LETTERS Biodiversity and biogeography of phages in modern stromatolites and thrombolites Christelle Desnues 1 , Beltran Rodriguez-Brito 1,2 , Steve Rayhawk 1,2 , Scott Kelley 1,3 , Tuong Tran 1 , Matthew Haynes 1 , Hong Liu 1 , Mike Furlan 1 , Linda Wegley 1 , Betty Chau 1 , Yijun Ruan 4 , Dana Hall 1 , Florent E. Angly 1 , Robert A. Edwards 1,2,3,5 , Linlin Li 1 , Rebecca Vega Thurber 1 , R. Pamela Reid 6 , Janet Siefert 7 , Valeria Souza 8 , David L. Valentine 9 , Brandon K. Swan 9 , Mya Breitbart 10 & Forest Rohwer 1,3 Viruses, and more particularly phages (viruses that infect bac- teria), represent one of the most abundant living entities in aquatic and terrestrial environments. The biogeography of phages has only recently been investigated and so far reveals a cosmopolitan distribution of phage genetic material (or genotypes) 1–4 . Here we address this cosmopolitan distribution through the analysis of phage communities in modern microbialites, the living represen- tatives of one of the most ancient life forms on Earth. On the basis of a comparative metagenomic analysis of viral communities associated with marine (Highborne Cay, Bahamas) and freshwater (Pozas Azules II and Rio Mesquites, Mexico) microbialites, we show that some phage genotypes are geographically restricted. The high percentage of unknown sequences recovered from the three metagenomes (.97%), the low percentage similarities with sequences from other environmental viral (n 5 42) and microbial (n 5 36) metagenomes, and the absence of viral genotypes shared among microbialites indicate that viruses are genetically unique in these environments. Identifiable sequences in the Highborne Cay metagenome were dominated by single-stranded DNA micro- phages that were not detected in any other samples examined, including sea water, fresh water, sediment, terrestrial, extreme, metazoan-associated and marine microbial mats. Finally, a marine signature was present in the phage community of the Pozas Azules II microbialites, even though this environment has not been in contact with the ocean for tens of millions of years. Taken together, these results prove that viruses in modern microbialites display biogeographical variability and suggest that they may be derived from an ancient community. Microbialites are organosedimentary structures accreted by sedi- ment trapping, binding and in situ precipitation due to the growth and metabolic activities of microorganisms 5 . Stromatolites and thrombolites are morphological types of microbialites classified by their internal mesostructure: layered and clotted, respectively 5 . Microbialites first appeared in the geological record ,3.5 billion years ago, and for more than 2 billion years they are the main evi- dence of life on Earth 6,7 . Whether modern microbialites are proxies of ancient ecosystems is a major outstanding question 6 . Viruses, and more specifically phages, are the most abundant bio- logical entities in the world’s oceans 8 . Phages influence microbial growth rates, genetic exchange, diversity and adaptation, and thus evolution 8 . Current biogeographical studies of phages suggest that they are cosmopolitan in distribution, unlike some examples of highly endemic populations of bacteria and archaea 9–12 . Metagenomic analysis of viral communities from four major ocean regions using the same pyrosequencing technology has shown that essentially all marine viruses are spread widely throughout the oceans 1 . Identical phage-encoded exotoxin genes, T7-like DNA poly- merase genes and T4-like structural genes are found in disparate terrestrial, aquatic and extreme environments 2–4 . Phages from soil, sediments and fresh water can productively infect marine microbes 13,14 , showing that viruses move between major biomes. Our metagenomic analysis of viral communities associated with a marine stromatolite (Highborne Cay, Bahamas) and two neighbour- ing (30 km) freshwater thrombolites and stromatolites (Pozas Azules II and Rio Mesquites, Mexico; Supplementary Fig. 1) showed that most of the sequences (98.8, 99.3 and 97.7% for Highborne Cay, Pozas Azules and Rio Mesquites, respectively) were unique when compared with the sequences in the non-redundant GenBank/ SEED databases (BLASTx, E-value ,10 22 ). This proportion is much higher than any other previously sequenced viral meta- genome (70–90% unknowns 1,15 ). A comparison of microbialite metagenomic sequences with 42 viral and 36 microbial metagenomic libraries generated using the same pyrosequencing technology (Tables 1 and 2, respectively; Supplementary Tables 1 and 2 for details), showed that they were less than 5% similar (BLASTn, E-value ,10 23 ), further confirming that these are largely unrelated viral communities. Using the approach developed by Angly et al. 1 , random subsets of 10,000 sequences from each virome were assembled against each other to identify cross-contigs (that is, sequence overlaps between two samples). A read from one metagenome that assembled with a read from another metagenome indicated an overlap between these two metagenomes 1 . Only contigs produced by sequences from dif- ferent metagenomes were taken into account to assess how many species were common to the two communities (percentage shared) 1 . Comparisons between Highborne Cay and Pozas Azules II and between Highborne Cay and Rio Mesquites did not produce any cross-contigs, indicating that none of the viruses was shared between these microbialites. The Pozas Azules II-Rio Mesquites comparison produced a very small average cross-contig spectrum, again indi- cating that essentially nothing is shared between these samples, even though they were taken from microbialites located 30 km from each other. A Monte Carlo analysis of the cross-contig spectra showed that the percentage of genome shared between Pozas Azules II, Highborne 1 Department of Biology, 2 Computational Sciences Research Center, 3 Center for Microbial Sciences, San Diego State University, San Diego, California 92182, USA. 4 Genome Institute of Singapore, Singapore 138672, Singapore. 5 Mathematics and Computer Science Division, Argonne National Laboratory, Argonne, Illinois 60439, USA. 6 Rosenstiel School of Marine and Atmospheric Science, University of Miami, Miami, Florida 33149, USA. 7 Department of Statistics, Rice University, Houston, Texas 77251, USA. 8 Departamento de Ecologı ´a Evolutiva, Instituto de Ecologı ´a, Universidad Nacional Auto ´noma de Me ´xico AP 70-275 Coyoaca´n, 04510 Mexico D.F., Mexico. 9 Department of Earth Science, University of California Santa Barbara, Santa Barbara, California 93106, USA. 10 College of Marine Science, University of South Florida, St Petersburg, Florida 33701, USA. doi:10.1038/nature06735 1 Nature Publishing Group ©2008

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

LETTERS
Biodiversity and biogeography of phages in modernstromatolites and thrombolitesChristelle Desnues1, Beltran Rodriguez-Brito1,2, Steve Rayhawk1,2, Scott Kelley1,3, Tuong Tran1, Matthew Haynes1,Hong Liu1, Mike Furlan1, Linda Wegley1, Betty Chau1, Yijun Ruan4, Dana Hall1, Florent E. Angly1,Robert A. Edwards1,2,3,5, Linlin Li1, Rebecca Vega Thurber1, R. Pamela Reid6, Janet Siefert7, Valeria Souza8,David L. Valentine9, Brandon K. Swan9, Mya Breitbart10 & Forest Rohwer1,3
Viruses, and more particularly phages (viruses that infect bac-teria), represent one of the most abundant living entities in aquaticand terrestrial environments. The biogeography of phages hasonly recently been investigated and so far reveals a cosmopolitandistribution of phage genetic material (or genotypes)1–4. Here weaddress this cosmopolitan distribution through the analysis ofphage communities in modern microbialites, the living represen-tatives of one of the most ancient life forms on Earth. On the basisof a comparative metagenomic analysis of viral communitiesassociated with marine (Highborne Cay, Bahamas) and freshwater(Pozas Azules II and Rio Mesquites, Mexico) microbialites, weshow that some phage genotypes are geographically restricted.The high percentage of unknown sequences recovered from thethree metagenomes (.97%), the low percentage similarities withsequences from other environmental viral (n 5 42) and microbial(n 5 36) metagenomes, and the absence of viral genotypes sharedamong microbialites indicate that viruses are genetically unique inthese environments. Identifiable sequences in the Highborne Caymetagenome were dominated by single-stranded DNA micro-phages that were not detected in any other samples examined,including sea water, fresh water, sediment, terrestrial, extreme,metazoan-associated and marine microbial mats. Finally, a marinesignature was present in the phage community of the Pozas AzulesII microbialites, even though this environment has not been incontact with the ocean for tens of millions of years. Takentogether, these results prove that viruses in modern microbialitesdisplay biogeographical variability and suggest that they may bederived from an ancient community.
Microbialites are organosedimentary structures accreted by sedi-ment trapping, binding and in situ precipitation due to the growthand metabolic activities of microorganisms5. Stromatolites andthrombolites are morphological types of microbialites classified bytheir internal mesostructure: layered and clotted, respectively5.Microbialites first appeared in the geological record ,3.5 billionyears ago, and for more than 2 billion years they are the main evi-dence of life on Earth6,7. Whether modern microbialites are proxies ofancient ecosystems is a major outstanding question6.
Viruses, and more specifically phages, are the most abundant bio-logical entities in the world’s oceans8. Phages influence microbialgrowth rates, genetic exchange, diversity and adaptation, and thusevolution8. Current biogeographical studies of phages suggest thatthey are cosmopolitan in distribution, unlike some examples of
highly endemic populations of bacteria and archaea9–12.Metagenomic analysis of viral communities from four major oceanregions using the same pyrosequencing technology has shown thatessentially all marine viruses are spread widely throughout theoceans1. Identical phage-encoded exotoxin genes, T7-like DNA poly-merase genes and T4-like structural genes are found in disparateterrestrial, aquatic and extreme environments2–4. Phages from soil,sediments and fresh water can productively infect marinemicrobes13,14, showing that viruses move between major biomes.
Our metagenomic analysis of viral communities associated with amarine stromatolite (Highborne Cay, Bahamas) and two neighbour-ing (30 km) freshwater thrombolites and stromatolites (Pozas AzulesII and Rio Mesquites, Mexico; Supplementary Fig. 1) showed thatmost of the sequences (98.8, 99.3 and 97.7% for Highborne Cay,Pozas Azules and Rio Mesquites, respectively) were unique whencompared with the sequences in the non-redundant GenBank/SEED databases (BLASTx, E-value ,1022). This proportionis much higher than any other previously sequenced viral meta-genome (70–90% unknowns1,15). A comparison of microbialitemetagenomic sequences with 42 viral and 36 microbial metagenomiclibraries generated using the same pyrosequencing technology(Tables 1 and 2, respectively; Supplementary Tables 1 and 2 fordetails), showed that they were less than 5% similar (BLASTn,E-value ,1023), further confirming that these are largely unrelatedviral communities.
Using the approach developed by Angly et al.1, random subsets of10,000 sequences from each virome were assembled against eachother to identify cross-contigs (that is, sequence overlaps betweentwo samples). A read from one metagenome that assembled with aread from another metagenome indicated an overlap between thesetwo metagenomes1. Only contigs produced by sequences from dif-ferent metagenomes were taken into account to assess how manyspecies were common to the two communities (percentage shared)1.Comparisons between Highborne Cay and Pozas Azules II andbetween Highborne Cay and Rio Mesquites did not produce anycross-contigs, indicating that none of the viruses was shared betweenthese microbialites. The Pozas Azules II-Rio Mesquites comparisonproduced a very small average cross-contig spectrum, again indi-cating that essentially nothing is shared between these samples, eventhough they were taken from microbialites located 30 km from eachother. A Monte Carlo analysis of the cross-contig spectra showed thatthe percentage of genome shared between Pozas Azules II, Highborne
1Department of Biology, 2Computational Sciences Research Center, 3Center for Microbial Sciences, San Diego State University, San Diego, California 92182, USA. 4Genome Institute ofSingapore, Singapore 138672, Singapore. 5Mathematics and Computer Science Division, Argonne National Laboratory, Argonne, Illinois 60439, USA. 6Rosenstiel School of Marine andAtmospheric Science, University of Miami, Miami, Florida 33149, USA. 7Department of Statistics, Rice University, Houston, Texas 77251, USA. 8Departamento de Ecologıa Evolutiva,Instituto de Ecologıa, Universidad Nacional Autonoma de Mexico AP 70-275 Coyoacan, 04510 Mexico D.F., Mexico. 9Department of Earth Science, University of California SantaBarbara, Santa Barbara, California 93106, USA. 10College of Marine Science, University of South Florida, St Petersburg, Florida 33701, USA.
doi:10.1038/nature06735
1Nature Publishing Group©2008

Cay and Rio Mesquites was zero (Supplementary Fig. 5) and there-fore that the viruses are genetically unique in all three microbialites.
The small number of ‘known’ phage sequences in the microbialitemetagenomes was assigned taxonomical designations based on thetop BLAST similarities (Fig. 1, right panel). Their relative abundanceswere plotted onto the Phage Proteomic Tree16 (PPT; Fig. 1, leftpanel). Microphages (icosahedral single-stranded DNA phagesinfecting Escherichia coli, Bdellovibrio, Chlamydia and Spiroplasmaspecies17, Supplementary Fig. 3) were the most common phages in
the Highborne Cay and Pozas Azules II phage communities, repre-senting 93.1% and 13.5% of the known phage sequences, respec-tively. In contrast, microphages were absent in Rio Mesquites, andthe phage community was dominated by Shewanella oneidensis pro-phages (MuSo2 and LambdaSo) and Burkholderia cepacia phagesequences (54.6% of the total number of phage reads). At the taxo-nomic resolution of the PPT, the Highborne Cay and Pozas Azules IIviral communities resembled each other and a previously describedmarine virome from the Sargasso Sea, which also contained high
Table 1 | Similarity among the microbialite viral metagenomes and other environmental viral metagenomes
Average percentage similarity (BLASTn, E-value ,1023)*
Highborne Cay viral metagenome Pozas Azules II viral metagenome Rio Mesquites viral metagenome
Highborne Cay 100 1.140 0.910
Pozas Azules II 4.020 100 1.100
Rio Mesquites 0.970 0.700 100
Freshwaters (n 5 4) 1.154 6 0.240 0.477 6 0.031 0.916 6 0.278
Coral reef waters (n 5 4) 1.462 6 0.285 0.840 6 0.032 0.808 6 0.043
Marine waters (n 5 4) 1.770 6 0.573 0.585 6 0.116 0.543 6 0.098
Fish (n 5 4) 0.701 6 0.156 0.279 6 0.015 0.387 6 0.061
Mosquito (n 5 1) 0.731 0.273 0.683
Coral (n 5 6) 0.735 6 0.150 0.290 6 0.027 0.243 6 0.024
Human (n 5 2) 0.881 6 0.336 0.377 6 0.019 0.375 6 0.019
Saltern waters (n 5 11) 0.690 6 0.145 0.439 6 0.059 0.445 6 0.058
Marine sediments (n 5 3) 0.654 6 0.079 0.568 6 0.057 0.401 6 0.089
*Average percentage similarity 6 s.e.m.
Table 2 | Similarity among the microbialite viral metagenomes and other environmental microbial metagenomes
Average percentage similarity (BLASTn, E-value ,1023)*
Highborne Cay viral metagenome Pozas Azules II viral metagenome Rio Mesquites viral metagenome
Highborne Cay 47.104 0.400 0.230
Pozas Azules II 4.310 3.742 0.410
Rio Mesquites 1.021 0.637 0.541
Freshwaters (n 5 4) 1.853 6 0.609 0.466 6 0.083 0.559 6 0.091
Coral reef waters (n 5 4) 0.903 6 0.256 0.340 6 0.050 0.276 6 0.022
Fish (n 5 4) 0.288 6 0.015 0.252 6 0.007 0.331 6 0.038
Coral (n 5 7) 0.805 6 0.167 0.255 6 0.016 0.252 6 0.031
Saltern waters (n 5 11) 0.655 6 0.122 0.419 6 0.034 0.398 6 0.037
Subterranean (n 5 2) 0.959 6 0.377 0.442 6 0.045 0.470 6 0.122
Marine sediments (n 5 1) 1.168 0.432 0.321
*Average percentage similarity 6 s.e.m.
Highborne CayRio
Mesquites
+ – + –
B. bacteriovorusphi MH2K (27.8%)
P. marinusphi P-SSM2
(0.8%)
P. marinusphi P-SSM2
(3.5%)
B. cepaciaphi BcepNazgul
(18.9%)
S. oneidensis phiMuSo2 prophage
(12.9%)
S. oneidensis phiLambdaSoprophage(22.8%)
6,832 460 5,788 4,997 31,182
Chlamydia phi Chp1 (3.4%)
Mycobacteriaphages
Chp1-likemicrophage 93.1% 13.5%
P. marinusphi P-SSM4 (7.1%)
SargassoSea
27,950
B. bacteriovorusphi MH2K (7.1%)
Chlamydia phi 3 (11.0%)
P. marinusphi P-SSM2
(17.9%)
P. marinusphi P-SSP7 (18.4%)
29.6%
Synechococcusphi S-PM2 (4.0%)
Chlamydia
-proteobacterium
Synechococcusphi S-PM2 (1.6%)
Chlamydia phi 3 (5.3%) phi 3 (34.3%)
phi Jl001 (2.0%)α
Pozas Azules II
MicrophagesNumber of phage reads
(with and without microphages)
Figure 1 | The phage proteomic tree. The tree(left) shows the similarities of the viralmetagenomic sequences to completely sequencedphage genomes. The presence and abundance ofphage reads (right; abundance is proportional toline length) are presented in green for HighborneCay, red for Pozas Azules II, blue for RioMesquites and grey for the Sargasso Sea samples.The total number of reads with significantsimilarity to phages (plus and minusmicrophages) is also indicated for Highborne Cayand Pozas Azules II. The name of the phageassociated with the most abundant reads of eachmetagenome is given as well as the percentage ofthe total represented by these reads.
LETTERS NATURE
2Nature Publishing Group©2008

abundances of microphages (29.6%), Prochlorococcus phagesP-SSM2 and P-SSM4 and Synechococcus phage S-PM2 (ref. 1) (Fig. 1).
Genetic distances of the microphages in Highborne Cay, PozasAzules II and the Sargasso Sea were calculated using global align-ments of the viral capsid protein (Vp1) reconstructed from the meta-genomes (Fig. 2). The microphages from these three environmentsclustered together and were branched to the group of phages infect-ing Chlamydia. However, cross-assembly of the microphage nucleic-acid sequences did not produce a single cross-contig, indicating thatamino-acid-level functionality is maintained but the nucleic acidshave significantly diverged. On the basis of each consensus sequencerecovered from the Highborne Cay, Pozas Azules II and Sargasso Seametagenomes (Supplementary Information part 2), primers target-ing the Vp1 genes were designed (Supplementary Table 4). The capsidgenes were successfully amplified from these metagenomes. Nopolymerase chain reaction (PCR) products were obtained whenone sample was tested with the two other primer sets (for example,PCR of Highborne Cay viral DNA with the Pozas Azules II or theSargasso Sea primer sets). Phylogenetic analysis of PCR productsfrom the Highborne Cay sample showed that the similarity betweenclones and cultured microphage capsid sequences ranged from 47.5to 61.2% at the nucleic-acid level and from 37.2 to 69.3% at theprotein level, respectively (Supplementary Figs 8A and 8B).
We previously recovered cosmopolitan, essentially identical, T7-like podophage DNA polymerase sequences in the major biomes onEarth, including: marine, freshwater, sediment, terrestrial, extremeand metazoan-associated3. These environmental samples, as wellas other marine microbial mats from different parts of the world(11 samples—from France, Israel, Bahamas, Puerto Rico andConnecticut, USA), were tested for the presence of the HighborneCay microphages (Supplementary Table 5). No such microphageswere detected in all the environmental samples tested, even thoughour PCR was sensitive enough to amplify fewer than 100 copies of theVp1 gene (Supplementary Fig. 6). New Highborne Cay stromatolitesamples (July 2007) tested positive for the presence of the micro-phages, further confirming that these phages are native to theHighborne Cay stromatolites and persistent across time. To ourknowledge, this is the first evidence of endemism in phages.
A ‘marine signature’ of the microbes from the Cuatro CienegasBasin was recently described by Souza et al.18, implying that the wholeecosystem may be derived from an ancient marine community.
Similarly, weighted and unweighted Unifrac analyses of the PPT(Supplementary Figs 4A, B) showed a genetic overlap between theGulf of Mexico, the Sargasso Sea and the Pozas Azules II phagecommunities, even though these environments have not been incontact since the late Jurassic. This observation supports the hypo-thesis that phages in modern microbialites may be relicts froman ancient community. An alternative hypothesis that we cannotexclude is that there was a recent marine phage introduction, possiblythrough aerial vectors such as birds or airborne particles. However,the observation that these microbialite phages are extremely divergedfrom the global virome and from its nearest neighbour is more con-gruent with our ancient phage hypothesis.
METHODS SUMMARY
Microbialites were collected from the Pozas Azules II (PAII) pool and the Rio
Mesquites (RM) River located in the Cuatro Cienegas Basin (Mexico) and from
the Highborne Cay (HC) marine waters (Bahamas). The viral particles were
resuspended and purified using a combination of filtration and caesium chloride
density gradient centrifugation15. Viral DNA was isolated by a formamide/CTAB
extraction19 and amplified with GenomiPhi (GE Healthcare) following the
manufacturer’s recommendations. Approximately 10 mg purified DNA was
sequenced using pyrosequencing technology20 (454 Life Sciences).
The sequences from each metagenome were compared to the SEED non-
redundant database, our in-house phage database and 78 other metagenomes
(using BLAST). The presence and the abundance of the sequences that have the
phage databases were mapped onto the PPT (Fig. 1) using Bio-Metamapper
(http://scums.sdsu.edu/Mapper). The diversity of the viral community and the
percentage of viral genomes shared among samples were determined as pre-
viously described1. The genetic distances were calculated using the online
UniFrac tool21. The Isolation by Distance web service22 was used to test the
correlation of the geographical distance and the genetic divergence between
two viral communities.
Microphage capsid consensus sequences were reconstructed from the HC,
PAII and Sargasso Sea1 metagenomes and replaced onto a phylogenetic tree
(Fig. 2). Primers were designed on the basis of these sequences (Supplemen-
tary Table 4) to retrospectively amplify the microphage capsid from the HC
stromatolites. These sequences were cloned, sequenced (8 clones) and replaced
in phylogenetic trees (Supplementary Figs 8A and 8B). PCR detection limit was
defined (Supplementary Fig. 6) and optimal conditions were used to test the
occurrence of the HC microphages in 63 different environmental samples
(Supplementary Table 5).
Full Methods and any associated references are available in the online version ofthe paper at www.nature.com/nature.
Received 5 December 2007; accepted 23 January 2008.Published online 2 March 2008.
1. Angly, F. E. et al. The marine viromes of four oceanic regions. PLoS Biol. 4, e368(2006).
2. Casas, V. et al. Widespread occurrence of phage-encoded exotoxin genes interrestrial and aquatic environments in Southern California. FEMS Microbiol. Lett.261, 141–149 (2006).
3. Breitbart, M., Miyake, J. H. & Rohwer, F. Global distribution of nearly identicalphage-encoded DNA sequences. FEMS Microbiol. Lett. 236, 249–256 (2004).
4. Short, C. M. & Suttle, C. A. Nearly identical bacteriophage structural genesequences are widely distributed in both marine and freshwater environments.Appl. Environ. Microbiol. 71, 480–486 (2005).
5. Walter, M. R. Stromatolites. (Elsevier, Amsterdam, 1976).
6. Allwood, A. C., Walter, M. R., Kamber, B. S., Marshall, C. P. & Burch, I. W.Stromatolite reef from the Early Archaean era of Australia. Nature 441, 714–718(2006).
7. Schopf, J. W. Fossil evidence of Archaean life. Phil. Trans. R. Soc. Lond. B 361,869–885 (2006).
8. Suttle, C. A. Viruses in the sea. Nature 437, 356–361 (2005).
9. Cho, J. C. & Tiedje, J. M. Biogeography and degree of endemicity of fluorescentPseudomonas strains in soil. Appl. Environ. Microbiol. 66, 5448–5456 (2000).
10. Papke, R. T., Ramsing, N. B., Bateson, M. M. & Ward, D. M. Geographical isolationin hot spring cyanobacteria. Environ. Microbiol. 5, 650–659 (2003).
11. Whitaker, R. J., Grogan, D. W. & Taylor, J. W. Geographic barriers isolate endemicpopulations of hyperthermophilic Archaea. Science 301, 976–978 (2003).
12. Whitaker, R. J. Allopatric origins of microbial species. Phil. Trans. R. Soc. Lond. B361, 1975–1984 (2006).
13. Sano, E., Carlson, S., Wegley, L. & Rohwer, F. Movement of viruses betweenbiomes. Appl. Environ. Microbiol. 70, 5842–5846 (2004).
phi alpha3 ( )Escherichia
phi X174 ( )Escherichia
phi SpV4 ( )Spiroplasma
phi MH2K ( )Bdellovibrio
phi Chp1 ( )Chlamydia
phi Chp2 ( )Chlamydia
phi Chp3 ( )Chlamydia
phi Chp4 ( )Chlamydia
phi CPAR39 ( )Chlamydia
Pozas Azules II
Highborne Cay
Sargasso Sea
0.1
1.00
1.00
1.00
1.00
0.92
0.92
1.00
1.00
1.00
Figure 2 | Phylogenetic relationships among viral capsid amino-acidsequences of microphages. The Bayes values represent the proportion ofsampled trees in which those sequences are clustered together.
NATURE LETTERS
3Nature Publishing Group©2008

14. Breitbart, M. & Rohwer, F. Here a virus, there a virus, everywhere the same virus?Trends Microbiol. 13, 278–284 (2005).
15. Breitbart, M. et al. Genomic analysis of uncultured marine viral communities. Proc.Natl Acad. Sci. USA 99, 14250–14255 (2002).
16. Rohwer, F. & Edwards, R. A. The phage proteomic tree: a genome-basedtaxonomy for phage. J. Bacteriol. 184, 4529–4535 (2002).
17. Fane, B. Microviridae, in Virus Taxonomy: Eighth Report of the InternationalCommittee on Taxonomy of Viruses. (eds Fauquet, M. A. M. C., Maniloff, J.,Desselberger, U. & Ball, L. A.) 289–299 (Elsevier Academic Press, San Diego,California, 2005).
18. Souza, V. et al. An endangered oasis of aquatic microbial biodiversity in theChihuahuan desert. Proc. Natl Acad. Sci. USA 103, 6565–6570 (2006).
19. Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning: A Laboratory Manual.(Cold Spring Harbor Laboratory Press, New York, 1989).
20. Margulies, M. et al. Genome sequencing in microfabricated high-density picolitrereactors. Nature 437, 376–380 (2005).
21. Lozupone, C., Hamady, M. & Knight, R. UniFrac - An online tool for comparingmicrobial community diversity in a phylogenetic context. BMC Bioinformatics 7,371 (2006).
22. Jensen, J., Bohonak, A. & Kelley, S. Isolation by distance, web service. BMC Genet.6, 13 (2005).
Supplementary Information is linked to the online version of the paper atwww.nature.com/nature.
Acknowledgements Logistical field support was provided by the crew of the RVWalton Smith, Highborne Cay management and personnel of the Area deProteccion de Flora y Fauna of Cuatro Cienegas. This work was supported by an
NSF grant to F.R. Support for B.K.S. and D.L.V. was provided by the NSF. M.B. wassupported by a grant from the University of South Florida’s Internal New ResearchAwards Program. V.S. was funded by the CONACYT 2002-C01-0237 project. Theauthors thank P. Visscher, K. Przekop, L. Rothschild, D. Rogoff, V. Michotey,P. Bonin, S. Norman and E. Bowlin for providing samples of marine microbial matsand M. Schaechter for a critical reading of the manuscript.
Author Contributions C.D. and F.R. designed the project. C.D. analysed most of thebioinformatic results, conducted the molecular biology and wrote the article. S.K.performed the bayesian analysis. S.R. implemented the cross-contig analyses. M.H.extracted viral DNAs. B.R.-B., H.L., F.E.A. and R.A.E. performed bioinformaticanalyses. R.V.T. and D.H. helped with the interpretation of the bioinformaticresults. V.S., M.B., J.S. and R.P.R. collected the samples. B.K.S., D.L.V., M.F., T.T., L.L.,Y.R., L.W. and B.C. provided metagenomic data. F.R. supervised the project andhelped with the writing. All authors edited and commented on the manuscript.
Author Information The microbialite viral metagenomes have been deposited intothe ftp server of the SEED public database ftp://ftp.theseed.org/metagenomes underthe project accession numbers 4440323.3 (Highborne Cay), 4440320.3 (PozasAzules II) and 4440321.3 (Rio Mesquites). The metagenomes are also publiclyaccessible in the CAMERA metagenomic database (http://camera.calit2.net) underthe project accession numbers HBCStromBahamasVir011105 (Highborne Cay),PAStromCCMexVir072205 (Pozas Azules II), and RMStromCCMexVir072205 (RioMesquites). The Vp1 cloned sequences from the Highborne Cay sample have beendeposited in GenBank under accession numbers EF679227 to EF679234. Reprintsand permissions information is available at www.nature.com/reprints.Correspondence and requests for materials should be addressed to C.D.([email protected]).
LETTERS NATURE
4Nature Publishing Group©2008

METHODSGeographical sampling. Microbialites were collected in November 2005 from
the Cuatro Cienegas Basin in Mexico and the Highborne Cay Island in the
Bahamas (Supplementary Fig. 1). In Mexico, thrombolite samples were collected
from a spring, thermally heated pool (Pozas Azules II, site 1) and a free flowing
river system (Rio Mesquites, site 2). These two spring sources are geographically
isolated by 30 km. Multiple subsamples were combined from the Highborne Cay
stromatolites (Highborne Cay, site 3) and used as one sample. The geologic
characteristics for the sampling sites were previously described in detail18,23.
Virus purification, viral DNA extraction and pyrosequencing. Approximately
5 g of microbialite were shaken in 30 ml of SM buffer (0.1 M NaCl, 1 mM
MgSO4, 0.2 M Tris pH 7.5, 0.01% gelatin) for Pozas Azules II and Rio
Mesquites samples and in 30 ml of 0.02 mm filtered seawater for Highborne
Cay sample for 1 hour. The viral particles were then purified using filtration
(0.22mm) combined with caesium chloride density gradient centrifugation15.
The absence of microbial and eukaryotic cells was verified under epifluorescence
microscopy after SYBR-Gold staining24 (Supplementary Figs 2A and 2B). For
electron microscopy, viral particles were stained with 1.0% uranyl acetate
and examined with a FEI Tecnai 12 transmission electron microscope
(Supplementary Fig. 2C). Viral DNA was isolated by a formamide/CTAB extrac-
tion19 and amplified with GenomiPhi (GE Healthcare) following the manufac-
turer’s recommendations. The resulting DNA was purified on silica columns
(Qiagen) and concentrated by ethanol precipitation. Approximately 10 mg DNA
was sequenced using pyrosequencing technology20 (454 Life Sciences). A total of
81,687,957 bp of DNA was generated from the three libraries (Pozas Azules II: 32
Mbp, Rio Mesquites: 35 Mbp and Highborne Cay: 15 Mbp). The 781,866
sequences had an average length of 104 bp. They have been deposited into the
ftp server of the SEED public database ftp://ftp.theseed.org/metagenomes under
the project accession numbers 4440323.3 (Highborne Cay), 4440320.3 (Pozas
Azules II) and 4440321.3 (Rio Mesquites).
Bioinformatics. The sequences from each metagenome were compared to
the SEED non-redundant (nr) database and environmental database using
BLASTx25 (E-value ,1022). The SEED database contains annotated protein
sequences from different databases such GenBank, Swiss-prot and KEGG. The
environmental database contains, among other things, sequences from acid
mine drainage, biofilm, soil or the Sargasso Sea. The best similarity for each
sequence that matched an annotated protein in the SEED or environmental
databases was automatically assigned as ‘known’ whereas ‘unknown’ describes
sequences that did not have similarity to anything. To define the inter-library
sequence similarities, the entire microbialite metagenomes were compared
(BLASTn, E-value ,1023) against each other and against other viral (Table 1)
and microbial (Table 2) metagenomes from different environments (details are
provided in Supplementary Tables 1 and 2, along with SEED accession num-
bers). All the metagenomes can be downloaded via the ftp server of the SEED
database (ftp://ftp.theseed.org/metagenomes).
Structure of the viral communities. A set of 10,000 random sequences was
extracted from each metagenome and assembled by the TIGR Assembler using
a minimum overlap of 35 bp and 98% of sequence identity. Twenty repetitions
were performed, leading to an average contig spectrum used to define the maxi-
mal likelihood community structure. Different rank-abundance models were
calculated (Supplementary Table 3) using PHACCS (PHAge Communities from
Contig Spectra) an online tool to analyse viral communities26 (http://biome.
sdsu.edu/phaccs/index.htm). As described previously1, rank-abundance models
as well as the cross-contig spectra generated between two metagenomes were
used to define the percentage of genotypes that are shared between two com-
munities (Supplementary Fig. 5). Even though the logarithmic rank-abundance
model was not the best model for Rio Mesquites and Highborne Cay, it gave
coefficients of errors close to those observed with the best models. To harmonize
the analysis and to limit the possible bias during the simulation, the same model
(logarithmic) was chosen for the three metagenomes (Supplementary Table 3).
Phage community taxonomy. The metagenome sequences from each library
were compared to the phage and prophage genome database using tBLASTx
(E-value ,1023). This database contains sequences from 510 complete genomes
of phages and prophages and was used to construct the Phage Proteomic Tree
version 4 (PPT, http://phage.sdsu.edu/,rob/PhageTree/v4). A previous version
of the tree detailing the construction steps was published in 2002 (ref. 16). The
presence and the abundance of sequences that have significant similarities to
those in the database were subsequently mapped onto the PPT (Fig. 1) using Bio-
Metamapper, an online metagenome mapper to the Phage Proteomic Tree
(http://scums.sdsu.edu/Mapper).
Genetic versus geographical distance of the phage community. UniFrac,
an online tool21, was used to measure the genetic differences in community
composition between microbialites and marine environments. The UniFrac
distance is calculated as the percentage of the branch length of the tree (in this
case, the Phage Proteomic Tree) that leads to descendants from either one
environment or the other, but not both. In this study, a weighted UniFrac
distance metric that also takes account of the relative abundance of sequences
in the different environments was used. Distances between the sets of sequences
from each pair of environments (stromatolites and marine environments) were
classified from lower quartile (red) to upper quartile (yellow); that is, a range
from complete similarity to complete differentiation in the phylogenetic
diversity of the samples (Supplementary Fig. 4). The Isolation by Distance
Web Service (IBDWS) was used to test for a correlation between the geographical
distance between two samples and the genetic divergence between viral com-
munities22. This online software uses Mantel tests to determine whether
phages in closer physical proximity have greater genetic similarity (as measured
by UniFrac), than those separated by large geographical distances (Supplemen-
tary Fig. 4).
Genetic divergence of the microphage sequences. The sequences that had
significant tBLASTx similarities (E-value ,1023) to microphages in the
Highborne Cay and the Pozas Azules II metagenomes were extracted into a
sublibrary. These microphage libraries were cross-compared at the nucleic-acid
level against themselves and against the microphages of the Sargasso Sea meta-
genome1 using Circonspect, an online tool to build contig-spectra (http://biome.
sdsu.edu/circonspect/index.php). The sublibraries were then assembled with
Sequencher 4.0 (Gene Codes) using a minimal match percentage of 98% and a
35 bp minimum overlap. When the largest contigs were compared with tBLASTx
against the nr database, most had similarities to the viral capsid protein (Vp1) of
sequenced microphage. Multiple alignments of Vp1 amino-acid sequences from
known microphages and from Pozas Azules II, Highborne Cay and Sargasso Sea
viral reconstructed Vp1 consensus sequences were performed using CLUSTAL
W27. The phylogenetic tree was generated using MrBayes 3.1 program28 (Fig. 2).
The protein evolutionary model (BLOSUM) used for this bayesian analysis was
chosen from among seven different models because it had the highest posterior
probability in an initial test of all models for the data. We ran four independent
Monte Carlo Markov chains for 1 million generations and the chains converged
after only 10,000 generations. To verify the assembly results, PCR primers were
designed on the basis of the Vp1 consensus sequences (Supplementary Table 4)
and PCRs were performed on each sample. The reaction mixture (50ml total)
contained target DNA, 1x Taq Buffer, 0.2 mM dNTPs, 1mM each primer, and 1 U
Taq DNA polymerase. The thermocycler conditions were: 5 min at 94 uC;
30 cycles of 1 min at 94 uC, 1 min at 52 uC, 1 min at 72 uC; and 10 min at
72 uC. Amplification products were checked for size on a 1% agarose gel. No
PCR product was obtained when one sample was tested with the two other
primer sets (for example, PCR of Highborne Cay viral DNA with the Pozas
Azules II or the Sargasso Sea primer sets; data not shown). PCR products from
the Highborne Cay sample were cloned into a TOPO TA vector (Invitrogen) and
transformed into Top 10 competent cells (Invitrogen). PCR was used to screen
positive colonies using primers M13F and M13R provided by the TOPO TA
cloning kit and following manufacturer’s instructions. PCR products from eight
clones were purified using a PCR clean-up kit (Mo Bio) and sequenced using the
M13F and M13R primers (sequences are in the Supplementary Information part
3, accession numbers EF679227 to EF679234). Multiple sequence alignments
of the clones and the known microphage Vp1 sequences were made using
CLUSTAL W27 (Supplementary Fig. 7). The nucleic-acid and protein-based
phylogenetic trees (Supplementary Figs 8A and 8B, respectively) were con-
structed using the neighbour-joining method29 and were plotted using the njplot
program30. Plasmid purifications were completed using PureLink Quick Plasmid
Miniprep Kit (Invitrogen).
Highborne Cay microphages in other environmental samples. The clone D4
was used to test the limit of the Vp1 gene concentration for PCR detection. Serial
dilutions were made to produce final concentrations ranging from 1 to 109
plasmid copies per microlitre (Supplementary Fig. 6). One microlitre of each
dilution was then amplified with the Vp1HC-F and Vp1HC-R set of primers
using touchdown PCR and a gradient of primer hybridization temperature
ranging from 47 uC to 57 uC. The thermocycler conditions giving optimal PCR
amplification (detection limit between 10 and 100 plasmid copies) were: 5 min
at 94 uC, 20 cycles of (1 min at 94 uC, 1 min at 65–0.5 uC per cycle, and 1 min at
72 uC) followed by 15 cycles of (1 min at 94 uC, 1 min at 55 uC, and 1 min at
72 uC); and 10 min at 72 uC. These PCR conditions were then used to test the
presence or absence of the Highborne Cay Vp1 gene in 63 different environ-
mental samples (Supplementary Table 5) including extreme, metazoan-
associated, freshwater, marine, sediment, terrestrial, other marine mats and
new viral DNA from the Highborne Cay stromatolites.
23. Reid, R. P., Macintyre, I. G. & Steneck, R. S. A microbialite/algal ridge fringing reefcomplex, Highborne Cay, Bahamas. Atoll Res. Bull. 465, 1–18 (1999).
doi:10.1038/nature06735
Nature Publishing Group©2008

24. Chen, F., Lu, J., Binder, B. J., Liu, Y. & Hodson, R. E. Application of digital imageanalysis and flow cytometry to enumerate marine viruses stained with SYBR Gold.Appl. Environ. Microbiol. 67, 539–545 (2001).
25. Altschul, S. F., Gish, W., Miller, W., Meyers, E. W. & Lipman, D. J. Basic LocalAlignment Search Tool. J. Mol. Biol. 215, 403–410 (1990).
26. Angly, F. et al. PHACCS, an online tool for estimating the structure and diversity ofuncultured viral communities using metagenomic information. BMCBioinformatics 6, 41 (2005).
27. Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving thesensitivity of progressive multiple sequence alignment through sequenceweighting, position-specific gap penalties and weight matrix choice. Nucleic AcidsRes. 22, 4673–4680 (1994).
28. Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetictrees. Bioinformatics 17, 754–755 (2001).
29. Saito, N. & Nei, M. The neighbour-joining method, a new method forreconstructing phylogenetic trees. Mol. Biol. Evol. 79, 426–434 (1987).
30. Perriere, G. & Gouy, M. WWW-Query: An on-line retrieval system for biologicalsequence banks. Biochimie 78, 364–369 (1996).
doi:10.1038/nature06735
Nature Publishing Group©2008

Contents: Part 1. Supplementary Figures (1 to 8), Tables (1 to 5), and legends. Part 2. Partial major viral capsid sequences assembled from Highborne Cay, Pozas Azules II and Sargasso Sea metagenomes. Part 3. Partial major viral capsid sequences from cloning experiment. Part 1. Supplementary Figures (1 to 8), Tables (1 to 5), and legends
- Description of the sampling sites: The Cuatro Ciénegas Basin, Mexico (Supplementary Figure 1, sites 1 and 2) and the Exuma Cays in the Bahamas (Figure 1, site 3) represent unique ecosystems. Both places are well-known hot spots of terrestrial and aquatic endemic biodiversity of higher organisms31 including unique species of plants, birds, snails, fishes, reptiles, turtles, and scorpions32. During the Pleistocene, these environments were geographically isolated oases and vicariance may explain this high level of endemism.
Supplementary Figure 1. Sampling sites (map from http://www.reefbase.org) and microbialites photos. Sites 1 (Rio Mesquites) and 2 (Pozas Azules II) are located in the Chihuahuan desert of Mexico and site 3 (Highborne Cay) is located in the Exuma Cays (Bahamas). Insets in picture 1, 2 and 3 show stromatolites collected in the Rio Mesquites River, thrombolites in Pozas Azules II and a stromatolite cross section from Highborne Cay, respectively.
SUPPLEMENTARY INFORMATION
doi: 10.1038/nature06735
www.nature.com/nature 1
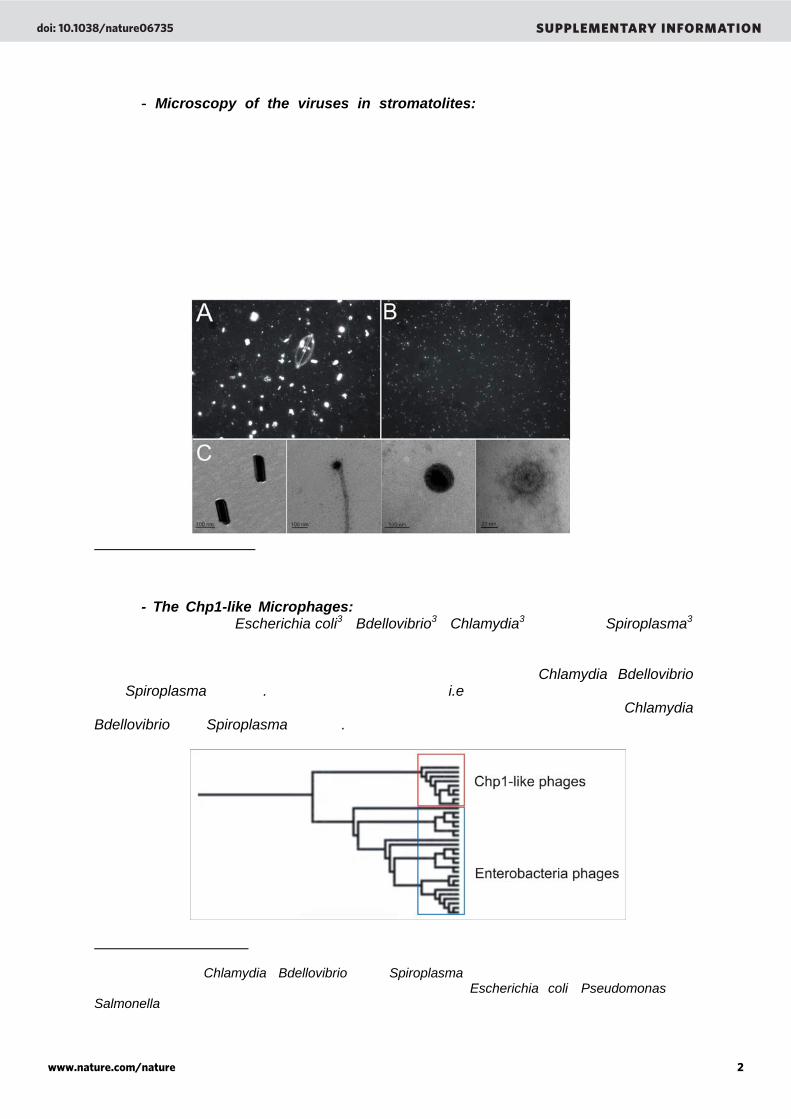
- Microscopy of the viruses in stromatolites: Viral particles were purified by cesium chloride (CsCl) gradient centrifugation (Supplementary figure A and B). Approximately 8 ml of viral concentrate, with CsCl added to create a density of 1.15 g ml-1, was layered onto a step gradient of CsCl solutions at 1.7 g ml-1, 1.5 g ml-1, and 1.25 g ml-1. CsCl solutions were made up with the same solutions than those used to resuspend the viruses from the microbialites (seawater for the Highborne Cay sample and SM buffer for Pozas Azules II and Rio Mesquites samples, see online Methods). The gradients were centrifuged at 22,000 rpm in an SW41 swinging bucket rotor at 4° C for 2 hours and the 1.5 ml corresponding to the 1.5 g ml-1 gradient step plus the interfaces above and below (fraction containing viruses), were withdrawn from the tubes. Purified virus-like particle were then visualized under epifluorescent microscopy and electron microscopy.
Supplementary Figure 2. Viral-like particles stained with SYBR-Gold and visualized under epifluorescence microscopy before (A) and after CsCl purification (B). Electron micrographs (C) of virus-like particles from Highborne Cay stromatolites (bars represent 100 nm and/or 20 nm).
- The Chp1-like Microphages: Microphages are icosahedral single-stranded DNA phages isolated from Escherichia coli33, Bdellovibrio34, Chlamydia35,36,37,38, and Spiroplasma39
species. Based on their genomic sequences, these phages form two distinct clusters in the Phage Proteomic Tree (Supplementary Figure 3). The first cluster grouped Microphages infecting Enterobacteria whereas the second contained phages of Chlamydia, Bdellovibrio, and Spiroplasma species. The same nomenclature (i.e., Chp1-like Microphages) that has been previously proposed40 will be used to characterize phages infecting Chlamydia, Bdellovibrio, and Spiroplasma species. Supplementary Figure 3. Inset of the Phage Proteomic Tree showing the genetic relationships between the Microphages and the division of this family. The Chp1-like phages cluster contains phages infecting Chlamydia, Bdellovibrio, and Spiroplasma species. The Enterobacteria phages cluster contains phages infecting Enterobacteria such as Escherichia coli, Pseudomonas and Salmonella species.
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 2
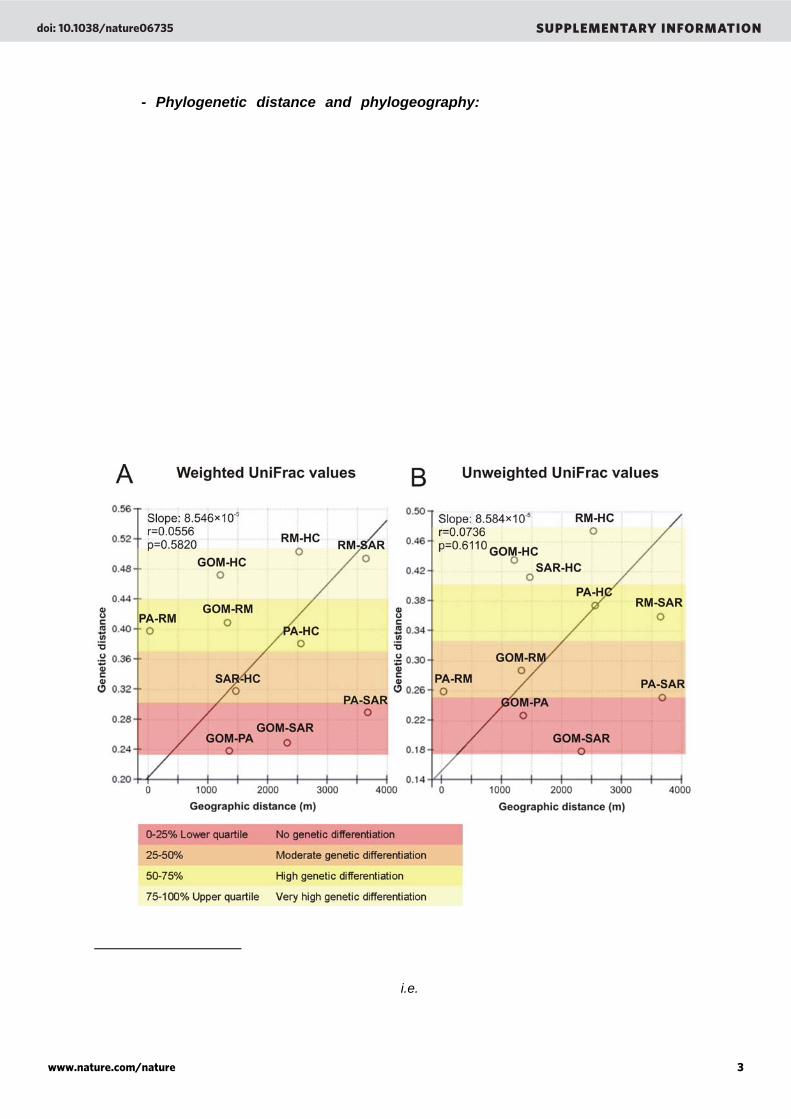
- Phylogenetic distance and phylogeography: To test whether the genetic divergence of viral communities in microbialites was a consequence of a spatial distance, we used the isolation by distance test41. This test assumes that levels of possible migrations between regions are linked to their spatial distance41; two very distant sites will support fewer migration events resulting in greater genetic divergence. Using this test, the geographic distance between microbialites was correlated to the genetic distance among their phage communities. Genetic distances were calculated by comparing the presence/absence and abundance of reads to each phage in the Phage Proteomic Tree using Unifrac (an online tool to compare microbial communities using phylogenetic information)42. Two marine samples (Sargasso Sea and Gulf of Mexico) were added to this test to verify the “marine-ness” of the Pozas Azules II sample. The geographic distance between samples did not explain the genetic divergence of the phage community (Supplementary Figure 4A, p = 0.5820). Nearby sites such Rio Mesquites and Pozas Azules II presented high phage community divergence that can be explained by a difference in extrinsic factors such as water chemistry and the hydrologic conditions (lotic and lentic ecosystems). In addition, abundance of the reads to one particular phage did not influence the results (Supplementary Figure 4B) since similar regression slopes were given by weighted and unweighted Unifrac values (8.546 × 10-5 and 8.584 × 10-5, respectively). Moreover, the marine quality of the Pozas Azules II phage community was confirmed by a close genetic overlap with both the Gulf of Mexico and the Sargasso Sea phage communities. Supplementary Figure 4. Mantel Test for matrix correlation between genetic distance (Unifrac values) and geographic distance (m) among stromatolites and marine samples. Weighted UniFrac values (left) or Unweighted UniFrac values (right) were used and the quartiles of UniFrac values were classified from lower quartile (red) to upper quartile (yellow) i.e., a range from complete similarity to complete differentiation in the genetic diversity of the samples.
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 3
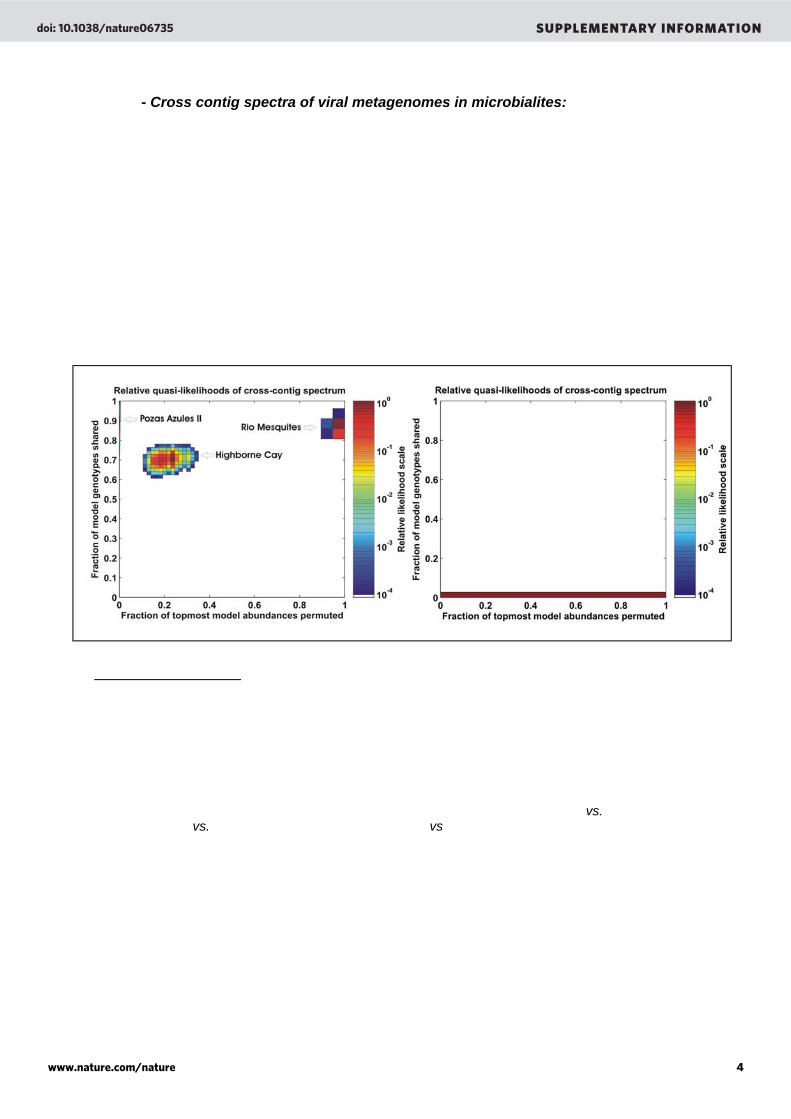
- Cross contig spectra of viral metagenomes in microbialites: A random subset of sequences (10,000) was extracted from each viral metagenome and assembled separately. The assembly process formed groups of overlapping sequences (contigs) based on their identities (98% identity over 35 bp). This process was repeated 20 times to produce an average contig spectrum. Each spectrum was then cross-compared among samples and the resulting cross-contig spectrum represented the sequence overlaps between a set of two samples at the nucleic acid level. A cross-contig spectrum ([0.3 0.1 0.1]) was obtained when Pozas Azules II and Rio Mesquites samples were compared. The cross-comparisons of Highborne Cay/Pozas Azules II and Highborne Cay/Rio Mesquites did not produce contig spectra (i.e., no overlaps were observed). A Monte Carlo simulation was generated to estimate the number of genomes shared between the samples. Results showed that the percent of genomes shared between Pozas Azules II, Highborne Cay and Rio Mesquites tended to zero (supplementary Figure 5B) proving that viruses are genetically unique in each microbialites.
Supplementary Figure 5. Cross-contig spectra of viral metagenomes in stromatolites. (A) controls, the metagenomic libraries where compared against themselves. The expected optimal models for the controls would have 100% shared genotypes and 0% permuted abundances. However, the optimal models obtained for our samples did not fit this expectation. In particular, in the best Rio Mesquites phage community model, 100% of the abundances were permuted. This is an artefact resulting from the limited flexibility of the population structure models in PHACCS and the low diversity of the Rio Mesquites sample. Models with 100% of abundances randomly permuted have large uncertainties in the predicted cross-contig spectrum. These uncertainties are needed to cover for the discrepancy between the observed cross-contig spectrum and the nearest cross-contig spectrum expected from a logarithmic rank-abundance model. (B) cross-contig spectrum of Pozas Azules II vs. Rio Mesquites, Pozas Azules II vs. Highborne Cay, and Rio Mesquites vs. Highborne Cay. The probability of having shared species between samples tended to zero.
A B
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 4
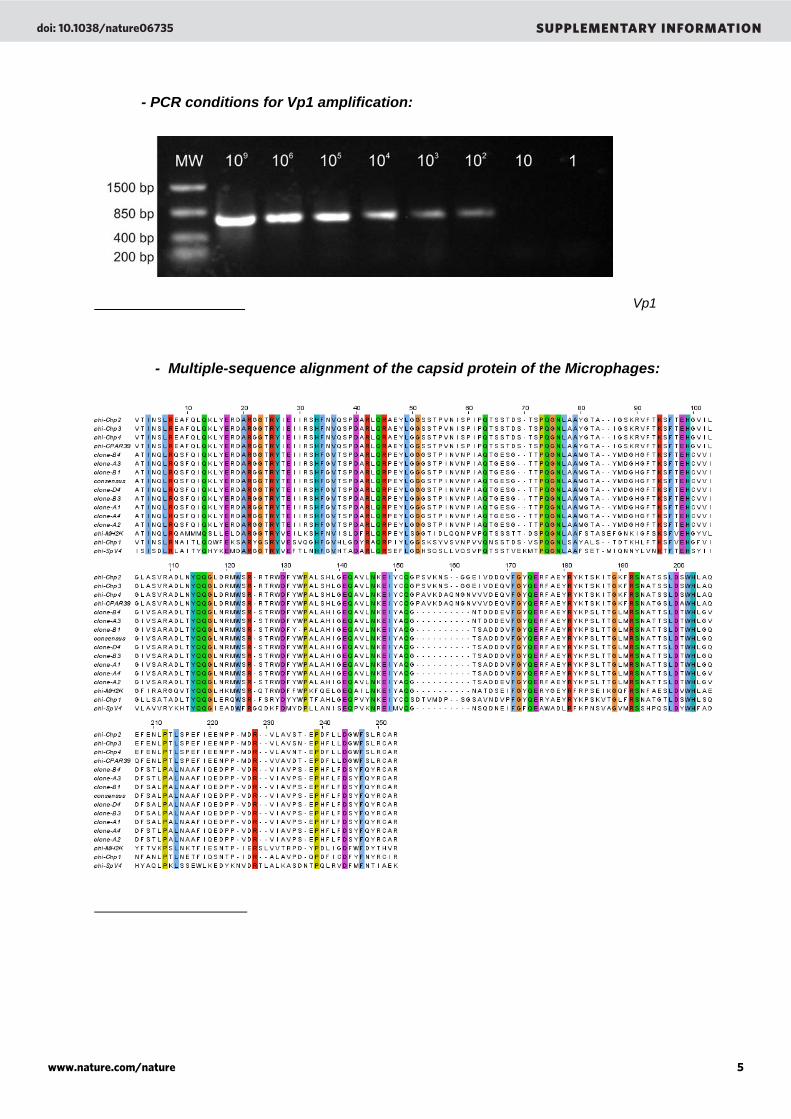
- PCR conditions for Vp1 amplification:
Supplementary Figure 6. Sensitivity of PCR to amplify the Highborne Cay Microphage Vp1 locus. Amplifications were carried out from 1 to 109 copies of a plasmid.
- Multiple-sequence alignment of the capsid protein of the Microphages:
Supplementary Figure 7. Multiple-sequence alignment of the capsid protein of the Microphages. Partial sequences of capsid protein of Chp1 (NP_044312), Chp2 (NP_054647), Chp3 (YP_022479) Chp4 (YP_338238), MH2K (NP_073538), SpV4 (NP_598320), CPAR39 (NP_063895), clones A1 to D4 (EF679227 to EF679234, this work) and the consensus sequence reconstructed from the Highborne Cay metagenome were aligned using CLUSTAL W43. Alignment file was visualized using Jalview44. Residues that are identical are boxed and gaps are indicated by dots.
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 5

- Phylogeny of the Vp1 cloned genes:
Supplementary Figure 8. Phylogenetic trees showing the relationships among the Vp1 sequences of the Highborne Cay sample (clones A1 to D4) and the known Chp1-like Microphages at the nucleic (A) and protein (B) levels. Trees were constructed with the Neighbour Joining method45 and were plotted using the njplot program46. The clone to clone divergence ranged from 0.1% to 7.0% of at the nucleic level and from 0% to 4.6% at the protein level. Clone nucleic sequences and their corresponding accession numbers are provided in the Supplementary Information part 3.
A B
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 6
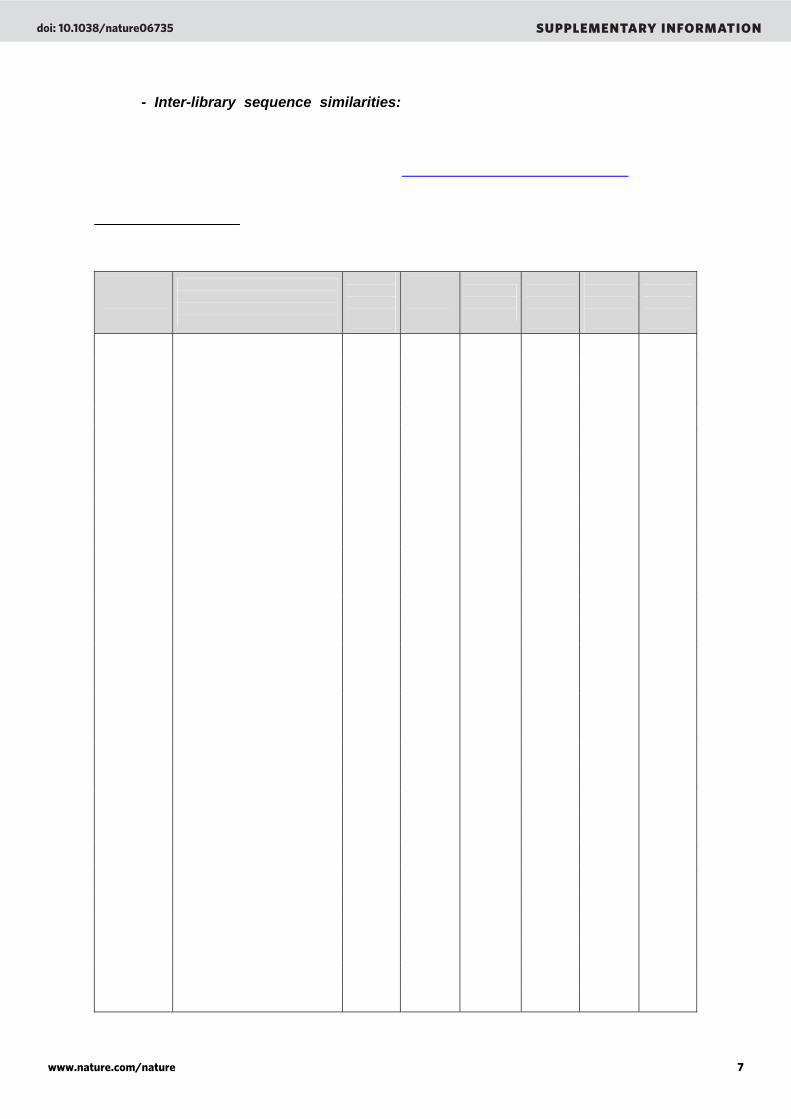
- Inter-library sequence similarities: the entire microbialite metagenomes were compared (BLASTn, E-value < 10-3) against each other and against other viral (Supplementary table 1, see also Table 1) and microbial (Supplementary Table 2, see also Table 2) metagenomes from different environments. The metagenomes can be downloaded via the ftp server of the SEED database (ftp://ftp.theseed.org/metagenomes/) and are publicly available at GenBank.
Supplementary Table 1. Percentage of similarity obtained after comparison of the microbialite viral metagenomes (BLASTn, E-value < 10-3) to each others and to other viral metagenomes. The similarity is always under 5% except when metagenomes are compared against themselves.
Environment
Viral Metagenome Name (All these metagenomes were done
using pyrosequencing technology)
GenBank Accession numbers
(locus tag)
SEED ID Total
Number of sequences
Percent of similarity (E < 10-3) Highborne
Cay
Percent of similarity (E < 10-3)
Pozas Azules II
Percent of similarity (E < 10-3)
Rio Mesquites
1. Freshwater TpondKentSTVir1105 28361 (AGY) 4440439.3 267640 1.584 0.555 1.643
2. Freshwater TilPondKentSTVir050406 28409 (AKA) 4440412.3 60319 1.418 0.430 0.577
3. Freshwater PrePondKentSTVir050406 28411 (AKC) 4440414.3 67988 1.119 0.499 1.045
4. Freshwater TilPondKentSTVir0806 - 4440424.3 57134 0.494 0.425 0.401 5. Coral reef waters KingLIVir082105 28345
(AGI) 4440036.3 94915 0.982 0.787 0.722
6. Coral reef waters XmasLIVir080505 28349
(AGM) 4440038.3 283390 1.118 0.924 0.830
7. Coral reef waters PalmLIVir081805 28365
(AI3) 4440040.3 320397 2.253 0.793 0.918
8. Coral reef waters FannLIVir081105 28369
(AI7) 4440280.3 380355 1.496 0.856 0.763
9. Marine waters SARVir063005 17771 4440322.3 399343 2.905 0.838 0.712
10. Marine waters GOMVir94to01 17765 4440304.3 263908 1.458 0.492 0.424
11. Marine waters BBCVir96to04 17767 4440305.3 416456 2.410 0.699 0.707
12. Marine waters ArcticVir2002 17769 4440306.3 688590 0.308 0.308 0.330
13. Fish associated FishHealGutKentSTVir050406 28397
(AIY) 4440062.3 47139 0.806 0.310 0.307
14. Fish associated FishMorGutKentSTVir050406 28399
(AK1) 4440063.3 53750 0.629 0.291 0.300
15. Fish associated FishHealSlimKentSTVir050406 28401
(AK3) 4440065.3 61476 1.054 0.274 0.380
16. Fish associated FishMorSlimKentSTVir050406 28403
(AK5) 4440064.3 60111 0.314 0.241 0.560
17. Mosquito associated MosqISDVir01252006 28413
(AKE) 4440052.3 340098 0.731 0.273 0.683
18. Coral associated T0PortComHawVir022306 28415
(AKG) 4440376.3 39270 0.871 0.374 0.216
19. Coral associated ConPorCompHawVir0206 28417
(AKI) 4440374.3 39340 1.163 0.285 0.340
20. Coral associated TempPorCompHawVir0206 28419
(AKK) 4440375.3 39036 0.295 0.193 0.191
21. Coral associated DOCPorCompVirHaw0206 28421
(AKM) 4440370.3 35680 0.599 0.341 0.288
22. Coral associated pHPorCompHawVir0206 28423
(AKO) 4440371.3 50364 0.381 0.245 0.222
23. Coral associated NutPorCompHawVir0206 28425
(AKQ) 4440377.3 34433 1.099 0.299 0.203
24. Human associated HealSputSDRep1Vir070706 28439
(AM7) 4440025.3 770739 1.217 0.357 0.394
25. Human associated CFLungSDPat001Rep1Vir050506 28441
(AM9) 4440026.3 776754 0.546 0.396 0.356
26. Microbialite HBCStromBahamasVir011105 28381
(AII) 4440323.3 150223 100 1.140 0.910
27. Microbialite PAStromCCMexVir072205 28355
(AGS) 4440320.3 302987 4.020 100 1.100
28. Microbialite RMStromCCMexVir072205 28357
(AGU) 4440321.3 328656 0.970 0.700 100
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 7
29. Saltern waters LowSalternSDbayVir111005 28373
(AIA) 4440432.3 110511 1.602 0.818 0.742
30. Saltern waters MedSalternSDbayVir111005 28375
(AIC) 4440431.3 39578 0.464 0.381 0.372
31. Saltern waters LowSalternSDbayPla112205 28443
(AMA) 4440090.3 111431 1.657 0.757 0.691
32. Saltern waters MedSalternSDbayVir112205 28445
(AMC) 4440417.3 55903 0.419 0.345 0.351
33. Saltern waters HighSalternSDbayVir120705 28447
(AME) 4440145.4 47587 0.312 0.209 0.206
34. Saltern waters HighSalternSDbayVir112805 28451
(AMI) 4440144.4 4645 0.433 0.285 0.382
35. Saltern waters LowSalternSDbayVir112805 28455
(AMM) 4440420.3 62685 0.545 0.424 0.428
36. Saltern waters MedSalternSDbayVir112805 28463
(AMU) 4440427.3 39943 0.640 0.320 0.246
37. Saltern waters MedSalternSDbayVir111605 28465
(AMW) 4440428.3 58735 0.485 0.362 0.414
38. Saltern waters LowSalternSDbayVir0704 28353 4440436.3 268534 0.725 0.588 0.747
39. Saltern waters HighSalternSDbayVir111605 28457
(AMO) 4440421.3 154167 0.312 0.344 0.311
40. Marine sediments/mat SaltonSeaVirOne082308 - 4440327.3 55787 0.518 0.458 0.290
41. Marine sediments/mat SaltonSeaVirTwo082308 - 4440328.3 29970 0.654 0.650 0.337
42. Marine sediments/mat SkanBayAKVir092706 - 4440330.3 31375 0.791 0.595 0.576
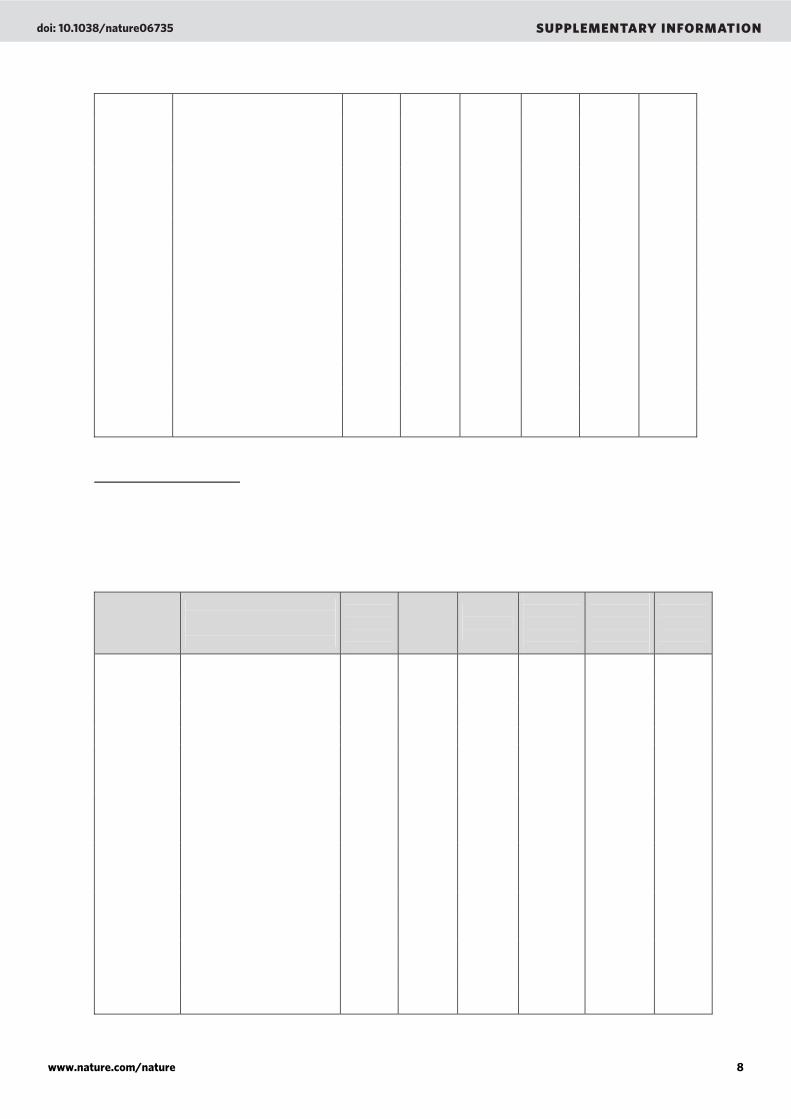
Supplementary Table 2. Percentage of similarity obtained after comparison of the microbialite viral metagenomes (BLASTn, E-value < 10-3) to each others and to other microbial metagenomes. The similarity is always under 5% except for the comparison between the Highborne Cay viral and the Highborne Cay Microbial metagenomes. About 47% of the viral sequences have homology to the sequences on the microbial fraction (lane 20, raw 5). However, these 47.104% of the sequences have homology to only 3.180% of the sequences of the microbial fraction showing that those are prophages that have been expressed in the environment.
Environment
Microbial Metagenome Name (All these metagenomes were done using pyrosequencing
technology)
GenBank Accession numbers
(locus tag)
SEED ID Total
Number of sequences
Percent of similarity (E < 10-3) Highborne
Cay
Percent of similarity (E < 10-3)
Pozas Azules II
Percent of similarity (E < 10-3)
Rio Mesquites
1. Freshwater TilPondKentSTMic1105 28387 (AIO) 4440440.3 381076 3.658 0.711 0.830
2. Freshwater TilPondKentSTMic050406 28405 (AK7) 4440413.3 63978 1.469 0.415 0.475
3. Freshwater PrePondKentSTMic050406 28407 (AK9) 4440411.3 44094 0.997 0.356 0.442
4. Freshwater TilPondKentSTMic0806 - 4440422.3 67612 1.287 0.380 0.487 5. Coral reef waters KingLIMic082105 28343
(AGG) 4440037.3 188445 0.485 0.308 0.282
6. Coral reef waters XmasLIMic080505 28347
(AGK) 4440041.3 227542 1.359 0.450 0.300
7. Coral reef waters PalmLIMic081805 28363
(AI1) 4440039.3 289723 1.334 0.387 0.309
8. Coral reef waters FannLIMic081105 28367
(AI5) 4440279.3 290844 0.435 0.217 0.212
9. Fish associated FishHealGutKentSTMic050406 28389
(AIQ) 4440055.3 51498 0.260 0.240 0.361
10. Fish associated FishMorGutKentSTMic050406 28391
(AIS) 4440056.3 60311 0.266 0.238 0.224
11. Fish associated FishHealSlimKentSTMic050406 28393
(AIU) 4440059.3 66066 0.310 0.267 0.400
12. Fish associated FishMorSlimKentSTMic050406 28395
(AIW) 4440066.3 82442 0.318 0.261 0.341
13. Coral associated BocasPAMic092105 28371
(AI9) 4440319.3 316279 1.314 0.322 0.312
14. Coral associated T0PortComHawMic022306 28427
(AKS) 4440380.3 53473 0.321 0.196 0.210
15. Coral associated ConPorCompHawMic0206 28429
(AKU) 4440378.3 65191 0.328 0.219 0.194
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 8

16. Coral associated TempPorCompHawMic0206 28431
(AKW) 4440373.3 61356 0.379 0.290 0.263
17. Coral associated DOCPorCompHawMic0206 28433
(AKY) 4440372.3 62959 1.056 0.232 0.173
18. Coral associated pHPorCompHawMic0206 28435
(AM3) 4440379.3 67994 1.207 0.274 0.408
19. Coral associated NutPorCompHawMic0206 28437
(AM5) 4440381.3 65008 1.028 0.249 0.206
20. Microbialite HBCStromBahamasMic011105 28383 (AIK) 4440061.3 257573 47.104 0.400 0.230
21. Microbialite PAStromBahamasMic072205 28385 (AIM) 4440067.3 326146 4.310 3.742 0.410
22. Microbialite RMStromCCMexMic072205 28351 (AGO) 4440060.3 124694 1.021 0.637 0.541
23. Saltern waters LowSalternSDbayMic0704 28359
(AGW) 4440437.3 268206 1.272 0.416 0.438
24. Saltern waters Bacteria pond 5 attempt 1 - 4440430.3 78524 0.347 0.350 0.233
25. Saltern waters Bacteria pond 5 attempt 2 - 4440429.3 39553 1.206 0.340 0.273
26. Saltern waters Bacteria pond 5 attempt 3 - 4440433.3 123879 0.317 0.425 0.372
27.Saltern waters MedSalternSDbayMic111005 28377
(AIE) 4440435.3 38929 0.438 0.383 0.419
28. Saltern waters MedSalternSDbayMic111105 28379
(AIG) 4440434.3 23261 0.374 0.383 0.385
29. Saltern waters MedSalterSDbayMic112805 28449
(AMG) 4440416.3 8062 0.407 0.392 0.521
30. Saltern waters MedSalternSDbayMic111605 28459
(AMQ) 4440425.3 120987 1.074 0.430 0.365
31. Saltern waters LowSalternSDbayMic112805 28461
(AMS) 4440426.3 34296 1.087 0.740 0.685
32. Saltern waters Bacteria pond 5 attempt 4 - 4440438.3 340725 0.302 0.332 0.309
33. Saltern waters HighSalternSDbayMic112805 28453
(AMK) 4440419.3 35446 0.379 0.424 0.378
34. Terrestrial, subterranean RedSoudMineMic033105 17633 4440281.3 334386 0.582 0.486 0.348
35. Terrestrial, subterranean BlackSoudMineMic033105 17635 4440282.3 388627 1.337 0.397 0.592
36. Marine sediments/mat SaltonSeaMic082308 - 4440329.3 178407 1.168 0.432 0.321
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 9

- Viral diversity in microbialites: The viral diversity in each microbialite was predicted using the online tool PHACCS47 (http://biome.sdsu.edu/phaccs/). The diversity was measured by the Shannon-Wiener index (H’nats) which takes into account the number of species and the distribution of individuals within each species48. Based on these two variables, the diversity estimate will increase either by having more species (higher richness) or by having a greater evenness of these species48. In microbialites, viral diversity ranged from 2.9 for Rio Mesquites to 3.8 for Highborne Cay and 8.9 for Pozas Azules II (Supplementary Table 3). The diversity of the viral community in the Pozas Azules II was extremely high and similar to values previously observed in marine waters40. The viral community in the Pozas Azules II microbialite also harboured the highest predicted richness (19,520 genotypes) and evenness (0.90). The high richness in the Pozas Azules II sample could be explained by the internal mesostructure of the thrombolite (i.e., a clotted fabric) which may offer different microniches. Supplementary Table 3. Summary of PHACCS rank-abundance model predictions Sample Model Error Richness (number
of genotypes) % of the most abundant genotype Evenness Shannon-Wiener
Index (H’nats) Best model Power 616 161 16.5 0.80 4.1 Highborne
Cay Model used Logarithmic 967 72 16.5 0.89 3.8
Best model Logarithmic 768 19520 8.6 0.90 8.9
Poza Azul Model used Logarithmic 768 19520 8.6 0.90 8.9
Best model Lognormal 1816 33 19.6 0.85 3.0 Rio
Mesquites Model used Logarithmic 2792 23 19.4 0.92 2.9
- Primer sequences: Primer sets were designed on conserved regions of the Vp1
consensus sequences reconstructed from each metagenome (Highborne Cay, Pozas Azules II and Sargasso Sea). Supplementary Table 4. Primers used to amplify the capsid gene (Vp1) in Highborne Cay, Pozas Azules II and Sargasso Sea metagenomes. Primers were designed based on the Vp1 consensus sequences reconstructed from each metagenome (sequences are provided in the Supplementary Information part 2). Sample Name Primer Name Sense Sequence (5’ 3’) Fragment size
Vp1HC-F Forward GCAACAATCAATCAGCTTCG Highborne Cay (HC1) Vp1HC-R Reverse GTCGAGCACAGCGATATTGA
703 bp
Vp1PA-F1 Forward GACCATCAGAAGTGATCCAT Pozas Azules II Vp1PA-R1 Reverse CCGGATTTCAGGCTACAG
666 bp
Vp1SAR-F Forward TACACGGGCATAGGTCTGGT Sargasso Sea Vp1SAR-R Reverse AAATGCAACAGCTGCAACAA
729 bp
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 10

- Location and type of environmental sample tested for the presence of the Vp1 gene: List of environmental viral communities that were analyzed by PCR to detect the Microphage Vp1 genes. Detailed protocol for viral isolation and DNA extraction was given elsewhere49. Supplementary Table 5. List of environmental samples tested for the presence of the Highborne Cay Microphages. Sampling date as and PCR results using the Highborne Cay Vp1 Microphage primers are indicated.
Environment Sampling
date PCR
result Environment Sampling date
PCR result
Extreme Marine
1. Salt Lake Marina, Utah 05/02 - 29. Antarctic 1, 500 m 10/01 -
2. Little hot Creek Hot Springs, California 07/02 - 30. Antarctic 8, 1 m 10/01 -
31. Bermuda Atlantic Time Series, 100 m 09/99 -
Metazoan-associated 32. Bermuda Atlantic Time Series, 3 m 09/99 -
3. Mohtastraea franski, Bermuda 06/00 - 33. Makapuu, Hawaii 02/02 -
4. Porites asteroides, Hawaii 06/05 - 34. Melbourne Beach, Florida 04/02 -
5. Porites compressa, Hawaii 11/05 - 35. Mission Bay, California 06/01 -
6. Cow rumen, Idaho 06/02 - 36. Puerto Rico nearshore surface sample 2 02/02 -
7. Fish gut aquaculture pond, California 04/06 - 37. Puerto Rico, 30 ft deep 02/02 -
8. Fish slime aquaculture pond, California 04/06 - 38. Scripps Pier, California 04/02 -
9. Human feces 07/02 - 39. Weimea Bay, Hawaii 02/02 -
Freshwaters Microbial mats
10. African stream 07/02 - 40. Berre lagoon intern mat white layer (BIB), France 06/00 -
11. Antarctic Ice 10/01 - 41. Berre lagoon intern mat black layer (BIN), France 06/00 -
12. Aquaculture pond, California 04/06 - 42. Berre lagoon mat extern, France 06/00 -
13. Colorado River, Colorado 07/02 - 43. Eilat microbial mat 0-1 mm, Israel 05/01 -
14. Hemet Well # 27, California 07/02 - 44. Eilat microbial mat 1-5 mm, Israel 05/01 -
15. Hemet Well # 29, California 07/02 - 45. Camargue microbial mat, 0-5 mm, France 06/01 -
16. Hemet Well # 34, California 07/02 - 46. Salt Pan crusty mat, hypersaline pond, Bahamas 07/07 -
17. Idaho Deep Well 06/02 - 47. Big Pond Flat mat, hypersaline pond, Bahamas 07/07 -
18. Idaho Shallow Well 06/02 - 48. Big Pond snake mat, hypersaline pond, Bahamas 07/07 -
19. Lake Havasu, Arizona 05/02 - 49. Puerto Rico microbial mat, Puerto Rico 07/07 -
20. Rio Grande, Arizona 07/02 - 50. Barn Island mat, salt marsh, Connecticut, USA 07/07 -
21. Saddle Creek, California 07/01 -
22. Utah Lake, Utah 05/02 - Microbialites
51. Green lake stromatolite, New-york USA 06/07 -
52. Rio Mesquites, Mexico 11/05 -
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 11

Terrestrial 53. Pozas Azules II, Mexico 11/05 -
23. Coastal Sage Scrub, California 05/02 - 54. Highborne Cay 1 (multiple samples as one), Bahamas 11/05 +
24. Cultivated Land, Idaho 06/02 - 55. Site 4, pudding, Highborne Cay, Bahamas 07/07 -
25. Desert Sand, New Mexico 07/02 - 56. Site 8 type 2, Highborne Cay, Bahamas 07/07 +
26. Rhizosphere, Idaho 06/02 - 57. Site 8 thrombolite, Highborne Cay, Bahamas 07/07 -
27. Sky Oaks Chapparal, California 02/02 - 58. Site 8 thrombolite pink, Highborne Cay, Bahamas 07/07 -
59. Site 10 type 1, Highborne Cay, Bahamas 07/07 +
Sediment 60. Site 10 type 3, Highborne Cay, Bahamas 07/07 +
28. La Parguera Mangroves, Puerto Rico 04/01 - 61. Site 12 type 1, Highborne Cay,
Bahamas 07/07 -
62. Site 12 yellow fur, Highborne Cay, Bahamas 07/07 -
63. Site 12 Pustular blankets, Highborne Cay, Bahamas 07/07 -
References 31. Myers, N., Mittermeier, R.A., Mittermeier, C.G., da Fonseca, G.A.B. & Kent, J.
Biodiversity hotspots for conservation priorities. Nature 403, 853-858 (2000). 32. Fritsch, P.W. & McDowell, T.D. Biogeography and phylogeny of caribbean plants-
introduction. Syst. Bot. 28, 376-377 (2003). 33. Hayashi, M., Aoyama, A., Richardson, D.L. & Hayashi, M.N. Biology of the
bacteriophage phi X174, in The bacteriophages, Vol. 2. (ed. R. Calendar) (Plenum Press, New York, N.Y; 1988).
34. Brentlinger, K.L. et al. Microviridae, a family divided: isolation, characterization, and genome sequence of {phi}MH2K, a bacteriophage of the obligate intracellular parasitic bacterium Bdellovibrio bacteriovorus. J. Bacteriol. 184, 1089-1094 (2002).
35. Storey, C.C., Lusher, M. & Richmond, S.J. Analysis of the complete nucleotide sequence of Chp1, a phage which infects avian Chlamydia psittaci J. Gen. Virol. 70, 3381-3390 (1989).
36. Hsia, R.C., Ting, L.M. & Bavoil, P.M. Microvirus of Chlamydia psittaci strain guinea pig inclusion conjunctivitis: isolation and molecular characterization. Microbiology 146, 1651-1660 (2000).
37. Everson, J.S. et al. Biological properties and cell tropism of Chp2, a bacteriophage of the obligate intracellular bacterium Chlamydophila abortus. J. Bacteriol. 184, 2748-2754 (2002).
38. Garner, S.A., Everson, J.S., Lambden, P.R., Fane, B.A. & Clarke, I.N. Isolation, molecular characterisation and genome sequence of a bacteriophage (Chp3) from Chlamydophila pecorum. Virus Genes 28, 207-214 (2004).
39. Chipman, P.R., Agbandje-McKenna, M., Renaudin, J., Baker, T.S. & McKenna, R. Structural analysis of the Spiroplasma virus, SpV4: implications for evolutionary variation to obtain host diversity among the Microviridae. Structure 6, 135-145 (1998).
40. Angly, F.E. et al. The marine viromes of four oceanic regions. PLoS Biol. 4, e368 (2006).
41. Jensen, J., Bohonak, A. & Kelley, S. Isolation by distance, web service. BMC Genetics 6, 13 (2005).
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 12

42. Lozupone, C., Hamady, M. & Knight, R. UniFrac - An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7, 371 (2006).
43. Thompson, J.D., Higgins, D.G. & Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673-4680 (1994).
44. Clamp, M., Cuff, J., Searle, S.M. & Barton, G.J. The Jalview Java alignment editor. Bioinformatics 20 (2004).
45. Saito, N. & Nei, M. The neighbour-joining method, a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 79, 426-434 (1987).
46. Perriere, G. & Gouy, M. WWW-Query: An on-line retrieval system for biological sequence banks. Biochimie 78, 364-369 (1996).
47. Angly, F. et al. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics 6, 41 (2005).
48. Shannon, C.E. & Weaver, W. The mathematical theory of communication. (University of Illinois Press, Urbana, Illinois; 1949).
49. Breitbart, M., Miyake, J.H. & Rohwer, F. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol. Lett. 236, 249-256 (2004).
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 13

Part 2. Partial major viral capsid sequences assembled from Highborne Cay, Pozas Azules II and Sargasso Sea metagenomes and used to design the primers (Supplementary Table 2) >Pozas Azules II GATCCGAAGCTGCATGCGGATCTGACTGGCGCGACGGCTGCGACGATCAACCAGCTGCGGCAGGCCTTCCAAATCCAGAAGCTCTATGAGCGCGATGCCCGCGGCGGCACGCGATACACCGAGATTGTTCGGTCTCACTTCGGCGTCGTGTCGCCGGACTCCCGGTTGCAGCGGCCGGAATACCTGGGCGGTGGCCAGTCGCCGGTGAACATTCACCAGGTCGAGCAGACTTCGGCGTCGGCGTATGGCTCGCCGGCGGACACGCCTCAGGGGAACCTGGCTGCCTTCGGCACGGCTGTGATGTCCGGTCACGGCTTCACCAAGAGCTTCACGGAGCATTGCGTGTTGCTCGGCCTGGTGTGCGTGCGGGCTGATCTGAATTACCAGCAGGGTCTCCCGCGCATGTGGAGTCGTCGCGGGCGGTTCGACTTCTACTGGCCAGTCCTCAGTCACATTGGCGAGCAGGCGGTCTTGTCAAAGGAGATTTACTGCGACGGGACTGCTGCCGACGAAGACGTGTGGGGCTATCAGGAGCGGTATGCGGAGTATCGCTACAAGCCCTCTATGATCACCGGCCAGATGCGGTCGCAGCATGCGACCTCGCTCGACACCTGGCACTGGGCGCAGGACTTCGGGTCTACTCGTCCTCTTCTCAACGATGTCTTCATTGAGGAGGCGCCGCCGATTGCGCGGACTATCGCGGTCAATaCGGAGCCTCACTTCATTGCGGACTTCTACTTCCGGATGCGTTGTGCGAGGCCCATGCCGGTTTAcGGCGTGCCTGGCTTGATAGACCACTTCTGATCTGGGAA >Highborne Cay CCATMGAGGTCGACCCAYTGGACGGCGACCGACCTTATATCTACGCYGATCTAACGGCTGCAACGGCAGCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACACGATACACAGAGATCATMCGATCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTACAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAACGTCAACCCCATCGCCCAGACCGGAGAATCCGGAACAACCCCACAGGGCAACCTTGCCGCCATGGGCACTGCCTATATGGACGGCCACGGCTTCACGAAATCATTCACGGAGCACTGCGTCGTGATCGGCATCGTYTCRGCCCGAGCCGATCTCACMTAYCAGCAGGGTCTCAACCGcATGTGGAGYAGATCGACCAGGTGGGACTTCTACTGGCCCGCCCTGGCACACATCGGTGAGCAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGAACMTCAGCCGATGACGACGTCTTCGGCTATCAAGAGCGCTTCGCGGAATACCGCTACAAACCGAGCCTCACTACCGGCCTTATGCGGTCAAACGCCACGACATCGCTCGACACTTGGCATCTTGGCCAAGACTTTTCGGCCTTACCGGCCCTGAATGCCGCGTTCATCCAAGAGGACCCCCCCGTTGACCGCGTCATTGCTGTCCCATCCGAACCTCACTTCTTGTTCGACAGCTACTTTCAATATCGCTGTGCTCGACCGATGCCCATGTACAGCGTCCCCGGCCTCATCGACCACTTCTGAGGTCGCCATAGGCCCCCCcTCCCCAGCCGCCTTTtCCGGCcTGGCAGCCCCTGAACAAACGGAGTTCAGAT >Sargasso Sea ACCAATGCCAATGATAaTSgTcCACTTRAATCGATCCATGTTTCACCTAAAAagTGATCTATTAGACCAGGKACAGARTACACGGGCATAGGTCTGGTTGTTTTGAGATCGAAATACCAATCCCAGATAAATTCTGGTTCTGARGGTACTGCTATTACTCGATCTACTGGTGGGTTTTCCTCGATRAACGATGCGTTAAGAGCGGGCAGCGCAGTGAAATCCTGCGCCAGATGCCACGCATCCAAGGTTCCAGTTGCGTTTGAACGCATCTTTCCGGTTATTTGTGAGGGCTTRTATCTRTATTCTGCAAACCTYTCCTGATATCCGAAGGTTTGTGTATCGGCRGATGTRCCTTGTGTGTAGATTTCTTGGTTAAGTACGGCCTGTTCGCCTAAATGCGCTAGTGAAGGCCAATAGAAATCCCACCGATCACGTCTTGACCACATTCGGTTCATACCTTGCTGRTAYGTYARGTCTGCAAATACACACGCCAAACCAATTAATACGCCATGCTCGACAAATGATTTTGAGAAACCGCCCCTCGAGGTTGCGGTACCTAAAGCTGCTAGGTTACCCTGCGGTGATGTCGAGTCAGTGCTGCTTGTTTGCGGTACTGTCTGCATCATTACTTCTGTTTTCTGTCCGCCCAAATATTCTGGGCGTTGTAGTCTTGCGTCGGGTGACGTTACTCCGAAATGTGATTGTAGAATTTCGGTATATCTTGTACCGCCTCGAGCGTCTTTTTCATACAGTCTCTGAATTTGAAACGCTTCGCGTAACTGATTTATTGTTGCAGCTGTTGCATTTGATAGATCTGCAAACATtCTTGTTTGTTTCAGGTGGAGTGCCACCGCC
Part 3. Partial major viral capsid sequences recovered from the Highborne Cay sample after cloning and sequencing. The GenBank accession numbers are associated with each sequence. >A1 (EF679227) GCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACACGATACACAGAGATCATACGATCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTACAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAACGTCAACCCCATCGCCCAGACCGGAGAATCCGGAACAACCCCACAGGGCAACCTTGCCGCCATGGGCACTGCCTATATGGACGGCCACGGCTTCACGAAATCATTCACGGAGCACTGCGTCGTGATCGGC ATCGTCTCAGCCCGAGCCGATCTCACATACCAGCAGGGTCTCAACCGCATGTGGAGCAGATCGACCAGGTGGGACTTCTACTGGCCCGCCCTGGCACACATCGGTGAGCAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGAACCTCAGCCGATGACGACGTCTTCGGCTATCAAGAGCGTTTCGCGGAGTACCGCTACAAACCGAGCCTAACTACCGGCCTTATGCGGTCAAACGCCACCACCAGCCTTGACACTTGGCATCTTGGTCAAGACTTTTCGGCCTTACCGGCCCTGAAT
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 14

GCCGCGTTCATCCAAGAGGACCCCCCAGTTGACCGCGTCATTGCTGTCCCATCCGAACCTCACTTCTTGTTCGACAGCTACTTTCAATATCGCTGTGCTCGAC >A2 (EF679228) GCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACACGATACACAGAGATCATACGATCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTACAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAACGTCAACCCCATCGCCCAGACCGGAGAATCCGGAACAACCCCACAGGGCAACCTTGCCGCCATGGGCACTGCCTATATGGACGGCCACGGCTTCACGAAATCATTCACGGAGCACTGCGTCGTGATCGGC ATCGTCTCAGCCCGAGCCGATCTCACATACCAGCAGGGTCTCAACCGCATGTGGAGCAGATCGACCAGGTGGGACTTCTACTGGCCCGCCCTGGCACACATCGGTGAGCAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGAACCTCAGCCGATGACGAAGTCTTCGGCTATCAAGAGCGCTTCGCGGAATACCGATACAAACCGAGCCTTACCACCGGCCTTATGCGGTCAAACGCCACAACCAGCCTCGACACTTGGCATCTTGGTGTAGACTTTTCGACCTTACCGGCCCTGAAT GCCGCGTTCATCCAAGAAGACCCCCCGGTTGACCGCGTCATTGCTGTCCCATCCGAACCACACTTCTTGTTCGACAGCTATTTTCAATATCGCTGTGCTCGAC >A3 (EF679229) GCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACACGATACACAGAGATCATACGATCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTACAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAACGTCAACCCCATCGCCCAGACCGGAGAATCCGGAACAACCCCACAGGGCAACCTTGCAGCCATGGGCACTGCCTACATGGACGGCCACGGCTTCACGAAATCATTTACGGAGCACTGCGTCGTGATCGGC ATCGTTTCAGCCCGCGCCGATCTCACCTATCAGCAGGGTCTCAACCGGATGTGGAGCAGATCGACCAGGTGGGACTTCTATTGGCCCGCCCTGGCACACATCGGTGAACAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGCAACACTGACGACGACGAAGTCTTCGGCTATCAAGAGCGCTTCGCGGAATACCGATACAAACCGAGCCTTACCACCGGCCTTATGCGGTCAAACGCCACAACCAGCCTCGACACTTGGCATCTTGGTGTAGACTTTTCGACCTTACCGGCCCTGAAT GCCGCGTTCATCCAAGAAGACCCCCCGGTTGACCGCGTCATTGCTGTCCCATCCGAACCACACTTCTTGTTCGACAGCTATTTTCAATATCGCTGTGCTCGAC >A4 (EF679230) GCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACACGATACACAGAGATCATACGATCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTCCAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAATGTTAACCCCATCGCCCAAACAGGCGAATCGGGAACAACCCCACAGGGCAACCTTGCCGCCATGGGCACCGCCTACATGGACGGCCACGGCTTCACGAAGTCATTCACGGAGCACTGCGTCGTGATCGGC ATCGTCTCAGCCCGAGCCGATCTCACATACCAGCAGGGTCTCAACCGCATGTGGAGCAGATCGACCAGGTGGGACTTCTACTGGCCCGCCCTGGCACACATCGGTGAGCAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGAACCTCAGCCGATGACGACGTCTTCGGCTATCAAGAGCGCTTCGCGGAATACCGCTACAAACCGAGCCTTACCACCGGCCTTATGCGGTCAAACGCCACAACCAGCCTCGACACTTGGCATCTTGGTGTAGACTTTTCGACCTTACCGGCCCTGAAT GCCGCGTTCATCCAAGAAGACCCCCCGGTTGACCGCGTCATTGCTGTCCCATCCGAACCTCACTTCTTGTTCGACAGCTACTTTCAATATCGCTGTGCTCGAC >B1 (EF679231) GCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACAAGATACACAGAGATCATCAGGTCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTCCAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAATGTCAACCCCATCGCCCAAACAGGCGAATCGGGAACAACCCCACAGGGCAACCTTGCCGCCATGGGCACCGCCTACATGGACGGCCACGGCTTCACGAAGTCATTCACGGAGCACTGCGTTGTGATCGGT ATCGTCTCAGCCCGAGCCGATCTCACATACCAGCAGGGTCTCAACCGCATGTGGAGCAGATCGACCAGGTGGGACTTCTACTGACCCGCCTTGGCACACATCGGTGAGCAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGAACCTCAGCCGATGACGACGTCTTCGGCTATCAAGAGCGTTTCGCGGAATACCGCTACAAACCGAGCCTCACTACCGGCCTTATGCGGTCAAACGCCACGACATCGCTCGACACTTGGCATCTTGGCCAAGACTTTTCGGCCTTACCGGCCCTGAAT GCCGCGTTCATCCAAGAGGACCCCCCCGTTGACCGCGTCATTGCTGTCCCATCCGAACCTCACTTCTTGTTCGACAGCTACTTTCAATATCGCTGTGCTCGAC >B3 (EF679232) GCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACACGATACACAGAGATCATACGATCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTCCAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAATGTCAACCCCATCGCCCAAACAGGCGAATCGGGAACAACCCCACAGGGCAACCTTGCCGCCATGGGCACTGCCTATATGGACGGCCACGGCTTCACGAAATCATTCACGGAGCACTGCGTCGTGATCGGC ATCGTCTCAGCCCGAGCCGATCTCACATACCAGCAGGGTCTCAACCGCATGTGGAGCAGATCGACCAGGTGGGACTTCTACTGGCCCGCCCTGGCACACATCGGTGAGCAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGAACCTCAGCCGATGACGACGTCTTCGGCTATCAAGAGCGCTTCGCGGAATACCGCTACAAACCGAGCCTCACTACCGGCCTTATGCGGTCAAACGCCACGACATCGCTCGACACTTGGCATCTTGGCCAAGACTTTTCGGCCTTACCGGCCCTGAAT
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 15

GCCGCGTTCATCCAAGAGGACCCCCCCGTTGACCGCGTCATTGCTGTCCCATCCGAACCTCACTTCTTGTTCGACAGCTACTTTCAATATCGCTGTGCTCGAC >B4 (EF679233) GCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACACGATACACAGAGATCATCCGATCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTACAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAACGTCAACCCCATCGCCCAGACCGGAGAATCCGGAACAACCCCACAGGGCAACCTTGCAGCCATGGGCACTGCCTACATGGACGGCCACGGCTTCACGAAATCATTTACGGAGCACTGCGTCGTGATCGGC ATCGTTTCAGCCCGCGCCGATCTCACCTATCAGCAGGGTCTCAACCGGATGTGGAGCAGATCGACCAGGTGGGACTTCTATTGGCCCGCCCTGGCACACATCGGTGAACAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGCAACACTGACGACGACGAAGTCTTCGGCTATCAAGAGCGCTTCGCGGAATACCGATACAAACCGAGCCTTACCACCGGCCTTATGCGGTCAAACGCCACAACCAGCCTCGACACTTGGCATCTTGGTGTAGACTTTTCGACCTTACCGGCCCTGAAT GCCGCGTTCATCCAAGAAGACCCCCCGGTTGACCGCGTCATTGCTGTCCCATCCGAACCACACTTCTTGTTCGACAGCTATTTTCAATATCGCTGTGCTCGAC >D4 (EF679234) GCAACAATCAATCAGCTTCGGCAATCGTTCCAAATTCAGAAGCTGTACGAACGTGACGCCCGAGGCGGCACACGATACACAGAGATCATACGATCTCATTTTGGTGTCACGTCACCGGACGCCCGCCTACAGCGTCCGGAATATCTCGGAGGCGGTAGCACTCCGATCAACGTCAACCCCATCGCCCAGACCGGAGAATCCGGAACAACCCCACAGGGCAACCTTGCCGCCATGGGCACTGCCTATATGGACGGCCACGGCTTCACGAAATCATTCACGGAGCACTGCGTCGTGATCGGC ATCGTCTCAGCCCGAGCCGATCTCACATACCAGCAGGGTCTCAACCGCATGTGGAGCAGATCGACCAGGTGGGACTTCTACTGGCCCGCCCTGGCACACATCGGTGAGCAAGCCGTCCTCAACAAAGAAATCTACGCTCAGGGAACCTCAGCCGATGACGACGTCTTCGGCTATCAAGAGCGCTTCGCGGAATACCGCTACAAACCGAGCCTCACTACCGGCCTTATGCGGTCAAACGCCACGACATCGCTCGACACTTGGCATCTTGGCCAAGACTTTTCGGCCTTACCGGCCCTGAAT GCCGCGTTCATCCAAGAGGACCCCCCCCGTTGACCGCGTCATTGCTGTCCCATCCGAACCTCACTTCTTGTTCGACAGCTACTTTCAATATCGCTGTGCTCGAC
doi: 10.1038/nature06735 SUPPLEMENTARY INFORMATION
www.nature.com/nature 16

Editor's Summary 20 March 2008
Living fossils
Stromatolites are living, layered structures formed in shallow waters by a combination of microbial biofilms — usually of blue-green algae — and granular deposits. They are rare today but for about 2 billion years, following their arrival in the fossil record 3.5 billion years ago, they are the main evidence of life on Earth. Modern stromatolites still look like their fossilized forebears. But are the modern microbes remnants of ancient ecosystems or just latecomers following a similar lifestyle? A metagenomic study of the bacteriophage communities in modern stromatolites and thrombolites (like stromatolites but with an irregular internal structure) shows that stromatolite-associated phages are very different from each other and from any other ecosystem studied so far. This finding strengthens the hypothesis that modern stromatolites are remnants of ancient ecosystems.
Related Documents











