ARTHRITIS & RHEUMATISM Vol. 62, No. 7, July 2010, pp 1911–1920 DOI 10.1002/art.27460 © 2010, American College of Rheumatology Breaking T Cell Tolerance Against Self Type II Collagen in HLA–DR4–Transgenic Mice and Development of Autoimmune Arthritis Tsvetelina Batsalova, Balik Dzhambazov, Patrick Merky, Alexandra Ba ¨cklund, and Johan Ba ¨cklund Objective. To establish a new animal model in DRB1*0401 (DR4)–transgenic mice in which T cell tolerance to self type II collagen (CII) can be broken and allow for the development of autoimmune arthritis, to investigate the role of posttranslational modifications of the CII 259–273 epitope in the induction and breaking of tolerance of DR4-restricted T cells, and to characterize DR4-restricted T cell recognition of the immunodomi- nant CII 259–273 epitope. Methods. DR4-transgenic mice expressing either the entire human CII protein (HuCII) or only the immunodominant T cell epitope of heterologous CII (MMC) in joint cartilage were established on different genetic backgrounds, and susceptibility to collagen- induced arthritis (CIA) was tested. Results. HuCII mice displayed stronger T cell tolerance to heterologous CII than did MMC mice. On the B10 background, arthritis developed only in MMC mice with a defective oxidative burst. However, MMC mice on the C3H background were susceptible to arthri- tis also with a functional oxidative burst. Significant recall responses in tolerized mice were detected only against the nonglycosylated CII 259–273 epitope. Recogni- tion of the CII 259–273 epitope was heterogeneous, but the majority of T cells in DR4 mice specifically recognized the nonglycosylated side chain of lysine at position 264. Conclusion. It is possible to break tolerance to self CII and induce arthritis in DR4 mice. However, arthritis susceptibility is tightly controlled by the ge- netic background and by the source of the transgenic element for expressing the heterologous CII peptide as a self CII protein in the joint. In contrast to CIA in A q -expressing mice, the nonglycosylated CII 259–273 epitope is clearly immunodominant in both tolerized and nontolerized DR4 mice. Rheumatoid arthritis (RA) is a chronic inflam- matory disease affecting the peripheral joints. The role of T cells in RA is supported by the occurrence of activated CD4 T cells within the inflamed synovium and by immunogenetic data linking RA with shared epitope class II major histocompatibility complex (MHC) genes and with PTPN22, as well as by the approval of abatacept as a treatment of RA (1). Type II collagen (CII) is the major component of hyaline cartilage and has been suggested as a possible autoantigen in RA, since autoimmunity to CII is com- monly detected in RA. In addition, an RA-like disease, collagen-induced arthritis (CIA), can be induced in mice after immunization with CII. Susceptibility to CIA is associated with the murine class II MHC molecule A q , but also mice with transgenic expression of the RA- associated class II MHC molecule HLA–DRB1*0401 (DR4) or *0101 (DR1) can develop CIA following immunization with CII. Interestingly, A q , DR1, and Supported by the O ¨ sterlund Foundation, the Anna Greta Crafoord Foundation for Rheumatology Research, the Åke Wiberg Foundation, the Greta and Johan Kock Foundation, King Gustaf V’s 80-Year Foundation, the Magnus Bergvall Foundation, the Nanna Svartz Foundation, the Royal Physiographic Society in Lund, the Tore Nilson Foundation, the Swedish Association Against Rheumatism, the Swedish Research Council, the European Union Sixth Framework Programme (projects AutoCure [LSHB-CT-2006-018661] and Euro-RA [MRTN-CT-2004-0005693]), and the European Union Sev- enth Framework Programme (project Masterswitch; HEALTH-F2- 2008-223404). Tsvetelina Batsalova, MSc, Balik Dzhambazov, PhD, Patrick Merky, MSc, Alexandra Ba ¨cklund, PhD, Johan Ba ¨cklund, PhD: Karo- linska Institute, Stockholm, Sweden. Ms Batsalova and Dr. Dzhambazov contributed equally to this work. Address correspondence and reprint requests to Johan Ba ¨ck- lund, PhD, Division of Medical Inflammation Research, Department of Medical Biochemistry and Biophysics, Karolinska Institute, 171 77 Stockholm, Sweden. E-mail: [email protected]. Submitted for publication August 22, 2009; accepted in revised form March 12, 2010. 1911

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ARTHRITIS & RHEUMATISMVol. 62, No. 7, July 2010, pp 1911–1920DOI 10.1002/art.27460© 2010, American College of Rheumatology
Breaking T Cell Tolerance Against Self Type II Collagen inHLA–DR4–Transgenic Mice and Development of
Autoimmune Arthritis
Tsvetelina Batsalova, Balik Dzhambazov, Patrick Merky,Alexandra Backlund, and Johan Backlund
Objective. To establish a new animal model inDRB1*0401 (DR4)–transgenic mice in which T celltolerance to self type II collagen (CII) can be brokenand allow for the development of autoimmune arthritis,to investigate the role of posttranslational modificationsof the CII259–273 epitope in the induction and breaking oftolerance of DR4-restricted T cells, and to characterizeDR4-restricted T cell recognition of the immunodomi-nant CII259–273 epitope.
Methods. DR4-transgenic mice expressing eitherthe entire human CII protein (HuCII) or only theimmunodominant T cell epitope of heterologous CII(MMC) in joint cartilage were established on differentgenetic backgrounds, and susceptibility to collagen-induced arthritis (CIA) was tested.
Results. HuCII mice displayed stronger T celltolerance to heterologous CII than did MMC mice. Onthe B10 background, arthritis developed only in MMCmice with a defective oxidative burst. However, MMC
mice on the C3H background were susceptible to arthri-tis also with a functional oxidative burst. Significantrecall responses in tolerized mice were detected onlyagainst the nonglycosylated CII259–273 epitope. Recogni-tion of the CII259–273 epitope was heterogeneous, but themajority of T cells in DR4 mice specifically recognizedthe nonglycosylated side chain of lysine at position 264.
Conclusion. It is possible to break tolerance toself CII and induce arthritis in DR4 mice. However,arthritis susceptibility is tightly controlled by the ge-netic background and by the source of the transgenicelement for expressing the heterologous CII peptide asa self CII protein in the joint. In contrast to CIA inAq-expressing mice, the nonglycosylated CII259–273
epitope is clearly immunodominant in both tolerizedand nontolerized DR4 mice.
Rheumatoid arthritis (RA) is a chronic inflam-matory disease affecting the peripheral joints. The roleof T cells in RA is supported by the occurrence ofactivated CD4� T cells within the inflamed synoviumand by immunogenetic data linking RA with sharedepitope class II major histocompatibility complex(MHC) genes and with PTPN22, as well as by theapproval of abatacept as a treatment of RA (1).
Type II collagen (CII) is the major component ofhyaline cartilage and has been suggested as a possibleautoantigen in RA, since autoimmunity to CII is com-monly detected in RA. In addition, an RA-like disease,collagen-induced arthritis (CIA), can be induced in miceafter immunization with CII. Susceptibility to CIA isassociated with the murine class II MHC molecule Aq,but also mice with transgenic expression of the RA-associated class II MHC molecule HLA–DRB1*0401(DR4) or *0101 (DR1) can develop CIA followingimmunization with CII. Interestingly, Aq, DR1, and
Supported by the Osterlund Foundation, the Anna GretaCrafoord Foundation for Rheumatology Research, the Åke WibergFoundation, the Greta and Johan Kock Foundation, King Gustaf V’s80-Year Foundation, the Magnus Bergvall Foundation, the NannaSvartz Foundation, the Royal Physiographic Society in Lund, the ToreNilson Foundation, the Swedish Association Against Rheumatism, theSwedish Research Council, the European Union Sixth FrameworkProgramme (projects AutoCure [LSHB-CT-2006-018661] andEuro-RA [MRTN-CT-2004-0005693]), and the European Union Sev-enth Framework Programme (project Masterswitch; HEALTH-F2-2008-223404).
Tsvetelina Batsalova, MSc, Balik Dzhambazov, PhD, PatrickMerky, MSc, Alexandra Backlund, PhD, Johan Backlund, PhD: Karo-linska Institute, Stockholm, Sweden.
Ms Batsalova and Dr. Dzhambazov contributed equally to thiswork.
Address correspondence and reprint requests to Johan Back-lund, PhD, Division of Medical Inflammation Research, Departmentof Medical Biochemistry and Biophysics, Karolinska Institute, 171 77Stockholm, Sweden. E-mail: [email protected].
Submitted for publication August 22, 2009; accepted inrevised form March 12, 2010.
1911

DR4 molecules present almost the identical immuno-dominant epitope, which has been localized to aminoacid positions 259–273 on CII (2–7).
Posttranslational modification has been sug-gested as a possible mechanism whereby immunologictolerance to self antigens can be broken. This has alsobeen highlighted in RA, in which autoantibodies specificfor citrullinated self antigens are highly specific for RAand can be detected several years before the onset ofclinical disease (1). CII is also subjected to citrullination;autoantibodies specific for citrullinated CII have beenobserved in 40% of RA patients (8), and antibodiesspecific for citrullinated CII are pathogenic in mice (9).With regard to T cells, the immunodominant CII259–273
peptide harbors 2 lysine residues, at positions 264 and270, which can be posttranslationally modified by hy-droxylation and subsequent glycosylation. Remarkably,the CII259–273 epitope has been found to be uniformlyglycosylated in normal cartilage but modified by arthritisin rats and humans (10,11).
Although the role of CII as a relevant autoanti-gen in RA is unclear, CII may still serve as an excellentmodel autoantigen for understanding how the immunesystem interacts with joint-derived self antigens in orderto establish immunologic tolerance and for understand-ing how posttranslational modifications may influencethese processes. We previously investigated T cell toler-ance to self CII in DR4-expressing mice (12). By com-paring the recall response in DR4-transgenic mice,which either did or did not express human CII in acartilage-specific manner, we could show that DR4-restricted T cells exhibit strong tolerance to self CII, anddespite considerable efforts, we were not able to induceCIA in CII-tolerized HuCII.DR4-transgenic mice. Theaim of the present investigation therefore was to estab-lish a new animal model in which the T cell tolerance toself CII could be broken in order to initiate an auto-immune response to self CII and the development ofarthritis. We also wanted to investigate the role ofposttranslational modifications of the CII259–273 epitopein the induction and breaking of tolerance of DR4-restricted T cells, since this has been found to be ofmajor importance in mice expressing the murine Aq
molecule (13).
MATERIALS AND METHODS
Antigens. Rat CII was prepared from Swarm rat chon-drosarcoma by pepsin digestion or from lathyritic rat chondro-sarcoma and was further purified as described previously (14).We used the following CII peptides, containing the 259–273
sequence of rat CII, with or without modifications of the K264
and/or K270 residue: K264 (unmodified lysine at position 264);K264R, K270R, and K264/270R (where K264, K270, and bothresidues are substituted for arginine); HyK ([5R]-5-hydroxy-L-lysine at position 264); and Gal264, Gal270, and Gal264/270
([�]-D-galactopyranosyl residue on L-hydroxylysine at position264, position 270, or both residues, respectively). The CIIpeptides were synthesized, purified, and characterized as pre-viously described (7,15,16). Both CII and CII peptides weredissolved and stored in 0.1M acetic acid at 4°C. Purifiedprotein derivative (PPD) was from Statens Serum Institut.
Mice. Transgenic mice coexpressing DR4 and humanCD4 as well as human CII (HuCII) while lacking endogenousmurine class II MHC have been described previously (12,17–19). These humanized mice were backcrossed for 10 genera-tions onto the C3H.Q and B10.Q backgrounds and subse-quently intercrossed before being used in experiments. Beforeintercrossing, the HuCII transgene was also exchanged for theMMC transgene (20) by crossing with MMC.C3H.Q orMMC.B10.Q.Ncf1*/* mice (21,22) (i.e., MMC-transgenic micethat also lack a functional Ncf1 gene, therefore lacking afunctional phagocytic NADPH oxidase complex 2). The MMCtransgene is a mutated mouse CII gene in which the amino acidat position 266 has been changed from an aspartic acid to aglutamic acid, thereby expressing the rat/human CII259–273
epitope in a CII-specific manner. Thus, while the HuCIItransgene will lead to expression of the entire human CIIprotein, MMC-transgenic mice only express the heterologous(rat/human) CII259–273 epitope in the cartilage. The strainsdescribed above were maintained by repeated intercrossing atthe animal facility at the Division of Medical InflammationResearch, Lund University. MMC- and HuCII-expressingmice were kept as heterozygotes, and only nontransgeniclittermates were used as controls. All animal experiments wereapproved by the Lund–Malmo laboratory animal ethics com-mittee.
CII immunization and arthritis evaluation. For arthri-tis experiments, mice were immunized at the base of the tailwith 100 �g of CII emulsified in 100 �l Freund’s completeadjuvant (CFA; Difco). Mice were boosted 5 weeks later with50 �g of CII in 50 �l Freund’s incomplete adjuvant (Difco).Arthritis development was followed through visual scoring 3times a week, starting 2 weeks after the primary immunization.Arthritis was evaluated using an extended protocol (23); eachpaw received a score of 1–15, with a maximum score of 60 permouse.
Antibody responses. Blood samples were collected 5and 10 weeks after primary immunization, and isolated serawere investigated for total anti-CII IgG, IgG1, IgG2b, andIgG2a/c levels through quantitative enzyme-linked immu-nosorbent assay (ELISA) as described previously (24), whereserum was titrated (1:10–1:106) in parallel to the standard, andtiter values were interpolated within the linear range andrelated to the standard curve. Biotinylated rat anti-mouseIgG� (prepared in-house; clone 187.1) or mouse anti-mouseIgG2c (BD Biosciences) or peroxidase-conjugated goat anti-mouse antibodies specific for IgG1, IgG2a, or IgG2b (South-ern Biotechnology) were used as detecting antibodies. Bindingof biotinylated antibodies was revealed by ExtrAvidin Peroxi-dase (Sigma-Aldrich). Of note, mice of the C3H and B10background express different, but related, IgG2 alleles: IgG2a
1912 BATSALOVA ET AL

and IgG2c, respectively (25). Plates were developed usingABTS (Roche Diagnostic Systems) as substrate and measuredat 405 nm (Synergy-2; BioTek Instruments). Total anti-CII IgGlevels were measured as �g/ml using purified polyclonal anti-CII IgG antibodies of a known concentration as a standard.Isotype levels were measured as arbitrary concentrations usingour purified monoclonal anti-CII antibodies of the IgG1 (cloneCB20), IgG2a (clone CII-CI), and IgG2b (clone M2139)isotypes or pooled sera from arthritic mice for the IgG2cELISA.
Cellular assays. Mice were immunized with CII inCFA (3 injections of 60 �g at 3 different locations around thebase of the tail), and 10 days later cells were prepared from thedraining lymph nodes and restimulated with CII peptides invitro in flat-bottomed 96-well plates (700,000 cells/well) for 96hours, whereupon the culture supernatants were assayed forinterferon-� (IFN�) and interleukin-17 (IL-17) content byELISA. Cells were cultured in Dulbecco’s modified Eagle’smedium plus Glutamax I (Gibco) supplemented with 5%heat-inactivated fetal calf serum and penicillin/streptomycin.For IFN� detection, R46A2 (5 �g/ml) and biotin-conjugatedAN18.17.24 (0.6 �g/ml; Mabtech) were used as capture anddetection antibodies, respectively. For IL-17 detection, TC11-18H10 (1 �g/ml; BD Biosciences) and biotin-conjugated TC11-8H4.1 (0.5 �g/ml; BD Biosciences) were used as capture anddetection antibodies, respectively. Binding of biotinylated an-tibodies was revealed by europium-labeled streptavidin, andplates (Costar) were analyzed using a Victor 1420 multilabelcounter (PerkinElmer). Cytokine levels were measured asarbitrary concentrations using supernatant from concanavalinA (Con A)–stimulated B10.Q mouse splenocytes as standard.
For enzyme-linked immunospot (ELISpot) assays,plates (Millipore) were used according to the manufacturer’sinstructions, using the same capture and detection antibodiesas for the ELISA, although at higher concentrations (10 �g/mland 2 �g/ml, respectively, for IFN� and 5 �g/ml and 1 �g/ml,respectively, for IL-17A). Five hundred thousand cells per wellwere incubated with antigen for 24 hours, and cytokine spotswere visualized using Sigma Fast BCIP/nitroblue tetrazoliumand subsequently enumerated using an ImmunoScan ELISpotAnalyzer (CTL Europe). Establishment of T cell hybridomaclones and determination of antigen specificity were per-formed essentially as described previously (7) by immunizationof mice with CII in CFA followed 10 days later by in vitrorestimulation of lymph node cells with lathyritic rat CII (pepsinfree) before fusing the cells with the T cell receptor (TCR)–negative BW5147 thymoma cell line variant (26).
MHC peptide binding assay. Peptide binding assayswere performed by measuring the ability of synthetic peptidesto inhibit the binding of biotinylated CLIP peptide to purifiedDR4 molecules as described (27). Briefly, a mixture of a fixedconcentration of purified soluble recombinant DR4 molecules(0.1 �M), biotinylated CLIP peptide (2.5 �M), and increasingconcentrations of competitor peptides was incubated for 48hours at room temperature in the presence of a proteaseinhibitor cocktail (Complete; Roche Diagnostics). Recombi-nant DR4 molecules were then captured on microtiter assayplates that had been precoated with L243 monoclonal antibodyand blocked with phosphate buffered saline (PBS) containing2% low-fat milk. To remove the excess of nonbound peptides,plates were washed with PBS, and the amount of biotinylated
CLIP peptide bound to the recombinant DR4 molecules wasmeasured with europium-labeled streptavidin.
RESULTS
B10 and C3H backgrounds have different influ-ences on arthritis susceptibility in mice displaying T celltolerance to self CII. To study T cell tolerance to self CIIand its impact on arthritis susceptibility in DR4-expressing mice, we backcrossed DR4 mice to MMC-transgenic mice on the B10 background (yieldingMMC.B10.DR4 mice) and subsequently immunizedthese mice with CII. MMC-transgenic mice express amutated mouse CII protein in joint cartilage that differsfrom the CII protein in their nontransgenic littermatesat a single amino acid within the DR4-restricted immu-nodominant T cell epitope (D266 in mouse CII versusE266 in heterologous CII). Hence, the MMC transgeneallows CII-specific T cells to interact with self CII andacquire tolerance to the heterologous T cell epitope inthe naive mouse. However, it was not possible to ad-dress tolerance to self CII in these mice, since bothMMC.B10.DR4 mice and B10.DR4 mice were found tobe highly resistant to CIA, with only few mice developingvery mild arthritis (Table 1 and Figure 1A).
We have previously reported that MMC-expressing B10.Q mice are completely protected fromCIA (28), while MMC-derived tolerance to self CII was
Table 1. CIA susceptibility in DR4 mice on different geneticbackgrounds*
Strain,gene
Arthritis,no. with/no.
without (%)†
Maximumscore,
mean � SEM‡
Day ofonset,
mean � SEM‡
B10.DR4Lm 3/20 (15) 2 � 1 44 � 3MMC 1/20 (5) 1 42
B10.DR4.Ncf1*Lm 6/11 (55) 34 � 10 45 � 3HuCII 0/11 (0)§ – –
B10.DR4.Ncf1*Lm 25/34 (74) 15 � 3 38 � 2MMC 16/29 (55) 7 � 2¶ 44 � 3
C3H.DR4Lm 6/11 (55) 19 � 6 45 � 6MMC 3/15 (20) 8 � 2 66 � 4¶
* Collagen-induced arthritis (CIA) susceptibility in DR4 mice express-ing either the wild-type or mutated Ncf1 gene (Ncf1*) and the humantype II collagen (CII) transgene (HuCII) or the MMC transgene(MMC). Lm � transgenic-negative littermates.† Includes all mice that developed arthritis among all animals through-out the experiment.‡ Arthritic mice only.§ P � 0.05 versus nontolerized littermates, by Fisher’s exact test.¶ P � 0.05 versus nontolerized littermates, by Mann-Whitney U test.
BREAKING T CELL TOLERANCE IN A MURINE ARTHRITIS MODEL 1913
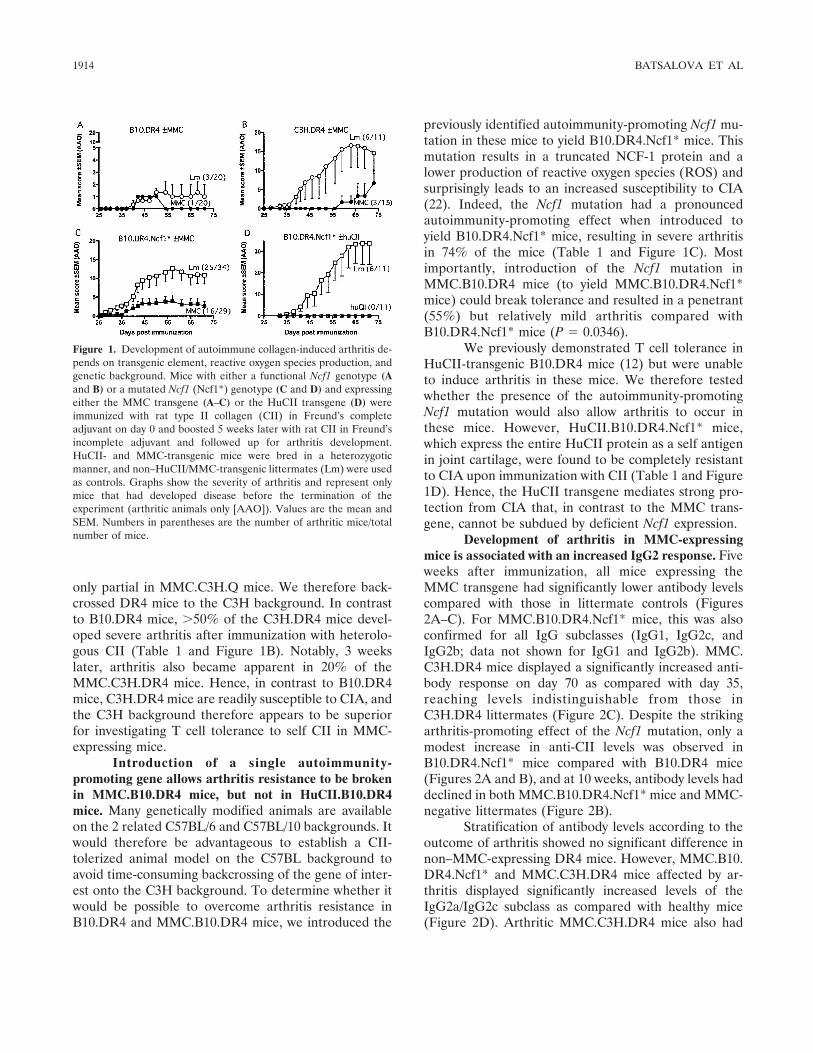
only partial in MMC.C3H.Q mice. We therefore back-crossed DR4 mice to the C3H background. In contrastto B10.DR4 mice, �50% of the C3H.DR4 mice devel-oped severe arthritis after immunization with heterolo-gous CII (Table 1 and Figure 1B). Notably, 3 weekslater, arthritis also became apparent in 20% of theMMC.C3H.DR4 mice. Hence, in contrast to B10.DR4mice, C3H.DR4 mice are readily susceptible to CIA, andthe C3H background therefore appears to be superiorfor investigating T cell tolerance to self CII in MMC-expressing mice.
Introduction of a single autoimmunity-promoting gene allows arthritis resistance to be brokenin MMC.B10.DR4 mice, but not in HuCII.B10.DR4mice. Many genetically modified animals are availableon the 2 related C57BL/6 and C57BL/10 backgrounds. Itwould therefore be advantageous to establish a CII-tolerized animal model on the C57BL background toavoid time-consuming backcrossing of the gene of inter-est onto the C3H background. To determine whether itwould be possible to overcome arthritis resistance inB10.DR4 and MMC.B10.DR4 mice, we introduced the
previously identified autoimmunity-promoting Ncf1 mu-tation in these mice to yield B10.DR4.Ncf1* mice. Thismutation results in a truncated NCF-1 protein and alower production of reactive oxygen species (ROS) andsurprisingly leads to an increased susceptibility to CIA(22). Indeed, the Ncf1 mutation had a pronouncedautoimmunity-promoting effect when introduced toyield B10.DR4.Ncf1* mice, resulting in severe arthritisin 74% of the mice (Table 1 and Figure 1C). Mostimportantly, introduction of the Ncf1 mutation inMMC.B10.DR4 mice (to yield MMC.B10.DR4.Ncf1*mice) could break tolerance and resulted in a penetrant(55%) but relatively mild arthritis compared withB10.DR4.Ncf1* mice (P � 0.0346).
We previously demonstrated T cell tolerance inHuCII-transgenic B10.DR4 mice (12) but were unableto induce arthritis in these mice. We therefore testedwhether the presence of the autoimmunity-promotingNcf1 mutation would also allow arthritis to occur inthese mice. However, HuCII.B10.DR4.Ncf1* mice,which express the entire HuCII protein as a self antigenin joint cartilage, were found to be completely resistantto CIA upon immunization with CII (Table 1 and Figure1D). Hence, the HuCII transgene mediates strong pro-tection from CIA that, in contrast to the MMC trans-gene, cannot be subdued by deficient Ncf1 expression.
Development of arthritis in MMC-expressingmice is associated with an increased IgG2 response. Fiveweeks after immunization, all mice expressing theMMC transgene had significantly lower antibody levelscompared with those in littermate controls (Figures2A–C). For MMC.B10.DR4.Ncf1* mice, this was alsoconfirmed for all IgG subclasses (IgG1, IgG2c, andIgG2b; data not shown for IgG1 and IgG2b). MMC.C3H.DR4 mice displayed a significantly increased anti-body response on day 70 as compared with day 35,reaching levels indistinguishable from those inC3H.DR4 littermates (Figure 2C). Despite the strikingarthritis-promoting effect of the Ncf1 mutation, only amodest increase in anti-CII levels was observed inB10.DR4.Ncf1* mice compared with B10.DR4 mice(Figures 2A and B), and at 10 weeks, antibody levels haddeclined in both MMC.B10.DR4.Ncf1* mice and MMC-negative littermates (Figure 2B).
Stratification of antibody levels according to theoutcome of arthritis showed no significant difference innon–MMC-expressing DR4 mice. However, MMC.B10.DR4.Ncf1* and MMC.C3H.DR4 mice affected by ar-thritis displayed significantly increased levels of theIgG2a/IgG2c subclass as compared with healthy mice(Figure 2D). Arthritic MMC.C3H.DR4 mice also had
Figure 1. Development of autoimmune collagen-induced arthritis de-pends on transgenic element, reactive oxygen species production, andgenetic background. Mice with either a functional Ncf1 genotype (Aand B) or a mutated Ncf1 (Ncf1*) genotype (C and D) and expressingeither the MMC transgene (A–C) or the HuCII transgene (D) wereimmunized with rat type II collagen (CII) in Freund’s completeadjuvant on day 0 and boosted 5 weeks later with rat CII in Freund’sincomplete adjuvant and followed up for arthritis development.HuCII- and MMC-transgenic mice were bred in a heterozygoticmanner, and non–HuCII/MMC-transgenic littermates (Lm) were usedas controls. Graphs show the severity of arthritis and represent onlymice that had developed disease before the termination of theexperiment (arthritic animals only [AAO]). Values are the mean andSEM. Numbers in parentheses are the number of arthritic mice/totalnumber of mice.
1914 BATSALOVA ET AL
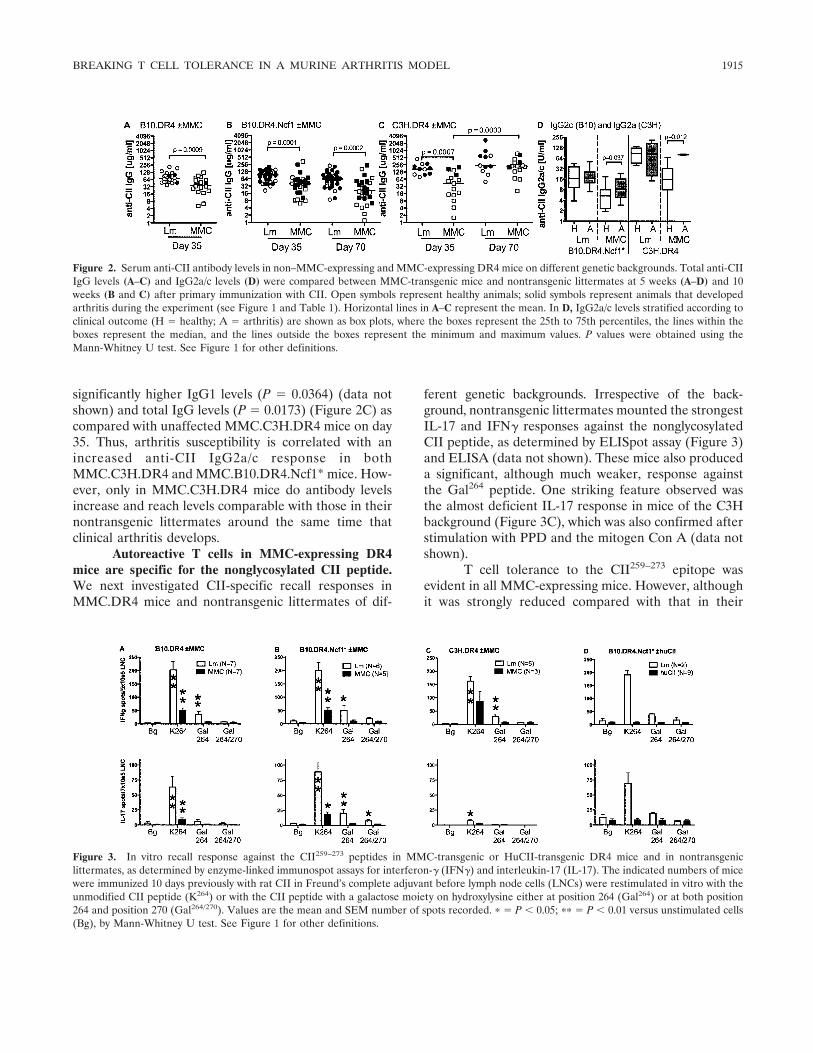
significantly higher IgG1 levels (P � 0.0364) (data notshown) and total IgG levels (P � 0.0173) (Figure 2C) ascompared with unaffected MMC.C3H.DR4 mice on day35. Thus, arthritis susceptibility is correlated with anincreased anti-CII IgG2a/c response in bothMMC.C3H.DR4 and MMC.B10.DR4.Ncf1* mice. How-ever, only in MMC.C3H.DR4 mice do antibody levelsincrease and reach levels comparable with those in theirnontransgenic littermates around the same time thatclinical arthritis develops.
Autoreactive T cells in MMC-expressing DR4mice are specific for the nonglycosylated CII peptide.We next investigated CII-specific recall responses inMMC.DR4 mice and nontransgenic littermates of dif-
ferent genetic backgrounds. Irrespective of the back-ground, nontransgenic littermates mounted the strongestIL-17 and IFN� responses against the nonglycosylatedCII peptide, as determined by ELISpot assay (Figure 3)and ELISA (data not shown). These mice also produceda significant, although much weaker, response againstthe Gal264 peptide. One striking feature observed wasthe almost deficient IL-17 response in mice of the C3Hbackground (Figure 3C), which was also confirmed afterstimulation with PPD and the mitogen Con A (data notshown).
T cell tolerance to the CII259–273 epitope wasevident in all MMC-expressing mice. However, althoughit was strongly reduced compared with that in their
Figure 2. Serum anti-CII antibody levels in non–MMC-expressing and MMC-expressing DR4 mice on different genetic backgrounds. Total anti-CIIIgG levels (A–C) and IgG2a/c levels (D) were compared between MMC-transgenic mice and nontransgenic littermates at 5 weeks (A–D) and 10weeks (B and C) after primary immunization with CII. Open symbols represent healthy animals; solid symbols represent animals that developedarthritis during the experiment (see Figure 1 and Table 1). Horizontal lines in A–C represent the mean. In D, IgG2a/c levels stratified according toclinical outcome (H � healthy; A � arthritis) are shown as box plots, where the boxes represent the 25th to 75th percentiles, the lines within theboxes represent the median, and the lines outside the boxes represent the minimum and maximum values. P values were obtained using theMann-Whitney U test. See Figure 1 for other definitions.
Figure 3. In vitro recall response against the CII259–273 peptides in MMC-transgenic or HuCII-transgenic DR4 mice and in nontransgeniclittermates, as determined by enzyme-linked immunospot assays for interferon-� (IFN�) and interleukin-17 (IL-17). The indicated numbers of micewere immunized 10 days previously with rat CII in Freund’s complete adjuvant before lymph node cells (LNCs) were restimulated in vitro with theunmodified CII peptide (K264) or with the CII peptide with a galactose moiety on hydroxylysine either at position 264 (Gal264) or at both position264 and position 270 (Gal264/270). Values are the mean and SEM number of spots recorded. � � P � 0.05; �� � P � 0.01 versus unstimulated cells(Bg), by Mann-Whitney U test. See Figure 1 for other definitions.
BREAKING T CELL TOLERANCE IN A MURINE ARTHRITIS MODEL 1915

nontransgenic littermates, MMC mice still mounted asignificant recall response to the nonglycosylated CIIpeptide. Again, despite the arthritis-promoting effect ofthe Ncf1 mutation, only minimal differences were ob-served in IL-17 responses between MMC.B10.DR4 andMMC.B10.DR4.Ncf1* mice. The immunodominance ofthe nonglycosylated CII peptide in MMC mice initiallysurprised us, since we have previously reported analmost completely deficient T cell response in HuCII-transgenic DR4 mice (12). However, by investigating therecall response in CII-primed HuCII.B10.DR4.Ncf1*mice, we could confirm our previous data, sinceHuCII.B10.DR4.Ncf1* mice also failed to mount asignificant response to both nonglycosylated and glyco-sylated self CII peptides (Figure 3D).
Collectively, these data show that the nonglyco-sylated CII peptide is immunodominant in DR4-expressing mice and that the choice of transgenic elementstrongly affects the level of achieved T cell tolerance toself CII. Moreover, tolerance to self CII in MMC-expressing DR4 mice is strong but incomplete, and theremaining autoreactive T cells are exclusively specific forthe nonglycosylated CII peptide and appear to be criticalfor autoimmune arthritis to develop in these mice.
T cell recognition of the nonglycosylated CII259–273
epitope is heterogeneous, but K264 is a critical TCRcontact point for the majority of responding cells. BothDR4 and Aq molecules present the CII259–273 peptide toCII-specific T cells. However, while the glycosylated CIIpeptide is immunodominant in Aq mice, our presentdata show that the nonglycosylated peptide dominates inDR4 mice. To investigate the reason for this discrep-ancy, we wanted to explore to what extent K264 and K270
would influence T cell recognition of the heterologousCII259–273 peptide. We therefore generated T cell clonesfrom CII-immunized B10.DR4.Ncf1* mice in order totest these against CII peptides in which K264 and/or K270
had been substituted for arginine (R), since this hasbeen found to be the preferred amino acid at position P2for peptides binding to DRB1*0401 (29). We also per-formed binding studies, which confirmed that substitu-tions of lysine for arginine residues had minimal conse-quences for class II MHC peptide binding (Figure 4A).Similarly, binding studies of nonglycosylated, hydroxy-lated, and glycosylated CII peptides showed that allposttranslationally modified peptides, with the possibleexception of Gal264/270, bound with affinity comparableto that of the unmodified CII259–273 peptide (Figure 4B).
All T cell clones tested responded to the nongly-cosylated K264 peptide. However, these could be dividedinto 4 groups according to their stringency in terms of
additional peptides recognized (Table 2). Three of 4groups were highly dependent on K264, since the subtlealteration from K264 to HyK264 resulted either in anabolished (group I) or in a reduced (group II–III)response, which could not be attributed to a reducedbinding of the HyK264 peptide to the DR4 molecule(Figure 4B). In contrast, only 1 group (group IV) displayeda more promiscuous pattern, with responses to the K264Rand K264/270R peptides as well as to the corresponding
Figure 4. HLA–DR4 binding, clonotypic T cell recognition, andDR4-restricted antigenicity of wild-type and modified type II collagen259–273 (CII259–273) peptides. A and B, Inhibition of the biotinylatedCLIP peptide in binding to soluble HLA–DR4 molecules with arginine-substituted peptides at position 264 (K264R) or position 270 (K270R) orposition 264 and position 270 (K264/270R) (A) or with CII259–273
peptides harboring a hydroxylysine at position 264 (HyK264) or with agalactose moiety on hydroxylysine either at position 264 (Gal264) orat both position 264 and position 270 (Gal264/270) (B). The binding ofthe nonbiotinylated CLIP peptide as well as that of the unmodifiedCII259–273 peptide (K264/270) is included as a reference. C, Specificrecognition of modified CII259–273 variants by the DR4-restricted T cellhybridoma clone 7C4H1 (group IV in Table 2), as determined byinterleukin-2 (IL-2) production. D, Investigation of antigenicity ofselected variants of the CII259–273 peptides and of cross-reactivitybetween these variants in B10.DR4.Ncf1* mice by immunization withthe wild-type (K264/270)– or arginine-substituted versions of the pep-tide, followed by in vitro restimulation 10 days later with the samepeptides as well as with the Gal264 peptide at 10 �g/ml. After 4 days ofstimulation, supernatants were collected and investigated forinterferon-� (IFN�) content. Values are the mean and SEM. Bg �unstimulated cells.
1916 BATSALOVA ET AL

mouse CII peptide (E266D) (Figure 4C), which has lowbinding affinity to the DR4 molecule (4,30).
Importantly, galactosylation of K264 (Gal264) re-sulted in a completely abrogated response, even forclones belonging to group IV and which appeared to beless dependent on K264 for their activation. Similarly,galactosylation of K270 (Gal270) inhibited or severelyimpaired the response in 2 of the 4 groups, even thoughall clones could respond to the K270R peptide. Thissuggests that the galactose moiety on either hydroxy-lysine residue could potentially act as a steric hindrancefor T cells that are more dependent on residues otherthan either of the 2 lysine residues for their activation.
The preferential recognition of K264 over K270
was also confirmed at the polyclonal level (Figure 4D),where immunization of DR4 mice with either the wild-type CII259–273 peptide or the K270R peptide resulted inan almost identical recall response against both pep-tides. In contrast, immunization with K264-modified pep-tides (K264R or K264/270R) resulted in a strongly reducedrecall response against the wild-type CII259–273 peptide.Finally, no recall response was detected against theGal264 peptide, irrespective of the immunizing peptide.Thus, DR4-restricted T cell recognition of the CII259–273
epitope is heterogeneous, but the majority of T cells
specific for the nonglycosylated CII peptide are criticallydependent on an unmodified K264 for their activation.
DISCUSSION
The first objective of the current investigationwas to establish an animal model in DR4 mice in whichT cell tolerance to self CII could be broken. Classic CIAin Aq mice is induced by immunization with CII ofnon-mouse origin, and the developing arthritis is highlydependent on activation of T cells specific for theheterologous CII259–273 peptide. Therefore, to studytolerance to self CII, we used the MMC-transgenicmouse, which expresses the immunodominant T cellepitope of heterologous CII in a mutated mouse CIIprotein. Because of this point mutation (D3E at posi-tion 266), only autoreactive T cells will become activatedin MMC mice upon immunization with rat CII, anddifferences observed between MMC and control micewill be either directly or indirectly related to T celltolerance.
We show herein that T cell tolerance is clearlypresent, since the recall response to rat CII is stronglyreduced in MMC-transgenic DR4 mice. However, T celltolerance is not complete, since MMC mice can stillmount a significant recall response. Furthermore, weshow that incomplete T cell tolerance to self CII mayrender mice susceptible to development of CIA, but thisis highly dependent on the genetic background, sinceMMC.C3H.DR4 mice, but not MMC.B10.DR4 mice,are susceptible. Still, by introducing the Ncf1 mutation, itis possible to increase susceptibility even in MMC miceon the B10 background. The exact mechanism by whichROS regulates autoreactive T cells in the CIA model iscurrently unknown, but it is believed to involve oxidationby antigen-presenting cells of the cell membrane and itsassociated proteins on interacting T cells (for review, seeref. 31). In the absence of a normal ROS production, thethreshold for activating autoreactive T cells may belower and/or cells will survive longer.
We observed a deficient IL-17 response inMMC.C3H.DR4 mice as compared with both MMC.B10.DR4 and MMC.B10.DR4.Ncf1* mice. Our C3H miceare Toll-like receptor 4 competent (32), and the weakIL-17 response is probably an intrinsic feature of theC3H background and has also been reported previouslyin other models (33). Still, the reduced IL-17 response inC3H mice is unlikely to explain the difference in arthritissusceptibility between B10.DR4 and C3H.DR4 mice,since IL-17 responses are generally assumed to promoteautoimmunity.
Table 2. T cell hybridoma responses to modified type II collagen259–273 (CII259–273) peptides*
Group
I II III IV
No. of clones 2 1 3 2K264 ���� ����� ���� ����HyK264 – �� ��� ���K264R – – – ��E266D (mouse) – – – ���K270R �� � ��� ���K264/270R – – – �Gal264 – – – –Gal270 ��� – ���� �Gal264/270 – – – –
* Shown is semiquantitative scoring of the T cell hybridoma responsefollowing stimulation with the wild-type heterologous nonglycosylatedCII259–273 collagen peptide (K264) or with modified versions thereof, inwhich lysine residues at position 264, position 270, or both positionshad been substituted for arginines (R), hydroxylysine (HyK), or agalactosylated hydroxylysine (Gal), as well as with the correspondingmouse peptide (E266D). Clones are categorized into 4 groups, whereeach group signifies a unique response pattern in terms of peptidesrecognized. Sensitivity of the T cell hybridomas was determined by theamount of peptide required for the induction of interleukin-2 produc-tion above background levels: � � no response; � � �50 �g/ml; ��� �10 �g/ml; ��� � �2 �g/ml; ���� � �0.4 �g/ml; ����� ��0.08 �g/ml.
BREAKING T CELL TOLERANCE IN A MURINE ARTHRITIS MODEL 1917

MMC.C3H.DR4 mice also displayed an alteredanti-CII antibody response as compared with MMC.B10.DR4.Ncf1* mice, where the anti-CII IgG response in-creased over time in MMC.C3H.DR4 mice to levelsindistinguishable from those in C3H.DR4 littermatecontrols. Meanwhile, antibody levels tended to decreasein MMC.B10.DR4.Ncf1* mice. The MMC-derived re-duction in anti-CII antibody responses is not likelyexplained by B cell tolerance to self CII, since B cellepitopes are identical between MMC mice and litter-mate controls. In fact, most anti-CII antibodies in bothrats and mice are germline encoded (34–36). They alsocross-react with CII of other species, but not with othercollagen types, such as type IV or type I collagen.Instead, low-affinity CII-specific T cells that have es-caped tolerance or that have acquired tolerance but havenot been deleted become sufficiently activated to help Bcells to produce anti-CII antibodies. However, such Tcells will be less frequent and less efficient in supplyingB cell help, thereby delaying the anti-CII antibodyresponse in reaching arthritogenic levels.
A stronger anti-CII IgG response was also ob-served in nontransgenic C3H.DR4 mice as comparedwith B10.DR4 and B10.DR4.Ncf1* mice. This has alsobeen observed in the C3H.Q strain (28) and probablyreflects an intrinsic phenotype of the genetic back-ground. It could be speculated that introduction of theNcf1 mutation will lead to a more efficient T cellresponse, possibly via IL-17, that could stimulate theanti-CII B cell response to reach arthritogenic levelsearlier in MMC.B10.DR4.Ncf1* mice compared withMMC.C3H.DR4 mice. Alternatively, the Ncf1 mutationmay lower the critical level of arthritogenic antibodiesrequired for arthritis to develop. Such a hypothesis cannow be investigated using these models.
The source of the CII-transgenic element had adramatic effect on the robustness of tolerance, where theHuCII transgene induced considerably stronger T celltolerance to the CII259–273 peptide than did the MMCtransgene. This difference probably relates to the ex-pression levels and availability of the transgenic CII fortolerance induction in vivo. This has also been shown forother model autoantigens (37,38) and may be due todifferences in transgenic promoter expression or num-ber of transgene copies inserted. In fact, by comparingdifferent lines (founders) of the HuCII transgene in Aq
mice, an expression level–dependent T cell tolerance tothe CII259–273 epitope could be observed (18).
A second objective of the study was to investigatethe extent to which T cells recognize posttranslationalmodifications of CII in DR4 mice. We show that the
nonglycosylated CII259–273 epitope is clearly immuno-dominant in both MMC.DR4 mice and littermate con-trols. This is in strong contrast to Aq-expressing mice, inwhich the galactosylated peptide dominates. Our earlierdata show that the K264 residue within the immunodom-inant CII259–273 epitope is uniformly O-glycosylatedwhen CII is isolated from healthy cartilage of humansand rats (10,11). Thus, T cell tolerance should primarilyaffect T cells specific for glycosylated CII if it is regu-lated through peripheral presentation of self CII derivedfrom joint cartilage, as suggested previously (39).
There is also evidence for ectopic expression ofCII in the thymus (40,41), but it is not known whetherthis includes presentation of either nonglycosylated orglycosylated CII peptides or both. Biased T cell toler-ance against the nonglycosylated T cell epitope inMMC-expressing mice of the H-2q haplotype could beexplained by a sufficient presentation of this peptide,leading to strong central tolerance. In contrast, peri-pheral tolerization of glycopeptide-specific T cells maybe less efficient, possibly as a result of an inferiorprocessing and presentation of the glycosylated CIIpeptide by macrophages and dendritic cells, as has beenshown in DR1-transgenic mice (42,43). It is reasonableto assume that the MMC transgene operates similarly inboth DR4- and Aq-expressing mice. However, nonphysi-ologic expression (low or toxic) of the DR4 transgenecould influence the selection of the nonglycosylated CIIpeptide as immunodominant in these mice, perhaps dueto a reduced presentation of the nonglycosylated CIIpeptide within the thymus.
The Aq and DR4 molecules present the CII259–273
peptide in a slightly different manner, in which residuesI260 and F263 bind to the P1 and P4 pockets, respectively,of Aq and in which residues F263 and E266 bind to the P1and P4 pockets, respectively, of DR4. Hence, the galac-tose moiety of K264 will be more centrally located (i.e.,P5) on the Aq molecule than on the DR4 molecule (P2).Meanwhile, the lysine at position 270 will be shiftedtoward the center (P8) on DR4, as compared with itsperipheral placement (P11) on Aq. The K264 residueclearly constitutes a critical TCR contact point forAq-restricted CII-specific T cells (44,45). However, theimportance of the K264 residue as a DR4-restricted TCRcontact or class II MHC anchor point has been ques-tioned, whereas K270 should be exposed toward the TCR(2,4,46,47).
It could be speculated that the nonglycosylatedCII peptide is immunodominant in DR4 mice becausethe majority of CII-specific T cells do not rely on theK264 residue, but rather, on residues located between P3
1918 BATSALOVA ET AL

and P7 of the peptide–DR4 complex. However, our datashow that this is not the case and that in fact K264
appears to be more important than the K270 residue forinteraction with the TCR. Instead, our data suggest thepossibility that the physiologic and uniform glycosylationof self CII at K264 (10,11) could act to sterically hinderthe acquisition of tolerance by T cells specific for thenonglycosylated epitope. If so, then traumatic alter-ations in the level of glycosylation of self CII could leadto peripheral presentation of nonglycosylated CII pep-tides. These could then act as neoepitopes and promoteautoimmunity toward joint-specific antigens in an in-flamed environment.
ACKNOWLEDGMENTS
We thank Rikard Holmdahl for support and discus-sions enabling the study and for critically reading the manu-script. We also thank Carlos and Kristina Palestro, JohannaEkelund, and Isabella Bohlin for taking care of the animals,Emma Mondoc and Malin Neptin for CII preparation, andKatrin Dilja Jonsdottir and Erik Jansson for technical supportof cell culture assays.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising itcritically for important intellectual content, and all authors approvedthe final version to be published. Dr. J. Backlund had full access to allof the data in the study and takes responsibility for the integrity of thedata and the accuracy of the data analysis.Study conception and design. Batsalova, Dzhambazov, Merky, J.Backlund.Acquisition of data. Batsalova, Dzhambazov, Merky, A. Backlund, J.Backlund.Analysis and interpretation of data. Batsalova, Dzhambazov, Merky,A. Backlund, J. Backlund.Project supervision. J. Backlund.
REFERENCES
1. Imboden JB. The immunopathogenesis of rheumatoid arthritis.Annu Rev Pathol 2009;4:417–34.
2. Rosloniec EF, Whittington KB, Zaller DM, Kang AH. HLA-DR1(DRB1*0101) and DR4 (DRB1*0401) use the same anchorresidues for binding an immunodominant peptide derived fromhuman type II collagen. J Immunol 2002;168:253–9.
3. Fugger L, Rothbard JB, Sonderstrup-McDevitt G. Specificity of anHLA-DRB1*0401-restricted T cell response to type II collagen.Eur J Immunol 1996;26:928–33.
4. Andersson EC, Hansen BE, Jacobsen H, Madsen LS, AndersenCB, Engberg J, et al. Definition of MHC and T cell receptorcontacts in the HLA-DR4 restricted immunodominant epitope intype II collagen and characterization of collagen-induced arthritisin HLA-DR4 and human CD4 transgenic mice. Proc Natl Acad SciU S A 1998;95:7574–9.
5. Rosloniec EF, Brand DD, Myers LK, Esaki Y, Whittington KB,Zaller DM, et al. Induction of autoimmune arthritis in HLA-DR4(DRB1*0401) transgenic mice by immunization with human andbovine type II collagen. J Immunol 1998;160:2573–8.
6. Rosloniec EF, Brand DD, Myers LK, Whittington KB, Gu-manovskaya M, Zaller DM, et al. An HLA-DR1 transgene conferssusceptibility to collagen-induced arthritis elicited with humantype II collagen. J Exp Med 1997;185:1113–22.
7. Michaelsson E, Andersson M, Engstrom A, Holmdahl R. Identi-fication of an immunodominant type-II collagen peptide recog-nized by T cells in H-2q mice: self tolerance at the level ofdeterminant selection. Eur J Immunol 1992;22:1819–25.
8. Snir O, Widhe M, von Spee C, Lindberg J, Padyukov L, LundbergK, et al. Multiple antibody reactivities to citrullinated antigens insera from patients with rheumatoid arthritis: association withHLA-DRB1 alleles. Ann Rheum Dis 2009;68:736–43.
9. Uysal H, Bockermann R, Nandakumar KS, Sehnert B, Bajtner E,Engstrom A, et al. Structure and pathogenicity of antibodiesspecific for citrullinated collagen type II in experimental arthritis.J Exp Med 2009;206:449–62.
10. Dzhambazov B, Holmdahl M, Yamada H, Lu S, Vestberg M,Holm B, et al. The major T cell epitope on type II collagen isglycosylated in normal cartilage but modified by arthritis in bothrats and humans. Eur J Immunol 2005;35:357–66.
11. Yamada H, Dzhambazov B, Bockermann R, Blom T, Holmdahl R.A transient post-translationally modified form of cartilage type IIcollagen is ignored by self-reactive T cells. J Immunol 2004;173:4729–35.
12. Backlund J, Carlsen S, Hoger T, Holm B, Fugger L, Kihlberg J, etal. Predominant selection of T cells specific for the glycosylatedcollagen type II epitope (263-270) in humanized transgenic miceand in rheumatoid arthritis. Proc Natl Acad Sci U S A 2002;99:9960–5.
13. Backlund J, Treschow A, Bockermann R, Holm B, Holm L,Issazadeh-Navikas S, et al. Glycosylation of type II collagen is ofmajor importance for T cell tolerance and pathology in collagen-induced arthritis. Eur J Immunol 2002;32:3776–84.
14. Andersson M, Holmdahl R. Analysis of type II collagen-reactive Tcells in the mouse. I. Different regulation of autoreactive vs.non-autoreactive anti-type II collagen T cells in the DBA/1 mouse.Eur J Immunol 1990;20:1061–6.
15. Broddefalk J, Backlund J, Almqvist F, Johansson M, Holmdahl R,Kihlberg J. T cells recognize a glycopeptide derived from type IIcollagen in a model for rheumatoid arthritis. J Am Chem Soc1998;120:7676–83.
16. Holm B, Broddefalk J, Flodell S, Wellner E, Kihlberg J. Animproved synthesis of a galactosylated hydroxylysine buildingblock and its use in solid-phase glycopeptide synthesis. Tetrahe-dron 2000;56:1579–86.
17. Fugger L, Michie SA, Rulifson I, Lock CB, McDevitt GS. Expres-sion of HLA-DR4 and human CD4 transgenes in mice determinesthe variable region �-chain T-cell repertoire and mediates anHLA-DR-restricted immune response. Proc Natl Acad Sci U S A1994;91:6151–5.
18. Malmstrom V, Ho KK, Lun J, Tam PP, Cheah KS, Holmdahl R.Arthritis susceptibility in mice expressing human type II collagenin cartilage. Scand J Immunol 1997;45:670–7.
19. Cosgrove D, Gray D, Dierich A, Kaufman J, Lemeur M, BenoistC, et al. Mice lacking MHC class II molecules. Cell 1991;66:1051–66.
20. Malmstrom V, Michaelsson E, Burkhardt H, Mattsson R, VuorioE, Holmdahl R. Systemic versus cartilage-specific expression of atype II collagen-specific T-cell epitope determines the level oftolerance and susceptibility to arthritis. Proc Natl Acad Sci U S A1996;93:4480–5.
21. Hultqvist M, Backlund J, Bauer K, Gelderman KA, Holmdahl R.Lack of reactive oxygen species breaks T cell tolerance to collagentype II and allows development of arthritis in mice. J Immunol2007;179:1431–7.
22. Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J,Holmdahl R. Enhanced autoimmunity, arthritis, and encephalo-
BREAKING T CELL TOLERANCE IN A MURINE ARTHRITIS MODEL 1919

myelitis in mice with a reduced oxidative burst due to a mutationin the Ncf1 gene. Proc Natl Acad Sci U S A 2004;101:12646–51.
23. Holmdahl R, Carlsen S, Mikulowska A, Vestberg M, Brunsberg U,Hansson A, et al. Genetic analysis of mouse models for rheuma-toid arthritis. New York: CRC Press; 1998.
24. Holmdahl R, Klareskog L, Andersson M, Hansen C. High anti-body response to autologous type II collagen is restricted to H-2q.Immunogenetics 1986;24:84–9.
25. Morgado MG, Cam P, Gris-Liebe C, Cazenave PA, Jouvin-Marche E. Further evidence that BALB/c and C57BL/6 �2a genesoriginate from two distinct isotypes. EMBO J 1989;8:3245–51.
26. White J, Blackman M, Bill J, Kappler J, Marrack P, Gold DP, etal. Two better cell lines for making hybridomas expressing specificT cell receptors. J Immunol 1989;143:1822–5.
27. Kjellen P, Brunsberg U, Broddefalk J, Hansen B, Vestberg M,Ivarsson I, et al. The structural basis of MHC control of collagen-induced arthritis; binding of the immunodominant type II collagen256-270 glycopeptide to H-2Aq and H-2Ap molecules. Eur J Im-munol 1998;28:755–67.
28. Backlund J, Nandakumar KS, Bockermann R, Mori L, HolmdahlR. Genetic control of tolerance to type II collagen and develop-ment of arthritis in an autologous collagen-induced arthritismodel. J Immunol 2003;171:3493–9.
29. Hammer J, Belunis C, Bolin D, Papadopoulos J, Walsky R,Higelin J, et al. High-affinity binding of short peptides to majorhistocompatibility complex class II molecules by anchor combina-tions. Proc Natl Acad Sci U S A 1994;91:4456–60.
30. Sakurai Y, Brand DD, Tang B, Rosloniec EF, Stuart JM, KangAH, et al. Analog peptides of type II collagen can suppressarthritis in HLA-DR4 (DRB1*0401) transgenic mice. ArthritisRes Ther 2006;8:R150.
31. Hultqvist M, Olsson LM, Gelderman KA, Holmdahl R. Theprotective role of ROS in autoimmune disease. Trends Immunol2009;30:201–8.
32. Nandakumar KS, Svensson L, Holmdahl R. Collagen type II-specific monoclonal antibody-induced arthritis in mice: descriptionof the disease and the influence of age, sex, and genes. Am JPathol 2003;163:1827–37.
33. Lewkowich IP, Lajoie S, Clark JR, Herman NS, Sproles AA,Wills-Karp M. Allergen uptake, activation, and IL-23 productionby pulmonary myeloid DCs drives airway hyperresponsiveness inasthma-susceptible mice. PLoS One 2008;3:e3879.
34. Mo JA, Bona CA, Holmdahl R. Variable region gene selection ofimmunoglobulin G-expressing B cells with specificity for a definedepitope on type II collagen. Eur J Immunol 1993;23:2503–10.
35. Mo JA, Holmdahl R. The B cell response to autologous type IIcollagen: biased V gene repertoire with V gene sharing andepitope shift. J Immunol 1996;157:2440–8.
36. Wernhoff P, Unger C, Bajtner E, Burkhardt H, Holmdahl R.Identification of conformation-dependent epitopes and V geneselection in the B cell response to type II collagen in the DA rat.Int Immunol 2001;13:909–19.
37. Akkaraju S, Ho WY, Leong D, Canaan K, Davis MM, GoodnowCC. A range of CD4 T cell tolerance: partial inactivation toorgan-specific antigen allows nondestructive thyroiditis or insulitis.Immunity 1997;7:255–71.
38. McLachlan SM, Nagayama Y, Pichurin PN, Mizutori Y, Chen CR,Misharin A, et al. The link between Graves’ disease and Hashi-moto’s thyroiditis: a role for regulatory T cells. Endocrinology2007;148:5724–33.
39. Malmstrom V, Backlund J, Jansson L, Kihlberg J, Holmdahl R. Tcells that are naturally tolerant to cartilage-derived type II colla-gen are involved in the development of collagen-induced arthritis.Arthritis Res 2000;2:315–26.
40. Campbell IK, Kinkel SA, Drake SF, van Nieuwenhuijze A, HubertFX, Tarlinton DM, et al. Autoimmune regulator controls T cellhelp for pathogenetic autoantibody production in collagen-in-duced arthritis. Arthritis Rheum 2009;60:1683–93.
41. Chin RK, Zhu M, Christiansen PA, Liu W, Ware C, Peltonen L, etal. Lymphotoxin pathway-directed, autoimmune regulator-inde-pendent central tolerance to arthritogenic collagen. J Immunol2006;177:290–7.
42. Von Delwig A, Altmann DM, Charlton FG, McKie N, Isaacs JD,Holmdahl R, et al. T cell responses to a non-glycosylated epitopepredominate in type II collagen-immunised HLA-DRB1*0101transgenic mice. Ann Rheum Dis 2007;66:599–604.
43. Von Delwig A, Altmann DM, Isaacs JD, Harding CV, HolmdahlR, McKie N, et al. The impact of glycosylation on HLA–DR1–restricted T cell recognition of type II collagen in a mouse model.Arthritis Rheum 2006;54:482–91.
44. Holm B, Baquer SM, Holm L, Holmdahl R, Kihlberg J. Role ofthe galactosyl moiety of collagen glycopeptides for T-cell stimula-tion in a model for rheumatoid arthritis. Bioorg Med Chem2003;11:3981–7.
45. Michaelsson E, Broddefalk J, Engstrom A, Kihlberg J, HolmdahlR. Antigen processing and presentation of a naturally glycosylatedprotein elicits major histocompatibility complex class II-restricted,carbohydrate-specific T cells. Eur J Immunol 1996;26:1906–10.
46. Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC. X-raycrystal structure of HLA-DR4 (DRA*0101, DRB1*0401) com-plexed with a peptide from human collagen II. Immunity 1997;7:473–81.
47. Rosloniec EF, Whittington KB, He X, Stuart JM, Kang AH.Collagen-induced arthritis mediated by HLA-DR1 (*0101) andHLA-DR4 (*0401). Am J Med Sci 2004;327:169–79.
1920 BATSALOVA ET AL
Related Documents











