cAMP increases mitochondrial cholesterol transport through the induction of arachidonic acid release inside this organelle in Leydig cells Ana Fernanda Castillo, Fabiana Cornejo Maciel, Rocı´o Castilla, Alejandra Duarte, Paula Maloberti, Cristina Paz and Ernesto J. Podesta ´ Department of Biochemistry, School of Medicine, University of Buenos Aires, Argentina Arachidonic acid (AA) is a fatty acid with 20 carbons and four cis double bonds that are the source of its flexibility and its reactivity with molecular oxygen. The oxidation can happen nonenzymatically or through the action of three types of oxygenases: cyclooxygenase, lipoxygenase and cytochrome P450. Most of the effects of AA are attributable to its conversion by those enzymes to prostaglandins, leukotrienes and other bio- active products [1]. AA itself also has biological activ- ity; however, the number of its described actions is reduced compared to the effects described for the AA metabolites. Moreover, it is not very well documented whether nonmetabolized AA is released and elicits spe- cial functions in a specific cellular compartment [2]. Transport of long-chain fatty acids in cells definitely occurs when they are tightly linked to CoA by esterifi- cation catalyzed by acyl-CoA synthetases [3]. In mam- malian and yeast cells [4] it appears that the acyl-CoA synthetases merely enhance uptake indirectly. Thus, formation of the polar CoA-ester effectively traps the fatty acid in the cell and functions as part of a facilita- ted distribution in different cellular compartments. The mechanisms involved in the compartmentaliza- tion of long-chain acyl-CoA esters and free fatty acids Keywords acyl-CoA synthetase; acyl-CoA thioesterase; arachidonic acid compartmentalization; Leydig cells; steroidogenesis Correspondence E. J. Podesta ´ , Department of Biochemistry, School of Medicine, University of Buenos Aires, Paraguay 2155–5th, C1121ABG, Buenos Aires, Argentina Fax: +54 11 45083672 ext. 31 Tel: +54 11 45083672 ext. 36 E-mail: [email protected] (Received 27 July 2006, revised 23 August 2006, accepted 12 September 2006) doi:10.1111/j.1742-4658.2006.05496.x We have investigated the direct effect of arachidonic acid on cholesterol transport in intact cells or isolated mitochondria from steroidogenic cells and the effect of cyclic-AMP on the specific release of this fatty acid inside the mitochondria. We show for the first time that cyclic-AMP can regulate the release of arachidonic acid in a specialized compartment of MA-10 Leydig cells, e.g. the mitochondria, and that the fatty acid induces choles- terol transport through a mechanism different from the classical pathway. Arachidonic acid and arachidonoyl-CoA can stimulate cholesterol trans- port in isolated mitochondria from nonstimulated cells. The effect of arach- idonoyl-CoA is inhibited by the reduction in the expression or in the activity of a mitochondrial thioesterase that uses arachidonoyl-CoA as a substrate to release arachidonic acid. cAMP-induced arachidonic acid accu- mulation into the mitochondria is also reduced when the mitochondrial thioesterase activity or expression is blocked. This new feature in the regu- lation of cholesterol transport by arachidonic acid and the release of arachidonic acid in specialized compartment of the cells could offer novel means for understanding the regulation of steroid synthesis but also would be important in other situations such as neuropathological disorders or oncology disorders, where cholesterol transport plays an important role. Abbreviations AA, arachidonic acid; AA-CoA, arachidonoyl-CoA; Acot2, mitochondrial acyl-CoA thioesterase; ACS4, acyl-CoA synthetase 4; BPB, 4-bromophenacyl bromide; 8Br-cAMP, 8-bromo-cAMP; CHX, cycloheximide; CPT1, carnitine-palmitoyl transferase 1; DBI, diazepam-binding inhibitor; NDGA, nordihydroguaiaretic acid; P450scc, cholesterol side-chain cleavage cytochrome P-450 enzyme; PBR, peripheral benzodiazepan receptor; StAR, steroidogenic acute regulatory protein. FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS 5011

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

cAMP increases mitochondrial cholesterol transportthrough the induction of arachidonic acid release insidethis organelle in Leydig cellsAna Fernanda Castillo, Fabiana Cornejo Maciel, Rocıo Castilla, Alejandra Duarte, Paula Maloberti,Cristina Paz and Ernesto J. Podesta
Department of Biochemistry, School of Medicine, University of Buenos Aires, Argentina
Arachidonic acid (AA) is a fatty acid with 20 carbons
and four cis double bonds that are the source of its
flexibility and its reactivity with molecular oxygen. The
oxidation can happen nonenzymatically or through the
action of three types of oxygenases: cyclooxygenase,
lipoxygenase and cytochrome P450. Most of the effects
of AA are attributable to its conversion by those
enzymes to prostaglandins, leukotrienes and other bio-
active products [1]. AA itself also has biological activ-
ity; however, the number of its described actions is
reduced compared to the effects described for the AA
metabolites. Moreover, it is not very well documented
whether nonmetabolized AA is released and elicits spe-
cial functions in a specific cellular compartment [2].
Transport of long-chain fatty acids in cells definitely
occurs when they are tightly linked to CoA by esterifi-
cation catalyzed by acyl-CoA synthetases [3]. In mam-
malian and yeast cells [4] it appears that the acyl-CoA
synthetases merely enhance uptake indirectly. Thus,
formation of the polar CoA-ester effectively traps the
fatty acid in the cell and functions as part of a facilita-
ted distribution in different cellular compartments.
The mechanisms involved in the compartmentaliza-
tion of long-chain acyl-CoA esters and free fatty acids
Keywords
acyl-CoA synthetase; acyl-CoA thioesterase;
arachidonic acid compartmentalization;
Leydig cells; steroidogenesis
Correspondence
E. J. Podesta, Department of Biochemistry,
School of Medicine, University of Buenos
Aires, Paraguay 2155–5th, C1121ABG,
Buenos Aires, Argentina
Fax: +54 11 45083672 ext. 31
Tel: +54 11 45083672 ext. 36
E-mail: [email protected]
(Received 27 July 2006, revised 23 August
2006, accepted 12 September 2006)
doi:10.1111/j.1742-4658.2006.05496.x
We have investigated the direct effect of arachidonic acid on cholesterol
transport in intact cells or isolated mitochondria from steroidogenic cells
and the effect of cyclic-AMP on the specific release of this fatty acid inside
the mitochondria. We show for the first time that cyclic-AMP can regulate
the release of arachidonic acid in a specialized compartment of MA-10
Leydig cells, e.g. the mitochondria, and that the fatty acid induces choles-
terol transport through a mechanism different from the classical pathway.
Arachidonic acid and arachidonoyl-CoA can stimulate cholesterol trans-
port in isolated mitochondria from nonstimulated cells. The effect of arach-
idonoyl-CoA is inhibited by the reduction in the expression or in the
activity of a mitochondrial thioesterase that uses arachidonoyl-CoA as a
substrate to release arachidonic acid. cAMP-induced arachidonic acid accu-
mulation into the mitochondria is also reduced when the mitochondrial
thioesterase activity or expression is blocked. This new feature in the regu-
lation of cholesterol transport by arachidonic acid and the release of
arachidonic acid in specialized compartment of the cells could offer novel
means for understanding the regulation of steroid synthesis but also would
be important in other situations such as neuropathological disorders or
oncology disorders, where cholesterol transport plays an important role.
Abbreviations
AA, arachidonic acid; AA-CoA, arachidonoyl-CoA; Acot2, mitochondrial acyl-CoA thioesterase; ACS4, acyl-CoA synthetase 4; BPB,
4-bromophenacyl bromide; 8Br-cAMP, 8-bromo-cAMP; CHX, cycloheximide; CPT1, carnitine-palmitoyl transferase 1; DBI, diazepam-binding
inhibitor; NDGA, nordihydroguaiaretic acid; P450scc, cholesterol side-chain cleavage cytochrome P-450 enzyme; PBR, peripheral
benzodiazepan receptor; StAR, steroidogenic acute regulatory protein.
FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS 5011

are important unresolved issues [5]. The simple struc-
ture of AA and the natural occurrence of so many
close chemical analogues are, not surprisingly, associ-
ated with a lack of specificity. The selective actions of
free AA may be explained simply by its specific release
under physiological conditions and by the absence of
such mechanisms for releasing other long-chain fatty
acids, compounds which might otherwise share its bio-
chemical effects [2]. Thus, the accessibility of AA to a
specific cellular compartment and the specificity of its
action are certainly linked.
The enzymes involved in the release of AA have
been well characterized, with the phospholipase A2
being the most important [6]. However, it remains
unclear as to how exactly AA is released in a specific
compartment of the cells under physiological condi-
tions [2]. Recently, using steroidogenic cells as an
experimental system, we described an alternative
releasing mechanism for AA as a mediator of hormone
action with the participation of an acyl-CoA synthe-
tase (ACS4) and a mitochondrial acyl-CoA thioest-
erase (Acot2) [7,8]. ACS4 has been described as an
AA-preferring acyl-CoA synthetase [9], while Acot2 is
a member of a new thioesterase family with long-chain
acyl-CoA thioesterase activity and it is associated with
the inner mitochondrial membrane [10–13].
In the steroidogenic cells, the step that determines
the rate of steroid synthesis (the rate-limiting step) is
the transport of cholesterol to the inner mitochondrial
membrane [14], a process in which ACS4 and Acot2
play a key role. In this mechanism, it has been sugges-
ted that ACS4 and Acot2 may constitute a system to
deliver AA into a specific intracellular compartment,
e.g. the mitochondria [8].
AA plays a crucial role in the steroidogenic cells,
mediating the induction of the steroidogenic acute reg-
ulatory (StAR) protein, one of the proteins involved in
cholesterol transport [15–19]. Although it is clear from
previous publications that AA plays its role in the pro-
cess through the conversion to its lipoxygenated
metabolites [15,17,20], a direct action of this fatty acid
on cholesterol transport into the mitochondria cannot
be ruled out.
In this context, a positive action of nonesterified
fatty acids on cholesterol metabolism has been des-
cribed in the mitochondrial membrane [21]. It was also
demonstrated that cholesterol binding to the enzyme
that transforms it into pregnenolone (P450scc) in lipid
vesicles is greatly potentiated when the local membrane
is rendered more fluid by the addition of nonesterified
fatty acids [22].
All the evidence described above led us to propose
the hypothesis that AA might have a direct action on
cholesterol transport into the mitochondria via a speci-
fic release in this organelle. This knowledge would be
important for the understanding of cholesterol trans-
port in the classical steroidogenic as well as in neuro-
logical systems, since changes in cholesterol transport
in the central nervous system are part of the phenotype
seen in the neuropathology and neurological disorders
such as Alzheimer’s, Parkinson’s and Huntington’s dis-
eases, and brain injury and inflammation, as well as in
animal models of epilepsy [23]. This is also valid for
cholesterol transport and metabolism in tumors such
as glioma and mammary tumor cells [24,25].
For these reasons, the objective of the present work
was to study the release of AA into the mitochondria
and a possible direct role of fatty acids on cholesterol
transport in this organelle.
Results
It is known that the acute response of steroidogenesis
to hormonal stimulation has an absolute requirement:
de novo protein synthesis [26,27]. This conclusion is
based on the fact that hormone stimulated steroid syn-
thesis is totally inhibited by cycloheximide (CHX), a
protein synthesis inhibitor. The two proteins required
in this step are ACS4 [28] and StAR [29]. ACS4 works
in the release of AA, which, in turn, acts on StAR pro-
tein induction.
Because exogenous AA stimulates steroidogenesis in
cells, the first experiment was carried out to study the
direct effect of AA on cholesterol transport in the
absence of newly synthesized StAR protein. For this
purpose, MA-10 cells were incubated with exogenous
AA either with or without submaximal concentration
of 8-bromo, 5¢-cAMP (8Br-cAMP) in the presence or
absence of CHX. Progesterone production as measure-
ment of cholesterol transport was evaluated in the
culture media after 1 h of incubation, as described
in Experimental procedures (Fig. 1). Exogenous AA
alone stimulated progesterone production, reaching
50–60% of the maximal value obtained with 8Br-
cAMP (Fig. 1A). Submaximal doses of 8Br-cAMP in
combination with AA stimulated steroid production at
the same level as the stimulation produced by satur-
ating doses of 8Br-cAMP. 8Br-cAMP-stimulated pro-
gesterone production was completely abolished in the
presence of CHX. However, AA-induced steroid pro-
duction was only partially blocked by the protein syn-
thesis inhibitor. Again, CHX did not totally reduce the
synergistic effect of AA on steroid production, either
(Fig. 1A). Protein synthesis inhibition did not affect
progesterone production supported by the water-
soluble derivative of cholesterol, 22(R)-OH-cholesterol,
AA release in a specific compartment of the cells A. F. Castillo et al.
5012 FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS

which travels freely across the membranes to reach the
inner mitochondrial cholesterol side-chain cleavage
cytochrome P-450 enzyme (P450scc) (Fig. 1B).
The widely known fact that cAMP cannot stimulate
steroidogenesis in the absence of protein synthesis is
due to the absence of two crucial proteins, ACS4 and
StAR. ACS4 is induced by hormones and it is neces-
sary for AA release [28], which participates in StAR
protein induction. Therefore, in the absence of ACS4,
no AA or StAR protein induction occurs, as previ-
ously described [28]. This is the reason why CHX com-
pletely abolished cAMP-stimulated steroidogenesis.
When exogenous AA is used in the presence of CHX,
the fatty acid bypasses the absence of ACS4 but not
the absence of StAR. Then, the partial inhibition pro-
duced by CHX on AA stimulated steroidogenesis was
unexpected. The stimulatory effect of AA on steroid
synthesis in the absence of protein synthesis suggests
that AA can per se enhance the cholesterol transport
and steroidogenesis in mitochondria of steroidogenic
cells without de novo protein synthesis. In order to test
this hypothesis, firstly, we tested whether AA exogen-
ously added to intact cells could reach the mitochon-
dria. Second, we studied the effect of exogenous AA
on cholesterol transport in isolated mitochondria from
nonstimulated MA-10 steroidogenic cells.
For the first approach, MA-10 cells were labeled with
[1-14C] AA during 5 h. After this period, the cells were
incubated in the presence or in the absence of 8Br-
cAMP. After this incubation, free AA was measured in
the mitochondria, as described in the Experimental pro-
cedures. Figure 2 shows the uptake of AA into the mito-
chondria in basal and stimulated conditions. As can
- CHX + CHX
AA Control 8Br-cAMP0.2 mM+ AA
8Br-cAMP0.2 mM
8Br-cAMP0.5 mM
A
b b
a a
a
a
b,c b,c
0
2
4
6
8
10
12
14
B
22(R)OH- cholesterol
0 10
20 30 40
50 60
Prog
este
rone
(ng
/ml)
Prog
este
rone
(ng
/ml)
Fig. 1. Effect of cAMP, AA and CHX on progesterone production
by MA-10 cells. MA-10 cells were incubated in the presence or
absence of 10 lgÆmL)1 CHX for 30 min and then stimulated for 1 h
with 8Br-cAMP (0.2 mM or 0.5 mM) and ⁄ or 300 lM AA (A), or 5 lM
22(R)-OH-cholesterol (B) in serum-free culture medium containing
0.1% fatty acid-free bovine serum albumin. Progesterone concen-
trations were measured by RIA and data are shown as progester-
one production (ngÆmL)1) in the incubation medium. Results are
expressed as the mean ± SD from five independent experiments.
(a) P < 0.001 versus control cells without CHX treatment; (b)
P < 0.001 versus respective treated cells in absence of CHX; and
(c) P < 0.01 versus control cells treated with CHX.
B 17
16
15
4
3
2
1
0 Control 8Br-cAMP
A
AA
Nuclei Mitochondria
Control 8Br-cAMP
***
a i r d n o h c o t i m
) s t i n u y r a r t i b r a (
A
i e l c u n
a i r d n o h c o t i m i e l c u n
Fig. 2. Effect of cAMP on mitochondrial and nuclear AA content.
MA-10 cells were labeled for 5 h at 37 �C with [1-14C] AA
(1 lCiÆmL)1 per 2 · 106 cells) in serum-free media containing 0.5%
fatty acid-free bovine serum albumin. Then, cells were incubated in
either the presence or absence of 1 mM 8Br-cAMP for 30 min.
After washing the cells, they were scraped and nuclear and mitoch-
ondrial fractions were obtained as described in the Experimental
procedures. The fractions were sonicated and lipids were extracted
with ethyl acetate. The organic phase was collected and dried
under nitrogen. The dried extracts were dissolved in chloro-
form:methanol (9 : 1, v ⁄ v) and analyzed by thin-layer chromato-
graphy on silica gel plates. (A) Representative autoradiography
showing AA spots in nuclear and mitochondrial fractions. (B) Auto-
radiography spots quantification by densitometry. The autoradio-
graphies were quantified by densitometry and the data were
normalized against the intensity of the signal of unlabeled AA
stained with iodine. Bars denote levels (in arbitrary units) of AA in
mitochondria and nuclei. Results are expressed as the mean ± SD
from three independent experiments. *** P < 0.001 versus control
mitochondria.
A. F. Castillo et al. AA release in a specific compartment of the cells
FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS 5013
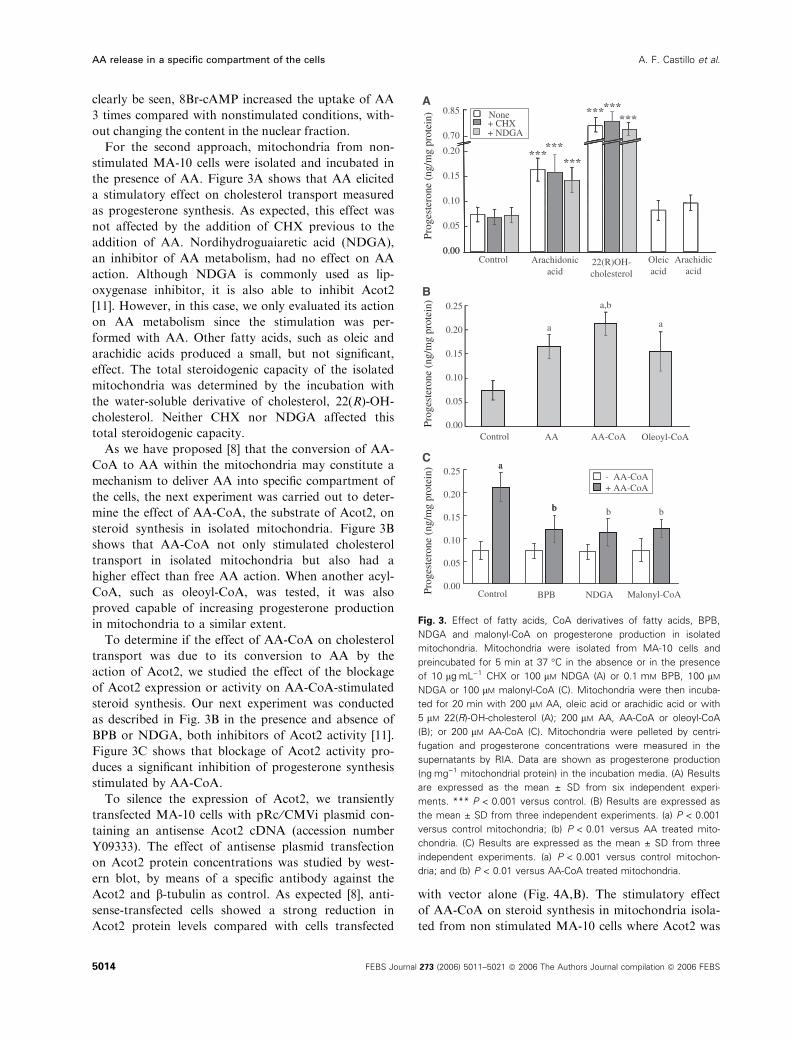
clearly be seen, 8Br-cAMP increased the uptake of AA
3 times compared with nonstimulated conditions, with-
out changing the content in the nuclear fraction.
For the second approach, mitochondria from non-
stimulated MA-10 cells were isolated and incubated in
the presence of AA. Figure 3A shows that AA elicited
a stimulatory effect on cholesterol transport measured
as progesterone synthesis. As expected, this effect was
not affected by the addition of CHX previous to the
addition of AA. Nordihydroguaiaretic acid (NDGA),
an inhibitor of AA metabolism, had no effect on AA
action. Although NDGA is commonly used as lip-
oxygenase inhibitor, it is also able to inhibit Acot2
[11]. However, in this case, we only evaluated its action
on AA metabolism since the stimulation was per-
formed with AA. Other fatty acids, such as oleic and
arachidic acids produced a small, but not significant,
effect. The total steroidogenic capacity of the isolated
mitochondria was determined by the incubation with
the water-soluble derivative of cholesterol, 22(R)-OH-
cholesterol. Neither CHX nor NDGA affected this
total steroidogenic capacity.
As we have proposed [8] that the conversion of AA-
CoA to AA within the mitochondria may constitute a
mechanism to deliver AA into specific compartment of
the cells, the next experiment was carried out to deter-
mine the effect of AA-CoA, the substrate of Acot2, on
steroid synthesis in isolated mitochondria. Figure 3B
shows that AA-CoA not only stimulated cholesterol
transport in isolated mitochondria but also had a
higher effect than free AA action. When another acyl-
CoA, such as oleoyl-CoA, was tested, it was also
proved capable of increasing progesterone production
in mitochondria to a similar extent.
To determine if the effect of AA-CoA on cholesterol
transport was due to its conversion to AA by the
action of Acot2, we studied the effect of the blockage
of Acot2 expression or activity on AA-CoA-stimulated
steroid synthesis. Our next experiment was conducted
as described in Fig. 3B in the presence and absence of
BPB or NDGA, both inhibitors of Acot2 activity [11].
Figure 3C shows that blockage of Acot2 activity pro-
duces a significant inhibition of progesterone synthesis
stimulated by AA-CoA.
To silence the expression of Acot2, we transiently
transfected MA-10 cells with pRc ⁄CMVi plasmid con-
taining an antisense Acot2 cDNA (accession number
Y09333). The effect of antisense plasmid transfection
on Acot2 protein concentrations was studied by west-
ern blot, by means of a specific antibody against the
Acot2 and b-tubulin as control. As expected [8], anti-
sense-transfected cells showed a strong reduction in
Acot2 protein levels compared with cells transfected
with vector alone (Fig. 4A,B). The stimulatory effect
of AA-CoA on steroid synthesis in mitochondria isola-
ted from non stimulated MA-10 cells where Acot2 was
a
b
0.15
0.05
0.00
Control AA Oleoyl-CoAAA-CoA
0.25
0.20
0.15
0.10
0.05
0.00
B
)nietorpg
m/gn(enoretsegorP
a
a,b
a
a
b
a0.25
0.20
0.15
0.10
0.05
0.00Malonyl-CoABPB NDGA
- AA-CoA+ AA-CoA
Control
C
)nietorpg
m/gn(enoretsegorP
b b b
0.70
0.20
0.10
0.00
)nietorpg
m/gn(enoretsegorP
0.85
Arachidonicacid
Arachidicacid
Oleicacid
A
Control
+ NDGA+ CHXNone
22(R)OH-cholesterol
Fig. 3. Effect of fatty acids, CoA derivatives of fatty acids, BPB,
NDGA and malonyl-CoA on progesterone production in isolated
mitochondria. Mitochondria were isolated from MA-10 cells and
preincubated for 5 min at 37 �C in the absence or in the presence
of 10 lgÆmL)1 CHX or 100 lM NDGA (A) or 0.1 mM BPB, 100 lM
NDGA or 100 lM malonyl-CoA (C). Mitochondria were then incuba-
ted for 20 min with 200 lM AA, oleic acid or arachidic acid or with
5 lM 22(R)-OH-cholesterol (A); 200 lM AA, AA-CoA or oleoyl-CoA
(B); or 200 lM AA-CoA (C). Mitochondria were pelleted by centri-
fugation and progesterone concentrations were measured in the
supernatants by RIA. Data are shown as progesterone production
(ngÆmg)1 mitochondrial protein) in the incubation media. (A) Results
are expressed as the mean ± SD from six independent experi-
ments. *** P < 0.001 versus control. (B) Results are expressed as
the mean ± SD from three independent experiments. (a) P < 0.001
versus control mitochondria; (b) P < 0.01 versus AA treated mito-
chondria. (C) Results are expressed as the mean ± SD from three
independent experiments. (a) P < 0.001 versus control mitochon-
dria; and (b) P < 0.01 versus AA-CoA treated mitochondria.
AA release in a specific compartment of the cells A. F. Castillo et al.
5014 FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS
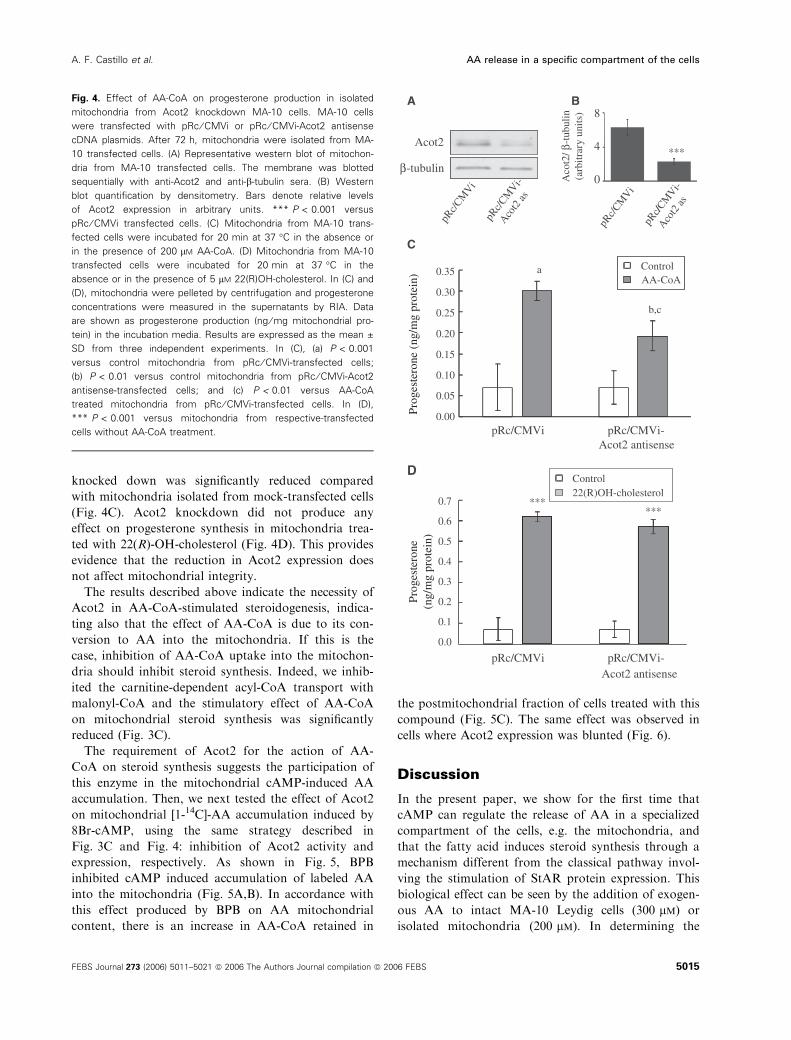
knocked down was significantly reduced compared
with mitochondria isolated from mock-transfected cells
(Fig. 4C). Acot2 knockdown did not produce any
effect on progesterone synthesis in mitochondria trea-
ted with 22(R)-OH-cholesterol (Fig. 4D). This provides
evidence that the reduction in Acot2 expression does
not affect mitochondrial integrity.
The results described above indicate the necessity of
Acot2 in AA-CoA-stimulated steroidogenesis, indica-
ting also that the effect of AA-CoA is due to its con-
version to AA into the mitochondria. If this is the
case, inhibition of AA-CoA uptake into the mitochon-
dria should inhibit steroid synthesis. Indeed, we inhib-
ited the carnitine-dependent acyl-CoA transport with
malonyl-CoA and the stimulatory effect of AA-CoA
on mitochondrial steroid synthesis was significantly
reduced (Fig. 3C).
The requirement of Acot2 for the action of AA-
CoA on steroid synthesis suggests the participation of
this enzyme in the mitochondrial cAMP-induced AA
accumulation. Then, we next tested the effect of Acot2
on mitochondrial [1-14C]-AA accumulation induced by
8Br-cAMP, using the same strategy described in
Fig. 3C and Fig. 4: inhibition of Acot2 activity and
expression, respectively. As shown in Fig. 5, BPB
inhibited cAMP induced accumulation of labeled AA
into the mitochondria (Fig. 5A,B). In accordance with
this effect produced by BPB on AA mitochondrial
content, there is an increase in AA-CoA retained in
the postmitochondrial fraction of cells treated with this
compound (Fig. 5C). The same effect was observed in
cells where Acot2 expression was blunted (Fig. 6).
Discussion
In the present paper, we show for the first time that
cAMP can regulate the release of AA in a specialized
compartment of the cells, e.g. the mitochondria, and
that the fatty acid induces steroid synthesis through a
mechanism different from the classical pathway invol-
ving the stimulation of StAR protein expression. This
biological effect can be seen by the addition of exogen-
ous AA to intact MA-10 Leydig cells (300 lm) or
isolated mitochondria (200 lm). In determining the
Acot2
β-tubulin***
D
cRp/
iV
MC cRp/
-iV
MC
sa 2tocA
A B
22(R)OH-cholesterolControl
pRc/CMVi pRc/CMVi-
pRc/CMVi pRc/CMVi-
Acot2 antisense
C
a
b,c
0.35
0.30
0.25
0.20
0.15
0.10
0.05
0.00
8
4
0
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
Acot2 antisense
******
AA-CoAControl
cRp/
iV
MC cRp/
-iV
MC
sa 2tocA
/2tocA
βnil ub ut -)stinu
yrartibra(
/
)nietorpg
m/gn(enoretsegor
Penoretsegor
P)nietorp
gm/gn(
Fig. 4. Effect of AA-CoA on progesterone production in isolated
mitochondria from Acot2 knockdown MA-10 cells. MA-10 cells
were transfected with pRc ⁄ CMVi or pRc ⁄ CMVi-Acot2 antisense
cDNA plasmids. After 72 h, mitochondria were isolated from MA-
10 transfected cells. (A) Representative western blot of mitochon-
dria from MA-10 transfected cells. The membrane was blotted
sequentially with anti-Acot2 and anti-b-tubulin sera. (B) Western
blot quantification by densitometry. Bars denote relative levels
of Acot2 expression in arbitrary units. *** P < 0.001 versus
pRc ⁄ CMVi transfected cells. (C) Mitochondria from MA-10 trans-
fected cells were incubated for 20 min at 37 �C in the absence or
in the presence of 200 lM AA-CoA. (D) Mitochondria from MA-10
transfected cells were incubated for 20 min at 37 �C in the
absence or in the presence of 5 lM 22(R)OH-cholesterol. In (C) and
(D), mitochondria were pelleted by centrifugation and progesterone
concentrations were measured in the supernatants by RIA. Data
are shown as progesterone production (ng ⁄ mg mitochondrial pro-
tein) in the incubation media. Results are expressed as the mean ±
SD from three independent experiments. In (C), (a) P < 0.001
versus control mitochondria from pRc ⁄ CMVi-transfected cells;
(b) P < 0.01 versus control mitochondria from pRc ⁄ CMVi-Acot2
antisense-transfected cells; and (c) P < 0.01 versus AA-CoA
treated mitochondria from pRc ⁄ CMVi-transfected cells. In (D),
*** P < 0.001 versus mitochondria from respective-transfected
cells without AA-CoA treatment.
A. F. Castillo et al. AA release in a specific compartment of the cells
FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS 5015

effective concentration of AA, we also assess AA’s bio-
activity. Although in some cases concentrations
between 1 and 10 lm AA are sufficient to detect a bio-
logical effect [2], other effects require 100–300 lm [2].
However, at this high concentration, AA may not even
be in solution. Previous studies performed with con-
centrations of AA of the same magnitude as ours
(200 lm) observed either none [30] or a small but signi-
ficant [31,32] effect of exogenously added AA on basal
steroid synthesis in MA-10 and rat Leydig cells,
respectively. In the present paper, we show the stimu-
lation of steroidogenesis using 300 lm AA in the pres-
ence of albumin. These different results can be
explained by the fact that protein binding can increase
the overall concentration of fatty acids present in an
aqueous environment by effectively decreasing the
insoluble fraction [2]. Albumin, in particular, binds
specifically to fatty acids [33]. In our experiments, we
added albumin in the preparation of the AA solution,
which allowed us to detect a significant stimulatory
effect of exogenous AA on steroid production of non-
stimulated steroidogenic cells (50–60% compared with
cAMP). The stimulation of steroid synthesis by exog-
enously added AA was lower than the stimulation
reached by cAMP or 22(R)-OH-cholesterol. These
results are similar to those obtained by us in rat Ley-
dig cells and by other authors in the same or other
steroidogenic tissues [20,32,34].
When exogenous AA is added together with sub-
maximal doses of 8Br-cAMP, there is a synergistic
a
b
Acot2 antisense
B
AA
8Br-cAMPControl
pRc/CMVi pRc/CMVi-
pRc/CMVi pRc/CMVi-
Acot2 antisense
A
a
b
0
1
2
3
4
5
6
7
control 8Br-cAMP control 8Br-cAMP
)stinuyrartibra(
AFig. 6. Effect of Acot2 knockdown on AA accumulation into mito-
chondria. MA-10 cells were transfected with pRc ⁄ CMVi or
pRc ⁄ CMVi-Acot2 antisense cDNA plasmids as described in Fig. 5.
After 72 h, MA-10 transfected cells were labeled and stimulated as
described in Fig. 2. (A) Representative autoradiography showing AA
spots in mitochondrial fractions. (B) Autoradiography spots quantifi-
cation by densitometry. Bars denote levels (in arbitrary units) of AA
in mitochondria from MA-10 transfected cells treated with or with-
out 8Br-cAMP. Results are expressed as the mean ± SD from
three independent experiments. (a) P < 0.01 versus mitochondria
from control pRc ⁄ CMVi-transfected cells; and (b) P < 0.001 versus
mitochondria from 8Br-cAMP treated pRc ⁄ CMVi antisense trans-
fected cells.
b
a
0
4
8
12
16
20
A Control 8Br-cAMP 8Br-cAMP+ BPB
B
Control 8Br-cAMP 8Br-cAMP+ BPB
AA
b
a)stinuyrartibra(
A
8Br-cAMP+ BPB
0
25
Control 8Br-cAMP
[14C
]AA
-CoA
(cpm
x10
–3/m
g pr
otei
n)
C 50 ***
Fig. 5. Effect of Acot2 activity inhibition on AA accumulation into
mitochondria and AA-CoA accumulation in the postmitochondrial
fraction. MA-10 cells were labeled as described in Fig. 2. When
indicated, cells were incubated with 0.1 mM BPB for 30 min prior
to the stimulation with 8Br-cAMP. (A) Representative autoradiogra-
phy showing AA spots in mitochondrial fractions. (B) Autoradiogra-
phy spots quantification by densitometry. The autoradiographies
were quantified by densitometry and the data were normalized
against the intensity of the signal of unlabeled AA stained with iod-
ine. Bars denote levels (in arbitrary units) of AA in mitochondria.
Results are expressed as the mean ± SD from three independent
experiments. (a) P < 0.001 versus mitochondria from control cells;
(b) P < 0.05 versus mitochondria from 8Br-cAMP-treated cells. (C)
AA-CoA content in the postmitochondrial fraction. Data are shown
as 14C-AA-CoA in cpm ⁄ mg protein in the postmitochondrial fraction.
Results are expressed as the mean ± SD from three independent
experiments. ***P < 0.001 versus control.
AA release in a specific compartment of the cells A. F. Castillo et al.
5016 FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS

effect in which steroid synthesis reaches maximal
activation (Fig. 1). This result agrees with that
obtained by other authors [35], who suggested that a
critical threshold of cAMP or cAMP-dependent pro-
tein kinase activation is required for the synergistic
effect of AA on cAMP-stimulated StAR protein
expression and steroidogenesis. The fact that the cells
treated with exogenous AA can be stimulated with
8Br-cAMP indicates that mitochondrial function
regarding the regulation of cholesterol metabolism
remains intact. Moreover, it is known that any distur-
bance or swelling of the mitochondria produces a total
loss of regulation, rendering mitochondria that pro-
duce full steroidogenesis. In our case, treatment with
AA also does not cause loss of the regulation of ster-
oid biosynthesis supported by 22(R)-OH-cholesterol
(data not shown). This explanation rules out the possi-
bility that AA or bovine serum albumin would have
effect on mitochondrial integrity. This consideration is
also valid when fatty acids or AA-CoA are used in iso-
lated mitochondria, as both treatments can still be
increased by stimulation with 22(R)-OH-cholesterol
(data not shown).
As is already known [26,27], the cAMP-dependent
transport of cholesterol from the mitochondrial outer
to inner membrane can be blocked by a protein syn-
thesis inhibitor such as CHX. However, this protein
synthesis inhibitor is not totally able to abolish the sti-
mulation produced by exogenously added AA. These
results strongly suggest that AA can exert a role on
cholesterol transport without the induction of StAR
protein.
The demonstration that AA and ⁄or AA-CoA stimu-
late cholesterol transport in isolated mitochondria sug-
gests that the accumulation of AA can occur by direct
uptake of AA itself inside the mitochondria or by the
previous esterification to AA-CoA by ACS4 and subse-
quent action of Acot2 to render free AA in the mito-
chondria. The fact that cAMP increases AA uptake
into the mitochondria and that this effect on AA accu-
mulation is reduced when Acot2 activity or expression
are blocked strongly indicates that the operating
mechanism is dependent on the concerted action
of ACS4 ⁄Acot2. In this mechanism, cAMP acts to
increase AA-CoA formation in the cytosol. The CoA
derivative enters the mitochondria through the CPT1-
dependent pathway. The specificity of this mechanism
to release AA inside the mitochondria is shown by the
fact that the content of labeled AA in another organ-
elle such as the nucleus is neither increased by cAMP
nor reduced by the inhibition of Acot2 (Fig. 2). This is
the first time that AA incorporation into a specific
subcellular compartment (the mitochondria) has been
shown as a consequence of the action of this second
messenger. Steroidogenic cells express Acot2 and also
a cytosolic isoform, Acot1, which is 92.5% homolog-
ous to the mitochondrial enzyme. We have ruled out
the possibility that mitochondria would uptake AA
produced by the action of Acot1 outside the mitochon-
dria, as part of our experiments were performed with
isolated mitochondria where Acot1 was not present.
Moreover, while the overexpression of Acot2 results in
an increase of hormone induced steroid synthesis [8],
overexpression of Acot1 does not produce this effect;
conversely, it produced a slight inhibition of the pro-
cess (data not shown). This last result supports the
notion that Acot2 is the thioesterase involved in the
release of AA inside the mitochondria.
Our model explaining how AA is released into the
mitochondria also supports the concept that the select-
ive actions of free AA may be explained simply by its
specific release under physiological conditions and by
the absence of such mechanisms for releasing other
long-chain fatty acids, compounds that might otherwise
share its biochemical effects. This is demonstrated by
the fact that when the mitochondria are stimulated
with other fatty acids, the response is lower than with
AA; however, there is a significant response of steroid-
ogenesis to a different Acyl-CoA (oleoyl-CoA, Fig. 4).
Thus, the specificity of the action is not due to the fatty
acid itself but to the acyl-CoA available to the mito-
chondrial Acot2. In our case, AA-CoA is formed pref-
erentially because of the specificity of ACS4 on AA [9].
The mitochondrial inner membrane is not permeable
to acyl-CoAs [3]; we wanted to know how AA-CoA
reaches the mitochondrial Acot2. The experiment using
malonyl-CoA (Fig. 5) indicates that AA-CoA follows
the usual pathway involving carnitine-palmitoyl transf-
erase 1 (CPT1) [3]. This enzyme plays a central role in
mitochondrial fatty acid oxidation. However, in our
case, it seems that CPT1 directs AA to another func-
tion. In this context, it has been proposed that a
potential route for long-chain acyl-CoAs to cross the
mitochondrial outer membrane could be the voltage-
dependent anion selective channel, also called mitoch-
ondrial porin and located in the contact sites [36]. It is
very interesting that a protein obligatory for choles-
terol transport in steroidogenic cells, the peripheral
benzodiazepine receptor (PBR), is also located in the
mitochondrial contact sites and includes the voltage-
dependent anion selective channel in its structure
together with the adenine nucleotide carrier [37]. PBR
is involved in cholesterol transport to the cytochrome
P450 side chain cleaving enzyme localized on the outer
surface of the mitochondrial inner membrane [37]. The
endogenous ligand of this receptor is an acyl-CoA
A. F. Castillo et al. AA release in a specific compartment of the cells
FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS 5017

binding protein known also as diazepam-binding inhib-
itor (DBI) [37,38]. It can be postulated that the role of
DBI is to facilitate the transport of fatty acids through
the mitochondrial outer membrane. The homology
between the DBI and an acyl-CoA binding protein cer-
tainly enhances this possibility. The specific interaction
between DBI and its endogenous receptor, the PBR
located on the outer ⁄ inner mitochondrial membrane
contact sites [37,38], may direct the AA-CoA to this
organelle. Contact sites between the mitochondrial outer
and inner membrane could represent the microenviron-
ment for bringing the machinery together to transport
AA-CoA [37] into the mitochondria and facilitate AA
release, which in turn facilitates cholesterol transport.
How intramitochondrial AA could stimulate cholesterol
transfer from the outer to the inner mitochondrial mem-
brane can also be explained by the action of AA on the
membrane permeability in the contact sites. This sugges-
tion is also in line with experiments showing that AA
induces the specific membrane permeability in heart and
liver mitochondria by opening the mitochondrial per-
meability transition pore [39,40]. The pore is a multiple
protein complex located in the mitochondrial contact
sites [41] and, in mitochondria of steroidogenic cells, it
participates in cholesterol transport.
In the present paper, we demonstrate that 20–30% of
total steroid production can be elicited without the
necessity for StAR synthesis. This is in accordance with
the pathological situation where deletions or mutations
of StAR are detected in humans born with the steroid
deficiency disease, lipoid adrenal congenital hyperplasia
[17,20,42–44]. Disruptions of the StAR gene in mice
produce similar phenotypes [18,45]. The effect of these
deletions establishes that StAR is necessary for 80–90%
of adrenal cholesterol metabolism [19,46]. In other
words, our results may explain the mechanism by
which in these situations there is a remaining 20% of
steroid synthesis, due to the direct effect of AA ⁄AA-CoA produced within the mitochondria by the
action of ACS4 ⁄Acot2 together with DBI ⁄PBR.
Thus, it can be postulated that in the acute phase
(early response) of steroid synthesis, the release of AA
into the mitochondria is the first stimulator of choles-
terol transport. The sustained phase of the acute
response will then need the induction of StAR. We
cannot exclude that an extraordinarily small amount
of intramitochondrial StAR present in resting condi-
tions and not detectable by current techniques can
contribute to the effect of AA on cholesterol transport
in mitochondria.
The absence of hormone ⁄ cAMP-induced steroid
synthesis when protein synthesis is inhibited can be
explained now by the inhibition in the induction of
ACS4 [28] during the early response and the inhibi-
tion of ACS4 and StAR inductions during the sus-
tained phase. In both phases, the presence of
DBI ⁄PBR may be necessary. This new feature in the
regulation of cholesterol transport by AA and the
release of AA in a specialized compartment of
the cells could offer novel means for understanding
the regulation of steroid synthesis, but would also be
important in other situations such as the neurosteroid
biosynthesis or oncology disorders, where cholesterol
transport, ACS4 and PBR play an important role
[23–25,47].
Experimental procedures
Materials
Fatty acid-free bovine serum albumin, AA, arachidic and
oleic acids, 4-bromophenacyl bromide (BPB), oleoyl-CoA,
malonyl-CoA, 8Br-cAMP, 22(R)-OH-cholesterol, cyclohexi-
mide (CHX) and Waymouth MB752 ⁄ 1 cell culture media
were purchased from Sigma Chemical Co. (St Louis, MO,
USA). Nordihydroguaiaretic acid (NDGA) and AA-CoA
were from Fluka (Buchs, Switzerland). Sera, antibiotics and
trypsin-EDTA were from Gibco-Life Technologies Inc.
(Gaithersburg, MD, USA). All other reagents were of the
highest grade available.
Cell culture
The MA-10 cell line is a clonal strain of mouse Leydig
tumor cells that produce progesterone rather than testoster-
one as the major steroid. The cells were generously provi-
ded by M. Ascoli, University of Iowa, College of Medicine
(Iowa City, IA, USA) and were handled as originally des-
cribed [48].
Cells were incubated in the presence or absence of
10 lgÆmL)1 CHX for 30 min and then stimulated with 8Br-
cAMP (0.2 mm or 0.5 mm), 300 lm AA or 5 lm 22(R)-OH-
cholesterol in the culture medium containing 0.1% fatty
acid-free bovine serum albumin. Progesterone production
was measured by radioimmunoanalysis (RIA) [7], and data
are shown as progesterone production (ngÆmL)1) in the
incubation medium.
Preparation of mitochondrial fraction
Mitochondria were obtained as previously described [17].
Briefly, all MA-10 cell cultures were washed with phos-
phate-buffered saline, scraped in 10 mm Tris ⁄HCl (pH 7.4),
250 mm sucrose, 0.1 mm EDTA (TSE buffer), homogenized
with a Pellet pestle motor homogenizer (Kontes) and centri-
fuged at 800 g during 15 min. A second centrifugation at
16 000 g during 15 min rendered a mitochondrial pellet and
AA release in a specific compartment of the cells A. F. Castillo et al.
5018 FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS

a supernatant (postmitochondrial fraction). The mitochond-
rial pellet was resuspended in TSE buffer.
Progesterone production in isolated
mitochondria
Thirty microliters of mitochondrial fraction (�200 lg of
protein) were added to 165 lL of medium consisting of
34 mm Tris ⁄HCl (pH 7.4), 20 mm KCl, 4 mm MgCl2 and
108 mm mannitol, containing 0.3% fatty acid-free bovine
serum albumin. When indicated, 200 lm AA, 200 lm AA-
CoA or 5 lm 22(R)-OH-cholesterol were added. The mix-
ture was completed by adding TSE buffer to complete a
final reaction volume of 500 lL (fatty acid-free bovine
serum albumin final concentration 0.1%). The incubations
were carried out at 37 �C for 20 min with gently shaking
and were stopped by cooling the tubes in an ice ⁄water bath.As indicated in each figure, inhibitors such as 10 lgÆmL)1
CHX, 100 lm NDGA, 0.1 mm BPB or 100 lm malonyl-
CoA were added to the reaction mixture and preincubated
5 min prior to the addition of AA, AA-CoA or 22(R)-OH-
cholesterol.
After the incubation time, mitochondria were pelleted by
centrifugation 16 000 g for 15 min and progesterone con-
centrations were measured in the supernatants by RIA.
Data are shown as progesterone production per mg of
mitochondrial protein (ngÆmg)1 protein).
[1-14C]Arachidonic acid incorporation
in MA-10 cells
MA-10 cells were labeled following a previously described
methodology [17], with minor modifications. [14C]-AA
(New England Nuclear, Boston, MA, USA; specific activity
53.0 mCiÆmmol)1) was added to the cultures in a concentra-
tion of 1 lCiÆmL)1 per well (2 · 106 cells) in serum-free
Waymouth MB752 ⁄ 1 containing 0.5% fatty acid-free
bovine serum albumin [17]. After 5 h of incubation at
37 �C in a humidified atmosphere containing 5% CO2, the
cells were incubated in the presence or absence of 1 mm
8Br-cAMP for 30 min. When indicated, cells were incuba-
ted with 0.1 mm BPB for 30 min prior to the stimulation
with 8Br-cAMP.
After these treatments, the cells were washed with serum-
free Waymouth medium containing 0.5% fatty acid-free
bovine serum albumin. Nuclear and mitochondrial pellets
were obtained as previously described [17] and resuspended
in 20 mm Hepes ⁄KOH (pH 7.4), 250 mm sucrose, 1 mm
EDTA, 10 mm KCl and 1.5 mm MgCl2 containing 500 ng
of unlabeled AA, and were then sonicated. Protein concen-
tration was measured and lipids were extracted from equal
amounts of nuclear or mitochondrial proteins (500 lg in
both cases) from each treatment. Lipid extraction was per-
formed twice with ethyl acetate (six volumes per one vol-
ume of nuclear or mitochondrial fraction). The organic
phase was then collected and dried under nitrogen at 25 �Cand analyzed by two successive thin-layer chromatographies
on silica gel. Radioactive spots were developed using a
Storm Phosphorimager (Amersham Biosciences, Sweden)
after 1 week of exposition. The postmitochondrial fraction
was treated as described for the mitochondria and the
[14C]-AA-CoA formation was evaluated by extraction from
the aqueous phase according to the literature [49].
Plasmid transfection
MA-10 cells were transiently transfected by electroporation
as previously described [8,50]. Transfection efficiency varied
from 40 to 50% and was estimated by counting fluorescent
cells transfected with pRc ⁄CMVi plasmid [51] containing
the enhanced form of green fluorescent protein (EGFP) [8].
MA-10 cells were transfected either with pRc ⁄CMVi plas-
mid containing the Acot2 antisense cDNA [8] or with the
empty vector as control. Approximately 72 h after transfec-
tion, cells were used as described in the respective figures.
SDS/PAGE and immunoblot assay
Mitochondrial proteins were separated by SDS ⁄PAGE
(10% acrylamide gels) and electrophoretically transferred to
poly(vinylidene difluoride) membranes (Bio-Rad Laborator-
ies Inc., Hercules, CA, USA) as described previously [8].
Acot2 protein was detected using anti-Acot2 antibodies [11]
and immunoreactive bands were detected by chemilumines-
cence using enhanced chemiluminescence reagents (GE
Healthcare, Chalfont St Giles, UK).
Protein quantification and statistical analysis
Protein was quantitated by the method of Bradford [52]
using bovine serum albumin as a standard. Statistical ana-
lysis was performed by t-test or anova followed by the
Student–Newman–Kuels test.
Acknowledgements
This work was supported in part by National Research
Council (CONICET), University of Buenos Aires
(UBA) and National Agency of Scientific and Techno-
logical Promotion (ANPCyT). Thanks are due to the
technical assistance provided by F. Meuli.
References
1 Sigal E (1991) The molecular biology of mammalian
arachidonic acid metabolism. Am J Physiol 260, L13–
L28.
2 Brash AR (2001) Arachidonic acid as a bioactive mole-
cule. J Clin Invest 107, 1339–1345.
A. F. Castillo et al. AA release in a specific compartment of the cells
FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS 5019

3 Kerner J & Hoppel C (2000) Fatty acid import into
mitochondria. Biochim Biophys Acta 1486, 1–17.
4 Klingenberg M & Huang SG (1999) Structure and func-
tion of the uncoupling protein from brown adipose tis-
sue. Biochim Biophys Acta 1415, 271–296.
5 Faergeman NJ & Knudsen J (1997) Role of long-chain
fatty acyl-CoA esters in the regulation of metabolism
and in cell signalling. Biochem J 323, 1–12.
6 Irvine RF (1982) How is the level of free arachidonic
acid controlled in mammalian cells?. Biochem J 204,
3–16.
7 Maloberti P, Lozano RC, Mele PG, Cano F, Colonna
C, Mendez CF, Paz C & Podesta EJ (2002) Concerted
regulation of free arachidonic acid and hormone-
induced steroid synthesis by acyl-CoA thioesterases and
acyl-CoA synthetases in adrenal cells. Eur J Biochem
269, 5599–5607.
8 Maloberti P, Castilla R, Castillo F, Maciel FC, Mendez
CF, Paz C & Podesta EJ (2005) Silencing the expression
of mitochondrial acyl-CoA thioesterase I and acyl-CoA
synthetase 4 inhibits hormone-induced steroidogenesis.
FEBS J 272, 1804–1814.
9 Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki
N, Nagura H, Iijima H & Yamamoto TT (1997) A
novel arachidonate-preferring acyl-CoA synthetase is
present in steroidogenic cells of the rat adrenal, ovary,
and testis. Proc Natl Acad Sci USA 94, 2880–2884.
10 Paz C, Dada L, Cornejo Maciel F, Mele P, Cymeryng
C, Neuman I, Mendez C, Finkielstein C, Solano A,
Minkiyu P et al. (1994) Purification of a novel 43-kDa
protein (p43) intermediary in the activation of steroido-
genesis from rat adrenal gland. Eur J Biochem 224, 709–
716.
11 Finkielstein C, Maloberti P, Mendez CF, Paz C,
Cornejo Maciel F, Cymeryng C, Neuman I, Dada L,
Mele PG et al. (1998) An adrenocorticotropin-regulated
phosphoprotein intermediary in steroid synthesis is sim-
ilar to an acyl-CoA thioesterase enzyme. Eur J Biochem
256, 60–66.
12 Svensson LT, Engberg ST, Aoyama T, Usuda N,
Alexson SE & Hashimoto T (1998) Molecular cloning
and characterization of a mitochondrial peroxisome
proliferator-induced acyl-CoA thioesterase from rat
liver. Biochem J 329, 601–608.
13 Hunt MC, Yamada J, Maltais LJ, Wright MW, Podesta
EJ & Alexson SE (2005) A revised nomenclature for
mammalian acyl-CoA thioesterases ⁄ hydrolases. J Lipid
Res 46, 2029–2032.
14 Crivello JF & Jefcoate CR (1980) Intracellular move-
ment of cholesterol in rat adrenal cells: kinetics and
effects of inhibitors. J Biol Chem 255, 8144–8151.
15 Walton KM & Dixon JE (1993) Protein tyrosine phos-
phatases. Annu Rev Biochem 62, 101–120.
16 Solano AR, Dada LA, Luz Sardanons M, Sanchez ML
& Podesta EJ (1987) Leukotrienes as common inter-
mediates in the cyclic AMP dependent and independent
pathways in adrenal steroidogenesis. J Steroid Biochem
27, 745–751.
17 Solano AR, Dada L & Podesta EJ (1988) Lipoxygenase
products as common intermediates in cyclic AMP-
dependent and -independent adrenal steroidogenesis in
rats. J Mol Endocrinol 1, 147–154.
18 Wang X & Stocco DM (1999) Cyclic AMP and arachi-
donic acid: a tale of two pathways. Mol Cell Endocrinol
158, 7–12.
19 Wang XJ, Dyson MT, Jo Y, Eubank DW & Stocco
DM (2003) Involvement of 5-lipoxygenase metabolites
of arachidonic acid in cyclic AMP-stimulated steroido-
genesis and steroidogenic acute regulatory protein gene
expression. J Steroid Biochem Mol Biol 85, 159–166.
20 Mele PG, Dada LA, Paz C, Neuman I, Cymeryng CB,
Mendez CF, Finkielstein CV, Cornejo Maciel F &
Podesta EJ (1997) Involvement of arachidonic acid and
the lipoxygenase pathway in mediating luteinizing hor-
mone-induced testosterone synthesis in rat Leydig cells.
Endocr Res 23, 15–26.
21 Jefcoate C (2002) High-flux mitochondrial cholesterol
trafficking, a specialized function of the adrenal cortex.
J Clin Invest 110, 881–890.
22 Dhariwal MS & Jefcoate CR (1989) Cholesterol meta-
bolism by purified cytochrome P-450scc is highly stimu-
lated by octyl glucoside and stearic acid exclusively in
large unilamellar phospholipid vesicles. Biochemistry 28,
8397–8402.
23 Papadopoulos V, Lecanu L, Brown RC, Han Z & Yao
ZX (2006) Peripheral-type benzodiazepine receptor in
neurosteroid biosynthesis, neuropathology and neurolo-
gical disorders. Neuroscience 138, 749–756.
24 Liang YC, Wu CH, Chu JS, Wang CK, Hung LF,
Wang YJ, Ho YS, Chang JG & Lin SY (2005) Involve-
ment of fatty acid-CoA ligase 4 in hepatocellular carci-
noma growth: roles of cyclic AMP and p38 mitogen-
activated protein kinase. World J Gastroenterol 11,
2557–2563.
25 Papadopoulos V, Guarneri P, Kreuger KE, Guidotti A
& Costa E (1992) Pregnenolone biosynthesis in C6–2B
glioma cell mitochondria: regulation by a mitochondrial
diazepam binding inhibitor receptor. Proc Natl Acad Sci
USA 89, 5113–5117.
26 Garren LD, Gill GN, Masui H & Walton GM (1971)
On the mechanism of action of ACTH. Recent Prog
Horm Res 27, 433–478.
27 Crivello JF & Jefcoate CR (1978) Mechanisms of corti-
cotropin action in rat adrenal cells. I. The effects of
inhibitors of protein synthesis and of microfilament for-
mation on corticosterone synthesis. Biochim Biophys
Acta 542, 315–329.
28 Cornejo Maciel F, Maloberti P, Neuman I, Cano F,
Castilla R, Castillo F, Paz C & Podesta EJ (2005) An
arachidonic acid-preferring acyl-CoA synthetase is a
AA release in a specific compartment of the cells A. F. Castillo et al.
5020 FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS

hormone-dependent and obligatory protein in the signal
transduction pathway of steroidogenic hormones. J Mol
Endocrinol 34, 655–666.
29 Artemenko IP, Zhao D, Hales DB, Hales KH &
Jefcoate CR (2001) Mitochondrial processing of newly
synthesized steroidogenic acute regulatory protein
(StAR), but not total StAR, mediates cholesterol trans-
fer to cytochrome P450 side chain cleavage enzyme in
adrenal cells. J Biol Chem 276, 46583–46596.
30 Wang X, Walsh LP, Reinhart AJ & Stocco DM (2000)
The role of arachidonic acid in steroidogenesis and ster-
oidogenic acute regulatory (StAR) gene and protein
expression. J Biol Chem 275, 20204–20209.
31 Lopez-Ruiz MP, Choi MS, Rose MP, West AP &
Cooke BA (1992) Direct effect of arachidonic acid on
protein kinase C and LH-stimulated steroidogenesis in
rat Leydig cells; evidence for tonic inhibitory control of
steroidogenesis by protein kinase C. Endocrinology 130,
1122–1130.
32 Romanelli F, Valenca M, Conte D, Isidori A &
Negro-Vilar A (1995) Arachidonic acid and its metabo-
lites effects on testosterone production by rat Leydig
cells. J Endocrinol Invest 18, 186–193.
33 Spector AA (1986) Structure and lipid binding proper-
ties of serum albumin. Methods Enzymol 128, 320–339.
34 Mele PG, Dada LA, Paz C, Cymeryng CB, Cornejo
Maciel MF, Neuman MI, Finkielstein CV, Mendez CF
& Podesta EJ (1996) Site of action of proteinases in
the activation of steroidogenesis in rat adrenal gland.
Biochim Biophys Acta 1310, 260–268.
35 Wang XJ, Dyson MT, Mondillo C, Patrignani Z,
Pignataro O & Stocco DM (2002) Interaction between
arachidonic acid and cAMP signaling pathways
enhances steroidogenesis and StAR gene expression in
MA-10 Leydig tumor cells. Mol Cell Endocrinol 188,
55–63.
36 Benz R (1994) Permeation of hydrophilic solutes
through mitochondrial outer membranes: review on
mitochondrial porins. Biochim Biophys Acta 1197,
167–196.
37 Papadopoulos V (1993) Peripheral-type benzodiazep-
ine ⁄diazepam binding inhibitor receptor: biological role
in steroidogenic cell function. Endocr Rev 14, 222–240.
38 Knudsen J, Hojrup P, Hansen HO, Hansen HF &
Roepstorff P (1989) Acyl-CoA-binding protein in the
rat. Purification, binding characteristics, tissue concen-
trations and amino acid sequence. Biochem J 262, 513–
519.
39 Di Paola M, Zaccagnino P, Oliveros-Celis C & Lorusso
M (2006) Arachidonic acid induces specific membrane
permeability increase in heart mitochondria. FEBS Lett
580, 775–781.
40 Petronilli V, Penzo D, Scorrano L, Bernardi P & Di
Lisa F (2001) The mitochondrial permeability transition,
release of cytochrome c and cell death. Correlation with
the duration of pore openings in situ. J Biol Chem 276,
12030–12034.
41 Halestrap AP, McStay GP & Clarke SJ (2002) The
permeability transition pore complex: another view.
Biochimie 84, 153–166.
42 Lin D, Sugawara T, Strauss JF, 3rd Clark BJ, Stocco
DM, Saenger P, Rogol A & Miller WL (1995) Role of
steroidogenic acute regulatory protein in adrenal and
gonadal steroidogenesis. Science 267, 1828–1831.
43 Rao RM, Jo Y, Leers-Sucheta S, Bose HS, Miller WL,
Azhar S & Stocco DM (2003) Differential regulation of
steroid hormone biosynthesis in R2C and MA-10 Ley-
dig tumor cells: role of SR-B1-mediated selective choles-
teryl ester transport. Biol Reprod 68, 114–121.
44 Mikami K, Omura M, Tamura Y & Yoshida S (1990)
Possible site of action of 5-hydroperoxyeicosatetraenoic
acid derived from arachidonic acid in ACTH-stimulated
steroidogenesis in rat adrenal glands. J Endocrinol 125,
89–96.
45 Caron KM, Soo SC, Wetsel WC, Stocco DM, Clark BJ
& Parker KL (1997) Targeted disruption of the mouse
gene encoding steroidogenic acute regulatory protein
provides insights into congenital lipoid adrenal hyper-
plasia. Proc Natl Acad Sci USA 94, 11540–11545.
46 Clark BJ, Soo SC, Caron KM, Ikeda Y, Parker KL &
Stocco DM (1995) Hormonal and developmental regula-
tion of the steroidogenic acute regulatory protein. Mol
Endocrinol 9, 1346–1355.
47 Guarneri P, Papadopoulos V, Pan B & Costa E (1992)
Regulation of pregnenolone synthesis in C6–2B glioma
cells by 4¢-chlorodiazepam. Proc Natl Acad Sci USA 89,
5118–5122.
48 Ascoli M (1981) Characterization of several clonal lines
of cultured Leydig tumor cells: gonadotropin receptors
and steroidogenic responses. Endocrinology 108, 88–95.
49 Sakuma S, Fujimoto Y, Kitao A, Sakamoto H, Nishida
H & Fujita T (1999) Simultaneous measurement of
prostaglandin and arachidonoyl CoA formed from ara-
chidonic acid in rabbit kidney medulla microsomes: the
roles of Zn2+ and Cu2+ as modulators of formation of
the two products. Prostaglandins Leukot Essent Fatty
Acids 61, 105–112.
50 Li H, Degenhardt B, Tobin D, Yao ZX, Tasken K &
Papadopoulos V (2001) Identification, localization, and
function in steroidogenesis of PAP7: a peripheral-type
benzodiazepine receptor- and PKA (RIalpha)-associated
protein. Mol Endocrinol 15, 2211–2228.
51 Leibiger B, Moede T, Schwarz T, Brown GR, Kohler
M, Leibiger IB & Berggren PO (1998) Short-term regu-
lation of insulin gene transcription by glucose. Proc
Natl Acad Sci USA 95, 9307–9312.
52 Bradford MM (1976) A rapid and sensitive method for
the quantitation of microgram quantities of protein util-
izing the principle of protein-dye binding. Anal Biochem
72, 248–254.
A. F. Castillo et al. AA release in a specific compartment of the cells
FEBS Journal 273 (2006) 5011–5021 ª 2006 The Authors Journal compilation ª 2006 FEBS 5021
Related Documents











