Bioinformatics and Evolutionary Bioinformatics and Evolutionary Genomics Genomics Gene Trees, Gene Duplications Gene Trees, Gene Duplications ( ( I I ), and Orthology ), and Orthology

Bioinformatics and Evolutionary Genomics Gene Trees, Gene Duplications ( I ), and Orthology
Dec 31, 2015
Bioinformatics and Evolutionary Genomics Gene Trees, Gene Duplications ( I ), and Orthology. Gene Trees, Gene Duplications and Orthology. Phylogenetic gene trees: how to make them. Homology: are two pieces of sequence related; Trees: when did they diverge ( how are they related) - PowerPoint PPT Presentation
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Bioinformatics and Evolutionary Bioinformatics and Evolutionary GenomicsGenomics
Gene Trees, Gene Duplications (Gene Trees, Gene Duplications (II), and ), and OrthologyOrthology
Bioinformatics and Evolutionary Bioinformatics and Evolutionary GenomicsGenomics
Gene Trees, Gene Duplications (Gene Trees, Gene Duplications (II), and ), and OrthologyOrthology

Gene Trees, Gene Duplications and Gene Trees, Gene Duplications and OrthologyOrthology
Gene Trees, Gene Duplications and Gene Trees, Gene Duplications and OrthologyOrthology

Phylogenetic gene trees: how to make themPhylogenetic gene trees: how to make themPhylogenetic gene trees: how to make themPhylogenetic gene trees: how to make them
• Homology: Homology: areare two pieces of sequence related; two pieces of sequence related; Trees: when did they diverge (Trees: when did they diverge (howhow are they related) are they related)
• Start from a multiple sequence alignmentStart from a multiple sequence alignment• All multiple sequence programs alignments make a All multiple sequence programs alignments make a
global alignment, thus feed it regions that you know global alignment, thus feed it regions that you know are homologous → Domains !are homologous → Domains !
• MUSCLE / clustal / t_coffeeMUSCLE / clustal / t_coffee• Visual inspection of alignments (gaps, Visual inspection of alignments (gaps,
fragments/complete sequences, weird things e.g. A)fragments/complete sequences, weird things e.g. A)
• Homology: Homology: areare two pieces of sequence related; two pieces of sequence related; Trees: when did they diverge (Trees: when did they diverge (howhow are they related) are they related)
• Start from a multiple sequence alignmentStart from a multiple sequence alignment• All multiple sequence programs alignments make a All multiple sequence programs alignments make a
global alignment, thus feed it regions that you know global alignment, thus feed it regions that you know are homologous → Domains !are homologous → Domains !
• MUSCLE / clustal / t_coffeeMUSCLE / clustal / t_coffee• Visual inspection of alignments (gaps, Visual inspection of alignments (gaps,
fragments/complete sequences, weird things e.g. A)fragments/complete sequences, weird things e.g. A)

Put homologs in the alignmentPut homologs in the alignmentPut homologs in the alignmentPut homologs in the alignment
• Even if they are not homologous MUSCLE will align Even if they are not homologous MUSCLE will align them (muscle/clustalw implicitly “assumes” that the them (muscle/clustalw implicitly “assumes” that the sequences you feed it are homologous)sequences you feed it are homologous)
• And in a phylogeny program, non-homologous And in a phylogeny program, non-homologous sequences sequences will bewill be clustered clustered
• Even if they are not homologous MUSCLE will align Even if they are not homologous MUSCLE will align them (muscle/clustalw implicitly “assumes” that the them (muscle/clustalw implicitly “assumes” that the sequences you feed it are homologous)sequences you feed it are homologous)
• And in a phylogeny program, non-homologous And in a phylogeny program, non-homologous sequences sequences will bewill be clustered clustered
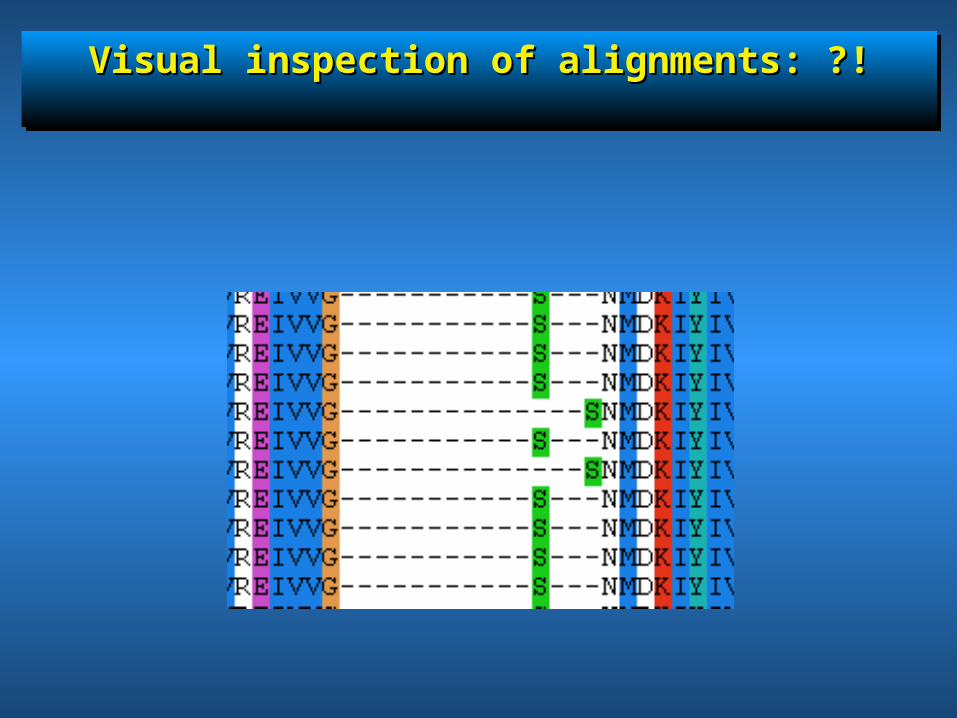
Visual inspection of alignments: ?!Visual inspection of alignments: ?!Visual inspection of alignments: ?!Visual inspection of alignments: ?!

An additive tree which is wrongly reconstructed by An additive tree which is wrongly reconstructed by UPGMAUPGMA
An additive tree which is wrongly reconstructed by An additive tree which is wrongly reconstructed by UPGMAUPGMA
11
66
33
22
55AA
BB
CC
DD
AABB
CCDD
44
333311
66
A B C DA x 12 9 9B 12 x 9 7C 9 9 x 6D 9 7 6 x
A B C DA x 12 9 9B 12 x 9 7C 9 9 x 6D 9 7 6 x
A BCDA x 10BCD 10 x
A BCDA x 10BCD 10 x
BBAA
4455
11
11 A B CDA x 12 9B 12 x 8CD 9 8 x
A B CDA x 12 9B 12 x 8CD 9 8 x
CC
DD
33
33

Neighbour-Joining (Saitou and Nei, 1987)Neighbour-Joining (Saitou and Nei, 1987)Neighbour-Joining (Saitou and Nei, 1987)Neighbour-Joining (Saitou and Nei, 1987)
• Global measure. keeps total branch length minimal• At each step, join two nodes such that distances are
minimal (criterion of minimal evolution)• Leads to unrooted tree
• Global measure. keeps total branch length minimal• At each step, join two nodes such that distances are
minimal (criterion of minimal evolution)• Leads to unrooted tree

Neighbour-JoiningNeighbour-Joining
At each step all possible “neighbour joinings” are checked and the one corresponding to the minimal total tree length (calculated by adding all branch lengths) is taken.
At each step all possible “neighbour joinings” are checked and the one corresponding to the minimal total tree length (calculated by adding all branch lengths) is taken.

Neighbour-JoiningNeighbour-Joining
A B C D rA x 12 9 9 30B 12 x 9 7 28C 9 9 x 6 24D 9 7 6 x 22
A B C D rA x 12 9 9 30B 12 x 9 7 28C 9 9 x 6 24D 9 7 6 x 22
Mab = dab – (ra+rb)/(N-2)
A B C D A x -17 -18 -17B x -17 -18C x -17D x
A B C D A x -17 -18 -17B x -17 -18C x -17D x
dau = dac/2 + (ra-rc)/(2(N-2)) = 9/2 + (30-24)/(2*2) = 6
dcu = dac - dau = 9 – 6 = 3
dbu = (dab + dbc – dac ) / 2 = (12 + 9 – 9 ) / 2 = 6
ddu = (dad + dcd – dac ) / 2 = (9+ 6 – 9) / 2 = 3
AC → U
6
A
3C
U
B
D
6
3
Mab = 12 – (30+28)/(4-2)) = -17
r= netdivergence

U B D rU x 6 3 9B 6 x 7 13D 3 7 x 10
U B D rU x 6 3 9B 6 x 7 13D 3 7 x 10
U B DU x -16 -16B x -16D x
U B DU x -16 -16B x -16D x
e.g. UB →V
Dvu = dub / 2 + (ru – rb )/ (2(N-2)) = 6/2 + (9-13)/(2*1) = 3 – 2 = 1Dvb = dub – duv = 6 – 1 = 5
Ddv = (dud +dbd –dub)/2 = (3+7-6)/2 = 2
1
6
3
2
5A
B
C
DUV

Unequal rates between speciesUnequal rates between speciesare a very real phenomenonare a very real phenomenon
Unequal rates between speciesUnequal rates between speciesare a very real phenomenonare a very real phenomenon
Character based: parsimony and maximum likelihoodCharacter based: parsimony and maximum likelihoodCharacter based: parsimony and maximum likelihoodCharacter based: parsimony and maximum likelihood
• Two way classification in phylogeny distance based Two way classification in phylogeny distance based vs character basedvs character based
• character state method. Searches “directly” (i.e. character state method. Searches “directly” (i.e. without defining distances) for a tree that fits best to without defining distances) for a tree that fits best to the data (the alignment)the data (the alignment)
• Two way classification in phylogeny distance based Two way classification in phylogeny distance based vs character basedvs character based
• character state method. Searches “directly” (i.e. character state method. Searches “directly” (i.e. without defining distances) for a tree that fits best to without defining distances) for a tree that fits best to the data (the alignment)the data (the alignment)

Maximum likelihoodMaximum likelihoodMaximum likelihoodMaximum likelihood
• Search the tree with the highest maximum likelihood Search the tree with the highest maximum likelihood • one searches for the maximum likelihood (ML) value one searches for the maximum likelihood (ML) value
for the character state configurations among the for the character state configurations among the sequences under study for each possible tree and sequences under study for each possible tree and chooses the one with the largest ML value as the chooses the one with the largest ML value as the preferred tree. preferred tree.
• Search the tree with the highest maximum likelihood Search the tree with the highest maximum likelihood • one searches for the maximum likelihood (ML) value one searches for the maximum likelihood (ML) value
for the character state configurations among the for the character state configurations among the sequences under study for each possible tree and sequences under study for each possible tree and chooses the one with the largest ML value as the chooses the one with the largest ML value as the preferred tree. preferred tree.

Maximum likelihoodMaximum likelihoodMaximum likelihoodMaximum likelihood
• have to specify a model of sequence evolutionhave to specify a model of sequence evolution• likelihood for all sites is the product of the likelihoods for likelihood for all sites is the product of the likelihoods for
individual sites individual sites assumingassuming all the nucleotide sites evolve all the nucleotide sites evolve independently.independently.
• maximum likelihood method computes the probabilities for all maximum likelihood method computes the probabilities for all possible combinations of ancestral states!possible combinations of ancestral states!
• ML methods evaluate phylogenetic hypotheses n terms of the ML methods evaluate phylogenetic hypotheses n terms of the probability that a proposed probability that a proposed modelmodel of the evolutionary process of the evolutionary process and the proposed unrooted tree (and the proposed unrooted tree (hypothesishypothesis) would give rise to ) would give rise to the observed the observed data data (the alignment). The tree found to have the (the alignment). The tree found to have the highest (log)ML value is considered to be the preferred tree. highest (log)ML value is considered to be the preferred tree.
• have to specify a model of sequence evolutionhave to specify a model of sequence evolution• likelihood for all sites is the product of the likelihoods for likelihood for all sites is the product of the likelihoods for
individual sites individual sites assumingassuming all the nucleotide sites evolve all the nucleotide sites evolve independently.independently.
• maximum likelihood method computes the probabilities for all maximum likelihood method computes the probabilities for all possible combinations of ancestral states!possible combinations of ancestral states!
• ML methods evaluate phylogenetic hypotheses n terms of the ML methods evaluate phylogenetic hypotheses n terms of the probability that a proposed probability that a proposed modelmodel of the evolutionary process of the evolutionary process and the proposed unrooted tree (and the proposed unrooted tree (hypothesishypothesis) would give rise to ) would give rise to the observed the observed data data (the alignment). The tree found to have the (the alignment). The tree found to have the highest (log)ML value is considered to be the preferred tree. highest (log)ML value is considered to be the preferred tree.

Interpreting treesInterpreting treesInterpreting treesInterpreting trees
(recurring theme)(recurring theme)(recurring theme)(recurring theme)

Interpreting the treeInterpreting the treeInterpreting the treeInterpreting the tree
• Taxonomic findingsTaxonomic findings
• ParaphylyParaphyly• MonophylyMonophyly
• Taxonomic findingsTaxonomic findings
• ParaphylyParaphyly• MonophylyMonophyly

Interpreting the treeInterpreting the treeInterpreting the treeInterpreting the tree
• Outgroup. place root between distant homologouss sequence and rest group (b)
• Midpoint. place root at midpoint of longest path (sum of branches between any two leafs) NB njplot
• Gene duplication. Place root between paralogous gene copies (b)
• NB all affected by rates !
• Outgroup. place root between distant homologouss sequence and rest group (b)
• Midpoint. place root at midpoint of longest path (sum of branches between any two leafs) NB njplot
• Gene duplication. Place root between paralogous gene copies (b)
• NB all affected by rates !
b
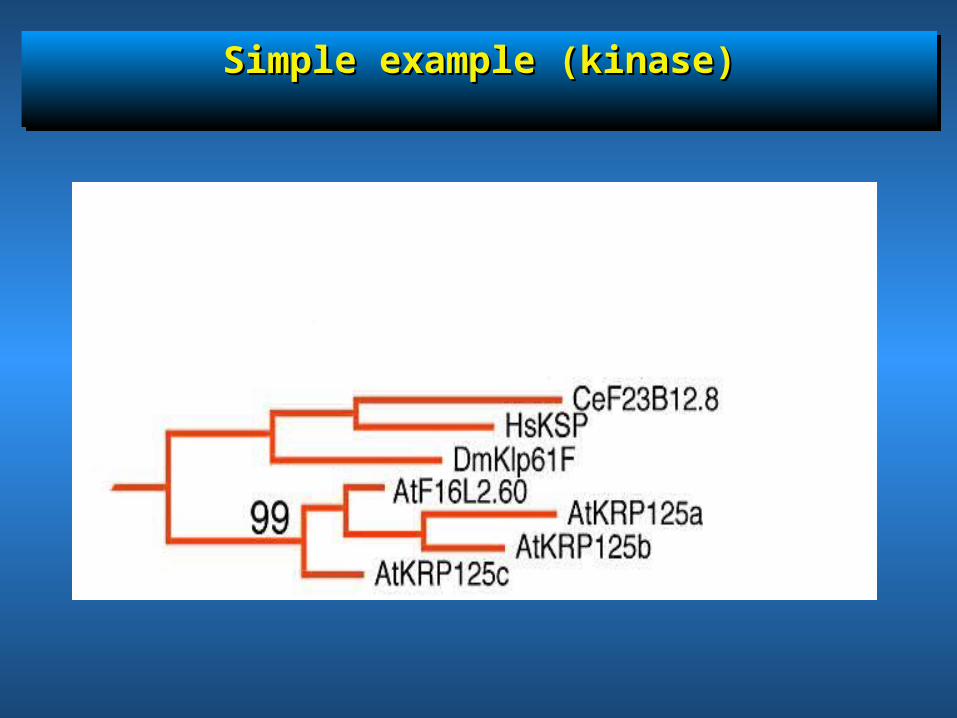
Simple example (kinase)Simple example (kinase)Simple example (kinase)Simple example (kinase)

Two genes per species: how to Two genes per species: how to differentiate between one ancient differentiate between one ancient
or two recent duplications?or two recent duplications?
Two genes per species: how to Two genes per species: how to differentiate between one ancient differentiate between one ancient
or two recent duplications?or two recent duplications?
• Two genes in Human chromosomes ( Human A & Two genes in Human chromosomes ( Human A & Human B) & two genes in mouse chromosomes Human B) & two genes in mouse chromosomes (Mouse A & Mouse B)(Mouse A & Mouse B)
• Two genes in Human chromosomes ( Human A & Two genes in Human chromosomes ( Human A & Human B) & two genes in mouse chromosomes Human B) & two genes in mouse chromosomes (Mouse A & Mouse B)(Mouse A & Mouse B)

?
32
1
Duplications, SpeciationsDuplications, SpeciationsDuplications, SpeciationsDuplications, Speciations

Interpreting the tree: duplications vs speciations, going Interpreting the tree: duplications vs speciations, going pseudo 3Dpseudo 3D
Interpreting the tree: duplications vs speciations, going Interpreting the tree: duplications vs speciations, going pseudo 3Dpseudo 3D
Gene
Gene
Duplic
ation
Duplic
ation
Gene
Gene
Duplic
ation
Duplic
ation
SpeciationSpeciationSpeciationSpeciation

Interpreting the tree: gene trees vs species treesInterpreting the tree: gene trees vs species treesInterpreting the tree: gene trees vs species treesInterpreting the tree: gene trees vs species trees

Interpreting the tree Interpreting the tree Example: vertebrate Example: vertebrate duplicationsduplications
Interpreting the tree Interpreting the tree Example: vertebrate Example: vertebrate duplicationsduplications
• Tetraploidy?Tetraploidy?• Tetraploidy?Tetraploidy?

Interpreting the tree: Horizontal Gene Transfer ( HGT )Interpreting the tree: Horizontal Gene Transfer ( HGT )Interpreting the tree: Horizontal Gene Transfer ( HGT )Interpreting the tree: Horizontal Gene Transfer ( HGT )
Bacteria
EukaryaArchaea

Jargon for interpretation: Orthology (and paralogy) as a Jargon for interpretation: Orthology (and paralogy) as a specification of homology when discussing two speciesspecification of homology when discussing two speciesJargon for interpretation: Orthology (and paralogy) as a Jargon for interpretation: Orthology (and paralogy) as a specification of homology when discussing two speciesspecification of homology when discussing two species
human1human1 mouse1mouse1 human2human2
DuplicationDuplication
Speciation, orSpeciation, or
Fitch 1970Fitch 1970Two genes in two species are Two genes in two species are orthologous if they derive from one orthologous if they derive from one gene in their gene in their lastlast common ancestor common ancestor
““Gene duplication by cell division”Gene duplication by cell division”
““the corresponding gene”the corresponding gene”
implied to have the same functionimplied to have the same function
Genes can diverge byGenes can diverge by

Orthology ~ annotating internal nodesOrthology ~ annotating internal nodesas duplications or speciations as duplications or speciations
Orthology ~ annotating internal nodesOrthology ~ annotating internal nodesas duplications or speciations as duplications or speciations
Because of the definition, how does that translate to a tree
With or without species phylogeny?
Because of the definition, how does that translate to a tree
With or without species phylogeny?

Terminology: inparalogs, outparalogs, co-Terminology: inparalogs, outparalogs, co-orthologsorthologs
Terminology: inparalogs, outparalogs, co-Terminology: inparalogs, outparalogs, co-orthologsorthologs
InparalogsInparalogs
Co-orthologsCo-orthologs
OutparalogsOutparalogs

Importance of orthology for comparative genomics: more Importance of orthology for comparative genomics: more resolutionresolution
Importance of orthology for comparative genomics: more Importance of orthology for comparative genomics: more resolutionresolution
EcEc AfAf AfAfHiHiBsBs EcEc BsBs MgMg Gene family present in Gene family present in Ec Hi Bs Mg AfEc Hi Bs Mg AfOrthologs 1 present inOrthologs 1 present inEc Hi Bs AfEc Hi Bs AfOrthologs 2 present in Orthologs 2 present in Ec Bs Mg Af Ec Bs Mg Af
Phenotype ~ gene correlationPhenotype ~ gene correlationFunc prediction if Hi is only biochem characterized enzymeFunc prediction if Hi is only biochem characterized enzymeFunc prediction by co-ocFunc prediction by co-ocEvolution of gene content: loss vs duplEvolution of gene content: loss vs dupl

Heurisitcs for orthology definitionHeurisitcs for orthology definitionHeurisitcs for orthology definitionHeurisitcs for orthology definition
• Needed becauseNeeded because– Speed (MSA plus reliable tree building is slow)Speed (MSA plus reliable tree building is slow)– Difficulty in deciding of which things you should Difficulty in deciding of which things you should
make a tree in the first place (PFAM?)make a tree in the first place (PFAM?)– Difficulty in operationalizing nuanced tree Difficulty in operationalizing nuanced tree
orthology into group orthologyorthology into group orthology
• Historically bidirectional blast hits BBHHistorically bidirectional blast hits BBH
• Needed becauseNeeded because– Speed (MSA plus reliable tree building is slow)Speed (MSA plus reliable tree building is slow)– Difficulty in deciding of which things you should Difficulty in deciding of which things you should
make a tree in the first place (PFAM?)make a tree in the first place (PFAM?)– Difficulty in operationalizing nuanced tree Difficulty in operationalizing nuanced tree
orthology into group orthologyorthology into group orthology
• Historically bidirectional blast hits BBHHistorically bidirectional blast hits BBH

BBHBBHBBHBBH
Ec1Ec1 AfAf AfAfHiHiBs1Bs1 Ec2Ec2Bs2Bs2 MgMgExtracting tree-like Extracting tree-like information from pairwise information from pairwise similaritiessimilarities
Ec1Bs1 50% Ec1Bs1 50% Ec1Bs2 35%Ec1Bs2 35%Ec2 Bs1 33%Ec2 Bs1 33%Ec2 Bs2 48%Ec2 Bs2 48%
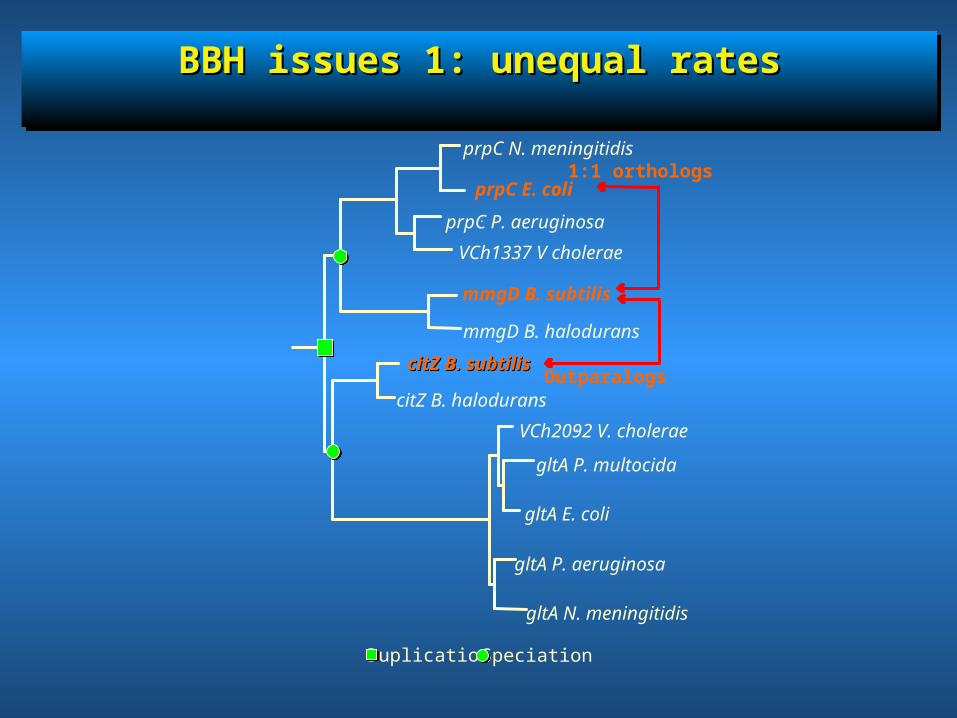
BBH issues 1: unequal ratesBBH issues 1: unequal ratesBBH issues 1: unequal ratesBBH issues 1: unequal rates
Outparalogs
1:1 orthologs
gltA P. multocida
gltA N. meningitidis
gltA P. aeruginosa
gltA E. coli
VCh2092 V. cholerae.
citZ B. halodurans
citZ B. subtiliscitZ B. subtilis
mmgD B. halodurans
mmgD B. subtilis
VCh1337 V cholerae.
prpC P. aeruginosa.
prpC E. coli
prpC N. meningitidis
Duplication Speciation

BBH issues 2: ignores inparalogsBBH issues 2: ignores inparalogsBBH issues 2: ignores inparalogsBBH issues 2: ignores inparalogs
Ec1Ec1 AfAf AfAfHiHiBs1Bs1 Ec2Ec2Bs2Bs2 Bs3Bs3
Ec2 Bs2 48%Ec2 Bs2 48%Ec2 Bs3 51%Ec2 Bs3 51%
(Bs2 Bs3 70%)(Bs2 Bs3 70%)
Ec1 Hi 70%Ec1 Hi 70%Ec2 Hi 38%Ec2 Hi 38%
Prevalence? Depends Prevalence? Depends on e.g. evo distance, on e.g. evo distance, group vs pairwise group vs pairwise orthologyorthologyAt least 16% At least 16% prokaryotesprokaryotes
INPARANOIDINPARANOID

BBH issues 3: differential gene lossBBH issues 3: differential gene lossBBH issues 3: differential gene lossBBH issues 3: differential gene loss
Ec1Ec1 AfAf AfAfHiHiBs1Bs1 Ec2Ec2Bs2Bs2 MgMg
Mg Hi 35%Mg Hi 35%

Other Large Scale orthology schemes: InparanoidOther Large Scale orthology schemes: InparanoidOther Large Scale orthology schemes: InparanoidOther Large Scale orthology schemes: Inparanoid
Eric SonnhammerEric SonnhammerEric SonnhammerEric Sonnhammer

Orthologous groupsOrthologous groupsOrthologous groupsOrthologous groups
• Solution to the non-transitivity of the concept of orthology sensu stricto is: “Group orthology”
• Conceptually: all proteins that are directly descended from one protein in the last common ancestor are considered orthologous to each other
• Operationally: Combine all connected “best triangular hits” into Clusters of Orthologous Groups (COGs, Tatusov et al, 1997). WWW.NCBI.NLM.GOV (Watch out for fusion/fission though !!!)
• Solution to the non-transitivity of the concept of orthology sensu stricto is: “Group orthology”
• Conceptually: all proteins that are directly descended from one protein in the last common ancestor are considered orthologous to each other
• Operationally: Combine all connected “best triangular hits” into Clusters of Orthologous Groups (COGs, Tatusov et al, 1997). WWW.NCBI.NLM.GOV (Watch out for fusion/fission though !!!)

Large Scale orthology schemes: COGLarge Scale orthology schemes: COG Large Scale orthology schemes: COGLarge Scale orthology schemes: COG
• 1. Perform the all-against-all protein sequence 1. Perform the all-against-all protein sequence comparison. comparison.
• 2. Detect and collapse obvious paralogs, that is, 2. Detect and collapse obvious paralogs, that is, proteins from the same genome that are more similar proteins from the same genome that are more similar to each other than to any proteins from other species. to each other than to any proteins from other species.
• 3. Detect triangles of mutually consistent, genome-3. Detect triangles of mutually consistent, genome-specific best hits (BeTs), taking into account the specific best hits (BeTs), taking into account the paralogous groups detected at step 2. paralogous groups detected at step 2.
• 4. Merge triangles with a common side to form COGs. 4. Merge triangles with a common side to form COGs.
• 5. A case-by-case analysis of each COG. This analysis serves to eliminate false-positives and to 5. A case-by-case analysis of each COG. This analysis serves to eliminate false-positives and to identify groups that contain multidomain proteins by examining the pictorial representation of the identify groups that contain multidomain proteins by examining the pictorial representation of the BLAST search outputs. The sequences of detected multidomain proteins are split into single-BLAST search outputs. The sequences of detected multidomain proteins are split into single-domain segments and steps 1–4 are repeated with these sequences, which results in the domain segments and steps 1–4 are repeated with these sequences, which results in the assignment of individual domains to COGs in accordance with their distinct evolutionary assignment of individual domains to COGs in accordance with their distinct evolutionary affinities. affinities.
• 6. Examination of large COGs that include multiple members from all or several of the genomes 6. Examination of large COGs that include multiple members from all or several of the genomes using phylogenetic trees, cluster analysis and visual inspection of alignments; as a result, some using phylogenetic trees, cluster analysis and visual inspection of alignments; as a result, some of these groups are split into two or more smaller ones that are included in the final set of COGs. of these groups are split into two or more smaller ones that are included in the final set of COGs.
• 1. Perform the all-against-all protein sequence 1. Perform the all-against-all protein sequence comparison. comparison.
• 2. Detect and collapse obvious paralogs, that is, 2. Detect and collapse obvious paralogs, that is, proteins from the same genome that are more similar proteins from the same genome that are more similar to each other than to any proteins from other species. to each other than to any proteins from other species.
• 3. Detect triangles of mutually consistent, genome-3. Detect triangles of mutually consistent, genome-specific best hits (BeTs), taking into account the specific best hits (BeTs), taking into account the paralogous groups detected at step 2. paralogous groups detected at step 2.
• 4. Merge triangles with a common side to form COGs. 4. Merge triangles with a common side to form COGs.
• 5. A case-by-case analysis of each COG. This analysis serves to eliminate false-positives and to 5. A case-by-case analysis of each COG. This analysis serves to eliminate false-positives and to identify groups that contain multidomain proteins by examining the pictorial representation of the identify groups that contain multidomain proteins by examining the pictorial representation of the BLAST search outputs. The sequences of detected multidomain proteins are split into single-BLAST search outputs. The sequences of detected multidomain proteins are split into single-domain segments and steps 1–4 are repeated with these sequences, which results in the domain segments and steps 1–4 are repeated with these sequences, which results in the assignment of individual domains to COGs in accordance with their distinct evolutionary assignment of individual domains to COGs in accordance with their distinct evolutionary affinities. affinities.
• 6. Examination of large COGs that include multiple members from all or several of the genomes 6. Examination of large COGs that include multiple members from all or several of the genomes using phylogenetic trees, cluster analysis and visual inspection of alignments; as a result, some using phylogenetic trees, cluster analysis and visual inspection of alignments; as a result, some of these groups are split into two or more smaller ones that are included in the final set of COGs. of these groups are split into two or more smaller ones that are included in the final set of COGs.

Large Scale orthology schemes: COGLarge Scale orthology schemes: COGLarge Scale orthology schemes: COGLarge Scale orthology schemes: COG
• 5. A case-by-case analysis of each COG. This analysis serves 5. A case-by-case analysis of each COG. This analysis serves to eliminate false-positives and to identify groups that contain to eliminate false-positives and to identify groups that contain multidomain proteins by examining the pictorial representation multidomain proteins by examining the pictorial representation of the BLAST search outputs. The sequences of detected of the BLAST search outputs. The sequences of detected multidomain proteins are split into single-domain segments and multidomain proteins are split into single-domain segments and steps 1–4 are repeated with these sequences, which results in steps 1–4 are repeated with these sequences, which results in the assignment of individual domains to COGs in accordance the assignment of individual domains to COGs in accordance with their distinct evolutionary affinities. with their distinct evolutionary affinities.
• 6. Examination of large COGs that include multiple members 6. Examination of large COGs that include multiple members from all or several of the genomes using phylogenetic trees, from all or several of the genomes using phylogenetic trees, cluster analysis and visual inspection of alignments; as a result, cluster analysis and visual inspection of alignments; as a result, some of these groups are split into two or more smaller ones some of these groups are split into two or more smaller ones that are included in the final set of COGs. that are included in the final set of COGs.
• 5. A case-by-case analysis of each COG. This analysis serves 5. A case-by-case analysis of each COG. This analysis serves to eliminate false-positives and to identify groups that contain to eliminate false-positives and to identify groups that contain multidomain proteins by examining the pictorial representation multidomain proteins by examining the pictorial representation of the BLAST search outputs. The sequences of detected of the BLAST search outputs. The sequences of detected multidomain proteins are split into single-domain segments and multidomain proteins are split into single-domain segments and steps 1–4 are repeated with these sequences, which results in steps 1–4 are repeated with these sequences, which results in the assignment of individual domains to COGs in accordance the assignment of individual domains to COGs in accordance with their distinct evolutionary affinities. with their distinct evolutionary affinities.
• 6. Examination of large COGs that include multiple members 6. Examination of large COGs that include multiple members from all or several of the genomes using phylogenetic trees, from all or several of the genomes using phylogenetic trees, cluster analysis and visual inspection of alignments; as a result, cluster analysis and visual inspection of alignments; as a result, some of these groups are split into two or more smaller ones some of these groups are split into two or more smaller ones that are included in the final set of COGs. that are included in the final set of COGs.

Other Large Scale orthology schemes: Ortho MCLOther Large Scale orthology schemes: Ortho MCLOther Large Scale orthology schemes: Ortho MCLOther Large Scale orthology schemes: Ortho MCL

The too ambitious comparative genomics dilemma: The too ambitious comparative genomics dilemma: duplication/speciation vs domainsduplication/speciation vs domains
The too ambitious comparative genomics dilemma: The too ambitious comparative genomics dilemma: duplication/speciation vs domainsduplication/speciation vs domains
~orthologs
Single structural elements?
homologs Distant homologs
Domain composition, accretionDomain composition, accretion
Sequence divergenceSequence divergence
Gene fusion Domain cassettes Domains
i.e. genome comparison between close species:no domain considerations, sub-sub-ortholog. Between distant Homologs, loads of domain considerations
TIMETIME
Gene
Gene Trivial orthologs
present Verydistantpast

Implication of coupling between duplication & domain Implication of coupling between duplication & domain accretion for evolution and function predictionaccretion for evolution and function prediction
Implication of coupling between duplication & domain Implication of coupling between duplication & domain accretion for evolution and function predictionaccretion for evolution and function prediction
• for some genes life is easy 1:1:1 orthologs, no for some genes life is easy 1:1:1 orthologs, no fusion / domains, couple of losses. But a minority of fusion / domains, couple of losses. But a minority of families but a large proportion of proteins is a families but a large proportion of proteins is a formidable challenge, domains permutations and formidable challenge, domains permutations and duplications make life complicatedduplications make life complicated
• for some genes life is easy 1:1:1 orthologs, no for some genes life is easy 1:1:1 orthologs, no fusion / domains, couple of losses. But a minority of fusion / domains, couple of losses. But a minority of families but a large proportion of proteins is a families but a large proportion of proteins is a formidable challenge, domains permutations and formidable challenge, domains permutations and duplications make life complicatedduplications make life complicated

Orthology & Orthology & function function
predictionprediction
Blast with a newly Blast with a newly sequenced globin sequenced globin
from frogfrom frog
Orthology & Orthology & function function
predictionprediction
Blast with a newly Blast with a newly sequenced globin sequenced globin
from frogfrom frog
What kind of globin is it?

GlobinsGlobinsGlobinsGlobins
Blast query
Orthologous & function prediction Orthologous & function prediction vs vs
homologous that are not orthologous & functionhomologous that are not orthologous & function
Orthologous & function prediction Orthologous & function prediction vs vs
homologous that are not orthologous & functionhomologous that are not orthologous & function
• Orthologs tend to have the exact same molecular Orthologs tend to have the exact same molecular function, mere HTANO’s not function, mere HTANO’s not
• and operate in the same “pathway”. and operate in the same “pathway”.
• Orthologs mostly have the same domain Orthologs mostly have the same domain composition;composition;
• Orthologs tend to have the exact same molecular Orthologs tend to have the exact same molecular function, mere HTANO’s not function, mere HTANO’s not
• and operate in the same “pathway”. and operate in the same “pathway”.
• Orthologs mostly have the same domain Orthologs mostly have the same domain composition;composition;
… … but inparalogs: fate after duplication: but inparalogs: fate after duplication: neofunctionalization or subfunctionalizationneofunctionalization or subfunctionalization
… … but inparalogs: fate after duplication: but inparalogs: fate after duplication: neofunctionalization or subfunctionalizationneofunctionalization or subfunctionalization
• Even evolutionary true orthologs can have “different Even evolutionary true orthologs can have “different functions”functions”
• Both co-orthologs have taken over some aspect of Both co-orthologs have taken over some aspect of the ancestral function and have lost other aspectsthe ancestral function and have lost other aspects
• Acquiring of new function or loss-of-function: one of Acquiring of new function or loss-of-function: one of co-orthologs does something different now.co-orthologs does something different now.
• Even evolutionary true orthologs can have “different Even evolutionary true orthologs can have “different functions”functions”
• Both co-orthologs have taken over some aspect of Both co-orthologs have taken over some aspect of the ancestral function and have lost other aspectsthe ancestral function and have lost other aspects
• Acquiring of new function or loss-of-function: one of Acquiring of new function or loss-of-function: one of co-orthologs does something different now.co-orthologs does something different now.
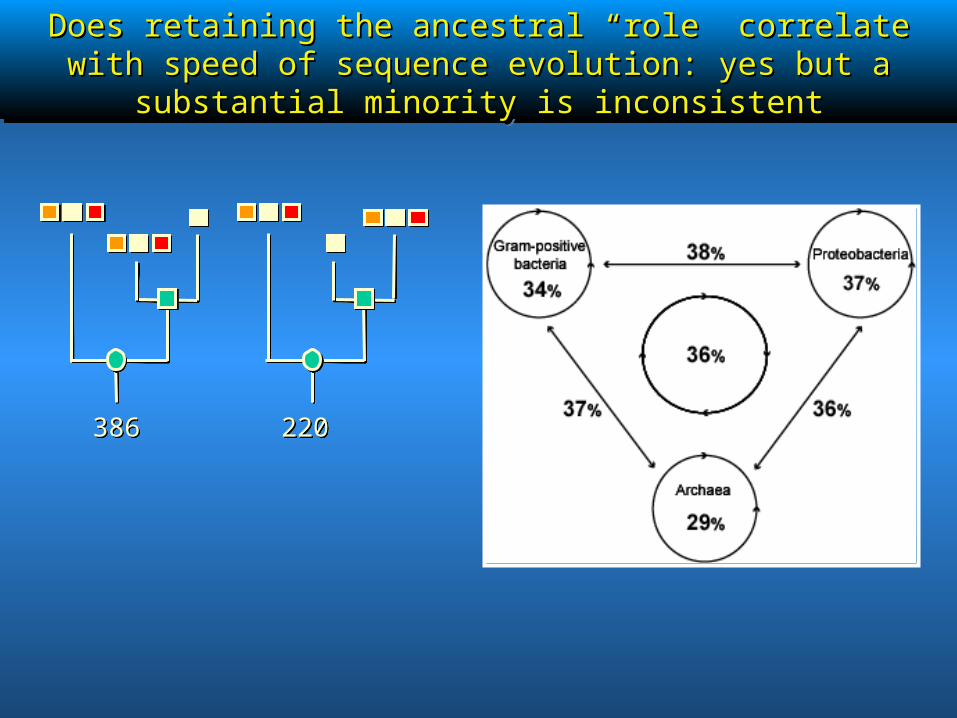
Does retaining the ancestral “role” correlate with speed of Does retaining the ancestral “role” correlate with speed of sequence evolution: yes but a substantial minority is inconsistentsequence evolution: yes but a substantial minority is inconsistent
Does retaining the ancestral “role” correlate with speed of Does retaining the ancestral “role” correlate with speed of sequence evolution: yes but a substantial minority is inconsistentsequence evolution: yes but a substantial minority is inconsistent
386386 220220

rfbBrfbB / / rffGrffGrfbBrfbB / / rffGrffG
RfbB and RffG catalyze the same reaction, but are involved in two different biological processes. rfb gene cluster: biosynthesis of O-specific polysaccharides (inner membrane). rff gene cluster: complex biosynthesis of enterobacteria common antigen (outer membrane).
RfbB and RffG catalyze the same reaction, but are involved in two different biological processes. rfb gene cluster: biosynthesis of O-specific polysaccharides (inner membrane). rff gene cluster: complex biosynthesis of enterobacteria common antigen (outer membrane).

Why do observe inconsistencies?Why do observe inconsistencies?Why do observe inconsistencies?Why do observe inconsistencies?
11 00 11 55 2020 2525 3030 3535 4040 4545 5050 5555 6060 6565 7070 7575 8080 8585 9090 9595 0 0 55 100100
Fre
que
ncy
(# c
ase
s)F
requ
enc
y (#
ca
ses)
0 0
10 10
30 30
20 20
40 40
60 60
50 50
70 70
Sequence identity between inparalogs (%)Sequence identity between inparalogs (%)
ConsistentConsistent
InconsistentInconsistent
Not because of chance due to lack of divergence time Not because of chance due to lack of divergence time

Why do observe inconsistencies?Why do observe inconsistencies?Why do observe inconsistencies?Why do observe inconsistencies?
Similar sequence divergence of inparalogs relative to their single-ortholog, molecular function similar?
Any inconsistencies are then a chance outcome: both duplicates have diverged, but at (roughly) the same evolutionary speed (most amino acids substitutions are only been subject to purifying selection and not to adaptive selection)
Similar sequence divergence of inparalogs relative to their single-ortholog, molecular function similar?
Any inconsistencies are then a chance outcome: both duplicates have diverged, but at (roughly) the same evolutionary speed (most amino acids substitutions are only been subject to purifying selection and not to adaptive selection)

• In certain orthology scheme gene order is given In certain orthology scheme gene order is given prevalence above most similarityprevalence above most similarity
• Gene at conserved position is considered the Gene at conserved position is considered the “original” and the other duplicate the “copy”“original” and the other duplicate the “copy”
• In certain orthology scheme gene order is given In certain orthology scheme gene order is given prevalence above most similarityprevalence above most similarity
• Gene at conserved position is considered the Gene at conserved position is considered the “original” and the other duplicate the “copy”“original” and the other duplicate the “copy”
Related Documents











