1 J ANUARY 2020 IVT Network Compiled by: Stacey Bruzzese Journal of Validation Technology Best of JVT 2019

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
JANUARY 2020
IVT Network
Compiled by: Stacey Bruzzese
Journal of Validation Technology Best of JVT 2019

2

3
2019 Best of JVT A Compilation of Peer Reviewed Journal Articles, Conference Compendia, Blog Posts and Podcasts
Our valued members recognize that our Journals provide much needed guidance and
regulatory insights for validating and remaining compliant while working in the medical
device, pharmaceutical and biotech industries.
Whether FDA regulations, EU GMP practices or Japanese GMP guidelines, the resources
provided in JVT and GXP publications allow professionals to stay current on existing and
emerging regulations, as well as learn from colleagues on best practices and audit
expectations.
Thanks to an expert team of authors, brilliantly lead by our Editorial Advisory Board, the
IVT Network content is always fresh and engaging. So, we are proud to provide this
"look back" on some of the best from 2019.
Unlock the Key to Validation Excellence
With IVT Network, members gain access to
innovative content, industry research, lifelong
learning and opportunities for networking
on a global level. IVT gives you the tools you
need to succeed in your profession.

4
Table of Contents
Journal of Validation Technology Best of JVT 2019 ....................................................................... 1
2019 Best of JVT Introduction ................................................................................................. 3
Pharmaceutical Industry: Best of .................................................................................................. 4
Periodic Review of Validated Systems ............................................................................. 4
Appendix A ............................................................................................................. 14
Appendix B ............................................................................................................. 21
Auditing And Assessing The Quality Control Laboratory ............................................................ 28
Cleaning Agent Selection And Cycle Development ..................................................................... 46
Computerized System Validation Industry: Best of ............................................................ 50
Agile Data-Centric Validation ........................................................................................ 50
Points to be Considered When Validating Big Data ..................................................................... 56
Medical Device Industry: Best of ................................................................................................. 61
Cleaning Validation for Medical Devices Exposed to Large Numbers of Processing
Agents ......................................................................................................................................... 61
Process Parameters and Range Settings for Medical Device Process Validation ......................... 66
PQ Forums: Best of ...................................................................................................................... 84
PQ Forum #12 – Validation Approvers and Documents .............................................................. 84
PQ Forum #13 – Validation Lexicon .............................................................................. 233
PQ Forum #14 - Numbers .............................................................................................. 23
Fan Favorites: Best of .................................................................................................................. 23
Liquid Cryongenic Storage Chamber Qualification ......................................................... 23
Effective Knowledge Transfer During Biopharmaceutical Technology Transfer ............. 120
Risk Considerations for the Use of Unidirectional Airflow Devices in Microbiology
Laboratories ................................................................................................................. 133
Bridging the Gaps in Data Integrity: Assessing Risk to Streamline Audit Trail Review .... 143

5
Pharmaceutical Industry
PERIODIC REVIEW OF VALIDATED SYSTEMS By: Donncadh J. Nagle, Validation Coordinator, Avara Pharmaceutical Services Ltd,
ABSTRACT The periodic review of validated systems in the pharmaceutical industry has come into the spotlight in recent
years due to an increased focus on data integrity.
A key fundamental of any pharmaceutical quality system is that companies must fully understand how their
critical data is performing. Evidence suggests that most companies do have reasonable periodic review
strategies in place for Computer System Validation and Process Validation. However, the challenge now exists
to ensure periodic review procedures also assess Equipment, Utilities, Cleaning and Analytical Method
validation.
This paper presents the results and findings of a research study which adopted the following research
methodologies:
• Literature review of the current regulations and guidelines relating to periodic review
• Interview with a HPRA regulator to gain insight into what the competent authorities have observed in
industry to-date with respect to periodic review and to gain some guidance on their expectations
• An industry study conducted with ten pharmaceutical companies (small molecule & large molecule).
The research conducted identified that there is a need within the pharmaceutical industry for more detailed
guidelines on what to assess during periodic reviews including instructions and how to conduct these reviews.
The author has developed the following templates to address this:
• Periodic review template for Computer System Validation (CSV)
• Periodic review template for Equipment and Utilities.
• Templates are appended and links to electronic versions are provided.
INTRODUCTION Compliance in the pharmaceutical industry is critical to business success, product quality and patient safety.
Periodic review programs are a key component of a company’s pharmaceutical quality system that help ensure
compliance. Periodic reviews should be used as an aid to check that validated systems remain compliant with
the appropriate quality and safety regulations as well as ensuring that the validated systems remain fit for
their intended use.
The International Society for Pharmaceutical Engineering (ISPE) define periodic review as follows: “a
documented assessment of documentation, procedures, records, and performance to ensure that facilities,
equipment, and systems continue to be fit for purpose” (1).
While the definition refers to periodic reviews as being a “documented assessment,” it is important that we do
not see periodic reviews as being routine exercises. Instead we must look to implementing effective periodic

6
review programs that can be compiled efficiently, that add value to the business, give real-time feedback on
critical quality metrics, and provide important business information.
This paper summarizes the results of an extensive research program which investigated periodic review of
validated systems within the pharmaceutical industry. This research was conducted in conjunction with the
Technological University Dublin (TU Dublin) as part of the Master of Science program in Pharmaceutical
Validation Technology (2). Research was conducted between May and November 2018 with the support of TU
Dublin and BioPharmaChem Ireland (BPCI) (3) using the following research methodology:
• Literature review of the current regulations and guidelines relating to periodic review
• An interview with a HPRA regulator carried out to get an insight into what the competent authorities
have observed in industry to date with respect to Periodic Review of Validated Systems, and to gain
insight into regulatory expectations.
• An industry study conducted with ten pharmaceutical companies (small molecule & large molecule).
In addition, this research demonstrates how effective collaboration within the pharmaceutical industry can
and does work. Collaboration and knowledge sharing amongst several leading pharmaceutical companies
within the BPCI group has allowed the author to create a best practice template that can be used by any
company within the Pharmaceutical industry. Two templates have been developed and are appended.
• A periodic review template for Computer Systems Validation (CSV)
• A periodic review template for Equipment and Utilities.
LITERATURE REVIEW OF REGULATIONS AND GUIDELINES This study reviewed the literature currently available to the pharmaceutical industry to understand regulations
and guidelines regarding periodic review. Periodic review of validated systems is a mandatory requirement.
There is no shortage of regulatory and industry guidance within the pharmaceutical industry confirming this.
The following are some brief excerpts from this review that highlight regulatory requirements and industry
expectations.
European Regulatory
The EU Good Manufacturing Practices (GMP) rules clearly state that “systems and processes should be
periodically evaluated” (4). The guidelines give further detail on specific systems by stating that “equipment,
facilities, utilities and systems should be evaluated at an appropriate frequency to confirm that they remain in
a state of control” (5).
US Regulatory
In its 2011 guideline on Process Validation, the FDA highlight the need for periodic reviews in industry by
stating that “the equipment and facility qualification data should be assessed periodically to determine
whether re-qualification should be performed and the extent of that re-qualification” (6). Also, the FDA’s code
of federal regulations (CFR) requires that “information and data about product quality and manufacturing
experience be periodically reviewed to determine whether any changes to the established process are
warranted” (7).

7
Industry Guidelines
For over twenty-five years the International Council for Harmonization Technical Requirements for
Pharmaceuticals for Human Use (ICH) has successfully brought regulatory and industry experts together to
produce key guideline documents available in the pharmaceutical industry today. These guidelines highlight
the need to conduct periodic review of validated systems most notably within ICH Q10 and ICHQ7. ICH Q10
emphasizes how management should “assess the conclusions of periodic reviews of process performance and
product quality and of the pharmaceutical quality system” (8). Regarding the good manufacture of active
pharmaceutical ingredients (APIs) ICH Q7 highlights the need to conduct periodic reviews by stating that
“systems and processes should be periodically evaluated to verify that they are still operating in a valid
manner” (9).
GAMP 5, published in 2008, makes direct reference to
periodic reviews 42 times in its landmark guideline for the
pharmaceutical industry. This highlights that periodic reviews
are not a new initiative for CSV systems. GAMP 5 also clearly
states that “periodic reviews are used throughout the
operational life of systems to verify that they remain
compliant with regulatory requirements, fit for intended use,
and meet company policies and procedures” (10).
The Pharmaceutical Inspection Co-operation Scheme (PIC/S)
aide-memoire for inspectors who conduct inspections of
pharmaceutical facilities who manufacture active
pharmaceutical ingredients states that “there should be a
periodic review of systems and processes with respect to
validation status” (11).
Guidelines for conducting periodic review are also referenced
with within ICH Q9, ISPE GAMP® DI Guide and the Recent
MHRA Data Integrity Guidance (12,13,14).
Figure 1. GAMP 5 (9)
INTERVIEW WITH HPRA REGULATOR An interview with a regulator from the Irish competent body, the Health Products Regulatory Authority (HPRA)
was conducted (15). This interview was conducted in October 2018, and a summary of the key items discussed
are as follows:
• The regulator highlighted the EU GMP requirements in this area, as set out primarily in Annex 15
(paragraphs 4.1 and 4.2) and in Part II to the EU GMPs (section 12.6). But he also referred to the
general requirements for QRM to be applied as part of the various GMP activities at manufacturing
sites, as set out in Chapter 1 of the EU GMP Guide and in the early sections of Part II. He explained
how these QRM requirements also apply to the periodic review of validated systems in that the design
and frequency of such reviews should be based on QRM principles. He also referred to the
requirement in Chapter 1 for Product Quality Reviews to include a review of the qualification status of
relevant equipment and utilities, e.g. HVAC, water, compressed gases, etc. (15).

8
• The regulator commented that there is a “wide variety of approaches in use by the industry in relation
to the periodic review of validated systems, and that these range greatly in terms of their frequency,
scope and depth” (15).
• Regarding content, it was discussed that companies should perform meaningful and holistic reviews of
change controls -- not just compiling a line listing of change controls for the periodic review report.
Often the “cumulative effect of small changes made to a system over time can impact upon its
validated state, but this can sometimes be missed during periodic review activities unless holistic
reviews are performed” (15).
• In relation to deviations and unexpected maintenance activities which relate to a previously validated
system, companies sometime fail to make a big picture assessment of those deviations and
maintenance interventions to see what those problem issues are indicating in an overall context (15).
• Regarding how product quality reviews (PQRs) work in tandem with periodic reviews of validated
systems, the regulator stressed “the importance of companies examining the value being derived from
both types of review.” He felt that PQRs could leverage off periodic reviews of validated systems in a
more efficient manner than they sometimes do at this time. Periodic reviews of validated systems are
usually more comprehensive when done outside of the PQR process, and they are “better off being
made separately, but the learnings of such reviews should feed into the PQR process as important
inputs.” He said that it was important during PQR work that companies review the results of periodic
reviews of validated systems in conjunction with the other data that are in PQR reports in order to
again extract the big picture message about the manufacturing process and the state of control for the
product. This aligned with the author’s thoughts that there is an expectation within the industry to
conduct separate periodic reviews for all validated systems (15).
• The discussion looked at how the regulatory authority sees the industry’s current status in relation to
performing risk assessments that support periodic reviews and the requirements for revalidation. The
feeling was that “only a small number of companies have really effective risk-based processes in place”
to support periodic review activities, and more could be done in this area, especially given the work
and cost involved in doing such reviews. This highlighted another area for improvement within the
pharmaceutical industry. (15)
INDUSTRY STUDY - COMPANIES WITH EXISTING PERIODIC REVIEW PROGRAMS A research study was carried out with ten companies, each of which shared their sites periodic review
approach and strategy (3). These policies, procedures, and templates were made available to the author in
confidence and as such are confidential proprietary information. No company names shall be used in this
paper. The author is keen to stress that this was an excellent example of how effective collaboration is
possible within the pharmaceutical industry – and most importantly it is the patient who will ultimately benefit
most from this type of group co-operation.
The first significant observation from the study was that each company who participated in the study did have
a periodic review program in place with site approved procedures. Each company had periodic review
programs in place specifically for computer systems validation (CSV).
9
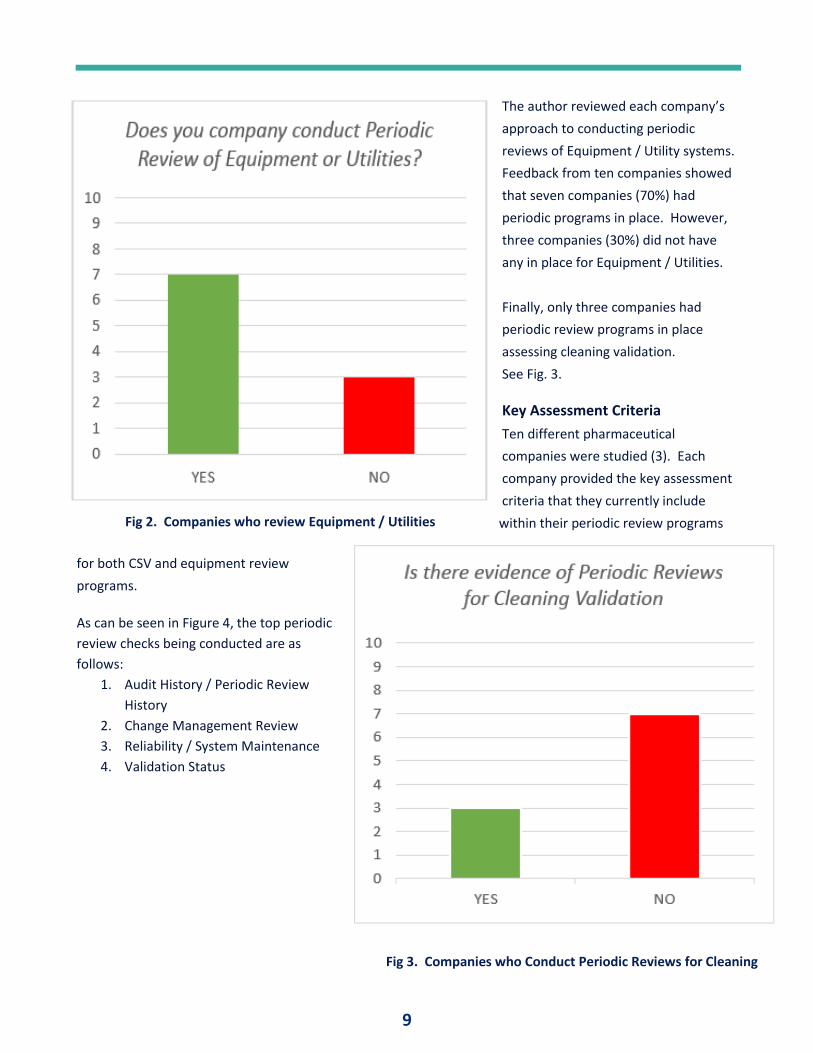
The author reviewed each company’s
approach to conducting periodic
reviews of Equipment / Utility systems.
Feedback from ten companies showed
that seven companies (70%) had
periodic programs in place. However,
three companies (30%) did not have
any in place for Equipment / Utilities.
Finally, only three companies had
periodic review programs in place
assessing cleaning validation.
See Fig. 3.
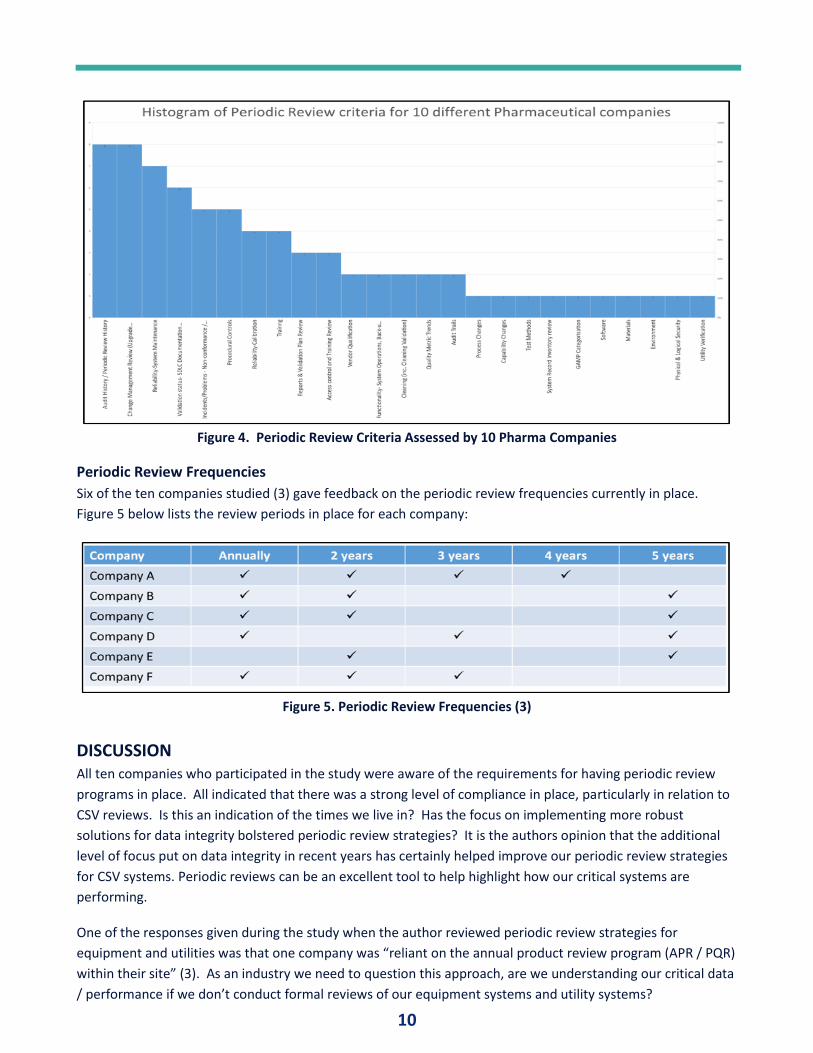
Key Assessment Criteria
Ten different pharmaceutical
companies were studied (3). Each
company provided the key assessment
criteria that they currently include
within their periodic review programs
for both CSV and equipment review
programs.
As can be seen in Figure 4, the top periodic
review checks being conducted are as
follows:
1. Audit History / Periodic Review
History
2. Change Management Review
3. Reliability / System Maintenance
4. Validation Status
Fig 2. Companies who review Equipment / Utilities
Fig 3. Companies who Conduct Periodic Reviews for Cleaning
10
Figure 4. Periodic Review Criteria Assessed by 10 Pharma Companies
Periodic Review Frequencies
Six of the ten companies studied (3) gave feedback on the periodic review frequencies currently in place.
Figure 5 below lists the review periods in place for each company:
Figure 5. Periodic Review Frequencies (3)
DISCUSSION All ten companies who participated in the study were aware of the requirements for having periodic review
programs in place. All indicated that there was a strong level of compliance in place, particularly in relation to
CSV reviews. Is this an indication of the times we live in? Has the focus on implementing more robust
solutions for data integrity bolstered periodic review strategies? It is the authors opinion that the additional
level of focus put on data integrity in recent years has certainly helped improve our periodic review strategies
for CSV systems. Periodic reviews can be an excellent tool to help highlight how our critical systems are
performing.
One of the responses given during the study when the author reviewed periodic review strategies for
equipment and utilities was that one company was “reliant on the annual product review program (APR / PQR)
within their site” (3). As an industry we need to question this approach, are we understanding our critical data
/ performance if we don’t conduct formal reviews of our equipment systems and utility systems?

11
All ten companies who participated in the study indicated that they had product quality review (PRR or APR)
programs in place for their processes on site. PQR’s are an ideal tool for assessing cumulative change and
therefore most companies would prefer to use their PQR procedures to conduct periodic review of their
processes. The author did not request to see the formal periodic review programs for process validation for
each company.
An additional observation that was made was that only 3 of 10 companies had implemented verification
checks for cleaning validation within their periodic review procedures / templates. Could this be an indication
of where the industry is at in relation to implementing review programs for cleaning validation? This is a
concerning statistic considering all the focus and effort that cleaning validation has received in recent years.
Periodic Review Frequencies
The industry study highlighted that there is a significant variance in review frequencies across six of the
companies assessed in the study. Six companies stated that they have annual periodic review programs in
place – a surprising frequency. Conducting annual reviews sounds excessive, labor intensive, and time
consuming. While the regulations and guidelines highlight the need for performing reviews, there is no strict
requirement for conducting these annually. Periodic review frequencies should be based on risk. This is spirit
of the guidance delivered in ICHQ9, where it is stated that “the frequency of any review should be based upon
the level of risk” (12). The author has included a simple frequency calculation tool within the best practice
periodic review templates which are based on risk. This calculation tool can be used as an aid for users to
determine when the next periodic review should be conducted. This calculation tool is based on a risk scoring
algorithm and makes its determination from evaluations made by the team who conducted the periodic
review exercise. This tool can be used within any periodic review template for all validated systems.
Further investigation into how companies schedule periodic reviews is also needed. Figure 5 illustrates the
variance in the frequency companies set for conducting periodic reviews. Risk assessment rational for
defining these frequencies should be reviewed to propose an industry standard for scheduling periodic
reviews.
Who should be involved?
Pharmaceutical companies need to pay particular attention to who conducts periodic reviews. In order to get
true value and proper compliance from periodic reviews, companies need to involve the correct subject matter
experts in periodic reviews. Approval of these reviews should be performed by management in accordance
with the guidelines stipulated within ICH Q10 (8).
CONCLUSION There is a clear expectation for companies to implement periodic review programs for all validated systems
within the pharmaceutical industry. This should not be looked upon as a just another layer of compliance. It is
not a case of tying up key site resources for extended time to enforce adherence to rigid predefined templates
and SOP’s. Companies need to implement smart solutions. The true challenge is to implement effective
working solutions that will be of value to the company.
If there is one thing, we have learned from the additional focus the pharmaceutical industry has placed on data
integrity in recent years is that companies need to know where their critical data are. Periodic review tools
provide an effective means to understanding how critical systems are performing.

12
Two periodic review templates have been proposed to support CSV system reviews and Equipment / Utilities
system reviews (appended). These are guideline tools that may be edited or adapted to suit any company
within the industry. However, companies need to find effective methods to generate, trend, and review
critical data. A recommendation from this study would be to conduct further research to create generic
templates for cleaning validation, analytical method validation and process validation. Also, additional
research could be conducted into the linkage between on how periodic reviews can be integrated into annual
product reviews.
Understanding the impact of cumulative change on a system was one of key points raised by the industry
regulator during the interview conducted to support this research. Process performance and quality
monitoring are key to this, and that is where effective periodic review programs have their rightful place.
We also know from ICH Q10 that we need to facilitate continual improvement. Knowledge management is a
key enabler of continual improvement (8). Periodic reviews are essentially toolkits that help provide
knowledge about a system – if designed in the right manner! So, we need to find a way to use our periodic
review programs to allow for more continuous feedback. This makes business sense and meets our regulatory
commitments.
Periodic reviews are a key focus area that should be addressed by all pharmaceutical companies. Effective
solutions come from effective collaboration. The project described in this paper has shown that the industry
can work together to deliver effective tools which can integrate nicely into the organization quality system.
These solutions will ultimately help us to deliver better results for our companies, patients, and customers.
REFERENCES 1. ISPE Glossary https://ispe.org/fr/glossary/p?title_contains=period&langcode=All
2. Master of Science program in Pharmaceutical Validation Technology, School of Chemical and
Pharmaceutical Sciences, Technological University Dublin, Dublin Ireland.
3. BioPharmaChem Ireland. IBEC CLG, Ireland. www.biopharmachemireland.ie
4. EudraLex GMP Vol.4, Part II, section 12.6
5. EudraLex GMP Annex 15, section 4.1
6. Code of Federal Regulations, Food and Drugs (Government Printing Office, Washington, DC) FDA
Process Validation Guidance (2011)
7. Code of Federal Regulations, Title 21, Food and Drugs (Government Printing Office, Washington, DC)
Part 211, Section 211.180(e)
8. ICH Harmonised Tripartite Guideline, Pharmaceutical Quality System, ICH Q10 (2008)
9. ICH Harmonised Tripartite Guideline, Good Manufacturing Practice Guide for Active Pharmaceutical
Ingredients, ICH Q7 (2000)
10. GAMP5, A Risk-Based Approach to Compliant GxP Computerized Systems, Section 4.3.5, ISPE (2008)
11. Pharmaceutical Inspection Co-operation Scheme (PIC/S), Aide-Memoire for API’s (2009)
12. ICH Q9, Quality Risk Management (2005)
13. ISPE GAMP® Guide: Records and Data Integrity (2017)
14. MHRA GXP Data Integrity Guidance and Definitions; (March 2018)
15. Dr. Kevin O’Donnell, Market Compliance Manager, HPRA, Ireland.
Editor’s note: This research was conducted in conjunction with the Technological University Dublin (TU
Dublin) as part of requirements for the Master of Science degree in Pharmaceutical Validation Technology.

13
APPENDIX A PERIODIC REVIEW TEMPLATE FOR COMPUTER SYSTEM VALIDATION (CSV)
TABLE OF CONTENTS
1 INTRODUCTION ............................................................................................................................. 23
1.1 Purpose ....................................................................................................................................... 23
1.2 Scope ........................................................................................................................................... 23
2 SYSTEM DETAILS ............................................................................................................................ 23
2.1 Validation Status and Review...................................................................................................... 23
2.2 Periodic Review History .............................................................................................................. 24
2.3 Audit History ............................................................................................................................... 24
2.4 Change Management .................................................................................................................. 24
2.5 Incident Management ................................................................................................................. 25
2.6 Environment ................................................................................................................................ 25
2.7 Risk Analysis ................................................................................................................................ 25
2.8 Capability Analysis ....................................................................................................................... 25
2.9 Matric Trends .............................................................................................................................. 25
2.10 Procedural Controls .................................................................................................................... 26
2.11 Training ....................................................................................................................................... 26
3 ANALYSIS & RECOMMENDATIONS ................................................................................................. 26
4 CONCLUSION ................................................................................................................................. 27
5 NEXT PERIODIC REVIEW ................................................................................................................. 27
6 REFERENCES .................................................................................................................................. 28
7 ATTACHMENTS .............................................................................................................................. 28
<Note for the Author>:
- All comments in blue aim to help with completing the document.
- The requirements included in the document are only examples.
- The information included in the document are given as examples only.
- Dedicated areas for entering information are marked as “[Enter content here]” and must be removed if not used.
- Blue Text boxes are not printed
- Please ensure removing instructions displayed in blue before initiating document review/approval.

14
1 INTRODUCTION
1.1 Purpose
[Enter content here]
This report provides the Periodic Review (PR) result for <System Name>, as a means to evaluate the
effectiveness of the applied Operational Phase procedures, based on which a conclusion is drawn in regard
to the continued controlled and compliant state of the system(s).
1.2 Scope
[Enter content here]
The scope of this document is limited to <System Name>,
• Security and access control
• Backup, Restore, Archive and Retrieval Performance monitoring
• Change management (change control and configuration management)
• Training
• Periodic Review
• Configuration Management
• Incident Management
• Problem Management
• IT Compliance Investigation and CAPA Management
• Security and Account Administration
1.3 Concept Overview
[Enter content here]
The Periodic Review effort, as a component of the overall risk based Periodic Review is based on following key principles:
• Consistent application of procedures assures the continued controlled and validated state of computer and automated systems. (i.e. Security, Change Management, etc.).
• The effectiveness of a single procedure is established based on a review of the expected procedure output (e.g. is a current backup present, is security up to date) in line with industry standard best practices.
• For a group of systems following the same procedures, the effectiveness of the applied and shared controls is evaluated by reviewing the output of a minimum of one representative sample system within this group.
• Periodic security checks, verifying up-to-date security / access controls settings
• Trending as part of regulatory incidents / problems
• Trending as part of calibration In support of this periodic review process, the following additional controls are in place as part of the overall site periodic review approach and site Quality Management System (i.e. prerequisite):
• A verification and confirmation of the applied key procedures per system is performed on a periodic basis as part of this document.
• An internal audit and/or self-assessment process to assure correct application of site procedures.
• An event management process, including appropriate Corrective Action / Preventative Action (CAPA) mechanisms to continually reduce human errors. The process must cover trending of events.
15
• A training process to assure correct and complete understanding of required controls and activities, as well as assuring a consistent execution of the procedures. (Including retraining as appropriate).
• The system use and system administration topics are assured implemented and maintained in system SOP’s with the existence of the initial validation process to guarantee initial implementation, the change process to the evaluate SOP impact due to implementation of a change, the SOP management process requiring SOP review every three years to reconfirm SOP topics and scope, and the incident and problem management process requiring documentation of incidents with SOP application and execution.
2 GENERAL INFORMATION
Year of PR effort <<XXXX>>
System Components included in the cluster: (including system component ID) <<if applicable, XXXX>>
System Procedures
Topic Procedure ID and Title Version Effective Date
Change Control //Describe the procedure ID and title for this topic. //
<<XX>> <<DD-MMM-YYYY>>
Configuration
Management
//Describe the procedure ID and title for this topic. //
<<XX>> <<DD-MMM-YYYY>>
Incident / Problem Mgmt. //Describe the procedure ID and title for this topic. //
<<XX>> <<DD-MMM-YYYY>>
Security / Account Mgmt. //Describe the procedure ID and title for this topic. //
<<XX>> <<DD-MMM-YYYY>>
System Use //Describe the procedure ID and title for this topic. //
<<XX>> <<DD-MMM-YYYY>>
Backup/Restore &
Archival/Retrieval
//Describe the procedure ID and title for this topic. //
<<XX>> <<DD-MMM-YYYY>>
Training //Describe the procedure ID and title for this topic. //
<<XX>> <<DD-MMM-YYYY>>
Periodic Review //Describe the procedure ID and title for this topic. //
<<XX>> <<DD-MMM-YYYY>>
Performance Monitoring //Describe the procedure ID and title for this topic. //
<<X.X>> <<DD-MMM-YYYY>>
Other //Describe the procedure ID and title for this topic. //
<<X.X>> <<DD-MMM-YYYY>>
Are all required SOP topics addressed or explained with appropriate
rationale? Yes No
Are SOP's up to date and within the required review cycle? Yes No
Comments/Observations including any action steps:
// Describe, if applicable //

16
3 SAMPLE SYSTEM
3.1 General Information - <<System Name >>
//Repeat this section for as many as Sample Systems are included //
System Name <<System Name>> Components ID <<Component
ID>>
Location <<System Location>>
Last Monitoring
Period From
<<DD-MMM-
YYYY>> To
<<DD-MMM-
YYYY>>
Current Monitoring
Period From
<<DD-MMM-
YYYY>> To
<<DD-MMM-
YYYY>>
Frequency/Duration
of Use Continuous/Frequent Occasional Infrequent
System Description <<System Description and intended use>>
3.2 System Data Collection - <<System Name >>
//Repeat this section for as many as Sample Systems are included //
Previous Periodic Review Report:
Were actions identified as a result of the last periodic review? Yes No N/A
If yes, provide a description and status:
//Provide the description and status of any actions identified as a result of the last periodic review report//
System Logs and Tracking Mechanisms (Hardcopy or Electronic)
Record the system logs and other applicable tracking mechanisms:
I. SYSTEM DEVELOPMENT LIFE CYCLE DOCUMENTATION
// Provide the description of the system log or applicable tracking mechanism for the System Life Cycle documentation generated during the review period // II. CONFIGURATION MANAGEMENT INFORMATION
// Provide the description of the system log or applicable tracking mechanism for the system configuration during the review period // III. UPGRADE HISTORY/CHANGE CONTROL INFORMATION
// Provide the description of the system log or applicable tracking mechanism for the system changes during the review period // IV. PROBLEM REPORTING INFORMATION
// Provide the description of the system log or applicable tracking mechanism for the system incidents / problems//
Were any issues identified? Yes No N/A
17
Comments / Observations including action plan if yes:
// Provide comments and action plan for the issues identified //
Backups
Code Yes No N/A Data Yes No N/A
Configuration Yes No N/A Other Yes No N/A
Comments / Observations including action steps for each no:
// Provide comments for the backup process //
User Access
Has the user access been reviewed for this period? Yes No N/A
Observations from the review and any action steps:
// Describe any observations from the review process and any action steps. Also include Managers review of user accounts access that should be done yearly. //
If no, provide description of action steps:
// Provide description of the actions to be taken if user access has not been reviewed //
System Use and Administration
Record the system use SOP: // Provide the description of the system use SOP //
Record the system administration SOP: // Provide the description of the system administration SOP//
Calibration and Preventative Maintenance
Is the system currently enrolled in a Calibration Program? Yes No N/A
Is the system currently enrolled in a Preventative Maintenance Program? Yes No N/A
Are there any unresolved issues from either program? Yes No N/A
Comments / Observations:
// Provide comments on any unresolved issues //
Change Controls (during THIS monitoring period – excluding Pre-Approved Changes)
# Change Controls Opened <<XX>>
# Change Controls Closed <<XX>>
# Change Controls Remaining Open <<XX>>
Summary and impact of the changes include observations and any action items:
// Provide a summary of the changes including any action items //
Incident Reporting / Problem Reporting - Corrective Actions and Preventative Actions
# Problems Opened <<XX>> # CAPAs Generated <<XX>>
# Events/Deviations Opened <<XX>>
Summary and observations including any action items:
// Provide a summary of the incidents/problems reported, including any resulted action items //
18
4 ANALYSIS & RECOMMENDATIONS – GROUP
Question 1: Actions
Are there any actions required as a result of this periodic review? Yes No
Question 2: Action Category
What is the overall nature of the required actions?
Documentation Performance/Functionality N/A (no actions required)
Provide summary of action(s): // Provide a summary of the required actions //.
Question 3: In-Service Status
Based on the nature of the required actions, can the system remain in
service with the required actions open?
Yes No N/A
Justification: // Provide the justification for maintaining the system in operation with the required actions
open //
Question 4: Periodic Review – Re-Execution
Based on the investigation(s) issued detected during this Periodic
Review, is PR re-execution during next year needed for any of the
selected sample systems? Identify relevant events and / or CAPAs.
Identify for each CAPA whether it is system specific or process related for
all systems in this group.
Yes No N/A
5 CONCLUSION
Case Question 1
Actions
Question 2
Action Category
Question 3
In-Service
Status
Question 4
Periodic
Review
Re-Execution
System Status and Action
Guidance
1 No N/A N/A N/A
Systems in this Cluster under
control. Validation Status under
control.
2 Yes Documentation Yes No
Inadequate procedures or out-of-
date deliverables require
investigation to assess structural
nature of this issue with definition
of corresponding CAPA.
19
3 Yes Performance /
Functionality Yes Yes
Issue related to system
performance/ functionality issues
require investigation to assess
structural nature of this issue with
definition of corresponding CAPA.
PR of related sample system must
be re-execution within the
following year.
4 Yes
Documentation
&
Performance /
Functionality
Yes Yes
Actions Plans related to the issues
required immediately for ongoing
control of system with required
investigation to assess structural
nature of this issue with definition
of corresponding CAPA. PR of
related sample system must be
re-execution within the following
year.
5 Yes
Documentation
&
Performance /
Functionality
No Yes
System not in a state of control.
Take related sample system out of
service, with required
documented assessment of
impact towards other systems in
the cluster.
6 NEXT PERIODIC REVIEW
Individual risk scores (High, Medium, Low) are overall evaluations made by the Periodic Review Team.
Scoring
Low = 1
Med = 2
High = 3
The recommended periodic review frequency is auto-calculated from the 7 risk scores using the embedded
Excel sheet at the end of the PR checklist and may vary between 1 to 4 years.
The final decision on the next Periodic Review due date is documented in the conclusion section and the
corresponding VMP is updated accordingly
Scoring
0 to 9 = 4 years
10 to 13 = 3 years
14 to 17 = 2 years
18 or more = Annual
20
Risk Area Score Value Information From:
Number & Severity of changes Low 1
Number & Severity of incidents Low 1
Supplier issues & related risks Low 1
Business criticality (Product approval &
market availability) Low 1
Complexity & Scope (System Difficulties &
User Base)Low 1
Level of the regulated activities (GMP,
GCP, GPvP) etc.. Low 1
Impact of patient safety, product quality,
record integrityLow 1
Recommended Frequency 4 Year 7
Decision Date Next Periodic Review TBD
Directly related to the
Periodic Review findings
Based on CSV Risk
Management documents
These documents may be
adapted as a result of the
Periodic Review findings
7 REFERENCES
Reference Identification / Description Source / Location
<Enter site periodic review SOP title and number> <Enter SOP location details>
8 ATTACHMENTS
Attachment ID Attachment Title
Attachment I Software/Hardware Change Requests for the Monitoring Period
Attachment II Computer Systems Problems Evaluation for the Monitoring Period
Attachment III Corrective Actions and Preventive Actions
Attachment <XX>> // Provide the Attachment Title //
Attachment I: Software/Hardware Change Requests for the Monitoring Period
<<System Name>>
Req No. System Proposed Change
Regulatory
Impact?
(Yes/No)
Status
[Open/Closed]
<<System
Name>>
// Describe the Proposed Change
// <<Yes/No>> <<Open/Closed>>
21
Attachment II: Computer Systems Problems Evaluation for the Monitoring Period
<<System Name>>
Computer System
Identification /
Problem Report
Number [Atypical No.
if applicable]
Recurrent
Problem?
(Yes/No)
Problem
Evaluation
[Also indicate the
cause for this
problem]
Corrective Action
[Indicate the action
taken and any
pending action]
Status
[Completed or
Pending]
[Include Date]
<<Problem Report
Number>> <<Yes/No>>
//Provide the
Problem
Evaluation
Description//
//Describe the
Problem Corrective
Action//
<<Open/Closed>>
Attachment III: Corrective Actions and Preventive Actions
CAPA Number Description Expected Completion Date Date Completed
<<CAPA
Number>> //Provide the CAPA Description// <<DD-MMM-YYYY>>
<<DD-MMM-
YYYY>>

22
APPENDIX B PERIODIC REVIEW TEMPLATE FOR EQUIPMENT AND UTILITIES.
TABLE OF CONTENTS
1 INTRODUCTION ............................................................................................................................. 23
1.1 Purpose ....................................................................................................................................... 23
1.2 Scope ........................................................................................................................................... 23
2 SYSTEM DETAILS ............................................................................................................................ 23
2.1 Validation Status and Review...................................................................................................... 23
2.2 Periodic Review History .............................................................................................................. 24
2.3 Audit History ............................................................................................................................... 24
2.4 Change Management .................................................................................................................. 24
2.5 Incident Management ................................................................................................................. 25
2.6 Environment ................................................................................................................................ 25
2.7 Risk Analysis ................................................................................................................................ 25
2.8 Capability Analysis ....................................................................................................................... 25
2.9 Matric Trends .............................................................................................................................. 25
2.10 Procedural Controls .................................................................................................................... 26
2.11 Training ....................................................................................................................................... 26
3 ANALYSIS & RECOMMENDATIONS ................................................................................................. 26
4 CONCLUSION ................................................................................................................................. 27
5 NEXT PERIODIC REVIEW ................................................................................................................. 27
6 REFERENCES .................................................................................................................................. 28
7 ATTACHMENTS .............................................................................................................................. 28
<Note for the Author>:
- All comments in blue aim at providing assistance for completing the document.
- The requirements included in the document are only examples.
- The information included in the document are given as examples only.
- Dedicated areas for entering information are marked as “[Enter content here]” and must be
removed if not used.
- Blue Text boxes are not printed
- Please ensure removing instructions displayed in blue before initiating document review/approval.
23
INTRODUCTION
Purpose
This report provides the periodic review (PR) result for <System Name>. This periodic review is the process whereby <Company Name> assesses its GMP equipment, utilities, facilities and systems to determine if the system remains in a qualified state and, as such, are fit for their intended use.
Scope
This periodic review report relates to <System Name> located within <provide system location / area> at <Company Name>
SYSTEM DETAILS
Validation Status and Review
System Name: <<System Name>>
Location: <<System Location>>
System Number: <<System ID Number>>
System Validation Number: <<System Validation Number>>
System Owner <<System Owner Name & Dept>>
Impact Assessments <<Systems and Component Impact Assessments>>
Qualification Status: Qualified Not Qualified Decommissioned N/A
User Requirements Spec <<List approved URS>>
Design Qualification <<List approved DQ>>
Installation Qualification <<List approved IQ>>
Operational Qualification <<List approved OQ>>
Qualification Lifecycle <<List approved lifecycle documents >>
Last Monitoring Period: From <DD-MMM-YYYY> To <DD-MMM-YYYY>
Current Monitoring Period: From <DD-MMM-YYYY> To <DD-MMM-YYYY>
Frequency/Duration of Use:
Continuous/Frequent Occasional Infrequent
System Description: <<System Description and intended use>>
Validation Plans / Reports:
24
Were there any validation plans & reports approved during the review period?
Yes No N/A
If yes, provide a description and status:
//Provide the description and status of any actions/recommendations identified //
Periodic Review History
Previous Periodic Review Report:
Were actions identified as a result of the last periodic review? Yes No N/A
If yes, provide a description and status:
//Provide the description and status of any actions identified as a result of the last periodic review report//
Audit History
Audit History
List of any audit findings/observations related to the equipment/system/process under review.
<<XX>>
Status of actions on these findings/observations. <<XX>>
Provide a description and status:
//Provide an update on the status of any actions still open and impact upon qualification / validation of the system//
Change Management
Change Controls (approved during THIS monitoring period – excluding Pre-Approved Changes)
# Change Controls Opened <<XX>> # Change Controls Closed <<XX>> # Change Controls Remaining Open <<XX>> Summary and impact of the changes include observations and any action items:
// Provide a summary of the changes including any action items //
Change No. System Change Detail / Description Regulatory Impact? (Yes/No)
Status
[Open/Closed]
<Change Number>
<System Name>
// Describe the Proposed Change //
<Yes/No> <Open/Closed>
Validation File Updates (approved during THIS monitoring period – excluding Pre-Approved Changes)
List the validation file (qualification) updates conducted during this review period?
<<Approval Date>>
How were the updates managed? Memo to File Validation Protocol Both
Summary and impact of the changes / modifications and include observations and any action items:
// Provide a summary of the modifications / changes including any action items //
25
Incident Management
Incident Reporting (Problem Reporting - Corrective Actions and Preventative Actions)
# Problems Opened <<XX>> # CAPAs Generated <<XX>>
# Events/Deviations Opened <<XX>>
Summary and observations including any action items:
// Provide a summary of the incidents/problems reported, including any resulted action items //
Problem Report Number
Recurrent Problem?
(Yes/No)
Problem Evaluation
[Also indicate the cause for this problem]
Corrective Action
[Indicate the action taken and any pending action]
Status
[Completed or Pending]
[Include Date]
<<Problem Report Number>>
<<Yes/No>>
//Provide the Problem Evaluation Description//
//Describe the Problem Corrective Action//
<Open/Closed>
Environment
Environmental impacts (Environment i.e. buildings, facilities, and utilities) in which the system operates can affect system performance.)
# Problems Opened <<XX>> # CAPAs Generated <<XX>>
# Events/Deviations Opened <<XX>>
Summary and observations including any action items:
// Provide a summary of the incidents/problems reported, including any resulted action items //
// Conduct a physical assessment of the area / location to ensure compliance //
Risk Analysis
Risk Analysis (The risk analysis for the system should be reviewed to ensure any changes detailed above are captured.)
Risk Assessment Number: <<XX>>
Summary and observations including any action items:
// Review a section of the Risk Assessment (s) to ensure it is up to date.//
Capability Analysis
Capability Analysis (Used to show that there has been no significant change in system performance.)
Capability assessment: <<XX>>
Summary and observations including any action items:
// Review results and statistical conclusion from most recent capability analysis //
Matric Trends
Metrics (Plant metrics may be used as good indicators of system performance.)

26
Matric Report: <<XX>>
Summary and observations including any action items:
// Review management reviews that may highlight issues with systems //
Procedural Controls
Operational Instructions / Guidelines (Ensuring that systems procedures are available an in date)
Summary and observations including any action items:
// Confirm that relevant procedure(s) are in place for operating the system under review //
Training
Operator Training Records (A check of the training records for those who operate the system under review)
Summary and observations including any action items:
// Audit the current users versus the procedure(s) in place for operating the system under review … also, review system security for current users and updates //
ANALYSIS & RECOMMENDATIONS
Question 1: Actions
Are there any actions required as a result of this periodic review? Yes No
Question 2: Action Category
What is the overall nature of the required actions?
Documentation Performance/Functionality N/A (no actions required)
Provide summary of action(s): // Provide a summary of the required actions //.
Question 3: In-Service Status
Based on the nature of the required actions, can the system remain in service with the required actions open?
Yes No N/A
Justification: // Provide the justification for maintaining the system in operation with the required actions open //
27
Question 4: Periodic Review – Re-Execution
Based on the investigation(s) issued detected during this Periodic Review, is periodic review re-execution during next year needed for any of the selected sample systems?
Identify relevant events and / or CAPAs. Identify for each CAPA whether it is system specific or process related for all systems in this group.
Yes No N/A
CONCLUSION
Case
Question 1 Actions
Question 2 Action Category
Question 3 In-Service Status
Question 4 Periodic Review Re-Execution
System Status and Action Guidance
1 No N/A N/A N/A System(s) in under control. Validation Status under control.
2 Yes Documentation Yes No Inadequate procedures or out-of-date deliverables require investigation to assess structural nature of this issue with definition of corresponding CAPA.
3 Yes Performance /
Functionality Yes Yes
Issue related to system performance/ functionality issues require investigation to assess structural nature of this issue with definition of corresponding CAPA.
PR of related sample system must be re-execution within the following year.
4 Yes
Documentation &
Performance / Functionality
Yes Yes
Actions Plans related to the issues required immediately for ongoing control of system with required investigation to assess structural nature of this issue with definition of corresponding CAPA. PR of related sample system must be re-execution within the following year.
5 Yes
Documentation &
Performance /
Functionality
No Yes
System not in a state of control. Take related sample system out of service, with required documented assessment of impact towards other systems affected.
NEXT PERIODIC REVIEW
Individual risk scores (High, Medium, Low) are overall evaluations made by the Periodic Review Team.
Scoring Low = 1 Med = 2 High = 3
The recommended periodic review frequency is auto calculated from the 7 risk scores using the embedded Excel sheet at the end of the periodic review checklist and may vary between 1 to 4 years.
28
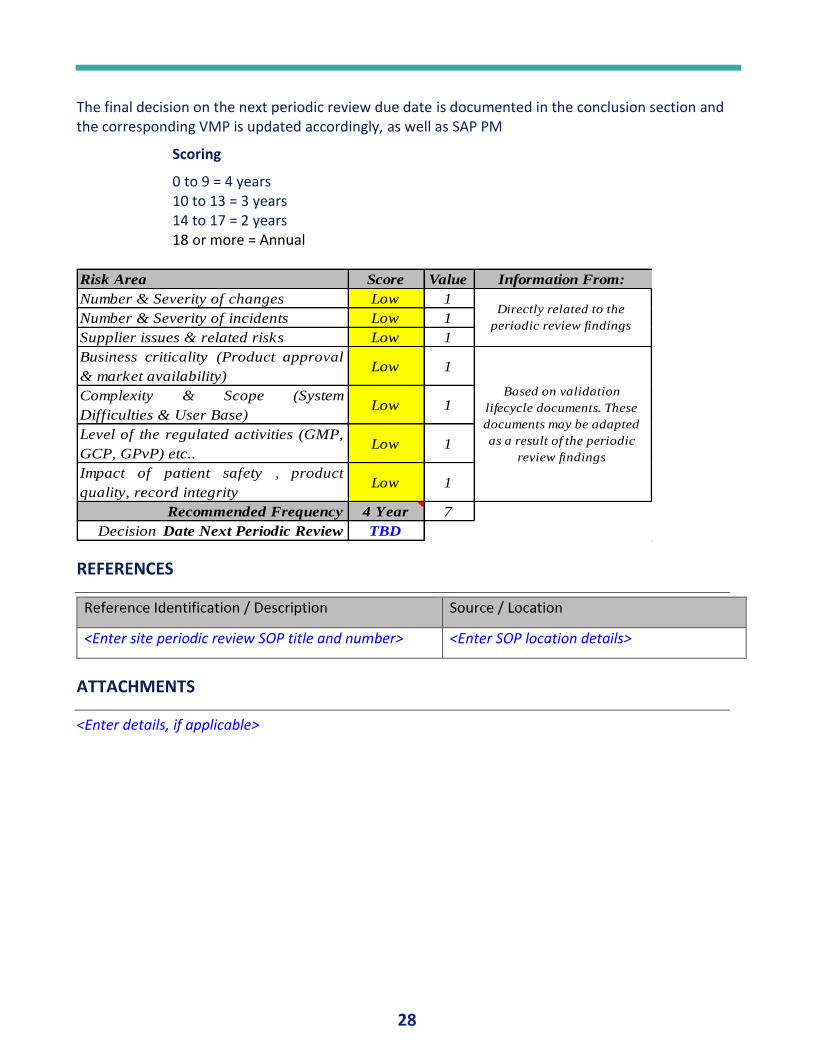
The final decision on the next periodic review due date is documented in the conclusion section and the corresponding VMP is updated accordingly, as well as SAP PM
Scoring
0 to 9 = 4 years 10 to 13 = 3 years 14 to 17 = 2 years 18 or more = Annual
Risk Area Score Value Information From:
Number & Severity of changes Low 1
Number & Severity of incidents Low 1
Supplier issues & related risks Low 1
Business criticality (Product approval
& market availability) Low 1
Complexity & Scope (System
Difficulties & User Base)Low 1
Level of the regulated activities (GMP,
GCP, GPvP) etc.. Low 1
Impact of patient safety , product
quality, record integrityLow 1
Recommended Frequency 4 Year 7
Decision Date Next Periodic Review TBD
Directly related to the
periodic review findings
Based on validation
lifecycle documents. These
documents may be adapted
as a result of the periodic
review findings
REFERENCES
Reference Identification / Description Source / Location
<Enter site periodic review SOP title and number> <Enter SOP location details>
ATTACHMENTS
<Enter details, if applicable>

29
AUDITING AND ASSESSING THE QUALITY CONTROL LABORATORY By Tim Sandle, Ph.D., Pharmaceutical Microbiologist, Head of Microbiology and Sterility Assurance Bio Products Laboratory Limited
INTRODUCTION The concept of quality is central to the delivery of laboratory services and this is achieved through the
incorporation of quality systems, quality control and quality assurance in all aspects of laboratory practice.
Essential to all aspects of laboratory results is to ensure that they are accurate, reliable and delivered in a
timely fashion. To ensure that these requirements are in place and that they are consistently being met, audits
should be regularly undertaken. Quality audits play an essential role in the Quality Management System and
these are typically a systematic examination of a system, discrete operate, product or process. In
pharmaceuticals and healthcare, the analytical laboratory function plays an important role in testing products
and samples against defined acceptance criteria and this information is used for release purposes. Such
laboratories tend organized along specific disciplines (such as chemistry or microbiology) and fall within a
generalized control laboratory (or quality control laboratory) unit.
Audits of the laboratory will be performed at predefined time intervals, assessing whether the laboratory
complies with the defined quality system processes and this can involve procedural or results-based
assessment criteria. Such audits (sometimes called ‘assessments’) can be internal (from within the company)
or external (such as conducted by customers or inspectors from regulator bodies or standards / certification
agencies for accreditation purposes or where inspections are performed by regulatory agencies).
Audits, as set out in ISO 9001: 2015, function to (1):
• Verify objective evidence of processes
• Assess how successfully processes have been implemented
• Assess the effectiveness of achieving defined target levels
• Provide evidence concerning reduction and elimination of problem areas
During audits of a laboratory function, information is gathered about:
• Suitability of processes and operating procedures
• Staff competence and training
• Reliability and accuracy of equipment
• Suitability of the laboratory environment
• The handling of samples
• Quality control and verification of results
• Recording and reporting practices
Audits therefore enable the laboratory to understand how well it is performing when compared to a
benchmark or standard. To be effective, laboratory auditing should report both non-conformances and
corrective actions, and to highlight areas of good practice so that other laboratories or departments can
exchange information and review working practices (as part of a culture of continuous improvement).

30
This article considers the approach required for auditing the laboratory and provides a general checklist
against which a laboratory could be audited, ort which can be adapted for such a purpose or, where checklists
already exist, to be used to benchmark against and to compare best practice. The article is aimed more
towards the analytical laboratory rather than the microbiological; however, there will be parts of the text of
general interest to any laboratory that falls within the quality function.
What makes for the good laboratory?
The key role of the laboratory is to ensure that the final laboratory results, as reported, are correct. To ensure
this, appropriate systems need to be in place that ensure planned and systematic laboratory activities which
ensure the accuracy and defensibility of test results. This rests on the effectiveness of the laboratory quality
system. The main elements of such a system are:
• Having a laboratory quality manual or the laboratory featuring in an overall organization quality
manual
• Standard Operating Procedures (SOPs)
• Reporting methods, either on controlled forms or via computerized information systems
• Suitable supplementary records (e.g., instrument logbooks)
In the controlled environment of a testing laboratory everything needs to be captured and documented. If the
activity is not documented, then the activity never happened.
In addition, the laboratory also needs to:
• Choose the correct methods for testing
• Establish protocols to detect errors and initiate corrective actions
• Have in place validated methods
• Be able to demonstrate that methods are fit for their intended purpose, with established accuracy,
precision, calibration and limits of detection and quantification
The good laboratory will have a clear and identifiable pathway – or what the World Health Organization refers
to as the “path to workflow.” (2) This captures the entire set of operations that occur in testing. The path of
workflow starts with the registering of a sample and ends in reporting and results interpretation. If the
pathway is not clear or followed correctly, then errors can arise, and quality is compromised (3). For example,
a sample that is damaged or altered as a result of improper collection or transport cannot provide a reliable
result. A medical report that is delayed or lost, or poorly written, can negate all the effort of performing the
test well.
AUDITING THE LABORATORY Laboratories should be audited regularly and at least once per year. Auditing is an independent activity and
separate to self-inspection. Auditing activities could include reviewing SOPs, worksheets, laboratory
notebooks, balance calibration records, working control data, pipette calibration records, equipment
monitoring logs and other related items for producing test results.

31
The auditor should use a checklist to determine the auditing scope and content (and example of such a
checklist is detailed below). The audit results then initiate corrective and/or preventive action to ensure
continuous improvement of the quality system. Based on past results and a review of laboratory performance
(such as the number of deviations or out-of-specification reports), the auditor may elect to focus on specific
areas (such as a test method) or a more general review of the laboratory, i.e. tracking the path of a sample
from receipt through testing and final reporting. These different approaches will often rest upon risk and a
risk-based approach to auditing (4).
It is important that the person tasked with auditing the laboratory understands laboratories is general and
Ideally of the specific function of the laboratory. During the audit the auditor should observe, check
documentation, and listen to what is being said (interestingly, the word audit is derived from a Latin word
"audire" which means "to hear").
The laboratory itself should prepare for the audit in advance, such as by:
• Planning thoroughly and carefully
• Organizing everything ahead of time, including documents and records, to save valuable time during
the audit
• Making sure all staff aware of the audit and arranging schedules so that all staff needed for the audit
will be available
SPECIFIC AREAS OF FOCUS
Laboratory Management
The laboratory must be managed correctly, with the appropriate trained and qualified people in place. In
addition, there must be a clear management function, with personnel and different levels of responsibility and
seniority. Management should have a focus on laboratory process control, which should include quality control
for testing, appropriate management of the sample, including collection and handling, and method verification
and validation. The laboratory should establish, implement and maintain a management system appropriate to
the scope of its activities. The laboratory must document its policies, systems, programs, procedures and
instructions to the extent necessary to assure the quality of the test and/or calibration results. The system's
documentation needs to be communicated to, understood by, available to, and implemented by the
appropriate personnel.
With management, the laboratory should have managerial and technical personnel who have the authority
and resources needed to carry out their duties including the implementation, maintenance and improvement
of the management system and to identify the occurrence of departures from the management system or
from the procedures for performing tests and/or calibrations, and to initiate actions to prevent or minimize
such departures. Furthermore, laboratory management should provide adequate supervision of testing and
calibration staff, including trainees, by persons familiar with methods and procedures, purpose of each test
and/or calibration, and with the assessment of the test or calibration results. In addition, they should have
technical management which has overall responsibility for the technical operations and the provision of the
resources needed to ensure the required quality of laboratory operations.

32
Good Laboratory Design
The laboratory must be appropriately designed laboratory to enable appropriate workflow and to avoid cross-
contamination or mix-up of samples. This will include dedicated work areas and materials of construction, such
as work benches being built of materials that are durable and easy to disinfect. Furthermore, access to rooms
where manipulation or analysis of samples takes place, or where hazardous chemicals or other materials are
stored (5).
As part of good design principles, the laboratory must have systems to monitor, control and record
environmental conditions as required by the relevant specifications, methods and procedures or where thy
influence the quality of the results. Due attention should be paid, for example, to biological sterility, dust,
electromagnetic disturbances, radiation, humidity, electrical supply, temperature, and sound and vibration
levels, as appropriate to the technical activities concerned. Management need to ensure that tests and
calibrations are stopped when the environmental conditions jeopardize the results of the tests and/or
calibrations.
There should be effective separation between neighbouring areas in which there are incompatible activities.
Measures must be taken to prevent cross-contamination and access to and from laboratory areas dealing with
the tests and/or calibrations need to be controlled. The laboratory must determine the extent of control
based on its circumstances. Further, measures need be taken to ensure good housekeeping in the laboratory.
Laboratory Personnel
The laboratory management should have mechanisms in place to ensure the competence of all who operate
specific equipment, perform tests and/or calibrations, evaluate results, and sign test reports and calibration
certificates. When using staff who are undergoing training, appropriate supervision needs to be provided.
Personnel performing specific tasks are to be qualified based on appropriate education, training, experience
and/or demonstrated skills, as required (6).
Instrumentation
Well operated equipment and instruments reduces variation in test results and improves the laboratory’s
confidence in the accuracy of testing results (7). Hence, it is important that laboratory results are of required
quality and this will often rest upon the suitability of the instrumentation, and whether instrument calibration
has been conducted. In addition to calibration, regular verifications are required to ensure that measurements
are accurate, such as verification of balances or pipettors. To support these requirements, an equipment
management program should be in place to address equipment selection, preventive maintenance and
procedures for troubleshooting and repair.
Sample Management
Sample management is a key part of process control. Importantly, the quality of the work a laboratory
produces is only as good as the quality of the samples it uses for testing. The laboratory needs to be proactive
in ensuring that the samples it receives meet all the requirements for producing accurate test results. This
means there needs to be sample records that show chain-of-custody. Records can be manual, e.g., forms or
logbooks, or electronic, e.g., LIMS (laboratory information management system) (8). In addition, samples need
to be stored in an area that maintains their quality. This includes areas that are properly identified, clean and

33
orderly, and is adequate to prevent mix-up and contamination from other samples, from chemicals and
reagents, and from spillage.
An important area is with sample labelling. Each sample should be clearly labelled with:
• Unique identification numbers
• The test that has been requested
• The time and date of collection
• The sample expiry time
• Sample storage conditions
• Identification of the person who collected the sample
This leads into sample receipt:
• Verifying the sample is properly labelled, adequate in quantity, in good condition and appropriate for
the test requested. The test request must be complete and include all necessary information
• Recording sample information into a register or log
• Enforce procedures for storing the sample prior to test (location, time, temperature, ensuring sample
segregation etc.)
Logging in the sample, recording either manually or electronically:
• Date and time of collection
• Date and time the sample was received in the laboratory
• Sample type
• Tests to be performed
With storage:
• Description of what samples should be stored
• Retention time
• Location
• Conditions for storage, such as atmospheric and temperature requirements
• System for storage organization—one method is to store samples by day of receipt or accession
number
The laboratory must have a system in place to allow for tracking a sample throughout the laboratory from the
time it is received until results are reported.
There should also be a process in place for rejecting samples. Samples could be rejected, for instance, due to:
• An unlabelled sample
• Broken or leaking tube/container
• Insufficient information
• Sample label and accompanying record do not match
• Sample collected in wrong tube/container
• Sample stored incorrectly
• Sample time expired

34
• Inadequate volume
• Poor handling during transport
Traceability, Uncertainty and Proficiency Testing
Traceability, uncertainty and proficiency testing are the three important areas to be addressed in an audit.
Traceability is about gaining the assurance that the measurement results can be related to a reference through
a documented, unbroken chain of comparisons. For example, the bias, precision and accuracy of testing can be
determined by testing a certified reference material and comparing the laboratory results with the certified
value. The certified value of the reference material is generally reported with uncertainty such that the
comparison is of statistical significance (9).
Proficiency tests serve as the external quality assurance, assuming the laboratory participates in a proficiency
sample test program organized outside of the laboratory (10). Proficiency testing is an interlaboratory
comparison, in which several laboratories conduct testing methods within their own lab on the same material
and report the results to the organizing party. Each individual laboratory is then evaluated for performance
based on statistical calculations (11). To establish required proficiency, the laboratory needs to maintain an
appropriate schedule to participate in a proficiency test program.
Reference Standards and Controls
The laboratory must establish a quality control program for all quantitative tests. Evaluating each test run in
this way allows the laboratory to determine if patient results are accurate and reliable (12). This will be
achieved using appropriate reference standards, and the suitability and verification of reference standards
should be assessed as part of an audit.
The audit should also consider the controls run with each test. Controls are substances that contain an
established amount of the substance being tested—the analyte. Controls are tested at the same time and in
the same way as test samples. The purpose of the control is to validate the reliability of the test system and
evaluate the operator’s performance and environmental conditions that might impact results.
The auditor should consider:
• If controls are appropriate for the targeted diagnostic test—the substance being measured in the test
must be present in the control in a measurable form.
• The amount of the analyte present in the controls should be close to the decision points of the test;
this means that controls should check both low values and high values.
• Controls should have the same matrix as the test sample.
• For quantitative testing, statistical analysis is often used for the monitoring process, such as the use of
Levey–Jennings charts.
The source of the control material should be considered: control materials may be purchased, obtained from a
central or reference laboratory, or made in-house.

35
Purchasing and Supplier Approval
The laboratory should have a policy and procedure(s) for the selection and purchasing of services and supplies
it uses that affect the quality of the tests and/or calibrations. Procedures should exist for the purchase,
reception and storage of reagents and laboratory consumable materials relevant for the tests and calibrations.
The laboratory needs to ensure that purchased supplies and reagents and consumable materials that affect the
quality of tests and/or calibrations are not used until they have been inspected or otherwise verified as
complying with standard specifications or requirements defined in the methods for the tests and/or
calibrations concerned. These services and supplies used need to comply with specified requirements and
records of actions taken to check compliance are to be maintained.
Documentation
An efficient laboratory requires good quality records and procedures. In this context "document" could be
policy statements, procedures, specifications, calibration tables, charts, textbooks, posters, notices,
memoranda, software, drawings, plans, etc. These may be on various media, whether hard copy or electronic,
and they may be digital, analog, photographic or written. All documents issued to personnel in the laboratory
as part of the management system should have been reviewed and approved for use by authorized personnel
prior to issue.
With standard operating procedures these must be clear instructions as to ensure consistency: everyone
should perform the tests the same way so that the same result can be expected from all staff. Such
procedures should also ensure accuracy, since following written procedures helps laboratory staff produce
more accurate results than relying on memory alone because they will not forget steps in the process. The
objective is to achieve a consistent (reliable) and accurate result.
Records should be available for all aspects of the laboratory function. These can be manual or computerized
and they will contain are the collected information produced by the laboratory in the process of performing
and reporting a laboratory test (13). Characteristics of records are that they need to be easily retrieved or
accessed and that they contain information that is permanent and does not require updating. Examples
include:
• Sample logbooks
• Sample registers
• Laboratory workbooks or worksheets
• Instrument printouts
• Equipment maintenance records
• Quality control data
• External quality assessment or proficiency testing records
• Training records
• Results of internal and external audits
• Out-of-specification reports
• Incident reports

36
Test records must contain key information, such as (14):
• Identification of the test
• Identification of laboratory
• Unique identification and location the sample
• Date and time of collection, and time of receipt in laboratory
• Date and time of release of report
• Primary sample type
• Results reported in SI units or units traceable to SI units, where applicable
• Reference materials, where applicable
• The test result, reported in appropriate units of measurement
• Interpretation of results, where appropriate
• Applicable comments relating to quality or adequacy of sample, methodology limitations or other
issues that affect interpretation
• Identification and signature of the person authorizing release of the report
• If relevant, notation of original and corrected results
• If the record is a copy, that it is a certified copy of the original
With manual forms these should be authorized in advance and serialized, so that each test record had a unique
identifier.
All laboratory documents need to be controlled. A system must be established for managing them so that
current versions are always available. A document control system provides procedures for formatting and
maintaining documents. The auditor should assess whether there are outdated documents in circulation, if
there are any document distribution problems, and if there are any external documents that are not being
properly controlled. Control also extends to storage and archiving.
Data and Computerized Systems
Data integrity is an important area, applying both to paper records and automated systems (15). Information
systems include the management of data and records contained in both computer and non-computerized
systems. Laboratory management must ensure that documented procedures for sample collection, indexing,
access, storage, maintenance, amendment and safe disposal of quality and technical records are in place,
among other more specific test activities. Such procedures must guarantee that the laboratory has access to
the data and information necessary to provide a service which meets the needs and requirements of the user
(16).
Electronic systems should also have:
• Permanence—backup systems are essential in case the main system fails. Additionally, regular
maintenance of the computer system will help to reduce system failures and loss of data
• Security—password protection with multiple user levels of access
• Traceability—electronic record systems should be designed in a way that allows for tracing the sample
throughout the entire process in the laboratory
• Audit trails

37
Trending and Controls
Much of quality control is concerned with measurements or at least an assessment of a result against
predetermined limits. Where discrete results are produced, the laboratory should be undertaken trend
analysis (such as by using statistical process control) (17). Such approaches involve the application of statistical
methods to evaluate variability in the laboratory testing. Control charts provide the best means by which to
monitor the testing procedures. In analytical laboratories, control charts are produced by calculating the long-
term mean and range by averaging multiple sets of experimental duplicates over time (18). Through this, the
laboratory can put in place a system to establish an expected average and variation for future comparison.
Control charts thus provide a standard against which the stability of the laboratory performance can be
evaluated, and they should feature within the audit.
Laboratory Non-Conformances
The laboratory must have a policy and procedures that that is be implemented when any aspect of its testing
and/or calibration work, or the results of this work, do not conform to its own procedures or the agreed
requirements of the customer. The policy and procedures must ensure that:
The responsibilities and authorities for the management of nonconforming work are designated and actions
(including halting of work and withholding of test reports and calibration certificates, as necessary) are defined
and taken when nonconforming work is identified
• An evaluation of the significance of the nonconforming work is made
• Correction is taken immediately, together with any decision about the acceptability of the
nonconforming work
• Where necessary, the customer is notified, and work is recalled
• The responsibility for authorizing the resumption of work is defined
In addition, the laboratory would have needed to establish a policy and procedure and should designate
appropriate authorities for implementing corrective action when nonconforming work or departures from the
policies and procedures in the management system or technical operations have been identified. A problem
with the management system or with the technical operations of the laboratory can be identified through a
variety of activities, such as control of nonconforming work, internal or external audits, management reviews,
feedback from customers and from staff observations. Furthermore, when improvement opportunities are
identified or if preventive action is required, action plans should be developed implemented and monitored to
reduce the likelihood of the occurrence of such nonconformities and to take advantage of the opportunities
for improvement.
Self-Inspection / Self-Audit
The laboratory should periodically, and in accordance with a predetermined schedule and procedure, conduct
internal audits of its activities to verify that its operations continue to comply with the requirements of the
management system. The internal audit program should address all elements of the management system,
including the testing and/or calibration activities. It is the responsibility of the quality manager to plan and
organize audits as required by the schedule and requested by management. Such audits need to be carried

38
out by trained and qualified personnel who are, wherever resources permit, independent of the activity to be
audited.
OTHER CONSIDERATIONS Other important considerations for the audit include the use of reference materials, repeated analyses, and
sample and reagent blank analyses.
Example Checklist for Auditing the Laboratory
Below is an example checklist that can be used or adapted for auditing a laboratory function. The checklist is
aimed at the quality control laboratory function. When considering the checklist, the following framing
questions may be useful:
• What procedures and processes are being followed in the laboratory
• What is being done at the time and against which procedure?
• Do the current procedures and processes comply with written policies and procedures?
• Are there written policies and procedures for each activity?
• Do written policies and procedures comply with appropriate standards, regulations, and
requirements?
The checklist below consists of example area are audit questions that could be asked.
LABORATORY MANAGEMENT
Example Questions:
• Are there a nominated manager & deputy who are suitably qualified and experienced?
• Is there a suitably qualified quality control manager responsible for all quality control activities in the
laboratory?
• Is the laboratory appropriately accredited?
• Are the methods for analysis of parameters of interest accredited?
• With laboratory management, the system should cover work carried out in the laboratory's permanent
facilities, at sites away from its permanent facilities, or in associated temporary or mobile facilities (if
applicable).
Staff Competency
• Is the laboratory manager supported by an adequate number of qualified staff, trained in the
principles and practice of relevant areas of analysis;
• Is a training procedure in place for laboratory staff? (This procedure should cover both analytical
procedures and the relevant principles and practice of analysis, including calibration and internal and
external analytical quality control)
• Do the training procedures set criteria and method of assessment of the competence of staff to
conduct analysis?
• Are staff training records in place and kept up to date? (a training record should set out clearly those
procedures and practices in which staff have been trained, the dates and results (competency) of that

39
training, the dates and results of audits of training and any re-training and the results of any annual
review)
Laboratory Quality Systems
• Is a documented quality manual in place?
• Is the quality manual based on appropriate the requirements (according to a local or international at
standard)?
• Does the quality manager conduct audits to assess compliance with systems and methods? (these
audits should be reviewed)
• Sampling
• The laboratory should have a sampling plan and procedures for sampling when it carries out sampling
of substances, materials or products for subsequent testing or calibration.
• The sampling plan as well as the sampling procedure must be available at the location where sampling
is undertaken. Sampling plans must, whenever reasonable, be based on appropriate statistical
methods. The sampling process will address the factors to be controlled to ensure the validity of the
test and calibration results.
• Sampling procedures should describe the selection, sampling plan, withdrawal and preparation of a
sample or samples from a substance, material or product to yield the required information.
• Does the laboratory have procedures for recording relevant data and operations relating to sampling
that forms part of the testing or calibration that is undertaken? (These records should include the
sampling procedure used, the identification of the sampler, environmental conditions (if relevant) and
diagrams or other equivalent means to identify the sampling location as necessary and, if appropriate,
the statistics the sampling procedures are based upon).
Sample Shipment, Receipt and Storage
• Is there a SOP for sample receipt, shipment and storage of materials and test samples?
• Does the SOP contain a chain of custody procedure?
• Is the sample receipt area maintained separate from the sample processing area?
Reference Materials
• Reference materials should be traceable to SI units of measurement, or to certified reference
materials.
• Internal reference materials must be checked. Checks are needed to maintain confidence in the
calibration status of reference, primary, transfer or working standards and reference materials.
Testing
• Are tests conducted according to authorized procedures?
• Are tests appropriate for the sample being tested?
• Are appropriate controls in place?
• Are results independently checked?
• Are there an assay validation, re-validation and limited validation process outlined in a SOP?
• Is there a written procedure for repeat testing or invalidating lab data? Is there a repeat decision tree?

40
• How are results that fail specifications investigated or non-conformances investigated?
• Are there validated methods and acceptance criteria for each test method?
• Is there a SOP for significant figures?
• Is there a SOP that outlines good documentation practices?
Reagent and Solution Labelling and Qualification
• Is there an SOP that outlines how reagents are labelled, how expiration dates are established?
• Are reagents qualified for use? Is parallel testing of reagents performed?
• Is there a current inventory of all reagents and solutions?
Test Records
• Does a documentation control system exist and is it functional?
• Is raw laboratory data recorded in lab notebooks, electronically, or controlled data sheets?
• Are laboratory final reports generated for clinical studies?
• Who reviews the reports?
• Is there a SOP that outlines the content of the final report?
• Is there a SOP or a system for the retention, storage, and destruction of records?
• Test reports should, where necessary for the interpretation of the test results, include the following:
o deviations from, additions to, or exclusions from the test method, and information on specific
test conditions, such as environmental conditions
o where relevant, a statement of compliance/non-compliance with requirements and/or
specifications
o where applicable, a statement on the estimated uncertainty of measurement; information on
uncertainty is needed in test reports when it is relevant to the validity or application of the test
results, when a customer’s instructions so require, or when the uncertainty affects compliance
to a specification limit
o where appropriate and needed, opinions and interpretations (see 5.10.5)
o additional information which may be required by specific methods, customers or groups of
customers
• Test reports containing results of sampling should include the following, where necessary for the
interpretation of test results:
o the date of sampling
o unambiguous identification of the substance, material or product sampled (including the name
of the manufacturer, the model or type of designation and serial numbers as appropriate)
o the location of sampling, including any diagrams, sketches or photographs
o a reference to the sampling plan and procedures used
o details of any environmental conditions during sampling that may affect the interpretation of
the test results
o any standard or other specification for the sampling method or procedure, and deviations,
additions to or exclusions from the specification concerned

41
Archiving
• Is there a dedicated facility/area for the archival of records?
• Is there control access to the archival facility?
• Is the environment of the facility monitored and controlled?
• Is the procedure for archiving records outlined in an SOP?
• Is the retention time for records stated in the SOP?
• Is there a method of electronic data archive?
Computer Systems
• Is access to computers limited by an individual username and password system (lab members cannot
share a username)?
• How is the computer network and computer systems maintained, if applicable?
• Are there a computer validation master plan and/or SOPs?
• List computers systems and software utilized. Validated?
• Are changes to computer systems controlled and documented?
• Are records of computer system errors maintained and investigated?
• Are records of hardware maintenance and repairs maintained?
• Are computers backed up routinely to prevent loss of data? Is there a backup log?
• Is there a preventative maintenance program for computer systems?
Equipment & Calibration
• Is a documented calibration program in place for all necessary equipment? (As well as major pieces of
instrumentation this should include small laboratory items e.g. pipettes, ovens)
• Are calibration records current for all equipment and maintained on file?
• Has traceability of the calibration been established to relevant SI units of measurement?
• Is a documented maintenance program in place in accordance with manufacture’s/supplier’s
recommendations for equipment utilized?
• Are laboratory facilities appropriate for the range of tests carried out?
• Is laboratory equipment located and utilized in an appropriate manner?
• Is the equipment and its software used for testing, calibration and sampling capable of achieving the
accuracy required and must comply with specifications relevant to the tests and/or calibrations
concerned?
• Have calibration programs been established for key quantities or values of the instruments where
these properties have a significant effect on the results?
• Before being placed into service, has equipment (including that used for sampling) been calibrated or
checked to establish that it meets the laboratory's specification requirements and complies with the
relevant standard specifications?
• Has equipment been operated only by authorized personnel?
• Are up-to-date instructions on the use and maintenance of equipment (including any relevant manuals
provided by the manufacturer of the equipment) available for use by the appropriate laboratory
personnel?

42
• Has each item of equipment and its software used for testing and calibration and significant to the
result should, when practicable, been uniquely identified?
• Are records maintained for each item of equipment and its software significant to the tests and/or
calibrations performed? The auditor should check that the records include at least the following:
o the identity of the item of equipment and its software
o the manufacturer’s name, type identification, and serial number or other unique identification
o check that equipment complies with the specification (see 5.5.2)
o the current location, where appropriate
o the manufacturer's instructions, if available, or reference to their location
o dates, results and copies of reports and certificates of all calibrations, adjustments, acceptance
criteria, and the due date of next calibration
o the maintenance plan, where appropriate, and maintenance carried out to date
o any damage, malfunction, modification or repair to the equipment
• Has equipment that has been subjected to overloading or mishandling, gives suspect results, or has
been shown to be defective or outside specified limits, been taken out of service? (equipment should
be isolated to prevent its use or clearly labelled or marked as being out of service until it has been
repaired and shown by calibration or test to perform correctly).
Calibration Certificates
• Do calibration certificates include:
o the conditions (e.g., environmental) under which the calibrations were made that have an
influence on the measurement results
o the uncertainty of measurement and/or a statement of compliance with an identified
metrological specification or clauses thereof
o evidence that the measurements are traceable
• Ensure the calibration certificate relate only to quantities and the results of functional tests. If a
statement of compliance with a specification is made, this must identify which clauses of the
specification are met or not met.
• If an instrument for calibration has been adjusted or repaired, the calibration results before and after
adjustment or repair, if available, should be reported.
Analytical Methods
• Are documented standard operating procedures in place for each test method?
• Are all relevant procedures based on reference standard methods (as defined in the license)?
• Is a copy of relevant standard available on-site?
• For analytical laboratories, does the laboratory have in place procedures for estimating uncertainty of
measurement?
Control of Data
• Are calculations and data transfers e subject to appropriate checks in a systematic manner?

43
• When computers or automated equipment are used for the acquisition, processing, recording,
reporting, storage or retrieval of test or calibration data, has the laboratory ensured that computer
software developed by the user is documented in enough detail and is suitably validated as being
adequate for use?
• Have procedures been established and implemented for protecting data; such procedures should
include, but not be limited to, integrity and confidentiality of data entry or collection, data storage,
data transmission and data processing?
• Are computers and automated equipment maintained to ensure proper functioning and are provided
with the environmental and operating conditions necessary to maintain the integrity of test and
calibration data?
Sample Storage
• Ask how samples are logged in and stored.
• Does the sample logbook (or other record) provide spaces for who delivered the sample and who then
took it for testing (chain of custody)?
• What type of samples might be temporarily stored while awaiting testing?
• What method validation or compendial reference supports the sample storage conditions (e.g.,
water)?
• What site SOP governs what happens when a sample time point is missed? (should be a deviation)?
• If LIMS is used for tracking all samples and activities, check if pen raw data precedes computer and
whether the former is properly retained
Transport
• Where applicable, has laboratory in place procedures for safe handling, transport, storage, use and
planned maintenance of measuring equipment to ensure proper functioning and in order to prevent
contamination or deterioration?
LABORATORY QUALITY CONTROL
Internal Controls
• Does the Laboratory have a documented internal quality control procedure in place?
• Are all relevant methods subject to internal quality control?
• Are quality control outputs subject to evaluation (such as charts maintained, are actions taken upon
failure)?
• Are acceptance criteria set for quality control fit for purpose?
External Controls
• Is the laboratory a participant in a laboratory proficiency scheme e.g. Aqua Check or EPA proficiency
scheme?
• The results of any analysis submitted has part of a proficiency should be checked, if any samples failed
the criteria set by the scheme, procedures should be in place to investigate)
• Are procedures in place to deal with proficiency scheme failures?
44
Method Qualification
• Laboratory method validation is the confirmation by examination and the provision of objective
evidence that the requirements for a specific intended use are fulfilled.
• Is a written methodology in place to determine the performance characteristics of test methods under
the following headings?
• Is method qualification assessed for: Limit of Quantitation, Accuracy, Precision, Uncertainty of
Measurement, Range & Linearity and System Suitability?
• Is a documented procedure in place to determine the suitability of the procedure for test matrices?
• Have the relevant test methods been assessed for their suitability to the test matrix?
• Is the introduction of test and calibration methods developed by the laboratory for its own use a
planned activity?
• Are validation activities assigned to qualified personnel equipped with adequate resources?
For recording findings. an example form for this purpose is:
Action Type Criteria Priority
Finding
Root Cause
Proposed Action
Due Date Task Assigned
To
Completion Date Task Verified By
Final Action
Action
Effectiveness
Evaluation Date Task Verified By
Table 1: Example laboratory audit action plan record

45
SUMMARY Each laboratory is a complex system, involving many steps of activity and many people. The complexity of the
system requires that many processes and procedures be performed properly, and this requires the adoption of
a quality management system model -one that considers the entire system, and which is focused on achieving
good laboratory performance. To assess the effectiveness of this system, periodic quality audits are required.
Quality audits of the laboratory help senior management to measure each different aspect of a laboratory
quality management system, tracking those measurements, convert the analysis results to opportunities to
produce optimized quality standards, processes and can add value to the overall organization. Effective audits
are also enabling, allowing the laboratory to understand its performance for planning and implementing the
quality system; monitoring effectiveness of the quality system; correcting any deficiencies that are identified;
and for working toward continuous improvement.
An audit of the laboratory represents a systematic examination of some part (or sometimes all) of the quality
management system to demonstrate to all concerned that the laboratory is meeting regulatory, accreditation
and customer requirements. This article has considered some of the essentials for the analytical function and
has provides some guidance against which current systems can be prepared or from which audit checklists can
be developed.
REFERENCES 1. ISO 9001:2015 Quality management systems — Requirements, International Standards Organization,
Geneva, Switzerland
2. WHO (2011) Laboratory Quality Management System, World Health Organization, WHO Lyon Office –
International Health Regulations Coordination, Lyon, France
3. Imoh LC, Mutale M, Parker CT, Erasmus RT, Zemlin AE (2016) Laboratory-based clinical audit as a tool for
continual improvement: an example from CSF chemistry turnaround time audit in a South-African teaching
hospital, Biochem Med (Zagreb). 26(2):194-201
4. Sandle, T. (2016) Risk-Based Approach to Internal Quality Auditing, Journal of Validation Technology, 22
(1): 1-10
5. Kobewka DM, Ronksley PE, McKay JA, Forster AJ, van Walraven C. (2015) Influence of educational, audit
and feedback, system based, and incentive and penalty interventions to reduce laboratory test utilization:
a systematic review, Clin Chem Lab Med.;53(2):157-83
6. Sandle, T. (2014) Best Practices in Microbiology Laboratory Training. In Handlon, G. and Sandle, T. (Eds.)
Industrial Pharmaceutical Microbiology: Standards & Controls, Euromed Communications, Passfield, UK,
2.1-2.24
7. McDowall, R.D. (1992) Strategic approaches to laboratory automation. Chemometrics and Intelligent
Laboratory Systems, Laboratory Information Management, 17: 265–282.
8. Conti, T.J. (1992) LIMS and quality audits of a quality control laboratory. Chemometrics and Intelligent
Laboratory Systems, Laboratory Information Management, 17: 301–304.
9. Ceriotti, F, Brugnoni, D, Mattioli, S. (2015) How to define a significant deviation from the expected internal
quality control result. Clin Chem Lab Med., 53: 913–918

46
10. Wadhwa V, Rai S, Thukral T, Chopra M. (2012) Laboratory quality management system: road to
accreditation and beyond, Indian J Med Microbiol.;30(2):131-40
11. Westgard, J.O. (2013) Statistical quality control procedures. Clin Lab Med 2013; 33: 111–124
12. Westgard, J.O. (2003) Internal quality control: planning and implementation strategies. Ann Clin Biochem.,
40: 593–611
13. Miller, S.D. (1992) Removing guesswork from the selection of laboratory information management
systems. Chemometrics and Intelligent Laboratory Systems: Laboratory Information Management, 17:
259–264
14. Wagar EA (2006) Patient safety in the clinical laboratory: a longitudinal analysis of specimen identification
errors. Archives of Pathology and Laboratory Medicine, 130(11):1662–1668
15. Sandle, T. (2016) Data Integrity Considerations for the Pharmaceutical Microbiology Laboratory, Journal of
GXP Compliance, 20 (6): 1-12
16. Weiss RB, Vogelzang NJ, Peterson BA, et al. (1993) A successful system of scientific data audits for clinical
trials. JAMA, 270:459-464
17. Kinns, H, Pitkin, S, Housley, D. (2013) Internal quality control: best practice. J Clin Pathol; 66: 1027–1032
18. Deng, H.; Runger, G.; Tuv, E. (2012). System monitoring with real-time contrasts, Journal of Quality
Technology. 44 (1). pp. 9–27

47
CLEANING AGENT SELECTION AND CYCLE DEVELOPMENT By Ivan Soto, Director Validation, Emergent BioSolutions
INTRODUCTION The selection of cleaning agents is one of the most crucial factors in developing cleaning cycles for
biopharmaceutical manufacturing equipment. Most companies typically address this element following the
implementation of a validated cleaning cycle, only to encounter issues such as visual inspection failures and
other cleaning related issues.
Cleaning validation is a critical department that is always scrutinized during regulatory inspections. Deviations
related to cleaning failures are records that regulatory agencies routinely request during inspections. Cleaning
failures related to visual inspections and residue left in manufacturing equipment are key indicators of
potential issues with the cleaning cycle.
One of the principal areas frequently overlooked is the selection of cleaning agents. Typically, most validation
engineers and subject matter experts confronting cleaning failures, concentrate their efforts on the cleaning
cycle and related parameters instead of the cleaning agents. One of the primary reasons that companies
ignore the possibility of failures related to the cleaning agent is due to either a lack of process knowledge or to
circumvent the need to revalidate the cleaning cycle.
Unfortunately cleaning failures that are related to selecting inadequate cleaning agents have a negative impact
on productivity, timely equipment releases for manufacturing operations and higher operational cost. This
article will discuss strategies about how to select adequate cleaning agents.
CONSIDERATIONS FOR SELECTING A CLEANING AGENT The selection of cleaning agents requires careful consideration of several factors including chemistry of
cleaning, historical cleaning data, and lab evaluation.
Cleaning Chemistry
The chemistry of cleaning is one critical factor that needs to be taken into consideration when developing a
cleaning cycle. Different cleaning agents have different chemical properties that have an impact on the ability
to develop an effective cleaning cycle. All cleaning agents have different cleaning mechanisms that can
effectively clean a variety of different contaminants. The table below describes the function of each
component found in cleaning agents.

48
Table 1: Cleaning agent component and function
Alkalinity and acidity are among the most important of properties to be considered when selecting an
adequate cleaning agent.
Alkaline cleaning agents. Alkaline agents typically consist of a specifically formulated mixture of chemical
blends containing potassium hydroxide, sodium hydroxide, other alkaline salts, wetting agents, and chelating
agents that dissolve or emulsify fats, oils, and greases. CIP-100 is a widely use alkaline cleaning agent in the
pharmaceutical and biotech industry. Alkaline cleaning agents are effective in removing successfully different
type of contaminants including the following:
• Organic acids
• Tableting excipients
• Protein residues
• Fermentation residues
• Oils, waxes, and fats
• Grease
• Polysaccharides
Acidic cleaning agents. Acids typically consist of a specifically formulated mixture of chemical blends
containing phosphoric acid, surfactants, and chelating agents. CIP-200 is a widely use acidic cleaning agents
used in the pharmaceutical and biotech industry.
The selection of cleaning agents is a critical factor during the development and design of an effective cleaning
cycle that can successfully validated and that it provides consistent cleaning during the entire lifecycle of the
product.
DESIGNING AN EFFECTIVE CLEANING CYCLE Designing a cleaning cycle requires a science-based approach that takes into considerations the following key
elements:
• Soils to be removed
• Equipment to be cleaned
• Manufacturing conditions
COMPONENT FUNCTION
Bases Alkalinity source, hydrolysis
Acids Acidity source, hydrolysis
Water Solvent
Surfactants Wet, solubilize, emulsify, disperse
Chelants Tie up calcium, iron, magnesium
Builders Assist in detergency
Antimicrobials Kill, reduce microbes
Oxidants Oxidize, kill microbes

49
• Cleaning methods
• Cleaning mechanisms
• Cleaning parameters
• Cleaning agents.
Soils
The first consideration in the development of an effective cleaning cycle is to understand the process residue
details. The details that need to be clearly understood about the process residue includes the following:
• Aqueous
• Oil based
• Solids
• Powder
• Suspension
• Cell based
• Organic
• Inorganic
The physical chemical properties of the soils to be removed need to be well understood to enable the selection
of adequate cleaning agents and development of a successful cleaning cycle. Different soils have unique
physical chemical characteristics that require cleaning agents that are adequate to breakdown and remove
soils during cleaning. Biological process soils are quite different in nature to small molecule pharmaceutical
products and they require cleaning agents that are capable to breakdown macromolecular structures. It is very
critical to understand the worst-case soil generated during the process as this will be used as the basis for
cleaning cycle development and validation.
Equipment
Equipment design, size, dimensions, and materials of construction are important. The design of the equipment
is a critical factor that needs to be well understood to develop and effective cleaning cycle. Physical
characteristics such as the size and dimensions of the equipment have an impact on cleanability and cycle
development. The materials of construction are also another critical factor that needs to be considered during
cleaning cycle development. The equipment surface materials of construction are another critical
characteristic that need to be well understood during cycle development. The type and grade of stainless steel
have an impact on cleanability and the removal of process residues. Glass surfaces have distinct characteristics
than metal which have an impact on cleaning cycle development and ensuring adequate removal of process
residues.
Manufacturing Process
The manufacturing conditions also have an impact on the selection of a cleaning agent and development of an
adequate cycle that successfully removes process soils. Sterile manufacturing conditions require cleaning
agents that clean and kill microbes generated during the manufacturing process. The details of the
manufacturing process including critical parameters such as temperature, agitation speed and pressure have

50
an impact on cleanability and the removal of process residues. Manufacturing equipment that is subject to
elevated temperatures and cool downs tend to create hard to remove residues during the process. Dirty hold
times and decontamination steps performed during sterile manufacturing operations create additional
cleaning challenges that need to be well understood during cycle development.
Cleaning Cycle
Cleaning cycle parameters are an output of the development process that need to be based on the
understanding of all the factors and characteristics discussed above in this article. Cleaning cycle parameters
that are critical to the process includes the following:
• Time
• Action
• Cleaning chemistry
• Concentration
• Temperature
• Mixing/flow/turbulence
• Water quality
• Rinsing
Restrictions and constraints such as the manufacturing schedule, waste, and facility limitations also have an
impact on process residue removal and cycle development.
Small scale studies must be conducted during cycle development to facilitate an understanding recovery
factors and limit of quantification values for process residues. Small scale studies should emulate process
conditions to develop an early understanding of cleanability and process parameters.
SUMMARY Selecting a cleaning agent is one of the many critical steps that need to be define during cycle development.
Selecting the right cleaning agent requires a science-based approach that begins with understanding the
following factors:
• Understanding not only the active but all potential sources of contributing residues
• Understanding equipment design issues and process issues
• Understanding limitations of cleaning process.
Process knowledge including constraints along with the factors discussed above are critical to develop an
adequate cleaning cycle that meets cleaning process requirements.

51
Computerized Systems Validation Industry
AGILE DATA-CENTRIC VALIDATION A Dynamic New Approach to Regulatory Compliance: Part 1. Overview
By Eric Toburen, Managing Partner / co-founder, Tx3 Services
ABSTRACT Computer System Validation (CSV) is a critical requirement for any life sciences company. CSV verifies that
computer systems and their associated applications operate in a consistent fashion and yield reproducible
results based on their intended use. Such validation is aimed at ensuring patient safety, product quality, and
data integrity when manufacturing regulated products. CSV is a stringent requirement of the US Food and
Drug Administration (FDA).
While documents and documentation have been the mainstay of compliance in the pharmaceutical industry
for years, the advent of new digital technologies is opening vistas to a better approach to validation…one that
does not rely on documents (paper or electronic), but instead focuses on the data, providing a more
comprehensive, integrated, and collaborative effort.
Recent statements by the FDA tout a renewed focus on “making risk-based regulatory decisions more modern,
more scientifically rigorous, and more efficient.” This paper will focus on how a data-centric approach extends
this focus to Computer System Validation.
INTRODUCTION Global pharmaceuticals and medical device markets are growing at a frenetic pace. Towards the end of 2018,
the FDA was on pace to approve 48 medical devices compared to 27 in all of 2017, and 60 novel drugs
compared to 46 in 2017 (1,2). This growth is driven by industry trends such as mergers and globalization, and
by new technologies and streamlined regulatory processes.
As a result of this growth, many life science companies find it increasingly difficult to demonstrate regulatory
compliance from discovery through commercialization. They maintain a fine balancing act, seeking to develop
and market their products quickly and cost-effectively, while at the same time strictly adhering to all
compliance requirements and regulations. Over the years, the validation process -- notably Computer System
Validation -- has been a critical factor for maintaining compliance.
REGULATORY CHALLENGES The importance of CSV cannot be understated. It is a complex exercise that must cover the entire lifecycle of
an application. Validation is aimed at a host of IT assets – from ERP systems, to lab data capture systems,
manufacturing, and clinical systems. And it is a process that is undergoing major changes.

52
Once the mainstay of the industry, the reign of paper-based document systems, has been steadily declining
over the last couple of decades. Geographically dispersed teams, storage limitations, and access/maintenance
challenges have helped fuel the transition to electronic-based documents.
In response to this trend, the FDA promulgated Code of Federal Title 21, Part 11 (better known as 21 CFR Part
11) to address the use and control of electronic records and electronic signatures. The European Union
followed suit with its Annex 11 regulation, which also addressed computerized systems as part of the Good
Manufacturing Process (GMP). Both major regulations mandate that all computerized systems and
applications adhere to good manufacturing, laboratory, documentation, and clinical practices (GxP).
In the US, failure to follow these guidelines can show up in FDA audits, which can then result in FDA warning
letters, longer regulatory approval timelines, and increased product development costs. Failure to take
corrective action can lead to further legal action, significant fines, and other penalties. FDA regulations
describe what is expected but are written in a way that does not prescribe how to comply. It is the
responsibility of the regulated company to interpret those regulations, and then define the internal procedures
needed to achieve compliance.
INFLUENCERS OF CHANGE The real drivers for change in validation and compliance are coming about from the rapidly evolving industry
landscape. Several industry trends are forcing companies to take a different look – and approach -- to
compliance challenges. These include:
Globalization
As companies grow and expand beyond national borders, they need to adapt their validation procedures to
the requirements of not just the FDA or other local body, but to a variety of different regulatory agencies. In
addition, validation teams are now geographically dispersed making communication and collaboration critical
elements to providing a consistent, defendable validation deliverable.
Mergers and Acquisitions
While the last couple of years may not measure up to the mega-merger deals of previous years (Pfizer and
Wyeth in 2009, Johnson & Johnson and Synthes in 2012; Bayer and Monsanto in 2016), continued merger
activity can be problematic in terms of multiple --and often redundant-- systems, non-integrated processes,
and differing procedures/processes.
Accelerated Pace
Novel drugs and biologicals are being approved quickly due to innovative efforts and efficiency improvements
by the FDA such as adaptive clinical trials. Better understanding of disease and drug mechanisms has bolstered
development of new targeted therapies. And approval of medical devices has surged at an unprecedented
level as consumers embrace wearables of all sorts: contact lenses to measure ocular pressure, patches that
measure blood glucose, and other innovations. Today even the applications in your cell phone might be
classified as FDA-regulated medical applications. These products are coming to market quickly but must first
undergo rigorous validation efforts before patients can enjoy their benefits.

53
Systems Which Lack an Enterprise-Wide View of Compliance Risk
Pharma – just like most other industries – has grown up with a “siloed” infrastructure -- one that is filled with
separate, individual business units, and operational areas. Many of these departments do not talk to each
other, let alone collaborate with one another. Data-centric CSV tools permit a broader, more integrated
compliance perspective.
Economic Pressures
Faced with increasing pressure to maintain competitiveness by shortening development timelines and
reducing costs, companies are finding they can no longer afford the inefficiency of lengthy, resource-
constrained, and manually-laden validation processes.
Outsourcing and Sub-Contracting
An increased number of companies are outsourcing major corporate functions such as research, product
development and manufacturing, which further strains the ability of current infrastructures and systems to
integrate test procedures while maintaining control and consistency across multiple partners/multiple sites.
Changing Regulatory Environment
Life science companies are already heavily regulated by several organizations including FDA, the European
Medicines Agency (EMA), the International Organization of Standardization (ISO), and many others. The
industry is also affected by non-FDA type regulations such as Sarbanes Oxley and General Data Protection
Regulation (GDPR). All these regulations routinely evolve to protect the interests of industry and consumers.
Limitations of Documents
Over time, it has become clear that current industry reliance on document-based validation (both paper and
electronic) is inefficient, costly, and risky. Some tools being used for CSV today – including MS Word and Excel
spreadsheets – aid in the documentation but not in the management of the underlying compliance process.
Supplementary tools can assist with the process by leveraging templates and electronic documents but are still
constrained by the inherent inefficiencies of using documents.
Inefficiencies abound when using documents for CSV. Paper documents require hand-written signatures and
are not easily routed or shared – especially among multiple locations. Paper documents are often stored in
binders and housed in different facilities, making timely retrieval difficult. The review and approval process is
prone to human error, and company manual processes are hard to enforce leading to inconsistencies.
Moreover, it is difficult to query data across multiple projects to reveal trends and to benefit from a risk-based
approach to CSV. Electronic documents (such as PDFs) offer some advantages over paper, but not much.
Electronic documents do allow for easier review and approval, document retrieval, and sharing. However, the
use of electronic documents does not allow you to unlock the value of the individual validation elements. Data
integrity concerns also arise because the documents often lack important metadata and their associated
electronic signatures often lack specificity. Electronic documents were not designed to improve the overall
compliance process; they simply facilitate the use of inefficient and antiquated systems.

54
DATA-CENTRIC CSV A data-centric approach to CSV moves away from the structure of static documents. It instead breaks the
information down into more granular data elements which are managed and approved along with supporting
metadata for use in myriad ways. While you can certainly create the familiar reports typically required by
auditors, this modern approach to CSV provides for a more rigorous use of testing data, such as comparing test
results and defects across multiple projects and multiple business units.
With the disruptive force of digital transformation hitting every industry, life science companies are beginning
to embrace the benefits of a comprehensive, integrated, data-driven approach to CSV. Thanks to digitization,
a host of new technologies, better tools, and increased automation capabilities are helping to fuel this change.
The benefits are many:
• Real-time insight and access: Gone are the days of “reactive” discovery and analysis to identify
compliance issues. Real-time access allows users to monitor and take corrective action before simple
compliance concerns become significant compliance breakdowns.
• Repeatable process: Quality control is paramount in all pharma business processes. Using
configurable workflows, data-centric CSV helps to provide repeatability and efficiency, greatly reducing
the risk of human error while enforcing the correct process.
• Test automation: Test automation can be easily incorporated into an application lifecycle
management (ALM) tool and leveraged along with manual testing. These test elements and their
associated execution results can be sent through a formal review and approval process while being
audit trailed. In addition, historical information such as number of executions, execution times, and
defect trends can be leveraged as future releases of the application are being considered.
• Comprehensive Reporting: The data-centric approach truly provides a panoramic view of the entire
CSV landscape – and captures all necessary information and data for rigorous, comprehensive
reporting. Real-time reports can show exactly where you are in a validation project, including what
reviews/approvals might be stuck and how you can re-assign elements to keep things moving.
• Analytics and analytical reporting: Life science companies are intense collectors and users of data;
however, they have historically been unable to extract intelligence from the underlying validation
data. A data-centric approach provides the ability to capture relevant and meaningful data at each
critical step of the process. This means users can analyze the data along the entire ALM cycle. For
example, analytics can help create “what-if” scenarios or highlight productivity (or lack thereof) with
different out-sourced teams.
• Traceability: The data-centric approach enables users to trace data across the full lifecycle –
regardless of the tools that are used (requirements tool, agile tool, testing tool, ITSM).
• Improved efficiency: It streamlines processes to help reduce processing time, while ensuring accurate
tracking.
• Reduced risk: A systematic, iterative approach, applied throughout a computer systems lifecycle,
helps drive better decision-making, ensure product quality, and minimize supply chain disruptions.
Factors that can reduce risk include repeatability, electronic signature security, visibility and an audit
trail.

55
• Reduced cost: Automating workflows, reducing unnecessary testing, and eliminating onerous,
inefficient tools are just some of the ways a data-centric approach can lower costs and keep pharma
companies competitive in the marketplace.
AGILE VS. WATERFALL METHODOLOGIES When it comes to a data-centric approach to compliance, it is necessary to discuss two different software
development methodologies.
Pharma companies have traditionally utilized the “waterfall” methodology,” a serial process where software
development is segregated into a sequence of pre-defined phases. From a project management perspective,
these phases would include feasibility, conception, requirement analysis, design, build, system testing,
production, deployment, and maintenance. Each of these phases represents a distinct stage of software
development, and each stage generally finishes before the next one begins.
In contrast, “Agile” methodology is an iterative, team-based approach. It emphasizes a flexible framework
designed to drive rapid delivery of an application in complete functional components. Delivery of new code
can happen in a matter of days or even hours instead of the months required by waterfall methods. Cross-
functional teams work on iterations of a product over a period of time (called “sprints”). More importantly,
Agile is open to requirements which change over time (even late in development) and encourages continued
feedback from the end users. Only a data-centric approach to CSV can accommodate this level of flexibility.
Due in part to the limitations of document-based validation, Agile methodology has not worked well in the
regulated environments of life sciences companies. However, changing the perspective from documents to
individual validation elements allows an Agile team to execute sprints and save validation specific data. Armed
with data-centric validation processes, more pharma companies are able to adopt Agile for its inherent
flexibility, continuous improvement, and speed.
Agile methods, combined with a data-centric approach to CSV compliance is a modern, rigorous, and efficient
combination. The Agile approach is driving greater success in life sciences companies around the globe.
FUTURE DISCUSSIONS More detailed discussions on specific considerations on this topic are planned for 2020.
SUMMARY The life sciences industry is undergoing major change. Digital transformation, increasing competitive pressures,
and need for more streamlined, cost-efficient processes is helping to drive that change.
Paper-based document validation is slow and inefficient. Electronic-based validation (using PDFs for example)
is somewhat better but still misses the mark and never fully leverages the intrinsic value of the validation data.
By focusing on data-centric validation, pharma companies can reap the rewards of real-time access to the
entire ALM process.

56
Data-centric validation, in combination with an agile methodology for software development, helps pharma
companies dramatically improve the compliance/validation process with lower costs, increased efficiency, and
improved accuracy. It allows IT professionals, compliance officers, and business analysts to manage and quickly
deliver better products to users the first time around.
REFERENCES 1. FDA. Medical Device Approvals.
https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceAp.... Accessed 12-20-
18.
2. FDA. Novel Drug Approvals.
https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm5.... Accessed 12-20-
2018.
3. FDA. Statement by FDA Commissioner Scott Gottlieb, M.D., on FDA's new steps to modernize drug
development, improve efficiency and promote innovation of targeted therapies," 10/15/2018.
https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm623411.htm. Accessed 12-
20-18.

57
POINTS TO BE CONSIDERED WHEN VALIDATING BIG DATA ENVIRONMENTS By Orlando Lopez, E-Compliance and E-Records Integrity, Subject Matter Expert
INTRODUCTION Big Data (1) is the method of collecting substantial amounts of data and immediate analysis to find hidden
information, recurring patterns, new correlations, and so on. Inherent are the following challenges for
consideration:
• The data set is so large and complex making traditional processing techniques ineffective.
• There’s difficulty analyzing, capturing, collecting, searching, sharing, storing, transferring, and
visualizing the data.
• Protection of personal data.
The volume of data has been steadily increasing making it difficult to collect and integrate all the information.
In Figure 1, electronic records (e-records) stored in the Big Data Level are central repositories (databases) of
data from one or more disparate sources. These sources are depicted in the figure as Operational Applications.
Figure 1: Sample Big Data Environments
From the context of the Big Data, the raw data (2) is extracted from its Operational Application or source
repositories locations. The raw data may be processed or transformed by applying rules or executing
programmatic functions. The raw data is converted to data (3) and then loaded into a final set of tables, Data
Marts, for the consumption of users.
The relevance of Big Data has motivated the European Medicines Agency (EMA) to establish a task force on
March 23, 2017 to evaluate its use in support of pharmaceutical research, innovation, and development (4).

58
WHAT CAN GO WRONG DURING THE VALIDATION PROCESS? From the context of Big Data functionality, the infrastructure, repository mappings, and integration sequencing
are critical elements to consider during the functionality design process.
The e-records acquisition process for Big Data storage uses standard technologies and procedures that
automatically replicate automatically each byte of data in several locations. An error in the manipulation of
data can generate errors affecting results loaded into Data Marts. Even worse, errors can result in serious
production issues and potential distribution of adulterated or misbranded products. Data Integrity for e-
records is also a critical topic to consider, but not discussed in this article.
Infrastructure
The capability to manage substantial amounts of bulk of data is provided by the technology infrastructure
which demands a lot of memory and data server capacity. In this case, critical issues in Big Data environments
are performance, usage demands, scalability requirements for analysis, integration with distributed storage,
and data processing.
In this case, a critical issue in Big Data environments withstands the performance, usage, and scalability
requirements for big data analyses and integrations with the distributed storage and processing of big data
infrastructures.
Some topics to be considered during the design are (5):
• Technology specific – network architecture, etc.
• Monitoring tools
• Logical, physical, and security configurations critical to proper system usage and maintenance
management
• Operating parameters
• Interfaces, connectivity, and interoperability between components and modules of infrastructure
• Assumptions and constraints of the infrastructure
• Support skills, training, and reporting requirements
• Backup and restore requirements (Recovery Time Objective and Recovery Point Objective, RTO and
RPO respectively)
• Capacity planning such as storage and memory needs
• Performance requirements
• Security and regulatory compliance
• Identification of suppliers
• Evaluation of the risk assessment
To demonstrate the suitability of a given infrastructure component, product, and/or service to the intended
use and, consequently, the qualification state of the infrastructure the main work products that are expected
include:

59
• Written design specification that describes what the technology infrastructure is intended to do and
how it is intended to do it
• A written test plan and procedures and practiced on Design Specification
• Test results and an evaluation of how these results demonstrate that the predetermined design
specification has been met
• Leveraging the test plan and associated procedure, it’s necessary to establish written procedures
covering equipment calibration, maintenance, monitoring, and control. It is pertinent to highlight that
automated systems exchanging data with other systems should include appropriate built-in checks for
the correct processing of data. (EU Annex 11-5)
The primary concern of the computer infrastructure engineers is to design reliable infrastructure supporting
the Big Data environment. Developing an unreliable infrastructure results in a poor performing solution that
does not meet performance requirements.
Repository Mappings
Big Data requires consolidation of various structures and sources into one cohesive dataset for analysis.
This consolidation requires defining a plan, requirements, designing an e-records model, and functionality to
manage the model. Planning provides the information requirements of functions necessary to collect the e-
records along with technology-independent techniques to arrive at a set of e-records and activity models that
represent the cGMP process.
Data mapping is used as the first step for a wide variety of data integration tasks. It is a process of defining how
individual fields are mapped, modified, joined, filtered, aggregated, and so on to produce the final desired
output. Database mappings can have a varying degree of complexity depending upon the relational database
schema (including primary and foreign keys) and data sources.
A Data Mapping Specification is a special type of data dictionary that shows how data from one source
repository location maps to one-or-more target data repositories. This document is developed during the data
loads design. As part of the design verification process, the Data Mapping Specification is qualified. The results
of the Design Qualification (DQ) to the Data Mapping Specification must be documented. It focuses on
ensuring that the proposed design is suitable for the intended purpose.
After clearly understanding file structure and their relationships, the e-records integrity risk assessment must
be re-visited (6). It is pertinent to highlight that automated systems exchanging data with other systems should
include appropriate built-in checks for the correct processing of data. (EU Annex 11-5)
The primary concern of Database Administrators in this environment is solving the specific problem of getting
the right data from point A to point B with the appropriate transformations. Not developing a Data Mapping
Specification and not performing the associated DQ can cause a prolonged period of debugging the data
mappings.

60
Integration Sequencing
Data integration involves combining and transforming data residing in various sources and providing users with
a unified view of them.
A sequential integration provides a real-time transformation of dissimilar data types from multiple sources
inside and outside a regulated Good Practice entity. This real-time integration provides a universal data access
layer (e.g., Data Marts) using pull technology or on-demand capabilities.
The Extract Transform Load (ETL) is an integration method tool for managing real-time data integration. An ETL
can collect, operate upon, move, and subsequently load large bulk sets of data. The target for ETL technology
is a database such as a data warehouse or data mart.
The capability to manage large body of data is provided by the infrastructure, including a great amount of
memory, robust databases, and a large capacity of data servers. ETL is not the only integration method for
managing big data. ETL is the integration method that applies in large volumes, complex transformations,
efficiency, and periodic runs.
The ETL is an application often estimated to consume 70% of the time and effort of building a data warehouse.
The complexity of the ETL is the reason why the system is so resource intensive. The complexity of ETL
products includes over forty (40) subsystems that are needed in almost every data warehouse.
These subsystems are configurable software connected to the databases individual record fields designed in
the Data Mapping Specification.
Two typical transformations are Derivation and Aggregations
Derivation comprises the creation of a new field whose value is calculated from one or more fields within an
input record. The algorithm that outlines derivation should be completely defined.
Aggregation involves the creation of a new field whose value is calculated to form one or more fields across
several input records. As in the derivation, the algorithm that defines the aggregation should be completely
defined.
Many of these transformations require assurance that the operations are completed following the proper
sequence, as applicable. The correct e-records integration sequencing provides the correct information to be
stored at the corresponding visualization repository intelligence repository. The use of operational checks is
required to enforce permitted sequencing of events, as appropriate (21 CFR Part 11.10(f).) The operational
checks usually fall in the category of operation sequencing and are built into the software.
An acceptable method for the validation operational checks is (7):
• The documentation of the program, including a requirements specification which describes what the
software is intended to do.
• The performance of inspections and testing so that no step or specification can be missed or poorly
executed/assigned.
• Documentation of the initial and final steps.

61
• The primary concern of integrating and transforming erroneously is to obtain incorrect data value(s)
and deviations of executed protocols
REFERENCES: 1. López, O., “Electronic Records Integrity in a Data Warehouse and Business Intelligence,” Journal of
Validation Technology, Volume 22 Number 2, Apr 2016
2. Raw Data - Raw data is defined as the original record (data) which can be described as the first capture
of information, whether recorded on paper or electronically. Information that is originally captured in
a dynamic state should remain available in that state (MHRA)
3. Data - Facts, figures and statistics collected together for reference or analysis. All original records and
true copies of original records, including source data and metadata and all subsequent transformations
and reports of these data, that are generated or recorded at the time of the GXP activity and allow
complete reconstruction and evaluation of the GXP activity (MHRA)
4. http://www.pharmtech.com/ema-creates-taskforce-big-data-0;
http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2...
5. López, O., “Infrastructure Lifecycle Approach,” in Computer Infrastructure Qualification for FDA
Regulated Industries, Eds (Davis Healthcare International Publishing, LLC., River Grove, IL, 1st ed.,
2006), pp. 5-23.
6. MHRA, “MHRA GxP Data Integrity Definitions and Guidance for Industry,” March 2018,
https://mhrainspectorate.blog.gov.uk/2018/03/09/mhras-gxp-data-integrity...
7. FDA, “Guide to Inspection of Computerized Systems in Drug Processing,” February 1983

62
Medical Device Industry
CLEANING VALIDATION FOR MEDICAL DEVICES EXPOSED TO A LARGE NUMBER OF PROCESSING AGENTS By Kurt Moyer, President, Pine Lake Laboratories
INTRODUCTION Medical devices are exposed to a wide range of processing agents and materials during manufacturing.
Depending upon the medical device, residual levels of these processing agents and materials pose a potential
toxicological risk to patients. Manufacturers of medical devices need to identify and properly control for
contamination of the medical device from processing agents and materials encountered during the
manufacturing of the device. This is done by validation of the cleaning of the medical device.
For the validation of the cleaning of a medical device, a cleaning limit needs to be established for each residual
processing agent. The cleaning limit is the level below which the residual processing agents pose no risk to the
patient. Traditionally this is done by performing a toxicological assessment following ISO 10993-12 on each
individual processing agent (and each component of a processing agent if it is a mixture) and setting a cleaning
limit for each chemical. When the total number of possible chemicals from the processing agents is small (less
than 10) and all have toxicological data available, this is the preferred approach. Unfortunately, this is
commonly not the case. If the manufacturer is faced with a large number of potential chemical residues from
the processing agents (greater than 10), the time and cost of a complete toxicological assessment to set the
cleaning limit of each component may be prohibitive. Also, complete toxicological data may not be available in
the literature which could prevent the determination of the cleaning limit for a specific compound.
We propose an approach to assist medical device manufacturers when faced with the challenging task of
setting cleaning limits for a large number of potential contaminants from process agents or when complete
toxicological data is not available. Our approach is to set a worst case scenario cleaning limit and then test the
medical device by a battery of sensitive analytical techniques to determine which of the potential
contaminants are actually present on the device at a level that could potentially present a risk to the patient
and eliminating from further evaluation any contaminants that are present at levels that present no risk to the
patient.
REGULATORY REQUIREMENTS The Quality System Regulations from the US FDA capture the requirement for medical device manufacturers to
validate the cleaning of the medical device by stating that each manufacturer shall (1,2):
1. Establish and maintain procedures to prevent contamination of product by substances that could be
expected to have an adverse effect on product quality.
2. Establish and maintain procedures for the use and removal of manufacturing materials to ensure that it is
removed or limited to an amount that does not adversely affect the device’s quality.

63
As should be obvious, any residual processing agent that presents a toxicological risk to the patient would
adversely affect the quality of the device.
In addition to the FDA QSR, ISO 13485 requires the establishment of documented cleaning specifications for a
medical device if the any of the following apply (3):
• Product is cleaned by the organization prior to sterilization and/or its use
• Product is supplied non-sterile to be subjected to a cleaning process prior to sterilization and/or its use
• Product is supplied to be used non-sterile and its cleanliness is of significance in use
• Process agents are to be removed from product during manufacture
As can be seen, both sets of regulations require cleaning validation for certain types of medical devices and
manufacturing practices.
REVIEW PROCESS FLOW TO IDENTIFY THE MANUFACTURING MATERIALS Before starting cleaning validation, the manufacturer needs to identify all of the manufacturing materials and
processing agents that contact the medical device. This is done by evaluating the complete manufacturing
process from beginning to finished product.
Manufacturing materials and processing agents can be broadly categorized into the following groups:
• Organic residuals. Examples: lubricants, detergents and disinfectants.
• Inorganic residuals. Examples: metals and metal ions.
• Particulates. Examples: Metallic particles from a cutting process.
The result of the process review should be a complete list of all possible contaminants that could be on the
device.
The manufacturer can perform a risk analysis to determine the likelihood of each processing agent remaining
as a residue on the medical device. For example, consider the following hypothetical process. A medical
device is exposed to chemical A in the first step of the manufacturing process and chemical Z is introduced in
the last step. After the medical device is exposed to chemical A, the medical device undergoes 3 steps in the
manufacturing process that are likely to wash off chemical A before the device is exposed to chemical Z.
Therefore, chemical Z is much more likely to remain as a residual than chemical A. In this case, the
manufacturer could use this risk assessment to focus more on chemical Z during the cleaning validation
following proper documentation of the risk assessment.
DETERMINATION OF WORSE CASE SCENARIO CLEANING LIMITS After the potential hazardous contaminants from the manufacturing process have been identified during the
risk assessment, the acceptable level for the contaminants is determined. Ideally, a toxicological assessment
should be done for each potential contaminant. However, if many potential contaminants have been
identified, the time and cost of a complete toxicological assessment may be prohibitive. In this case, the
determination of a worst case scenario cleaning limit is recommended.

64
To determine a worst case scenario cleaning limit, a level needs to be determined below which even the most
toxic contaminants would not present a risk to the patient. Unfortunately, the Quality System Regulations and
guidances from the US FDA do not provide any assistance in evaluating the toxicological risk of contaminants
on a medical device. However, guidelines from the pharmaceutical industry do pertain to establishing
thresholds for patient exposure and can be applied to this situation.
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use
(ICH) has issued a guideline titled “Assessment and Control of DNA Reactive (Mutagenic) Impurities in
Pharmaceuticals to Limit Potential Carcinogenic Risk” (M7). The ICH M7 guideline describes a consistent
approach to identify, categorize, and control DNA reactive and mutagenic impurities taken in by patients to
limit the carcinogenic risk. In ICH M7 can be found the Threshold of Toxicological Concern defined as follows
(4):
“A Threshold of Toxicological Concern (TTC) concept was developed to define
an acceptable intake for any unstudied chemical that poses a negligible risk of
carcinogenicity or other toxic effects.”
The TTC (expressed as µg/day) represents the level below which intake of a potential compound would not
present a significant carcinogenic risk to a patient. In ICH M7, the guidance also accounts for acceptable higher
daily intakes for shorter exposure periods. The recommended TTC by duration of treatment from the ICH M7
guideline are as follows (4):
Acceptable Intakes for an Individual Impurity
Duration of treatment ≤ 1 month >1 – 12 months >1 – 10 years >10 years to lifetime
Daily Intake (ug/day) 120 20 10 1.5
These acceptable intake levels are based upon exposure and are determined for mutagenic compounds.
Mutagenic compounds represent the worst case scenario for toxicity; therefore, any compound below these
levels for the listed duration of treatment would present no toxicological risk.
Although these recommendations were developed for pharmaceuticals, the acceptable daily intakes are only
based upon toxicity and the daily intake by the patient. Therefore, a manufacturer of a medical device could
use these levels based upon the intended exposure of the patient to the medical device.
The TTC could then be used to calculate a worst case scenario cleaning limit (CLwc) for all individual potential
contaminants as follows:
CLwc =TCC (ug/day)×(elution time (day) )/(# of devices)
where elution time is the time required for 100% of the individual potential contaminant to elute from the
device into the patient and the number of devices is the expected total number of devices used on the patient.
Since the elution time is most likely unknown, the worst case scenario would be for entire amount of the
potential contaminant to enter the patient on first exposure, so 1 day is used as the elution time for the CLwc.
If the number of devices is variable, a realistic estimate of the largest number of devices is used. The final units

65
for the CLwc are µg /device. Instead of the number of devices, an amount of device (i.e. weight, surface area,
length, etc.) can also be used.
Once the CLwc had been calculated, the medical device can be tested for residual process contaminants from
the manufacturing process.
ANALYTICAL TESTING FOR RESIDUAL PROCESSING AGENTS An analytical testing strategy needs to be developed that will detect all of the processing agents identified in
the risk assessment of the manufacturing process. While the analytical testing strategy will need to be specific
to the manufacturing process and the medical device, each analytical testing strategy will include the following
steps: Washing the residues from the medical device, testing the washes for the processing agents, evaluating
the results against the CLwc, and setting compound specific cleaning limits.
1 . Washing the medical device
The first step is to wash the medical device in solvents that are expected to dissolve the processing agent. The
residual processing agents are likely to have varying polarities; therefore, more than one solvent will probably
be needed. Usually water and an alcohol such as isopropanol will be sufficient but other solvents may be
appropriate based upon the expected residual processing agents. The solvents selected and the conditions
should be appropriate for the purpose of washing contaminants from the surface without being so aggressive
as to dissolve or cause leaching from the medical device.
2. Analysis for Residual Processing Agents
The washes are then analyzed for the residual processing agents. The analytical methods will be selected
based upon the residual processing agents with the following analytical methods being commonly used:
• Headspace GC-MS for volatile organic residual contaminants
• Direct Inject GC-MS for semi-volatile organic residual contaminants
• LC-UV/MS for non-volatile residual organic contaminants
• ICP-MS for inorganic residual contaminants
3. Evaluation of Results
At the end of the analytical analyses, the level of each residual processing agent is evaluated against the CLwc
with the following potential outcomes:
• The level of the residual processing agent is below the CLwc. The residual processing agent would not
present a risk to the patient and therefore the device would be considered clean from this processing
agent.
• The level of the residual processing agent is at or above CLwc. The residual processing agent should be
submitted for a toxicological evaluation to set a compound specific cleaning limit as described in the
next section.
4. Determine Compound Specific Cleaning Limit
The manufacturer should follow ISO 10993-17 to set the cleaning limits for each processing agent above the
CLwc. When limited toxicological data is available, at a minimum the LD50 values (readily obtained from the
MSDS) can be used to calculate the compound specific cleaning limit.

66
The compound specific cleaning limit will likely be higher than the CLwc. If the level of the residual processing
agent observed from the analytical analyses is below the compound specific cleaning limit, the device would
be considered clean from residual for the processing agent. If the level of the residual processing agent is
above the compound specific cleaning limit, the device would not be clean from that processing agent.
CONCLUSION Manufacturers of medical devices are required to demonstrate that their medical devices do not present a
threat to patient health from residual processing agents. An experimental strategy was presented that allows
the manufacturer to test for all the potential residual processing agents on the medical device based upon the
determination of a worst case scenario cleaning limit. For medical devices where there is a risk of a large
number of residual processing agents, the manufacturer may find this approach to be more efficient and less
costly than determining the cleaning limit for each compound from a toxicological assessment.
REFERENCES 1. Code of Federal Regulations, Title 21, Quality System Regulations, Part 820.70(e), 2013
2. Code of Federal Regulations, Title 21, Quality System Regulations, Part 820.70(h), 2013
3. ISO 13485:2003, Medical devices -- Quality management systems -- Requirements for regulatory purposes,
section 7.5.1.2.1.
4. The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human
Use (ICH) guideline titled “Assessment and Control of DNA Reactive (Mutagenic) Impurities in
Pharmaceuticals to Limit Potential Carcinogenic Risk” (M7) 31 March 2017

67
PROCESS PARAMETERS AND RANGE SETTINGS FOR MEDICAL DEVICE PROCESS VALIDATION By Yeong-Lin Chen, Process Validation Manager, SHL Group Taiwan (retired)
ABSTRACT During medical device process validation, its Operational Qualification (OQ) requires worst case testing for
upper and lower process parameter limits (or process parameter ranges). For mechanical-based medical
device products, since their production practice is in 2 levels (component production level and device assembly
level), it makes the process parameters and range settings study a big burden. This article describes the
process and process parameters characteristics of the mechanical-based medical devices, illustrates the
general approach for process parameters and range settings study, and suggest a simplified approach for the
study to relieve the burden.
INTRODUCTION For process validation practitioners in medical device industry, one of the most difficult validation activities is
equipment/process Operational Qualification (OQ), which needs to identify key operating process parameters
and set their ranges (or upper and lower limits) for OQ worst case (or OQ window) testing according to GHTF
process validation guidance (1) developed for medical device industry. As the GHTF suggestions, Screening
Experiment of Design of Experiment (DOE) should be adopted to identify the key input variables (or key
process parameters), and then Response Surface Study of DOE should be conducted, and an equation can be
established that fits to the data to model the effects of the inputs (process parameters) on the outputs (quality
attributes). This equation can be used to find optimal targets and operating windows (or ranges) for key
process parameters.
Medical device industry encompasses a wide range of technologies and applications, and a major portion of
medical devices are mechanical-based products with mechanical in nature. For this kind of devices, their
production is a two-level practice, which means that device production consists of component production
(e.g., plastic component, metal component) and final device assembly production, and each production is with
their own batch number. For pharmaceutical production, it is chemical in nature and generally comprises one
level production, which means that the finished product (i.e., drug) is manufactured from the raw materials
and covered by one batch number. The differences of production level (2-level vs 1-level) and production
technology nature (mechanical vs chemical) bring out different requirements and implementation approaches
for DOE study between medical device and pharmaceutical industries.
While compared with pharmaceutical DOE study, mechanical-based medical device process parameter effects
on the product quality attributes are more obvious since the quality attributes for each component and device
finished product are clearly defined, and the parameter effects on the products (both component and finished
device) are more direct and science predictable, which means that we only need to understand the process
parameters effects on their direct product, either component or device assembly product. Two-level

68
production makes a mechanical-based medical device needs more DOE study items. For example: Each
medical device component and finished product could need a DOE study, so there are many DOE study items
in total due to multiple components in a device. However, for a pharmaceutical product, it generally needs
only one DOE study item which is for the finished drug product. Based on the above, each DOE study item
(e.g., a component DOE study) for medical device can be justified to spend less efforts than pharmaceutical
DOE study. Therefore, how to simplify each DOE study item, to relieve the mechanical-based medical device’s
heavy burden on too many DOE study items needed, is crucial to medical device process validation.
Despite the above, before starting to plan process validation for any process, we should determine whether
the process should be validated or is verification/testing sufficient to provide assurance of the required quality.
All of this was described in GHTF process validation guidance Section 3.1 - Process Validation Decision. For
example: Limited verifications in lieu of full validation at the component production level for select
components should be considered based on patient risk analysis, especially while there is company history on
similar components/processes, which will yield the desired benefits. Even though it may not yield fully
optimized results, but if component robustness is sufficient, there is no need to proceed process validation. If
such a determination cannot be made and documented, then the process should be validated.
MEDICAL DEVICE AND PHARMACEUTICAL PROCESS VALIDATION GUIDANCE
DOCUMENTS ON PROCESS PARAMETERS AND RANGE SETTINGS Both medical device and pharmaceutical process validation guidance documents describe that the
equipment’s key process parameters and their upper and lower operating limits (or “worst case” conditions)
should be studied, identified and tested in OQ phase, and the parameter limits range is considered as
anticipated operating range in routine production. The main process validation guidance documents for both
medical device and pharmaceutical industries are listed below.
Medical Device Process Validation Guidance
• GHTF/SG3/N99-10, Quality Management Systems - Process Validation Guidance (January 2004) (1)
Pharmaceutical Process Validation Guidance
• FDA Guidance for Industry, Process Validation: General Principles and Practices (January 2011) (2)
• EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use,
Annex 15: Qualification and Validation (March 2015) (3)
• PIC/S - Recommendation on Validation Master Plan, Installation and Operational Qualification, Non-
Sterile Process Validation, Cleaning Validation (September 2007) (4)
To facilitate the understanding of the process parameters and range settings requirements/approaches in the
validation guidance documents, all the related key points are extracted and analyzed in the tables below.
69
Section No.
Key Points
5.4 Operational Qualification (OQ)
• The use of statistically valid techniques such as screening experiments to establish key process parameters and statistically designed experiments to optimize the process can be used during this phase.
• In this phase the process parameters should be challenged to assure that they will result in a product that meets all defined requirements under all anticipated conditions of manufacturing, i.e., worst case testing. OQ should include short term stability and capability of the process.
• During routine production and process control, it is desirable to measure process parameters and/or product characteristics to allow for the adjustment of the manufacturing process at various action level(s) and maintain a state of control.
Annex A.3
Design of Experiment (DOE)
• The term DOE is a general term that encompasses screening experiments, response surface studies, and analysis of variance. In general, a designed experiment involves purposely changing one or more inputs and measuring resulting effect on one or more outputs.
Author’s Analysis: Medical device process validation should follow the validation guidance since it is created for medical device industry. This guidance describes 1) The key process parameters (or critical process parameters) should be identified and optimized through statistic approach (i.e., Design of Experiment), 2) Worst case testing for all anticipated conditions (operating ranges of key process parameters) should be challenged in OQ with a shorter period of time (compared with PQ test) to make sure the process is capable (e.g., process capability index Cpk or Ppk should meet the acceptance criteria ≧1.0 or 1.33), 3) During routine production, the key process parameters are allowed for adjustment in the range to maintain a state of control.
Table 1 - GHTF/SG3/N99-10, Quality Management Systems - Process Validation Guidance (January 2004)
Section
No. Key Points
IV, A General Considerations for Process Validation
• The terms attribute(s) (e.g., quality, product, component) and parameter(s) (e.g., process, operating, and equipment) are not categorized with respect to criticality in this guidance. With a lifecycle approach to process validation that employs risk-based decision making throughout that lifecycle, the perception of criticality as a continuum rather than a binary state is more useful. All attributes and parameters should be evaluated in terms of their roles in the process and impact on the product or in-process material and reevaluated as new information becomes available. The degree of control over those attributes or parameters should be commensurate with their risk to the process and process output. In other words, more control is appropriate for attributes or parameters that pose a higher risk. The Agency recognizes that terminology usage can vary and expects that each manufacturer will communicate the meaning and intent of its terminology and categorization to the Agency.
IV, B, 1 Building and Capturing Process Knowledge and Understanding
• Designing an efficient process with an effective process control approach is dependent on the process knowledge and understanding obtained. Design of Experiment (DOE) studies can help develop process knowledge by revealing relationships, including multivariate interactions, between the variable inputs (e.g., component characteristics or process parameters) and the resulting outputs (e.g., in-process material, intermediates, or the final product).
• Risk analysis tools can be used to screen potential variables for DOE studies to minimize the total number of experiments conducted while maximizing knowledge gained. The results of DOE studies can provide justification for establishing ranges of incoming component quality, equipment parameters, and in-process material quality attributes. FDA does not usually expect manufacturers to develop/test the process until it fails.
70
IV, C, 1 Design of a Facility and Qualification of Utilities and Equipment
• Verifying that utility systems and equipment operate in accordance with the process requirements in all anticipated operating ranges. This should include challenging the equipment or system functions while under load comparable to that expected during routine production.
Author’s Analysis: The validation guidance is created for pharmaceutical industry. It provides the perception of process parameter’s criticality as a continuum rather than a binary state (critical or not critical) and mentions that the DOE study should be conducted in process design/development stage. Process parameter’s criticality classification as a continuum is a risk-based approach, which is a very normal requirement recently in health care industry. However, if process parameters are classified into too many risk levels (e.g., 5 levels: critical, high, medium, low, neglect), it could make the process validation exercises and routine production more complex and not easy to follow. Except for these points, all the others regarding process parameters and range settings are the same as the GHTF guidance.
Table 2 - FDA Guidance for Industry, Process Validation: General Principles and Practices (January 2011)
Section
No. Key Points
3.11 Operational Qualification (OQ) should include but is not limited to the following:
• Tests that have been developed from the knowledge of processes, systems and equipment to ensure the system is operating as designed.
• Tests to confirm upper and lower operating limits, and /or “worst case” conditions.
5.21 Process Validation
• A process validation protocol should be prepared which defines the critical process parameters (CPP), critical quality attributes (CQA) and the associated acceptance criteria which should be based on development data or documented process knowledge.
12 Glossary
• Design Space: The multidimensional combination and interaction of input variables, e.g. material attributes, and process parameters that have been demonstrated to provide assurance of quality. Working within the design space is not considered as a change. Movement out of the design space is considered a change and would normally initiate a regulatory post approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval.
• Quality by design (QbD): A systematic approach that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.
Author’s Analysis: The validation guidance is created for pharmaceutical industry. It categorizes process parameter and quality attribute criticality as binary state (i.e., critical or not critical). Design space concept is important to process control change management. DOE is the core of Quality by Design (QbD) for process understanding. The basic concept regarding process parameters and range settings are the same as the GHTF guidance.
Table 3 - EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary
Use, Annex 15: Qualification and Validation (March 2015)
Section
No. Key Points
5.4 Operational Qualification (OQ) - Overview Statement
• Operational Qualification is an exercise oriented to the engineering function, generally referred to as commissioning. Studies on the critical variables (parameters) of the operation of the equipment or systems will define the critical characteristics for operation of the system or sub-system. All testing equipment should be identified and calibrated before use. Test methods should be authorized, implemented and resulting data collected and evaluated.
• It is expected that during the Operational Qualification stage the manufacturer should develop draft

71
standard operating procedures (SOPs) for the equipment and services operation, cleaning activities, maintenance requirements and calibration schedules.
5.5 Operational Qualification - Essential Elements
• The critical operating parameters for the equipment or the plant should be identified at the Operational Qualification (OQ) stage.
• Studies on the critical variables should include a condition or a set of conditions encompassing upper and lower processing or operating limits and circumstances; commonly referred to as "worst case" conditions. Such conditions should not necessarily induce product or process failure.
• The completion of a successful Operational Qualification should allow the finalization of operating procedures and operator instructions documentation for the equipment.
Author’s Analysis: The validation guidance is created for pharmaceutical industry. Since the guidance only requires equipment IQ (Installation Qualification) & OQ, there is no requirement for the equipment PQ. Under such a condition, it mentions that the equipment SOP should be finalized after the equipment OQ completion (instead of equipment PQ completion). The basic concept regarding process parameters and range settings are the same as the GHTF guidance.
Table 4 - PIC/S - Recommendation on Validation Master Plan, Installation and Operational Qualification,
Non-Sterile Process Validation, Cleaning Validation (September 2007)
PROCESS PARAMETERS CATEGORIZATION AND DEFINITIONS It is well known that quality needs to be built into, rather than tested into, a product. Product quality should be
achieved and assured by design of an effective manufacturing process and by effective control of the process,
which depend on the process knowledge. Design of Experiment (DOE) study can help develop process
knowledge by revealing relationships, including multivariate interactions, between the process parameters
(process inputs) and the quality attributes (process outputs). In addition, both medical device and
pharmaceutical validation guidance documents emphasize that the upper and lower operating limits should be
challenged in OQ (i.e., worst-case testing for process parameter ranges) to assure that they will result in a
product that meets all defined requirements under all anticipated conditions of manufacturing. Therefore,
process parameters studies are critical to process control and validation. How to decide which process
parameters have a significant impact on the product quality attributes and set their operating ranges are
important to medical device industry, which are usually considered difficult and time/resources consuming
activities.
Before starting process parameters studies, process parameters categorization should be clearly defined. In
the paper scope, process parameter is defined as an input variable or condition (e.g., time, temperature,
pressure, pH) of the manufacturing process/equipment that can be directly controlled. To simplify the
categorization, two widely used terms (critical process parameter, critical quality attribute) are adopted, and
their definitions by ICH Q8 (5) are introduced below:
Critical Process Parameter (CPP)
A process parameter whose variability has an impact on a critical quality attribute and therefore should be
monitored or controlled to ensure the process produces the desired quality.
Critical Quality Attribute (CQA)
A physical, chemical, biological or microbiological property or characteristic that should be within an
appropriate limit, range, or distribution to ensure the desired product quality.

72
In medical device industry, since there is no standardized categorization for process parameters, also the
categorization could have a significant impact on medical device manufacturer’s process validation and routine
operation, each device company should have their own process parameter categorization clearly defined in
their Standard Operating Procedures (SOPs) and/or Validation Master Plans (VMPs). Quality related process
parameters (i.e., Critical Process Parameter, Non-Critical Process Parameter) and process performance (or
business) related process parameter (i.e., Non-Quality Process Parameter) are proposed with definitions
shown below.
Critical Process Parameter (CPP)
• A process parameter whose variability has a significant impact on a critical quality
attribute and therefore should be monitored or controlled to ensure the process
produces the desired product quality.
Non-Critical Process Parameter (NCPP)
• A process parameter whose variability has a minor impact on a critical quality attribute
but still need to be monitored or controlled (with less efforts while compared with CPP)
to ensure the process produces the desired product quality.
Non-Quality Process Parameter (NQPP)
• A process parameter whose variability has no effect on critical quality attributes but has a
significant impact on a process performance (e.g., yield, cycle time, machine smooth
running) and therefore should be monitored to ensure the process performance.
All three-type parameter setting requirements (including target and range where applicable) should be
stipulated in the equipment SOP so that the operators can follow and operate the machine correctly.
PROCESS PARAMETER RANGE AND OPERATION SETTING REQUIREMENTS For the three process parameter types, their process parameter range study and operating parameter setting
requirements are described below.
Critical Process Parameter (CPP)
• According to the process validation guidance documents, Critical Process Parameter (CPP)
and its range setting should be obtained with a DOE study and qualified during the OQ
stage by worst case testing. Since the CPP variability has a significant impact on critical
quality attribute, also the range setting has been qualified, the equipment/process
routine operation should set the parameter within the range according to the equipment
SOP.
Non-Critical Process Parameter (NCPP)
• During the screening experiment, Non-Critical Process Parameter (NCPP) is identified,
which does not need a range study/setting because its variability has a minor impact on
critical quality attributes, and therefore a single value is set and qualified in the OQ and
PQ stages. Since the NCPP is a quality related parameter and has been qualified, the
equipment/process routine operation should set the parameter according to the single
value setting shown in the equipment SOP.

73
Non-Quality Process Parameter (KPP)
• There is no requirement to conduct DOE for the Non-Quality Process Parameter (KPP)
because it is not quality related. However, to keep the equipment/process running at
good performance is important to business, the parameter study could be necessary but
not required. For operation flexibility, the parameter setting value can be described in the
equipment SOP as reference. In other words, while in routine operation, the parameter
setting can be adjusted according to the production needs without a binding requirement
by the SOP.
BENEFITS AND CONCERNS OF PROCESS PARAMETER RANGE STUDIES &
QUALIFICATION Process parameter range study and qualification can enhance the degree of process/product quality
assurance. However, it will always bring some concerns to business. In a real world “quality is a goal, and
resources are limited,” how to trade off the benefits and concerns is important and should be based on science
and risk considerations.
BENEFITS OF PROCESS PARAMETER RANGE STUDIES AND QUALIFICATION
Maximizing Parameter Effect Knowledge
• Design of experiment (DOE), a core component in Quality by Design (QbD), is a
multipurpose tool that is widely used for identifying important input factors (critical
process parameters), understanding how they are related to the outputs (critical quality
attributes), and optimizing the process parameters and ranges.
Establishing Process Control
• Many product quality nonconformities are not the result of errors, instead they are the
result of excessive variation and off-target processes. Reducing variation and proper
targeting of a process requires process controls on these input process parameters to
ensure that the outputs (product quality) conform to requirements.
Process Capability and Repeatability Understanding
• During the process validation, the process capability (Cpk or Ppk) for worst-case settings is
challenged by short-term OQ runs, while the process capability & repeatability are
challenged for parameter nominal set points (generally, the midpoint of the range) by
long-term PQ runs. Both the qualification runs can prove the process performance can
consistently/repeatedly meet the process capability requirement (e.g., Ppk ≧1.0 or ≧
1.33 depending on the quality risk evaluation).
Operational Flexibility
• Once the parameter range settings are qualified, the equipment/process operating
conditions can have a freedom to adjust within the range settings, which is important
during routine production because the variability of process input materials (or critical
material attributes) could happen.

74
CONCERNS OF PROCESS PARAMETER RANGE STUDIES AND QUALIFICATION
Parameter Range Studies Concern
• Design of Experiments (DOE) is a statistical tool for key variable screening (screening
experiments) and range optimization study (response surface study) that involves
planning experiments, conducting experiments, and analyzing/interpreting the data. The
difficult parts of DOE include how to select variable level for variable screening test, how
to maintain all experimental conditions at the same except the variable change, what
parameters should be selected as critical process parameters by data analysis and
interpretation, how many data points are needed and how to set the interest region in
the response surface study, and what statistical methods (e.g., regression) to use for
establishing the relationship equation between input parameters and output quality
attributes.
Parameter Range Qualification Concern
• During OQ/PQ qualification, the wider the parameter range is set, the more likely the
qualification test will fail. However, if a narrower parameter range is set, it will make the
process capability demonstration less meaningful because parameter variation could
happen, and calibration of parameter monitored gauge is always with allowed tolerance.
In addition, narrowing the range setting will reduce the operational flexibility in routine
production. Therefore, how to determine an appropriate parameter range for OQ test is
challenging.
Time and Resources Concern
• Because comprehensive experiments/sample inspection & testing/data analysis are
needed for both parameter range study and parameter range qualification, it will cost
massive time period and resources, which could be challenges for business, especially
process validation is considered the last quality check event prior to commercial
production.
MEDICAL DEVICE MANUFACTURING PROCESS AND PROCESS PARAMETERS
CHARACTERISTICS While compared with pharmaceutical manufacturing process characteristics, medical device manufacturing
process parameters are generally less complex and related to the product quality, and therefore do not need
to conduct DOE study as much as pharmaceutical process. Medical device manufacturing process and process
parameters characteristics compared with pharmaceuticals are described below.
Medical Device Manufacturing Process Characteristics
Except for chemical-based (e.g., lens care solutions) and software-based (e.g., mobile apps for processing
ECGs) medical devices, most of the medical devices are mechanical in nature. The process characteristics of
mechanical-based medical devices are described as follows.

75
• Mechanical-based medical device production process generally includes plastic component
manufacturing process (e.g., plastic injection molding), metal component manufacturing process
(e.g., forming, machining, polishing, thermal treatment, cleaning), and device assembly process.
• Mechanical-based medical device production process generally is classified as continuous (or semi-
continuous) process instead of batch (or semi-batch) process used for pharmaceutical production.
• Mechanical-based medical device production generally consists of two-level production (i.e.
component production level & device assembly level with different batch numbers) instead of one-
level production used for pharmaceutical production (i.e. only one batch number for all production
steps).
• Mechanical-based medical device production process technologies from different production scale
machines are generally different and cannot be shared, while pharmaceutical process technologies
between different production scale equipment (e.g., pilot-scale, commercial scale) are generally
similar and can be shared.
Medical Device Process Parameters and Range Settings Characteristics
• For pharmaceutical process parameters understanding, it is much more complex than medical devices.
Pharmaceutical process parameters understanding is through relating process unit operations
(including process parameters for each unit operation) to drug product critical quality attributes
(CQAs) using a cause-and-effect matrix, conducting DOE and comparing the parameter’s impact on the
CQAs to assign parameter risk level (e.g., high risk, medium risk, low risk), and then deciding the CPPs.
Compared with medical device two-level production (component production level & device assembly
level with different batch numbers), pharmaceutical one-level production is with a longer chain effect
between process parameters and final product CQAs (for example: If a drug product manufacturing
process consists of 5 unit operation steps, and one product CQA could be affected by unit operation 2,
4, & 5, then the relationship between each unit operation’s process parameters and the final product
CQA will be difficult to establish.), and also the cause-and-effect matrix relationship between process
parameters and CQAs are not obvious (e.g., CQA-drug degradation effected by moisture content,
temperature, particle size; CQA-drug dissolution effected by excipient attribute, water amount,
granule size). Except for the injection molding process (with complex parameter effects on the plastic
component), medical device process parameters are generally more direct and easier to assess their
impact levels to product CQAs (both component product CQAs and final device product CQAs).
Therefore, medical device process parameters DOE studies generally should be simpler than
pharmaceuticals.
• Pharmaceutical process is generally batch (or semi-batch) process, while medical device process is
more like continuous (or semi-continuous) process. For batch process, the process material conditions,
and process parameters change with time during the process, which makes the parameter settings
more complex. While for continuous process, the process parameter settings are simpler since they
generally remain constant during the processing time.
• For medical device process parameters, since they can be controlled at one set point, there should be
no need to provide ranges for them by initial thinking. However, in order to secure a high degree of
product quality and provide operational flexibility, range studies are still necessary for CPPs and this is
because any process parameter is measured/calibrated by instrument or gauge, which always is

76
accompanied by the measured value uncertainty (e.g., calibration allowed tolerance). In addition,
medical device production process could be sensitive to the process input materials (e.g., critical
material attributes, CMAs), which needs the parameter range to adjust and cope with batch-to-batch
variability of the CMAs.
• For pharmaceutical process parameters understanding, conduct DOE response surface study can
develop an equation that models the relationship between the input parameters and output quality
attributes. This equation is then used to determine the design space region of the input parameters
where the output quality attributes will meet specifications. Working within the design space is not
considered as a change, and the design space is proposed by the pharmaceutical applicant and is
subject to regulatory assessment and approval. Generally, normal operating range (or parameter range
setting) is within the Design Space to enhance the product quality assurance (see ICH Q8). However,
for medical device industry, there is no such a regulatory requirement, and generally no need to
develop the mathematic equation for process parameter design space identification. The reasons
could be the following: 1) Design space establishment is generally through massive experiments to get
multi-dimensional combination and interaction of process parameters on quality attributes presented
by mathematical model equations/graphs that have been demonstrated to provide assurance of
quality, and the studies are costly in time and resources; 2) Pharmaceutical production is in one level,
however, medical device production is in two-level, which means for a finished device production,
there are numerous components (appx. more than 10 components) need to be manufactured first and
then the device is assembled from them. If design space is required, every component plus finished
device product could need the design space study, which is too much and almost impossible; 3)
Pharmaceutical process is chemical in nature, the design space study results could be shared with
different production scale equipment (more worthy to do), while medical device manufacturing is
mechanical in nature, design space study results can only be applicable to the studied machine itself
and cannot be shared with another production ramp-up machine. However, under special conditions
(e.g., for an extremely critical plastic molding component), a detailed response surface study for
accurate model equation establishment could still be needed.
PROCESS PARAMETERS AND RANGE SETTINGS APPROACH FOR MEDICAL DEVICE
PROCESS VALIDATION Before equipment validation OQ, generally, we need to set the worst-case process parameter range (upper
and lower limits) for testing. GHTF Process Validation Guidance provides a general approach for how to obtain
optimal process parameters and their range settings: “In general, one should, identify the key input variables
(or critical process parameters), understand the effect of these inputs on the output, understand how the
inputs behave and finally, use this information to establish targets (nominals) and tolerances (windows) for the
inputs. Following techniques can be used: One type of designed experiment called a screening experiment can
be used to identify the key inputs. Another type of designed experiment called a response surface study can be
used to obtain a detailed understanding of the effects of the key inputs on the outputs. Capability studies can
be used to understand the behavior of the key inputs. Armed with this knowledge, optimal targets for the
inputs and tolerance analysis can be used to establish operating windows or control schemes that ensure the
output consistently conforms to requirements.”

77
To be specific and detailed, the general approach for the optimal process parameters and range settings is
broken down step-by-step as follows.
• Identify critical quality attribute (response variable): This is the test samples measurable outcome
(average, standard deviation) of the experiment, which should be identified at the first stage.
• Identify potential process parameters (input variables): Process knowledge and scientific knowledge
are applied to identify potential process parameters (e.g., develop an Ishikawa fishbone diagram)
which can have an impact on the critical quality attribute.
• Design and proceed screening experiment: Choose the region of interest (should be relatively large)
for each input variable as the variable high (+) / low (‒) limits, proceed full factorial design tests with
number of combinations (e.g., for 2 input variables, the test (or trial) number is four (22); for 3 input
variables, the test number is eight (23)). At the screening stage, the objective is to eliminate as many
parameters from the potential list of CPPs as possible so that the subsequent response surface study
for CPPs can be effectively conducted. Therefore, to save experimental resources, we can cut down
the test number by using fractional factorial design. For example: Use combination test number of “2 +
number of input variables” (e.g. 5 tests for 3 input variables, 6 tests for 4 input variables), which is
detailed in the section of Case Study.
• Screening experiment data statistical analysis: Data can be analyzed with the help of suitable statistical
software (e.g., Minitab, Excel) to demonstrate which input variable effect is not significant to the
response variable, and they (non-critical process parameters, NCPP) should be screened out of further
response surface study (only for CPPS). Often, we find that simple graphical methods play an
important role in data analysis and interpretation (see the section of Case Study).
• Design and conduct response surface study: Keep all the NCPPs at constant, choose the region of
interest (can be narrowed down while compared with screening experiment if necessary), use at least
3 points in the region (high (+), medium (0), low (‒) limits) to conduct full factorial design tests or a
simpler test approach (e.g., central composite design, with number of test to perform is expressed by
the formula 2k+2k+1, where k is the number of monitored variables). To further simplify the response
surface study, we can make the regions of interest for both screening experiment and response
surface study at the same (to be able to share the data) and insert the range middle point data for
response surface study (see the section of Case Study).
• Response surface study data statistical analysis: Data can be analyzed with the help of suitable
statistical software and develop an equation that models the relationship between the input variables
and response variable that can be used to locate input variable targets and ranges with optimization
and tolerance analysis, respectively. Within the range variation, it will not result in unacceptable
variation of the response variable (the quality attribute).
PROCESS PARAMETERS AND RANGE SETTINGS CONSIDERATIONS FOR MEDICAL
DEVICE PROCESS VALIDATION Process parameters and range settings are generally difficult and complex issues in medical device process
validation. The following exemplifies typical process parameters and range settings considerations for
mechanical medical devices.

78
Process Parameters and Range Settings Considerations for Plastic Component Production
• Plastic component production (or plastic injection molding process) is a complex process since there
are many process parameters, such as injection speed, injection pressure, melt temperature, mold
temperature, holding time, holding pressure and cooling time, that could affect the molding process
outcome (part dimension, cosmetic, function).
• According to the article “Medical Molding, How to Validate the Process” (6), it mentions that before
DOE study, process development, consisting of short-shot study, in-mold rheology, a stability or cavity-
to cavity balance run, gate-seal analysis, and a pack-pressure study, should be conducted to establish
suitable parameter window for the DOE study. DOE can define the optimum dimensional process
window and its specific influence on each dimensional response, and these process limits (or worst-
case conditions) will then be challenged in the subsequent operational qualification (OQ) test.
• For plastic parts with complex geometry, it could be with many critical dimensions (generally 5 ~ 10),
and all the dimensions could be influenced by change of each parameter, so the parameter effects are
overly complex. In addition, except the part should meet its specification (dimensional, cosmetic,
functional), molding cycle time needs to be considered as well, because it affects the molding capacity
directly. Therefore, DOE study could be a huge work including experiment, sample
inspection/testing/metrology, and data analysis, which makes the plastic molding process DOE
exceedingly difficult.
• Sometimes due to resources constraint, molding process parameters and range settings are identified
based on trial shots data and molding experts’ knowledge and experience instead of by formal DOE
study. Since all the process parameters and range settings will be challenged in the subsequent
operational qualification (OQ) and Performance Qualification (PQ), the product quality risk by
inappropriate process parameters and range settings is low.
• During process scale-up, for pharmaceutical, its DOE results are basically considered sharable between
different scales of the production equipment, while for different molding tools (e.g., 1-cavity vs 4-
cavity, cold runner vs hot runner) or different molding machines used for medical device, their DOE
study results could be quite different. Therefore, how to effectively simplify the molding process DOE
study is crucial to medical device process validation.
Process Parameters and Range Settings Considerations for Metal Component Production
• Metal component manufacturing equipment/processes are diversified, however, for metal product
manufacturing, generally the process parameters are few and their effects on the product quality
(dimension, no greasy dirt/burrs, function) are not difficult to identify.
• If the equipment is with pneumatic operation, air pressure target (or set) point (e.g., 5 bar) and range
(e.g., +/- 1 bar) can be easily identified and used for OQ window test. Except for parameter limits
challenge, OQ window test can help verify the equipment/process operation performance in a shorter
time period which is important and a prerequisite for the subsequent PQ test.
• To be more precise, the skills of identifying the process parameter set points and ranges for metal
component production depends on the process knowledge, science and risk considerations. For a

79
special condition, a formal DOE still could be needed, but generally the formal DOE study is not
required.
Process Parameters and Range Settings Considerations for Device Assembly
• The main purpose of device assembly is to assemble all the plastic and metal components into a
finished device without component damage and dislocation. Therefore, no part damage and no part
dislocation are the quality attributes of all assembly machines.
• For pharmaceutical production, process parameters are more closely related to drug quality attributes.
However, for medical device finished products, their variable-based quality (i.e., use specification
range limits to make accept or reject decisions) are more determined by the component design and
manufacturing rather than by the device assembly. Despite this, device assembly process is still
considered critical since it will affect device attribute-based quality (i.e., use the count of defects to
make accept or reject decisions). Any part damage and part dislocation could cause device mechanical
malfunction.
• For some assembly operations (e.g., ultrasonic welding, gluing, pouch hot sealing), since the
welding/gluing/sealing strength is critical to device quality (e.g., no parts separation, pouch seal
integrity), and there are many process parameters that could manipulate the operation results, more
in-depth DOE study for parameters and range settings could be needed.
• For advanced assembly machines with servo motor, there are many operation step parameters (e.g.,
position, speed) need to be set for the machine. Only the critical step parameter which affects
assembly quality directly should be considered CPP and with a range setting and challenged in OQ.
• General assembly machines are pneumatically driven, air pressure set point (including pressure alarm
setting) and range can be easily identified (without DOE) and used for OQ window test. But we should
know that air pressure just provides pneumatic cylinder to produce enough force to assemble the
components to the right positions (e.g., snap fit), and the assembly performance results are either in
right position or not in right position.
• Another important assembly equipment parameter is optical sensor parameter setting, the setting
purpose is to judge the object (either component or subassembly) on the machine is in good status or
in bad status by the sensor threshold value setting. The setting value needs to be tried and optimized
(by using the average and standard deviation data of known-good objects and known-bad objects) to
get the best differentiability between known-good and known-bad objects. Parameter range setting
could be needed but not required, and the decision depends on science and risk considerations.
• For product assembly machines, no part damage and no part dislocation are the machine performance
requirements (or the product quality attributes), which are attributes-based quality data (e.g.,
pass/fail) instead of variables-based quality data (numerical values). In statistics, to maintain the same
quality assurance level (i.e., same confidence level and same probability content level), the test sample
size of attribute-based test (i.e., test results are shown in defect counts) is much larger than the
variable-based test (i.e., test results are shown in numerical values), and thus more test samples are

80
needed for inspection/testing to make sure that the assembly product is with no damage and no part
dislocation.
Parameter Range Setting General Considerations
• If the parameter range is enlarged, the positive point is the larger range gives more equipment
operational flexibility (by parameter manipulation) to cope with the variability of the process input
materials, equipment condition, or environment condition. However, the negative point is the chance
of a successful OQ test result becomes less likely. So, it is a tradeoff issue.
• The parameter range setting should be larger than the parameter monitoring gauge’s allowed
calibration tolerance. If a larger parameter range setting is proven workable, the equipment
performance against the parameter uncertainty is more assured.
• If the DOE study is more formal, detailed and accurate, a larger parameter range could be set.
• While the parameter high (+) and low (‒) limits of DOE study can meet the equipment, performance
needs (e.g., process capability requirement), the two limit values can be used for the range setting.
• Good DOE study is generally very resources/time consuming. Sometimes, we need to simplify the DOE
study and combine the equipment/process experience to set the range. Under such a condition, a
pretest for the range setting should be conducted before the formal OQ window test runs to boost the
confidence that the range setting can pass the OQ test.
• There is no rule for how to finalize the range setting, and it all depends on the science data and risk
evaluation. If the range setting is too large, it may not pass the OQ test acceptance criteria. If the range
is set too small, it may cause problems during the routine production because there is no room to
manipulate the parameter. For these two conditions, we have to re-study the range setting and re-
validate the new range setting.
PROCESS PARAMETERS AND RANGE SETTINGS CASE STUDY FOR MEDICAL DEVICE PROCESS
VALIDATION
As mentioned earlier, the DOE study for process parameters and range settings is an effort intensive work. In
addition, for medical device production, it is with a two-level production practice, which means that a lot of
DOE study items could be needed because each component and finished device may need to have some sort
of DOE study. To relieve the DOE burden for mechanical-based medical device process validation, we may
simplify the DOE study with examples shown in the following two case studies.
Case 1 – Injection Molding Part Flash Formation Process DOE Study
In the article “Design of Experiments Application, Concepts, Examples: State of the Art” (7), its DOE study is
applied on injection molding process with the aim of improving product quality such as excessive flash. Factors
considered as affecting for flash formation are: pack pressure (A), pack time (B), injection speed (C), and screw
RPM (D), while clamping pressure, injection pressure and melting temperature were under control. Each factor
affecting flash formation is considered at low and high levels as level 1 and 2, respectively. Since full factorial
design was used, the test design matrix for 24 factorial design is with 16 trials. In order to simplify the DOE
study, total 6 trials (2 + number of input variables) are proposed, and the 6 trials test data, comes from the
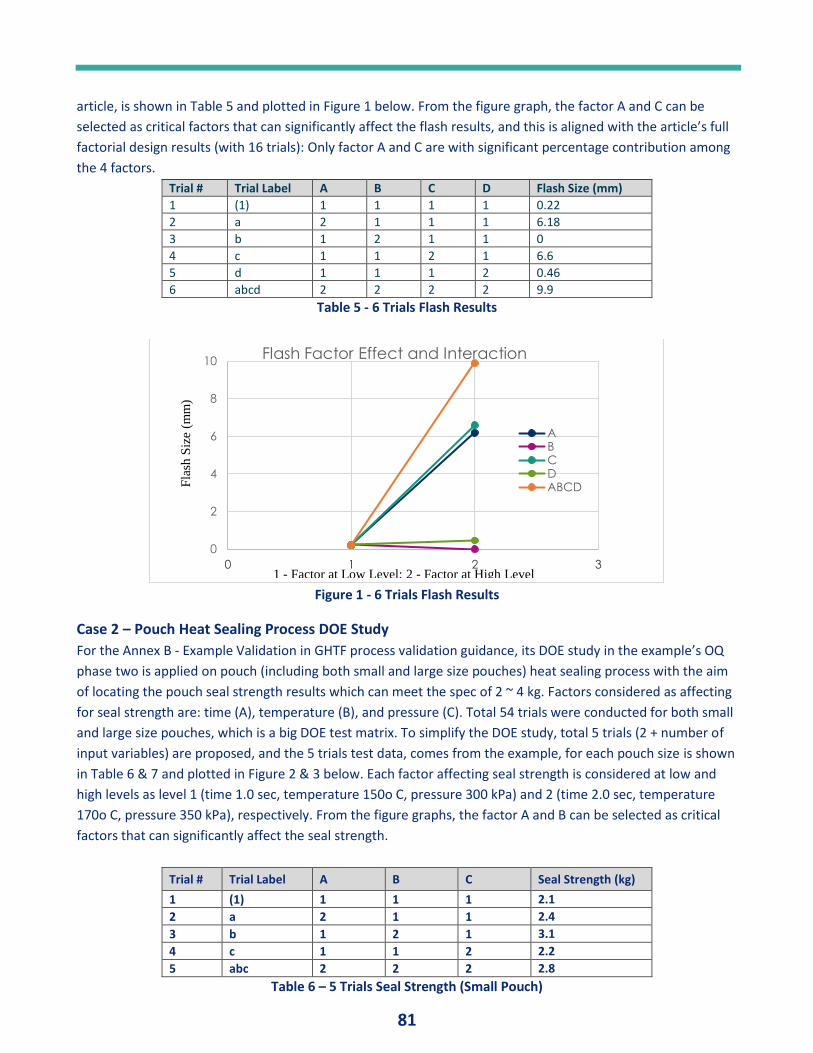
81
article, is shown in Table 5 and plotted in Figure 1 below. From the figure graph, the factor A and C can be
selected as critical factors that can significantly affect the flash results, and this is aligned with the article’s full
factorial design results (with 16 trials): Only factor A and C are with significant percentage contribution among
the 4 factors.
Trial # Trial Label A B C D Flash Size (mm)
1 (1) 1 1 1 1 0.22
2 a 2 1 1 1 6.18
3 b 1 2 1 1 0
4 c 1 1 2 1 6.6
5 d 1 1 1 2 0.46
6 abcd 2 2 2 2 9.9
Table 5 - 6 Trials Flash Results
Figure 1 - 6 Trials Flash Results
Case 2 – Pouch Heat Sealing Process DOE Study
For the Annex B - Example Validation in GHTF process validation guidance, its DOE study in the example’s OQ
phase two is applied on pouch (including both small and large size pouches) heat sealing process with the aim
of locating the pouch seal strength results which can meet the spec of 2 ~ 4 kg. Factors considered as affecting
for seal strength are: time (A), temperature (B), and pressure (C). Total 54 trials were conducted for both small
and large size pouches, which is a big DOE test matrix. To simplify the DOE study, total 5 trials (2 + number of
input variables) are proposed, and the 5 trials test data, comes from the example, for each pouch size is shown
in Table 6 & 7 and plotted in Figure 2 & 3 below. Each factor affecting seal strength is considered at low and
high levels as level 1 (time 1.0 sec, temperature 150o C, pressure 300 kPa) and 2 (time 2.0 sec, temperature
170o C, pressure 350 kPa), respectively. From the figure graphs, the factor A and B can be selected as critical
factors that can significantly affect the seal strength.
Trial # Trial Label A B C Seal Strength (kg)
1 (1) 1 1 1 2.1
2 a 2 1 1 2.4
3 b 1 2 1 3.1
4 c 1 1 2 2.2
5 abc 2 2 2 2.8
Table 6 – 5 Trials Seal Strength (Small Pouch)
0
2
4
6
8
10
0 1 2 3
Flash Factor Effect and Interaction
ABCDABCDF
lash
Siz
e (m
m)
1 - Factor at Low Level; 2 - Factor at High Level
82
Trial # Trial Label A B C Seal Strength (kg)
6 (1) 1 1 1 2.3
7 a 2 1 1 2.8
8 b 1 2 1 3.2
9 c 1 1 2 2.3
10 abc 2 2 2 3
Table 7 - 5 Trials Seal Strength (Large Pouch)
Figure 2 - 5 Trials Seal Strength (Small Pouch)
Figure 3 - 5 Trials Seal Strength (Large Pouch)
For response surface study, if 2 factors with full factorial design study with 3 data points in the range (or 3
levels), the trial number will be 32 = 9. While using a simplified central composite design, with number of trials
to perform is expressed by the formula 2k+2k+1, the trial number is still 9. In order to simplify the study, total
7 trials are proposed. Since we already have the data for 4 trials with trial label (1), a, b, abc, we only need to
add 3 more trials data (time 1.5 sec, temperature 160 oC, and Pressure 325 kPA for Level 1.5) and the result
graphs are shown in Figure - 4 & 5 below (data comes from the example). From the graphs, they show that the
factor combination effect on the seal strength is not linear and the range settings for parameter of A & B
should be roughly located between the level 1.3 to 2 (i.e., temperature range of 156 ~ 170 oC, and time range
of 1.3 ~ 2 sec). The simplified DOE study with total 16 trials for both small and large pouch size results optimal
2
2.5
3
3.5
0 1 2 3
Sea
l S
tren
gth
(kg)
1 - Factor at Low Level; 2 - Factor at High Level
Seal Strength Factor Effect and Interaction (Small Pouch)
ABC
2
2.5
3
3.5
0 1 2 3
Sea
l S
tren
th (
kg)
1 - Factor at Low Level; 2 - Factor at High Level
Seal Strength Factor Effect and Intercation (Large Pouch)
A
B
C
83
temperature range setting at 156 ~ 170 oC, which is aligned with the example’s 54 trials for both small and
large pouch size window test range 155 ~ 170 oC set for the OQ phase three test.
Figure 4 – 3 Levels Seal Strength (Small Pouch)
Figure 5 – 3 Levels Seal Strength (Large Pouch)
CONCLUSION Based on the analysis and description above, the following conclusions can be drawn:
• In FDA process validation guidance for pharmaceutical industry, it uses criticality as a continuum rather
than a binary state (critical, not critical), which is a risk-based approach stressing that control should be
commensurate with the risk. Risk approach is a standard requirement recently in health care industry.
For mechanical-based medical devices, criticality classification (for either process parameters or quality
attributes) by using the binary state may be more appropriate and is still deemed a risk approach.
Otherwise, multi-level criticality classification (e.g., 3 or 5 levels) will cause process parameter control
and documentation for both process validation as well as routine production more complex and not
easy to follow.
2
2.5
3
3.5
0 0.5 1 1.5 2 2.5 3
Sea
l S
tren
gth
(kg)
1 - Factor at Low Level; 1.5 - Factor at Middle Level; 2 - Factor at High Level
Seal Srength Linearity Study (Small Pouch)
A
B
2
2.5
3
3.5
0 0.5 1 1.5 2 2.5 3
Sea
l S
tren
gth
(kg)
1- Factor at Low Level; 1.5 - Factor at Middle Level; 2 - Factor at High Level
Seal Strength Linearity Study (Large Pouch)
ABABC

84
• Process parameter category should be defined clearly in company SOPs, since it will have a significant
impact on process validation exercises and routine production practice.
• Process parameters and range settings study (or DOE study) for mechanical-based medical device
process validation is an effort intensive work. How to relieve the burden of DOE study is crucial to
medical device process validation.
• DOE study is a kind of process understanding, and it does not have predetermined acceptance criteria
which is a prerequisite for process validation. To simplify the DOE documentation and approval, it is
proposed that DOE study should be separate from process validation OQ exercise. In other words,
manufacturers may choose to incorporate this DOE study during developmental work in separate
documentation packages prior to OQ.
• A simplified DOE study approach, such as the suggestions in the Case Study, can be justified for
mechanical-based medical device process validation based on the following reasons: 1) Too many DOE
study items could be needed for both component production and device assembly, 2) Equipment DOE
study result is only dedicated to the equipment itself, and cannot be shared between different scale
machines, 3) All the process parameters and range settings obtained during the simplified DOE study
will be challenged in OQ and PQ, and therefore the risk of inappropriate settings for both process
parameters and ranges is low.
• Despite the above, the issue of “how detailed a DOE study (e.g., full factorial design vs partial factorial
design, 3-level vs 5-level data points in the interest region should be tested, whether we should use
regression software to establish model equation) should be conducted to identify the critical process
parameters and optimize their ranges should be based on risk approach. In other words, under a
special condition with a very high-risk concern, a detailed DOE study still should be conducted.
REFERENCES 1. GHTF/SG3/N99-10, Quality Management Systems - Process Validation Guidance (January 2004)
2. FDA Guidance for Industry, Process Validation: General Principles and Practices (January 2011)
3. EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use,
Annex 15: Qualification and Validation (March 2015)
4. PIC/S - Recommendation on Validation Master Plan, Installation and Operational Qualification, Non-Sterile
Process Validation, Cleaning Validation (September 2007)
5. ICH Harmonized Tripartite Guideline, Pharmaceutical Development Q8 (R2) (August 2009)
6. “Medical Molding: How to Validate the Process,” Molding Technologies, Premiere Issue (July 2010)
7. “Design of Experiments Application, Concepts, Examples: State of the Art,” Periodicals of Engineering and
Natural Sciences, Vol 5, No 3 (December 2017)

85
PQ Forums
PQ FORUM #12 - VALIDATION APPROVERS
AND DOCUMENTS By: Paul L. Pluta, Editor in Chief, Journal of Validation Technology and Journal of GxP Compliance
ABSTRACT This discussion addresses two major problems associated with the site Validation Approval Committee (VAC) and the documents they approve. These include:
• VAC approvers. The number and expertise of required validation document approvers
• Documents reviewed. The types and numbers of validation documents reviewed, and their respective approval processes.
Validation documents are critical documents that must demonstrate technical excellence, compliance with regulatory requirements, and grammatical correctness. Document writers and approvers must ensure these requirements are met. Proposals to limit VAC document approvers based on technical expertise and identify which documents warrant VAC approval are described. Proposals addressing related documents that identify authors, require functional management approval, describe relationships to other validation documents, and recommend document storage/retention are also described. Successful integration and implementation of
“PQ Forum” provides a mechanism for validation practitioners to share information related to Stage 2
Process Qualification PPQ in the validation lifecycle. Information about supporting activities such as
design and development, equipment, and analytical validation will also be shared. The information
provided should be helpful and practical to enable application in actual work situations. Our objective:
Useful information.
Previous PQ topics discussed in this series include the following:
1. Is it Validated? published in Journal of Validation Technology (JVT), V 16, #3, Summer 2010.
2. Validation Equals Confirmation, JVT, V 16, #4, Autumn, 2010.
3. Responsibilities of the Validation Approval Committee, JVT, V 17, #1, Winter 2011.
4. Lifecycle Approach to Process Validation, JVT, V 17, #2, Spring 2011.
5. PQ Documentation – Three Simple Rules, JVT, V 17, # 3, Summer 2011.
6. Original Data Supporting PQ, JVT, V 17, # 4, Autumn 2011.
7. Sampling Pages, JVT, V 18, #1, Winter 2012.
8. Results Pages, JVT V 18, #4, Autumn 2012
9. PQ Initiation – What, Why, How, and What Else? JVT V 20, #4, 2015.
10. Technical Writing for Validation. JVT V24, #6, 2018.
11. Validation Document Writing Sequence. JVT V 25, #1, 2019.
12. Validation Approvers and Documents. JVT V 25, #3, 2019.
13. Validation Lexicon. JVT V 25, #4, 2019.
14. Numbers. JVT V 25, #5, 2019
Comments from readers are needed to help us fulfill our objective for this column. Suggestions for future
discussion topics are invited. Readers are also invited to participate and contribute manuscripts for this
column – please share your successful practices with others. We need your help to make “PQ Forum” a
useful resource. Please contact column coordinators Paul Pluta at [email protected] or Stacey
Bruzzese at [email protected] with comments, suggestions, or topics for discussion.

86
these proposals should improve document quality, increase efficiency, minimize approval times, and reduce workplace frustration.
INTRODUCTION The topic of validation problems was discussed with several validation and quality managers from multiple companies at IVT Validation Week in San Diego, CA, USA, October 2018. Several problems related to the Validation Approval Committee (VAC) were identified. Most comments addressed the specific problems of too many VAC approval signatures required to approve validation documents. The second most frequently mentioned problem was excessive numbers of validation documents requiring VAC approval. Site policies often identified organization members of the VAC and then required all VAC members to approve all validation documents. – the site had no flexibility in following policy. Some site policies were written by high level corporate Quality or Regulatory functions. These policies implied that more approval signatures would provide improved document quality, but without realizing the downsides of too many reviewers. More approvers resulted in more time needed to complete the approval process, usually with no document quality benefits. Some approvers did not have the technical expertise to critically evaluate document topics; for example, a Regulatory representative with background in clinical studies was not qualified to evaluate new manufacturing equipment qualification. Discussions with unqualified individuals also caused prolonged Q&A and time delays. These members also requested excessive changes in validation documents based on their limited understanding, again causing time delays and general VAC frustration. Related to the above is the topic of too many validation documents required to be approved by the VAC. There are many validation documents – specific PQ validation documents, directly-supportive documents, and more distant ancillary documents such as calibration, PM, drawings, and similar documents. Not all of these documents must be approved by the VAC. Some are more appropriately approved and retained by their internal functional departments. The scope and ramifications of these problems as described by managers is potentially overwhelming – too many approvers, approvers lacking technical expertise, busy approvers with excessive job responsibilities, too many documents to be approved, and combinations thereof – all leading to substandard documents, time delays, increased business pressure, and general work frustration.
Discussion Topics The following topics associated with the above described problems are discussed:
• Validation documents overview. Categories of validation documents
• Validation Approval Committee. Primary members and invited members
• Validation approver responsibilities. What is the primary work of the VAC?
• Validation document approval. An approach to simplify document approval requirements.
VALIDATION DOCUMENTS OVERVIEW The range of validation documentation has greatly expanded in the recent past. Validation documents were previously considered to be primarily protocols and results documents associated with the manufacturing process, equipment, utilities, computer system, or other specific validation. Other associated documents such as preventive maintenance schedules, system drawings, and calibration requirements were also required validation documents. A typical VAC would include all these documents as validation documents requiring VAC approval with retention in the validation library. Publication of the FDA validation guidance in January 2011 (1) greatly expanded the scope of validation activities and validation documents to include a staged lifecycle approach. Validation now included the following:

87
• Stage 1 – Process Design. Development of the validated process. This included topics described in ICH Q8/QbD (2).
• Stage 2 -- Process Qualification. These comprise traditional validation documents; however, protocol and results requirements were expanded and more thoroughly detailed;
• Stage 3 -- Continued Process Verification. Documents verifying post-validation performance were also now required validation documents.
While the FDA guidance specifically addressed manufacturing process validation, its concepts are now being applied to equipment, facilities, utilities, computers, and other validated systems – these too must be properly designed, tested to confirm performance, and continually monitored to verify acceptable ongoing operation. Some organizations have expanded VAC review to include even more documents such as User Requirements Specifications (URS), Design Requirements, Piping and Instrumentation Diagrams (P&IDs), User Manuals, and so on. The following proposes a general categorization of validation documents and describes their document origination, content, approval requirements, and retention storage. Three categories are proposed: Major Documents, Support Documents, and Ancillary Documents. Separation of these categories is integral to the proposed document approval process.
Major Documents Major documents are traditional validation documents. These include validation initiation documents, validation plans, protocols (PPQ, PV, PQ, others), IQ, OQ, and PQ equipment/facilities/utilities qualification documents, cleaning validation, computer system validation, analytical method validation, and other system validation documents. These documents are originated by the document technical author and have a broad range of technical content depending on the subject validation. The document author is primarily responsible for this document, and the author’s functional manager should approve the document affirming document quality. The approved document is then submitted to the VAC for site validation review and approval. Site approval of these documents is the primary responsibility of the VAC. The approved validation document is then retained in the validation library either as hard copy or as an electronic file. This describes typical validation document practice that has been in place in some organizations for 40+ years. This category of validation documents are the most frequent validation documents to be reviewed by regulatory auditors.
Support Documents Validation support documents are technical documents developed in organizations that directly support major validation documents and are vital to major document content. These documents are originated by a technical author who is responsible for content; the author’s functional manager should approve the document. The document is appended to the validation protocol or otherwise included with major validation documents. For example, a manufacturing process validation protocol should include a document describing critical process parameters (CPP) and critical quality attributes (CQA) for respective unit operations; this listing must be consistent with the process testing described in the validation protocol. Another example: Equipment qualification IQ/OQ/PQ protocols should include a listing of product operating conditions such as process temperatures at which equipment will be used; the range of process temperatures must be consistent with the equipment testing ranges in qualification protocols. Another example: Statistical analysis of experimental data that determines processing parameters must agree with that tested in the manufacturing process protocol. These examples illustrate a direct connection between support document content and protocol testing. Since these documents are appended and retained with the primary validation documents, their consistency with the protocol must be confirmed by the VAC. Document content, which may be complex and beyond the expertise of the VAC, has already been approved by the author and functional manager.
88
Ancillary Documents Ancillary validation documents are documents that are referenced in major validation documents. Example Ancillary Documents are preventive maintenance (PM) programs, calibration requirements, training documents, operating procedures, and similar documents. These documents are written by an appropriate author and approved by the author’s management. For example, an equipment PM program may be written by a manufacturing engineer and approved by Manufacturing Engineering management; it is then transferred to the site Preventive Management department for the recommended PM execution. These documents are usually retained within the respective related department under their control. They are referenced in the validation protocol confirming their availability but are not included in the actual protocol document. Ancillary documents are not approved by the VAC and may be beyond the expertise of the VAC.
Documents. Author, Management, and VAC Approval The above provides a structured approach to categorizing validation and validated-related documents. Implementing this approach should enable the VAC to better focus on major validation documents and fulfil VAC responsibilities. All documents are assumed to be written by expert functional authors and approved by their corresponding expert management. Table I summarizes the proposed document categories and approval signature requirements: .
DOCUMENT CATEGORY
DESCRIPTION DOCUMENT EXAMPLE
AUTHOR APPROVAL
AUTHOR MANAGEMENT APPROVAL
STORAGE LOCATION
VALIDATION APPROVAL
Major Validation documents
Plan, Protocol, IQ/OQ/ PQ, Results
Yes Yes Validation Library
VAC
Support Major document direct support
CPP, CQA, Test Justification
Yes Yes Validation Library
VAC approves consistency with major document. Author/Management approves technical content.
Ancillary Major document requirement
PM, Calibration, URS, Operating Procedures
Yes Yes Function Department
No document approval. Reference in protocol.
Table I. Document Categories and Approval Requirements
Category Terminology The above document terminology – “Major”, “Support,” and “Ancillary” were selected to hopefully be unique and to prevent multiple site usage of the same word for different applications. For example, “Primary” and “Secondary” are often used in connection with original data and transcribed data. “Level 1,” “Level 2,” and “Level 3” may be confused with validation “Stage 1”, “Stage 2”, and “Stage 3” terms. Organizations may prefer other descriptive words. Word choices should be described in a site lexicon containing definitions of all words as used at the site. The point of these categories was to distinguish between types of documents and identify their initiation, approval, and retention in the document approval process.

89
VALIDATION APPROVAL COMMITTEE MEMBERSHIP The site Validation Approval Committee (VAC) is a multidisciplinary committee with responsibility for approving all validation documents processed at the sight. The following proposes a general categorization of VAC members – permanent and invited -- and describes their appropriate function and application. This categorization is integral to the proposed document approval process.
Permanent Members Permanent members of the VAC are designated members of the major functional organizations at the site who are directly involved in major validation projects. Permanent members of the VAC are representatives of Manufacturing, Packaging, Product Technical Support, Engineering, Analytical, Environmental Monitoring, and other major site groups depending on organizational structure and functions. Even though these individuals are permanent members of the VAC, they are not required to approve all major validation document. They approve documents for which they have technical expertise and are able to critically evaluate content. Validation management and Quality Assurance management are permanent members of the VAC who must approve all major validation documents.
Invited Members Invited members of the VAC are designated members of the major organizations at the site who may not have frequent validation involvement but are invited as needed (ad hoc members) for specific validation projects. These may include representatives from Regulatory, various R&D functions, Microbiology, Statistics, and other groups who are consulted and asked to approve specific validation documents for which they have expertise. For example, a new product process validation being validated in advance of a pre-approval inspection would likely include Regulatory CMC and Product R&D as VAC approvers.
VALIDATION APPROVAL COMMITTEE RESPONSIBILITIES The site VAC has a vital responsibility – review and approval of site validation documents – critical to the success of the site validation program. The VAC must be an experienced and competent group and have a performance record for completing validation activities. Their responsibilities must be clearly defined and accepted by all members. They must have the time to execute these responsibilities. The vital roles and responsibilities of the VAC at the manufacturing site is briefly described as follows:
• Technical / Scientific Justification. The VAC must ensure that the validation / qualification and all associated activities are conducted with consideration for scientific and technical principles and utilize a generally consistent approach based on risk to patient, product, and process.
• Compliance with Regulations. The VAC must ensure that the validation / qualification is compliant with relevant policies, regulatory standards, local regulations, and industry expectations.
• Document Quality. The VAC must ensure that validation documentation must demonstrate the technical and compliance aspects of the validation in a logical and grammatically correct manner.
• Internal Consultant. The VAC should also serve as an internal consultant to site personnel working on validation projects and should help site staff to prepare acceptable documents. The VAC will be the ultimate approvers of these documents.
• Internal Regulatory Auditor. The VAC must also function as an internal regulatory agency auditor. They must critically evaluate validation documents in the manner of a regulatory auditor and not automatically approve documents through the system -- a key site responsibility. If a regulatory auditor finds deficiencies in a validation document, VAC members have not done their job.
Importance of VAC Responsibilities Members of the VAC must have the wisdom, knowledge, experience, foresight, maturity, interpersonal communication skills, and training to fulfil responsibilities of the VAC position. Further, they must have tie time to thoroughly evaluate validation documentation without distraction or other interfering regular job

90
responsibilities. When the number of VAC approvers are limited as proposed above, all VAC members must accept and embrace their specific responsibilities. Validation management must advocate for VAC members if significant member overwork is noticed. Senior management must respect the responsibilities of the VAC and not treat VAC work as extra work on top of the regular job responsibilities. VAC should not be used to train individuals- rather it can be considered a logical extension for those who have proven their validation skills to expand their capabilities. Case Study: One manager described a situation in her VAC in which several members were not proving critical input due to new facility construction and excessive regular site job responsibilities. One VAC member provided essentially all critical input to validation projects. Errors in protocol documentation eventually were unnoticed because they were assumed to have been checked by other members. When errors were ultimately determined, several protocol amendments were required, and corrected testing was repeated – an embarrassing situation resulting in substandard validation documentation. When VAC review personnel are limited as is proposed above, each member must provide critical evaluation and review. “Rubber stamp” approvals without thorough review by each VAC member in their area of expertise must not happen in a well-functioning VAC. There must be clear understanding of roles and responsibilities of VAC members.
VALIDATION DOCUMENT APPROVAL PROCESS The following validation document approval process is straightforward, streamlined, simple, and logical. Its effectiveness has been demonstrated. It proposes the following:
• All documents - major, support, and ancillary -- are written by competent authors.
• All documents - major, support, and ancillary -- are reviewed and approved by the author’s functional management.
• Major approved documents (plans, protocols, results) are approved by the VAC members with relevant expertise and responsibility associated with the validation. Their review should mirror the criticality at of a regulatory auditor.
• Support approved documents (QbD, CQA, CPP) are confirmed to be consistent with major validation documents by the VAC. No additional specific document approval by VAC – the author and functional manager have responsibility for support document content and quality.
• Ancillary approved documents are referenced in major documents. No specific document approval by VAC - the author and functional manager have responsibility for ancillary document content and quality.
Implementing the above document approval process should result in high quality documents written and approved by appropriate expert people. The approval process should be reasonably rapid and with minimal frustration. The VAC reviews and approvals major documents and confirms major document consistency with support documents. Major document approval is by competent people – not by many people who do not have relevant expertise. Approvals are from appropriate permanent VAC members and invited members with relevant expertise or responsibility. All documents are written by competent authors and approved by their respective management; there should be no need for additional VAC approval of support and ancillary documents. All personnel must fulfil their respective responsibilities – no one should assume errors will be detected by another reviewer.
Identification of Specific Project VAC Personnel Implied in the above proposal is the identification of appropriate permanent VAC and invited VAC personal to be approvers for specific documents. Identification of the appropriate people – but not an excessive number of people – is key to developing an efficient VAC approval process. For example, the VAC for an NDA tablet product/process validation utilizing new equipment technology would likely require personnel representing
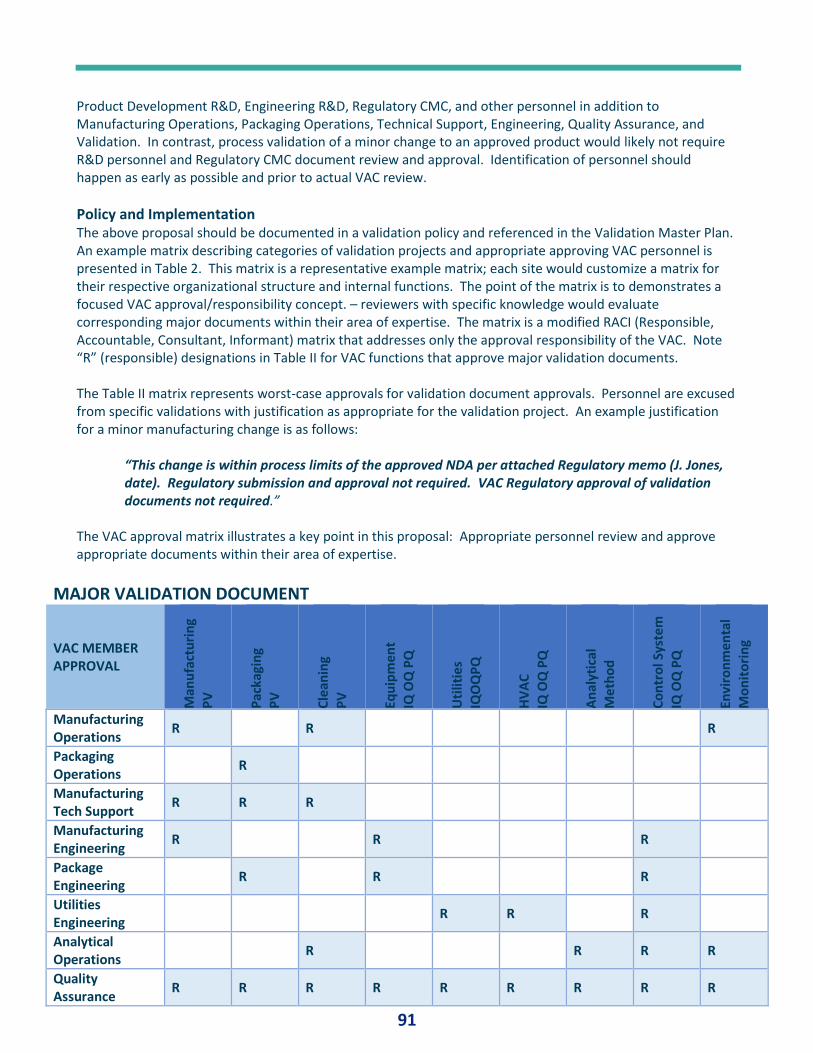
91
Product Development R&D, Engineering R&D, Regulatory CMC, and other personnel in addition to Manufacturing Operations, Packaging Operations, Technical Support, Engineering, Quality Assurance, and Validation. In contrast, process validation of a minor change to an approved product would likely not require R&D personnel and Regulatory CMC document review and approval. Identification of personnel should happen as early as possible and prior to actual VAC review.
Policy and Implementation The above proposal should be documented in a validation policy and referenced in the Validation Master Plan. An example matrix describing categories of validation projects and appropriate approving VAC personnel is presented in Table 2. This matrix is a representative example matrix; each site would customize a matrix for their respective organizational structure and internal functions. The point of the matrix is to demonstrates a focused VAC approval/responsibility concept. – reviewers with specific knowledge would evaluate corresponding major documents within their area of expertise. The matrix is a modified RACI (Responsible, Accountable, Consultant, Informant) matrix that addresses only the approval responsibility of the VAC. Note “R” (responsible) designations in Table II for VAC functions that approve major validation documents. The Table II matrix represents worst-case approvals for validation document approvals. Personnel are excused from specific validations with justification as appropriate for the validation project. An example justification for a minor manufacturing change is as follows:
“This change is within process limits of the approved NDA per attached Regulatory memo (J. Jones, date). Regulatory submission and approval not required. VAC Regulatory approval of validation documents not required.”
The VAC approval matrix illustrates a key point in this proposal: Appropriate personnel review and approve appropriate documents within their area of expertise.
MAJOR VALIDATION DOCUMENT
VAC MEMBER APPROVAL
Man
ufa
ctu
rin
g
PV
Pac
kagi
ng
P
V
Cle
anin
g P
V
Equ
ipm
en
t
IQ O
Q P
Q
Uti
litie
s
IQO
QP
Q
HV
AC
IQ
OQ
PQ
An
alyt
ical
M
eth
od
Co
ntr
ol S
yste
m
IQ O
Q P
Q
Envi
ron
me
nta
l M
on
ito
rin
g
Manufacturing Operations
R R R
Packaging Operations
R
Manufacturing Tech Support
R R R
Manufacturing Engineering
R R R
Package Engineering
R R R
Utilities Engineering
R R R
Analytical Operations
R R R R
Quality Assurance
R R R R R R R R R
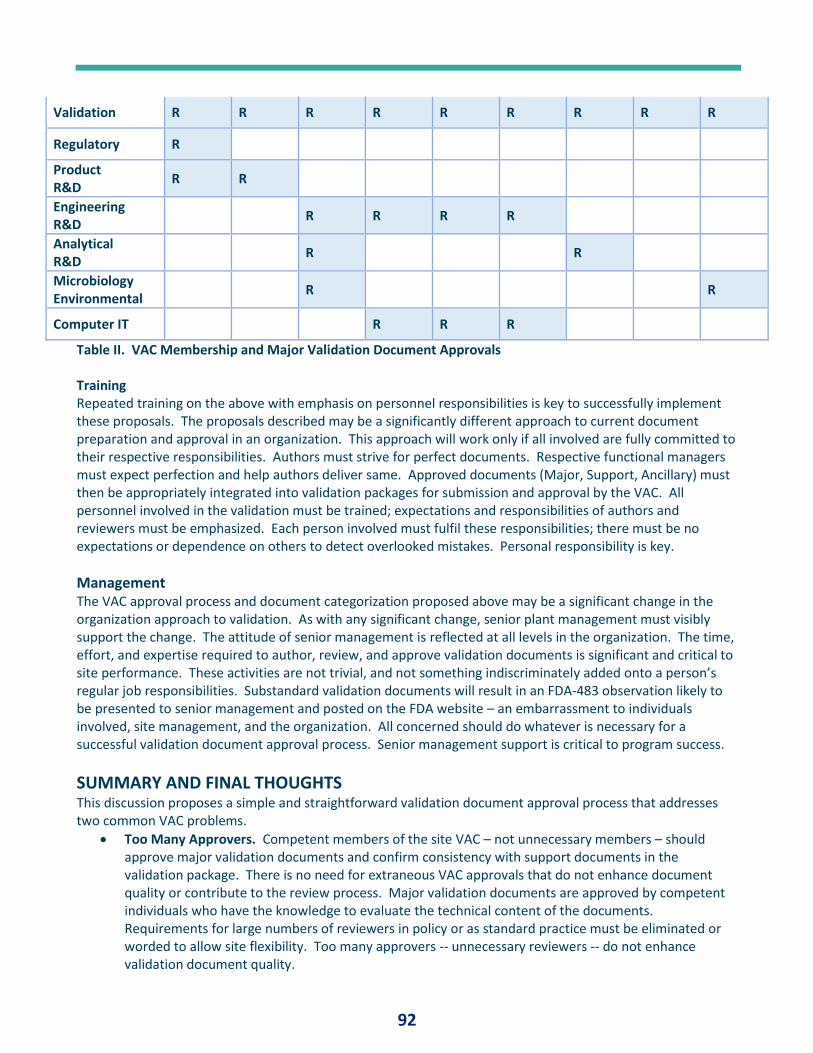
92
Validation R R R R R R R R R
Regulatory R
Product R&D
R R
Engineering R&D
R R R R
Analytical R&D
R R
Microbiology Environmental
R R
Computer IT R R R
Table II. VAC Membership and Major Validation Document Approvals Training Repeated training on the above with emphasis on personnel responsibilities is key to successfully implement these proposals. The proposals described may be a significantly different approach to current document preparation and approval in an organization. This approach will work only if all involved are fully committed to their respective responsibilities. Authors must strive for perfect documents. Respective functional managers must expect perfection and help authors deliver same. Approved documents (Major, Support, Ancillary) must then be appropriately integrated into validation packages for submission and approval by the VAC. All personnel involved in the validation must be trained; expectations and responsibilities of authors and reviewers must be emphasized. Each person involved must fulfil these responsibilities; there must be no expectations or dependence on others to detect overlooked mistakes. Personal responsibility is key.
Management The VAC approval process and document categorization proposed above may be a significant change in the organization approach to validation. As with any significant change, senior plant management must visibly support the change. The attitude of senior management is reflected at all levels in the organization. The time, effort, and expertise required to author, review, and approve validation documents is significant and critical to site performance. These activities are not trivial, and not something indiscriminately added onto a person’s regular job responsibilities. Substandard validation documents will result in an FDA-483 observation likely to be presented to senior management and posted on the FDA website – an embarrassment to individuals involved, site management, and the organization. All concerned should do whatever is necessary for a successful validation document approval process. Senior management support is critical to program success.
SUMMARY AND FINAL THOUGHTS This discussion proposes a simple and straightforward validation document approval process that addresses two common VAC problems.
• Too Many Approvers. Competent members of the site VAC – not unnecessary members – should approve major validation documents and confirm consistency with support documents in the validation package. There is no need for extraneous VAC approvals that do not enhance document quality or contribute to the review process. Major validation documents are approved by competent individuals who have the knowledge to evaluate the technical content of the documents. Requirements for large numbers of reviewers in policy or as standard practice must be eliminated or worded to allow site flexibility. Too many approvers -- unnecessary reviewers -- do not enhance validation document quality.

93
• Too Many Documents to Approve. All documents – major, support, and ancillary -- must be written by competent authors within their area of expertise and approved by their respective management. All documents must be high-quality documents – Not marginal document s with “let’s see if VAC will accept this.” If high quality documents are written by competent authors and approved by their management, there is no need for additional VAC approval. Expectations and standards for written documents must be clearly stated and embraced – personal responsibility is key.
Revising site policy and approach to validation document approval to include concepts described above will significantly benefit the organization. However, implementation will not be successful without repeated training and communication of expectations for associated personnel as well as visible senior management support. Competent people with a clear understanding of responsibilities and expectations and who are supported by site management are key to successful change in any organization.
REFERENCES 1. FDA. Guidance for Industry. Process Validation: General Principles and Practices. January 2011. 2. ICH. Pharmaceutical Development Q8(R2). August 2009.
ACKNOWLEDGMENT Helpful comments from Rich Poska and Alan M. Mancini are greatly appreciated.

94
PQ FORUM #13 - VALIDATION LEXICON By: Paul L. Pluta, Editor in Chief, Journal of Validation Technology and Journal of GxP Compliance
ABSTRACT This discussion addresses problems with usage of varied validation terminology within a pharmaceutical
manufacturing site. Inconsistent use of validation terminology is an ongoing problem for validation managers.
The development of a Validation Lexicon to standardize validation terminology and provide other benefits to
the validation quality system is proposed. A Validation Lexicon is a compilation of words associated with
validation practice at an individual manufacturing site. Document structure options and content are described.
Relation to the Validation Master Plan and other site validation documents is discussed. Initiation,
development, and ongoing management of the Validation Lexicon are described. The site Validation Approval
Committee is key to implementation of standardized terminology. Initiation of a Validation Lexicon project is a
difficult but worthwhile endeavour with expected frustration. A completed Validation Lexicon at a
manufacturing site demonstrates control of the site validation program, attention to regulatory requirements,
attention to the details of terminology, and control of validation content – all of which are characteristics of a
well-managed validation quality system that will create a positive impression on internal and external
regulatory auditors.
INTRODUCTION This discussion addresses problems with usage of validation terminology within a pharmaceutical
manufacturing site. It proposes the development of a Validation Lexicon to standardize validation terminology
and provide other benefits to the validation quality system. Each manufacturing site should have its own
Validation Lexicon to standardize and govern validation terminology. This discussion does not address
harmonization of validation terminology between multiple sites within a company – a much more complex
problem especially within global organizations and with mergers and acquisitions.
The general topic of validation problems was discussed with several validation and quality managers from
multiple companies at IVT Validation Week in San Diego, CA, USA, October 2018. Problems with validation
terminology were mentioned by all managers at the session. The major terminology problem identified was
the range of words used by distinct functions at their respective sites; specifically, the same words with
different meanings or different words with same meanings used within their sites. Validation managers were
unanimous in expressing frustration with these problems. Different functions at the site use different
terminology for the same category of document. For example, process validations may be termed process
validation by one group, verification by another group, and qualification by another group, all within the same
site. Individuals within the same function were sometimes inconsistent in their choice of terminology.
Validation authors may create combination documents; for example, a combined IQ and OQ will be written as
an IOQ. Consistent use of terminology with a single manufacturing is an ongoing problem; when multiple site
with a corporation are involved, the problem becomes much more complex. Communication between
companies is even more complicated. One manager described a group of eight managers from different
companies and several different industries discussing Validation Master Plans (VMP); each manager described
their respective site VMP – all eight names were different, and several had significant differences in content.

95
Development of a Validation Lexicon is proposed to unify personnel use of validation terminology in validation
documents within a given site. Many of the problems cited by Validation and Quality manages are addressed
by implementation of a Validation Lexicon.
Lexicon Definition
A lexicon is the vocabulary of a body of knowledge, such as a medical lexicon, baseball lexicon, computer
lexicon, cooking lexicon, or other specialized compilation of words associated with a specific knowledge topic –
a dictionary containing limited words connected to a specific topic. Lexicons are very useful documents. A site
lexicon focuses the user on specific related terms. A lexicon is especially useful as a learning tool for new
workers who are somewhat unfamiliar with the language of a given field.
Site Validation Lexicon
A site Validation Lexicon is a compilation of words associated with validation practice at an individual
manufacturing site. A site with a Validation Lexicon to which all functions have agreed should use the same
technical terminology on all validation documents at the site. The Validation Lexicon is an approved document
that standardizes terminology and is available to all site employees for reference and guidance. It also is
available to regulatory auditors to define validation terminology that will be reviewed in an audit.
Validation Terminology Evolution. Validation terminology has changed over the 40+ years of validation
performance. Different wording has been used to describe the same validation activity. For example, a
manufacturing process validation protocol has been known as Process Validation (PV), Process Qualification
(PQ), Process Performance Qualification (PPQ), Validation Protocol (VP), Qualification Protocol (QP), Process
Verification, and other terms. The 1987 FDA Validation Guidance (1) described Equipment: Installation
Qualification, Process: Performance Qualification, and Product: Performance Qualification. This document
addressed both pharmaceutical and medical device validation at that time. Terminology for medical device
validation has been addressed in the 2004 Global Harmonization Task Force (GHTF) (2) guidance and proposed
IQ for equipment, OQ for worst case product process testing, and PQ Performance Qualification for typical
product process validation. The 2011 FDA Process Validation Guidance (3) for pharmaceutical products
described a Process Performance Qualification (PPQ) protocol.
Global agencies have also stated preferred validation terminology. EU Annex 15 (4) describes IQ, OQ, and PQ
as part of equipment qualification but acknowledges that PQ may be performed with Process Validation. EMA
(5) defines Process Validation but does not discuss equipment validation or recommend terminology. The
Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S) (6)
describes Design Qualification (DQ), Installation Qualification (IQ). and Operational Qualification (OQ) for
equipment and Process Validation or Performance Qualification (PQ) for processes; The Australian Therapeutic
Good Administration (7) utilizes PIC/S terminology. Health Canada (8) utilizes similar wording as PIC/S, but with
slightly different definitions.
Validation Lexicon Benefits. Managers agree that a Validation Lexicon has several positive benefits.
• Standardized site terminology. All validation documents at a site uses the same terminology.
• Author resource. Document authors have an approved resource for selection of terminology for
documents they are preparing.
• Site training. New employees may read lexicon for training on site terminology and exposure to other
terms to help learn the “language” of validation.

96
• Audit reference. Regulatory auditors may read lexicon terms for site terminology orientation prior to
audit. Auditors adapt to different terminology used at different companies; however, they have
expressed concern when different functions within the same site use inconsistent terminology.
• Reference document for technology transfer between sites and contract organizations.
Implementing a Validation Lexicon. This discussion addresses preparation of a Validation Lexicon within an
individual manufacturing site. Implementing a Validation Lexicon is a complex endeavour that will likely
require substantial time and work. It requires agreement from function groups within the site to use the same
terminology for a given validation action. Some groups must concede that their terms are not preferred. After
agreements are reached, groups must then develop definition wording; regulatory-based definitions should be
used whenever possible. Getting staff to understand and use new terminology in their work, especially when
staff may not agree with changes to terminology, is a final and difficult hurdle.
When multiple sites and global sites within the same company are tasked with unifying terminology, the effort
is more complex. Mergers and acquisitions and other business transactions involving multiple manufacturing
sites may be handled in diverse ways. An acquiring company may mandate use of its terminology in all
manufacturing sites; mergers of equal partners may utilize a joint harmonization effort; still others may ignore
terminology differences between sites and allow the status quo. When companies undergo multiple corporate
changes, they may find themselves in a constant state of change. When a company utilizes multiple contract
manufacturing organizations, complexity increases exponentially. Some companies incorporate lexicons into
Quality Agreements with contract organizations to ensure consistent use of terminology.
Discussion Topics
The following topics associated with the above described problems are discussed:
• Validation Lexicon description, structure, and content. Basic design and options
• Development and administration of a Validation Lexicon. Role of the Validation Approval Committee
(VAC).
• Problem examples. Terminology problems descried by validation managers.
VALIDATION LEXICON DESCRIPTION, STRUCTURE, AND CONTENT A Validation Lexicon is a compilation of words and definitions associated with validation performance at a
specific manufacturing site including useful supplementary explanations.
Structure Options. There are two basic structure types for a Validation Lexicon – the dictionary approach and
the function approach.
Dictionary approach. The dictionary approach for the structure of a Validation Lexicon comprises an
alphabetical listing of selected words in the same manner as a standard dictionary. Sections are divided A to Z
as in a standard dictionary. This is the most straightforward approach to developing a site lexicon. Sites
developing their initial Validation Lexicon often utilize this approach to initiate the development of the lexicon
and the compilation and ordering of words.
Function approach. The other approach to structuring the Validation Lexicon is to develop function sections
containing validation terms specific to each function. For example, the lexicon would have sections such as

97
Process Validation, Cleaning Validation, Equipment Qualification, HVAC Qualification, Utilities Qualification,
and similar sections depending on site organization. Each section would contain words relevant to the function
with minimal overlap. For example, the equipment qualification section would be much different in content
than the cleaning validation section. This approach has the advantage of easy comparison of words specific to
a function. For example, an auditor wanting to compare site use of DQ, IQ, OQ, and PQ could easily look in the
Equipment Qualification section and see all definitions in close proximity; this same activity in a dictionary
format would require looking in the respective D, I, O, and P sections of the lexicon. The same words may be
repeated in different function areas with identical definitions for uniformity of use. For example, DQ, IQ, OQ,
and PQ would be listed in Equipment Qualification, HVAC, Utilities, Analytical, and other sections with identical
definitions.
Validation Master Plan (VMP) and Other Site Documents
If the site uses a function approach to the structuring of their VMP, the function order of sections in the
Validation Lexicon should be consistent with the site VMP. Similarly, if the site uses a function approach to list
completed validation and qualification documents, i.e., Validation Document Library, this too should be
consistent with the sections of the Validation Lexicon and the site VMP. All terminology in the site Validation
Master Plan and other site documents must be consistent with terminology in the Validation Lexicon.
Definition Content
Site validation definitions should be thorough and complete. Many definitions may be obtained from published
regulatory guidance or other technical references. Regulatory authority-based definitions are preferred to
definitions created by site personnel. The reference document should be cited along with the definition. See
Figure 1.
Figure 1. Validation Lexicon Definition – Process Validation
Definition Supplementary information. Site validation definitions will be more useful to users of the lexicon if
relevant supplementary information is provided. For example, a site using “process performance qualification”
Process validation: The collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality product. Process validation involves a series of activities taking place over the lifecycle of the product and process.
Process validation activities are described in three stages:
• Stage 1 – Process Design: The commercial process is defined during this stage based on knowledge gained through development and scale-up activities.
• Stage 2 – Process Qualification: During this state, the process design is confirmed as being capable of reproducible commercial manufacturing.
• Stage 3 – Continued Process Verification: Ongoing assurance is gained during routine production that the process remains in a state of control.
Reference: FDA. Guidance for Industry. Process Validation: General Principles and Practices. January 2011.

98
as terminology for manufacturing process validation protocols should provide reference for their preference.
The site should also list unacceptable terms that must not be used. See Figure 2.
Figure 2. Validation Lexicon Definition -- Process Performance Qualification (PPQ) Protocol
Another example: After defining User Requirements Specifications (URS), Functional Requirements
Specifications (FRS), Technical Requirements Specifications (TRS), IQ, OQ, and PQ, a site could provide
discussion and the Qualification V model diagram to demonstrate the relationships between equipment design
requirements and corresponding qualification testing (Figure 3), i.e., specific testing should confirm respective
requirement specifications of the constructed system. Validation definitions in the lexicon need not be limited
to very minimal statements but should provide useful and helpful information.
Figure 3. Equipment Qualification - Relationship Between Equipment Design and Qualification Testing
Table of Contents and Index
The Validation Lexicon should have a Table of Contents for sections and an Index with page numbers for all
definitions.
INITIATION, DEVELOPMENT, AND MANAGEMENT OF THE VALIDATION LEXICON
The site validation group should be responsible for development and management of the site Validation
Lexicon. The Validation function should be the site owners of the document and be responsible for maintaining
its content and usefulness.
Process Performance Qualification (PPQ) Protocol: The process validation protocol required in Stage 2 – Process Qualification that specifies the manufacturing conditions, controls, testing, and expected outcomes for this stage of process validation.
Reference: FDA. Guidance for Industry. Process Validation: General Principles and Practices. January 2011.
• Use Process Performance Qualification (PPQ) protocol as the title for all process validation protocols.
• Do not use Process Validation, Process Qualification, Performance Qualification, Validation Protocol, Qualification Protocol, Process Verification, or other terms as titles for process validation protocols.

99
Corporate Governing Documents
The first step in initiation Validation Lexicon development is to consult corporate validation policy documents
to which all site documents must comply. Corporate documents may require use of certain terms or may limit
other choices of terms. Corporate word lists may be extensive or may be minimal, only addressing major
categories of terms. Corporate documents take precedence over site documents. A site Validation Lexicon
must be consistent with corporate documents.
Word List Compilation
Management of site functions should be requested to provide words, definitions, and supplementary
information relevant to their function. SMEs or experts in each function should provide key terms, confused
terms, and other terms needing clarification for their respective function. Regulatory-based definitions from
published references should be used whenever possible.
Validation Approval Committee (VAC) Responsibility
While the site validation group is responsible for the maintenance of the Validation Lexicon, the site VAC is
responsible for technical correctness in the same manner as for site validation documents. All documents to be
incorporated into the Validation Lexicon are submitted to the site VAC for review and approval as any
validation document. The site VAC has several essential functions related to the Validation Lexicon.
• Content approval and change management. The site VAC approves all word selections, definitions, and
supplementary explanations in the Validation Lexicon. VAC approvals should be obtained from all
primary VAC members including functions that have conflicting word definitions. The Quality
Assurance and Validation members of the VAC must also approve all content. All changes to the
Validation Lexicon content should be submitted to the VAC for discussion and approval. This activity is
equivalent to VAC responsibility for validation plans, protocols, and other validation documents at the
site.
• Use of correct terminology in validation documents. The site VAC reviews and approves all validation
documents processed at the site. Their review must also include inclusion of approved wording as
listed in the Validation Lexicon. Allowing use of non-approved wording in validation documents
defeats one of the objectives of the Validation Lexicon – uniformity in site validation terminology.
Additions and Changes
Additions and changes to the site Validation Lexicon to improve the word content and increase consistent
language usage are encouraged. Word additions should be submitted to the site VAC for approval to include
into the lexicon and to evaluate the proposed word, definition, and use. Major wording changes to the
Validation Lexicon should be considered with good justification; changes disrupt the continuity of validation
terminology at the site.
Training
All involved with validation documents, especially protocol and results authors, must be trained and so
documented on the content and application of content in validation documents. Retraining must occur when
new words are added, or changes are implemented.

100
PROBLEM TERMINOLOGY Validation managers described several frequent validation terminology problems including Validation Master
Plan terms, process validation terms, and equipment qualification terms. Examples below are major term
categories. These will be the primary terms to be standardized in a Validation Lexicon effort.
Validation Master Plan
Validation Master Plan (VMP) is a universally known term. Essentially all pharmaceutical and related industry
manufacturing sites have some type of VMP in their facility. Despite widespread awareness, there is great
diversity regarding structure, content, and application. Meetings with validation professionals at
pharmaceutical meetings indicate much different understandings of the VMP -- the VMP to certain individuals
is not the same as the VMP to others. There are at least four distinct applications of the VMP term comprising
site overview documents and project documents:
Overview Documents
1. VMP – Site document. This document comprises all fundamental information about the site. It includes
specific individual sections about functions at the site.
2. VMP – Function document. This document comprises all fundamental information about a specific
function at the site. A VMP function document is one of the sections in the above-described VMP site
document.
Project Documents
3. VMP -- Major project document. This document comprises all information about a specific major
validation project at the site such as a new facility addition with multiple validation activities and
protocols.
4. VMP -- Single project document. This document comprises all information about a specific individual
validation project at the site. This document is one of the sections in the above-described VMP major
project document
The above four categories are the most widely used terms associated with VMP. There are numerous lesser-
used terms also used within organizations to describe equivalent information. Other terms include validation
plan, qualification master plan, qualification plan, project plan, validation project master plan, qualification
project master plan, validation approach, qualification approach, validation strategy, qualification strategy,
project strategy, project approach, and many others. Some organizations do not have a specific VMP, but
simply list validation documents in a spreadsheet. Whichever VMP approach(s) at the site is used should be
identified in the Validation Lexicon.
Process Validation Terminology
The list of terms used to describe validation of manufacturing, cleaning, packaging, and other processes is
extensive. Process Validation, Process Qualification, Validation Protocol, Process Verification, Performance
Validation, Performance Verification, Process Performance Qualification, and other terms have been used over
the years.
The most recent FDA guidance on Process Validation (2011) identifies Stage 2 of the Lifecycle Approach to
Process Validation as Process Qualification, and the process validation protocols in this stage as Process
Performance Qualification protocols (PPQ). Global agencies have also stated preferred process validation

101
terminology as either Process Qualification (PQ) or Process Validation (PV). This use of PQ overlaps with
equipment qualification PQ terminology which has been commonly used for many years. Whatever process
validation term is used at the site should be specified in the Validation Lexicon.
Qualification Terminology
The list of terms used to describe the respective stages of equipment, facilities, utilities, and other
qualifications is extensive. The theoretical sequence of activities is presented in Figure 4. These terms should
be defined in the Validation Lexicon. Validation manages from multiple companies commented that validation
practitioners often do not understand the objectives of stages, do not understand relationships between
sections, and do not assign appropriate testing to sections; URS, TRS, and FRS are not universal terms. When
inconsistent documents are approved by the site VAC, these problems worsen as erroneous documents
become institutionalized and are copied by other authors. Confusion may be due in part to use of the same
qualification terms in other validation areas. For example, IQ, OQ, and PQ equipment activities described in the
V Model are combined in the IQ for medical devices; the medical device OQ and PQ have product/process
applications. PIC/S recognizes IQ and OQ as part of equipment qualification; PQ is an alternate term for
process validation. Commissioning is another confused term regarding testing – when done, objective of
testing, types of testing, and so on. These distinctions should be explained in the Validation Lexicon.
Whichever terms are assigned for the various stages of qualification at the manufacturing site should be
carefully chosen to be in general alignment with regulatory guidelines. Their content should then be clearly
defined in the Validation Lexicon. All site organizations must support the defined content. The site VAC who
approve validation documents must then reject or modify documents that are not consistent with lexicon
terminology. Specific terms chosen for use are less important than consistent acceptance and use by site
personnel.
Figure 4. Stages of Qualification Process
SUMMARY AND FINAL THOUGHTS This discussion addresses problems with validation terminology within a pharmaceutical manufacturing site.
Each manufacturing site should have a Validation Lexicon to standardize and govern validation terminology at
DESIGN REQUIREMENTS 1. User Requirements Specification (URS)
2. Functional Requirements Specification (FRS)
3. Technical Requirements Specification (TRS)
4. Build Equipment
QUALIFICATION TESTING 5. Factory Acceptance Testing (FAT) Commissioning
6. Site Acceptance Testing (SAT) Commissioning
7. Installation Qualification (IQ)
8. Operation Qualification (OQ)
9. Performance Qualification (PQ)
Note: Some organizations use Design Qualification (DQ) in place of some or all design requirements (1-3 above) for commercially available systems.
.

102
the site. A Validation Lexicon provides several benefits to validation document authors and others connected
to validation. It also helps regulatory auditors become oriented to site validation terminology prior to audit.
Validation Lexicon contents are described and an approach to development is discussed. Examples of
widespread validation terms with differing usage – Validation Master Plan, process validation terms, and
equipment validation terms - identified by validation managers are described.
Many manufacturing sites do not have a Validation Lexicon. Designing and developing a Validation Lexicon is
an arduous task requiring a sustained effort. Depending on site size, scope of validation activities and the
number of technical functions involved with validation, this effort may involve significant frustration. Function
areas may need to completely change their typical validation approaches to standardize with other areas.
Working with function leaders whose initial response is to defend their way of doing things and argue against
change is not easy.
A strong and committed Validation function demonstrating leadership at the manufacturing site is vital to
initiation, development, and implementation of a Validation Lexicon. The site Validation Approval Committee
(VAC) lead by the Validation function is also critical. The VAC will review and approve lexicon content in the
same manner as done for validation documents. Support of respected site leaders, function management and
senior management is also necessary.
Initiation of a Validation Lexicon project is a difficult but worthwhile endeavour. A completed Validation
Lexicon at a manufacturing site demonstrates control of the site validation program, attention to regulatory
requirements, attention to the details of terminology, and control of validation content – all of which are
characteristics of a well-managed validation quality system. Again, frustration should be expected. But the final
Validation Lexicon, implemented and utilized in daily performance, will be well worth the effort.
REFERENCES 1. FDA. Guideline on General Principles of Process Validation. May 1987. Available at: http://fdaguidance.net/wp-
content/uploads/2016/04/FDA-1987-Guideline-on-General-Principles-of-Process-Validation.pdf Accessed Aug 6 2019
2. Global Harmonization Task Force SG3. Quality Management Systems – Process Validation Guidance. Edition 2,
January 2004. Available at: http://www.imdrf.org/docs/ghtf/final/sg3/technical-docs/ghtf-sg3-n99-10-2004-qms-
process-guidance-04010.pdf Accessed Aug 6 2019
3. FDA. Guidance for Industry. Process Validation: General Principles and Practices. January 2011. Available at:
https://www.fda.gov/media/71021/download Accessed Aug 6, 2019
4. European Commission. Eudralex Volume 4. EU Guideline for Good Manufacturing Practice for Medicinal Products for
Human and Veterinary Use. Annex 15: Qualification and Validation. Mar 30, 2015. Available at:
https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2015-10_annex15.pdf Accessed Aug 6, 2019
5. EMA. Guideline on process validation for finished products – information and data to be provided in regulatory
submissions. November 2016. Available at: https://www.ema.europa.eu/en/documents/scientific-
guideline/guideline-process-validation-manufacture-biotechnology-derived-active-substances-data-be-
provided_en.pdf#page=1 Accessed August 12, 2018.
6. PIC/S. Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process
Validation, Cleaning Validation. Sep 25, 2007. Available at: http://academy.gmp-
compliance.org/guidemgr/files/PICS/PI%20006-
3%20RECOMMENDATION%20ON%20VALIDATION%20MASTER%20PLAN.PDF Accessed Aug 6 2019
7. Therapeutics Goods Administration, Department of Health, Australian Government. Process Validation for Listed and
Complementary Medicines. Technical Guidance on the Interpretation of PIC/S Guide to GMP. Ver 2. Jan 2019.

103
Available at: https://www.tga.gov.au/sites/default/files/process-validation-listed-and-complementary-medicines.pdf
Accessed Aug 6, 2019.
8. Health Canada. Validation Guidelines for Pharmaceutical Dosage Forms (GUI-0029), 2009. Available at:
https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/good-
manufacturing-practices/validation/validation-guidelines-pharmaceutical-dosage-forms-0029.html#a4 Accessed Aug
6 2019.

104
PQ Forum #14: Numbers By: Paul L. Pluta, Editor in Chief, Journal of Validation Technology and Journal of GxP Compliance
ABSTRACT This discussion addresses the use of numbers in validation and other technical documents. The writing of
numbers in these documents may violate technical writing norms, be inconsistently presented within the
document, and have grammatical errors. Numbers in technical documents differ from numbers in non-
technical applications; technical writing favors use of Arabic numeric digits rather than spelled words. Basic
technical writing rules for numbers are described; areas for author judgment are identified. Fractions,
decimals, and sentence use are discussed. Rules for writing numbers are not globally consistent. Miscellaneous
topics related to use of numbers including numbering of tables and figures, Roman numerals including rules
for reading, and use of numbers versus bullets in lists are discussed. A simple stepwise approach to verifying
the correct use of numbers in technical documents is presented. Consistent use of the selected number format
throughout the document is key. Organizations may identify preferred number formats to foster consistency
among all authors at their site in the same manner as specifying preferred technical validation terminology.
INTRODUCTION Many validation documents contain numbers in protocols, in tables of test data, and in narrative discussions
of results. The writing of numbers and their associated units of measure in these documents is often
problematic. Validation managers from multiple companies comment that numbers in technical documents
may violate technical writing norms, be inconsistently presented within the document, and have grammatical
errors.
Authors of validation documents should have a basic familiarity with general rules for writing numbers. Some
rules are obvious and straightforward; others are more obscure or complex; still others require careful
attention to detail. This discussion provides accepted rules that address common writing errors with numbers.
Rules presented should be useful to authors and should help to standardize writing norms in the organization.
Units of measure such as feet, milliliters, pounds/square inch, and other terms are another source of error
related to numbers. A future PQ Forum will discuss technical writing rules for units of measure.
The topic of validation documents was discussed among several validation and quality managers from multiple
industry companies at Validation Week in San Diego, CA, USA, in October 2018. The quality of validation
documents is a widespread problem. Managers described errors in content, inadequate explanations, illogical
document structures, and errors in grammar and punctuation including problems with numbers and units of
measure. Previous issues of PQ Forum have initiated discussion of technical writing principles and approaches
(1, 2). We now continue with focus on numbers. Manager’s comments, suggestions, and actual experiences
with problems were the impetus for this series of PQ Forum discussions.
Number Terms and Definitions Before discussing specific number-writing rules, we clarify related language used in technical and grammar
references as well as our use in the following discussion.

105
What is a Number, and What is a Numeral? Some references describe “number” as a theoretical concept -- an
idea in our heads (3,4). We imagine a quantity of some object such as imagining five apples. We may then
express the concept of the quantity “fiveness” in many ways. It may be expressed as a written word “five,”
Arabic figure “5,” Roman numeral “V,” or other representations. “Five,” “5,” and “V” are termed “numerals.”
Numerals are the written expression of the fiveness imaginary concept.
Despite the distinct difference in definition, we use number and numeral as interchangeable words in daily
spoken and written language. We say number when we should say numeral. To many of us, number or
numeral means numeric figures or symbols, even though the dictionary definition of numeral includes the
spelled word. We do not usually think of written words as numerals.
The proper and appropriate writing of numbers as Arabic symbols or as words in validation and other technical
documents is the objective of this discussion. We will use the words “number” and “numeral” interchangeably
as in common language use even though they are not technically the same. We distinguish between Arabic
figures (numbers and numerals) and spelled words in this discussion.
Discussion Topics The following addresses the writing of numerals in validation and other technical documents as recommended
in several writing references (5,6,7,8). The fundamental question to be answered is the following: When
should numerals be written as Arabic digit symbols and when should they be written as spelled words? The
writing of numbers in technical documents is different than writing numbers in non-technical documents.
Technical documents tend to present numbers as Arabic digits; non-technical documents more frequently use
spelled words. Rules discussed in this paper for technical documents will favor Arabic numerals. Basic technical
writing rules and recommended variations are described. Fractions, decimals, and sentence use are discussed.
Related topics such as numbers in tables and figures are addressed. Roman numeral calculations are
presented. Logical and reasonable guidelines for use of numbers should help to highlight proper use of
numbers, improve readability, and reduce grammatical errors.
NUMBERS IN TECHNICAL DOCUMENTS The following are basic approaches associated with writing numerals in technical documents. Rules for writing
numbers are complex. Some rules may be modified or approached in diverse ways, i.e., number rules are not
universally consistent.
Contradictory rules may apply in a single sentence. Number-writing practices are different in different
countries. When reasonable recommended options exist, authors should select their approach and then
consistently use that same format in the entire document. Consistency in writing is key element in writing a
successful technical document containing numbers.
Symbols and Words 1. Use spelled words for numbers ten and below including zero; use Arabic symbols for numbers eleven and
above.
This is a generally accepted rule for writing numbers in technical documents. Some references expand this
general rule to include spelled words divisible by ten, i.e., the “Rule of Tens;” twenty, thirty, forty, and so on
may thus be written as words. Others recommend that all one-word numerals be written as words; thus,
eleven through nineteen are written as words. Some style manuals specify numbers through ninety-nine be
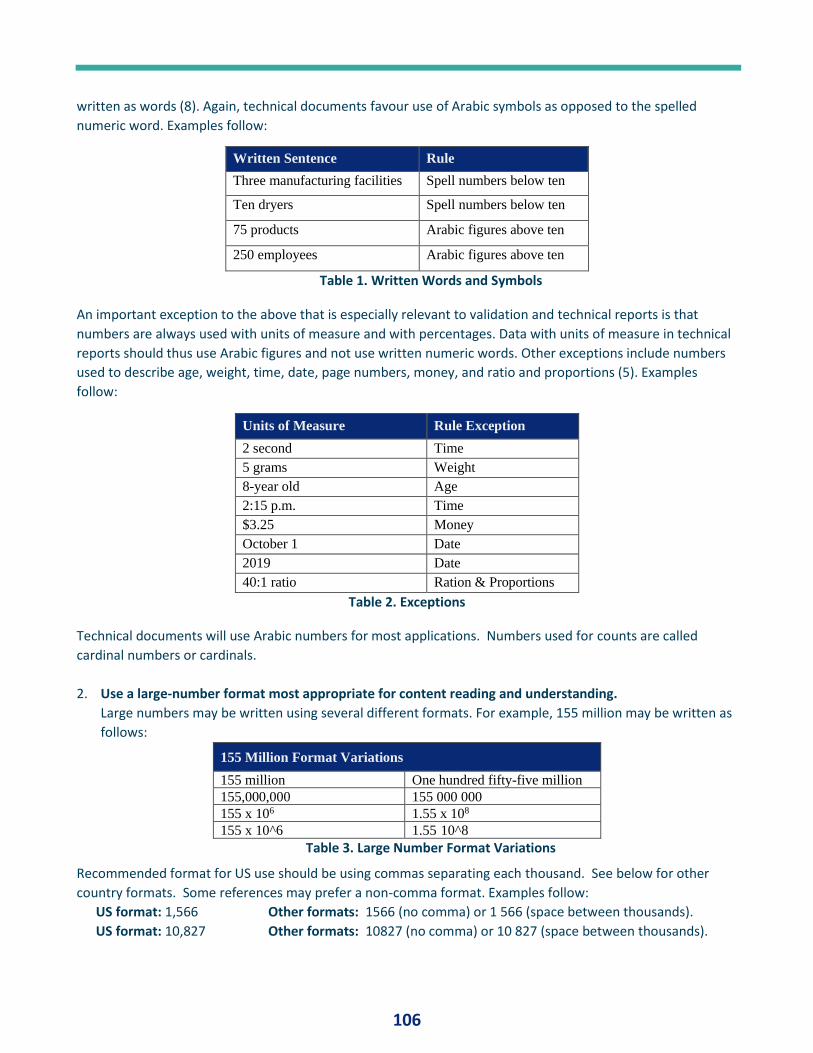
106
written as words (8). Again, technical documents favour use of Arabic symbols as opposed to the spelled
numeric word. Examples follow:
Written Sentence Rule
Three manufacturing facilities Spell numbers below ten
Ten dryers Spell numbers below ten
75 products Arabic figures above ten
250 employees Arabic figures above ten
Table 1. Written Words and Symbols
An important exception to the above that is especially relevant to validation and technical reports is that
numbers are always used with units of measure and with percentages. Data with units of measure in technical
reports should thus use Arabic figures and not use written numeric words. Other exceptions include numbers
used to describe age, weight, time, date, page numbers, money, and ratio and proportions (5). Examples
follow:
Units of Measure Rule Exception
2 second Time
5 grams Weight
8-year old Age
2:15 p.m. Time
$3.25 Money
October 1 Date
2019 Date
40:1 ratio Ration & Proportions
Table 2. Exceptions
Technical documents will use Arabic numbers for most applications. Numbers used for counts are called
cardinal numbers or cardinals.
2. Use a large-number format most appropriate for content reading and understanding.
Large numbers may be written using several different formats. For example, 155 million may be written as
follows:
155 Million Format Variations
155 million One hundred fifty-five million
155,000,000 155 000 000
155 x 106 1.55 x 108
155 x 10^6 1.55 10^8
Table 3. Large Number Format Variations
Recommended format for US use should be using commas separating each thousand. See below for other
country formats. Some references may prefer a non-comma format. Examples follow:
US format: 1,566 Other formats: 1566 (no comma) or 1 566 (space between thousands).
US format: 10,827 Other formats: 10827 (no comma) or 10 827 (space between thousands).

107
The chosen format should be based on ease of reading, understanding by readers, and to facilitate
comparisons between numbers. The selected format must then be consistently used throughout the entire
document.
3. Use commas and periods in numerals according to country norms.
The use of commas and periods in numbers is not universal. In the US, periods are used in decimals and
commas are used to separate thousands in large numbers; for example, 27.5 and 1,255, respectively. In
some global countries, opposite use is true: 27,5 and 1.255 for the same numbers. Global authors should
follow the conventional practice of their country. The international system of units (SI) uses a space (not a
period or comma) between each 1000 digits on each side of a decimal (9). Examples follow in Table 4:
Number Separator Examples Country Rule
US decimal: 87.52 Use period in US decimal format
US large number: 1,899 Use comma in US large-number format
Global decimal: 27,82 Use comma in global decimal format
Global large number: 1.399.422 Use period in global large-number format
International large-number and decimal: 5 275.333 23 Use space to group each three digits on
each side of decimal point
Table 4. Commas, Periods, and Spaces
4. Use words for numbers that are an approximation. Applications that do not require exact numerical amounts should use written words. Examples follow in Table 5:
Recommended Not Recommended
Fill tank half full Fill tank ½ full, or fill tank to 0.5 of capacity
Energy levels are reduced by one-third reduced by 1/3, or reduced by 0.333
Pressure was doubled Pressure increased 2x original, or increased by 2.0
Reduce vacuum to half of set point 1/2 of set point, or 0.5 of set point
Table 5. Number Approximations
5. Inclusive numbers. Use Arabic numbers with hyphens to indicate a range of numbers.
A range of included numbers is expressed by using Arabic numbers joined by hyphens. Do not include
spaces between numbers and the hyphen. Do not use spelled words. Examples follow in Table 6:
Recommended Not Recommended
Pages 2-5 Pages two to five
Tests 12-22 Tests twelve through twenty-two
Items 105-16 Items one hundred five to one hundred sixteen
Table 6. Inclusive Numbers
6. Ordinal numbers follow rules of cardinal numbers.
Ordinal numbers tell the position of something such as first, second, third, and so on. Numbers discussed
above that describe data and counts are called cardinal numbers. Rules discussed above (cardinal
numbers) also apply to ordinal numbers. Table 7 describes cardinal numbers and corresponding ordinal
numbers. Examples follow in Table 7:
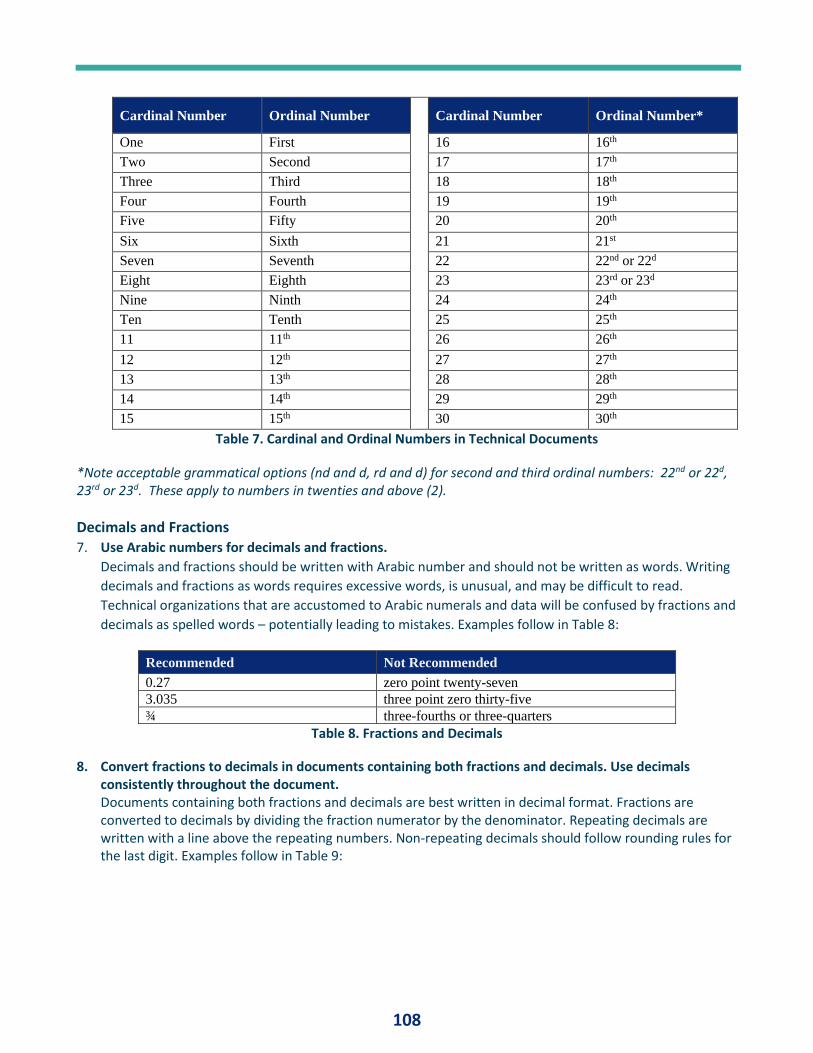
108
Cardinal Number Ordinal Number
Cardinal Number Ordinal Number*
One First 16 16th
Two Second 17 17th
Three Third 18 18th
Four Fourth 19 19th
Five Fifty 20 20th
Six Sixth 21 21st
Seven Seventh 22 22nd or 22d
Eight Eighth 23 23rd or 23d
Nine Ninth 24 24th
Ten Tenth 25 25th
11 11th 26 26th
12 12th 27 27th
13 13th 28 28th
14 14th 29 29th
15 15th 30 30th
Table 7. Cardinal and Ordinal Numbers in Technical Documents
*Note acceptable grammatical options (nd and d, rd and d) for second and third ordinal numbers: 22nd or 22d, 23rd or 23d. These apply to numbers in twenties and above (2).
Decimals and Fractions 7. Use Arabic numbers for decimals and fractions.
Decimals and fractions should be written with Arabic number and should not be written as words. Writing
decimals and fractions as words requires excessive words, is unusual, and may be difficult to read.
Technical organizations that are accustomed to Arabic numerals and data will be confused by fractions and
decimals as spelled words – potentially leading to mistakes. Examples follow in Table 8:
Recommended Not Recommended
0.27 zero point twenty-seven
3.035 three point zero thirty-five
¾ three-fourths or three-quarters
Table 8. Fractions and Decimals
8. Convert fractions to decimals in documents containing both fractions and decimals. Use decimals consistently throughout the document. Documents containing both fractions and decimals are best written in decimal format. Fractions are converted to decimals by dividing the fraction numerator by the denominator. Repeating decimals are written with a line above the repeating numbers. Non-repeating decimals should follow rounding rules for the last digit. Examples follow in Table 9:
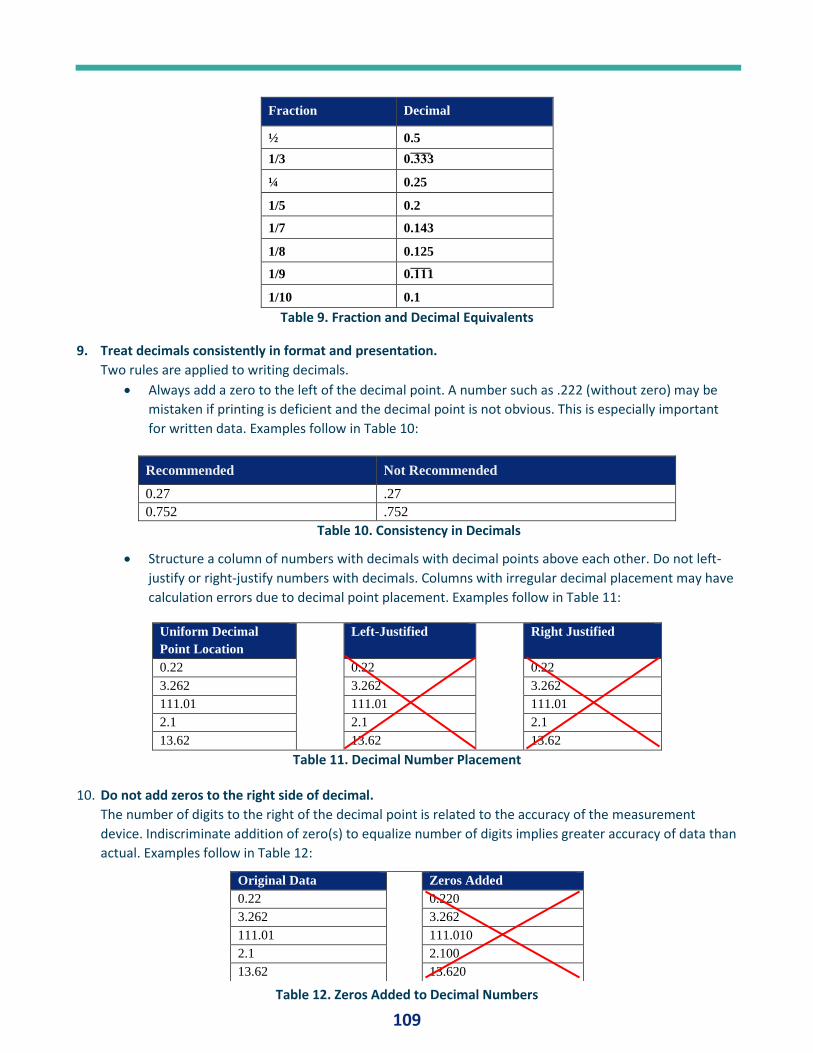
109
Fraction Decimal
½ 0.5
1/3 0.̅3̅3̅3
¼ 0.25
1/5 0.2
1/7 0.143
1/8 0.125
1/9 0.̅1̅1̅1
1/10 0.1
Table 9. Fraction and Decimal Equivalents
9. Treat decimals consistently in format and presentation.
Two rules are applied to writing decimals.
• Always add a zero to the left of the decimal point. A number such as .222 (without zero) may be
mistaken if printing is deficient and the decimal point is not obvious. This is especially important
for written data. Examples follow in Table 10:
Recommended Not Recommended
0.27 .27
0.752 .752
Table 10. Consistency in Decimals
• Structure a column of numbers with decimals with decimal points above each other. Do not left-
justify or right-justify numbers with decimals. Columns with irregular decimal placement may have
calculation errors due to decimal point placement. Examples follow in Table 11:
Uniform Decimal
Point Location
Left-Justified Right Justified
0.22 0.22 0.22
3.262 3.262 3.262
111.01 111.01 111.01
2.1 2.1 2.1
13.62 13.62 13.62
Table 11. Decimal Number Placement
10. Do not add zeros to the right side of decimal.
The number of digits to the right of the decimal point is related to the accuracy of the measurement
device. Indiscriminate addition of zero(s) to equalize number of digits implies greater accuracy of data than
actual. Examples follow in Table 12:
Table 12. Zeros Added to Decimal Numbers
Original Data Zeros Added
0.22 0.220
3.262 3.262
111.01 111.010
2.1 2.100
13.62 13.620
110
11. Spell one of two consecutive numbers as words – usually the shorter number.
Phrases in technical documents may require use of consecutive numbers. These phrases are inherently
problematic. For example, equipment has 2 3-inch valves, or cleaning requires 3 3-person teams, or 6 3-
liter vessels. Each of these may be confusing to the reader. They may be clarified by writing one number,
usually the shorter number of the two, in spelled word form. Examples follow in Table 13:
Recommended Not Recommended
Two 3-inch valves 2 3-inch valves
Three 3-person teams 3 3-person teams
Six 3-liter vessels 6 3-liter vessels
Table 13. Handling Consecutive Numbers
Sentences, Sections, and Documents 12. Do not begin a sentence with a numeric symbol.
Another universally accepted rule for writing numbers is that sentences are not written starting with an
Arabic number. Sentences are expected to begin with words. Sentences that begin with a number are
confusing and are unusual in appearance. Sentences that begin with a numeral must use a spelled numeric
word to begin the sentence. Sentences may also be reworded to contain the Arabic number within the
sentence. Examples follow in Table 14:
Acceptable Not Recommended Recommended
Fifty mL of buffer is added to
the vessel.
50 mL of buffer is added to the
vessel.
A volume of 50 mL is added
to the vessel.
Two thousand patients
participated in the study.
2,000 patients participated in the
study.
Clinical study participation
comprised 2,000 patients.
Table 14. Starting Sentences with Numbers
13. Do not mix numerals and words in the same sentence, same section, or same document.
Rule #1 above suggested use of words for numbers ten and less, and numerals for larger numbers above
ten. What should be done when a sentence contains numbers both less than ten and greater than ten?
When this occurs, consistency using Arabid numerals is preferred. Consistency using Arabic numerals
should extend to all sentences in a section and to the entire document. Consistent number format makes a
document easier to read. If all numbers in a document were ten or less, writing all numbers as words could
be considered.
Example: Original sentence: This site manufactures more than 200 products comprising more than 25 different
active drugs and ten different dosage forms. However, only four different cleaning methods are
required for cleaning. Two of these are automated methods and two are manual methods.
Revision: This site manufactures more than 200 products comprising more than 25 different active
drugs and 10 different dosage forms. However, only 4 different cleaning methods are required for
cleaning. Of these, 2 are automated methods and 2 are manual methods.
The revised sentence uses Arabid numbers for all numbers. The last sentence was reworded to start
with the word “Of” rather than a number.

111
RELATED TOPICS Tables and Figures Tables are compilations of data in the form of columns and rows. Figures contain graphs, images, or other
visuals.
Tables generally have titles at the top of the table. Tables have titles for columns and rows. Figures may have
titles at the top or bottom of the figure. Figures usually contain a brief explanation of the content in the figure.
Tables and figures are numbered consecutively, each within their respective group. For example, a document
containing three tables and three figures would be numbered Table 1, Table 2, Table 3, Figure 1, Figure 2,
Figure 3, regardless of their order in the document. The document would not be labelled Table 1, Table 2,
Figure 3, Table 4, Figure 5, Figure 6. Label tables consecutively within all tables; label figures consecutively
within all figures; do not intermix numbering of tables and figures.
Books and documents with sections may number Tables and Figures consecutively within each section. For
example, Table 1.1, 1.2, 1.3 in the first chapter, Table 2.1, 2.2, and 2.3 in the second chapter, and so on.
Roman Numerals Roman numerals may be used in Validation documents as part of proprietary names, personal names, version names, and other applications. The following illustrates the relationship between Arabic and Roman numerals. Document writers should know how to read Roman numerals, and then must maintain consistency in their use when writing validation documents. Writing Version III and Version 3 in the same document is not acceptable. See Table 15 for equivalent Roman and Arabic Numerals.
Roman Numerals Arabic Numerals
I 1
V 5
X 10
L 50
C 100
D 500
M 1000
Table 15. Roman and Arabic Numerals
The following are rules associated with writing Roman numerals (10):
• If a Roman numeral is repeated, its value is added. V, L, and D are not repeated. A Roman numeral is
not repeated more than three times. Examples follow:
→ III = 3
→ XX = 20
• A smaller Roman numeral written to the right of a large numeral is added to the large number.
Examples follow:
→ VI = 6
→ XI = 11
• A smaller Roman numeral written to the left of a large numeral is subtracted from the large number.
Examples follow:
→ IV = 4

112
→ IX = 9
→ XLIV = 44
→ XCIX = 99
→ V, L, and D are not subtracted. I can only be subtracted from V or X. X can only be
subtracted from L, M, or C.
• Only one symbol may be subtracted from a number which is of greater value. Examples follow:
→ XCVIII = 98. IIC for 98 is incorrect.
• A line above a Roman numeral multiplies the value of the numeral by 1000. Examples follow:
→ XXX = 30,0̅00̅ ̅ CCC = 300,0̅̅00̅ ̅
→ A double line above a Roman numeral multiplies the value of the numeral by 100,000.
Numerals Versus Bullets in Lists Lists of items are often described in validation documents. Lists that describe activities that must be performed
in chronological order should be listed using numerals; for example, a stepwise equipment cleaning procedure
should be written using numerals. Lists of items for probable future reference should use numerals to facilitate
reference; for example, rules for validation documentation practices should use numerals. In contrast, lists of
items that are essentially equivalent, do not describe prioritization, or do not require chronological
performance should be bulleted; for example, short lists of items required for an Installation Qualification that
are not prioritized should be bulleted.
• Use Arabic numbers for list to be addressed in chronological order or for long lists.
• Use bullets for randomized lists with no prioritization (7).
FINAL THOUGHTS This discussion has identified generally accepted rules for the writing of numerals in validation and technical
documents as compiled from several references. The following is recommended as a stepwise approach to
writing documents with numbers:
1. Document numbers review. Fundamental writing must be correct and follow technical-writing rules.
When the document has been essentially completed, the author should review usage of numbers to
confirm that basic technical rules as described above have not been violated.
2. Readability evaluation. The document is then reviewed for clarity and readability. Evaluation may
indicate instances where certain number rules may be interpreted in different ways and may be
contradictory or inconsistent within the document. Authors must use good judgment to reconcile
differences and select the best writing approach. Changes to rules for reasons of ease in reading,
consistency within the document, eliminating confusion, or other logical reasons should be
implemented.
3. Consistency. After all number formats have been decided, the entire document should be again
reviewed to confirm consistent formats. Consistency with numerals within the entire document while
following basic rules of format and grammar will yield a successful document.
Site Application and Implementation. Organizations may identify preferred number formats to foster
consistency among all authors at the site. Guidelines should be documented in an SOP or other approved
document. A previous issue of PQ Forum (11) discussed the use of a site lexicon to define and standardize

113
validation terminology and language at the site; recommended number usage may be added to the site
lexicon. Consistent rules for writing numbers should be considered similarly to consistency in use of technical
terminology.
REFERENCES 1. Pluta, Paul L. Technical Writing for Validation. Journal of Validation Technology, Volume 24, #6, 2018.
2. Pluta, Paul L. Validation Document Writing Sequence. Journal of Validation Technology, Volume 25, #1,
2019.
3. Numbers, Numerals, and Digits. Math is Fun. https://www.mathsisfun.com/numbers/numbers-numerals-
digits.htm Accessed 10-21-19.
4. What is the Difference Between a Number and a Numeral? Reference.com.
https://www.reference.com/math/difference-between-number-numeral-f92a563db3262a17. Accessed
10-21-19.
5. Blake, Gary, and Robert W. Bly. The Elements of Technical Writing. Longman Publishing, New York, NY.
1993.
6. Baugh, L. Sue. Essentials of English Grammar, third edition. The McGraw-Hill Companies, Inc. New York,
New York, NY. 2005.
7. Lindsell-Roberts, Sheryl. Technical Writing for Dummies. Wiley Publishing, Hoboken NJ. 2001.
8. Fogarty, Mignon. How to Write Numbers. Quick and Dirty Tips, The New York Times.
https://www.quickanddirtytips.com/education/grammar/how-to-write-numbers. Accessed 10-21-19.
9. National Institute of Standards and Technology (NIST). The International System of Units (NS).
https://physics.nist.gov/cuu/pdf/sp811.pdf. Accessed 10-21-19.
10. Roman Numerals. Toppr, https://www.toppr.com/guides/maths/knowing-our-numbers/roman-
numerals/. Accessed 10-21-19.
11. Pluta, Paul L. Validation Lexicon. Journal of Validation Technology, Volume 25, #4, August 2019.

114
Fan Favorites
LIQUID CRYOGENIC STORAGE CHAMBER QUALIFICATION By John Orange, Director of Validation, Masy BioServices
ABSTRACT Cold chain storage of biopharmaceutical products and associated materials is required to maintain product
stability and is a regulatory expectation. Critical areas of concern in cold chain practice related to liquid
nitrogen cryogenic storage include container selection, method of temperature monitoring, and qualification
approach based on risk management. Qualification of liquid nitrogen cryogenic storage equipment is described
in a three-step process:
1. Understanding cryogenic storage chambers
2. Using your cryogenic storage chamber
3. Qualifying your cryogenic storage chamber.
INTRODUCTION Cold chain storage of biopharmaceutical products and associated materials is a vital concern in regulated
industries. Temperature control of cold chain storage throughout the entire cold chain process is required to
maintain product stability. Cold chain storage practices have come under scrutiny from auditors and
regulatory agency inspectors in recent years. Chamber qualification for temperature-controlled equipment
intended for storage or for vessels intended to be used during shipping is now a regulatory expectation.
Cold chain storage begins at the source of manufacturing where products or materials are kept cool or frozen
until delivery to their final destination for administration to the patient. References to cold chain storage
range from refrigerated conditions to cryogenic conditions. While refrigerated (2-8°C) transport conditions are
always a concern, significant attention is also being paid to cryogenic conditions (-196°C), specifically liquid
nitrogen vapor-phase material storage. Critical areas of concern in cold chain practice related to cryogenic
storage chambers include transportation, method of temperature monitoring during transport, and
qualification approach for permanent storage solutions.
Cryogenic Transportation
Companies involved in cold chain storage under cryogenic conditions must be attentive to several factors from
how material is packed to the method of transportation used for transport. Finding the correct vessel or
container and appropriate packing materials to control temperature for an extended period can prove
challenging. The International Safe Transit Association (ISTA) provides guidelines identifying test scenarios for
packaging. Many companies are embracing the recommendations of the ISTA and are identifying more robust
safeguards for their cold chain shipping by vetting their processes and performing rigorous testing on their
packaging and shipping practices.
Temperature Monitoring During Transport

115
The monitoring of products and materials during transport is also a critical piece of the cold chain process.
Traditionally, a temperature recording datalogger is placed with the shipment and data is downloaded upon
arrival at its final destination. This is a reactive approach that leaves the end user questioning whether their
product remained within specifications during transport. Advances in technology are allowing product owners
to be proactive with the use of live data-monitoring sensors. Monitoring sensors connect to cellular networks
to transmit data to product owners to track the environmental conditions and locations during real time
through the entire shipping route. If the conditions deviate from the specification range, the problem can be
addressed during transportation by reacting to potential temperature threats long before the product arrives
at the final destination.
In addition to ground transportation, air transportation is being impacted with respect to identifying or
reinforcing cold chain storage strategies as well. Storage locations in airports for cold chain storage are held to
standards defined by the International Air Transit Association (IATA). The IATA has developed a Centre for
Excellence of Independent Validators which is starting a global standard for air shipment compliance in
pharmaceutical storage practices.
Until recently, the focus on biopharmaceutical storage and transport has been limited to pharmaceutical
companies. As cold chain best practices evolve, the focus has grown to include transportation and distribution
strategies across the globe. While maintaining temperature control during transport is a vital part of the cold
chain, it is just as important to consider permanent storage before and after transport.
Controlled temperature unit qualification is a key part of demonstrating compliance in cGMP regulated cold
chain applications. Since the operation of a permanent cryogenic storage chamber differs from that of a typical
controlled temperature unit, such as a refrigerator, this white paper was created to help identify typical
operation and standardized qualification of cryogenic storage chambers in accordance with validation best
practices.
The following information applies to cryogenic storage chambers installed in a permanent or semi-permanent
location.
Qualification of Cryogenic Storage Equipment
The qualification of liquid nitrogen cryogenic storage equipment may be considered to be a three-step process
as follows:
1. Understanding cryogenic storage chambers
2. Using Your Cryogenic Storage Chamber
3. Qualifying Your Cryogenic Storage Chamber.
UNDERSTANDING CRYOGENIC STORAGE CHAMBERS Understanding how your particular cryogenic or Liquid Nitrogen (LN2) chamber works along with some general
facts will help determine how to qualify your LN2 chamber. Cryogenic storage chambers are used to store
material in a suspended or inactive state. Cell banks, cell tissues, and other types of human or animal samples
are commonly stored in these types of chambers. For example, cell banks are stored in these cryogenic
conditions long-term until needed for manufacturing. An approach to mitigating complete loss is to store this
type of material at two separate sites to support a robust disaster recovery strategy.

116
LN2 Liquid Level Control
In general, LN2 chambers for cryogenic storage are liquid-level controlled – they are not temperature
controlled. The LN2 operation is controlled by at least two liquid level settings, either by using pressure
sensors or thermistors to determine liquid level measurement. For reference purposes, the liquid level
settings will be designated “High Level LN2” and “Low Level LN2” settings. When the “Low Level LN2” setting
is triggered, the controller will call for a LN2 fill, a process where LN2 fills the bottom of the chamber until the
“High Level LN2” setting is triggered. Temperature stratification will be widest before a LN2 fill begins and
narrowest after a LN2 fill is completed. This process will repeat indefinitely during normal operation. The
duration of time between LN2 fills is dependent upon the size of the chamber, load conditions,
environmental/ambient conditions, and the distance between the active High and Low Level LN2 settings. This
duration of time is considered a full cycle.
Figure 1. Cryogenic Storage Vessel
Temperature Zones Within the Chamber
Liquid nitrogen is contained at the bottom of the chamber and temperatures within the chamber are colder
near the LN2 and are warmer away from the liquid. With that information, you can assume that temperature
zones within the unit are planar and exhibit stratification, trending colder at the bottom and warmer (worst
case) at the top of the unit.

117
Figure 2. Materials for Cryogenic Storage
USING YOUR CRYOGENIC STORAGE CHAMBER Determining how you intend to use your chamber will provide you with the blueprint to move forward into
qualification. This should be a combination of considerations between both the use of the chamber and the
product requirements.
LN2 Liquid Level Determination
Prior to qualification, the product SME / organization should determine if product should be stored in liquid
phase or vapor phase within the LN2 chamber. The Liquid Nitrogen phase changes at approximately -195.79°C
of atmospheric pressure. This is the expected product temperature if product is stored in liquid phase. The
liquid level settings can be modified to fit storage requirements to target liquid or vapor phase storage. The
larger the gap between fill/stop reduces the amount of internal storage space as liquid level will increase. The
settings can also be modified to increase or decrease the LN2 fill frequency - LN2 fill duration increases as
distance between the active liquid level settings increase. The same is true for the amount of LN2 in the
chamber. Generally, larger units can contain more LN2 and as such, will take more time to evaporate as
opposed to a smaller unit.
Identify Your Temperature Range
LN2 chambers can come equipped with alarm functionality that will trigger when the controller/display RTD
reaches a high temperature set point. It is important to know what the high temperature set point is of your
unit (e.g. -135°C). This set point is determined by the technical function and is directly correlated to product
storage conditions of the intended contents. This temperature will also ultimately define the temperature
acceptance criteria for validation.
Safety Considerations
Adequate Personal Protective Equipment (PPE) must be worn when working with LN2. These include
cryogenic-rated gloves, safety glasses, face shield, apron, and related equipment as applicable. Oxygen
monitors are typically installed in rooms with LN2 chambers because oxygen is displaced by nitrogen gas and

118
can cause asphyxiation. Doors to these rooms are left open when occupied placing the vent away from
operators.
Figure 3. Personal Protective Equipment
QUALIFYING YOUR CRYOGENIC STORAGE CHAMBER Qualification of the LN2 chamber can be performed once the initial setup parameters have been identified and
implemented. The acceptance criteria for temperature distribution should be based on product storage
conditions of tended contents identified by the technical experts.
Study Types and Test Durations
Once acceptance criteria are established, the study types and test durations can be determined. It is
recommended that at least one full cycle of temperature data is recorded (e.g., 3 days); however, three full
cycles would demonstrate more data points and complies with a best practice approach. Contrary to typical
controlled temperature unit validation, capturing data for a set period of time (e.g., 24 hours) will likely only
reveal data from a segment of a full cycle and is not suggested as a best practice as it may not capture worst
case data just prior to the next fill cycle. Technical experts can determine if the qualification should capture
temperature mapping under empty chamber and/or loaded chamber conditions and whether an open-door
study should be performed. A LN2 failure study can also be conducted to determine how long the chamber
stays within range after a LN2 fill triggers and does not receive LN2. This test is usually performed to
demonstrate temperature conformity in the event of LN2 shortage or an extended-time power loss.
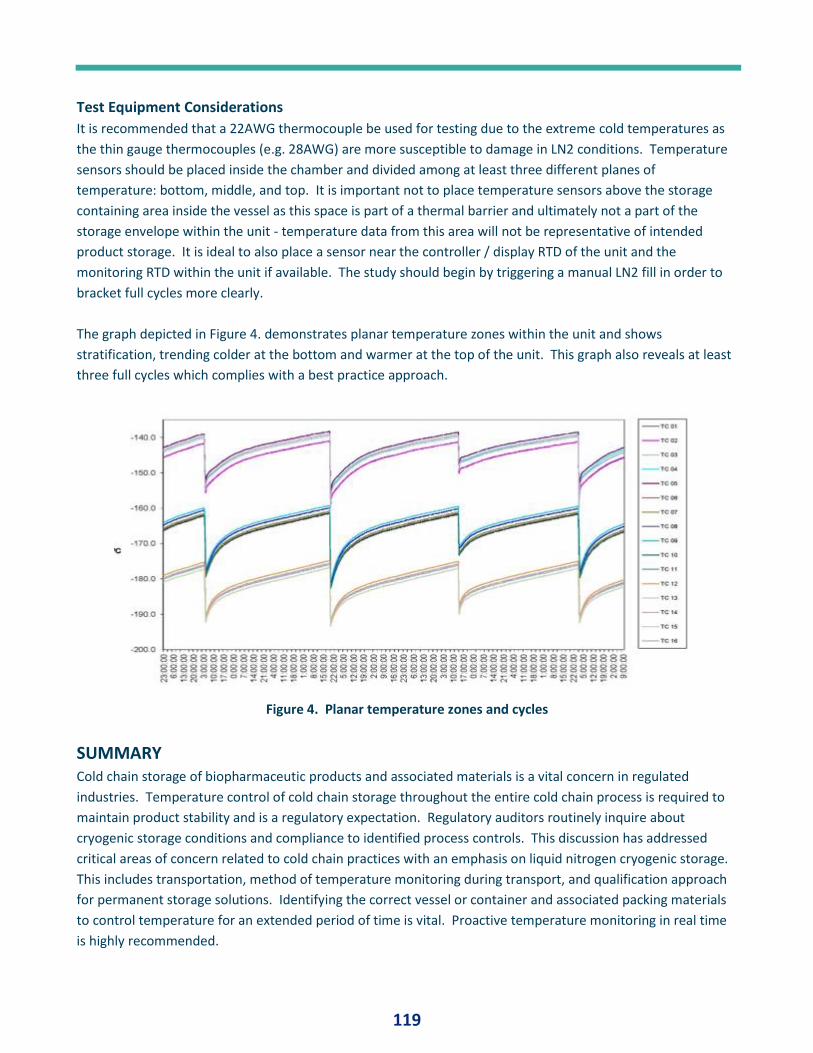
119
Test Equipment Considerations
It is recommended that a 22AWG thermocouple be used for testing due to the extreme cold temperatures as
the thin gauge thermocouples (e.g. 28AWG) are more susceptible to damage in LN2 conditions. Temperature
sensors should be placed inside the chamber and divided among at least three different planes of
temperature: bottom, middle, and top. It is important not to place temperature sensors above the storage
containing area inside the vessel as this space is part of a thermal barrier and ultimately not a part of the
storage envelope within the unit - temperature data from this area will not be representative of intended
product storage. It is ideal to also place a sensor near the controller / display RTD of the unit and the
monitoring RTD within the unit if available. The study should begin by triggering a manual LN2 fill in order to
bracket full cycles more clearly.
The graph depicted in Figure 4. demonstrates planar temperature zones within the unit and shows
stratification, trending colder at the bottom and warmer at the top of the unit. This graph also reveals at least
three full cycles which complies with a best practice approach.
Figure 4. Planar temperature zones and cycles
SUMMARY Cold chain storage of biopharmaceutic products and associated materials is a vital concern in regulated
industries. Temperature control of cold chain storage throughout the entire cold chain process is required to
maintain product stability and is a regulatory expectation. Regulatory auditors routinely inquire about
cryogenic storage conditions and compliance to identified process controls. This discussion has addressed
critical areas of concern related to cold chain practices with an emphasis on liquid nitrogen cryogenic storage.
This includes transportation, method of temperature monitoring during transport, and qualification approach
for permanent storage solutions. Identifying the correct vessel or container and associated packing materials
to control temperature for an extended period of time is vital. Proactive temperature monitoring in real time
is highly recommended.

120
A three-step LN2 qualification process is described:
1. Understanding cryogenic storage chambers. This involves how your particular cryogenic chamber works.
LN2 chambers for cryogenic storage are generally liquid-level controlled.
2. Using your cryogenic storage chamber. Determining how you intend to use your chamber will provide you
with the blueprint to move forward into qualification. Product storage in the liquid phase or vapor phase
is determined. Qualification temperature range is then determined. Adequate ventilation with oxygen
monitoring is mandatory and Personal Protective Equipment (PPE) must be worn when working with LN2.
3. Qualifying your cryogenic storage chamber. Once acceptance criteria are established, the study types and
test durations can be determined. At least one full cycle of temperature data must be recorded; three full
cycles would demonstrate more data points and complies with a best practice approach. Temperature
mapping under empty chamber and/or loaded chamber conditions and whether an open-door study may
also be performed in validation.

121
EFFECTIVE KNOWLEDGE TRANSFER DURING BIOPHARMACEUTICAL TECHNOLOGY TRANSFER
How Well Do We Do It? By Martin Lipa, Paige Kane and Anne Greene
ABSTRACT While knowledge management (KM) has been widely applied in other sectors, the international
biopharmaceutical sector has struggled with the meaningful and sustained application of effective KM
practices. This is evident even though KM has been highlighted in regulatory guidance for over 10 years, and
the positive business impact of KM is well recognized in other sectors. This paper focuses on the topic of KM as
applied to biopharmaceutical technology transfer, introducing new research that explores the importance and
effectiveness of knowledge transfer as an integral component of a biopharmaceutical product technology
transfer. Results from multiple sources explored in this paper are well aligned in recognizing that knowledge
transfer is particularly important to enable technology transfer, yet the biopharmaceutical sector is not
remarkably effective at this knowledge transfer. This is especially true of tacit knowledge transfer which is
often reported to be ineffective. Additional research will further define the barriers to improve knowledge
transfer effectiveness and how the biopharmaceutical sector might improve in this area.
INTRODUCTION This paper presents a case for the need to improve knowledge transfer (more broadly knowledge
management) as part the technology transfer stage in the pharmaceutical product lifecycle. The importance of
effective knowledge transfer to enabling successful technology transfer is established and the current
effectiveness is characterized using multiple inputs which are assessed, reported, and discussed.
While knowledge management (KM) approaches have been widely applied in other sectors, the international
biopharmaceutical sector has struggled with meaningful and sustained application of effective KM practices.
Furthermore, recent research carried out by the TU Dublin Pharmaceutical Regulatory Science Team [1] has
identified that technology transfer occurs over many phases of the product lifecycle and that knowledge
transfer is underappreciated and undervalued during such transfers. This paper seeks to further understand
the barriers to improve knowledge transfer for enabling successful technology transfer and knowledge
management application for the biopharmaceutical sector.
BACKGROUND
Pharmaceutical Regulatory Context
In 2008 The International Council for Harmonisation (ICH) published a guideline on Pharmaceutical Quality
System Q10 [2], commonly referred to as “ICH Q10.” The objectives of ICH Q10 are:
• to achieve product realization
• to establish and maintain a state of control
• to facilitate continual improvement

122
ICH Q10 positioned knowledge management (KM) as an enabler to the Pharmaceutical Quality System (PQS)
(Figure 1) suggesting that effective knowledge management is required to realize an effective PQS, and
therefore to achieve the objectives of ICH Q10. This regulatory guidance marked the first time that knowledge
management was identified as an expectation for the sector. However, minimal guidance on what is required
or how this might be achieved is provided in ICH Q10. Although the sector has struggled with KM adoption, no
further regulatory guidance has been published beyond the Q&A document [3] since the release of ICH Q10.
However, the Q&A document also discusses what KM is not to be. It is not viewed as an information
technology (IT) system. Rather, the ‘what’ and ‘how’ for KM were left up to individual organizations. The
absence of further guidance, such as models for best practices, guiding principles, or measures of progress or
realization is a contributory factor as to why progress in KM has been slow and elusive in the sector.
Figure 1 - ICH Q10 Pharmaceutical Quality System
Formal research on knowledge management in the biopharmaceutical sector was undertaken by Kane in 2014
[1]. At that time, little guidance existed to describe how KM might enable a more effective pharmaceutical
quality system. Kane’s research has led to the establishment of a model, known as the Pharma KM Blueprint
[1] which consists of four key elements one of which is the premise of this paper: The Pharmaceutical Product
Knowledge Lifecycle (PPKL) Model. The PPKL addresses the challenge of enabling knowledge flow to increase
visibility, access and use of product and process knowledge assets across the product lifecycle. Specifically, this
model asserts the pharmaceutical product lifecycle diagram depicted in ICH Q10 [2] does not account for the
multiple instances of technology transfer that would typically occur over the lifecycle of a product, nor the
generation and capture of tacit knowledge generated during technology transfer or continual improvement
activities. Kane’s model presented in Figure 2 substitutes the ICHQ10 Technology Transfer lifecycle stage with
an enhanced lifecycle stage entitled New Product Introduction and highlights the need for technology and
knowledge transfer along the full lifecycle of the product.

123
Figure 2 - Pharmaceutical Product Knowledge Lifecycle (PPKL) highlighting technology and knowledge
transfer in multiple points along the product lifecycle [1]
While the PPKL model develops the concept of Technology and Knowledge Transfer (as highlighted in the
orange bar in Figure 2, it is acknowledged that future research opportunities are warranted in the area of new
product introduction and technology transfer. This paper outlines the first of a series of research to addresses
this.
Currently ICH Q12, Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle
Management, Q12 [4], is in draft. Q12 is intended to further advance the expectation that improved product
and process knowledge can contribute to a reduction in the number post-approval change submissions, as the
accumulated knowledge gained during development and implementation of changes will manage risks to
product quality. These regulatory expectations should further increase the importance and urgency for the
sector to be more effective at the practice of knowledge management.
Technology Transfer’s Dependency on Knowledge Transfer A scan of current literature suggests the importance of successful technology transfer, and of knowledge to the
success of technology transfer. The PDA Technical Report No. 65 on Technology Transfer [5] states “technology
transfer can affect drugs and patients”, clearly highlighting the importance of an effective technology transfer
to ensure product outcomes and protect patients. And Millili [6] outlines examples where insufficient process
knowledge result in a poorly scaled-up process, along with other undesirable outcomes including:
• Non-robust processes (decreased process capability, i.e. Cpk)
• Decreased reliability
• Reduced production rates
• Increased number of atypical events (e.g. defects, elegance issues, etc.)

124
• Difficulty handling variations (raw materials, process controls, …)
• Inefficient validation.
Further examples in the literature refer to other areas where the sector struggles with transferring knowledge
during technology transfer, such as contamination control and sterilization technology risks [7]. Consider the
following issues and shortcomings cited on knowledge transfer effectiveness during technology transfer:
• “…assays were transferred but the sending party did not provide complete information and some of
the information was out-of-date…” [8]
• “…poor process understanding, coupled with incomplete documentation (i.e. codification) of all the
required process parameters…” [9]
• “The third mistake is not arranging for scientist-to-scientist interaction during the transfer process.
Scientists from similar departments at both the transferring company and the receiving company need
to get acquainted, understand the transfer process, and then work side by side at the bench or in the
plant. Without that personal interaction, your transfer is risky” [10]
• “…incomplete knowledge transfer…is a consistent problem…” [11]
• “…there was no master document to track all the information and it was sent out piecemeal to
different points of contact…” [8]
• “…providing incomplete information about the nature of the biopharmaceutical or protein molecule
such as its properties, its activities, and its stability under different conditions. Often, companies know
this information, but don’t pass it on…” [10]
There is a clear opportunity to improve the effectiveness of knowledge transfer during technology transfer,
which in turn will improve technology transfer outcomes and associated patient outcomes. In better leveraging
the knowledge of the organizations involved – and ensuring that knowledge is available and accessible, such
improvements will also address, at least in part, the regulatory expectations emerging from ICH Q12.
New Research to Advance Knowledge Transfer Understanding and Effectiveness Building on the foundational research by Kane, and the advancing expectation to better manage product and
process knowledge highlighted in Q12 [4] and other business contexts [12], further research on knowledge
management during at technology transfer has commenced by Lipa. The research will explore elements of
both explicit and tacit knowledge management during technology transfer. Lipa’s preliminary research
hypothesis is as follows:
a. The sector does not have a holistic end-to-end view of what it knows about its products across the
product lifecycle, nor how to best ensure this knowledge ‘flows’ to ensure the best possible
product outcomes. These outcomes include product realization through a readily available, cost
effective and high-quality product to patients, as well as additional outcomes of operational
efficiency and a workforce that has the knowledge it needs to do its best work.
b. Further, tacit knowledge is critical but is not effectively managed or transferred during key
activities in the product lifecycle, including key processes such as technology transfer.
To raise awareness and to provide guidance on how to improve knowledge transfer associated with
technology transfer, and to ultimately improve technology transfer outcomes, this research commences by
characterizing the current state of how KM enables technology transfer, including perceived importance and
effectiveness for explicit and tacit knowledge.
The research approach is to gather input from multiple sources to establish a baseline on knowledge transfer
effectiveness within technology transfer.

125
Three distinct research activities were undertaken to gather input as follows: 1. Literature review of industry guidance on technology transfer.
2. Survey, from an audience survey conducted in April 2019, on the importance and effectiveness of
knowledge transfer as part of technology transfer
3. Expert interviews from international industry and health authorities
Further additional research may include: 1. Benchmark other industries on processes and proven effectiveness of knowledge transfer.
2. Develop a model to describe the maturity of knowledge transfer.
3. Develop recommendations for enhancing knowledge transfer during technology transfer, including
any supporting tools, assessments, or models to accelerate post-research uptake.
RESULTS Characterization of Current State Knowledge Transfer Literature Review: Industry Guidance on Technology Transfer - Initial research included a review of common
industry guidance on technology transfer, to assess the extent to which knowledge transfer, knowledge
management and tacit knowledge concepts are presented and explained, along with the extent of illustrative
examples and guidance or tips on the ‘how.’ The following technology transfer guidance was reviewed, and
the frequency of these concepts was tabulated and summarized in Table 1.
• WHO Guidelines on Transfer of Technology in Pharmaceutical Manufacturing [13]
• ISPE Good Practice: Technology Transfer, 2nd Edition [14]
• ISPE Good Practice: Technology Transfer, 3rd Edition [15]
• PDA Technical Report, No. 65, Technology Transfer [5]
• PDA Tech Transfer Interest Group Report Out, PDA 2019 Annual Meeting (presentation) [16]
A qualitative assessment was conducted on how well these guidance documents introduced the knowledge
transfer concepts above, including how well they are collectively explained, whether they provided illustrative
examples, and whether they provided guidance / tips on ‘how.’ These results are also provided along with
author commentary in Table 1.
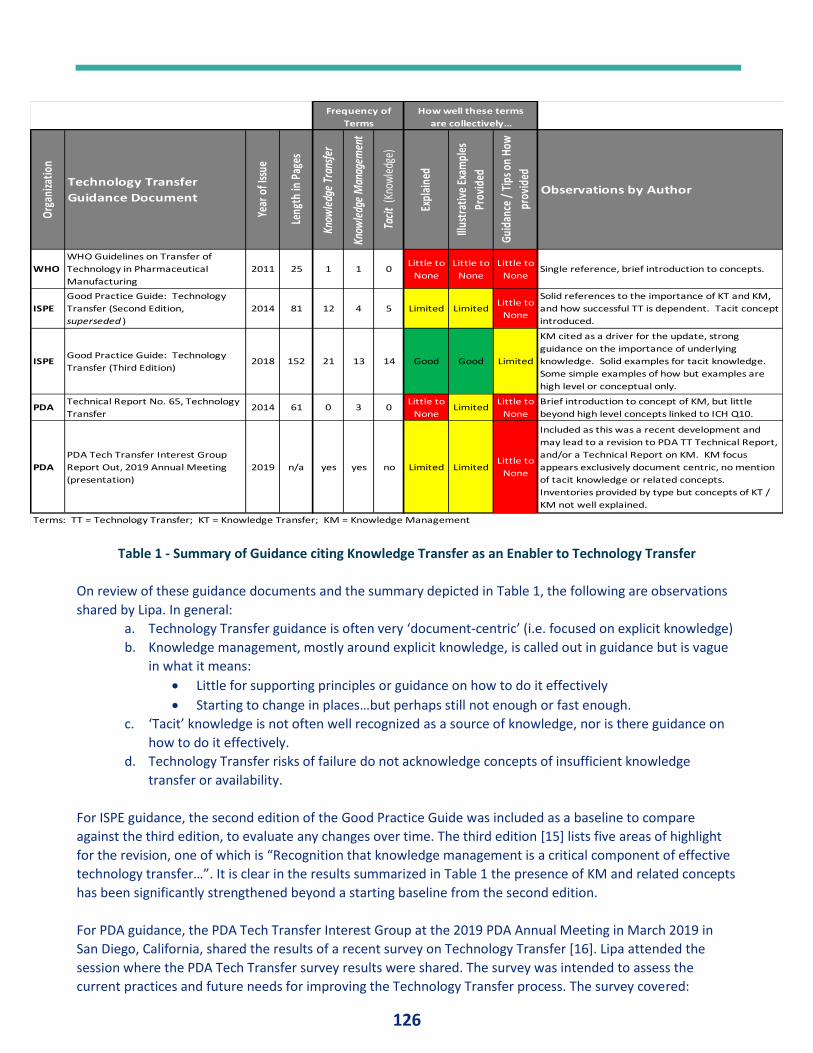
126
Table 1 - Summary of Guidance citing Knowledge Transfer as an Enabler to Technology Transfer
On review of these guidance documents and the summary depicted in Table 1, the following are observations
shared by Lipa. In general:
a. Technology Transfer guidance is often very ‘document-centric’ (i.e. focused on explicit knowledge)
b. Knowledge management, mostly around explicit knowledge, is called out in guidance but is vague
in what it means:
• Little for supporting principles or guidance on how to do it effectively
• Starting to change in places…but perhaps still not enough or fast enough.
c. ‘Tacit’ knowledge is not often well recognized as a source of knowledge, nor is there guidance on
how to do it effectively.
d. Technology Transfer risks of failure do not acknowledge concepts of insufficient knowledge
transfer or availability.
For ISPE guidance, the second edition of the Good Practice Guide was included as a baseline to compare
against the third edition, to evaluate any changes over time. The third edition [15] lists five areas of highlight
for the revision, one of which is “Recognition that knowledge management is a critical component of effective
technology transfer…”. It is clear in the results summarized in Table 1 the presence of KM and related concepts
has been significantly strengthened beyond a starting baseline from the second edition.
For PDA guidance, the PDA Tech Transfer Interest Group at the 2019 PDA Annual Meeting in March 2019 in
San Diego, California, shared the results of a recent survey on Technology Transfer [16]. Lipa attended the
session where the PDA Tech Transfer survey results were shared. The survey was intended to assess the
current practices and future needs for improving the Technology Transfer process. The survey covered:
Org
aniza
tion
Technology Transfer
Guidance Document
Year
of I
ssue
Leng
th in
Pag
es
Know
ledg
e Tr
ansf
er
Know
ledg
e M
anag
emen
t
Taci
t (K
now
ledg
e)
Expl
aine
d
Illus
trat
ive
Exam
ples
Prov
ided
Guid
ance
/ T
ips o
n Ho
w
prov
ided
Observations by Author
WHO
WHO Guidelines on Transfer of
Technology in Pharmaceutical
Manufacturing
2011 25 1 1 0Little to
None
Little to
None
Little to
NoneSingle reference, brief introduction to concepts.
ISPE
Good Practice Guide: Technology
Transfer (Second Edition,
superseded )
2014 81 12 4 5 Limited LimitedLittle to
None
Solid references to the importance of KT and KM,
and how successful TT is dependent. Tacit concept
introduced.
ISPEGood Practice Guide: Technology
Transfer (Third Edition)2018 152 21 13 14 Good Good Limited
KM cited as a driver for the update, strong
guidance on the importance of underlying
knowledge. Solid examples for tacit knowledge.
Some simple examples of how but examples are
high level or conceptual only.
PDATechnical Report No. 65, Technology
Transfer2014 61 0 3 0
Little to
NoneLimited
Little to
None
Brief introduction to concept of KM, but little
beyond high level concepts linked to ICH Q10.
PDA
PDA Tech Transfer Interest Group
Report Out, 2019 Annual Meeting
(presentation)
2019 n/a yes yes no Limited LimitedLittle to
None
Included as this was a recent development and
may lead to a revision to PDA TT Technical Report,
and/or a Technical Report on KM. KM focus
appears exclusively document centric, no mention
of tacit knowledge or related concepts.
Inventories provided by type but concepts of KT /
KM not well explained.
Terms: TT = Technology Transfer; KT = Knowledge Transfer; KM = Knowledge Management
Frequency of
Terms
How well these terms
are collectively…

127
• Demographics
• Types of Technology Transfer Performed
• The Technology Transfer Process
• Use of Multi‐Disciplinary Teams
• Technology Transfer Tools
• Challenges.
The results indicated that KM would be an area where additional PDA guidance would be helpful. The
subsequent discussion on KM in session focused heavily on a ‘master plan’ for knowledge management which
primarily focused on documents and information. Also, a set of KM “soft skills” was identified as required,
although in the opinion of Lipa, these are primarily good business communication and team leadership skills,
rather than traditional KM skills as described elsewhere [17].
In general, across any of the guidance documents, there does not appear to be a measure for the effectiveness
or completeness of knowledge transfer associated with technology transfer, except for document turnover
lists. This will be further investigated during subsequent research.
Survey: Audience Survey on Knowledge Transfer Enabling Successful Technology Transfer Once the literature review was complete and based on the review findings and the researcher’s own
experiences, a survey was developed to further support the hypothesis problem statement by testing the
opinion of a naïve audience. The survey was designed to solicit their perspectives on the importance and
effectiveness of knowledge transfer to enable an effective and efficient technology transfer. This survey was
deployed at a recent seminar, An Audience with Regulatory, Academia and Industry on The Role of Effective
QRM & KM in Product Realization for Patients in the 21st Century on 04-April-2019 at Technological University
Dublin.
The survey was distributed to the audience of approximately 120 attendees, and 56 responses were received.
It is important to note results from this survey are considered directional in nature due to the qualitative
nature of the questions provided, although useful comparisons can be made within the response data. A
detailed review of the complete survey results can be found in the monograph of the proceeding from the
seminar [18].
A key focus of the survey was to evaluate the perceived importance of both explicit and tacit knowledge to an
effective and efficient technology transfer.
• Explicit knowledge was defined as: Documents and other ‘codified’ knowledge that takes no
explanation or dialog to fully understand.
• Tacit knowledge was defined as: Knowledge associated with experience, subject matter expertise,
decision rationale, observation, undocumented history, and other knowledge “in people’s heads.”
The survey also solicited opinions on the corresponding effectiveness for each explicit and tacit knowledge
transfer. The results are summarized in Figure 3.
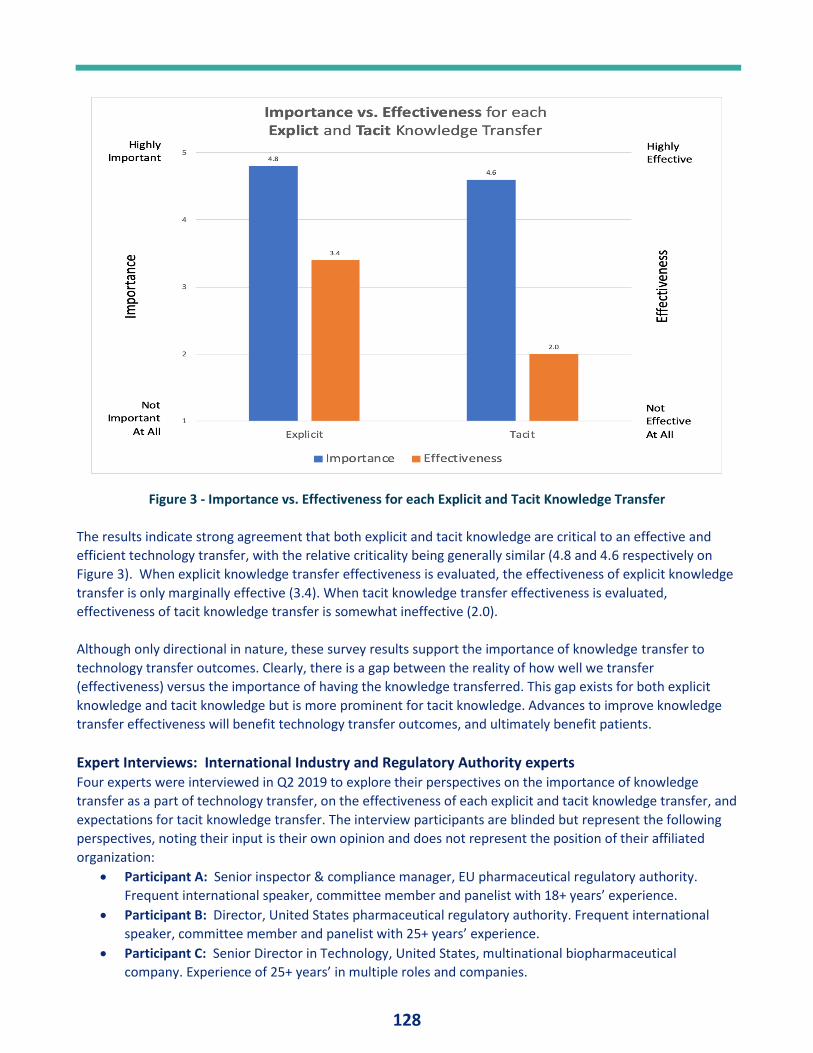
128
Figure 3 - Importance vs. Effectiveness for each Explicit and Tacit Knowledge Transfer
The results indicate strong agreement that both explicit and tacit knowledge are critical to an effective and
efficient technology transfer, with the relative criticality being generally similar (4.8 and 4.6 respectively on
Figure 3). When explicit knowledge transfer effectiveness is evaluated, the effectiveness of explicit knowledge
transfer is only marginally effective (3.4). When tacit knowledge transfer effectiveness is evaluated,
effectiveness of tacit knowledge transfer is somewhat ineffective (2.0).
Although only directional in nature, these survey results support the importance of knowledge transfer to
technology transfer outcomes. Clearly, there is a gap between the reality of how well we transfer
(effectiveness) versus the importance of having the knowledge transferred. This gap exists for both explicit
knowledge and tacit knowledge but is more prominent for tacit knowledge. Advances to improve knowledge
transfer effectiveness will benefit technology transfer outcomes, and ultimately benefit patients.
Expert Interviews: International Industry and Regulatory Authority experts Four experts were interviewed in Q2 2019 to explore their perspectives on the importance of knowledge
transfer as a part of technology transfer, on the effectiveness of each explicit and tacit knowledge transfer, and
expectations for tacit knowledge transfer. The interview participants are blinded but represent the following
perspectives, noting their input is their own opinion and does not represent the position of their affiliated
organization:
• Participant A: Senior inspector & compliance manager, EU pharmaceutical regulatory authority.
Frequent international speaker, committee member and panelist with 18+ years’ experience.
• Participant B: Director, United States pharmaceutical regulatory authority. Frequent international
speaker, committee member and panelist with 25+ years’ experience.
• Participant C: Senior Director in Technology, United States, multinational biopharmaceutical
company. Experience of 25+ years’ in multiple roles and companies.

129
• Participant D: Senior Director, EU, multinational pharmaceutical company. Experience of 30+ years’ in
multiple roles and companies, including health authority and academic experience in the
biopharmaceutical sector.
The interviews followed a structured set of questions and were typically an hour long. The interviews were
transcribed, coded and responses summarized in Table 2.
Considering the end-to-end (E2E) product lifecycle depicted in ICH Q10 [2],
To what extent do you agree that knowledge transfer could be improved for technology transfer, leading to better
outcomes?
Regulatory Authority Perspectives Industry Perspectives
The ‘Big Picture’
• Lots of opportunity to improve
• Several companies do a good job
• Initial technology transfer is critical
• Starts with taking learning from development
• Need honesty and transparency
• Understand how much variability
• Residual latent risk remains
Business Process Challenges
• Ceremonial writing of report
• Many companies capture only part
• Knowledge gets lost between development and
commercial manufacturing
• Deep investigations were eventually uncovered still
in place at the old facility
• Don’t lose in translation
Document Challenges
• Knowledge gets lost or buried in documents
• Documents may not be in usable format
• Documents may be long
The ‘Big Picture’
• Knowledge transfer is essential
• Tech transfer as opportunity to give another group of
people the ability and skill to do what you have been
doing adequately
• Could be improved deeper understanding and benefits
would accrue, including cost, quality, and availability
• Technology transfer sometimes driven by a compliance
need, not a knowledge need
• Technology transfer sometimes seen as a tedious task
that must be done
Business Process Challenges
• Many functions work in a vacuum
• Not everyone knows what everyone else is doing
• Delays due to needing to purchase or modify equipment
not planned ahead
• Ensure quality systems can handle new process
• Approval delay for insufficient quality systems
• Know how may not be transferred
On a scale of 1 to 10 (10 = exceptional) - How would you rate the range and average effectiveness of knowledge transfer
of explicit knowledge during technology transfer?
Regulatory Authority Perspectives Industry Perspectives
Participant A:
Average: 6 out of 10
Range: 3 to 8
Participant B:
Average: 7 out of 10
Range: 3 to 9
Participant C:
Average: 6 out of 10
Range: 3 to 10
Participant D:
Average/ Range: “in the upper half, with wide standard
deviation”
On a scale of 1 to 10 (10 = exceptional) - How would you rate the range and average effectiveness of knowledge transfer
of tacit knowledge during technology transfer?
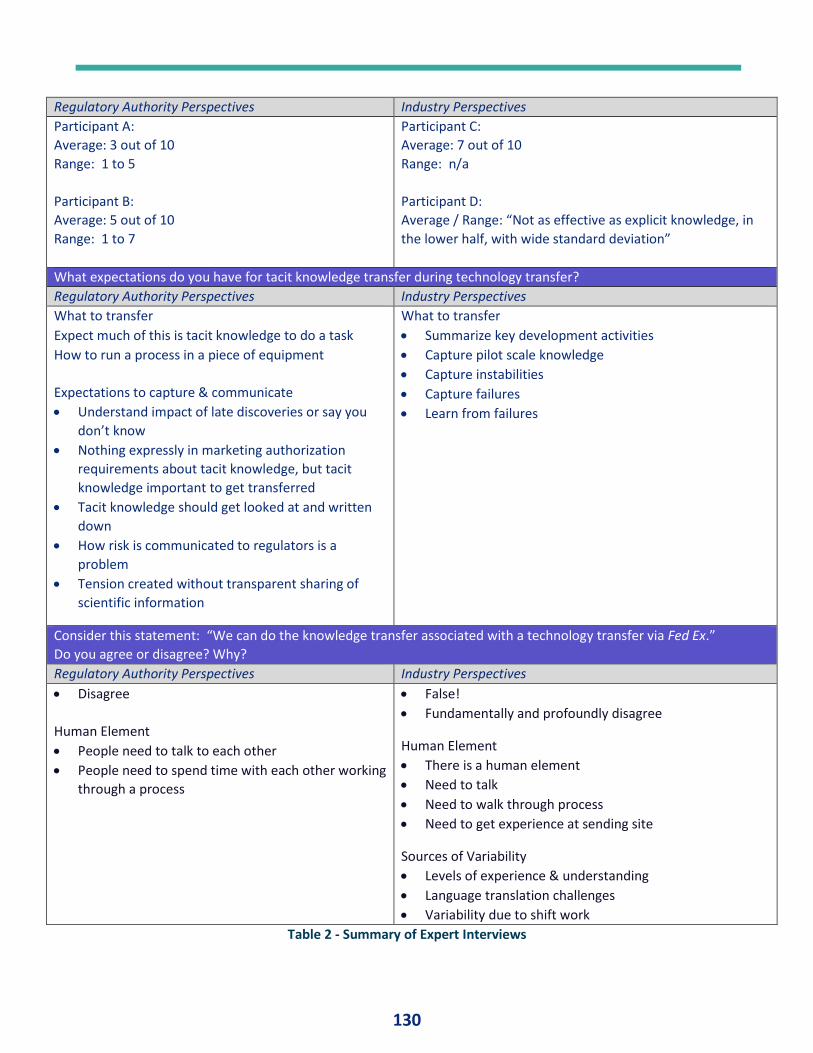
130
Regulatory Authority Perspectives Industry Perspectives
Participant A:
Average: 3 out of 10
Range: 1 to 5
Participant B:
Average: 5 out of 10
Range: 1 to 7
Participant C:
Average: 7 out of 10
Range: n/a
Participant D:
Average / Range: “Not as effective as explicit knowledge, in
the lower half, with wide standard deviation”
What expectations do you have for tacit knowledge transfer during technology transfer?
Regulatory Authority Perspectives Industry Perspectives
What to transfer
• Expect much of this is tacit knowledge to do a task
• How to run a process in a piece of equipment
Expectations to capture & communicate
• Understand impact of late discoveries or say you
don’t know
• Nothing expressly in marketing authorization
requirements about tacit knowledge, but tacit
knowledge important to get transferred
• Tacit knowledge should get looked at and written
down
• How risk is communicated to regulators is a
problem
• Tension created without transparent sharing of
scientific information
What to transfer
• Summarize key development activities
• Capture pilot scale knowledge
• Capture instabilities
• Capture failures
• Learn from failures
Consider this statement: “We can do the knowledge transfer associated with a technology transfer via Fed Ex.”
Do you agree or disagree? Why?
Regulatory Authority Perspectives Industry Perspectives
• Disagree
Human Element
• People need to talk to each other
• People need to spend time with each other working
through a process
• False!
• Fundamentally and profoundly disagree
Human Element
• There is a human element
• Need to talk
• Need to walk through process
• Need to get experience at sending site
Sources of Variability
• Levels of experience & understanding
• Language translation challenges
• Variability due to shift work
Table 2 - Summary of Expert Interviews

131
These results speak for themselves; a summary is as follows: 1. Knowledge transfer can be improved and would have meaningful positive impact to technology
transfer outcomes, including cost, quality, and product availability.
2. Some companies appear to do well but this is the exception, not the norm.
3. Transparency on the level of process understanding is critical to a productive regulatory dialog.
4. Often knowledge gets ‘stuck,’ often based on process or people barriers (e.g. judgement it is not
important, buried in long documents, may be in an unusable format.
5. On average, knowledge transfer effectiveness of explicit knowledge is marginal and there is wide
variation.
6. On average, knowledge transfer effectiveness of tacit knowledge is ineffective to marginal and there is
wide variation.
7. Successful technology transfer required human to human interactions, preferably face to face and time
to walk through the details of a process to explore details, sensitivities, what is not known, etc.
8. There is a clear desire that we must get better at this as a sector.
DISCUSSION The three independent research activities and resulting data correlate well and suggest these key findings:
1. Overall, knowledge transfer is critical to a successful and sustainable technology transfer. Ineffective
knowledge transfer can have a long-lasting impact on the ability of the receiving site to provide cost-
effective, high-quality product with the desired availability.
2. Knowledge to be transferred associated with technology transfer is biased toward explicit knowledge
(e.g. documents). This explicit knowledge is critical to the success of the transfer yet we as a sector are
only marginally effective at it – it is clearly not a strength. There is some supporting guidance on
explicit knowledge that should be transferred, but not prescriptive means on how to do this or how to
measure effectiveness.
3. Tacit knowledge associated with technology transfer is not widely recognized as an asset to be
transferred, nor is there evidence to suggest we as a sector do it effectively. There is limited
understanding on what tacit knowledge is, why it is important and how it can be transferred, including
how to measure effectiveness of transfer. There is little acknowledgement of tacit knowledge in
common industry guidance for technology transfer, although there has been an upward trend very
recently on calling out tacit knowledge categorically.
4. Regulatory authorities and industry are generally well aligned on these issues and their impact. Both
recognize the opportunity – and the need – to improve for the good of patients.
These findings support the problem statements which are being explored, namely, that knowledge does not
‘flow’ readily through technology transfer, and that tacit knowledge is critical but is not effectively managed or
transferred. The subsequent research activities to benchmark other industries, develop a knowledge transfer
maturity model and associated recommendations to improve knowledge transfer will proceed with the aim to
address this opportunity.
CONCLUSION In conclusion, knowledge management is still a relatively immature practice in the biopharmaceutical sector,
especially when compared to Quality Risk Management, Change Management, and other practice domains.
The need for improved knowledge transfer for technology transfer, as a key focus point of knowledge
management in the biopharmaceutical sector, is evident given the findings presented in this paper, supported
by the broad alignment and recognition of the issue across different cohorts presented herein. This KM focus
first and foremost to protect the patient through availability of a high quality, cost effective product, and

132
present the opportunity pursue other business drivers which ensure the continued competitiveness of the
organizations in the sector [12].
The next phases of research by Lipa as introduced in this paper intend to provide practical advice to help the
sector apply good KM practices to improve technology transfer outcomes through the following:
a. Benchmark other industries on processes and proven effectiveness of knowledge transfer.
b. Develop a model to describe the maturity of knowledge transfer.
c. Develop recommendations for enhancing knowledge transfer during technology transfer, including
any supporting tools, assessments, or models to accelerate post-research uptake.
The initial findings presented within this paper well justify the planned efforts in this area.
REFERENCES
[1] P. Kane, A Blueprint for Knowledge Management in the Biopharmaceutical Sector, Dublin: Dublin
Institute of Technology, 2018.
[2] International Council for Harmonisation, "Pharmaceutical Quality System Q10," ICH, Geneva,
Switzerland, 2008.
[3] International Council for Harmonisation, "Quality Implementation Working Group on Q8, Q9 and Q10 -
Questions & Answers (R4)," ICH, Geneva, Switzerland, 2010.
[4] International Council for Harmonisation, "Technical and Regulatory Considerations for Pharmaceutical
Product Lifecycle Management," ICH, Geneva, Switzerland, 2017.
[5] Parenteral Drug Association, Inc., "Technical Report No. 65 - Technology Transfer," PDA, Bethesda, 2014.
[6] G. Millili, Scale-up & Technology Transfer as a Part of Pharmaceutical Quality Systems, Arlington, VA:
ISPE, 2011.
[7] T. Sandle, "Sterility Assurance in Early Phase Development," in Phase Appropriate GMP for Biological
Processes: Pre-clinical to Commercial Production, Arlington Heights, IL, USA, DHI/PDA Books, 2018, pp.
500-520.
[8] S. Perry, "Tech Transfer: Do It Right the First Time," 06 January 2010. [Online]. Available:
https://www.pharmamanufacturing.com/articles/2010/007/. [Accessed 01 February 2019].
[9] W. Schmidt and I. Uydess, "Keys to Executing a Successful Technology Transfer," Pharmaceutical
Technology, vol. 2011 Supplement, no. 2, pp. 1-5, 2011.
[10] A. Shanley, "Getting Biopharmaceutical Tech Transfer Right the First Time," in Outsourcing Resources
2017, London, BioPharm International, 2017, pp. 27-31.
[11] A. Shanley, "Tech Transfer: Tearing Down the Wall," Pharmaceutical Technology, vol. 42, no. 12, pp. 24-
25, 48, 2018.
[12] N. Calnan, M. Lipa, P. Kane and J. Menezes, "Why Knowledge Management is Good Business," in A
Lifecycle Approach to Knowledge Excellence in the Biopharmaceutical Industry, Boca Raton, CRC Press,
2018, pp. 3-18.
[13] World Health Organization, "WHO guidelines on transfer of technology in pharmaceutical manufacturing,
WHO Technical Report Series, No. 961," WHO, Washington, 2011.
[14] ISPE, "Good Practice Guide: Technology Transfer, Second Edition," ISPE, Tampa, 2014.
[15] ISPE, "Good Practice Guide: Technology Transfer, Third Edition," ISPE, 2018, Tampa.
[16] B. Haas, "Technology Transfer (TT) IG," in 2019 PDA Annual Meeting, San Diego, 2019.

133
[17] N. Calnan, M. Lipa, P. Kane and J. Menezes, "The House of Knowledge Excellence - A Framework for
Success," in A Lifecycle Approach to Knowlege Excellence in the Biopharmaceutical Industry, Boca Raton,
CRC Press, 2018, pp. 181-224.
[18] A. Greene, K. O'Donnell, A. Murphy and E. Harris, "An Audience with Pharmaceutical Regulators,
Academia and Industry - The role of Quality Risk Management (QRM) and Knowledge Management (KM)
in Medicinal Product Realisation for Patients in the 21st century," TU Dublin Academic Press, Dublin,
2019.

134
RISK CONSIDERATIONS FOR THE USE OF UNIDIRECTIONAL AIRFLOW DEVICES IN MICROBIOLOGY LABORATORIES By Tim Sandle, Ph.D., Pharmaceutical Microbiologist, Bio Products Laboratory Limited
INTRODUCTION Unidirectional airflow devices and microbiological safety cabinets are a common feature in many laboratories, including applications to achieve contamination control. Such devices are suitable for a variety of applications and especially where a demarcated clean air environment is required for processing smaller items and for practicing aseptic technique.
Unidirectional airflow, as defined by the World Health Organization, is a rectified airflow over the entire cross-sectional area of a clean zone with a steady velocity and approximately parallel streamlines. Conventional unidirectional airflow systems, where an ISO 14644 class 5/ WHO and EU GMP Grade A condition is required, have a guidance airflow velocity of 0.36 to 0.54 m/s. This form of airflow contrasts to turbulent flow, which is air distribution that is introduced into the controlled space and then mixes with room air by means of induction (1).
Such devices work using in-flow unidirectional air drawn through one or more high efficiency particulate air (HEPA) filters, where air control designed to create a particle-free working environment and provide product protection (2). Air is taken through a filtration system and then exhausted across the work surface as part of the airflow process. Commonly, the filtration system comprises of a pre-filter and a HEPA filter. The device is enclosed on the sides and constant positive air pressure is maintained to prevent the intrusion of contaminated room air.
Such devices are subject to physical problems and increased microbiological risks if they are not maintained correctly, especially in relation to air control. This paper assesses the most common risks affecting these laboratory devices. Prior to examining the devices and undertaking a model assessment, the paper discusses risk assessment in general, and looks at a generalized approach for conducting qualitative Failure Modes and Effects Analysis.
RISK ASSESSMENT Risk assessment can be expressed as a formal process, and here such an activity based on a series of key steps. In short, these involve:
• Establishing the context and environment that could present a risk.
• Identifying the hazards and considering the risks these hazards present.
• Analyzing the risks, including an assessment of the various contributing factors.
• Evaluating and prioritizing the risks in terms of further actions required.
• Identifying the range of options available to tackle the risks and deciding how to implement risk mitigation strategies.
To perform this exercise there are a range of risk methodologies available and some will be more applicable to a given situation than others. Importantly, risk assessment does not need to be a complicated process and should be appropriate to the organization, process, product or scenario.

135
Often the term risk, as the outcome of an assessment, is confused with the word hazard. In fact, risk is the expression of a hazard. A hazard is the potential source of harm, and one that either exists or does not exist (it is the assessment of risk that quantified or qualifies a hazard). Some hazards can cause more harm than others (different degrees of severity). Hazards exist everywhere but this does not mean they are a problematic risk; most hazards are dormant, with the potential to cause harm depending upon circumstances. Here the hazard concept connects with the probability of the hazard causing harm, and when this occurs the hazard causes an incident. Think of it as stored energy waiting to be released (3). What is of concern with hazards are consequences; these are the potential outcomes resulting from a hazard when it occurs. Consequences shape risks based on the potential severity of the hazard and the likelihood or probability that the hazard will cause something to occur. This may be a simple ‘risk’ or feeling, or with one risk compared another or against a benchmark, when expressed qualitatively, as low, medium or high; or semi-quantitatively as a number (4). Thus, risk and hazard form the following relationship (5):
So, risk is an expression of the probability that a given hazard will cause harm. Here a hazard will pose low risk if the likelihood of its exposure is minimized. Risk identification is concerned with selecting the hazards of concern in relation to a product or process. Hazard tend to be classified into different categories. Examples of hazard classification are (6):
• Physical hazards. These are factors that can cause physical harm to a person or damage to equipment.
• Chemical hazards are substances that can cause harm to a person or product.
• Biological hazards are biological agents that can cause harm to the human body or adulterate a product. This includes microorganisms and microbial toxins.
• Psychological hazards. These would include psychological factors or personnel fatigue.
• Ergonomic hazards. This would include factors such as access to equipment. As an aid to clearly defining the risk(s) for risk assessment purposes, four fundamental questions are often helpful:
1. What might go wrong? 2. What is the likelihood (probability) it will go wrong? 3. What are the consequences (severity)? 4. What is the detectability?
Thus, the components of quality risk management are:
• Severity of harm ‐ a measure of the possible consequences or degree of harm,
• Probability that harm will occur ‐ frequency or likelihood of occurrence of the hazard,
• Detection of risk ‐ the ability to discover or determine the existence, or presence of the hazard. There are a range of different risk models and risk tools. The example used in this paper is a variant for Failure Modes and Effects Analysis (FMEA).
Failure Modes and Effects Analysis (FMEA) There are a range of different approaches to FMEA, some using number scoring systems (the most common) to produce a risk priority number, and others using qualitative terms, as used in this paper. There is no right, or
Risk = Hazard x Vulnerability (or severity)
Capacity (or likelihood)

136
wrong approach and the model used in this paper is purely illustrative, and the FMEA could be approached in different ways (7). In general, the FMEA approach requires the identification of the following basic information (8):
• Item(s)
• Function(s)
• Failure(s)
• Effect(s) of Failure
• Cause(s) of Failure
• Current Control(s)
• Recommended Action(s) This is achieved by (9):
• Assembling a team
• Establishing common rules
• Gathering and reviewing relevant information
• Identifying the item(s) or process(es) to be analyzed
• Identifying the function(s), failure(s), effect(s), cause(s) and control(s)
• Evaluate the risk
• Prioritizing and assigning corrective actions,
• Performing corrective actions and re-evaluating risks
• Distributing, reviewing and updating the analysis as appropriate From then on, approaches vary, and one possible approach is discussed later in this paper. While most organizations use a quantitative FMEA, where a scoring system is used to generate a risk priority number (10), an equaling legitimate approach is to use a qualitative methodology. This can avoid the need haggle over numbers and can help to conceptualize risks more meaningfully when the team can only select from categories like ‘low,’ ‘medium’ and ‘high.’ To use qualitative criticality analysis to evaluate risk and prioritize corrective actions, the team must rate the severity of the potential effects of failure and rate the likelihood of occurrence for each potential failure mode. It is then possible to compare failure modes via a Criticality Matrix, which identifies severity on the horizontal axis and occurrence on the vertical axis.
UNIDIRECTIONAL AIRFLOW DEVICES Unidirectional airflow refers to air that flows in a straight, unimpeded path. Unidirectional flow is maintained using cabinets that direct air jets downward in a straight path. Such devices utilize HEPA filters to filter and clean all air entering the environment. The filter housings are often composed of stainless steel (for cleanability); with the filter composed of non-shedding materials to ensure the number of particles that enter the facility remains low. There are two common types of devices – vertical and horizontal. These are distinguished:
1. Horizontal devices receive their name due to the direction of air flow which comes from above but then changes direction and is processed across the work in a horizontal direction. The constant flow of filtered air provides material and product protection.
2. Vertical devices function equally well as horizontal devices with the unidirectional air directed vertically downwards onto the working area. The air can leave the working area via holes in the base. Vertical flow cabinets can provide greater operator protection.

137
Figure 1: Microbiologist using a Type II MSC (Image: Tim Sandle) With vertical and horizontal UDAF devices, there is no significant difference in terms of microbial contamination for the testing of samples for bioburden, although there are differences in the placement of items in order to achieve a clean airflow. However, there may be some differences in terms of application, and this will affect the choice of the unit air direction design (11). For aseptic operations, where the objective is the avoidance of product contamination horizontal airflows offer an advantage (12). An additional concern is with operator safety, where powders are handled. Although horizontal unidirectional flow, with air traveling from the rear of the hood and exiting the front opening, may not encounter large obstructions inside the hood, it does eventually encounter the person performing the work. Substances, such as fumes, microorganisms or fine powders, may be blown into the operator’s face (13). While this collision may not compromise the unidirectional flow where work is performed, it may pose a health risk. In such cases, vertical flow is probably preferable or, preferably, a safety cabinet. Such concerns should be highlighted in individual sample handling risk assessments. In addition to standard devices, biological safety cabinets (biosafety cabinets) must be used when additional protection of the user and the environment is also required. This paper outlines a general risk assessment approach that can be considered in relation to the use of a cabinet.
DESCRIPTION AND FUNCTION OF THE EQUIPMENT Unidirectional Air Flow (UDAF) units (or Class I devices) and Class II Microbiological Safety Cabinets (MSC) are located within a Microbiology laboratory (operated as per EN 12469) (14). The UDAF units and the MSC are designed to protect microbiological testing from the surrounding environment. With these (15):
• A Class I cabinet protects the product as air is pulled into the cabinet and exhausted (usually out the top) out away from the user. A standard unidirectional (laminar) airflow cabinet would fall under this category.

138
• Class II biological safety cabinets protect the product or specimen, the user, and the environment from contamination. This is especially crucial for applications that require sterile or particulate-free conditions, such as cell culture experiments, drug preparations, or toxicology when working with fumes and gases is required. All types of Class II biological safety cabinet have HEPA filters at their air supply and exhaust points. A standard laboratory microbiological safety cabinet would fall under this category.
• Class III biological safety cabinets, also known as glove boxes or barrier isolators, provide maximum protection for work in biosafety level 4 containment labs. A class III biosafety cabinet is crucial for working with any biosafety level 4 agents, or other dangerous materials, such as aerosols of pathogens or toxins. These biological safety cabinets are hermetically sealed glove boxes with access through a dunk tank or an adjacent, double-door chamber that allows for decontamination. Typically, a class III biosafety cabinet has two HEPA filters in the exhaust system for additional environmental protection. Manipulations within the hermetically-sealed chamber of the glove box are performed with built-in arm-length rubber gloves. Class III biosafety cabinets with carbon filtration systems are also available for working with hazardous chemicals.
These devices are separate from fume cupboards. A fume cupboard is a key protective and control device in laboratories where chemicals are used. It is primarily a protective ventilated enclosure (partial containment device) designed such that hazardous concentrations of quantities of airborne contaminants are prevented from escaping from the fume cupboard into the work room or laboratory by means of a protective air barrier between the user and the materials placed within the enclosure.
Importantly MSCs are designed to protect the operator from microbiological contamination when handling microorganisms in biohazard groups I and II. The main components of the units are enclosed within paneled body. The units are directly connected to a standard electrical supply. Each unit consists of:
• Synthetic pre-filters
• Electric fans to produce the required air flow
• Panel High Efficiency Particulate Air (HEPA) filters
• Differential pressure gauge, mounted in a control panel
Within the cabinets sufficient space exists beneath or within each type of unit to allow access for operators to conduct their required task.
The siting of the devices had previously been accounted for. This is extremely important. Air currents and movement of people in the laboratory can adversely affect the performance (operator protection) of a cabinet. Factors to be considered include the proximity of cabinets to doors, windows, ventilation ducts and to movement routes.
• A second factor in place was correct use of the cabinets. Here the following factors were addressed:
• Users should avoid sudden and sweeping movement of arms to minimize disturbance of the air flow patterns.
• Large and bulky equipment should not be placed in the devices, nor should equipment be placed on air grilles as both these will disturb air flow patterns.
• Centrifuges, including microfuges, should not be placed in a Microbiological Safety Cabinet unless an operator protection factor (KI Discus) test has been carried out with it running in situ and has been proven not to compromise operator protection.
• Bunsen burners should not be used in devices, particularly Class II cabinets. This is because of the concern about the effect of the heat rising from the flame on the laminar downflow of air in the cabinet. However, if they are used, they should be placed towards the back of the cabinet and a low-profile type used. If the Bunsen is used in conjunction with alcohol etc. for flaming, then the alcohol pot should always be placed to the far side of the Bunsen in order that any drips from the item being flamed do not drop in the pot and ignite it.

139
• Cabinets should always be installed in appropriate locations to ensure any traffic movement within the laboratory does not cause draughts to disturb the airflow patterns at the front of the cabinet and affect performance. Users should be aware of this requirement and should ensure the standard 1 meter clear behind rule is observed when they are using the cabinet.
Such devices must be certified by a qualified individual. Certification is either against a national standard for a safety cabinet and / or against the appropriate ISO 14644 standard (to assess air cleanliness and other factors). Assessment is typically undertaken at the following times:
• when newly installed
• when moved or relocated
• after major repair such as replacement of HEPA filters or motor
• annually (minimum) but preferably every 6 months Prior to an inspection, maintenance or service event takes place, decontamination of the cabinet may be required. The level of decontamination required should be appropriate to the type of maintenance event taking place and must be the subject of a suitable and sufficient risk assessment. Service engineers, contractors etc. should be informed of any potential risks include the contamination status of the cabinet prior to the service.
RISK ASSESSMENT CRITERIA The assessment outlined in this paper determines some key risks associated with both the operation of units and the function it serves; this will ensure the air produced from the units is suitable for their current purpose. The main functions of the units are to (16):
• Maintain and control particle levels within the UDAF (of which some may be microbial carrying particles).
• Maintain air velocity and unidirectional air flow within the UDAF.
• Air temperature limits are of a low impact to Microbiology laboratory operations. Equipment is infrequently used within the units, except for a membrane filtration system for water testing.
• With safety cabinets, operator protection from microbial cultures. Microbiological safety cabinets are not designed to protect the user from all hazards, e.g. radioactive, toxic or corrosive hazards, and the exhaust HEPA filters will not remove these types of contaminants from the exhaust air.
Considering the above, the units were assessed for:
1. Temperature 2. Particle counts 3. Air-velocity 4. General breakdown 5. Filter failure 6. Blockages 7. Safety
Implicit in each of the above was the risk of microbiological contamination. The risk assessment aimed to highlight any work to be undertaken to confirm that the required unit function is maintained.
Risk assessment approach For the risk assessment a standard approach, assessing severity, likelihood and detection was used. This translated as:
Severity of Impact
• High = Test samples may become contaminated.
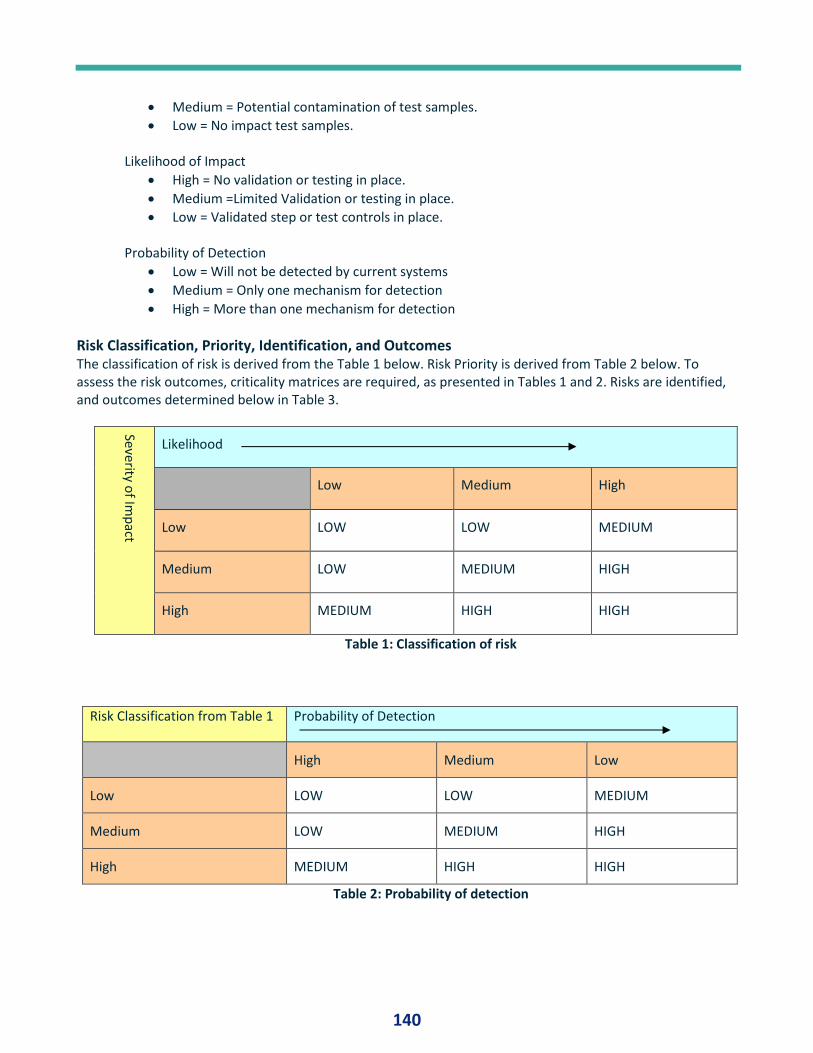
140
• Medium = Potential contamination of test samples.
• Low = No impact test samples. Likelihood of Impact
• High = No validation or testing in place.
• Medium =Limited Validation or testing in place.
• Low = Validated step or test controls in place. Probability of Detection
• Low = Will not be detected by current systems
• Medium = Only one mechanism for detection
• High = More than one mechanism for detection
Risk Classification, Priority, Identification, and Outcomes The classification of risk is derived from the Table 1 below. Risk Priority is derived from Table 2 below. To assess the risk outcomes, criticality matrices are required, as presented in Tables 1 and 2. Risks are identified, and outcomes determined below in Table 3.
Severity of Im
pact
Likelihood
Low Medium High
Low LOW LOW MEDIUM
Medium LOW MEDIUM HIGH
High MEDIUM HIGH HIGH
Table 1: Classification of risk
Risk Classification from Table 1 Probability of Detection
High Medium Low
Low LOW LOW MEDIUM
Medium LOW MEDIUM HIGH
High MEDIUM HIGH HIGH
Table 2: Probability of detection
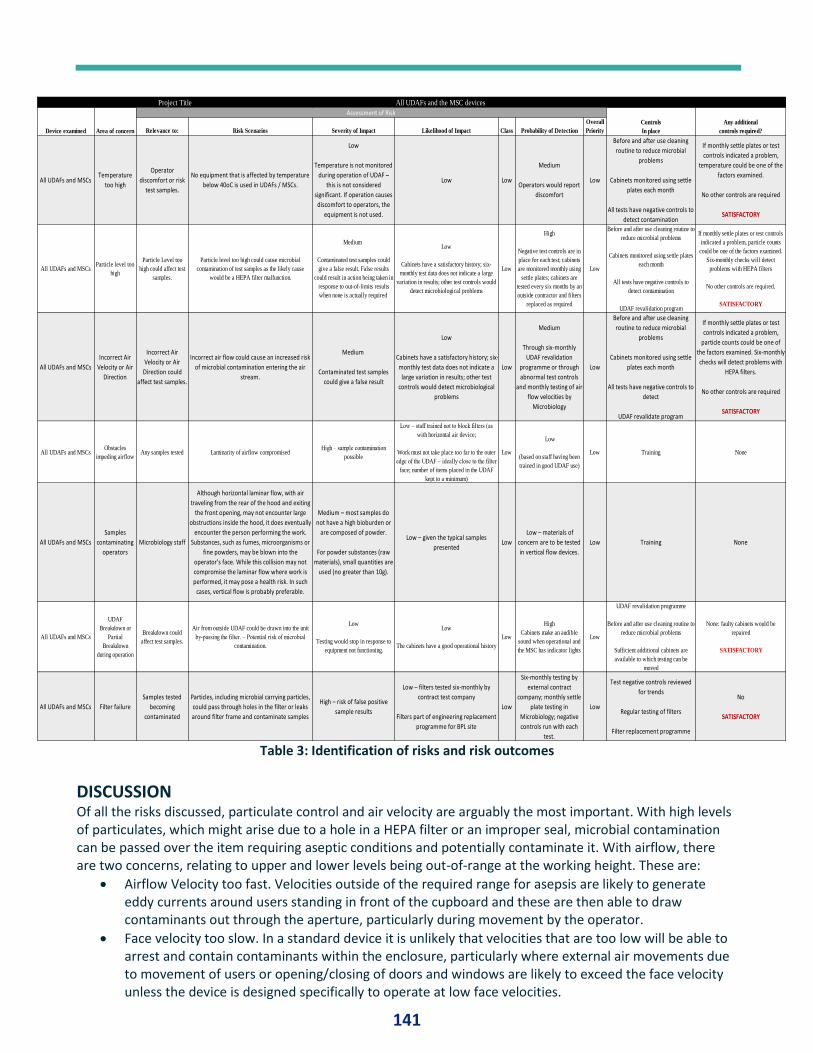
141
Table 3: Identification of risks and risk outcomes
DISCUSSION Of all the risks discussed, particulate control and air velocity are arguably the most important. With high levels of particulates, which might arise due to a hole in a HEPA filter or an improper seal, microbial contamination can be passed over the item requiring aseptic conditions and potentially contaminate it. With airflow, there are two concerns, relating to upper and lower levels being out-of-range at the working height. These are:
• Airflow Velocity too fast. Velocities outside of the required range for asepsis are likely to generate eddy currents around users standing in front of the cupboard and these are then able to draw contaminants out through the aperture, particularly during movement by the operator.
• Face velocity too slow. In a standard device it is unlikely that velocities that are too low will be able to arrest and contain contaminants within the enclosure, particularly where external air movements due to movement of users or opening/closing of doors and windows are likely to exceed the face velocity unless the device is designed specifically to operate at low face velocities.
Relevance to: Risk Scenarios Severity of Impact Likelihood of Impact Class Probability of Detection
Overall
Priority
All UDAFs and MSCsTemperature
too high
Operator
discomfort or risk
test samples.
No equipment that is affected by temperature
below 40oC is used in UDAFs / MSCs.
Low
Temperature is not monitored
during operation of UDAF –
this is not considered
significant. If operation causes
discomfort to operators, the
equipment is not used.
Low Low
Medium
Operators would report
discomfort
Low
Before and after use cleaning
routine to reduce microbial
problems
Cabinets monitored using settle
plates each month
All tests have negative controls to
detect contamination
If monthly settle plates or test
controls indicated a problem,
temperature could be one of the
factors examined.
No other controls are required
SATISFACTORY
All UDAFs and MSCsParticle level too
high
Particle Level too
high could affect test
samples.
Particle level too high could cause microbial
contamination of test samples as the likely cause
would be a HEPA filter malfunction.
Medium
Contaminated test samples could
give a false result. False results
could result in action being taken in
response to out-of-limits results
when none is actually required
Low
Cabinets have a satisfactory history; six-
monthly test data does not indicate a large
variation in results; other test controls would
detect microbiological problems
Low
High
Negative test controls are in
place for each test; cabinets
are monitored monthly using
settle plates; cabinets are
tested every six months by an
outside contractor and filters
replaced as required
Low
Before and after use cleaning routine to
reduce microbial problems
Cabinets monitored using settle plates
each month
All tests have negative controls to
detect contamination
UDAF revalidation program
If monthly settle plates or test controls
indicated a problem, particle counts
could be one of the factors examined.
Six-monthly checks will detect
problems with HEPA filters
No other controls are required.
SATISFACTORY
All UDAFs and MSCs
Incorrect Air
Velocity or Air
Direction
Incorrect Air
Velocity or Air
Direction could
affect test samples.
Incorrect air flow could cause an increased risk
of microbial contamination entering the air
stream.
Medium
Contaminated test samples
could give a false result
Low
Cabinets have a satisfactory history; six-
monthly test data does not indicate a
large variation in results; other test
controls would detect microbiological
problems
Low
Medium
Through six-monthly
UDAF revalidation
programme or through
abnormal test controls
and monthly testing of air-
flow velocities by
Microbiology
Low
Before and after use cleaning
routine to reduce microbial
problems
Cabinets monitored using settle
plates each month
All tests have negative controls to
detect
UDAF revalidate program
If monthly settle plates or test
controls indicated a problem,
particle counts could be one of
the factors examined. Six-monthly
checks will detect problems with
HEPA filters.
No other controls are required
SATISFACTORY
All UDAFs and MSCsObstacles
impeding airflowAny samples tested Laminarity of airflow compromised
High – sample contamination
possible
Low – staff trained not to block filters (as
with horizontal air device;
Work must not take place too far to the outer
edge of the UDAF – ideally close to the filter
face; number of items placed in the UDAF
kept to a minimum)
Low
Low
(based on staff having been
trained in good UDAF use)
Low Training None
All UDAFs and MSCs
Samples
contaminating
operators
Microbiology staff
Although horizontal laminar flow, with air
traveling from the rear of the hood and exiting
the front opening, may not encounter large
obstructions inside the hood, it does eventually
encounter the person performing the work.
Substances, such as fumes, microorganisms or
fine powders, may be blown into the
operator’s face. While this collision may not
compromise the laminar flow where work is
performed, it may pose a health risk. In such
cases, vertical flow is probably preferable.
Medium – most samples do
not have a high bioburden or
are composed of powder.
For powder substances (raw
materials), small quantities are
used (no greater than 10g).
Low – given the typical samples
presentedLow
Low – materials of
concern are to be tested
in vertical flow devices.
Low Training None
All UDAFs and MSCs
UDAF
Breakdown or
Partial
Breakdown
during operation
Breakdown could
affect test samples.
Air from outside UDAF could be drawn into the unit
by-passing the filter. – Potential risk of microbial
contamination.
Low
Testing would stop in response to
equipment not functioning.
Low
The cabinets have a good operational history
Low
High
Cabinets make an audible
sound when operational and
the MSC has indicator lights
Low
UDAF revalidation programme
Before and after use cleaning routine to
reduce microbial problems
Sufficient additional cabinets are
available to which testing can be
moved
None: faulty cabinets would be
repaired
SATISFACTORY
All UDAFs and MSCs Filter failure
Samples tested
becoming
contaminated
Particles, including microbial carrying particles,
could pass through holes in the filter or leaks
around filter frame and contaminate samples
High – risk of false positive
sample results
Low – filters tested six‐monthly by
contract test company
Filters part of engineering replacement
programme for BPL site
Low
Six-monthly testing by
external contract
company; monthly settle
plate testing in
Microbiology; negative
controls run with each
test.
Low
Test negative controls reviewed
for trends
Regular testing of filters
Filter replacement programme
No
SATISFACTORY
Assessment of RiskControls
In place
Any additional
controls required?
Project Title
Device examined Area of concern
All UDAFs and the MSC devices

142
The correct placement of items is also of importance. Items should be placed as close to the back as possible for horizontal devices, and in the center for vertical devices. Items, and testing, should never be conducted at the edge of the unidirectional airflow device. Moreover, users should avoid placing large objects inside the device as these may have an adverse effect on performance and block the rear slots. If large items must be placed inside the device, these should be raised approximately 50 mm to allow air to pass beneath the object and placed near the rear of the cupboard taking care not to block the rear slots / baffle openings, especially at the base of the device. The impact of airflow can also be assessed by airflow visualization. This is important since even with airflow readings within range, air may not be moving ideally, and it can be disrupted by eddying. An airflow visualization or ‘smoke test’ can be conducted using a smoke pellet or a smoke-tube. The technique is to ignite a smoke pellet placed in a suitable container within the fume cupboard and note the behavior of the smoke to assess the turbulence, clearing the base of the device, and the efficiency of general contaminant removal without dead spots.
CONCLUSION This paper has considered some of the hazards and an approach to assessing risks for unidirectional airflow devices and microbiological safety cabinets. While the examples are specific, the general approach can be applied to other laboratories. In looking at risks it is important to note that a unidirectional airflow is only part of the contamination control system: it has imitations, especially in that protection is afforded mainly by nothing more than airflow which can be relatively easily disturbed. It remains important to practice aseptic technique and abide by effective disinfection processes. With this risk assessment, the risk in each case was assessed as low and the outcomes were satisfactory. This is because sufficient controls and suitable test were in place to ensure:
1. The units are operating in a way to reduce microbiological contamination 2. The tests in place will detect any problems with the operation of a unit and allow any necessary
corrective or preventative action to be taken This was supported by the following good practices:
• All work areas being cleaned and disinfected before and after use with an approved disinfectant.
• All cabinets being cleaned and disinfected once per month with an alternative disinfectant.
• All tests having negative controls, which will detect contamination.
• Each cabinet being tested for airborne microbial contamination once per month using settle plates.
• Each cabinet being examined for air-velocity once per month by.
• Each cabinet being tested every six-months as part of routine re-qualification (as per a re-validation program). This testing examines:
o Filter Inspection o Installation Leak Test o Airborne Particle Cleanliness Test o Pressure Differentials Across HEPA Filter o Air Velocity o UDAF condition
In addition to the points noted above, it is also important that those using the cabinets have been suitably trained. Training should cover:
• Principles of how the different classes of cabinets work including airflow patterns
• Suitability of different cabinets for certain types of work
• Principles of airflow, operator protection factor and filter penetration tests
• Limitations of cabinet performance

143
• How to work at cabinets safely
• Operation and function of all controls and indicator;
• How to decontaminate the cabinet after use (routine cleaning)
• Requirements for fumigation and, where appropriate, how to do this
REFERENCES 1. WHO Technical Report Series, No. 961, 2011. Annex 5 Supplementary guidelines on good
manufacturing practices for heating, ventilation and air conditioning systems for non-sterile pharmaceutical dosage forms, pp231-235
2. Robertson, P., Whyte, W. and Bailey, P.V. (1983) A unidirectional aerodynamic class I microbiological safety cabinet, Med Lab Sci.;40(2):159-64
3. Kasperson, R.E., Renn, O., Slovic, P., Brown, H.S., Emel, J., Goble, R., Kasperson, J.X., Ratick, S. (1988). The social amplification of risk: A conceptual framework. Risk Analysis. 8 (2): 177–187
4. Tay K. M.; Lim C.P. (2008) On the use of fuzzy inference techniques in assessment models: part II: industrial applications. Fuzzy Optimization and Decision Making. 7: 283–302
5. Whyte W and Eaton T. (2004) Microbial risk assessment in pharmaceutical cleanrooms. European Journal of Parenteral and Pharmaceutical Sciences;9(1):16–23
6. Sandle, T. (2013). Contamination Control Risk Assessment in Masden, R.E. and Moldenhauer, J. (Eds.) Contamination Control in Healthcare Product Manufacturing, Volume 1, DHI Publishing, River Grove: USA, pp423-474
7. Sandle, T. (2003) The use of a risk assessment in the pharmaceutical industry – the application of FMEA to a sterility testing isolator: a case study, European Journal of Parenteral and Pharmaceutical Sciences; 8(2): 43-49
8. McCollin, C. (1999) Working Around Failure. Manufacturing Engineer, 78 (1): 37-40 9. Kececioglu, D. (1991) Reliability Engineering Handbook Volume 2. Prentice-Hall Inc., Englewood Cliffs,
New Jersey, pp. 473-506 10. Jordan, W. E. (1972) Failure modes effects and criticality analyses In Proc. Ann. Reliability &
Maintainability Symp., IEEE Press, pp. 30-37 11. Ritter MA, Eitzen HE, French M. (1977) Comparison of horizontal and vertical unidirectional (laminar)
air-flow systems in orthopedic surgery, Clin Orthop Relat Res.;(129):205-8 12. McDadde, J., Sabel, F.L., Akers, R. L. and Walker, R. W. (1968) Microbiological Studies on the
Performance of a Laminar Airflow Biological Cabinet, Applied Microbiology, 16 (7): 1068-1092 13. Schulte, H. F., E. C. Hyatt, H. S. Jordan, and R. N. Mitchell. (1954) Evaluation of laboratory fume hoods.
Am. Ind. Hyg. Assoc. Quart. 15:195-202 14. EN 12469 (2000) Biotechnology - Performance criteria for microbiological safety cabinets, European
Commission, Brussels, Belgium 15. Cesard, V., Belut, E., Prevost, C., Taniere, A. and Rembert N. (2013) Assessing the containment
efficiency of a microbiological safety cabinet during the simultaneous generation of a nanoaerosol and a tracer gas, Ann Occup Hyg.;57(3):345-59
16. Sandle, T. (2012). An Air of Safety: The application of cleanrooms and clean air devices within the hospital setting, European Medical Hygiene, Issue 1, pp11-17

144
BRIDGING THE GAPS IN DATA INTEGRITY: ASSESSING RISK TO STREAMLINE AUDIT TRAIL REVIEW By Denise Diehr-New, Computer Systems Validation Engineer, Hikma Pharmaceuticals
ABSTRACT Data Integrity is a timely and well-discussed topic in the pharmaceutical industry. The regulatory agencies demand it, the pharma companies aim to provide it, and the patient depends upon it. Although it may seem straightforward, there are many factors impeding the zero-findings review. Computerized Systems Validation (CSV) teams have a large responsibility in the data integrity of the organization. This paper, which is based on a presentation given at IVT’s 2019 CSV conference, will discuss data integrity, common violations, audit trails, and risk-based audit trail review strategies to assist in the development of gap assessments and the implementation of remediation activities.
INTRODUCTION This discussion defines data integrity as “The assurance that data records are complete, accurate, intact and original. Measures are in place to ensure that GxP systems and data are under continuous care, custody, and control to ensure integrity, availability, and confidentiality.” Data Integrity is EVERYONE’S responsibility. All authors, executors, reviewers, and approvers of CSV records are required to have the necessary knowledge to create, review/approve and/or execute CSV records according to established policies, procedures, and regulations. Individuals in these roles should understand the regulations and guidances that drive data integrity efforts, as well as the tools and frameworks that help ensure compliance.
UNDERSTANDING EXPECTATIONS ALCOA is a well-known acronym associated with data integrity. This acronym is a context for defining data integrity created by FDA to further define regulatory requirements and expectations relative to pharmaceutical research, manufacturing, testing, the supply chain, and Good Manufacturing Practices (GMP). The acronym stands for:
• Attributable: All data generated are attributable to a person. This is done via a paper log or audit trails. Any changes to data are required to be logged with the person’s name, date, time, and old and new values.
• Legible: Data records must be readable and permanent throughout the data lifecycle.
• Contemporaneous: Data are generated real time and never back-dated. Time and date stamps should reflect order of execution.
• Original: Data are the original or primary data recorded. Additions or changes should be captured via audit trail or version histories.
• Accurate: Data that are recorded exactly matches the data being captured. No error correction or editing without documented evidence.
o Complete: Data record is the original, captured data with all modifications made to include repeat or reanalysis.
o Consistent: Elements such as injection sequence and time and date stamps form a sequence of events. Data is recorded only if it conforms to data rules (example – established date format).

145
o Enduring: Data are recorded permanently to a durable medium in Human Readable Format for the duration of the data’s useful life or the required retention period.
o Available: Data are available and accessible for review for the life of the data record.
DATA INTEGRITY REGULATIONS The FDA expects that all data be reliable and accurate. cGMP regulations and guidance allow for flexible and risk-based strategies to prevent and detect data integrity issues. Some of the FDA regulations that pertain to data integrity in GMP facilities are presented in Table 1.
Table 1. FDA Regulations
FDA’s DATA INTEGRITY GUIDANCE In December 2018, the FDA published a guidance for data integrity titled ‘‘Data Integrity and Compliance with Drug CGMP: Questions and Answers.’’. The guidance clarified key terms as they relate to cGMP records and addressed the invalidation of results, audit trails, access restrictions, retention of paper printouts or static records, handling of quality issues, training on data integrity, and FDA audits of electronic records. In addition, the guidance gives recommendations for addressing data integrity problems. The Guidance Recommends asking the following questions:
• “Are controls in place to ensure that data is complete?”
• “Are activities documented at the time of performance?”
• “Are activities attributable to a specific individual?”
• “Can only authorized individuals make changes to records?”
• “Is there a record of changes to data?”
• “Are records reviewed for accuracy, completeness, and compliance with established standards?”
• “Are data maintained securely from data creation through disposition after the record’s retention period?”

146
DATA INTEGRITY Data Integrity is critical to the pharmaceutical industry as it directly impacts product quality, reliability, company reputation, and most importantly, patient safety. As the pharmaceutical industry relies heavily on the collection, processing, and usage of important data, then the collection of such data must be guarded from accidental or intentional modifications, duplication, deletion, and falsification to ensure the product quality and consistency. Data Integrity is not a new concept, but it is getting more attention for many reasons including high profile regulatory cases, increased use of mobile applications, and cloud technologies. To ensure data accuracy and consistency over its lifecycle, companies are encouraged to have a plan or strategy for maintaining data integrity. Electronic data integrity includes system monitoring, backup, recovery / restoration, disaster recovery, data retention and archival system security, user account management, user training, and audit trail review. Problems with Data Integrity can result in product recalls, product holds, and non-approvals -- all of which can be extremely costly. During an audit, problems with data integrity will result in regulatory findings which could yield warning letters or consent decrees. Issues can occur, whether accidental or intentional, but understanding data integrity requirements and having procedures in place for aspects of data integrity, such as audit trail review, is critical to managing a compliant program which produces successful inspections.
AUDITING DATA INTEGRITY The intent of a data integrity audit is to recognize any problems with existing data that might have gone unnoticed. Data integrity issues often may occur, whether accidental or intentional, such as deleted data, manipulated data, or data being misused or withheld in any way. By identifying risk factors within your process, you can begin to map out a strategy to identify, monitor, and remediate high-risk activities which may lead to future data integrity breaches.
When developing a strategy and planning for internal controls, the following should be considered:
• Validation documentation and test results
• Data integrity requirements in design documents
• Audit trail reviews and other procedural controls
• Backup and restoration process and testing
• Disaster recovery process and testing
• Service level agreements
• Original data and data availability
• Data security (example: external access policy - VPN)
• Access security: Are personnel authorized, do they have the correct role with documented training, is there a procedure in place for periodic security review
• Testing into compliance
Examples of Data Integrity Violations in FDA Warning Letters Data Integrity related FDA warning letters have become increasingly common over the past few years. Lapses in data integrity are most often cited as the FDA deems this to be a serious risk to patient safety. The following may be considered a violation by the FDA:
• Falsifying data
• Lack of control in systems to prevent deletion of data
• Data manipulation
• Backdating
• Destruction of ‘original’ or true copies
• Not documenting activities ‘real time’

147
• Security controls (shared username and passwords)
• No audit trails
• Insufficient audit trails
• No audit train review
Recent guidance documents were released with recommendation for drug makers to ensure manufacturing data sets are complete, consistent, and accurate. Non-compliance may not only be cause for warning letters but could also result in the regulatory agency imposing import alerts or denying approval of new applications. The following is an excerpt from an FDA warning letter:
“Your quality system does not adequately ensure the accuracy and integrity of data to support the safety, effectiveness, and quality of the drugs you manufacture…”
“In response to this letter, provide the following:
a. A comprehensive investigation into the extent of the inaccuracies in data records and reporting. Your investigation should include: a detailed investigation protocol and methodology, a summary of all laboratories, manufacturing operations…
b. An assessment of the extent of data integrity deficiencies at your facility… c. A comprehensive retrospective evaluation of the nature of the testing and
manufacturing data integrity deficiencies…”
Nobody wants to be remediating data integrity issues after being issued a warning letter from the FDA. The best course of action is to identify your gaps internally and implement a remediation plan to set your company up for successful agency audits.
AUDIT TRAILS An Audit Trail is a secure, computer-generated, time-stamped electronic record of a computerized system activity of application processes that run within it and a record of the activity of the system users. The audit trail allows for reconstruction of the course of events relating to the creation, modification, or deletion of the electronic record.
Audit Trails: 1. Track creations, modifications, and deletions 2. Record the identity of the individual making the changes 3. Include the system ID 4. Are automated - computer generated 5. Function independently (can’t be disabled by admins or users) 6. Are secure – protected from intentional or accidental modification or alteration 7. Remain operational at all times 8. Record the reason for changes 9. Convert to intelligible form 10. Are record linking – bound to raw data, backed up, archived, and retrievable
AUDIT TRAIL REVIEWS Audit Trail Reviews are conducted to ensure product quality and corresponding patient safety. Audit trail review is conducted to ensure the control of all product relevant processes and related records, and that decisions are based on available data that are complete, accurate, and trustworthy. Audit Trail Review falls into two main categories:
• Manufacturing process or application level audit trails (Business Audit Trail Review)
• System related audit trails (IT Audit Trail Review)

148
Manufacturing Audit Trail Review for the manufacturing process (or business data) is the responsibility of the business, typically the quality department. The primary goal is to ensure the integrity of the reported data for product manufacturing, product testing, investigations, change control process, etc. The Business Audit Trail Review should:
• Ensure that the product is manufactured as per specifications
• Focus on events during manufacturing such as addition of ingredients and the occurrence and acknowledgement of alarms and events that impact product quality
• Focus on events that include critical process parameters (CPPs) and critical quality attributes (CQAs)
• Focus on data manipulation (Example: Excessive/manual peak integration in chromatography systems)
• Look for deletion of data
System Audit Trial Review for the system related controls (system security or IT data) is the responsibility of IT. The primary focus is to ensure that there are no security violations and data integrity is maintained in the system. System Administrators (IT) should perform audit trail review for the following:
• Account logon / logoff (attempts to authenticate account data)
• Account management (changes to user and computer accounts and groups)
• Incorrect login attempts
• Unsuccessful login attempts (expired passwords)
• Anomalies in login/logoff
• Detailed tracking (activities of individual applications and users on the systems) Automation of monitoring services for security breaches may help IT manage their audit trail review. The diagram at left is an example of a potential automation strategy:
Access Batch Job
with stored connection and
query info to multiple
System DBs
Job Fires Daily
For each connection, run
custom query to return user ID
and Email for invalid attempts
exceeding the established
threshold over the established
time period
System 1
System 2
System n
For each result found create a
record in the Access job
system to track the incident to
its conclusion
System sends auto-generated
email to user of the event
including system name, date
or date range and the number
of invalid attempts.
User is instructed to click one
of two links:
A link acknowledging and
accepting the invalid attempts
as their own
A link denying or unsure if the
login attempts were their own
User admits Login
Errors
User Denies
Login Errors
Simple custom Web
Application processes query
string parameters to update
the Access Job record with
response date and the yes/no
acknowledgement info
Second job fires daily to
evaluate status of all open
records
Open records where user
admits login errors are closed
Open records where user
denies login errors or user
does not respond in more than
two days are emailed to
system admin(s) for
investigation and resolution
with link to close record when
investigation completed
Admin confirms
non-responses as
necessary
For confirmed
Denies:
Ticket opened for
operations group to
reset domain
password
Admin investigation
is completed and
clicks daily email
notification link to
close
Figure 1. Example automated program to detect excessive invalid login
attempts

149
DEVELOPING A PROCESS What do we look at during audit trail review? Primarily, we need to ensure accuracy and integrity of the data to include:
• Changes to test parameters
• Changes to data processing parameters
• Data deletion
• Data modifications
• Analyst actions
• Excessive integration of chromatography
• Security breaches related to data
• Changes to established pass/fail criteria
• Change history of test results
• Changes to run sequences
• Changes to sample identification
• Changes to critical process parameters.
When, Who, and How? Audit Trail Reviews should be conducted as follows:
1. Audit trail review should be completed for all high-risk systems (risk considerations defined below) before the final approval of the record.
2. The personnel responsible for the record review under cGMP (for production data, this means review and approval by quality) should review the audit trail.
3. Reviewers should be reviewing all the critical data changes and manipulations as defined above in the section “What do we look at during audit trail review?”
What if a potential data integrity breach is discovered?
• Escalate the potential data integrity breach to the appropriate management / quality group
• Management should investigate further according to internal procedures
• Management should take appropriate action if a true data integrity breach occurred o Product investigation and corrective action o Personnel investigation and corrective action.
RISK CONSIDERATIONS According to the FDA Guidance Document from December 2018:
“FDA expects that all data be reliable and accurate. CGMP regulations and guidance allow for flexible and risk-based strategies to prevent and detect data integrity issues. Firms should implement meaningful and effective strategies to manage their data integrity risks based on their process understanding and knowledge management of technologies and business models.”(2)
When defining your risk-based approach strategy for audit trail review, it is important to consider:
• Impact on patient safety, product quality, data integrity
• Supported business processes
• CQAs for systems that monitor or control CPPs
• Regulatory requirements
• System components and architecture
• System functions
• Probability of occurrence
• Probability of detection
• GxP relevance.

150
Risk Based Approach A risk-based approach provides a way of substantiating or weighing data-based decisions and the impact on quality of product and patient safety, while not resulting in negative impact on costs and resources. To take a risk-based approach to audit trail reviews, the system risk level needs to be identified. Prioritizing the audit trail assessment based on the level of risk is critical to prioritization of assessments and implementation.(3) To effectively prioritize you must first evaluate audit trail review risk, looking at high-risk versus low-risk systems. High Risk systems. Audit trail review is mandatory to ensure data integrity compliance. The compliance cost is too high to assume the risk. High risk systems:
• Have a likely probability of a possible data integrity breach
• Have low detectability of a breach
• Have a high severity of impact in the case of a breach
• Generate GxP relevant data
• Enforce Regulatory requirements
• Have an impact on product quality or patient safety
• Support a business process that has a direct impact on CQAs
• Control CPPs
• Have architecture that is designed to enforce CQAs and CPPs Low Risk Systems. Audit trail review is not mandatory and can be done on a periodic basis or as defined by the business (for example, at the time of related investigations, etc.); therefore, the cost to the company is minimized. Low Risk Systems:
• Have a low probability that a data integrity issue would occur
• Have a high probability of a data integrity breach detection outside of audit trail review
• Audit trail review would not show a data integrity issue
• Have a low severity of impact in the case of a breach
• Do not generate GxP relevant data
• Do not enforce regulatory requirements Figure 2 illustrates a flow chart based on the critical questions that should be asked to assess the audit trail review risk for each GxP systems.
GxP Systems with non-GxP Functionality How do we implement effective audit trail review for a GxP system with non-GxP Functionality; what functionality is GxP relevant?
1. Focus audit trail review on GxP relevant functionality. 2. Identify and document GxP relevant functionality so that audit trail review is efficient and focused.
Example: SAP
• Financial transactions are considered non-GxP (Low Risk)– audit trail review would not be required
• Product Quality release transactions are GxP (High Risk)– audit trail review would be required 3. Clear documentation of your audit trail review strategy is key!

151
IMPLEMENTATION OF AUDIT TRAIL REVIEW Effective Implementation of Audit Trail Review requires that policies and standard operating procedures give clear direction and are compliant to current cGMP regulations. The following should be included in the SOPs:
• Ethics and Code of Conduct
• Quality Systems processes
• Data integrity training as part of yearly cGMP
• Additional data integrity training for audit trail review for all quality reviewer/approvers and other responsible parties
Strategy should be a risk-based approach. The system requirements should be used as a basis for determining frequency and range of audit trail review. For high-risk systems the review should include test data to ensure integrity of test data (4). The who, what, when, and what ifs, should also be addressed in SOPs which includes clearly defining mandatory audit trail review. Most importantly the business should understand the time investment and ensure appropriate implementations to maintain compliance to regulations.
Figure 2 - Assessing Risk for Audit Trail Review of GxP Systems

152
CONDUCTING A GAP ASSESSMENT Gap assessments should be conducted for each GxP computer system against data integrity regulations. Documented evidence should be included in this assessment with explanations in entirety. Simple “pass/fail” is not adequate. As appropriate, remediation activities should be identified, tracking progress utilizing the CAPA quality process. The following steps can be followed in designing your gap assessment:
• Outline your purpose
• Identify your scope
• Identify roles and responsibilities
• What is the current process / procedure?
• What are the gaps to the current regulations / expectations?
• Define a risk-based approach
• Identify high-risk systems (for audit trail review)
• List remediation activities
• Set a remediation schedule with clear deliverables and timelines o Prioritize high risk systems
• Ensure remediation due dates are met using CAPA, etc. A Data Integrity Checklist is available at IVTNetwork.com. https://cdn2.hubspot.net/hubfs/200783/IVT_Promo%20Materials/IVT_Blog%20Materials/Ginsbury_GMP%20Checklist%20for%20DI%20Audit.doc
REMEDIATION ACTIVITIES Developing Remediation Activities - CAPA (corrective action / preventative action) procedures can be used to ensure remediation is completed within the desired timeframe and as defined in SOPs. During remediation it is important to develop and document the criteria for business process controls, including controls for high risk systems that have non-meaningful audit trail review. Key points for Audit Trail Review remediation:
• Split responsibilities for audit trail review: security (IT) and business data (Quality)
• Define who is responsible
• Define what constitutes a data integrity issue
• Define the escalation process in the event of a data integrity breach
• Train employees
• Implement and enforce.
Implementation and Enforcement Implementation should include purpose, as outlined in the system policies; principles cited in ethics plan and code of conduct; processes (SOPs); people citing roles and responsibilities; and performance metrics. Enforcement consists of data integrity controls, to include management controls – ethics, code of conduct, data governance; procedural controls – audit trail review, data backup and recovery and technical controls; security, audit trail design.
ADDITIONAL TOOLS Implement an Application Administration Plan (AAP) that defines all administrative processes to operate and maintain the system in a compliant manner and maintain the validated state of the system. The AAP should address the following, including all the aspects of data integrity:

153
• User account management (new accounts, assigning and revoking access, changing accounts, disabling accounts, resetting passwords and locked accounts, and periodic review of system access)
• Training
• User support
• Audit trail review
• Backup and recovery
• Disaster recovery
• Business continuity process
• Data archival
• Routing changes (changes that can be made outside of change control, for example, additions to dropdown lists).
The AAP is a place to document and define the audit trail review strategy based on risk assessment. You should clearly state whether your system is high risk or low risk and how you determined the risk rating. Additionally, for high risk systems, you should document the ‘who, what, when and what if’ for audit trail review or alternatively point to the procedure that defines this for that system.
CONCLUSION Data integrity can be a scary subject for companies. The regulations are vague and bring many challenges on how best to implement procedures to ensure compliance. Ultimately the goal is to implement a process that will ensure data integrity in a cost effective, efficient manner that is value-added for the business and compliant to the regulations. Effective implementation of an audit trail review process remains a challenge for many companies due to inherent resource / time constraints. Utilizing a risk-based approach for audit trail review will ensure data integrity compliance while minimizing resource constraints and reducing associated costs. Your risk-based approach needs to properly define who, what, when, and what if. Ensure your strategy is properly documented for each GxP system and that your logic and justifications are in place and scientifically sound. Procedures should be in place and employees should know the expectations and have the appropriate training. Performing a data integrity gap assessment and defining remediation activities is a first step in ensuring your company will have successful data integrity audits. Implementing a strong data integrity foundation at your company will build employee and customer confidence and ultimately ensure product quality and patient safety.
REFERENCES 1. Data Integrity and Compliance with Drug CGMP, Questions and Answers, Guidance for Industry; U.S.
Department of Health and Human Services Food and Drug Administration; December 2018. https://www.fda.gov/media/119570/download
2. Data Integrity and Compliance with Drug CGMP, Questions and Answers, Guidance for Industry; U.S. Department of Health and Human Services Food and Drug Administration; December 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/data-integrity-and-compliance-drug-cgmp-questions-and-answers-guidance-industry
3. Audit Trails Reviews for Data Integrity; Peer-Reviewed: Data Integrity; IVT Network; October 19, 2015; http://www.ivtnetwork.com/article/audit-trails-reviews-data-integrity
4. Unger, Barbara; Tips For Identifying And Correcting Data Integrity Deficiencies In Your Organization; Pharmaceutical Online; May 3, 2016; https://www.pharmaceuticalonline.com/doc/tips-for-identifying-and-correcting-data-integrity-deficiencies-in-your-organization-0001

154

155

156
Related Documents











