (0.025 mmol, 10.5 mg), Me3NO (0.05 mmol,
3.75 mg) and cyclopentyl methyl ether (solvent, 2 ml). The solid starting materials
were added into the Schlenk tube under air, the Schlenk tube was subsequently
connected to an argon line and a vacuum-argon exchange was performed three
times. Liquid starting materials and solvent were charged under an argon stream
followed by addition of 35.0–45.0 mg activated molecular sieves 4A. The Schlenk
tube was capped and the mixture was rapidly stirred at room temperature for 1
minute, then was placed into a pre-heated oil bath at the appropriate temperature
and stirred for a given time. The reaction mixture was cooled down to room
temperature and the crude mixture was filtered through celite, eluted with ethyl
acetate, and concentrated in vacuo. The residue was purified by flash column
chromatography to provide the pure amine product.
Upscaling procedure for N-alkylation of p-methoxyaniline An oven-dried
100 ml Schlenk tube, equipped with a stirring bar, was charged with p-
methoxyaniline (7.40 mmol, 0.910 g, 1 equiv), iron complex Cat 3 (0.37 mmol,
155 mg, 0.05 equiv) and Me3NO (0.74 mmol, 55.5 mg, 0.10 equiv) under air and
the Schlenk tube was subsequently connected to an argon line and vacuum-argon
exchange was performed three times. Then 1-pentanol (14.8 mmol, 1.62 ml, 2
equiv), cyclopentylmethylether (solvent, 30 ml), 3Å molecular sieves (800 mg)
were charged under an argon stream. The Schlenk tube was capped and the
mixture was rapidly stirred at room temperature for 3 min, then was placed into a
pre-heated oil bath at 130 °C and stirred for 18 h. After cooling down to room
temperature, the crude mixture was concentrated in vacuo. The residue was
purified by flash column chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5)
to provide 1.085 gram of p-methoxy-N-pentylaniline in 76% isolated yield.
General procedure for in situ 1H NMR study of the N-alkylation of p-
methoxyaniline (1a) with 1-pentanol (2a) in toluene-d8 at 100 °C. 0.12 mmol p-
anisidine (1a), 0.24 mmol 1-pentanol (2a), 0.012 mmol Cat 3, 0.024 mmol Me3NO
and 0.6 ml toluene-d8 were added in a J-Young NMR tube under argon. The tube
was sealed in an argon flow and placed in the pre-heated (100 °C) NMR machine,
shimming was performed at reaction temperature. In the following 8 h, 16 spectra
were recorded.

Alkylation of amines with alcohols
37
Spectral data of isolated compounds
4-methoxy-N-pentylaniline (3aa): Synthesized according to
General procedure A. p-Anisidine (0.062 g, 0.500 mmol) affords
3aa (0.088 g, 91% yield). Yellow oil obtained after column
chromatography (SiO2, N-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 6.78 (d, J = 8.9 Hz, 2H), 6.59 (d, J = 8.8 Hz,
2H), 3.75 (s, 3H), 3.06 (t, J = 7.2 Hz, 2H), 1.55 - 1.69 (m, 2H),
1.30 - 1.50 (m, 4H), 0.92 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz,
CDCl3) δ 151.93, 142.79, 114.85, 114.00, 55.78, 44.99, 29.34, 22.50, 14.01. The
physical data were identical in all respects to those previously reported.[33]
2-methoxyl-N-pentylaniline (3ba): Synthesized according to
General procedure B. o-Anisidine (0.056 ml, 0.5 mmol) affords
3ba (0.041 g, 42% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 6.90 (td, J = 7.6, 1.1 Hz, 1H), 6.79 (d, J =
7.9 Hz, 1H), 6.58 – 6.74 (m ,2H), 4.05 – 4.35 (br.s, 1H), 3.87
(s, 3H), 3.14 (t, J = 7.1 Hz, 2H), 1.61 – 1.75 (m, 2H), 1.32 –
1.50 (m, 4H), 0.95 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 146.68, 138.47,
121.26, 116.02, 109.69, 109.29, 55.33, 43.67, 29.40, 29.23, 22.52, 14.02. HRMS
(APCI+, m/z): calculated for C12H20NO [M+H]+:194.15394; found: 194.15387.[34]
2-methyl-N-pentylaniline (3ca): Synthesized according to
General procedure B. o-Toluidine (0.053 ml, 0.5 mmol) affords 3ca
(0.043 g, 49% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.15 (m, J = 7.7 Hz, 1H), 7.07 (d, J = 7.2, 1H),
6.60 – 6.70 (m, 2H), 3.35 – 3.60 (br.s, 1H), 3.17 (t, J = 7.1 Hz,
2H), 2.16 (s, 3H), 1.64 – 1.74 (m, 2H), 1.35 – 1.50 (m, 4H), 0.96
(t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 146.36, 129.96,
127.10, 121.63, 116.58, 109.58, 43.93, 29.41, 29.30, 22.52, 17.42, 14.04. HRMS
(APCI+, m/z): calculated for C12H20N [M+H]+: 178.15903; found: 178.15896.[34]
3-methyl-N-pentylaniline (3da): Synthesized according to
General procedure A. m-Toluidine (0.054 ml, 0.5 mmol) affords
3da (0.074 g, 84% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.05 – 7.15 (m, 1H), 6.51 – 6.62 (m, 1H),
6.38 – 6.51 (m, 2H), 3.32 – 3.85 (br.s, 1H), 3.13 (t, J = 7.2 Hz,
2H), 2.32 (s, 3H), 1.57 – 1.72 (m, 2H), 1.32 – 1.52 (m, 4H), 0.96
(t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 148.52, 138.89, 129.03, 117.97,
113.42, 109.82, 43.95, 29.32, 29.26, 22.48, 21.59, 14.02. HRMS (APCI+, m/z):
calculated for C12H19N [M+H]+: 178.15903; found: 178.15874.[35]

Chapter 2
38
4-methyl-N-pentylaniline (3ea): Synthesized according to
General procedure A. p-Toluidine (0.054 g, 0.5 mmol) affords 3ea
(0.081 g, 91% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 6.99 (d, J = 8.1 Hz, 2H), 6.56 (d, J = 8.2 Hz,
2H), 3.08 (t, J = 7.1 Hz, 2H), 2.24, (s, 3H), 1.56 – 1.67 (m, 2H),
1.27 – 1.50 (m, 4H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz,
CDCl3) δ 146.26, 129.65, 126.23, 112.87, 44.34, 29.34, 29.30, 22.50, 20.33,
14.02.[33]
4-(pentylamino)phenol (3fa): Synthesized according to General
procedure A. 4-Aminophenol (0.055 g, 0.5 mmol) affords 3fa (0.084
g, 94% yield).Yellow oil obtained after column chromatography
(SiO2, n-pentane/EtOAc 90:10 to 60:40). 1H NMR (400 MHz, CDCl3)
δ 6.68 (d, J = 8.0 Hz, 2H), 6.55 (d, J = 8.2 Hz, 2H), 3.90 – 1.69
(br.s, 2H), 3.05 (t, J = 6.8 Hz, 2H), 1.52 – 1.69 (m, 2H), 1.28 –
1.45 (m, 4H), 0,91 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ
148.12, 142.23, 116.23, 114.83, 45.42, 29.30, 22.47, 13.99. HRMS
(APCI+, m/z): calculated for C11H25N [M+H]+: 180.13829; found: 180.13823.
4-fluoro-N-pentylaniline (3ha): Synthesized according to
General procedure A. 4-fluoroaniline (0.047 ml, 0.5 mmol) affords
3ha (0.070 g, 77% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 6.80 – 6.95 (m, 2H), 6.47 – 6.58 (m, 2H), 3.27
– 3.59 (br.s, 1H), 3.06 (t, J = 7.1 Hz, 2H), 1.55 - 1.67 (m, 2H),
1.31 - 1.44 (m, 4H), 0.88 – 0.99 (m, 3H). 13C NMR (100 MHz,
CDCl3) δ 155.61 (d, J = 234.4 Hz), 144.86, 115.53 (d, J = 22.3 Hz), 113.49 (d, J
= 7.4 Hz), 44.63, 29.30, 29.20, 22.48, 14.00.[36]
4-Chloro-N-pentylaniline (3ia): Synthesized according to
General procedure A. 4-Chloroaniline (0.063 g, 0.5 mmol)
affords 3ia (0.075 g, 76% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.11 (d, J = 8.6 Hz, 2H), 6.54 (d, J = 8.7 Hz,
2H), 3.75 (s, 3H), 3.07 (t, J = 7.2 Hz, 2H), 1.55 - 1.67 (m, 2H),
1.27 - 1.44 (m, 4H), 0.92 (t, J = 6.7 Hz, 3H). 13C NMR (100
MHz, CDCl3) δ 147.02, 128.93, 121.45, 113.63, 44.03, 29.25,
29.08, 22.45, 13.98.[33]
4-bromo-3-methyl-N-pentylaniline (3ja): Synthesized
according to General procedure B. 4-Bromo-3-methylaniline
(0.092 g, 0.5 mmol) affords 3ja (0.074 g, 58% yield). Yellow
oil obtained after column chromatography (SiO2, n-
pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ
7.26 (d, J = 8.6 Hz, 1H), 6.48 (d, J = 2.8 Hz, 1H), 6.31 (dd, J
= 8.6, 2.8 Hz, 1H), 3.35 – 3.80 (br.s, 1H), 3.06 (t, J = 7.1 Hz,
2H), 2.31 (s, 3H), 1.50 – 1.65 (m, 2H), 1.30 – 1.42 (m, 4H), 0.92 (t, J = 6.8 Hz,

Alkylation of amines with alcohols
39
3H). 13C NMR (100 MHz, CDCl3) δ 147.74, 138.14, 132.56, 114.93, 111.83, 111.32,
43.99, 29.27, 29.12, 23.05, 22.47, 14.01. HRMS (APCI+, m/z): calculated for
C12H19NBr [M+H]+: 258.06954; found: 258.06738.
N-Pentylaniline (3oa): Synthesized according to General
procedure A. Aniline (0.046 ml, 0.5 mmol) affords 3oa (0.073 g,
90% yield). Yellow oil obtained after column chromatography (SiO2,
n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.15
– 7.25 (m, 2H), 6.68 – 6.67 (m, 1H), 6.59 – 6.67 (m, 2H), 3.45 –
3.80 (br.s, 1H), 3.13 (t, J = 7.2 Hz, 2H), 1.57 – 1.71 (m, 2H), 1.32
– 1.48 (m, 4H), 0.95 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 148.45,
129.16, 117.03, 112.65, 43.94, 29.31, 29.22, 22.49, 14.02. The physical data
were identical in all respects to those previously reported.[33]
4-Methyl-N1-pentylbenzene-1,3-diamine (3qa): Synthesized
according to General procedure A. 2,4-Diaminotoluene (0.061 g, 0.5
mmol) affords 3qa (0.056 g, 58% yield). Yellow oil obtained after
column chromatography (SiO2, n-pentane/EtOAc 90:10 to 50:50). 1H
NMR (400 MHz, CDCl3) δ 6.85 (d, J = 8.0, 1H), 6.05 (dd, J = 8.0, 2.2
Hz, 1H), 6.00 (d, J = 2.2 Hz, 1H), 3.25 – 3.65 (br.s, 3H), 3.07 (t, J =
7.2 Hz, 2H), 2.08 (s, 3H), 1.55 – 1.66 (m, 2H), 1.33 – 1.43 (m, 4H),
0.94 (t, J = 7.0, 3H). 13C NMR (100 MHz, CDCl3) δ 147.93, 145.17,
130.91, 111.44, 103.87, 99.64, 44.21, 29.29, 29.27, 22.46, 16.33, 14.00. NOESY-
NMR spectrum see figure 7. HRMS (APCI+, m/z): calculated for C12H21N2 [M+H]+:
193.16993; found: 193.16962.
1,2,3,4-Tetrahydroquinoxaline (3pi): Synthesized according to
General procedure A. o-Phenylenediamine (0.054 g, 0.5 mmol) affords
3pi (0.030 g, 45% yield). Orange solid obtained after column
chromatography (SiO2, n-pentane/EtOAc 90:10 to 50:50). 1H NMR (400
MHz, CDCl3) δ 6.55 – 6.63 (m, 2H), 6.46 – 6.54 (m, 2H), 3.42 (s, 4H), 3.25 –
3.54 (br.s, 2H). 13C NMR (100 MHz, CDCl3) δ 133.57, 118.77, 114.72, 41.34. The
physical data were identical in all respects to those previously reported.[37]
4-Methoxy-N-octylaniline (3ab): Synthesized according
to General procedure A. p-Anisidine (0.062 g, 0.5 mmol)
affords 3ab (0.081 g, 69% yield). Yellow oil obtained after
column chromatography (SiO2, n-pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CDCl3) δ 6.68 (d, J = 8.7 Hz, 2H),
6.60 (d, J = 8.7 Hz, 2H), 3.75 (s, 3H), 3.06 (t, J = 7.2 Hz,
2H), 1.55 – 1.65 (m, 2H), 1.34 – 1.44 (m, 2H), 1.16 – 1.44
(m, 8H). 13C NMR (100 MHz, CDCl3) δ 151.96, 142.81, 114.88, 114.03, 55.81,
45.05, 31.81, 29.67, 29.42, 29.25, 27.19, 22.64, 14.08. The physical data were
identical in all respects to those previously reported.[38]

Chapter 2
40
4-Methoxy-N-ethylaniline (3ac): Synthesized according to General
procedure A. p-Anisidine (0.062 g, 0.5 mmol) affords 3ac (0.064 g, 85%
yield). Yellow oil obtained after column chromatography (SiO2, n-
pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 6.79 (d, J =
7.1 Hz, 2H), 6.60 (d, J = 7.3 Hz, 2H), 3.75 (s, 3H), 3.02 – 3.23 (m, 2H),
1.24 (t, J = 6.8, 3H). 13C NMR (100 MHz, CDCl3) δ 152.04, 142.70, 114.86,
114.10, 55.79, 39.44, 14.97. The physical data were identical in all respects to
those previously reported.[39]
4-Methoxy-N-phenethylaniline (3ah): Synthesized according to
General procedure A. p-Anisidine (0.062 g, 0.5 mmol) affords 3ah
(0.085 g, 75% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5).1H NMR
(400 MHz, CDCl3) δ 7.28 - 7.46 (m, 2H), δ 7.12 – 7.28 (m, 3H),
6.79 (d, J = 8.8 Hz, 2H), 6.61 (d, J = 8.6 Hz, 2H), 3.75 (s, 3H),
3.37 (t, J = 7.0 Hz, 2H), 2.91 (t, J = 6.9 Hz, 2H). 13C NMR (100
MHz, CDCl3) δ 152.15, 142.16, 139.35, 128.73, 128.52, 126.32,
114.88, 114.36, 55.74, 46.01, 35.54. The physical data were identical in all
respects to those previously reported.[40]
2-((4-Methoxyphenyl)amino)ethanol (3ai): Synthesized
according to General procedure A. p-Anisidine (0.062 g, 0.5 mmol)
affords 3ai (0.062 g, 74% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 90:10 to 50:50). 1H NMR
(400 MHz, CDCl3) δ 6.74 – 6.84 (m, 2H), 6.58 – 6.66 (m, 2H), 3.79
(t, J = 5.2 Hz, 2H), 3.75 (s, 3H), 3.23 (t, J = 5.2 Hz, 2H), 2.86 – 3.08
(br.s, 1H). 13C NMR (100 MHz, CDCl3) δ 152.49, 142.14, 114.86,
114.76, 61.18, 55.75, 47.16. The physical data were identical in all respects to
those previously reported.[41]
6-((4-Methoxyphenyl)amino)hexan-1-ol (3ak):
Synthesized according to General procedure A. p-Anisidine
(0.062 g, 0.5 mmol) affords 3ak (0.048 g, 43% yield). Yellow
oil obtained after column chromatography (SiO2, n-
pentane/EtOAc 90:10 to 50:50). 1H NMR (400 MHz, CDCl3) δ
6.72 – 6.82 (m, 2H), 6.52 – 6.62 (m, 2H), 3.74 (s, 3H), 3.63
(t, J = 6.6 Hz, 2H), 3.06 (t, J = 7.1 Hz, 2H), 2.31 – 2.51 (br.s,
2H), 1.51 – 1.68 (m, 4H), 1.31 – 1.43 (m, 4H). 13C NMR (100
MHz, CDCl3) δ 151.99, 142.64, 114.85, 114.10, 62.75, 55.79, 44.94, 32.61, 29.56,
26.92, 25.55. HRMS (APCI+, m/z): calculated for C13H21NO2 [M+H]+: 224.16451;
found: 224.16428.
N,N-Dipentylfurfurylamine (6caa): Synthesized
according to General procedure B. Furfurylamine (0.044 ml,
0.5 mmol) affords 6caa (0.075 g, 63% yield). Yellow oil
obtained after column chromatography (SiO2, n-
pentane/EtOAc 95:5 to 60:40). 1H NMR (400 MHz, CDCl3) δ
7.35 (d, J = 2.0, 1H), 6.30 (dd, J = 2.8, 2.0 Hz, 1H), 6.15 (d, J = 3.0, 1H), 3.64

Alkylation of amines with alcohols
41
(s, 2H), 2.40 (m, J = 7.6, 2H), 1.40 – 1.62 (m, 4H), 1.15 – 1.40 (m, 8H), 0.88
(t, J = 7.0, 6H). 13C NMR (100 MHz, CDCl3) δ 152.88, 141.66, 109.88, 108.11,
53.84, 49.97, 29.71, 26.74, 22.60, 14.06. HRMS (APCI+, m/z): calculated for
C15H25NO [M+H]+: 238.21666; found: 238.21654.
1-Phenethylpiperidine (6bh): Synthesized according to General
procedure B. Piperidine (0.049 ml, 0.5 mmol) affords 6bh (0.050 g, 52%
yield). Yellow oil obtained after column chromatography (Al3O2, n-
pentane/EtOAc 100:0 to 90:10). 1H NMR (400 MHz, CD2Cl2) δ 7.23 –
7.34 (m, 2H), 7.10 – 7.34 (m, 3H), 2.72 – 2.80 (m, 2H), 2.48 – 2.56
(m, 2H), 2.35 – 2.48 (m, 4H), 1.52 – 1.62 (m, 4H), 1.33 – 1.48 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 140.60, 128.66, 128.29, 125.88, 61.43,
54.52, 33.62, 25.97, 24.39. The physical data were identical in all respects to
those previously reported.[42]
N-Pentyl-phenethylamine (6ah or 6da): Synthesized
according to General procedure A. Phenethylamine (0.063 ml,
0.5 mmol) affords 6da (0.031 g, 32% yield). Amylamine (0.058
ml, 0.5 mmol) affords 6ah (0.014 g, 15% yield). Yellow oil
obtained after column chromatography (SiO2, MeOH/EtOAc
0:100 to 5:95). 1H NMR (400 MHz, CD2Cl2) δ 7.24 – 7.33 (m,
2H), 7.12 – 7.24 (m, 3H), 2.79 – 2.91 (m, 2H), 2.68 – 2.79 (m, 2H), 2.58 (t, J =
7.2 Hz), 1.38 – 1.48 (m, 2H), 1.22 – 1.32 (m, 4H), 0.89 (t, J = 7.0 Hz, 3H). 13C
NMR (100 MHz, CD2Cl2) δ 141.03, 129.09, 128.68, 126.29, 51.65, 50.19, 36.83,
30.21, 29.98, 23.03, 14.24. The physical data were identical in all respects to
those previously reported.[43]
N-Pentylbenzylamine (6ag or 8aa): Synthesized according
to General procedure A. Benzylamine (0.055 ml, 0.5 mmol)
affords 8aa (0.055 g, 62% yield). Amylamine (0.058 ml, 0.5
mmol) affords 6ag (0.059 g, 67% yield). Yellow oil obtained
after column chromatography (SiO2, n-pentane/EtOAc 50:50 to
0:100). 1H NMR (400 MHz, CD2Cl2) δ 7.26 – 7.37 (m, 4H), 7.16
– 7.26 (m, 1H), 3.77 (s, 2H), 2.61 (t, J = 7.2 Hz, 2H), 1.57 – 1.80 (br.s, 1H), 1.45
– 1.55 (m, 2H), 1.27 – 1.36 (m, 4H), 0.91 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz,
CD2Cl2) δ 141.46, 128.62, 128.45, 127.07, 54.33, 49.89, 30.24, 30.01, 23.07,
14.27. The physical data were identical in all respects to those previously
reported.[43]
3-Chloro-N-pentylbenzylamine (8ba): Synthesized
according to General procedure A. 3-Chlorobenzylamine (0.061
ml, 0.5 mmol) affords 8ba (0.084 g, 80% yield). Yellow oil
obtained after column chromatography (SiO2, n-pentane/EtOAc
50:50 to 0:100). 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 7.06
– 7.25 (m, 3H), 3.76 (s, 2H), 2.60 (t, J = 7.2 Hz, 2H), 1.59 – 1.70 (br.s, 1H), 1.45
– 1.55 (m, 2H), 1.27 – 1.35 (m, 4H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz,
CDCl3) δ 142.50, 134.17, 129.56, 128.14, 126.98, 126.16, 53.40, 49.37, 29.67,

Chapter 2
42
29.46, 22.56, 14.01. HRMS (APCI+, m/z): calculated for C12H19ClN [M+H]+:
212.12005; found: 212.11983.[44]
3-Fluoro-N-pentylbenzylamine (8ca): Synthesized
according to General procedure A. 3-Fluorobenzylamine (0.057
ml, 0.5 mmol) affords 8ca (0.085 g, 87% yield). Yellow oil
obtained after column chromatography (SiO2, n-pentane/EtOAc
50:50 to 0:100). 1H NMR (400 MHz, CDCl3) δ 7.20 – 7.34 (m,
1H), 6.98 – 7.18 (M, 2H), 6.93 (td, J = 8.4, 2.1 Hz, 1H), 3.79
(s, 2H), 2.61 (t, J = 7.3 Hz, 2H), 1.76 – 2.04 (br.s, 1H), 1.45 – 1.61 (m, 2H), 1.22
– 1.37 (m, 4H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 162.93 (d,
J = 245.5 Hz), 142.96 (d, J = 7.3 Hz), 129.73 (d, J = 8.2 Hz), 129.63 (d, J = 2.8
Hz), 114.86 (d, J = 21.2 Hz), 113.70 (d, J = 21.2 Hz), 53.35, 49.31, 29.62, 29.47,
22.56, 14.00. HRMS (APCI+, m/z): calculated for C12H19FN [M+H]+: 196.14960;
found: 196.14938.[44]
3-Trifluoromethyl-N-pentylbenzylamine (8da):
Synthesized according to General procedure A. 3-
Trifluorobenzylamine (0.072 ml, 0.5 mmol) affords 8da (0.115
g, 94% yield). Yellow oil obtained after column chromatography
(SiO2, n-pentane/EtOAc 50:50 to 0:100). 1H NMR (400 MHz,
CDCl3) δ 7.60 (s, 1H), 7.46 – 7.56 (m, 2H), 7.37 – 7.46 (m,
1H), 3.84 (s, 2H), 2.62 (t, J = 7.2 Hz, 2H), 2.06 – 2.21 (br.s, 1H), 1.45 – 1.57 (m,
2H), 1.26 – 1.35 (m, 4H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ
141.31, 131.44, 130.66 (q, J = 32.3 Hz), 128.72, 124.79 (q, J = 3.7 Hz), 124.20
(d, J = 272.2 Hz), 123.73 (q, J = 3.8 Hz), 53.41, 49.39, 29.61, 29.46, 22.55,
13.98. HRMS (APCI+, m/z): calculated for C12H29F3N [M+H]+: 246.14641; found:
246.14594.[44]
3-Fluoro-5-trifluoromethyl-N-pentylbenzylamine
(8ea): Synthesized according to General procedure A. 3-
Fluoro-5-trifluoromethylbenzylamine (0.073 ml, 0.5
mmol) affords 8ea (0.125 g, 95% yield). Yellow oil
obtained after column chromatography (SiO2, n-
pentane/EtOAc 50:50 to 0:100). 1H NMR (400 MHz, CDCl3) δ 7.40 (s, 1H), 7.28
(d, J = 9.1 Hz, 1H), 7.19 (d, J = 8.0 Hz, 1H), 3.88 – 4.04 (br.s, 1H), 3.48 (s, 2H),
2.61 (t, J = 7.1 Hz, 2H), 1.43 – 1.59 (m, 2H), 1.25 – 1.37 (m, 4H), 0.88 (t, J =
6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 162.56 (d, J = 248.5 Hz), 143.59 –
143.81 (m), 132.39 (dd, J = 33.1, 7.8 Hz), 123.30 (dd, J = 272.4, 2.7 Hz), 120.52
– 120.74 (m), 118.50 (d, J = 21.6 Hz), 111.39 (dq, J = 24.5, 3.8 Hz), 52.62,
49.08, 29.35, 29.23, 22.48, 13.92. HRMS (APCI+, m/z): calculated for C13H18F4N
[M+H]+: 264.13699; found: 264.13668.
3-Chloro-4-fluoro-N-pentylbenzylamine (8fa): Synthesized according to
General procedure A. 3-Chloro-4-fluorobenzylamine (0.069 ml, 0.5 mmol) affords
8fa (0.050 g, 43% yield). Yellow oil obtained after column chromatography (SiO2,
n-pentane/EtOAc 50:50 to 0:100). 1H NMR (400 MHz, CDCl3) δ 7.33 – 7.43 (m,

Alkylation of amines with alcohols
43
1H), 7.14 – 7.22 (m, 1H), 7.02 – 7.11 (m, 1H), 3.73 (s,
2H), 2.59 (t, J = 7.5 Hz, 2H), 1.93 – 2.08 (br.s, 1H), 1.43
– 1.58 (m, 2H), 1.25 – 1.37 (m, 4H), 0.83 – 0.93 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 157.07 (d, J = 247.5 Hz),
137.34, 130.15, 127.69 (d, J = 7.0 Hz), 120.69 (d, J = 17.8
Hz), 116.29 (d, J = 20.9 Hz), 52.71, 49.26, 29.56, 29.45,
22.54, 13.99.
1-(3-fluorobenzyl)pyrrolidine (9cm): Synthesized according to
General procedure A. 3-Fluorobenzylamine (0.057 ml, 0.5 mmol)
affords 9cm (0.054 g, 60% yield). Yellow oil obtained after column
chromatography (Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CD2Cl2) δ 7.21 – 7.32 (m, 1H), 7.02 – 7.15 (m, 2H), 6.93
(td, J = 8.6, 2.2 Hz, 1H), 3.59 (s, 2H), 2.32 – 2.60 (m, 4H), 1.67 –
1.82 (m, 4H). 13C NMR (100 MHz, CD2Cl2) δ 162.87 (d, J = 245.1 Hz), 142.06 (d,
J = 6.2 Hz), 129.56 (d, J = 8.4 Hz), 124.27 (d, J = 2.8 Hz), 115.57 (d, J = 21.4
Hz), 113.70 (d, J = 21.4 Hz), 60.14, 54.14, 23.45. The physical data were identical
in all respects to those previously reported.[45]
1-(3-fluorobenzyl)piperidine (9cl): Synthesized according to
General procedure A. 3-Fluorobenzylamine (0.057 ml, 0.5 mmol)
affords 9cl (0.060 g, 62% yield). Yellow oil obtained after column
chromatography (Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400
MHz, CD2Cl2) δ 7.25 – 7.37 (m, 1H), 7.07 – 7.25 (m, 2H), 6.90 – 7.07
(m, 1H), 3.60 (s, 2H), 2.30 – 2.72 (m, 4H), 1.56 – 1.77 (m, 4H), 1.39
– 1.55 (m, 2H). 13C NMR (100 MHz, CD2Cl2) δ 163.34 (d, J = 244.1 Hz),
142.76 (d, J = 7.0 Hz), 129.87 (d, J = 8.3 Hz), 124.86 (d, J = 2.7 Hz), 115.81 (d,
J = 21.2 Hz), 113.80 (d, J = 21.2 Hz), 63.48 (d, J = 1.9 Hz), 54.93, 53.84, 26.51,
24.82. The physical data were identical in all respects to those previously
reported.[46]
1-(3-fluorobenzyl)azepane (9ck): Synthesized according to
General procedure A. 3-Fluorobenzylamine (0.057 ml, 0.5 mmol)
affords 9ck (0.069 g, 67% yield). Yellow oil obtained after column
chromatography (Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.18 – 7.30 (m, 1H), 7.00 – 7.18 (m, 2H), 6.84
– 6.98 (m, 1H), 3.63 (s, 2H), 2.45 – 2.72 (m, 4H), 1.43 – 1.84 (m,
8H). 13C NMR (100 MHz, CDCl3) δ 162.94 (d, J = 244.9 Hz), 143.01 (d, J = 6.0
Hz), 129.40 (d, J = 8.1 Hz), 124.06 (d, J = 2.7 Hz), 115.31 (d, J = 21.2 Hz),
113.49 (d, J = 21.3 Hz), 62.24 (d, J = 1.9 Hz), 55.61, 28.26, 26.97. HRMS (APCI+,
m/z): calculated for C12H19FN [M+H]+: 208.14960; found: 208.14928.
1-(3-chlorobenzyl)azepane (9bk) : Synthesized according to
General procedure A. 3-Chlorobenzylamine (0.061 ml, 0.5 mmol)
affords 9bk (0.095 g, 85% yield). Yellow oil obtained after column
chromatography (Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.36 (s, 1H), 7.14 – 7.25 (m, 3H), 3.61 (s, 2H),
2.55 – 2.66 (m, 4H), 1.58 – 1.70 (m, 8H). 13C NMR (100 MHz, CDCl3)
Chapter 2
44
δ 142.36, 134.01, 129.32, 128.66, 126.82, 126.72, 62.15, 55.58, 28.20, 26.98.
HRMS (APCI+, m/z): calculated for C13H19ClN [M+H]+: 224.12005; found:
224.11979.
1-(3-(trifluoromethyl)benzyl)azepane (9dk): Synthesized
according to General procedure A. 3-Trifluoromethylbenzylamine
(0.072 ml, 0.5 mmol) affords 9dk (0.087 g, 68% yield). Yellow oil
obtained after column chromatography (Al2O3, n-pentane/EtOAc
100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 7.55 (d, J
= 7.5 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.41 (m, 1H), 3.69 (s, 2H),
2.51 – 2.70 (m, 4H), 1.53 – 1.72 (m, 8H). 13C NMR (100 MHz, CDCl3) δ 143.31,
131.91, 130.43 (q, J = 21.7 Hz), 128.48, 125.25 (q, J = 3.8 Hz), 124.33 (d, J =
272.4 Hz), 123.53 (q, J = 3.8 Hz), 62.22, 55.60, 28.27, 26.98. HRMS (APCI+,
m/z): calculated for C14H19F3N [M+H]+: 258.14641; found: 258.14615.
1-(3-fluoro-5-(trifluoromethyl)benzyl)azepane (9ek): Synthesized
according to General procedure A. 3-Fluoro-5-
trifluoromethylbenzylamine (0.073 ml, 0.5 mmol) affords 9ek
(0.091 g, 66% yield). Yellow oil obtained after column
chromatography (Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.40 (s, 1H), 7.32 (d, J = 9.7 Hz, 1H), 7.18 (d,
J = 8.2 Hz, 1H), 3.67 (s, 2H), 2.53 – 2.69 (m, 4H), 1.52 – 1.73
(m, 8H). 13C NMR (100 MHz, CDCl3) δ 162.58 (d, J = 247.7), 144.78 (d, J = 7.1
Hz), 132.08 (dd, J = 33.0, 8.3 Hz), 123.46 (dd, J = 272.5, 3.1 Hz), 120.67 –
120.86 (m), 118.65 (d, J = 21.2 Hz), 111.04 (dq, J = 24.9, 3.8 Hz), 61.90 (d, J =
1.6 Hz), 55.68, 28.36, 26.95. HRMS (APCI+, m/z): calculated for C14H18F4N
[M+H]+: 267.13699; found: 276.13666.
Piribedil (12): Synthesized according to General
procedure B. 1-(2-Pyrimidyl)piperazine (10) (0.082 g,
0.5 mmol) and 4 eq. piperonyl alcohol (11) affords 12
(0.080 g, 54% yield). Yellow oil obtained after column
chromatography (Al2O3, n-pentane/EtOAc 90:10 to
50:50). 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 4.7Hz, 2H), 6.88 (s, 1H), 6.75 (s,
2H), 6.45 (t, J = 4.7Hz, 1H), 5.93 (s, 2H), 3.81 (t, J = 4.9 Hz, 4H), 3.44 (s, 2H),
2.47 (t, J = 5.0Hz, 4H). 13C NMR (100 MHz, CDCl3) δ 161.60, 157.62, 147.62,
146.62, 131.74, 122.19, 109.66, 109.45, 107.83, 100.85, 62.80, 52.77, 43.62.
The physical data were identical in all respects to those previously reported.[47,48]
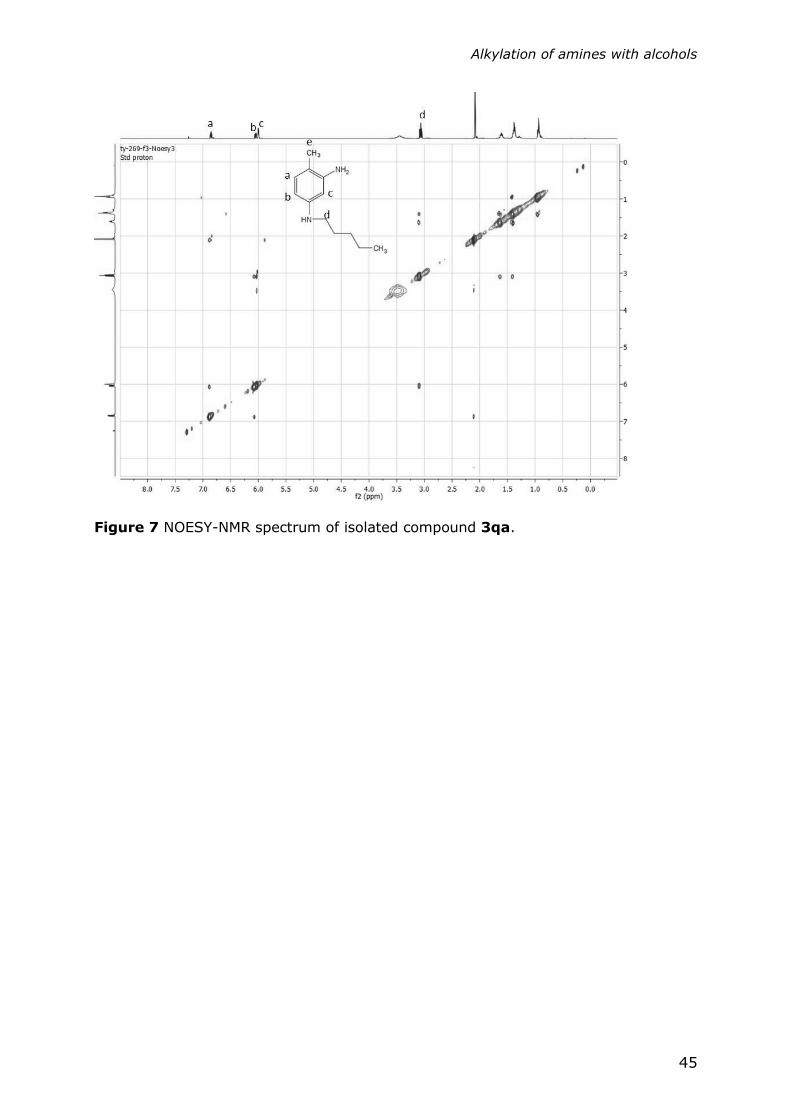
Alkylation of amines with alcohols
45
Figure 7 NOESY-NMR spectrum of isolated compound 3qa.

Chapter 2
46
References
[1] G. Guillena, D. J. Ramon, M. Yus, Chem. Rev., 2010, 110, 1611–1641.
[2] a) A. Hager, N. Vrielink, D. Hager, J. Lefranc, D. Trauner, Nat. Prod. Rep., 2016, 33,
491–522; b) J. L. McGuire, Pharmaceuticals: Classes, Therapeutic agents, areas of
Application, Vol. 1–4, Wiley-VCH, 2000.
[3] S. Bähn, S. Imm, L.Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem, 2011,
3, 1853–1864.
[4] A. J. A. Watson, J. M. J. Williams, Science, 2010, 329, 635–636.
[5] C. Gunanathan, Y. Ben-David, D. Milstein, Science, 2007, 317, 790-792.
[6] P. J. Deuss, K. Barta, J. G. de Vries, Catal. Sci. Technol., 2014, 4, 1174-1196.
[7] K. Barta, P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512.
[8] B. M. Trost, Science, 1991, 254, 1471–1477.
[9] P. T. Anastas, J. C. Warner, Green Chemistry: Theory and Practice, Oxford University
Press, 1998.
[10] R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit, N. J. Tongpenyai, J. Chem. Soc. Chem.
Commun., 1981, 611.
[11] M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A.
Watson, J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774.
[12] S-I. Murahashi, K. Kondo, T. Hakata, Tetrahedron Lett., 1982, 23, 229–232.
[13] K.-I. Fujita, T. Fujii, R. Yamaguchi, Org. Lett., 2004, 6, 3525–3528.
[14] M. H. S. A. Hamid, P. A. Slatford, J. M. J. Williams, Adv. Synth. Catal., 2007, 349,
1555–1575.
[15] C. Gunanathan, D. Milstein, Science, 2013, 341, 1229712.
[16] G. E. Dobereiner, R. H. Crabtree, Chem. Rev., 2010, 110, 681–703.
[17] R. Martinez, D. J. Ramon, M. Yus, Org. Biomol. Chem., 2009, 7, 2176–2181.
[18] S. Enthaler, K. Junge, M. Beller, Angew. Chem. Int. Ed., 2008, 47, 3317–3321.
[19] M. Bala, P. K. Verma, U. Sharma, N. Kumar, B. Singh, Green Chem., 2013, 15,
1687–1693.
[20] Y. Zhao, S. W. Food, S. Saito, Angew. Chem. Int. Ed., 2011, 50, 3006–3009.
[21] H.-J. Knölker, E. Baum, H. Goesmann, R. Klauss, Angew. Chem. Int. Ed., 1999, 38,
2064-2066.
[22] C. P. Casey, H. Guan, J. Am. Chem. Soc., 2007, 129, 5816-5817.
[23] A. Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard, J.-L. Renaud, Angew.
Chem. Int. Ed., 2012, 51, 4976-4980.
[24] a) T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal., 2012, 354,
597-601; b) M. G. Coleman, A. N. Brown, B. A. Bolton, H. Guan, Adv. Synth. Catal.,
2010, 352, 967–970.
[25] a) S. Zhou, S. Fleischer, K. Junge, M. Beller, Angew. Chem. Int. Ed., 2011, 50, 5120–
5124; b) A. Quintard, T. Constantieux, J. Rodriguez, Angew. Chem. Int. Ed., 2013,
52, 12883–12887.
[26] a) 1H NMR spectrum of commercially available pentane-1-al in toluene-d8 at 100 °C.
The chemical shift of the indicative aldehyde proton (HA) is at 9.40 ppm; b) 1H NMR
spectrum of imine 4aa, in situ generated from 0.12 mmol p-methoxy-aniline (1a)
and 0.50 mmol pentane-1-al. The compounds were solubilized in 0.6 ml toluene-d8
at room temperature, after 0.5 hour the 1H NMR spectrum was recorded at 100 °C.
The indicative aldehyde proton HA is found at 9.40 ppm. The indicative imine proton
HI is found at 7.67 ppm, in agreement with the literature: J. C. Anderson, G. P. Howell,
R. M. Lawrence, C. S. Wilson, J. Org. Chem., 2005, 70, 5665–5670 (7.63 ppm for
4-methoxy-N-hexylaniline in C6D6 at r.t.).
[27] C. P. Casey, H. Guan, J. Am. Chem. Soc., 2009, 131, 2499–2507.
[28] K. Watanabe, N. Yamagiwa, Y. Torisawa, Org. Process Res. Dev., 2007, 11, 251–
258.
[29] M. A. Berliner, S. P. A. Dubant, T. Makowski, K. Ng, B. Sitter, C. Wager, Y. Zhang,
Org. Process Res. Dev., 2011, 15, 1052–1062.
[30] Although the benzene ring is not conjugated to the imine, the effect from the
substitutions to the reduction rate of imine might be non-linear.

Alkylation of amines with alcohols
47
[31] M. Jaber, S. W. Robinson, C. Missale, M. G. Caron, Neuropharmacology, 1996, 35,
1503–1519.
[32] T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal., 2012, 354, 597-
601.
[33] D. Jiang, H. Fu, Y. Jiang, Y. Zhao, J. Org. Chem., 2007, 72, 672-674.
[34] E. J. Debeer, J. S. Buck, W. S. Ide, A. M. Hjort, J. Pharm. Exp. Ther., 1936, 57, 19-
33.
[35] J. Barluenga, A. M. Bayón, G. Asensio, J. Chem. Soc. Chem. Commun., 1983, 1109-
1110.
[36] J. Lau, J. T. Kodra, M. Guzel, K. C. Santosh, A. M. M. Mjalli, R. C. Andrews, D. R.
Polisetti, PCT Int. Appl. WO 2003055482 A1, 2003.
[37] J. Wu, C. Wang, W. Tang, A. Pettman, J. Xiao, Chem. Eur. J., 2012, 18, 9525-9529.
[38] Q. Shen, S. Shekhar, J. P. Stambuli, J. F. Hartwig, Angew. Chem. Int. Ed., 2005, 44,
1371-1375.
[39] O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden, J. M. J. Williams, Angew. Chem.
Int. Ed., 2009, 48, 7375-7378.
[40] J. C.-H. Yim, J. A. Bexrud, R. O. Ayinla, D. C. Leitch, L. L. Schafer, J. Org. Chem.,
2014, 79, 2015-2028.
[41] M. Yang, F. Liu, J. Org. Chem., 2007, 72, 8969-8971.
[42] M. Utsunomiya, J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 2702-2703.
[43] S. Lu, J. Wang, X. Cao, X. Li, H. Gu, Chem. Comm., 2014, 50, 3512-3515.
[44] W. R. Meindl, E. V. Angerer, H. Schoenenberger, G. Ruckdeschel, J. Med. Chem.,
1984, 27, 1111-1118.
[45] S. D. Banister, L. M. Rendina, M. Kassiou, Bioorg. Med. Chem. Lett., 2012, 22, 4059-
4063.
[46] L. G. Erwan, D. Alexandre, M. Thierry, T. Michel, Synthesis, 2010, 249-254.
[47] M. Haniti, S. A. Hamid, J. M. J. Williams, Tetrahedron Lett., 2007, 48, 8263–8265.
[48] X. Cui, X. Dai, Y. Deng, F. Shi, Chem. Eur. J., 2013, 19, 3665-3675.

Chapter 2
48

49
Chapter 3
Benzylamines via iron catalyzed direct amination of benzyl alcohols
Benzylamines play a prominent role in numerous pharmaceutically active
compounds. Thus, the development of novel, sustainable catalytic methodologies
to provide access to these privileged structural motifs is of central importance.
Herein we describe the use of a well-defined homogeneous iron-complex for the
construction of a large variety of benzylamines. The methodology consists of the
direct coupling of readily available benzyl alcohols with simple amines through the
borrowing hydrogen methodology. A variety of substituted secondary and tertiary
benzylamines are obtained in moderate to excellent yields. Furthermore, we
explore the versatility of this methodology in the one-pot synthesis of asymmetric
tertiary amines, sequential functionalization of diols and the synthesis of N-benzyl
piperidines, for the first time with an iron catalyst. In addition, direct conversion
of renewable building block 2,5-furan-dimethanol to pharmaceutically relevant
compounds is achieved.
Part of this chapter was published:
T. Yan, B. L. Feringa, K. Barta, ACS Catal., 2016, 6, 381−388.

Chapter 3
50
Introduction
Benzylamines are frequently encountered motifs in biological systems, and are
highly valuable targets due to their versatility[1] (Scheme 1). Many
pharmaceuticals contain a benzylamine moiety. Prominent examples include
Rivastigmine[1b], a cholinergic agent for treating dementia due to Parkinson’s
disease; Ezetimibe[1c], a drug that helps reducing plasma cholesterol levels; and
Emend[1d], an aprepitant that blocks the neurokinin 1 (NK1) receptor. Developing
efficient pathways that allow for selective synthesis of benzylamines, especially
starting from readily available substrates using sustainable catalysts based on non-
toxic and inexpensive metals is an important goal[2-4].
Scheme 1: Benzylamine based bio-active compounds.
Methods for the synthesis of benzylamine derivatives are shown in Scheme 2, A
and B. Catalytic hydroamination of alkenes or alkynes is an efficient way to access
1-methyl-benzylamines.[2] Furthermore, benzylation of amines with benzyl-halides
via nucleophilic substitution[3] leads to the formation of stoichiometric amounts of
waste. Reductive amination[4] is a catalytic and atom-economic alternative,
however here the aldehyde reaction partner is usually unstable and frequently
generates side-products.
Among these methods, direct amination of benzyl alcohols is a preferred method
because these substrates are readily available and can even be obtained from
renewable resources[5,6], making this route a highly sustainable alternative.
However, direct nucleophilic substitution of the hydroxyl group of benzyl alcohols
with amines is an energy and cost intensive process[7,8], while, installing a good
leaving group instead of the hydroxyl functionality will suffer from low atom-
efficiency. The desired, catalytic way to perform the direct coupling of benzyl
alcohols with amines involves the borrowing hydrogen[9] strategy (Scheme 2, C).
In this case, a specific sequence of reaction steps will occur: dehydrogenation of
the alcohol to aldehyde (step a), imine formation (step b) reduction of imine (step
c), which maintains high atom-efficiency[10] while requiring much lower activation

Benzylamines
51
energy[9c]. Additionally, this method provides innocuous water as the only side
product.
Scheme 2: Comparison of synthetic pathway to access benzylamine derivatives.
A Hydroamination of styrene with amine. B Comparison of amination of benzyl alcohols,
benzyl halides and benzyl aldehydes. C Catalytic amination of alcohols through borrowing
hydrogen.
Since the first examples of catalytic amination of alcohols through borrowing
hydrogen reported by Grigg[11] and Watanabe[12] in 1981, considerable progress
has been made in this area[13]. However, mostly precious metal catalysts containing
ruthenium[14] or iridium[15] were used. Cheap, low-toxic and abundant transition
metals like iron, have been only scarcely used for this transformation[16,18,19].
In Chapter 2, the first example of direct amination of alcohols catalyzed by a well-
defined iron[17] complex through the borrowing hydrogen strategy is described.[18]
This work focused on the use of diverse aromatic amines and aliphatic alcohols
and diols. Two examples were also included using benzyl alcohol as substrate,
however, these reactions suffered from rather low yields. Very recently, Wills and
coworkers also reported on the iron catalyzed amination of alcohols[19a]. They
observed no product formation when benzylamine was employed as the reaction
partner. Later, Zhao and coworkers reported iron catalyzed amination of secondary
alcohols with assistance of 0.4 equiv AgF[19b]. However, only a limited number of
examples have been reported on the use of benzyl alcohols.
Here, a highly versatile method for the synthesis of a large variety of benzylamines
through direct iron catalyzed amination of benzyl alcohols is presented. In addition
to the impressive scope, important novel aspects are the one-pot synthesis of
asymmetric tertiary amines as well as uncovering the reactivity trends in the
sequential functionalization of benzyl alcohols. Moreover, a fully sustainable, two

Chapter 3
52
step pathways from a cellulosic platform chemical to a pharmaceutically active
compound is described.
Results and discussion
Optimization of reaction conditions 4-Methylbenzyl alcohol (1a) and
morpholine (2a) were selected as the starting materials for optimization of the
reaction conditions for direct coupling of benzyl alcohols with secondary amines
(Table 1). Using the previously reported conditions[18] with additional assistance of
molecular sieves, only 39% conversion was obtained (Table 1, entry 1). A solvent
screening showed, that etherate solvents like tetrahydrofuran (THF) and dioxane
gave low conversions (33% and 30%, respectively, Table 1, entry 2-3).
Dichloroethane (DCE) gave full conversion but the desired product (3a) was not
detected presumably due to nucleophilic substitution of the solvent (DCE) with
morpholine (2a) (Table 1, entry 4). More polar solvents like acetonitrile and
dimethylformamide (DMF) gave very poor conversion (Table 1, entry 5-6). In
toluene, 64% conversion was obtained (Table 1, entry 7). When other iron sources
such as FeCl3, Fe2(CO)9, and iron(II) phtalocyanine were applied instead of Cat 3,
the conversions were unsatisfactory (Table 1, entries 8−10). Using Cat 3, the
conversion improved to 87% when the temperature was increased to 135 °C in
toluene, probably due to the acceleration of imine reduction (Table 1, entry 11).
Similar results were obtained in CPME at 135 °C (82%, Table 1, entry 12).
Increasing the loading of 1a to 2 mmol in toluene gave full conversion and an 87%
isolated yield of 3a (Table 1, entry 13).
Table 1: Optimization of reaction conditions for amination of 4-methylbenzyl
alcohol (1a) with morpholine (2a).
Entry 1a [mmol] Solvent T [°C] Conversion [%]
1 1 CPME 130 39
2 1 THF 130 33
3 1 Dioxane 130 30
4 1 DCE 130 >95a
5 1 CH3CN 130 <5
6 1 DMF 130 <5
7 1 Toluene 130 64
8b 1 Toluene 130 <5
9c 1 Toluene 130 <5
10d 1 Toluene 130 <5
11 1 Toluene 135 87
12 1 CPME 135 82 13 2 Toluene 135 >95 (87)
General reaction conditions: General procedure, 0.5 mmol 2a, 1 or 2 mmol 1a, 0.02 mmol
Cat 3, 0.04 mmol Me3NO, 2 ml solvent, 18 h, 130 or 135 °C, 95-105 mg molecular sieves,
unless otherwise specified, isolated yield shown in parenthesis, conversion was determined
by GC-FID using decane as the internal standard; aNo 3a has been observed based on GC-
MS, high conversion of amine probably due to the reaction between amine and solvent; b4

Benzylamines
53
mol% FeCl3 instead of Cat 3 and Me3NO; c2 mol% Fe2(CO)9 instead of Cat 3 and Me3NO; d4 mol% Iron(II) phthalocyanine instead of Cat 3 and Me3NO.
Reactions of benzyl alcohols with secondary amines Next, under optimized
reaction conditions, a variety of secondary amines and benzyl alcohols were tested
(Table 2). Benzyl alcohol 1b with an electron-donating –OCH3 substituent reacted
smoothly with 2a providing full conversion and 88% isolated yield of 3b (Table 2,
entry 1). When less electron-rich substrates 1c and 1d were employed, lower
reactivity was observed; 60% of 3c and 40% of 3d were isolated after 18 and 24
h reaction time, respectively (Table 2, entry 2-3). Interestingly, also for 2-
thiophenemethanol (1e), a high isolated yield (74%) of 3e was obtained (Table 2,
entry 4). For other secondary amines, such as 1-methyl-piperazine (2b),
piperazine (2c) and di-n-butyl-amine (2d), the corresponding products were also
obtained in good to excellent yields (Table 2, entry 5-9). Interestingly, when
piperazine (2c), which has two reactive -NH sides, was tested with 2 mmol (4
equiv) of 1c under the general conditions (Table 2), 35% of mono N-benzylation
and 55% of di-N-benzylation product was obtained. By increasing the amount of
1c to 3 mmol (6 equiv), the di-N-benzylation product (3h) was obtained in 90%
isolated yield (Table 2, entry 7).
Table 2: Amination of benzyl alcohols with secondary amines.
Entry Substrate 1 Product 3 Yield [%]
1 1b
3b
88
2 1c
3c
60
3a 1d
3d
40
4a 1e
3e
74
5b 1a
3f
78
6 1f
3g
69
7cde 1c
3h
90
8cde 1e
3i
80
9a 1a
3j
65
10 1a
3k
91

Chapter 3
54
11a 1d
3l
63
12a 1g
3m
69
13 1a
3n
89
14a 1e
3o
79
15a 1h
3p
59
General reaction conditions: General procedure, 0.5 mmol 2, 2 mmol 1a, 0.02 mmol Cat
3, 0.04 mmol Me3NO, 2 ml toluene, 18 h, 135 °C, 95-105 mg molecular sieves, isolated
yields are shown; a24 h; b0.03 mmol Cat 3 and 0.06 mmol Me3NO were employed; c3
mmol 1 was employed.
Next, secondary benzylic amines, which are less basic compared to secondary
aliphatic amines[20], were used as the substrate (Table 2). N-methyl benzylamine
(2e) reacted smoothly with 1a, 1d, 1g and the corresponding products were
obtained in good to excellent yields (Table 2, entry 10–12). Similarly the reaction
of 1,2,3,4-tetrahydroisoquinoline (2f) with alcohols 1a, 1e, 1h provided the
corresponding products in high yields (Table 2, entry 13–15). Remarkably, alcohols,
which possess hetero-aromatic moieties such as 1e and 1h, which have the
possibility to act as chelating ligands[21], could also be used as shown in various
entries in Table 2, and products 3e, 3i, 3o and 3p were obtained in good to
excellent yield.
Reactions of benzyl alcohols with primary amines The synthesis of mono-N-
alkylated benzylamines from the corresponding primary benzylamines and
aliphatic alcohols was described in our previous work[18], which is also shown in
chapter 2. Here we demonstrate a new alternative route to the same products,
starting from benzyl alcohols and aliphatic amines (Table 3), which to the best of
our knowledge has not been previously reported with any iron catalyst and allows
great synthetic flexibility for the selection of the substrates and more insight into
the reaction mechanism.
First, n-pentylamine (4a) was selected for the synthesis of a variety of
functionalized benzyl alcohols (Table 3, entry 1–6). Selectivity towards the mono-
alkylation products was sensitive to the amount of alcohol substrate added. For
example, 2 equiv loading of 4-methoxybenzyl alcohol (1b) lead to preferential
formation and 54% isolated yield of 5a (Table 3, entry 1), however further
increasing 1b loading lead to more dialkylation product. On the other hand, only
using 1.5 eq. of 1b, the corresponding imine was detected as the major product.
The same behavior was observed with 4-methylbenzyl alcohol (1a). In addition,
increasing Cat 3 loading to 6 mol% provided 61% isolated yield of 5b (Table 3,
entry 2).

Benzylamines
55
Table 3: Amination of benzyl alcohols with primary amines.
Entry Substrat 1 Product 5 Yield [%]b
1a 1b
5a
54
2a 1a
5b
61
3 1c
5c
59
4 1g
5d
53
5bc 1i
5e
42d
6bc 1j
5f
22e
7 1c
5g
60
8 1c
5h
61
9bc 1f
5i
60
10b 1a
5j
70
11bc 1a
5k
66
12bc 1c
5l
56
General reaction conditions: General procedure, 0.5 mmol 2, 2 mmol 1a, 0.02 mmol Cat
3, 0.04 mmol Me3NO, 2 ml toluene, 18 h, 135 °C, 95-105 mg molecular sieves, isolated
yields are shown. a1 mmol 1 was employed; b0.03 mmol Cat 3 and 0.06 mmol Me3NO
were employed; c24 h; d41% of corresponding was observed based on GC-FID integration; e58% of corresponding was observed based on GC-FID integration.
Interestingly, when less electron-rich substrates such as benzyl alcohol (1c) and
4-fluorobenzyl alcohol (1g) were examined, less di-N-benzylation product was
observed and 59% and 53% of mono-N-benzylation products were isolated,
respectively (Table 3, entry 3-4).
The reaction of 3-chlorobenzyl alcohol (1i) and 3-trifluoromethylbenzyl alcohol (1j)
with n-pentylamine (4a) (Table 3, entry 5-6) was examined as comparison with
our previous approach[18] that used 3-chloro and 3-trifluoromethyl substituted
benzylamines. In the present case, both reactions provided preferentially imine.
Increasing the catalyst loading to 6 mol% and reaction time to 24 h, provided more
amine product 5e, but still low amount of desired amine 5f (Table 3, entry 5-6).
Increasing the reaction temperature to 140 °C in order to facilitate imine reduction
resulted in catalyst decomposition.

Chapter 3
56
Scheme 3: Reactivity difference through different substrates.
In our previous study[18], electron-deficient benzylamines were more reactive
towards mono-alkylation than their electron rich analogues. The results shown
here, however, conclude that N-alkylated benzylamines are more readily obtained
starting from electron-rich benzyl alcohols than from electron poor benzyl alcohols
(Scheme 3). The reason for this reactivity difference can likely be attributed to
differences in the rate of the imine reduction step.[22] It has to be noted that the
imine intermediates formed from benzyl alcohols will possess a double bond in
conjugation with the aromatic system (Scheme 3, Pathway A) and those formed
from benzyl amines will not (Scheme 3, Pathway B). These reactivity differences
under similar conditions, also show that isomerization of the imine double bond is
not likely. The detailed understanding of these mechanistic details and rate limiting
steps will be subject of future in depth studies.
Next, a series of other primary amines were tested as substrates (Table 3, Entry
7-12). Long chain amines like n-nonylamine (4b) and 2-phenylethamine (4c) were
benzylated with benzyl alcohol (1c) providing 5g and 5h in good isolated yields
(Table 3, entry 7-8). Furthermore, primary benzylamines and anilines could be
applied to readily provide 5k and 5l (Table 3, entry 9–12).
Three component synthesis of asymmetric benzylic tertiary amines Taking
advantage of the differences in reactivity between aliphatic and benzyl alcohols,
we have developed a straightforward approach for the direct three component
synthesis of asymmetric benzylic tertiary amines (Table 4). To this end, a method
using benzyl alcohol (1c), n-pentylamine (4a) and n-butanol (8a) was
implemented with 6 mol% Cat 3 loading, at 135 °C (Table 4, entry 1-3).
Gratifyingly, the desired non-symmetric N-n-butyl-N-n-pentylbenzylamine (9a)
was predominant in the reaction mixture that also contained smaller amounts of
the expected di-N-benzyl-n-pentylamine (10a), and di-N-n-butyl-n-pentylamine
(11a). The desired tertiary amine with three distinctly different alkyl moieties 9a
was isolated in 51% yield (Table 4, entry 1). The one-pot procedure was extended
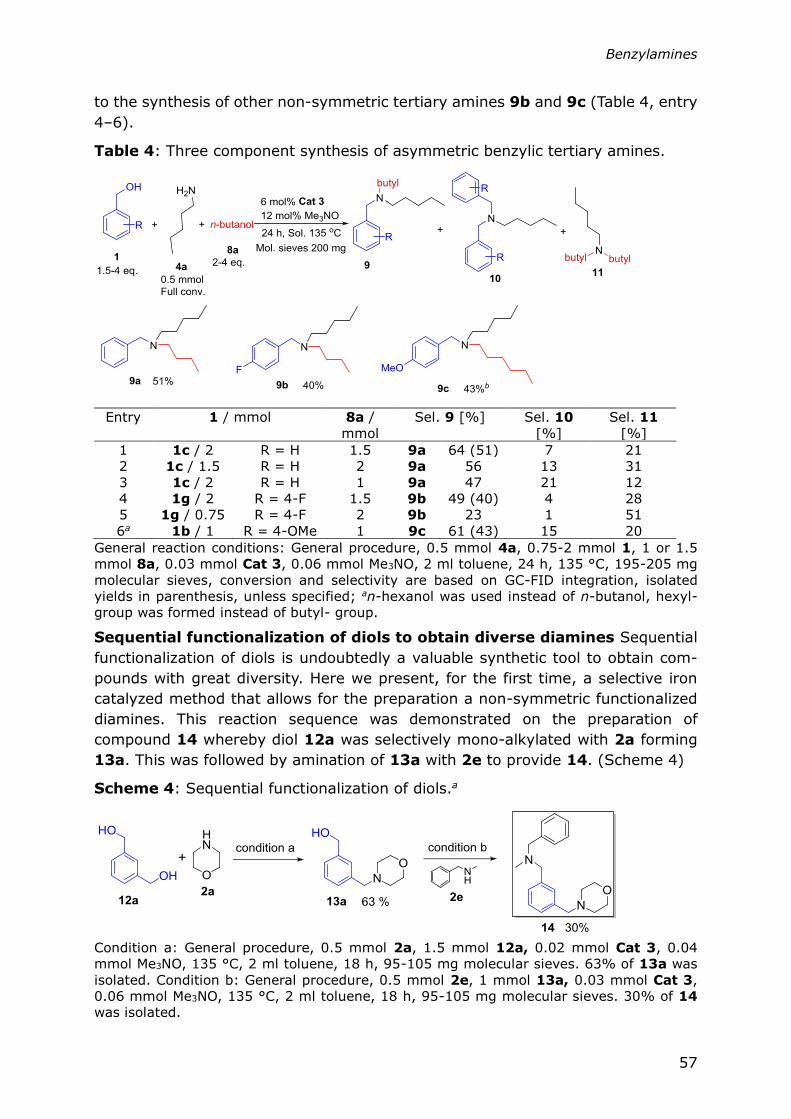
Benzylamines
57
to the synthesis of other non-symmetric tertiary amines 9b and 9c (Table 4, entry
4–6).
Table 4: Three component synthesis of asymmetric benzylic tertiary amines.
Entry 1 / mmol
8a /
mmol
Sel. 9 [%] Sel. 10
[%]
Sel. 11
[%]
1 1c / 2 R = H 1.5 9a 64 (51) 7 21
2 1c / 1.5 R = H 2 9a 56 13 31
3 1c / 2 R = H 1 9a 47 21 12
4 1g / 2 R = 4-F 1.5 9b 49 (40) 4 28
5 1g / 0.75 R = 4-F 2 9b 23 1 51
6a 1b / 1 R = 4-OMe 1 9c 61 (43) 15 20
General reaction conditions: General procedure, 0.5 mmol 4a, 0.75-2 mmol 1, 1 or 1.5
mmol 8a, 0.03 mmol Cat 3, 0.06 mmol Me3NO, 2 ml toluene, 24 h, 135 °C, 195-205 mg
molecular sieves, conversion and selectivity are based on GC-FID integration, isolated
yields in parenthesis, unless specified; an-hexanol was used instead of n-butanol, hexyl-
group was formed instead of butyl- group.
Sequential functionalization of diols to obtain diverse diamines Sequential
functionalization of diols is undoubtedly a valuable synthetic tool to obtain com-
pounds with great diversity. Here we present, for the first time, a selective iron
catalyzed method that allows for the preparation a non-symmetric functionalized
diamines. This reaction sequence was demonstrated on the preparation of
compound 14 whereby diol 12a was selectively mono-alkylated with 2a forming
13a. This was followed by amination of 13a with 2e to provide 14. (Scheme 4)
Scheme 4: Sequential functionalization of diols.a
Condition a: General procedure, 0.5 mmol 2a, 1.5 mmol 12a, 0.02 mmol Cat 3, 0.04
mmol Me3NO, 135 °C, 2 ml toluene, 18 h, 95-105 mg molecular sieves. 63% of 13a was
isolated. Condition b: General procedure, 0.5 mmol 2e, 1 mmol 13a, 0.03 mmol Cat 3,
0.06 mmol Me3NO, 135 °C, 2 ml toluene, 18 h, 95-105 mg molecular sieves. 30% of 14
was isolated.
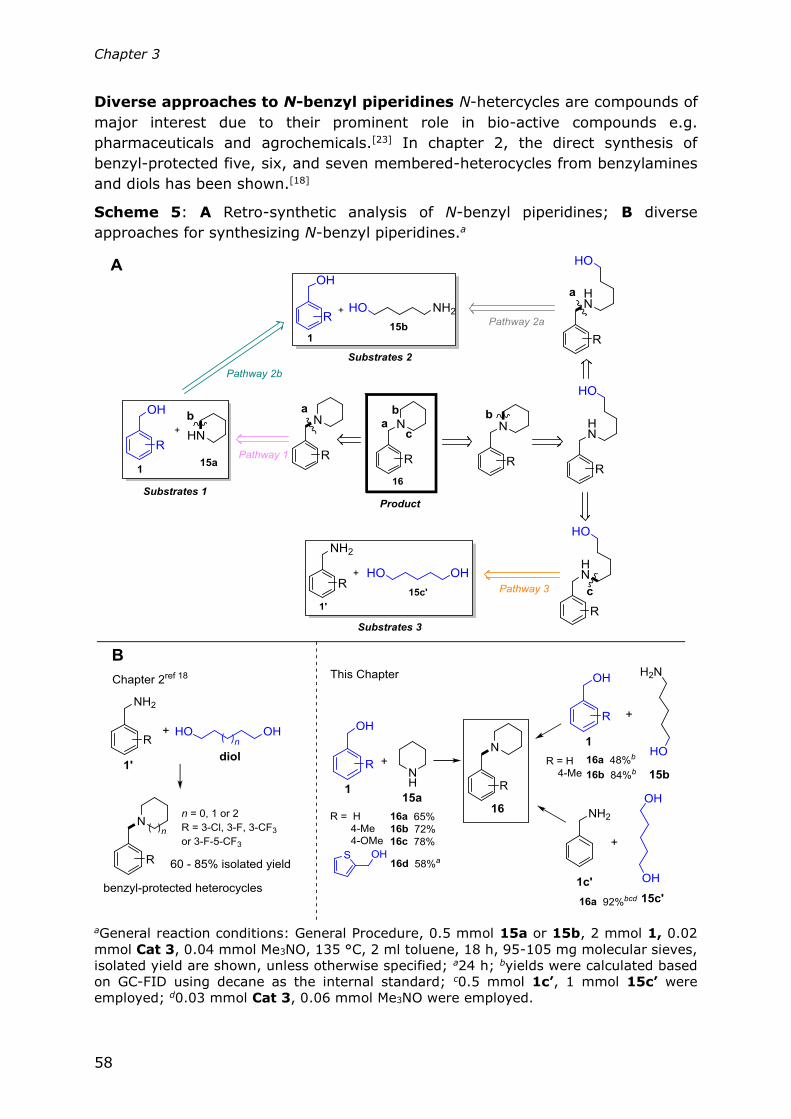
Chapter 3
58
Diverse approaches to N-benzyl piperidines N-hetercycles are compounds of
major interest due to their prominent role in bio-active compounds e.g.
pharmaceuticals and agrochemicals.[23] In chapter 2, the direct synthesis of
benzyl-protected five, six, and seven membered-heterocycles from benzylamines
and diols has been shown.[18]
Scheme 5: A Retro-synthetic analysis of N-benzyl piperidines; B diverse
approaches for synthesizing N-benzyl piperidines.a
aGeneral reaction conditions: General Procedure, 0.5 mmol 15a or 15b, 2 mmol 1, 0.02
mmol Cat 3, 0.04 mmol Me3NO, 135 °C, 2 ml toluene, 18 h, 95-105 mg molecular sieves,
isolated yield are shown, unless otherwise specified; a24 h; byields were calculated based
on GC-FID using decane as the internal standard; c0.5 mmol 1c’, 1 mmol 15c’ were
employed; d0.03 mmol Cat 3, 0.06 mmol Me3NO were employed.

Benzylamines
59
Here, we have investigated versatile synthesis routes for the preparation of N-
benzyl piperidines as representative example for benzyl protected N-heterocycles,
illustrated in Scheme 5. The same N-benzyl piperidine 16 can be obtained via three
distinct routes, starting from different substrates. For example, 16 can be
synthesized from three sets of substrates through four pathways (Scheme 5, A).
N-benzyl piperidine 16 can be obtained from benzyl alcohol (1) and piperidine
(15a) through the formation of bond a (Scheme 5, A, Pathway 1). Alternatively,
compound 16 can be obtained from alcohol 1 and 5-amino-1-pentanol (15b)
through the sequential formation of bonds a and b (Scheme 5, A, Pathway 2a), or
b and a (Scheme 5, A, Pathway 2b). Also, N-benzyl piperidine 16 can be
synthesized starting from 1,5-pentanediol (15c’) and benzyl amine 1c’ during
which bonds b and c are formed (Scheme 5, A, Pathway 3). These substrate
variations allow for choosing the most suitable pathway[24] taking into account
optimal balance of reactivity, selectivity and substrate abundance. In order to show
the power of this method, N-benzyl piperidines 16a, 16b, 16c and 16d were
synthesized through different pathways, with good to excellent yields (Scheme 5,
B).
Scheme 6: Synthesis of key intermediate to muscarinic agonist, N-[5-([l'-
substituted-acetoxy)methyl]-2-furfurylldialkylamines.
General reaction condition: General Procedure, 0.5 mmol 2e, 2 mmol 12b, 0.02 mmol
Cat 3, 0.04 mmol Me3NO, 135 °C, 2 ml toluene, 24 h, 95-105 mg molecular sieves. 60%
of 13b was isolated.
From cellulose derived platform chemicals to pharmaceutically active
molecules It was reported that furanic compounds of the general structure 18
shown on Scheme 6 are a class of pharmaceutically active compounds possessing
potential antimuscarinic activity. Pharmaceutical studies have especially focused
on systematic modifications on variations in the ester- and amine side chains.[25]
Here we show an efficient and fully sustainable synthetic strategy towards
obtaining key intermediate 13b. This compound can be prepared directly from
benzyl amine derivative 2e and diol 12b using our iron catalyzed methodology.
Diol 12b used in this reaction can be obtained in high yield from 5-
(hydroxymethyl)furfural (HMF, 17), via a sustainable pathway we have recently
reported[26]. In this procedure, HMF, which is one of the most important cellulose
derived platform chemicals undergoes catalytic hydrogenation using robust, CuZn
alloy nanopowder catalysts[26b]. This novel combination of methods allows for the

Chapter 3
60
easy, waste free synthesis of key bioactive intermediate 13b in 60% yield from
renewable resources, using sustainable catalysis.
Conclusion
In conclusion, we have established, for the first time, general methodology for the
catalytic formation of value-added benzylamines through amination of benzyl
alcohols using a well-defined iron catalyst that operates through a hydrogen
borrowing mechanism. Many synthetically challenging routes were systematically
explored, starting from readily accessible substrates that do not require prior
alcohol activation by stoichiometric methods. This included the one-pot synthesis
of asymmetric tertiary amines, the sequential functionalization of diols, and the
synthesis of important synthons, for example, N-benzylpiperidines, through
diverse synthetic pathways. In addition, direct conversion of the renewable
building block 2,5-furan-dimethanol to pharmaceutically relevant compounds was
achieved with unprecedented simplicity.
Experimental section
General methods
Chromatography: Merck silica gel type 9385 230-400 mesh or Merck Al2O3 90
active neutral, TLC: Merck silica gel 60, 0.25 mm or Al2O3 60 F254 neutral.
Components were visualized by UV, Ninhydrin or I2 staining. Progress of the
reactions was determined by GC-MS (GC: HP 6890, MS: HP 5973) with an HP012
column (Agilent Technologies, Palo Alto, CA). Mass spectra were recorded on an
AEI-MS-902 mass spectrometer (EI+) or a LTQ Orbitrap XL (ESI+). Conversions
were determined by GC-FID (GC: HP 6890) with an HP-5 column (Agilent
Technologies, Palo Alto, CA). GC-MS and GC-FID analysis method: 60 °C 5 min,
180 °C 5 min (10 °C/min), 260 °C 5 min (10 °C/min). 1H- and 13C NMR spectra
were recorded on a Varian AMX400 (400 and 100.59 MHz, respectively) using
CDCl3, CD3OD, or CD2Cl2 as solvent. Chemical shift values are reported in ppm with
the solvent resonance as the internal standard (CDCl3: 7.26 for 1H, 77.00 for 13C;
CD3OD: 3.31 for 1H, 49.00 for 13C; CD2Cl2: 5.32 for 1H, 53.84 for 13C). Data are
reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t =
triplet, q = quartet, br. = broad, m = multiplet), coupling constants (Hz), and
integration. All reactions were carried out under an Argon atmosphere using oven
(110 °C) dried glassware and using standard Schlenk techniques. THF and toluene
were collected from a MBRAUN solvent purification system (MB SPS-800). Dioxane
(99.5%, extra dry), dichloroethane (DCE, 99.8%, extra dry), N,N-
dimethylformamide (DMF, 99.8%, extra dry) and acetonitrile (CH3CN, 99.9%,
extra dry) were purchased from Acros without further purification. Molecular sieves
4A were purchased from Acros, and heated in a Schlenck flask under 180 °C in
vacuo overnight for activation before using. All other reagents were purchased
from Sigma or Acros in reagent or higher grade and were used without further
purification. Complex Cat 3 was synthesized according to literature procedures[27]
with slight modifications. The synthesis of Cat 3 was carried out as described in
Chapter 2.

Benzylamines
61
Representative procedures
General procedure An oven-dried 20 ml Schlenk tube, equipped with a stirring
bar, was charged with amine (0.5 mmol, 1 equiv), alcohol (given amount), iron
complex Cat 3 (4–6 mol%), Me3NO (8–12 mol%) and toluene (solvent, 2 ml). The
solid starting materials were added into the Schlenk tube under air, the Schlenk
tube was subsequently connected to an argon line and a vacuum-argon exchange
was performed three times. Liquid starting materials and solvent were charged
under an argon stream followed by addition of 95–105 mg activated molecular
sieves 4A. The Schlenk tube was capped and the mixture was rapidly stirred at
room temperature for 1 min, then it was placed into a pre-heated oil bath at the
appropriate temperature and stirred for a given time. The reaction mixture was
cooled down to room temperature and the crude mixture was filtered through celite,
eluted with ethyl acetate, and concentrated in vacuo. The residue was purified by
flash column chromatography to provide the pure amine product.
Reagents and characterization methods Reagents were of commercial grade
and used as received, unless stated otherwise. Chromatography: Merck silica gel
type 9385 230-400 mesh or Merck Al2O3 90 active neutral, TLC: Merck silica gel
60, 0.25 mm or Al2O3 60 F254 neutral. Components were visualized by UV,
Ninhydrin or I2 staining. 1H- and 13C NMR spectra were recorded on a Varian
AMX400 (400 and 100.59 MHz, respectively) using CDCl3, CD3OD, or CD2Cl2 as
solvent. Chemical shift values are reported in ppm with the solvent resonance as
the internal standard (CDCl3: 7.26 for 1H, 77.00 for 13C; CD3OD: 3.31 for 1H, 49.00
for 13C; CD2Cl2: 5.32 for 1H, 53.84 for 13C). Data are reported as follows: chemical
shifts, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br. = broad,
m = multiplet), coupling constants (Hz), and integration.
Procedure of amination of 4-methoxybenzyl alcohol (1b) with morpholine
(2a) provides 3b: An oven-dried 20 ml Schlenk tube, equipped with a stirring
bar, was charged with 4-methoxybenzyl alcohol (2 mmol, 0.276 g), iron complex
Cat 3 (4 mol%, 8 mg) and Me3NO (8 mol%, 3 mg) under air. The Schlenk tube
was subsequently connected to an argon line and a vacuum-argon exchange was
performed three times. Morpholine (0.5 mmol, 0.044 g), and toluene (solvent, 2
ml) were charged under an argon stream followed by addition of 95–105 mg
activated molecular sieves 4A. The Schlenk tube was capped and the mixture was
rapidly stirred at room temperature for 1 min, then was placed into a pre-heated
oil bath at 135 °C and stirred for 18 h. The reaction mixture was cooled down to
room temperature and the crude mixture was filtered through celite, eluted with
ethyl acetate, and concentrated in vacuo. The residue was purified by flash column
chromatography (SiO2, CH2Cl2/EtOAc 80:20 to 50:50) to provide the pure amine
product 3b (0.091 g, 88% isolated yield).

Chapter 3
62
Spectral data of isolated compounds
4-(4-Methylbenzyl)morpholine (3a): Synthesized according to
General procedure. Morpholine (0.044 g, 0.50 mmol) affords 3a (0.083
g, 87% yield). Light yellow solid obtained after column chromatography
(SiO2, CH2Cl2/EtOAc 80:20 to 50:50). 1H NMR (400 MHz, CDCl3) δ 7.21
(d, J = 8.0 Hz, 2H), 7.13 (d, J = 7.9 Hz, 2H), 3.70 (t. J = 7.9 Hz, 4H),
3.47 (s, 2H), 2.36 – 2.51 (m, 4H), 2.34 (s, 3H). 13C NMR (100 MHz, CD3OD) δ
138.22, 134.90, 130.71, 129.93, 67.64, 64.08, 54.55, 21.15. The physical data
were identical in all respects to those previously reported.[28]
4-(4-Methoxybenzyl)morpholine (3b): Synthesized according to
General procedure. Morpholine (0.044 g, 0.50 mmol) affords 3b (0.091
g, 88% yield). Yellow oil obtained after column chromatography (SiO2,
CH2Cl2/EtOAc 80:20 to 50:50). 1H NMR (400 MHz, CDCl3) δ 7.18 – 7.26
(m, 2H), 6.18 – 6.90 (m, 2H), 3.79 (s, 3H), 3.70 (t, J = 4.7 Hz, 4H),
3.44 (s, 2H), 2.42 (t, J = 4.5 Hz, 4H. 13C NMR (100 MHz, CDCl3) δ
158.73, 130.35, 129.52, 113.55, 66.91, 62.76, 55.18, 53.44. The physical data
were identical in all respects to those previously reported.[28]
4-Benzylmorpholine (3c): Synthesized according to General
procedure. Morpholine (0.044 g, 0.50 mmol) affords 3c (0.053 g, 60%
yield). Yellow oil obtained after column chromatography (SiO2,
CH2Cl2/EtOAc 80:20 to 50:50). 1H NMR (400 MHz, CD2Cl2) δ 7.10 – 7.45
(m, 5H), 3.65 (t, J = 4.7 Hz, 4H), 3.48 (s, 2H), 2.35 – 2.46 (m, 4H). 13C NMR (100 MHz, CD2Cl2) δ 138.57 129.50, 128.53, 127.37, 67.33, 63.68, 54.06.
The physical data were identical in all respects to those previously reported.[29]
4-(4-Chlorobenzyl)morpholine (3d): Synthesized according to
General procedure. Morpholine (0.044 g, 0.50 mmol) affords 3d (0.042
g, 40% yield). Yellow oil obtained after column chromatography (SiO2,
CH2Cl2/EtOAc 80:20 to 50:50). 1H NMR (400 MHz, CD2Cl2) δ 7.21 – 7.37
(m, 4H), 3.57 – 3.74 (m, 4H), 3.45 (s, 2H), 2.26 – 2.51 (m, 4H). 13C
NMR (100 MHz, CDCl3) δ 136.17, 132.89, 130.42, 128.40, 66.89, 62.58,
53.50. The physical data were identical in all respects to those previously
reported.[29]
4-(Thiophen-2-ylmethyl)morpholine (3e): Synthesized according
to General procedure. Morpholine (0.044 g, 0.50 mmol) affords 3e
(0.068 g, 74% yield). Yellow oil obtained after column chromatography
(SiO2, CH2Cl2/EtOAc 80:20 to 50:50). 1H NMR (400 MHz, CDCl3) δ 7.19
– 7.25 (m, 1H), 6.86 – 7.00 (m, 2H), 3.69 – 3.74 (m, 6H), 2.44 – 2.53 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 141.15, 128.05, 127.53, 126.47, 67.72, 58.07, 54.25.
The physical data were identical in all respects to those previously reported.[29]

Benzylamines
63
1-Methyl-4-(4-methylbenzyl)piperazine (3f): Synthesized
according to General procedure. 1-Methylpiperazine (0.050 g, 0.50
mmol) affords 3f (0.081 g, 79% yield).Yellow oil obtained after column
chromatography (SiO2, EtOAc/MeOH 100:0 to 90:10). 1H NMR (400
MHz, CDCl3) δ 7.20 (d, J = 7.8 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 3.46
(s, 2H), 2.20 – 2.70 (br.s, 8H), 2.33 (s, 3H), 2.28 (s, 3H). 13C NMR (100 MHz,
CDCl3) δ 136.52, 134.97, 129.11, 128.80, 62.71, 55.05, 52.97, 45.96, 21.05. The
physical data were identical in all respects to those previously reported.[30] HRMS
(APCI+, m/z): calculated for C13H21N2 [M+H]+: 205.16993; found: 205.16999.
1-Methyl-4-piperonyl piperazine (3g): Synthesized according to
General procedure. 1-Methylpiperazine (0.050 g, 0.50 mmol) affords
3g (0.081 g, 69% yield). Yellow oil obtained after column
chromatography (SiO2, EtOAc/MeOH 100:0 to 90:10). 1H NMR (400
MHz, CDCl3) δ 6.83 (s, 1H), 6.58 – 6.79 (m, 2H), 5.91 (s, 2H), 3.39
(s, 2H), 2.15 – 2.70 (br.s, 8H), 2.27 (s, 3H). 13C NMR (100 MHz, CDCl3)
δ 147.51, 146.47, 132.00, 122.14, 107.74, 100.77, 62.64, 55.02, 52.80, 45.91.
The physical data were identical in all respects to those previously reported.[31]
1,4-Dibenzylpiperazine (3h): Synthesized according to
General procedure. Piperazine (0.043 g, 0.50 mmol) affords 3h
(0.120 g, 90% yield). Yellow solid obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H
NMR (400 MHz, CD2Cl2) δ 7.27 – 7.55 (m, 8H), 7.07 – 7.27 (m,
2H), 3.49 (s, 4H), 2.26 – 2.63 (br.s, 8H). 13C NMR (100 MHz, CDCl3) δ 138.02,
129.19, 128.13, 126.95, 63.02, 53.00. The physical data were identical in all
respects to those previously reported.[32]
1,4-Bis(thiophen-2-ylmethyl)piperazine (3i): Synthesized
according to General procedure. Piperazine (0.043 g, 0.50 mmol)
affords 3i (0.095 g, 68% yield). Brown solid obtained after
column chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.18 - 7.26 (m, 2H), 6.93 – 9.97 (m,
2H), 6.88 – 6.93 (m, 2H), 3.73 (s, 4H), 2.27 – 2.83 (br.s, 8H). 13C NMR (100 MHz,
CDCl3) δ 141.23, 126.38, 126.13, 124.99, 56.94, 52.60. HRMS (APCI+, m/z):
calculated for C14H19N2S2 [M+H]+: 279.09842; found: 279.09850.
Di-N-n-butyl-4-methylbenzylamine (3j): Synthesized according
to General procedure. Di-n-butylamine (0.065 g, 0.50 mmol) affords
3j (0.076 g, 65% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.23 (d, J = 7.9 Hz, 2H), 7.12 (d, J = 7.6 Hz,
2H), 3.53 (s, 2H), 2.41 (t, J = 7.3 Hz, 4H), 2.35 (s, 3H), 1.40 – 1.55
(m, 4H), 1.25 – 1.37 (m, 4H), 0.90 (t, J = 7.3 Hz, 6H). 13C NMR (100
MHz, CDCl3) δ 136.93, 136.03, 128.74, 128.70, 58.16, 53.39, 29.15, 21.07, 20.07,
20.61, 14.09. HRMS (APCI+, m/z): calculated for C16H28N [M+H]+: 234.22163;
found: 234.22173.

Chapter 3
64
N-benzyl-N-methyl-4-methylbenzylamine (3k):
Synthesized according to General procedure. N-
Methylbenzylamine (0.061 g, 0.50 mmol) affords 3k (0.102 g, 91% yield). Yellow
oil obtained after column chromatography (Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.28 – 7.44 (m, 4H), 7.23 – 7.28 (m, 3H), 7.08 – 7.17
(m, 2H), 3.52 (s, 2H), 3.51 (s, 2H), 2.34 (s, 3H), 2.19 (s, 3H). 13C NMR (100 MHz,
CDCl3) δ 139.17, 136.50, 135.99, 128.95, 128.91, 128.90, 128.19, 126.91, 61.69,
61.55, 42.11, 21.10. The physical data were identical in all respects to those
previously reported.[33]
N-benzyl-N-methyl-4-chlorobenzylamine (3l):
Synthesized according to General procedure. N-
Methylbenzylamine (0.061 g, 0.50 mmol) affords 3l (0.077 g, 63% yield). Orange
solid obtained after column chromatography (Al2O3, n-pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CDCl3) δ 7.13 – 7.50 (m, 2H), 3.54 (s, 2H), 3.50 (s, 2H),
2.19 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 138.90, 137.73, 132.55, 130.15, 128.86,
128.24, 128.23, 127.02, 61.77, 60.95, 42.12. HRMS (APCI+, m/z): calculated for
C15H17ClN [M+H]+: 246.10440; found: 246.10451.
N-benzyl-N-methyl-4fluorobenzylamine (3m):
Synthesized according to General procedure. N-
Methylbenzylamine (0.061 g, 0.50 mmol) affords 3m (0.079
g, 69% yield). Yellow oil obtained after column chromatography (Al2O3, n-
pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.31 – 7.52 (m, 6H),
7.23 – 7.31 (m, 1H), 6.95 – 7.10 (m, 2H), 3.54 (s, 2H), 3.50 (s, 2H), 2.20 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.88 (d, J = 244.6 Hz), 139.12, 134.96 (d, J = 3.1
Hz), 130.29 (d, J = 7.7 Hz), 128.85, 128.21, 126.96, 114.95 (d, J = 21.1 Hz),
61.77, 60.95, 42.10. The physical data were identical in all respects to those
previously reported.[33]
2-(4-methylbenzyl)-1,2,3,4-tetrahydroisoquinoline
(3n): Synthesized according to General procedure. 1,2,3,4-
Tetrahydroisoquinoline (0.067 g, 0.50 mmol) affords 3n (0.106
g, 89% yield). Yellow oil obtained after column chromatography (Al2O3, n-
pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.27 – 7.35 (m, 2H),
7.15 – 7.21 (m, 2H), 7.03 – 7.15 (m, 3H), 6.97 – 7.03 (m, 1H), 3.67 (s, 2H), 3.65
(s, 2H), 2.91 (t, J = 6.0 Hz, 2H), 2.76 (t, J = 5.9 Hz, 2H), 2.37 (s, 3H). 13C NMR
(100 MHz, CDCl3) δ 136.68, 135.15, 134.85, 134.35, 129.05, 128.95, 128.65,
126.58, 126.03, 125.51. 62.46, 56.04, 50.51, 29.09, 21.12. The physical data
were identical in all respects to those previously reported.[34]
2-(thiophen-2-ylmethyl)-1,2,3,4-tetrahydroisoquinoline
(3o): Synthesized according to General procedure. 1,2,3,4-
Tetrahydroisoquinoline (0.067 g, 0.50 mmol) affords 3o (0.090 g, 79% yield).
Yellow oil obtained after column chromatography (Al2O3, n-pentane/EtOAc 100:0
to 95:5).1H NMR (400 MHz, CDCl3) δ 7.24 - 7.32 (m, 1H), δ 7.05 – 7.23 (m, 3H),
6.90 – 7.04 (m, 3H), 3.93 (s, 2H), 3.72 (s, 2H), 2.93 (t, J = 6.0 Hz, 2H), 2.81 (t,
J = 5.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 141.78, 134.61, 134.26, 128.65,

Benzylamines
65
126.57, 126.41, 126.08, 125.94, 125.56, 125.03, 56.76, 55.68, 50.22, 29.04. The
physical data were identical in all respects to those previously reported.[35]
2-((5-methylfuran-2-yl)methyl)-1,2,3,4-
tetrahydroisoquinoline (3p): Synthesized according to
General procedure. 1,2,3,4-Tetrahydroisoquinoline (0.067 g, 0.50
mmol) affords 3p (0.067 g, 59% yield). Yellow oil obtained after column
chromatography (Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3)
δ 7.05 – 7.20 (m, 3H), 6.96 – 7.04 (m, 1H), 6.15 (d, J = 3.0 Hz, 1H), 5.80 – 6.03
(m, 1H), 3.67 (s, 4H), 2.93 (t, J = 6.0 Hz, 2H), 2.79 (t, J = 5.9 Hz, 2H), 2.30 (s,
3H). 13C NMR (100 MHz, CDCl3) δ 151.94, 149.72, 134.57, 134.13, 128.55, 126.53,
126.01, 125.48, 109.64, 105.88, 77.32, 76.68, 55.47, 54.62, 50.26, 28.89, 13.68.
HRMS (APCI+, m/z): calculated for C15H18NO [M+H]+: 228.13829; found:
228.13844.
N-n-Pentyl-4-methoxybenzylamine (5a): Synthesized
according to General procedure. n-Pentylamine (0.044 g, 0.50
mmol) affords 5a (0.056 g, 54% yield). Yellow oil obtained after
column chromatography (SiO2, n-pentane/EtOAc 50:50 to 0:100). 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.5 Hz, 2H), 6.86 (d, J =
8.6 Hz, 2H), 3.79 (s, 3H), 3.73 (s, 2H), 2.61 (t, J = 7.3 Hz, 2H), 1.87 – 2.03 (br.s,
1H), 1.44 – 1.61 (m, 2H), 1.17 – 1.39 (m, 4H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR
(100 MHz, CDCl3) δ 158.57, 132.27, 129.30, 113.71, 55.18, 53.31, 49.23, 29.57,
29.50, 22.55, 13.99. The physical data were identical in all respects to those
previously reported.[36]
N-n-Pentyl-4-methylbenzylamine (5b): Synthesized according
to General procedure. n-Pentylamine (0.044 g, 0.50 mmol) affords
5b (0.058 g, 61% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 50:50 to 0:100). 1H NMR
(400 MHz, CDCl3) δ 7.22 (d, J = 7.9 Hz, 2H), 7.14 (d, J = 7.8 Hz,
2H), 3.76 (s, 2H), 2.63 (t, J = 7.3 Hz, 2H), 2.34 (s, 3H), 2.10 – 2.20 (br.s, 1H),
1.46 – 1.60 (m, 2H), 1.24 – 1.38 (m, 4H), 0.90 (t, J = 6.80 Hz, 6H). 13C NMR (100
MHz, CDCl3) δ 136.92, 136.45, 129.00, 128.10, 53.57, 49.21, 29.52, 29.48, 22.54,
21.02, 13.99. The physical data were identical in all respects to those previously
reported.[36]
N-n-Pentyl-benzylamine (5c): Synthesized according to General
procedure. n-Pentylamine (0.044 g, 0.50 mmol) affords 5c (0.052
g, 59% yield). Yellow oil obtained after column chromatography
(SiO2, n-pentane/EtOAc 50:50 to 0:100). 1H NMR (400 MHz,
CD2Cl2) δ 7.26 – 7.37 (m, 4H), 7.16 – 7.26 (m, 1H), 3.77 (s, 2H), 2.61 (t, J = 7.2
Hz, 2H), 1.57 – 1.80 (br.s, 1H), 1.45 – 1.55 (m, 2H), 1.27 – 1.36 (m, 4H), 0.91
(t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CD2Cl2) δ 141.46, 128.62, 128.45, 127.07,
54.33, 49.89, 30.24, 30.01, 23.07, 14.27. The physical data were identical in all
respects to those previously reported.[36]

Chapter 3
66
N-n-Pentyl-4-fluorobenzylamine (5d): Synthesized according
to General procedure. n-Pentylamine (0.044 g, 0.50 mmol) affords
5d (0.052 g, 52% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 50:50 to 0:100). 1H NMR
(400 MHz, CDCl3) δ 7.18 – 7.42 (m, 2H), 6.80 – 7.15 (m, 2H), 3.75
(s, 2H) 2.60 (t, J = 7.3 Hz, 2H), 1.53 – 1.65 (br.s, 1H), 1.42 – 1.57 (m, 4H), 1.20
– 1.40 (m, 4H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 161.8 (d, J
= 244.5 Hz) 136.1 (d, J = 3.4 Hz), 129.6 (d, J = 7.9 Hz), 115.1 (d, J = 21.1 Hz),
77.00, 53.26, 49.38, 29.69, 29.51, 22.58, 14.03. HRMS (APCI+, m/z): calculated
for C12H19FN [M+H]+: 196.14960; found: 196.14961.
N-n-Pentyl-3-chloro-benzylamine (5e): Synthesized
according to General procedure. n-Pentylamine (0.044 g, 0.50
mmol) affords 5e (0.044 g, 42% yield). Yellow oil obtained after
column chromatography (SiO2, n-pentane/EtOAc 50:50 to 0:100). 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 7.06 – 7.25 (m, 3H), 3.76 (s, 2H), 2.60
(t, J = 7.2 Hz, 2H), 1.59 – 1.70 (br.s, 1H), 1.45 – 1.55 (m, 2H), 1.27 – 1.35 (m,
4H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 142.50, 134.17, 129.56,
128.14, 126.98, 126.16, 53.40, 49.37, 29.67, 29.46, 22.56, 14.01. The physical
data were identical in all respects to those previously reported.[18]
N-n-Pentyl-3-trifluoromethylbenzylamine (5f): Synthesized
according to General procedure. n-Pentylamine (0.044 g, 0.50
mmol) affords 5f (0.27 g, 22% yield). Yellow oil obtained after
column chromatography (SiO2, n-pentane/EtOAc 50:50 to 0:100). 1H NMR (400 MHz, CDCl3) δ 7.60 (s, 1H), 7.46 – 7.56 (m, 2H), 7.37 – 7.46 (m,
1H), 3.84 (s, 2H), 2.62 (t, J = 7.2 Hz, 2H), 2.06 – 2.21 (br.s, 1H), 1.45 – 1.57 (m,
2H), 1.26 – 1.35 (m, 4H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ
141.31, 131.44, 130.66 (q, J = 32.3 Hz), 128.72, 124.79 (q, J = 3.7 Hz), 124.20
(d, J = 272.2 Hz), 123.73 (q, J = 3.8 Hz), 53.41, 49.39, 29.61, 29.46, 22.55,
13.98. The physical data were identical in all respects to those previously
reported.[18]
N-Benzyl-n-nonylamine (5g): Synthesized according to General
procedure. n-Nonylamine (0.072 g, 0.50 mmol) affords 5g (0.070 g,
60% yield). Yellow oil obtained after column chromatography (SiO2, n-
pentane/EtOAc 50:50 to 0:100). 1H NMR (400 MHz, CDCl3) δ 7.29 –
7.43 (m, 4H), 7.22 – 7.28 (m, 1H), 3.81 (s, 2H), 2.63 (t, J = 7.4 Hz, 2H), 2.32 –
2.42 (br.s, 1H), 1.46 – 1.60 (m, 2H), 1.05 – 1.42 (m, 14H), 0.87 (t, J = 6.9 Hz,
3H). 13C NMR (100 MHz, CDCl3) δ 139.72, 128.35, 128.22, 126.97, 53.73, 49.18,
31.83, 29.72, 29.51, 29.49, 29.23, 27.28, 22.62, 14.06. The physical data were
identical in all respects to those previously reported.[37]
N-Benzyl-2-phenylethamine (5h): Synthesized according to
General procedure. 2-Phenylethamine (0.061 ml, 0.50 mmol)
affords 5h (0.064 g, 61% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 50:50 to 0:100). 1H NMR
(400 MHz, CDCl3) δ 7.15 – 7.40 (m, 10H), 3.83 (s, 2H), 2.89 – 2.96 (m, 2H), 2.82

Benzylamines
67
– 2.89 (m, 2H), 2.64 – 2.73 (br.s, 1H). 13C NMR (100 MHz, CDCl3) δ 139.78, 139.62,
128.69, 128.44, 128.38, 128.17, 127.01, 126.15, 77.32, 77.00, 76.68, 53.59,
50.28, 36.05. The physical data were identical in all respects to those previously
reported.[38]
N-Piperonyl-3-trifluoromethylbenzylamine (5i):
Synthesized according to General procedure. 3-
Trifluoromethylbenzylamine (0.088 ml, 0.50 mmol) affords 5i
(0.093 g, 60% yield). Yellow oil obtained after column
chromatography (SiO2, n-pentane/EtOAc 90:10 to 50:50). 1H
NMR (400 MHz, CDCl3) δ 7.61 (s, 1H), 7.48 – 7.58 (m, 2H), 7.40 – 7.47 (m, 1H),
6.87 (s, 1H), 6.72 – 6.80 (m, 2H), 5.95 (s, 2H), 3.84 (s, 2H), 3.72 (s, 2H), 1.75
– 1.87 (br.s, 1H). 13C NMR (100 MHz, CDCl3) δ 1.47.77, 146.64, 141.14, 133.76,
131.47 (q, J = 1.3 Hz), 130.69 (q, J = 32.0 Hz), 128.77, 124.83
(q, J = 3.8 Hz), 124.19 (d, J = 272.2 Hz), 123.82 (q, J = 3.9 Hz),
121.28, 108.65, 108.10, 100.92, 52.95, 52.36. HRMS (APCI+,
m/z): calculated for C16H15F3NO2 [M+H]+: 310.10494; found:
310.10516.
N-(4-Methylbenzyl)-4-fluorobenzylamine (5j): Synthesized according to
General procedure. 4-Fluorobenzylamine (0.063 g, 0.50 mmol) affords 5j (0.080
g, 70% yield). Yellow oil obtained after column chromatography (SiO2, n-
pentane/EtOAc 90:10 to 50:50). 1H NMR (400 MHz, CDCl3) δ 7.28 – 7.38 (m, 2H),
7.20 – 7.26 (m, 2H), 7.12 – 7.20 (m, 2H), 6.95 – 7.09 (m, 2H), 3.78 (s, 4H), 2.36
(s, 3H), 1.75 – 1.81 (br.s, 1H). 13C NMR (100 MHz, CDCl3) δ 161.87 (d, J = 244.6
Hz), 137.00, 136.55, 135.95 (d, J = 3.1 Hz), 129.63 (d, J = 8.0 Hz), 129.06,
128.06, 115.08 (d, J = 21.1 Hz), 52.75, 52.23, 21.05. HRMS (APCI+, m/z):
calculated for C14H19N2S2 [M+H]+: 230.13395; found: 230.13404.
N-(4-Methylbenzyl)-4-methoxyaniline (5k): Synthesized
according to General procedure. 4-Methoxyaniline (0.062 g, 0.50
mmol) affords 5k (0.076 g, 67% yield). Yellow oil obtained after
column chromatography (SiO2, n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 7.8 Hz, 2H), 7.17 (d, J =
7.8 Hz, 2H), 6.80 (d, J = 8.9 Hz, 2H), 6.63 (d, J = 8.9 Hz, 2H), 4.25 (s, 2H), 3.76
(s, 3H), 2.36(s, 3H). 13C NMR (100 MHz, CDCl3) δ 152.16, 142.38, 136.75, 136.50,
129.22, 127.52, 114.86, 114.13, 55.76, 49.00, 21.06. The physical data were
identical in all respects to those previously reported.[39]
N-Benzyl-4-methoxyaniline (5l): Synthesized according to General
procedure. 4-Methoxyaniline (0.062 g, 0.50 mmol) affords 5l (0.060
g, 56% yield). Yellow oil obtained after column chromatography (SiO2,
n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.20 –
7.45 (m, 5H), 6.81 (d, J = 8.7 Hz, 2H), 6.63 (d, J = 8.7 Hz, 2H), 4.30 (s, 2H),
3.76 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 152.17, 142.32, 139.59, 128.53, 127.51,
127.12, 114.86, 114.11, 55.75, 49.22. The physical data were identical in all
respects to those previously reported.[39]

Chapter 3
68
N-n-Butyl-N-n-pentylbenzylamine (9a): Synthesized
according to General procedure. n-Pentylamine (0.044 g, 0.50
mmol) affords 9a (0.059 g, 51% yield). Yellow oil obtained after
column chromatography (Al2O3, n-pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CD2Cl2) δ 7.26 – 7.45 (m, 4H), 7.15 – 7.26 (m,1H), 3.56
(s, 2H), 2.32 – 2.50 (m, 4H), 1.40 – 1.55 (m, 4H), 1.20 – 1.38 (m, 6H), 0.80 –
1.00 (m, 6H). 13C NMR (100 MHz, CD2Cl2) δ 140.42, 128.76, 127.92, 126.48, 58.55,
29.60, 29.17, 26.62, 22.59, 20.53, 13.84, 13.79. HRMS (APCI+, m/z): calculated
for C16H28N [M+H]+: 234.22163; found: 234.22160.
N-n-Butyl-N-n-pentyl-4-fluorobenzylamine (9b):
Synthesized according to General procedure. n-Pentylamine
(0.044 g, 0.50 mmol) affords 9b (0.050 g, 40% yield). Yellow
oil obtained after column chromatography (Al2O3, n-
pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.20 – 7.42 (m, 2H),
6.85 – 7.10 (m, 2H), 3.51 (s, 2H), 2.29 – 2.51 (m, 4H), 1.37 – 1.57 (m, 4H), 1.15
– 1.37 (m, 6H), 0.77 – 1.01 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 161.25 (d, J =
243.1 Hz), 135.96, 129.75 (d, J = 7.8 Hz), 114.12 (d, J = 21.0 Hz), 57.37, 29.18,
28.79, 26.24, 22.17, 20.11, 13.44, 13.38. HRMS (APCI+, m/z): calculated for
C16H27FN [M+H]+: 252.21220; found: 252.21233.
N-n-Hexyl-N-n-pentyl-4-methoxylbenzylamine
(9c): Synthesized according to General procedure. n-
Pentylamine (0.044 g, 0.50 mmol) affords 9c (0.063 g,
43% yield). Yellow oil obtained after column
chromatography (Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H),
3.80 (s, 2H), 3.51 (s, 3H), 2.30 – 2.48 (m, 4H), 1.39 – 1.55 (m, 4H), 1.15 – 1.38
(m, 10H), 0.80 – 1.00 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 158.46, 130.01,
113.45, 57.76, 55.20, 53.52, 31.77, 29.66, 27.12, 26.74, 26.46, 22.65, 22.60,
14.08, 14.05. HRMS (APCI+, m/z): calculated for C19H34NO [M+H]+: 292.26349;
found: 292.26360.
(3-(morpholinomethyl)phenyl)methanol (13a): Synthesized
according to General procedure. Morpholine (0.044 g, 0.50 mmol)
affords 13a (0.065 g, 63% yield). Yellow solid obtained after
column chromatography (SiO2, EtOAc/MeOH 100:0 to 90:10). 1H
NMR (400 MHz, CD2Cl2) δ 7.15 – 7.45 (m, 4H), 4.65 (s, 2H), 3.65 (t, J = 4.7 Hz,
4H), 3.49 (s, 2H), 2.42 (t, J = 4.7 Hz, 4H), 2.28 – 2.47 (br.s, 1H) 13C NMR (100
MHz, CD2Cl2) δ 141.80, 138.59, 128.68, 128.65, 128.08, 126.04, 67.25, 65.23,
63.65, 54.04. HRMS (APCI+, m/z): calculated for C12H18NO2 [M+H]+: 208.13321;
found: 208.13327.

Benzylamines
69
N-Benzyl-N-methyl-5-hydroxymethyl-furfurylamine (13b):
Synthesized according to General procedure. N-Methylbenzylamine
(0.061 g, 0.50 mmol) affords 13b (0.069 g, 60% yield). Yellow solid
obtained after column chromatography (SiO2, EtOAc/MeOH 100:0 to
90:10). 1H NMR (400 MHz, CDCl3) δ 7.27 – 7.47 (m, 4H), 7.20 –
7.28 (m, 1H), 6.00 – 6.33 (m, 2H), 4.55 (s, 2H), 3.45 – 3.64 (m,
4H), 2.90 – 3.08 (br.s, 1H), 2.22 (s, 3H) 13C NMR (100 MHz, CDCl3) δ 153.81,
151.99, 138.07, 129.16, 128.19, 127.06, 109.44, 108.08, 61.12, 57.31, 53.27,
41.88. HRMS (APCI+, m/z): calculated for C14H18NO2 [M+H]+: 232.13321; found:
232.13323.
N-Benzyl-N-methyl-3-morpholinomethyl-benzylamine
(14): Synthesized according to General procedure. N-
Methylbenzylamine (0.061 g, 0.50 mmol) affords 14 (0.047 g,
30% yield). Yellow solid obtained after column chromatography
(Al2O3, n-pentane/EtOAc 90:10 to 80:20). 1H NMR (400 MHz,
CDCl3) δ 7.29 – 7.42 (m, 5H), 7.24 – 7.29 (m, 3H), 7.18 – 7.24
(m, 1H), 3.71 (t, J = 4.7 Hz, 4H), 3.53 (s, 2H), 3.48 – 3.53 (m, 4H), 2.45 (t, J =
4.7 Hz, 4H), 2.20 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 139.18, 139.16, 137.60,
129.74, 128.91, 128.19, 128.12, 127.88, 127.80, 126.94, 66.99, 63.42, 61.78,
61.69, 53.60, 42.25. HRMS (APCI+, m/z): calculated for C20H27N2O [M+H]+:
311.21195; found: 311.21179.
1-Benzylpiperidine (16a): Synthesized according to General
procedure. Piperidine (0.043 g, 0.50 mmol) affords 17a (0.057 g, 65%
yield). Yellow oil obtained after column chromatography (Al2O3, n-
pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.05 – 7.55
(m, 5H), 3.46 (s, 2H), 2.27 – 2.49 (m, 4H), 1.52 – 1.66 (m, 4H), 1.40
– 1.51 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 139.58, 129.45, 128.43, 127.11,
64.10, 54.93, 26.51, 24.89. The physical data were identical in all respects to
those previously reported.[31]
1-(4-Methylbenzyl)piperidine (16b): Synthesized according to
General procedure. Piperidine (0.043 g, 0.50 mmol) affords 17b (0.068
g, 72% yield). Yellow solid obtained after column chromatography (Al2O3,
n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J
= 7.8 Hz, 2H), 7.13 (d, J = 7.8 Hz, 2H), 3.46 (s, 2H), 2.32 – 2.46 (m,
4H), 2.34 (s, 3H), 1.53 – 1.63 (m, 4H), 1.39 – 1.48 (m, 2H). 13C NMR (100 MHz,
CDCl3) δ 136.37, 135.26, 129.22, 128.74, 63.53, 54.36, 25.91, 24.35, 21.06. The
physical data were identical in all respects to those previously reported.[31]
1-(4-Methoxybenzyl)piperidine (16c): Synthesized according to
General procedure. Piperidine (0.043 g, 0.50 mmol) affords 17c (0.080
g, 78% yield). Yellow solid obtained after column chromatography (Al2O3,
n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J
= 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 3.79 (s, 3H), 3.42 (s, 2H), 2.20
– 2.50 (m, 4H), 1.50 – 1.65 (m, 4H), 1.35 – 1.50 (m, 2H) 13C NMR (100

Chapter 3
70
MHz, CDCl3) δ 158.52, 130.49, 130.34, 113.40, 63.18, 55.16, 54.30, 25.93, 24.38.
The physical data were identical in all respects to those previously reported.[28]
1-(thiophen-2-ylmethyl)piperidine (16d): Synthesized according to
General procedure. Piperidine (0.043 g, 0.50 mmol) affords 17d (0.053
g, 58% yield). Yellow solid obtained after column chromatography
(Al2O3, n-pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ
7.18 – 7.25 (m, 1H), 6.92 – 6.98 (m, 1H), 6.86 – 6.92 (m, 1H), 3.70 (s, 2H), 2.34
– 2.51 (m, 4H), 1.54 – 1.63 (m, 4H), 1.36 – 1.47 (m, 2H). 13C NMR (100 MHz,
CDCl3) δ 141.79, 126.31, 125.92, 124.71, 57.74, 54.06, 25.89, 24.24. The
physical data were identical in all respects to those previously reported.[40]

Benzylamines
71
References
[1] a) D. Lednicer, The organic chemistry of drug synthesis, Vol. 2, John Wiley & Sons:
New Jersey, 2008; b) M. W. Jann, Pharmacotherapy, 2000, 20, 1; c) M. Sniezek, S.
Stecko, I. Panfil, B. Furman, M. Chmielewski, J. Org. Chem., 2013, 78, 7048-7057;
d) K. M. Brands, J. et al. J. Am. Chem. Soc., 2003, 125, 2129-2135.
[2] a) S.-L. Shi, S. L. Buchwald, Nature Chem., 2015, 7, 38-44; b) S. Zhu, N. Niljianskul,
S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746-15749; c) Y. Miki, K. Hirano,
T. Satoh, M. Miura, Angew. Chem. Int. Ed., 2013, 52, 10830-10834; d) T. E. Muller,
K. C. Hultzsch, M. Yus, F. Foubelo, M. Tada, Chem. Rev., 2008, 108, 3795-3892; e)
R. Severin, S. Doye, Chem. Soc. Rev., 2007, 36, 1407-1420.
[3] M. B. Smith, March, J. March’s advanced organic chemistry: reactions, mechanisms,
and structure, chap. 10, 6th ed., John Wiley & Sons, Inc., Hoboken, New Jersey,
2007.
[4] a) S. Pisiewicz, T. Stemmler, A.-E. Surkus, K. Junge, M. Beller, ChemCatChem, 2015,
7, 62-64; b) P. N. Kolesnikov, N. Z. Yagafarov, D. L. Usanov, V. I. Maleev, D. Chusov,
Org. Lett., 2015, 17, 173-175; c) C. Guyon, E. Da Silva, Lafon, R.; E. Metay, M.
Lemaire, RSC Adv., 2015, 5, 2292-2298; d) S. Zhou, S. Fleischer, H. Jiao, K. Junge,
M. Beller, Adv. Synth. Catal., 2014, 356, 3451-3455; e) D. Chusov, B. List, Angew.
Chem. Int. Ed., 2014, 53, 5199-5201; f) A. Pagnoux-Ozherelyeva, N. Pannetier, M.
D. Mbaye, S. Gaillard, J.-L. Renaud, Angew. Chem. Int. Ed., 2012, 51, 4976-4980.
[5] Benzyl alcohol is produced naturally by many plants, see: The Merck Index: An
Encyclopedia of Chemicals, Drugs, and Biologicals, 11th ed., Merck, 1989, ISBN
091191028X, p1138.
[6] Benzyl alcohols may be produced by hydrogenation of benzyl aldehydes which are
derivated from oxidative depolymerization of lignin, see: a) A. Rahimi, A. Azarpira,
H. Kim, J. Ralph, S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415-6418; b) P. J.
Deuss, K. Barta, J. G. de Vries, Catal. Sci. Technol., 2014, 4, 1174-1196.
[7] Methylamines can be produced under 350-500 °C and 15-30 bar pressure using
aluminum-based heterogeneous catalyst from ammonia and methanol. See: K.
Weissermel, H.-J. Arpe, Industrial Organic Chemistry, 4th ed., Wiley-VCH: Weinheim,
2003, p. 51.
[8] One promising example showed the direct amination of alcohols catalyzed by iron-
amino acid through a nucleophilic substitution pathway under relatively mild
condition (160–200 °C). See: Y. Zhao, S. W. Foo, S. Saito, Angew. Chem. Int. Ed.,
2011, 50, 3006-3009.
[9] a) M. H. S. A. Hamid, P. A. Slatford, J. M. J. Williams, Adv. Synth. Catal., 2007, 349,
1555-1575; b) T. D. Nixon, M. K. Whittlesey, J. M. J. Williams, Dalton Trans., 2009,
753-762; c) A. J. A. Watson, J. M. J. Williams, Science, 2010, 329, 635-636; d) G.
Guillena, D. J. Ramon, M. Yus, Chem. Rev., 2010, 110, 1611-1641; e) S. Bahn, S.
Imm, L. Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem, 2011, 3, 1853-
1864; f) G. E. Dobereiner, R. H. Crabtree, Chem. Rev., 2010, 110, 681-703; g) C.
Gunanathan, D. Milstein, Science, 2013, 341, 1229712.
[10] a) B. M. Trost, Science, 1983, 219, 245-250; b) B. M. Trost, Science, 1991, 254,
1471-1477.
[11] R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit, N. Tongpenyai, J. Chem. Soc. Chem.
Commun., 1981, 611-612.
[12] Y. Watanabe, Y. Tsuji, Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667-2670.
[13] Selected examples: a) K.-i. Fujita, K. Yamamoto, R. Yamaguchi, Org. Lett., 2002, 4,
2691–2694; b) K.-i. Fujita, T. Fujii, R. Yamaguchi, Org. Lett., 2004, 6, 3525–3528;
c) D. Gnanamgari, E. L. O. Sauer, N. D. Schley, C. Butler, C. D. Incarvito, R. H.
Crabtree, Organometallics, 2009, 28, 321-325; d) B. Blank, M. Madalska, R. Kempe,
Adv. Synth. Catal., 2008, 350, 749-758; e) M. H. S. A. Hamid, J. M. J. Williams,
Chem. Commun., 2007, 725-727; f) S. Bahn, S. Imm, K. Mevius, L. Neubert, A.

Chapter 3
72
Tillack, J. M. J. Williams, M. Beller, Chem. Eur. J., 2010, 16, 3590-3593; g) M. Haniti,
S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson,
J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766-1774; h) C. Gunanathan, D.
Milstein, Angew. Chem. Int. Ed., 2008, 47, 8661-8664; i) R. Kawahara, K.-i. Fujita,
R. Yamaguchi, J. Am. Chem. Soc., 2010, 132, 15108-15111; j) X. Ye, P. N. Plessow,
M. K. Brinks, M. Schelwies, T. Schaub, F. Rominger, R. Paciello, M. Limbach, P.
Hofmann, J. Am. Chem. Soc., 2014, 136, 5923-5929.
[14] Recent examples with Ru: a) V. R. Jumde, L. Gonsalvi, A. Guerriero, M. Peruzzini, M.
Taddei, Eur. J. Org. Chem., 2015, 1829-1833; b) V. R. Jumde, E. Cini, A. Porcheddu,
M. Taddei. Eur. J. Org. Chem., 2015, 1068-1078; c) E. Balaraman, D. S. Y. Diskin-
Posner, D. Milstein, Catal. Lett., 2015, 145, 139-144; d) P. S. Siah, X. Xie, G.
Boopathy, T. D. Tuan, R. Balamurugan, V. H. Han, M. Abdul, S. RSC Adv., 2015, 5,
4434-4442; e) N. J. Oldenhuis, V. M. Dong, Z. Guan, J. Am. Chem. Soc., 2014, 136,
12548-12551; f) A. B. Enyong, B. Moasser, J. Org. Chem., 2014, 79, 7553-7563; g)
M. Chen, M. Zhang, F. Xie, X. Wang, H. Jiang, ChemCatChem, 2014, 6, 2993-2997;
h) S. Demir, F. Coskun, I. Özdemir, J. Organomet. Chem., 2014, 755, 134-140.
[15] Recent examples with Ir: a) S. Wöckel, P. Plessow, M. Schelwies, M. K. Brinks, F.
Rominger, P. Hofmann, M. Limbach, ACS Catal., 2014, 4, 152-161; b) Y. Zhang, C.-
S. Lim, D. S. B. Sim, H.-J. Pan, Y. Zhao, Angew. Chem. Int. Ed., 2014, 53, 1399-
1403; c) S. Ruch, T. Irrgang, R. Kempe, Chem. Eur. J., 2014, 20, 13279-13285; d)
P. Qu, C. Sun, J. Ma, F. Li, Adv. Synth. Catal., 2014, 356, 447-459; e) Y.-H. Chang,
Y. Nakajima, F. Ozawa, Organometallics, 2013, 32, 2210-2215.
[16] a) M. Bala, P. K. Verma, U. Sharma, N. Kumar, B. Singh, Green Chem., 2013, 15,
1687-1693; b) X. Cui, F. Shi, Y. Zhang, Y. Deng, Tetrahedron Lett., 2010, 51, 2048-
2051.
[17] Selected examples with the use of Knölker complex for other transformations: a) H.-
J. Knölker, E. Baum, H. Goesmann, Klauss, R. Angew. Chem. Int. Ed., 1999, 38,
2064-2066; b) C. P. Casey, H. Guan, J. Am. Chem. Soc., 2007, 129, 5816-5817; c)
C. P. Casey, H. Guan, J. Am. Chem. Soc., 2009, 131, 2499-2507; d) A. Pagnoux-
Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard, J.-L. Renaud, Angew. Chem. Int.
Ed., 2012, 51, 4976-4980; e) A. Quintard, T. Constantieux, J. Rodriguez, Angew.
Chem. Int. Ed., 2013, 52, 12883-12887; f) M. G. Coleman, A. N. Brown, B. A. Bolton,
H. Guan, Adv. Synth. Catal., 2010, 352, 967-970; g) S. Zhou, S. Fleischer, K. Junge,
M. Beller, Angew. Chem. Int. Ed., 2011, 50, 5120-5124; h) A. Quintard, J. Rodriguez,
Angew. Chem. Int. Ed., 2014, 53, 4044-4055.
[18] T. Yan, B. L. Feringa, K. Barta, Nat. Commun., 2014, 5, 5602.
[19] During the preparation of our manuscript, two related studies have been published,
see: a) A. J. Rawlings, L. J. Diorazio, M. Wills, Org. Lett., 2015, 17, 1086-1089. For
amination of benzylalcohols, only the use of anilines has been described. b) H.-J. Pan,
T. W. Ng, Y. Zhao, Chem. Commun., 2015, 51, 11907-11910. With the assistance of
40 mol% AgF, the reactivity of the amination of secondary alcohol has been
significantly improved.
[20] For instance, Pka of N-methylbenzylamine is 9.58, of N-methylphenethylamine is
10.15. See: F. Barbato, G. di Martino, L. Grumetto, M. I. La Rotonda, Eur. J. Pharm.
Sci., 2004, 22, 261-269.
[21] M. D. Joesten, K. G. Claus, K. P. Lannert, J. Inorg. Nucl. Chem., 1967, 29, 1421-
1426.
[22] S. E. Clapham, A. Hadzovic, R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201-2237.
[23] L. D. Quin, J. A. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in
Nature and in the Synthesis of Pharmaceuticals; John Wiley & Sons, Inc.: Hoboken,
New Jersey, 2010.
[24] a) E. J. Corey, Chem. Soc. Rev., 1988, 17, 111-133; b) E. J. Corey, Angew. Chem.
Int. Ed. Engl., 1991, 30, 455-465.
[25] A. Feriani, G. Gaviraghi, G. Toson, M. Mor, A. Barbieri, E. Grana, C. Boselli, M.

Benzylamines
73
Guarneri, D. Simoni, S. Manfredini, J. Med. Chem., 1994, 37, 4278-4287.
[26] a) A. J. Kumalaputri, G. Bottari, P. M. Erne, H. J. Heeres, K. Barta, ChemSusChem,
2014, 7, 2266-2275; b) G. Bottari, A. J. Kumalaputri, K. K. Krawczyk, B. L. Feringa,
H. J. Heeres, K. Barta, ChemSusChem, 2015, 8, 1323-1327; c) R.-J. van Putten, J.
C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres, J. G. de Vries, Chem.
Rev., 2013, 113, 1499-1597.
[27] T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal., 2012, 354, 597-
601.
[28] Q. Li, C. W. Liskey, J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 8755 –8765.
[29] X. Cui, X. Dai,; Y. Deng, F. Shi, Chem. Eur. J., 2013, 19, 3665–3675.
[30] P. Chaudhary, R. Kumar, A. K. Verma, D. Singh, V. Yadav, A. K. Chhillar, G. L. Sharmab,
R. Chandra, Bioorg. Med. Chem., 2006, 14, 1819-1826.
[31] S. P. Shan, T. T. Dang, A. M. Seayad, B. Ramalingam, ChemCatChem, 2014, 6, 808-
814.
[32] L. U. Nordstrøm, R. Madsen, Chem. Commun., 2007, 5034-5036.
[33] K. Yamaguchi, J. He, T. Oishi, N. Mizuno, Chem. Eur. J., 2010, 16, 7199-7207.
[34] P. Kowalski, K. Mitka, J. Jaskowska, B. Duszynska, A. J. Bojarski, Arch. Pharm. Chem.
Life Sci., 2013, 346, 339-348.
[35] E. Le Gall, A. Decompte, T. Martens, M. Troupel, Synthesis, 2010, 249-254.
[36] S. Lu, J. Wang, X. Cao, X. Li, H. Gu, Chem. Commun., 2014, 50, 3512-3515.
[37] S. Das, B. Join, K. Junge, M. Beller, Chem. Commun., 2012, 48, 2683-2685.
[38] J. Zheng, T. Roisnel, C. Darcel, J.-B. Sortais, ChemCatChem, 2013, 5, 2861-2864.
[39] Q. Lei, Y. Wei, D. Talwar, C. Wang, D. Xue, J. Xiao, Chem. Eur. J., 2013, 19, 4021-
4029.
[40] S. Das, D. Addis, K. Junge, M. Beller, Chem. Eur. J., 2011, 17, 12186-12192.

Chapter 3
74

75
Chapter 4
Pyrroles via Iron-Catalyzed N-Heterocyclization from
Unsaturated Diols and Primary Amines
Pyrroles are prominent scaffolds in pharmaceutically active compounds and play
an important role in medicinal chemistry. Therefore, the development of novel,
atom-economic and sustainable catalytic strategies to obtain these moieties is
highly desired. Recently, direct catalytic pathways have been established that
utilize readily available alcohol substrates. These approaches rely on the use of
noble metals such as ruthenium or iridium. Here we report on the direct synthesis
of pyrroles with a catalyst based on the earth-abundant and inexpensive iron. The
method uses 2-butyne-1,4-diol or 2-butene-1,4-diol, which can be directly coupled
with anilines, benzyl amines and aliphatic amines to obtain a variety of pyrroles in
moderate to very good isolated yields.
Part of this chapter was published:
T. Yan, K. Barta, ChemSusChem, 2016, 9, 2321–2325.

Chapter 4
76
Introduction
Pyrroles are important building blocks in medicinal chemistry[1] since many
pharmaceutically active compounds contain these moieties. For example,
aloracetam[2] was been previously used in studies for treating Alzheimer’s disease,
isamoltane[3] was shown to exhibit anxiolytic effects on rodents, elopiprazole[4] is
an antipsychotic drug, and Lipitor is a drug for treating cardiovascular disease
(Figure 1).
Figure 1: Bioactive compounds containing pyrrole moieties.
Owing to the importance of pyrroles, many classical synthetic pathways such as
the Hantzsch[5], Knorr[6], and Paal-Knorr[7] synthesis (Scheme 1, A) as well as
related multicomponent reactions[8] have already been established. These
stoichiometric routes however, may suffer from poor substrate accessibility, harsh
reaction conditions and multi-step syntheses that lead to the formation of waste
and low atom economy[9]. Thus the development of novel catalytic methods, to
create the pyrrole scaffold efficiently is subject of intensive research.[10] Several
elegant approaches, broadly related to the borrowing hydrogen strategy,[11] have
been recently reported that rely on the catalytic dehydrogenation of easily
accessible alcohol substrates, (Scheme 1, A). In 2013, Beller[12] and coworkers
reported on the ruthenium-catalyzed three-component pyrrole synthesis where
secondary alcohols, diols and primary amines were coupled in analogy to the
classical Hantzsch synthesis. Michlik and Kempe[13] achieved the direct iridium-
catalyzed coupling of alcohols and amino alcohols to obtain pyrroles, and Milstein[14]
and coworkers presented a similar, ruthenium-catalyzed method. In 2011,
Crabtree et al.[15] described the formation of pyrroles from 1,4-diols and amines.
In the course of these reactions, the alcohol substrates undergo dehydrogenation
to the corresponding carbonyl-compounds, which further react with the amine to
form the desired pyrrole product and the hydrogen equiv borrowed from the
alcohol substrate are concomitantly liberated from the catalyst.[12-15]
In the studies by the groups of Watanabe[16] and Williams[17] it was shown that
pyrroles can also be directly obtained from amines and unsaturated diols, such as
2-butyne-1,4-diol with an appropriate ruthenium catalyst. In this case it was
proposed that the reaction likely proceeds through an internal hydrogen transfer

Pyrroles
77
isomerization to the corresponding saturated dicarbonyl compounds that
subsequently undergoes pyrrole formation with an amine reaction partner.
Our group has previously established the first iron-catalyzed formation of
pyrrolidines[18] from amines and 1,4-butane-diol using Knölker’s complex[19,20]
(Scheme 1, B), which was described in chapter 2. Based our previous results[18,21]
and the reports of Watanabe and Williams, we envisioned the possibility of the iron
catalyzed direct pyrrole formation starting from primary amines and unsaturated
diols. A reactivity similar to the ruthenium-based system was expected, since the
iron-complex is capable of alcohol dehydrogenation.
Scheme 1: A Classic and modern synthetic pathways to access pyrroles; B iron
catalyzed direct synthesis of pyrrolidines and pyrroles from amines and diols.
A
Classic pyrrole synthesis
B
Catalytic pyrrole synthesis
C
Results and discussion
We started our investigation using 2-butyne-1,4-diol (2a) and 4-(N,N-
dimethylamino)-aniline (1a) to establish the novel iron-catalyzed methodology
towards pyrroles. Similarly to our previous work[18,21], Cat 3 was selected as the
pre-catalyst, while Me3NO was used to generate the catalytically active iron
complex. A variety of solvents were screened and the reaction temperature varied
between 110-130 °C. The first attempts at 110 °C in solvents tetrahydrofuran
(THF), dioxane, acetonitrile (CH3CN) and dimethylformamide (DMF) resulted in
very similar conversion values of up to 70% and moderate product selectivities
(Table 1, entry 1-4). Conversion of 1a and product selectivity slightly improved in

Chapter 4
78
CPME and toluene at 110 °C (Table 1, entry 5-6). The results could be further
improved to full conversion of 1a and near perfect selectivity of 3a at 130 °C in
toluene and a good 83% isolated yield of 3a was achieved (Table 1, entry 7).
Similarly, full conversion but slightly lower 3a yield (76%) was obtained when the
reaction was conducted at 120 °C (Table 1, entry 8).
Table 1: Optimization of reaction conditions to obtain N-(4-dimethylamino-
phenyl)-pyrrole (3a) from 4-(N,N-dimethylamino)-aniline (1a) and 1,4-diols (2).
Entry 2 Solvent T [oC] Conv. 1a [%]a Sele. 3a [%]a
1 2a THF 110 67 65
2 2a dioxane 110 70 68
3 2a CH3CN 110 68 67
4 2a DMF 110 67 66
5 2a CMPE 110 71 69
6 2a toluene 110 73 71
7 2a toluene 130 >99 98 (83)
8 2a toluene 120 >99 95 (76)
9 2b toluene 110 94 44
10 2b CPME 110 90 25
11 2b toluene 120 >99 63 (59)
General reaction conditions: General Procedure, 0.5 mmol 1a, 1 mmol 2, 0.02 mmol Cat
3, 0.04 mmol Me3NO, 2 ml solvent, 18 h, 110-130 °C, isolated yield in parenthesis. aValues
based on GC-FID selectivity.
After having established that diol 2a can be successfully used as the substrate to
form 3a, we explored the use of cis-2-butene-1,4-diol (2b) as the starting material.
Catalytic runs conducted with 2b in toluene and CPME at 110 °C resulted in 94%
and 90% conversion but only 44 and 25% selectivity for 3a, respectively (Table 1,
entry 9-10). At 120 °C full 1a conversion and a good, 59% isolated yield of 3a
was obtained (Table 1, entry 11) without significant over-reduction to
pyrrolidines.[16] Therefore 2b can be regarded as alternative reaction partner to
2a, despite the slightly lower product yields obtained.
With the optimized reaction conditions in hand, a variety of anilines were tested
(Table 2). Electron-rich 4-methoxy-aniline (1b) reacted smoothly with 2-butyne-
1,4-diol (2a), leading to 80% isolated yield of 3b (Table 2, entry 1). When 4-
methyl-aniline (1c) was used as substrate, much lower conversion and 36%
isolated yield of 3c was obtained. Similar behavior was observed in the reaction of
electron poor 4-fluoro-aniline (1d) with 2-butyne-1,4-diol (2a), that yielded 30%
of 3d at 33% substrate conversion. Interestingly, in both of these cases almost
full conversion and much higher product selectivity were achieved when cis-2-
butene-1,4-diol was employed as coupling partner instead of 1a, providing 59%

Pyrroles
79
and 47% isolated yields of 3c and 3d, respectively (Table 2, entry 2-3). According
to these results, 2b was selected for further reactions with 4-chloro-aniline (1e),
4-bromo-aniline (1f) and 3,4-(methenedioxy)-aniline (1g). In all these cases
excellent substrate conversions and very good product selectivity was observed
and the desired products 3e, 3f and 3g were obtained in 45%, 44% and 37%
isolated yields, respectively (Table 2, entry 4-6). It was also shown that product
selectivity and isolated yields could be improved in the coupling of 1e with 4 equiv
of diol 2b. Ortho-substituted anilines (1h–1j) could also be successfully used
whereby electron-donating substituents gave better product yields (Table 2, entry
7–9).
Table 2: Direct synthesis of N-substituted pyrroles from anilines and 1,4-diols.
Entry 1 2 Product 3 Conv. 1 [%]a Sele. 3 [%]a
1 1b 2a 3b 92 90 (80)
2 1c 2a
3c 42 36
2b >99 90 (59)
3 1d 2a
3d 33 30
2b 91 75 (47)
4 1e 2b
3e 85 63 (45)
2bb 98 72 (54)
5 1f 2b 3f 94 61 (44)
6 1g
2b 3g >99 68 (37)
7 1h
2b 3h 70 42 (36)
8 1i
2b 3i 90 52 (25)
9 1j
2b 3j 56 36 (15)
General reaction conditions: General Procedure, 0.5 mmol 1a, 1 mmol 2a, 0.02 mmol Cat
3, 0.04 mmol Me3NO, 2 ml toluene, 18 h, 130 °C, isolated yield in parenthesis, unless
otherwise specified. aConversion is based on GC-FID selectivity; bThe reaction was operated
in a sealed 20 ml vial with 4 equiv 2b in 22 h.
After aniline derivatives were also converted to N-phenyl-pyrroles successfully,
primary aliphatic amines were explored under standard reaction conditions using
2-butyne-1,4-diol (2a) as reaction partner. Interestingly, a series of benzyl amines
with varying electron density all afforded full substrate conversions, excellent
product selectivity and good isolated yields of the desired N-benzylpyrroles. The
coupling of benzylamine (5a) with 2a leads to the formation of N-benzylpyrrole
(6a) with 61% isolated yield (Table 3, entry 1). Similarly, good results were
obtained with a variety of halogenated benzyl amines, such as 4-chloro-
benzylamine (5b), 4-fluoro-benzylamine (5c), 3-trifluoromethyl-benzylamine (5d)
and 3-fluoro-4-chloro-benzylamine (5e), which afforded the corresponding N-(4-

Chapter 4
80
chlorobenzyl)-pyrrole (6b), N-(4-fluorobenzyl)-pyrrole (6c), N-(3-
trifluoromethylbenzyl)-pyrrole (6d) and N-(3-fluoro-4-chlorobenzyl)-pyrrole (6e)
products in 57%, 52%, 55% and 65% isolated yields, respectively (Table 3, entry
2-5). With electron-rich benzylamines 5f and 5g, the desired N-(4-methylbenzyl)-
pyrrole (5f) and N-piperonyl-pyrrole (5g) were obtained in good, 65% and 60%
isolated yields, respectively (Table 3, entry 6-7). Interestingly, even 3-picolylamine
(5h) reacted smoothly with 2-butyne-1,4-diol (2a) forming N-(3-picolyl)pyrrole in
76% isolated yield, although pyridine is a potential ligand that may coordinate to
iron[22] (Table 3, entry 8). Product N-furfuryl-pyrrole (6i), which has already been
proposed as food additive[23], was obtained in 43% yield from furfurylamine (5i)
(Table 3, entry 9). For other aliphatic amines such as 2-phenylethylamine (5g) and
dodecylamine (5k), the corresponding pyrrole products were obtained in 41% and
42% isolated yield (Table 3, entry 10-11). Cyclohexylamine (5l) reacted with 2a
providing N-cyclohexylpyrrole (6l) in 33% yield (Table 3, entry 12).
Table 3: Direct synthesis of N-substituted pyrroles from anilines and 1,4-diols.
Entry 5 Product 6 Sele. 3 [%]a
1 5a
6a 90 (61)
2 5b
6b 85 (57)
3 5c 6c 73 (52)
4 5d
6d 88 (55)
5 5e
6e 87 (65)
6 5f 6f 87 (65)
7 5g
6g 83 (55)
8 5h 6h 92 (76)
9 5i 6i 71 (43)
10 5j 6j (41)
11 5k 6k 86 (42)
12 5l 6l 85 (33)
General reaction conditions: General Procedure, 0.5 mmol 5, 1 mmol 2a, 0.02 mmol Cat
3, 0.04 mmol Me3NO, 2 ml toluene, 18 h, 130 °C. In all cases, full conversion was obtained. aValues shown are GC-FID selectivity, numbers in brackets are isolated yields.
Scheme 2: GPC analysis of N-benzyl pyrrole formation from benzylamine and 2-
butyne-1,4-diol.
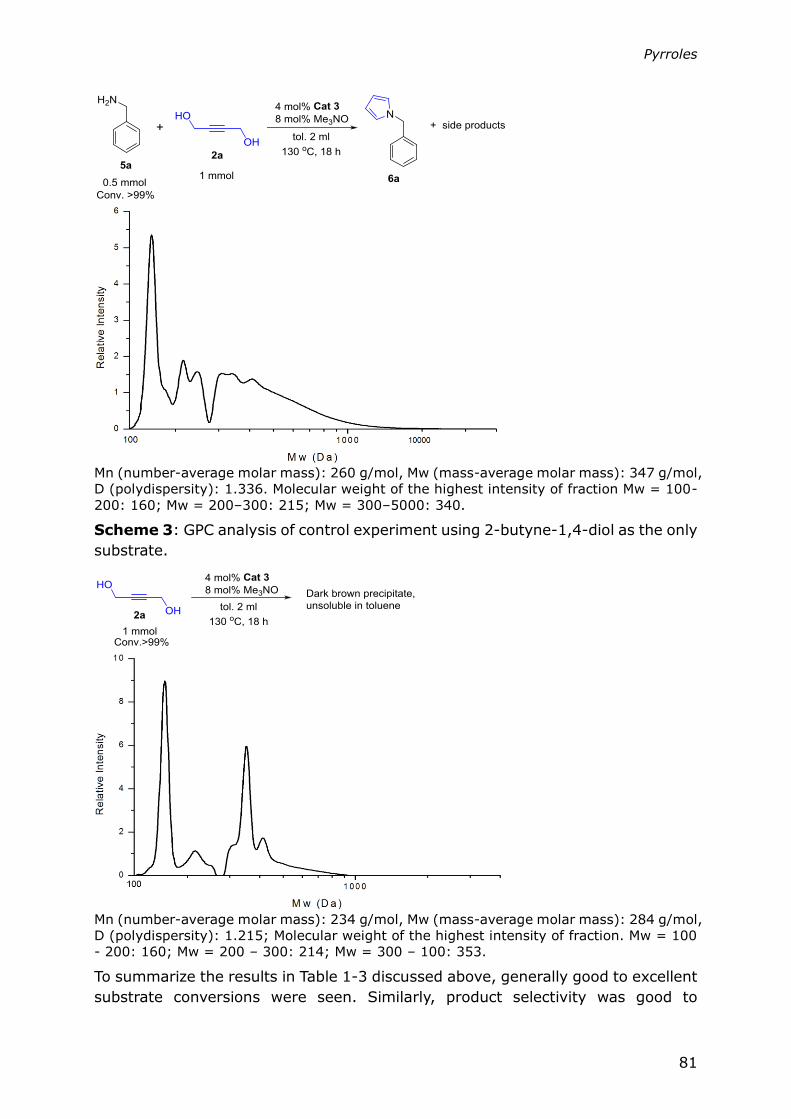
Pyrroles
81
Mn (number-average molar mass): 260 g/mol, Mw (mass-average molar mass): 347 g/mol,
D (polydispersity): 1.336. Molecular weight of the highest intensity of fraction Mw = 100-
200: 160; Mw = 200–300: 215; Mw = 300–5000: 340.
Scheme 3: GPC analysis of control experiment using 2-butyne-1,4-diol as the only
substrate.
Mn (number-average molar mass): 234 g/mol, Mw (mass-average molar mass): 284 g/mol,
D (polydispersity): 1.215; Molecular weight of the highest intensity of fraction. Mw = 100
- 200: 160; Mw = 200 – 300: 214; Mw = 300 – 100: 353.
To summarize the results in Table 1-3 discussed above, generally good to excellent
substrate conversions were seen. Similarly, product selectivity was good to

Chapter 4
82
excellent based on GC-FID and GC-MS measurements, albeit the isolated product
yields were lower. This may be an indication of side reactions involving species not
detectable by these GC measurements. Indeed, gel permeation chromatography
(GPC) measurements of the crude product mixture confirmed the presence of
oligomeric side products for a typical run with 2-butyne-1,4-diol (2a) and
benzylamine (5a) (Scheme 2) in the broad molecular weight (Mw) range of 300–
5000 g/mol.
In addition, when 2a alone was subjected to standard catalytic conditions, full
substrate conversion was observed alongside the formation of a dark brown
unidentified precipitate and no volatiles were detected by GC measurement
(Scheme 3). Furthermore, GPC measurement confirmed the formation of
oligomeric side products. Thus, the most likely source of such competing side
reactions is the isomerization of substrate 2a to the corresponding ,β-unsaturated
aldehyde as shown in Scheme 4.
Scheme 4: Possible pathways for pyrrole formation.
Other reaction pathways such as the formation of secondary or tertiary amines
that may be a result of over-alkylation and imine reduction were not observed,
indicating the preference for intramolecular pyrrole formation. Also, the
corresponding pyrrolidine analogues were only sparingly observed when 2b was
used as substrate. More mechanistic and spectroscopic insights are required to
understand the sequence of reaction steps occurring. This may lead to
improvement of product yields. Future research should also address a broader
substrate scope, especially different substitution patterns on the pyrrole ring.
Conclusion
In conclusion, in this chapter, the first iron-catalyzed direct method for the catalytic
formation of pyrroles by coupling of unsaturated diols with primary amines has
been described. Various derivatives of anilines, and benzyl amines as well as other
aliphatic primary amines were successfully used in the construction of pyrrole
moieties, which are important scaffolds in medicinal chemistry. The desired
product yields range from high to moderate and future studies will address further
mechanistic details of this interesting noble metal free transformation. The
presented catalytic strategy is direct, straightforward, and allows for the use of a
wide range of amines. In addition, this new catalytic method relies on the use of

Pyrroles
83
inexpensive and abundant catalyst for the construction of scaffolds that are very
important in the pharmaceutical industry.
Experimental section
General methods
Chromatography: Merck silica gel type 9385 230-400 mesh or Merck Al2O3 90
active neutral, TLC: Merck silica gel 60, 0.25 mm or Al2O3 60 F254 neutral.
Components were visualized by UV, Ninhydrin or I2 staining. Progress of the
reactions was determined by GC-MS (GC: HP 6890, MS: HP 5973) with an HP012
column (Agilent Technologies, Palo Alto, CA). Mass spectra were recorded on an
AEI-MS-902 mass spectrometer (EI+) or a LTQ Orbitrap XL (ESI+). Conversions
were determined by GC-FID (GC: HP 6890) with an HP-5 column (Agilent
Technologies, Palo Alto, CA). GC-MS and GC-FID analysis method: 60 °C 5 min,
180 °C 5 min (10 °C/min), 260 °C 5 min (10 °C/min). 1H- and 13C NMR spectra
were recorded on a Varian AMX400 (400 and 100.59 MHz, respectively) using
CDCl3, CD3OD, or CD2Cl2 as solvent. Chemical shift values are reported in ppm with
the solvent resonance as the internal standard (CDCl3: 7.26 for 1H, 77.00 for 13C;
CD3OD: 3.31 for 1H, 49.00 for 13C; CD2Cl2: 5.32 for 1H, 53.84 for 13C). Data are
reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t =
triplet, q = quartet, br. = broad, m = multiplet), coupling constants (Hz), and
integration. All reactions were carried out under an Argon atmosphere using oven
(110oC) dried glassware and using standard Schlenk techniques. THF and toluene
were collected from a MBRAUN solvent purification system (MB SPS-800). Dioxane
(99.5%, extra dry), dichloroethane (DCE, 99.8%, extra dry), N,N-
dimethylformamide (DMF, 99.8%, extra dry) and acetonitrile (CH3CN, 99.9%,
extra dry) were purchased from Acros without further purification. Molecular sieves
4A were purchased from Acros, and heated in Schlenck under 180 °C in vacuo
overnight for activation before using. All other reagents were purchased from
Sigma or Acros in reagent or higher grade and were used without further
purification. Complex Cat 3 was synthesized according to literature procedures[24]
with slightly modification. The synthesis of Cat 3 was carried out as described in
Chapter 2.
Representative procedures
General procedure: An oven-dried 20 ml Schlenk tube, equipped with stirring
bar, was charged with amine (0.5 mmol, 1 equiv), alcohol (given amount), iron
complex Cat 3 (4 mol%), Me3NO (8 mol%) and Toluene (solvent, 2 ml). The solid
starting materials were added into the Schlenk tube under air, the Schlenk tube
was subsequently connected to an argon line and a vacuum-argon exchange was
performed three times. Liquid starting materials and solvent were charged under
an argon stream. The Schlenk tube was capped and the mixture was rapidly stirred
at room temperature for 1 min, then was placed into a pre-heated oil bath at the
appropriate temperature and stirred for a given time. The reaction mixture was
cooled down to room temperature and concentrated in vacuo. The residue was
purified by flash column chromatography to provide the pure amine product.

Chapter 4
84
Procedure of synthetizing N-(4-dimethyl-phenyl)-pyrrole from 4-
dimethylamino aniline and butyn-1,4-diol: An oven-dried 20 ml Schlenk tube,
equipped with stirring bar, was charged with 4-dimethylamino aniline (0.5 mmol,
0.068 g), butyn-1,4-diol (0.5 mmol, 0.086 g), iron complex Cat 3 (4 mol%, 8 mg)
and Me3NO (8 mol%, 3 mg) under air. The Schlenk tube was subsequently
connected to a vacuum/argon Schlenk line and a vacuum-backfill cycle was
performed three times. Toluene (solvent, 2 ml) were charged under an argon
stream. The Schlenk tube was sealed with a screw cap and the mixture was rapidly
stirred at room temperature for 1 min, then was placed into a pre-heated oil bath
at 130 °C and stirred for 18 h. The reaction mixture was cooled down to room
temperature and the crude mixture was filtered through celite, eluted with ethyl
acetate, and concentrated in vacuum. The residue was purified by flash column
chromatography (SiO2, pentane/EtOAc 100:0 to 80:20) to provide the pure
product N-(4-dimethyl-phenyl)-pyrrole (0.077 g, 83% isolated yield).

Pyrroles
85
Spectral data of isolated compounds
N-(4-dimethyl-phenyl)-pyrrole (3a): Synthesized according to General
procedure. 4-Dimethylamino aniline (0.068 g, 0.50 mmol) affords 3a
(0.077 g, 83% yield). Brown solid was obtained after column
chromatography (SiO2, pentane/EtOAc 100:0 to 80:20). 1H NMR (400 MHz,
CDCl3) δ 7.27 – 7.31 (m, 2H), 6.99 – 7.05 (m, 2H), 6.77 – 6.81 (m, 2H),
6.30 – 6.38 (m, 2H), 3.00 (s. 6H). 13C NMR (100 MHz, CDCl3) δ 148.86, 131.18,
122.12, 119.68, 113.04, 109.27, 40.78. The physical data were identical in all
respects to those previously reported[25].
N-(4-Methoxy-phenyl)-pyrrole (3b): Synthesized according to General
procedure. 4-Methoxy aniline (0.062 g, 0.50 mmol) affords 3b (0.070 g,
80% yield). Light yellow solid was obtained after column chromatography
(SiO2, Pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.29 –
7.37 (m, 2H), 6.99 – 7.06 (m, 2H), 6.91 – 6.99 (m, 2H), 6.30 – 6.38 (m,
2H), 3.84 (s. 3H). 13C NMR (100 MHz, CDCl3) δ 157.60, 134.45, 122.13, 119.64,
114.57, 109.79, 55.51. The physical data were identical in all respects to those
previously reported[26].
N-(4-Methyl-phenyl)-pyrrole (3c): Synthesized according to General
procedure. 4-Methyl aniline (0.054 g, 0.50 mmol) affords 3c (0.028 g, 36%
yield with the use of 2a; 0.047 g, 59% yield with the use of 2b). Light yellow
solid was obtained after column chromatography (SiO2, Pentane/EtOAc
100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.29 – 7.37 (m, 2H), 7.19 –
7.29 (m, 2H), 7.04 – 7.14 (m, 2H), 6.32 – 6.42 (m, 2H), 2.41 (s. 3H). 13C NMR
(100 MHz, CDCl3) δ 138.44, 135.30, 129.98, 120.48, 119.33, 110.02, 20.80. The
physical data were identical in all respects to those previously reported[27].
N-(4-Fluoro-phenyl)-pyrrole (3d): Synthesized according to General
procedure. 4-Fluoro aniline (0.056 g, 0.50 mmol) affords 3d (0.024 g, 30%
yield with the use of 2a; 0.038 g, 47% yields with use of 2b). Light yellow
oily liquid was obtained after column chromatography (SiO2, Pentane/EtOAc
100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.30 – 7.40 (m, 2H), 7.08 –
7.18 (m, 2H), 6.96 – 7.06 (m, 2H), 6.28 – 6.44 (m, 2H), 2.41 (s. 3H). 13C NMR
(100 MHz, CDCl3) δ 160.61 (d, J = 244.98 Hz), 137.15 (d, J = 2.86 Hz), 122.26
(d, J = 8.39 Hz), 119.60, 116.25 (d, J = 22.77 Hz), 110.42, . The physical data
were identical in all respects to those previously reported[28].
N-(4-Chloro-phenyl)-pyrrole (3e): Synthesized according to General
procedure. 4-Chloro aniline (0.064 g, 0.50 mmol) affords 3e (0.040 g, 45%
yield). Light yellow solid was obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.44 (m,
2H), 7.30 – 7.36 (m, 2H), 7.00 – 7.11 (m, 2H), 6.30 – 6.42 (m, 2H), 2.41
(s. 3H). 13C NMR (100 MHz, CDCl3) δ 139.28, 131.00, 129.58, 121.57, 119.23,
110.79. The physical data were identical in all respects to those previously
reported[29].

Chapter 4
86
N-(4-Bromo-phenyl)-pyrrole (3f): Synthesized according to General
procedure. 4-Bromo aniline (0.085 g, 0.50 mmol) affords 3f (0.048 g, 44%
yield). Light yellow solid was obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.50 – 7.60 (m,
2H), 7.22 – 7.35 (m, 2H), 7.00 – 7.12 (m, 2H), 6.30 – 6.45 (m, 2H), 2.41
(s. 3H). 13C NMR (100 MHz, CDCl3) δ 139.73, 132.54, 121.88, 119.15, 118.65,
110.86. The physical data were identical in all respects to those previously
reported[30].
N-(3,4-Methylenedioxy-phenyl)-pyrrole (3g): Synthesized according
to General procedure. 3,4-(Methylenedioxy)aniline (0.069 g, 0.50 mmol)
affords 3g (0.035 g, 37% yield). Light yellow solid was obtained after
column chromatography (SiO2, Pentane/EtOAc 90:10 to 70:30). 1H NMR
(400 MHz, CDCl3) δ 6.95 – 7.02 (m, 2H), 6.88 – 6.94 (m, 1H), 6.80 – 6.88
(m, 2H), 6.27 – 6.38 (m, 2H), 6.01 (s. 2H). 13C NMR (100 MHz, CDCl3) δ 148.30,
145.62, 135.68, 119.82, 114.01, 109.93, 108.42, 103.01, 101.57, 77.00. HRMS
(APCI+, m/z): calculated for C11H10NO2 [M+H]+: 188.07061; found: 188.07078.
N-(2-Methoxy-phenyl)-pyrrole: Synthesized according to General
procedure. 2-Methoxy aniline (0.062 g, 0.50 mmol) affords 3c (0.031
g, 36% yield with the use of 2b). Light yellow liquid was obtained after
column chromatography (SiO2, pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.20 – 7.38 (m, 2H), 6.95 – 7.10 (m, 4H), 6.25 – 6.40 (m,
2H), 3.85 (s. 3H). 13C NMR (100 MHz, CDCl3) δ 152.67, 130.21, 127.42, 125.72,
122.03, 120.85, 112.24, 108.72, 55.75. The physical data were identical in all
respects to those previously reported[31].
N-(2-Methyl-phenyl)-pyrrole: Synthesized according to General
procedure. 2-Methyl aniline (0.054 g, 0.50 mmol) affords 3c (0.020 g,
25% yield with the use of 2b). Light yellow liquid was obtained after
column chromatography (SiO2, pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.23 – 7.33 (m, 4H), 6.78 – 6.82 (m, 2H), 6.30 – 6.35 (m,
2H), 2.22 (s. 3H). 13C NMR (100 MHz, CDCl3) δ 140.55, 133.79, 131.01, 127.45,
126.58, 126.49, 122.01, 108.64, 17.85. The physical data were identical in all
respects to those previously reported[32].
N-(2-fluoro-phenyl)-pyrrole: Synthesized according to General
procedure. 2-fluoro aniline (0.056 g, 0.50 mmol) affords 3c (0.012 g,
15% yield with the use of 2b). Light yellow liquid was obtained after
column chromatography (SiO2, Pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.35 – 7.45 (m, 1H), 7.16 – 7.29 (m, 3H), 7.02 – 7.08 (m,
2H), 6.30 – 6.40 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 154.47 (d, J = 249.3 Hz),
129.0 (d, J = 10.5 Hz), 127.11 (d, J = 7.7 Hz), 124.93 (d, J = 1.5 Hz), 124.73 (d,
J = 3.8 Hz), 121.35 (d, J = 3.6 Hz), 117.03 (d, J = 20.6 Hz), 109.92. The physical
data were identical in all respects to those previously reported[33].

Pyrroles
87
N-benzyl-pyrrole (6a): Synthesized according to General procedure.
Benzylamine (0.054 g, 0.50 mmol) affords 6a (0.048 g, 61% yield).
Light yellow oily liquid was obtained after column chromatography (SiO2,
Pentane/EtOAc 95:5 to 90:10). 1H NMR (400 MHz, CDCl3) δ 7.27 – 7.38
(m, 3H), 7.10 – 7.17 (m, 2H), 6.66 – 6.76 (m, 2H), 6.16 – 6.26 (m, 2H), 5.09 (s.
2H). 13C NMR (100 MHz, CDCl3) δ 138.13, 128.67, 127.58, 126.94, 121.12, 108.45,
53.28. The physical data were identical in all respects to those previously
reported[34].
N-(4-Chloro-benzyl)-pyrrole (6b): Synthesized according to General
procedure. 4-Chloro benzylamine (0.044 g, 0.50 mmol) affords 6b
(0.055 g, 57% yield). Light yellow oily liquid obtained after column
chromatography (SiO2, Pentane/EtOAc 95:5 to 90:10). 1H NMR (400
MHz, CDCl3) δ 7.27 – 7.40 (m, 3H), 7.10 – 7.13 (m, 2H), 6.64 – 6.78
(m, 2H), 6.16 – 6.30 (m, 2H), 5.05 (s. 2H). 13C NMR (100 MHz, CDCl3) δ 136.65,
133.42, 128.81, 128.24, 121.02, 108.73, 52.57. The physical data were identical
in all respects to those previously reported[35].
N-(4-Fluoro-benzyl)-pyrrole (6c): Synthesized according to General
procedure. 4-Fluoro benzylamine (0.063 g, 0.50 mmol) affords 6c
(0.046 g, 52% yield). Light yellow oily liquid was obtained after column
chromatography (SiO2, Pentane/EtOAc 95:5 to 90:10). 1H NMR (400
MHz, CDCl3) δ 7.07 – 7.15 (m, 3H), 6.96 – 7.06 (m, 2H), 6.65 – 6.75
(m, 2H), 6.17 – 6.27 (m, 2H), 5.05 (s. 2H). 13C NMR (100 MHz, CDCl3) δ 162.22
(d, J = 245.87 Hz), 133.88 (d, J = 3.11 Hz), 128.65 (d, J = 8.11 Hz), 120.97,
115.55 (d, J = 21.74 Hz), 108.64, 52.58. The physical data were identical in all
respects to those previously reported[36].
N-(3-Trifluoromethyl-benzyl)-pyrrole (6d): Synthesized according
to General procedure. 3-Trifluoromethyl benzylamine (0.088 g, 0.50
mmol) affords 6d (0.062 g, 55% yield). Light yellow oily liquid obtained
after column chromatography (SiO2, Pentane/EtOAc 95:5 to 90:10). 1H
NMR (400 MHz, CDCl3) δ 7.52 – 7.62 (m, 1H), 7.43 – 7.49 (m, 1H),
7.38 – 7.43 (m, 1H), 7.23 – 7.29 (m, 1H), 6.65 – 6.75 (m, 2H), 6.20 – 6.30 (m,
2H), 5.14 (s. 2H). 13C NMR (100 MHz, CDCl3) δ 139.28, 131.09 (d, J = 22.29 Hz),
130.19 (d, J = 1.41 Hz), 129.30, 124.54 (quat, J = 3.84 Hz), 129.95 (d, J = 272.50
Hz), 123.64 (quat, J = 3.83 Hz), 121.12, 109.04, 52.81. HRMS (APCI+, m/z):
calculated for C12H11F3N [M+H]+: 226.08381; found: 226.08415.
N-(3-Fluoro-4-chloro-benzyl)-pyrrole (6e): Synthesized according
to General procedure. 3-Fluoro-4-chloro benzylamine (0.080 g, 0.50
mmol) affords 6e (0.068 g, 65% yield). Light yellow oily liquid was
obtained after column chromatography (SiO2, Pentane/EtOAc 95:5 to
90:10). 1H NMR (400 MHz, CDCl3) δ 7.13 – 7.21 (m, 1H), 7.05 – 7.13
(m, 1H), 6.93 – 7.02 (m, 1H), 6.63 – 6.73 (m, 2H), 6.18 – 6.28 (m, 2H), 5.03 (s.
2H). 13C NMR (100 MHz, CDCl3) δ 157.49 (d, J = 248.78 Hz), 135.25 (d, J = 3.85
Hz), 129.08, 126.59 (d, J = 7.30 Hz), 121.28 (d, J = 17.87 Hz), 120.96, 116.76

Chapter 4
88
(d, J = 21.31 Hz), 108.99, 52.10. HRMS (APCI+, m/z): calculated for C11H10ClFN
[M+H]+: 210.04803; found: 210.04824.
N-(4-Methyl-benzyl)-pyrrole (6f): Synthesized according to General
procedure. 4-Methyl benzylamine (0.061 g, 0.50 mmol) affords 6f (0.052
g, 60% yield). Light yellow oily liquid was obtained after column
chromatography (SiO2, Pentane/EtOAc 95:5 to 90:10). 1H NMR (400
MHz, CDCl3) δ 7.13 – 7.22 (m, 2H), 7.03 – 7.10 (m, 2H), 6.66 – 6.76
(m, 2H), 6.16 – 6.26 (m, 2H), 5.06 (s. 2H), 2.37 (s, 2H). 13C NMR (100 MHz, CDCl3)
δ 137.29, 135.06, 129.32, 127.02, 121.01, 108.34, 53.07, 21.04. The physical
data were identical in all respects to those previously reported[35].
N-Piperonyl-pyrrole (6g): Synthesized according to General
procedure. Piperonylamine (0.076 g, 0.50 mmol) affords 6g (0.056 g,
55% yield). Light yellow oily liquid was obtained after column
chromatography (SiO2, Pentane/EtOAc 90:10 to 60:40). 1H NMR (400
MHz, CDCl3) δ 6.74 – 6.79 (m, 1H), 6.66 – 6.72 (m, 2H), 6.63 – 6.66
(m, 1H), 6.60 – 6.63 (m, 1H), 6.16 – 6.22 (m, 2H), 5.94 (s. 2H), 4.97 (s, 2H). 13C
NMR (100 MHz, CDCl3) δ 148.00, 147.09, 131.88, 120.88, 120.48, 108.46, 108.23,
107.72, 101.07, 53.12. The physical data were identical in all respects to those
previously reported[35].
N-(3-Picolyl)-pyrrole (6h): Synthesized according to General
procedure. 3-Picolylamine (0.054 g, 0.50 mmol) affords 6h (0.060 g,
76% yield). Light yellow oily liquid was obtained after column
chromatography (SiO2, Pentane/EtOAc 80:20 to 40:60). 1H NMR (400
MHz, CDCl3) δ 8.40 – 8.60 (m, 2H), 7.32 – 7.40 (m, 1H), 7.20 -7.29 (m, 1H), 6.62
– 6.74 (m, 2H), 6.15 – 6.25 (m, 2H), 5.09 (s. 2H). 13C NMR (100 MHz, CDCl3) δ
149.05, 148.28, 134.66, 133.68, 123.68, 120.92, 109.02, 50.70. The physical
data were identical in all respects to those previously reported[35].
N-(2-Furfuryl)-pyrrole (6i): Synthesized according to General
procedure. Furfurylamine (0.049 g, 0.50 mmol) affords 6i (0.032 g, 43%
yield). Light yellow oily liquid was obtained after column chromatography
(SiO2, Pentane/EtOAc 80:20 to 40:60). 1H NMR (400 MHz, CDCl3) δ 7.32–
7.42 (m, 1H), 6.68 – 6.76 (m, 2H), 6.30 – 6.38 (m, 1H), 6.22 – 6.30 (m, 1H),
6.15 – 6.22 (m, 2H), 5.03 (s. 2H). 13C NMR (100 MHz, CDCl3) δ 150.74, 142.65,
120.67, 110.36, 108.50, 108.09, 46.05. The physical data were identical in all
respects to those previously reported[35].
N-(2-Phenylethyl)-pyrrole (6g): Synthesized according to General
procedure. 2-Phenyl ethylamine (0.061 g, 0.50 mmol) affords 6g (0.035
g, 41% yield). Light yellow oily liquid was obtained after column
chromatography (SiO2, Pentane/EtOAc 100:0 to 90:10). 1H NMR (400
MHz, CDCl3) δ 7.29– 7.39 (m, 2H), 7.26 – 7.29 (m, 1H), 7.10 – 7.18 (m, 2H),
6.58 – 6.68 (m, 2H), 6.12 – 6.22 (m, 2H), 4.14 (t, J = 7.65 Hz, 2H), 3.09 (t, J =
7.61 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 138.39, 128.62, 128.50, 126.55,

Pyrroles
89
120.42, 107.95, 51.12, 38.35. The physical data were identical in all respects to
those previously reported[37].
N-Dodecyl-pyrrole (6k): Synthesized according to General procedure.
Dodecylamine (0.093 g, 0.50 mmol) affords 6k (0.050 g, 42% yield).
Light yellow oily liquid was obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 90:10). 1H NMR (400 MHz, CDCl3) δ 6.62– 6.70 (m, 2H),
6.12 – 6.18 (m, 2H), 3.87 (t, J = 7.20 Hz, 2H), 1.70 – 1.83 (m, 2H), 1.18 – 1.38
(m, 18H), 0.90 (t, J = 6.63 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 120.41, 107.71,
49.62, 31.90, 31.57, 29.61, 29.60, 29.55, 29.48, 29.32, 29.20, 26.76, 22.67,
14.10. The physical data were identical in all respects to those previously
reported[38].
N-Cyclohexyl-pyrrole (6l): Synthesized according to General procedure.
Cyclohexylamine (0.050 g, 0.50 mmol) affords 6l (0.025 g, 33% yield).
Light yellow oily liquid was obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 90:10). 1H NMR (400 MHz, CDCl3) δ 6.70– 6.78
(m, 2H), 6.10 – 6.18 (m, 2H), 3.75 – 3.87 (m , 2H), 2.05 – 2.15 (m, 2H), 1.82 –
1.94 (m, 2H), 1.56 – 1.69 (m, 2H), 1.33 – 1.48 (m , 2H), 1.19 – 1.32 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 118.39, 107.31, 58.65, 34.66, 25.72, 25.49. The
physical data were identical in all respects to those previously reported[39].

Chapter 4
90
References
[1] V. Bhardwaj, D. Gumber, V. Abbot, S. Dhimana, P. Sharmaa, RSC Adv., 2015, 5,
15233–15266.
[2] F. Fischer, M. Matthisson, P. Herrling, Neurodegener. Dis., 2004, 1, 50–70.
[3] P. C. Waldmeier, M. Williams, P. A. Baumann, S. Bischoff, M. A. Sills, R. F. Neale,
Naunyn-Schmiedeberg’s Arch. Pharmacol., 1988, 337, 609–620.
[4] I. Van Wijngaarden, C. G. Kruse, R. Van Hes, J. A. M. Van der Heyden, M. T. M. Tulp,
J. Med. Chem., 1987, 30, 2099–2104.
[5] A. Hantzsch, Ber. Dtsch. Chem. Ges., 1890, 23, 1474–1476.
[6] L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635–1642.
[7] C. Paal, Ber. Dtsch. Chem. Ges., 1885, 18, 367–371.
[8] V. Estévez, M. Villacampa, J. C. Menendez, Chem. Soc. Rev., 2014, 43, 4633–4657.
[9] a) B. M. Trost, Science, 1991, 254, 1471; b) P. T. Anastas, J. C. Warner, Green
Chemistry: Theory and Practice; Oxford University Press, Oxford, England, 1998.
[10] a) I. Nakamura, Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198; b) N. T. Patil, Y.
Yamamoto, ARKIVOC, 2007, 121–141; c) X. Xin, D. Wang, X. Li, B. Wang, Angew.
Chem. Int. Ed., 2012, 51, 1693–1697; d) Y. Jiang, W. C. Chan, C. M. Park, J. Am.
Chem. Soc., 2012, 134, 4104–4107; e) M. P. Huestis, L. Chan, D. R. Stuart, K.
Fagnou, Angew. Chem. Int. Ed., 2011, 50, 1338–1341; f) S. Rakshit, F. W. Patureau,
F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585–9587; g) D. R. Stuart, P. Alsaben,
M. Kuhn, K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326–18339; h) E. Lourdusamy,
L. Yao, C. M. Park, Angew. Chem. Int. Ed., 2010, 49, 7963–7967.
[11] a) A. J. A. Watson, J. M. J. Williams, Science, 2010, 329, 635–636; b) G. Guillena,
D. J. Ramon, M. Yus, Chem. Rev., 2010, 110, 1611–1641; c) S. Bähn, S. Imm, L.
Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem, 2011, 3, 1853–1864; d)
G. E. Dobereiner, R. H. Crabtree, Chem. Rev., 2010, 110, 681–703; e) C.
Gunanathan, D. Milstein, Science, 2013, 341, 1229712; f) Q. Yang, Q. Wang, Z. Yu,
Chem. Soc. Rev., 2015, 44, 2305–2329; g) A. Quintard, J. Rodriguez,
ChemSusChem, 2016, 9, 28–30.
[12] a) M. Zhang, H. Neumann, M. Beller, Angew. Chem. Int. Ed., 2013, 52, 597–601; b)
M. Zhang, X. Fang, H. Neumann, M. Beller, J. Am. Chem. Soc., 2013, 135, 11384–
11388.
[13] S. Michlik, R. Kempe, Nat. Chem., 2013, 5, 140–144.
[14] D. Srimani, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed., 2013, 52, 4012–4015.
[15] N. D. Schley, G. E. Dobereiner, R. H. Crabtree, Organometallics, 2011, 30, 4174–
4179.
[16] Y. Tsuji, Y. Yokoyama, K.-T. Huh, Y. Watanabe, Bull. Chem. Soc. Jpn., 1987, 60, 3456–
3458.
[17] a) S. J. Pridmore, P. A. Slatford, A. Daniel, M. K. Whittlesey, J. M. J. Williams,
Tetrahedron Lett., 2007, 48, 5115–5120; b) S. J. Pridmore, P. A. Slatford, J. E. Taylor,
M. K. Whittlesey, J. M. J. Williams, Tetrahedron, 2009, 65, 8981–8986.
[18] T. Yan, B. L. Feringa, K. Barta, Nat. Commun., 2014, 5, 5602.
[19] H.-J. Knölker, E. Baum, H. Goesmann, R. Klauss, Angew. Chem. Int. Ed., 1999, 38,
2064–2066.
[20] a) A. Quintard, T. Constantieux, J. Rodriguez, Angew. Chem. Int. Ed., 2013, 52,
12883–12887; b) A. Quintard, J. Rodriguez, Angew. Chem. Int. Ed., 2014, 53, 4044–
4055; c) A. J. Rawlings, L. J. Diorazio, M. Wills, Org. Lett., 2015, 17, 1086–1089; d)
H.-J. Pan, T. W. Ng, Y. Zhao, Chem. Commun., 2015, 51, 11907–11910; e) S.
Elangovan, J.-B. Sortais, M. Beller, C. Darcel, Angew. Chem. Int. Ed., 2015, 54,
14483–14486.
[21] T. Yan, B. L. Feringa, K. Barta, ACS Catal., 2016, 6, 381–388.

Pyrroles
91
[22] C. P. Casey, H. Guan, J. Am. Chem. Soc., 2009, 131, 2499–2507.
[23] P. Y. Nikolov, V. A. Yaylayan, J. Agric. Food Chem., 2012, 60, 10155–10161.
[24] T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal., 2012, 354, 597-
601.
[25] J. C. Antilla, Baskin, M. Jeremy, T. E. Barder, S. L. Buchwald, J. Org. Chem., 2004,
69, 5578–5587.
[26] A. K. Verma, T. Kesharwani, J. Singh, V. Tandon, R. C. Larock, Angew. Chem. Int.
Ed., 2009, 48, 1138–1143.
[27] Z.-L. Xu, H.-X. Li, Z.-G. Ren, W.-Y. Du, W.-C. Xu, J.-P. Lang, Tetrahedron, 2011, 67,
5282–5288.
[28] T. Niwa, H. Ochiai, Y. Watanabe, T. Hosoya, J. Am. Chem. Soc., 2015, 137, 14313–
14318.
[29] H.-C. Ma, X.-Z. Jiang J. Org. Chem., 2007, 72, 8943-8946.
[30] W. Chen, J. Wang, Organometallics, 2013, 32, 1958–1963.
[31] G. Pai, A. P. Chattopadhyay, Tetrahedron Lett., 2014, 55, 941–944.
[32] W. Chen, J. Wang, Organometallics, 2013, 32, 1958−1963.
[33] P. Wang, F.-P. Ma, Z.-H. Zhang, J. Mol. Liq., 2014, 198, 259–262.
[34] G. A. Molander, D. W. Ryu, M. Hosseini-Sarvari, R. Devulapally, D. G. Seapy, J. Org.
Chem., 2013, 78, 6648–6656.
[35] I. Deb, D. J. Coiroa, D. Seidel, Chem. Commun., 2011, 47, 6473–6475.
[36] Z. Zou, Z. Deng, X. Yu, M. Zhang, S. Zhao, T. Luo, X. Yin, H. Xu, W. Wang, Sci. China
Chem., 2012, 55, 43–49.
[37] A. R. Katritzky, H. Lang, X. Lan, Tetrahedron, 1993, 49, 2829-2838.
[38] T. W. Brockmann, J. M. Tour, J. Am. Chem. Soc., 1995, 117, 4437–4447.
[39] M. Abid, S. M. Landge, B. Torok, Org. Prep. Proc. Int., 2006, 38, 495-500.

Chapter 4
92

93
Chapter 5
Direct N-alkylation of unprotected amino acids with alcohols
-Amino acids are the most abundant chiral amine sources in nature and basic
units of proteins. Their derivatives are widely used in catalysis, fine chemical
synthesis or as building blocks in functional materials. Therefore, developing
efficient ways for the derivatization of -amino acids is highly desired. N-alkylation
of -amino acids is one of the most common ways of derivatization. The traditional
pathways including reductive alkylation with aldehydes and nucleophilic
substitution of alkyl halides face a number of limitations such as the formation of
stoichiometric amount of side products and complex procedures for purification. In
this chapter, an unprecedented methodology for the direct N-alkylation of
unprotected -amino acids with alcohols is described. Notably, this methodology
allows for good retention of the optical purity in the final products. The described
method is direct, catalytic and highly selective, only producing water as by-product
and allows for an extremely simple purification process. The scope includes N-
alkylation of natural -amino acids and simple peptides with simple alcohols, fatty
alcohols and diols. Using fatty alcohols and amino acids as only reaction partners
result in the formation of mono-N-alkyl amino acids using a molecular iron catalyst.
Finally, a fully sustainable catalytic system for the production of renewable
surfactants is presented.
Part of this chapter will be issued as a patent:
N-alkylated amino acids and oligopeptides, uses thereof and methods for providing
them, P114699EP00.
Part of this chapter has been submitted for publication:
T. Yan, B. L. Feringa, K. Barta, submitted.
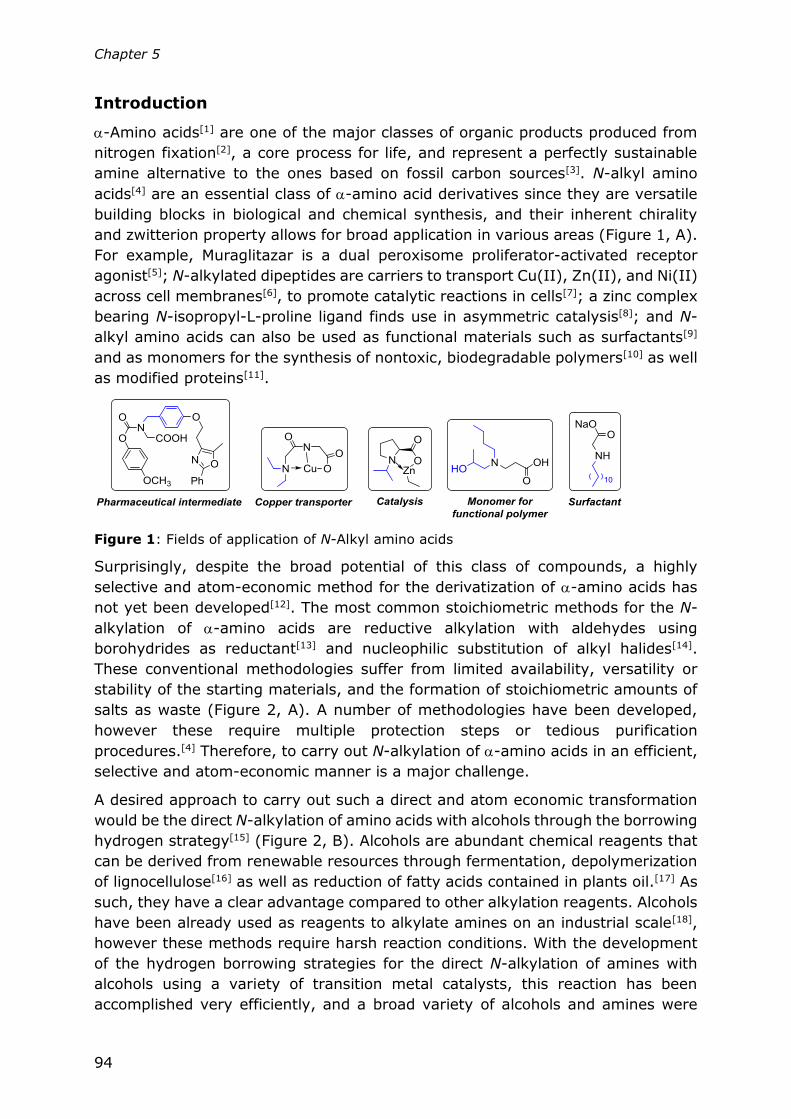
Chapter 5
94
Introduction
-Amino acids[1] are one of the major classes of organic products produced from
nitrogen fixation[2], a core process for life, and represent a perfectly sustainable
amine alternative to the ones based on fossil carbon sources[3]. N-alkyl amino
acids[4] are an essential class of -amino acid derivatives since they are versatile
building blocks in biological and chemical synthesis, and their inherent chirality
and zwitterion property allows for broad application in various areas (Figure 1, A).
For example, Muraglitazar is a dual peroxisome proliferator-activated receptor
agonist[5]; N-alkylated dipeptides are carriers to transport Cu(II), Zn(II), and Ni(II)
across cell membranes[6], to promote catalytic reactions in cells[7]; a zinc complex
bearing N-isopropyl-L-proline ligand finds use in asymmetric catalysis[8]; and N-
alkyl amino acids can also be used as functional materials such as surfactants[9]
and as monomers for the synthesis of nontoxic, biodegradable polymers[10] as well
as modified proteins[11].
Figure 1: Fields of application of N-Alkyl amino acids
Surprisingly, despite the broad potential of this class of compounds, a highly
selective and atom-economic method for the derivatization of -amino acids has
not yet been developed[12]. The most common stoichiometric methods for the N-
alkylation of -amino acids are reductive alkylation with aldehydes using
borohydrides as reductant[13] and nucleophilic substitution of alkyl halides[14].
These conventional methodologies suffer from limited availability, versatility or
stability of the starting materials, and the formation of stoichiometric amounts of
salts as waste (Figure 2, A). A number of methodologies have been developed,
however these require multiple protection steps or tedious purification
procedures.[4] Therefore, to carry out N-alkylation of -amino acids in an efficient,
selective and atom-economic manner is a major challenge.
A desired approach to carry out such a direct and atom economic transformation
would be the direct N-alkylation of amino acids with alcohols through the borrowing
hydrogen strategy[15] (Figure 2, B). Alcohols are abundant chemical reagents that
can be derived from renewable resources through fermentation, depolymerization
of lignocellulose[16] as well as reduction of fatty acids contained in plants oil.[17] As
such, they have a clear advantage compared to other alkylation reagents. Alcohols
have been already used as reagents to alkylate amines on an industrial scale[18],
however these methods require harsh reaction conditions. With the development
of the hydrogen borrowing strategies for the direct N-alkylation of amines with
alcohols using a variety of transition metal catalysts, this reaction has been
accomplished very efficiently, and a broad variety of alcohols and amines were

N-alkylation of unprotected amino acids
95
used.[15] However, the direct and selective N-alkylation of amino acids with
alcohols has not yet been demonstrated using this method, probably due to the
poor solubility of amino acids in organic solvents and their zwitterionic property,
which makes the substrates sensitive to changes of pH and basic or acidic reagents.
Moreover, racemization under the reaction conditions may be a serious problem,
since most hydrogen borrowing methods require the addition of base.[15]
Figure 2: A Comparison of different methodologies of N-alkylation of -amino acids; B
proposed mechanism of alkylation of amines with alcohols.
To the best of our knowledge, only 2 examples[19ab] of N-alkylation of free amino
acids with alcohols have been reported using heterogeneous catalysts and showing
limited substrate scope (Scheme 1, A). Notably, palladium catalyzed N-allylation
of unprotected amino acids with allylic alcohols was recently reported. A palladium
π-allyl species was involved in the catalytic cycle (Scheme 1, B)[19c]. In Chapter 2,
the first direct N-alkylation of amines with alcohols with a well-defined iron
complex was described[20] (Scheme 1, C). This methodology is efficient and base-
free, thus we envisioned that it would also allow for the direct N-alkylation of
unprotected -amino acids with alcohols, possibly without racemization.
Results and discussion
Alkylation with EtOH With the above mentioned goal in mind, the investigation
started using proline (1a) as the substrate, ethanol (2a) as the alkylation reagent
and Knölker’s complex[21a] Cat 3 as the catalyst (Table 1, entry 1). Quantitative
yield of N-ethyl proline (3aa) was obtained with 4 mol% of Cat 3, at 90 °C for 16
h (Table 1, entry 2). However, when leucine (1b) was used as the substrate, at
90 °C for 15 h, 15% conversion of 1b was observed. The dramatic drop of
reactivity of using 1b as the substrate probably due to the low solubility of 1b in
ethanol (Table 1, entry 3). Then the loading of Cat 3 was increased to 10 mol%,

Chapter 5
96
at 110 °C, 70% conversion of 1b was observed (Table 1, entry 4). When CF3CH2OH
was selected as the solvent, at 100 °C for 24 h, Cat 3 and its analogue Cat 3b
were employed and gave 30% and 70% conversion of 1b, respectively. (Table 1,
entry 5 and 6). Using Cat 3b gave higher conversion of leucine (2b) comparing to
using Cat 3, probably because Cat 3b was more efficient in generating the
catalytically active species.[22] Employing 4 mol% Cat 3b as the catalyst,
CF3CH2OH as the solvent, at 110 °C for 42 h, full conversion of 1b and 49% isolated
yield of diethyl leucine (3ba) was obtained.
Next, the ruthenium analogue of Cat 3, the Shvo catalyst[21b] (Cat 1) was also
investigated. Surprisingly, quantitative yields of both 3aa and 3ba from 1a and
1b were observed, respectively (Table 1, entry 8 and 9). Interestingly, the Shvo
catalyst (Cat 1) has mainly been used in hydrogen transfer reactions[21c] and only
very few examples have shown its ability to promote N-alkylation of amines[23].
The conditions described in entry 7 and 9 (Table 1) were taken as starting point
for further investigation.
One crucial requirement for a method designed for the modification of amino acids
is the retention of the chiral information contained in the starting material. Thus,
the enantiomeric excess (ee) of the N-alkylated products was investigated (Table
2). In this case, Cat 3b gave quantitative yield of 3aa with 94% retention of ee,
and 45% yield of 3ba with 80% ee retention (Table 2, entry 2 and 4). In the
meantime, Cat 1 gave both 3aa and 3ba in quantitative yields as described, with
retention of ee of 93% and 99%, respectively (Table 2, entry 1 and 3).
Phenylalanine (1d) was selected to react with 2a, catalyzed by both Cat 3b and
Cat 1 for further comparison. Cat 3b gave 3da in 55% yield with 72% retention
of ee, and Cat 1 gave 3da in quantitative yield with 97% retention of ee (Table 2,
Scheme 1: A N-Methylation of unprotected amino acids with MeOH catalyzed by
heterogeneous catalysts; B palladium catalyzed N-allylation of tryptophan with 2-
methyl-3-buten-2-ol; C base-free iron catalyzed alkylation of amines with alcohols.

N-alkylation of unprotected amino acids
97
entry 7 and 8). Based on these preliminary results, Cat 1 was chosen for the
further study of the reaction scope.
Then, other -amino acids including valine (1c), serine (1e) and alanine (1f) were
examined and quantitative yields of the corresponding diethylation products 3ca,
3ea and 3fa were obtained, with ee values of 99, 86 and 84%, respectively (Table
2, entry 6, 9 and 10). The partial racemization in the latter cases was probably
due to the activity of the Shvo catalyst in the dehydrogenation of amines to imines
and rehydrogenation of imines back to amines, which were described previously
by the groups of Beller[23] and Casey[24]. The lower steric hindrance of the
stereogenic center of 3ea and 3fa comparing to 3ca allows for easier racemization.
In order to improve the retention of stereochemistry in 3fa, the temperature was
lowered to 60 °C while prolonging the reaction time. Quantitative yield of 3fa was
obtained, however the ee of the final product was only improved slightly to 86%
(Table 2, entry 11).
The simplest -amino acid glycine (1g) was used and gave a quantitative yield of
N,N-diethyl-glycine (3ga) (Table 2, entry 12). On the other hand, lysine (3h) was
unreactive, whereas N6-acetyl-lysine (3i) gave 74% yield of the corresponding
N2,N2-diethylation product 3ia (Table 2, entry 13-16). The above results clearly
showed that all primary amino acids selectively gave N,N-diethylated products in
excellent yields when reacted with 2a under neat condition.
Table 2: Direct N-ethylation of unprotected amino acid (1) with ethanol (2a).
Table 1: Optimization of reaction conditions for direct N-ethylation of proline (1a)
or leucine (1b) with ethanol (2a).
Entry 1
[mmol]
Cat. [mol%] 2a
[ml]
T.
[h]
Temp.
[oC]
Conv. 1
[%]a
Sel. 3
[%]a
1 1a / 0.5 Cat 3 / 4, Me3NO / 8 2 18 110 >99 >99
2 1a / 0.5 Cat 3 / 4, Me3NO / 8 2 16 90 >99 >99 (99)
3 1b / 0.5 Cat 3 / 4, Me3NO / 8 4 15 90 15 n.d.
4 1b / 0.2 Cat 3 / 10, Me3NO / 20 5 18 110 70 n.d.
5b 1b / 0.5 Cat 3 / 4, Me3NO / 8 4 24 100 30 n.d.
6b 1b / 0.5 Cat 3b / 4 4 24 100 70 n.d.
7b 1b / 0.5 Cat 3b / 4 4 42 110 >95 (49)
8 1a / 0.5 Cat 1 / 0.5 5 18 90 >99 >99 (99)
9 1b / 0.2 Cat 1 / 1 5 18 90 >99 >99 (99)
General reaction conditions: general procedure, 0.2 or 0.5 mmol 1a (or 1b), 2–5 ml 2a,
Cat 3 + Me3NO, Cat 3b, or Cat 1 (given amount), neat, 14-24 h, 90-110 °C , isolated
yields in parenthesis. aConversion and selectivity are based on 1H NMR; b1 ml CF3CH2OH
was added.
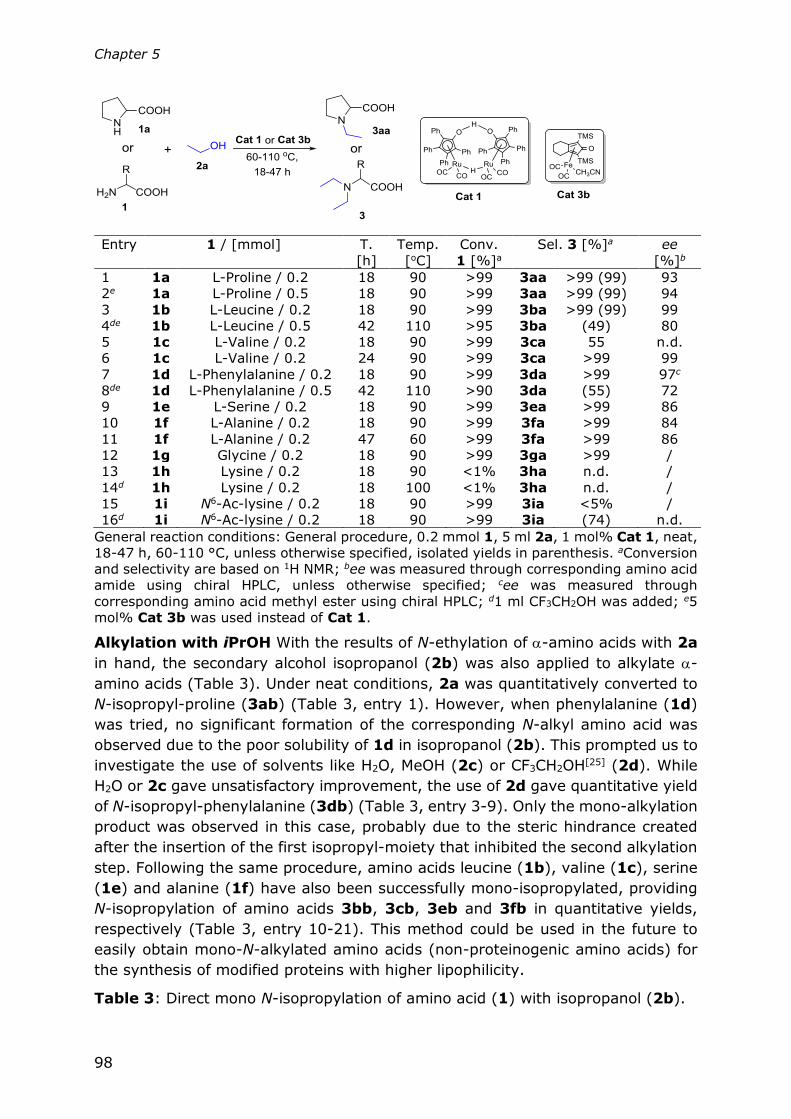
Chapter 5
98
Entry 1 / [mmol] T.
[h]
Temp.
[oC]
Conv.
1 [%]a
Sel. 3 [%]a ee
[%]b
1 1a L-Proline / 0.2 18 90 >99 3aa >99 (99) 93
2e 1a L-Proline / 0.5 18 90 >99 3aa >99 (99) 94
3 1b L-Leucine / 0.2 18 90 >99 3ba >99 (99) 99
4de 1b L-Leucine / 0.5 42 110 >95 3ba (49) 80
5 1c L-Valine / 0.2 18 90 >99 3ca 55 n.d.
6 1c L-Valine / 0.2 24 90 >99 3ca >99 99
7 1d L-Phenylalanine / 0.2 18 90 >99 3da >99 97c
8de 1d L-Phenylalanine / 0.5 42 110 >90 3da (55) 72
9 1e L-Serine / 0.2 18 90 >99 3ea >99 86
10 1f L-Alanine / 0.2 18 90 >99 3fa >99 84
11 1f L-Alanine / 0.2 47 60 >99 3fa >99 86
12 1g Glycine / 0.2 18 90 >99 3ga >99 /
13 1h Lysine / 0.2 18 90 <1% 3ha n.d. /
14d 1h Lysine / 0.2 18 100 <1% 3ha n.d. /
15 1i N6-Ac-lysine / 0.2 18 90 >99 3ia <5% /
16d 1i N6-Ac-lysine / 0.2 18 90 >99 3ia (74) n.d.
General reaction conditions: General procedure, 0.2 mmol 1, 5 ml 2a, 1 mol% Cat 1, neat,
18-47 h, 60-110 °C, unless otherwise specified, isolated yields in parenthesis. aConversion
and selectivity are based on 1H NMR; bee was measured through corresponding amino acid
amide using chiral HPLC, unless otherwise specified; cee was measured through
corresponding amino acid methyl ester using chiral HPLC; d1 ml CF3CH2OH was added; e5
mol% Cat 3b was used instead of Cat 1.
Alkylation with iPrOH With the results of N-ethylation of -amino acids with 2a
in hand, the secondary alcohol isopropanol (2b) was also applied to alkylate -
amino acids (Table 3). Under neat conditions, 2a was quantitatively converted to
N-isopropyl-proline (3ab) (Table 3, entry 1). However, when phenylalanine (1d)
was tried, no significant formation of the corresponding N-alkyl amino acid was
observed due to the poor solubility of 1d in isopropanol (2b). This prompted us to
investigate the use of solvents like H2O, MeOH (2c) or CF3CH2OH[25] (2d). While
H2O or 2c gave unsatisfactory improvement, the use of 2d gave quantitative yield
of N-isopropyl-phenylalanine (3db) (Table 3, entry 3-9). Only the mono-alkylation
product was observed in this case, probably due to the steric hindrance created
after the insertion of the first isopropyl-moiety that inhibited the second alkylation
step. Following the same procedure, amino acids leucine (1b), valine (1c), serine
(1e) and alanine (1f) have also been successfully mono-isopropylated, providing
N-isopropylation of amino acids 3bb, 3cb, 3eb and 3fb in quantitative yields,
respectively (Table 3, entry 10-21). This method could be used in the future to
easily obtain mono-N-alkylated amino acids (non-proteinogenic amino acids) for
the synthesis of modified proteins with higher lipophilicity.
Table 3: Direct mono N-isopropylation of amino acid (1) with isopropanol (2b).
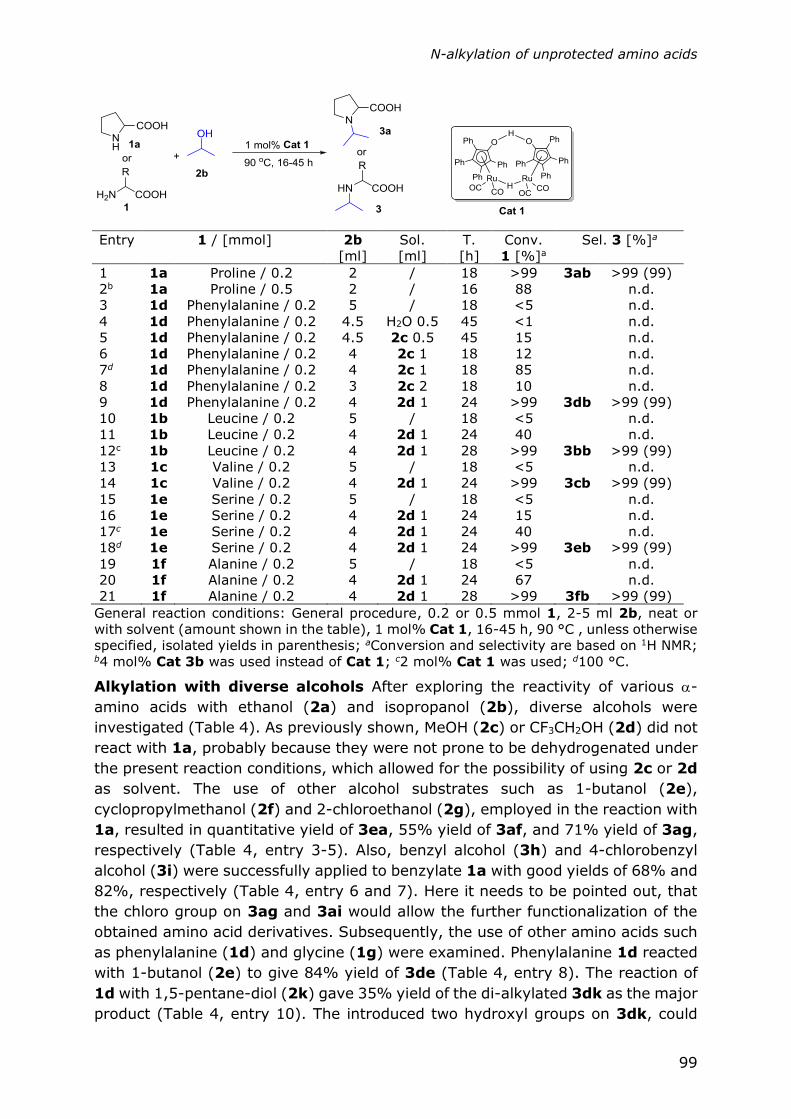
N-alkylation of unprotected amino acids
99
Entry 1 / [mmol] 2b
[ml]
Sol.
[ml]
T.
[h]
Conv.
1 [%]a
Sel. 3 [%]a
1 1a Proline / 0.2 2 / 18 >99 3ab >99 (99)
2b 1a Proline / 0.5 2 / 16 88 n.d.
3 1d Phenylalanine / 0.2 5 / 18 <5 n.d.
4 1d Phenylalanine / 0.2 4.5 H2O 0.5 45 <1 n.d.
5 1d Phenylalanine / 0.2 4.5 2c 0.5 45 15 n.d.
6 1d Phenylalanine / 0.2 4 2c 1 18 12 n.d.
7d 1d Phenylalanine / 0.2 4 2c 1 18 85 n.d.
8 1d Phenylalanine / 0.2 3 2c 2 18 10 n.d.
9 1d Phenylalanine / 0.2 4 2d 1 24 >99 3db >99 (99)
10 1b Leucine / 0.2 5 / 18 <5 n.d.
11 1b Leucine / 0.2 4 2d 1 24 40 n.d.
12c 1b Leucine / 0.2 4 2d 1 28 >99 3bb >99 (99)
13 1c Valine / 0.2 5 / 18 <5 n.d.
14 1c Valine / 0.2 4 2d 1 24 >99 3cb >99 (99)
15 1e Serine / 0.2 5 / 18 <5 n.d.
16 1e Serine / 0.2 4 2d 1 24 15 n.d.
17c 1e Serine / 0.2 4 2d 1 24 40 n.d.
18d 1e Serine / 0.2 4 2d 1 24 >99 3eb >99 (99)
19 1f Alanine / 0.2 5 / 18 <5 n.d.
20 1f Alanine / 0.2 4 2d 1 24 67 n.d.
21 1f Alanine / 0.2 4 2d 1 28 >99 3fb >99 (99)
General reaction conditions: General procedure, 0.2 or 0.5 mmol 1, 2-5 ml 2b, neat or
with solvent (amount shown in the table), 1 mol% Cat 1, 16-45 h, 90 °C , unless otherwise
specified, isolated yields in parenthesis; aConversion and selectivity are based on 1H NMR; b4 mol% Cat 3b was used instead of Cat 1; c2 mol% Cat 1 was used; d100 °C.
Alkylation with diverse alcohols After exploring the reactivity of various -
amino acids with ethanol (2a) and isopropanol (2b), diverse alcohols were
investigated (Table 4). As previously shown, MeOH (2c) or CF3CH2OH (2d) did not
react with 1a, probably because they were not prone to be dehydrogenated under
the present reaction conditions, which allowed for the possibility of using 2c or 2d
as solvent. The use of other alcohol substrates such as 1-butanol (2e),
cyclopropylmethanol (2f) and 2-chloroethanol (2g), employed in the reaction with
1a, resulted in quantitative yield of 3ea, 55% yield of 3af, and 71% yield of 3ag,
respectively (Table 4, entry 3-5). Also, benzyl alcohol (3h) and 4-chlorobenzyl
alcohol (3i) were successfully applied to benzylate 1a with good yields of 68% and
82%, respectively (Table 4, entry 6 and 7). Here it needs to be pointed out, that
the chloro group on 3ag and 3ai would allow the further functionalization of the
obtained amino acid derivatives. Subsequently, the use of other amino acids such
as phenylalanine (1d) and glycine (1g) were examined. Phenylalanine 1d reacted
with 1-butanol (2e) to give 84% yield of 3de (Table 4, entry 8). The reaction of
1d with 1,5-pentane-diol (2k) gave 35% yield of the di-alkylated 3dk as the major
product (Table 4, entry 10). The introduced two hydroxyl groups on 3dk, could
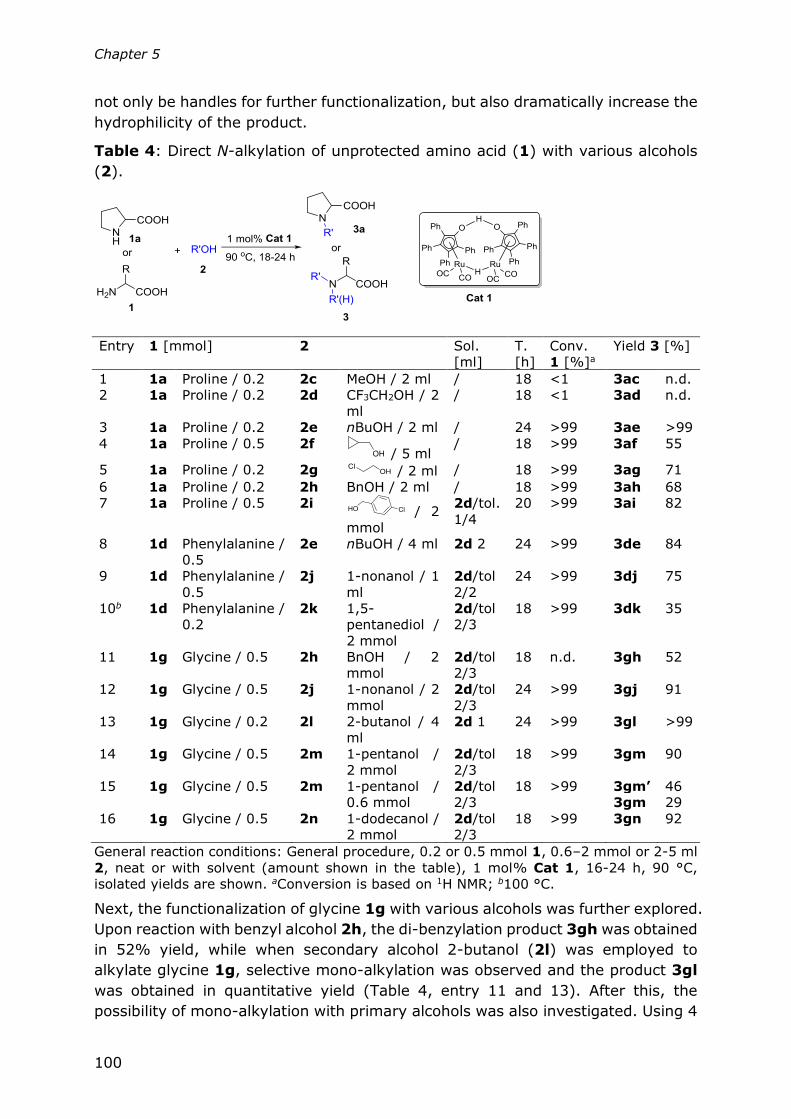
Chapter 5
100
not only be handles for further functionalization, but also dramatically increase the
hydrophilicity of the product.
Table 4: Direct N-alkylation of unprotected amino acid (1) with various alcohols
(2).
Entry 1 [mmol] 2 Sol.
[ml]
T.
[h]
Conv.
1 [%]a
Yield 3 [%]
1 1a Proline / 0.2 2c MeOH / 2 ml / 18 <1 3ac n.d.
2 1a Proline / 0.2 2d CF3CH2OH / 2
ml
/ 18 <1 3ad n.d.
3 1a Proline / 0.2 2e nBuOH / 2 ml / 24 >99 3ae >99
4 1a Proline / 0.5 2f / 5 ml
/ 18 >99 3af 55
5 1a Proline / 0.2 2g / 2 ml / 18 >99 3ag 71
6 1a Proline / 0.2 2h BnOH / 2 ml / 18 >99 3ah 68
7 1a Proline / 0.5 2i / 2
mmol
2d/tol.
1/4
20 >99 3ai 82
8 1d Phenylalanine /
0.5
2e nBuOH / 4 ml 2d 2 24 >99 3de 84
9 1d Phenylalanine /
0.5
2j 1-nonanol / 1
ml
2d/tol
2/2
24 >99 3dj 75
10b 1d Phenylalanine /
0.2
2k 1,5-
pentanediol /
2 mmol
2d/tol
2/3
18 >99 3dk 35
11 1g Glycine / 0.5 2h BnOH / 2
mmol
2d/tol
2/3
18 n.d. 3gh 52
12 1g Glycine / 0.5 2j 1-nonanol / 2
mmol
2d/tol
2/3
24 >99 3gj 91
13 1g Glycine / 0.2 2l 2-butanol / 4
ml
2d 1 24 >99 3gl >99
14 1g Glycine / 0.5 2m 1-pentanol /
2 mmol
2d/tol
2/3
18 >99 3gm 90
15 1g Glycine / 0.5 2m 1-pentanol /
0.6 mmol
2d/tol
2/3
18 >99 3gm’ 46
3gm 29
16 1g Glycine / 0.5 2n 1-dodecanol /
2 mmol
2d/tol
2/3
18 >99 3gn 92
General reaction conditions: General procedure, 0.2 or 0.5 mmol 1, 0.6–2 mmol or 2-5 ml
2, neat or with solvent (amount shown in the table), 1 mol% Cat 1, 16-24 h, 90 °C,
isolated yields are shown. aConversion is based on 1H NMR; b100 °C.
Next, the functionalization of glycine 1g with various alcohols was further explored.
Upon reaction with benzyl alcohol 2h, the di-benzylation product 3gh was obtained
in 52% yield, while when secondary alcohol 2-butanol (2l) was employed to
alkylate glycine 1g, selective mono-alkylation was observed and the product 3gl
was obtained in quantitative yield (Table 4, entry 11 and 13). After this, the
possibility of mono-alkylation with primary alcohols was also investigated. Using 4

N-alkylation of unprotected amino acids
101
eq. of 1-pentanol (1m), glycine 1g was successfully di-alkylated giving 90% of
3gm. When the amount of 1m was decreased to 1.2 eq., 46% mono-alkylated
product 3gm’ and 29% of the di-alkylated product 3gm were obtained (Table 4,
entry 14 and 15).
Scheme 2: N-alkylation of amino acids with various alcohols.
General reaction conditions: General procedure, 0.2 or 0.5 mmol 1, 1-5 ml or 0.6–2 mmol
2, 1 mol% Cat 1, neat when using ethanol (2a), 1 ml CF3CH2OH (2d) was added when
using iPrOH (2b), 18-28 h, 90 °C, isolated yields are shown, ee was measured through
corresponding amino acid amide using chiral HPLC, unless otherwise specified. #neat;
^CF3CH2OH was used as solvent; aee was measured through corresponding amino acid
methyl ester using chiral HPLC; b2 mol% Cat 1 was used; c100 °C. For details see Table
2-4.
The isolated yields of N-alkylated amino acids and their retention of ee under
optimized conditions are shown in Scheme 2.
Alkylation of peptides Encouraged by the promising results regarding the
functionalization of amino acids, we attempted the extension of this novel method
to simple free peptides, which possess similar physical properties and functional

Chapter 5
102
groups as the corresponding amino acids (Table 5). First, the simple dipeptide
glycylalanine (4a) was chosen to react with 2a and gratifyingly, the corresponding
di-ethylated product 5aa was obtained quantitatively (Table 5, entry 1). Based on
this protocol, the hydrophobicity or hydrophylicity of peptides can be modulated
by introducing either longer chain alkyl groups or more polar moieties bearing
hydroxyl groups. Indeed, when 1-dodecanol (2n) was reacted with dipeptide 4a,
the corresponding dialkylated product 5an was obtained in 82% yield. This type
of lipophilic dipeptide has been used for transporting metal ions across
membranes[6] (Table 5, entry 2). On the other hand, the reaction of 1,5-pentane-
diol 2k with 4a, afforded 36% 5ak, bearing additional hydroxyl groups (Table 5,
entry 3).
Table 5: Alkylation of N-terminus of oligo-peptides with alcohols.
Entry 4 [mmol] 2 Sol.
[ml]
T.
[h]
Conv.
4 [%]a
Yield 5 [%]
1 4a
/ 0.2
2a ethanol / 5
ml
/ 24 >95 5aa >95
2 4a
/ 0.5
2n dodecanol
/ 2 mmol
2d/tol
2/3
18 n.d. 5an 82
3 4a
/ 0.5
2k 1,5-
pentanediol
/ 2 mmol
2d/tol
2/3
24 n.d. 5ak 36
4 4b
/ 0.2
2a ethanol / 5
ml
/ 24 n.d. 5ba 50
5 4b
/ 0.5
2a ethanol / 5
ml
2d 2 18 n.d. 5ba 67
General reaction conditions: General procedure, 0.2 or 0.5 mmol 4, 2 mmol or 5 ml 2,
neat or with solvent (amount shown in the table), 1 mol% Cat 1, 18 or 24 h, 90 °C, isolated
yields are shown. aConversion is based on 1H NMR.
Following the successful and diverse functionalization of a dipeptide, a tri-peptide
leucylglycylglycine (4b) was tested in the reaction with 2a, which lead to the
formation of the corresponding di-ethylated product 5ba with 67% isolated yield
(Table 5, entry 4 and 5). The above reactions represent the first example of the
selective di-alkylation of peptide substrates on their N-terminus using simple
alcohols, allowing for good product yields and easy purification[26]. This
methodology can potentially be used for protein N-terminus modification, thereby

N-alkylation of unprotected amino acids
103
affecting protein activation, conversion and degradation, allowing further
diversifying of biological functions[27].
Sustainable surfactants Amino acids attract increasing interest in recent years
as starting material for synthetizing bio-based surfactants, among which N-
alkylated amino acids derived surfactants are not well studied because they are
relatively difficult to be synthetized[9]. Envisioning the possibility of easily
synthetizing long-chain N-alkylated amino acids with our methodology, 1-nonanol
(2j) was selected for the N-alkylation of phenylalanine 1d and glycine 1g. Di-
alkylated compounds 3dj and 3gj were selectively obtained with yields of 75%
and 91%, respectively (Scheme 2, Table 4, entry 9 and 12). The reaction also
readily proceeded with 1-dodecanol (2n) and glycine 1g as substrate, obtaining
the corresponding product 3gn in 92% yield (Table 4, entry 16). For all the cases,
selective dialkylated products were obtained.
Realizing that chirality is not an essential property requirement for a surfactant,
here the iron based Cat 3b was employed for the direct synthesis of a surfactant
from natural amino acids and fatty alcohols (Scheme 3). Glycine (1g) and 1-
dodecanol (2n) were reacted under 110 °C for 24 h with 5 mol% Cat 3b.
Surprisingly, 54% mono-N-dodecylglycine (3gn’) and 8% N,N-didodecylglycine
(3gn) were isolated. The use of Cat 3b leads to the preferential formation of
mono-N-alkyl amino acids, which have already shown surfactant properties[9].
After adding KOH and H2O to 3gn’, a rich foam formation was clearly seen
(Scheme 3), indicating its amphiphilic property. Subsequently, various fatty
alcohols that can be potentially derived from biomass, including 1-nonanol (2j),
1-decanol (2o), 1-tetradecanol (2p), 1-hexadecanol (2q) and 1-octadecanol (2n)
were reacted with 1g, and gave 32–69% isolated yields of the corresponding
mono-N-alkyl glycine derivatives 3gj', 3go', 3gp', 3gq' and 3gr' (Scheme 3).
Alanine (1f) and proline (1a) were reacted with 1-dodecanol (2n) and 1-nonanol
(2j), with 5 mol% Cat 3b, 49% of 3fn’ and 52% of 3aj were isolated, respectively
(Scheme 3). Long-chain alcohols can be produced from natural fats and oils[23].
This opens possibilities for the fully sustainable production of long-chain N-alkyl
amino acid based surfactants entirely derived from biomass, with non-precious
iron based catalyst[28].

Chapter 5
104
Scheme 3: Iron catalyzed N-alkylation of amino acids with fatty alcohols.
General reaction conditions: General procedure, 0.5 mmol 1, 1 ml 2, 3 ml CF3CH2OH, 5
mol% Cat 3b, 24 h, 110 °C, isolated yields are shown. aThe products was transformed to
corresponding methyl ester before isolation, 2 mmol 2; bneat.
Conclusion
In conclusion, the first direct N-alkylation of free -amino acids and simple
peptides with a variety of alcohols using 0.5-1 mol% of a homogeneous Ru catalyst
was demonstrated. The presented atom-economic transformations only result in
water as byproduct, thereby significantly simplifying the purification procedure.
The reaction is highly selective, and most products were obtained in quantitative
yield. Reaction temperature as low as 60 °C can be used with several substrates.
Particularly, the use of long chain alcohols and amino acids as only reaction
partners to obtain mono-N-alkyl amino acids, especially with a molecular iron
catalyst was established, which holds great potential for the fully sustainable
production of completely bio-based surfactants.
This work is not only a significant addition to sustainable homogeneous catalysis
related to the atom economic modification of challenging substrates such as amino
acids, but will likely open new possibilities for material science for the production
of surfactant as well as for the easy and selective chemical modification of proteins
or peptides in biochemistry. Future research will focus on exploring the potential
of this intriguing reaction under mild conditions as well as in aqueous solutions.
Experimental section
General methods
Chromatography: Merck silica gel type 9385 230-400 mesh or Merck Al2O3 90
active neutral, TLC: Merck silica gel 60, 0.25 mm. Components were visualized by

N-alkylation of unprotected amino acids
105
UV, Ninhydrin or I2 staining. Progress of the reactions was determined by NMR.
Mass spectra were recorded on an AEI-MS-902 mass spectrometer (EI+) or a LTQ
Orbitrap XL (ESI+). 1H- and 13C NMR spectra were recorded on a Varian AMX400
(400 and 100.59 MHz, respectively) using CDCl3, CD3OD, D2O or DMSO-d6 as
solvent. Chemical shift values are reported in ppm with the solvent resonance as
the internal standard (CDCl3: 7.26 for 1H, 77.00 for 13C; CD3OD: 3.31 for 1H, 49.00
for 13C; D2O: 4.79 for 1H; DMSO-d6: 2.50 for 1H, 39.52 for 13C). Data are reported
as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t = triplet, q =
quartet, br. = broad, m = multiplet), coupling constants (Hz), and integration. All
reactions were carried out under an Argon atmosphere using oven (110 °C) dried
glassware and using standard Schlenk techniques. Toluene were collected from a
MBRAUN solvent purification system (MB SPS-800). CF3CH2OH (>99.0%) was
purchased from TCI without further purification. The synthesis of Cat 3 and Cat
3b was carried out as described in Chapter 2. Cat 1 was purchased from Strem.
All other reagents were purchased from Sigma, TCI or Acros in reagent or higher
grade and were used without further purification.
Representative procedures
General procedure: An oven-dried 20 ml Schlenk tube, equipped with stirring
bar, was charged with amino acid (or peptide, given amount), Cat 1 or Cat 3
(given amount) and alcohol (given amount), solvent (or neat). Amino acid (or
peptide) and catalyst were added into the Schlenk tube under air, the Schlenk tube
was subsequently connected to an argon line and a vacuum-backfill cycle was
performed three times. Alcohol and solvent was charged under an argon stream.
The Schlenk tube was sealed with screw cap. The mixture was rapidly stirred at
room temperature for 1 min, then was placed into a pre-heated oil bath at the
appropriate temperature and stirred for a given time. The reaction mixture was
cooled down to room temperature and concentrated in vacuum. The residue was
characterized by 1H NMR spectroscopy to determine conversion. Further
purification was conducted through flash column chromatography or crystallization
to provide the pure N-alkyl amino acid (or peptide) product.
Esterification procedure (for preparation of methyl esters of long-chain N-alkyl
amino acids 3gp’, 3gq’ and 3gr’): General procedure was performed and
continued until the stage when the reaction mixture was cooled down to room
temperature. Subsequently, 3 ml benzene was added, and TMSCHN2 (2M in
toluene) was added under stirring. The progress of the reaction can be monitored
by TLC (SiO2, mono-N-alkyl amino acid, Rf = 0.3 in ethylacetate/MeOH = 1/1;
methyl mono-n-alkyl amino acid ester, Rf = 0.3 in Et2O). Then the corresponding
methyl ester was purified by flash chromatography (SiO2, tol/Et2O 50/50 – 0/100).
Procedure of ethylation of proline (1a) with ethanol (2a): An oven-dried 20
ml Schlenk tube, equipped with stirring bar, was charged with proline (0.5 mmol,
58 mg) and Cat 1 (0.0025 mmol, 2.7 mg) under air. The Schlenk tube was
subsequently connected to a vacuum/argon Schlenk line and a vacuum-backfill
cycle was performed three times. Then 5 ml ethanol was charged under an argon
stream. The Schlenk tube was sealed with a screw cap. The mixture was rapidly

Chapter 5
106
stirred at room temperature for 1 min, then was placed into a pre-heated oil bath
at 90 °C and stirred for 18 h. The reaction mixture was cooled down to room
temperature, concentrated and dried in high vacuum for 2 h. The residue was
weighed (72 mg) and characterized by 1H- and 13C NMR spectrospcopy. The NMR
spectrums of the residue show pure ethyl proline (3aa) without detectable
impurity and this indicates that the desired product was obtained in quantitative
yield. Ethyl proline 3aa was transformed to the corresponding amino acid amide
6aa for ee determination (93%) measured by chiral HPLC.
Procedure of iron catalyzed mono-alkylation of glycine (1g) with 1-
decanol (2o): An oven-dried 20 ml Schlenk tube, equipped with stirring bar, was
charged with glycine (0.5 mmol, 38 mg) and Cat 3b (0.025 mmol, 10 mg) under
air. The Schlenk tube was subsequently connected to a vacuum/argon Schlenk line
and a vacuum-backfill cycle was performed three times. Then 1 ml 1-decanol, 3
ml CF3CH2OH were charged under an argon stream. The Schlenk tube was sealed
with a screwed cap. The mixture was rapidly stirred at room temperature for 1
min, then was placed into a pre-heated oil bath at 110 °C and stirred for 24 h. The
reaction mixture was cooled down to room temperature, concentrated in vacuum.
The residue was purified by flash chromatography (SiO2, EtOAc/MeOH 70:30 to
50:50) to provide mono-decyl-glycine (3go’) (74 mg, 69% isolated yield).

N-alkylation of unprotected amino acids
107
Spectral data of isolated compounds
N-ethyl-proline (3aa): Synthesized according to General
procedure. Quantitative yield of 3aa was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400 MHz,
D2O) δ 3.83 – 3.96 (m, 1H), 3.64 – 3.78 (m, 1H), 3.15 – 3.33 (m,
2H), 3.05 – 3.15 (m, 1H), 2.35 – 2.52 (m, 1H), 1.99 – 2.16 (m, 2H), 1.85 – 1.99
(m, 1H), 1.27 (t, J = 7.28 Hz, 3H). 13C NMR (100 MHz, MeOD) δ 173.46, 70.13,
55.45, 51.51, 30.32, 24.34, 11.31. HRMS (APCI+, m/z): calculated for C7H14NO2
[M+H]+: 144.10191; found: 144.10181. The physical data are identical to those
previously reported.[19a] The ee (93% when using Cat 1, 94% when using Cat 3b)
were measured through corresponding amino acid amide 6aa using chiral HPLC,
for details see Determination of enantiomeric excesses retention.
N,N-diethyl-leucine (3ba): Synthesized according to General
procedure. Quantitative yield of 3ba was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400
MHz, D2O) δ 3.58 – 3.67 (m, 1H), 3.08 – 3.30 (m, 4H), 1.67 –
1.78 (m, 1H), 1.51 – 1.67 (m, 2H), 1.26 (t, J = 5.24 Hz, 6H), 0.83
– 1.00 (m, 6H). 13C NMR (100 MHz, D2O) δ 176.34, 68.56, 48.14 (br.s), 38.83,
27.77, 25.52, 23.04, 11.13 (br.s). HRMS (APCI+, m/z): calculated for C10H20NO2
[M-H]-: 186.14886; found: 186.15012. The ee (99% when using Cat 1, 80% when
using Cat 3b) were measured through corresponding amino acid amide 6ba using
chiral HPLC, for details see Determination of enantiomeric excesses retention.
N,N-diethyl-valine (3ca): Synthesized according to General
procedure. Quantitative yield of 3ca was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400
MHz, D2O) δ 3.45 – 3.52 (m, 1H) 3.05 – 3.35 (m, 4H), 2.22 –
2.40 (m, 1H), 1.15 – 1.40 (m, 6H), 0.99 – 1.10 (m, 3H), 0.86 – 0.99 (m, 3H) 13C
NMR (100 MHz, D2O) δ 174.31, 74.85, 49.85, 45.30, 28.02, 22.22, 18.81, 11.73,
9.62. HRMS (APCI+, m/z): calculated for C9H20NO2 [M+H]+: 174.14886; found:
174.14879. The physical data are identical to those previously reported.[29] The ee
(99%) was measured through corresponding amino acid amide 6ca using chiral
HPLC, for details see Determination of enantiomeric excesses retention.
N,N-diethyl-phenylalanine (3da): Synthesized according to
General procedure. Quantitative yield of 3da was obtained after
removing the volatiles of reaction residue by high vacuum. 1H
NMR (400 MHz, D2O) δ 7.12 – 7.43 (m, 5H), 3.80 – 3.93 (m,
1H), 3.08 – 3.40 (m, 4H), 2.98 – 3.31 (m, 2H), 1.17 – 1.33 (m, 6H). 13C NMR
(100 MHz, D2O) δ 172.37, 135.44, 129.06, 128.80, 127.28, 67.81, 45.70 (br.s),
33.44, 8.42 (br.s). HRMS (APCI+, m/z): calculated for C13H18NO2 [M-H]-:
220.13321; found: 220.13433. The ee (97% when using Cat 1, 72% when using
Cat 3b) were measured through corresponding amino acid amide 6da using chiral
HPLC, for details see Determination of enantiomeric excesses retention.

Chapter 5
108
N,N-diethyl-serine (3ea): Synthesized according to General
procedure. Quantitative yield of 3ea was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400
MHz, D2O) δ 3.98 – 4.18 (m, 2H), 3.79 – 3.91 (m, 1H), 3.37 –
3.52 (m, 1H), 3.18 – 3.37 (m, 3H), 1.18 – 1.40 (m, 6H). 13C NMR
(100 MHz, D2O) δ 173.91, 69.38, 60.55, 50.29, 47.45, 11.81, 10.96. HRMS (APCI+,
m/z): calculated for C7H14NO3 [M-H]-: 160.09682; found: 160.09811. The ee was
measured through corresponding amino acid amide 6ea using chiral HPLC, see
Determination of enantiomeric excesses retention.
N,N-diethyl-alanine (3fa): Synthesized according to General
procedure. Quantitative yield of 3fa was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400
MHz, D2O) δ 3.77 – 3.92 (m, 1H), 3.19 – 3.39 (m, 2H), 3.00 –
3.20 (m, 2H), 1.38 – 1.52 (m, 3H), 1.17 – 1.37 (m, 6H). 13C NMR (100 MHz, D2O)
δ 174.31, 61.46, 47.04, 44.97, 11.47, 9.29, 8.32. HRMS (APCI+, m/z): calculated
for C7H16NO2 [M+H]+: 146.11756; found: 146.11746. The physical data are
identical to those previously reported.[30] The ee (84% at 90 °C, 86% at 60 °C)
was measured through corresponding amino acid amide 6fa using chiral HPLC, see
Determination of enantiomeric excesses retention.
N,N-diethyl-glycine (3ga): Synthesized according to General
procedure. Quantitative yield of 3ga was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400
MHz, D2O) δ 3.66 (s, 2H). 3.22 (q. J = 7.31 Hz, 4H), 1.26 (t. J = 7.32 Hz, 6H). 13C
NMR (100 MHz, D2O) δ 173.50, 57.36, 52.14, 52.11, 11.13. HRMS (APCI+, m/z):
calculated for C6H14NO2 [M+H]+: 132.10191; found: 132.10180. The physical data
are identical to those previously reported.[31]
N6-acetyl-N2,N2-di-ethyl-lysine (3ha): Synthesized
according to General procedure. N6-acetyl-lysine (0.094 g,
0.50 mmol) affords 3ha (0.090 g, 75% yield). White solid
was obtained after column chromatography (SiO2,
EtOAc/MeOH 80:20 to 50:50). 1H NMR (400 MHz, D2O) δ
4.02 – 4.13 (m, 1H), 2.96 – 3.08 (m, 4H), 2.82 – 2.96 (m, 2H), 1.97 (s, 3H), 1.70
– 1.84 (m, 1H), 1.51 – 1.70 (m, 3H), 1.25 – 1.38 (m, 2H), 1.07 – 1.24 (m, 6H). 13C NMR (100 MHz, D2O) δ 181.63, 176.06, 57.47, 54.04, 49.59, 33.75, 25.86,
25.15, 24.49, 11.12. HRMS (APCI+, m/z): calculated for C12H25N2O3 [M+H]+:
245.18597; found: 245.18593.
N-isopropyl-proline (3ab): Synthesized according to General
procedure. Quantitative yield of 3ab was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400 MHz,
D2O) δ 3.93 – 4.05 (m, 1H), 3.62 – 3.72 (m, 1H), 3.47 – 3.61 (m,
1H), 3.13 – 3.26 (m, 1H), 2.29 – 2.44 (m, 1H), 2.00 – 2.15 (m, 2H), 1.79 – 1.97
(m, 1H), 1.20 – 1.40 (m, 6H). 13C NMR (100 MHz, D2O) δ 174.53, 65.80, 56.99,
52.28, 29.59, 23.59, 17.39, 17.26. HRMS (APCI+, m/z): calculated for C8H16NO2

N-alkylation of unprotected amino acids
109
[M+H]+: 158.11756; found: 158.11747. The physical data are identical to those
previously reported.[8]
N-isopropyl-phenylalanine (3db): Synthesized according
to General procedure. Quantitative yield of 3db was obtained
after removing the volatiles of reaction residue by high
vacuum. 1H NMR (400 MHz, D2O) δ 6.87 – 7.24 (m, 5H), 3.24
– 3.34 (m, 1H), 2.71 – 2.80 (m, 1H), 2.45 – 2.65 (m, 2H), 0.74 – 0.93 (m, 6H). 13C NMR (100 MHz, D2O) δ 181.45, 137.84, 129.08, 128.33, 126.40, 62.55, 45.96,
39.09, 22.57, 19.72. HRMS (APCI+, m/z): calculated for C12H18NO2 [M+H]+:
208.13321; found: 208.13312. The physical data are identical to those previously
reported.[32]
N-isopropyl-leucine (3bb): Synthesized according to General
procedure. Quantitative yield of 3bb was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400 MHz,
D2O-NaOH) δ 3.08 – 3.21 (m, 1H), 2.52 – 2.66 (m, 1H), 1.37 – 1.54
(m, 1H), 1.16 – 1.35 (m, 2H), 0.86 – 1.05 (m, 6H), 0.68 – 0.86 (m,
6H). 13C NMR (100 MHz, D2O) δ 177.15, 61.65, 53.02, 42.23, 27.09, 24.64, 23.83,
21.72, 20.32. HRMS (APCI+, m/z): calculated for C9H20NO2 [M+H]+: 174.14886;
found: 174.14872. The physical data are identical to those previously reported.[33]
N-isopropyl-valine (3cb): Synthesized according to General
procedure. Quantitative yield of 3cb was obtained after removing the
volatiles of reaction residue by high vacuum. 1H NMR (400 MHz, D2O)
δ 3.09 – 3.20 (m, 1H), 2.82 – 2.96 (m, 1H), 1.81 – 2.00 (m, 1H),
0.99 – 1.25 (m, 6H), 0.85 – 0.98 (m, 6H). 13C NMR (100 MHz, D2O) δ 181.45,
69.24, 51.18, 32.88, 23.89, 21.59, 21.17, 20.60. HRMS (APCI+, m/z): calculated
for C8H18NO2 [M+H]+: 160.13321; found: 160.13307. The physical data are
identical to those previously reported.[34]
N-isopropyl-serine (3eb): Synthesized according to General
procedure. Quantitative yield of 3eb was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400 MHz,
D2O) δ 3.85 – 4.01 (m, 2H), 3.72 – 3.79 (m, 1H), 3.38 – 3.52 (m,
1H), 1.24 – 1.36 (m, 6H). 13C NMR (100 MHz, D2O) δ 174.36, 63.67, 62.42, 53.03,
21.26, 21.24, 20.58. HRMS (APCI+, m/z): calculated for C6H14NO3 [M+H]+:
148.09682; found: 148.09671.
N-isopropyl-alanine (3fb): Synthesized according to General
procedure. Quantitative yield of 3fb was obtained after removing the
volatiles of reaction residue by high vacuum. 1H NMR (400 MHz, D2O)
δ 3.71 – 3.80 (m, 1H), 3.38 – 3.50 (m, 1H), 1.42 – 1.53 (m, 3H),
1.26 – 1.37 (m, 6H). 13C NMR (100 MHz, D2O) δ 177.76, 57.76, 52.17, 21.17,
20.71, 18.14. HRMS (APCI+, m/z): calculated for C6H14NO2 [M+H]+: 132.10191;
found: 132.10181. The physical data are identical to those previously reported.[35]

Chapter 5
110
N-n-butyl-proline (3ae): Synthesized according to General
procedure. Quantitative yield of 3ae was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400 MHz,
MeOD) δ 3.78 – 3.90 (m, 1H), 3.65 – 3.78 (m, 1H), 3.15 – 3.27
(m, 1H), 2.95 – 3.15 (m,2H), 2.30 – 2.46 (m, 1H), 1.99 – 2.15
(m, 2H), 1.82 – 1.98 (m, 1H), 1.56 – 1.76 (m, 2H), 1.30 – 1.47 (m, 2H), 0.83 –
1.04 (m, 3H). 13C NMR (100 MHz, MeOD) δ 173.50, 70.60, 56.38, 55.95, 30.30,
28.94, 24.34, 20.88, 13.96. HRMS (APCI+, m/z): calculated for C9H16NO2 [M-H]-:
170.11756; found: 170.11876. The physical data are identical to those previously
reported.[36]
N-cyclopropylmethyl-proline (3fb): Synthesized according to
General procedure. Proline (0.058 g, 0.50 mmol) affords 3fb (0.046
g, 55% yield). White solid was obtained after column
chromatography (SiO2, EtOAc/MeOH 80:20 to 80:20). 1H NMR (400
MHz, CDCl3) δ 3.83 – 4.01 (m, 1H), 3.67 – 3.83 (m, 1H), 2.98 -
3.14 (m, 1H), 2.70 – 2.95 (m, 2H), 2.22 – 2.38 (m, 1H), 2.05 – 2.22 (m, 1H),
1.86 – 2.05 (m, 2H), 0.95 – 1.09 (m, 1H), 0.49 – 0.70 (m, 2H), 0.18 – 0.40 (m,
2H). 13C NMR (100 MHz, CDCl3) δ 170.64, 68.63, 59.14, 53.96, 29.01, 23.06, 6.63,
4.45, 4.06. HRMS (APCI+, m/z): calculated for C9H14NO2 [M-H]-: 168.10191;
found: 168.10321.
N-(2-chloroethyl)-proline (3ag): Synthesized according to
General procedure. Proline (0.058 g, 0.50 mmol) affords 3ag (0.063
g, 71% yield). White solid was obtained after crystallization in
MeOH/Et2O. 1H NMR (400 MHz, D2O) δ 4.45 – 4.65 (m, 3H), 3.76 –
3.93 (m, 2H), 3.33 – 3.54 (m, 2H), 2.38 – 2.55 (m, 1H), 2.15 – 2.32
(m, 1H), 1.97 – 2.14 (m, 2H). 13C NMR (100 MHz, D2O) δ 169.50, 66.38, 59.35,
46.16, 41.59, 28.06, 23.07. HRMS (APCI+, m/z): calculated for C7H13ClNO2
[M+H]+: 178.06293; found: 178.06289.
N-benzyl-proline (3ah): Synthesized according to General
procedure. Proline (0.019 g, 0.20 mmol) affords 3ah (0.028 g,
68% yield). White solid was obtained after crystallization in Et2O. 1H NMR (400 MHz, CDCl3) δ 9.33 (br.s, 1H), 7.28 – 7.52 (m, 5H),
4.11 – 4.37 (m, 2H), 3.74 – 3.90 (m, 1H), 3.59 – 3.74 (m, 1H),
2.78 – 2.94 (m, 1H), 2.17 – 2.38 (m, 2H), 1.79 – 2.08 (m, 2H). 13C NMR (100
MHz, CDCl3) δ 171.02, 130.73, 130.53, 129.40, 129.04, 67.31, 57.61, 53.31,
28.89, 22.89. HRMS (APCI+, m/z): calculated for C12H16NO2 [M+H]+: 206.11756;
found: 206.11742. The physical data are identical to those previously reported.[37]
N-(4-chloro-benzyl)-proline (3ai): Synthesized according to
General procedure. Proline (0.058 g, 0.50 mmol) affords 3ai
(0.098 g, 82% yield). White solid was obtained after column
chromatography (SiO2, EtOAc/MeOH 90:10 to 50:50). 1H NMR
(400 MHz, D2O) δ 7.28 – 7.48 (m, 4H), 4.12 – 4.34 (m, 2H), 3.75
– 3.88 (m, 1H), 3.41 – 3.54 (m, 1H), 3.03 – 3.18 (m, 1H), 2.31 – 2.48 (m, 1H),
1.80 – 2.09 (m, 3H). 13C NMR (100 MHz, D2O) δ 176.56, 137.60, 134.51, 131.87,

N-alkylation of unprotected amino acids
111
131.63, 70.76, 59.87, 56.86, 31.30, 25.17. HRMS (APCI+, m/z): calculated for
C12H15ClNO2 [M+H]+: 240.07858; found: 240.07854. The physical data are
identical to those previously reported.[38]
N,N-(di-n-butyl)-phenylalanine (3de): Synthesized
according to General procedure. Phenylalanine (0.083 g, 0.50
mmol) affords 3de (0.116 g, 84% yield). White solid was
obtained after column chromatography (SiO2, EtOAc/MeOH
90:10 to 80:20). 1H NMR (400 MHz, CDCl3) δ 9.36 (br.s, 1H),
7.10 – 7.35 (m, 5H), 3.83 – 3.94 (m, 1H), 3.42 – 3.56 (m, 1H), 2.88 – 3.07 (m,
3H), 2.70 – 2.85 (m, 2H), 1.52 – 1.67 (m, 2H), 1.36 – 1.52 (m, 2H), 1.05 – 1.27
(m, 4H). 13C NMR (100 MHz, CDCl3) δ 170.49, 137.88, 128.76, 128.55, 126.65,
67.49, 51.45, 33.82, 26.82, 19.90, 13.49. HRMS (APCI+, m/z): calculated for
C17H26NO2 [M-H]-: 276.19581; found: 276.19703.
N,N-(di-n-nonyl)-phenylalanine (3dj): Synthesized
according to General procedure. Phenylalanine (0.083 g, 0.50
mmol) affords 3dj (0.147 g, 75% yield). White solid was
obtained after column chromatography (SiO2, Pentane/EtOAc
50:50 to 0:100, then EtOH/MeOH 90/10). 1H NMR (400 MHz, CDCl3) δ 8.68 (br.s,
1H), 7.13 – 7.31 (m, 5H). 3.84 – 3.93 (m, 1H), 3.50 – 3.58 (m, 1H), 2.88 – 3.05
(m, 3H), 2.67 – 2.79 (m, 2H), 1.53 – 1.67 (m, 2H), 1.38 – 1.52 (m, 2H), 0.98 –
1.35 (m, 24H), 0.75 – 0.89 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 170.49, 138.02,
128.75, 128.59, 126.67, 67.41, 51.76, 33.71, 31.67, 29.30, 29.05, 29.03, 26.70,
25.07, 22.50, 13.94. HRMS (APCI+, m/z): calculated for C27H48NO2 [M-H]-:
418.36796; found: 418.36763. The physical data are identical to those previously
reported.[32]
N,N-di-(5-hydroxypentyl)-phenylalanine (3dk):
Synthesized according to General procedure.
Phenylalanine (0.083 g, 0.50 mmol) affords 3dk
(0.059 g, 35% yield). White solid was obtained after
column chromatography (SiO2, EtOAc/MeOH 50:50 to 30:70). 1H NMR (400 MHz,
D2O) δ 7.23 – 7.45 (m, 5H), 3.91 – 4.03 (m, 1H), 3.47 – 3.65 (m, 4H), 3.13 –
3.32 (m, 4H), 2.95 – 3.13 (m, 2H), 1.58 – 1.79 (m, 4H), 1.45 – 1.58 (m, 4H),
1.21 – 1.43 (m, 4H). 13C NMR (100 MHz, D2O) δ 172.40, 135.73, 128.94, 128.85,
127.27, 68.29, 61.15, 33.38, 30.56, 23.33, 22.13. HRMS (APCI+, m/z): calculated
for C19H32NO4 [M+H]+: 338.23258; found: 338.23176.
N,N-di-n-nonyl-glycine (3gj): Synthesized according to General
procedure. Glycine (0.038 g, 0.50 mmol) affords 3gj (0.149 g, 91%
yield). White solid was obtained after column chromatography (SiO2,
EtOAc/MeOH 90:10 to 70:30). 1H NMR (400 MHz, CDCl3) δ 8.78 (br.s,
1H), 3.48 (s, 2H), 2.95 – 3.15 (m, 4H), 1.52 – 1.75 (m, 4H), 1.03 – 1.42 (m,
24H), 0.65 – 0.95 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 167.42, 55.99, 53.92,
31.63, 29.26, 29.03, 28.99, 26.66, 23.71, 22.47, 13.90. HRMS (APCI+, m/z):
calculated for C20H44NO2 [M+H]+: 328.32155; found: 328.32095.

Chapter 5
112
N,N-di-benzyl-glycine (3gh): Synthesized according to
General procedure. Glycine (0.038 g, 0.50 mmol) affords 3gh
(0.066 g, 52% yield). White solid was obtained after
precipitation in MeOH/Et2O. 1H NMR (400 MHz, DMSO-d6) δ
7.20 – 7.40 (m, 10H), 3.73 (s, 4H), 3.15 (s, 2H). 13C NMR (100
MHz, DMSO-d6) δ 172.19, 139.00, 128.53, 128.24, 126.98, 56.79, 53.00. HRMS
(APCI+, m/z): calculated for C16H18NO2 [M+H]+: 256.13321; found: 256.13325.
The physical data are identical to those previously reported.[39]
N-2-butyl-glycine (3gl): Synthesized according to General
procedure. Quantitative yield of 3gl was obtained after removing
the volatiles of reaction residue by high vacuum. 1H NMR (400
MHz, D2O) δ 3.48 – 3.67 (m, 2H), 3.14 – 3.27 (m, 1H), 1.68 – 1.84 (m, 1H), 1.47
– 1.66 (m, 1H), 1.22 – 1.35 (m, 3H). 0.88 – 1.03 (m, 3H). 13C NMR (100 MHz,
D2O) δ 174.11, 58.44, 49.00, 28.27, 17.51, 11.47. HRMS (APCI+, m/z): calculated
for C6H14NO2 [M+H]+: 132.10191; found: 132.10181.
N,N-di-(n-pentyl)-glycine (3gm): Synthesized
according to General procedure. Glycine (0.038 g, 0.50
mmol) affords 3gm (0.097 g, 90% yield). White solid was
obtained after column chromatography (SiO2, EtOAc/MeOH 90:10 to 70:30). 1H
NMR (400 MHz, D2O) δ 3.68 (s, 2H), 3.05 – 3.25 (m, 4H), 1.56 – 1.80 (m, 4H),
1.20 – 1.40 (m, 8H), 0.75 – 1.00 (m, 6H). 13C NMR (100 MHz, D2O) δ 173.36,
58.39, 57.49, 30.46, 25.61, 24.08, 15.65. HRMS (APCI+, m/z): calculated for
C12H26NO2 [M+H]+: 216.19581; found: 216.19574.
N-n-pentyl-glycine (3gm’): Synthesized according to
General procedure. Glycine (0.038 g, 0.50 mmol) affords
3gm’ (0.033 g, 46% yield). White solid was obtained after column
chromatography (SiO2, EtOAc/MeOH 60:40 to 30:70). 1H NMR (400 MHz, D2O) δ
3.57 (s, 2H), 2.91 – 3.13 (m, 2H), 1.54 – 1.80 (m, 2H), 1.20 – 1.46 (m, 4H), 0.75
– 0.99 (m, 3H). 13C NMR (100 MHz, D2O) δ 174.07, 51.74, 50.13, 30.40, 27.75,
24.05, 15.62. HRMS (APCI+, m/z): calculated for C7H16NO2 [M+H]+: 146.11756;
found: 146.11751. The physical data are identical to those previously reported.[40]
N,N-di-n-dodecyl-glycine (3gn): Synthesized according to
General procedure. Glycine (0.038 g, 0.50 mmol) affords 3gn (0.189
g, 92% yield). White solid was obtained after column
chromatography (SiO2, EtOAc/MeOH 90:10 to 70:30). 1H NMR (400
MHz, CDCl3) δ 8.36 (br.s, 1H), 3.49 (s, 2H), 2.95 – 3.15 (m, 4H),
1.52 – 1.75 (m, 4H), 1.03 – 1.42 (m, 36H), 0.75 – 0.95 (m, 6H). 13C NMR (100
MHz, CDCl3) δ 167.51, 56.23, 54.01, 31.80, 29.51, 29.42, 29.38, 29.23, 29.11,
26.73, 23.74, 22.57, 13.99. HRMS (APCI+, m/z): calculated for C26H54NO2 [M+H]+:
412.41491; found: 412.41482. The physical data are identical to those previously
reported.[31]

N-alkylation of unprotected amino acids
113
N-dodecyl-glycine (3gn’): Synthesized according to General
procedure. Glycine (0.038 g, 0.50 mmol) affords 3gn’ (0.066 g,
54% yield). White solid was obtained after column chromatography (SiO2,
EtOAc/MeOH 70:30 to 40:60). 1H NMR (400 MHz, KOH, D2O) δ 3.00 – 3.20 (m,
2H), 2.38 – 2.58 (m, 2H), 1.36 – 1.57 (m, 2H), 1.12 – 1.35 (m, 18H), 0.73 – 0.90
(m, 3H). 13C NMR (100 MHz, KOH, D2O) δ 180.96, 55.23, 51.69, 34.63, 32.69,
32.61, 32.54, 32.40, 32.22, 31.93, 30.15, 25.28, 16.42. HRMS (APCI+, m/z):
calculated for C14H30NO2 [M+H]+: 244.22711; found: 244.22711. The physical
data are identical to those previously reported.[41]
N-nonyl-glycine (3gj’): Synthesized according to General
procedure. Glycine (0.038 g, 0.50 mmol) affords 3gj’ (0.052 g,
51% yield). White solid was obtained after column chromatography (SiO2,
EtOAc/MeOH 70:30 to 40:60). 1H NMR (400 MHz, KOH, D2O) δ 2.90 – 3.18 (m,
2H), 2.30 – 2.56 (m, 2H), 1.28 – 1.55 (m, 2H), 1.02 – 1.38 (m, 12H), 0.65 – 0.91
(m, 3H). 13C NMR (100 MHz, KOH, D2O) δ 181.71, 55.18, 51.50, 34.34, 32.09,
31.96, 31.80, 31.74, 29.78, 25.05, 16.31. HRMS (APCI+, m/z): calculated for
C11H24NO2 [M+H]+: 202.18016; found: 202.18010.
N-decyl-glycine (3go’): Synthesized according to General
procedure. Glycine (0.038 g, 0.50 mmol) affords 3go’ (0.075
g, 69% yield). White solid was purified by column
chromatography (SiO2, EtOAc/MeOH 70:30 to 50:50). 1H NMR (400 MHz, KOH,
D2O) δ 3.00 – 3.15 (m, 2H), 2.36 – 2.58 (m, 2H), 1.36 – 1.54 (m, 2H), 1.05 –
1.35 (m, 14H), 0.73 – 0.90 (m, 3H). 13C NMR (100 MHz, KOH, D2O) δ 181.42,
55.34, 51.67, 34.49, 32.45, 32.30, 32.22, 32.05, 31.99, 30.04, 25.18, 16.35.
HRMS (APCI+, m/z): calculated for C12H26NO2 [M+H]+: 216.19581; found:
216.19589. The physical data are identical to those previously reported.[41]
methyl N-tetradecylglycinate (methyl ester of 3gp’: 3gp’-
Me): Synthesized according to General procedure and
Esterification procedure. Glycine (0.038 g, 0.50 mmol) affords
3gp’-Me (0.046 g, 32% yield). Oily compound 3gp’-Me was purified by column
chromatography (SiO2, tol/Et2O 50/50 – 0/100). 1H NMR (400 MHz, CDCl3) δ 3.72
(s, 3H), 3.41 (s, 2H), 2.52 – 2.64 (m, 2H), 1.42 – 1.54 (m, 2H), 1.15 – 1.36 (m,
22H), 0.83 – 0.91 (m, 3H) 13C NMR (100 MHz, CDCl3) δ 173.02, 51.71, 50.84,
49.67, 31.91, 30.04, 29.68, 29.66, 29.65, 29.64, 29.60, 29.57, 29.51, 29.34,
27.21, 22.68, 14.10. HRMS (APCI+, m/z): calculated for C17H36NO2 [M+H]+:
286.27406; found: 286.27430.
methyl N-hexadecylglycinate (methyl ester of 3gq’: 3gq’-
Me): Synthesized according to General procedure and
Esterification procedure. Glycine (0.038 g, 0.50 mmol) affords 3gq’-Me (0.059 g,
38% yield). Oily compound 3gq’-Me was purified by column chromatography
(SiO2, tol/Et2O 50/50 – 0/100). 1H NMR (400 MHz, CDCl3) δ 3.73 (s, 3H), 3.43 (s,
2H), 2.52 – 2.64 (m, 2H), 1.42 – 1.58 (m, 2H), 1.13 – 1.37 (m, 26H), 0.80 – 0.94
(m, 3H) 13C NMR (100 MHz, CDCl3) δ 172.71, 51.78, 50.65, 49.60, 31.92, 29.85,

Chapter 5
114
29.68, 29.67, 29.65, 29.60, 29.57, 29.49, 29.35, 27.19, 22.68, 14.11. HRMS
(APCI+, m/z): calculated for C19H40NO2 [M+H]+: 314.30536; found: 314.30540.
methyl N-octadecylglycinate (methyl ester of 3gr’: 3gr’-
Me): Synthesized according to General procedure and
Esterification procedure. Glycine (0.038 g, 0.50 mmol) affords
3gr’-Me (0.067 g, 39% yield). Oily compound 3gr’-Me was purified by column
chromatography (SiO2, tol/Et2O 50/50 – 0/100). 1H NMR (400 MHz, CDCl3) δ 3.71
(s, 3H), 3.40 (s, 2H), 2.53 – 2.64 (m, 2H), 1.42 – 1.52 (m, 2H), 1.17 – 1.36 (m,
30H), 0.80 – 0.91 (m, 3H) 13C NMR (100 MHz, CDCl3) δ 173.01, 51.68, 50.83,
49.67, 31.90, 30.04, 29.67, 29.66, 29.64, 29.59, 29.57, 29.50, 29.34, 27.21,
22.67, 14.09. HRMS (APCI+, m/z): calculated for C21H44NO2 [M+H]+: 342.33666;
found: 342.33681.
N-dodecyl-alanine (3gn’): Synthesized according to General
procedure. Alanine (0.045 g, 0.50 mmol) affords 3gn’ (0.063
g, 49% yield). Compound 3gn’ was purified by column
chromatography (SiO2, EtOAc/MeOH 70:30 to 40:60). 1H NMR (400 MHz, KOH,
D2O) δ 3.00 – 3.20 (m, 2H), 2.38 – 2.58 (m, 2H), 1.36 – 1.57 (m, 2H), 1.12 –
1.35 (m, 18H), 0.73 – 0.90 (m, 3H). 13C NMR (100 MHz, KOH, D2O) δ 180.96,
55.23, 51.69, 34.63, 32.69, 32.61, 32.54, 32.40, 32.22, 31.93, 30.15, 25.28,
16.42. HRMS (APCI+, m/z): calculated for C15H32NO2 [M+H]+: 258.24276; found:
258.24302.
N-nonyl-proline (3aj): Synthesized according to
General procedure. Proline (0.053 g, 0.50 mmol) affords
3aj (0.063 g, 52% yield). White solid was obtained after
column chromatography (SiO2, EtOAc/MeOH 90:10 to
50:50). 1H NMR (400 MHz, CDCl3) δ 3.92 – 3.06 (m, 1H), 3.61 – 3.78 (m, 1H),
3.06 – 3.22 (m, 1H), 2.90 – 3.06 (m, 1H), 2.71 – 2.89 (m, 1H), 2.27 – 2.43 (m,
1H), 2.13 – 2.27 (m, 1H), 1.88 – 2.09 (m, 2H), 1.60 – 1.79 (m, 2H), 1.05 – 1.43
(m, 12H), 0.72 – 0.95 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 170.24, 69.66, 55.60,
54.77, 31.68, 29.38, 29.27, 29.05, 26.64, 25.66, 23.48, 22.53, 13.97. HRMS
(APCI+, m/z): calculated for C14H28NO2 [M+H]+: 242.21146; found: 242.21144.
N,N-diethyl-glycyl-alanine (5aa): Synthesized according
to General procedure. Quantitative yield of 5aa was
obtained after removing the volatiles of reaction residue by
high vacuum. 1H NMR (400 MHz, D2O) δ 4.08 – 4.22 (m,
1H), 3.88 – 4.03 (m, 2H), 3.17 – 3.31 (m, 4H), 1.30 – 1.36 (m, 3H), 1.21 – 1.30
(m, 6H). 13C NMR (100 MHz, D2O) δ 179.44, 164.77, 53.19, 51.17, 49.43, 16.92,
8.30. HRMS (APCI+, m/z): calculated for C9H19N2O3 [M+H]+: 203.13902; found:
203.13895.
N,N-di-(5-hydroxy-pentyl)-glycyl-alanine
(5ak): Synthesized according to General
procedure. Glycyl-alanine (0.073 g, 0.50 mmol)
affords 5ak (0.036 g, 36% yield). White solid was obtained after column

N-alkylation of unprotected amino acids
115
chromatography (SiO2, EtOAc/MeOH 60:40 to 30:70). 1H NMR (400 MHz, D2O) δ
4.15 – 4.23 (m, 1H), 3.55 – 3.67 (m, 4H), 3.19 – 3.34 (m, 2H,), 2.52 – 2.71 (m,
4H), 1.47 – 1.66 (m, 8H), 1.28 – 1.44 (m, 7H). 13C NMR (100 MHz, D2O) δ 182.21,
175.12, 64.25, 59.67, 57.15, 53.19, 33.76, 28.20, 25.58, 20.44. HRMS (APCI+,
m/z): calculated for C15H31N2O5 [M+H]+: 319.22275; found: 319.22278.
N,N-di-(n-dodecyl)-glycyl-alanine (5an):
Synthesized according to General procedure. Glycyl-
alanine (0.073 g, 0.50 mmol) affords 5an (0.199 g,
82% yield). White solid was obtained after column
chromatography (SiO2, EtOAc/MeOH 90:10 to 50:50). 1H NMR (400 MHz, CDCl3) δ
8.20 (br.s, 1H), 4.17 – 4.38 (m, 1H), 3.24 – 3.60 (m, 2H), 2.52 – 2.95 (m, 4H),
1.45 – 1.64 (m, 4H), 1.11 – 1.42 (m, 39H), 0.70 – 0.95 (m, 6H). 13C NMR (100
MHz, CDCl3) δ 177.93, 167.98, 56.32, 54.16, 49.95, 31.87, 29.64, 29.61, 29.58,
29.37, 29.31, 27.15, 25.34, 22.63, 18.39, 14.04. HRMS (APCI+, m/z): calculated
for C29H59N2O3 [M+H]+: 483.45202; found: 483.45170.
N,N-diethyl-glycyl-alanine (5ba): Synthesized
according to General procedure. Leucyl-glycyl-
glycine (0.123 g, 0.50 mmol) affords 5ba (0.101
g, 67% yield). White solid was obtained after
column chromatography (SiO2, EtOAc/MeOH 60:40
to 30:70). 1H NMR (400 MHz, D2O) δ 3.86 – 4.02 (m, 2H), 3.75 (s, 2H), 3.38 –
3.48 (m, 1H), 2.71 – 2.88 (m, 2H), 2.46 – 2.62 (m, 2H), 1.68 – 1.83 (m, 1H),
1.32 – 1.54 (m, 2H), 0.97 – 1.13 (m, 6H), 0.83 – 0.96 (m, 6H). 13C NMR (100
MHz, D2O) δ 179.02, 177.83, 173.32, 64.90, 46.57, 45.81, 44.84, 40.43, 27.43,
25.57, 23.61, 14.04. HRMS (APCI+, m/z): calculated for C14H28N3O4 [M+H]+:
302.20743; found: 302.20747.

Chapter 5
116
Determination of enantiomeric excesses retention
N-(4-bromophenyl)-1-ethylpyrrolidine-2-
carboxamide (6aa): According to a literature procedure[42],
6aa was prepared from 3aa (28.6 mg, 0.20 mmol), 4-
bromoaniline (38.1 mg, 0.22 mmol), EDC•HCl (46.3 mg,
0.24 mmol), DMAP (29.3 mg, 0.24 mmol), and HOBt•H2O (30.8 mg, 0.20 mmol),
in 3ml CH3CN at room temperature overnight. Then aq. NaHCO3 was added, and
the mixture was extracted 3 times with EtOAc. The organic phase was
concentrated under vacuo and the residue was purified by column chromatography
(SiO2, Pentane/EtOAc 50:50 to 0:100) to afford 6aa as a light yellow solid. 1H NMR
(400 MHz, CDCl3) δ 9.57 (s, 1H), 7.35 – 7.57 (m, 4H), 3.21 – 3.31 (m, 1H), 3.09
– 3.19 (m, 1H), 2.65 – 2.78 (m, 1H), 2.52 – 2.65 (m, 1H), 2.33 – 2.47 (m, 1H),
2.14 – 2.31 (m, 1H), 1.92 – 2.02 (m, 1H), 1.68 – 1.89 (m, 2H), 1.12 (t, J = 7.22
Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.46, 136.84, 131.85, 120.78, 116.35,
67.61, 53.86, 49.85, 30.66, 24.39, 14.28. HRMS (APCI+, m/z): calculated for
C13H16BrN2O [M-H]-: 295.04405; found: 295.04510.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99:1); flow rate: 0.5 ml/min; detection:
UV 232 nm; retention times 22.2 min (major) and 26.8 min (minor).
2-(diethylamino)-N-(4-methoxyphenyl)-4-
methylpentanamide (6ba): According to a literature
procedure[42], 6ba was prepared from 3ba (37.4 mg,
0.20 mmol), 4-methoxyaniline (27 mg, 0.22 mmol),
EDC•HCl (46.3 mg, 0.24 mmol), NEt3 (56 ul, 0.4 mmol),
and HOBt•H2O (30.8 mg, 0.20 mmol), in 3 ml CH3CN at
room temperature overnight. Then aq. NaHCO3 was added, and the mixture was
extracted 3 times with EtOAc. The organic phase was concentrated under vacuo
and the residue was purified by column chromatography (SiO2, Pentane/EtOAc
50:50 to 20:80) to afford 6ba as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ
9.44 (s, 1H), 7.42 – 7.52 (m, 2H), 6.79 – 6.91 (m, 2H), 3.78 (s, 3H), 3.30 – 3.43
(m, 1H), 2.38 – 2.75 (m, 4H), 1.73 – 1.98 (m, 2H), 1.28 – 1.37 (m, 1H), 1.06 –
1.15 (m, 6H), 0.95 – 1.01 (m, 3H), 0.90 – 0.95 (m, 3H). 13C NMR (100 MHz, CDCl3)
δ 155.95, 131.42, 125.62, 120.66, 114.12, 55.48, 44.48, 34.47, 26.43, 23.44,
21.98, 13.79. HRMS (APCI+, m/z): calculated for C14H23N2O3 [M+H]+: 293.22235;
found: 293.22245.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99:1); flow rate: 0.5 ml/min; detection:
UV 250 nm; retention times 20.2 min (minor) and 29.0 min (major).
2-(diethylamino)-N-(4-methoxyphenyl)-3-
methylbutanamide (6ca): Compound 6ca was
prepared from 3ca (35.0 mg, 0.20 mmol), 4-
methoxyaniline (27 mg, 0.22 mmol), COMU[43] (257 mg,
0.60 mmol) and N,N-diisopropylethylamine (70 ul, 0.4
mmol) in 1 ml DMF, at room temperature for 1 h. DMF was removed under vacuo

N-alkylation of unprotected amino acids
117
and purified by column chromatography (SiO2, Pentane/EtOAc 50:50 to 20:80) to
afford 6ca as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.58 (br.s, 1H), 7.40
– 7.53 (m, 2H), 6.79 – 6.90 (m, 2H), 3.78 (s, 3H), 2.91 – 3.12 (m, 1H), 2.58 –
2.80 (m, 4H), 2.12 – 2.28 (m, 1H), 1.09 (d, J = 6.9 Hz, 3H), 1.00 – 1.07 (m, 6H),
0.98 (d, J = 6.7 Hz, 3H). HRMS (APCI+, m/z): calculated for C16H27N2O2 [M+H]+:
279.20670; found: 279.20673.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (98:2); flow rate: 0.5 ml/min; detection:
UV 250 nm; retention times 23.7 min (minor) and 25.4 min (major).
methyl diethylphenylalaninate (6da): According to a
literature procedure[44], 6da was prepared from 3da. To a
stirred solution of the 3da (44.2 mg, 0.2 mmol) in
toluene/MeOH (1/1 ml), TMSCHN2 (0.4 mmol, 2M in toluene)
was added. The mixture was stirred for 1 h at room temperature and concentrated
in vacuo to give 6da as a transparent oily compound. 1H NMR (400 MHz, CDCl3) δ
7.12 – 7.40 (m, 5H), 3.64 – 3.74 (m, 1H), 3.65 (s, 3H), 3.06 – 3.17 (m, 1H), 2.89
– 3.02 (m, 1H), 2.75 – 2.89 (m, 2H), 2.52 – 2.65 (m, 2H), 1.08 (t, J = 7.17 Hz,
6H). 13C NMR (100 MHz, CDCl3) δ 173.06, 138.68, 129.21, 128.18, 126.20, 64.95,
51.04, 44.50, 35.99, 13.66. The physical data were identical in all respects to
those previously reported.[45]
The ee was determined by chiral HPLC analysis. Chiralcel OZ-H column,
Phenomenex, Ltd; heptane/isopropanol (99.7:0.3); flow rate: 0.5 ml/min;
detection: UV 190 nm; retention times 18.1 min (major) and 19.9 min (minor).
2-(diethylamino)-3-hydroxy-N-(4-methoxy-
phenyl)propanamide (6ea): According to a literature
procedure[42], 6ea was prepared from 3ea (32.0 mg,
0.20 mmol), 4-methoxyaniline (27 mg, 0.22 mmol),
EDC•HCl (46.3 mg, 0.24 mmol), NEt3 (56 ul, 0.4 mmol),
and HOBt•H2O (30.8 mg, 0.20 mmol), in 3 ml CH3CN at room temperature
overnight. Then aq. NaHCO3 was added, the mixture was extracted 3 times with
EtOAc. The organic phase was concentrated under vacuo and the residue was
purified by column chromatography (SiO2, Pentane/EtOAc 50:50 to 20:80) to
afford 6ea as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 9.42 (s, 1H), 7.40
– 7.50 (m, 2H), 6.83 – 6.92 (m, 2H), 4.02 – 4.10 (m, 1H), 3.81 – 3.89 (m, 1H),
3.79 (s, 3H), 3.48 – 3.57 (m, 1H), 2.56 – 2.82 (m, 4H), 1.14 (t, J = 7.07 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 156.45, 130.40, 121.15, 114.21, 101.59, 63.98,
58.56, 55.48, 44.87, 14.11. HRMS (APCI+, m/z): calculated for C14H23N2O3
[M+H]+: 267.17032; found: 267.17031.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (97:3); flow rate: 0.5 ml/min; detection:
UV 250 nm; retention times 42.5 min (minor) and 45.1 min (major).

Chapter 5
118
N-(4-bromophenyl)-2-(diethylamino)propanamide
(6fa): According to a literature procedure[42], 6fa was
prepared from 3fa (29.0 mg, 0.20 mmol), 4-bromoaniline
(38.1 mg, 0.22 mmol), EDC•HCl (46.3 mg, 0.24 mmol),
DMAP (29.3 mg, 0.24 mmol), and HOBt•H2O (30.8 mg, 0.20 mmol), in 3 ml CH3CN
at room temperature overnight. Then aq. NaHCO3 was added, the mixture was
extracted 3 times with EtOAc. The organic phase was concentrated under vacuo
and the residue was purified by column chromatography (SiO2, Pentane/EtOAc
50:50 to 20:80) to afford 6fa as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ
9.65 (s, 1H), 7.35 – 7.57 (m, 4H), 3.39 – 3.58 (m, 1H), 2.35 – 2.74 (m, 4H), 1.22
– 1.31 (m, 3H), 1.04 – 1.15 (m ,6H). HRMS (APCI+, m/z): calculated for
C13H20BrN2O [M+H]+: 299.07535; found: 299.07614.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99:1); flow rate: 0.5 ml/min; detection:
UV 254 nm; retention times 17.0 min (minor) and 19.8 min (major).

N-alkylation of unprotected amino acids
119
References
[1] a) T.M. Lammensa, M.C.R. Franssenb, E.L. Scotta, J.P.M. Sandersa, Biomass
Bioenergy, 2012, 44 168-181; b) I. Wagner, H. Musso, Angew. Chem. Int. Ed. Engl.,
1983, 22, 816-828.
[2] G. Xu, X. Fan, A. J. Miller, Annu. Rev. Plant Biol., 2012, 63, 153–182.
[3] A) C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon, M. Poliakoff, Science, 2012,
337, 695-699; b) E. Scott, F. Peter, J. Sanders, Appl. Microbiol. Biotechnol., 2007,
75, 751–762.
[4] L. Aurelio, A. B. Hughes, Amino Acids, Peptides and Proteins in Organic Chemistry,
Vol.1, Origins and Synthesis of Amino Acids, chapter 6, Synthesis of N-Alkyl Amino
Acids, 2010, Wiley-VCH Verlag GmbH & Co. KGaA.
[5] C. R. Waites, M. A. Dominick, T. P. Sanderson, B. E. Schilling, Toxicol. Sci., 2007,
100, 248–258.
[6] M. C. Cleij, P. Scrimin, P. Tecilla, U. Tonellato, J. Org. Chem., 1997, 62, 5592–5599.
[7] P. K. Sasmal, C. N. Streu, E. Meggers, Chem. Commun., 2013, 49, 1581-1587.
[8] D. R. Fandrick et al., J. Org. Chem., 2013, 78, 3592–3615.
[9] a) Y. Li, K. Holmberg, R. Bordes, J. Colloid Interface Sci., 2013, 411, 47–52; b) R.
Bordes, K. Holmberg, Adv. Colloid Interface Sci., 2015, 222, 79–91.
[10] S. Santra, J. M. Perez, Biomacromolecules, 2011, 12, 3917−3927.
[11] M. Y. Pavlov, R. E. Watts, Z. Tan, V. W. Cornish, M. Ehrenberg, A. C. Forster, Proc.
Natl. Acad. Sci. U.S.A., 2009, 106, 50–54.
[12] A recent example derived protected amino acids through C-H activation, see: T. J.
Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan, M. C. White, Nature, 2016, 537,
214–219.
[13] Y. Ohfune, N. Kurokawa, N. Higuchi, M. Saito, M. Hashimoto, T. Tanaka, Chem. Lett.,
1984, 13, 441-444.
[14] G. Barrett, Chemistry and Biochemistry of the Amino Acids, chapter 11, reactions of
amino acids, page 360, Chapman and hall ltd, London, 1985.
[15] a) M. H. S. A. Hamid, P. A. Slatford, J. M. J. Williams, Adv. Synth. Catal., 2007, 349,
1555−1575; b) T. D. Nixon, M. K. Whittlesey, J. M. J. Williams, Dalton Trans., 2009,
753−762; c) G. Guillena, D. J. Ramon, M. Yus, Chem. Rev., 2010, 110, 1611−1641;
d) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem,
2011, 3, 1853−1864; e) G. E. Dobereiner, R. H. Crabtree, Chem. Rev., 2010, 110,
681−703. f) C. Gunanathan, D. Milstein, Science, 2013, 341, 1229712; g) Q. Yang,
Q. Wang, Z. Yu, Chem. Soc. Rev., 2015, 44, 2305—2329.
[16] K. Barta, P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512.
[17] U. R. Kreutzer, J. Am. Oil. Chem. Soc., 1984, 61, 343–348.
[18] Methylamines can be produced under 350-500 °C and 15-30 bar pressure using
aluminum-based heterogeneous catalyst from ammonia and methanol. See: K.
Weissermel, H.-J. Arpe, Industrial Organic Chemistry, Fourth Completely Revised
Edition, Wiley-VCH: Weinheim, 2003, page 51.
[19] a) C.-P. Xu, Z.-H. Xiao, B.-Q. Zhuo, Y.-H. Wang, P.-Q. Huang, Chem. Commun.,
2010, 46, 7834–7836; b) V. N. Tsarev, Y. Morioka, J. Caner, Q. Wang, R. Ushimaru,
A. Kudo, H. Naka, S. Saito, Org. Lett., 2015, 17, 2530−2533; c) H. Hikawa, Y.
Yokoyama, Org. Biomol. Chem., 2011, 9, 4044-4050.
[20] T. Yan, B. L. Feringa, K. Barta, Nat. Commun., 2014, 5, 5602.
[21] a) H.-J. Knölker, E. Baum, H. Goesmann, R. Klauss, Angew. Chem. Int. Ed., 1999,
38, 2064-2066; b) Y. Shvo, D. Czarkie, Y. Rahamim, J. Am. Chem. Soc., 1986, 108,
7400-7402; c) B. L. Conley, M. K. Pennington-Boggio, E. Boz, T. J. Williams, Chem.
Rev., 2010, 110, 2294–2312.
[22] T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal., 2012, 354, 597–
601.
[23] a) For N-alkylation of anilines with amine with Shvo Catalyst, see D. Hollmann, S.
Bähn, A. Tillack, M. Beller, Angew. Chem. Int. Ed., 2007, 46, 8291–8294; b) for C-
3-alkylation of indoles with alcohols, see: S. Imm, S. Bähn, A. Tillack, K. Mevius, L.
Neubert, M. Beller, Chem. Eur. J., 2010, 16, 2705–2709; c) for alkylation of

Chapter 5
120
ammonium salts with primary alcohols, see: C. Segarra, E. Mas-Marza, J. A. Mata, E.
Peris, Adv. Synth. Catal., 2011, 353, 2078–2084.
[24] C. P. Casey, J. B. Johnson, J. Am. Chem. Soc., 2005, 127, 1883-1894.
[25] I. A. Shuklov, N. V. Dubrovina, A. Börner, Synthesis, 2007, 19, 2925–2943.
[26] a) E. Baslé, N. Joubert, M. Pucheault, Chem. Biol., 2010, 17, 213-227; b) C. D.
Spicer, B, G. Davis, Nat. Commun., 2014, 5, 4740; c) O. Boutureira, G. J. L.
Bernardes, Chem. Rev., 2015, 115, 2174−2195.
[27] Z. W Lai, A. Petrera, O. Schilling, Curr. Opin. Chem. Biol., 2015, 24, 71–79.
[28] These results will be issued as a patent, filed reference: P114699EP00.
[29] O. D. Lopez, Q. Chen, M. Belema, L. G. Hamann, US2010/249190 A1, 2010.
[30] K. P. Shawn, T. Steven, P. P. Bharat, J. N. Kenneth, B. Makonen, US 2013317213 A1,
2013.
[31] M. C. Cleij, P. Scrimin, P. Tecilla, U. Tonellato J. Org. Chem., 1997, 62, 5592–5599.
[32] G. Verardo, P. Geatti, E. Pol, A. G. Giumanini, Can. J. Chem., 2002, 80, 779–788.
[33] Y. Song, A. D. Sercel, D. R. Johnson, N. L. Colbry, K.-L. Sun, B. D. Roth, Tetrahedron
Lett., 2000, 41, 8225–8230.
[34] D. Papaioannou, C. Athanassopoulos, V. Magafa, N. Karamanos, G. Stavropoulos, A.
Napoli, G. Sindona, D. W. Aksnes, G. W. Francis, Acta Chem. Scand., 1994, 48, 324–
333.
[35] A.-H. Liu, R. Ma, C. Song, Z.-Z. Yang, A. Yu, Y. Cai, L.-N. He, Y.-N. Zhao, B. Yu, Q.-
W. Song, Angew. Chem. Int. Ed., 2012, 51, 11306–11310.
[36] T. Wein, M. Petrera, L. Allmendinger, G. Hofner, J. Pabel, K. T. Wanner,
ChemMedChem, 2016, 11, 509–518.
[37] M. Jorres, X. Chen, J. L. Acena, C. Merkens, C. Bolm, H. Liu, V. A. Soloshonok, Adv.
Synth. Catal., 2014, 356, 2203–2208.
[38] S. J. Barraza, P. C. Delekta, J. A. Sindac, C. J. Dobry, J. Xiang, R. F. Keep, D. J. Miller,
S. D. Larsen, Bioorg. Med. Chem., 2015, 23, 1569–1587.
[39] R. A. Breitenmoser, H. Heimgartner, Helv. Chim. Acta, 2001, 84, 786–796.
[40] M. Ferrer-Casal, C. Li, M. Galizzi, C. A. Stortz, S. H. Szajnman, R. Docampo, S. N. J.
Moreno, J. B. Rodriguez, Bioorg. Med. Chem., 2014, 22, 398–405.
[41] F. Wessendorf, B. Grimm, D. M. Guldi, A. Hirsch, J. Am. Chem. Soc., 2010, 132,
10786–10795.
[42] V. H. Thorat, T. S. Ingole, K. N. Vijayadas, R. V. Nair, S. S. Kale, V. V. E. Ramesh,
H. C. Davis, P. Prabhakaran, R. G. Gonnade, R. L. Gawade, V. G. Puranik, P. R.
Rajamohanan, G. J. Sanjayan, Eur. J. Org. Chem., 2013, 3529–3542.
[43] A. El-Faham, R. S. Funosas, R. Prohens, F. Albericio, Chem. Eur. J., 2009, 15, 9404–
9416.
[44] A. Presser, A. Hufner, Monatsh. Chem., 2004, 135, 1015–1022.
[45] N. Matsuda, K. Hirano, T. Satoh, M. Miura, Angew. Chem. Int. Ed., 2012, 51, 11827–
11831.

121
Chapter 6
Ruthenium catalyzed N-alkylation of amino acid esters with
alcohols
-Amino acids are the most abundant chiral amine sources in nature. Selective
functionalization of amino acids and their derivatives is a highly-desired
transformation. In Chapter 5, the direct N-alkylation of unprotected amino acids is
described. As common amino acid derivatives, the functionalization of amino acid
esters holds vast interest, due to their better solubility in organic solvents
compared to their acid analogues. This Chapter describes, for the first time, the
direct N-alkylation of amino acid esters with alcohols, producing water as the only
waste. The scope includes direct N-alkylation of phenylalanine, alanine, valine,
proline esters and prolinamide with 1-pentanol and benzyl alcohols, with good to
excellent yields and stereochemistry retention.
Part of this chapter will be submitted for publication:
T. Yan, B. L. Feringa, K. Barta, to be submitted.

Chapter 6
122
Introduction
N-alkyl amino acid esters are prominent chiral moieties in bio-active compounds
(Figure 1). For example, Cilazapril[1] and Enalapril[2] are angiotensin-converting
enzyme inhibitors; Ximelagatran[3] is an anticoagulant that has been investigated
as a replacement for warfarin.
Figure 1: N-alkyl amino acid ester contained pharmaceutical compounds.
An attractive way to synthesize N-alkyl amino acid esters is the direct alkylation
of amino acid esters with alcohols through the borrowing hydrogen strategy. The
advantage of using alcohols as alkylation reagents has been extensively discussed
in previous chapters (Chapter 2, 3 and 5). In this Chapter we explore the possibility
of the direct alkylation of N-alkyl amino acid esters with alcohols. With the
development of the borrowing hydrogen strategy[4], the direct alkylation of amines
with alcohols has been achieved under relatively mild reaction conditions[5].
However, this method still has not been applied to direct N-alkylation of amino
acid esters bearing acidic α-proton[5g]. Probably this is due to the possibility of
competing transesterification[6] and unsatisfactory ee retention[7], as common
catalyst systems frequently require the addition of a strong base for activation of
the catalyst or substrates[5].
Figure 2: A Alkylation of amines catalyzed by Cat 1 or Cat 3; B generation of active
species Cat-O.
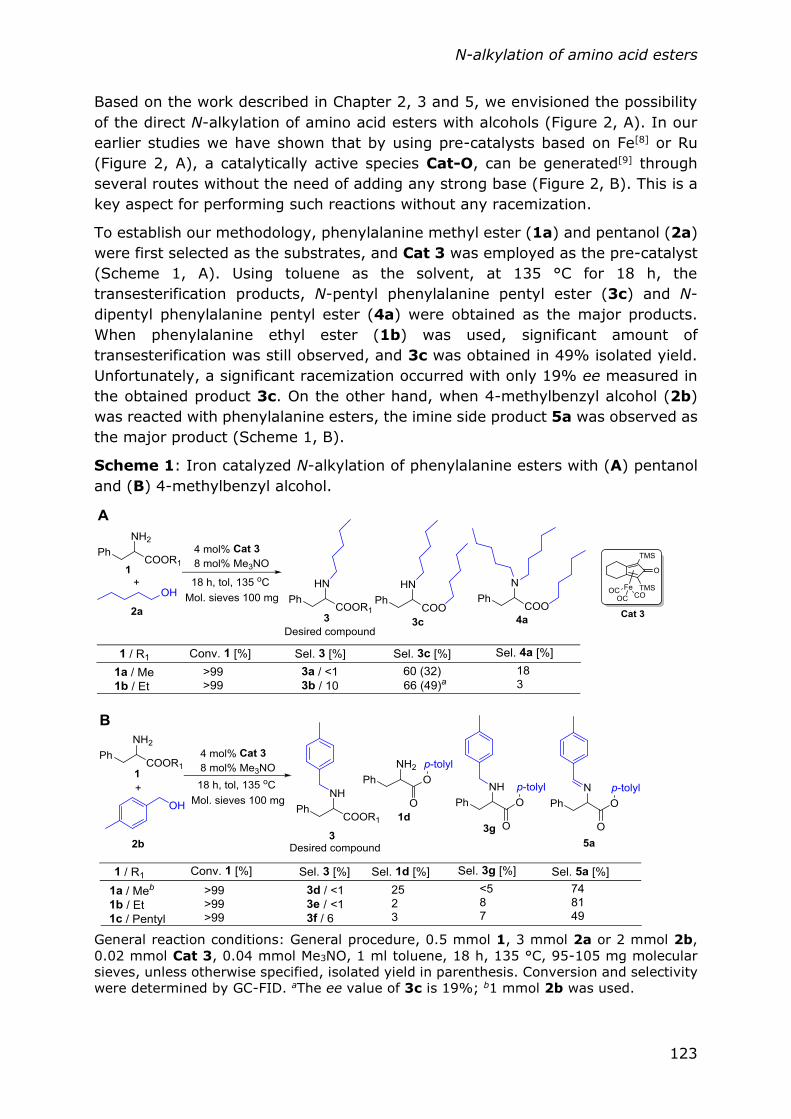
N-alkylation of amino acid esters
123
Based on the work described in Chapter 2, 3 and 5, we envisioned the possibility
of the direct N-alkylation of amino acid esters with alcohols (Figure 2, A). In our
earlier studies we have shown that by using pre-catalysts based on Fe[8] or Ru
(Figure 2, A), a catalytically active species Cat-O, can be generated[9] through
several routes without the need of adding any strong base (Figure 2, B). This is a
key aspect for performing such reactions without any racemization.
To establish our methodology, phenylalanine methyl ester (1a) and pentanol (2a)
were first selected as the substrates, and Cat 3 was employed as the pre-catalyst
(Scheme 1, A). Using toluene as the solvent, at 135 °C for 18 h, the
transesterification products, N-pentyl phenylalanine pentyl ester (3c) and N-
dipentyl phenylalanine pentyl ester (4a) were obtained as the major products.
When phenylalanine ethyl ester (1b) was used, significant amount of
transesterification was still observed, and 3c was obtained in 49% isolated yield.
Unfortunately, a significant racemization occurred with only 19% ee measured in
the obtained product 3c. On the other hand, when 4-methylbenzyl alcohol (2b)
was reacted with phenylalanine esters, the imine side product 5a was observed as
the major product (Scheme 1, B).
Scheme 1: Iron catalyzed N-alkylation of phenylalanine esters with (A) pentanol
and (B) 4-methylbenzyl alcohol.
General reaction conditions: General procedure, 0.5 mmol 1, 3 mmol 2a or 2 mmol 2b,
0.02 mmol Cat 3, 0.04 mmol Me3NO, 1 ml toluene, 18 h, 135 °C, 95-105 mg molecular
sieves, unless otherwise specified, isolated yield in parenthesis. Conversion and selectivity
were determined by GC-FID. aThe ee value of 3c is 19%; b1 mmol 2b was used.

Chapter 6
124
Since unsatisfactory results were obtained with the Fe based Cat 3, next the Ru-
based Shvo catalyst[10] (Cat 1), was investigated (Table 1). Phenylalanine pentyl
ester (1c) and 4-methylbenzyl alcohol (2b) were selected as substrates and
toluene as solvent. The use of only 0.5 mol% Cat 1, at 120 °C for 18 h, gave full
conversion of 1c and 55% selectivity of N-(4-methyl)-benzyl phenylalanine pentyl
ester (3f), although significant transesterification side-products were also
observed (Table 1, entry 1). However, when phenylalanine benzyl ester (1f) was
used as starting material, 69% conversion of 1f and 60% selectivity of N-(4-
methyl)-benzyl phenylalanine benzyl ester (3g) were observed, showing lower
reactivity but higher stability of the benzyl ester compared to the pentyl ester
(Table 1, entry 2). Gratifying, when the reaction temperature was lowered to
100 °C, no significant trans-esterification was observed and 39% conversion of 1c
and 38% selectivity for 3f were obtained (Table 1, entry 3). Next, in order to
improve substrate conversion under milder reaction temperatures, we used 4 mol%
of diphenyl phosphate (A1) based on previous studies by the group of Noyori in
Brønsted acid assisted ruthenium catalyzed hydrogenation of ketones[11]. Indeed,
conversion of 1c was improved to 55%, and 54% selectivity of 3f was achieved
(Table 1, entry 4). While phenylalanine methyl ester (1a) gave 99% conversion
and 69% yield of 3d, phenylalanine ethyl ester (1b) has shown 47% conversion
and 45% selectivity of 3e (Table 1, entry 5-6). The positive results with phosphoric
acid are likely due to the facilitation of the imine formation[12] step. Also, the
formation of ruthenium-hydride-phosphonate complex (Figure 3, C) bearing a
more acidic proton comparing to the original Cat 2-H (Figure 3, A and B),
facilitates the imine reduction step.[11,13]
Figure 3: A Generation of active species Cat 2-H; B reduction of imine intermediate with
Cat 2-H; C reduction of imine intermediate with Cat 2-H cooperated by diphenyl
phosphate.
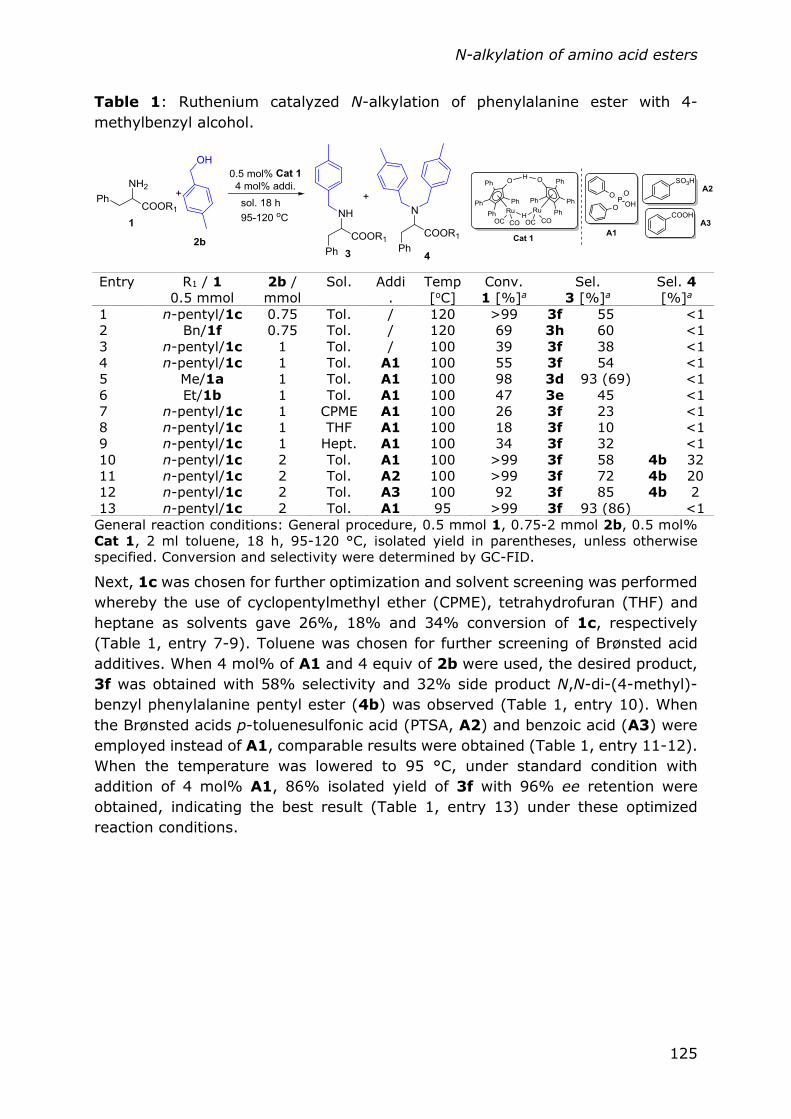
N-alkylation of amino acid esters
125
Table 1: Ruthenium catalyzed N-alkylation of phenylalanine ester with 4-
methylbenzyl alcohol.
Entry R1 / 1
0.5 mmol
2b /
mmol
Sol. Addi
.
Temp
[oC]
Conv.
1 [%]a
Sel.
3 [%]a
Sel. 4
[%]a
1 n-pentyl/1c 0.75 Tol. / 120 >99 3f 55 <1
2 Bn/1f 0.75 Tol. / 120 69 3h 60 <1
3 n-pentyl/1c 1 Tol. / 100 39 3f 38 <1
4 n-pentyl/1c 1 Tol. A1 100 55 3f 54 <1
5 Me/1a 1 Tol. A1 100 98 3d 93 (69) <1
6 Et/1b 1 Tol. A1 100 47 3e 45 <1
7 n-pentyl/1c 1 CPME A1 100 26 3f 23 <1
8 n-pentyl/1c 1 THF A1 100 18 3f 10 <1
9 n-pentyl/1c 1 Hept. A1 100 34 3f 32 <1
10 n-pentyl/1c 2 Tol. A1 100 >99 3f 58 4b 32
11 n-pentyl/1c 2 Tol. A2 100 >99 3f 72 4b 20
12 n-pentyl/1c 2 Tol. A3 100 92 3f 85 4b 2
13 n-pentyl/1c 2 Tol. A1 95 >99 3f 93 (86) <1
General reaction conditions: General procedure, 0.5 mmol 1, 0.75-2 mmol 2b, 0.5 mol%
Cat 1, 2 ml toluene, 18 h, 95-120 °C, isolated yield in parentheses, unless otherwise
specified. Conversion and selectivity were determined by GC-FID.
Next, 1c was chosen for further optimization and solvent screening was performed
whereby the use of cyclopentylmethyl ether (CPME), tetrahydrofuran (THF) and
heptane as solvents gave 26%, 18% and 34% conversion of 1c, respectively
(Table 1, entry 7-9). Toluene was chosen for further screening of Brønsted acid
additives. When 4 mol% of A1 and 4 equiv of 2b were used, the desired product,
3f was obtained with 58% selectivity and 32% side product N,N-di-(4-methyl)-
benzyl phenylalanine pentyl ester (4b) was observed (Table 1, entry 10). When
the Brønsted acids p-toluenesulfonic acid (PTSA, A2) and benzoic acid (A3) were
employed instead of A1, comparable results were obtained (Table 1, entry 11-12).
When the temperature was lowered to 95 °C, under standard condition with
addition of 4 mol% A1, 86% isolated yield of 3f with 96% ee retention were
obtained, indicating the best result (Table 1, entry 13) under these optimized
reaction conditions.
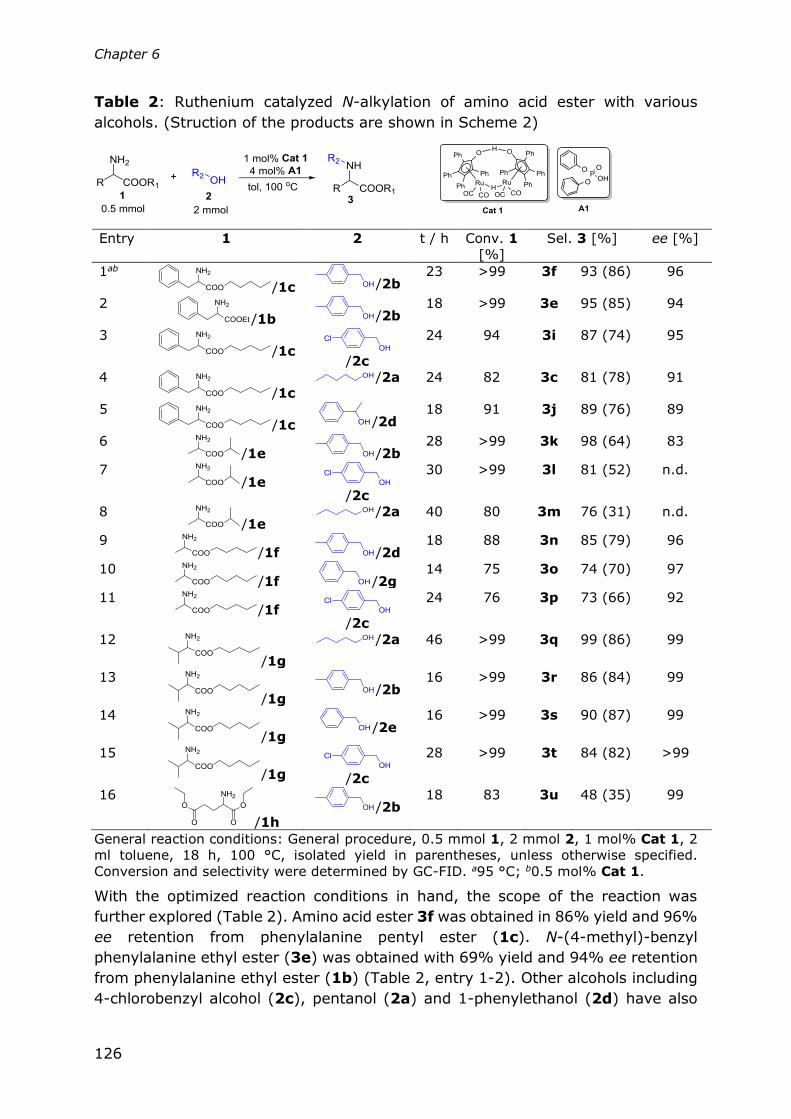
Chapter 6
126
Table 2: Ruthenium catalyzed N-alkylation of amino acid ester with various
alcohols. (Struction of the products are shown in Scheme 2)
Entry 1 2 t / h Conv. 1
[%]
Sel. 3 [%] ee [%]
1ab
/1c /2b 23 >99 3f 93 (86) 96
2 /1b /2b
18 >99 3e 95 (85) 94
3 /1c
/2c
24 94 3i 87 (74) 95
4 /1c
/2a 24 82 3c 81 (78) 91
5 /1c /2d
18 91 3j 89 (76) 89
6 /1e /2b
28 >99 3k 98 (64) 83
7 /1e
/2c
30 >99 3l 81 (52) n.d.
8 /1e
/2a 40 80 3m 76 (31) n.d.
9 /1f /2d
18 88 3n 85 (79) 96
10 /1f /2g
14 75 3o 74 (70) 97
11 /1f
/2c
24 76 3p 73 (66) 92
12
/1g
/2a 46 >99 3q 99 (86) 99
13
/1g /2b
16 >99 3r 86 (84) 99
14
/1g /2e
16 >99 3s 90 (87) 99
15
/1g /2c
28 >99 3t 84 (82) >99
16
/1h /2b
18 83 3u 48 (35) 99
General reaction conditions: General procedure, 0.5 mmol 1, 2 mmol 2, 1 mol% Cat 1, 2
ml toluene, 18 h, 100 °C, isolated yield in parentheses, unless otherwise specified.
Conversion and selectivity were determined by GC-FID. a95 °C; b0.5 mol% Cat 1.
With the optimized reaction conditions in hand, the scope of the reaction was
further explored (Table 2). Amino acid ester 3f was obtained in 86% yield and 96%
ee retention from phenylalanine pentyl ester (1c). N-(4-methyl)-benzyl
phenylalanine ethyl ester (3e) was obtained with 69% yield and 94% ee retention
from phenylalanine ethyl ester (1b) (Table 2, entry 1-2). Other alcohols including
4-chlorobenzyl alcohol (2c), pentanol (2a) and 1-phenylethanol (2d) have also
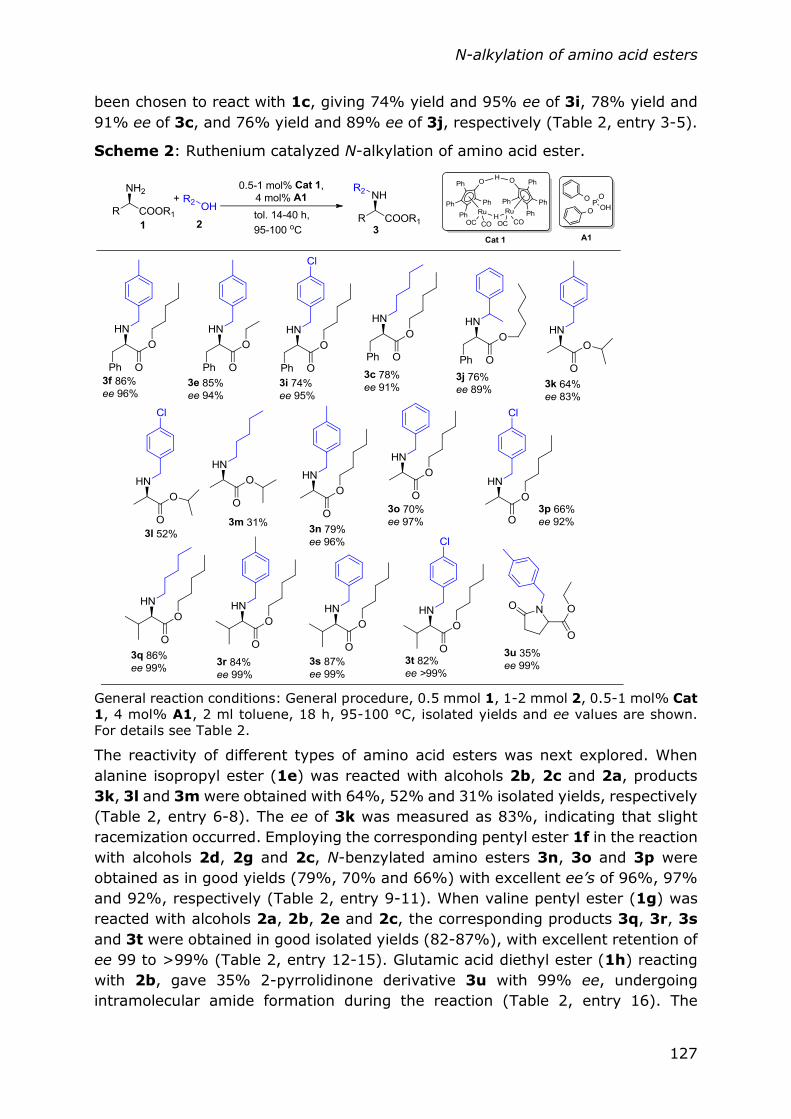
N-alkylation of amino acid esters
127
been chosen to react with 1c, giving 74% yield and 95% ee of 3i, 78% yield and
91% ee of 3c, and 76% yield and 89% ee of 3j, respectively (Table 2, entry 3-5).
Scheme 2: Ruthenium catalyzed N-alkylation of amino acid ester.
General reaction conditions: General procedure, 0.5 mmol 1, 1-2 mmol 2, 0.5-1 mol% Cat
1, 4 mol% A1, 2 ml toluene, 18 h, 95-100 °C, isolated yields and ee values are shown.
For details see Table 2.
The reactivity of different types of amino acid esters was next explored. When
alanine isopropyl ester (1e) was reacted with alcohols 2b, 2c and 2a, products
3k, 3l and 3m were obtained with 64%, 52% and 31% isolated yields, respectively
(Table 2, entry 6-8). The ee of 3k was measured as 83%, indicating that slight
racemization occurred. Employing the corresponding pentyl ester 1f in the reaction
with alcohols 2d, 2g and 2c, N-benzylated amino esters 3n, 3o and 3p were
obtained as in good yields (79%, 70% and 66%) with excellent ee’s of 96%, 97%
and 92%, respectively (Table 2, entry 9-11). When valine pentyl ester (1g) was
reacted with alcohols 2a, 2b, 2e and 2c, the corresponding products 3q, 3r, 3s
and 3t were obtained in good isolated yields (82-87%), with excellent retention of
ee 99 to >99% (Table 2, entry 12-15). Glutamic acid diethyl ester (1h) reacting
with 2b, gave 35% 2-pyrrolidinone derivative 3u with 99% ee, undergoing
intramolecular amide formation during the reaction (Table 2, entry 16). The

Chapter 6
128
isolated yields and optical purities of the obtained N-alkylated amino acid esters
are described in Table 2 and are illustrated in Scheme 2.
Table 3: Ruthenium catalyzed N-alkylation of proline ester.
Entry t [h] Conv. 1 [%] Sel. 3v [%] ee [%]
1a 24 82 62 (42) 70
2b 15 73 62 71
3c 15 70 51 62
4d 15 53 47 78
General reaction conditions: General procedure, 0.5 mmol 1, 2 mmol 2b, 0.5-1 mol% Cat
1, 2 ml toluene, 18 h, 95-100 °C, isolated yield in parentheses, unless otherwise specified.
Conversion and selectivity were determined by GC-FID. a95 °C; b2 mol% A1; c2 mol% A2
instead of A1; dno A1.
Next, proline pentyl ester (1j), comprising a secondary amine moiety was chosen
as substrate (Table 3). When 1j reacted with 2b, N-(4-methyl)-benzyl proline
pentyl ester (3v) was obtained with 42% yield and 70% ee retention (Table 3,
entry 1). The ee dropped significantly compared to the previously used substrates
that have a primary amine moiety. When decreasing the amount of Brønsted acid
additives A1 and A2 to 2 mol%, 71% and 62% ee for N-benzylated product 3v
were obtained, respectively (Table 3, entry 2-3). Without using any Brønsted acid,
78% ee of 3v and lower conversion of proline pentyl ester 1j were observed (Table
3, entry 4).
Scheme 3: Catalytic N-alkylation of prolinamide with 4-methylbenzyl alcohol.
Further, we explored the possibility of using an amide, prolinamide (6), instead of
the amino acid ester as the substrate, the corresponding product, N-(4-methyl)-
benzyl prolinamide (7) was obtained in 83% yield and ee of 59% (Scheme 3).
Interestingly, when Cat 3 was employed in the reaction between 6 and 2b, only
the corresponding imidazolidin-4-one derivative 8 was obtained, indicating a

N-alkylation of amino acid esters
129
slower imine reduction step that allowed for an intramolecular addition of the
formed imine with the amide (Scheme 3).
In the present case, with both the proline ester and prolinamide, the corresponding
N-alkylated products were obtained with ee values of 70% and 59%, respectively
(Table 3, entry 1; Scheme 3). However, the direct N-alkylation of the amino acid
itself (L-proline), gave excellent retention of ee (Figure 2, A) as described in
Chapter 5.
Conclusion
In summary, this chapter describes a general method for the direct N-alkylation of
amino acid esters with alcohols through borrowing hydrogen strategy, with good
to excellent yields and retention of ee. Only proline pentyl ester and prolinamide
show significant racemization under the established reaction conditions for N-
alkylation with alcohols. Future work should focus on the development of new
catalytic systems that would allow for even milder reaction conditions, which could
lead to higher ee retention and prevent transesterification. Future efforts should
also focus on extending the scope of the reaction to achieve the selective N-
functionalization of a broad variety of amino acid derivatives.
Experimental section
General methods
Chromatography: Merck silica gel type 9385 230-400 mesh or Merck Al2O3 90
active neutral, TLC: Merck silica gel 60, 0.25 mm or Al2O3 60 F254 neutral.
Components were visualized by UV, Ninhydrin or I2 staining. Progress of the
reactions was determined by GC-MS (GC: HP 6890, MS: HP 5973) with an HP012
column (Agilent Technologies, Palo Alto, CA). Mass spectra were recorded on an
AEI-MS-902 mass spectrometer (EI+) or a LTQ Orbitrap XL (ESI+). Conversions
were determined by GC-FID (GC: HP 6890) with an HP-5 column (Agilent
Technologies, Palo Alto, CA). GC-MS and GC-FID analysis method: 60 °C 5 min,
180 °C 5 min (10 °C /min), 260 °C 5 min (10 °C/min). 1H- and 13C NMR spectra
were recorded on a Varian AMX400 (400 and 100 MHz, respectively) using CDCl3,
CD3OD, or CD2Cl2 as solvent. Chemical shift values are reported in ppm with the
solvent resonance as the internal standard (CDCl3: 7.26 for 1H, 77.00 for 13C;
CD3OD: 3.31 for 1H, 49.00 for 13C; CD2Cl2: 5.32 for 1H, 53.84 for 13C). Data are
reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t =
triplet, q = quartet, br. = broad, m = multiplet), coupling constants (Hz), and
integration. All reactions were carried out under an Argon atmosphere using oven
(110 °C) dried glassware and using standard Schlenk techniques. THF and toluene
were collected from a MBRAUN solvent purification system (MB SPS-800). Dioxane
(99.5%, extra dry), dichloroethane (DCE, 99.8%, extra dry), N,N-
dimethylformamide (DMF, 99.8%, extra dry) and acetonitrile (CH3CN, 99.9%,
extra dry) were purchased from Acros without further purification. Molecular sieves
4A were purchased from Acros, and heated in a Schlenck under 180 °C in vacuo
overnight for activation before use. The synthesis of Cat 3 was carried out as
described in Chapter 2. Cat 1 was purchased from Strem. All other reagents were

Chapter 6
130
purchased from Sigma, TCI or Acros in reagent or higher grade and were used
without further purification.
Representative procedure
General procedure: An oven-dried 20 ml Schlenk tube was equipped with a
stirring bar. The solid starting materials were added into the Schlenk tube under
air, the Schlenk tube was subsequently connected to a vacuum/argon Schlenk line
and a vacuum-backfill cycle was performed three times. Liquid starting materials
and solvent were charged under an argon stream. The Schlenk tube was sealed
with a screw cap. The mixture was rapidly stirred at room temperature for 1 min,
then it was placed into a pre-heated oil bath at the appropriate temperature and
the mixture was stirred for a given time. The reaction mixture was cooled down to
room temperature and the crude mixture was concentrated in vacuum and purified
by flash column chromatography to provide the product. The enantiomeric excess
(ee) was measured by chiral HPLC.
Procedure of N-alkylation of valine pentyl ester (1g) with 4-methyl benzyl
alcohol (2b): An oven-dried 20 ml Schlenk tube, equipped with a stirring bar,
was charged with 4-methylbenzyl alcohol (2 mmol, 0.244 g) and Cat 1 (1 mol%,
5.4 mg) under air. The Schlenk tube was subsequently connected to a
vacuum/argon Schlenk line and a vacuum-backfill cycle was performed three times.
Then valine pentyl ester (0.5 mmol, 0.094 g) and 2 ml toluene were charged under
an argon stream. The Schlenk tube was sealed with a screw cap. The mixture was
rapidly stirred at room temperature for 1 min, then was placed into a pre-heated
oil bath at 100 °C for 18 h. The reaction mixture was cooled down to room
temperature, concentrated in vacuum. The residue was purified by flash
chromatography (SiO2, pentane/EtOAc 100:0 to 95:5) to provide the product N-
(4-methylbenyl) valine penyl ester 3r (0.124 g, 84% yield) with ee value of 99%
measured by chiral HPLC.

N-alkylation of amino acid esters
131
Spectral data and ee determination of isolated compounds
N-pentyl-phenylalanine pentyl ester (3c): Synthesized
according to General procedure. Phenylalanine penyl ester (0.118
g, 0.50 mmol) affords 3c (0.119 g, 78% yield). Light yellow oil
compound obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.14 –
7.32 (m, 5H), 3.94 – 4.08 (m, 2H), 3.47 – 3.52 (m, 2H), 2.94 – 3.02 (m, 1H),
2.86 – 2.94 (m, 1H), 2.52 – 2.62 (m, 1H), 2.42 – 2.52 (m, 1H), 1.38 – 1.55 (m,
4H), 1.16 – 1.32 (m, 8H), 0.80 – 0.92 (m, 6H). 13C NMR (100 MHz, CDCl3) δ
174.80, 137.34, 129.15, 128.34, 126.62, 64.70, 63.20, 48.15, 39.81, 29.71,
29.35, 28.20, 27.96, 22.49, 22.24, 13.99, 13.90. The physical data were identical
in all respects to those previously reported HRMS (APCI+, m/z): calculated for
C19H32NO2 [M+H]+: 306.24276; found: 306.24298.
The ee was determined by chiral HPLC analysis. Chiralcel OZ-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 221 nm; retention times 21.3 min (minor) and 24.1 min (major).
N-(4-methylbenzyl)-phenylalanine methyl ester (3d):
Synthesized according to General procedure. Phenylalanine methyl
ester (0.090 g, 0.50 mmol) affords 3d (0.098 g, 69% yield). Light
yellow oil compound obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.05 –
7.34 (m, 9H), 3.73 – 3.84 (m, 1H), 3.66 (s, 3H), 3.59 – 3.65 (m, 1H), 3.53 – 3.59
(m, 1H), 2.93 – 3.04 (m, 2H), 2.34 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 174.95,
137.25, 136.52, 136.41, 129.15, 128.95, 128.30, 128.03, 126.60, 61.94, 51.65,
51.55, 39.65, 21.02. The physical data were identical in all respects to those
previously reported HRMS (APCI+, m/z): calculated for C18H22NO2 [M+H]+:
284.16451; found: 284.16520.
N-(4-methylbenzyl)-phenylalanine ethyl ester (3e):
Synthesized according to General procedure. Phenylalanine ethyl
ester (0.097 g, 0.50 mmol) affords 3e (0.126 g, 85% yield). Light
yellow oil compound obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.05 –
7.34 (m, 9H), 4.06 – 4.17 (m, 2H), 3.75 – 3.83 (m, 1H), 3.58 – 3.67 (m ,1H),
3.50 – 3.57 (m, 1H), 2.90 – 3.03 (m, 2H), 2.33 (s, 3H). 13C NMR (100 MHz, CDCl3)
δ 174.54, 137.34, 136.53, 136.51, 129.23, 128.96, 128.26, 128.06, 126.56,
62.01, 60.52, 51.65, 39.73, 21.04, 14.15. The physical data were identical in all
respects to those previously reported HRMS (APCI+, m/z): calculated for
C19H24NO2 [M+H]+: 298.18053; found: 298.18016.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.2:0.8); flow rate: 0.5 ml/min;
detection: UV 190 nm; retention times 24.5 min (minor) and 27.1 min (major).
N-(4-methylbenzyl)-phenylalanine pentyl ester (3f): Synthesized according
to General procedure. Phenylalanine pentyl ester (0.118 g, 0.50 mmol) affords 3f
(0.146 g, 86% yield). Light yellow oil compound obtained after column

Chapter 6
132
chromatography (SiO2, Pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.05 – 7.34 (m, 9H), 3.97 – 4.10 (m, 2H), 3.73
– 3.84 (m, 1H), 3.58 – 3.66 (m, 1H), 3.50 – 3.57 (m, 1H), 2.90 –
3.03 (m, 2H), 2.33 (s, 3H), 1.48 – 1.60 (m, 2H), 1.17 – 1.37 (m,
4H), 0.82 – 0.96 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 174.69,
137.36, 136.53, 129.22, 128.97, 128.28, 128.07, 126.57, 77.32,
77.00, 76.68, 64.74, 62.07, 51.67, 39.79, 28.22, 27.97, 22.24, 21.05, 13.91. The
physical data were identical in all respects to those previously reported HRMS
(APCI+, m/z): calculated for C22H30NO2 [M+H]+: 340.22711; found: 340.22764.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.7:0.3); flow rate: 0.5 ml/min;
detection: UV 230 nm; retention times 61.5 min (major) and 69.4 min (minor).
N-(4-chlorobenzyl)-phenylalanine pentyl ester (3i):
Synthesized according to General procedure. Phenylalanine pentyl
ester (0.118 g, 0.50 mmol) affords 3i (0.133 g, 74% yield). Light
yellow oil compound obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.10 –
7.34 (m, 9H), 3.99 – 4.12 (m, 2H), 3.75 – 3.84 (m, 1H), 3.55 –
3.65 (m, 1H), 3.44 – 3.53 (m, 1H), 2.89 – 3.03 (m, 2H), 1.46 – 1.63 (m, 2H),
1.16 – 1.39 (m, 4H), 0.83 – 0.96 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 174.55,
138.08, 137.22, 132.55, 129.34, 129.17, 128.32, 128.26, 126.59, 64.80, 61.90,
51.11, 39.76, 28.16, 27.92, 22.20, 13.89. HRMS (APCI+, m/z): calculated for
C21H27ClNO2 [M+H]+: 360.17248; found: 360.17287.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.7:0.3); flow rate: 0.5 ml/min;
detection: UV 223 nm; retention times 49.7 min (minor) and 54.8 min (major).
N-(1-phenylethyl)-phenylalanine pentyl ester (3j): Synthesi-
zed according to General procedure. Phenylalanine pentyl ester
(0.118 g, 0.50 mmol) affords 3j (0.129 g, 76% yield). Light yellow
oil compound obtained after column chromatography (SiO2,
Pentane/EtOAc 100:0 to 95:5). 1H NMR (400 MHz, CDCl3) δ 7.17 –
7.33 (m, 6H), 7.05 – 7.16 (m, 4H), 3.97 – 4.12 (m, 2H), 3.65 – 3.75 (m, 1H),
3.22 – 3.32 (m, 1H), 2.80 – 2.95 (m, 2H), 1.48 – 1.60 (m, 2H), 1.18 – 1.37 (m,
7H), 0.83 – 0.95 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 175.25, 144.78, 137.38,
129.28, 128.25, 128.14, 126.84, 126.71, 126.46, 77.00, 64.64, 60.30, 56.52,
40.14, 28.22, 27.96, 25.34, 22.23, 13.93. HRMS (APCI+, m/z): calculated for
C22H30NO2 [M+H]+: 340.22711; found: 340.22739.
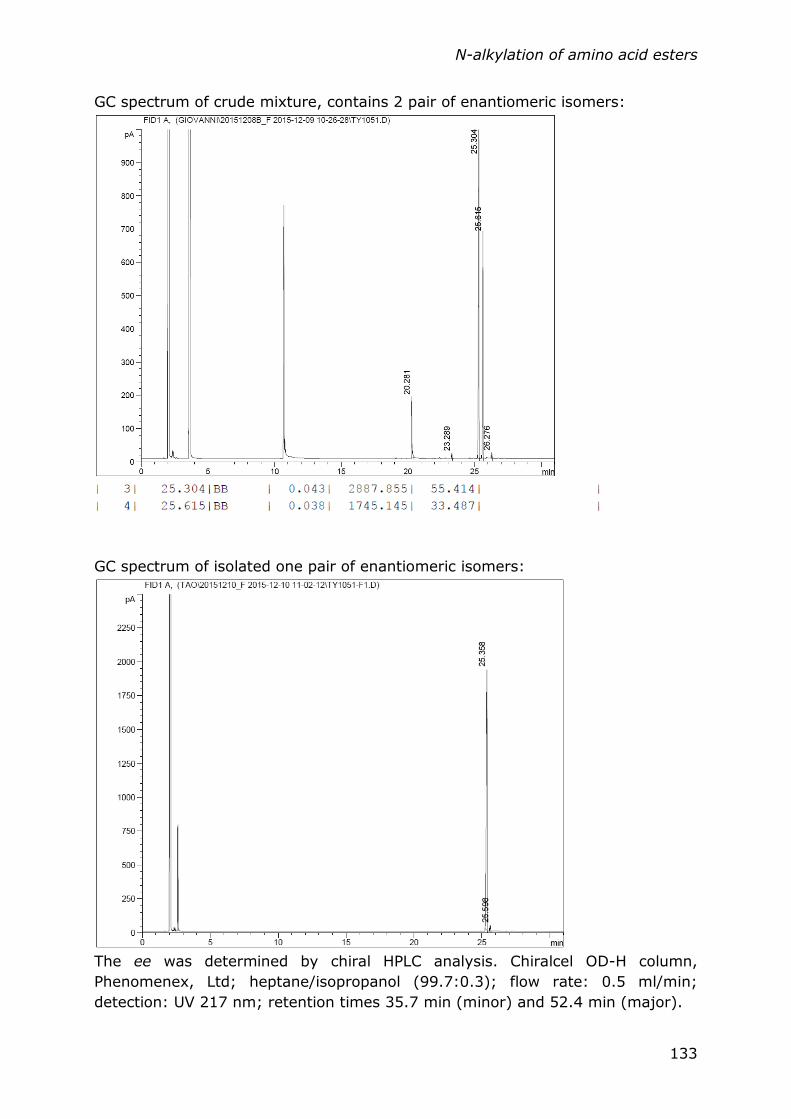
N-alkylation of amino acid esters
133
GC spectrum of crude mixture, contains 2 pair of enantiomeric isomers:
GC spectrum of isolated one pair of enantiomeric isomers:
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.7:0.3); flow rate: 0.5 ml/min;
detection: UV 217 nm; retention times 35.7 min (minor) and 52.4 min (major).

Chapter 6
134
N-(4-methylbenzyl)-alanine isopropyl ester (3k): Synthesized
according to General procedure. Alanine isopropyl ester (0.066 g, 0.50
mmol) affords 3k (0.075 g, 64% yield). Light yellow oil compound
obtained after column chromatography (SiO2, Pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CDCl3) δ 7.12 – 7.32 (m, 4H), 5.03 – 5.19 (m,
1H), 3.74 – 3.86 (m, 1H), 3.60 – 3.73 (m, 1H), 3.30 – 3.42 (m, 1H),
2.37 (s, 3H), 1.23 – 1.38 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 175.19, 136.66,
136.52, 129.00, 128.16, 77.00, 67.95, 55.96, 51.56, 21.84, 21.71, 21.00, 18.98.
HRMS (APCI+, m/z): calculated for C14H22NO2 [M+H]+: 236.16451; found:
236.16467.
The ee was determined by chiral HPLC analysis. Chiralcel OZ-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 217 nm; retention times 28.0 min (minor) and 32.3 min (major).
N-(4-chlorobenzyl)-alanine isopropyl ester (3l): Synthesized
according to General procedure. Alanine isopropyl ester (0.066 g, 0.50
mmol) affords 3l (0.075 g, 52% yield). Light yellow oil compound
obtained after column chromatography (SiO2, Pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CDCl3) δ 7.24 – 7.35 (m, 4H), 4.98 – 5.12
(m, 1H), 3.70 – 3.82 (m, 1H), 3.54 – 3.68 (m, 1H), 3.24 – 3.33 (m,
1H), 1.05 – 1.45 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 175.11, 138.21, 132.73,
129.55, 128.47, 68.17, 64.28, 55.99, 51.13, 21.87, 21.73, 19.02. HRMS (APCI+,
m/z): calculated for C13H19ClNO2 [M+H]+: 256.10988; found: 256.11062.
N-pentyl-alanine isopropyl ester (3m): Synthesized according to
General procedure. Alanine isopropyl ester (0.066 g, 0.50 mmol) affords
3m (0.31 g, 31% yield). Light yellow oil compound obtained after column
chromatography (SiO2, Pentane/EtOAc 100:0 to 95:5). 1H NMR (400
MHz, CDCl3) δ 4.96 – 5.10 (m, 1H), 3.21 – 3.32 (m, 1H), 2.50 – 2.59
(m, 1H), 2.41 – 2.50 (m, 1H), 1.38 – 1.55 (m, 2H), 1.17 – 1.37 (m, 13H), 0.80 –
0.95 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 175.39, 67.93, 56.86, 47.98, 29.87,
29.44, 22.51, 21.86, 21.73, 19.05, 13.97.
N-(4-methylbenzyl)-alanine pentyl ester (3n): Synthesized
according to General procedure. Alanine pentyl ester (0.080 g, 0.50
mmol) affords 3n (0.104 g, 79% yield). Light yellow oil compound
obtained after column chromatography (SiO2, Pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CDCl3) δ 7.17 – 7.25 (m, 2H), 7.07 – 7.16 (m,
2H), 4.07 – 4.17 (m, 2H), 3.72 – 3.82 (m, 1H), 3.57 – 3.67 (m, 1H),
3.32 – 3.42 (m, 1H), 2.33 (s, 3H), 1.58 – 1.73 (m, 2H), 1.26 – 1.42 (m, 7H), 0.84
– 0.98 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 175.82, 136.62, 136.61, 129.05,
128.20, 64.80, 55.89, 51.64, 28.33, 28.03, 22.28, 21.07, 19.14, 13.95. HRMS
(APCI+, m/z): calculated for C16H26NO2 [M+H]+: 264.19581; found: 264.19614.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 223 nm; retention times 20.6 min (major) and 25.5 min (minor).

N-alkylation of amino acid esters
135
N-benzyl-alanine pentyl ester (3o): Synthesized according to
General procedure. Alanine pentyl ester (0.080 g, 0.50 mmol) affords
3o (0.087 g, 70% yield). Light yellow oil compound obtained after
column chromatography (SiO2, Pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.05 – 7.55 (m, 5H), 4.03 – 4.18 (m, 2H), 3.75 –
3.85 (m, 1H), 3.61 – 3.71 (m, 1H), 3.31 – 3.42 (m, 1H), 1.57 – 1.72 (m, 2H),
1.22 – 1.42 (m, 7H), 0.82 – 0.97 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 175.79,
139.69, 128.38, 128.23, 127.04, 77.00, 64.82, 55.98, 51.93, 28.33, 28.03, 22.27,
19.15, 13.94. HRMS (APCI+, m/z): calculated for C15H24NO2 [M+H]+: 250.18016;
found: 250.18030.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 211 nm; retention times 22.2 min (major) and 28.0 min (minor).
N-(4-chlorobenzyl)-alanine pentyl ester (3p): Synthesized
according to General procedure. Alanine pentyl ester (0.080 g, 0.50
mmol) affords 3p (0.093 g, 66% yield). Light yellow oil compound
obtained after column chromatography (SiO2, Pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CDCl3) δ 7.15 – 7.30 (m, 4H), 4.02 – 4.16
(m, 2H), 3.70 – 3.79 (m, 1H), 3.53 – 3.64 (m, 1H), 3.25 – 3.36 (m,
1H), 1.54 – 1.67 (m, 2H), 1.22 – 1.38 (m, 7H), 0.80 – 0.95 (m, 3H). 13C NMR
(100 MHz, CDCl3) δ 13C NMR (101 MHz, cdcl3) δ 175.68, 138.21, 132.69, 129.51,
128.44, 64.85, 55.88, 51.15, 28.29, 27.99, 22.24, 19.16, 13.93. HRMS (APCI+,
m/z): calculated for C15H23ClNO2 [M+H]+: 284.14118; found: 284.14156.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 190 nm; retention times 17.3 min (minor) and 18.1 min (major).
N-pentyl-valine pentyl ester (3q): Synthesized according to
General procedure. Valine pentyl ester (0.094 g, 0.50 mmol) affords
3q (0.111 g, 86% yield). Light yellow oil compound obtained after
column chromatography (SiO2, Pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.09 – 7.28 (m, 4H), 4.03 – 4.15 (m, 2H), 2.88 –
2.97 (m, 1H), 2.47 – 2.58 (m, 1H), 2.32 – 2.43 (m, 1H), 1.78 – 1.94 (m, 1H),
1.55 – 1.68 (m, 1H), 1.36 – 1.53 (m, 3H), 1.17 – 1.36 (m, 8H), 0.77 – 0.98 (m,
12H). 13C NMR (100 MHz, CDCl3) δ 175.45, 67.56, 64.37, 48.74, 31.64, 29.88,
29.41, 28.37, 28.07, 22.53, 22.23, 19.09, 18.82, 13.99, 13.89. HRMS (APCI+,
m/z): calculated for C15H32NO2 [M+H]+: 258.24276; found: 258.24287. The ee
was determined by chiral HPLC analysis. Chiralcel OZ-H column, Phenomenex, Ltd;
heptane/isopropanol (99.8:0.2); flow rate: 0.5 ml/min; detection: UV 223 nm;
retention times 13.6 min (major) and 15.6 min (minor).
N-(4-methylbenzyl)-valine pentyl ester (3r): Synthesized
according to General procedure. Valine pentyl ester (0.094 g, 0.50
mmol) affords 3r (0.124 g, 84% yield). Light yellow oil compound
obtained after column chromatography (SiO2, Pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CDCl3) δ 4.03 – 4.15 (m, 2H), 2.88 – 2.97

Chapter 6
136
(m, 1H), 2.47 – 2.58 (m, 1H), 2.32 – 2.43 (m, 1H), 1.78 – 1.94 (m, 1H), 1.55 –
1.68 (m, 1H), 1.36 – 1.53 (m, 3H), 1.17 – 1.36 (m, 8H), 0.77 – 0.98 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 175.45, 67.56, 64.37, 48.74, 31.64, 29.88, 29.41,
28.37, 28.07, 22.53, 22.23, 19.09, 18.82, 13.99, 13.89. HRMS (APCI+, m/z):
calculated for C18H30NO2 [M+H]+: 292.22711; found: 292.22741.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 221 nm; retention times 14.1 min (major) and 20.9 min (minor).
N-benzyl-valine pentyl ester (3s): Synthesized according to
General procedure. Valine pentyl ester (0.094 g, 0.50 mmol) affords
3s (0.120 g, 87% yield). Light yellow oil compound obtained after
column chromatography (SiO2, Pentane/EtOAc 100:0 to 95:5). 1H NMR
(400 MHz, CDCl3) δ 7.20 – 7.45 (m, 5H), 4.07 – 4.20 (m, 2H), 3.78 –
3.89 (m, 1H), 3.54 – 3.68 (m, 1H), 2.95 – 3.05 (m, 1H), 1.85 – 1.99 (m, 1H),
1.58 – 1.73 (m, 2H), 1.27 – 1.43 (m, 4H), 0.83 – 1.02 (m, 9H). 13C NMR (100
MHz, CDCl3) δ 175.32, 140.13, 128.24, 128.22, 126.91, 66.61, 64.51, 52.52,
31.69, 28.39, 28.10, 22.26, 19.31, 18.61, 13.94. HRMS (APCI+, m/z): calculated
for C17H28NO2 [M+H]+: 278.21146; found: 278.21182.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 221 nm; retention times 13.7 min (major) and 19.3 min (minor).
N-(4-chlorobenzyl)-valine pentyl ester (3t): Synthesized
according to General procedure. Valine pentyl ester (0.094 g, 0.50
mmol) affords 3t (0.128 g, 87% yield). Light yellow oil compound
obtained after column chromatography (SiO2, Pentane/EtOAc 100:0 to
95:5). 1H NMR (400 MHz, CDCl3) δ 7.18 – 7.36 (m, 4H), 4.05 – 4.17
(m, 2H), 3.75 – 3.83 (m, 1H), 3.47 – 3.58 (m, 1H), 2.90 – 2.98 (m,
1H), 1.84 – 1.97 (m, 1H), 1.57 – 1.70 (m, 2H), 1.25 – 1.42 (m, 4H), 0.75 – 1.03
(m, 9H). 13C NMR (100 MHz, CDCl3) δ 175.21, 138.62, 132.56, 129.52, 128.32,
77.32, 77.00, 76.68, 66.44, 64.56, 51.73, 31.65, 28.36, 28.07, 22.24, 19.33,
18.50, 13.92. HRMS (APCI+, m/z): calculated for C17H27ClNO2 [M+H]+: 312.17248;
found: 312.17291.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 221 nm; retention times 12.3 min (major) and 13.3 min (minor).
ethyl N-(4-methylbenzyl)-5-oxopyrrolidine-2-carboxylate (3u):
Synthesized according to General procedure. Glutamic acid diethyl
ester (0.102 g, 0.50 mmol) affords 3u (0.038 g, 35% yield). Light
yellow oil compound obtained after column chromatography (SiO2,
Pentane/EtOAc 70:30 to 50:50). 1H NMR (400 MHz, CDCl3) δ 7.03 –
7.15 (m, 4H), 4.94 – 5.04 (m, 1H), 4.05 – 4.20 (m, 2H), 3.88 – 3.97 (m, 2H),
2.48 – 2.62 (m, 1H), 2.34 – 2.45 (m, 1H), 2.31 (s, 3H), 2.13 – 2.28 (m, 1H), 1.98
– 2.09 (m, 1H), 1.18 – 1.28 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 174.93, 171.74,
137.42, 132.67, 129.31, 128.46, 77.00, 61.36, 58.70, 45.23, 29.57, 22.75, 21.05,

N-alkylation of amino acid esters
137
14.08. HRMS (APCI+, m/z): calculated for C15H20NO3 [M+H]+: 262.14377; found:
262.14421.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (96:4); flow rate: 0.5 ml/min; detection:
UV 223 nm; retention times 31.3 min (minor) and 40.0 min (major).
N-(4-methylbenzyl)-proline pentyl ester (3v): Synthesized
according to General procedure. Proline pentyl ester (0.093 g, 0.50
mmol) affords 3u (0.061 g, 42% yield). Light yellow oil compound
obtained after column chromatography (Al2O3, Pentane/EtOAc 95:5 to
90:10). 1H NMR (400 MHz, CDCl3) δ 7.03 – 7.28 (m, 4H), 3.98 – 4.18
(m, 2H), 3.83 – 3.94 (m, 1H), 3.45 – 3.58 (m, 1H), 3.17 – 3.31 (m, 1H), 2.96 –
3.08 (m, 1H), 2.30 – 2.44 (m, 1H), 2.32 (s, 3H), 2.04 – 2.18 (m, 1H), 1.83 – 2.01
(m, 2H), 1.70 – 1.81 (m, 1H), 1.55 – 1.69 (m, 2H), 1.21 – 1.43 (m, 4H), 0.78 –
1.02 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 174.17, 136.53, 135.19, 129.12,
128.78, 65.13, 64.61, 58.13, 52.99, 29.27, 28.29, 28.01, 22.85, 22.27, 21.07,
13.93. HRMS (APCI+, m/z): calculated for C18H28NO2 [M+H]+: 290.21146; found:
290.21177.
The ee was determined by chiral HPLC analysis. Chiralcel OZ-H column,
Phenomenex, Ltd; heptane/isopropanol (99.5:0.5); flow rate: 0.5 ml/min;
detection: UV 225 nm; retention times 23.1 min (minor) and 26.8 min (major).
N-(4-methylbenzyl)-prolinamide (7): Synthesized according to
General procedure. Prolinamide (0.057 g, 0.50 mmol) affords 3v
(0.090 g, 83% yield). Light yellow oil compound obtained after column
chromatography (SiO2, Pentane/EtOAc 20:80 to 0:100). 1H NMR (400
MHz, CDCl3) δ 7.20 – 7.35 (br.s, 1H), 7.07 – 7.20 (m, 4H), 6.20 – 6.40
(br.s, 1H), 3.84 – 3.97 (m, 1H), 3.35 – 3.50 (m, 1H), 3.09 – 3.23 (m, 1H), 2.94
– 3.04 (m, 1H), 2.26 – 2.40 (m, 1H), 2.33 (s, 3H), 2.14 – 2.25 (m, 1H), 1.85 –
1.97 (m, 1H), 1.64 – 1.82 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 178.30, 136.78,
135.38, 129.02, 128.54, 67.15, 59.31, 53.62, 30.50, 23.93, 21.02. HRMS (APCI+,
m/z): calculated for C13H19NO2 [M+H]+: 219.14919; found: 219.14921.
The ee was determined by chiral HPLC analysis. Chiralcel OD-H column,
Phenomenex, Ltd; heptane/isopropanol (94:6); flow rate: 0.5 ml/min; detection:
UV 190 nm; retention times 34.8 min (minor) and 51.0 min (major).
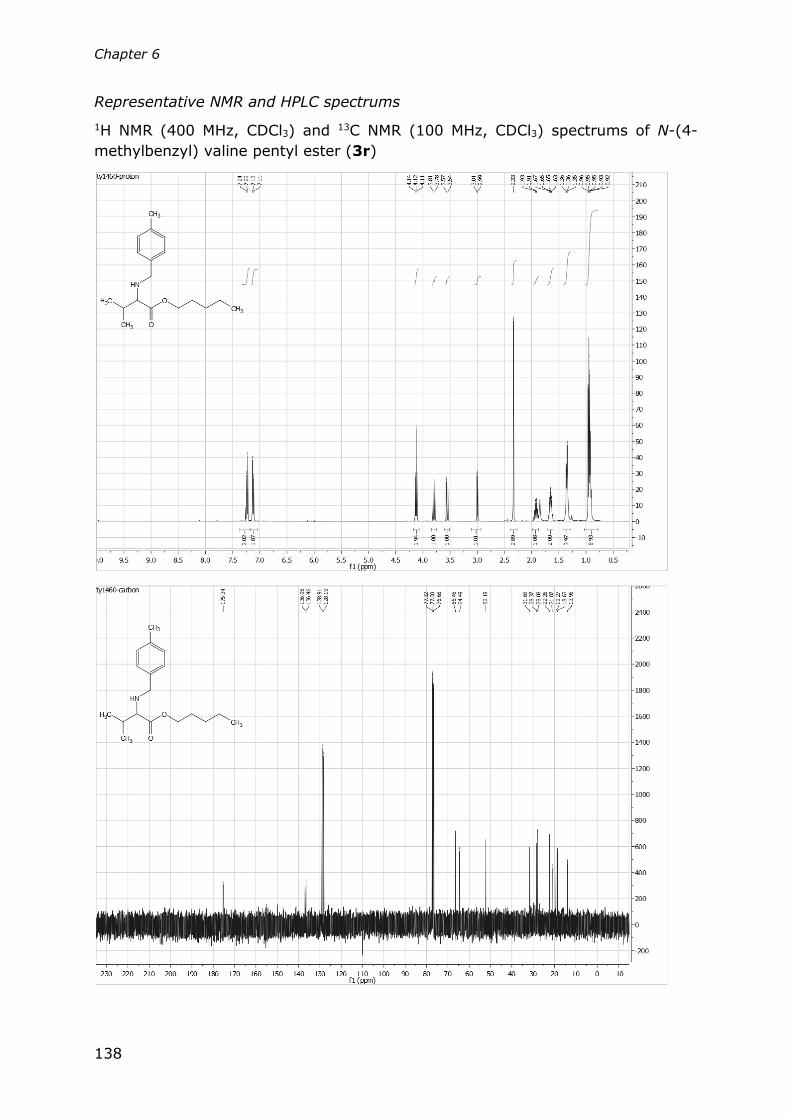
Chapter 6
138
Representative NMR and HPLC spectrums
1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) spectrums of N-(4-
methylbenzyl) valine pentyl ester (3r)
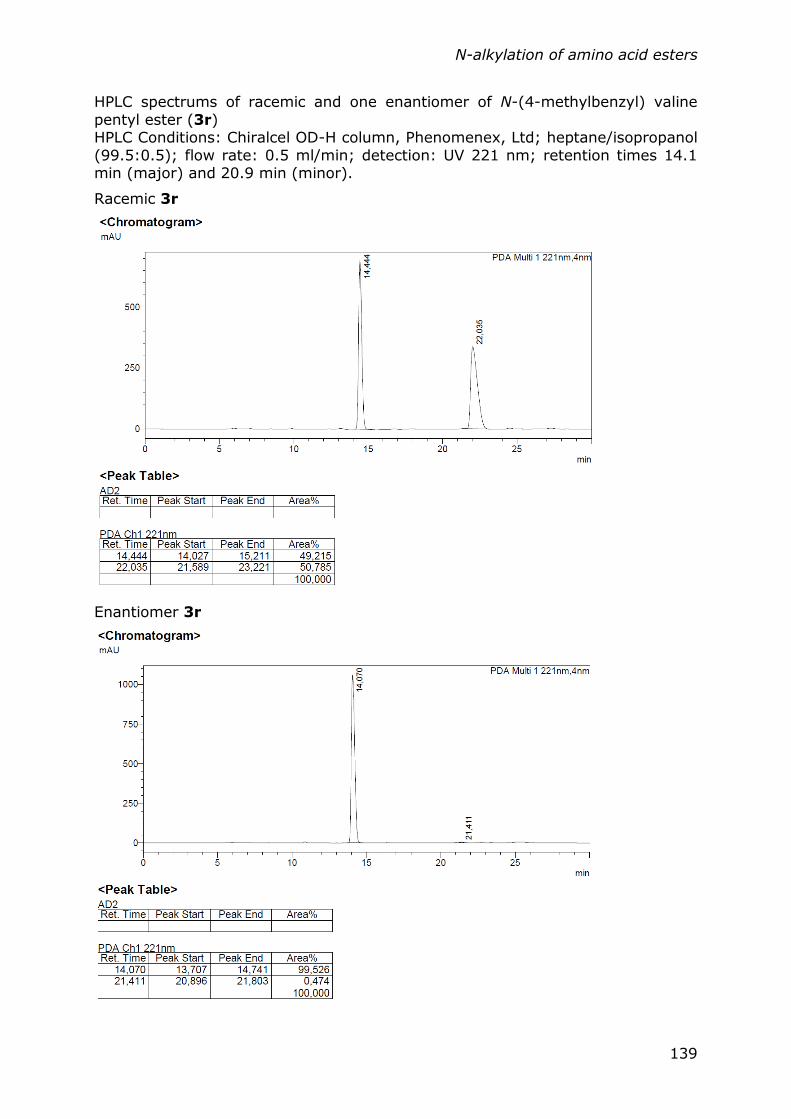
N-alkylation of amino acid esters
139
HPLC spectrums of racemic and one enantiomer of N-(4-methylbenzyl) valine
pentyl ester (3r) HPLC Conditions: Chiralcel OD-H column, Phenomenex, Ltd; heptane/isopropanol
(99.5:0.5); flow rate: 0.5 ml/min; detection: UV 221 nm; retention times 14.1 min (major) and 20.9 min (minor).
Racemic 3r
Enantiomer 3r

Chapter 6
140
References
[1] T. Szucs, Drugs, 1991, 41, 18-24.
[2] W. J. Greenlee, P. L. Allibone, D. S. Perlow, A. A. Patchett, E. H. Ulm, T. C. Vassil, J.
Med. Chem., 1985, 28, 434–442.
[3] S.-J. Ho, T. A. Brighton, Vasc. Health Risk Manag., 2006, 2, 49–58.
[4] M. G. Edwards, R. F. R. Jazzar, B. M. Paine, D. J. Shermer, M. K. Whittlesey, J. M. J.
Williams, D. D. Edney, Chem. Commun., 2004, 90–91.
[5] a) M. H. S. A. Hamid, P. A. Slatford, J. M. J. Williams, Adv. Synth. Catal., 2007, 349,
1555−1575; b) T. D. Nixon, M. K. Whittlesey, J. M. J. Williams, Dalton Trans., 2009,
753−762; c) G. Guillena, D. J. Ramon, M. Yus, Chem. Rev., 2010, 110, 1611−1641;
d) S. Bahn, S. Imm, L. Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem,
2011, 3, 1853−1864; e) G. E. Dobereiner, R. H. Crabtree, Chem. Rev., 2010, 110,
681−703; f) C. Gunanathan, D. Milstein, Science, 2013, 341, 1229712; g) Q. Yang,
Q. Wang, Z. Yu, Chem. Soc. Rev., 2015, 44, 2305—2329; g) for alkylation of an
acidic proton free amino acid ester see: J. Leonard, A. J. Blacker, S. P. Marsden, M.
F. Jones, K. R. Mulholland, R. Newton, Org. Process Res. Dev., 2015, 19, 1400−1410.
[6] One example shows ester group intolerance: M. Zhang, S. Imm, S. Bahn, H.
Neumann, M. Beller, Angew. Chem. Int. Ed., 2011, 50, 11197–11201.
[7] Selected factors leading to amino acid esters racemization: a) K. Kaiser, R. Benner,
Limnol. Oceanogr.: Methods, 2005, 3, 318–325; b) L. G. Barry, M. Pugniere, B.
Castro, A. Previero, Int. J. Peptide Protein Res., 1993, 41, 323-325; c) M. Pugniere,
C. San Juan, A. Previero, Biotechnol. Lett., 1985, 7, 31-36; d) M. Bodanszky, A.
Bodanszky, Chem. Commun., 1967, 591-593.
[8] T. Yan, B. L. Feringa, K. Barta, Nat. Commun., 2014, 5, 5602.
[9] a) T. Yan, B. L. Feringa, K. Barta, ACS Catal., 2016, 6, 381–388; b) T. Yan, K. Barta,
ChemSusChem, 2016, 9, 2321–2325; c) A. J. Rawlings, L. J. Diorazio, M. Wills, Org.
Lett., 2015, 17, 1086−1089; d) H.-J. Pan, T. W. Ng, Y. Zhao, Chem. Commun.,
2015, 51, 11907-11910.
[10] Selected review and reactivities with Shvo catalyst: a) B. L. Conley, M. K. Pennington-
Boggio, E. Boz, T. J. Williams, Chem. Rev., 2010, 110, 2294–2312; b) D. Hollmann,
S. Bahn, A. Tillack, M. Beller, Angew. Chem. Int. Ed., 2007, 46, 8291–8294; c) S.
Imm, S. Bahn, A. Tillack, K. Mevius, L. Neubert, M. Beller, Chem. Eur. J., 2010, 16,
2705–2709; d) C. Segarra, E. Mas-Marza, J. A. Mata, E. Peris, Adv. Synth. Catal.,
2011, 353, 2078–2084.
[11] The ruthenium hydride-phosphonate interaction was initially reported by M.
Tokunaga, PhD Thesis, Nagoya University, 1995, later on cited in review: R. Noyori,
T. Ohkuma, Angew. Chem. Int. Ed., 2001, 40, 40-73.
[12] One example shows the addition of acid facilitates the formation of an imine
intermediate in the catalytic reductive amination of aldehydes with amines, see: A.
Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard, J.-L. Renaud, Angew.
Chem. Int. Ed., 2012, 51, 4976–4980.
[13] Two recent examples of metal-phosphoric acid interaction in catalytic redox
chemistry, see: a) S. Zhou, S. Fleischer, K. Junge, M. Beller, Angew. Chem. Int. Ed.,
2011, 50, 5120–5124; b) Y. Zhang, C.-S. Lim, D. S. B. Sim, H.-J. Pan, Y. Zhao,
Angew. Chem. Int. Ed., 2014, 53, 1399–1403.

Nederlandse samenvatting
141
Nederlandse samenvatting
Dit proefschrift beschrijft de ontwikkeling van nieuwe methodes voor het vormen
van koolstof-stikstof verbindingen via katalytische alcohol activatie met de nadruk
op de directe aminering van alcoholen via een cyclus van het onttrekken en
toevoegen van waterstof.
Alcoholen zijn goedkope, atoxische en gemakkelijk verkrijgbare stoffen. Ze worden
in de organische synthese veel gebruikt als alkylerende. Daarentegen is de directe
aminering van alcoholen bijzonder uitdagend omdat de hydroxylgroep (–OH) een
slecht vertrekkende groep is. Alcoholen worden daarom meestal geactiveerd door
de –OH groep om te zetten in een beter vertrekkende groep zoals een tosylaat (–
OTs), mesylaat (–OMs) of halogenide (–X) groep. Dit is een strategie die
stoichiometrische hoeveelheden potentieel schadelijk afval produceert.
De beschreven katalytische methodologie overkomt de typische beperking van de
klassieke amine synthese die een redox-actieve katalysator gebruikt. De
katalysator dehydrogeneert alcoholen tot de afgeleide carbonyl moleculen die
daarna gekoppeld worden met een amine met vorming van een iminogroep. Het
in de dehydrogenering van het alcohol gevormde waterstofmolecuul wordt
opgeslagen op de katalysator en hergebruikt voor de hydrogenering van de
verkregen iminogroep tot het gewenste aminoproduct. De reactie is direct,
gebruikt geen stoichiometrische reagentia en is relatief atoom-efficiënt met alleen
één molecuul water als bijproduct.
Hoofdstuk 1 is een inleiding op het werk in dit proefschrift. Het begint met een
algemeen overzicht van de rol en het belang van katalyse. Daarna presenteert het
de achtergrond van metaal-ligand bifunctionele complexen inclusief de twee
belangrijkste gebruikte katalysatoren in dit onderzoek: Knölkers complex en
Shvo’s katalysator. Daarna worden de nieuwste ontwikkelingen in de katalyse die
gebruikt maakt van een waterstofcyclus beschreven. Tot slot wordt een conclusie
gepresenteerd gebaseerd op het voorafgaande werk en worden de
onderzoekvragen en aanpak van het onderzoek in dit proefschrift geformuleerd.
Hoofdstuk 2 beschrijft het eerste voorbeeld van een goed-gedefinieerd
ijzercomplex dat geschikt is voor de katalytische alkylering van amines met
alcoholen. Voor de directe katalytische alkylering van amines met alcoholen via
een strategie die gebruikt maakt van een waterstof cyclus werden tot nu toe alleen
edelmetalen gebruikt, zoals op ruthenium en iridium gebaseerde complexen. De
pioniers in dit veld verklaarden dat de volgende uitdaging zou zijn om dit soort
omzettingen te realiseren met goedkope metalen, zoals ijzer. Het onderhavige
werk toont aan dat amines selectief gealkyleerd kunnen worden met alcohol via
een homogene ijzerkatalysator, Knölkers complex, waarbij vergelijkbare
reactiviteit wordt behaald als via edelmetalen. Tot de mogelijkheden van deze
nieuwe methode behoren ook de selectieve mono-alkylering van primaire anilines
en benzylische amines met alcoholen met daarbij de synthese van stikstof-
houdende heterocyclische structuren met 5, 6 en 7 atomen van benzylische amines
en diolen, een soort verbindingen die zeer belangrijk zijn voor de farmaceutische

Nederlandse samenvatting
142
industrie. Zo wordt de één staps-synthese van een anti-Parkinson medicijn vanuit
twee commerciële grondstoffen beschreven. De aldehyde- en imine-houdende
tussenproducten worden aangetoond met in situ 1H NMR.
Hoofdstuk 3 focust op de synthese van benzylische amines via de directe aminering
van benzylische alcoholen. Tijdens het werk gepresenteerd in hoofdstuk 2 bleken
benzylische alcoholen uitdagende substraten te zijn voor de daar gepresenteerde
reactie, omdat de imino groepen in de tussenverbindingen geconjugeerd zijn met
de aromatische ring structuur en dus moeilijk te hydrogeneren zijn met Knölkers
complex. Daarom laat dit hoofdstuk specifieke reactiecondities en
substraatcombinaties zien waarbij benzylische alcoholen efficiënt geamineerd
kunnen worden met een grote verscheidenheid aan amines. Verschillende
synthetisch uitdagende verbindingen zijn hierbij relatief eenvoudig verkregen.
In hoofdstuk 4 wordt de ijzer-gekatalyseerde pyrrool synthese vanuit primaire
amines en onverzadigde diolen beschreven. Hoofdstuk 2 liet zien dat benzylische
amines en 1,4-butaandiol leiden tot pyrrolidines, terwijl dit hoofdstuk laat zien dat
1,4-buteendiol of 1,4-butyndiol resulteren in pyrrolen. Deze reactie vindt
waarschijnlijk plaats via ijzer-gekatalyseerde isomerisatie van de onverzadigde
diolen naar de bijbehorende aldehydes gevolgd door een in situ Paal-Knorr pyrrool
synthese. Met deze methode worden een grote hoeveelheid verschillende primaire
anilines, benzylische amines en alifatische amines succesvol omgezet in pyrrolen
met matige tot goede opbrengsten. Gelpermeatiechromatografie laat zien dat
oligomeren en polymeren, die waarschijnlijk het resultaat zijn van condensatie van
de onstabiele carbonyl-houdende tussenproducten, het belangrijkste bijproduct
zijn van deze reacties.
Selectieve N-alkylering van amines in aminozuren en korte eiwitketens zonder de
noodzaak voor het gebruik van beschermende groepen is een essentiële chemische
omzetting voor de farmaceutische- en materiaalindustrie. Hoofdstuk 5 introduceert
een nieuwe, algemeen toepasbare methode voor de directe N-alkylering van
onbeschermde aminozuren en korte eiwitketens met alcoholen in hoge
opbrengsten en met behoud van de stereochemie onder toepassing van zowel
ijzer- als rutheniumkatalysatoren. In de meeste gevallen kunnen de N-
gealkyleerde aminozuren verkregen worden in kwantitatieve opbrengst na het
afdampen van de vluchtige stoffen van het reactiemengsel. Selectieve di-alkylering
van de N-terminus van di- en tripeptides met alcoholen wordt ook beschreven. Ten
slotte wordt de ijzer gekatalyseerde mono-N-alkylering van aminozuren met
vetalcoholen aangetoond. De verkregen N-gealkyleerde aminozuren met lange
vetketens kunnen gebruikt worden als oppervlakte-actieve stoffen.

English Sumary
143
English Sumary
This thesis describes the development of novel methodologies for carbon-nitrogen
bond formation through catalytic alcohol activation, mainly focusing on the direct
amination of alcohols through borrowing hydrogen strategy.
Alcohols are cheap, low-toxic and abundant chemicals. They are common
alkylation reagents used in organic synthesis. However, the direct amination of
alcohols is highly challenging, as the hydroxyl group (-OH) is a poor leaving group.
Alcohols are conventionally activated through converting the –OH to a better
leaving group, such as a tosylate (-OTs), mesylate (-OMs) or halide (-X), which
produces stoichiometric amounts of potentially toxic salts as waste. An alternative
pathway is oxidizing the alcohols to the corresponding carbonyl compounds,
followed by reductive amination, which requires stoichiometric amount of oxidants
and reductants and also produces large amounts of waste.
The presented catalytic method, avoids the common limitations encountered by
the classical amine formation reactions through employing a redox-active catalyst.
The catalyst dehydrogenates alcohols to the corresponding carbonyl compounds
which undergo imine formation with the amine reaction partner. The hydrogen
“borrowed” from the alcohol is temporarily stored on the metal catalyst and
returned back to the formed imine to obtain the desired amine product. The
reaction is direct, applies no stoichiometric reagents and produces water as the
only byproduct.
Chapter 1 is an introduction to this thesis. It starts with an overview of the role
and importance of catalysis in general. Then, it presents the literature background
of metal-ligand bifunctional complexes including the Knölker complex and the Shvo
catalyst, which are the two main catalysts employed in this thesis. The following
content shows the current state-of-the-art in borrowing hydrogen type catalysis.
The last part of this chapter is a conclusion of the above parts and contains the
outline of this thesis.
Chapter 2 describes the first example of a well-defined iron complex catalyzed
alkylation of amines with alcohols. Direct alkylation of amines with alcohols
through borrowing hydrogen strategy is an area dominated by noble metals, e.g.
ruthenium and iridium based catalytic systems. The pioneers of this field stated
that the next challenge was replacing noble metals with cheap metals, such as iron.
This work shows that amines can be selective alkylated with alcohols using a
homogeneous iron catalyst, the Knölker complex, achieving comparable reactivity
as noble metals did in this transformation. The scope of this novel method includes
the selective mono-alkylation of primary anilines and benzylamines with alcohols
as well as the formation of 5, 6 and 7 membered nitrogen containing heterocycles
from benzylamines and diols – moieties highly relevant for the pharmaceutical
industry. Moreover, anti-Parkinson drug has been synthetized in one step from 2
commercially available substrates in good yield. The aldehyde and imine
intermediates have been observed in an in situ 1H NMR study.

English Sumary
144
Chapter 3 focuses on the direct amination of benzyl alcohols to form benzylamines.
In Chapter 2 it was found that benzyl alcohols are rather challenging substrates
since the corresponding imines that are conjugated to aromatic rings are more
difficult to be reduced using the Knölker complex. Therefore this chapter’s focus is
devoted to finding reaction conditions and substrate combinations which allow for
benzyl alcohols to be aminated efficiently with a large variety of amines. Otherwise
synthetically challenging products were obtained this way.
In chapter 4, iron catalyzed pyrrole formation from primary amines and
unsaturated diols is described. Chapter 2 shows that benzylamines and 1,4-
butandiol provide pyrrolidines, while this chapter shows that primary amines and
1,4-butendiol or 1,4-butyndiol provide pyrroles. The reaction is believed to occur
via iron catalyzed isomerization of unsaturated diols to the corresponding
aldehydes followed by in situ Paal-Knorr pyrrole synthesis. Using this method, a
large variety of primary anilines, benzylamines and aliphatic amines have been
converted to pyrroles in moderate to good yields. Gel permeation chromatography
(GPC) showed that oligomers and polymers, presumably formed via the
condensation of instable carbonyl intermediates, are the main side products during
these transformations.
Selective N-alkylation of amines of amino acids and oligopeptides without the need
for protecting groups is an essential chemical transformation in the pharmaceutical
and materials industry. Chapter 5 demonstrates a novel, general method for the
direct N-alkylation of unprotected amino acids and oligo-peptides with alcohols in
excellent yields and retention of stereochemistry. In this work, both iron and
ruthenium based catalysts are used. For most cases, N-alkyl amino acids can be
obtained in quantitative yields upon removing the volatiles from the reaction
mixture. Selective dialkylation of N-terminus of di- and tripeptides with alcohols
are also presented. Finally, iron catalyzed mono-N-alkylation of amino acids with
fatty alcohols are shown. The formed long-chain N-alkyl amino acids were shown
to act as surfactants.
Chapter 6 presents the ruthenium (Shvo catalyst) catalyzed N-alkylation of amino
acid esters with alcohols. It provides a general method for direct mono-N-
alkylation of amino acid esters with a variety of benzyl alcohols and 1-pentanol in
excellent yields and retention of stereochemistry. When a proline ester or
prolinamide is employed, significant racemization is observed.
To conclude, this thesis demonstrates that metal-ligand bifunctional catalysts are
able to promote N-alkylation of amine with alcohols. The work has addressed
highly challenging goals within this research field; the development of efficient
methods employing iron-based catalytic system as well as functionalizing highly
challenging substrates such as unprotected amino acids. Due to these
achievements that are well in-line with the principles of green-chemistry the
presented methods offer new sustainable catalytic strategies for the
pharmaceutical and chemical industries.

Acknowlegments
145
Acknowlegments
On September 2nd 2013, a rainy early morning, I took a train in northern Germany
and arrived in Groningen in the afternoon. This was the day I started my PhD.
Time flies and now it comes to the end. My six years European journey, at this
time, will also going to the end. Sometimes, when I recalled this period, it was just
like a long movie playing in my head. I am so happy that I made the decision to
explore Europe, another side of the world. I want to thank every people I have
met, who have helped me to grow.
First, I would like to thank Ben and Katalin, for giving me this opportunity to do
my PhD research in Groningen. Thanks for your patient and strong support during
the last four years. I am glad I have worked with both of you. Katalin, I am really
appreciated that you have never lost your patient on me and always want to
encourage me. Ben, if I need to find a role model for my future career, it must be
you. Thanks for your inspiration and support.
I also would like to thank all other Stratingh staff members, thanks for the
discussions during group seminars. Because of you all, we have such a nice
working environment.
Thanks Prof. Beller, Prof. de Bruin and Hans for being the members of my thesis
reading committee. Peter, thank you (and your father) for the translation of this
thesis abstract and summary.
Peter and Giovanni, thanks for being my paranymphs. It was such a great time we
have spent together, especially the fantastic trips to Iceland and California, pizza
Friday, board games weekends (with delicious food from Marzia), etc.
Martin, you are also almost finishing your PhD now, you are always energetic and
brought so much fun to the group. I hope I can manage getting to your wedding.
Zhuohua, you are always quite, but once talking with you, everyone will know you
are a great guy. Anand, how are you doing now, got married? Laurie, I did not
know we have overlapped staying in Rennes. It was really great to meet another
cheerful French girl in Groningen. Anaϊs, sorry I said your home-made sweets were
too sweet, haha. Hopefully you are soon finishing your Bachelor in Brazil and
starting your PhD in Europe. I look forward to meeting you in Brazil or States in
the future. Ciaran, you are a big fan of board game, hopefully we will play it again
soon. Chris, thanks for hosting Peter and me in Santa Barbara, it was a really great
memory for me. Jurjen, it always fun to talk with you, especially when you are
eating your “delicious sandwich”. Marilena, it always happy to talk with you, and
thanks for your pasta. I heard you will start your Master in Amsterdam, so see you
soon in my promotion. Balint, you are a highly motivated guy and always want to
improve yourself. I believe all your dreams will come true. Nastya, a strong
independent Russian girl, I am glad to meet you and sometimes be your mentor.

Acknowlegments
146
I believe you will be more relaxed, and I am afraid probably I cannot manage to
speak Russian. Saravana, we both did Master in Rennes, I was shocked when you
told me you would come to our group for a postdoc when I was still in my last year
of PhD. Ale, welcome to the group, unfortunately I am just going to leave,
otherwise we will have a lot of fun for sure. Walle, probably you cannot read my
writing, but please remember the good days when I was feeding you.
Tilde, you have special power, as every time I talk with you, I just feel happier.
Thanks for inviting me to lots of events. Pablo, you are a cool guy, you always
know how to make parties. One of the greatest things I have done in Groningen
was cycling to Maastricht with you. Ravi, we are always having funny conversion,
I hope you will find a very good position in India. Juan Fer, I cannot believe I am
drinking beer and discussing about politics with a Spanish, haha. I normally do not
drink, but with you guys I have to. And other guys, Mamen, Ciccio, Simone,
Xingchen, Jiafei, it was great time that I hanged out with you.
Jiawei, it was a bit sad when you left Groningen. I really had great time with you;
especially we had Scuba diving together. Depeng, thanks for your all suggestions
during my research. Now, you have started your independent scientific career in
Guangzhou, good luck for you. Jiajia, I “followed” you from Rennes to Groningen,
but now not to Delft, haha. Thanks for all the random talks.
Giulia, you always collect people for Borrel. Now you found a real job, bought a
house and a car, just waiting for getting retired, haha. Boris, you are definitely a
fun guy when you got drunk. Stefano, I enjoyed a lot when we were snowboarding
together, I hope I can be as good as you one day. Diederik, it was a great fun that
we organized workweek together. Massimo, it was always great to talk with you,
thanks for all your suggestions. Annika, probably the first time I talked with you
was in the Dutch class. Although I do not remember what I have learned, I am so
glad to be your friend. Ivana, thanks for your explanation about the defence
process, otherwise I will get delayed. I wish you good luck for your grant proposal
and hopefully see you in the States. Raquel, you are a typical Spanish girl, as you
always give everybody warm greeting. I hope you enjoying happy life with Matt in
Spain.
I would like to thank you all, Michael, Sander, Valentín, Anne, Hugo, Lilianna, Manu,
Cora, Lara, Andreas, Ruben, Ana, Nathalie, Yans, Arjan, Sara, Duenpen, Sandeep,
Juan, Pat, Davide, Francesco, Jonas, Steven, Francesca, Wiktor, Tom, Ani, Marco,
Jana, Kaja, Dusan, Erik, Jinling, Jiawen, Yuchen, Wojtek, Matea, krzysztof, Carlos,
Tati, Wenhao, Varsha, Bert, Shermin, etc. Well, Probably I have not counted
everybody.
Also, I would like to thank the guys I have met and had fun with outside of
Stratingh Institute, from Groningen, Rennes, Rostock and other random spots on
the map where I have travelled in.

Acknowlegments
147
Last, I want to thank my parents for your endless support. (感谢父母多年来的支持)
Now, I am waiting for the visa to States. It will be a new journey. I believe a lot
of fun experiences, interesting people and challenges are waiting for me. Of course,
I am ready for you.
See you guys! Take care!
Sincerely,
Tao
August 9th 2017, Groningen
Related Documents











