52 La nota 90 61 “Deregulation” del trattamento farmacologico del sovrappeso e rischi correlati ai disturbi del comportamento alimentare 64 Somministrazione di vaccini contenenti tiomersal e sviluppo neuropsicologico del bambino 83 Reazioni di fotosensibilizzazione da ketoprofene Anno XVI N.2 Marzo-aprile 2009 Bimestrale Bollettino d’Informazione sui Farmaci 2/09 Bimestrale dell’Agenzia Italiana del Farmaco

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

52 La nota 90
61 “Deregulation” del trattamentofarmacologico del sovrappeso e rischi correlati ai disturbi del comportamento alimentare
64 Somministrazione di vaccinicontenenti tiomersal e svilupponeuropsicologico del bambino
83 Reazioni di fotosensibilizzazioneda ketoprofene
Ann
o X
VI
N.2
Mar
zo-a
prile
200
9 B
imes
tral
e
Bollettino d’Informazione sui Farmaci
2/09
Bimestrale dell’Agenzia Italiana del Farmaco

© Agenzia Italianadel FarmacoLa riproduzione e la divulgazione dei contenuti del Bif sono consentite fatti salvi la citazione della fonte e il rispetto dell’integrità dei dati utilizzati.
Autorizzazione del Tribunale di Roma n. 401 del 20/11/2008.
Questo numero è stato chiuso a luglio 2009.
come autoreG. AngielloF. FirenzuoliS. GiacomelliE. MarottaE. MatarangoloF. NoninoI. PaganoV. PatriarcaL. PierattiniC. SantuccioV. SeveriL. SottosantiA. TozziM. Venegoni
come collaboratoreM.C. BarberaM. CoriM. DelbòM.P. LucantonioF. Mannino
Direttore responsabile e scientificoAntonio Addis
Comitato scientificoMarco BobbioFausto BodiniFranca De LazzariAlbano Del FaveroNicola MontanaroLuigi PagliaroPaolo PreziosiAlessandro RosselliAlessandro TagliamonteGianni TognoniFrancesca TosoliniMassimo Valsecchi
RedazioneGabriele AngielloElisabetta NeriLinda PierattiniFrancesca RocchiCarmela SantuccioValeria Severi
Segreteria di RedazioneMonica Pirri
Redazione editorialeIl Pensiero Scientifico EditoreVia Bradano, 3/c 00199 RomaTel. 06 862 82 335Fax 06 862 82 [email protected]
ResponsabileManuela Baroncini
Comunicazioni e osservazioni al Bollettino dovranno essere inoltrate presso:RedazioneBollettino d’Informazionesui FarmaciAgenzia Italianadel FarmacoVia della Sierra Nevada, 6000144 RomaFax 06 597 84 [email protected]
Bollettino d’Informazione sui FarmaciBimestrale dell’Agenzia Italiana del Farmaco
Tra gli autori del n. 1/09 vanno annoverati ancheC. Colombo, S. Donati, A. Liberati, A. Mele, R. Satolli.
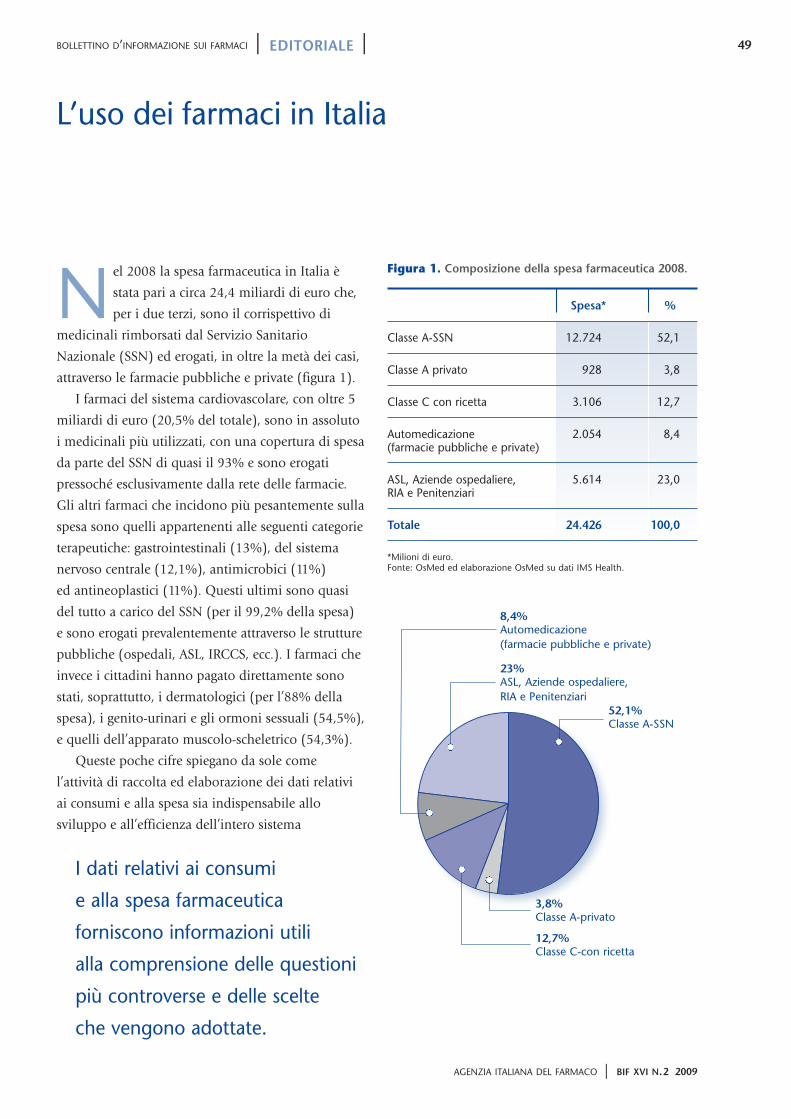
49
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
N el 2008 la spesa farmaceutica in Italia è
stata pari a circa 24,4 miliardi di euro che,
per i due terzi, sono il corrispettivo di
medicinali rimborsati dal Servizio Sanitario
Nazionale (SSN) ed erogati, in oltre la metà dei casi,
attraverso le farmacie pubbliche e private (figura 1).
I farmaci del sistema cardiovascolare, con oltre 5
miliardi di euro (20,5% del totale), sono in assoluto
i medicinali più utilizzati, con una copertura di spesa
da parte del SSN di quasi il 93% e sono erogati
pressoché esclusivamente dalla rete delle farmacie.
Gli altri farmaci che incidono più pesantemente sulla
spesa sono quelli appartenenti alle seguenti categorie
terapeutiche: gastrointestinali (13%), del sistema
nervoso centrale (12,1%), antimicrobici (11%)
ed antineoplastici (11%). Questi ultimi sono quasi
del tutto a carico del SSN (per il 99,2% della spesa)
e sono erogati prevalentemente attraverso le strutture
pubbliche (ospedali, ASL, IRCCS, ecc.). I farmaci che
invece i cittadini hanno pagato direttamente sono
stati, soprattutto, i dermatologici (per l’88% della
spesa), i genito-urinari e gli ormoni sessuali (54,5%),
e quelli dell’apparato muscolo-scheletrico (54,3%).
Queste poche cifre spiegano da sole come
l’attività di raccolta ed elaborazione dei dati relativi
ai consumi e alla spesa sia indispensabile allo
sviluppo e all’efficienza dell’intero sistema
BOLLETTINO D’INFORMAZIONE SUI FARMACI | EDITORIALE |
L’uso dei farmaci in Italia
I dati relativi ai consumi
e alla spesa farmaceutica
forniscono informazioni utili
alla comprensione delle questioni
più controverse e delle scelte
che vengono adottate.
52,1% Classe A-SSN
3,8% Classe A-privato
12,7% Classe C-con ricetta
8,4% Automedicazione(farmacie pubbliche e private)
23% ASL, Aziende ospedaliere, RIA e Penitenziari
Figura 1. Composizione della spesa farmaceutica 2008.
Spesa* %
Classe A-SSN 12.724 52,1
Classe A privato 928 3,8
Classe C con ricetta 3.106 12,7
Automedicazione 2.054 8,4(farmacie pubbliche e private)
ASL, Aziende ospedaliere, 5.614 23,0RIA e Penitenziari
Totale 24.426 100,0
*Milioni di euro. Fonte: OsMed ed elaborazione OsMed su dati IMS Health.
EDITORIALE | L’USO DEI FARMACI IN ITALIA50
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
dell’assistenza farmaceutica. Conoscere per capire
e per governare: anche quest’anno il contributo
fornito dalla pubblicazione del Rapporto nazionale
su L’uso dei Farmaci in Italia risulterà
particolarmente prezioso per tutti i soggetti
che si occupano del problema.
Ma anche per i comuni cittadini che potranno
derivarne informazioni utili alla comprensione
delle questioni più controverse e delle scelte,
non sempre facili, che vengono adottate in materia.
Nel Rapporto si è ricomposto un quadro
complessivo della spesa per farmaci in Italia grazie
alla disponibilità di dati che riguardano sia l’acquisto
dalle farmacie (privato e a carico del SSN),
sia l’acquisto da parte delle strutture pubbliche
(ospedali, ASL, IRCCS, ecc.) sia, infine, i dati di spesa
derivanti dalla distribuzione diretta e per conto
(questi ultimi per le 14 Regioni che hanno trasmesso
centralmente i dati per l’anno 2008 secondo quanto
previsto dal DM di fine luglio 2007).
La struttura generale del Rapporto è quella che si è
andata consolidando negli anni, organizzata per
argomenti tra loro coerenti: i dati generali di
consumo, le categorie terapeutiche e i principi attivi,
i farmaci equivalenti, le note AIFA, il consumo
privato, la distribuzione diretta e per conto.
È stata ulteriormente strutturata l’analisi degli
atteggiamenti prescrittivi della Medicina Generale
relativamente alla appropriatezza di trattamento di
alcune problematiche cliniche di rilievo
(la prevenzione del rischio cardiovascolare,
la prevenzione ed il trattamento di eventi
tromboembolici, il trattamento dell’ulcera peptica
e della malattia da reflusso gastroesofageo,
il trattamento dell’osteoporosi, il ricorso ad
antibiotici ad ampio spettro). Questo tipo di
valutazioni ha una crescente rilevanza nel contesto
del Rapporto, in quanto aspetto fondamentale della
qualità assistenziale, fortemente correlato con altre
componenti della qualità quali: la sicurezza,
l’efficacia, l’equità, la continuità assistenziale,
il coinvolgimento del cittadino, l’efficienza.
Per l’insieme dei cinque problemi clinici
analizzati quest’anno nel Rapporto, sono stati
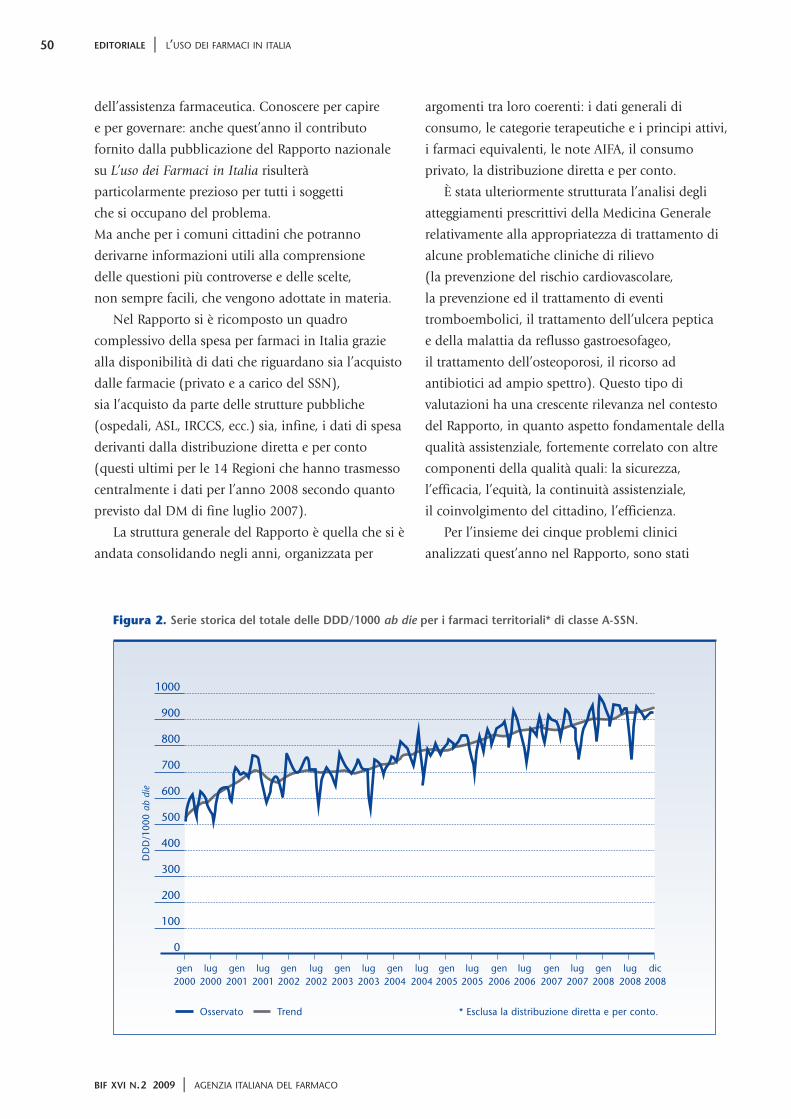
Figura 2. Serie storica del totale delle DDD/1000 ab die per i farmaci territoriali* di classe A-SSN.
1000
900
800
700
600
500
400
300
200
100
0
Osservato Trend * Esclusa la distribuzione diretta e per conto.
gen lug gen lug gen lug gen lug gen lug gen lug gen lug gen lug gen lug dic2000 2000 2001 2001 2002 2002 2003 2003 2004 2004 2005 2005 2006 2006 2007 2007 2008 2008 2008
DD
D/1
000
ab d
ie

EDITORIALE | L’USO DEI FARMACI IN ITALIA 51
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
identificati e formalizzati 15 indicatori di
appropriatezza mirati a valutare la copertura
terapeutica dei pazienti che si presentano con queste
problematiche (attraverso la prevalenza d’uso) e/o
l’adesione dei pazienti ad un trattamento cronico.
La principale finalità di questo tipo di analisi è quella
di offrire dei possibili spunti nell’ambito di iniziative
di informazione/formazione rivolte ai medici.
La regolarità con la quale sono stati raccolti e
validati dall’OsMed i dati negli ultimi 9 anni
consente di introdurre nelle analisi una dimensione
temporale, sempre più solida, utile a fotografare la
dinamica della prescrizione farmaceutica in diversi
aspetti quali, per citare un esempio, l’efficacia degli
interventi informativi da parte delle Autorità
regolatorie quando, per alcuni trattamenti, occorre
che i medici adottino delle cautele d’uso.
Dai dati 2008 emerge come l’atto di acquistare-
assumere un farmaco sia uno dei più consueti per un
cittadino medio italiano il quale consuma, ogni
giorno, 1,5 dosi di farmaco con un andamento che è
andato crescendo negli anni (dal 2000 l’aumento di
consumo è stato quasi del 60%; figura 2). Siamo
evidentemente di fronte ad una crescita di natura
strutturale dovuta a molteplici fattori (tra i quali
certamente il crescente peso delle patologie croniche
legato all’invecchiamento della popolazione, ma
anche aspetti di natura più squisitamente socio-
culturale) ed è complicato ricercare specifici punti di
singolarità in un fenomeno che si presenta con una
caratteristica di disarmante regolarità nel tempo.
La diffusione dell’informazione agli operatori
sanitari, ai mass media, ai cittadini in generale, nel
dare trasparenza ai meccanismi decisionali adottati
dalle Autorità regolatorie, costituisce la necessaria
premessa ad iniziative tese a promuovere un uso
migliore dei farmaci. Ed è questa la logica di lavoro
che sottende le molte e complesse analisi che (in
continuità con la redazione dei Rapporti degli anni
precedenti) compongono il Rapporto 2008; una
logica tesa ad offrire in un unico contesto le diverse
sfaccettature di un fenomeno complesso, che non
può essere ridotto unicamente alla dimensione della
contabilità di spesa.

Con la nota 90 l’AIFA
ha voluto razionalizzare l’impiego
del medicinale limitandone
la rimborsabilità ai casi trattati
con oppiacei da almeno
2 settimane e con resistenza
ai lassativi osmotici
da oltre 3 giorni.
Nota 90
52
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
N ello scorso mese di aprile l’EMEA ha concessol’autorizzazione all’immissione in commercio alRelistor, un medicinale a base di metilnaltrexone
bromuro. Relistor è indicato per il “Trattamento della costipazione
indotta da oppioidi in pazienti con malattia avanzata che ri-cevono cure palliative, nel caso in cui la risposta alla terapialassativa usuale non sia sufficiente”. L’impiego del medici-nale è attualmente riservato ai pazienti adulti (età supe-riore a 18 anni), in quanto non sono disponibili dati diefficacia e sicurezza per i bambini e gli adolescenti.
Come il naltrexone, il metilnaltrexone bromuro è unantagonista selettivo del recettore μ per gli oppioidi ma,contrariamente al primo, ha scarsa capacità di attraversarela barriera emato-encefalica. Pertanto, Relistor, bloccandoi soli recettori μ intestinali, contrasta in modo efficace lacostipazione da oppiacei, spesso resistente ai comuni las-sativi, mentre non riduce l’efficacia analgesica degli op-piacei stessi (assenza di azione centrale).
Relistor viene somministrato per via sottocutanea agiorni alterni, fatto che rappresenta un evidente vantaggiorispetto alla somministrazione rettale di clismi o alle fa-stidiose manovre di svuotamento manuale.
Con la nota 90 l’AIFA ha voluto razionalizzare l’im-piego del medicinale nell’ambito dell’indicazione auto-rizzata, limitandone la prescrivibilità a carico del SSN aicasi in cui il trattamento con oppiacei sia iniziato da al-meno 2 settimane e il paziente mostri resistenza ai lassa-
La nota 90
BOLLETTINO D’INFORMAZIONE SUI FARMACI | PANORAMI E PERCORSI |
tivi osmotici da oltre 3 giorni. Si tratta, per altro, di limi-tazioni che rispecchiano i criteri di inclusione utilizzatinegli studi clinici che hanno consentito l’autorizzazionedel medicinale.
BackgroundL’uso degli oppiacei nel trattamento del dolore mode-
rato-severo è limitato dall’insorgenza di costipazione, ef-fetto secondario sfavorevole del trattamento con questaclasse di farmaci. La costipazione riduce notevolmente laqualità della vita di questi pazienti a causa del frequentericorso a lassativi per via rettale e/o manovre di svuota-mento manuale.
Diversi lassativi (osmotici, lubrificanti, da contatto eprocinetici) sono stati utilizzati nel trattamento della co-stipazione da oppiacei ma i loro effetti non sono specificie molti pazienti non rispondono a tali terapie.
Il metilnaltrexone, amina quaternaria e antagonista deirecettori µ per gli oppioidi, ha una ristretta capacità di at-traversare la barriera emato-encefalica limitando i propri
metilnaltrexone
La prescrizione a carico del SSN è limitata alle seguenti condizioni:
costipazione indotta da oppiacei in soggetti con malattia in statoterminale che rispondanocontemporaneamente alle seguenticaratteristiche:· terapia continuativa con oppiacei
della durata di almeno di 2 settimane;
· resistenza al trattamento con lassativi ad azione osmotica per più di 3 giorni.

PANORAMI E PERCORSI | LA NOTA 90 53
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
effetti alla periferia. La co-somministrazione del farmacocon gli oppiacei ne ridurrebbe l’effetto costipante, senzainterferire con la loro azione a livello del sistema nervosocentrale.
Evidenze disponibiliL’efficacia e la sicurezza del farmaco nel trattamento
della costipazione indotta da oppiacei in pazienti che ri-cevono cure palliative sono state dimostrate in due studiclinici randomizzati, in doppio cieco, placebo-controllati.Gli studi sono stati condotti per un periodo di quattromesi su un totale di 287 pazienti (età media di 68 anni;51% donne), con malattia in fase terminale ed un’aspet-tativa di vita limitata. Per la maggior parte di questi pa-zienti, la diagnosi primaria era malattia cancerosa.
Prima del trattamento con metilnaltrexone, i pazientiavevano ricevuto oppiacei per almeno 2 settimane ed unregime stabile di lassativi per almeno 3 giorni prima del-l’ingresso nello studio. L’eleggibilità è stata valutata sullabase di una costipazione definita sia come un numero dievacuazioni inferiore a tre nella settimana precedente al-l’inizio del trattamento con metilnaltrexone sia comeun’evacuazione clinicamente irrilevante (come determi-nato dall’investigatore) nelle 48 ore precedenti al tratta-mento. In entrambi gli studi nessuna prova ha suggeritoeffetti differenti in funzione dell’età o del sesso sulla sicu-rezza o l’efficacia del farmaco. In questi studi, non è stata
riscontrata alcuna significativa relazione tra la dose di op-piacei alla valutazione basale e la risposta clinica in pa-zienti trattati con metilnaltrexone. Inoltre, la dose dioppiacei media giornaliera non variava significativamenteda quella al basale sia nei pazienti trattati con metilnal-trexone sia nei pazienti trattati con placebo. Non ci sonostate modifiche clinicamente rilevanti dei punteggi del do-lore rispetto a quelli rilevati al basale nei pazienti trattaticon metilnaltrexone o con placebo.
Gli studi hanno dimostrato che il metilnaltrexone pervia sottocutanea induce rapidamente defecazione in pa-zienti con patologie in stadio avanzato e costipazione in-dotta da oppiacei: l’effetto insorge entro 30 minuti nellametà dei pazienti ed entro un’ora nella maggior parte diquesti.
Il trattamento è riservato a pazienti con aspettativa divita non superiore a 6 mesi.
Il farmaco non deve essere usato in pazienti di età in-feriore ai 18 anni poiché non c’è esperienza sul suo uso intali pazienti (vedi scheda tecnica).
Bibliografia di riferimento1. Thomas J, et al. Methylnaltrexone for opioid-induced
constipation in advanced illness. N Engl J Med 2008;358: 2332-43.
2. Slatkin N, et al. Methylnaltrexone for treatment ofopioid-induced constipation in advanced illness patients.J Support Oncology 2009; 7: 39-46.
Il modulo AIFA1 per la prescrizione dell’isotretinoinadeve essere consegnato solo alla paziente alla primaprescrizione del dermatologo. In seguito il modulorimarrà alla paziente che lo dovrà presentare almedico (dermatologo o medico di medicinagenerale, MMG) alle successive prescrizioni.Il modulo non deve essere compilato in caso ditrattamento con isotretinoina per i pazienti di sessomaschile per i quali la prima prescrizione deve essere
comunque effettuata dal dermatologo mentre quellesuccessive anche dal MMG.Il dermatologo deve acquisire il consenso informatodei pazienti sia di sesso maschile sia femminile econsegnare la guida alla terapia. Per le pazienti lospecialista deve altresì consegnare la guida allacontraccezione.Inoltre, il farmacista non deve controllare il moduloall’atto della dispensazione.
1. Determinazione 12 febbraio 2009. Modifica delle modalità di prescrizione dei medicinali contenenti isotretinoina ad usosistemico. GU n°43/09.
IsotretinoinaA proposito di…

54
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
L a degenerazione maculare correlata all’età o senile(AMD, age-related macular degeneration) è una delleprincipali cause di perdita della capacità visiva nel-
l’anziano e si manifesta nella forma “secca” o non essu-dativa e in quella “umida” o essudativa o neovascolare. Iltrattamento della AMD essudativa si basa soprattutto sul-l’uso di farmaci inibitori del fattore di crescita dell’endo-telio vascolare (anti-VEGF, vascular endothelial growth fac-tor), proteina responsabile della neovascolarizzazionecoroidale, che è la principale causa della patologia1.
Negli ultimi anni, l’armamentario farmacologico uti-lizzabile in questa malattia angiogenetica retinica si è ar-ricchito di due importanti risorse, specificamente indicateper il “trattamento della degenerazione maculare neova-scolare (essudativa) correlata all’età (AMD)”: il ranibizu-mab – un anticorpo monoclonale capace di inibire ilVEGF legandosi alla sua forma attiva – ed il pegaptanib –un oligonucleotide modificato (pegilato) che, grazie allasua struttura tridimensionale, è in grado di aderire al VEGFextracellulare impedendone l’interazione con il relativo re-cettore e permette di sopprimere, quindi, la neovascola-rizzazione patologica.
I due corrispondenti prodotti medicinali, Lucentis (ra-nibizumab) e Macugen (pegaptanib), la cui commercia-lizzazione è stata approvata dall’Agenzia Europea dei Me-dicinali (EMEA) con procedura centralizzata, sono statirecentemente riclassificati, ai fini della rimborsabilità, dal-l’Agenzia Italiana del Farmaco (AIFA) che ne ha stabilitoil passaggio dalla classe C alla classe H con la propria de-terminazione del 4 dicembre 20082.
Il rimborso a carico del Servizio Sanitario Nazionale(SSN) è vincolato all’adesione da parte dei centri utilizza-tori alle condizioni previste dal “Registro dei farmaci of-talmici sottoposti a monitoraggio”, previa registrazione alsito http://oftalmologici.agenziafarmaco.it/. In partico-lare, le schede di arruolamento del Registro identificano ipazienti eleggibili al trattamento con ranibizumab e pe-gaptanib come soggetti affetti da degenerazione maculareneovascolare attiva con acuità visiva ≥ 2/10. Il trattamentoprevisto è per occhio singolo, in pazienti non trattati pre-cedentemente con altri inibitori della neovascolarizza-zione per via intravitreale.
Prima dell’inserimento di questi due medicinali nell’e-
lenco dei farmaci erogati dal SSN una ulteriore possibilitàterapeutica, per i pazienti affetti da AMD neovascolare, eraofferta da un altro anticorpo monoclonale approvato dal-l’EMEA, il bevacizumab, che è però autorizzato solo perl’uso come antineoplastico. Nonostante ciò – grazie alfatto che questo è un anticorpo direttamente correlato alranibizumab (quest’ultimo, più precisamente, è un fram-mento anticorpale del bevacizumab, con alcune modifichenella sequenza aminoacidica che ne aumentano il legameal VEGF) e in virtù della documentata esperienza clinicaaccumulata con l’impiego off-label del farmaco sommi-nistrato per via intravitreale – l’AIFA, con determinazionedel 23 maggio 20073, su richiesta della Società Oftalmo-logica Italiana, ha inserito il bevacizumab (Avastin) nell’e-lenco previsto dalla legge 648/96 per l’indicazione “Trat-tamento delle maculopatie essudative e del glaucomaneovascolare”. In questo modo l’impiego del bevacizu-mab era a totale carico del SSN per quest’indicazione, conmonitoraggio sia a livello clinico che di spesa.
Dopo la riclassificazione di Lucentis (ranibizumab) eMacugen (pegaptanib) – e considerato che un farmaconon può più essere inserito nell’elenco dei medicinali ero-gabili ai sensi della legge 648/96 per indicazioni per lequali esistono farmaci approvati – è stata recentementemodificata la citata determinazione del 23 maggio 2007che aveva previsto l’inserimento dell’Avastin in tale elenco.
La determinazione AIFA del 4 marzo 2009 ha quindistabilito che bevacizumab (Avastin) è ora erogabile, aisensi della legge 648/96, solo per le seguenti indicazioni,
BOLLETTINO D’INFORMAZIONE SUI FARMACI | PANORAMI E PERCORSI |
Il trattamento della degenerazione maculareneovascolare: le regole del gioco
Bevacizumab può essere
prescritto, ai sensi della legge
648/96, per il trattamento
delle maculopatie essudative
non correlate all’età
o per il trattamento
del glaucoma neovascolare.

PANORAMI E PERCORSI | IL TRATTAMENTO DELLA DEGENERAZIONE MACULARE NEOVASCOLARE: LE REGOLE DEL GIOCO 55
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
non riportate nel Riassunto delle Caratteristiche del Pro-dotto del medicinale4:
trattamento delle maculopatie essudative non correlate all’età;trattamento delle maculopatie essudative correlateall’età già in trattamento con bevacizumab;trattamento del glaucoma neovascolare.Le condizioni sopra elencate rappresentano anche i cri-
teri di inclusione per l’accesso al trattamento, mentre co-stituiscono criteri di esclusione la degenerazione maculareneovascolare (essudativa) correlata all’età, le maculopatienon essudative e le patologie oculari non caratterizzate daneovascolarizzazione.
Ai fini del rimborso a carico del SSN è prevista la ste-sura di un Piano Terapeutico che, in accordo con gli studipubblicati in letteratura, preveda l’iniezione intravitreale didosaggi dipendenti dalle caratteristiche del paziente e cioèpari a 1,0, 1,25, 1,5, 2,0 o 2,5 mg. Tali quantità sonosomministrate a cadenza mensile e con regimi di sommi-nistrazione variabili: una o tre iniezioni iniziali e succes-sive iniezioni sulla base di criteri anatomo-funzionali dipersistenza o recidiva della lesione neovascolare.
Altre condizioni da osservare ai fini del rimborso a ca-rico del SSN sono il rispetto delle modalità previste dagliarticoli 4, 5 e 6 del provvedimento CUF del 20 luglio2000, e cioè:
art. 4: istituzione del registro, rilevamento e trasmissione dei dati di monitoraggio clinico ed informazioni riguardo a sospensioni del tratta-mento;art. 5: acquisizione del consenso informato, modalitàdi prescrizione e di dispensazione del medicinale;art. 6: rilevamento e trasmissione dei dati di spesa.Numerose richieste di chiarimenti sono pervenute ne-
gli ultimi tempi all’AIFA sia da parte di pazienti che di cen-tri specialistici e sono state generate, in larga misura, da al-cune inesattezze contenute nel materiale informativoattinente al trattamento della degenerazione maculareneovascolare recentemente distribuito dalla Società Of-
talmologica Italiana. Ciò rende ragione della pubblica-zione di questi brevi cenni sulla situazione regolatoriadei medicinali utilizzabili in questa patologia che, in con-clusione, può essere così sintetizzata: il trattamento con pe-gaptanib e ranibizumab è indicato per i pazienti affetti dadegenerazione maculare essudativa correlata all’età, men-
tre il bevacizumab può essere prescritto, ai sensi dellalegge 648/96, per il trattamento delle maculopatie essu-dative non correlate all’età o per il trattamento del glau-coma neovascolare; mentre solo i pazienti che erano già interapia con bevacizumab prima del 17 marzo 2009 (datadi entrata in vigore della determinazione AIFA del 4 marzo2009) possono proseguire la terapia per le maculopatie es-sudative correlate all’età con questo farmaco.
Solo i pazienti che erano già
in terapia con bevacizumab prima
del 17 marzo 2009 possono
proseguire la terapia per le
maculopatie essudative correlate
all’età con questo farmaco.
Bibliografia1. Anonimo. Terapia della
degenerazione macularecorrelata all’età.Bollettinod’Informazione suiFarmaci 2007; 14: 103-7.
2. Determinazione AIFA 4 dicembre 2008,pubblicata nella GazzettaUfficiale 18 dicembre2008, n. 295.
3. Determinazione AIFA 23maggio 2007, pubblicata nella Gazzetta Ufficiale28 maggio 2007, n. 122.
4. Determinazione AIFA 4marzo 2009, pubblicata nella Gazzetta Ufficiale16 marzo 2009, n. 62.

Riassunto La fibromialgia (FM) è un disordine di eziologia
ignota caratterizzato da dolore diffuso, disturbi delsonno, stanchezza e, spesso, problemi psicologici, la cuidiagnosi è basata sulla presenza del dolore da almeno3 mesi e sulla dolorabilità di punti caratteristici. Le co-noscenze attuali inducono a considerare la FM un di-sordine neuropsichiatrico ed orientano la terapia versoi meccanismi coinvolti nella percezione del dolore everso le condizioni morbose associate. Un’altra carat-teristica della FM è la mole di pareri contrastantiespressi da fonti autorevoli ai quali, ultimamente, si èaggiunta la valutazione divergente di FDA ed EMEA ri-guardo alla terapia con antiepilettici e antidepressivi.Ciò, unitamente alla facilità di accesso alle informazionie alla promozione di qualche farmaco, può indurre uninappropriato uso off-label di alcuni medicinali nonscevro da rischi per i pazienti.
AbstractFibromyalgia (FM) is a disorder of unknown aetiol-
ogy, characterized by chronic widespread pain, sleep dis-turbances, fatigue and often psychological problems.The clinical diagnosis of FM is based on the presence ofa history of pain lasting more than three months and ona painful response to touch in typical points of the body.According to current knowledge FM is a neuropsychiatricdisorder and its treatment is mainly aimed at manage-ment of the mechanisms involved in pain perception andrelated morbid conditions. Furthermore, several contro-versial opinions on FM exist, with regard to the use ofantiepileptics and antidepressants expressed by reliablesources, among which also the divergent assessment onthis issue made by the FDA and the EMEA. This, togetherwith the easily accessable information and promotion ofsome drugs, may determine an inappropriate, off-labeluse of some drugs which may be harmful for patients.
Il quadro clinico La fibromialgia (FM)1,2 è un disordine di eziologia
ignota caratterizzato da dolore diffuso, disturbi del
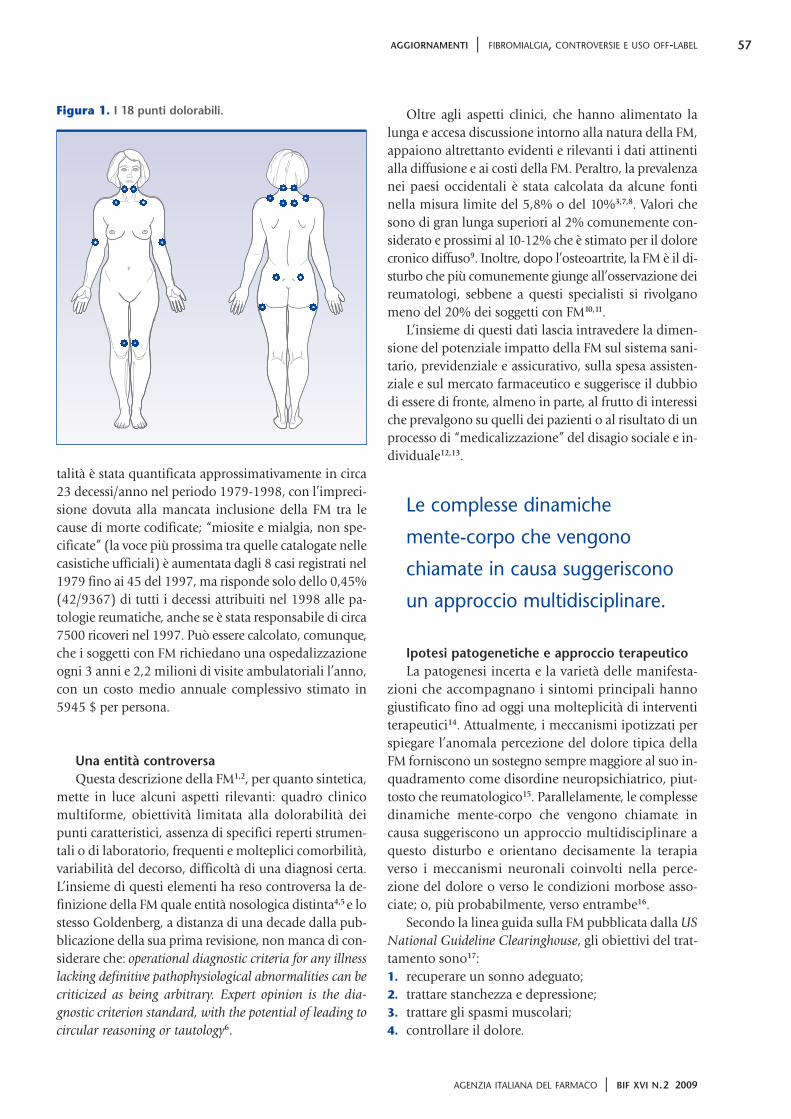
sonno, stanchezza e, spesso, problemi di ordine psi-cologico. Possono essere presenti anche altri sintomivariamente combinati: rigidità mattutina, formicolio ointorpidimento delle mani o dei piedi, varie forme dicefalea compresa l’emicrania, turbe dell’ideazione odella memoria, dismenorrea. Inoltre, in una elevatapercentuale di casi (25-65%), la FM può coesistere conmalattie reumatiche quali artrite reumatoide (AR), lu-pus eritematoso sistemico (LES) e spondilite anchilo-sante (SA). È comune, però, anche l’associazione conla sindrome dell’intestino irritabile, la sindrome pre-mestruale, la sindrome sicca, la sindrome delle gambesenza riposo oppure con il fenomeno di Raynaud, conil dolore dell’articolazione temporomandibolare o conil dolore toracico non-cardiaco. Nel 1990 l’AmericanCollege of Rheumatology (ACR) ha definito i criteri ne-cessari alla diagnosi: presenza di dolore diffuso da al-meno 3 mesi e dolorabilità alla pressione di almeno 11di 18 punti caratteristici esattamente localizzati (figura1). L’obiettività muscoloscheletrica e neurologica ènormale e non sono presenti tipiche alterazioni degliesami di laboratorio o della diagnostica strumentale.
Le cause o i fattori di rischio sono sconosciuti ma al-cuni eventi sono labilmente correlati con l’esordio dellamalattia: situazioni stressanti o fatti traumatici (per es.incidenti automobilistici, lesioni o ferite ripetute), ma-lattie intercorrenti (per es. infezioni virali), particolaripatologie (per es. AR, LES o sindrome da fatica cronica);né può essere esclusa una predisposizione genetica.
Il decorso è variabile, con un andamento sponta-neamente oscillante (“va-e-vieni”) in alcuni pazientimentre, in altri, dolore e stanchezza persistono nono-stante il trattamento.
L’epidemiologia e i costiNegli Stati Uniti2, la FM ha una prevalenza di circa il
2% e nel 2005 sono stati stimati 5 milioni di adulti af-fetti, in maggioranza donne (3,4% vs 0,5%; rapportofemmine:maschi di 7:1). La maggior parte delle dia-gnosi di FM, che non risparmia i bambini, viene postadurante l’età media e la prevalenza aumenta con l’età(nelle donne tra i 60 e i 79 anni supera il 7%3). La mor-
Fibromialgia, controversie e uso off-label
BOLLETTINO D’INFORMAZIONE SUI FARMACI | AGGIORNAMENTI |
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
56
AGGIORNAMENTI | FIBROMIALGIA, CONTROVERSIE E USO OFF-LABEL
Oltre agli aspetti clinici, che hanno alimentato lalunga e accesa discussione intorno alla natura della FM,appaiono altrettanto evidenti e rilevanti i dati attinentialla diffusione e ai costi della FM. Peraltro, la prevalenzanei paesi occidentali è stata calcolata da alcune fontinella misura limite del 5,8% o del 10%3,7,8. Valori chesono di gran lunga superiori al 2% comunemente con-siderato e prossimi al 10-12% che è stimato per il dolorecronico diffuso9. Inoltre, dopo l’osteoartrite, la FM è il di-sturbo che più comunemente giunge all’osservazione deireumatologi, sebbene a questi specialisti si rivolganomeno del 20% dei soggetti con FM10,11.
L’insieme di questi dati lascia intravedere la dimen-sione del potenziale impatto della FM sul sistema sani-tario, previdenziale e assicurativo, sulla spesa assisten-ziale e sul mercato farmaceutico e suggerisce il dubbiodi essere di fronte, almeno in parte, al frutto di interessiche prevalgono su quelli dei pazienti o al risultato di unprocesso di “medicalizzazione” del disagio sociale e in-dividuale12,13.
Ipotesi patogenetiche e approccio terapeuticoLa patogenesi incerta e la varietà delle manifesta-
zioni che accompagnano i sintomi principali hannogiustificato fino ad oggi una molteplicità di interventiterapeutici14. Attualmente, i meccanismi ipotizzati perspiegare l’anomala percezione del dolore tipica dellaFM forniscono un sostegno sempre maggiore al suo in-quadramento come disordine neuropsichiatrico, piut-tosto che reumatologico15. Parallelamente, le complessedinamiche mente-corpo che vengono chiamate incausa suggeriscono un approccio multidisciplinare aquesto disturbo e orientano decisamente la terapiaverso i meccanismi neuronali coinvolti nella perce-zione del dolore o verso le condizioni morbose asso-ciate; o, più probabilmente, verso entrambe16.
Secondo la linea guida sulla FM pubblicata dalla USNational Guideline Clearinghouse, gli obiettivi del trat-tamento sono17: 1. recuperare un sonno adeguato;2. trattare stanchezza e depressione; 3. trattare gli spasmi muscolari;4. controllare il dolore.
Figura 1. I 18 punti dolorabili.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
57
Le complesse dinamiche
mente-corpo che vengono
chiamate in causa suggeriscono
un approccio multidisciplinare.
talità è stata quantificata approssimativamente in circa23 decessi/anno nel periodo 1979-1998, con l’impreci-sione dovuta alla mancata inclusione della FM tra lecause di morte codificate; “miosite e mialgia, non spe-cificate” (la voce più prossima tra quelle catalogate nellecasistiche ufficiali) è aumentata dagli 8 casi registrati nel1979 fino ai 45 del 1997, ma risponde solo dello 0,45%(42/9367) di tutti i decessi attribuiti nel 1998 alle pa-tologie reumatiche, anche se è stata responsabile di circa7500 ricoveri nel 1997. Può essere calcolato, comunque,che i soggetti con FM richiedano una ospedalizzazioneogni 3 anni e 2,2 milioni di visite ambulatoriali l’anno,con un costo medio annuale complessivo stimato in5945 $ per persona.
Una entità controversaQuesta descrizione della FM1,2, per quanto sintetica,
mette in luce alcuni aspetti rilevanti: quadro clinicomultiforme, obiettività limitata alla dolorabilità deipunti caratteristici, assenza di specifici reperti strumen-tali o di laboratorio, frequenti e molteplici comorbilità,variabilità del decorso, difficoltà di una diagnosi certa.L’insieme di questi elementi ha reso controversa la de-finizione della FM quale entità nosologica distinta4,5 e lostesso Goldenberg, a distanza di una decade dalla pub-blicazione della sua prima revisione, non manca di con-siderare che: operational diagnostic criteria for any illnesslacking definitive pathophysiological abnormalities can becriticized as being arbitrary. Expert opinion is the dia-gnostic criterion standard, with the potential of leading tocircular reasoning or tautology6.

AGGIORNAMENTI | FIBROMIALGIA, CONTROVERSIE E USO OFF-LABEL
Il diverso punto di vista di EMEA e FDANel 2007 la Food and Drug Administration (FDA) ha
autorizzato per la prima volta l’inserimento del tratta-mento della FM tra le indicazioni terapeutiche di un far-maco: l’antiepilettico pregabalin; nel 2008 è stato ap-provato l’uso dell’antidepressivo duloxetina, un inibitoreselettivo della ricaptazione della serotonina e della no-radrenalina (SSNRI) e, quindi, nel 2009 è stato il turnodel milnacipran, un altro SSNRI18. A queste scelte dellaFDA non hanno fatto seguito analoghe decisioni dellaEuropean Medicines Agency (EMEA), anzi: fino ad oggi,l’Agenzia europea non ha espresso alcun giudizio favo-revole al riguardo.
In particolare, nell’ottobre del 2008, l’EMEA ha resonote le ragioni del rifiuto opposto alla richiesta di esten-sione delle indicazioni al trattamento della FM presen-tata per due prodotti a base di duloxetina (Cymbalta eXeristar)19. Ha destato preoccupazione nel Committeefor Medicinal Products for Human Use (CHMP) dell’E-MEA il fatto che l’efficacia della duloxetina nel tratta-mento della FM non fosse stata sufficientemente dimo-strata. È stato considerato che l’effetto del farmaco, neglistudi a breve termine, fosse troppo ridotto per essere cli-nicamente rilevante per i pazienti; inoltre, per il CHMP,non è stata prodotta la chiara dimostrazione del mi-glioramento dei sintomi ed il modesto effetto osser-vato potrebbe, in realtà, essere dovuto alla nota capacitàdella duloxetina di migliorare l’umore del paziente. IlCHMP ha anche ritenuto che i dati a lungo terminenon fossero sufficienti per dimostrare l’efficacia del far-maco e che ulteriori studi sono necessari. Nel tratta-mento della FM, per il CHMP, i benefici della duloxetinanon superano i rischi.
Un giudizio sostanzialmente sovrapponibile è statorecentemente espresso nei confronti dell’analoga ri-chiesta presentata per il prodotto “Lyrica”, a base di pre-gabalin20. Anche in questo caso, il beneficio del tratta-mento non risulta dimostrato né a breve né a lungotermine. In particolare, gli studi a breve termine non
hanno evidenziato un miglioramento del dolore o de-gli altri sintomi, costante o direttamente correlato allaterapia. Inoltre, l’effetto del farmaco si è rivelato indi-pendente dal dosaggio somministrato e, negli studi alungo termine, non è stato possibile verificarne la per-sistenza nel corso del tempo. Il CHMP ha mostratopreoccupazione, altresì, per il fatto che i dati di sicurezzaed efficacia non fossero relativi a pazienti europei.Come per la duloxetina, il CHMP ha ritenuto che, neltrattamento della FM, i benefici del pregabalin non su-perano i rischi.
Una metanalisi relativa all’uso degli antidepressivinella FM, pubblicata successivamente alla decisionedell’EMEA, fornisce elementi che possono aiutare a ri-flettere sulle differenti conclusioni di FDA ed EMEA21;non è disponibile, naturalmente, una simile revisionecondotta successivamente al recentissimo parereespresso sul pregabalin. La metanalisi ha considerato 18studi clinici randomizzati e controllati (RCT) per un to-tale di 1427 pazienti, di cui 3 relativi alla duloxetina,con durata media di 8 settimane (range 4-28). Secondogli autori risultano “forti evidenze” in favore della te-rapia con antidepressivi, duloxetina compresa, perquanto attiene al dolore, all’umore depresso e alla qua-lità del sonno.
A fronte di queste conclusioni, tuttavia, l’articolopone in evidenza numerosi aspetti che meritano di es-sere tenuti in adeguata considerazione. Innanzitutto, ladimensione dell’effetto del trattamento è, per tutti que-sti sintomi, “piccola” mentre è “insignificante” per lastanchezza. Inoltre: nessuno degli studi ha previsto unaverifica degli outcome dopo la cessazione del tratta-mento; i livelli sierici degli antidepressivi non sonostati misurati in alcuno degli studi; in tutti, è stata con-sentita la somministrazione addizionale di paraceta-molo che è stato associato, in 8 studi, con acido acetil-salicilico, antinfiammatori non-steroidei o codeina; innessuno studio, tuttavia, è stato verificato il consumodegli analgesici, il dosaggio utilizzato o i loro effetti in-desiderati; la somministrazione addizionale di altri far-maci non è stata controllata in alcuno studio e in nes-suno sono state fornite informazioni dettagliate circaeventuali terapie non-farmacologiche o circa gli even-tuali controlli sulle stesse, oppure dati sulla presenza dieventuali patologie non-psichiatriche. Ulteriori limitidegli RCT esaminati erano: la dimensione ridotta delcampione studiato e la durata contenuta del periodo diosservazione; l’assenza di un follow-up successivo al-l’interruzione del trattamento e, quindi, di informa-zioni sugli eventuali benefici a lungo termine e, altresì,la mancanza di dati sui pazienti di età superiore a 65anni o su quelli affetti da malattie somatiche gravi,
Nel 2007 la FDA ha autorizzato
l’uso nella fibromialgia
del pregabalin e poi di duloxetina
e milnacipran; ad oggi l’EMEA
non ha invece espresso
alcun giudizio favorevole.
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
58

AGGIORNAMENTI | FIBROMIALGIA, CONTROVERSIE E USO OFF-LABEL
comprese quelle infiammatorie articolari, oppure, in-fine, sugli individui di sesso maschile. Per quanto at-tiene specificamente alla duloxetina, gli autori segna-lano, inoltre, che: la sequenza di somministrazioneutilizzata negli studi (placebo per una settimana seguitodal farmaco attivo) può aver pregiudicato il requisito dicecità; i dati sugli individui di sesso maschile sono pre-senti ma, in uno studio, non si è avuta alcuna rispostasignificativa, sia per gli outcome primari che secondari,mentre, in un altro, la riduzione del dolore era simile tramaschi e femmine; l’effetto sul dolore non era differentetra i pazienti affetti da depressione, o meno; in due studisu tre, sono stati esclusi i pazienti giudicati dai ricerca-tori come refrattari alla terapia e, in questo modo, èstato probabilmente influenzato l’esito in favore deltrattamento.
Il rischio di un uso off-label inappropriatoIl punto di vista divergente delle autorità statunitensi
ed europee si aggiunge alla già notevole mole dei pareriautorevoli e contrastanti espressi sulla FM e contribuiscead aumentare i dubbi e le incertezze dei pazienti. Inol-tre, poiché ogni tentativo terapeutico ha buone proba-bilità di risultare deludente per il paziente e frustranteper il medico, non può essere trascurata l’importanza
che può assumere, per chi soffre di FM, la ricerca di unrimedio, qualunque sia.
D’altra parte, la quantità di informazioni offerte, so-prattutto attraverso internet, a chiunque desideri docu-mentarsi su un problema di salute che lo riguardi èenorme: digitando “fibromyalgia” su Google si otten-gono 7.990.000 risultati, mentre sono ben 1.760.000 iblog che hanno trattato l’argomento. Tali informazioni,peraltro, sono spesso accompagnate da forme più omeno esplicite di pubblicità che possono indurre pa-zienti sfiduciati a sollecitare la prescrizione di un deter-minato farmaco (l’argomento è stato trattato nell’edi-toriale dell’ultimo numero del Bif: L’informazione suifarmaci definisce un nuovo orizzonte).
In questa situazione, il ricorso all’uso off-label dipregabalin o duloxetina – farmaci che in Italia non sonoutilizzabili nella FM – può apparire come la sola possi-bilità di alleviare le sofferenze di questi pazienti. Occorreconsiderare, tuttavia, che in questo caso non possono es-sere completamente rispettati i criteri che consentonol’uso off-label e, in particolare, come si è visto, manca ilconsenso unanime e consolidato sulla validità della te-rapia. Ciò dovrebbe suggerire una prudente valutazionedelle possibili conseguenze negative per i pazienti anchealla luce dei gravi rischi che sono stati segnalati in asso-ciazione ad antiepilettici e antidepressivi22,23.
Bibliografia1. Langford CA, Gilliland BC.
Fibromyalgia. In: Fauci AS, KasperDL, Longo DL, et al., eds. Harrison’sprinciples of internal medicine. 17thed. New York: McGraw-Hill, 2008.
2. Centers for Disease Control and Prevention. Fibromyalgia.www.cdc.gov/arthritis/arthritis/fibromyalgia.htm (accesso verificato il 16/04/2009).
3. Wolfe F, Ross K, Anderson J, et al. The prevalence and characteristics of fibromyalgia in the generalpopulation. Arthritis Rheum 1995;38: 19-28.
4. Cohen ML. Is fibromyalgia a distinctclinical entity? The disapprovingrheumatologist’s evidence. BaillieresBest Pract Res Clin Rheumatol 1999;13: 421-5.
5. Gordon DA. Fibromyalgia. Real or Imagined? J Rheumatol 2003; 30: 1665.
6. Goldenberg DL. Fibromyalgiasyndrome a decade later: what havewe learned? Arch Intern Med 1999;159: 777-85.
7. Gran JT. The epidemiology of chronicgeneralized musculoskeletal pain.Best Pract Res Clin Rheumatol 2003;17: 547-61.
8. Forseth KO, Gran JT. The occurrenceof fibromyalgia-like syndromes in a general female population. Clin Rheumatol 1993; 12: 23-7.
9. Croft P, Rigby AS, Boswell R,Schollum J, Silman A. The prevalenceof chronic widespread pain in thegeneral population. J Rheumatol1993; 20: 710-3.
10. Marder WD, Meenan RF, Felson DT,et al. Editorial: the present and futureadequacy of rheumatologymanpower: a study of health careneeds and physician supply. Arthritis Rheum 1991; 34: 1209-17.
11. Crofford LJ, Clauw DJ. Fibromyalgia:where are we a decade after theAmerican College of Rheumatologyclassification criteria were developed?Arthritis Rheum 2002; 46: 1136-8.
12. Ehrlich GE. Fibromyalgia, a virtualdisease. Clin Rheumatol 2003; 22: 8-11.
13. Hadler NM. “Fibromyalgia” and themedicalization of misery [Editorial]. J Rheumatol 2003; 30: 1668-70.
14. Goldenberg DL, Burckhardt C,Crofford L. Management offibromyalgia syndrome. JAMA 2004;292: 2388-95.
15. Shir Y, Fitzcharles MA. Shouldrheumatologists retain ownership offibromyalgia? [Editorial]. J Rheumatol 2009; 36: 667-70.
16. Abeles AM, Pillinger MH, Solitar BM,Abeles M. Narrative review: thepathophysiology of fibromyalgia.Ann Intern Med 2007; 146: 726-34.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
59

AGGIORNAMENTI | FIBROMIALGIA, CONTROVERSIE E USO OFF-LABEL60
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
17. National Guideline Clearinghouse.Fibromyalgia treatment guideline.www.guideline.gov/summary/summary.aspx?ss=15&doc_id=7352&nbr=004350&string=fibromyalgia (accesso verificato il 16/04/2009).
18. MedlinePlus. Medical encyclopedia.Fibromyalgia. www.nlm.nih.gov/MEDLINEPLUS/ency/article/000427.htm#Treatment (accesso verificato il 16/04/2009).
19. European Medicines Agency.Questions and answers onrecommendation for the refusal of a change to the marketingauthorisation for Cymbalta/Xeristar.(London, 23 October 2008)www.emea.europa.eu/humandocs/PDFs/EPAR/cymbalta/Cymbalta_55118108en.pdf (accesso verificato il 16/04/2009).
20. European Medicines Agency.Questions and answers onrecommendation for the refusal of a change to the marketingauthorisation for Lyrica. (London, 23 April 2009)http://www.emea.europa.eu/pdfs/human/opinion/LyricaQ&A_23113109en.pdf (accesso verificato il 29/04/2009).
21. Häuser W, Bernardy K, Üceyler N,Sommer C. Treatment of fibromyalgiasyndrome with antidepressants. A meta-analysis. JAMA 2009; 301: 198-209.
22. US Food and Drug Administration.Suicidal behavior and ideation andantiepileptic drugs.www.fda.gov/cder/drug/infopage/antiepileptics/default.htm (accesso verificato il 16/04/2009).
23. US Food and Drug Administration.FDA Alert: SSRIs/SNRI/Triptan andSerotonin Syndrome.www.fda.gov/cder/drug/infopage/duloxetine/default.htm (accesso verificato il 16/04/2009).
In Italia la terapia del dolore è resa difficoltosa dagliobblighi previsti per la prescrizione degliantidolorifici contenenti oppiacei. In attesa di unarevisione della legislazione relativa agli stupefacenti,il Vice Ministro Prof. Ferruccio Fazio ha emanato il 16 giugno 2009 una Ordinanza che semplifica, in alcuni casi, l’accesso a questi farmaci.L’Ordinanza, è vigente dal 20 giugno scorso e rimane tale fino all’entrata in vigore delledisposizioni di revisione del Testo Unico (DPR 309del 1990) o comunque non oltre i dodici mesi.In particolare, è stata resa più semplice laprescrizione di alcuni farmaci oppiacei, mediante laloro ricollocazione in una diversa sezione delletabelle allegate al Testo Unico, consentendo quindial medico di utilizzare il ricettario normale anzichéquello speciale ed eliminando così le difficoltàburocratiche che spesso scoraggiano tali prescrizioni.Le composizioni medicinali, per le quali vienetemporaneamente adottata la ricetta semplice, sonotutte composizioni ad uso diverso da quelloparenterale, utilizzate nella terapia del dolore grave
di qualsiasi origine e sono specificate nell’Ordinanzastessa. Sono esclusi i medicinali indicati nella terapiadella disassuefazione degli stati di tossicodipendenza.Da tale ricollocazione (dalla sezione A a quella Ddella tabella II) è inoltre derivato un nuovo regime di fornitura di questi medicinali che è da riportare in etichetta come: “medicinale iscritto nella tabella II,sezione D del testo unico di cui al D.P.R. 309/90 - da vendersi dietro presentazione di ricetta medicautilizzabile una sola volta“. La conseguente modificadelle etichette è attuata dalle aziende interessantesenza specifica autorizzazione dell’AIFA e ha validitàper la durata temporale dell’Ordinanza.I medicinali già presenti nel canale distributivo al momento dell’entrata in vigore dell’Ordinanza, e quindi con etichette conformi al precedenteregime di fornitura, possono essere dispensati daifarmacisti secondo le previsioni dell’Ordinanza stessa,senza necessità di aggiornare le etichette;analogamente procederanno i farmacisti ospedalieri,per quanto riguarda la distribuzione di tali medicinalialle singole unità operative.
Terapia del doloreA proposito di…

61
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
RiassuntoI drammatici dati epidemiologici relativi all’anores-
sia e agli altri disturbi del comportamento alimentareesigono che la società intera ponga attenzione alle in-numerevoli sfaccettature del problema. Particolarmenteimportante appare la necessità di contrastare la diffu-sione di modelli estetici negativi o di informazioni scor-rette su alimentazione, controllo del peso e questionicorrelate. Nei giovani, in particolare, strumenti socio-culturali spesso insufficienti si aggiungono ai cambia-menti adolescenziali nel determinare una forterecettività alle suggestioni dei media. Ciò, unitamenteall’elevata probabilità di superare facilmente i limiti delproprio peso “idealizzato”, li rende i soggetti maggior-mente esposti al rischio di ricevere, attraverso una co-municazione non adeguatamente controllata, messaggipotenzialmente pericolosi per la salute. In questo con-testo, preoccupa l’eventualità che anche la pubblicità deimedicinali indicati per il trattamento del sovrappesopossa diventare un possibile veicolo di informazioni fa-cilmente suscettibili di essere non correttamente inter-pretate, soprattutto dai soggetti più fragili.
AbstractEpidemiological data concerning anorexia nervosa
and other disorders of eating behaviour require that so-ciety pays attention to the problem. It is extremely im-portant to contrast the spreading of aesthetic models orincorrect information on diet, weight control, and ques-tions related to the problem. In young people, insuffi-cient socio-cultural tools and adolescent changes oftenwork together to make the young conditioned by mediasuggestions. It makes young people more exposed to therisk of receiving potential dangerous health messagesthrough uncontrolled communication. Under this re-spect, the possibility that advertising of products indi-cated for the treatment of fatness may turn into a vehicleof information susceptible of being incorrectly inter-preted, mainly by weaker people, is worrying.
Introduzione“Ragazze, anoressia e bulimia sono le prime cause di
morte” (La Repubblica del 13 marzo 2009); “Anoressia,ora tocca agli uomini. Triplicato il numero di quelli chene soffrono. Un giovane su quattro è ossessionato dalpeso” (Corriere della Sera del 14 febbraio 2009).
Quando i dati epidemiologici compaiono ripetuta-mente sulle pagine dei giornali è segno che un problemamedico ha travalicato i confini della medicina per esigereattenzione dalla società intera. E questo è certamente ilcaso dei disturbi del comportamento alimentare (DCA).
Le cifre sempre più drammatiche rendono evidentel’urgenza di un intervento efficace e coordinato da partedi tutti i soggetti che sono in qualche modo coinvolti eresponsabili. Infatti, se da un lato occorre migliorare leattività assistenziali e di sostegno, dall’altro è necessariocontrastare la diffusione di modelli sociali ed estetici ne-gativi, così come è altresì impossibile trascurare l’impor-tanza della divulgazione di informazioni corrette edadeguate su alimentazione, controllo del peso e que-stioni correlate. Binge, dieting o vigoressia non dovrebberopiù essere termini sconosciuti per la famiglia, il medicoo la scuola. Né dovrebbero rappresentare solo realtà in-teramente delegate all’intervento specialistico e tutte lecomponenti sociali, dalle istituzioni alle associazioni dicategoria, dovrebbero farsi carico delle innumerevolisfaccettature del problema.
Alcuni passi in questa direzione, tuttavia, sono giàstati fatti e tra le varie iniziative si segnalano quelle chedimostrano l’impegno ed il coinvolgimento diretto delleautorità sanitarie nazionali e che appaiono particolar-mente attente agli aspetti legati alla comunicazione e allapromozione di stili di vita salutari e di modelli esteticinaturali1-3.
La popolazione maggiormente a rischio: i giovaniSecondo i dati dell’Organizzazione Mondiale della
Sanità (OMS) la prevalenza dell’obesità in Europa sta cre-scendo rapidamente e si prevede che interesserà, entro il2010, 150 milioni di adulti e 15 milioni di bambini4. Idati più allarmanti riguardano i bambini e gli adole-
“Deregulation” del trattamento farmacologicodel sovrappeso e rischi correlati ai disturbi del comportamento alimentare
BOLLETTINO D’INFORMAZIONE SUI FARMACI | AGGIORNAMENTI |

AGGIORNAMENTI | “DEREGULATION” DEL TRATTAMENTO FARMACOLOGICO DEL SOVRAPPESO E RISCHI CORRELATI AI DCA
scenti: il tasso attuale è 10 volte più alto di quello deglianni ’70. In Italia, in particolare, il 24% dei bambinidella classe terza elementare è in sovrappeso, mentre il12% può essere già definito obeso2. L’obesità, inoltre, èdiffusa nel nostro continente soprattutto nelle classi so-ciali più svantaggiate4.
Quindi, strumenti socio-culturali spesso insufficientisi aggiungono ai cambiamenti adolescenziali nel deter-minare una forte recettività alle suggestioni dei media.Ciò, unitamente all’elevata probabilità di superare facil-mente i limiti del proprio peso “idealizzato”, contribui-sce a fare dei giovani la popolazione più fragile emaggiormente esposta al rischio di ricevere, attraversouna comunicazione non adeguatamente controllata,messaggi potenzialmente pericolosi per la salute.
Trattamento del sovrappeso e dell’obesità: cosa dicono le linee guidaLa linea guida NICE sottolinea che la prevenzione ed
il trattamento dell’obesità e del sovrappeso sono pro-blemi complessi che non prevedono risposte facili5. Perl’OMS4, un approccio corretto al sovrappeso-obesità nonpuò prescindere dall’approfondimento diagnostico deifattori di rischio associati e delle comorbilità, nonchédall’eventuale intervento terapeutico su di essi. Il tratta-mento dietetico, inoltre, ha un ruolo primario e insosti-tuibile ma, comunque, comporta rischi che, soprattuttonei giovani, comprendono la perdita di massa magra e ilpossibile peggioramento dei DCA. Infatti, non dovrebbemai essere sottovalutato che anche un modesto sovrap-peso può essere indicativo della presenza di un DCA po-tenzialmente in grado di evolvere sino agli esiti più gravi,specialmente se gestito in solitudine dal paziente condiete-fai-da-te o ricorrendo all’assunzione non corretta diintegratori o di farmaci. Medici e pazienti, viene sottoli-neato, devono essere consapevoli che il trattamento del-l’obesità non si esaurisce nel breve periodo ma che si pro-trae nel tempo ed è, di fatto, una terapia “cronica” alungo termine.
Il trattamento del sovrappeso e dell’obesità può pre-vedere anche il ricorso a farmaci che si sono rivelati ef-ficaci nell’indurre un decremento ponderale; ilvantaggio, rispetto al placebo, è però di entità contenutae limitato nel tempo. Inoltre questi medicinali presen-tano, come di norma, un certo numero di effetti indesi-derati e producono il loro effetto solo se associati alladieta. Il trattamento farmacologico è da prendere in con-siderazione solo dopo aver iniziato una dieta adeguata,accompagnata dall’incremento dell’esercizio fisico edalle necessarie modifiche del comportamento, e dopoaverne valutato i risultati a distanza di tempo4,5.
L’orlistat, in particolare, dovrebbe essere prescrittodal medico come parte di uno schema di gestione glo-bale dell’obesità solo in adulti5:
che hanno un indice di massa corporea (BMI, bodymass index) di 28 kg/m2 o superiore e con fattori dirischio associati;o che hanno un BMI di 30 kg/m2 o più.
Inoltre:la terapia dovrebbe essere continuata oltre i 3 mesisolo se il calo ponderale ottenuto dall’inizio dellasomministrazione del farmaco è pari ad almeno il5% del peso iniziale;la decisione di prolungare il trattamento per più di 12mesi dovrebbe essere presa dal medico solo dopoaver discusso e valutato con il paziente i potenzialibenefici e i limiti della terapia;non è raccomandata la co-prescrizione di orlistat conaltri farmaci utilizzati per ottenere una riduzione delpeso.
Secondo i dati riportati nella linea guida NICE ilvantaggio ottenuto con orlistat (più dieta), rispetto alplacebo (più dieta), in termini di decremento ponde-rale è stato di: 3,27 kg dopo 12 mesi e di 3,26 kg dopo24 mesi, mentre la differenza tra chi aveva assunto orli-stat per 48 mesi rispetto a quelli che avevano ricevutoplacebo era di 2,80 kg. Ancora più contenuto il benefi-cio evidenziato confrontando l’effetto del farmaco e delplacebo sul mantenimento del peso: dopo 12 mesi ilvantaggio di orlistat (più dieta) vs placebo (più dieta) èpari a 0,19 kg (IC 95% da 0,97 a 0,58)5.
La decisone dell’EMEA In questo contesto – e considerato quanto è stabi-
lito dalla direttiva europea che regola la materia (tabellaI) – ha fatto discutere la decisione della European Me-dicines Agency (EMEA) di eliminare l’obbligo di ricettamedica per un prodotto medicinale* a base di orlistat6.Forti perplessità al riguardo sono state espresse in uneditoriale dell’autorevole Lancet: “… una migliore ac-cessibilità di orlistat potrebbe non necessariamente es-sere nell’interesse principale dei cittadini … l’aver resoquesto medicinale accessibile senza ricetta aggiungeràuna falsa credibilità alla nozione che basti ingoiare unapillola come facile rimedio per l’obesità invece di af-frontare faticosi cambiamenti a lungo termine dellostile di vita … il mancato controllo medico significa chel’abuso del farmaco potrebbe condurre a deficit nutri-zionali non diagnosticati … c’è un urgente bisogno
*Alli, Glaxo Group Limited.
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
62

AGGIORNAMENTI | “DEREGULATION” DEL TRATTAMENTO FARMACOLOGICO DEL SOVRAPPESO E RISCHI CORRELATI AI DCA
della dimostrazione che le decisioni relative ai medici-nali senza prescrizione sono prese nell’interesse princi-pale dei cittadini”7.
Dalla ricetta medica alla pubblicità?La decisione dell’EMEA di classificare come non
soggetto a prescrizione questo medicinale è valida intutta l’Unione Europea e gli stati membri sono tenutiad implementarla.
I medicinali non soggetti a prescrizione sono defi-
niti, per negazione, come quelli che non rispondono aicriteri che obbligano a classificare un prodotto comesoggetto a prescrizione (tabella I). Questi medicinalirecano un bollino di riconoscimento che ne permette lachiara individuazione da parte del consumatore e su diessi il farmacista può dare consigli al cliente.
Solo alcuni, però, possono essere oggetto di pub-blicità presso il pubblico “se hanno i requisiti stabilitidalle norme vigenti in materia”, ovvero: “possono es-sere oggetto di pubblicità presso il pubblico i medici-nali che, per la loro composizione e il loro obiettivoterapeutico, sono concepiti e realizzati per essere uti-lizzati senza intervento di un medico per la diagnosi, laprescrizione o la sorveglianza nel corso del tratta-mento”8. In tal caso, i medicinali sono identificati inetichetta dalla dicitura “medicinale di automedica-zione” e sono definiti come “medicinali da banco o diautomedicazione” o “farmaci non soggetti a ricetta me-dica con accesso alla pubblicità al pubblico (OTC)”9.Essi possono inoltre essere “oggetto di accesso direttoda parte dei clienti in farmacia e nei punti vendita” au-torizzati10.
La classificazione di un medicinale come non sog-getto a prescrizione, quindi, non implica automatica-mente la facoltà di pubblicizzarlo presso il pubblico,ma rappresenta semplicemente una delle pre-condi-zioni perché ciò possa realizzarsi.
Le decisioni in materia competono all’autorità na-zionale e non c’è motivo di dubitare che siano assuntedall’Agenzia Italiana del Farmaco nell’interesse princi-pale dei cittadini. Lo scopo principale delle norme relativealla produzione, alla distribuzione e all’uso di medicinalideve essere quello di assicurare la tutela della sanità pub-blica11.
Tabella I. Direttiva 2001/83/CE del ParlamentoEuropeo e del Consiglio del 6 novembre 2001 e successive modificazioni.
Articolo 71
1. I medicinali sono soggetti a prescrizionemedica in uno dei casi seguenti:
possono presentare un pericolo, direttamente o indirettamente, anche in condizioni normali di utilizzazione, se sono usati senza controllomedico;
sono utilizzati spesso, e in larghissima misura,in condizioni anormali di utilizzazione e ciò rischia di mettere in pericolo direttamente o indirettamente la salute;
contengono sostanze o preparazioni a base di tali sostanze, di cui è indispensabile approfondire l’attività e/o gli effetti collaterali negativi;
salvo eccezioni, sono prescritti da un medicoper essere somministrati per via parenterale.
Bibliografia
1. Progetto “Le buone pratiche di cura e la pre venzione sociale dei disturbidel comportamento alimentare”.www.ministerosalute.it/imgs/C_17_pubblicazioni_767_allegato.pdf(accesso verificato il 17/03/2009).
2. Progetto “Tim Shel, chi si ama, ci segua”.www.ministerosalute.it/stiliVita/paginaInternaMenuStiliVita.jsp?id=1220&menu=progetti (accesso verificato il 17/03/2009).
3. Pe(n)sa differente. www.pensa-differente.it/home.php(accesso verificato il 17/03/2009).
4. Branca F, Nikogosian H, Lobstein T,eds. La sfida dell’obesità nella
Regione europea dell’OMS e lestrategie di risposta. Compendio.Organizzazione Mondiale dellaSanità, 2007 – Centro Nazionale per la Prevenzione e il Controllodelle malattie (CCM), Ministero della Salute, Italia, 2008.
5. Obesity: the prevention,identification, assessment andmanagement of overweight andobesity in adults and children.London, National Institute for Health and Clinical Excellence,December 2006.
6. EMEA. European Medicines Agencyrecommends first switch fromprescription-only to non-prescriptionfor a centrally authorised medicine.
Press Release. London, 23 October2008.
7. Anonymous. Over-the-countermedicines: in whose best interests?Lancet 2009; 373: 354.
8. Decreto legislativo 24 aprile 2006, n. 219, articoli 96 e 115.
9. Decreto legislativo 24 aprile 2006, n. 219, articolo 87 e Legge 24dicembre 1993, n. 537, articolo 8,comma 10.
10. Decreto legislativo 24 aprile 2006, n. 219, articolo 96.
11. Direttiva 2001/83/CE del ParlamentoEuropeo e del Consiglio del 6novembre 2001 e successivemodificazioni.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
63

RiassuntoIn passato è stata espressa preoccupazione circa il
possibile effetto neurotossico del tiomersal, un compo-sto del mercurio utilizzato come conservante per i vac-cini somministrati durante l’infanzia. Un recente studioha confrontato lo sviluppo neuropsicologico di duegruppi di bambini che avevano ricevuto differenti quan-tità di tiomersal attraverso le vaccinazioni durante iprimi 12 mesi di vita. È stato selezionato un sotto-gruppo dei soggetti arruolati originariamente in un cli-nical trial randomizzato sull’efficacia dei vaccini acellu-lari contro la pertosse iniziato nel 1992-93. Durante ilclinical trial i bambini randomizzati a diversi vaccini ri-sultavano esposti in modo casuale a una dose cumula-tiva di 62,5 o 137,5 mcg di etilmercurio nel primo annodi vita. È stato confrontato l’outcome di 11 test neuro-psicologici standardizzati che venivano somministrati aibambini durante l’orario scolastico. Sono stati analizzatii punteggi medi dei test neuropsicologici nei dominidella memoria e dell’apprendimento, attenzione, fun-zioni esecutive, funzioni visuo-spaziali, linguaggio eabilità motoria per dose di esposizione al tiomersal esesso. Per valutare l’effetto dell’esposizione al tiomersalè stata applicata una regressione lineare multipla e cal-colati i coefficienti di regressione standardizzati. Hannopartecipato allo studio 1403 bambini. Solo due deglioutcome neuropsicologici esaminati sono risultati as-sociati in modo significativo all’esposizione a tiomersal:le bambine con una esposizione a dosi maggiori di tio-mersal avevano punteggi più bassi al test del finger tap-ping e al Boston naming Test. Le associazioni significativetrovate non sono coerenti con quelle trovate da altristudi e sono comunque relative a piccole differenze.Sulla base dei risultati dello studio l’associazione travaccinazioni e deficit neuropsicologici del bambino èimprobabile o clinicamente non rilevante.
AbstractConcern has been expressed on the neurotoxical ef-
fect of thiomersal, a mercury compound used as a pre-servative in vaccines administered during infancy. Wecompared the neuropsychological performance, 10 years
after vaccination, of 2 groups of children exposed ran-domly to different amounts of thiomersal through im-munization. We selected a subset of children from a co-hort of individuals who were enrolled in an efficacytrial of pertussis vaccines in 1992-1993. Children wererandomized during the trial to different vaccines result-ing in two groups randomized to a ethylmercury expo-sure of 62.5 or 137.5 mcg in the first year of life. Wecompared the neuropsychological outcomes of elevenstandardized neuropsychological tests which were ad-ministered to children during school hours. Mean scoresof neuropsychological tests in the domains of memoryand learning, attention, executive functions, visuospatialfunctions, language, and motor skills were compared ac-cording to thiomersal exposure and gender. Standard re-gression coefficients obtained through multivariate lin-ear regression analyses were used as a measure of effect.A total of 1403 children participated in the study.Among the neuropsychological outcomes that were eval-uated, only 2 were significantly associated with thiome-rsal exposure: girls with higher thiomersal intake hadlower mean scores in the finger-tapping Test with thedominant hand and in the Boston naming Test. Wefound two significant associations, in females only,which are not consistent with the results of other stud-ies. The associations found were based on small differ-ences in mean test scores, and their clinical relevance isuncertain and remains to be determined.
IntroduzioneIl tiomersal, un conservante utilizzato ampiamente
nella preparazione dei vaccini, è un composto organicodel mercurio che dopo idrolisi dà luogo ad etilmercu-rio. Alcuni studi condotti sulla progenie di donne espo-ste durante la gravidanza a quantità variabili di metil-mercurio, attraverso la dieta, avevano suggerito chequesta forma organica del mercurio poteva rappresen-tare un rischio per lo sviluppo neuropsicologico delbambino1,2. Assumendo una similitudine tra i due com-posti organici del mercurio, sono stati condotti altristudi su bambini esposti a diverse quantità di tiomer-sal attraverso la somministrazione di vaccini durante
Somministrazione di vaccini contenenti tiomersal e sviluppo neuropsicologico del bambino
BOLLETTINO D’INFORMAZIONE SUI FARMACI | AGGIORNAMENTI |
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
64

AGGIORNAMENTI | SOMMINISTRAZIONE DI VACCINI CONTENENTI TIOMERSAL E SVILUPPO NEUROPSICOLOGICO DEL BAMBINO
l’infanzia per indagarne il possibile effetto sullo svi-luppo neuropsicologico. Nonostante questi studi ab-biano raggiunto risultati contrastanti, l’opinione pub-blica (quella americana in particolare) è stata colpitadalle conclusioni di alcuni di essi che hanno attribuitoal tiomersal contenuto nei vaccini una relazione causalecon autismo, tic motori, ritardo mentale, ritardo del lin-guaggio e sindrome da deficit dell’attenzione ed ipe-rattività3-7. A seguito del vivace dibattito sviluppatosi,numerose agenzie regolatorie, incluse l’EMEA (Agenziaeuropea dei medicinali) per l’Europa e la FDA (Foodand Drug Administration) per gli USA, hanno racco-mandato l’eliminazione di questo conservante dallepreparazioni vaccinali come misura precauzionale8,9.
Nel 1992 era stato condotto in Italia uno studio cli-nico controllato randomizzato per valutare l’efficacia ela sicurezza di due vaccini contro difterite, tetano e per-tosse acellulari (DTaP)10. La composizione dei due vac-cini era differente per il tipo di conservante utilizzato. Ilvaccino DTaP prodotto da Chiron Biocine incluso nellostudio conteneva tiomersal, mentre quello prodotto daGlaxoSmithKline conteneva come conservante 2-poli-fenossietanolo. Durante lo studio clinico randomiz-zato venivano somministrati anche altri vaccini, checontenevano tiomersal, secondo il calendario racco-mandato. I bambini che avevano partecipato a questostudio erano quindi stati esposti in modo randomizzatoa quantità diverse di tiomersal durante l’infanzia. Ap-profittando di questa circostanza è stato condotto un ul-teriore studio sui bambini originariamente parte deltrial di efficacia e sicurezza dei vaccini contro la pertosse,che aveva l’obiettivo di confrontare lo sviluppo neuro-psicologico dei bambini 10 anni dopo l’esposizionerandomizzata a due differenti quantità di tiomersal.
MetodiDegli originari 15.601 bambini partecipanti allo
studio per la valutazione dell’efficacia e della sicurezzadei vaccini contro la pertosse, ne sono stati consideratieleggibili per lo studio 3399 residenti nella Regione
Veneto. La combinazione di vaccini che avevano rice-vuto, che comprendeva 3 dosi di vaccino contro epatiteB e una dose addizionale di difto-tetano all’età di 11mesi, entrambi contenenti tiomersal, ha permesso di di-stinguere due gruppi, uno dei quali era stato esposto aduna dose cumulativa di etilmercurio di 137,5 mcg e l’al-tro di 62,5 mcg entro i primi 12 mesi di vita. I bambinipartecipanti allo studio non hanno ricevuto ulteriorivaccini fino ai 6 anni di vita.
La valutazione neuropsicologica è stata effettuatatra il 2003 e il 2005 quando i bambini che partecipa-vano originariamente allo studio clinico avevano un’etàcompresa tra 10 e 12 anni. La valutazione è stata con-dotta durante l’orario scolastico da psicologi apposita-mente addestrati. La batteria di test neuropsicologici uti-lizzata era composta da 11 test, selezionati sulla base deiprecedenti studi sulla neurotossicità dei composti delmercurio e della presenza di dati normativi italiani ap-propriati per la fascia d’età considerata (10-12 anni).Sono stati esaminati i seguenti domini cognitivi: a. memoria e apprendimento, per mezzo del Ca-
lifornia Verbal Learning Test for Children11 e della Me-moria di cifre (WISC-R)12, che richiedono l’appren-dimento di liste di parole o numeri;
b. attenzione, per mezzo del Continuous PerformanceTest13, un test computerizzato che valuta le abilità diattenzione sostenuta e inibizione;
c. funzioni esecutive, per mezzo del Test del Cifrario(WISC-R) e del Test di fluenza fonemica14, che va-lutano rispettivamente la messa in atto di strategiedi ricerca visiva e l’abilità di accedere al lessico at-traverso un indizio fonemico;
d. funzioni visuo-spaziali, attraverso il Test del Di-segno con cubi (WISC-R), che esamina la capacità diricostruire tridimensionalmente delle immagini bi-dimensionali;
e. linguaggio, per mezzo del Test del Vocabolario edelle Somiglianze (WISC-R), del Boston NamingTest15, e del test di fluenza semantica, che valutanorispettivamente il grado di padronanza lessicale, lefunzioni espressive, la capacità di individuare rela-zioni concettuali, l’abilità di accedere al lessico permezzo di un cue semantico;
f. abilità motorie fini, attraverso il test di velocità discrittura16 e il Finger Tapping Test17.
Durante l’esecuzione dei test lo psicologo ha inoltrerilevato la presenza di tic motori e/o vocali. La pre-senza di disturbi dello spettro autistico è stata indagatadurante l’intervista ai genitori e al pediatra di famiglia,sulla base dei criteri del DSM-IV18.
La dimensione dello studio (700 bambini per
Per il dibattito sviluppatosi, molte
agenzie regolatorie, tra cui EMEA
e FDA, hanno raccomandato
l’eliminazione del tiomersal
dai vaccini in via precauzionale.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
65

AGGIORNAMENTI | SOMMINISTRAZIONE DI VACCINI CONTENENTI TIOMERSAL E SVILUPPO NEUROPSICOLOGICO DEL BAMBINO
gruppo di esposizione, per un totale di 1400 bambini)è stata determinata assumendo che i bambini nelgruppo di esposizione più basso avessero valori medi edeviazioni standard uguali ai corrispondenti valori di ri-ferimento, per ciascuno dei parametri neuropsicolo-gici esaminati, e con l’obiettivo di individuare in un testa due code una differenza tra i due gruppi di almeno il5% del valore medio nel gruppo di controllo, con unaprobabilità di errore di I tipo =0,05 e una potenza deltest -1 =0,90.
Le differenze tra i due gruppi di esposizione per levariabili quantitative sono state valutate con il test diStudent, utilizzando il test di Mann-Whitney per vali-dare i risultati. Le differenze per le variabili categorichesono state analizzate con il test del chi quadrato e il testdella probabilità esatta di Fisher.
Per valutare l’effetto dell’esposizione sul valore delsingolo parametro neuropsicologico, è stata applicata inanalisi separate la regressione lineare multipla, conside-rando come potenziali confondenti: l’età del bambinoal momento del test, il sesso, il peso alla nascita, l’etàgestazionale, il tipo di parto, la durata dell’allattamentoal seno, la composizione familiare, l’età della madre allanascita del bambino, il livello di istruzione e la profes-sione dei genitori, la presenza di malattie del SNC o dialtre malattie croniche nel bambino, l’assunzione di far-maci antistaminici o antiepilettici al momento del test,la somministrazione di immunoglobuline Rh allamadre durante la gravidanza e l’immunizzazione anti-epatite B al momento del parto. I coefficienti di regres-sione standardizzati (SRC) sono stati utilizzati comemisura dell’effetto. Le stesse analisi sono state poi con-dotte anche separatamente nei due sessi.
Secondariamente, i punteggi ai test neuropsicologicisono stati categorizzati in “normali”/“anormali”, e l’ef-fetto dell’esposizione e dei potenziali confondenti su diessi è stato valutato con la regressione logistica.
L’analisi statistica è stata condotta in cieco: la codi-fica dei due gruppi è stata resa nota solo ad analisi ul-timate. Non è stata applicata alcuna correzione per con-fronti multipli, per non ridurre la sensibilità dell’analisi.
RisultatiDei 3399 bambini eleggibili, sono stati invitati 1979
selezionati casualmente. In totale sono stati valutati1403 bambini (697 esposti al livello più alto di etil-mercurio e 706 a quello più basso). La tabella I illustrale caratteristiche sociodemografiche dei due gruppi cheappaiono del tutto simili, fatta eccezione per il peso allanascita, leggermente più basso per il gruppo esposto aduna dose più alta di etilmercurio.
I punteggi medi dei test neuropsicologici erano com-presi nei range normali per tutte le valutazioni. All’a-nalisi univariata i bambini esposti a maggiore quantitàdi etilmercurio avevano una performance peggiore altest del finger tapping, anche se la differenza nel pun-teggio era modesta (differenza media 1,01; IC 95%0,30 -1,73; P=0,006). Non è stata osservata invece al-cuna differenza tra i punteggi medi dei test neuropsi-cologici o nella frequenza dei tic all’analisi multivariata.Nell’analisi multivariata condotta separatamente persesso, è stata osservata una performance peggiore nelBoston naming test (coefficiente di regressione standar-dizzato: -0,16; P=0,025) e nel test del finger tapping conla mano dominante (coefficiente di regressione stan-dardizzato: -0,16; P=0,029), nelle sole femmine. I ri-sultati dell’analisi multivariata per i test neuropsicolo-gici per sesso e nella popolazione totale sono illustratinella tabella II (in grassetto le associazioni significative).
Tutti i punteggi aumentano con il migliorare dellaperformance, fatta eccezione per il test CPT (attenzionesostenuta) nel quale un punteggio minore corrispondead una prova migliore. Un SRC > 0 indica un valore me-dio del punteggio del test più alto nel gruppo espostoa una maggiore dose di tiomersal in confronto algruppo esposto ad una dose minore. Un SRC < 0 indicaun valore medio del punteggio del test più basso nelgruppo esposto a una maggiore dose di tiomersal inconfronto al gruppo esposto ad una dose minore.
L’analisi effettuata categorizzando i punteggi deitest, i valori adeguati al range per l’età e quelli nonadeguati non hanno messo in evidenza alcuna associa-zione significativa con le dosi di tiomersal alle quali ibambini erano stati esposti.
DiscussioneLa valutazione degli effetti del tiomersal sullo svi-
luppo neuropsicologico del bambino a grande distanzadi tempo dall’esposizione è molto complessa. Neglistudi osservazionali è quasi impossibile controllare com-pletamente l’effetto confondente di una serie di variabilie quello di altre distorsioni. Lo studio aveva il vantaggiodi osservare un gruppo di bambini randomizzati a di-
La relazione causale
tra tiomersal presente nei vaccini
e alcuni deficit neuropsicologici
appare improbabile.
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
66
AGGIORNAMENTI | SOMMINISTRAZIONE DI VACCINI CONTENENTI TIOMERSAL E SVILUPPO NEUROPSICOLOGICO DEL BAMBINO
verse quantità di tiomersal in una situazione unica inquesto tipo di studi. Lo scopo principale era quello diesaminare le differenze relative tra i due gruppi espostia diverse quantità di tiomersal, e per questo sono staticonfrontati i punteggi medi ottenuti ai diversi test attra-verso un’analisi multivariata. Con questo approccio ipunteggi ottenuti in un test incluso nell’area del lin-guaggio (Boston naming Test), e in uno nell’area dellamotricità fine (Finger Tapping Test) sono risultati lieve-mente inferiori nei bambini esposti a maggiori quantitàdi tiomersal solo nel sesso femminile. I risultati ottenuti
non sono tuttavia coerenti con il resto della letteraturadisponibile5,6,19.
Nonostante lo studio avesse un elevato livello di va-lidità, il livello di esposizione a etilmercurio dei parte-cipanti non era elevato quanto si poteva osservare negliUSA dove l’uso dei vaccini combinati era modesto, e nonera disponibile un gruppo con esposizione al tiomersaluguale a zero. Inoltre lo studio è stato condotto in unapopolazione sana in accordo con i criteri di eleggibilitàdello studio originario. Per questo motivo la prevalenzadi bambini con peso alla nascita inferiore a 2500 g era
Tabella I. Caratteristiche dei pazienti.
Minore Maggiore Totalequantità quantitàdi tiomersal di tiomersal
Media (DS) o % Media (DS) o % Media (DS) o % P
Età anni 11,76 (0,47) 11,76 (0,46) 11,76 (0,47) 0,9634
Femmine, n (%) 348 (49,29) 345 (49,50%) 693 (49,39%) 0,957
Peso alla nascita, g 3375 (481) 3323 (484) 3349 (483) 0,0414
Età gestazionale, settimane 39,43 (1,48) 39,47 (1,48) 39,45 (1,48) 0,6183
Parto operativo, n (%) 148 (21,20) 159 (22,94) 307 (22,07) 0,439
Durata allattamento al seno, mesi 2,97 (2,60) 2,78 (2,57) 2,88 (2,59) 0,1643
Ordine di nascita 1,57 (0,79) 1,54 (0,72) 1,55 (0,76) 0,5244
Numero di fratelli 2,12 (0,84) 2,06 (0,78) 2,09 (0,81) 0,1748
Genitore single, n (%) 62 (8,78) 57 (8,18) 119 (8,48) 0,702
Scolarità, n (%) 0,268
· Elementare, 4a 4 (0,57) 0 (0,00) 4 (0,29)
· Elementare, 5a 61 (8,66) 56 (8,05) 117 (8,36)
· Media inferiore, 1a 444 (63,07) 443 (63,65) 887 (63,36)
· Media inferiore, 2a 194 (27,56) 197 (28,30) 391 (27,93)
· Media inferiore, 3a 1 (0,14) 0 (0,00) 1 (0,07)
Età della madre alla nascita, anni 30,18 (4,58) 30,12 (4,36) 30,15 (4,47) 0,7956
Madri diplomate, n (%) 68 (9,63) 60 (8,62) 128 (9,13) 0,518
Padri diplomati, n (%) 76 (10,81) 65 (9,42) 141 (10,12) 0,424
Malattie del sist. nervoso centrale, n (%) 13 (1,84) 7 (1,00) 20 (1,43) 0,260
Altre malattie croniche, n (%) 35 (4,96) 28 (4,02) 63 (4,49) 0,440
Trattamento con antistaminici, n (%) 14 (1,98) 13 (1,87) 27 (1,92) 1,000
Trattamento con antiepilettici, n (%) 3 (0,42) 1 (0,14) 4 (0,29) 0,624
Immunoglobuline anti Rh 85 (12,27) 81 (11,84) 166 (12,06) 0,869in gravidanza, n (%)
Vaccinazione epatite B 3 (0,42) 7 (1,00) 10 (0,71) 0,222alla nascita, n (%)
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
67
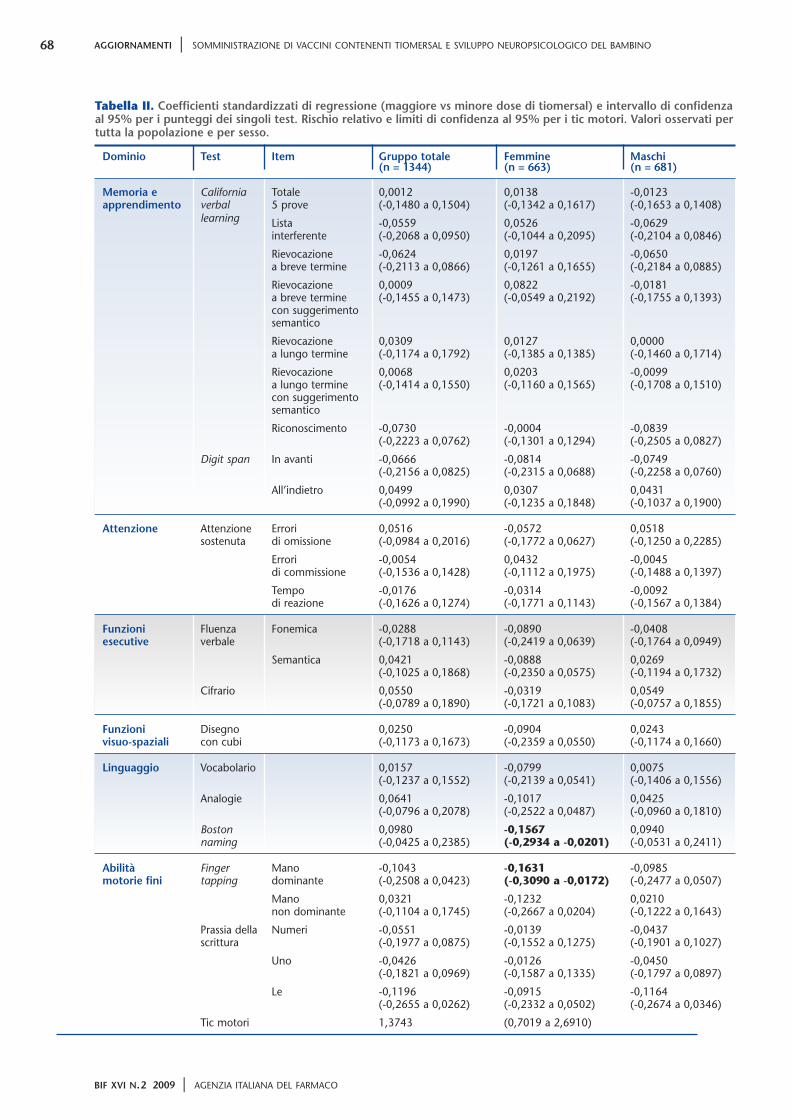
AGGIORNAMENTI | SOMMINISTRAZIONE DI VACCINI CONTENENTI TIOMERSAL E SVILUPPO NEUROPSICOLOGICO DEL BAMBINO
Tabella II. Coefficienti standardizzati di regressione (maggiore vs minore dose di tiomersal) e intervallo di confidenzaal 95% per i punteggi dei singoli test. Rischio relativo e limiti di confidenza al 95% per i tic motori. Valori osservati pertutta la popolazione e per sesso.
Dominio Test Item Gruppo totale Femmine Maschi(n = 1344) (n = 663) (n = 681)
Memoria e California Totale 0,0012 0,0138 -0,0123apprendimento verbal 5 prove (-0,1480 a 0,1504) (-0,1342 a 0,1617) (-0,1653 a 0,1408)
learning Lista -0,0559 0,0526 -0,0629interferente (-0,2068 a 0,0950) (-0,1044 a 0,2095) (-0,2104 a 0,0846)
Rievocazione -0,0624 0,0197 -0,0650a breve termine (-0,2113 a 0,0866) (-0,1261 a 0,1655) (-0,2184 a 0,0885)
Rievocazione 0,0009 0,0822 -0,0181a breve termine (-0,1455 a 0,1473) (-0,0549 a 0,2192) (-0,1755 a 0,1393)con suggerimentosemantico
Rievocazione 0,0309 0,0127 0,0000a lungo termine (-0,1174 a 0,1792) (-0,1385 a 0,1385) (-0,1460 a 0,1714)
Rievocazione 0,0068 0,0203 -0,0099a lungo termine (-0,1414 a 0,1550) (-0,1160 a 0,1565) (-0,1708 a 0,1510)con suggerimentosemantico
Riconoscimento -0,0730 -0,0004 -0,0839(-0,2223 a 0,0762) (-0,1301 a 0,1294) (-0,2505 a 0,0827)
Digit span In avanti -0,0666 -0,0814 -0,0749(-0,2156 a 0,0825) (-0,2315 a 0,0688) (-0,2258 a 0,0760)
All’indietro 0,0499 0,0307 0,0431(-0,0992 a 0,1990) (-0,1235 a 0,1848) (-0,1037 a 0,1900)
Attenzione Attenzione Errori 0,0516 -0,0572 0,0518sostenuta di omissione (-0,0984 a 0,2016) (-0,1772 a 0,0627) (-0,1250 a 0,2285)
Errori -0,0054 0,0432 -0,0045di commissione (-0,1536 a 0,1428) (-0,1112 a 0,1975) (-0,1488 a 0,1397)
Tempo -0,0176 -0,0314 -0,0092di reazione (-0,1626 a 0,1274) (-0,1771 a 0,1143) (-0,1567 a 0,1384)
Funzioni Fluenza Fonemica -0,0288 -0,0890 -0,0408esecutive verbale (-0,1718 a 0,1143) (-0,2419 a 0,0639) (-0,1764 a 0,0949)
Semantica 0,0421 -0,0888 0,0269(-0,1025 a 0,1868) (-0,2350 a 0,0575) (-0,1194 a 0,1732)
Cifrario 0,0550 -0,0319 0,0549(-0,0789 a 0,1890) (-0,1721 a 0,1083) (-0,0757 a 0,1855)
Funzioni Disegno 0,0250 -0,0904 0,0243visuo-spaziali con cubi (-0,1173 a 0,1673) (-0,2359 a 0,0550) (-0,1174 a 0,1660)
Linguaggio Vocabolario 0,0157 -0,0799 0,0075(-0,1237 a 0,1552) (-0,2139 a 0,0541) (-0,1406 a 0,1556)
Analogie 0,0641 -0,1017 0,0425(-0,0796 a 0,2078) (-0,2522 a 0,0487) (-0,0960 a 0,1810)
Boston 0,0980 -0,1567 0,0940naming (-0,0425 a 0,2385) (-0,2934 a -0,0201) (-0,0531 a 0,2411)
Abilità Finger Mano -0,1043 -0,1631 -0,0985motorie fini tapping dominante (-0,2508 a 0,0423) (-0,3090 a -0,0172) (-0,2477 a 0,0507)
Mano 0,0321 -0,1232 0,0210non dominante (-0,1104 a 0,1745) (-0,2667 a 0,0204) (-0,1222 a 0,1643)
Prassia della Numeri -0,0551 -0,0139 -0,0437scrittura (-0,1977 a 0,0875) (-0,1552 a 0,1275) (-0,1901 a 0,1027)
Uno -0,0426 -0,0126 -0,0450(-0,1821 a 0,0969) (-0,1587 a 0,1335) (-0,1797 a 0,0897)
Le -0,1196 -0,0915 -0,1164(-0,2655 a 0,0262) (-0,2332 a 0,0502) (-0,2674 a 0,0346)
Tic motori 1,3743 (0,7019 a 2,6910)
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
68

AGGIORNAMENTI | SOMMINISTRAZIONE DI VACCINI CONTENENTI TIOMERSAL E SVILUPPO NEUROPSICOLOGICO DEL BAMBINO
Bibliografia1. Grandjean P, Weihe P, White RF,
et al. Cognitive deficit in 7-yearchildren with prenatal exposure tomethylmercury. Neurotoxicol Teratol1997; 19: 417-28.
2. Davidson PW, Myers GJ, Cox C, et al.Effects of prenatal and postnatalmethylmercury exposure from fishconsumption on neurodevelopment:outcomes at 66 months of age in theSeychelles child development study.JAMA 1998; 280: 701-7.
3. Institute of Medicine. ImmunizationSafety Review: thimerosal-containingvaccines and neurodevelopmentaldisorders. Washington, DC: NationalAcademy Press; 2001.
4. Geier DA, Geier MR. An assessmentof downward trends inneurodevelopmental disorders in theUnited States following removal ofthimerosal from childhood vaccines.Med Sci Monit 2006; 12: CR231–CR9.
5. Verstraeten T, Davis RL, DeStefano F,et al. Safety of thimerosal-containingvaccines: a two-phased study ofcomputerized health maintenanceorganization databases. Pediatrics2003; 112: 1039-48.
6. Andrews N, Miller E, Grant A, Stowe J, Osborne V, Taylor B.Thimerosal exposure in infants and developmental disorders: a retrospective cohort study in theUnited Kingdom does not support a causal association. Pediatrics 2004;114: 584-91.
7. Geier D, Geier MR.Neurodevelopmental disordersfollowing thimerosal-containingchildhood immunizations: a follow-up analysis. Int J Toxicol 2004; 23: 369-76.
8. European Agency for the Evaluationof Medicinal Products. EMEA publicstatement on thiomersal containingmedicinal products, July 8, 1999.www.emea.europa.eu/pdfs/human/press/pus/2096299EN.pdf (accesso verificato il 25/03/2009).
9. Centers for Disease Control andPrevention. Thimerosal in vaccines: a joint statement of the AmericanAcademy of Pediatrics and the PublicHealth Service. MMWR Morb MortalWkly Rep 1999; 48: 563-5.
10. Greco D, Salmaso S, Mastrantonio P,et al. A controlled trial of twoacellular vaccines and one whole-cellvaccine against pertussis. N Engl J Med 1996; 334: 341-8.
11. Delis DC, Kramer JH, Kaplan E, OberBA. The California verbal learningtest-children version. New York, NY:Psychological Corp; 1994.
12. Wechsler D. WISC-R: scala diintelligenza Wechsler per bambiniriveduta: manuale. Firenze:Organizzazioni Speciali; 1998.
13. Conners CK; Multi-Health SystemsStaff. Conners’ ContinuousPerformance Test. Toronto, Canada:Multi-Health Systems; 2000.
14. Bisiacchi PS, Cendron M, GugliottaM, Tressoldi P, Vio C. Batteria divalutazione neuropsicologica per l’etàevolutiva. Trento: Erickson; 2005.
15. Kaplan E, Goodglas H, Weintraub S.Boston naming test. Philadelphia, PA: Lea & Febinger; 1983.
16. Tressoldi PE, Cornoldi C. Batteria perla valutazione della scrittura e dellacompetenza ortografica nella scuoladell’obbligo. Firenze: OrganizzazioniSpeciali; 1991.
17. Nussbaum NL, Bigler ED. Halstead-Reitan neuropsychological testbatteries for children. In: Reynolds C,ed. Handbook of clinical childneuropsychology. 2nd ed. New York,NY: Plenum Press; 1997.
18. American Psychiatric Association.Diagnostic and statistical manual of mental disorders. 4th ed.Washington, DC: AmericanPsychiatric Association; 1994.
19. Thompson WW, Price C, Goodson B,et al. Early thimerosal exposure andneuropsychological outcomes at 7 to 10 years. N Engl J Med 2007; 357: 1281-92.
modesta. È anche possibile che alcune famiglie abbianodeclinato l’invito a partecipare allo studio per la presenzadi problemi cognitivi del bambino senza dichiararloesplicitamente. Questa circostanza potrebbe aver dimi-nuito la prevalenza di difetti cognitivi ma non vi è ra-gione di pensare che la loro distribuzione sia stata sbi-lanciata per gruppo di esposizione al tiomersal. Infineera bassa anche la proporzione di bambini che avevanoricevuto la vaccinazione contro l’epatite B alla nascita.
Questo studio si aggiunge alle evidenze disponibilicirca l’effetto del tiomersal contenuto nei vaccini sullosviluppo neuropsicologico del bambino. La mancatacoerenza dei risultati dello studio con quelli degli altridisponibili e le piccole differenze osservate suggerisconoche una relazione causale tra etilmercurio sommini-strato attraverso le vaccinazioni dell’infanzia e alcuni de-ficit neuropsicologici del bambino sia improbabile op-pure clinicamente non rilevante.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
69

RiassuntoL’uso di preparati ormonali estro-progestinici a scopo
contraccettivo si è dimostrato sicuro ed efficace anche se,naturalmente, non è del tutto esente da rischi. In parti-colare, la presenza di alcuni tipi di trombofilia e la con-temporanea assunzione di preparati anticoncezionaliaumenta di molto il rischio di eventi trombotici. La di-sponibilità di dati scientifici troppo scarsi e imprecisi perconsentire comportamenti omogenei e condivisi tra imedici ha fatto sentire la necessità di promuovere unaconferenza di consenso durante la quale affrontare gliaspetti più rilevanti di questo tema. Gruppi di esperti in-vitati dal Comitato Promotore hanno prodotto una sin-tesi delle prove scientifiche, mettendola a disposizionedi una giuria multidisciplinare composta da rappresen-tanti della comunità scientifica e delle donne. Ogni rac-comandazione della giuria è accompagnata – nella ver-sione integrale del documento – da un testo esplicativoconsultabile sul sito dell’Istituto Superiore di Sanità.
La giuria ha concluso che la contraccezione con estro-progestinici è un metodo ampiamente studiato, e il cuibilancio benefici/rischi è decisamente favorevole. Il ri-schio di trombosi nelle donne in età fertile che assu-mono la pillola è molto basso, ed è variabile in rapportoa diversi fattori individuali e ambientali. L’esecuzione ditest genetici e di test della coagulazione non è racco-mandata di routine, mentre è fortemente auspicabile for-nire alle donne che intendono assumere la pillola unaadeguata informazione riguardo a benefici e rischi.
AbstractMany large studies showed that the use of contra-
ceptive pill is effective and that adverse events are rela-tively rare. The most common adverse event that maycause potentially serious consequences on a woman’shealth is venous thrombosis. Women affected by somethrombophylic syndromes may especially have an in-creased risk of venous thrombosis if they take the pill.Available data are not robust enough to support consis-tent behaviours among doctors with respect to this mat-ter, therefore the Istituto Superiore di Sanità promoteda consensus conference about it. Groups of experts in-
vited by the Promoting Committee produced docu-ments summarising available evidence, which were ap-praised by a multidisciplinary jury including healthprofessionals and women’s representatives. Each rec-ommendation comes with an explanatory comment,available on the Istituto Superiore di Sanità website.
The jury’s conclusions were that the estroprogestinpill is a widely tested contraceptive method, and its ben-efit/risk balance is largely favourable. The risk of venousthrombosis among fertile women taking the pill is verylow, and may vary depending on individual as well asenvironmental factors. Routine use of coagulation testsand of genetic testing is not recommended, while it isstrongly recommended to provide appropriate infor-mation about benefits and risks to women who want totake the contraceptive pill.
L’uso di preparati ormonali estro-progestinici (EP) ascopo contraccettivo viene considerato tra gli interventi disalute pubblica più importanti del ventesimo secolo, tantoche la contraccezione e la pianificazione familiare sonodiventate parte integrante della salute della donna.
La contraccezione ormonale si è ripetutamente dimo-strata come un intervento dotato di grande costo-efficaciarelativamente all’allocazione delle risorse sanitarie1. Negliultimi 30 anni vi sono stati significativi progressi nello svi-luppo di nuove tecnologie contraccettive efficaci, ottima-mente tollerate e con sempre meno rischi per la donna,tra cui una transizione dai contraccettivi orali ad alto do-saggio verso quelli a minore dosaggio ormonale2.
I benefici dell’utilizzazione di EP a scopo contraccet-tivo riguardano non solo le gravidanze indesiderate evi-tate, ma la salute della donna in generale, in quanto tra ivantaggi non contraccettivi degli EP si annoverano la ri-duzione dei sintomi associati al ciclo mestruale, della du-rata del sanguinamento e delle perdite ematiche, e unariduzione del rischio di patologie della sfera riproduttiva1,oltre che la riduzione della frequenza di neoplasie mali-gne (45 ogni 100.000 donne/anno nelle donne tra i 50 ei 59 anni)3 e di carcinoma ovarico e dell’endometrio (ri-spettivamente del 40% e del 50%)4.
In Italia circa il 16% delle donne tra i 15 e i 44 anni
Il punto sulle complicanze trombotiche associate all’uso dei contraccettivi estro-progestinici
BOLLETTINO D’INFORMAZIONE SUI FARMACI | AGGIORNAMENTI |
BIF XVI N.3 2009 | AGENZIA ITALIANA DEL FARMACO
70

AGGIORNAMENTI | IL PUNTO SULLE COMPLICANZE TROMBOTICHE ASSOCIATE ALL’USO DEI CONTRACCETTIVI ESTRO-PROGESTINICI
(2,2 milioni) assume EP a scopo anticoncezionale e, tra il1982 e il 2007, parallelamente all’incremento nell’utilizzodi EP (dal 5,2% al 16,1% delle donne in età fertile) si èavuto un decremento delle interruzioni volontarie di gra-vidanza del 45,9%5.
I contraccettivi orali EP sono stati oggetto di valuta-zioni a lungo termine e sono probabilmente tra i farmacimaggiormente studiati. Le loro tollerabilità e sicurezzasono ampiamente provate, specie in relazione ai rischiconnessi con una gravidanza indesiderata. Si può affer-mare che la contraccezione ormonale combinata può es-sere ininterrottamente utilizzata senza grossi rischi indonne sane non fumatrici per tutta l’età riproduttiva, finoalla menopausa1.
I principali rischi associati all’uso cronico di contrac-cettivi orali consistono in un aumento della frequenza ditumore al seno (3 casi in più ogni mille donne che fannouso di contraccettivi orali per 8 anni), al collo dell’utero (2casi per mille in più) e al fegato (0,7 casi per mille in più)6.I dati disponibili mostrano, tuttavia, che il rischio di ma-lattie neoplastiche associato all’uso di contraccettivi oraliè molto basso, e comunque non condiziona la sopravvi-venza delle donne che ne fanno uso rispetto a quelle chenon li assumono6,7.
Tra le rare complicanze associate all’uso di EP, la piùcomune è la trombosi venosa (TV). I dati epidemiologiciin nostro possesso per quantificare tale rischio sono tut-tavia molto scarsi. Le casistiche a nostra disposizione ri-guardo alla frequenza naturale della TV tra le donne in etàfertile provengono da studi su popolazioni del nord Eu-ropa e del nord America, le cui caratteristiche le rendonoscarsamente trasferibili alla nostra realtà8,9. Pur essendonoto che l’assunzione di EP aumenta di molto (in terminirelativi) il rischio di TV, è difficile stimare con precisionein termini assoluti il numero atteso di eventi tromboticivenosi nell’ambito delle donne italiane che assumono EPa scopo contraccettivo, anche in considerazione del fattoche si tratta di un evento molto raro.
ControversieUn aspetto emergente e controverso legato all’utilizzo
di contraccettivi EP riguarda l’utilità di sottoporre ledonne in procinto di assumere la pillola anticoncezionalea test diagnostici – anche genetici – con lo scopo di ac-certare l’eventuale presenza di una anomalia trombofilica,intesa come condizione congenita o acquisita predispo-nente a trombosi. Se da un lato il rischio assoluto di TV as-sociato all’uso di EP in età riproduttiva è basso, lapossibilità di un tale evento in giovane età comporta rica-dute non trascurabili in termini di successiva morbilità,oltre alla necessità di eseguire una profilassi antitrombo-
tica in caso di situazioni a rischio nel corso della vita (peresempio una gravidanza). D’altro canto l’opportunità dieseguire uno screening di laboratorio per identificareeventuali anomalie trombofiliche presenta delle incer-tezze, non essendo chiaro il bilancio tra benefici (ridu-zione dell’incidenza di TV in corso di terapia con EP,identificazione di soggetti a rischio) e inconvenienti (ec-cessiva medicalizzazione delle donne, controindicazioneingiustificata all’uso della pillola, oneri economici nonproporzionati al risultato). Infine, la disponibilità di testche consentono una diagnosi genetica solleva importantiquestioni di carattere etico per le implicazioni che una talediagnosi comporta per la donna e per i suoi familiari.
Per affrontare una problematica come questa, ricca diimplicazioni non limitate solamente a un ambito stretta-mente scientifico, è stata promossa una Conferenza diconsenso, metodo che consiste nella stesura di racco-mandazioni da parte di un panel/giuria al termine di undibattito pubblico, nel corso del quale gruppi di espertisintetizzano le conoscenze scientifiche disponibili.
La Conferenza Nazionale di Consenso La Conferenza Nazionale di Consenso “Prevenzione
delle complicanze trombotiche associate all’uso di estro-progestinici nelle donne in età riproduttiva” si è svolta il18 e 19 settembre 2008 a Roma, presso l’Istituto Superioredi Sanità (ISS), promossa dal Sistema Nazionale LineeGuida dell’ISS e dal CeVEAS (Centro per la Valutazionedell’Efficacia della Assistenza Sanitaria) della Azienda USLdi Modena. I lavori preparatori della Conferenza sono ini-ziati nel novembre del 2007 e si sono svolti secondo loschema riportato in figura 1.
Il documento di consenso si compone di 8 capitoli,corrispondenti alle aree tematiche più rilevanti nell’am-bito dell’argomento affrontato, strutturati in forma di do-mande alle quali fanno seguito gli “statements” dellagiuria. Al testo di ciascuna raccomandazione qui sotto ri-portato fa seguito, nella versione integrale del documento,un testo di accompagnamento in cui vengono discusse leprove dalle quali le raccomandazioni sono state ricavate ele problematiche affrontate nelle relazioni degli esperti in-vitati alla conferenza.
La versione integrale del documento, con bibliografiacompleta, è scaricabile dal sito del Piano Nazionale LineeGuida dell’Istituto Superiore di Sanità: www.snlg-iss.it
� Quali sono i rischi associati all’uso di estro-progestinici?
1.1 La contraccezione con EP, utilizzata su scala mondialeda milioni di donne da oltre 40 anni, è efficace e bentollerata, con bassa frequenza di complicanze.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.3 2009
71

AGGIORNAMENTI | IL PUNTO SULLE COMPLICANZE TROMBOTICHE ASSOCIATE ALL’USO DEI CONTRACCETTIVI ESTRO-PROGESTINICI
1.2 Tra le rare complicanze associate alla contraccezionecon EP, la più rilevante è la TV. Nelle donne in età fer-tile si tratta di un evento molto raro. Pur non essendodisponibili dati certi per l’Italia, è stimabile comples-sivamente attorno a 4-7 casi l’anno ogni 10.000 donne,1-2 dei quali sono attribuibili alla pillola EP.
1.3 La contraccezione EP produce anche un aumentomolto piccolo del rischio di trombosi arteriosa, sti-mabile tra 0,06 e 0,4 casi/anno ogni 10.000 donne ri-spetto a un rischio di base di circa 2 casi ogni 10.000donne in età fertile.
� Quali sono i fattori che condizionano l’entitàdel rischio trombotico venoso nelle donne che assumono contraccettivi EP?
2.1 I principali fattori che aumentano il rischio di TV, nelledonne che assumono contraccettivi EP, sono:fattori individuali: età, precedenti episodi trombo-tici, familiarità, obesità (BMI, body mass index ≥30),trombofilia (aumento della tendenza alla trombosicausata da ipercoagulabilità del sangue di origine con-genita o acquisita);fattori intercorrenti: interventi chirurgici, immobilitàprolungata, traumi, puerperio (le prime 4-6 settimane).
2.2 Il rischio di TV con i contraccettivi EP di “terza gene-razione” (contenenti desogestrel o gestodene comeprogestinico) è circa doppio rispetto a quelli di “se-conda generazione” (contenenti levonorgestrel).
2.3 Il rischio di TV associato all’uso di EP è maggiore du-rante il primo anno in cui una donna inizia per laprima volta ad assumere un qualunque tipo di con-traccettivo EP.
� Quali strategie raccomandare per ridurre le complicanze trombotiche?
3.1 Prima di iniziare una contraccezione con EP si racco-manda:
la raccolta di una anamnesi personale e familiare conparticolare attenzione agli eventi trombotici;l’offerta attiva di informazione e la discussione conla donna sul rischio individuale di trombosi e sullapossibilità di ridurlo.
3.2 Non si raccomanda, né prima di prescrivere un con-traccettivo EP né durante l’uso, l’esecuzione routinariadi:
esami ematochimici generici;test generici di coagulazione;test specifici per trombofilia (compresi i test genetici).
3.3 Anche in presenza di anamnesi familiare positiva pereventi trombotici, al fine di prescrivere una contracce-zione EP, non si raccomanda l’esecuzione dei test spe-cifici per trombofilia.
Figura 1. Attività della Conferenza di Consenso.
Novembre 2007-Aprile 2008
• Scrittura del protocollo
• Identificazione dei quesiti
• Scrittura del regolamento della giuria
• Individuazione dei membri della giuria e degli esperti
• Produzione di una metodologia predefinita per il reperimento, la selezione e la sintesi delle prove a supporto delle relazioni da presentare alla Conferenza
• Convocazione degli esperti e loroorganizzazione in gruppi di lavoro sui temi da affrontare
Aprile-Giugno 2008
• Produzione da parte del Comitato promotore di revisioni della letteratura su ciascun argomento oggetto dei quesiti
• Invio del materiale bibliografico agli esperti
• Produzione delle relazioni da parte degli esperti
30 Luglio 2008
• Consegna delle relazioni degli esperti alla giuria
18-19 Settembre 2008(Istituto Superiore di Sanità, Roma)
• Incontro pubblico di presentazione delle relazioni degli esperti presso l’Istituto Superiore di Sanità, Roma
• Riunione a porte chiuse della giuria
• Presentazione pubblica delle raccomandazioni (Roma, Istituto Superiore di Sanità - 19 settembre 2008)
BIF XVI N.3 2009 | AGENZIA ITALIANA DEL FARMACO
72

AGGIORNAMENTI | IL PUNTO SULLE COMPLICANZE TROMBOTICHE ASSOCIATE ALL’USO DEI CONTRACCETTIVI ESTRO-PROGESTINICI
3.4 Al momento della prescrizione, si raccomanda un con-traccettivo a minor rischio trombotico (progestinicodi II generazione con 20-30 mcg di estrogeno)
� Quali test sono disponibili per l’identificazionedel rischio trombotico e con quale validità?
4.1 Rispetto ai più comuni test ematochimici, quelli spe-cifici di trombofilia presentano globalmente una mag-giore complessità che si associa a una più alta variabi-lità dei loro risultati.Nella pratica corrente è stato riscontrato un elevatotasso di errore diagnostico per ridurre il quale se ne rac-comanda l’esecuzione solo in laboratori qualificati.
4.2 In aggiunta a questi limiti, lo scarso valore predittivodei test di predisposizione genetica o acquisita è il mo-tivo per cui non se ne raccomanda l’uso di routine. Inconsiderazione dell’alta utilizzazione, il più delle volteinappropriata, dei test di predisposizione genetica sene elencano i tipi e le loro caratteristiche principali.
4.3 I seguenti test identificano una predisposizione geneticaalla trombosi:1. mutazione del fattore V Leiden (eterozigote/omozi-
gote);2. mutazione della protrombina G20210A (eterozi-
gote/omozigote);3. deficit di proteina C;4. deficit di proteina S;5. deficit di antitrombina.
Le prime due alterazioni sono relativamente frequentinella popolazione generale (3% circa ciascuna) maaumentano di poco il rischio assoluto di trombosi. Lealtre tre alterazioni sono invece molto più rare (preva-lenza dallo 0,02% allo 0,5%) ma aumentano più de-cisamente il rischio assoluto.Altri test disponibili non identificano condizioni ge-netiche per le quali sia documentato un aumentato ri-schio trombotico e sono pertanto in ogni caso sconsi-gliati.
4.4 I seguenti test identificano una predisposizione acquisitaalla trombosi:1. anticoagulante tipo lupico (LAC);2. anticorpi antifosfolipidi;3. elevati livelli di fattore VIII;4. omocisteinemia.
Questi fattori di rischio sono più frequenti di quelliprecedenti e comportano un aumento di rischio va-riabile, maggiore per i primi due e minore per l’ipero-mocisteinemia e il fattore VIII alto.
� Quali sono le implicazioni cliniche, psicologichee organizzative in caso di una applicazione su vasta scala dei test per la valutazione del rischio trombotico?
5.1 Sul piano clinico, la loro scarsa predittività li rende arischio di sovradiagnosi e di eccessiva medicalizza-zione, in caso di applicazione su vasta scala.Un risultato negativo potrebbe essere falsamente ras-sicurante. Un risultato positivo potrebbe scoraggiarel’uso di contraccezione EP in donne che potrebberogiovarsene.
5.2 In particolare, la mutazione eterozigote del fattore VLeiden o della protrombina (alterazioni che rappre-sentano le mutazioni più frequenti) e la carenza diproteina C costituiscono un rischio da confrontare coipotenziali benefici della contraccezione EP da valutarecaso per caso.Invece, il deficit congenito di antitrombina, la omozi-gosi e la doppia eterozigosi (fattore V e protrombina)nonché i difetti multipli costituiscono alterazionimolto rare che comportano un rischio trombotico piùalto, tale da sconsigliare l’uso di EP. Infine, per quanto riguarda la carenza congenita diproteina S, non ci sono informazioni sufficienti peruna chiara raccomandazione.
5.3 Sul piano psicologico, l’esecuzione e l’esito positivodei test sembrano non avere un impatto avverso nellamaggioranza delle donne in contesti nei quali era for-nito un adeguato counselling pre e post test.Pertanto, è fortemente raccomandato offrire un coun-selling adeguato nel momento precedente al test e almomento della comunicazione del risultato.
5.4 Un medico che applicando le raccomandazioni diquesta conferenza di consenso prescriva la contracce-zione EP senza richiedere l’esecuzione di test di predi-sposizione genetica segue una buona pratica clinica.Sul piano organizzativo, l’inappropriata diffusione diquesti test comporta costi non giustificati per il SSN eper i cittadini.
� Che informazione dare alle donne che intendonoiniziare una contraccezione EP sul rischio di TV?
6.1 Occorre informare la donna sui seguenti argomenti:la TV in età fertile è molto rara;la contraccezione EP aumenta il rischio relativo dicirca 2 volte ma il rischio assoluto rimane molto pic-colo (1-2 casi all’anno ogni 10.000 donne; a titolodi confronto, il rischio di TV in gravidanza è intornoa 6 casi ogni 10.000 gravidanze);il rischio non è uguale in tutte le donne;il rischio è più elevato durante il primo anno d’uso;il rischio non è uguale per tutti i contraccettivi EP;
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.3 2009
73

AGGIORNAMENTI | IL PUNTO SULLE COMPLICANZE TROMBOTICHE ASSOCIATE ALL’USO DEI CONTRACCETTIVI ESTRO-PROGESTINICI
interventi chirurgici, traumi e immobilità prolungataaumentano il rischio e richiedono che la donna se-gnali al medico che sta assumendo EP;gli stili di vita salutari riducono il rischio trombotico;i test per individuare la predisposizione al rischiosono molto usati ma non sono raccomandati.
6.2 Tutte queste informazioni andrebbero date con l’in-tento di facilitare una decisione consapevole da partedella donna riguardo alla prescrizione sia di EP che dieventuali test di predisposizione.
� Quale informazione è necessaria per le donnepositive ai test specifici per trombofilia?
7.1 In caso di avvenuta esecuzione di test specifici pertrombofilia, occorre informare la donna sui seguentiargomenti:
complessità, variabilità e possibilità di errore dia-gnostico;scarso valore predittivo;rischio di rinuncia alla contraccezione EP;possibilità di falsa rassicurazione e di sovradiagnosi;misure di prevenzione della trombosi.
7.2 Si raccomanda in ogni caso che tutte le informazionisiano offerte attivamente prima della eventuale esecu-zione del test.
� Quali misure di politica sanitaria sono auspicabili per la prevenzione delle complicanze trombotichenelle utilizzatrici di EP?
8.1 Considerata la carenza di conoscenze epidemiologi-che, in particolare italiane, su molti aspetti affrontatidalla Conferenza di Consenso, si raccomanda l’esecu-zione di studi epidemiologici sul rischio tromboticovenoso e sui fattori che lo influenzano e in particolaresulla contraccezione EP.Tali studi dovrebbero rappresentare una delle prioritàdel sistema sanitario per il finanziamento della ricercaindipendente.
8.2 In considerazione del minor rischio trombotico attri-buibile ai contraccettivi EP a basso contenuto estroge-nico e contenenti levonorgestrel, si raccomanda larevisione della politica di rimborsabilità con l’inseri-mento in fascia A dei prodotti a minor rischio.
8.3 In considerazione della grande variabilità dei risultatie del tasso di errore dei test di trombofilia, si racco-manda l’attivazione di politiche di accreditamento e dicontrollo di qualità dei laboratori a livello regionale.
8.4 È fondamentale che le autorità sanitarie italiane si im-pegnino in una campagna di comunicazione per infor-mare i professionisti sanitari sulle presenti raccomanda-zioni, in particolare in relazione all’uso inappropriato deitest specifici di predisposizione genetica e acquisita.
Comitato promotoreIstituto Superiore di Sanità, Roma; CeVEAS - Centro per la Valutazione dell’Efficaciadell’Assistenza Sanitaria, AUSL di Modena
Comitato scientificoI. Martinelli (Milano), E. Arisi (Trento), S. Coccheri (Bologna), V. De Stefano (Roma), E. Grandone (S. Giovanni Rotondo), P. Simioni (Padova), F. Facchinetti (Modena)
Gruppo di lavoroInquadramento epidemiologicoRelatori: E. Arisi (Trento); I. Martinelli, (Milano).Coautori: A. Volpe (Modena); R. Abbate (Firenze); F. Parazzini (Milano); A. Tosetto (Vicenza); P. Bucciarelli (Milano); G. Castaman (Vicenza); F. Facchinetti (Modena).Quali i possibili/auspicabili criteri generali perridurre l’incidenza di complicanze tromboticheRelatori: B. Cosmi (Bologna); E. D’Aloia(Cagliari); V. De Stefano (Roma). Coautori: A.M. Paoletti (Cagliari); E. Grandone (S. Giovanni Rotondo); F. Nonino (Modena); M. R. Di Tommaso (Firenze); M. Marietta(Modena); C. Benedetto (Torino); G. B. Allais (Torino)Vantaggi e limiti della diagnostica per trombofiliaRelatori: V. Bruni (Firenze); C. Legnani(Bologna). Coautori: A. Tripodi (Milano); D. Tormene (Padova); F. Fruzzetti (Pisa); L. Del Pup (Aviano); A. Cagnacci (Modena); A. Zambon (Padova); S. Pagliani (Milano)Implicazioni di una eventuale applicazione sularga scala di diagnostica di laboratorio pertrombofiliaRelatori: P.E. Ricci-Bitti (Bologna); S. Panico(Napoli). Coautori: M. Margaglione (Foggia);T. Frusca (Brescia); P. Simioni (Padova); P. Martinelli (Napoli); R. Nappi (Pavia); P. Mandich (Genova); G. Cavalli (Milano); A. Tranquilli (Ancona).
GiuriaR. Satolli (Presidente, Milano); S. Coyaud (Milano); P. Crosignani (Milano); G. Domenighetti (Canton Ticino, CH); S. Donati (Roma); S. Fargion (Milano); P. Iaccarino (Napoli); L. Luzzatto (Firenze); M. Maggini (Roma); N. Magrini (Modena); P. M. Mannucci (Milano); A.M. Messa (Roma); R. Michieli (Mestre); G. Palareti (Bologna); S. Quadrino (Torino)
BIF XVI N.3 2009 | AGENZIA ITALIANA DEL FARMACO
74

Bibliografia1. Blumenthal PD, Edelman A.
Hormonal contraception. ObstetGynecol 2008; 112: 670-84.
2. WHO Medical Eligibility Criteria for Contraceptive Use (WHO Third Edition, 2004) World Health Organization, Genevawww.who.int/reproductivehealth/publications/mec/ (accesso verificato il 20/05/2009).
3. Hannaford PC, Selvaraj S, Elliott AM,et al. Cancer risk among users of oral contraceptives: cohort datafrom the Royal College of GeneralPractitioner’s oral contraceptionstudy. BMJ 2007; 335: 651-8.
4. Sherif K. Benefits and risks of oralcontraceptives. Am J Obstet Gynecol1999; 180: S343-8.
5. Relazione del Ministro della Salutesulla attuazione della leggecontenente norme per la tutela socialedella maternità e per l’interruzione volontaria di gravidanza (Legge 194/78)www.ministerosalute.it/imgs/C_17_pubblicazioni_804_allegato.pdf(accesso verificato il 20/05/2009).
6. Burkman R, Schlesselman JJ, Zieman M. Safety concerns andhealth benefits associated with oralcontraception. Am J Obstet Gynecol2004; 190 (4 Suppl): S5-22.
7. Vessey M, Painter R, Yeates D.Mortality in relation to oralcontraceptive use and cigarettesmoking. Lancet 2003; 362: 185-91.
8. Samuelsson E, Hagg S. Incidence ofvenous thromboembolism in youngSwedish women and possiblypreventable cases among combinedoral contraceptive users. Acta ObstetGynecol Scand 2004; 83: 674-81.
9. Silverstein MD, Heit JA, Mohr DN, et al. Trends in the incidence of deepvein thrombosis and pulmonaryembolism: a 25-year population-based study. Arch Intern Med 1998;158: 585-93.
AGGIORNAMENTI | IL PUNTO SULLE COMPLICANZE TROMBOTICHE ASSOCIATE ALL’USO DEI CONTRACCETTIVI ESTRO-PROGESTINICI
Una recente sentenza della Corte di giustiziaeuropea ha chiarito ulteriormente quale sia lacorretta interpretazione della definizione dimedicinale che è stata stabilita dalle direttivecomunitarie e poi recepita a livello nazionale nel“Codice sui medicinali”.Decreto legislativo 24 aprile 2006, n. 219 – Articolo 1,comma 1a. prodotto medicinale o medicinale, di seguito
indicato con il termine «medicinale»:1. ogni sostanza o associazione di sostanze
presentata come avente proprietà curative o profilattiche delle malattie umane;
2. ogni sostanza o associazione di sostanze che può essere utilizzata sull’uomo o somministrataall’uomo allo scopo di ripristinare, correggere o modificare funzioni fisiologiche, esercitandoun’azione farmacologica, immunologica o metabolica, ovvero di stabilire una diagnosimedica.
La Corte precisa che:la norma di salvaguardia (in caso di dubbio, iprodotti di incerta classificazione sono anch’essisoggetti alla disciplina sui medicinali; vd. d.lgs.219/06, art. 2, comma2) non si applica ad unprodotto la cui qualità di medicinale per funzione non sia scientificamente dimostrata, pur non potendoessere esclusa;i criteri delle modalità d’uso di un prodotto,dell’ampiezza della sua diffusione, della conoscenzache ne hanno i consumatori e dei rischi che possonoderivare dalla sua utilizzazione sono ancora rilevantiper stabilire se tale prodotto sia compreso nelladefinizione di medicinale per funzione;
esclusi i casi di sostanze o composizioni destinate astabilire una diagnosi medica, un prodotto non puòessere considerato come medicinale ai sensi di taledisposizione quando, tenuto conto della suacomposizione – compreso il dosaggio di sostanzeattive – e in condizioni normali di uso, non è idoneoa ripristinare, correggere o modificare in modosignificativo funzioni fisiologiche dell’uomo,esercitando un’azione farmacologica, immunologica o metabolica.
La sentenza è stata pronunciata a seguitodell’istanza presentata dalla Corte amministrativafederale tedesca al fine di dirimere una controversiarelativa ad un prodotto che la ditta titolare intendevacome integratore alimentare e che, viceversa, era stato considerato quale medicinale dalle autoritàcompetenti. Nel dispositivo della sentenza la Corte rammenta cheper stabilire se un prodotto rientri nella definizione dimedicinale per funzione … le autorità nazionali …devono decidere caso per caso, tenendo conto di tuttele caratteristiche del prodotto, tra le quali, inparticolare, la composizione, le proprietàfarmacologiche, immunologiche o metaboliche qualirisultano allo stato attuale delle conoscenze scientifiche,le modalità d’uso, l’ampiezza della sua diffusione, laconoscenza che ne hanno i consumatori e i rischi chepossono derivare dalla sua utilizzazione mentre daparte dell’Avvocato generale viene sottolineatal’esigenza che le autorità nazionali applichino ledisposizioni sui medicinali solo qualora abbianoaccertato positivamente, alla luce del rispettivo statodell’arte in campo scientifico, che il prodotto inquestione sia effettivamente un medicinale.
Definizione di medicinaleA proposito di…
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.3 2009
75

Effetti renali e retinici di enalapril e losartan nel diabete di tipo 1 La nefropatia e la retinopatia sono importanti com-
plicanze del diabete di tipo 1 (DT1). Non è chiaro tutta-via se la loro progressione sia rallentata dalla sommini-strazione precoce di farmaci attivi sul sistemarenina-angiotensina (SRA). Per cercare di rispondere aquesta domanda Mauer et al. hanno condotto uno studiomulticentrico, controllato verso placebo, su 285 soggettinormotesi con DT1 e normoalbuminuria. I pazienti, as-segnati in modo casuale a ricevere losartan (LOS; 100mg/die), enalapril (ENA; 20 mg/die) o placebo (PLA),sono stati seguiti per 5 anni. Come end point primarioper la nefropatia è stata considerata la variazione della fra-zione di volume glomerulare occupata dal mesangio(FVGM) determinata su campioni bioptici. Per la retino-patia, l’end point è stato la progressione misurata su unascala di gravità di due o più gradi. Le biopsie renali e laraccolta dei reperti retinici sono state completate, rispet-tivamente, nel 90% e nel 82% dei casi. Le variazionidella FVGM nei 5 anni di follow-up non hanno mostratodifferenze significative tra il gruppo PLA (0,016 U) ri-spetto a quello ENA (0,005 U; P=0,38) o LOS (0,026 U;P=0,26); né il trattamento ha mostrato benefici per altrevariabili della struttura renale prese in considerazione. Lamicroalbuminuria ha avuto un’incidenza cumulativa a 5anni del 6% nel gruppo PLA mentre è stata superiore nelgruppo LOS (17%; P=0,01) ma non in quello ENA (4%;P=0,96). La progressione della retinopatia si è invece ri-dotta, in confronto al placebo, del 65% con ENA (OR0,35; IC 95% 0,14-0,85) e del 70% con LOS (OR 0,30; IC95% 0,12-0,73), indipendentemente dal cambiamentodei valori pressori.
Conclusioni. Il blocco precoce del SRA nei pa-zienti con DT1 rallenta la progressione della retino-patia ma non quella della nefropatia. Nell’editorialeche accompagna l’articolo viene evidenziato comequesti dati modifichino una convinzione consoli-data (l’effetto favorevole dell’inibizione del SRA intutti gli stadi della nefropatia diabetica) e siano innetto contrasto rispetto a quanto accade negli stadiavanzati delle complicanze diabetiche; fase in cui l’i-nibizione del SRA attenua il deterioramento della
funzione renale senza influenzare la progressionedella retinopatia. Riguardo alla prevenzione dellanefropatia, quindi, i risultati di Mauer et al. dovreb-bero sconsigliare il trattamento con farmaci attivisul SRA nei pazienti con DT1 normotesi e normoal-buminurici. In questi stessi soggetti è possibile otte-nere, invece, un effetto favorevole sull’evoluzionedelle alterazioni retiniche, se esse sono inizialmenteminime o del tutto assenti.
Mauer M, Zinman B, Gardiner R, et al. Renal and retinaleffects of enalapril and losartan in type 1 diabetes. N Engl J Med 2009; 361: 40-51.
Perkins BA, Aiello LP, Krolewski AS. Diabetescomplications and the renin-angiotensin system. N Engl J Med 2009; 361: 83-5.
Valsartan nella prevenzione delle recidivedella fibrillazione atriale Nessuna delle terapie correntemente utilizzate nella fi-
brillazione atriale (FA) può essere considerata il mezzoideale per controllare questa che è l’aritmia più comune.Tuttavia, i risultati di alcuni studi suggeriscono che gli ini-bitori dell’angiotensina II (ARB) potrebbero essere ingrado di influenzare il rimodellamento atriale e di pre-venire la FA. Per verificare se valsartan, un ARB, possa ef-fettivamente ridurre le recidive di FA è stato condotto lostudio GISSI-AF (Gruppo Italiano per lo Studio della So-pravvivenza nell’Infarto Miocardico-Atrial Fibrillation).Nello studio, randomizzato, prospettico, controllatoverso placebo e multicentrico, sono stati arruolati 1442pazienti, in ritmo sinusale, ma con 2 o più episodi di FAnei 6 mesi precedenti o nei quali era stata eseguita, consuccesso, una cardioversione nelle 2 settimane antece-denti. L’intervallo di tempo che precedeva la prima reci-diva e la proporzione di soggetti con più di un episodiodi FA nell’arco di 1 anno di osservazione, rappresenta-vano i due end point primari. I risultati non hanno evi-denziato differenze tra i due bracci di trattamento.
Conclusioni. Gli autori concludono che l’uso divalsartan non si associa ad una riduzione dell’inci-denza di recidive di FA. L’editoriale di commentoallo studio richiama l’attenzione sugli aspetti chepossono limitare queste conclusioni, segnalandocome particolarmente importante il dubbio relativoalla possibilità che gli ARB riescano a modificare ilsubstrato anatomo-fisio-patologico della FA nel corsodel breve periodo di follow-up. Tanto più, alla lucedel fatto che circa la metà degli end point primari siverifica entro due mesi dalla randomizzazione. Se-condo Gillis, i risultati dello studio sottolineano an-
Bif watch
BOLLETTINO D’INFORMAZIONE SUI FARMACI | AGGIORNAMENTI |
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
76

cora una volta come i percorsi che conducono rapi-damente dalle promettenti intuizioni biomedichealla pratica clinica possano essere irti di insidie.
The GISSI-AF Investigators. Valsartan for prevention of recurrent atrial fibrillation. N Engl J Med 2009; 360: 1606-17.
Gillis A. Angiotensin-receptor blockers for prevention of atrial fibrillation. A matter of timing or target? N Engl J Med 2009; 360: 1669-71. [Editorial]
Esposizione fetale a farmaci antiepilettici e funzione cognitiva a 3 anni d’etàIn modelli animali, la somministrazione di farmaci
antiepilettici a dosi più basse di quelle necessarie a de-terminare malformazioni congenite può produrre alte-razioni cognitive; rimane da chiarire l’effetto dell’espo-sizione fetale di questi farmaci nell’uomo.
Gli autori di questo studio hanno arruolato tra il1999 e il 2004, in 25 centri degli Stati Uniti e del RegnoUnito, 303 donne in gravidanza affette da epilessia chehanno assunto farmaci antiepilettici quali carbamaze-pina, lamotrigina, fenitoina o valproato, al fine di con-frontare gli esiti neuroevolutivi all’età di 6 anni neibambini esposti in utero ai differenti farmaci.
Il report pubblicato è un’analisi intermedia, pro-grammata all’età di 3 anni, che ha valutato gli esiti del-l’esposizione fetale a farmaci antiepilettici sulla fun-zione cognitiva di 309 bambini esposti.
Gli autori hanno rilevato che i bambini le cui madriin gravidanza avevano assunto acido valproico mostra-vano valori di QI (Quoziente Intellettivo) significativa-mente più bassi di quelli esposti ad altri farmaci antie-pilettici. Dopo aggiustamento statistico le analisi hannorilevato che il QI medio dei bambini esposti alla la-motrigina era di 101, alla fenitoina 99, alla carbamaze-pina 98 e all’acido valproico 92.
L’associazione tra assunzione di acido valproico e va-riazione del QI è risultata essere dose-dipendente.
Conclusioni. L’esposizione fetale a valproato ri-spetto ad altri antiepilettici è associata ad un maggiorrischio di alterazioni cognitive nei bambini di treanni d’età. Gli stessi autori hanno riconosciuto qualilimiti dello studio la mancanza della randomizza-zione e di un gruppo di controllo e la scarsità deidati, per cui, come suggerisce l’autore dell’editorialedi accompagnamento, sono necessari ulteriori studiper confermare i dati e affinare le valutazioni sui ri-schi associati all’assunzione di questi farmaci.
Meador KJ, Baker GA, Browning N, et al. Cognitivefunction at 3 years of age after fetal exposure to
antiepileptic drugs. N Engl J Med 2009; 360: 1597-605.
Tomson T. Which drug for the pregnant woman withepilepsy? Editorial. N Engl J Med 2009; 360: 1667-9.
Qual è il trattamento migliore per le sindromi coronariche acute?Nei paesi occidentali milioni di persone sono ricove-
rate ogni anno per una sindrome coronarica acuta (SCA)manifestatasi come angina instabile o infarto miocardicosenza sopraslivellamento del tratto-ST. In questi pazientiè possibile ridurre l’incidenza di decessi, infarto o reinfartoe di recidive ischemiche grazie ai diversi trattamenti che sisono rivelati efficaci e che includono la terapia medica in-tensiva e l’angiografia coronarica (AC) seguita da rivasco-larizzazione, se indicata. D’altra parte la disponibilità dinumerose alternative valide può, in alcuni casi, generareincertezza. Due articoli pubblicati sul New England forni-scono utili indicazioni riguardo al tipo di approccio e allarelativa tempistica. In uno studio, il confronto tra l’inter-vento invasivo precoce (AC a 14 h [mediana]) e quello ri-tardato (AC a 50 h [mediana]) non ha mostrato nessunadifferenza nei due gruppi di pazienti con SCA, global-mente considerati. Nei soggetti ad alto rischio, però, lastrategia precoce ha prodotto esiti migliori mostrando, inparticolare, un minore rischio di ischemia refrattaria.
Gli inibitori della glicoproteina IIb/IIIa sono usati abi-tualmente nei pazienti con SCA sottoposti a procedure in-vasive ma il tempo ottimale di somministrazione è ignoto.Nel secondo studio, l’uso precoce e routinario di eptifi-batide non si è dimostrato migliore dell’uso ritardato edeventuale, risultando peraltro associato ad un maggiore ri-schio di sanguinamento.
Conclusioni. L’editoriale che accompagna questidue studi sottolinea come il trattamento dei pazienticon SCA possa essere considerato ottimale quando laterapia, per ciascun paziente, viene modulata sulla basedel rischio di incorrere in un evento ischemico o in unacomplicanza iatrogena. Nei soggetti a più alto rischio,il trattamento ottimizzato può produrre una sensibileriduzione dell’incidenza di infarto miocardico e ische-mia recidivante (dell’ordine del 20-40%) ed un mode-sto calo dei decessi (approssimativamente del 10%).L’entità del beneficio è correlata con il livello di rischiodel paziente.
Mehta SR, Granger CB, Boden WE, et al. Early versusdelayed invasive intervention in acute coronarysyndromes. N Engl J Med 2009; 360: 2165-75.
Giugliano RP, White JA, Bode C, et al. Early versusdelayed, provisional eptifibatide in acute coronarysyndromes. N Engl J Med 2009; 360: 2176-90.
Hillis LD, Lange RA. Optimal management of acutecoronary syndromes. N Engl J Med. 2009; 360: 2237-40.
AGGIORNAMENTI | BIF WATCH
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
77

nei nuovi casi sviluppatisi, la prevalenza di casi di tu-bercolosi multi-resistente (MDR, multidrug-resistant) èaumentata sostanzialmente nella Corea del Sud e nelleregioni russe dell’Oblast’ di Tomsk e dell’Oblast’ diOrël, ma è rimasta invariata in Estonia e Lettonia.
In generale, la prevalenza di TBC MDR è diminuitaa Hong Kong e negli Stati Uniti. Trentasette paesi hannoriportato dati rappresentativi di forme di TBC estrema-mente resistente ai farmaci (XDR, extensively drug-resi-stant). Cinque paesi, tutti appartenenti alla ex UnioneSovietica, hanno riportato ognuno 25 o più casi di TBCXDR, con una prevalenza tra i casi di TBC MDR che va-riava dal 6,6% al 23,7%.
Conclusioni. La TBC resistente ai farmaci conti-nua a rappresentare una minaccia per il controllodella TBC in Cina e in diversi paesi della ex UnioneSovietica. I dati sulla resistenza ai farmaci non sonodisponibili in molti paesi, soprattutto africani. Ciòevidenzia la necessità di sviluppare metodi più sem-plici di sorveglianza della farmaco-resistenza nella tu-bercolosi.
Wright A, Zignol M, Van Deun A, et al. for the GlobalProject on Anti-Tubercolosis Drug ResistanceSurveillance. Epidemiology of antitubercolosis drugresistance 2002-07: an updated analysis of the GlobalProject on Anti-Tubercolosis Drug ResistanceSurveillance. Lancet 2009; doi: 10.1016/S0140-6736(09)60331-7.
Epidemiologia della farmaco-resistenzaantitubercolare, 2002-2007Il ‘Global Project on Anti-tubercolosis Drug Resi-
stance’ raccoglie dati, a partire dal 1994, sull’esten-sione, a livello mondiale, della farmaco-resistenza an-titubercolare.
I dati di questo studio sulla sensibilità al farmacosono stati raccolti in 90.726 pazienti di 83 differentipaesi tra il 2002 e il 2007. La raccolta standardizzata deirisultati ha consentito di effettuare confronti sia trapaesi diversi sia all’interno degli stessi paesi. Quando èstato possibile, sono stati ottenuti anche dati sull’even-tuale coesistenza di una infezione HIV e sulla resistenzaa farmaci di seconda linea. La qualità dei dati di labo-ratorio è stata garantita dal Supranational TubercolosisReference Laboratory Network.
La prevalenza mediana della resistenza a qualsiasifarmaco nei nuovi casi di tubercolosi (TBC) è stata di11,1% (IQR 7,0-22,3). La prevalenza della resistenza apiù farmaci nei nuovi casi di TBC è stata dello 0% inotto paesi, 7% in due province della Cina, 11,1% nelleIsole Marianne del nord (anche se si tratta di solo duecasi), e tra il 6,8% e il 22,3% in nove paesi della exUnione Sovietica, incluso il 19,4% della Moldavia e il22,3% di Baku, Azerbaijan (mediana per i paesi oggettodell’indagine 1,6%, IQR 0,6-3,9).
L’analisi dei dati ha rivelato che tra il 1994 e il 2007,
AGGIORNAMENTI | BIF WATCH
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
78
I medicinali per le terapie avanzate sono prodottibasati sulla terapia genica, sulla terapia cellularesomatica o sull’ingegneria tessutale. La complessità di questi medicinali e la rapidità con cui si sviluppanole conoscenze scientifiche in questo settore spessoprovocano incertezze riguardo alla loroclassificazione; anche perché, non di rado, questiprodotti sono al confine tra i medicinali ed altrecategorie, come ad esempio i dispositivi medici.Per questi motivi la nuova legislazione europeasulle terapie avanzate ha previsto una particolareprocedura per consentire alle aziende che sviluppanomedicinali innovativi di verificare se un proprioprodotto possa essere effettivamente inquadrato inquesto ambito. Il Committee for Advanced Therapiesdella European Medicines Agency, responsabile delprocedimento, ha annunciato il 30 giugno 2009 diaver adottato la sua prima raccomandazione sulla
classificazione di un prodotto medicinale per le terapie avanzate.Si tratta di un medicinale per la terapia cellularesomatica utilizzabile per il trattamento delle ulcerevenose croniche degli arti inferiori composto da fibroblasti e cheratinociti umani allogenicisomministrati congiuntamente a fibrina ed altricomponenti strutturali.È opportuno sottolineare che la procedura che portaall’espressione del parere del Committee for AdvancedTherapies sulla classificazione di un particolareprodotto è facoltativa e preliminare allapresentazione della relativa domanda diautorizzazione all’immissione in commercio.Pertanto, non c’è da attendersi che all’annuncio di questa importante novità in un settore di frontieraquale quello delle terapie avanzate, faccia seguito intempi brevi la commercializzazione del medicinale.
Medicinali per le terapie avanzateA proposito di…
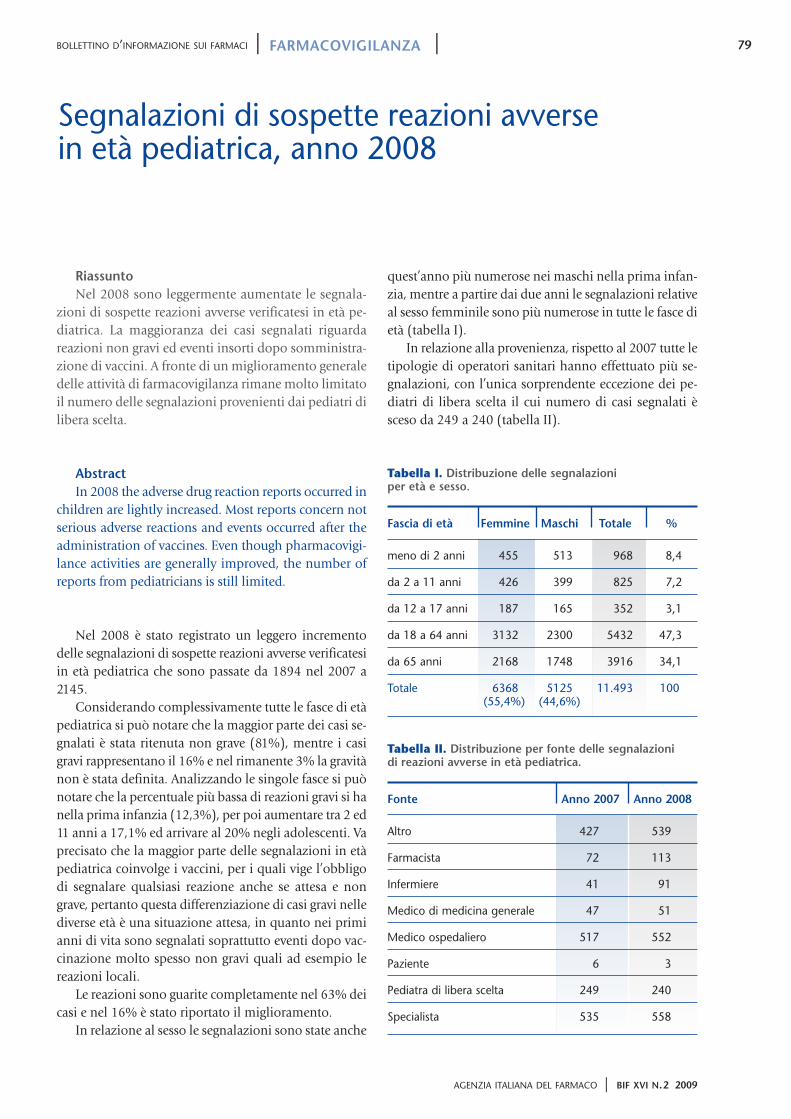
RiassuntoNel 2008 sono leggermente aumentate le segnala-
zioni di sospette reazioni avverse verificatesi in età pe-diatrica. La maggioranza dei casi segnalati riguardareazioni non gravi ed eventi insorti dopo somministra-zione di vaccini. A fronte di un miglioramento generaledelle attività di farmacovigilanza rimane molto limitatoil numero delle segnalazioni provenienti dai pediatri dilibera scelta.
AbstractIn 2008 the adverse drug reaction reports occurred in
children are lightly increased. Most reports concern notserious adverse reactions and events occurred after theadministration of vaccines. Even though pharmacovigi-lance activities are generally improved, the number ofreports from pediatricians is still limited.
Nel 2008 è stato registrato un leggero incrementodelle segnalazioni di sospette reazioni avverse verificatesiin età pediatrica che sono passate da 1894 nel 2007 a2145.
Considerando complessivamente tutte le fasce di etàpediatrica si può notare che la maggior parte dei casi se-gnalati è stata ritenuta non grave (81%), mentre i casigravi rappresentano il 16% e nel rimanente 3% la gravitànon è stata definita. Analizzando le singole fasce si puònotare che la percentuale più bassa di reazioni gravi si hanella prima infanzia (12,3%), per poi aumentare tra 2 ed11 anni a 17,1% ed arrivare al 20% negli adolescenti. Vaprecisato che la maggior parte delle segnalazioni in etàpediatrica coinvolge i vaccini, per i quali vige l’obbligodi segnalare qualsiasi reazione anche se attesa e nongrave, pertanto questa differenziazione di casi gravi nellediverse età è una situazione attesa, in quanto nei primianni di vita sono segnalati soprattutto eventi dopo vac-cinazione molto spesso non gravi quali ad esempio lereazioni locali.
Le reazioni sono guarite completamente nel 63% deicasi e nel 16% è stato riportato il miglioramento.
In relazione al sesso le segnalazioni sono state anche
quest’anno più numerose nei maschi nella prima infan-zia, mentre a partire dai due anni le segnalazioni relativeal sesso femminile sono più numerose in tutte le fasce dietà (tabella I).
In relazione alla provenienza, rispetto al 2007 tutte letipologie di operatori sanitari hanno effettuato più se-gnalazioni, con l’unica sorprendente eccezione dei pe-diatri di libera scelta il cui numero di casi segnalati èsceso da 249 a 240 (tabella II).
Segnalazioni di sospette reazioni avverse in età pediatrica, anno 2008
BOLLETTINO D’INFORMAZIONE SUI FARMACI | FARMACOVIGILANZA |
Tabella I. Distribuzione delle segnalazioni per età e sesso.
Fascia di età Femmine Maschi Totale %
meno di 2 anni 455 513 968 8,4
da 2 a 11 anni 426 399 825 7,2
da 12 a 17 anni 187 165 352 3,1
da 18 a 64 anni 3132 2300 5432 47,3
da 65 anni 2168 1748 3916 34,1
Totale 6368 5125 11.493 100(55,4%) (44,6%)
Tabella II. Distribuzione per fonte delle segnalazioni di reazioni avverse in età pediatrica.
Fonte Anno 2007 Anno 2008
Altro 427 539
Farmacista 72 113
Infermiere 41 91
Medico di medicina generale 47 51
Medico ospedaliero 517 552
Paziente 6 3
Pediatra di libera scelta 249 240
Specialista 535 558
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
79
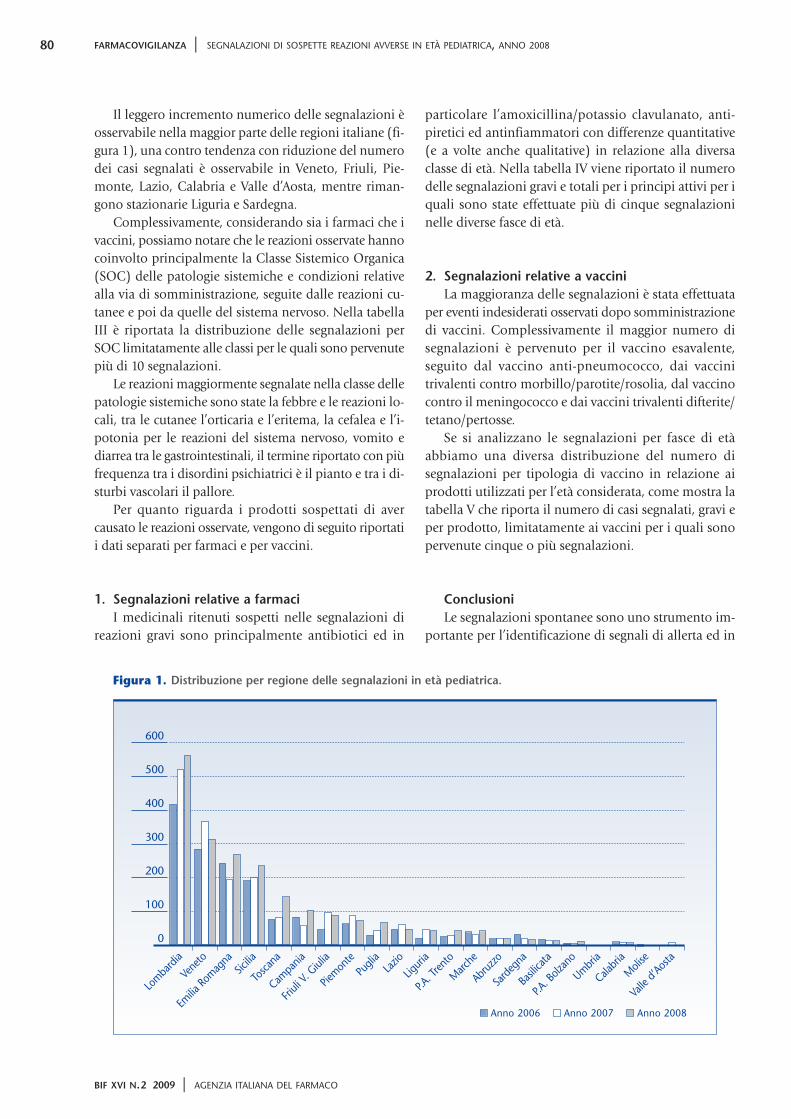
Figura 1. Distribuzione per regione delle segnalazioni in età pediatrica.
600
500
400
300
200
100
0
Lomba
rdia
Vene
to
Emilia
Romag
naSic
ilia
Tosca
na
Campa
nia
Friuli
V. G
iulia
Piemon
tePu
glia
Lazio
Liguri
a
P.A. T
rento
Marche
Abruzzo
Sarde
gna
Molise
Calabri
a
P.A. B
olzan
o
Umbria
Basili
cata
Valle
d’Aos
ta
Anno 2006 Anno 2007 Anno 2008
FARMACOVIGILANZA | SEGNALAZIONI DI SOSPETTE REAZIONI AVVERSE IN ETÀ PEDIATRICA, ANNO 2008
Il leggero incremento numerico delle segnalazioni èosservabile nella maggior parte delle regioni italiane (fi-gura 1), una contro tendenza con riduzione del numerodei casi segnalati è osservabile in Veneto, Friuli, Pie-monte, Lazio, Calabria e Valle d’Aosta, mentre riman-gono stazionarie Liguria e Sardegna.
Complessivamente, considerando sia i farmaci che ivaccini, possiamo notare che le reazioni osservate hannocoinvolto principalmente la Classe Sistemico Organica(SOC) delle patologie sistemiche e condizioni relativealla via di somministrazione, seguite dalle reazioni cu-tanee e poi da quelle del sistema nervoso. Nella tabellaIII è riportata la distribuzione delle segnalazioni perSOC limitatamente alle classi per le quali sono pervenutepiù di 10 segnalazioni.
Le reazioni maggiormente segnalate nella classe dellepatologie sistemiche sono state la febbre e le reazioni lo-cali, tra le cutanee l’orticaria e l’eritema, la cefalea e l’i-potonia per le reazioni del sistema nervoso, vomito ediarrea tra le gastrointestinali, il termine riportato con piùfrequenza tra i disordini psichiatrici è il pianto e tra i di-sturbi vascolari il pallore.
Per quanto riguarda i prodotti sospettati di avercausato le reazioni osservate, vengono di seguito riportatii dati separati per farmaci e per vaccini.
1. Segnalazioni relative a farmaciI medicinali ritenuti sospetti nelle segnalazioni di
reazioni gravi sono principalmente antibiotici ed in
particolare l’amoxicillina/potassio clavulanato, anti-piretici ed antinfiammatori con differenze quantitative(e a volte anche qualitative) in relazione alla diversaclasse di età. Nella tabella IV viene riportato il numerodelle segnalazioni gravi e totali per i principi attivi per iquali sono state effettuate più di cinque segnalazioninelle diverse fasce di età.
2. Segnalazioni relative a vacciniLa maggioranza delle segnalazioni è stata effettuata
per eventi indesiderati osservati dopo somministrazionedi vaccini. Complessivamente il maggior numero disegnalazioni è pervenuto per il vaccino esavalente,seguito dal vaccino anti-pneumococco, dai vaccinitrivalenti contro morbillo/parotite/rosolia, dal vaccinocontro il meningococco e dai vaccini trivalenti difterite/tetano/pertosse.
Se si analizzano le segnalazioni per fasce di etàabbiamo una diversa distribuzione del numero disegnalazioni per tipologia di vaccino in relazione aiprodotti utilizzati per l’età considerata, come mostra latabella V che riporta il numero di casi segnalati, gravi eper prodotto, limitatamente ai vaccini per i quali sonopervenute cinque o più segnalazioni.
ConclusioniLe segnalazioni spontanee sono uno strumento im-
portante per l’identificazione di segnali di allerta ed in
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
80
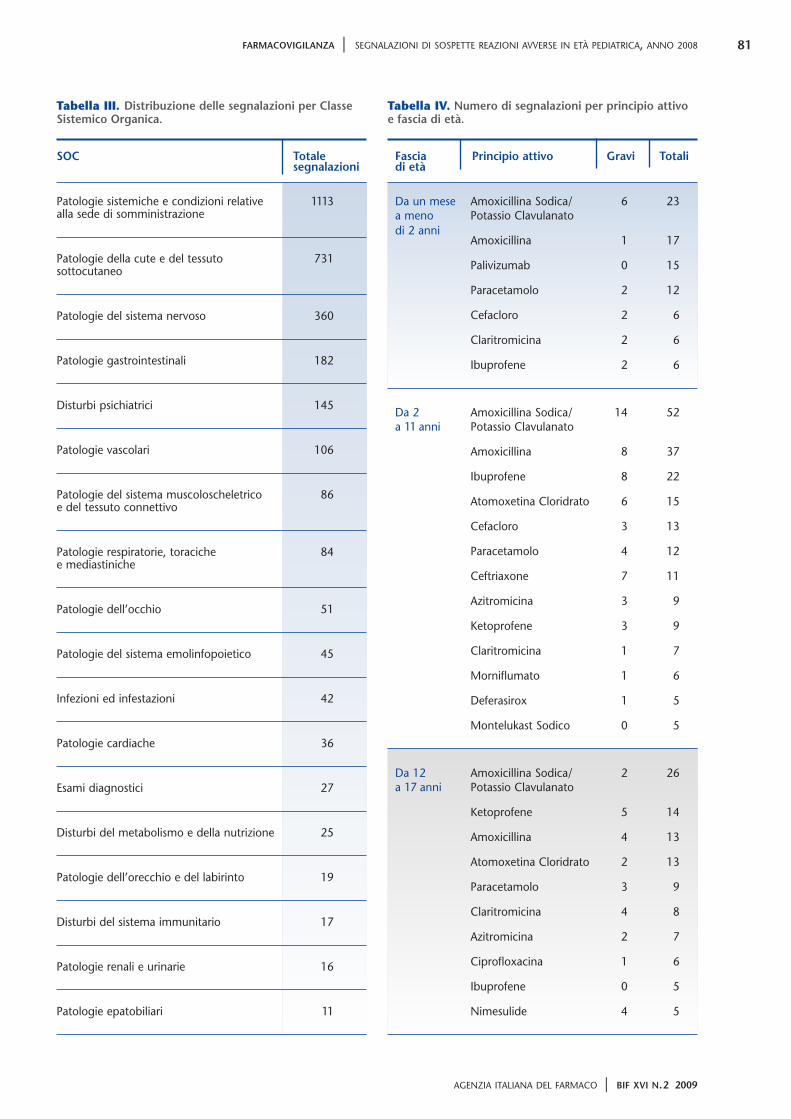
FARMACOVIGILANZA | SEGNALAZIONI DI SOSPETTE REAZIONI AVVERSE IN ETÀ PEDIATRICA, ANNO 2008
Patologie sistemiche e condizioni relative 1113alla sede di somministrazione
Patologie della cute e del tessuto 731sottocutaneo
Patologie del sistema nervoso 360
Patologie gastrointestinali 182
Disturbi psichiatrici 145
Patologie vascolari 106
Patologie del sistema muscoloscheletrico 86e del tessuto connettivo
Patologie respiratorie, toraciche 84e mediastiniche
Patologie dell’occhio 51
Patologie del sistema emolinfopoietico 45
Infezioni ed infestazioni 42
Patologie cardiache 36
Esami diagnostici 27
Disturbi del metabolismo e della nutrizione 25
Patologie dell’orecchio e del labirinto 19
Disturbi del sistema immunitario 17
Patologie renali e urinarie 16
Patologie epatobiliari 11
Tabella III. Distribuzione delle segnalazioni per ClasseSistemico Organica.
SOC Totale segnalazioni
Da un mese Amoxicillina Sodica/ 6 23a meno Potassio Clavulanatodi 2 anni
Amoxicillina 1 17
Palivizumab 0 15
Paracetamolo 2 12
Cefacloro 2 6
Claritromicina 2 6
Ibuprofene 2 6
Da 2 Amoxicillina Sodica/ 14 52a 11 anni Potassio Clavulanato
Amoxicillina 8 37
Ibuprofene 8 22
Atomoxetina Cloridrato 6 15
Cefacloro 3 13
Paracetamolo 4 12
Ceftriaxone 7 11
Azitromicina 3 9
Ketoprofene 3 9
Claritromicina 1 7
Morniflumato 1 6
Deferasirox 1 5
Montelukast Sodico 0 5
Da 12 Amoxicillina Sodica/ 2 26a 17 anni Potassio Clavulanato
Ketoprofene 5 14
Amoxicillina 4 13
Atomoxetina Cloridrato 2 13
Paracetamolo 3 9
Claritromicina 4 8
Azitromicina 2 7
Ciprofloxacina 1 6
Ibuprofene 0 5
Nimesulide 4 5
Tabella IV. Numero di segnalazioni per principio attivoe fascia di età.
Fascia Principio attivo Gravi Totalidi età
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
81

FARMACOVIGILANZA | SEGNALAZIONI DI SOSPETTE REAZIONI AVVERSE IN ETÀ PEDIATRICA, ANNO 2008
ambito pediatrico sono ancora più rilevanti per la limi-tata disponibilità di informazioni di sicurezza relative aifarmaci usati nei bambini. Infatti è dalle segnalazioni ri-cevute che, recentemente, si è potuto intervenire conazioni regolatorie sui colliri per la midriasi contenentifenilefrina, e sull’oxatomide.
Negli ultimi anni le attività di farmacovigilanza sonomigliorate nella maggior parte delle regioni italiane:
sono aumentati i segnalatori (in particolare farmacisti edinfermieri), è aumentato il numero di segnalazioni, èmigliorata la qualità dei dati forniti, ma purtroppo ri-mane limitato il numero delle segnalazioni provenientidai pediatri. Gli operatori sanitari, ed i pediatri in par-ticolare, devono essere maggiormente sensibilizzati sullanecessità di segnalare le reazioni avverse che osservanonei bambini.
Tabella V. Segnalazioni per vaccino e per fascia di età.
Fascia Principio attivo Gravi Totalidi età
Da un mese Vaccino difterico/Epatitico B ricombinante/Haemofilus Influenzae B 44 529a meno coniugato e adiuvato/Pertossico acellulare/Poliomielitico inattivato/di 2 anni Tetanico
Vaccino Pneumococcico saccaridico coniugato adsorbito 30 326
Vaccino Morbillo/Parotite/Rosolia 29 160
Vaccino Meningococcico gruppo C coniugato 9 85
Vaccino Morbillo/Parotite/Rosolia/Varicella 9 46
Vaccino Meningococcico Polisaccaridico 1 31
Vaccino Difterico/Pertossico Acellulare/Tetanico 2 30
Vaccino Varicella Vivo 3 22
Vaccino Influenzale 1 5
Da 2 Vaccino Difterico Adsorbito/Pertossico Adsorbito/Tetanico Adsorbito 12 210a 11 anni Vaccino Morbillo/Parotite/Rosolia 19 125
Vaccino Papillomavirus Umano 8 101
Vaccino Meningococcico Gruppo C 5 68
Vaccino Meningococcico Polisaccaridico 2 32
Vaccino Influenzale 0 17
Vaccino Varicella Vivo 0 17
Vaccino Pneumococcico Saccaridico Coniugato Adsorbito 4 15
Vaccino Difterico/Pertossico/Poliomielitico/Tetanico 2 6
Vaccino Difterico/Epatitico B Ricombinante/Haemofilus Influenzae B 1 5Coniugato E Adiuvato/Pertossico Acellulare/Poliomielitico Inattivato/Tetanico
Da 12 Vaccino Papillomavirus umano 7 56a 17 anni Vaccino Meningococcico Gruppo C coniugato con Tossoide Difterico 3 42
Vaccino Difterico Adsorbito/Tetanico Adsorbito 1 29
Vaccino Meningococcico Polisaccaridico 2 28
Vaccino Difterico Adsorbito/Pertossico Adsorbito/Tetanico Adsorbito 3 27
Vaccino Morbillo/Parotite/Rosolia 3 23
Vaccino Varicella Vivo 0 6
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
82

Il segnaleI farmaci antinfiammatori non steroidei (FANS) sono
comunemente usati per via sistemica nel trattamento deldolore e delle condizioni infiammatorie. Tuttavia il lorouso non è esente da effetti indesiderati soprattutto a li-vello gastrointestinale e renale. Per questo motivo nelleforme moderate di dolore muscoloscheletrico se ne con-siglia l’uso per via topica. I FANS, però, possono anche in-durre reazioni di fotosensibilità, definite come reazionicutanee esagerate o anormali alla luce. Questa tipologiadi reazioni sembra essere riportata più comunemente conle formulazioni topiche a causa della più alta concentra-zione di farmaco a livello cutaneo.
Tra i FANS, quello con il più alto numero di reazionidi fotosensibilità è il ketoprofene e si stima che la gravitàpuò essere tale da richiedere un ricovero ospedaliero in 1caso su 101.
Le reazioni di fotosensibilizzazione da ketoprofenesono da attribuire alla struttura chimica (funzione carbo-linica) che, in seguito a fotoesposizione, subisce una de-gradazione chimica che porta alla formazione di radicalie di specie ossigenate reattive responsabili dei fenomenifototossici2. Studi di spettroscopia hanno infatti dimo-strato che in seguito ad esposizione alla luce il ketopro-fene subisce un processo di decarbossilazione, dal quale sioriginano radicali (il principale è il benzoilfeniletano) chehanno una azione litica a livello delle membrane cellularie che sono responsabili quindi degli effetti collaterali as-sociati alla somministrazione del ketoprofene, in partico-
lar modo quando questa è accompagnata dall’esposizioneai raggi UV2,3. Questi processi sono scarsamente attivi incondizioni di bassi livelli di luce, ma possono diventare ri-levanti con un’alta intensità di luce.
Le reazioni cutanee da ketoprofene possono anche es-sere di natura fotoallergica, alla base delle quali vi è unarisposta di ipersensibilità ritardata mediata da linfociti T.Questo tipo di reazione si verifica in individui preceden-temente sensibilizzati e richiede un periodo di latenza. Lelesioni sono polimorfe ed eczematose e, contrariamentealla fototossicità, sono dose indipendenti e possono esten-dersi anche ad aree non irradiate3.
I risultati di alcuni studi di fotoallergenicità eseguiti sucavie suggeriscono che uno dei fattori importanti per l’in-duzione della fotoallergia è la struttura chimica del ben-zofenone cromoforo nella molecola del ketoprofene. Taleipotesi si basa sul fatto che gli enantiomeri del ketopro-fene (loxoprofene e flurbiprofene) non possiedono que-sta struttura e, di conseguenza, non hanno questopotenziale fotoallergenico4.
La durata della risposta all’esposizione alla luce, dopola sospensione del ketoprofene, varia da poche settimanea diversi mesi, ma sono stati riportati anche casi di persi-stenza della fotosensibilizzazione superiore a un anno5.La reazione può essere limitata al sito di applicazione delfarmaco, estendersi anche al sito controlaterale, coinvol-gere altre zone del corpo esposte al sole e, in qualche rarocaso, anche zone contigue non esposte.
In più studi è stato confermato il ruolo di alcuni ecci-pienti sulla comparsa delle dermatiti da contatto: sostanzecome l’Aurantii amari floris etheroleum e la Lavandulaeetheroleum, presenti in alcuni medicinali a base di keto-profene, sono stati indicati come agenti scatenanti rea-zioni fotoallergiche o fototossiche6.
I dati della Rete Nazionale di FarmacovigilanzaNella Rete Nazionale di Farmacovigilanza (RNF), dal
gennaio del 2001 al marzo del 2009, sono state inserite206 segnalazioni di sospette reazioni avverse da ketopro-fene per uso cutaneo (dall’analisi sono state escluse le se-gnalazioni di ADR – adverse drug reactions – relative aicerotti transdermici). Il 26% delle segnalazioni ha riguar-
Reazioni di fotosensibilizzazione da ketoprofene
BOLLETTINO D’INFORMAZIONE SUI FARMACI | FARMACOVIGILANZA |
In seguito a fotoesposizione
la struttura chimica
del ketoprofene subisce una
degradazione che porta alla
formazione di radicali e di specie
ossigenate reattive responsabili
dei fenomeni fototossici.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.3 2009
83
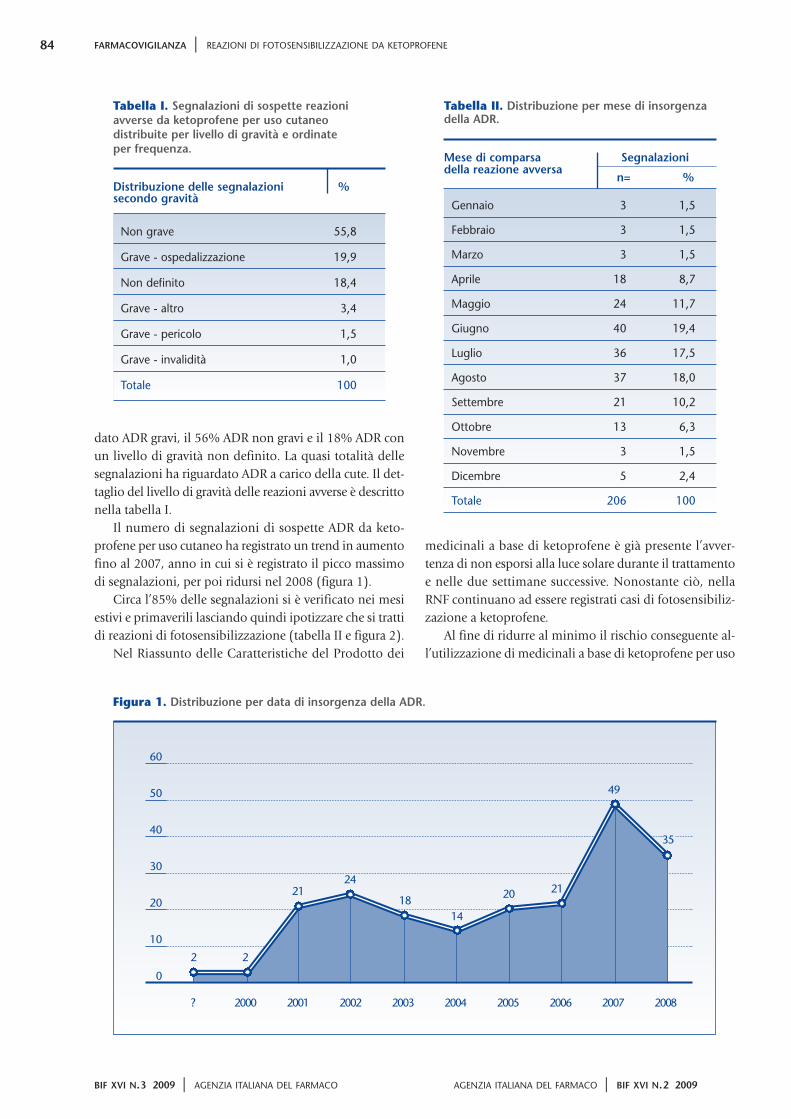
Figura 1. Distribuzione per data di insorgenza della ADR.
60
50
40
30
20
10
0
FARMACOVIGILANZA | REAZIONI DI FOTOSENSIBILIZZAZIONE DA KETOPROFENE
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
dato ADR gravi, il 56% ADR non gravi e il 18% ADR conun livello di gravità non definito. La quasi totalità dellesegnalazioni ha riguardato ADR a carico della cute. Il det-taglio del livello di gravità delle reazioni avverse è descrittonella tabella I.
Il numero di segnalazioni di sospette ADR da keto-profene per uso cutaneo ha registrato un trend in aumentofino al 2007, anno in cui si è registrato il picco massimodi segnalazioni, per poi ridursi nel 2008 (figura 1).
Circa l’85% delle segnalazioni si è verificato nei mesiestivi e primaverili lasciando quindi ipotizzare che si trattidi reazioni di fotosensibilizzazione (tabella II e figura 2).
Nel Riassunto delle Caratteristiche del Prodotto dei
medicinali a base di ketoprofene è già presente l’avver-tenza di non esporsi alla luce solare durante il trattamentoe nelle due settimane successive. Nonostante ciò, nellaRNF continuano ad essere registrati casi di fotosensibiliz-zazione a ketoprofene.
Al fine di ridurre al minimo il rischio conseguente al-l’utilizzazione di medicinali a base di ketoprofene per uso
Tabella I. Segnalazioni di sospette reazioniavverse da ketoprofene per uso cutaneodistribuite per livello di gravità e ordinate per frequenza.
Distribuzione delle segnalazioni %secondo gravità
Non grave 55,8
Grave - ospedalizzazione 19,9
Non definito 18,4
Grave - altro 3,4
Grave - pericolo 1,5
Grave - invalidità 1,0
Totale 100
Tabella II. Distribuzione per mese di insorgenzadella ADR.
Mese di comparsa Segnalazionidella reazione avversa
n= %
Gennaio 3 1,5
Febbraio 3 1,5
Marzo 3 1,5
Aprile 18 8,7
Maggio 24 11,7
Giugno 40 19,4
Luglio 36 17,5
Agosto 37 18,0
Settembre 21 10,2
Ottobre 13 6,3
Novembre 3 1,5
Dicembre 5 2,4
Totale 206 100
? 2000 2001 2002 2003 2004 2005 2006 2007 2008
2 2
2124
1814
20 21
49
35
BIF XVI N.3 2009 | AGENZIA ITALIANA DEL FARMACO
84
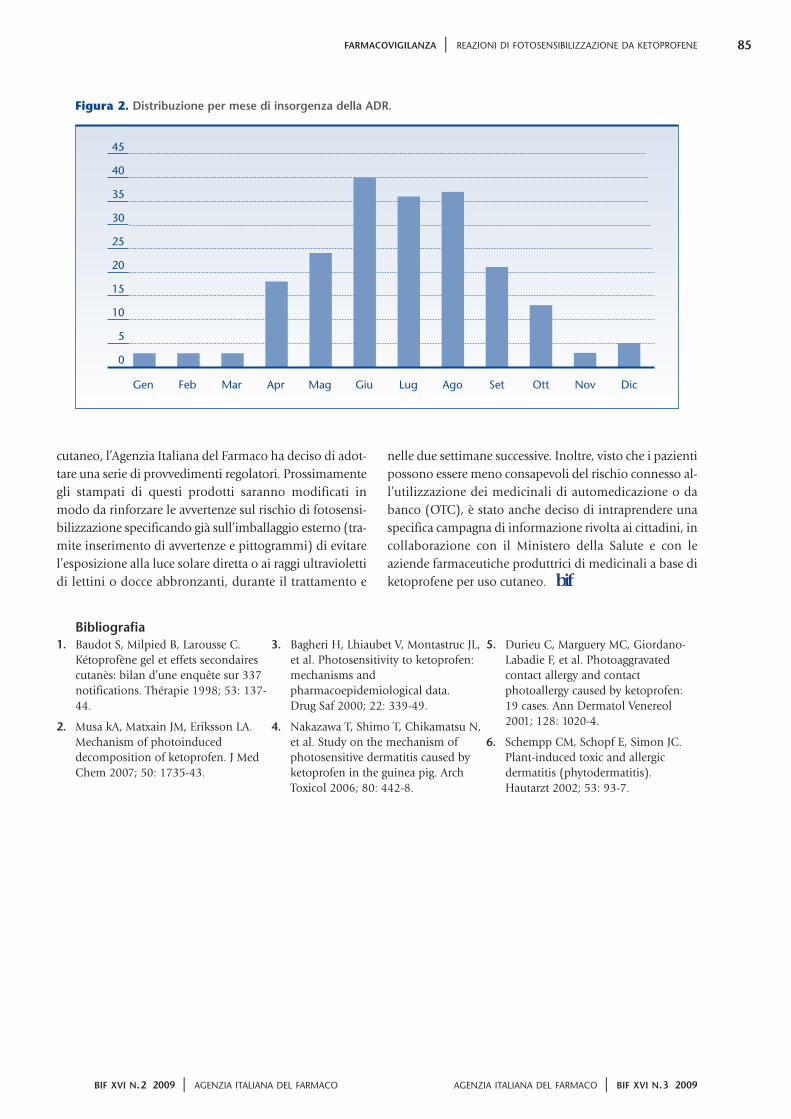
FARMACOVIGILANZA | REAZIONI DI FOTOSENSIBILIZZAZIONE DA KETOPROFENE
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
cutaneo, l’Agenzia Italiana del Farmaco ha deciso di adot-tare una serie di provvedimenti regolatori. Prossimamentegli stampati di questi prodotti saranno modificati inmodo da rinforzare le avvertenze sul rischio di fotosensi-bilizzazione specificando già sull’imballaggio esterno (tra-mite inserimento di avvertenze e pittogrammi) di evitarel’esposizione alla luce solare diretta o ai raggi ultraviolettidi lettini o docce abbronzanti, durante il trattamento e
nelle due settimane successive. Inoltre, visto che i pazientipossono essere meno consapevoli del rischio connesso al-l’utilizzazione dei medicinali di automedicazione o dabanco (OTC), è stato anche deciso di intraprendere unaspecifica campagna di informazione rivolta ai cittadini, incollaborazione con il Ministero della Salute e con leaziende farmaceutiche produttrici di medicinali a base diketoprofene per uso cutaneo.
Figura 2. Distribuzione per mese di insorgenza della ADR.
45
40
35
30
25
20
15
10
5
0
Gen Feb Mar Apr Mag Giu Lug Ago Set Ott Nov Dic
Bibliografia1. Baudot S, Milpied B, Larousse C.
Kétoprofène gel et effets secondairescutanès: bilan d’une enquête sur 337notifications. Thérapie 1998; 53: 137-44.
2. Musa kA, Matxain JM, Eriksson LA.Mechanism of photoinduceddecomposition of ketoprofen. J MedChem 2007; 50: 1735-43.
3. Bagheri H, Lhiaubet V, Montastruc JL,et al. Photosensitivity to ketoprofen:mechanisms andpharmacoepidemiological data. Drug Saf 2000; 22: 339-49.
4. Nakazawa T, Shimo T, Chikamatsu N,et al. Study on the mechanism ofphotosensitive dermatitis caused byketoprofen in the guinea pig. ArchToxicol 2006; 80: 442-8.
5. Durieu C, Marguery MC, Giordano-Labadie F, et al. Photoaggravatedcontact allergy and contactphotoallergy caused by ketoprofen:19 cases. Ann Dermatol Venereol2001; 128: 1020-4.
6. Schempp CM, Schopf E, Simon JC.Plant-induced toxic and allergicdermatitis (phytodermatitis).Hautarzt 2002; 53: 93-7.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.3 2009
85

Si pubblicano di seguito, in forma sintetica, Dear Doctor Letter (DDL) recentemente inviate ai medici per diffondere tempestivamentenuove evidenze sulla sicurezza di alcuni medicinali.Le DDL sono concordate con l’AIFA che
quindi ne condivide i contenuti; con la loro pubblicazione sul Bollettino d’Informazione sui Farmaci si intende sottolinearne l’importanza e facilitarne l’archiviazione. Le versioni integrali sono disponibili sul portale dell’AIFA (www.agenziafarmaco.it) nella sezione dedicata alla Farmacovigilanza.Si ricorda inoltre che per ulteriori informazioni ci si può rivolgere via fax all’Ufficio Farmacovigilanza:06 597 84 142 o al numero verde del Servizio d’Informazione sul Farmaco Farmaci-line: 800 571 661.
Dear Doctor Letter
Radiofarmaci contenentialbumina
Azienda: varieInformazioni di sicurezza:segnalazione di casi fatali direazione avversa di tipo anafilatticoassociati alla somministrazioneendovenosa di radiofarmacicontenenti albumina. L’occorrenza di reazionianafilattiche associate allasomministrazione di questiradiofarmaci è un fenomeno moltoraro ma rappresenta un problemaclinico che deve essere conosciutodagli operatori sanitari cheutilizzano tali medicinali. La possibilità di reazioni diipersensibilità, incluse reazionianafilattiche/anafilattoidi gravi ofatali, deve sempre essere presa inconsiderazione in seguito asomministrazioni singole oripetute di radiofarmaci contenentialbumina (soluzioni iniettabili di99mTc-macroaggregati dialbumina). Pertanto dovrebberoessere prontamente disponibiliattrezzature adeguate per larianimazione. Possono inoltreinsorgere reazioni allergiche localial sito di iniezione del medicinale.Febbraio 2009
Thalidomide Celgene
Principio attivo: talidomideAzienda: CelgeneIndicazioni: in associazione amelfalan e prednisone, è indicataper il trattamento di prima linea dipazienti con mieloma multiplonon trattato di età ≥ 65 anni o non idonei a chemioterapia adosi elevate.Informazioni di sicurezza:l’utilizzo clinico di ThalidomideCelgene è subordinato alla pienaconoscenza di alcune importantiinformazioni di sicurezza eall’attuazione di uno specificoPiano di Gestione del Rischio,stabilito in accordo con l’AgenziaEuropea dei Medicinali (EMEA) econ l’AIFA. Tra i rischimaggiormente rilevanti ci sono:teratogenicità, neuropatiaperiferica, eventi tromboembolici,sincope e bradicardia, reazionicutanee, sonnolenza e capogiri. Inparticolare, in considerazione deipotenti effetti teratogenici delprincipio attivo talidomide,Thalidomide Celgene ècontroindicato in gravidanza enelle donne potenzialmente fertili,a meno che non siano state attuatetutte le misure previste da unapposito Programma diPrevenzione della Gravidanza.Aprile 2009
Tarceva
Principio attivo: erlotinibAzienda: RocheIndicazioni: trattamento dipazienti affetti da carcinomapolmonare non a piccole cellulelocalmente avanzato o metastaticodopo il fallimento di almeno unprecedente regime chemioterapico.In associazione con gemcitabina,Tarceva è indicato anche neltrattamento dei pazienti affetti dacarcinoma pancreaticometastatico.Informazioni di sicurezza: ipazienti che assumono Tarcevapresentano un rischio più elevatodi sviluppare perforazionigastrointestinali, soprattutto seassumono insieme a Tarceva agentiantiangiogenici, corticosteroidi,FANS e/o chemioterapia a base ditaxani, o se risultano positiviall’anamnesi per ulcera peptica omalattia diverticolare; in caso diperforazione gastrointestinale, iltrattamento con Tarceva deveessere sospeso in manieradefinitiva. Inoltre, il trattamentocon Tarceva deve essere interrottoo sospeso se il paziente sviluppagravi disturbi cutanei di tipobolloso, vescicolare o esfoliativo, oqualora i pazienti dovesseropresentare disturbi oculari acuti oin peggioramento, come doloreoculare.Maggio 2009
BOLLETTINO D’INFORMAZIONE SUI FARMACI | FARMACOVIGILANZA NEWS |
Maasol, Macrotec,Nanocoll, Pulmocis,Technescan Lyomaa,Venticoll
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
86

News
Rasilez: nuove restrizioni d’uso
Che cosa è Rasilez. Rasilez è indicato per il tratta-mento dell’ipertensione essen-ziale e contiene il principio attivoaliskiren, un potente e selettivoinibitore diretto della reninaumana, non-peptidico e attivoper via orale. È in commercio nel-l’Unione Europea dall’agosto del2007 con i nomi commerciali diRasilez, Enviage, Sprimeo, Tek-turna e Riprazo.Il problema di sicurezza.Sono stati riportati casi di an-gioedema o di reazioni similariassociati all’uso di farmaci con-tenenti aliskiren. A seguito dellavalutazione di tutte le evidenzedisponibili, il Comitato per i me-dicinali per uso umano (CHMP)dell’EMEA ha concluso che il be-neficio correlato all’uso dei me-dicinali contenenti aliskiren neltrattamento della pressione es-senziale è ancora superiore al ri-schio associato al suo utilizzo,anche se con questi medicinalipuò manifestarsi angioedemaquale effetto avverso grave e raro.Quali azioni ha intrapresol’EMEA.L’EMEA ha raccomandato di ag-giungere una nuova controindi-cazione all’uso nei pazienti cheabbiano sviluppato angioedemadurante un precedente utilizzodei medicinali contenenti aliski-ren e una nuova avvertenza af-finché i pazienti che dovesserosviluppare segni di angioedemadurante il trattamento interrom-pano subito la terapia e infor-mino il medico curante.
Gardasil, un bilancio tra rischi e benefici
Che cosa è Gardasil.Gardasil è un vaccino per la pre-venzione del cancro della cervicee di altre malattie precancerosecausate dal papilloma virusumano (HPV). È stato autoriz-zato nell’Unione Europea dal set-tembre del 2006. Circa tre mi-lioni di ragazze sono state vac-cinate in Europa con questo me-dicinale da quando fu autorizzatoper la prima volta.Il problema di sicurezza.A seguito di due casi di stato epi-lettico con mioclono segnalati indue ragazze vaccinate in Spagna,il 9 febbraio 2009 le autorità spa-gnole della sanità pubblica han-no bloccato, per precauzione, lavaccinazione con il lotto interes-sato di Gardasil. Poco dopo an-che le autorità italiane hannobloccato la vaccinazione con que-sto lotto. La distribuzione del-l’intero lotto è stata bloccata il10 febbraio 2009.Quali azioni ha intrapresol’EMEA.Sulla base dei dati attuali, ilCHMP dell’EMEA ha conclusoche è improbabile che i due casisiano correlati alla vaccinazionecon Gardasil e che i benefici diquesto vaccino continuano a su-perare i suoi rischi. Perciò il Co-mitato raccomanda di continuarela vaccinazione con Gardasil inaccordo con i programmi vacci-nali nazionali negli Stati Mem-bri. Nell’ambito del continuomonitoraggio dei medicinali, ilCHMP e il suo Pharmacovigi-lance Working Party continue-
FARMACOVIGILANZA | NEWS
L’AIFA coglie l’occasione perricordare a tutti i medicil’importanza dellasegnalazione delle reazioniavverse da farmaci, qualestrumento indispensabile perconfermare un rapportobeneficio-rischio favorevolenelle loro reali condizioni diimpiego. Le segnalazioni disospetta reazione avversa dafarmaci devono essere inviateal Responsabile diFarmacovigilanza dellaStruttura di appartenenza.
Cellcept
Principio attivo: micofenolatomofetileAzienda: RocheIndicazioni: in associazione conciclosporina e corticosteroidi per laprofilassi del rigetto acuto diorgano trapiantato in pazientiadulti sottoposti a trapiantoallogenico di rene, cuore o fegato ein pazienti pediatrici e adolescenti(2-18 anni) sottoposti a trapiantodi rene.Informazioni di sicurezza: alivello mondiale sono statiassociati all’impiego di Cellcept 41casi di aplasia eritroide pura(pure red cell aplasia, PRCA).Alcuni dei pazienti stavanoassumendo anche altri farmaci chepotrebbero aver contribuito allosviluppo della PRCA(alemtuzumab, tacrolimus,azatioprina e cotrimoxazolo). In16 casi tra quelli riportati, lariduzione della dose di Cellcept (4casi) o la sua interruzione (12 casi)hanno portato alla risoluzionedella condizione. Non è noto ilmeccanismo attraverso cui Cellceptpuò causare PRCA. Una relazionedi causalità tra Cellcept e PRCAnon può essere esclusa.Giugno 2009
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
87
Continua

Continua
FARMACOVIGILANZA | NEWS
ranno ad indagare su questa situa-zione. È stato richiesto al titolaredell’autorizzazione all’immissionein commercio di fornire un’analisicompleta del lotto, come pure ulte-riori informazioni sugli effetti inde-siderati del vaccino o su qualsiasicaso simile e sulle possibili moda-lità con le quali il Gardasil potrebbeessere collegato ai casi osservati inSpagna. A seguito della valutazionedi tutti i dati disponibili, il CHMPdeciderà se sono necessarie ulterioriazioni.
Clopidogrel e inibitori di pompa protonica
Che cosa è clopidogrel. Clopidogrel è un antiaggreganteutilizzato per prevenire eventi ate-rosclerotici nelle arteriopatie peri-feriche o dopo infarto acuto delmiocardio o dopo ictus ischemico.Nell’Unione Europea il clopidogrelè autorizzato con i seguenti nomicommerciali: Plavix, Iscover, Clopi-dogrel BMS e Clopidogrel Winth-rop.Il problema di sicurezza.La valutazione di recenti studi, chehanno esaminato gli esiti clinicidei pazienti in terapia con clopi-dogrel, ha suggerito la possibilità diun’interazione significativa tra clo-pidogrel e inibitori di pompa pro-tonica (PPI); questa interazione de-termina una minore efficacia delclopidogrel quando assunto inconcomitanza con i PPI. Una possibile spiegazione di que-sta interazione potrebbe essere chealcuni PPI impediscono la conver-sione del clopidogrel nella sua
forma biologicamente attiva, ini-bendone gli effetti antiaggregantipiastrinici e aumentando il rischiodi infarto cardiaco. Quali azioni ha intrapreso l’EMEA.A seguito della rivalutazione di tuttii dati disponibili, il CHMP ed ilPharmacovigilance Working Partydell’EMEA hanno raccomandatoche il Riassunto delle Caratteristi-che del Prodotto di tutti i medici-nali contenenti clopidogrel vengaaggiornato e che vengano incluse leinformazioni relative all’intera-zione con i PPI, specificando chel’uso concomitante con i PPI do-vrebbe essere evitato a meno chenon sia strettamente necessario. Il CHMP ritiene, inoltre, che sianonecessarie ulteriori informazioni suquesta interazione e sull’implica-zione della variante genetica pre-sente in una piccola popolazionedi individui (denominati“CYP2C19 metabolizzatori scarsi”)incapaci di convertire completa-mente il clopidogrel nella suaforma attiva.
Cerezyme e Fabrazyme:una carenza da gestire
Che cosa sono Cerezyme e Fabrazyme.Cerezyme (imiglucerasi) e Fa-brazyme (agalsidasi beta) sono far-maci utilizzati in terapia enzima-tica sostitutiva a lungo termine inpazienti con diagnosi confermatadi malattia di Gaucher e malattiadi Fabry (deficit di alfa-galattosidasiA), rispettivamente .Cerezyme è stato autorizzato a no-vembre 1997 e Fabrazyme ad ago-
sto 2001, entrambi sono commer-cializzati in tutti gli Stati Membridell’Unione Europea.Il problema di sicurezza.Nel corso dell’anno l’azienda cheproduce Cerezyme e Fabrazyme èvenuta a conoscenza di una dimi-nuita produzione da parte dei bio-reattori usati per produrre i due me-dicinali nel sito di produzione diAllston Landing, negli Stati Uniti.L’azienda ha trovato che i bioreat-tori erano stati contaminati da unvirus (calicivirus del tipo Vesivirus2117), che non è causa di malattianell’uomo, ma può attaccare le cel-lule usate per produrre questi far-maci. La contaminazione ha un im-patto sulla crescita delle cellule eintacca la quantità, ma non la qua-lità degli enzimi prodotti. Tutti ilotti preparati usando enzimi pro-dotti prima della contaminazionesono stati analizzati e l’EMEA haconfermato che essi possono esseredistribuiti sul mercato. L’azienda hachiuso lo stabilimento in attesa chevenga decontaminato. Mentre que-sto processo è in corso, Genzymenon può produrre alcun nuovolotto di Cerezyme o Fabrazyme. Lescorte esistenti di Cerezyme an-dranno ad esaurimento non primadi agosto 2009, e di ottobre 2009quelle di Fabrazyme.Raccomandazioni durante il periodo di carenzaCerezymeDurante il periodo di carenza, leraccomandazioni per il tratta-mento, concordate con l’EMEA,sono le seguenti:
i neonati, i bambini e gli adole-scenti devono assumere Cerezymealla dose e al tempo di infusionestabiliti, dato che tali “pazienti con
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
88

FARMACOVIGILANZA | NEWS
esordio precoce” presentano unaprogressione più rapida della ma-lattia e un rischio di gravi problemia lungo termine;
i pazienti adulti, con progres-sione della malattia (e.g. iperten-sione polmonare, malattia schele-trica attiva, grave trombocitopeniao grave anemia), devono assumereCerezyme in base alla dose e altempo di infusione approvati;
i pazienti adulti senza evidenzecliniche di una progressione dellamalattia devono assumere Ce-rezyme a una dose ridotta (e.g. ri-duzione del 50% della dose unavolta ogni due settimane) o a unaridotta frequenza di infusione (e.g.una volta al mese alla dose cor-rente). Nessun paziente deve es-sere trattato ad una dose inferiorea 15 unità/kg ogni 2 settimane. In questi pazienti devono esseremonitorati, come appropriato, icambiamenti dei livelli di emoglo-bina, piastrine e chitotriosidasi(inizialmente i livelli basali ed inseguito quelli bimestrali). Fabrazyme Durante il periodo di ridotta di-sponibilità del farmaco, le racco-mandazioni per il trattamento,concordate con l’EMEA, sono le se-guenti:
i bambini, gli adolescenti (<18anni) e i pazienti adulti di sessomaschile devono assumere Fa-brazyme in base alla dose e allafrequenza raccomandate;
i pazienti adulti di sesso femmi-nile, senza l’evidenza di una di-sfunzione clinicamente significa-tiva o di danno d’organo, possonoassumere Fabrazyme con un ag-giustamento della dose che deverisultare compresa tra 0,3-0,5
mg/kg ogni due settimane. In que-sti pazienti, è necessario monito-rare i cambiamenti dei livelli uri-nari di GL3 (inizialmente i livellibasali ed in seguito quelli bime-strali).
Insulina glargine e rischio di cancro
Che cosa è l’insulina glargine. Insulina glargine è un analogo del-l’insulina a lunga durata d’azione,autorizzato nell’Unione Europeacome Lantus e Optisulin, per il trat-tamento del diabete negli adulti, ne-gli adolescenti e nei bambini di seianni e oltre, quando si ritiene ne-cessario il trattamento con insulina. Il problema di sicurezza.L’EMEA sta esaminando quattrostudi pubblicati di recente eseguitiper analizzare una possibile rela-zione tra l’insulina glargine e il ri-schio di cancro. I risultati degli studi si sono dimo-strati inconsistenti. In due studi(Scottish Diabetes ResearchNetwork Epidemiology Group e Jo-nasson et al.) è stata trovata un’as-sociazione con il cancro al seno nelgruppo di pazienti che assumevanoinsulina glargine come monotera-pia, ma non è stata riscontrata nel-l’altro gruppo di pazienti che usavainsulina glargine insieme ad altritipi di insulina. Non è emersa al-cuna associazione con altri tipi dicancro. In questi due studi non èstata valutata la dose-dipendenza.Il terzo studio (Hemkens et al.) hariportato un’associazione dose-di-pendente tra l’uso di insulina glar-gine e le neoplasie, ma non vi sonoinformazioni sui tipi di neoplasie
trovate in questo studio. Nel quartostudio (Currie et al.), non è statatrovata alcuna associazione tra can-cro (sia al seno che al colon-retto,che al pancreas o alla prostata) el’uso di insulina glargine, o altri tipidi insulina. Sulla base dei dati attualmente di-sponibili, non può essere confer-mata né esclusa una relazione trainsulina glargine e cancro. Tuttavia,i problemi sollevati dai quattrostudi richiedono una valutazionepiù approfondita. Quali sono le conclusioni delCHMP.Il Comitato dei medicinali per usoumano (CHMP) dell’EMEA effet-tuerà una dettagliata valutazione deirisultati degli studi e di ogni altrainformazione rilevante. Questa re-visione valuterà anche i seguentiaspetti: gli effetti dose-risposta, leimplicazioni della durata degli studirelativamente breve e l’influenza dialtri fattori sul rischio di cancro alseno e altri tipi di cancro (e.g. età,indice di massa corporea [BMI],stato menopausale, numerositàdella prole, stato socio-economico).
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
89

sere esposti al rischio di contesta-zioni dovute alla difficoltà di clas-sificare un particolare prodottopresente sullo scaffale della pro-pria farmacia (vedi “definizione dimedicinale”, pag. 75). Inoltre, nonsi può escludere che alle oggettivedifficoltà di classificazione sisommi l’interesse di chi vede nellainevitabile parziale sovrapposi-zione tra le varie categorie, e nel-l’ambiguità che ne deriva, unattraente vantaggio in terminicommerciali per prodotti che po-trebbero essere confusi con i me-dicinali ma che sono ben lontani
dal rispondere agli stessi requisiti di qualità, sicurezzaed efficacia.
I rischi per la saluteIl mercato dei prodotti a base di erbe è in forte
espansione, tanto da risultarne una esposizione dellapopolazione tale da giustificare qualche preoccupa-zione per la salute pubblica2.
Tra le ragioni di tale successo va senza dubbio an-noverata la convinzione fortemente radicata tra gli uti-lizzatori, ma anche tra alcuni operatori del settore, checiò che è “naturale” è per definizione anche innocuo. Iprodotti a base di erbe agiscono invece con meccanismid’azione del tutto analoghi a quelli dei prodotti di sin-tesi e possono quindi presentare una specifica attivitàfarmacologica, così come effetti tossici o interazionicon i medicinali. Le problematiche di qualità, sicurezzaed efficacia connesse con l’uso delle preparazioni abase di erbe sono state recentemente discusse sulle pa-gine del Bif3, ponendo in evidenza come questi aspettiassumano un rilievo del tutto particolare quando sitratta di prodotti diversi dai medicinali. Infatti, ricor-diamo brevemente che nella maggior parte dei casi ladocumentazione relativa agli effetti positivi delle erbeè solo di tipo aneddotico e, quindi, di limitata affida-bilità; altrettanto modeste, inoltre, sono le conoscenze
Di cosa parliamoUna grande varietà di
prodotti di origine vege-tale può essere utilizzataper finalità diverse: ascopo alimentare, volut-tuario, estetico o terapeu-tico. Da qui la distinzionein categorie merceologichedifferenti che, nel corso deltempo, sono state discipli-nate giuridicamente te-nendo conto della destina-zione d’uso e dell’esigenzadi salvaguardare la salutepubblica.
A livello europeo è in corso il tentativo di mettere or-dine in queste norme, collocando le piante destinate al-l’uso umano nell’ambito degli alimenti, dei farmaci odei cosmetici. Non è stata al momento stabilita, in-vece, una collocazione specifica per i prodotti erbori-stici, poiché non viene previsto il riconoscimento diprodotti diversi, per finalità e presentazione, da quelliclassificabili in una delle categorie sopra indicate1. Ciò,inevitabilmente, complica il recepimento dell’orienta-mento emerso in sede europea nella normativa nazio-nale poiché, in Italia, la categoria dei prodotti erboristicidi fatto esiste ed è regolata da un Regio Decreto risalenteal 1931 e tuttora vigente2.
Anche se le norme verranno più chiaramente defi-nite, tuttavia, la confusione sullo scaffale del farmacistapotrebbe non essere definitivamente risolta. I confinitra le varie categorie, infatti, spesso tendono ad essereincerti, come dimostra la circostanza che, sovente, unastessa pianta trova impiego in vari ambiti. Ciò dipendedal fatto che la distinzione nei diversi gruppi merceo-logici non è basata sulla natura o composizione deiprodotti bensì in base alla loro presentazione, finalitàd’uso e al quantitativo e modo con cui vengono as-sunti.
La mancanza di una distinzione netta può rappre-sentare un motivo di confusione e preoccupazione pergli stessi farmacisti che possono, in qualche caso, es-
I prodotti di origine vegetale
BOLLETTINO D’INFORMAZIONE SUI FARMACI | NELLO SCAFFALE DEL FARMACISTA |
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
90

NELLO SCAFFALE DEL FARMACISTA | I PRODOTTI DI ORIGINE VEGETALE
sugli effetti sfavorevoli e sulle interazioni con altre so-stanze farmacologicamente attive. Per quanto attienealla sicurezza occorre considerare, altresì, che per i pro-dotti non medicinali non c’è l’obbligo di raccogliere etrasmettere all’autorità sanitaria le segnalazioni di rea-zioni avverse e comunque, poiché i rimedi a base dierbe sono ritenuti pregiudizialmente sicuri, c’è unascarsa probabilità che una reazione avversa venga attri-buita al rimedio stesso. A tale riguardo è emblematicoche, proprio a causa della diffusa percezione della loroinnocuità, essi vengano frequentemente utilizzati nellecondizioni maggiormente a rischio, quali l’età pedia-trica e la gravidanza, quando in realtà le informazionirelative agli effetti sul feto e sul bambino sono di solitoparticolarmente carenti.
A queste perplessità, relative all’azione farmacolo-gica e tossicologica proprie del rimedio naturale, va ag-giunto – per quanto attiene alla qualità – che i prodottia base di erbe che non sono autorizzati come medici-nali sono soggetti a controlli meno rigorosi sia sulle ma-terie prime che sul procedimento di preparazione. Diconseguenza, il loro uso espone ad un maggiore ri-schio, ad esempio, di variabilità del titolo delle so-stanze attive o di presenza di contaminanti, con tutte leripercussioni che tali aspetti possono poi determinaresul piano della sicurezza e dell’efficacia3.
Il ruolo del farmacista Si stima che, in Italia, almeno la metà dei prodotti
di origine vegetale sia venduta attraverso la farmacia2;molti dei medicinali a base di erbe, inoltre, non richie-dono la prescrizione medica e tutti i prodotti non clas-sificati come medicinali sono di libera vendita. Il far-macista, pertanto, è un interlocutore fondamentale peril paziente che desideri ricorrere ad un rimedio naturale,
ed è importante che egli metta a disposizione le proprieconoscenze e la propria professionalità – anche nel ri-spetto degli obblighi previsti dal codice deontologico –per guidare il consumatore ad un uso appropriato deiprodotti a base di erbe. Per raggiungere questo obiettivoil farmacista non dovrebbe dimenticare di:
informare il paziente che anche i prodotti a base dierbe possono provocare effetti indesiderati ed invi-tarlo, nel caso si verifichi questa eventualità, adinformare il proprio medico o farmacista. Segnalare la reazione avversa tramite la scheda messaa punto dall’Istituto Superiore di Sanità in accordocon l’AIFA (disponibile su: www.epicentro.iss.it), nelcaso il paziente riferisca disturbi che possano essereassociati all’assunzione di un prodotto a base di erbe.
Chiedere al paziente se fa uso di farmaci e/o altriprodotti naturali e, a seconda del caso, sconsigliarel’associazione o richiamare la sua attenzione sullapossibilità di eventuali interazioni.Sconsigliare l’uso o, comunque, consigliare la mas-sima cautela se il prodotto è destinato ad una donnain gravidanza o in allattamento oppure ad un bam-bino.Informare il paziente che i prodotti diversi dai me-dicinali hanno prove di efficacia limitate e che il ri-corso ad essi può in alcuni casi limitare, ritardare osostituire una terapia medica realmente efficace, conconseguenze tanto più serie quanto maggiore è lagravità della malattia.
Bibliografia1. Guidotti M. La fitoterapia e
l’erboristeria: legge n.99/1931 esuccessive regolamentazionigiuridiche. www.galenotech.org/index.html (accesso verificatoil 20/02/2009).
2. Silano M, Silano V. Prodotti di origine vegetale in medicina,alimentazione, erboristeria ecosmetica. Aspetti normativi,scientifici e tecnici relativi a qualitàsicurezza ed efficacia dei diversi usi delle piante officinali in Italia e nell’Unione Europea. Milano: Editore Tecniche Nuove, 2006.
3. Anonimo. Medicinali di originevegetale tradizionali. Le nuove regole.Bif 2007; 14: 195-9.
Anche i prodotti a base
di erbe possono provocare
effetti indesiderati.
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
91

A pprezzato da sempre come tipica e piacevolebevanda nelle popolari abitazioni delle cam-pagne cinesi e nei salotti occidentali1,2, il tè è
oggi utilizzato come bevanda salutistica, e sempre piùinteressa i farmacologi per le sue caratteristiche e do-cumentate attività antiossidanti sfruttabili anche in fi-toterapia e in medicina3. La specie utilizzabile è laCamellia sinensis Kuntze, tipica pianta di origini in-diane oggi in uso in tutto il mondo con differenti pre-ferenze per le sue molteplici varietà e i suoi aromi. Ivari tipi di tè si differenziano fondamentalmente indue gruppi per la tecnica di preparazione delle foglie:quelle nelle quali i naturali processi di ossidazione efermentazione enzimatica sono bloccati durante le fasidi preparazione forniscono il cosiddetto tè verde, chein questo si distingue dal tè nero, nel quale le foglie du-rante la fase di essiccamento si ossidano, diventanoscure, e sottoposte a fermentazione enzimatica in am-biente umido vengono poi essiccate. Tra gli altri inol-tre: il tè oolong il cui grado di ossidazione è intermedio,aromatico e pregiato, a basso contenuto di caffeina; iltè bianco, più raro, pregiato, dal sapore delicato e otte-nuto dai germogli della pianta; il tè bancha, non fer-mentato, più diffuso in Giappone, a bassissimocontenuto di caffeina; i tè verdi aromatizzati, al gelso-mino, alla menta, ecc.
Vengono comunque sempre utilizzate le foglie dipiante di almeno tre anni di età per la loro maggioreconcentrazione dei numerosi principi attivi:
alcaloidi (metil-xantine: caffeina, teofillina,teobromina): sono molto più concen-trati nel tè nero (caffeina: 40-70 mgper tazza) piuttosto che nel tè verde(caffeina 15-25 mg per tazza);polifenoli (flavonoidi, cate-chine, epicatechine, epigal-locatechin-3-gallato,e p i c a t e c h i n a - 3 -gallato): sono moltopiù concentrati,di almeno 5volte, nel tè
verde piuttosto che nel tè nero;polifenoli ossidati (teoflavine e teorubicine): sonopresenti solo nel tè nero;vitamina C: è presente in particolare nel tè verde;sostanze aromatiche, teatina, saponine, acidi orga-nici e minerali: sono presenti in maniera similenelle due varietà.
Le foglie del tè possono essere utilizzate sostanzial-mente secondo tre modalità:a. come tali per la tipica bevanda, preparata secondo
varie modalità. Le differenti varietà di tè fornisconobevande più o meno aromatiche, con sfumature dicolore, profumo e gusto, talvolta ben percettibilianche ai meno esperti, e preferite l’una all’altra inbase al gusto personale e alle abitudini di popola-zione;
b. come materia prima per l’estrazione di singole mo-lecole da sempre utilizzate in medicina: in particolarela teofillina, capostipite dei farmaci broncodilatatori(anche nella forma più solubile dell’aminofillina) ela caffeina, utilizzata a scopo antiemicranico. A dif-ferenza della caffeina la quantità di teofillina pre-sente nelle foglie è molto bassa;
c. in forma di estratti fitoterapici, presenti in inte-gratori alimentari, ma utilizzabili anche per medi-cinali galenici, titolati e standardizzati in polifenoli,la frazione più significativa, presente anche nel caffè
e nel cacao, con o senza caffeina. Per l’usomedicinale del tè non è possibile infatti ri-
correre alla prescrizione di tisane per l’am-pia variabilità dei principi attivi, la pre-senza di tannini e soprattutto di caffeina,
responsabile di effetti collateralie controindicazioni.
Attività biologicheNumerosi sono gli
studi presenti inletteratura di
tipo epide-miologico
Tè verde, e non solo
BOLLETTINO D’INFORMAZIONE SUI FARMACI | ROASTBIF |
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
92

FARMACOVIGILANZA | ROASTBIF | TÈ VERDE, E NON SOLO
(di coorte o caso-controllo), condotti in particolare sul-l’uso della bevanda, così come alcuni RCT, studi pre-clinici su colture cellulari e modelli animali condottiinvece con singoli polifenoli o estratti standardizzati,che dimostrano un’attività antiossidante, endotelio-protettiva e chemiopreventiva a carico soprattutto dellafrazione polifenolica. Sperimentalmente è stata stu-diata anche la farmacocinetica dei polifenoli del tèverde, che vengono assorbiti molto facilmente, specie adigiuno e a stomaco vuoto, mentre l’aggiunta di lattepuò ridurne l’assorbimento.
Interessanti sono anche le proprietà neuroprotettive,antimicrobiche ed antinvecchiamento sfruttabili in am-bito neurologico, ma anche nella cosmesi e nella pre-venzione di alcune lesioni cutanee oltre che della cariedentaria. Ad oggi, di maggiore interesse medico rimanetuttavia il ruolo che il tè verde può giocare nell’ambitodella prevenzione cardiovascolare ed oncologica.
Apparato cardiovascolareIn particolare, i polifenoli del tè ad attività antiossi-
dante si sono dimostrati utili nella prevenzione deidanni endoteliali da aterosclerosi o altri fattori di ri-schio (fumo, radicali liberi, ipercolesterolemia, ecc.), estudi epidemiologici anche recenti hanno dimostratoil ruolo protettivo e significativo nella riduzione del ri-schio di stroke che origina dall’assunzione di tè verde4.Del resto sono ben note le proprietà antiossidanti delcaffè, del cacao e del tè per l’apparato cardiovascolare eper il distretto coronarico in particolare, relative nontanto alla presenza di caffeina quanto di polifenoli5.Recenti dati sperimentali confermano l’importanza deipolifenoli del tè nella protezione dell’endotelio: lasomministrazione in acuto di tè verde, ma non dell’e-quivalente dose di caffeina, apporta benefici all’endo-telio6. Il consumo di tè induce rapidi miglioramenti deiparametri emoreologici e della funzione delle celluleprogenitrici dell’endotelio e può rappresentare un im-portante fattore di prevenzione nei fumatori. Alcunesostanze hanno pure dimostrato una certa attività an-tiaggregante piastrinica e antitrombotica. In un RCT èstata pure dimostrata la capacità di ridurre la quota delcolesterolo LDL di un estratto di tè verde titolato in po-lifenoli e teoflavine.
OncologiaLa ricerca ha confermato l’attività antiossidante,
pro-apoptotica e antiangiogenetica degli estratti di tèverde, soprattutto nella componente polifenolica, e inparticolare dell’epigallocatechina-3-gallato, in grado di
inibire la promozione del tumore in vari modelli spe-rimentali. I polifenoli agiscono con molteplici mecca-nismi sia nella chemioprevenzione sia nella fase diprogressione e diffusione del tumore: pro-apoptosi,down-regulation della telomerasi, attività antiangioge-nica per inibizione delle COX-2, del fattore NF-KB, delVEGF, della proteina Bcl-2 e delle metallo-proteine. Larecente letteratura e la valutazione critica degli studiepidemiologici consentono di confermare l’attivitàprotettiva dei polifenoli del tè verde, indicati anchenella prevenzione in pazienti fumatori, e come possi-bili agenti inibitori della diffusione tumorale7. Le evi-denze più significative riguardano il ruolo protettivonei confronti dell’insorgenza del carcinoma prostaticoe della mammella.
Un recente studio epidemiologico prospettico con-dotto su circa 50.000 uomini dal 1990 al 2004 ha mo-strato che l’assunzione di almeno 5 tazze al giorno ditè verde riduce in modo significativo il rischio di can-cro prostatico8. Sperimentalmente è stata dimostratal’elevata concentrazione di polifenoli del tè verde neltessuto prostatico, mentre ricercatori italiani9 ne hannostudiato il meccanismo d’azione e l’efficacia clinica: inun RCT, 600 mg/die di polifenoli del tè verde vs pla-cebo in un anno di trattamento hanno ridotto signifi-cativamente la progressione di carcinomi intraepitelialiprostatici.
Per quanto riguarda la donna, interessante può es-sere il ruolo del tè verde nella prevenzione del tumoreal seno: una recente metanalisi10 di studi epidemiolo-gici caso-controllo e di coorte ha dimostrato come l’as-sunzione di almeno 5 tazze/die di tè verde ne riducain maniera significativa l’incidenza. Sperimentalmenteil tè verde ha dimostrato di migliorare l’effetto inibito-rio del tamoxifene sulla crescita tumorale e attenua laresistenza al tamoxifene. Un recente grande studio pro-spettico di coorte ha dimostrato che l’assunzione di tèverde protegge dall’insorgenza del cancro all’ovaio,anche se in letteratura permangono studi con dati con-trastanti.
Ampie divergenze tra i dati emergono anche tra i 25studi epidemiologici analizzati in una revisione siste-matica sul tumore del colon retto10. Per quanto con-cerne il cancro gastrico i risultati rimangono contrad-dittori e non consentono un giudizio definitivo. In ungruppo di 59 pazienti affetti da leucoplachia del cavoorale e partecipanti ad un trial clinico controllato versoplacebo, la somministrazione per 6 mesi di un estrattodi tè verde ha ridotto in modo significativo, rispetto alplacebo, le dimensioni delle lesioni del cavo orale. Percontro non possono essere trascurati i dati sull’au-mento dell’incidenza di tumore dell’esofago, proba-
AGENZIA ITALIANA DEL FARMACO | BIF XVI N.2 2009
93

FARMACOVIGILANZA | ROASTBIF | TÈ VERDE, E NON SOLO
bilmente dovuto all’alta temperatura della bevanda. In conclusione, in base ai dati disponibili relativi
alla ricerca epidemiologica, farmacologica e clinica, gliestratti di tè verde possono avere un ruolo anche nellaterapia complementare e preventiva del carcinoma pro-statico. È raccomandato l’uso di estratti purificati, tito-lati e standardizzati in polifenoli.
AvvertenzeI rischi da sovradosaggio di caffeina (tremori, in-
sonnia, palpitazioni, ecc.) si corrono solo nel caso diassunzione di molte tazze al giorno di tè, soprattuttoquello nero. È possibile anche una riduzione dell’as-sorbimento del ferro11.
Non ci sono evidenze circa l’epatotossicità di alcuniestratti di tè verde, attribuita a casualità12.
Non esistono interferenze clinicamente significativesulle varie isoforme del CYP 450, mentre importante è
la dimostrazione sperimentale di come il tè verde ri-duca l’efficacia del bortezomib, un farmaco utilizzatonella terapia del mieloma multiplo13.
Concludendo, dopo millenni di uso comune diquesta gradevolissima bevanda, sta ancora al medicostabilire se, quando e come sfruttare le proprietà bio-logiche del tè verde a scopo strettamente curativo o pre-ventivo, con l’obiettivo di trarne i maggiori benefici elimitarne al massimo i rischi e le interazioni farmaco-logiche.
Bibliografia1. Massi C. È l’ora del tè.
Milano: Guido TommasiEd., 2005.
2. AAVV. Tè, bevandaeuforizzante, infusoterapeutico, disciplinatopiacere. Firenze: BonechiEd., 2008.
3. Firenzuoli F. Fitoterapia. 4a ed. Milano: Masson Ed.,2008.
4. Kuriyama S, Shimazu T,Ohmori K, et al. Green tea consumption andmortality due tocardiovascular disease,cancer, and all causes inJapan: the Ohsaki study.JAMA 2006; 296: 1255-65.
5. Gensini GF, Lippi D, Conti AA. Storia e attualitàdel tè verde: da bevandavoluttuaria a farmaco incompresse? Recenti ProgrMed 2007; 98: 347-51.
6. Alexopoulos N,Vlachopoulos C,Aznaouridis K, et al. The acute effect of greentea consumption onendothelial function inhealthy individuals. Eur JCardiovasc Prev Rehabil2008; 15: 300-5.
7. Garbisa S, Sartor L, BigginS, et al. Tumor gelatinasesand invasion inhibited bythe green tea flavanolepigallocatechin-3-gallate.Cancer 2001; 91: 822-32.
8. Kurahashi N, Sasazuki S,Iwasaki M. Green teaconsumption and prostatecancer risk in Japanesemen: a prospective study.Am J Epidemiol 2008; 167: 71-7.
9. Bettuzzi S, Brausi M, RizziF, et al. Chemopreventionof human prostate cancerby oral administration ofgreen tea catechins involunteers with high-gradeprostate intraepithelialneoplasia: a preliminaryreport from a one-yearproof-of-principle study.Cancer Res 2006; 66: 1234-40.
10. Sun CL, Yuan JM, Koh WP,Yu MC. Green tea, blacktea and breast cancer risk:a meta-analysis ofepidemiological studies.Carcinogenesis 2006; 27: 1310-5.
11. Firenzuoli F. Interazioni tra erbe, alimenti efarmaci. 2a ed. Milano:Tecniche Nuove Ed., 2009.
12. Mazzanti G, Menniti-Ippolito F, Moro PA, et al.Hepatotoxicity from greentea: a review of theliterature and twounpublished cases. Eur JClin Pharmacol 2009; 65: 331-41.
13. Golden EB, Lam PY,Kardosh A, et al. Green teapolyphenols block theanticancer effects ofbortezomib and otherboronic acid-basedproteasome inhibitors.Blood 2009. Prepublishedonline Feb 3, 2009; DOI10.1182/blood-2008-07-171389.
Il tè verde riduce l’efficacia
del bortezomib, un farmaco
utilizzato nella terapia
del mieloma multiplo.
BIF XVI N.2 2009 | AGENZIA ITALIANA DEL FARMACO
94

La g
uida
alla
com
pila
zion
e è
cons
ulta
bile
on
line
all’i
ndir
izzo
ww
w.a
genz
iafa
rmac
o.it
Informazioni sul farmaco
Informazioni sulla segnalazione
Scheda unica di segnalazione di sospetta reazione avversa
Da compilarsi a cura dei medici o degli altri operatori sanitari e dainviare al Responsabile di farmacovigilanza della struttura sanitaria di appartenenza
Iniziali del paziente Data di nascita Sesso Data insorgenza reazione Origine etnica Codice segnalazione
Descrizione reazione ed eventuale diagnosi (se il segnalatore è un medico)
Eventuali esami di laboratorio rilevanti per ADR (riportare risultati e data degli accertamenti)
Azioni intraprese (specificare). In caso di sospensione compilare i campi da 16 a 19
Farmaco (i) sospetto (i). Nome della specialità medicinale. In caso di vaccini specificare il numero di dosi e/o di richiamo e ora di somministrazione
A. ............................................................................. 12. Lotto ............................................ 13. Dosaggio/die ..............................................
14. Via di somministrazione 15. Durata uso da ........./........./......... a ........./........./.........
B. ............................................................................. 12. Lotto ............................................ 13. Dosaggio/die ..............................................
14. Via di somministrazione 15. Durata uso da ........./........./......... a ........./........./.........
C. ............................................................................. 12. Lotto ............................................ 13. Dosaggio/die ..............................................
14. Via di somministrazione 15. Durata uso da ........./........./......... a ........./........./.........
16. Il farmaco è stato sospeso? A. sì no B. sì no C. sì no17. La reazione è migliorata dopo la sospensione? A. sì no B. sì no C. sì no18. Il farmaco è stato assunto nuovamente? A. sì no B. sì no C. sì no19. Sono ricomparsi i sintomi dopo la somministrazione? A. sì no B. sì no C. sì no
Indicazioni o altro motivo per cui il farmaco è stato utilizzato
A. .........................................................................................................................................................................................................................
B. .........................................................................................................................................................................................................................
C. .........................................................................................................................................................................................................................
Qualifica segnalatore
Medico di Medicina Generale Pediatra di Libera Scelta
Medico Ospedaliero Farmacista
Specialista Altro
Data compilazione Codice ASL
Firma segnalatore
......................................................................................................
Farmaco (i) concomitante (i), dosaggio, via di somministrazione, durata trattamento
Uso concomitante di altri prodotti a base di piante officinali, omeopatici, integratori alimentari ecc. (specificare)
Condizioni concomitanti predisponenti (se il farmaco sospetto è un vaccino, riportare anamnesi ed eventuali vaccini somministrati nelle 4 settimane precedenti la somministrazione
Gravità della reazione
Grave
Decesso
Ospedalizzazione o prolungamento osp.
Invalidità grave o permanente
Pericolo di vità
Anomalie congenite/deficit neonato
Non grave
Dati segnalatore
Nome e cognome .................................................................................................
Indirizzo ................................................................................................................
Tel e fax ................................................................................................................
E-mail ....................................................................................................................
Firma responsabile di Farmacovigilanza
..............................................................................................................................
Esito
Risoluzione completa ADR il ......./......./.......
Risoluzione con postumi
Miglioramento
Reazione invariata o peggiorata
Decesso il ......./......./.......
Dovuto alla reazione avversa
Il farmaco può avere contribuito
Non dovuto al farmaco
Causa sconosciuta
Non disponibile
1
6
8
10
11
20
21
22
23
24 25
2927
26 28
2 3 4 5
7
9

52 La nota 90
61 “Deregulation” del trattamentofarmacologico del sovrappeso e rischi correlati ai disturbi del comportamento alimentare
64 Somministrazione di vaccinicontenenti tiomersal e svilupponeuropsicologico del bambino
83 Reazioni di fotosensibilizzazioneda ketoprofene
Ann
o X
VI
N.2
Mar
zo-a
prile
200
9 B
imes
tral
e
Bollettino d’Informazione sui Farmaci
2/09
Bimestrale dell’Agenzia Italiana del Farmaco
La pubblicazione del Bif proseguirà regolarmente sul portale dell'AIFA (www.agenziafarmaco.it) e tramite il Bif online (www.bif-online.it).
AVVISO AI LETTORI
Si avvisano i lettori che il Bollettinod'Informazione sui Farmaci nonpotrà essere edito e recapitato come di consueto per problemi legati al servizio di stampa e distribuzione della rivista.

Il Bif raddoppia
È un’iniziativa dell’Agenzia Italiana del Farmacowww.agenziafarmaco.it
Visita il sito www.bif-online.it e iscriviti alla newsletter.
Bif online non è solo la versione online del Bollettino di Informazione sui Farmaci ma molto di più: commenti, interviste, notizie, sondaggi.

EDITORIALE 49 L’uso dei farmaci in Italia
Pubblicato il Rapporto OsMed 2008
PANORAMI E PERCORSI52 La nota 90
Definiti i casi in cui può essererimborsato il metilnaltrexone
54 Il trattamento della degenerazionemaculare neovascolare: le regole del gioco Condizioni e limiti per una prescrizione corretta
AGGIORNAMENTI56 Fibromialgia, controversie
e uso off-labelFDA ed EMEA divergono sull’uso di antiepilettici e antidepressivi nella fibromialgia
61 “Deregulation” del trattamentofarmacologico del sovrappeso e rischi correlati ai disturbi del comportamento alimentareL’importanza di una informazionecorretta sui farmaci per i DCA
64 Somministrazione di vaccinicontenenti tiomersal e svilupponeuropsicologico del bambinoImprobabile o clinicamente non rilevante l’associazione travaccinazioni e deficit neuropsicologici
70 Il punto sulle complicanzetrombotiche associate all’uso dei contraccettivi estro-progestiniciLe conclusioni della ConferenzaNazionale di Consenso
Bif watch76 Sintesi da NEJM e Lancet
FARMACOVIGILANZA79 Le segnalazioni
di sospette reazioni avverse in età pediatrica, anno 2008Aumentano le segnalazioni ma quelleprovenienti dai pediatri sono poche
83 Reazioni di fotosensibilizzazione da ketoprofeneUn effetto indesiderato frequente e spesso grave
Farmacovigilanza news86 Dear Doctor Letter87 Rasilez: nuove restrizioni d’uso87 Gardasil, un bilancio
tra rischi e benefici88 Clopidogrel e inibitori
di pompa protonica88 Cerezyme e Fabrazyme:
una carenza da gestire89 Insulina glargine
e rischio di cancro
NELLO SCAFFALE DEL FARMACISTA93 I prodotti di origine vegetale
L’importanza del ruolo del farmacistanel consumo dei prodotti a base di erbe
Roastbif90 Tè verde, e non solo
A proposito di...53 Isotretinoina60 Terapia del dolore75 Definizione di medicinale78 Medicinali per le terapie avanzate
Bollettino d’Informazione sui FarmaciBimestrale dell’Agenzia Italiana del Farmaco
www.agenziafarmaco.it
2/09
Related Documents











