Biodegradable injectable polyurethanes: Synthesis and evaluation for orthopaedic applications Raju Adhikari a, * , Pathiraja A. Gunatillake a , Ian Griffiths a , Lisa Tatai a , Malsha Wickramaratna b , Shadi Houshyar a , Tim Moore a , Roshan T.M. Mayadunne b , John Field c , Margaret McGee d , Tania Carbone d a PolyNovo Biomaterials Limited, Bayview Avenue, Clayton South 3169, Victoria, Australia b CSIRO Molecular Science and Health Technologies, Bayview Avenue, Clayton South 3169, Victoria, Australia c Comparative Orthopedic Research Surgical Facility, Flinders University, Adelaide, Australia d Department of Orthopedic and Trauma, Royal Adelaide Hospital and University of Adelaide, Adelaide, Australia article info Article history: Received 14 April 2008 Accepted 12 June 2008 Available online 15 July 2008 Keywords: Injectable polyurethanes In vitro degradation In vivo degradation Biocompatibility Mechanical properties Orthopaedic abstract Biodegradable polyurethanes offer advantages in the design of injectable or preformed scaffolds for tissue engineering and other medical implant applications. We have developed two-part injectable prepolymer systems (prepolymer A and B) consisting of lactic acid and glycolic acid based polyester star polyols, pentaerythritol (PE) and ethyl lysine diisocyanate (ELDI). This study reports on the formulation and properties of a series of cross linked polyurethanes specifically developed for orthopaedic applica- tions. Prepolymer A was based on PE and ELDI. Polyester polyols (prepolymer B) were based on PE and DL-lactic acid (PEDLLA) or PE and glycolic acid (PEGA) with molecular weights 456 and 453, respectively. Several cross linked porous and non-porous polyurethanes were prepared by mixing and curing pre- polymers A and B and their mechanical and thermal properties, in vitro (PBS/37 C/pH 7.4) and in vivo (sheep bi-lateral) degradation evaluated. The effect of incorporating b-tricalcium phosphate (b-TCP, 5 microns, 10 wt.%) was also investigated. The cured polymers exhibited high compressive strength (100– 190 MPa) and modulus (1600–2300 MPa). b-TCP improved mechanical properties in PEDLLA based polyurethanes and retarded the onset of in vitro and in vivo degradation. Sheep study results demon- strated that the polymers in both injectable and precured forms did not cause any surgical difficulties or any adverse tissue response. Evidence of new bone growth and the gradual degradation of the polymers were observed with increased implant time up to 6 months. Ó 2008 Elsevier Ltd. All rights reserved. 1. Introduction Recently, there has been a major drive to develop biodegradable synthetic polymers with properties tailored to meet the bio- chemical and biomechanical requirements for various orthopaedic applications. Biodegradable polymer implants can provide initial structural support for damaged bone and subsequently degrade within a timeframe appropriate for new bone growth and remod- elling process to take place. It is also relatively easy to tailor the mechanical properties, degradation times, and other properties of synthetic polymers to suit the application. More recently designing synthetic polymer systems as injectable liquids, gels or pastes has received considerable research interests because they have the advantage of employing minimally invasive procedures such as arthroscopic delivery [1]. Injectable polymer systems could help in complex fracture and bone defect repair where substantial amount of bone is lost due to trauma or disease. Further, the injectable polymers may be used to stabilise and reinforce the fixation of implants such as plates and screws, particularly in patients with osteoporotic bone. Ideally upon injection the liquid polymer should polymerise to form a cross linked network without detrimental effects to the surrounding tissue, maintain mechanical and physical integrity and facilitate cell attachment and growth. To date, very few synthetic polymer based injectable systems have been developed that possess suitable properties for ortho- paedic applications (e.g. high strength, and controlled degradation rates) [2–4]. Mikos et al. [1] and Anseth et al. [2,5] have reported in situ polymerisable systems based on polypropylene fumarate and polyanhydride chemistry, respectively. These systems can be cured in situ with low reaction exotherm and encourage cell attachment, proliferation, and differentiation of osteoblastic function in vivo [6– 8]. However, such polymer systems have limited mechanical properties and as such may have limitation in orthopaedic appli- cations. Guelcher et al. [9] have reported injectable polyurethane networks based on lysine polyisocyanate for orthopaedic application. * Corresponding author. Tel.: þ61 395 458 048; fax: þ61 395 458 050. E-mail address: [email protected] (R. Adhikari). Contents lists available at ScienceDirect Biomaterials journal homepage: www.elsevier.com/locate/biomaterials 0142-9612/$ – see front matter Ó 2008 Elsevier Ltd. All rights reserved. doi:10.1016/j.biomaterials.2008.06.021 Biomaterials 29 (2008) 3762–3770

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

lable at ScienceDirect
Biomaterials 29 (2008) 3762–3770
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomater ia ls
Biodegradable injectable polyurethanes: Synthesis and evaluationfor orthopaedic applications
Raju Adhikari a,*, Pathiraja A. Gunatillake a, Ian Griffiths a, Lisa Tatai a, Malsha Wickramaratna b,Shadi Houshyar a, Tim Moore a, Roshan T.M. Mayadunne b, John Field c, Margaret McGee d, Tania Carbone d
a PolyNovo Biomaterials Limited, Bayview Avenue, Clayton South 3169, Victoria, Australiab CSIRO Molecular Science and Health Technologies, Bayview Avenue, Clayton South 3169, Victoria, Australiac Comparative Orthopedic Research Surgical Facility, Flinders University, Adelaide, Australiad Department of Orthopedic and Trauma, Royal Adelaide Hospital and University of Adelaide, Adelaide, Australia
a r t i c l e i n f o
Article history:Received 14 April 2008Accepted 12 June 2008Available online 15 July 2008
Keywords:Injectable polyurethanesIn vitro degradationIn vivo degradationBiocompatibilityMechanical propertiesOrthopaedic
* Corresponding author. Tel.: þ61 395 458 048; faxE-mail address: [email protected] (R. Adhikari
0142-9612/$ – see front matter � 2008 Elsevier Ltd.doi:10.1016/j.biomaterials.2008.06.021
a b s t r a c t
Biodegradable polyurethanes offer advantages in the design of injectable or preformed scaffolds fortissue engineering and other medical implant applications. We have developed two-part injectableprepolymer systems (prepolymer A and B) consisting of lactic acid and glycolic acid based polyester starpolyols, pentaerythritol (PE) and ethyl lysine diisocyanate (ELDI). This study reports on the formulationand properties of a series of cross linked polyurethanes specifically developed for orthopaedic applica-tions. Prepolymer A was based on PE and ELDI. Polyester polyols (prepolymer B) were based on PE andDL-lactic acid (PEDLLA) or PE and glycolic acid (PEGA) with molecular weights 456 and 453, respectively.Several cross linked porous and non-porous polyurethanes were prepared by mixing and curing pre-polymers A and B and their mechanical and thermal properties, in vitro (PBS/37 �C/pH 7.4) and in vivo(sheep bi-lateral) degradation evaluated. The effect of incorporating b-tricalcium phosphate (b-TCP,5 microns, 10 wt.%) was also investigated. The cured polymers exhibited high compressive strength (100–190 MPa) and modulus (1600–2300 MPa). b-TCP improved mechanical properties in PEDLLA basedpolyurethanes and retarded the onset of in vitro and in vivo degradation. Sheep study results demon-strated that the polymers in both injectable and precured forms did not cause any surgical difficulties orany adverse tissue response. Evidence of new bone growth and the gradual degradation of the polymerswere observed with increased implant time up to 6 months.
� 2008 Elsevier Ltd. All rights reserved.
1. Introduction
Recently, there has been a major drive to develop biodegradablesynthetic polymers with properties tailored to meet the bio-chemical and biomechanical requirements for various orthopaedicapplications. Biodegradable polymer implants can provide initialstructural support for damaged bone and subsequently degradewithin a timeframe appropriate for new bone growth and remod-elling process to take place. It is also relatively easy to tailor themechanical properties, degradation times, and other properties ofsynthetic polymers to suit the application. More recently designingsynthetic polymer systems as injectable liquids, gels or pastes hasreceived considerable research interests because they have theadvantage of employing minimally invasive procedures such asarthroscopic delivery [1]. Injectable polymer systems could help incomplex fracture and bone defect repair where substantial amount
: þ61 395 458 050.).
All rights reserved.
of bone is lost due to trauma or disease. Further, the injectablepolymers may be used to stabilise and reinforce the fixation ofimplants such as plates and screws, particularly in patients withosteoporotic bone. Ideally upon injection the liquid polymer shouldpolymerise to form a cross linked network without detrimentaleffects to the surrounding tissue, maintain mechanical and physicalintegrity and facilitate cell attachment and growth.
To date, very few synthetic polymer based injectable systemshave been developed that possess suitable properties for ortho-paedic applications (e.g. high strength, and controlled degradationrates) [2–4]. Mikos et al. [1] and Anseth et al. [2,5] have reported insitu polymerisable systems based on polypropylene fumarate andpolyanhydride chemistry, respectively. These systems can be curedin situ with low reaction exotherm and encourage cell attachment,proliferation, and differentiation of osteoblastic function in vivo [6–8]. However, such polymer systems have limited mechanicalproperties and as such may have limitation in orthopaedic appli-cations. Guelcher et al. [9] have reported injectable polyurethanenetworks based on lysine polyisocyanate for orthopaedicapplication.

Table 1Formulation of synthesised polymers
Polymerdesignation
Prepolymer B Molar ratioof prepolymer A:prepolymer B
Filler % b-TCP(5 micron)
PN1 PEGA 1:1PN2 PEGA-F 1:1 10PN3 PEDLLA 1:1PN4 PEDLLA-F 1:1 10PN5 PEGA:PEDLLA 1:0.5:0.5PN6 PEGA:PEDLLA-F 1:0.5:0.5 10Implant samplesPN7 PEGA:PEDLLA:H2O 1:0.4:0.4:0.2PN8 PEGA:PEDLLA:H2O-F* 1:0.4:0.4:0.2 20PN9 PEGA:PEDLLA:H2O-I* 1:0.4:0.4:0.2PN10 PEGA:PEDLLA-I* 1:0.5:0.5
*All formulation contained PE-ELDI as prepolymer A. Prepolymer B MW:PEGA ¼ 532, PEDLLA ¼ 530. Abbreviation: PE ¼ pentaerythritol; ELDI: ethyl lysinediisocyanate; GA: glycolic acid; DLLA: DL-lactic acid. *I ¼ injectable, F ¼ filler (b-TCP,b-tricalcium phosphate).
R. Adhikari et al. / Biomaterials 29 (2008) 3762–3770 3763
Recently, we reported urethane based two-part prepolymersystems which can be cross linked in situ to form both rigid orelastomeric polymer networks useful for a range of biomedicalapplications including scaffolds for tissue engineering [10,11]. Theprepolymers are liquids at ambient temperature and above. Thefirst prepolymer (prepolymer A) is a reaction product of an aliphaticdiisocyanate such as ethyl lysine diisocyanate (ELDI) and a poly-hydroxy compound such as pentaerythritol (PE). The second pre-polymer is a difunctional or multifunctional polyester polyol. Theseprepolymers can be reacted along with other additives if needed inappropriate proportions to produce cross linked polymer networks.Further, the incorporation of a predetermined quantity of water into the prepolymer mixture allows the formation of a porousstructure due to release of carbon dioxide during curing. The me-chanical properties and degradation characteristics of the polymernetwork is determined by the chemical composition, molecularweight and functionality of the prepolymers. We have designeda series of prepolymer systems to evaluate their suitability asinjectable polymers for orthopaedic applications.
The prepolymers are prepared from compounds such that whenthe cross linked polymer network degrades, the degradationproducts are non-toxic, bioresorb or released from the body due totheir low molecular weight and water solubility. In this study theprepolymer A was based on ethyl 2,6-diisocyanato hexanoate(ELDI) and pentaerythritol (PE), whereas prepolymer B was a starpolyol based on PE and with either glycolic acid (GA) or DL-lacticacid (DLLA).
This paper reports on the effect of prepolymer B structure onmechanical properties and in vitro degradation of the polymernetworks formed by in situ cross linking. A selected polymer systemwas further investigated in a sheep implant study to assess bio-compatibility, biodegradation and surgical utility in both injectableand cross linked forms.
2. Materials and methods
Pentaerythritol, glycolic acid (70% w/v in water), DL-lactic acid (85% w/v in water)and stannous octoate were purchased from Sigma-Aldrich (Sydney, Australia) andused as received. Ethyl 2,6-diisocyanato hexanoate (Kyowa Hakko Kogyo Co Ltd,Japan) was distilled before use and b-tricalcium phosphate (Plasma Biotal Limited,UK) was used as received.
2.1. Hydroxyl number, acid number and molecular weight of polyol [12]
The number average molecular weight was calculated based on hydroxylnumbers of polyols. The hydroxyl and acid numbers were determined in accordancewith the ASTM method E 1899-02, Vol 6.03, 2002 and ASTM D1980-87, Vol 6.03,1998 respectively.
2.2. Polymer synthesis
A total of six polymer compositions were prepared to test mechanical proper-ties. The first three polymers were prepared by varying the structure of prepolymerB and other three were prepared by incorporating TCP at 10 wt.%. Polymer PEDLLA:PEGA was chosen for in vitro and in vivo experiments and prepared separately. Thefollowing general procedures describe the preparation of prepolymer A and B in thisstudy.
2.2.1. Prepolymer APredried pentaerythritol (4.818 g) (Aldrich, Sydney, Australia) was weighed into
a dry three-neck flask equipped with a magnetic stirrer, nitrogen inlet and dryingtube. Ethyl 2,6-diisocyanato hexanoate (ELDI) (32.04 g) was then added to the flaskfollowed by catalyst stannous-2-ethyl hexanoate (0.1 wt.%) under nitrogen. Thereaction mixture was then stirred and heated to 50 �C for 72 h under nitrogen at-mosphere to give prepolymer A. The instantaneous viscosity of the prepolymer wasfound to be 8.7 � 104 cP at 25 �C and GPC showed the number average molecularweight and polydispersity 1348 and 1.73, respectively.
2.2.2. Prepolymer BPredried pentaerythritol (27.23 g) was weighed in a dry three-neck flask
equipped with a magnetic stirrer, nitrogen inlet and air condenser attached to theflask through a distillation head. Glycolic acid (136.21 g, 70% w/v) was then added tothe flask. The flask was heated in an oil bath to 80 �C for about 3 h until all
pentaerythritol dissolved. The temperature of the flask was then increased to 160 �Cand heating continued. Nitrogen was bubbled through the reaction mixture for fastremoval of water formed during the reaction. The reaction was stopped after 24 hand the reaction flask taken off from the oil bath after carefully turning off nitrogenflow. The polyol was then transferred to a single-neck round bottom flask anddegassed at 120 �C under vacuum (w0.1 torr) using Kugelrohr and stored undernitrogen. Polyols PEDLLA was prepared using the same procedure using DL-lactic acidwhereas polyol PEGA: PEDLLA was prepared by blending PEGA and PEDLLA polyol in1:1 wt ratio. The prepolymers were characterised by GPC, Hydroxyl and Acidnumber determinations [11].
2.2.3. Polymer preparationPolymers for mechanical property testing were prepared by reacting prepoly-
mer A and B such that the mixture has equimolar amounts of isocyanate andhydroxyl functional groups according to the following general procedure.
Prepolymer B was dried and degassed under vacuum at 80 �C for few hoursbefore polymerisation and allowed to cool to ambient temperature. Prepolymer A(5.0 g) and prepolymer B (PEGA, 2.55 g) were mixed at ambient temperature withcatalyst stannous 2-ethyl hexanoate (0.007 g) for 3 min. The mixture was degassedunder vacuum (0.1 torr) for 3 min and the liquid mixture was taken in to a 10 mLsyringe and dispensed to fill cylindrical (6 mm D � 12 mm L) cavities in a Teflonmould. The mould was placed in an oven at 37 �C for 24 h to cure the polymer. Forsamples containing the filler, TCP was added to the prepolymer mixture and mixedat high speed (6000 rpm) for 3 min using a Sliverson L4RT high speed mixer.
Precured polymer samples were prepared similarly for in vitro and in vivoexperiments. The prepolymer B was a 50/50 mixture of PEGA and PEDLLA. Porouspolymer plugs were prepared by incorporating an amount of water equivalent to20 mol.% of the prepolymer B hydroxyl groups to generate carbon dioxide during thereaction. The amount of TCP incorporated was 20 wt.%. For evaluating injectableprepolymer samples, sterilised prepolymers (Gamma sterilisation, 25 KGy) wereprovided to the surgeon in syringes and the mixing was done in the theatre.
Polymer samples are designated starting with PN and controls with C,C1 ¼ control (no treatment), C2 ¼ control (PMMA) and C3 ¼ control (PURASORB�).Table 1 summarises the formulation details of polymers prepared for mechanicalproperty testing, and in vitro and in vivo evaluations.
2.3. Gel permeation chromatography and viscosity
The molecular weight and viscosity of the prepolymers were determined by gelpermeation chromatography (GPC) and Bohlin Rheometer (CRL110), respectively.GPC was performed on Waters Associates Liquid Chromatograph system (Waters717) equipped with a differential refractometer and four m-Styragel columns (105,104, 103 and 100 Å). The mobile phase was tetrahydrofuran (THF) at a flow rate of1 mL/min. Prepolymer was dissolved in THF by warming the solution at 50 �C for 1 hand filtered through 0.45 micron filter before analysis. The system was calibratedwith narrow disperse polystyrene standards and molecular weights are reported aspolystyrene equivalents.
The viscosity of the prepolymer was measured at 23 �C using a Bohlin Rheom-eter CRL 110.
2.4. Mechanical properties
The mechanical properties were determined using Instron Universal TestingSystem, model 5568 (Instron corporation, series IX Automated materials testingSystem) equipped with Merlin 2002 software. Five replicates of cylindrical testspecimens (6 mm � 12 mm) tested at a cross head speed of 1 mm/min (ASTM F451-95). Specimens were equilibrated for 24 h at ambient conditions (23 �C and 45%–
R. Adhikari et al. / Biomaterials 29 (2008) 3762–37703764
50% humidity) before testing. The average values for compression modulus, stress atyield and ultimate compressive strength (UCS) were calculated and reported.
The standard pull-out test was performed according to ASTM F543-02. A metalscrew (3.5 mm D � 17.5 mm L) was placed in mould and liquid prepolymer pouredand cured to form a solid polymer block to prepare the test specimen. Each specimenwas mounted in a custom made jig to ensure that the screw was pulled only along itslong axis. The screw was pulled using a materials testing machine (Instron 4468)under displacement control until failure at a rate of 10 mm/min. Load and dis-placement data were obtained, and the pull-out strength was determined as themaximum force on the load displacement curve by using the following equation [13]
s ¼ Ppdh
where P is ultimate load (N), d is the major diameter of screw (mm) and h is thelength of effective threads in the screw in the cancellous bone.
2.5. Thermal analysis
DSC analysis was carried out over the temperature range �50 �C to 200 �C usingMettler Toledo DSC821e. The experiments were carried out at a heating rate 10 �C/min under nitrogen. Sample weights ranged between 20 and 25 mg. The sample wasdried at 40 �C for 48 h under a vacuum (0.1 torr) prior to analysis.
2.6. Degradation
2.6.1. In vitro studyThe in vitro degradation study was carried out according to ASTM method F
1635-04. The degradation medium was 0.1 M PBS with a pH w7.4 � 0.2 and PBSreplenished when pH approached 7.2. The polymers were incubated at 37 �C and theresults were reported as an average of six replicates. Each specimen was placed in anindividual vial immersed in phosphate buffered saline solution and stored in a 37 �Coven in a static mode. Sample to PBS ratio was w1:300 and sampling time pointswere 1, 14, 42, 90, 180 days. Samples at different time intervals were removed fromthe degradation media, rinsed with deionised water to remove embedded saltswithin the pores of the polymer and dried on surface with Kimwipes wipers beforemeasuring the wet mass and dimension of the samples. The samples were thenfurther dried under vacuum for 5 days at 40 �C to determine dry mass.
Table 2Mechanical properties of cured polymers
Sample code Compression properties
UCS (MPa) Stress @ yield (MPa) Modulus (GPa)
PN1 136 � 14 84 � 15 2.0 � 0.3PN2 139 � 11 76 � 13 2.3 � 0.01PN3 145 � 40 85 � 6 2.1 � 0.2PN4 187 � 20 89 � 8 2.2 � 0.3PN5 179 � 12 71 � 10 1.9 � 0.2PN6 104 � 4 69 � 2 1.6 � 0.06PN7 44 � 11 5.6 � 1.1 0.27 � 0.09PN8 35 � 16 13.2 � 0.9 0.58 � 0.07Cortical Bone13 200 � 36 – 19.6 � 6.2Cancellous Bone13 9.3 � 4.5 – 0.9 � 71Acrylic bone cements13 35 � 3.2 – 3.0 � 1.5
– Not available, 13 ¼mechanical testing of bone and the bone-implant interfaceedited by Yuehuei H. An and Robert A. Draughn; CRC press, 1999.
0100020003000400050006000700080009000
PN1 Normal bone
Max L
oad
(N
) &
S
tiffn
ess (N
/m
m)
Ultimate loStiffness (NStrength (M
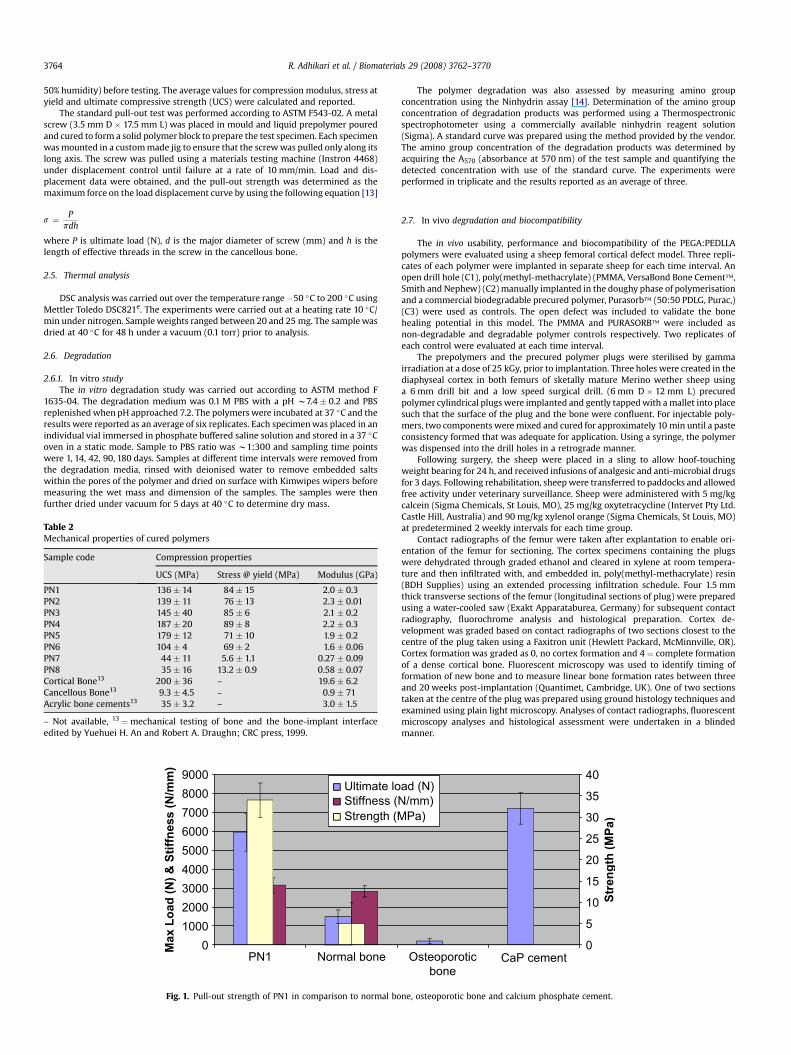
Fig. 1. Pull-out strength of PN1 in comparison to normal bo
The polymer degradation was also assessed by measuring amino groupconcentration using the Ninhydrin assay [14]. Determination of the amino groupconcentration of degradation products was performed using a Thermospectronicspectrophotometer using a commercially available ninhydrin reagent solution(Sigma). A standard curve was prepared using the method provided by the vendor.The amino group concentration of the degradation products was determined byacquiring the A570 (absorbance at 570 nm) of the test sample and quantifying thedetected concentration with use of the standard curve. The experiments wereperformed in triplicate and the results reported as an average of three.
2.7. In vivo degradation and biocompatibility
The in vivo usability, performance and biocompatibility of the PEGA:PEDLLApolymers were evaluated using a sheep femoral cortical defect model. Three repli-cates of each polymer were implanted in separate sheep for each time interval. Anopen drill hole (C1), poly(methyl-methacrylate) (PMMA, VersaBond Bone Cement�,Smith and Nephew) (C2) manually implanted in the doughy phase of polymerisationand a commercial biodegradable precured polymer, Purasorb� (50:50 PDLG, Purac,)(C3) were used as controls. The open defect was included to validate the bonehealing potential in this model. The PMMA and PURASORB� were included asnon-degradable and degradable polymer controls respectively. Two replicates ofeach control were evaluated at each time interval.
The prepolymers and the precured polymer plugs were sterilised by gammairradiation at a dose of 25 kGy, prior to implantation. Three holes were created in thediaphyseal cortex in both femurs of sketally mature Merino wether sheep usinga 6 mm drill bit and a low speed surgical drill. (6 mm D � 12 mm L) precuredpolymer cylindrical plugs were implanted and gently tapped with a mallet into placesuch that the surface of the plug and the bone were confluent. For injectable poly-mers, two components were mixed and cured for approximately 10 min until a pasteconsistency formed that was adequate for application. Using a syringe, the polymerwas dispensed into the drill holes in a retrograde manner.
Following surgery, the sheep were placed in a sling to allow hoof-touchingweight bearing for 24 h, and received infusions of analgesic and anti-microbial drugsfor 3 days. Following rehabilitation, sheep were transferred to paddocks and allowedfree activity under veterinary surveillance. Sheep were administered with 5 mg/kgcalcein (Sigma Chemicals, St Louis, MO), 25 mg/kg oxytetracycline (Intervet Pty Ltd.Castle Hill, Australia) and 90 mg/kg xylenol orange (Sigma Chemicals, St Louis, MO)at predetermined 2 weekly intervals for each time group.
Contact radiographs of the femur were taken after explantation to enable ori-entation of the femur for sectioning. The cortex specimens containing the plugswere dehydrated through graded ethanol and cleared in xylene at room tempera-ture and then infiltrated with, and embedded in, poly(methyl-methacrylate) resin(BDH Supplies) using an extended processing infiltration schedule. Four 1.5 mmthick transverse sections of the femur (longitudinal sections of plug) were preparedusing a water-cooled saw (Exakt Apparataburea, Germany) for subsequent contactradiography, fluorochrome analysis and histological preparation. Cortex de-velopment was graded based on contact radiographs of two sections closest to thecentre of the plug taken using a Faxitron unit (Hewlett Packard, McMinnville, OR).Cortex formation was graded as 0, no cortex formation and 4 ¼ complete formationof a dense cortical bone. Fluorescent microscopy was used to identify timing offormation of new bone and to measure linear bone formation rates between threeand 20 weeks post-implantation (Quantimet, Cambridge, UK). One of two sectionstaken at the centre of the plug was prepared using ground histology techniques andexamined using plain light microscopy. Analyses of contact radiographs, fluorescentmicroscopy analyses and histological assessment were undertaken in a blindedmanner.
Osteoporotic bone
CaP cement0
5
10
15
20
25
30
35
40
Stren
gth
(M
Pa
)
ad (N)/mm)Pa)
ne, osteoporotic bone and calcium phosphate cement.
Wg ^-10.2
min°C-50 -40 -30 -20 -10 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140
58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77
^exo
STARe SW 9.00Lab: METTLER
PEGA- Polyol
PEDLLA- Polyol
a
b^exo
STARe SW 9.00Lab: METTLER
mW2
min
°C-20 0 20 40 60 80 100 120 140
62 64 66 68 70 72 74 76
PN1
PN3
PN5
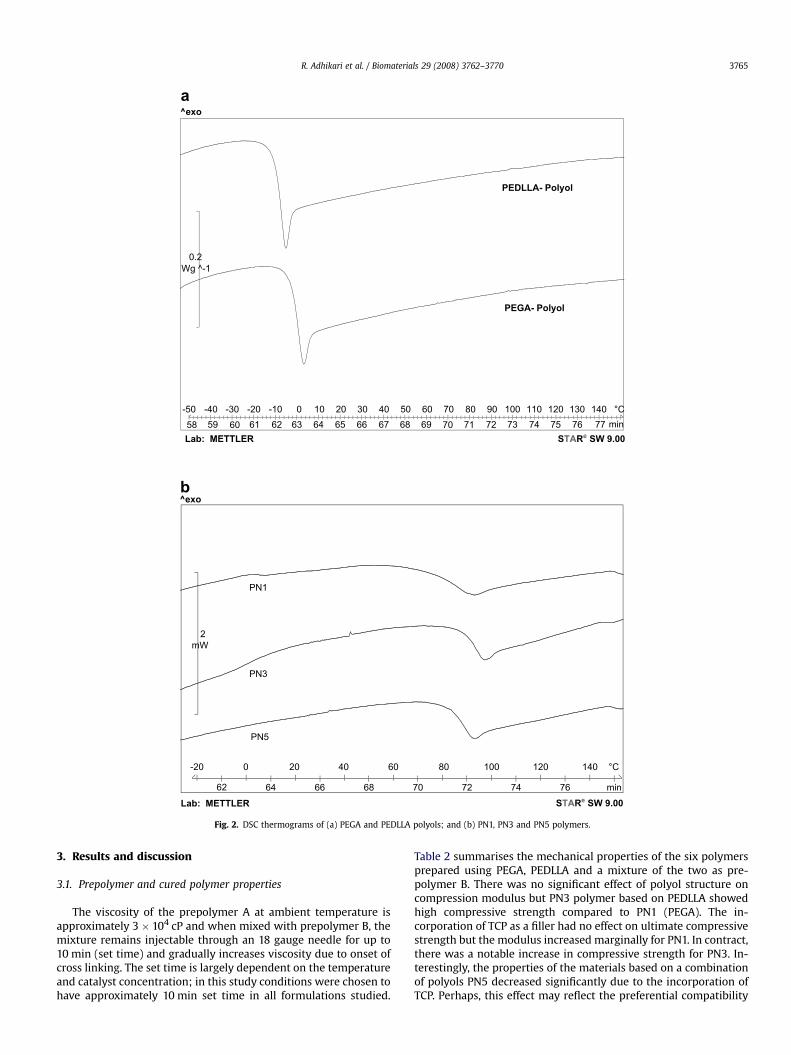
Fig. 2. DSC thermograms of (a) PEGA and PEDLLA polyols; and (b) PN1, PN3 and PN5 polymers.
R. Adhikari et al. / Biomaterials 29 (2008) 3762–3770 3765
3. Results and discussion
3.1. Prepolymer and cured polymer properties
The viscosity of the prepolymer A at ambient temperature isapproximately 3 � 104 cP and when mixed with prepolymer B, themixture remains injectable through an 18 gauge needle for up to10 min (set time) and gradually increases viscosity due to onset ofcross linking. The set time is largely dependent on the temperatureand catalyst concentration; in this study conditions were chosen tohave approximately 10 min set time in all formulations studied.
Table 2 summarises the mechanical properties of the six polymersprepared using PEGA, PEDLLA and a mixture of the two as pre-polymer B. There was no significant effect of polyol structure oncompression modulus but PN3 polymer based on PEDLLA showedhigh compressive strength compared to PN1 (PEGA). The in-corporation of TCP as a filler had no effect on ultimate compressivestrength but the modulus increased marginally for PN1. In contract,there was a notable increase in compressive strength for PN3. In-terestingly, the properties of the materials based on a combinationof polyols PN5 decreased significantly due to the incorporation ofTCP. Perhaps, this effect may reflect the preferential compatibility
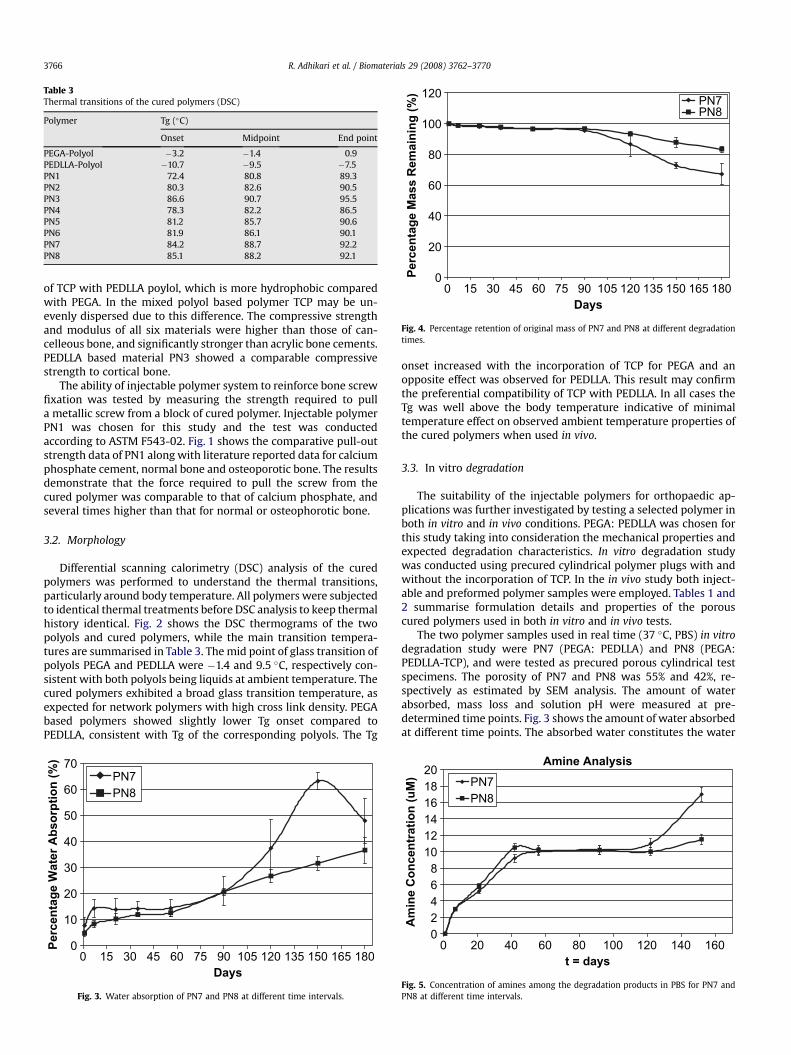
Table 3Thermal transitions of the cured polymers (DSC)
Polymer Tg (�C)
Onset Midpoint End point
PEGA-Polyol �3.2 �1.4 0.9PEDLLA-Polyol �10.7 �9.5 �7.5PN1 72.4 80.8 89.3PN2 80.3 82.6 90.5PN3 86.6 90.7 95.5PN4 78.3 82.2 86.5PN5 81.2 85.7 90.6PN6 81.9 86.1 90.1PN7 84.2 88.7 92.2PN8 85.1 88.2 92.1
0
20
40
60
80
100
120
0 15 30 45 60 75 90 105 120 135 150 165 180Days
Pe
rc
en
ta
ge
M
as
s R
em
ain
in
g (%
) PN7PN8
Fig. 4. Percentage retention of original mass of PN7 and PN8 at different degradationtimes.
R. Adhikari et al. / Biomaterials 29 (2008) 3762–37703766
of TCP with PEDLLA poylol, which is more hydrophobic comparedwith PEGA. In the mixed polyol based polymer TCP may be un-evenly dispersed due to this difference. The compressive strengthand modulus of all six materials were higher than those of can-celleous bone, and significantly stronger than acrylic bone cements.PEDLLA based material PN3 showed a comparable compressivestrength to cortical bone.
The ability of injectable polymer system to reinforce bone screwfixation was tested by measuring the strength required to pulla metallic screw from a block of cured polymer. Injectable polymerPN1 was chosen for this study and the test was conductedaccording to ASTM F543-02. Fig. 1 shows the comparative pull-outstrength data of PN1 along with literature reported data for calciumphosphate cement, normal bone and osteoporotic bone. The resultsdemonstrate that the force required to pull the screw from thecured polymer was comparable to that of calcium phosphate, andseveral times higher than that for normal or osteophorotic bone.
3.2. Morphology
Differential scanning calorimetry (DSC) analysis of the curedpolymers was performed to understand the thermal transitions,particularly around body temperature. All polymers were subjectedto identical thermal treatments before DSC analysis to keep thermalhistory identical. Fig. 2 shows the DSC thermograms of the twopolyols and cured polymers, while the main transition tempera-tures are summarised in Table 3. The mid point of glass transition ofpolyols PEGA and PEDLLA were �1.4 and 9.5 �C, respectively con-sistent with both polyols being liquids at ambient temperature. Thecured polymers exhibited a broad glass transition temperature, asexpected for network polymers with high cross link density. PEGAbased polymers showed slightly lower Tg onset compared toPEDLLA, consistent with Tg of the corresponding polyols. The Tg
0
10
20
30
40
50
60
70
0 15 30 45 60 75 90 105 120 135 150 165 180Days
Percen
tag
e W
ater A
bso
rp
tio
n (%
)
PN7PN8
Fig. 3. Water absorption of PN7 and PN8 at different time intervals.
onset increased with the incorporation of TCP for PEGA and anopposite effect was observed for PEDLLA. This result may confirmthe preferential compatibility of TCP with PEDLLA. In all cases theTg was well above the body temperature indicative of minimaltemperature effect on observed ambient temperature properties ofthe cured polymers when used in vivo.
3.3. In vitro degradation
The suitability of the injectable polymers for orthopaedic ap-plications was further investigated by testing a selected polymer inboth in vitro and in vivo conditions. PEGA: PEDLLA was chosen forthis study taking into consideration the mechanical properties andexpected degradation characteristics. In vitro degradation studywas conducted using precured cylindrical polymer plugs with andwithout the incorporation of TCP. In the in vivo study both inject-able and preformed polymer samples were employed. Tables 1 and2 summarise formulation details and properties of the porouscured polymers used in both in vitro and in vivo tests.
The two polymer samples used in real time (37 �C, PBS) in vitrodegradation study were PN7 (PEGA: PEDLLA) and PN8 (PEGA:PEDLLA-TCP), and were tested as precured porous cylindrical testspecimens. The porosity of PN7 and PN8 was 55% and 42%, re-spectively as estimated by SEM analysis. The amount of waterabsorbed, mass loss and solution pH were measured at pre-determined time points. Fig. 3 shows the amount of water absorbedat different time points. The absorbed water constitutes the water
Amine Analysis
02468
101214161820
0 20 40 60 80 100 120 140 160t = days
Am
in
e C
on
cen
tratio
n (u
M) PN7
PN8
Fig. 5. Concentration of amines among the degradation products in PBS for PN7 andPN8 at different time intervals.
0
1
2
3
4
5
6 12 24Implantation time (weeks)
Ave
ra
ge
g
ra
de
o
f c
orte
x d
ev
elo
pm
en
t
PN7PN8PN9PN10C1C2C3
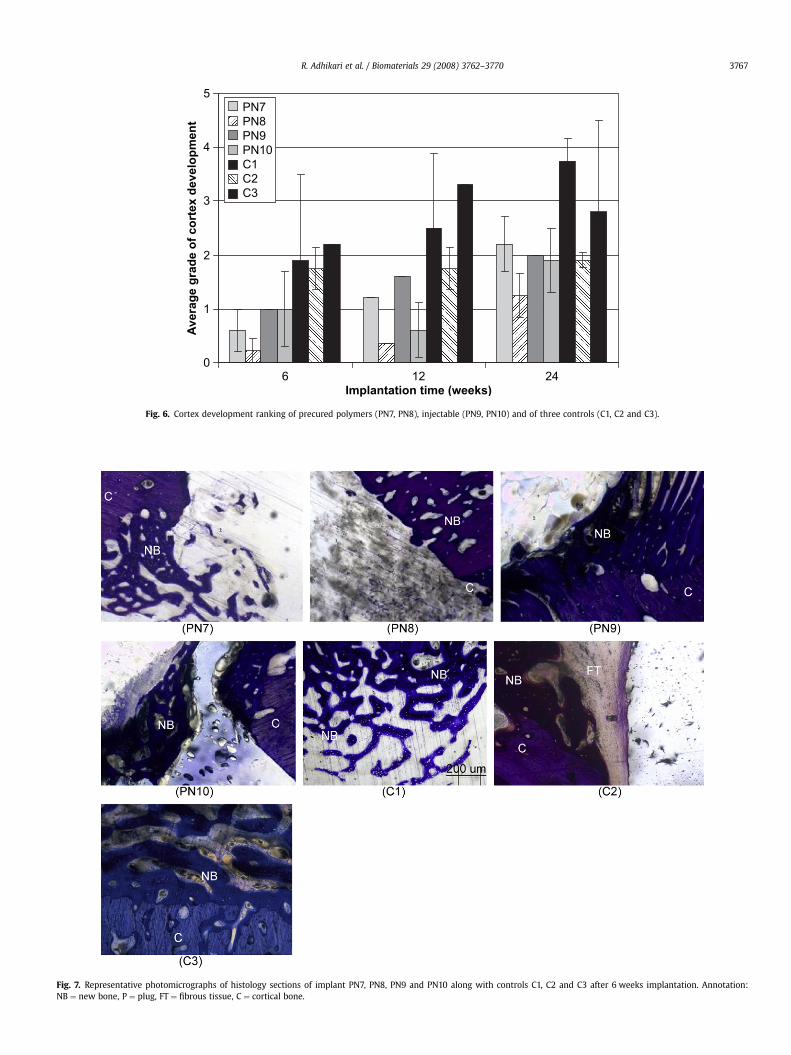
Fig. 6. Cortex development ranking of precured polymers (PN7, PN8), injectable (PN9, PN10) and of three controls (C1, C2 and C3).
Fig. 7. Representative photomicrographs of histology sections of implant PN7, PN8, PN9 and PN10 along with controls C1, C2 and C3 after 6 weeks implantation. Annotation:NB ¼ new bone, P ¼ plug, FT ¼ fibrous tissue, C ¼ cortical bone.
R. Adhikari et al. / Biomaterials 29 (2008) 3762–3770 3767
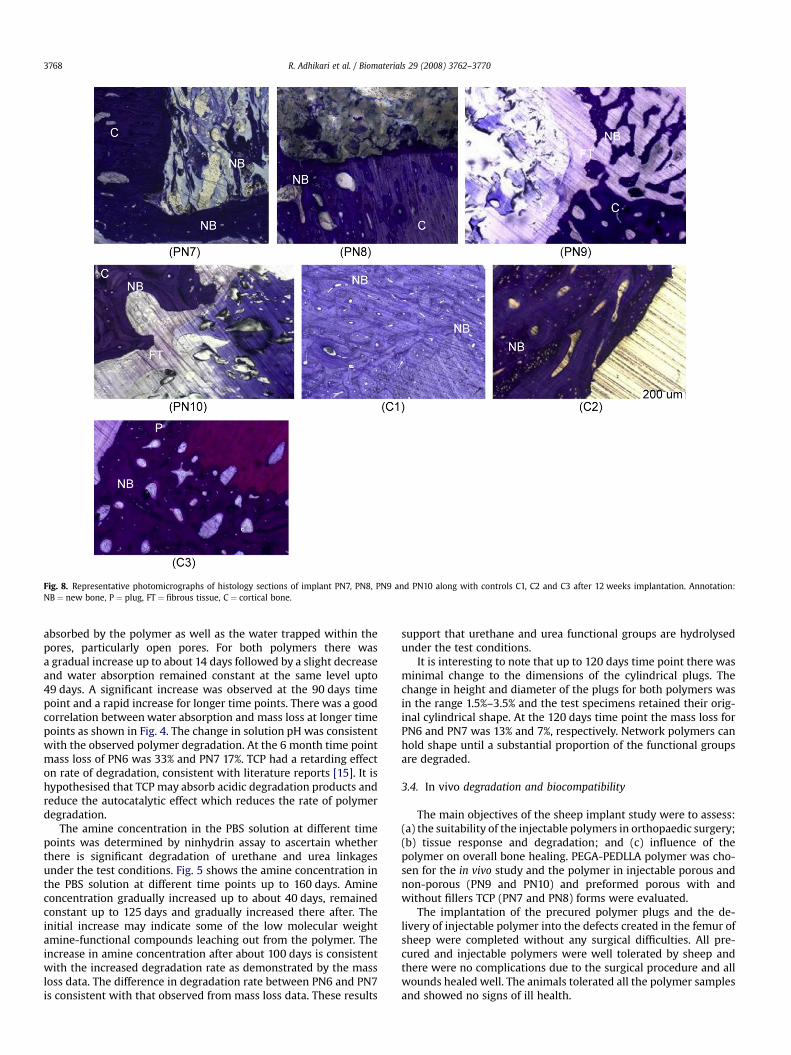
Fig. 8. Representative photomicrographs of histology sections of implant PN7, PN8, PN9 and PN10 along with controls C1, C2 and C3 after 12 weeks implantation. Annotation:NB ¼ new bone, P ¼ plug, FT ¼ fibrous tissue, C ¼ cortical bone.
R. Adhikari et al. / Biomaterials 29 (2008) 3762–37703768
absorbed by the polymer as well as the water trapped within thepores, particularly open pores. For both polymers there wasa gradual increase up to about 14 days followed by a slight decreaseand water absorption remained constant at the same level upto49 days. A significant increase was observed at the 90 days timepoint and a rapid increase for longer time points. There was a goodcorrelation between water absorption and mass loss at longer timepoints as shown in Fig. 4. The change in solution pH was consistentwith the observed polymer degradation. At the 6 month time pointmass loss of PN6 was 33% and PN7 17%. TCP had a retarding effecton rate of degradation, consistent with literature reports [15]. It ishypothesised that TCP may absorb acidic degradation products andreduce the autocatalytic effect which reduces the rate of polymerdegradation.
The amine concentration in the PBS solution at different timepoints was determined by ninhydrin assay to ascertain whetherthere is significant degradation of urethane and urea linkagesunder the test conditions. Fig. 5 shows the amine concentration inthe PBS solution at different time points up to 160 days. Amineconcentration gradually increased up to about 40 days, remainedconstant up to 125 days and gradually increased there after. Theinitial increase may indicate some of the low molecular weightamine-functional compounds leaching out from the polymer. Theincrease in amine concentration after about 100 days is consistentwith the increased degradation rate as demonstrated by the massloss data. The difference in degradation rate between PN6 and PN7is consistent with that observed from mass loss data. These results
support that urethane and urea functional groups are hydrolysedunder the test conditions.
It is interesting to note that up to 120 days time point there wasminimal change to the dimensions of the cylindrical plugs. Thechange in height and diameter of the plugs for both polymers wasin the range 1.5%–3.5% and the test specimens retained their orig-inal cylindrical shape. At the 120 days time point the mass loss forPN6 and PN7 was 13% and 7%, respectively. Network polymers canhold shape until a substantial proportion of the functional groupsare degraded.
3.4. In vivo degradation and biocompatibility
The main objectives of the sheep implant study were to assess:(a) the suitability of the injectable polymers in orthopaedic surgery;(b) tissue response and degradation; and (c) influence of thepolymer on overall bone healing. PEGA-PEDLLA polymer was cho-sen for the in vivo study and the polymer in injectable porous andnon-porous (PN9 and PN10) and preformed porous with andwithout fillers TCP (PN7 and PN8) forms were evaluated.
The implantation of the precured polymer plugs and the de-livery of injectable polymer into the defects created in the femur ofsheep were completed without any surgical difficulties. All pre-cured and injectable polymers were well tolerated by sheep andthere were no complications due to the surgical procedure and allwounds healed well. The animals tolerated all the polymer samplesand showed no signs of ill health.
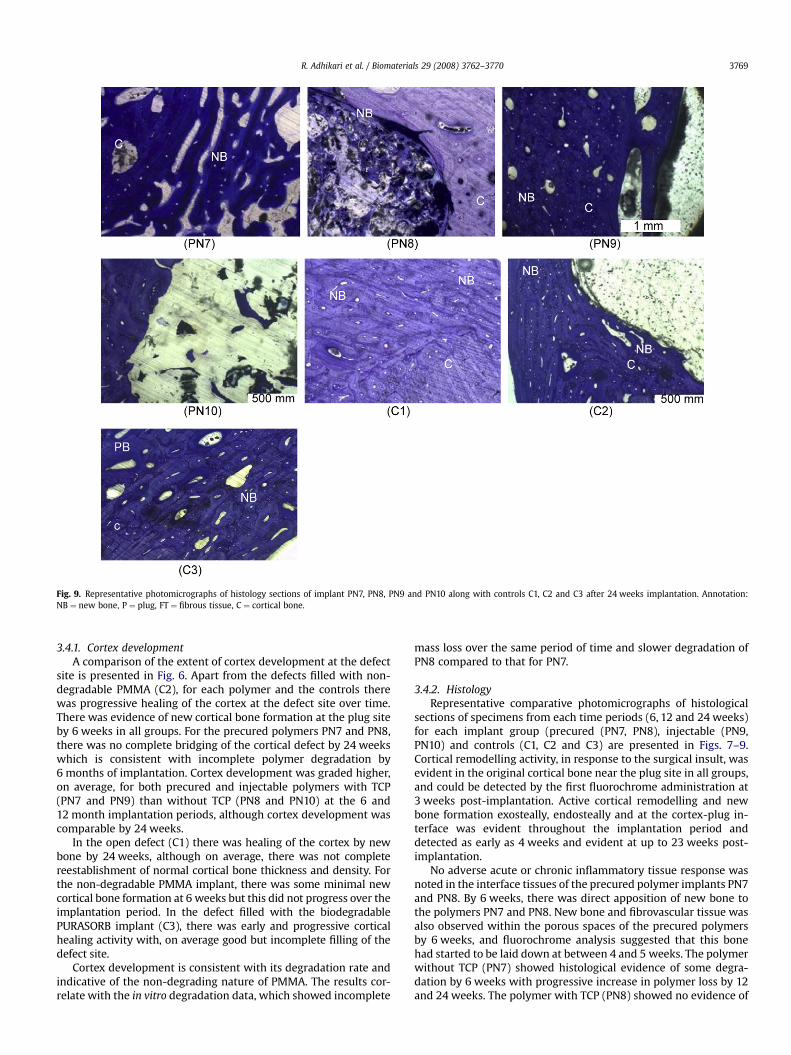
Fig. 9. Representative photomicrographs of histology sections of implant PN7, PN8, PN9 and PN10 along with controls C1, C2 and C3 after 24 weeks implantation. Annotation:NB ¼ new bone, P ¼ plug, FT ¼ fibrous tissue, C ¼ cortical bone.
R. Adhikari et al. / Biomaterials 29 (2008) 3762–3770 3769
3.4.1. Cortex developmentA comparison of the extent of cortex development at the defect
site is presented in Fig. 6. Apart from the defects filled with non-degradable PMMA (C2), for each polymer and the controls therewas progressive healing of the cortex at the defect site over time.There was evidence of new cortical bone formation at the plug siteby 6 weeks in all groups. For the precured polymers PN7 and PN8,there was no complete bridging of the cortical defect by 24 weekswhich is consistent with incomplete polymer degradation by6 months of implantation. Cortex development was graded higher,on average, for both precured and injectable polymers with TCP(PN7 and PN9) than without TCP (PN8 and PN10) at the 6 and12 month implantation periods, although cortex development wascomparable by 24 weeks.
In the open defect (C1) there was healing of the cortex by newbone by 24 weeks, although on average, there was not completereestablishment of normal cortical bone thickness and density. Forthe non-degradable PMMA implant, there was some minimal newcortical bone formation at 6 weeks but this did not progress over theimplantation period. In the defect filled with the biodegradablePURASORB implant (C3), there was early and progressive corticalhealing activity with, on average good but incomplete filling of thedefect site.
Cortex development is consistent with its degradation rate andindicative of the non-degrading nature of PMMA. The results cor-relate with the in vitro degradation data, which showed incomplete
mass loss over the same period of time and slower degradation ofPN8 compared to that for PN7.
3.4.2. HistologyRepresentative comparative photomicrographs of histological
sections of specimens from each time periods (6, 12 and 24 weeks)for each implant group (precured (PN7, PN8), injectable (PN9,PN10) and controls (C1, C2 and C3) are presented in Figs. 7–9.Cortical remodelling activity, in response to the surgical insult, wasevident in the original cortical bone near the plug site in all groups,and could be detected by the first fluorochrome administration at3 weeks post-implantation. Active cortical remodelling and newbone formation exosteally, endosteally and at the cortex-plug in-terface was evident throughout the implantation period anddetected as early as 4 weeks and evident at up to 23 weeks post-implantation.
No adverse acute or chronic inflammatory tissue response wasnoted in the interface tissues of the precured polymer implants PN7and PN8. By 6 weeks, there was direct apposition of new bone tothe polymers PN7 and PN8. New bone and fibrovascular tissue wasalso observed within the porous spaces of the precured polymersby 6 weeks, and fluorochrome analysis suggested that this bonehad started to be laid down at between 4 and 5 weeks. The polymerwithout TCP (PN7) showed histological evidence of some degra-dation by 6 weeks with progressive increase in polymer loss by 12and 24 weeks. The polymer with TCP (PN8) showed no evidence of

R. Adhikari et al. / Biomaterials 29 (2008) 3762–37703770
degradation at 6 weeks and only minimal loss at 12 weeks. Con-sistent with the degradation pattern observed, bone and osteogenictissue, within porous spaces and voids left by polymer PN7 deg-radation, progressively advanced towards the centre of the polymerand occupied the cortical defect site over the 24 week implantationperiod. Bone formation into the porous PN8 implant, was largelyconfined to the outer pores of the implant until 24 weeks by whichstage new bone had advanced deeper into the porous spaces of theplug as degradation occurred.
At 6 weeks there was no notable polymer degradation in eitherof the two injectable polymer implants PN9 and PN10. There wasdirect apposition of new bone onto the implant and new bone waspresent within the porous spaces. There was early cortical bonebridging of the defect over the outer aspect of the plug. There wasno adverse acute or chronic inflammatory tissue response noted inthe interface tissues of the injectable polymer implants at 6 weeks.By 12 weeks, there had been considerable degradation of thepolymers creating new space within the plugs although degrada-tion of polymer PN9 was more advanced compared to polymerPN10. Active bone formation was evident around and within poresof both implants. In some of the polymer specimens, there wassome reactive cellular activity, likely in response to the polymerdegradation, with some small pockets of neutrophils, lymphocytesand some necrotic tissue at the interface and within the porousspaces. By 24 weeks, polymer PN9 was completely degraded whilstpolymer PN10 was only partly degraded. New bone completely ormostly occupied the defect site previously occupied by the plug. By24 weeks only one specimen had a very localised infiltration ofneutrophil cells.
In controls, consistent with the radiographic analyses, by6 weeks of implantation there was cortical bone defect interfacebone formation and new bone present with the open defect in theC1 group. There was also partial outer bridging of the cortical gap.The C3 PURASORB plug, had almost completely degraded by6 weeks and replaced by an infiltrate of fibrovascular tissue. In re-sponse to the PMMA C2 implant, there was extensive fibrous tissueformation at the implant-bone interface and a notable reduction inthe density of the interface bone. At 12 weeks C1 showed nearcomplete filling of the gap with new bone and fibrovascular tissuein non-mineralised areas of the defect. Similarly in the PURASORBC3 specimens, there was almost complete outer bridging of thecortical gap at the defect site and complete polymer degradation. InC2 some new bone formation at the interface and bridging ofperiosteal cortical gap was observed. By 24 weeks the open defectwas completely or close to completely filled by new bone and therewas some resultant thickening of the cortical rim at the defect site.Similar histological appearances were found for the PURASORB C3specimens although in one specimen, pockets of lymphocyte richtissue were noted within the defect site occupied by non-miner-alised tissue. With no degradation of the PMMA C2 implant, therewas only progressive cortical bridging over the outer aspect of theimplant over the 24 week implantation period.
4. Conclusion
This study demonstrated that injectable polymer systems basedon isocyanate prepolymers and polyester polyols could be
formulated with properties suitable for orthopaedic applications.The chemical structure of the polyol influenced the mechanicalproperties and DL-lactic acid based polyols yielded polymers withhigh compressive strength, comparable to that of cortical bone. Thepolymer networks formed were amorphous and exhibited glasstransition temperatures in the range 80–90 �C. The in vitro degra-dation results confirmed PEGA based polymer degraded at a fasterrate compared to PEDLLA and detection of amines among degra-dation products confirmed the hydrolysis of urethane and ureagroups. Sheep implant study demonstrated that both injectable andpreformed polymers could be utilised to fill cavities in bone undersurgical conditions. Histology evaluations confirmed the absence ofany adverse tissue reaction to both injectable and preformedpolymers and new bone formation was observed apposed toimplants and within the pores of the polymer. PEGA based polymerdegraded faster than PEDLLA based polymers in consistent within vitro degradation results. The incorporation of b-tricalciumphosphate retarded the onset of degradation. The above polymersexhibited a good combination of mechanical and degradationproperties and have potential for a number of orthopaedicapplications ranging from bone substitutes to scaffolds and also asan in situ forming synthetic graft for bone defects.
References
[1] Temenoff JS, Mikos AG. Injectable biodegradable materials for orthopaedictissue engineering. Biomaterials 2000;21(23):2405–12.
[2] Burkoth AK, Anseth KS. A review of photocrosslinked polyanhydrides in-situforming degradable networks. Biomaterials 2000;21(23):2395–404.
[3] Burdick JA, Frankel D, Dernell WS, Anseth KS. An initial investigation ofphotocurable three-dimensional lactic acid based scaffolds in a critical-sizedcranial defect. Biomaterials 2003;24(9):1613–20.
[4] Gunatillake PA, Mayadunne RTM, Adhikari R. ‘‘Recent developments in bio-degradable synthetic polymers’’, biotechnology annual reviews, vol. 12.Elsevier B.V; 2006. pp. 301-347.
[5] Poshusta AK, Burdick JA, Mortisen DJ, Padera RF, Ruehlman D, Yaszemski MJ,et al. Histocompatibility of photocrosslinked polyanhydrides: a novel in-situforming orthopaedic biomaterial. J Biomed Mater Res Part A 2003;64(1):62–9.
[6] Muggli DS, Burkoth AK, Anseth KS. Crosslinked polyanhydrides for use inorthopaedic applications: degradation behaviour and mechanics. J BiomedMater Res 1999;46(2):271–8.
[7] Peter SJ, Kim P, Yasko AW, Yaszemski MJ, Mikos AG. Crosslinkingcharacteristics of an injectable poly(propylene fumarate)/beta-tricalciumphosphate paste and mechanical properties of the crosslinked compositefor use as a biodegradable bone cement. J Biomed Mater Res. 1999;44(3):314–21.
[8] Peter SJ, Miller ST, Zhu G, Yasko AW, Mikos AG. In vivo degradation of apoly(propylene fumarate)/beta-tricalcium phosphate injectable compositescaffold. J Biomed Mater Res 1998;41(1):1–7.
[9] Guelcher AS, Srinivisan A, Dumas JF, Didier JE, McBride S, Hollinger JO.Synthesis, mechanical properties, biocompatibility, and biodegradation ofpolyurethane networks from lysine polyisocyanates. Biomaterials 2008;29:1762–75.
[10] Adhikari R, Gunatillake PA. International PCT application PCT/AU2003/000935.[11] Bonzani IC, Adhikari R, Houshyar S, Mayadunne R, Gunatillake P, Stevens MM.
Synthesis of two-component injectable polyurethanes for bone tissueengineering. Biomaterials 2007;28:423–33.
[12] Annual book of ASTM Standards vol. 06.03 (E1899–02).[13] An YH, Draughn RA. Mechanical testing of bone and the bone-implant
interface. CRC Press; 1999.[14] Moore S. Amino acid analysis: aqueous dimethyl sulfoxide as solvent for the
ninhydrin reaction. J Biol Chem 1968;243(23):6281–3.[15] David D, Neal S, Topham S, Cristina M, Michael S, Wolfe KJ, et al.
Poly(propylene fumarate) and poly(DL-lactic-co-glycolic acid) as scaffoldmaterials for solid and foam-coated composite tissue-engineered constructsfor cranial reconstruction. Tissue Eng 2003;9(3):495–504.
Related Documents











