Academiejaar 2015-2016 BILATERALE PROGRESSIEVE PIJNLOZE OPTICUS NEUROPATHIE Deborah DE BRUYN Promotor: Dr. Julie De Zaeytijd Co-promotor: Prof. Dr. Bart Leroy Masterproef voorgedragen in de master in de specialistische geneeskunde OFTALMOLOGIE FACULT EIT GENEESKUNDE EN GEZONDHEIDSWETENSCHAPPEN

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Academiejaar 2015-2016
BILATERALE PROGRESSIEVE PIJNLOZE OPTICUS
NEUROPATHIE
Deborah DE BRUYN
Promotor: Dr. Julie De Zaeytijd
Co-promotor: Prof. Dr. Bart Leroy
Masterproef voorgedragen in de master in de specialistische geneeskunde
OFTALMOLOGIE
FACULTEIT GENEESKUNDE EN GEZONDHEIDSWETENSCHAPPEN


Academiejaar 2015-2016
BILATERALE PROGRESSIEVE PIJNLOZE OPTICUS
NEUROPATHIE
Deborah DE BRUYN
Promotor: Dr. Julie De Zaeytijd
Co-promotor: Prof. Dr. Bart Leroy
Masterproef voorgedragen in de master in de specialistische geneeskunde
OFTALMOLOGIE
FACULTEIT GENEESKUNDE EN GEZONDHEIDSWETENSCHAPPEN

Deze pagina is niet beschikbaar omdat ze persoonsgegevens bevat.Universiteitsbibliotheek Gent, 2021.
This page is not available because it contains personal information.Ghent University, Library, 2021.

Bilaterale progressieve pijnloze opticus neuropathie I
VOORWOORD
Ik bedank al mijn dierbaren maar in het bijzonder mijn man en zoontje die mij tijdens het
schrijven van deze masterproef veel liefde, begrip en steun hebben gegeven. Ik ben fier op de
realisatie van dit werk. Ik wens de lezer veel plezier toe.

Bilaterale progressieve pijnloze opticus neuropathie II
INHOUDSOPGAVE
Voorwoord ............................................................................................................................................................... I
Lijst met afkortingen .............................................................................................................................................. IV
I. Abstract ................................................................................................................................................................ 1
II. Inleiding ............................................................................................................................................................... 1
2.1. Klinische casuïstiek ...................................................................................................................................... 1
2.2. Doelstellingen en objectieven ..................................................................................................................... 3
III. Methodologie ..................................................................................................................................................... 4
IV. Resultaten .......................................................................................................................................................... 4
4.1. Differentiaal diagnose en bespreking pro’s en contra’s .............................................................................. 4
4.2. Overerfbare opticus neuropathieën ............................................................................................................ 6
4.2.1. Leber hereditaire opticus neuropathie (LHON) .................................................................................... 7
4.2.1.1. Inleiding ......................................................................................................................................... 7
4.2.1.2. Mitochondriale fysiologie ............................................................................................................. 7
4.2.1.3. Het mitochondriale genoom ......................................................................................................... 8
4.2.1.4. Mitochondriale overerving ......................................................................................................... 10
4.2.1.5. Mitochondriale DNA mutaties in LHON ...................................................................................... 10
4.2.1.6. Incomplete penetrantie van LHON ............................................................................................. 12
4.2.1.7. Epidemiologie ............................................................................................................................. 14
4.2.1.8. Klinische manifestaties en prognose........................................................................................... 14
4.2.1.9. Behandeling ................................................................................................................................ 15
4.2.1.9.1. Ubiquinone analogen .......................................................................................................... 16
4.2.1.9.2. Gentherapie ......................................................................................................................... 17
4.2.1.10. Genetische counseling .............................................................................................................. 17
4.2.2. Autosomaal dominante opticus atrofie (ADOA) ................................................................................. 17
4.2.2.1 Epidemiologie en klinische manifestaties .................................................................................... 17
4.2.2.2. Extra-oculaire manifestaties ....................................................................................................... 19

Bilaterale progressieve pijnloze opticus neuropathie III
4.2.2.3. Behandeling ................................................................................................................................ 19
4.2.3. Toxische opticus neuropathieën ........................................................................................................ 19
4.2.3.1. Inleiding ....................................................................................................................................... 19
4.2.3.2. Roken, alcohol en nutritionele deprivatie ................................................................................... 20
4.2.3.3. Toxische opticus neuropathie ..................................................................................................... 20
4.2.3.3.1. Ethambutol .......................................................................................................................... 20
4.2.3.3.2. Methanol ............................................................................................................................. 21
4.2.3.3.3. Vigabatrin ............................................................................................................................ 21
4.2.3.3.4. Disulfiram (Antabuse®) ........................................................................................................ 21
V. Discussie ............................................................................................................................................................ 22
VI. Referentielijst ................................................................................................................................................... 22
BIJLAGEN ...................................................................................................................................................................i

Bilaterale progressieve pijnloze opticus neuropathie IV
LIJST MET AFKORTINGEN
ADOA autosomale dominante opticus atrofie
ATP adenosine trifosfaat
CPEO chronisch progressieve externe oftalmoplegie
DNA desoxyribonucleïnezuur
LASIK Laser in-situ keratomileusis
LHON Leber hereditaire opticus neuropathie
MRI magnetische resonantie beeldvorming
mtDNA mitochondriale desoxyribonucleïnezuur
OCT optische coherentie tomografie
OXPHOS oxidatieve fosforylering
rRNA ribosomale ribonucleïnezuur
tRNA transfer ribonucleïnezuur
TON toxische opticus neuropathieën
VEP visuele geëvokeerde potentialen

Bilaterale progressieve pijnloze opticus neuropathie 1
I. ABSTRACT
Bilateraal progressief en pijnloos visusverlies heeft een brede differentiaal diagnose. De
meeste diagnoses kunnen door een grondige anamnese en een routine oogheelkundig
onderzoek reeds uitgesloten worden. Erfelijke en toxisch-nutritionele opticus neuropathieën
zijn enerzijds zeldzame en anderzijds uitsluitingsdiagnosen met een gelijkaardig klinisch
beeld. Er is evidentie in de literatuur dat omgevingsfactoren in het algemeen en toxische en
nutritionele factoren in het bijzonder een invloed kunnen hebben op de expressie van het
fenotype van Leber hereditaire opticus neuropathie. De bevestiging van de diagnose van een
erfelijke opticus neuropathie door middel van mutatie-analyse is noodzakelijk voor de
behandeling, genetische counseling en prognose.
II. INLEIDING
2.1. KLINISCHE CASUÏSTIEK
Een 58-jarige man consulteerde met de subjectieve klacht van wazig zicht ter hoogte van
beide ogen sinds 4 maanden. Het zicht zou in beide ogen geleidelijk verminderd zijn over een
periode van een aantal weken. Er was geen nachtblindheid, noch fotopsieën, noch pijn. De
meest optimale visus bedroeg 3.5/10 rechts en 2.5/10 links. In de oftalmologische
voorgeschiedenis weerhielden we een LASIK ingreep aan beide ogen, zes jaar eerder, in
kader van een myopie. Vroegere oogcontroles op school of bij arbeidsonderzoeken bleken
steeds normaal. In de familiale voorgeschiedenis waren er geen oogziekten te weerhouden.
Bij navragen vermeldde de patiënt dat hij vroeger 1.5 pakje sigaretten per dag rookte. Bij de
initiële presentatie rookte hij nog gemiddeld 3 à 4 sigaretten per dag en dronk hij ongeveer ½
fles wijn per dag.
De patiënt werd onderworpen aan een compleet oogheelkundig onderzoek bestaande uit een
biomicroscopie, oogdrukmeting, gedilateerd fundusonderzoek, autofluorescentie, Optische
Coherentie Tomografie (OCT) van de macula en retinale zenuwvezellaag, patroon VEP
(visuele geëvokeerde potentialen), Goldmann gezichtsveldonderzoek, kleurenonderzoek, en
tot slot een contrastonderzoek.
Bilaterale progressieve pijnloze opticus neuropathie 2
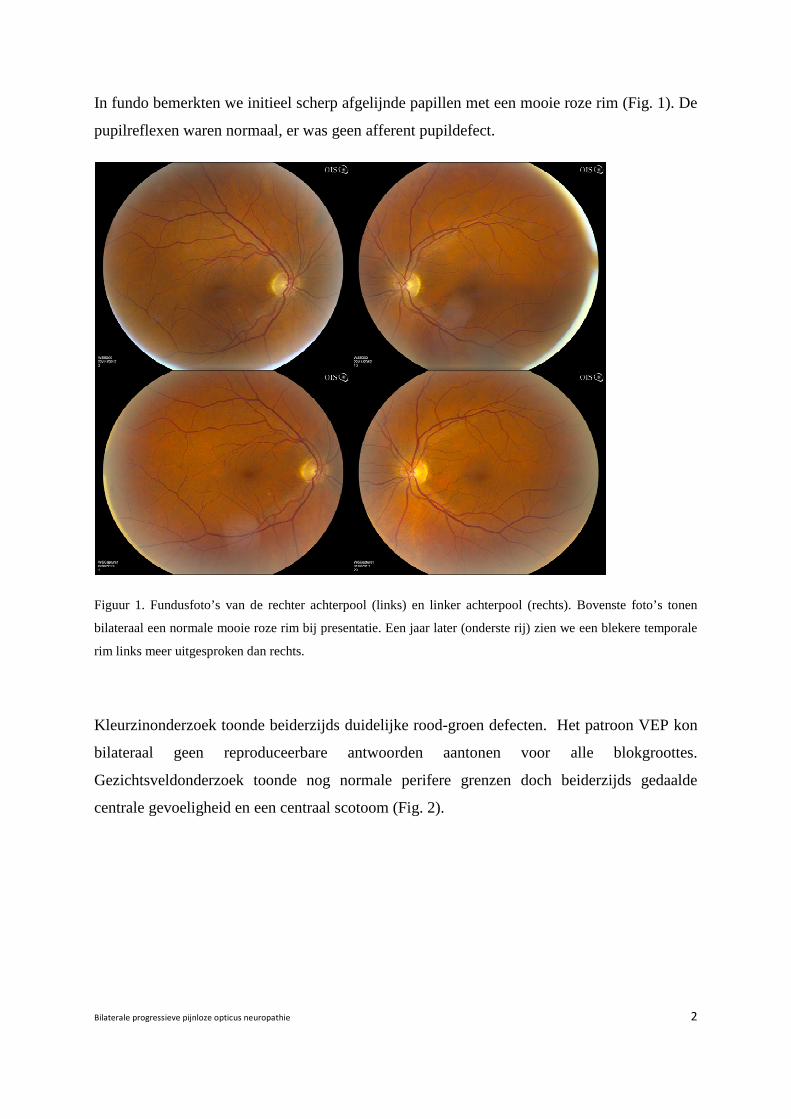
In fundo bemerkten we initieel scherp afgelijnde papillen met een mooie roze rim (Fig. 1). De
pupilreflexen waren normaal, er was geen afferent pupildefect.
Figuur 1. Fundusfoto’s van de rechter achterpool (links) en linker achterpool (rechts). Bovenste foto’s tonen
bilateraal een normale mooie roze rim bij presentatie. Een jaar later (onderste rij) zien we een blekere temporale
rim links meer uitgesproken dan rechts.
Kleurzinonderzoek toonde beiderzijds duidelijke rood-groen defecten. Het patroon VEP kon
bilateraal geen reproduceerbare antwoorden aantonen voor alle blokgroottes.
Gezichtsveldonderzoek toonde nog normale perifere grenzen doch beiderzijds gedaalde
centrale gevoeligheid en een centraal scotoom (Fig. 2).

Bilaterale progressieve pijnloze opticus neuropathie 3
Figuur 2. Goldmann gezichtsveldonderzoek bij presentatie (rechteroog rechts afgebeeld, linkeroog links) toonde
normale perifere grenzen doch beiderzijds een gedaalde centrale gevoeligheid en een centraal scotoom met
stimulus I2.
De klinische symptomatologie gecombineerd met de rood-groen defecten en de resultaten van
het patroon VEP, wezen in de richting van een opticus neuropathie. MRI hersenen en
cardiologisch onderzoek waren normaal. In eerste instantie dachten we bij deze patiënt aan
een toxisch-nutritionele opticus neuropathie op basis van alcohol- en tabak gebruik. We
raadden een volledige rook- en alcoholstop aan. Een jaar later was zijn zicht terug
gerecupereerd tot 7.5/10 bilateraal. Er was op dat moment wel een duidelijke temporale
atrofie van de beide oogzenuwen op te merken (Fig. 1). Genetische bloedanalyse voor Leber
hereditaire opticus neuropathie (LHON) kon een heteroplasmatische primaire mitochondriale
DNA mutatie aantonen (m.3460G>A). Twee jaar later was er een opmerkelijke volledige
recuperatie van het zicht tot 10/10 en een verbeterd kleurenzicht. Volledige rookstop bleek
echter moeilijk vol te houden voor onze patiënt.
2.2. DOELSTELLINGEN EN OBJECTIEVEN
Aan de hand van de casuïstiek willen we in deze masterproef een paar vragen beantwoorden
omtrent bilaterale progressieve en pijnloze opticus neuropathie. Vooreerst overlopen we de
differentiaal diagnose en bespreken alle pro’s en contra’s voor de verschillende diagnoses met
de gegevens uit de casus als achtergrond. Daarna focussen we meer op de pathologie,
overerving en klinische manifestaties van de erfelijke opticus neuropathieën en dan in het
bijzonder op Leber hereditaire opticus neuropathie. Tot slot vergelijken we de klinische
symptomatologie en manifestatie van LHON en toxisch-nutritionele opticus neuropathie

Bilaterale progressieve pijnloze opticus neuropathie 4
waarbij we in de literatuur evidentie zoeken voor een mogelijke interactie tussen de beide
pathologieën.
III. METHODOLOGIE
Er werd een systematisch literatuuronderzoek uitgevoerd via de zoekmachines Web of Science
en Pubmed met de volgende zoektermen:
- Bilateral progressive vision loss
- Optic neuropathy
- Mitochondrial optic neuropathies
- Toxic optic neuropathies
Op basis van de inhoud van het abstract werd dan een selectie gemaakt van te lezen artikels.
De referentielijst van elk gelezen artikel werd daarna uitgepluisd op zoek naar andere
relevante artikels. Deze artikels werden dan via Web of Science elektronisch opgezocht. Via
Web of Science werd ook de ‘times cited’ van bepaalde artikels geraadpleegd. Op deze
manier kunnen recentere artikels opgespoord worden die het oorspronkelijk artikel citeren en
die handelen over hetzelfde of een nauw verwant onderwerp. De laatste zoekactie gebeurde in
december 2015.
IV. RESULTATEN
4.1. DIFFERENTIAAL DIAGNOSE EN BESPREKING PRO’S EN CONTRA’S
De uitgebreide differentiaal diagnose van bilateraal progressief en pijnloos visusverlies omvat
de volgende ziekten (1):
- dominant overerfbare opticus neuropathieën
- Leber hereditaire opticus neuropathie
- toxisch-nutritionele opticus neuropathie
- compressieve opticus neuropathie

Bilaterale progressieve pijnloze opticus neuropathie 5
- infiltratieve opticus neuropathie
- bilateraal inflammatoire opticus neuropathie
- infectieuze oorzaken (bv. syfilitische opticus neuritis)
- demyeliniserende opticus neuropathie
- maculaire dystrofieën
- retinale degeneraties zoals kegeltjes dystrofie
- ziekte van Graves
- radiatie-geïnduceerde opticus neuropathie
- diabetische papillopathie
- niet-organisch visusverlies (hysterie, aggravatie)
Zowel de ziekte van Graves, radiatie-geïnduceerde opticus neuropathie en diabetische
papillopathie werden door een grondige algemene anamnese en een basis oogheelkundig
onderzoek reeds uitgesloten bij onze patiënt. Bij een compressieve opticus neuropathie
zouden we initieel een papiloedeem met een latere evolutie naar volledige papilatrofie
verwachten. MRI van de hersenen was tevens normaal bij onze patiënt. Ook bij een
infiltratieve opticus neuropathie zouden we initieel een papiloedeem verwachten. Bilaterale
aantasting bij de initiële presentatie is hierbij zeldzaam. Een bilateraal inflammatoire of
infectieuze opticus neuropathie zou ook eerder papiloedeem geven en een cellulaire reactie in
het achtersegment, wat bij onze patiënt niet het geval was. Een demyeliniserende opticus
neuropathie behoorde tot de mogelijkheden doch een simultane bilaterale aantasting is eerder
zeldzaam. Daarenboven kon de MRI hersenen geen demyeliniserende foci aantonen bij onze
patiënt. Maculaire dystrofieën en retinale degeneraties presenteren met maculaire en/of
retinale afwijkingen en zullen pas over het verloop van een aantal jaren leiden tot bleekheid
van de oogzenuw. Bij onze patiënt was de macula en de perifere fundus normaal. De
overblijvende diagnoses zijn dan nog erfelijke opticus neuropathieën of een toxisch-
nutritionele oorzaak. Bij onze patiënt was een erfelijke oorzaak minder waarschijnlijk gezien
de negatieve familiale anamnese en de vrij simultane visusdaling in beide ogen. Bij navraag
bleek onze patiënt vroeger een forse roker te zijn geweest met op het moment van presentatie

Bilaterale progressieve pijnloze opticus neuropathie 6
nog steeds een rookgedrag, alsook een alcoholabusus. De toxisch-nutritionele opticus
neuropathie leek ons het meest plausibel. Bloedanalyse om LHON uit te sluiten werd
zekerheidshalve toch verricht en was verrassend genoeg positief voor de primaire
mitochondriale DNA mutatie m.3460G>A in heteroplasmie.
Potentieel toxische drugs (bv. Ethambutol)
Toxische mitochondriale neuropathie?
Verworven bilateraal visusverlies
Refractie afwijking, media opaciteiten, maculair probleem? Trauma of hoge dosis radiotherapie?
Pijnlijke oogbewegingen?
Papiloedeem?
Peripapillaire telangiectasieën? Bewaarde pupilreflexen ondanks slechte visus?
Familiale voorgeschiedenis van onverklaard visusverlies?
Voorgeschiedenis van kanker
Ataxia, gehoorsverlies, diabetes
Anemie, polyneuropathie
Alcohol, roken, hoge transaminasen?
Ja
Ja
Ja
Ja
Ja
Ja
Ja
Ja
Ja
Ja
MRI
MRI
Adequate therapie
Neuritis optica?
Verhoogde ICD? Necrose van de basale ganglia? Bilaterale AION? Amiodarone?
Familiale voorgeschiedenisGenanalyse
LHON?
Onderzoek oudersKleurzinonderzoekGenanalyse
MRIFundus autofluorescentieElectrofysiologie
Genanalyse
Vitamine B12
Toxische mitochondriale neuropathie?
Overerfbare ziekte?ADOA (tritan defect)?
Infiltratieve of paraneoplastischeziekte (bv. MAR/CAR)?
Wolfram syndroom, andere overerfbare neuropathieën?
Nutritionele opticus neuropathie?
Stop nicotine en alcohol
Stop drugs indien mogelijk
Figuur 3. Flowchart voor de evaluatie van bilateraal visusverlies (1). MRI, magnetische resonantie
beeldvorming; ICD, intracraniële druk; AION, anterieure ischemische opticus neuropathie; LHON, Leber
hereditaire opticus neuropathie; ADOA, autosomaal dominante opticus atrofie; MAR, melanoma geassocieerde
retinopathie; CAR, kanker geassocieerde retinopathie.
4.2. OVERERFBARE OPTICUS NEUROPATHIEËN
De twee klassieke prototypes van overerfbare opticus neuropathieën zijn Leber hereditaire
opticus neuropathie (LHON) en autosomaal dominante opticus atrofie (ADOA) (2). Beiden
hebben een gelijkaardig klinisch beeld en zijn gekarakteriseerd door het unieke verlies van
retinale ganglion cellen met vroegtijdige schade aan de papillomaculaire bundel (2).

Bilaterale progressieve pijnloze opticus neuropathie 7
4.2.1. LEBER HEREDITAIRE OPTICUS NEUROPATHIE (LHON)
4.2.1.1. INLEIDING
Leber hereditaire opticus neuropathie is genoemd naar de Duitse oftalmoloog Dr. Theodor
Carl Gustav von Leber (1840-1917) (3). In zijn publicatie ‘Ueber hereditäre und congenital-
angelegte Sehnervenleiden’ uit 1871, beschreef hij vier stambomen en 15 patiënten met de
symptomen van LHON (4). Zijn observaties toonden reeds enkele belangrijke karakteristieken
van LHON aan zoals de maternele overerving, de predominantie van mannen en de bijna
exclusieve aantasting van de nervus opticus (3). Initieel dacht men dat LHON een recessieve
X-gebonden overerfbare ziekte was door de hierboven reeds vernoemde predominantie van
aangetaste mannen (5). Pas in 1988 kon Wallace et al. aantonen dat een mitochondriale
puntmutatie aan de basis lag van het niet-Mendeliaanse en maternele overervingspatroon bij
LHON (6). LHON was hiermee de eerst beschreven mitochondriale ziekte (3).
4.2.1.2. MITOCHONDRIALE FYSIOLOGIE
Mitochondria zijn tubulaire intracellulaire organellen, omringd door een dubbele membraan
(7). De binnenste membraan is sterk geplooid in cristae en bevat de ‘respiratory chain’
complexen (2). De ‘respiratory chain’ bestaat uit vijf complexen (I-V) die via oxidatieve
fosforylering (OXPHOS) adenosine trifosfaat (ATP) genereren (Fig. 4) (7). Verschillende co-
factoren zoals ubiquinone (Co-enzyme Q10) en cytochrome c zijn belangrijk in de overdracht
van elektronen tussen de verschillende complexen onderling (2, 7). Naast ATP productie zijn
ook detoxificatie van reactieve zuurstofradicalen, ijzermetabolisme, vetzuuroxidatie, de
biosynthese van aminozuren en de regulatie van cellulaire apoptose belangrijke taken van
mitochondria (7).

Bilaterale progressieve pijnloze opticus neuropathie 8
Figuur 4. Schematische illustratie van de gemuteerde genen (blauw) in mitochondriale opticus neuropathieën en
hun functies in de mitochondria. Vraagtekens duiden op een ongekende lokalisatie en/of mitochondriale functie
(8).
4.2.1.3. HET MITOCHONDRIALE GENOOM
Hetgeen mitochondria uniek maakt, is het bezit van hun eigen genoom (2). Dit bestaat uit een
circulaire, dubbelstrengige molecule (2, 7). Het mitochondriale genoom codeert voor 2
ribosomale RNA’s, 22 transfer RNA’s en 13 subunits van de ‘respiratory chain’ complexen
(2). De meerderheid van deze genen is gelokaliseerd op de H-streng (Fig. 5) (2). Meer dan 80
subunits van de ‘respiratory chain’ complexen worden gecodeerd door het nucleaire genoom
(7).

Bilaterale progressieve pijnloze opticus neuropathie 9
Figuur 5. Het mitochondriale genoom (2). Eiwit coderende (geel), rRNA (rood) en tRNA (paars) coderende
genen worden voorgesteld op de zware (‘heavy’, H, buitenste) en de lichte (‘light’, L, binnenste) streng.
Elk mitochondrium bevat meerdere kopieën van het mitochondriale genoom (2). Het aantal
mitochondriale DNA (mtDNA) moleculen per cel is afhankelijk van de energievereisten van
die specifieke cel en kan variëren van honderden tot duizenden exemplaren per cel (2). Indien
al deze kopieën identiek zijn aan elkaar, spreekt men over homoplasmie (7). In het geval van
heteroplasmie zijn er twee of meer mitochondriale DNA varianten aanwezig op een specifieke
nucleotide positie (2) met het ontstaan van een wild-type en gemuteerd mtDNA in één
mitochondrium (7).
In tegenstelling tot nucleair DNA kan mitochondriaal DNA continu repliceren, onafhankelijk
van de fase van de celcyclus en dit in zowel mitotische als postmitotische cellen (2, 7). Dit is
één van de redenen waarom het mitochondriaal genoom vlugger mutaties verwerft en er meer
polymorfismen bestaan (2, 3). Andere redenen zijn de afwezigheid van protectieve histonen,
het gebrek aan herstelmechanismen en de nabijheid van de ‘respiratory chain’ complexen met
hoge blootstelling aan vrije zuurstofradicalen tot gevolg (2).

Bilaterale progressieve pijnloze opticus neuropathie 10
4.2.1.4. MITOCHONDRIALE OVERERVING
Het mitochondriale genoom wordt materneel overgeërfd (2, 7). Het kleine aantal
mitochondria aanwezig in spermacellen wordt vernietigd door proteolytische enzymen in de
zygote op het moment van de conceptie (7, 9). Vaders kunnen de mutatie dus nooit doorgeven
aan hun kinderen (7).
Na conceptie worden de verschillende mtDNA-moleculen afkomstig van de moedercel at
random verdeeld tussen de dochtercellen, een proces genaamd replicatieve segregatie (7). Dit
heeft als gevolg dat bij heteroplasmie de verdeling van mutant en wild-type mtDNA tussen
dochtercellen ongelijk gebeurt (7). Een bepaalde hoeveelheid van het mutante mtDNA kan
echter nog gecompenseerd worden door de werking van het wild-type mtDNA (7). De
kritische drempel, waarbij het wild-type mtDNA de effecten van het mutante DNA niet langer
kan compenseren, is vaak weefselspecifiek (7). Vooral de retinale ganglioncellen hebben een
lagere mutatiedrempel dan minder metabool actieve weefsels (7).
De nadelige effecten van mtDNA-mutaties op de werking van de oxidatieve fosforylering
worden meestal merkbaar bij een aanwezigheid van meer dan 60-80% mutant DNA (2).
Daarnaast accumuleren postmitotische cellen zoals neuronen meer mutante mtDNA kopieën
over het verloop van de tijd waardoor mitochondriale ziekten vaker tot uiting komen op latere
leeftijd (7).
Een combinatie van de maternele overerving, de principes van heteroplasmie, replicatieve
segregatie en de kritische mutatiedrempel, zorgt voor een unieke overerving en penetratie van
primaire mitochondriale mutaties (7).
4.2.1.5. MITOCHONDRIALE DNA MUTATIES IN LHON
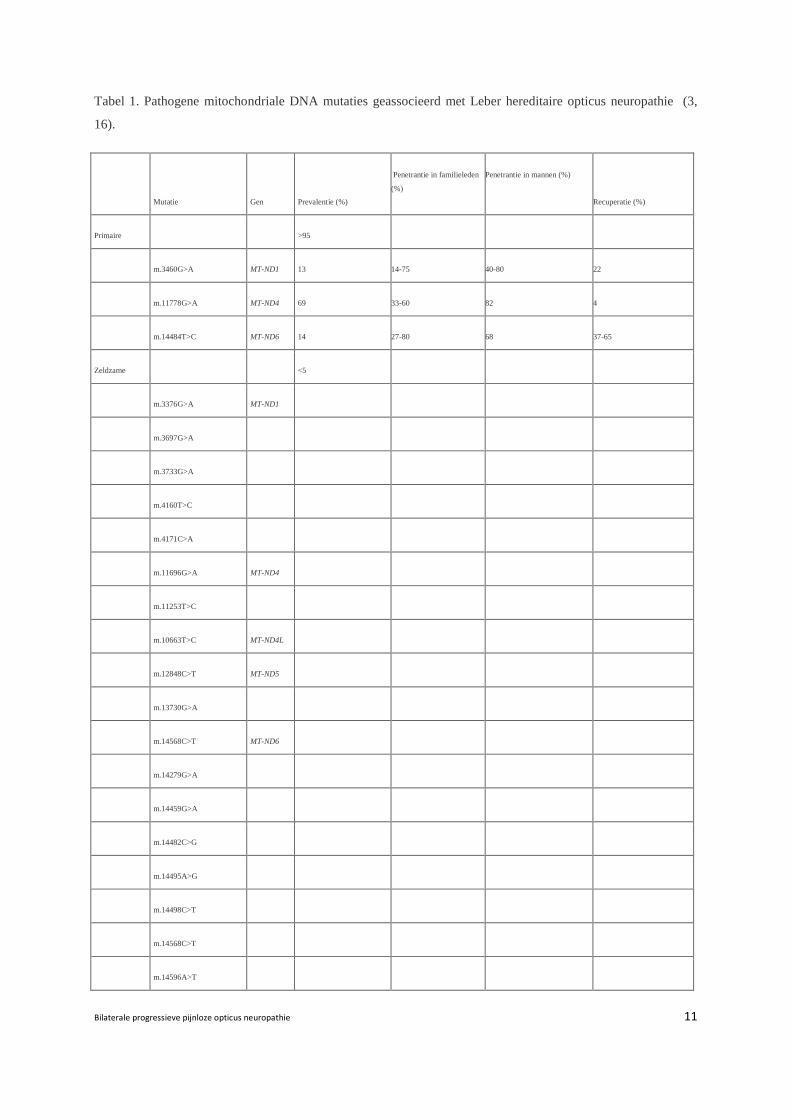
90-95% van de patiënten met LHON hebben één van de drie primaire puntmutaties in hun
mitochondriale DNA: m.3460G>A, m.11778G>A, m.14484T>C (2) (Tabel 1). De mutatie op
nucleotide positie 11778 werd als eerste geïdentificeerd door Wallace et al. in 1988 (6) en is
de meest prevalente LHON mutatie (70%) in Noord-Europa, Australië en het Verre Oosten (2,
7, 10-12). De m.14484T>C is de meest voorkomende mutatie (87%) bij Frans Canadezen als
gevolg van de kolonisatie (13). Alle primaire puntmutaties liggen in genen die de subunits
voor complex I van de ‘respiratory chain’ coderen, vooral in de ND1 en ND6 subunits (7, 14,
15).
Bilaterale progressieve pijnloze opticus neuropathie 11
Tabel 1. Pathogene mitochondriale DNA mutaties geassocieerd met Leber hereditaire opticus neuropathie (3,
16).
Mutatie Gen Prevalentie (%)
Penetrantie in familieleden
(%)
Penetrantie in mannen (%)
Recuperatie (%)
Primaire >95
m.3460G>A MT-ND1 13 14-75 40-80 22
m.11778G>A MT-ND4 69 33-60 82 4
m.14484T>C MT-ND6 14 27-80 68 37-65
Zeldzame <5
m.3376G>A MT-ND1
m.3697G>A
m.3733G>A
m.4160T>C
m.4171C>A
m.11696G>A MT-ND4
m.11253T>C
m.10663T>C MT-ND4L
m.12848C>T MT-ND5
m.13730G>A
m.14568C>T MT-ND6
m.14279G>A
m.14459G>A
m.14482C>G
m.14495A>G
m.14498C>T
m.14568C>T
m.14596A>T

Bilaterale progressieve pijnloze opticus neuropathie 12
4.2.1.6. INCOMPLETE PENETRANTIE VAN LHON
In 80-90% van de LHON patiënten wordt een homoplasmie van de pathogene mitochondriale
mutatie aangetroffen (3, 7). Een bijzondere eigenschap van LHON is echter dat slechts
ongeveer 50% van de mannen en ongeveer 10% van de vrouwen die drager zijn van één van
de drie primaire mitochondriale mutaties de ziekte tot expressie zullen brengen (3, 7). De
mitochondriale mutatie is dus noodzakelijk, maar niet voldoende om de pathologie tot
expressie te brengen (8). De mechanismen voor deze incomplete penetrantie en de mannelijke
predominantie bij LHON zijn nog niet volledig ontrafeld, maar complexe genetische- en
omgevingsfactoren zouden een rol spelen (Fig. 6) (3, 7).
Figuur 6. Secundaire factoren die kunnen interageren met de primaire mitochondriale DNA mutaties bij LHON
en zo tot visusverlies leiden (3). ATP, adenosine trifosfaat, ROS reactive oxygen species.
Als additionele genetische determinant wordt onder andere gedacht aan nucleaire
modificerende genen (8). In 2007 toonde Hudson et al. in een meta-analyse van 159 Europese
LHON stambomen aan dat het risico op visusverlies bij de drie primaire LHON mutaties
beïnvloed wordt door de mitochondriale DNA achtergrond of haplogroep (17).
Omdat mitochondriaal DNA materneel overgeërfd wordt en dus niet recombineert, zijn
mitochondriale polymorfismen geaccumuleerd langs vrouwelijke afstammingslijnen

Bilaterale progressieve pijnloze opticus neuropathie 13
duizenden jaren geleden (3). Als gevolg van deze evolutie ontstonden bepaalde stabiele
polymorfische varianten geclusterd in specifieke combinaties namelijk haplogroepen (3). De
Europese bevolking behoort tot één van de negen volgende haplogroepen: H, I, J, K, T, U, V,
W en X (3). Dragers van de m.11778G>A en de m.14484T>C mutaties bleken meer risico op
visusverlies te hebben wanneer ze behoorden tot haplogroep J (17). Daarentegen hadden
dragers van de m.3460G>A mutaties meer kans op een opticus neuropathie wanneer ze
behoorden tot haplogroep K (17). Een combinatie van haplogroep H en de m.11778G>A
mutatie had dan weer een lager risico op visusverlies (17). Ondanks deze resultaten, kon een
studie van Zuidoost Aziatische LHON stambomen geen associatie aantonen tussen specifieke
mtDNA haplogroepen en het risico op visusverlies (18).
Een andere hypothese die de mannelijke predominantie zou moeten verklaren is het bestaan
van een recessief X-gebonden susceptibiliteitsgen (3, 7). Daarbij zouden de vrouwelijke
draagsters die visusverlies ervaren ofwel homozygoot (40%) zijn voor de X-locus of
heterozygoot maar met een ongelukkige X-chromosoom inactivatie (60%) van het wild-type
allel (3, 7). Er zijn echter verschillende studies die deze X-chromosoom inactivatie in
vrouwelijke draagsters nog niet konden aantonen (3, 19-21). Twee studies vonden door
middel van linkage analyses een regio op de lange arm van het X-chromosoom (22, 23) doch
een exact causatief gen is nog niet geïdentificeerd (3, 7).
De geslachtsbias daarentegen zou ook kunnen resulteren uit een combinatie van anatomische,
hormonale en fysiologische verschillen tussen mannen en vrouwen (3). Zo zouden
oestrogenen de mitochondriale dysfunctie in LHON kunnen verbeteren door het teveel aan
vrije zuurstofradicalen tegen te gaan (24).
Tot slot werd een verschil in mitochondriale massa ook reeds beschreven als mogelijke
verklaring voor de incomplete penetrantie van LHON (7). Zo zouden onaangetaste dragers
een significant hoger kopijenaantal hebben van hun mitochondriaal DNA vergeleken met
aangetaste dragers (7, 25). Een vergelijking van fibroblasten van onaangetaste dragers,
aangetaste dragers en controlepatiënten toonde aan dat cellen van onaangetaste dragers meer
mitochondriale transcripten, eiwitten en enzymactiviteit tot expressie brengen vergeleken met
controles en aangetaste dragers (7, 25).
In de zoektocht naar de rol van omgevingsfactoren in de expressie van LHON, werden
observationele studies met twee paar monozygote tweelingen beschreven (26, 27). Daarbij

Bilaterale progressieve pijnloze opticus neuropathie 14
zou 1 zus of broer geen visusverlies ervaren hebben na langdurige opvolging. Verschillende
andere triggerende omgevingsfactoren zoals hoofdtrauma, psychologische stress,
werkgerelateerde blootstelling aan chemische toxines, antiretrovirale middelen en
antituberculostatica werden genoteerd in meestal klinische rapporteringen (2). Een causaal
verband is dus niet altijd met zekerheid vast te stellen.
4.2.1.7. EPIDEMIOLOGIE
LHON komt voor bij 1 op 31.000 mensen in het noordoosten van Engeland (28).
Gelijkaardige cijfers werden beschreven in andere Kaukasische populaties (3). In Nederland
bedraagt de LHON prevalentie 1 op 39.000 (29). In Finland is dit 1 op 50.000 (30).
4.2.1.8. KLINISCHE MANIFESTATIES EN PROGNOSE
De symptomatologie begint karakteristiek met een (sub)acuut, pijnloos verlies van het
centrale zicht in één oog (2, 7). Bilateraal visusverlies als initiële klacht is aanwezig in 25%
(2). Bij unilaterale gevallen is het tweede oog vaak betrokken binnen zes tot acht weken (2).
Binnen het jaar heeft 97% visusverlies in het tweede oog (7). De meerderheid van de
patiënten krijgen symptomen in de tweede of derde levensdecade (2). Visusdaling kan echter
optreden op elk moment gedurende de eerste tot zevende levensdecade (2). Het visusverlies
verslecht na de acute fase progressief over een periode van vier tot zes weken met uiteindelijk
ernstig visusverlies tot 6/60 of slechter (2). Deze visusdaling gaat ook gepaard met
kleurzinafwijkingen en een dens centraal of cecocentraal scotoom op gezichtsveld (2). De
pupilreflexen blijven meestal intact door de relatief mindere atrofie van de melanopsine
bevattende retinale ganglioncellen (7). Geslacht of aard van het mutatietype zou geen
significante invloed hebben op de timing of initiële ernst van het visusverlies (2).
In het acute stadium kan er in fundo vasculaire tortuositeit van de centrale retinale vaten,
zwelling van de retinale zenuwvezellaag en een telangieëctatische microangiopathie rondom
de papil gezien worden (2). Echter in 20% van de LHON gevallen ziet de nervus opticus er
volledig normaal uit (2). Deze patiënten worden vaak gecategoriseerd als functioneel
visusverlies (2).
Na zes weken treedt er bleekheid van de nervus opticus op (2). Initieel is dit temporaal meer
uitgesproken als gevolg van het axonenverlies in de papillomaculaire bundel (2).

Bilaterale progressieve pijnloze opticus neuropathie 15
Pathologische cupping van de papil kan gezien worden bij meer uitgesproken verlies van de
retinale ganglion cel axonen (2).
Visuele recuperatie is minimaal en afhankelijk van het mutatietype (2). De kans op enige
visuele recuperatie is het grootst bij de m.14484T>C mutatie (37-58%) en het minst bij de
m.11778G>A mutatie (4%) (Tabel 1) (2, 7).
Naast visusverlies zijn er nog andere extra-oculaire manifestaties mogelijk bij LHON, met
name cardiale arythmieën en neurologische afwijkingen zoals perifere neuropathie,
myopathie, dystonie en myoclonus (2). Deze fenotypes worden ‘LHON-plus syndromen’
genoemd en worden gelinkt aan mtDNA puntmutaties verschillend van de drie primaire
puntmutaties bij LHON (7).
Het selectieve verlies van vooral de retinale ganglioncellen in LHON is opmerkelijk (3). De
papillomaculaire bundel verantwoordelijk voor het centrale zicht wordt het eerst en meest
ernstig aangetast als gevolg van zijn dunne vezels en grote afhankelijkheid aan mitochondria
(7, 31). Het is nog onduidelijk waarom retinale ganglioncellen afsterven, maar men denkt aan
een lage ATP-productie, toegenomen productie van vrije zuurstofradicalen of een combinatie
van deze twee mechanismen (7).
4.2.1.9. BEHANDELING
De behandeling van mitochondriale ziekten in het algemeen en LHON in het bijzonder staat
op heden nog in zijn kinderschoenen (7). Er zijn geen algemeen aanvaarde therapieën die de
ziekte preventief tegengaan of kunnen uitstellen (3). Verschillende vitaminepreparaten (bv.
B2, B3, B12, C, E en foliumzuur) en andere supplementen blijken weinig effectief te zijn in
de behandeling van LHON (7). De voornaamste pijler in de behandeling van LHON bestaat
uit een supportieve behandeling (7). Visuele hulpmiddelen zijn bijvoorbeeld belangrijk voor
patiënten met centraal visusverlies. Daarnaast wordt zoveel mogelijk aangeraden om
schadelijke omgevingsfactoren te vermijden (7). Dit laatste heeft vooral betrekking op
rookstop, vermijden van zware alcoholabusus, vermijden van medicaties met mitochondriale
toxiciteit en blootstelling aan omgevingstoxines (7). Verder is een screening met
electrocardiogram en neurologisch onderzoek ter opsporing van extra-oculaire manifestaties
van LHON aangewezen (7).

Bilaterale progressieve pijnloze opticus neuropathie 16
Wat de rookstop betreft, is naast het peilen naar de rookstopmotivatie, helpen opstellen van
een rookstopplan en follow-upplan, het ook belangrijk om farmacologische hulp aan te bieden
(32). Alle vormen van nicotinesubstitutie bevorderen het rookstoppercentage en worden
aanbevolen bij alle nicotineafhankelijke rokers die gemotiveerd zijn om te stoppen met roken
(32). Ook bupropion is bewezen doeltreffend bij rookstop (32). Daarnaast moet de
begeleiding langdurig zijn omdat het risico op herval zeer groot is in de loop van het eerste
jaar (32).
Ook het aanpakken van problematisch alcoholgebruik is belangrijk en heeft, zoals bij
rookstop, alleen zin bij gemotiveerde patiënten (33). In de terugvalpreventie van
alcoholabusus is psychosociale ondersteuning met actieve deelname aan zelfhulpgroepen de
belangrijkste factor (34). Geneesmiddelen zoals acamprosaat, naltrexon en nalmefeen hebben
slechts een bescheiden effect in het behoud van alcoholabstinentie (34). Disulfiram wordt
gebruikt als aversietherapie zonder veel evidentie (34). Eén van de ernstige bijwerkingen bij
het gebruik van disulfiram is echter het ontstaan van een opticus neuropathie (34). Dit wordt
ook nog verder besproken bij het deel ‘Toxische opticus neuropathieën’.
4.2.1.9.1. UBIQUINONE ANALOGEN
Recent zijn er nieuwe studies die zich richten op de interessante mitochondriale cofactor
ubiquinone. Ubiquinone is gesitueerd in de mitochondriale membraan en is verantwoordelijk
voor het transport van electronen van de mitochondriale eiwitcomplexen I en II naar complex
III (7). Aangezien de drie primaire LHON-puntmutaties de functie aantasten van
eiwitcomplex I denkt men dat het ubiquinone analoog coenzyme Q10 de flow van electronen
naar complex III kan verbeteren (7). Coenzyme Q10 is echter moeilijk in de mitochondria te
krijgen door middel van orale toediening als gevolg van zijn uitgesproken lipofiele karakter
(7).
Er bestaan evenwel ubiquinone analogen met korte ketens zoals idebenone en EPI-743 die
beter penetreren in de mitochondria (7). De gerandomiseerde, dubbelblinde, placebo
gecontroleerde studie RHODOS (Rescue of Hereditary Optic Disease Outpatient Study)
onderzocht het effect van een behandeling met idebenone bij 85 LHON patiënten (35). Na een
behandeling van 24 weken met 900 mg idebenone per dag, bleek er een positieve trend te zijn
in de secundaire eindpunten namelijk verandering in best gecorrigeerde visus vergeleken met
baseline en verandering in visus voor beide ogen (35). Uit de observationele follow-up studie

Bilaterale progressieve pijnloze opticus neuropathie 17
(RHODOS-OFU) bij 59 van de oorspronkelijke patiënten kon men na 30 maanden nog
voordelige effecten zien bij degenen die behandeld waren met idebenone (36). Nog twee
andere studies konden respectievelijk een verbetering in zicht en een verbetering in blauw-
geel kleurenzicht vaststellen (37, 38). In de toekomst is wel nog verder onderzoek nodig om
de meest efficiënte dosis en behandelingsduur van idebenone te bepalen (7).
4.2.1.9.2. GENTHERAPIE
Gentherapie zou een veelbelovende optie zijn voor mitochondriale aandoeningen en dan
vooral voor LHON, gezien de goede bereikbaarheid van de retinale ganglioncellaag (7). De
dubbele membraan van mitochondria maakt het echter niet gemakkelijk om genen in het
mitochondriaal genoom te incorporeren (7). Om dit probleem te omzeilen, wordt de techniek
van nucleaire allotopische expressie van een mitochondriaal gen gebruikt (7). Hierbij wordt
door middel van een virale vector het desbetreffende gen (bij LHON het ND4 gen) in het
nucleaire genoom geïntegreerd (7). Het eiwit kan dan daarna naar de mitochondria
getransporteerd worden en zijn werking opnieuw uitvoeren (7). Op heden worden de eerste
klinische trials nog maar opgestart om de veiligheid van deze techniek na te gaan (7).
4.2.1.10. GENETISCHE COUNSELING
Door de hierboven reeds beschreven incomplete penetrantie bij LHON, is het onmogelijk om
exact te voorspellen of en wanneer dragers van de LHON-mutaties symptomen zullen krijgen
(3). De twee belangrijkste predictieve factoren voor visusverlies blijven geslacht en leeftijd
(3). De kans om symptomen te krijgen daalt met stijgende leeftijd en wordt minimaal na de
leeftijd van 50 jaar (3).
4.2.2. AUTOSOMAAL DOMINANTE OPTICUS ATROFIE (ADOA)
4.2.2.1 EPIDEMIOLOGIE EN KLINISCHE MANIFESTATIES
De prevalentie van ADOA is ongeveer vergelijkbaar met LHON (2). Het visusverlies begint
insidieus in de eerste twee levensdecaden (2). De gemiddelde leeftijd waarop de
symptomatologie start is 6-10 jaar (2). ADOA heeft een milder fenotype vergeleken met
LHON (2). Bij één op vier patiënten zijn er geen visusklachten en wordt de opticusatrofie
toevallig ontdekt bij routine oogheelkundig onderzoek of naar aanleiding van een gekend
familielid met ADOA (2). De gemiddelde visusscores bedragen 6/18 tot 6/60, waarbij de
Bilaterale progressieve pijnloze opticus neuropathie 18
visus gaat van 6/6 tot handbewegingen (2). Er is een duidelijke inter- en intrafamiliale
variabiliteit wat de ernst van het visusverlies betreft (3) en alhoewel de visuele prognose bij
ADOA beter is dan bij LHON, voldoet de helft van de ADOA patiënten niet aan de wettelijke
vereisten voor het besturen van een wagen (2).
Klinisch hebben de meeste patiënten een veralgemeende dyschromatopsia (2). De meest
voorkomende gezichtsvelddefecten zijn centrale, caecocentrale en paracentrale scotomen met
een bewaard perifeer veld als gevolg van de primaire aantasting van de papillomaculaire
bundel (2). Zoals bij LHON zijn de pupilreflexen bewaard (2). De bleekheid van de nervus
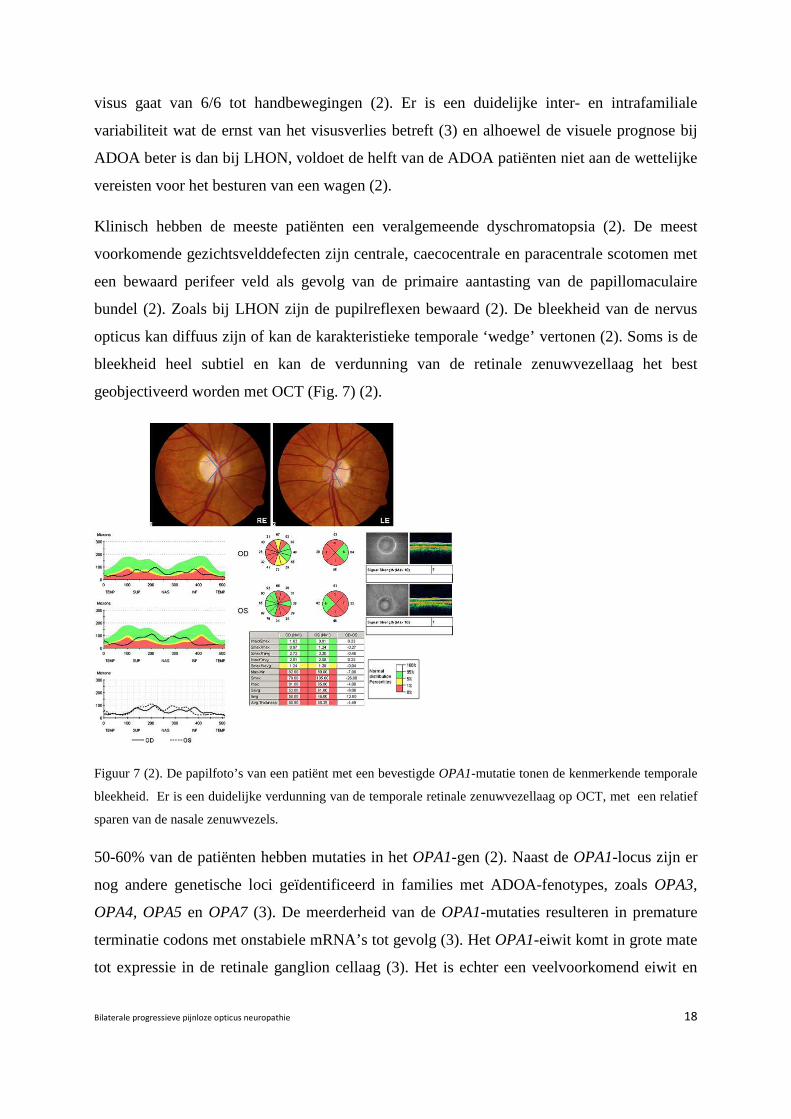
opticus kan diffuus zijn of kan de karakteristieke temporale ‘wedge’ vertonen (2). Soms is de
bleekheid heel subtiel en kan de verdunning van de retinale zenuwvezellaag het best
geobjectiveerd worden met OCT (Fig. 7) (2).
Figuur 7 (2). De papilfoto’s van een patiënt met een bevestigde OPA1-mutatie tonen de kenmerkende temporale
bleekheid. Er is een duidelijke verdunning van de temporale retinale zenuwvezellaag op OCT, met een relatief
sparen van de nasale zenuwvezels.
50-60% van de patiënten hebben mutaties in het OPA1-gen (2). Naast de OPA1-locus zijn er
nog andere genetische loci geïdentificeerd in families met ADOA-fenotypes, zoals OPA3,
OPA4, OPA5 en OPA7 (3). De meerderheid van de OPA1-mutaties resulteren in premature
terminatie codons met onstabiele mRNA’s tot gevolg (3). Het OPA1-eiwit komt in grote mate
tot expressie in de retinale ganglion cellaag (3). Het is echter een veelvoorkomend eiwit en

Bilaterale progressieve pijnloze opticus neuropathie 19
grote hoeveelheden zijn ook al geïdentificeerd in fotoreceptoren en andere niet-oculaire
weefsels zoals het binnenoor en de hersenen (3). OPA1 is een transmembranair mechano-
enzyme gelokaliseerd ter hoogte van de binnenste mitochondriale membraan (3). OPA1-
mutaties leiden tot mitochondriale fragmentatie en deficiënte oxidatieve fosforylering (3).
Recent is ook duidelijk geworden dat OPA1 een cruciale rol speelt in het bewaren van de
integriteit van het mitochondriale genoom (3).
4.2.2.2. EXTRA-OCULAIRE MANIFESTATIES
Zoals LHON+-syndromen, bestaan er ook ADOA+-fenotypes waarbij de meest voorkomende
extra-oculaire manifestatie bilaterale neurosensoriële doofheid is (3). Andere klinische
tekenen zoals ataxia, myopathie, perifere neuropathie en chronisch progressieve externe
oftalmoplegie (CPEO) werden ook beschreven (3). De visuele prognose bij het ADOA+-
fenotype is veel slechter (2).
4.2.2.3. BEHANDELING
Er is op heden nog geen behandeling die de natuurlijke evolutie van ADOA beïnvloedt (3).
Zoals bij LHON is de behandeling supportief (3). Ondanks de autosomaal dominante
overerving is genetische counseling voor mutatiedragers moeilijk en dit door de uitgesproken
intra- en interfamiliale variabiliteit (3).
4.2.3. TOXISCHE OPTICUS NEUROPATHIEËN
4.2.3.1. INLEIDING
De oogzenuw is gevoelig voor schade veroorzaakt door toxische substanties zoals drugs,
metalen, organische solventen, methanol, koolstofdioxide en tabak (1). Een toxische opticus
neuropathie kan getriggerd of versterkt worden door voedingsdeficieten (1). Het is algemeen
aanvaard dat toxische producten mitochondriale schade induceren en een onevenwicht creëren
in de homeostase van intracellulaire en extracellulaire vrije radicalen (1). Dit verklaart de
gelijkenis met LHON en is ook de reden dat men toxische opticus neuropathieën (TON)
verworven mitochondriale opticus neuropathieën noemt (1).

Bilaterale progressieve pijnloze opticus neuropathie 20
4.2.3.2. ROKEN, ALCOHOL EN NUTRITIONELE DEPRIVATIE
Patiënten met excessief alcohol- en tabaksmisbruik kunnen een bilaterale opticus neuropathie
ontwikkelen met traag progressief visusverlies, dyschromatopsieën en caecocentrale scotomen
(39). Een vroegere en foute benaming voor deze entiteit was ‘tobacco-alcohol amblypia’(39).
Het is mogelijk dat alcohol en tabak niet de enige causale factoren zijn die leiden tot een
toxisch effect op de retinale ganglioncellen. Ook gelijktijdige malnutritie en vitamine
deficiënties (thiamine (B1), cyanocobalamine (B12), riboflavine (B2), pyridoxine (B6),
foliumzuur (B11)) kunnen bijdragen tot de pathologie (1, 39). Vitamine deficiënties vooral
van vitamine B12 komen vaak voor bij patiënten met ernstige alcoholabusus. Interessant
genoeg werden bij patiënten met tobacco-alcohol opticus neuropathie mtDNA LHON
mutaties gevonden (40). Daarnaast blijkt roken een sterke risicofactor te zijn voor visusverlies
bij LHON mutatiedragers (41). Een grote Braziliaanse studie met 332 LHON patiënten toonde
een verdubbeling aan van het risico op LHON symptomen wanneer er een hoge consumptie
was van alcohol of tabak (1, 41, 42). Een andere studie toonde aan dat het effect van roken op
het risico op visusverlies dosisafhankelijk was waarbij het risico groter was bij hevige rokers
(43). Er zou ook een verhoogd risico op visusverlies bij ernstige alcoholgebruikers zijn, doch
deze associatie was niet zo sterk als bij rokers (43).
4.2.3.3. TOXISCHE OPTICUS NEUROPATHIE
TON is duur- en dosisafhankelijk (1). Het komt vaker voor bij behandelingen met
antituberculostatica (ethambutol en isoniazide), sommige antibiotica (linezolid, ciprofloxacin,
cimetidine en chlooramphenicol), antiepileptica (vigabatrin), disulfiram, gehalogeneerde
hydroquinolones, antimetabolieten (methotrexaat, cisplatin, carboplatin, vincristine en
cyclosporine), tamoxifen en sildenafil (1). In de volgende alinea’s zullen enkele frequentere
toxische producten meer in detail besproken worden.
4.2.3.3.1. ETHAMBUTOL
Opticus neuropathie is één van de meest ernstige bijwerkingen gerelateerd aan ethambutol-
inname en komt voor bij ongeveer 6% van de patiënten (1). De klinische symptomen zijn
gelijkaardig aan andere toxische opticus neuropathieën en omvatten vroegtijdige
dyschromatopsieën en gezichtsvelddefecten (1). Visusproblemen treden in het algemeen op
tussen de 4e en 12e maand na de start van de behandeling (1). De oculaire toxiciteit is duur –

Bilaterale progressieve pijnloze opticus neuropathie 21
en dosisafhankelijk (1). Vroegtijdige detectie van TON en onmiddellijk stoppen van de
medicatie zijn belangrijk om verder visusverlies te voorkomen (1).
4.2.3.3.2. METHANOL
Methanol wordt gemetaboliseerd tot de toxische component mierenzuur (1). De toxiciteit
wordt veroorzaakt door een combinatie van metabole acidose en de intrinsieke toxiciteit van
het formiaat anion zelf (1). Methanol veroorzaakt niet alleen schade aan de oogzenuw, maar
ook aan de retina (1). Oogsymptomen komen voor bij 50% van de patiënten (1). Ze kunnen
optreden vanaf zes uur na de inname (1). Methanol vergiftiging is levensbedreigend (1). De
prognose is eerder afhankelijk van de ernst van de acidose dan van de serumconcentraties van
methanol (1).
4.2.3.3.3. VIGABATRIN
Vigabatrin is een antiepilepticum waarbij gezichtsvelddefecten voorkomen bij 15-31% van de
baby’s, 15% van de kinderen en 25-30% van de volwassenen (44). In het begin stelt men
vooral bilaterale nasale gezichtsvelddefecten vast (1). Deze evolueren progressief naar
bilateraal concentrisch vernauwde gezichtsvelden met een bewaard centraal zicht en kunnen
irreversibel zijn (1). Het optreden van de gezichtsvelddefecten is leeftijds- en duurafhankelijk:
3 maand bij baby’s, 11 maand bij kinderen en 9 maand bij volwassenen (44).
4.2.3.3.4. DISULFIRAM (ANTABUSE®)
Disulfiram wordt gebruikt als aversietherapie bij alcoholontwenning (34). Het remt de afbraak
van alcohol met als gevolg hiervan een accumulatie aan acetaldehyde (45). Bilaterale opticus
neuropathie werd beschreven bij een patiënt die disulfiram nam voor alcoholontwenning (45).
De opticus neuropathie verdween bij het stopzetten van de behandeling en verscheen opnieuw
wanneer de behandeling herstart werd (45). Men moet er dus bedacht op zijn dat perifere
neuropathieën en opticus neuropathieën bij alcoholici soms optreden als gevolg van een
toxisch effect van behandeling met disulfiram eerder dan van de alcohol zelf (46). Bij direct
stoppen van de medicatie is de opticus neuropathie reversibel (45).

Bilaterale progressieve pijnloze opticus neuropathie 22
V. DISCUSSIE
Leber hereditaire opticus neuropathie is de meest voorkomende mitochondriale overerfbare
ziekte. Het is een uitsluitingsdiagnose met een brede differentiaal diagnose. Het progressieve
pijnloze bilaterale visusverlies gepaard gaande met een normale fundoscopie in 20% van de
patiënten kan leiden tot de verkeerde diagnose van functioneel visusverlies. De diagnose kan
enkel bevestigd worden door moleculair genetisch onderzoek.
Verder blijft het een groot vraagteken waarom nu juist de retinale ganglioncellen afsterven bij
LHON. Andere mitochondriale ziekten met mutaties die veel ernstigere dysfuncties van de
oxidatieve fosforylering veroorzaken, leiden niet allemaal tot opticus atrofie.
Daarnaast is er een interessante overlap in klinische symptomatologie tussen hereditaire
opticus neuropathieën en toxische opticus neuropathieën. Sommigen denken dat
verschillende (toxische) omgevingsfactoren een trigger kunnen zijn voor het tot expressie
komen van subklinische LHON patiënten.
LHON is dus op vele vlakken nog steeds een mysterieuze ziekte ondanks de eerste
beschrijving ervan in 1871. De overerving van LHON, die tot op heden nog niet volledig is
ontrafeld, is complex en verschillende factoren spelen een rol. De laatste jaren werd al veel
vooruitgang geboekt zowel op moleculair genetisch, biochemisch als therapeutisch vlak. De
behandeling met idebenone lijkt veelbelovend, maar verdere studies zijn nodig om de
efficiëntste dosis en behandelingsduur te bepalen.
VI. REFERENTIELIJST
1. Grzybowski A, Zulsdorff M, Wilhelm H, Tonagel F. Toxic optic neuropathies: an updated review. Acta
Ophthalmol. 2015;93(5):402-10.
2. Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - Disease mechanisms and
therapeutic strategies. Progress in Retinal and Eye Research. 2011;30(2):81-114.
3. Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. J Med
Genet. 2009;46(3):145-58.
4. Leber T. Ueber hereditaere und congenital angelegte sehnervenleiden. . Graefes Arch Ophthal.
1871;17:249-91.
5. Piotrowska A, Korwin M, Bartnik E, Tonska K. Leber hereditary optic neuropathy - Historical report in
comparison with the current knowledge. Gene. 2015;555(1):41-9.

Bilaterale progressieve pijnloze opticus neuropathie 23
6. Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, et al. Mitochondrial DNA mutation
associated with Leber's hereditary optic neuropathy. Science. 1988;242(4884):1427-30.
7. Meyerson C, Van Stavern G, McClelland C. Leber hereditary optic neuropathy: current perspectives.
Clin Ophthalmol. 2015;9:1165-76.
8. Maresca A, la Morgia C, Caporali L, Valentino ML, Carelli V. The optic nerve: A "mito-window" on
mitochondrial neurodegeneration. Molecular and Cellular Neuroscience. 2013;55:62-76.
9. Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm
mitochondria. Nature. 1999;402(6760):371-2.
10. Mackey DA, Oostra RJ, Rosenberg T, Nikoskelainen E, Bronte-Stewart J, Poulton J, et al. Primary
pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum
Genet. 1996;59(2):481-5.
11. Mashima Y, Kigasawa K, Hasegawa H, Tani M, Oguchi Y. High incidence of pre-excitation syndrome in
Japanese families with Leber's hereditary optic neuropathy. Clin Genet. 1996;50(6):535-7.
12. Yen MY, Wang AG, Chang WL, Hsu WM, Liu JH, Wei YH. Leber's hereditary optic neuropathy--the
spectrum of mitochondrial DNA mutations in Chinese patients. Jpn J Ophthalmol. 2002;46(1):45-51.
13. Macmillan C, Kirkham T, Fu K, Allison V, Andermann E, Chitayat D, et al. Pedigree analysis of French
Canadian families with T14484C Leber's hereditary optic neuropathy. Neurology. 1998;50(2):417-22.
14. Valentino ML, Barboni P, Ghelli A, Bucchi L, Rengo C, Achilli A, et al. The ND1 gene of complex I is a
mutational hot spot for Leber's hereditary optic neuropathy. Ann Neurol. 2004;56(5):631-41.
15. Chinnery PF, Brown DT, Andrews RM, Singh-Kler R, Riordan-Eva P, Lindley J, et al. The mitochondrial
ND6 gene is a hot spot for mutations that cause Leber's hereditary optic neuropathy. Brain. 2001;124(Pt
1):209-18.
16. MITOMAP A human mitochondrial genome database. Leber's hereditary optic neuropathy (LHON)
disease mutations. Online 15 september 2015;Opgehaald op 20 maart 2016 van http://www.mitomap.org.
17. Hudson G, Carelli V, Spruijt L, Gerards M, Mowbray C, Achilli A, et al. Clinical expression of Leber
hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet.
2007;81(2):228-33.
18. Tharaphan P, Chuenkongkaew WL, Luangtrakool K, Sanpachudayan T, Suktitipat B, Suphavilai R, et al.
Mitochondrial DNA haplogroup distribution in pedigrees of Southeast Asian G11778A Leber hereditary optic
neuropathy. J Neuroophthalmol. 2006;26(4):264-7.
19. Hudson G, Carelli V, Horvath R, Zeviani M, Smeets HJ, Chinnery PF. X-Inactivation patterns in females
harboring mtDNA mutations that cause Leber hereditary optic neuropathy. Mol Vis. 2007;13:2339-43.
20. Oostra RJ, Kemp S, Bolhuis PA, Bleeker-Wagemakers EM. No evidence for 'skewed' inactivation of the
X-chromosome as cause of Leber's hereditary optic neuropathy in female carriers. Hum Genet. 1996;97(4):500-
5.
21. Pegoraro E, Carelli V, Zeviani M, Cortelli P, Montagna P, Barboni P, et al. X-inactivation patterns in
female Leber's hereditary optic neuropathy patients do not support a strong X-linked determinant. Am J Med
Genet. 1996;61(4):356-62.
22. Hudson G, Keers S, Yu Wai Man P, Griffiths P, Huoponen K, Savontaus ML, et al. Identification of an X-
chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am J Hum
Genet. 2005;77(6):1086-91.

Bilaterale progressieve pijnloze opticus neuropathie 24
23. Shankar SP, Fingert JH, Carelli V, Valentino ML, King TM, Daiger SP, et al. Evidence for a novel x-linked
modifier locus for leber hereditary optic neuropathy. Ophthalmic Genet. 2008;29(1):17-24.
24. Giordano C, Montopoli M, Perli E, Orlandi M, Fantin M, Ross-Cisneros FN, et al. Oestrogens ameliorate
mitochondrial dysfunction in Leber's hereditary optic neuropathy. Brain. 2011;134(Pt 1):220-34.
25. Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, et al. Efficient mitochondrial
biogenesis drives incomplete penetrance in Leber's hereditary optic neuropathy. Brain. 2014;137(Pt 2):335-53.
26. Johns DR, Smith KH, Miller NR, Sulewski ME, Bias WB. Identical twins who are discordant for Leber's
hereditary optic neuropathy. Arch Ophthalmol. 1993;111(11):1491-4.
27. Biousse V, Brown MD, Newman NJ, Allen JC, Rosenfeld J, Meola G, et al. De novo 14484 mitochondrial
DNA mutation in monozygotic twins discordant for Leber's hereditary optic neuropathy. Neurology.
1997;49(4):1136-8.
28. Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF. The epidemiology of Leber
hereditary optic neuropathy in the North East of England. Am J Hum Genet. 2003;72(2):333-9.
29. Spruijt L, Kolbach DN, de Coo RF, Plomp AS, Bauer NJ, Smeets HJ, et al. Influence of mutation type on
clinical expression of Leber hereditary optic neuropathy. Am J Ophthalmol. 2006;141(4):676-82.
30. Puomila A, Hamalainen P, Kivioja S, Savontaus ML, Koivumaki S, Huoponen K, et al. Epidemiology and
penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet. 2007;15(10):1079-89.
31. Sadun AA, Win PH, Ross-Cisneros FN, Walker SO, Carelli V. Leber's hereditary optic neuropathy
differentially affects smaller axons in the optic nerve. Trans Am Ophthalmol Soc. 2000;98:223-32; discussion
32-5.
32. Gailly J. Stoppen met roken. Huisarts Nu. 2006;35(7):395-425.
33. Folia Pharmacotherapeutica. Geneesmiddelen bij alcoholmisbruik en alcoholafhankelijkheid deel 1: de
alcoholontwenning. Online maart 2016;Opgehaald op 24 april 2016 vanaf http://www.bcfi.be.
34. Folia Pharmacotherapeutica. Geneesmiddelen bij alcoholmisbruik en alcoholafhankelijkheid deel 2: de
terugvalpreventie. Online april 2016;Opgehaald 24 april 2016 vanaf http://www.bcfi.be.
35. Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, et al. A randomized placebo-
controlled trial of idebenone in Leber's hereditary optic neuropathy. Brain. 2011;134(Pt 9):2677-86.
36. Klopstock T, Metz G, Yu-Wai-Man P, Buchner B, Gallenmuller C, Bailie M, et al. Persistence of the
treatment effect of idebenone in Leber's hereditary optic neuropathy. Brain. 2013;136(Pt 2):e230.
37. Carelli V, La Morgia C, Valentino ML, Rizzo G, Carbonelli M, De Negri AM, et al. Idebenone treatment in
Leber's hereditary optic neuropathy. Brain. 2011;134(Pt 9):e188.
38. Rudolph G, Dimitriadis K, Buchner B, Heck S, Al-Tamami J, Seidensticker F, et al. Effects of idebenone
on color vision in patients with leber hereditary optic neuropathy. J Neuroophthalmol. 2013;33(1):30-6.
39. Chiotoroiu SM, Noaghi M, Stefaniu GI, Secureanu FA, Purcarea VL, Zemba M. Tobacco-alcohol optic
neuropathy--clinical challenges in diagnosis. J Med Life. 2014;7(4):472-6.
40. Amaral-Fernandes MS, Marcondes AM, Miranda PM, Maciel-Guerra AT, Sartorato EL. Mutations for
Leber hereditary optic neuropathy in patients with alcohol and tobacco optic neuropathy. Mol Vis.
2011;17:3175-9.
41. Sadun AA, Carelli V, Salomao SR, Berezovsky A, Quiros P, Sadun F, et al. A very large Brazilian pedigree
with 11778 Leber's hereditary optic neuropathy. Trans Am Ophthalmol Soc. 2002;100:169-78; discussion 78-9.

Bilaterale progressieve pijnloze opticus neuropathie 25
42. Sadun AA, Carelli V, Salomao SR, Berezovsky A, Quiros PA, Sadun F, et al. Extensive investigation of a
large Brazilian pedigree of 11778/haplogroup J Leber hereditary optic neuropathy. Am J Ophthalmol.
2003;136(2):231-8.
43. Kirkman MA, Yu-Wai-Man P, Korsten A, Leonhardt M, Dimitriadis K, De Coo IF, et al. Gene-
environment interactions in Leber hereditary optic neuropathy. Brain. 2009;132(Pt 9):2317-26.
44. Kedar S, Ghate D, Corbett JJ. Visual fields in neuro-ophthalmology. Indian J Ophthalmol.
2011;59(2):103-9.
45. Leibold JE. Drugs having a toxic effect on the optic nerve. Int Ophthalmol Clin. 1971;11(2):137-57.
46. Gardner-Thorpe C, Benjamin S. Peripheral neuropathy after disulfiram administration. J Neurol
Neurosurg Psychiatry. 1971;34(3):253-9.

Bilaterale progressieve pijnloze opticus neuropathie i
BIJLAGEN
I. Copyrighttoelating voor figuur 3
JOHN WILEY AND SONS LICENSE TERMS AND CONDITIONS Apr 23, 2016
This Agreement between Deborah De Bruyn ("You") and John Wiley and Sons ("John Wiley and Sons") consists of your license details and the terms and conditions provided by John Wiley and Sons and Copyright Clearance Center. License Number 3854820128474 License date Apr 23, 2016 Licensed Content Publisher
John Wiley and Sons
Licensed Content Publication
Acta Ophthalmologica
Licensed Content Title Toxic optic neuropathies: an updated review
Licensed Content Author Andrzej Grzybowski,Magdalena Zülsdorff,Helmut Wilhelm,Felix Tonagel
Licensed Content Date Aug 26, 2014 Pages 9 Type of use Dissertation/Thesis Requestor type University/Academic Format Print and electronic Portion Figure/table Number of figures/tables 1 Original Wiley figure/table number(s)
Figure 1
Will you be translating? Yes, without English rights Number of languages 1 Languages Dutch Title of your thesis / dissertation
Bilaterale progressieve pijnloze opticus neuropathie
Expected completion date
May 2016
Expected size (number of pages)
25
Requestor Location Deborah De Bruyn

Bilaterale progressieve pijnloze opticus neuropathie ii
Ottergemsesteenweg 446 Ghent, Belgium 9000 Attn: Deborah De Bruyn
Billing Type Invoice
Billing Address
Deborah De Bruyn Ottergemsesteenweg 446 Ghent, Belgium 9000 Attn: Deborah De Bruyn
Total 0.00 EUR
II. Copyrighttoelating voor figuur 4
Apr 23, 2016
This is an Agreement between Deborah De Bruyn ("You") and Elsevier ("Elsevier"). It consists of your order details, the terms and conditions provided by Elsevier, and the payment terms and conditions.
All payments must be made in full to CCC. For payment instructions, please see information listed at the bottom of this form.
Supplier Elsevier Limited The Boulevard,Langford Lane Kidlington,Oxford,OX5 1GB,UK
Registered Company Number
1982084
Customer name Deborah De Bruyn
Customer address Ottergemsesteenweg 446
Ghent, None 9000
License number 3830760007066
License date Mar 16, 2016
Licensed content publisher Elsevier
Licensed content publication Molecular and Cellular Neuroscience
Licensed content title The optic nerve: A “mito-window” on mitochondrial neurodegeneration
Licensed content author Alessandra Maresca,Chiara la Morgia,Leonardo Caporali,Maria Lucia Valentino,Valerio Carelli
Licensed content date July 2013
Licensed content volume 55

Bilaterale progressieve pijnloze opticus neuropathie iii
number
Licensed content issue number
n/a
Number of pages 15
Start Page 62
End Page 76
Type of Use reuse in a thesis/dissertation
Portion figures/tables/illustrations
Number of figures/tables/illustrations
1
Format both print and electronic
Are you the author of this Elsevier article?
No
Will you be translating? Yes
Number of languages 1
Languages Dutch
Original figure numbers figure 1
Title of your thesis/dissertation
Bilaterale progressieve pijnloze opticus neuropathie
Expected completion date May 2016
Estimated size (number of pages)
25
Elsevier VAT number GB 494 6272 12
Price 0.00 EUR
VAT/Local Sales Tax 0.00 EUR / 0.00 GBP
Total 0.00 EUR
III. Copyrighttoelating voor figuur 5 en figuur 7
Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - Disease mechanisms and therapeutic strategies. Progress in Retinal and Eye Research. 2011;30(2):81-114.
Open Access funded by Wellcome Trust
Under a Creative Commons license

Bilaterale progressieve pijnloze opticus neuropathie iv
Creative Commons Attribution License (CC BY)
This article is available under the terms of the Creative Commons Attribution License (CC BY). You may distribute and copy the article, create extracts, abstracts, and other revised versions, adaptations or derivative works of or from an article (such as a translation), to include in a collective work (such as an anthology), to text or data mine the article, including for commercial purposes without permission from Elsevier. The original work must always be appropriately credited. Permission is not required for this type of reuse.
IV. Copyrighttoelating voor figuur 6 en tabel 1
Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. J Med Genet. 2009;46(3):145-58.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Related Documents











