1 Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen Bioinformatik für Biochemiker Oliver Kohlbacher WS 2009/2010 13. Ab-initio-Vorhersage Gliederung • Ab-Initio-Vorhersage – Konformationsraum – Systematische Suche – Stochastische Methoden – Metropolis-Monte-Carlo-Methode • ROSETTA – Fragment Assembly – Bewertungsfunktion – Algorithmus – Ergebnisse • ROBETTA Ab-Initio-Vorhersage • Modellierung auf homologe Strukturen schlägt dann fehlt, wenn ein Protein einer völlig neuen Faltungsklasse angehört • Threading findet in diesem Fall keine passende Schablonenstruktur • Abhilfe schaffen ab-initio-Verfahren, die die Struktur „aus ersten Prinzipien“, d.h. ohne Zuhilfenahme homologer Strukturen vorhersagen • Dieses Problem ist wesentlich schwieriger als Threading, entsprechend sind die Erfolgsquoten schlechter • Ab-initio-Methoden müssen Konformationsraum der Sequenz durchmustern und darin die native (d.h. in der Natur vorliegende) Struktur identifizieren (über eine geeignete Energiefunktion)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard Karls Universität Tübingen
Bioinformatik für Biochemiker
Oliver Kohlbacher WS 2009/2010
13. Ab-initio-Vorhersage
Gliederung
• Ab-Initio-Vorhersage – Konformationsraum – Systematische Suche – Stochastische Methoden – Metropolis-Monte-Carlo-Methode
• ROSETTA – Fragment Assembly – Bewertungsfunktion – Algorithmus – Ergebnisse
• ROBETTA
Ab-Initio-Vorhersage • Modellierung auf homologe Strukturen schlägt dann fehlt, wenn
ein Protein einer völlig neuen Faltungsklasse angehört
• Threading findet in diesem Fall keine passende Schablonenstruktur
• Abhilfe schaffen ab-initio-Verfahren, die die Struktur „aus ersten Prinzipien“, d.h. ohne Zuhilfenahme homologer Strukturen vorhersagen
• Dieses Problem ist wesentlich schwieriger als Threading, entsprechend sind die Erfolgsquoten schlechter
• Ab-initio-Methoden müssen Konformationsraum der Sequenz durchmustern und darin die native (d.h. in der Natur vorliegende) Struktur identifizieren (über eine geeignete Energiefunktion)

2
Suche im Konformationsraum
• Energiehyperflächen beschreiben Energie des Proteins als Funktion der Konformation
• Minima – entsprechen günstigen Konformationen
(Konformeren)
– sind meist lokale Minima!
• Globales Minimum ist Lösung des Vorhersageproblems
• Kann man die Energiehyperfläche systematisch durchmustern?
Systematische Suche
• Geht nur für kleine Anzahl Freiheitsgrade (kombinatorische Explosion)
• Beispiel – Protein mit 1000 Atomen – Koordinaten in Würfel von 20 Å Seitenlänge – Diskretisierung mit 0.2 Å Abstand ) 100 mögliche Werte für jede der 3000 Koordinaten
) 106000 mögliche Energien (Das Universum enthält ca. 1080 Teilchen!)
Systematische Suche
• Unabhängige Betrachtung der
Koordinaten ist naiv
• Flexibilität wird überwiegend durch
Torsionen bestimmt
• Wesentlich geringere Anzahl
Freiheitsgrade (ca. 2-7 pro AS)
• Wenige Minima in den Torsionen
) grobe Rasterung (0/120/240°)
• Vermeidet Betrachtung physikal.
unsinniger Konformationen
Beispiel:
• 1000 Atome ~ 50 AS
) 200 Torsionen
• 3200 ¼ 1095 Konformationen
) immer noch viel!

3
Stochastische Methoden
• Naiver Ansatz: Zufälliges Abtasten des Konformationsraums • Problem
– Energetisch ungünstige Punkte werden in der Natur seltener angenommen, hier jedoch gleich oft betrachtet
) Betrachtung vieler völlig sinnloser Konformationen
A
Sampling des Konformationsraums • Protein kann per se beliebige
Punkte im Konformationsraum annehmen
• Fast alle sind energetisch sehr ungünstig
• In der Realität liegt ein Ensemble von Molekülen vor
• Einzelne Konformationen treten mit ihrer Energie gewichtet auf
Boltzmann-Statistik
• Gegeben ein System
– N Teilchen
– Konstante Gesamtenergie
– Zustände E0 ... Ek mit E0 < E1 < E2...
– Ni Teilchen sind in Ei
– Gesamtzahl ∑ Ni = N
• Im Gleichgewicht verteilen sich die
Teilchen auf die Zustände gemäß
einer Boltzmann-Verteilung

4
Wahrscheinlichkeitsdichte
• Boltzmann-Verteilung entspricht der
Wahrscheinlichkeitsdichte ρ im NVT-Ensemble
mit der Zustandssumme Q
• ρ(r, p) = Wahrscheinlichkeit ein Teilchen des
Ensembles im Zustand (r, p) zu finden.
Stochastische Methoden
• Wähle zufällige Stichproben im Konformationsraum aus
• Die Mehrzahl der betrachteten Punkte wird energetisch ungünstig sein
• Mit einer großen Anzahl Versuche steigt aber auch die
Wahrscheinlichkeit in Regionen niedriger Energie zu gelangen
A
Importance Sampling
• Für fast alle Punkte des Konformationsraums sind die Boltzmann-Faktoren vernachlässigbar (ρ ¼ 0)
• Idee – Stichproben bevorzugt dort, wo ρ > 0
– Erspart unnütze Berechnungen
• Probleme – Wie kann ich ρ vermeiden?
– Berechnung der Zustandssumme?

5
Monte-Carlo-Methode
• Die Monte-Carlo-Methode hat ihren
Namen aus der Verwendung von
Zufallszahlen
• 1949: Metropolis und Ulam
verwenden den Begriff zum ersten
Mal
• 1953 Metropolis-Algorithmus
• 1970 und 1995 von Hastings und
Green zur Metropolis-Hastings-Green-Methode generalisiert
Metropolis, Ulam. The Monte Carlo method, J. Am. Statist. Assoc. (1949), 44 (247), 335-341
Systematische Suche vs. MC
Frenkel, Smit: Understanding Molecular Simulation, p. 24
Metropolis-Monte-Carlo
• Zustandssumme (und damit ρk) ist aufwändig zu berechnen
• Leicht dagegen: ρi/ρj
• Metropolis-Monte-Carlo-Algorithmus erzeugt nun diese einzelnen Zustände mit ihren relativen Wahrscheinlichkeiten, d.h. energetisch günstigere Zustände werden häufiger durchlaufen als ungünstige

6
MMC-Algorithmus
Für k Iterationen Wähle Schritt von rk-1 ! rk im Konformationsraum Falls Ek · Ek-1:
Akzeptiere Schritt Falls Ek > Ek-1:
Wähle gleichverteilte Zufallszahl x 2 [0, 1] Falls x < exp(-(Ek – Ek-1)/(kBT))):
Akzeptiere Schritt Andernfalls:
Bleibe bei alter Konformation
Einfluss der Temperatur
• Temperatur entspricht Energie:
Ekin = 3/2 RT
• Mit der Gaskonstante R = 8.314 J/(K mol) ergibt sich für Raumtemperatur (298 K): Ekin = 3.7 kJ/mol
E
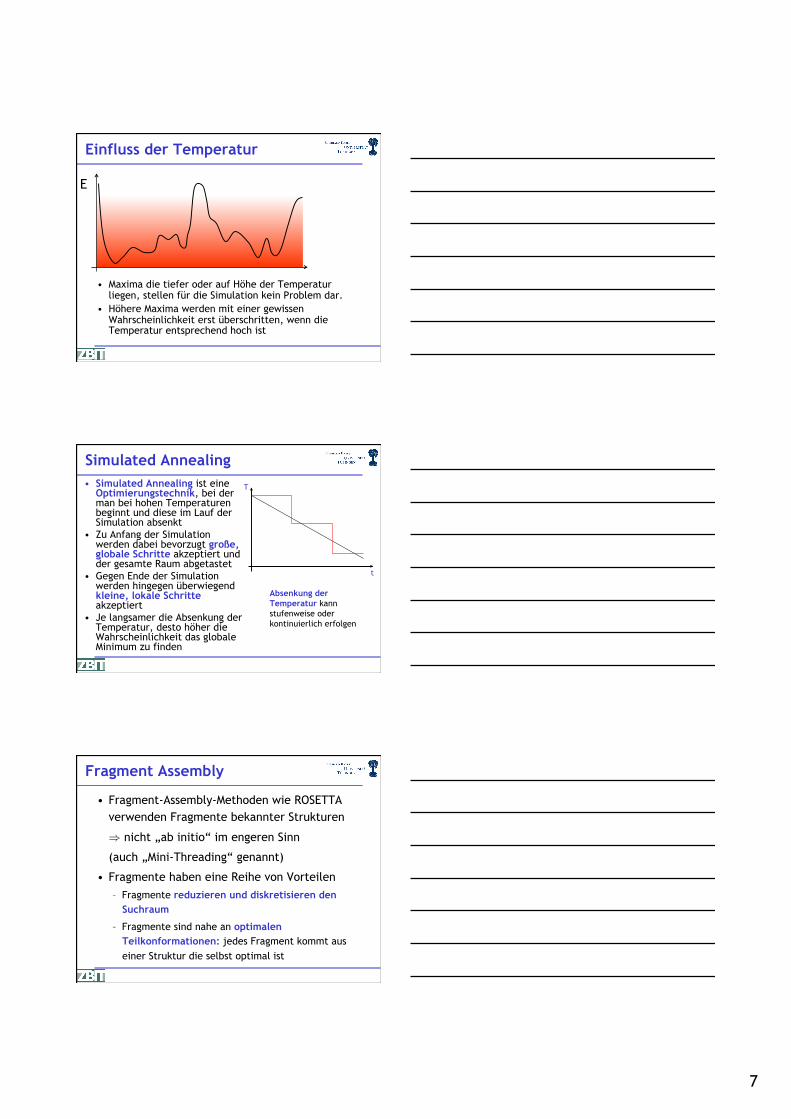
Einfluss der Temperatur
• Maxima die tiefer oder auf Höhe der Temperatur liegen, stellen für die Simulation kein Problem dar.
• Höhere Maxima werden mit einer gewissen Wahrscheinlichkeit erst überschritten, wenn die Temperatur entsprechend hoch ist.
E
7
Einfluss der Temperatur
• Maxima die tiefer oder auf Höhe der Temperatur liegen, stellen für die Simulation kein Problem dar.
• Höhere Maxima werden mit einer gewissen Wahrscheinlichkeit erst überschritten, wenn die Temperatur entsprechend hoch ist
E
Simulated Annealing • Simulated Annealing ist eine
Optimierungstechnik, bei der man bei hohen Temperaturen beginnt und diese im Lauf der Simulation absenkt
• Zu Anfang der Simulation werden dabei bevorzugt große, globale Schritte akzeptiert und der gesamte Raum abgetastet
• Gegen Ende der Simulation werden hingegen überwiegend kleine, lokale Schritte akzeptiert
• Je langsamer die Absenkung der Temperatur, desto höher die Wahrscheinlichkeit das globale Minimum zu finden
Absenkung der Temperatur kann stufenweise oder kontinuierlich erfolgen
T
t
Fragment Assembly
• Fragment-Assembly-Methoden wie ROSETTA verwenden Fragmente bekannter Strukturen
) nicht „ab initio“ im engeren Sinn
(auch „Mini-Threading“ genannt)
• Fragmente haben eine Reihe von Vorteilen – Fragmente reduzieren und diskretisieren den
Suchraum
– Fragmente sind nahe an optimalen Teilkonformationen: jedes Fragment kommt aus einer Struktur die selbst optimal ist

8
ROSETTA
Kernideen • Betrachtung des
Konformationsraums für Teilsequenzen
• Teilsequenzen nehmen nur geringe Anzahl energetisch günstiger Konformationen an
• Diese Konformationen werden durch einen Satz Fragmente äquivalent repräsentiert
• Konformationen der Fragmente überwiegend durch lokale WW bestimmt
Simons et al., J. Mol. Biol. (1997), 268, 209 Simons et al., Proteins (1999), 34, 82
ROSETTA
• Modell – Torsionswinkelraum, reduziert auf
Fragmente – Seitenketten auf Cβ reduziert
• Potentialfunktion – Wahrscheinlichkeitsbasiert (Bayes-Ansatz)
• Algorithmus – Simulated Annealing: MMC mit linear sinkender Temperatur
– Feste Anzahl Schritte (10000)
Fragmentbibliothek
• Abgeleitet aus nicht-redundantem Teilsatz der PDB
• Aus den Strukturen werden alle 9-mere und 3-mere gesammelt
• Zu jeder Teilsequenz der Zielsequenz werden daraus die 25 nächsten Fragmente ausgewählt
• ROSETTA verwendet Fragmente der Längen 3 und 9

9
Bewertungsfunktion • Rosetta verwendet eine Bewertungsfunktion basierend auf
bedingten Wahrscheinlichkeiten, abgeleitet aus Strukturdatenbanken
• Prinzipiell nur ein elegante Art ein paarweises Potential für eine Sequenz x in einer Struktur y zu schreiben: – P(x|y) : Wahrscheinlichkeit, dass Sequenz x Struktur y annimmt – P(xi, xj|rij): Wahrscheinlichkeit für Reste xi, xj für einen Abstand rij – P(xi, xj): A-priori-Wahrscheinlichkeiten der Reste xi, xj
Unabhängig von Struktur Sippl-Potential!
Algorithmus
Bestimme 25 nächste Nachbarn für jede Teilsequenz
Starte mit gestreckter Struktur
Für 10000 Iterationen:
Wähle zufällig eine Teilsequenz x´ aus x
Wähle zufällig x´´ aus den Fragmenten für x´
Ersetze die Torsionswinkel in x´ mit denen aus x´´
Wenn dadurch Atome überlappen, verwerfe Zug
Berechne Score
Akzeptiere Zug gemäß MMC-Kriterium
ROSETTA
ROSETTA-Trajektorie von 1UBI

10
ROSETTA – Ergebnisse CASP5
Verwendung von ROSETTA • ROSETTA erzeugt eine ganze Anzahl von Strukturen
• Diese Strukturen werden mit Hilfe der Scoring-Funktion bewertet: bester (negativster) Score = beste Struktur
• Je mehr Durchläufe (d.h. unabhängige Simulationen) durchgeführt werden, desto höher die Chance eine sehr gute Struktur zu finden
• Stochastische Methoden verwenden Zufallszahlen: jeder Lauf erzeugt andere Ergebnisse!
• Beispielausgabe (Details in der Übung):
filename score env pair vdw hs ss no_pdbfile_fail -71.86 -27.02 -18.88 1.99 -1.69 -22.96 aa2PTL0001.pdb -67.85 -24.50 -12.15 0.58 -4.43 -21.45 no_pdbfile_fail -64.30 -27.23 -16.53 1.07 -2.72 -17.13 aa2PTL0002.pdb -67.77 -18.90 -9.64 2.06 2.63 -33.28 no_pdbfile_fail -69.70 -31.18 -12.69 1.47 -1.76 -21.73 aa2PTL0003.pdb -91.94 -18.14 -13.14 2.01 -8.71 -40.83 no_pdbfile_fail -43.20 -11.10 -10.10 1.83 -7.51 -11.75 Beste Energie
Beste Struktur
ROSETTA – Ergebnis
ROSETTA-Ergebnis (rot) für 2PTL (gelb: PDB-Struktur)

11
ROBETTA
• ROBETTA ist ein vollautomatischer Online-Server zur Proteinstruktur-Vorhersage
• ROBETTA kombiniert – Domänenzerlegung
– Ab-initio-Vorhersage für kleine Domänen
– Threading für Domänen mit bekanntem Fold
• Diese Schritte werden bei ROBETTA vollautomatisch durchgeführt, heraus kommen recht passable Strukturen
• Nachteil: extrem lange Wartezeiten des Online-Servers machen diesen fast unbenutzbar
ROBETTA – Überblick
ROBETTA – Überblick

12
ROBETTA – Submission
ROBETTA – Status
Literatur + Links
Literatur • Simons, Kooperberg, Huang, Baker: Assembly of protein tertiary
structures from fragments with similar local sequences using simulate anealing and Bayesian scoring functions. J. Mol. Biol. (1997), 268, 209-25
• Kim, Chivian, Baker: Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. (2004), 32 Suppl 2, W526-31
Links • Bakerlab (Entwickler von ROSETTA) http://www.bakerlab.org • ROBETTA-Server
http://robetta.bakerlab.org
Related Documents











