J Cancer Res Clin Oncol (1992) 118:425-434 Cancer esearch and Cfinical Springer-Verlag 1992 Calcium channel blocker treatment of tumor cells induces alterations in the cytoskeleton, mobility of the integrin eUbfl3 and tumor-cell-induced platelet aggregation* Jozsef Timar 1, Hemi Chopra 1, **, X. Rong 1, James S. Hatfield 5, Suzanne E. G. Fligiel 3, 5, James M. Onoda 2, 4, John D. Taylor 1, 2, 4, and Kenneth V. Honn 1, 4 Departments of 1 Radiation Oncology, 2BiologicalSciencesand 3Pathology, Wayne State University, Detroit, MI 48202, USA, 4Gershenson Radiation OncologyCenter, Harper/Grace Hospitals, Detroit, MI 48201, USA s Laboratory Service, VA Medical Center, Allen Park, MI 48101, USA Received 7 August 1991 / Accepted 27 December 1991 Summary. Calcium channel blockers of the phenylalkyl- amine (i.e. verapamil), benzothiazepine (i.e. diltiazem) and dihydropyridine (i.e. nifedipine) classes were evalu- ated for effects on the tumor cell/platelet interactions us- ing Walker 256 carcinosarcoma cells (W256 cells). When W256 cells were pretreated for 15 rain with channel blockers at concentrations of 50-200 gM, macroscopic tumor-cell-induced platelet aggregation was inhibited (order of potency; nifedipine > diltiazem >> verapamil). However, ultrastructurat analysis revealed limited, focal platelet aggregates associated with tumor cell plasma membranes of verapamil- and diltiazem-treated cells. There was no evidence of platelet activation or platelet association with the tumor cell membrane in cells pre- treated with nifedipine. Walker 256 cells possess the inte- grin ~IIbfl3" Tumor cell ~IIbfl3 was shown to mediate tumor eeU/platelet interactions in vitro [Chopra et al. (1988) Cancer Res. 48:3787]. Patching and capping of surface 0qlbfl 3 were inhibited by nifedipine > diltiazem ~> verapa- rail. The degree of inhibition of ~Ilbfl3 receptor mobility parallels the inhibition of tumor-call-induced platelet ag- gregation. W256 cells are characterized by a well-devel- oped microfilament and intermediate filament network and by the absence of a distinct microtubular network. Calcium channel blockers had no effect on the low poly- merization level of tubulin. However, they induced rear- rangement of mierofilament stress fibers. Intermediate filaments were also rearranged but to varying degrees. The order of effectiveness for alteration of intermediate filament organization was nifedipine>diltiazem while verapamil was ineffective. We propose that the pre- * This work was supported by Public Health Service grant CA- 47115, a grant from Harper Hospital and a grant from the Veterans Administration ** Present address: Department of Biological Chemistry, School of Medicine, The Johns Hopkins University, Baltimore, MD 21205, USA Abbreviations: CCB, calcium channel blocker; Ctnbfl3 , platelet glyco- protein IIb/IIIa complex; TCIPA, tumor cell induced platelet aggre- gation; W256, Walker 256 carcinosarcoma Offprint requests to: J. Timar viously reported inhibition of tumor cell/platelet interac- tion and tumor cell metastasis by calcium channel blockers [Honn et al. (1984) Clin Exp Metastasis 1:61] is due not only to the effects of the Ca 2 + channel blockers on platelets, but also to their effect on the tumor cell cy- toskeleton resulting in an inhibition of the mobility and function of the ~Hd~3receptor. Key words: Walker 256 carcinosarcoma - ~IIbfl3 -- Platelet aggregation - Cytoskeleton - Diltiazem - Verapamil - Nifedipine Introduction Calcium channel blockers (CCB) inhibit the influx of cal- cium into several cell types, including neurons, and muscle cells (Fleckenstein 1977; Godfraid 1981; Braun- wald 1982). One cell type, the function of which is also af- fected by representatives of three chemical classes (i.e. phenylalkylamine, benzothiazepine, dihydropyridine) of CCB, is the platelet. There are numerous reports that CCB can inhibit the in vitro aggregation ofcitrated plate- let-rich plasma from humans, rabbits, and cats (Shinjo et al. 1978; Ribeiro et al. 1982; Schmunk and Lefer 1982; Dale et al. 1983; Ono and Kimura 1984) and that they af- fect platelet function in vivo, and in humans, rats, and mice (Ribeiro et al. 1982; Dale et al. 1983; Ortega et al. 1987). There are also reports that demonstrate CCB of the dihydropyridine class (i.e. nifedipine and nimopidine) inhibit agonist and tumor-cell-induced platelet aggrega- tion and can synergize with other anti-platelet agents (i.e. prostacyclin and thromboxane synthase inhibitors) (Okamatsu et al. 1981; Onoda et al. 1984; Honn et al. 1985 a, b; Ortega et al. 1987). However, platelets lack the classic Ca 2 channels, and the observed effects of CCB on platelet function have been attributed to inhibition of the intracellular release of Ca ~+ (Honn et al. 1985 b). Honn and colleagues (1983, 1984, 1985a, b, 1986) in- vestigated CCB for their potential inhibitory effects on

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

J Cancer Res Clin Oncol (1992) 118:425-434 Cancer esearch
and
Cfinical �9 �9 Springer-Verlag 1992
Calcium channel blocker treatment of tumor cells induces alterations in the cytoskeleton, mobility of the integrin eUbfl3 and tumor-cell-induced platelet aggregation* Jozsef Timar 1, Hemi Chopra 1, **, X. Rong 1, James S. Hatfield 5, Suzanne E. G. Fligiel 3, 5, James M. Onoda 2, 4, John D. Taylor 1, 2, 4, and Kenneth V. Honn 1, 4
Departments of 1 Radiation Oncology, 2 Biological Sciences and 3 Pathology, Wayne State University, Detroit, MI 48202, USA, 4Gershenson Radiation Oncology Center, Harper/Grace Hospitals, Detroit, MI 48201, USA s Laboratory Service, VA Medical Center, Allen Park, MI 48101, USA
Received 7 August 1991 / Accepted 27 December 1991
Summary. Calcium channel blockers of the phenylalkyl- amine (i.e. verapamil), benzothiazepine (i.e. diltiazem) and dihydropyridine (i.e. nifedipine) classes were evalu- ated for effects on the tumor cell/platelet interactions us- ing Walker 256 carcinosarcoma cells (W256 cells). When W256 cells were pretreated for 15 rain with channel blockers at concentrations of 50-200 gM, macroscopic tumor-cell-induced platelet aggregation was inhibited (order of potency; nifedipine > diltiazem >> verapamil). However, ultrastructurat analysis revealed limited, focal platelet aggregates associated with tumor cell plasma membranes of verapamil- and diltiazem-treated cells. There was no evidence of platelet activation or platelet association with the tumor cell membrane in cells pre- treated with nifedipine. Walker 256 cells possess the inte- g r i n ~IIbfl3" Tumor cell ~IIbfl3 w a s shown to mediate tumor eeU/platelet interactions in vitro [Chopra et al. (1988) Cancer Res. 48:3787]. Patching and capping of surface 0qlbfl 3 w e r e inhibited by nifedipine > diltiazem ~> verapa- rail. The degree of inhibition of ~Ilbfl3 receptor mobility parallels the inhibition of tumor-call-induced platelet ag- gregation. W256 cells are characterized by a well-devel- oped microfilament and intermediate filament network and by the absence of a distinct microtubular network. Calcium channel blockers had no effect on the low poly- merization level of tubulin. However, they induced rear- rangement of mierofilament stress fibers. Intermediate filaments were also rearranged but to varying degrees. The order of effectiveness for alteration of intermediate filament organization was nifedipine>diltiazem while verapamil was ineffective. We propose that the pre-
* This work was supported by Public Health Service grant CA- 47115, a grant from Harper Hospital and a grant from the Veterans Administration ** Present address: Department of Biological Chemistry, School of Medicine, The Johns Hopkins University, Baltimore, MD 21205, USA Abbreviations: CCB, calcium channel blocker; Ctnbfl3 , platelet glyco- protein IIb/IIIa complex; TCIPA, tumor cell induced platelet aggre- gation; W256, Walker 256 carcinosarcoma Offprint requests to: J. Timar
viously reported inhibition of tumor cell/platelet interac- tion and tumor cell metastasis by calcium channel blockers [Honn et al. (1984) Clin Exp Metastasis 1:61] is due not only to the effects of the Ca 2 + channel blockers on platelets, but also to their effect on the tumor cell cy- toskeleton resulting in an inhibition of the mobility and function of the ~Hd~3 receptor.
Key words: Walker 256 carcinosarcoma - ~IIbfl3 -- Platelet aggregation - Cytoskeleton - Diltiazem - Verapamil - Nifedipine
Introduction
Calcium channel blockers (CCB) inhibit the influx of cal- cium into several cell types, including neurons, and muscle cells (Fleckenstein 1977; Godfraid 1981; Braun- wald 1982). One cell type, the function of which is also af- fected by representatives of three chemical classes (i.e. phenylalkylamine, benzothiazepine, dihydropyridine) of CCB, is the platelet. There are numerous reports that CCB can inhibit the in vitro aggregation ofcitrated plate- let-rich plasma from humans, rabbits, and cats (Shinjo et al. 1978; Ribeiro et al. 1982; Schmunk and Lefer 1982; Dale et al. 1983; Ono and Kimura 1984) and that they af- fect platelet function in vivo, and in humans, rats, and mice (Ribeiro et al. 1982; Dale et al. 1983; Ortega et al. 1987). There are also reports that demonstrate CCB of the dihydropyridine class (i.e. nifedipine and nimopidine) inhibit agonist and tumor-cell-induced platelet aggrega- tion and can synergize with other anti-platelet agents (i.e. prostacyclin and thromboxane synthase inhibitors) (Okamatsu et al. 1981; Onoda et al. 1984; Honn et al. 1985 a, b; Ortega et al. 1987). However, platelets lack the classic Ca 2 channels, and the observed effects of CCB on platelet function have been attributed to inhibition of the intracellular release of Ca ~ + (Honn et al. 1985 b).
Honn and colleagues (1983, 1984, 1985a, b, 1986) in- vestigated CCB for their potential inhibitory effects on

426
tumor-cell-induced platelet aggregation (TCIPA), plate- let-enhanced tumor cell adhesion to endothelial cells and to subendothelial matrix and, finally, on experimental and spontaneous metastasis. They originally reported that platelets pretreated with the dihydropyridine CCB, nifedipine, were less sensitive to aggregation induced by B16a tumor cells (Honn et al. 1983). These results were extended to other compounds of the dihydropyridine class (nimodipine, felodipine) which, in addition to inhib- iting TCIPA, were found to inhibit platelet-enhanced tu- mor cell adhesion to endothelial cells (Honn et al. 1984) as well as experimental and spontaneous metastasis (Onoda et al. 1984; Honn et al. 1985a, b). Onoda et al. (1984) compared the relative effectiveness of CCB from the phenylalkylamine, benzothiazepine, and dihydro- pyridine classes of CCB for inhibition of T C I P A in vitro and metastasis in vivo. They found the dihydropyridines superior to other classes of CCB.
In these previous studies the reduction in spontaneous and experimental metastasis was interpreted as due to the direct effect of the CCB on platelet functions (Honn et al. 1983, 1984, 1985a). However, since calcium antagonists have been shown to disrupt the cytoskeleton of normal cells (Sasaki et al. 1987), they may equally affect tumor cell responses that are dependent upon an intact tumor cell cytoskeleton. Chopra et al. (1988) reported that TCIPA can be inhibited by disruption of tumor cell microfilaments or intermediate filaments. This effect was due to an inhibition of the plasma membrane mobility of the integrin (~iib]~3 receptor. The cqld~ 3 receptor was dem- onstrated to mediate platelet adhesion to the tumor cell plasma membrane prior to platelet activation (Chopra et al. 1988). In addition, the disruption of tumor cell micro- filaments or intermediate filaments results in decreased tumor cell/platelet interaction in vivo as well as decreased lung colony format ion (Chopra et al. 1990). Therefore, in the present study we investigated the effects o f pretreat- ment by one representative compound f rom each of three classes of CCB, (i.e. phenylalkylamine, benzothiazepine, and dihydropyridine) on tumor cell/platelet interaction by analyzing the alterations in the organization of the tu- mor cell cytoskeleton and alterations in the mobility of the tumor cell Cq~b/~ 3 receptor.
Materials and methods
Tumor cell culture
w256 carcinosarcoma cells were obtained from the Animal and Hu- man Tumor Bank (Division of Cancer Treatment, National Cancer Institute, Frederick, Md,) and passaged subcutaneously in female Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, Ind.). Subcutaneous tumors were removed aseptically, minced and placed in culture flasks containing minimal essential medium (MEM; Gibco, Grand Island, N.Y.) supplemented with 10% fetal bovine serum (Gibco). After 24 h, tumor pieces were removed and fresh medium was added to the cultures. Adherent tumor cells were grown to confuence, and serially passaged every 3 days in MEM containing 5% fetal bovine serum as previously described (Menter et al. 1987).
Calcium channel blockers
Verapamil (Searle Laboratories, Arlington Heights, Ill.), diltiazem (Marion Laboratories, Kansas City, Mo.) and nifedipine (Miles Laboratories, West Haven, Conn.) were solubilized in 95% etha- nol.
Antibodies
Antibodies to eytoskeletalproteins. Rabbit antisera to tubulin, goat antiserum to mouse vimentin, fluorescein-isothiocyanate(FITC)- conjugated secondary anti-(goat IgG) and anti-(rabbit IgG) were from ICN (Lisle, Ill.). Rhodamine-labeled phalloidin (Molecular Probes, Eugene, Ore.) was used to label polymerized actin.
Antibodies to platelet integrin 0~llbfl 3. Monoclonal antibody 10E5 (a generous gift from Dr. B.S. Coller, Stony Brook, N.Y.), an IgG, was raised in mice and affinity-purified from ascites. This mAb is di- rected against an epitope on the human platelet anbfl3 complex (Col- ler et al. 1983) and was used to identify tumor cell cqlbfl 3. The spec- ificity of this mAb for tumor cell ~nb/~3 and its ability to distinguish ~nJ~3 from the vitronectin receptor and the fibronectin receptor have been published previously (Chopra et al. 1988, 1991; Grossi et al. 1988). Goat whole serum (34-37 ng/ml total protein; Cooper Biomedical) was used as a blocking serum for Fc receptors in immu- nofluorescenee studies, and FITC, conjugated anti-(mouse IgG) or FITC-conjugated anti-(rabbit IgG) (both from goat) was used as the secondary antibody. Aliquots of all antibodies were stored at -20 ~ C prior to use.
Immunofluorescence localization o f cytoskeletal components
Vimentin intermediate filaments. W256 cells c/5 • 105 cells/ml) were grown on coverslips placed in 6-well culture plates (Falcon; Becton Dickinson; Oxnard, Calif.) for 48 h at 37 ~ C. Cells were incubated with veraparnil, diltiazem or nifedipine (150 ~tM) or appropriate sol- vent controls for 15 min at 20 ~ C, rinsed in phosphate-buffered sa- line (PBS; pH 7.2) and drained to remove excess buffer. To permea- bilize the cells prior to the localization of vimentin, a 20-min fixation in absolute methanol (4 ~ C) followed by a 5-min incubation in ace- tone ( -20 ~ C) was employed. Cells were then incubated (37 ~ C) with 50 ~tl diluted (1:20) primary antibody in a humidified atmo- sphere for 30 min, washed (2 • ) with distilled water and incubated (37 ~ C) for 30 rain with 50 gl appropriate FITC-labeled secondary antibody in the dark (37 ~ C). Following incubation, cells on cover- slips were washed (2 • ) with distilled water and mounted with a drop of glycerol placed on glass slides. Samples incubated only with the secondary antibody served as negative controls.
Polymerized-actin-containing microfilaments. To localize polymer- ized actin micro filaments, cells were fixed with 3.7% formaldehyde for 10rain, washed, permeabilized with acetone ( -20 ~ C) for 10 rain, incubated with rhodamine-labeled phalloidin (Molecular Probes, Eugene, Ore., 1:200), washed with PBS and mounted with glycerol. Samples incubated with unconjugated rhodamine served as negative controls.
Immunofluorescence
Effects o f calcium channel blockers on mobility of the ~nbfl3 receptor complex
W256 tumor cells were grown on coverslips as described above and incubated with verapamil, diltiazem, or nifedipine (150 gM) or sol-
vent alone for 15 rain (20 ~ C). The cells were then washed (I x ) in PBS, fixed in 1% paraformaldehyde/PBS for 10 rain and processed for immunofluorescence localization of %b/~3- First the samples were incubated with goat whole serum (40 ng/ml in PBS; they were then incubated with mAb 10E5 (5 ~tg/ml) for 1 h, washed (3 x ) in PBS and incubated with goat anti-mouse IgG-FITC conjugate (Cappel, West Chester, Penn.) for 1 h. Other cells were incubated with the monoclonal antibody 10E5 for 30 rain at 37 ~ C then fixed with 2% paraformaldehyde for 10 rain, washed 3 x with PBS and incubated with fiuorescein-conjugated goat anti-(mouse IgG). Samples incubated with anti-mouse IgG-FITC alone served as neg- ative controls. For specificity control of mAb 10E5, the primary antibody was replaced with mouse monoclonal anti-vimentin anti- body, epitopes of which are not present at the cell surface.
Images for all of the above experiments were recorded on a Ko- dak TMAX 400 panchromatic film using a Leitz Orthoplan micro- scope or Nikon Optiphot 108 microscope, using barrier filters ap- propriate for FITC or rhodamine.
Platelet-rich plasma
Blood was drawn from the inferior vena cava of rats anesthetized with sodium pentobarbital (50 mg/kg) into heparin sulfate (50 U/ml) and 4.8% dextrose in 0.9% saline (heparin: blood ratio = 1:9 v/v). Platelet-rich and platelet-poor plasma was prepared as pre- viously described (Menter et al. 1987).
427
plasma. A 250- ~1 aliquot of platelet-rich plasma was added to each cuvette equilibrated at 37 ~ C for 6 rain and stirred at 800 rpm. Tu- mor cells were harvested from 65%-75% confluent cultures, centri- fuged (150 g; 10 rain) and washed (t x ) with medium 199. Cells were enumerated with a model ZBI counter (Coulter Electronics; Hia- leah, Fla.) and resuspended in medium 199 at 7 • l06 cells/ml. Tu- mor cells were incubated (15 min) with nifedipine, diltiazem, or verapamil (50-200 ~t_M) and washed 3 x with MEM. Calcium-chan- nel-blocker-treated W256 cells were added to an aggregometer cuvette containing 4.7 • 107 normal platelets and the aggregation re- sponse was recorded for 10 rain. Experiments were repeated a mini- mum of three times.
Electron microscopy
The interactions between platelets and tumor cells pretreated with the various calcium channel blockers were examined in samples re- moved from aggregometer cuvettes 20 rain after addition of tumor cells. This technique has been described previously (Honn et al. 1986). Samples were fixed in 2.5 % glutaraldehyde in 0. l M cacodyl - ate buffer (pH 7.2). After fixation samples were pelleted in l-m1 Ep- pendorf microtubes and processed for electron microscopy as de- scribed previously (Menter et al. 1987). Thin sections were examined and photographed with a Zeiss EM109 transmission electron micro- scope. All experiments in this study were repeated a minimum of three times with comparable results.
Aggregometry
Aggregometry studies were performed with a model DP-247E dual- channel aggregometer (Seinco; Morrison, Colo.) as described pre- viously (Menter et al. 1987). Briefly, platelet-rich plasma concentra- tions were adjusted to 1.9x 108 platelets/ml with platelet-poor
100
90
80
70
6O r
.9 5 0
._~ E 40 O~
30
.~2o
10
CQN-i~OL
I I I l I I I I I I t I I I I I " -5 0 5 10 15 20
t ime (min)
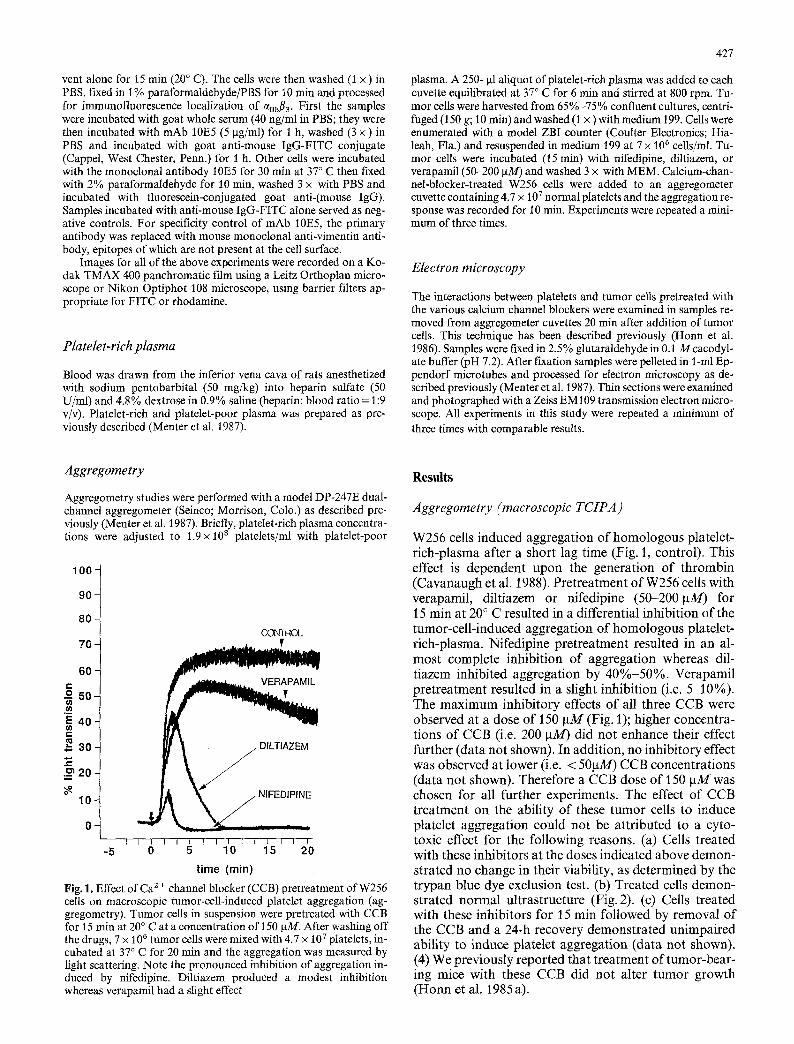
Fig. 1. Effect of Ca 2 + channel blocker (CCB) pretreatment of W256 ceils on macroscopic tumor-cell-induced platelet aggregation (ag- gregometry). Tumor cells in suspension were pretreated with CCB for 15 rain at 20 ~ C at a concentration of 150 ~tM. After washing off the drugs, 7 • 106 tumor cells were mixed with 4.7 x 10 7 platelets, in- cubated at 37 ~ C for 20 rain and the aggregation was measured by fight scattering. Note the pronounced inhibition of aggregation in- duced by nifedipine. Diltiazem produced a modest inhibition whereas verapamil had a slight effect
Results
Aggregometry (macroscopic TCIPA)
W256 cells induced aggregat ion o f h o m o l o g o u s platelet- r ich-plasma after a short lag time (Fig. 1, control) . This effect is dependent upon the generat ion o f th rombin (Cavanaugh et al. 1988). Pre t rea tment o f W256 cells with verapamil, diltiazem or nifedipine (50-200 laM) for 15 min at 20 ~ C resulted in a differential inhibit ion o f the tumor-cel l - induced aggregat ion o f h o m o l o g o u s platelet- r ich-plasma. Nifedipine pre t rea tment resulted in an al- mos t complete inhibit ion o f aggregat ion whereas dil- t iazem inhibited aggregat ion by 4 0 % - 5 0 % . Verapamil pre t rea tment resulted in a slight inhibit ion (i.e. 5-10%). The m a x i m u m inhibi tory effects o f all three CCB were observed at a dose o f 150 ~tM (Fig. 1); higher concentra- tions o f CCB (i.e. 200 ~tM) did no t enhance their effect fur ther (data not shown). In addit ion, no inhibi tory effect was observed at lower (i.e. < 50pM) CCB concentra t ions (data no t shown). Therefore a CCB dose o f 150 ~tM was chosen for all fur ther experiments. The effect o f CCB t rea tment on the ability o f these t u m o r cells to induce platelet aggregat ion could no t be at tr ibuted to a cyto- toxic effect for the fol lowing reasons. (a) Cells treated with these inhibitors at the doses indicated above demon- strated no change in their viability, as determined by the t rypan blue dye exclusion test. (b) Treated cells demon- strated normal ul t rastructure (Fig. 2). (c) Cells treated with these inhibitors for 15 min followed by removal o f the CCB and a 24-h recovery demons t ra ted unimpaired ability to induce platelet aggregat ion (data no t shown). (4) We previously reported that t rea tment o f tumor-bear - ing mice with these CCB did no t alter t u m o r growth ( H o n n et al. 1985a).
428
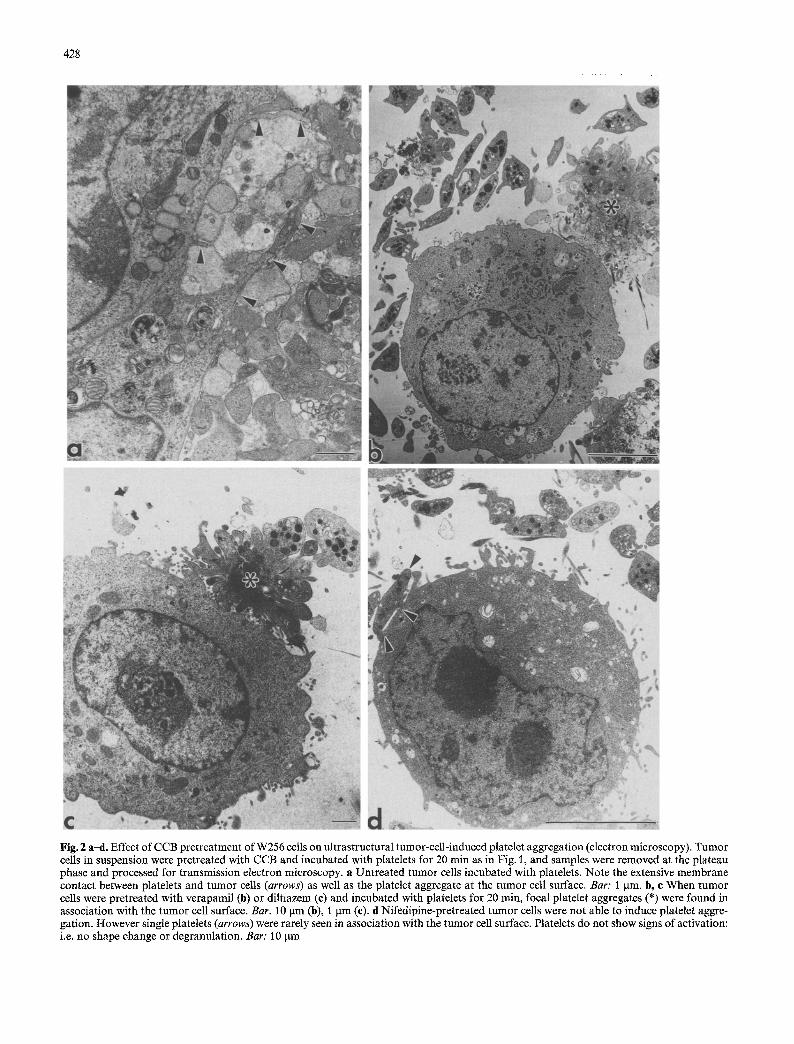
Fig. 2 a--d. Effect of CCB pretreatment of W256 cells on ultrastructural tumor-cell-induced platelet aggregation (electron microscopy). Tumor cells in suspension were pretreated with CCB and incubated with platelets for 20 min as in Fig. I, and samples were removed at the plateau phase and processed for transmission electron microscopy, a Untreated tumor cells incubated with platelets. Note the extensive membrane contact between platelets and tumor cells (arrows) as well as the platelet aggregate at the tumor cell surface. Bar: 1 lam. b, e When tumor cells were pretreated with verapamil (b) or diltiazem (c) and incubated with platelets for 20 min, focal platelet aggregates (*) were found in association with the tumor cell surface. Bar. 10 txm (b), 1 gm (c). d Nifedipine-pretreated tumor cells were not able to induce platelet aggre- gation. However single platelets (arrows) were rarely seen in association with the tumor cell surface. Platelets do not show signs of activation: i.e. no shape change or degranulation. Bar: 10 ltm
429
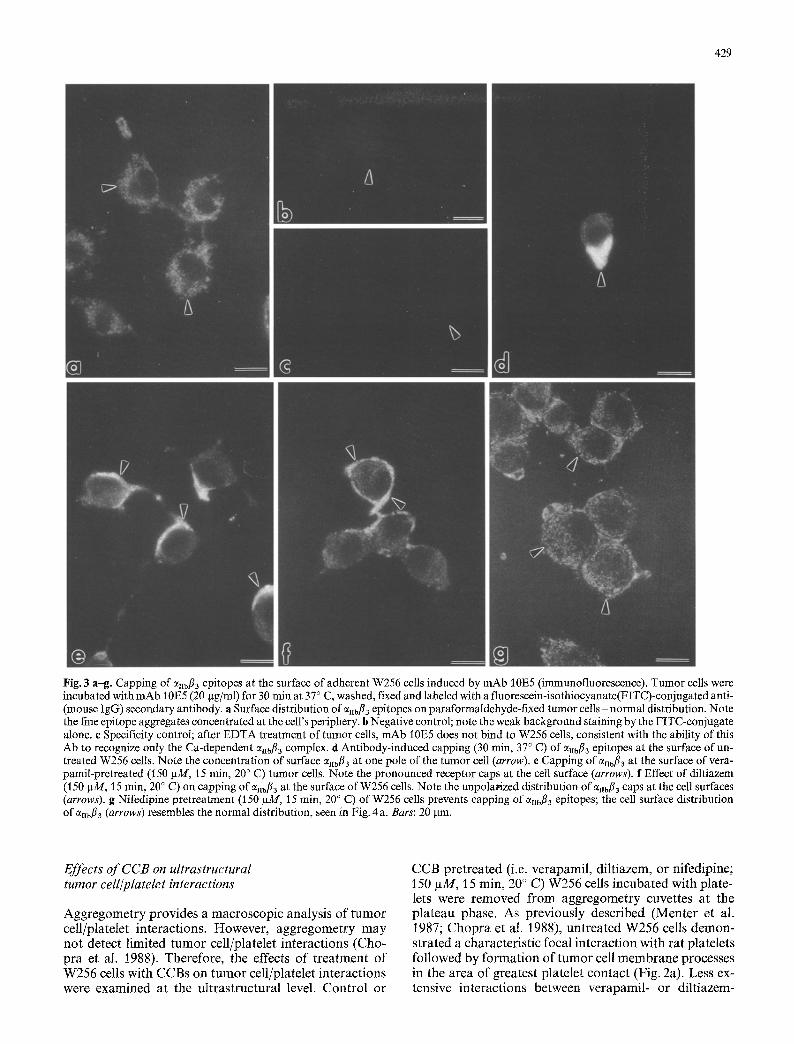
Fig. 3 a-g. Capping of ellbfl~ epitopes at the surface of adherent W256 cells induced by mAb 10E5 (immunofluorescence). Tumor cells were incubated with mAb 10E5 (20 Ixg/ml) for 30 min at 37 ~ C, washed, fixed and labeled with a fluorescein-isothiocyanate(FITC)-conjugated anti- (mouse IgG) secondary antibody, a Surface distribution of eHbfl3 epitopes on paraformaldehyde-fixed tumor cells- normal distribution. Note the fine epitope aggregates concentrated at the cell's periphery, b Negative control; note the weak background staining by the FITC-conjugate alone, e Specificity control; after EDTA treatment of tumor cells, mAb 10E5 does not bind to W256 cells, consistent with the ability of this Ab to recognize only the Ca-dependent %bf13 complex, d Antibody-induced capping (30 rain, 37 ~ C) of Cqlbfl 3 epitopes at the surface of un- treated W256 cells. Note the concentration of surface 7ttbfl3 at one pole of the tumor cell (arrow). e Capping of 0~ilbfl 3 a t the surface of vera- pamil-pretreated (t 50 gM, t 5 rain, 20 ~ C) tumor cells. Note the pronounced receptor caps at the cell surface (arrows). f Effect of diltiazem (150 IxM, 15 rain, 20 ~ C) on capping of enbfl3 at the surface of W256 cells. Note the unpolarized distribution of e~b/~3 caps at the cell surfaces (arrows). g Nifedipine pretreatrnent (150 gM, 15 rain, 20 ~ C) of W256 cells prevents capping of %bfl3 epitopes; the cell surface distribution of cqlbfl 3 (arrows) resembles the normal distribution, seen in Fig. 4 a. Bars: 20 p.m.
Effects o f CCB on ultrastructural tumor cell/platelet interactions
Aggregomet ry provides a macroscopic analysis o f t u m o r cell/platelet interactions. However , aggregomet ry may not detect limited t u m o r cell/platelet interactions (Cho- pra et al. 1988). Therefore, the effects o f t rea tment o f W256 cells with CCBs on t u m o r cell/platelet interactions were examined at the ul t ras t ructural level. Cont ro l or
CCB pretreated (i.e. verapamil, diltiazem, or nifedipine; 150 gM, 15 min, 20 ~ C) W256 cells incubated with plate- lets were removed f rom aggregomet ry cuvettes at the pla teau phase. As previously described (Menter et al. 1987; C h o p r a et al. 1988), unt rea ted W256 cells demon- strated a characterist ic focal interact ion with rat platelets followed by fo rma t ion o f t u m o r cell membrane processes in the area o f greatest platelet con tac t (Fig. 2a). Less ex- tensive interactions between verapamil- or diltiazem-
430
Effect of CCB on mobility of the tumor cell ellb/?3
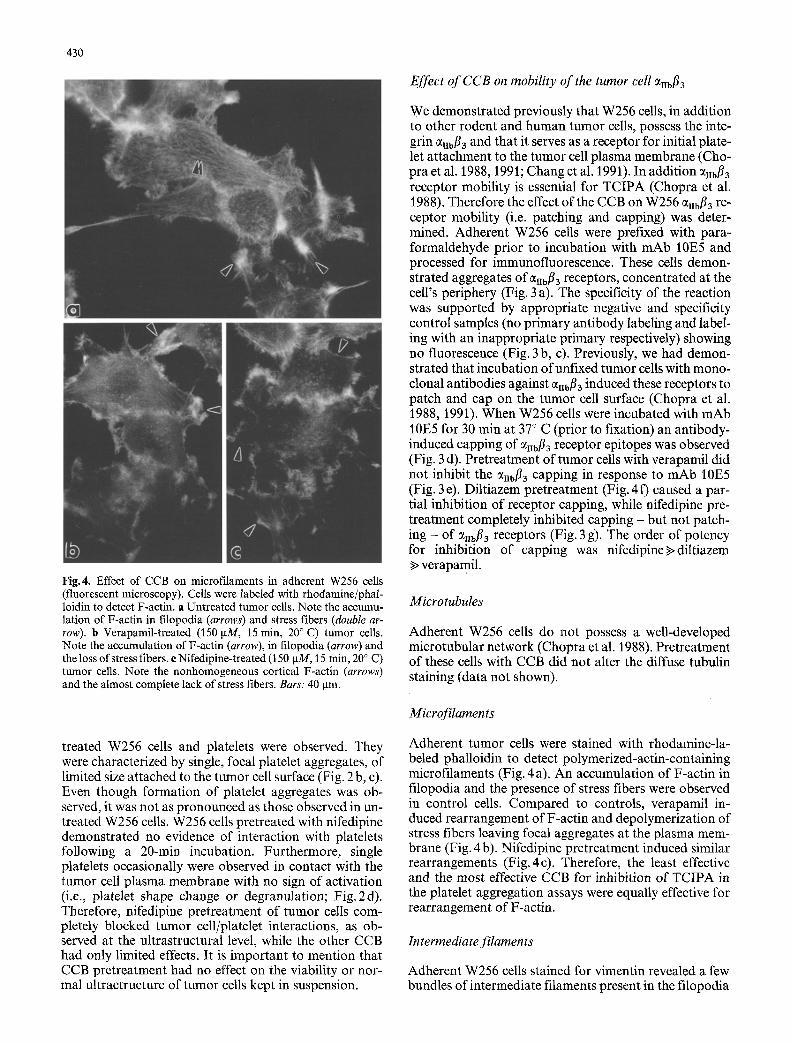
Fig.4. Effect of CCB on microfilaments in adherent W256 cells (fluorescent microscopy). Cells were labeled with rhodamine/phal- loidin to detect F-actin. a Untreated tumor cells. Note the accumu- lation of F-actin in filopodia (arrows) and stress fibers (double ar- row). b Verapamil-treated (150 IxM, 15 min, 20 ~ C) tumor cells. Note the accumulation of F-actin (arrow), in filopodia (arrow) and the loss of stress fibers, e Nifedipine-treated (150 p.M, 15 rain, 20 ~ C) tumor cells. Note the nonhomogeneous cortical F-actin (arrows) and the almost complete lack of stress fibers. Bars: 40 ]am.
We demonstrated previously that W256 cells, in addition to other rodent and human tumor cells, possess the inte- grin ~nbfl3 and that it serves as a receptor for initial plate- let attachment to the tumor cell plasma membrane (Cho- pra et al. 1988, 1991; Chang et al. 1991). In addition Cqibfl 3 receptor mobility is essential for TCIPA (Chopra et al. 1988). Therefore the effect of the CCB on W256 a~Ibfl3 re- ceptor mobility (i.e. patching and capping) was deter- mined. Adherent W256 cells were prefixed with para- formaldehyde prior to incubation with mAb 10E5 and processed for immunofluorescence. These cells demon- strated aggregates of aHbfl3 receptors, concentrated at the cell's periphery (Fig. 3 a). The specificity of the reaction was supported by appropriate negative and specificity control samples (no primary antibody labeling and label- ing with an inappropriate primary respectively) showing no fluorescence (Fig. 3 b, c). Previously, we had demon- strated that incubation of unfixed tumor cells with mono- clonal antibodies against enbfi3 induced these receptors to patch and cap on the tumor cell surface (Chopra et al. 1988, 1991). When W256 cells were incubated with mAb 10E5 for 30 min at 37 ~ C (prior to fixation) an antibody- induced capping of Cq~bfi 3 receptor epitopes was observed (Fig. 3 d). Pretreatment of tumor cells with verapamil did not inhibit the eUbfi3 capping in response to mAb 10E5 (Fig. 3 e). Diltiazem pretreatment (Fig. 4 f) caused a par- tial inhibition of receptor capping, while nifedipine pre- treatment completely inhibited capping - but not patch- ing - of anbfl3 receptors (Fig. 3 g). The order of potency for inhibition of capping was nifedipine~>diltiazem ~> verapamil.
Microtubules
Adherent W256 cells do not possess a well-developed microtubular network (Chopra et al. 1988). Pretreatment of these cells with CCB did not alter the diffuse tubulin staining (data not shown).
Micro filaments
treated W256 cells and platelets were observed. They were characterized by single, focal platelet aggregates, of limited size attached to the tumor cell surface (Fig. 2 b, c). Even though formation of platelet aggregates was ob- served, it was not as pronounced as those observed in un- treated W256 cells. W256 cells pretreated with nifedipine demonstrated no evidence of interaction with platelets following a 20-rain incubation. Furthermore, single platelets occasionally were observed in contact with the tumor cell plasma membrane with no sign of activation (i.e., platelet shape change or degranulation; Fig. 2d). Therefore, nifedipine pretreatment of tumor cells com- pletely blocked tumor cell/platelet interactions, as ob- served at the ultrastructural level, while the other CCB had only limited effects. It is important to mention that CCB pretreatment had no effect on the viability or nor- mal ultractructure of tumor cells kept in suspension.
Adherent tumor cells were stained with rhodamine-la- beled phalloidin to detect polymerized-actin-containing microfilaments (Fig. 4 a). An accumulation of F-actin in filopodia and the presence of stress fibers were observed in control cells. Compared to controls, verapamil in- duced rearrangement of F-actin and depolymerization of stress fibers leaving focal aggregates at the plasma mem- brane (Fig. 4 b). Nifedipine pretreatment induced similar rearrangements (Fig. 4c). Therefore, the least effective and the most effective CCB for inhibition of TCIPA in the platelet aggregation assays were equally effective for rearrangement of F-actin.
Intermediate filaments
Adherent W256 cells stained for vimentin revealed a few bundles of intermediate filaments present in the filopodia
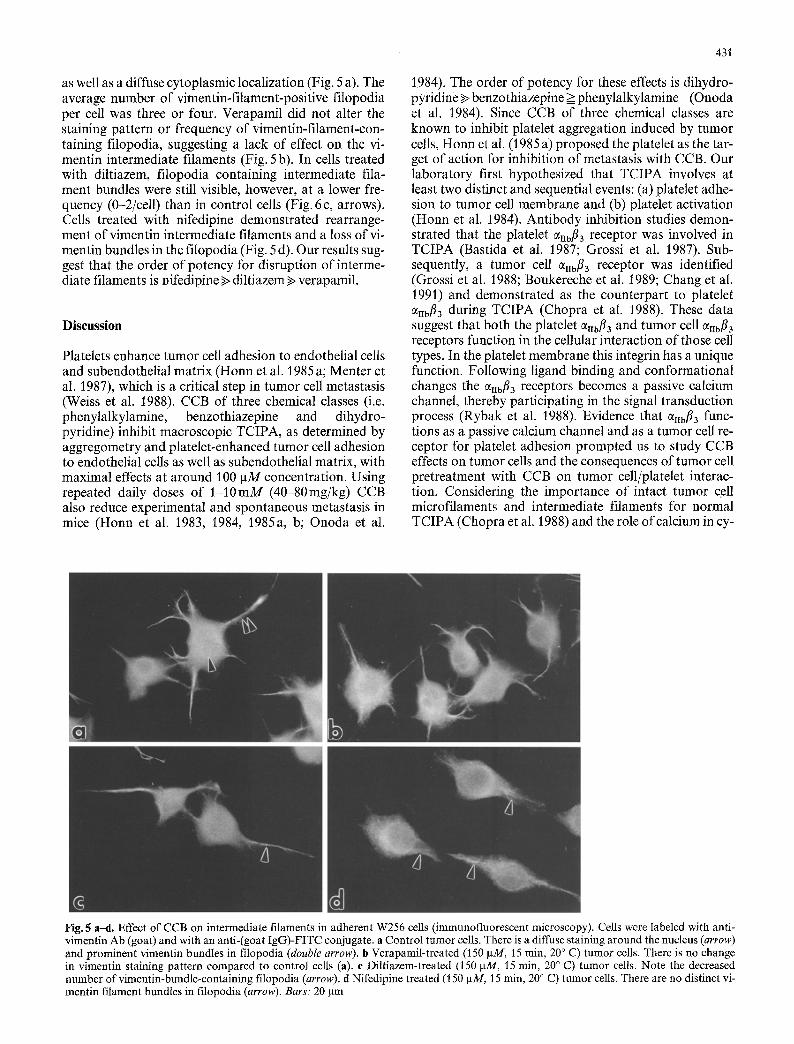
as well as a diffuse cytoplasmic localization (Fig. 5 a). The average number of vimentin-filament-positive filopodia per cell was three or four. Verapamil did not alter the staining pattern or frequency of vimentin-filament-con- taining filopodia, suggesting a lack of effect on the vi- mentin intermediate filaments (Fig. 5 b). In cells treated with diltiazem, filopodia containing intermediate fila- ment bundles were still visible, however, at a lower fre- quency (0-2/cell) than in control cells (Fig. 6 c, arrows). Cells treated with nifedipine demonstrated rearrange- ment of vimentin intermediate filaments and a loss of vi- mentin bundles in the filopodia (Fig. 5 d). Our results sug- gest that the order of potency for disruption of interme- diate filaments is nifedipine ~> diltiazem,> verapamil.
Discussion
Platelets enhance tumor cell adhesion to endothelial cells and subendothelial matrix (Honn et al. 1985 a; Menter et al. 1987), which is a critical step in tumor cell metastasis (Weiss et al. 1988). CCB of three chemical classes (i.e. phenylalkylamine, benzothiazepine and dihydro- pyridine) inhibit macroscopic TCIPA, as determined by aggregometry and platelet-enhanced tumor cell adhesion to endothelial cells as well as subendothelial matrix, with maximal effects at around 100 gM concentration. Using repeated daily doses of 1-10ram (40-80mg/kg) CCB also reduce experimental and spontaneous metastasis in mice (Honn et al. 1983, ~984, 1985a, b; Onoda et al.
431
1984 ). The order of potency for these effects is dihydro- pyridine ~> benzothiazepine > phenylalkylamine (Onoda et al. 1984). Since CCB of three chemical classes are known to inhibit platelet aggregation induced by tumor cells, Honn et al. (1985 a) proposed the platelet as the tar- get of action for inhibition of metastasis with CCB. Our laboratory first hypothesized that TCIPA involves at least two distinct and sequential events: (a) platelet adhe- sion to tumor cell membrane and (b) platelet activation (Honn et al. 1984). Antibody inhibition studies demon- strated that the platelet enb/~3 receptor was involved in TCIPA (Bastida et al. 1987; Grossi et al. 1987). Sub- sequently, a tumor cell %b/~3 receptor was identified (Grossi et al. 1988; Boukereche et al. 1989; Chang et al. 1991) and demonstrated as the counterpart to platelet %b/~3 during TCIPA (Chopra et al. 1988). These data suggest that both the platelet %J~3 and tumor cell ~Iib/~3 receptors function in the cellular interaction of those cell types. In the platelet membrane this integrin has a unique function. Following ligand binding and conformational changes the %b/~3 receptors becomes a passive calcium channel, thereby participating in the signal transduction process (Rybak et al. 1988). Evidence that %b/~3 func- tions as a passive calcium channel and as a tumor cell re- ceptor for platelet adhesion prompted us to study CCB effects on tumor cells and the consequences of tumor cell pretreatment with CCB on tumor cell/platelet interac- tion. Considering the importance of intact tumor cell microfilaments and intermediate filaments for normal TCIPA (Chopra et al. 1988) and the role of calcium in cy-
Fig. 5 a-d. Effect of CCB on intermediate filaments in adherent W256 cells (immunofluorescent microscopy). Ceils were labeled with anti- vimentin Ab (goat) and with an anti-(goat IgG)-FITC conjugate, a Control tumor cells. There is a diffuse staining around the nucleus (arrow) and prominent vimentin bundles in filopodia (double arrow), b Verapamil-treated (150 gM, 15 rain, 20 ~ C) tumor cells. There is no change in vimentin staining pattern compared to control cells (a). c Diltiazem-treated (150 gM, 15 rain, 20 ~ C) tumor cells. Note the decreased number of vimentin-bundle-containing filopodia (arrow). fl Nifedipine treated (150 gM, 15 rain, 20 ~ C) tumor cells. There are no distinct vi- mentin filament bundles in filopodia (arrow). Bars: 20 ~tm

432
Table 1. Summary of the effects of calcium channel blockers on W256 cells"
CCB Inhibition Rearrangement
TCIPAm TCIPAu Receptor M F IF mobility
Verapamil - - - + - Diltiazem _+ - + NT - Nifedipine + + + + +
" CCB, Ca 2 + channel blocker; TCIPAm, macroscopic tumor-cell- induced platelet aggregation; TCIPAu, tdtrastructural tumor-cell- induced platelet aggregation; RM, tumor cell enb/~ receptor mobil- ity; MF, microfilament; IF, intermediate filament. - , No effect; + , pronounced effect; + , weak effect; NT, not tested
toskeletal integrity, we focused our attention on the tu- mor cell cytoskeleton as well (Table I).
Scant information exists demonstrating an effect for CCB on the cytoskeletal elements of normal or neoplastic cells. Thompson et al. (1983) found that nifedipine in- hibits leukocyte mobility in response to a cancer cell ex- tract, but these authors did not examine the effect of ni- fedipine on the cytoskeleton. Sasaki et al. (1987) reported that the intracellular calcium antagonists TMB8 and HAl077 induced disruption of actin-containing microfi- laments in rabbit aortic smooth muscle cells. Moreover CCB (verapamil and diltiazem), applied at high concen- trations (300-400 gM), disrupted actin microfilaments. Effects on microtubules and intermediate filaments were not examined (Sasaki et al. 1987). In aggreement with those observations, CCB (administered at doses of 50- 100 gM) induced rearrangement of microfilaments (i.e. depolymerization of stress fibers) in W256 cells (Table 1). The mechanism responsible for this effect is presently un- known but may involve an alteration of intracellular Ca 2 + levels. A drop in intracellular C a 2 + which would inactivate actin-severing proteins (gelsolin, villin, frag- min) (Weeds 1982). In addition, the decreased intracellu- lar Ca 2 + may also inactivate Ca z +-dependent kinases, which phosphorylate and activate other actin-binding proteins such as talin, e-actinin and vinculin (Beckerle 1990). These proteins provide the necessary link of micro- filaments to plasma membrane receptors. This link is dis- rupted in the absence of Ca 2 +-dependent phosphoryla- tion. We suggest that the microfilament rearrangement observed in CCB-treated tumor cells is due to the altered function of those actin-binding proteins (i.e. talin, e-acti- nin, vinculin). Ca 2 + also stimulates disruption of micro- tubules (Keith et al. 1983), however, W256 cells do not posses a well-defined microtubular network. Therefore the effect of CCB on microtubules was difficult to study. In addition, microtubules were demonstrated not to play a role in tumor-cell-induced platelet aggregation (Chopra et al. 1988). Further CCB utilized in this study did not alter the already existing low polymerization level of microtubules (data not shown).
In contrast to their similar effects on microfilaments, there was a clear distinction between the effects of ni- fedipine and verapamil on intermediate filaments as well as on the capping of cq~b/~ 3 receptors induced by mAb
10E5 (Table 1). The sum of the rank orders of potency for the inhibitory effects of each compound on vimentin intermediate filaments and enb/~3 receptor mobility (i.e. nifedipine > diltiazem >> verapamil) paralleled the rank order of potency of these CCB for inhibition of macro- scopic TCIPA (i.e. aggregometry; Table 1). In addition the order of potency for inhibition of cq~d~ 3 receptor mo- bility correlated with the ultrastructural evidence for tu- mor cell/platelet interaction. Nifedipine completely in- hibited ~IIbfl3 capping and platelet adhesion to the W256 cell plasma membrane. In contrast, diltiazem only was partially effective and verapamil ineffective for inhibition of the end~3 receptor capping. W256 cells treated with ei- ther of these CCB demonstrated focal aggregates of acti- vated platelets attached to their plasma membranes. We previously demonstrated that disruption of tumor cell microfilaments or vimentin intermediate filaments in- hibits TCIPA in vitro (Chopra et al. 1988) and tumor cell/ platelet interaction in vivo (Chopra et al. 1990), suggest- ing that both cytoskeletal elements are necessary for suc- cessful tumor cell/platelet interaction. Therefore, the in- hibition of TCIPA by CCB may be caused - at least par- tially - by the alterations of the tumor cell cytoskeleton induced by CCB. It is known that receptor patching and capping are not only dependent on the polymerization of microfilaments but also on actin/myosin interactions (Taylor et al. 1971; Bourguignon and Bourguignon 1984). It seems that in W256 cells intermediate filaments may also be involved in this process (Chopra et al. 1988, 1991). Further, the function of myosin depends on its phosphorylation status by Ca 2 +-dependent kinase activ- ity (Kawamoto et al. 1989). Receptor capping is preceded by activation of signal transduction pathways with sub- sequent release of C a 2+. The absence of a capping re- sponse of end~3 receptors upon ligand stimulation (i.e. mAb 10E5) in nifedipine-pretreated tumor cells suggests that the mobility of these receptors involves Ca z + release. The differences in the effectiveness of various CCBs for disruption of the cytoskeletal integrity and inhibition of integrin e~b/~3 mobility may be attributed to their dif- ferential abilities to affect intracellular concentrations of C a 2 + in W256 cells. Studies are in progress to determine the effect of these CCB on intracellular Ca / + levels in tu- mor cells.
The potential use of CCB in the treatment of malig- nant tumors is suggested (Tsuruo 1983; Helson 1984). Verapamil was shown to be a specific inhibitor of P-gly- coprotein (Tsuruo 1983; Caro-Gauchi and Riosden 1987) responsible for multi-drug resistance of malignant tu- mors, and nifedipine enhances, in a synergistic manner, the chemotherapeutic effect of cisplatin against non-re- sistant as well as resistant tumor cells (Onoda et al. 1988, 1989, 1990). Nifedipine also is the most potent CCB for inhibition of tumor cell/platelet interaction in vitro (Honn et al. 1983, 1984, 1985 a) and metastasis formation in vivo (Onoda et al. 1984; Honn et al. 1985a, b). In the present study we demonstrated that nifedipine is the most potent CCB for alteration of cytoskeletal integrity and mobility of the enb/?3 receptor in W256 tumor cells. Since cytoskeletal integrity (i.e. microfilaments and intermedi- ate filaments) and e~ib/?3 receptor mobility are critical for

433
tumor cell/platelet interaction, an interaction that en- hances metastasis formation (Honn et al. 1991) we pro- pose that the antimetastatic effects previously reported for CCB, in particular the dihydropyridines (Onoda et al. 1984; Honn et al. 1985 a, b), are a result, in part, of their direct effect on the tumor cell cytoskeleton.
References
Bastida E, Almirall L, Ordinas A (1987) Tumor cell induced platelet aggregation is a glycoprotein dependent and lipoxygenase-asso- ciated process. Int J Cancer 39:760-763
Beckerle M (1990) The adhesion plaque protein, talin, is phos- phorylated in vivo in chicken embryo fibroblasts exposed to a tumor-promoting phorbol ester. Cell Regul 1:227-236
Bourguignon LYW, Bourguignon GJ (1984) Capping and the cy- toskeleton. Int Rev Cytol 87:195-224
Bourkereche H, Berthier-Vergues O, Tabone E, Dore J, Leung LLK, McGregor JL (1989) Platelet-melanoma cell interaction is mediated by the glycoprotein IIb/IIIa complex. Blood 74:658-- 663
Braunwald E (1982) Mechanism of addition of calcium channel blocking agents. J Engl J Med 307:1618-1627
Caro-Gauci DF, Riosden JR (1987) Action of calcium antagonists on multidrug resistant cells. Biochem Pharmacol 36:2115-2123
Cavanaugh PC, Sloane BF, Honn KV (1988) Role of the coagula- tion system in tumor cell induced platelet aggregation and me- tastasis. Hemostasis 18:37-46
Chang SY, Chen YQ, Fitzgerald L, Honn KV (1991) Analysis ofin- tegrin mRNA in human and rodent tumor cells. Biochem Bio- phys Res Commun 176:108-113
Chopra H, Hatfield JS, Chang YS, Grossi IM, Fitzgerald LA, O'Gara CY, Marnett LJ, Diglio CA, Taylor JD, Honn KV (1988) Role of tumor cell cytoskeleton and membrane glycopro- tein IRGPIIb/IIla in platelet adhesion to tumor cell membrane and tumor cell induced platelet aggregation. Cancer Res 48:3787-3800
Chopra H, Hatfield JS, Fligiel SEG, Finch CF, Fitzgerald LA, Tay- lor JD, Honn KV (1990) An in vivo study of the role of the tu- mor cell cytoskeleton in tumor cell-platelet interaction. Cancer Res 50:7686-7696
Chopra H, Timar J, Chen YQ, Rong X, Grossi IM, Fitzgerald LA, Taylor JD, Honn KV (1991) The lipoxygenase metabolite 12(S)- HETE induces a cytoskeleton-dependent increase in surface ex- pression of integrin cq~bfl 3 on melanoma cells. Int J Cancer 49:774-786
Coller BS, Peerschke El., Scudder LE, Sullivan CA (1983) A murine monoclonal antibody that completely blocks the binding of fi- brinogen to platelets and produces a thrombastenic-like state in normal platelets and binds to glycoproteins IIb and/or IIIa. J Clin Invest 72:325-338
Dale J, Landmark KH, Myhre E (1983) The effects of nifedipine, a calcium antagonist, on platelet function. Am Heart J 105:103- 105
Fleckenstein A (1977) Specific pharmacology of calcium in myocar- dium, cardiac pacemakers and vascular smooth muscle cells. Annu Rev Pharmacol Toxicol 17:149-166
Godfraind T (1981) Mechanism of action of calcium entry blockers. Fed Proc 40:2866-2871
Grossi IM, Fitzgerald LA, Kendall A, Taylor JD, Sloane BF, Honn KV (1987) Inhibition of human tumor cell induced platelet ag- gregation by antibodies to platelet glycoproteines Ib and IIb/ IIIa. Proc Soc Exp Biol Med 186:378-383
Grossi IM, Hatfield JS, Fitzgerald LA, Newcombe M, Taylor JD, Honn KV (1988) Role of glycoproteins immunologically related to Ib and IIb/IIIa in tumor cell-platelet and tumor cell matrix in- teractions. FASEB J 2:2385--2395
Helson L (1984) Calcium channel blocker enhancement of finti- cancer drug cytotoxicity - a review. Cancer Drug Deliv 1:353- 361
Honn KV, Onoda JM, Diglio CA, Sloane BF (t983) Calcium chan- nel blockers: potential antimetastatic agents. Proc Soc Exp Biol Med 174:16-19
Honn KV, Onoda JM, Diglio CA, Battaglia MM, Taylor JD, Sloane FB (1984) Inhibition of tumor cell-platelet interactions and tumor metastasis by the calcium channel blocker, nimo- dipine. Clin Exp Metastasis 1:61-72
Honn KV, Onoda JM, Pampalona K, Battaglia M, Neagos G, Tay- lor JD, Diglio CA, Sloane BF (1985 a) Inhibition by dihydro- pyridine class calcium channel blockers of tumor cell-platelet- endothelial cell interactions in vitro and metastasis in vivo. Bio- chem Pharmacol 34:235-241
Honn KV, Onoda JM, Taylor JD, Sloane BF (1985b) Antimeta- static therapy with calcium active compounds. In: Hellman K, Eccles SA (eds) Treatment of metastasis: problems and pro- spects. Taylor and Francis, London, pp 259-266
Honn KV, Cavanaugh PG, Crissman JD, Taylor JD, Sloane BF (1986) Platelet-tumor cells interactions as a target for antimeta- static therapy. In: Honn KV, Powers WE, Sloane BF (eds) Mechanisms of cancer metastasis. Potential therapeutic implica- tions. Nijhoff, Boston, pp 117-144
Honn KV, Grossi IM, Timar J, Chopra H, Taylor JD (1991) Plate- lets and cancer metastasis. In: Orr FW, Buchanan MR, Weiss L (eds) Microcirculation in cancer metastasis. CRC Press, Boca Raton, Fla, pp 93-110
Kawamoto S, Resai Bangur A, Sellers JR, Adelstein RS (1989) In situ phosphorylation of human platelet myosin heavy and light chains by protein kinase C. J Biol Chem 264:2258-2265
Keith C, di Paola M, Maxfield FR, Shebanski ML (1983) Microin- jection of Ca z +-calmodulin causes a localized depolymerization of microtubules. J Cell Biol 97:1918-1924
Menter DG, Hatfield JS, Harkins C J, Sloane BF, Taylor JD, Criss- man JD, Honn KV (1987) Tumor cell-platelet interactions in vi- tro and their relationship to in vivo arrest of hematogenous cir- culating tumor cells. Clin Exp Metastasis 5:65-78
Okamatsu S, Peck RC, Lefer AM (1981) Effects of calcium channel blockers on arachidonate-induced sudden death in rabbits. Proc Soc Exp Biol Med 166:551-555
Ono H, Kimura M (1984) Effect of Ca2+-antagonist vasodilators, diltiazem, nifedipine, perhexiline and verapamil on platelet ag- gregation in vitro. Arzneimittelforschung/Drug Res 31:1131- 1134
Onoda JM, Sloane BF, Taylor JD, Honn KV (1984) Calcium chan- nel blockers: inhibitors of tumor cell-platelet-endothelial cell.in- teractions. In: Honn KV, Sloane BF (eds) Hemostatic mecha- nisms and metastasis. Nijhoff, Boston, pp 244-258
Onoda, JM, Nelson KK, Taylor JD, Honn KV (1988) Cisplatin and nifedipine synergistic antitumor effects against inherently cis- platin-resistant tumor: Cancer Lett 40:39-47
Onoda JM, Nelson KK, Taylor JD, Honn KV (1989) In vivo char- acterization of combination antitumor chemotherapy with cal- cium channel blockers and cis-diammino-dichtoroplatinum (II). Cancer Res 49:2844-2850
Onoda JM, Nelson KK, Pilarsky SM, White NS, Mihu RG, Honn KV (1990) Combination chemotherapy with cisplatin and ni- fedipine: synergistic antitttmor effects against a cisplatin-resis- tant subline of the B I 6 amelanotic melanoma. Clin Exp Metast 8:59-73
Ortega MP, Sunkel C, Priego JG, Statkow PR (1987) The anti- thrombogenic in vivo effects of calcium channel blockers in ex- perimental thrombosis in mice. Thromb Haemost 57:283-285
Ribeiro LGT, Brandon TA, Horak JK, Ware JA, Miller RR, Soils RT (1982) Inhibition ofplatelet aggregation by verapamil: quan- tification by in vivo and in vitro techniques. J Cardiovasc Phar- macol 4:170-173
Rybak ME, Renzulli LA, Bruns M J, Calaly DP (I 988) Platelet gly- coproteins IIb and IIIa as a calcium channel in liposomes. Blood 72:714-720

434
Sasaki Y, Kanno K, Hidaka H (1987) Disorganization of calcium antagonists of actin microfilament in aortic smooth muscle cells. Am J Physiol 253:c71--c78
Scmunk GA, Lefer AM (1982) Antiaggregatory actions of calcium channel blockers in cat platelets. Res Commun Chem Pathol Pharmacol 35:179-187
Shinjo A, Sasaki Y, Inimasu M, Morita T (1978) In vitro effect of the coronary vasodilator diltiazem on human and rabbit plate- lets. Thromb Res 13:941-955
Taylor RB, Duffers PH, Raff MC,.De Petris S (1971) Redistribution and pinocytosis of lymphocyte surface immunoglobulin mole- cules induced by anti-immunoglobulin antibody. Nature 233:225-229
Thompson DMP, Phelan K J, Slanzano R (1983) Oxidative metab- olism, cytoskeletal system, and calcium entry of leukocytes in the phenomenon of sensitizing cancer extract-induced leukocyte ad- herence inhibition. Cancer Res 43:1066-1073
Tsuruo T (1983) Reversal of acquired resistance to Vinca alkaloids and anthracyclin antibiotics. Cancer Treat Rep 67:889-894
Weeds A (1982) Actin binding proteins-regulators of cell architec- ture and motility. Nature 296:811-816
Weiss L, Orr FW, Honn KV (1988) Interactions of cancer cells with the microvasculature during metastasis. FASEB J 2:12-21
Related Documents











