Gen. Pharmac. Vol. 27, No. 1, pp. 55-63, 1996 Copyright © 1996 Elsevier Science Inc. Printed in the USA. ELSEVIER ISSN 0306-3623/96 $15.00 + .00 SSDI 0306-3623(95)00028-3 All rights reserved REVIEW Blood Pressure Regulation By The Kallikrein-Kinin System* J. N. Sharma,? K. Urea, A. R. Noor and A. R. A. Rahman DEPARTMENTOF PHARMACOLOGY, SCHOOL OF MEDICAL SCIENCES,UNIVERSITI gAINSMALAYSIA, 16150 KUBANG KERIAN,KELANTAN, MALAYSIA ABSTRACT. 1. The kallikrein-kinin system has a significant role in regulating arterial blood pressure. 2. Reduced formation of the kinin compontents may cause hypertensive diseases. This is because of the fact that this system is responsible for vasodilatation, reduction in total peripheral resistance, natriuresis, diuresis, increasing renal blood flow and releasing various vasodilator agents. 3. Reduced kinin-kallikrein generation in hypertensive subjects may also be associated with genetic and environmental defects. 4. The kallikrein-kinin system when administered to hypertensive patients can lower their raised blood pressure to normotensive levels. 5. The mode of action of angiotensin-converting enzyme inhibitors principally may be dependent on the kinin system protection. OEN PHARMAC27;1:55-63, 1996. INTRODUCTION Kinins are potent vasorelaxantpeptides generated in arterial and venous circulation and cause an important contribution to blood pressure (BP) homeostasis (Sharma, 1984, 1988). Kinins can induce hypotension, diuresis, natriuresis, increased renal blood flow, vasodilatation and reduction in peripheral resistance (de Freitas et al., 1964; Willis et al., 1969; Mills 1982; Mohsin et al., 1992). The observation that urinary excretion of tissue kallikrein was highly reduced in untreated hyperten- sive patients was described as early as 1934 (Elliot and Nuzum, 1934) and confirmed more than three decades later (Margolius et al., 1971; 1974; Carretero and Scicli, 1971). In experimental hypertensive models, urinary kallikrein activity is also found to be reduced (Croxatto and San Martin, 1970; Lechi et al., 1978; Carretero et al., 1978; Arbeit and Serra, 1985). These findings are viewed as an index of reduced kinin formation in hypertensive conditions. A recent study involving 57 Utah subjects' pedigrees indicated that a dominant allele expressed as high urinary kallikrein excretion may be associated with decreased risk of essential hypertension (Berry et al., 1989). In fact, hypertensive patients can be treated by oral administra- tion of hog pancreatic kallikrein (Overlack et al., 1981; Ogawa et al., 1985). Tissue kallikrein has been associated with BP regulation by restriction fragment length polymorphisms (Woodly-Miller et al., 1989) and cosegregation of high BP with a restriction fragment length poly- morphism marking the kallikrein gene family in a hypertensive rat model (Pravenec et al., 1991). Also, human tissue kallikrein induces hypotension in transgenic mice (Wang et al., 1994). This finding raises the possibility of tissue kallikrein being a potent modulator of BP. The object of this review is to discuss the possible role of the kallikrein-kinin system (KKS) in relation to BP regulation. HISTORICAL ASPECT OF KKS The discovery of the KKS started when Abelous and Bardier (1909) detected the presence of a hypotensive substance in normal human urine that they called urohypotensin. However, investigation of the *This work is dedicated to honor Professor W. C. Bowman, a distinguished pharmacologist,University of Strathclyde,Glasgow,who willbe retiringin 1995. t To whom all correspondenceshould be addressed. Received 20 December 1994. kinin-forming system properly may be said to date from 1926, when Frey and his colleagues extensively studied intravenous injection of pancreatic extract, pancreatic secretion and urine in anesthetized nor- motensive dogs that produced a fall in arterial blood pressure (Frey, 1926; Frey et al., 1930; Frey and Werle, 1933). On the assumption that the active principle in urine was identical with that in pancreas, they named it kallikrein from the Greek word for pancreas. Later, it was demonstrated that kallikrein itself did not cause contractions of isolated guinea pig ileum (Werle et al., 1937). These investigators also observed that when kallikrein was incubated with plasma, a potent smooth muscle-stimulating substance was released. This active agent was of low molecular weight and thermostable. It was not a split product of kallikrein but a split product of a plasma protein. The agent, which Werle and co-workers (1937) thought to be a polypeptide, was initially called Darmkontrahierende substanz (gut-contracting substance or Substanz DK). Werle and Berek (1948) renamed the smooth muscle stimulant kallidin and suggested kallidinogenfor the inactive precursor protein present in the plasma. They concluded that the pharmacologi- cally active substance kallidin was released from its precursor(s) by the proteolytic action of the enzyme, kallikrein. Independently of the work of Werle in Germany, in Brazil, Rocha e Silva and colleagues (1949) found that incubating dog plasma with venom of certain snakes and with the enzyme trypsin produced an agent, probably a polypeptide, that also lowered BP and caused a slowly developing contraction of the guinea pig ileum in vitro. Because of the slow contraction of the guinea pig ileum, they named it bradykinin (BK) from the Greek word"slow moving." Because BK and kallidin were formed under similar conditions and had the same pharmacological actions, it was suspected that they were closely related and were derived from the same substrate (Werle et al., 1953). The purification of these substances 7 yr later confirmed this suspicion. Elliott et al. (1960) isolated BK formed by reacting trypsin with globulin, and it was synthesized by Boissonnas et al. (1960). KININS The kinin family mainly includes BK, kallidin and methionyl-lysyl-BK (Fig. 1). These are biologically active peptides derived from circulating precursors (kininogens) by the action of serine proteases, termed kalli-

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Gen. Pharmac. Vol. 27, No. 1, pp. 55-63, 1996 Copyright © 1996 Elsevier Science Inc. Printed in the USA.
ELSEVIER
ISSN 0306-3623/96 $15.00 + .00 SSDI 0306-3623(95)00028-3
All rights reserved
REVIEW Blood Pressure Regulation By The Kallikrein-Kinin System*
J. N. Sharma,? K. Urea, A. R. Noor and A. R. A. Rahman DEPARTMENT OF PHARMACOLOGY, SCHOOL OF MEDICAL SCIENCES, UNIVERSITI gAINS MALAYSIA,
16150 KUBANG KERIAN, KELANTAN, MALAYSIA
ABSTRACT. 1. The kal l ikrein-kinin system has a significant role in regulating arterial blood pressure. 2. Reduced formation of the kinin compontents may cause hypertensive diseases. This is because
of the fact that this system is responsible for vasodilatation, reduction in total peripheral resistance, natriuresis, diuresis, increasing renal blood flow and releasing various vasodilator agents.
3. Reduced kinin-kall ikrein generation in hypertensive subjects may also be associated with genetic and environmental defects.
4. The kall ikrein-kinin system when administered to hypertensive patients can lower their raised blood pressure to normotensive levels.
5. The mode of action of angiotensin-converting enzyme inhibitors principally may be dependent on the kinin system protection. OEN PHARMAC 27;1:55-63, 1996.
I N T R O D U C T I O N
Kinins are potent vasorelaxantpeptides generated in arterial and venous circulation and cause an important contribution to blood pressure (BP) homeostasis (Sharma, 1984, 1988). Kinins can induce hypotension, diuresis, natriuresis, increased renal blood flow, vasodilatation and reduction in peripheral resistance (de Freitas et al., 1964; Willis et al., 1969; Mills 1982; Mohsin et al. , 1992). The observation that urinary excretion of tissue kallikrein was highly reduced in untreated hyperten- sive patients was described as early as 1934 (Elliot and Nuzum, 1934) and confirmed more than three decades later (Margolius et al., 1971; 1974; Carretero and Scicli, 1971). In experimental hypertensive models, urinary kallikrein activity is also found to be reduced (Croxatto and San Martin, 1970; Lechi et al. , 1978; Carretero et al., 1978; Arbeit and Serra, 1985). These findings are viewed as an index of reduced kinin formation in hypertensive conditions.
A recent study involving 57 Utah subjects' pedigrees indicated that a dominant allele expressed as high urinary kallikrein excretion may be associated with decreased risk of essential hypertension (Berry et al., 1989). In fact, hypertensive patients can be treated by oral administra- tion of hog pancreatic kallikrein (Overlack et al., 1981; Ogawa et al., 1985). Tissue kallikrein has been associated with BP regulation by restriction fragment length polymorphisms (Woodly-Miller et al., 1989) and cosegregation of high BP with a restriction fragment length poly- morphism marking the kallikrein gene family in a hypertensive rat model (Pravenec et al., 1991). Also, human tissue kallikrein induces hypotension in transgenic mice (Wang et al. , 1994). This finding raises the possibility of tissue kallikrein being a potent modulator of BP. The object of this review is to discuss the possible role of the kallikrein-kinin system (KKS) in relation to BP regulation.
HISTORICAL ASPECT OF KKS
The discovery of the KKS started when Abelous and Bardier (1909) detected the presence of a hypotensive substance in normal human urine that they called urohypotensin. However, investigation of the
*This work is dedicated to honor Professor W. C. Bowman, a distinguished pharmacologist, University of Strathclyde, Glasgow, who will be retiring in 1995.
t To whom all correspondence should be addressed. Received 20 December 1994.
kinin-forming system properly may be said to date from 1926, when Frey and his colleagues extensively studied intravenous injection of pancreatic extract, pancreatic secretion and urine in anesthetized nor- motensive dogs that produced a fall in arterial blood pressure (Frey, 1926; Frey et al., 1930; Frey and Werle, 1933). On the assumption that the active principle in urine was identical with that in pancreas, they named it kallikrein from the Greek word for pancreas. Later, it was demonstrated that kallikrein itself did not cause contractions of isolated guinea pig ileum (Werle et al. , 1937). These investigators also observed that when kallikrein was incubated with plasma, a potent smooth muscle-stimulating substance was released. This active agent was of low molecular weight and thermostable. It was not a split product of kallikrein but a split product of a plasma protein. The agent, which Werle and co-workers (1937) thought to be a polypeptide, was initially called Darmkontrahierende substanz (gut-contracting substance or Substanz DK). Werle and Berek (1948) renamed the smooth muscle stimulant kallidin and suggested kallidinogen for the inactive precursor protein present in the plasma. They concluded that the pharmacologi- cally active substance kallidin was released from its precursor(s) by the proteolytic action of the enzyme, kallikrein.
Independently of the work of Werle in Germany, in Brazil, Rocha e Silva and colleagues (1949) found that incubating dog plasma with venom of certain snakes and with the enzyme trypsin produced an agent, probably a polypeptide, that also lowered BP and caused a slowly developing contraction of the guinea pig ileum in vitro. Because of the slow contraction of the guinea pig ileum, they named it bradykinin (BK) from the Greek word"slow moving." Because BK and kallidin were formed under similar conditions and had the same pharmacological actions, it was suspected that they were closely related and were derived from the same substrate (Werle et al., 1953). The purification of these substances 7 yr later confirmed this suspicion. Elliott et al. (1960) isolated BK formed by reacting trypsin with globulin, and it was synthesized by Boissonnas et al. (1960).
KININS
The kinin family mainly includes BK, kallidin and methionyl-lysyl-BK (Fig. 1). These are biologically active peptides derived from circulating precursors (kininogens) by the action of serine proteases, termed kalli-

56 J.N. Sharma et al.
I 2 3 4 5 6 7 8 {)
A r g - P r o - P r o - G l y - P h e - S e r - P r o - P h e - A r g Bradykin in
i , y s * A r g - P r o - P r o - G l y - P h e - S e t - P r o - P h e - A r g Kal l id in ( L y s y l - b r a d y k i n i n )
M e l - I ,ys - A r g - P r o - P r o - G l y - P h e - S e t - P r o - P h e - A r g M e l h i o n y l - l y s y l - b r a d y k i n i n
F I G U R E 1 . S t r u c t u r e o f i m p o r t a n t k i n i n s .
kreins (see Sharma, 1988a,b). Once released into the circulation, kinins are rapidly (< 15 sec) inactivated by a group of enzymes (Erdos, 1990) called kininases. BK and related kinins can act on four types of receptors, designated as BI, B2, B3 and B4 (Table 1), in inducing numerous physio- pathological processes. The kinin receptor stimulation can cause activa- tion of several second-messenger systems, such as arachidonic acid products, calcium, cyclic AMP and cyclic GMP (Freay et al., 1989; Burch, 1990; Schini et al., 1990). These systems have vital importance to the pharmacological activities ofkinins. The mode of kinin formation is presented in Fig. 2.
KALLIKREINS
Kinin-forming enzymes are known as kallikreins, which are divided into two types: plasma and tissue (organ). These two kallikreins differ in their molecular weights, origin, biochemical properties and biological actions on plasma kininogens (Schachter, 1980). Plasma prekallikrein circulates in an inactive state and is also known as the Fletcher factor, because the deficiency of prekallikrein was first noticed by Wuepper (I 973) in a patient named Fletcher. Prekallikrein is a single-chain glyco- protein synthesized in the liver. It is present in plasma as a complex bound with the high molecular weight kininogen (HMWK) (Mandle et al., 1976; Mandle and Kaplan, 1977). Inactive prekallikrein can be activated to form kallikrein by activated Hageman factor (HFa or factor XIIa), and HMWK is digested to release BK (Mandle et al., 1976; Kaplan et al., 1992). Also, inactive HFa becomes active by kallikrein through a positive feedback reaction (Cochrane et al. , 1973). Activated HFa and plasma thromboplastin antecedent (factor XI) circulate bound to HMWK (Thompson et al., 1977). Inactive factor XI is, therefore, con- verted to active factor XIa through HMWK to participate in the intrinsic coagulation pathway (Ratnoff et al., 1961).
Plasma kallikrein acts on HMWK to produce BK, whereas tissue kallikrein releases kallidin by the action on both HMWK and low molecular weight kininogen (LMWK) (Jacobsen, 1966; Pierce and Guim-
• - Ilqmm F.~or (Ime~)
Plasma l~lmllil~in Hagcun Faclm" (llmCli~) (Acfi'~)
~ HMW- Kinmogen I~ HMW- Kinmol~l
Plasma Kallik~in F*nclm Xl ~ Fac~" XIA (Active)
T c ~ HMW Kinmogen ~_
T Bmdlkinin ,.~
Bmdykinin Recept~'~ Activation ~ BiologicMActiom
(B,, B2, B:+md B4 )
T ~ I~kMli~.in (Imt~ive)
T ~ m e ~
!
FIGURE 2. Mode of k i n i n f o r m a t i o n i n t h e b o d y .
araes, 1977). Plasma kallikrein is a member of single gene code (Seidah et al., 1989).
Tissue kallikrein is a single-chain acidic glycoprotein that differs physicochemically and immunologically from plasma kallikrein. This kallikrein is found to be more widely distributed in the tissues (organs), such as kidney (urine), pancreas, salivary glands, intestine, prostate gland and the synovial tissue (Nustad et al., 1975; Zeitlin, 1971; Amund- sen and Nustad, •965; Sharma et al., 1983; A1-Haboubi et al., 1986).
Various organs that release tissue kallikreins are immunologically identical within a given species (Schachter, 1980). The pancreatic and kidney (urinary) kallikreins are detected in the inactive form as prokalli- krein (Fiedler and Werle, 1967; Matsas et al., 1981; Corthorn et al., 1979), whereas the submandibular tissue kallikrein exists in an active form (Brandtzaeg et al., 1976). The occurrence and distribution of the tissue kallikrein genes have been evaluated extensively (see Drinkwater et al., 1988). Investigations on the complementary DNA cloning and sequence analysis revealed that human pancreatic and renal kallikreins possess identical amino acid sequences and no more than three closely related genes (Baker and Shine, 1985; Schedlich etal . , 1987), in contrast with the situation in the mouse, in which the kallikrein group of serine proteases consist of 24 highly homologous genes (Evans et al., 1987). In human, tissue kallikrein gene family comprises the hRKALL, hGK- 1, and PAS. These are clustered on the long arm of chromosome 19 q13.3- 13.4 (Evans et al., 1988; Morris, 1989; Digby et al., 1989; Clements, 1994). However, in both mouse and rat, kallikrein activity is associated with the highly conserved members of a large multigene family located on chromosome 7 (Richards et al., 1982; Mason et al., 1983; Schedlich et al., 1988). Furthermore, research directed to investigate the structure
TABLE 1. B i o l o g i c a l p r o p e r t i e s o f B K r e c e p t o r s
B r a d y k i n i n r e c e p t o r F u n c t i o n A n t a g o n i s t s
Bi (formed by de novo synthesis in isolated arotic vascular smooth muscle and by patho- logical states in vivo)
B2
8 3
B:
Stimulation of smooth muscle, increased cell proliferation, increased collagen synthesis, contraction of venous and arterial preparation, in vitro and relaxation of peripheral resistance vessels in vivo, EDRF and PGI2 release from aortic endothelial cells.
Stimulation of rat uterus, cat and uinea pig ileum, mediation of pain and vasodilation, increased vascular permeability, hypotension, release of histamine and PGs, relaxation of arteries and contraction of vein, bronchoconstriction, EDRF and PGI2 release from aortic endo- thelial cells.
Contraction of airways, opossum esophageal longitudinal with rapid desensitization (action involves PGs).
Contraction of opossum esophageal longitudinal muscle with no tachy- phylaxis (action does not involve PGs).
Des-Arg%[LeuS]-BK
0 3 58 7 D-Arg -[Hyp -Thi ' ,D-Phe ]-BK [Th:.8,D-PheT]-BK D-Arg;[Hyp3,ThiS-D-TicT,Oic8]-BK
3 5 7 8 D-Arg[Hyp -Thi -D-Tic -Tic ]-BK
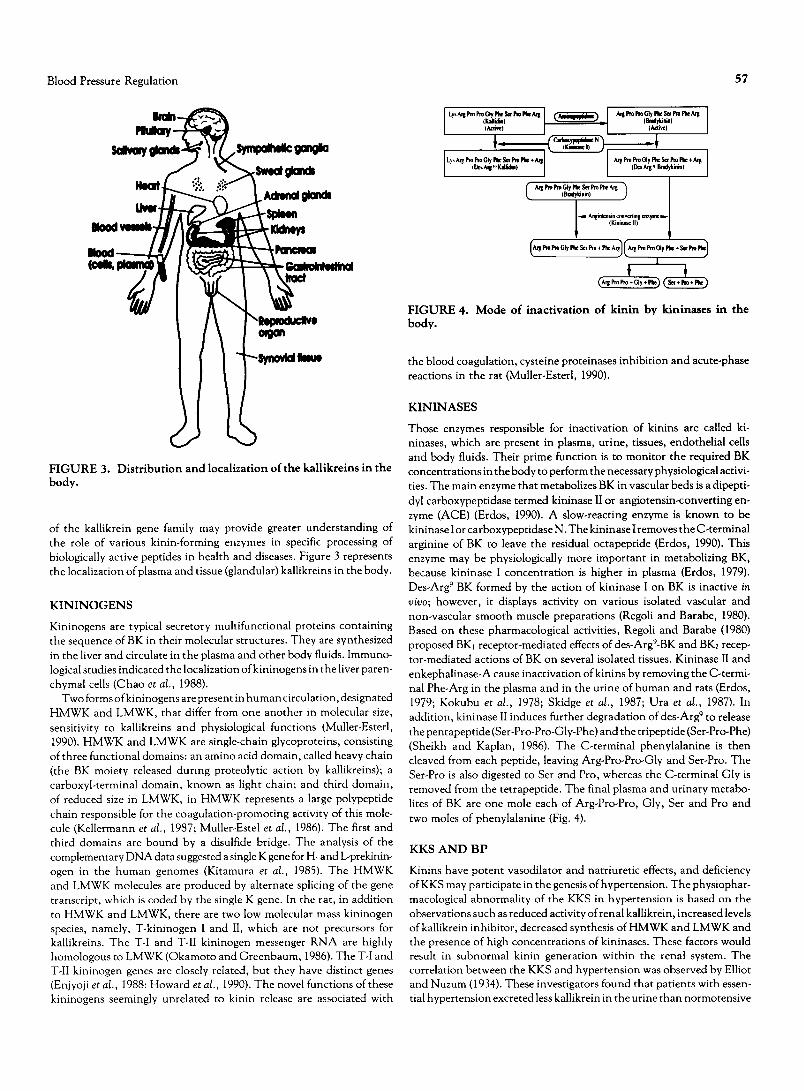
Blood Pressure Regulation 57
FIGURE 3. Distribution and localization of the kallikreins in the body.
of the kallikrein gene family may provide greater understanding of the role of various kinin-forming enzymes in specific processing of biologically active peptides in health and diseases. Figure 3 represents the localization of plasma and tissue (glandular) kallikreins in the body.
KININOGENS
Kininogens are typical secretory multifunctional proteins containing the sequence of BK in their molecular structures. They are synthesized in the liver and circulate in the plasma and other body fluids. Immuno- logical studies indicated the localization ofkininogens in the liver paren- chymal cells (Chao et al., 1988).
Two forms ofkininogens are present in human circulation, designated HMWK and LMWK, that differ from one another in molecular size, sensitivity to kallikreins and physiological functions (Muller-Esterl, 1990). HMWK and LMWK are single-chain glycoproteins, consisting of three functional domains: an amino acid domain, called heavy chain (the BK moiety released during proteolytic action by kallikreins); a carboxyl-terminal domain, known as light chain; and third domain, of reduced size in LMWK, in HMWK represents a large polypeptide chain responsible for the coagulation-promoting activity of this mole- cule (Kellermann et al., 1987; Muller-Estel et al., 1986). The first and third domains are bound by a disulfide bridge. The analysis of the complementary DNA data suggested a single K gene for H- and L-prekinin- ogen in the human genomes (Kitamura et al., 1985). The HMWK and LMWK molecules are produced by alternate splicing of the gene transcript, which is coded by the single K gene. In the rat, in addition to HMWK and LMWK, there are two low molecular mass kininogen species, namely, T-kininogen I and II, which are not precursors for kallikreins. The T-I and T-II kininogen messenger RNA are highly homologous to LMWK (Okamoto and Greenbaum, 1986). The T-I and T-II kininogen genes are closely related, but they have distinct genes (Enjyoji et al., 1988: Howard et al., 1990). The novel functions of these kininogens seemingly unrelated to kinin release are associated with
(IcJi~ l) ) I L~ Argl~o~oGly~$¢*~ Am ArsPmProGlyP,~SerProF~+A ~ ~ (De~r~.,KaJSdko +.-m L. (D.~us~ Bradykiil)
C I Bi'adykinin ) ,}
I
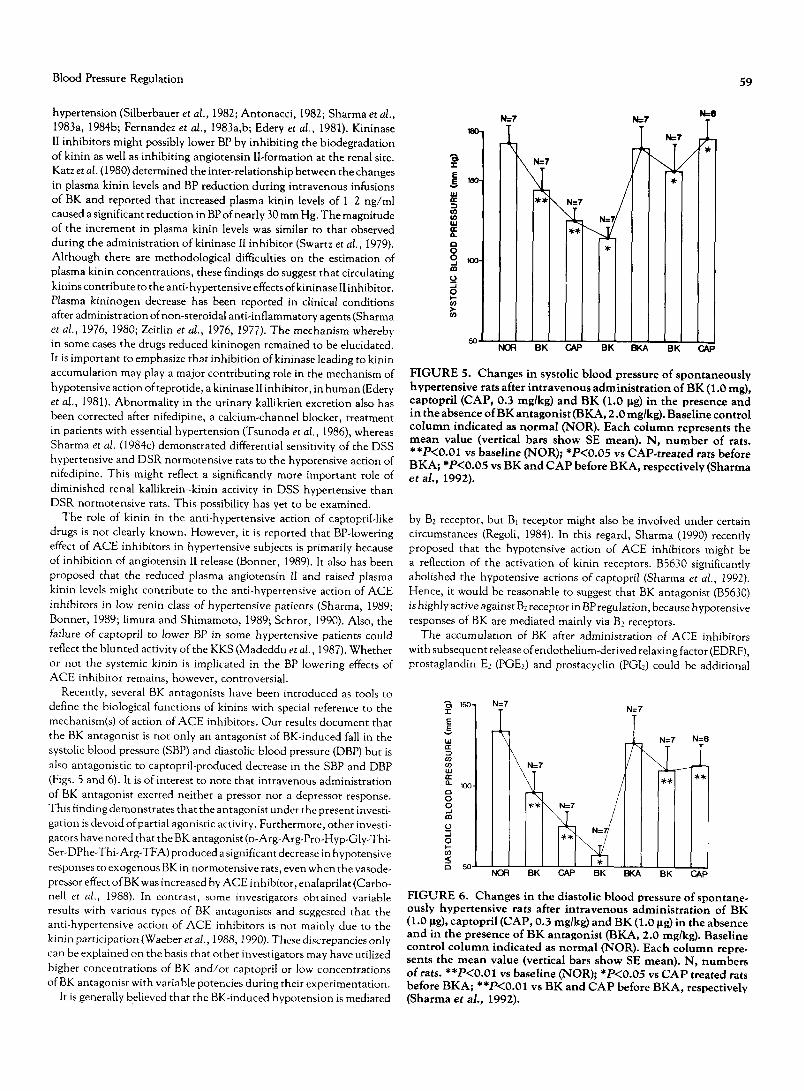
FIGURE 4. Mode of inactivation of kinin by kininases in the body.
the blood coagulation, cysteine proteinases inhibition and acute-phase reactions in the rat (Muller-Esterl, 1990).
KININASES
Those enzymes responsible for inactivation of kinins are called ki- ninases, which are present in plasma, urine, tissues, endothelial cells and body fluids. Their prime function is to monitor the required BK concentrations in the body to perform the necessary physiological activi- ties. The main enzyme that metabolizes BK in vascular beds is a dipepti- dyl carboxypeptidase termed kininase II or angiotensin-converting en- zyme (ACE) (Erdos, 1990). A slow-reacting enzyme is known to be kininase I or carboxypeptidase N. The kininase I removes the C-terminal arginine of BK to leave the residual octapeptide (Erdos, 1990). This enzyme may be physiologically more important in metabolizing BK, because kininase I concentration is higher in plasma (Erdos, 1979). Des-Arg 9 BK formed by the action of kininase I on BK is inactive in vivo; however, it displays activity on various isolated vascular and non-vascular smooth muscle preparations (Regoli and Barabe, 1980). Based on these pharmacological activities, Regoli and Barabe (1980) proposed BKl receptor-mediated effects of des-Argg-BK and BK2 recep- tor-mediated actions of BK on several isolated tissues. Kininase II and enkephalinase-A cause inactivation of kinins by removing the C-termi- nal Phe-Arg in the plasma and in the urine of human and rats (Erdos, 1979; Kokubu et al., 1978; Skidge et al., 1987; Ura et al., 1987). In addition, kininase II induces further degradation of des-Arg 9 to release the pentapeptide (Ser-Pro-Pro-Gly-Phe) and the tripeptide (Ser-Pro-Phe) (Sheikh and Kaplan, 1986). The C-terminal phenylalanine is then cleaved from each peptide, leaving Arg-Pro-Pro-Gly and Ser-Pro. The Ser-Pro is also digested to Ser and Pro, whereas the C-terminal Gly is removed from the tetrapeptide. The final plasma and urinary metabo- lites of BK are one mole each of Arg-Pro-Pro, Gly, Ser and Pro and two moles of phenylalanine (Fig. 4).
KKS AND BP
Kinins have potent vasodilator and natriuretic effects, and deficiency of KKS may participate in the genesis of hypertension. The physiophar- macological abnormality of the KKS in hypertension is based on the observations such as reduced activity of renal kallikrein, increased levels of kallikrein inhibitor, decreased synthesis of HMWK and LMWK and the presence of high concentrations of kininases. These factors would result in subnormal kinin generation within the renal system. The correlation between the K K S and hypertension was observed by Elliot and Nuzum (1934). These investigators found that patients with essen- tial hypertension excreted less kallikrein in the urine than normotensive

58 J.N. Sharma et al.
individuals. This observation was confirmed after 37 yr, when Margolius and co-investigators (1971, 1972, 1974) reported reduced urinary kalli- krein excretion in various types of hypertensive patients and in rats with hypertension. Reduced urinary kallikrein excretion would suggest that hypertension might result from a defect in the kinin generation.
The pharmacological effects of kinin in relation to the regulation of BP are vasodilatation in most areas of circulation, reduction in total pheripheral resistance and regulation of sodium excretion from the kidney (Adetuyibi and Mills, 1972; de Freitas et al., 1964; Marin-Grez et al., 1982; Mills, 1982; Maxwell et al., 1962). The injection of BK into the renal artery causes diuresis and natriuresis by increasing renal blood flow (Webster and Gilmore, 1964). These actions of BK have been attributed to PGS release in the renal circulation (McGiff et al., 1975, 1976). Nevertheless, renal KKS has been suggested as an intrarenal hormone system that controls water and electrolyte excretion and par- ticipates in the regulation of BP (Scicli and Carretero, 1986; Carretero and Scicli, 1981). Moreover, the concentration of kallikrein present in the urine may serve as an indicator of renal activity of KKS because it originates from the kidney (Nustad, 1970; Mills, 1979), although the underlying mechanisms are poorly understood. In this regard, genetical abnormalities and the loss of renal parenchyma might be of clinical significance.
It has been suggested that the race and sodium intake in hypertension have greater influence on kallikrein excretion (Zinner et al., 1976, •978; Levy et al., 1977). These investigators evaluated urinary kallikrein levels in large populations of hypertensive patients and their families. The results showed that whites excrete more kallikrein than blacks and white hypertensive patients excrete less kallikrein than white normotensive individuals. All test groups had higher kallikrein excretion when kept on low sodium intake. Black hypertensive patients excreted less kallikrein than black normotensive individuals during sodium reduction. Further- more, families with reduced kallikrein excretion had higher BP than those with increased urinary kallikrein excretion. This could suggest a genetic defect in the renal kallikrein and the presence of higher amounts of tissue kallikrein inhibitor in certain races. In this connection, Horl et al. (1982) stated that the inhibitory effect of cq-antitrypsin on kallikrein activity should be taken into account in studies related to the renal kallikrein.
Changes in the KKS also have been observed in genetically salt- sensitive hypertensive rats. This experimental hypertensive model is thought to have similar pathogenic mechanisms as human essential hypertension. Through selective inbreeding, Dahl et al. (•962) devel- oped two strains from Sprague-Dawley rats: Dahl salt-sensitive (DSS) hypertensive and Dahl salt-resistant (DSR) normotensive rats. It has been reported that urinary kallikrein activity is reduced in DSS hyper- tensive rats when compared with the DSR normotensive rats (Carretero et al., 1978). Arbeit and Serra (1985) also reported reduced urinary kallikrein excretion in DSS rat fed on 0.0064% and 0.4% sodium chlo- ride. However, these investigators did not report the BP of DSS rats. Hence, it is impossible to evaluate the correlation between the severity of hypertension and reduction in urinary kallikrein concentration in DSS rats. However, it has been suggested that altered sodium and water excretion due to reduced production of kinin-forming enzyme might be the cause of hypertension in DSS rats (Sustarsic et al. , 1980, 1981).
A similar mechanism might also prevail in human essential hyperten- sion. The mechanism of altered renal kallikrein activity in hypertensive patients remains unknown. It is speculated that the mode of decreased kallikrein levels in hypertensive patients might be due to decreased synthesis or an increased inhibition. Kallikrein excretion is also sup- pressed in renal parenchymal hypertension, experimental diabetic hy- pertension, hypertension after renal transplant and glucocorticoid in- duced hypertension in rats (Mitas et al., 1978; Hayashi et al., 1983;
O'Connor et al., 1982; Handa et al., 1983). These findings further support the role of renal kallikrein in the genesis of various forms of hypertension. Furthermore, it has been found that the rate of kinin inactivation is increased in one-kidney, one clip hypertension in rats (Salgado et al., 1986) and in the hypertension caused by acute renal artery constriction in the dog (Moore et al. , 1984). It has been indicated that a defect in prokallikrein activation rather than in kallikrein synthe- sis in the renal cortex may take place in spontaneously hypertensive rats at birth (Praddaude et al. , 1989; Mohsin et al., 1992). These data indicate once again that decreased circulating kinin might potentiate the vascoconstrictor action of other vasoconstrictor agents and contrib- ute to the development of hypertension.
However, urinary kallikrein excretion is increased in desoxycortico- sterone acetate ( D O C A ) - i n d u c e d hypertension in rats (Margolius et al., 1972) and in rabbits (Marchetti et al., 1984). Urinary kallikrein activity also has been observed in the majority of hypertensive patients who have primary aldosteronism (Margolius et al., 1971, 1974). This possibly could be due to mineral corticoid mediated regulation of renal kallikrein (Vince et al., 1979; Horwitz et al., 1978).
Research on the systemic changes in the KKS has provided further evidence regarding the mechanism of various hypertensive conditions. In this connection, it is known that kininogen levels and a kinin- potentiating factor have been found to be reduced in essential and malignant hypertensive patients (Sharma and Zeitlin, 1981; Almeida et al., 1981). It may be that the deficiency in plasma HMW might be related to the decrease in liver synthesis in individuals who develop hypertension after mild exercise (James and Donaldson, 1981). It seems possible that deficient kallikrein-kininogens-kinin formation might be a significant factor in physiopathology of hypertension. Further, it is suggested that reduced concentrations of kinin-forming system might be the cause of increased arterial vasoconstriction in clinical and experi- mental hypertension (Sharma and Zeitlin, 1981; Sharma, 1984; Sharma and Fernandez, 1982). The kinin system also participates in the regula- tion of vascular reactivity by opposing the vasoconstrictor actions of vasopressor agents and enhanced adrenergic activity (Carretero and Scicli, 1981). However, the role of KKS in the regulation of renal physiology remains incompletely understood. It may not be possible to understand distinctly the actions of KKS in the BP regulation and in the pathogenesis of hypertension, cardiovascular and renal diseases until systemic, renal, myocardial and vascular smooth muscle responsi- bilities of kallikrein-kinin are investigated.
TISSUE KALLIKREIN AND KININASE II (ACE INHIBITORS) IN TREATING HYPERTENSION
The Kallikrein might have a prime action in the regulation of systemic BP, because administration of tissue kallikrein to hypertensive patients can bring BP to normal levels. It has been shown that pig pancreatic kallikrein therapy lowered BP significantly and normalized reduced urinary kallikrein excretion in patients with essential hypertension (Overlack et al., 1980, 1981; Ogawa et al., 1985). Wang et al. (1994) reported that sustained high level of tissue kallikrein in the circulation induces chronic hypotension in transgenic mice. These data provide favorable evidence that the presence of subnormal activity of the kinin- generating system might be a prominent predisposing cause in the genesis of hypertension. Because the anti-hypertensive mechanism(s) of pancreatic kallikrein treatment remains unknown, the possibility exists that tissue kallikrein may have independent actions in regulating arterial BP. There is, however, no direct evidence in support of this hypothesis.
Kininase I1 (ACE) inhibitors such as captopril, enalapril and teprotide are currently used in the treatment of both clinical and experimental
Blood Pressure Regulation 59
hypertension (Silberbauer et al. , 1982; Antonacci, 1982; Sharma et al., 1983a, 1984b; Fernandez et al. , 1983a,b; Edery et a l , 1981). Kininase II inhibitors might possibly lower BP by inhibiting the biodegradation of kinin as well as inhibiting angiotensin II-formation at the renal site. Katz et al. (1980) determined the inter-relationship between the changes in plasma kinin levels and BP reduction during intravenous infusions of BK and reported that increased plasma kinin levels of 1-2 ng/ml caused a significant reduction in BP of nearly 30 mm Hg. The magnitude of the increment in plasma kinin levels was similar to that observed during the administration of kininase II inhibitor (Swartz et al., 1979). Although there are methodological difficulties on the estimation of plasma kinin concentrations, these findings do suggest that circulating kinins contribute to the anti-hypertensive effects ofkininase II inhibitor. Plasma kininogen decrease has been reported in clinical conditions after administration of non-steroidal anti-inflammatory agents (Sharma et al., 1976, 1980; Zeitlin et al. , 1976, 1977). The mechanism whereby in some cases the drugs reduced kininogen remained to be elucidated. It is important to emphasize that inhibition of kininase leading to kinin accumulation may play a major contributing role in the mechanism of hypotensive action ofteprotide, a kininase II inhibitor, in hum an (Edery et al., 1981). Abnormality in the urinary kallikrien excretion also has been corrected after nifedipine, a calcium-channel blocker, treatment in patients with essential hypertension (Tsunoda et al., 1986), whereas Sharma et al. (1984c) demonstrated differential sensitivity of the DSS hypertensive and DSR normotensive rats to the hypotensive action of nifedipine. This might reflect a significantly more important role of diminished renal kallikrein-kinin activity in DSS hypertensive than DSR normotensive rats. This possibility has yet to be examined.
The role of kinin in the anti-hypertensive action of captopril-like drugs is not clearly known. However, it is reported that BP-lowering effect of A C E inhibitors in hypertensive subjects is primarily because of inhibition of angiotensin II release (Bonnet, 1989). It also has been proposed that the reduced plasma angiotensin II and raised plasma kinin levels might contribute to the anti-hypertensive action of ACE inhibitors in low renin class of hypertensive patients (Sharma, •989; Bonner, 1989; Iimura and Shimamoto, 1989; Schror, 1990). Also, the failure of captopril to lower BP in some hypertensive patients could reflect the blunted activity of the KKS (Madeddu et al., 1987). Whether or not the systemic kinin is implicated in the BP lowering effects of ACE inhibitor remains, however, controversial.
Recently, several BK antagonists have been introduced as tools to define the biological functions of kinins with special reference to the mechanism(s) of action of ACE inhibitors. Our results document that the BK antagonist is not only an antagonist of BK-induced fall in the systolic blood pressure (SBP) and diastolic blood pressure (DBP) but is also antagonistic to captopril-produced decrease in the SBP and DBP (Figs. 5 and 6). It is of interest to note that intravenous administration of BK antagonist exerted neither a pressor nor a depressor response. This finding demonstrates that the antagonist under the present investi- gation is devoid of partial agonistic activity. Furthermore, other investi- gators have noted that the BK antagonist (D-Arg-Arg-Pro-Hyp-Gly-Thi- Ser-DPhe-Thi-Arg-TFA) produced a significant decrease in hypotensive responses to exogenous BK in normotensive rats, even when the vasode- pressor effect of BK was increased by ACE inhibitor, enalaprilat (Carbo- nell et al., 1988). In contrast, some investigators obtained variable results with various types of BK antagonists and suggested that the anti-hypertensive action of ACE inhibitors is not mainly due to the kinin participation (Waeber et al., 1988, 1990). These discrepancies only can be explained on the basis that other investigators may have utilized higher concentrations of BK and/or captopril or low concentrations of BK antagonist with variable potencies during their experimentation.
It is generally believed that the BK-induced hypotension is mediated
Z
E
Ilc
w
a .
0 q m
o p .
3-
5 0
N:7 N=6 N=7
I " = '
N=7
NOR BK CAP BK BKA BK
] CAP
FIGURE 5. Changes in systolic blood pressure of spontaneously hypertensive rats after intravenous administration of BK (1.0 mg), captopril (CAP, 0.3 mg/kg) and BK (1.0 gg) in the presence and in the absence of BK antagonist (BKA, 2.0 mg/kg). Baseline control column indicated as normal (NOR). Each column represents the mean value (vertical bars show SE mean). N, number of rats. **P<0.01 vs baseline (NOR); *P<0.05 vs CAP-treated rats before BKA; *P<0.05 vs BK and CAP before BKA, respectively (Sharma e t al . , 1992).
by B2 receptor, but B1 receptor might also be involved under certain circumstances (Regoli, 1984). In this regard, Sharma (1990) recently proposed that the hypotensive action of ACE inhibitors might be a reflection of the activation of kinin receptors. B5630 significantly abolished the hypotensive actions of captopril (Sharma et al., 1992). Hence, it would be reasonable to suggest that BK antagonist (B5630) is highly active against B2 receptor in BP regulation, because hypotensive responses of BK are mediated mainly via B2 receptors.
The accumulation of BK after administration of ACE inhibitors with subsequent release ofendothelium-derived relaxing factor (EDRF), prostaglandin E2 (PGE2) and prostacyclin (PGI2) could be additional
3-
E
w
I1.
0 q lID
0
0
<
150. N=7
\ 1130
N=7 ~ N=7 N=6
N=7
BK CAP BK BKA ,Sg
NOR BK CAP
FIGURE 6. Changes in the diastolic blood pressure of spontane. ously hypertensive rats after intravenous administration of BK (1.0 og), captopril (CAP, 0.3 mg/kg) and BK (1.0 ~tg) in the absence and in the presence of BK antagonist (BKA, 2.0 mg/kg). Baseline control column indicated as normal (NOR). Each column repre. sents the mean value (vertical bars show SE mean). N, numbers of rats. **P<0.01 vs baseline (NOR); */)<0.05 vs CAP treated rats before BKA; **/)<0.01 vs BK and CAP before BKA, respectively (Sharma e t al . , 1992).

60 J . N . Sharma et al.
Kallikreins
1 Kininogens
Kinins
Activation or Release ~)'~ Inactivation
ACEIs [1 l
l Kininase II (ACE) ~/ ACEIs [2]
Kinin Receptors Activation - -~ PGE2, PGh and EDRF
1 Hypotensive Effects (Antihypertensive Actions)
F I G U R E 7. A hypothetical explanation of the anti-hypertensive actions of A C E i n h i b i t o r s (ACEIs) med ia t ed through the KKS [ 1]. I n addition, kinin may cause hypotension by activating kinin receptor (Bz), and by releasing PGEz, PGIz a n d E D R F from the endothelial cells [2] (Sharma et al., 1992).
mediators released in the process of anti-hypertensive effects ofcaptopril- like drugs. Mullane and Moncada (1980) indicated tha t after ACE inhibit ion, the renal product ion of PGI2 can contr ibute significantly to the hypotensive effect of BK. It is interesting that PGI2 showed a more powerful hypotensive action than PGE2 in SHR (Pace-Asciak et al., 1978).
Exogenously added BK-induced release of EDRF and PGI2 from bovine aortic endothelial cells can be abolished by B2 receptor antagonist (D-Arg °, Hyp 3, Thi s's D-Phq)-BK (D'Orleans-Juste et al., 1989). Kinins have been shown to stimulate the synthesis of vasodilator PGE2 (Con- klin et al., 1988). Thus, the interactions between the BK and arachidonic acid products seem to be the most appropriate way of evaluating the pat tern of BP changes after the application of BK antagonist . The depressor effect of BK can be prevented by PG synthetase inhibi t ion (Sharma et al., 1984). Several investigators observed tha t captopril is able to increase the synthesis of PGI2 in hypertensive humans and rats (Vinci et al., 1979; Dusting et al., 1983). This is also in accord with the finding that t reatment of normotensive rats with BK antagonist inhibits vascular PGI2 release stimulated by enalaprilat (Beierwaltes and Carret- ero, 1989). This mechanism may contr ibute to the generalized vascular smooth muscle dilatation (Sharma, 1988). It is conceivable, therefore, tha t captopril-like drugs may act via accumulating kinins, stimulating B2 receptor and causing release ofPGE2, PGI2 and EDRF (Fig. 7) within the vascular smooth muscle in lowering high BP.
C O N C L U S I O N
The KKS plays an impor tan t role in the regulation of systemic BP. This system is involved in the physiology of sodium and water balance, renal hemodyn amics, cardiovascular protect ion and BP control. Hence, reduced formation of KKS due to genetic defects a n d / o r envi ronmenta l al ternations may cause genesis of high BP diseases. Unde r these situa- tions, hypertensive and cardiovascular disorders should be treatable with the help of naturally occurring kallikreins and by protecting kinins biodegradation in the body.
This work was supported by a Research and Development Grant from the Ministry of Science and Technology of Malaysia, Under 6th Malaysian Plan (IRPA) to J. N. Sharma. Secretarial assistance by Mrs. Memi A. Hamid is highly appreciated.
References Abelous J. E. and Bardier E. (1909) Les substances hypotensives des I'urine
humaine normale. C.R. Soc. Biol. (Paris) 66, 511-512. Adetuyibi A. and Mills I. H. (1972) Relationship between urinary kallikrein and
renal function, hypertension, and excretion of sodium and water in man. Lancet 2, 203-207.
AI-Haboubi H. A., Bennett D., Sharma J. N., Thomas G. R. and Zeitlin I. J. (1986) A synovial amidase acting on tissue kallikrein-selective substrate in clinical and experimental arthritis. Adv. exp. Med. Biol. 198B, 405-411.
Almeida F. A., Stella R. C. R., Voos A., Ajzen H. and Ribeiro A. B. (1981) Malignant hypertension: a syndrome associated with low plasma kininogen and kinin potentiating factor. Hypertension 3, 11-46-50.
Amundsen E. and Nustad K. (1965) Kinin-forming and destroying activities of cell homogenates. J. Physiol. Lond. 179, 497-488.
Antonaccio M. J. (1982) Angiotensin converting enzyme (ACE) inhibitors. Annu. Rev. Pharmac. Toxicol. 22, 57-87.
Arbeit L. A. and Serra S. R. (1985) Decreased total and active urinary kallikrein in normotensive Dahl salt susceptible rats. Kidney Int. 28, 440-446.
Baker A. R. and Shine J. (1985) Human kidney kallikrein: eDNA cloning and sequence analysis. DNA 4, 445-450.
Beierwaltes W. H. and Carretero O. A. (1989) Kinin antagonist reverses con- verting enzyme inhibitor-stimulated vascular prostaglandin I2 synthesis. Hy- pertension 13, 754-758.
Bery T. D., Hasstedt S. J., Hunt S. C., Wu L. L., Smith J. B., Ash K. O., Kuida H. and Williams R. R. (1989). A gene for high urinary kallikrein may protect against hypertension in Utah kindreds. Hypertension 13, 3-8.
Bonner G. (1988) Haben die Kinin eine Bedeutung fur die. 77(suppl.3), 23-27. Boissonnas R. A., Guttmann S. and Jaquenoud P. A. (1960) Synthese de la
r-arginyl-L-pr olyl-L-prolyl-r-glycyl-L - phenylalanyl - L - seryl - L - prolyl - r - phenyl- alanyl-r-arginine, un nonapeptide presentant les proprietes de la bradykinine. Helv. Chim. Acta 43, 1349-1355.
Brandtzaeg P., Gautvik K. M., Nustad K. and PierceJ. V. (1976) Rat submandibu- lar gland kallikreins: purification and cellular localization. Br. J. Pharmac. 56, 155-167.
Burch R. M. (1990) Kinin signal transduction: role of phosphoinositides and eicosanoids. J. cardiovasc. Pharmacol. 15(suppl.6), $44-$45.
CarbonelI L. F., Carretero O. A., Stewart J. M. and Scicli A. G. (1988) Effect of a kinin antagonist on the acute antihypertensive activity of enalaprilat in severe hypertension. Hypertension 11, 239-243.
Carretero O. A. and Scicli A. G. (1971) The renal kallikrein-kinin system in human and experimental essential hypertension. Clin. Wochenschr. 56 (suppl.1), 113-125.
Carretero O. A. and Scicli A. G. (1981) Possible role of kinins in circulatory homeostasis: state-of-the art review. Hypertension 3, 366-369.
Carretero O. A., Amin V. M., Ocholik T., Scicli A. G. and Koch J. (1978) Urinary kallikrein in rats bred for their susceptibility and resistance to the hypertensive effect of salt. A new radioimmunoassay for its direct determina- tion. Circ. Res. 42, 727-731.
Chao J., Swain C., Chao S., Xiong W. and Chao L. (1988) Tissue distribution and kininogen gene expression after acute phase inflammation. Biochim. biophys. Acta 964, 329-339.
Clement J. A. (I 994) The Human kallikrein gene family: a diversity of expression and function. MoL cell. Endocrinol. 99, C1-C6.
Cochrane C. G., Revak S. D. and Wuepper K. D. (1973) Activation of Hageman factor in solid and fluid phages. J. exp. Med. 138, 1564-1583.
Conklin B. R., Burch R. M., Steranka L. R. and Axelrod J. (1988) Distinct bradykinin receptors mediate stimulation of prostaglandin synthesis by endo- thelial cells and fibroblasts. J. Pharmacol. exp. Ther. 244, 646-649.
Corthorn J., Imanari T., Yoshida H., Kaizu T., Pierce J. V. and Pisano J. J. (1979) Isolation of prokallikrein from human urine. Adv. exp. Med. Biol. 120B, 575-579.
Croxatto H. R. and San Martin M. (1970) Kallikrein-like activity in the urine of renal hypertensive rats. Experientia 26, 1261-1217.
Dahl L. K., Heine M. and Tassinar L. (1962) Role of genetic factors in susceptibility to experimental to experimental hypertension due to chronic excess salt ingestion. Nature 194, 480-482.
de Freitas F. M., Farraco E. Z. and de Azevedo D. F. (1964) General circulatory alterations induced by intravenous infusion of synthetic bradykinin in man. Circulation 29, 66-70.
Digby M., Zhang X. Y. and Richards R. I. (1989) Human prostate specific antigen (PSA) gene: structure and linkage to the kallikrein-like gena, hGK-1. Nucleic Acids Res. 17, 2137.
D'Orleans-Juste P., de Nucci G. and Vane J. R. (1989) Kinins act on BI or B2 receptors to release conjointly endothelium-derived relaxing factor and prostacyclin from bovine aortic endotheliel cells. Br. J. Pharmac. 96, 920- 926.

Blood Pressure Regulat ion 61
Drinkwater C. C., Evans B. A. and Richards R. I. (1988) Kallikreins, kinins and growth factor biosynthesis. Trends Biochem. Sci. 13, 169-172.
Dusting R., Scherhag R., Landsberg G., Glanzer K. and Kramer H. J. (1983) The converting enzyme inhibitor captopril stimulates prostacyclin synthesis by isolated rat aorta. Eur. J. Pharmac. 91, 501-504.
Edery H., Rosenthal T., Amitzur G., Rubinstein A. and Stern N. (1981) The influence of SQ 20881 on the blood kinin system of renal hypertensive patients. Drugs exp. clin. Res. VII, 749-756.
Elliot A. H. and Nuzum F. R. (1934) Urinary excretion of a depressor substance (kallikrein of Frey and Kraut) in arterial hypertension. Endocrinology 18, 462- 474.
Elliot D. F., Lewis G. P. and Horton E. W. (1960) The structure of bradykinin-a plasma kinin from ox blood. Biochem. biophys. Res. Commun. 3, 87-91.
Enjyoji K., Kato H., Hayashi I., Oh-Ishi S. and lwanage S. (1988) Purification and characterization of rat T-kininogens isolated from plasma of adjuvant-treated rats. Identification of three types of T-kininogens. J. biol. Chem. 263, 973- 979.
Erdos E. G. (1979) Kininages. In: Bradykinin, Kallidin and Kallikrein (Edited by Erdos E. G.), pp. 427-487. Springer-Verlag, Berlin.
Erdos E. G. (1990) Some old and some new ideas on kinin metabolism. J. cardiovasc. Pharmac. 15(suppl.6), $20-$24.
Evans B. A., Drinkwater C. C. and Richards R. I. (1987) Mouse glandular kallikrein genes: structure and partial sequence analysis of the kaUikrein gene locus. J. biol. Chem. 262, 8027-8034.
Evans B. A., Yun Z. X., Close J. A., Tregear C. W., Kitamura N., Nakanishi S., Callen D. F., Baker E., Hyland V. J., Sutherland G. R. and Richards R. I. (1988) Structure and chromosomal localization of the hum an renal kallikrein gene. Biochemistry 27, 3124-3129.
Fernandez P. G., Kim B. K., Sharma J. N., Idikio H. and Triggle C. R. (1983a) Left ventricle regression (LVR) in association with blood pressure control in the Dahl model of hypertensive that (DS and DR) treated with enalapril malelate (MK-421, an angiotensin converting enzyme inhibitor) or hydrochlo- rothiazide (HTZ). Clin. invest. Med. 6 (suppl. 2), 55. (Abstr.)
Fernandez P. G., Sharma J. N., Kim B. K., Triggle C. R., Idikio H. and Laher I. (1983b) Left ventricular regression and blood
pressure control in Dahl (D) rat with MK-421 (an angiotensin I converting enzyme inhibitor, CEI) and hydrochlorothiazide (HTZ). Clin. Res. 31, 332A. (Abstr.).
Fiedler F. and Werle E. (1967) Two prekallikreins from porcine pancreas, Hoppe- Seyler's Z. Physiol. Chem. 348, 1087-1089.
Frey E. K. (1926) Zusammenhange zwischen Herzarbeit und Nierentatigkeit, Langenbeck's Arch. Klin. Chir. 142, 663-669.
Frey E. K., Kraut H. and Schultz F. (1930) Uber eine neue innersekretorische Funktion des Pankreas. Naunyn-Schmiedeberg's Arch. exp. Pathol. Pharmac. 158, 334-347.
Frey E. K. and Werle E. (1933) Kallikrein im innere n und au Beren P ankreassekret. Klin. Wschr. 12, 600.
Freay A., Johns A., Adam D. J., Ryan U. S. and Van Breemen C. (1989) Bradykinin and inositol 1,4,5-triphosphate-stimulated calcium release from intracellular stores in cultured bovine endothelial cells. Pflugers Arch. 414, 377-384.
Handa M., Kondo K., Suzuki H. and Saruta T. (1983) Urinary prostaglandin E2 and kallikrein excretion in glucocorticoide hypertension in rats. Clin. Sci. 65, 37-42.
Hayashi M., Senba S., Saito I., Kitajima W. and Saruta T. (1983) Changes in blood pressure, urinary kallikrein, and urinary prostaglandin E2 in rats with streptozotocin-induced diabetes. Naunyn-Schmiedeberg's Arch. Pharmac. 322, 290-294.
Horol W. H., Schafer R. M. and Heidland A. (1982) Role of Ixl-antitrypsin in padutin (kallikrein) inactivation. Eur. J. din. Pharmac. 22, 541-544.
Horwitz D., Margolius H. S. and Keiser H. R. (1978) Effect of dietary potassium and race on urinary excretion on kallikrein and aldosterone in man. J. clin. Endocrinol. Metab. 47, 269-299.
Howard E. F., Thompson Y. G., Lapp C. A. and Greenbaum L. M. (I990) Reduction of T-kininogen messenger RNA levels by dexamethasone in the adjuvant-treated rat. Life Sci. 46, 411-417.
Iimura O. and Shimamoto K. (1989) Role of kallikrein-kinin system in the hypotensive mechanisms of converting enzyme inhibitors in essential hyper- tension. J. cardiovasc. Pharmacol. 13(suppl.3), $63-66.
Jacobsen S. (1966) Separation of two different substrates for plasma kinin-forming enzyme. Nature 210, 98-99.
James F. W. and Donaldson V. H. (1981) Decreased exercise tolerance and hypertension in severe hereditary deficiency of plasma kininogens. Lancet 1,889.
Kaplan A. P., Reddigari S., Brunnee T., Nishikawa K., Kuna P. and Silverberg M.
(1992) Studies of the activation and inhibition of the plasma kallikrein-kinin system. Agents Actions 38(suppl. III), 317-328.
Katz J., Williams G. H. and Hollenberg N. K. (1980) Plasma concentration and the depressor response to bradykinin infusion. Life Sci. 27, 573-576.
Kellermann, J., Thelen C., Lottspeich F., Henschen A., Vogel R. and Muller- Esterl W. (1987) Arrangement of the disulphide bridges in human low-Mr kininogen. Biochem. J. 247, 15-21.
Kitamura N., Kitagawa H., Fukushima D., Takagaki Y., Miyata T., and Nakani- shi S. (1985) Structural organization of the human kininogen gene and a model for its evaluation. J. biol. Chem. 260, 8610-8617.
Kokubu T., Kato I., Nishimura K., Hiwada K., and Ueda E. (1978) Angiotensin l-converting enzyme in human urine. Clin. Chim. Acta 89, 375-379.
Lechi A., Covi G., Lechi C., Corgnati A., Arosio E., Zatti M. and Scuro L. A. (1978) Urinary kallikrein excretion and plasma renin activity in patients with essential hypertension and primary aldosteronism. Clin. Sci. molec. Med. 55, 51-55.
Levy S. B., Lilley J. J., Frigon R. P. and Stone R. A. (1977) Urinary kallikrein and plasma renin activity as determinants of renal blood flow: the influence of race and dietary sodium intake. J. din. Invest. 60, 129-138.
Madeddu P., Oppes M., Rubattu S., Dessi-fulgheri P., Gloriosa N., Soro A. and Rappelli A. (I 987) Role of renal kallikrein in modulating the antihypertensive effect of a single dose of captopril in normal- and low-renin antihypertensive effect of a single dose ofcaptopril in normal- and low-renin essential hyperten- sives. J. Hypertens. 5, 645-648.
Mandle R. Jr., Colman R. W. and Kaplan A. P. (1976) Identification of prekalli- krein and HMW-kininogen as a circulating complex in human plasma. Proc. ham. Acad. Sci. USA 73, 4179-4183.
Mandle R. Jr. and Kaplan A. P. (1977) Hageman factor substrates. II. Human plasma prekallikrein. Mechanism of activation by Hageman factor and partici- pation of Hageman factor dependent fibrinolysis. J. biol. Chem. 252, 6097- 6104.
Marchetti J., Imbert-Teboul M., Alhenc-Gelas F., Allegrini J., Menard J. and Morel F. (1984) Kallikrein along the rabbit microdissected nephron: a micro- method for its measurement, effect of adrenalectomy and D O C A treatment. Pflugers Arch. 401, 27-33.
Margolius H. S., Geller R., Pisano J. J. and Sjoerdsma A. (1971) Altered urinary kallikrein excretion in human hypertension. Lancet 2, 1063-1065.
Margolius, H. S., Geller R., deJong W., Pisano J. J. and Sjoerdsma A. (1972) Altered urinary kallikrien excretion in rats with hypertension. Circ. Res. 30, 358-362.
Margolius H. S., Horwitz D., Pisano J. J. and Keiser H. R. (1974) Urinary kallikrein excretion in hypertensive man: relationships to sodium intake and sodium-retaining steroids. Circ. Res. 35, 820-825.
Marin-Grez M., Schaechtelin G. and Bonner G. (1982) Relationship between the renal kallikrein activity and the urinary excretion of kallikrein in rats. Experientia 38, 941-943.
Mason A. J., Evans B. A., Cox D. R., Shine J. and Richard R. I. (1983) Structure of mouse kallikrein gene family suggests a role in specific processing of biological active peptides. Nature 303, 300-307.
Matsas R., Proud D., Nustad K., and Bailey G. S. (1981} Rapid purification of a prekallikrein from rat pancreas. Analyt. Biochem. 113, 264-270.
Maxwell G. M., Elliott R. B. and Kneebone G. M. (1962) Effect of bradykinin on system and coronary vascular bed on intact dog. Circ. Res. 10, 359-365.
McGiffJ. C., Itskovitz H. D. and Terragno N. A. (1975) The action ofbradykinin and eledoisin in the carline isolated kidney: relationship to prostaglandins. Clin. Sci. molec. Med. 49, 125-131.
McGiffJ. C., Itskovitz H. D., Terr ango A. and Wong P. Y. K. (1976) Modulation and mediation of the renal kallikrein-kinin system by prostaglandins. Fed. Proc. 35, 175-180.
Mills I. H. (1979) Kallikrein, kininogen and kinin in control of blood pressure. Nephron 23, 61-71.
Mills I. H. (1982) The renal kallikrein-kinin system and sodium excretion. Q. J. exp. Physiol. 67, 393-399.
Mitas J. A., Levy S. B., Holle R., Frigon R. P. and Stone R. A. (1978) Urinary kallikrein activity in the hypertension of renal parenchymal disease. New Engl. J. Med. 299, 162-165.
Mohsin S. S. J., Majima M., Katori M. and Sharma J. N. (1992) Important suppressive roles of the kallikrein-kinin system during the developmental stage of hypertension in spontaneously hypertensive rats. Asia Pacific J. Pharmac. 7, 73-82.
Moore J. Jr., Gagnon J. A., Verma P. R., Sander S. E. and Butkus D. E. (1984) Plasma kinin levels in acute renovascular hypertension in dogs. Renal Physiol. 7, 102-114.
Morris B. J. (1989) hGK-I: a kallikrein gene expressed in human prostate. Clin. exp. Pharmac. Physiol. 16, 345-351.
Mullane K. M. and Moncada S. (1980} Prostacyclin mediates the potentiated

62 J . N . Sharma et al.
hypotensive effect of bradykinin following captopril treatment. Eur. J. Phar- mac. 66, 355-365.
Muller-Esterl W. (1990) Kininogens, kinins, and kinships. J. Cardiovasc. Pharma- col. 15(suppl.6), S1-$6.
Muller-Esterl W., Iwanaga S. and Nakanishi S. (1986) Kininogens revisited. Trends biochem. Sci. 11, 336-339.
Nustad K. (1970) Relationship between kidney and urinary kininogenases. Br. J. Pharmac. 39, 73-86.
Nustad K., Vaaje K. and Pierce J. V. (1975) Synthesis of kallikreins by rat kidney slices. Br. J. Pharmac. 53, 229-234.
O'Conner D. T., Barg A. P., Amend W. and Vincenti F. (1982) Urinary kallikrein excretion after transplantation: relationship to hypertension, graft source and renal function. Am. J. Med. 73, 475-481.
Ogawa K., Ito T., Ban M., Mochizuki M. and Satake T. (1985) Effect of orally administered glandular kallikrein on urinary kallikrein and prostaglandin excretion, plasma immuno-reactive prostanoids and platelet aggregation in essential hypertension. Klin. Wochenschr. 63, 332-336.
Okamoto H. and Greenbaum L. M. (1986) Isolation properties of two rat plasma T-kininogens. Adv. exp. Med. Biol. 198, 69-75.
Overlack A., Stumpe K. O., Ressel C., Kollock R., Zywzock W. and Krueck F. (1980) Decreased urinary kallikrein activity and elevated blood pressure normalized by orally applied kallikrein in essential hypertension. Klin. Wsch. 58, 37-40.
Overlack A., Stumpe K. O., Kollock R., Ressel C. and Krueck F. (1981) Antihy- pertensive effect of orally administered glandular kallikrein in essential hyper- tension. Hypertension 3, 1-18-I-21.
Pace-Asciak C. R., Carrara M. C., Rangaraj G. and Nicoalou K. C. (1978) Prostaglandin Iz has more potent hypotensive properties than prostaglandin E2 in normal and spontaneously hypertensive rats. Prostaglandins 15, 999- 1010.
Pierce J. V. and Guimaraes J. A. (1977) Further characterization of highly purified human plasma kininogens. In: Chemistry and Biology of the Kallikrein-Kinin System in Health and Disease (Edited by Pisano J. J. and Austen K. F.), pp. 121-I27. U.S. Government Printing Office, Washington, D.C.
Praddaude F., Tran-van T. and Ader J. L. (1989) Renal kallikrein activity in rats developing spontaneous hypertension. Clin. Sci. 76, 311-315.
Pravenec M., Ken V., Kunes J., Sclicli G., Carretero O. A., Simonet L. and Kurtz T. W. (1991) Cosegregation of blood pressure with a kallikrein gene family polymorphism. Hypertension 17, 242-246.
Ramoff O. D., Davie J. W. and Mallet D. L. (1961) Studies on the action of Hageman factor. Evidence that activated Hageman factor in turn activates plasma thromboplastin antecedent. J. din. Invest. 40, 803-819.
Regoli D. (i 984) Neurohumoral regulation of precapillary vessels: the kallikrein- kinin system. J. Cardiovasc. Pharmacol. 6 (suppl.), S401-S412.
Regoli D. and Barabe S. (1980) Pharmacology of hradykinin and related kinins. Pharmacol. Rev. 31, 1-46.
Richards R. I., Catanzaro D. F., Mason A. J., Morris B. J., Baxter J. D. and Shine J. (1982) Mouse glandular kallikrein genes: nucleofide sequence of cloned cDNA coding for a member of the kallikrein arginyl esteropeptidase group of serine proteases. J. biol. Chem. 257, 2758-2761.
Rocha e Silva M., Beraldo W. T. and Rosenfeld G. (1949) Bradykinin, hypoten- sion and smooth muscle stimulating factor released from plasma globulin by snake venom and by tripsin. Am. J. Physiol. 156, 261-273.
Salgado M. C. O., Rabito S. F. and Carretero O. A. (1986) Blood kinin in one-kidney, one clip hypertensive rats. Hypertension 8(suppl. 1), I-110-I-113.
Schachter M. (1980) Kallikrein (kininogenases) a group of serine proteases with biological actions. Pharmac. Rev. 31, 1-17.
Schedlich L. J., Bennets B. H. and Morris B. J. (1987) Primary structure of a human glandular kallikrein gene. DNA 6, 429-437.
Schedlich L. J., Catanzaro D. F. and Morris B. J. (1988) Kallikrein genes: cloning in man and expression in rat renal hypertension. J. Hypertens. 6(suppl.4), $395-$398.
Schini V. B., Boulanger C., Regoli D. and Vonhoutte P. M. (1990) Bradykinin stimulates t he production of cyclic GMP via activation of B2 kinin receptors in cultured porcine aortic endothelial cells. J. Pharmac. exp. Ther. 252, 581- 585.
Schror K. (1990) Converting enzyme inhibitors and the interaction between kinins and eicosanoids. J. Cardiovasc. Pharmac. 15(suppl.6), $60-$68.
Scicli A. G. and Carretero O. A. (1986) Renal kallikrein-kinin system. Kidney Int. 29, I20-130.
Seidah N. G., Ladenheim R., Mbikay M., Hamelin J., Lutfalla G., Rougeon F., Lazure C. and Chretien M. (1989) The cDNA structure of rat plasma kallikrein. DNA 8, 563-574.
Sharma J. N. (1984) Kinin-forming system in the genesis of hypertension. Agents Actions 14, 200-205.
Sharma J. N. (1988a) Interrelationship between the kallikrein-kinin system and hypertension: a review. Gen. Pharmac. 19, 177-187.
Sharma J. N. (1988b) The kinin system and prostaglandins in the intestine. Pharmac. Toxicol. 63, 310-316.
Sharma J. N. (1988c) The kallikrein-kinin system in hypertension. In: Renal Function, Hypertension and Kallikrein-Kinin System (Edited by Iimura O. and Margolius H. S.), pp. 147-154. University of Tokyo Press, Tokyo.
Sharma J. N. (I 989) Contribution of kinin system to the antihypertensive action of angiotensin converting enzyme inhibitors. Adv. exp. Med. Biol. 247A, 197-205.
Sharma J. N. (1990) Does kinin mediate the hypotensive action of angiotensin converting enzyme (ACE) inhibitors? Gen. Pharmac. 21, 451-457.
Sharma J. N. and Fernandez P. G. (1982) Pharmacological abnormality of kalli- krein-kinin system in hypertension. Med. Hypoth. 9, 379-384.
Sharma J. N., Fernandez P. G., Kim B. K., Idikio H. and Triggle C. R. (1983a) Cardiac regression and blood pressure control in the Dahl rat treated with enalapril maleate (MK 421, an angiotensin converting enzyme inhibitor) and hydrochlorothiazide. J. Hypertension 1, 251-256.
Sharma J. N., Fernandez P. G., Kim B. K. and Triggle C. R. (1984a) Systolic blood pressure responses to enalapril maleate (MK 421, an angiotensin converting enzyme inhibitor) and hydrochlorthiazide in conscious Dahl salt-sensitive (S) and salt-resistant (R) rats. Can. J. Physiol. Pharmac. 62, 846-849.
Sharma J. N., Fernandez P. G., Laher I. and Triggle C. R. (1984b) Defferential sensitivity of Dahl salt-sensitive and salt-resistant rats to the hypotensive action of acute nifedipine administration. Can. J. Physiol. Pharmac. 62, 241- 243.
Sharma J. N., Fernandez P. G. and Triggle C. R. (1984c) The effect ofindemetha- cin on the duration of the hypotensive action of bradykinin in Dahl salt- resistant rats: role of cycloxygenase inhibition. Prost. Leukotri. Med. 14, 131- 135.
Sharma J. N., Stewart J. M., Mohsin S. S. J., Katori M. and Vavrek R (1992). Influence of a kinin antagonist on acute hypotensive responses induced by bradykinin and captopril in spontaneously hypertensive rats. Agents Actions 38 (Suppl.llI), 258-269.
Sharma J. N. and Zeitlin [. J. (1981) Altered plasma kininogen in clinical hyperten- sion. Lancet 1, 1259-1260.
Sharma J. N., Zeitlin I. J., Brooks P. M., Buchanan W. W. and Dick W. C. (1980) The action of asprin on plasma kininogen and other proteins in rheumatoid patients: relationship to disease activity. Clin. exp. Pharmac. Physiol. 7, 347-354.
Sharma J. N., Zeitlin I. J., Brooks P. M. and Dick W. C. (1976) A novel relationship between plasma kininogen and rheumatoid disease. Agents Actions 6, 148- 153.
Sharma J. N., Zeitlin I. J., Deodhar S. D. and Buchanan W. W. (1983b) Detection of kallikrein-like activity in inflamed synovial tissue. Arch. Int. Pharmac. Ther. 262, 279-286.
Sheikh I. A. and Kaplan A. P. (1986) Studies of the digestion of bradykinin, lysylbradykinin, and des-arg 9 bradykinin by angiotensin converting enzyme. Biochem. Pharmac. 35, 1951-1956.
Silberbauer K., Stanek B. and Temple H. (1982) Acute hypotensive effect of captopril in man modified by prostaglandin synthesis inhibition. Br. J. clin. Pharmac. 14, 87S-93S.
Skidge R. A., Schulz W. W., Tam L. T. and Erdos E. G. (1987) Human renal angiotensin-I converting enzyme and its helper enzyme. Kidney Int. 3 l(suppl.20), 45-48.
Sustarsic D. L., McPartland R. P. and Rapp J. P. (1980) Total land kallikrein arginine esterase activities in the urine of salt-hypertensive susceptible and resistant rats. Hypertension 2, 813-820.
Sustarsic D. L., McPartland R. P. and Rapp J. P. (1981) Developmental patterns of blood pressure and urinary protein, allikrein, and prostaglandin E2 in Dahl salt-hypertensive susceptible rats. J. Lab. clin. Med. 98, 599-606.
Swartz S. L., Williams G. H., Hollenberg N. K., Moore T. J. and Dluby R. C. (1979) Converting enzyme inhibition in essential hypertension: the hypotensive response does not reflect only reduced angiotensin II formation. Hypertension 1, 106-111.
Thompson R. E., Mandle R. Jr. and Kaplan A. P. (1977) Association of factor XI and high molecular weight kininogen in human plasma. J. clin. Invest. 60, 1376-1380.
Tsunoda K., Abe K., Omata K., Kudo K., Sato M., Kohzuki M., Tanno M., Seino M., Yasujima M. and Yoshinaga K. (1986) Hypotensive and natriuretic effects of nifedipine in essential hypertension. Role of renal kallikrein-kinin- prostaglandin and renin-angiotensin-aldosterone systems. J. clin. Hypertens. 2, 263-270.
Ura N., Carretero O. A. and Erdos E. G. (1987) Role of renal endopeptidase 24. I I in kinin metabolism in vitro and in vivo. Kidney Int. 32, 507-513.

Blood Pressure Regula t ion 63
Vavrek R. J. and Stewart J. M. (1985) Competitive antagonists of bradykinin. Peptides 6, 161-164.
Vinci J. M., Horowitz D., Zusman R. M., Pisano J. J., Cart K. J. and Keiser H. R. (1979a) The effect of converting enzyme inhibition with SQ 20881 on plasma and urinary kinins, prostaglandin E and angiotensin II in hypertensive man. Hypertension 1, 416--426.
Vinci J. M., Zusman R. M., Izzo J. L., Bowden R. E., Horwitz D., Pisano J. J. and Keiser H. R. (1979b) Human urinary and plasma kinins. Relationship to plasma sodium-retaining steroids and plasma renin activity. Circ. Res. 44, 228-237.
Waeber B., Aubert J. F., Fluckiger J. P., Nussberger J., Vavrek R. J., Stewart J. M. and Bru nner H. R. (1988) Role of endogenous bradykinin i n blood pressure control of conscious rats. Kidney Int. 34(suppl.26), $63-$68.
Waeber B., Niederberger M., Gavras H., Nussberger J. and Brunner H. R. (1990) Hemodynamic effects of a kinin antagonist. J. Cardiovasc. Pharmacol. 15(suppl. 6), $78-$82.
Wang J., Xiong W., Yang Z., Davis T., Dewey M. J., Chao J. and Chao L. (1994). Human tissue kallikrein induces hypotension in transgenic mice. Hypertension 23, 236-243.
Webster M. E. and Gilmore J. P. (1964) Influence of kallidin- 10 on renal function. Am. J. Physiol. 206, 714-718.
Werle E., Gotze W. and Keppler A. (1937) Uber die Wirkung des Kallikreins auf den isolierten darm und uber eine neue darmkontrahierende substanz. Biochem. Z. 289, 217-233.
Werle E. and Berek U. (1948) Zur kenntnis des Kallikreins. Z. Angew. Chem. 60A, 53.
Werle E., Maier L. and Ringelmann E. (1953) Hemmung yon Proteinasen durch kallikrein-Inaktivatoren. Naturwissenschaften 39, 328.
Willis L. R., Luden J. H., Hook J. B. and Williamson H. E. (1969) Mechanisam of natriuretic action of bradykinin. Am. J. Physiol. 217, 1-5.
Woodley-Miller C. M., Chao J. and Chao L. (1989) Restriction fragment length polymorphisms mapped in spontaneously hypertensive rats using kallikrein probes. J. Hypertens. 7, 865-871.
Wuepper K. D. (1973) Prekallikrein deficiency in man. J. exp. Med. 138, 1345- 1355.
Zeitlin I. J. (1971) Pharmacological characterization of kinin-forming activity in rat intestinal tissue. Br. J. Pharmacol. 42, 648-649P.
Zeitlin I. J., Sharma J. N., Brooks P. M. and Dick W. C. (1976) Raised plasma kininogen levels in rheumatoid arthritis-response to therapy with non- steroidal anti-inflammatory drugs. Adv. exp. Med. Biol. 70, 335-343.
Zeitlin I. J., Sharma J. N., Brooks P. M. and Dick W. C. (1977) An effect of indomethacin on raised plasma kininogen levels in rheumatoid patients. In: Chemistry and Biology of the Kallikrein-Kinin System in Health and Disease (Edited by Pisano J. J. and Austen K.F.), pp. 483-486. U.S. Government Printing Office, Washington, D.C.
Zinner S. H., Margolius H. S., Rosner B. and Kass E. H. (1976) Familiar aggrega- tion of urinary kallikrein concentration in childhood. Am. J. Epidemiol. 104, 124-132.
Zinner S. H., Margolius H. S., Rosener B. and Kass E. H. (1978) Stability of blood pressure rank and urinary kallikrein concentration in childhood: an eight-year follow-up. Circulation 58, 908-915.
Related Documents











