UNIVERSIDAD NACIONAL DE TRUJILLO ESCUELA DE INGENIERÍA QUÍMICA FACULTAD DE INGENIERÍA QUÍMICA DETERMINACIÓN DE PARÁMETROS CINÉTICOS EN LA OBTENCIÓN DE 1-FENILETANOL EMPLEANDO COMO BIOCATALIZADOR Diplogelasinospora grovesii IMI 171018 EN MEDIO ACUOSO”. TESIS PARA OPTAR EL TITULO DE: INGENIERO QUÍMICO AUTOR: Br. CARLOS JOHANN FALCÓN JAVE ASESOR: Dr. MEDARDO ALBERTO QUEZADA ALVAREZ TRUJILLO – PERÚ 2012 Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/ Biblioteca de Ingeniería Química UNT

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

UNIVERSIDAD NACIONAL DE TRUJILLO
ESCUELA DE INGENIERÍA QUÍMICA FACULTAD DE INGENIERÍA QUÍMICA
DETERMINACIÓN DE PARÁMETROS CINÉTICOS EN LA OBTENCIÓN DE 1-FENILETANOL EMPLEANDO COMO
BIOCATALIZADOR Diplogelasinospora grovesii IMI 171018 EN MEDIO ACUOSO”.
TESIS
PARA OPTAR EL TITULO DE:
INGENIERO QUÍMICO
AUTOR:
Br. CARLOS JOHANN FALCÓN JAVE
ASESOR:
Dr. MEDARDO ALBERTO QUEZADA ALVAREZ
TRUJILLO – PERÚ 2012
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

II
Presidente: Secretario
Dr. Luis Moncada Albitres Dr. José Luis Silva Villanueva
Dr. Alberto Quezada Alvarez
Asesor de Tesis
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III
DEDICATORIA
A mis padres: Adolfo y Diana
que con su amor, educación y ejemplo,
son inspiración de mi desarrollo
y mi superación.
A mis hermanos menores: Elizabeth y Franz
y para que sean perseverantes y
llenos de ímpetu en el alcance de sus metas.
.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV
AGRADECIMIENTO
A Dios por guiar mis pasos y por iluminar mi día a día.
A mis padres que me apoyaron en todo el transcurso de mi desarrollo
profesional. Todo gracias a su afecto, educación, trabajo y esfuerzo.
A los docentes de mi facultad, quienes me brindaron su orientación en la
adquisición de conocimientos que afianzaron mi vocación profesional.
Al Dr. Alberto Quezada por su asesoramiento, dedicación, tiempo y gestión en el
desarrollo y cumplimiento de la presente tesis.
Al Dr. José Vicente Sinisterra Gago por sus capacitaciones en sus constantes
visitas a nuestra facultad de Ingeniería Química, por su apoyo e interés mi
formación científica.
Al Ing. Cruz Monzón por su apoyo en los temas de análisis instrumental y
acceso a los equipos de instrumentación.
Al Ing. Wilson Reyes por facilitarnos en un inicio el laboratorio de Investigación
IV, para el desarrollo de las investigaciones respectivas.
Al grupo inicial de investigación: Diana, Pilar, Melva y Gonzalo con quienes
realicé los primeros ensayos para la ejecución de éstas investigaciones.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

V
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

VI
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

VII
ÍNDICE
Pág.
I. INTRODUCCIÓN
1.1. Problemática 2
1.2. La Química y el Desarrollo Sostenible 4
1.3. Química Sostenible 5
1.4. Química Sostenible y Biotecnología Blanca 7
1.5. Biotransformaciones 9
1.5.1. Diferencias entre Fermentación y Biotransformación 10
1.6. Ventajas de las Biotransformaciones 11
1.7. Reacciones de Reducción 13
1.7.1. Regeneración de Coenzima 14
1.8. Enzimas frente aisladas frente a células enteras:
Ventajas y Desventajas 16
1.9. Reducción de Aldehídos y Cetonas usando Células Enteras 18
1.9.1. ADH Dehidrogenasas 20
1.10. Screening de Nuevos Biocatalizadores 22
1.10.1. Screening a partir de banco de genes 22
1.10.2. Screening a partir de enzimas comerciales 22
1.10.3. Screening a partir de colecciones de microorganismos 23
1.11. Metodología de Screening 23
1.12. Diplogelasinospora grovesii 25
II. OBJETIVOS Y PLAN DE TRABAJO
2.1. Objetivos 27
2.1.1. Objetivo General 27
2.1.2. Objetivos Específicos 27
2.2. Plan de Trabajo 28
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

VIII
III. PARTE EXPERIMENTAL
3.1. Materiales 30
3.1.1. Materiales para vidrio 30
3.1.2. Materiales de plástico 30
3.1.3. Material Auxiliar 31
3.1.4. Instrumental de laboratorio 32
3.2. Métodos 33
3.2.1. Procedimiento para el Screening Global 33
3.2.1.1. Estado de Colección 33
3.2.1.2. Medios de Cultivo 33
a. Medio de Cultivo para Ascomicetos 34
b. Medio de Cultivo para Bacterias 34
c. Medio de Cultivo para Levaduras 35
3.2.1.3. Preparación del medio sólido de cultivo 35
3.2.1.4. Esterilización de medios de cultivo 35
Reacción test para el screening global 35
3.2.2. Cualificación de los E. de Instrumentación Analítica 36
3.2.2.1. Cualificación de las Balanzas Analíticas 36
3.2.2.2. Cualificación del Espectrofotómetro UV 37
3.2.2.3. Cualificación del HPLC 38
a. Cualificación de la Bomba del HPLC 38
b. Cualificación del Sistema de Inyección 39
c. Cualificación del Detector UV-Visble 40
d. Cualificación de la Columna Cromatográfica 41
3.2.3. Validación y Cualificación del Método de Análisis 43
3.2.3.1. Determinación de la Longitud de Onda Óptima 43
3.2.3.2. Validación del Método de Análisis Cromatográfico 44
3.2.3.3. Obtención de las Curvas de Calibración para
Acetofenona y 1-Feniletanol 45
3.2.3.4. Obtención de las Concentraciones de Acetofenona
y 1-Feniletanol durante la Reacción de Reducción 46
3.2.4. Metodología Experimental para el proceso de
biotransformación con Diplogelasinospora grovesii 48
3.2.4.1. Asentamiento de la Colección 48
3.2.4.2. Crecimiento de ascomiceto Diplogesinospora grovesii 49
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IX
a. Crecimiento de ascomicetos en medio sólido 49
b. Crecimiento de ascomicetos en medio líquido 51
3.2.4.3. Replicación del ascomiceto Diplogesinospora grovesii 52
3.2.4.4. Curva de crecimiento del ascomiceto 53
3.2.4.5. Preparación del Medio de Reacción 54
a. Preparación de la Solución Buffer 54
b. Preparación de la Solución Reacción 55
3.2.5. Estudio Cinético de Bioreducción 57
3.2.5.1. Velocidad inicial de reacción observada (Vo) 58
3.2.5.2. Orden de reacción (n) 61
3.2.5.3. Parámetros cinéticos de reacción 61
a. Conversión 61
b. Rendimiento 62
c. Productividad 62
IV. RESULTADOS Y DISCUSIONES
4.1. Cualificación de los Instrumentos Analíticos 64
4.1.1. Cualificación de Balanza Analítica 64
a. Primer Rango de Cualificación: 0 a 1g 65
b. Segundo Rango de Cualificación: 0 a 100g 66
4.1.2. Cualificación de Espetrofotómetro Ultravioleta-Visible 67
a. Absorbancia de Benzoato de Sodio a 236nm 68
b. Absorbancia de Benzoato de Sodio a 254nm 69
c. Absorbancia de Benzoato de Sodio a 270nm 70
4.1.3. Cualificación de Cromatógrafo de Líquidos
Alta Performancia 71
4.1.3.1. Cualificación de la Bomba Cuaternaria 71
a. Cualificación del Canal A 71
b. Cualificación del Canal B 72
c. Cualificación del Canal C 73
d. Cualificación del Canal D 74
4.1.3.2. Cualificación del Sistema de Inyección 74
a. Precisión del Inyector 75
b. Magnitud del efecto memoria (efecto “carryover”) 76
4.1.3.3. Cualificación del Detector UV-Visible 78
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

X
4.1.3.4. Cualificación de la Columna 80
4.2. Validación del Método de Análisis 82
4.2.1. Longitud de Onda Óptima de análisis 82
4.2.2. Validación de Método de Análisis Cromatográfico 83
4.2.3. Curvas de Calibración para Acetofenona y 1-Feniletanol 84
4.3. Proceso de Bioreducción con Diplogelasinospora grovesii 86
4.3.1. Screening Taxonómico 86
4.3.2. Curva de Crecimiento 88
4.3.3. Estudio Cinético 90
4.3.4. Orden de Reacción 95
4.3.5. Parámetros Cinéticos 97
a. Parámetros Cinéticos del sustrato Acetofenona 97
b. Parámetros Cinéticos del producto 1- Feniletanol 97
4.3.6. Productividad de la Biotransformación 99
V. CONCLUSIONES 101
VI. BIBLIOGRAFÍA 104
VII. ANEXOS 108
- Cualificación de la Balanza Sartorious AG Germany CPA 224S
- Cualificación del Equipo Espectrofotómetro UV- visible
Hewlett Packard Mod 8452 A
- Cualificación del Equipo HPLC Agilent 1100 Series.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

XI
RESUMEN
Es patente que las sociedades más avanzadas se han hecho muy
sensibles a todo aquello que pueda afectar la calidad medioambiental
como uno de los indicadores de la calidad de vida. En este contexto la
población señala a la industria química como la principal causante del
deterioro medio ambiental ocurrido en el último siglo, sin embargo, las
investigaciones realizadas en la ciencia química han contribuido a evitar o
a paliar estos males.
En esta tesis se muestran los resultados obtenidos después de un
screening de microorganismos en busca de nuevos biocatalizadores
activos a la reducción de cetonas en medio acuoso. El grupo taxonómico
más activo para la reducción de cetonas catalizadas por células fueron
los ascomicetos y entre ellos el mejor biocatalizador fue
Diplogelasinospora grovesii. La principal ventaja de la utilización de
biocatalizadores es el remplazo de catalizadores tradicionales para el
mismo propósito como metales y boranos, los cuales generan bajas
productividades, contaminación por subproductos que son incompatibles
con el medio ambiente. La actividad reductasa del microorganismo se
evaluó con la reacción test de reducción de la acetofenona. Asimismo se
muestran los resultados cinéticos obtenidos en esta reacción como:
velocidad inicial de reacción, constante especifica de velocidad,
productividad, conversión, rendimiento, etc. Finalmente, resaltar que
todos los análisis para este propósito se realizaron con equipos
cualificados (HPLC, UV-VIS, Balanzas analíticas, etc.) y métodos de
análisis validados.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

XII
ABSTRACT
It is clear that the most advanced societies have become very sensitive to
anything that might affect environmental quality as one of the indicators of
quality of life. In this context the population draws the chemical industry as
the main cause of environmental deterioration occurred in the last century,
however, research in chemical science have helped prevent or alleviate
these evils.
In this thesis the results are displayed after a screening of microorganisms
for new active biocatalysts to the reduction of ketones in aqueous media.
The most active for the reduction of ketones catalyzed by taxonomic
group cells were Ascomycetes and among them the best biocatalyst was
Diplogelasinospora grovesii. The main advantage of the use of
biocatalysts is the replacement of traditional catalysts for the same
purpose as metals and boranes, which generate low yields, contamination
by products that are incompatible with the environment. Reductase activity
of the microorganism was evaluated with the test reaction acetophenone
reduction. initial reaction rate constant specific speed, productivity,
conversion, yield, etc.: also the kinetic results obtained in this reaction as
shown Finally, note that all analyzes were conducted for this purpose with
qualified equipment (HPLC, UV-VIS, analytical balances, etc.) and
validated methods of analysis.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
1
I. INTRODUCCION
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
2
I. INTRODUCCION
1.1. Problemática
No corren buenos tiempos para la química. Aunque nuestra sociedad
occidental demanda cada vez una mayor cantidad de productos químicos,
la percepción que se tiene de lo químico es, de forma paralela, cada vez
más negativa. Es cierto que el sector químico ha tenido un elevado
crecimiento en la producción de compuestos químicos demandados por una
sociedad cada vez más depredadora de bienes de consumo, y además, las
previsiones indican que este crecimiento continuará en el futuro. Esta
presión incrementa la actividad productiva, pero también aumenta la
actividad contaminante y generadora de residuos molestos, dando lugar a
una imagen sucia de todo aquello que se relaciona con la química.1
En ese sentido desarrollar procesos en el campo de la Química Sostenible
(Green Chemistry & Engineering) hoy en día juega un rol muy importante
toda vez que utiliza un conjunto de principios que reducen o eliminan el uso
y generación de sustancias peligrosas en el diseño, manufactura y
aplicación de los productos químicos, manteniendo la calidad del producto
final y la rentabilidad económica del proceso; lo que lleva implícito, alcanzar
un proceso que sea inocuo en sí mismo y respetuoso con el medio
ambiente.2
La Biotecnología Blanca es un campo emergente dentro de la moderna
Biotecnología, que está implicado en procesos industriales de
producción. Su desarrollo viene determinado por el cumplimiento de las
nuevas normativas de la Unión Europea sobre control del medio ambiente y
producción de bienes y servicios en condiciones sostenibles. La
Biotecnología Blanca se suele definir como la producción de productos de
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
3
alto valor añadido o de servicios, en condiciones sostenibles tanto desde el
punto de vista del empleo de disolventes y de reactivos así como en la
obtención de productos no contaminantes y/o no agresivos con el medio
ambiente, utilizando biocatalizadores no patógenos.3,4,5
Los biocatalizadores (enzimas o células, libres o inmovilizadas) son ahora
instrumentos indispensables para químicos y biotecnólogos, y están siendo
cada vez más empleados en la síntesis asimétrica de productos
farmacéuticos, agroquímicos, vitaminas y fragancias.6,7 La reducción de
cetonas empleando biocatalizadores para obtener alcoholes constituye una
alternativa muy eficiente y atractiva, que opera a condiciones de pH y
temperatura mucho más suaves. La ventaja de las reacciones biocatalíticas
sobre las reacciones químicas tradicionales consiste en que aquellas son
estereoselectivas y pueden realizarse a presión atmosférica y temperatura
ambiente. Dado que muchos procesos biocatalíticos se pueden llevar a
cabo en medio acuoso u orgánico/acuoso empleando disolventes poco
tóxicos, justifica que la biocatálisis sea una de las líneas fundamentales en
el desarrollo de la Química Sostenible. Además, las enzimas y células
microbianas se pueden inmovilizar y reutilizar lo cual rebaja los costes de
producción.8
En el 2002, la producción de alcoholes quirales supuso unas ventas de 7
billones de dólares, estimándose un aumento de hasta 14,9 billones para el
2009.9 Debido a la enorme importancia de este tipo de compuestos en la
síntesis de moléculas bioactivas, la reducción estereoselectiva de cetonas
ha adquirido un enorme interés industrial, y a éste efecto se han descrito
numerosas rutas catalíticas; así, la hidrogenación asimétrica de cetonas
catalizada por metales10 o el empleo de boranos11,12 constituyen procesos
que han sido descritos de manera eficiente a escala industrial; no obstante,
se suelen precisar condiciones poco compatibles con la Química Sostenible
(Green Chemistry & Engineering)13-17, tales como temperaturas elevadas, el
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
4
empleo de disolventes y/o reactivos tóxicos y obtener productos
concomitantes.
En ese sentido, la presente tesis para optar el título de ingeniero químico,
pretende contribuir en la determinación de los parámetros cinéticos en
procesos biocatalíticos para obtener alcoholes a través de biorreducciones
de cetonas en condiciones sostenibles.
1.2. La Química y el Desarrollo Sostenible
Básicamente el término desarrollo sostenible tiene dos implicaciones:
- El uso de recursos naturales a ritmos suficientemente bajos como para
que no se agote el suministro de largo plazo.1,13
- La generación y disipación de los recursos y emisiones a velocidades
suficientemente bajas, de forma que puedan ser asimiladas por el medio
natural. 1,13
Así pues, para poner en práctica mejoras ambientales en el proceso
productivo, no solo se debe centrar el interés en la instalación concreta en
la que se realiza el proceso químico, sino que deben evaluarse los procesos
de forma global, en un amplio escenario temporal, y sobre grandes
dominios territoriales.15
Con esta perspectiva, el reto para el químico es desarrollar nuevos
productos y nuevos procesos de acuerdo con los beneficios del desarrollo
sostenible. Ello requiere una nueva aproximación consistente en la
reducción de la intensidad material y energética utilizada durante las
reacciones químicas, minimizando o eliminando la liberación de sustancias
químicas nocivas para la salud y el medio ambiente, y maximizando el uso
de recursos renovables y la durabilidad de los productos obtenidos.13-17
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
5
1.3. Química Sostenible
Esta rama de la química tiene como objetivo el diseño de compuestos y
procesos químicos que reduzcan o eliminen la generación de sustancias
peligrosas para la salud humana y el medio ambiente, haciendo un uso
sostenible de los recursos. Debe señalarse que, muchas de las acciones
que introduce la química sostenible (química verde) para la mejora
ambiental llevan implícitas unas mejoras económicas (ecoeficiencia) para
los sectores industriales involucrados, de factor positivo para su
implementación.13
Tal como está definida la química sostenible, ésta se centra en el diseño, la
investigación y la implementación de productos y procesos. Es decir, no
sólo su ámbito de acción es el diseño de estructuras moleculares para que
el compuesto no sea tóxico, sino que, además debe incluir todas las
transformaciones a lo largo de la manufactura del producto, las cuales
deberán evitar el uso de sustancias tóxicas, así como tampoco generarlas.13
Los retos de la química sostenible (química verde) se centran en cuatro
ámbitos: los recursos, los residuos, los reactivos, y las reacciones:13-17
1. Recursos: Utilización de recursos materiales y energéticos obtenidos de
fuentes renovables para la obtención de los productos básicos. Uso
eficiente de la energía, a través de un diseño apropiado de las
condiciones de reacción. Desmaterialización, a través del uso de
materiales nanométricos y técnicas que minimicen el uso de materiales,
con el objetivo de conseguir la función demandada por un determinado
material, intentando reducir la cantidad de recursos materiales.13-17
2. Residuos: Maximización de la eficiencia molecular durante las
transformaciones, evitando al máximo la obtención de subproductos y
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
6
residuos que incrementen el coste ambiental y económico del
proceso.13-17
3. Reactivos: Disminución del uso de reactivos, a través de catalizadores
duraderos. Diseño de compuestos químicos inocuos, mediante
manipulación de la estructura molecular y el conocimiento de su
mecanismo de acción toxicológica.13-17
4. Reacciones: Reducción del uso de disolventes por medio de sistemas
reactivos sin disolventes o del uso de disolventes alternativos a los
tradicionales, más fácilmente reciclables o de un menor impacto
ambiental, como por ejemplo, el uso de fluidos supercríticos. O líquidos
iónicos. Aplicación de técnicas analíticas in situ, con el fin de llevar a
cabo un control de las condiciones de reacción a tiempo real.13-17
El desarrollo de la Química Sostenible se basa una serie de principios que
se enumeran a continuación (Mestres, 2003):17
1. Es mejor prevenir la formación de residuos que tratarlos o eliminarlos
tras su formación.
2. Los métodos sintéticos deben ser diseñados para conseguir la máxima
incorporación en el producto final de todas las materias usadas en el
proceso.
3. En tanto sea posible, se deben diseñar metodologías sintéticas para el
uso y la generación de substancias de escasa toxicidad humana y
ambiental.
4. Se deben diseñar productos químicos que, preservando la eficacia de su
función, presenten una toxicidad escasa.
5. Las substancias auxiliares (disolventes, agentes de separación, etc.)
deben resultar innecesarias en lo posible o cuanto menos deben ser
inocuas.
6. Las necesidades energéticas deben ser consideradas en relación a sus
impactos ambientales y económicos, y deben ser minimizadas. Los
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
7
métodos de síntesis deben llevarse a cabo en condiciones de
temperatura ambiente y presión atmosférica.
7. Las materias de partida deber ser renovables y no extinguibles, en la
medida en que esto sea posible técnica y económicamente.
8. La formación innecesaria de derivados (bloqueo de grupos,
protección/desprotección, modificación temporal de propiedades
químicas o físicas debe ser evitada en tanto sea posible.
9. Los reactivos catalíticos deben ser tan selectivos como sea posible.
10. Los productos químicos han de ser diseñados de manera que al final de
su función no persistan en el medio ambiente, sino que se fragmenten
en productos inocuos para el medio ambiente.
11. Se deben desarrollar las metodologías analíticas que permitan el
seguimiento en tiempo real del proceso y el control previo de la posible
formación de substancias peligrosas.
12. Las sustancias y las formas de su uso en un proceso químico deben ser
elegidas de manera que resulte mínima la posibilidad de accidentes
químicos incluyendo emisiones, explosiones e incendios.
1.4. Química Sostenible y Biotecnología Blanca
Anastas P.T y Warner J.C. (1998) definen la Química Sostenible (Green
Chemistry) como la utilización de un conjunto de principios que reducen o
eliminan el uso y generación de sustancias peligrosas en el diseño en el
diseño, manufacturación y aplicación de los productos químicos,
manteniendo la calidad del producto final y la rentabilidad económica del
proceso; esto lleva implícito el alcanzar un proceso que sea inocuo en sí
mismo y respetuoso con el medio ambiente. La aplicación industrial de
estos criterios basados en Química Sostenible se conoce como
Biotecnología Blanca.13 Además, cabe resaltar que mientras la Química
medioambiental tiene como objetivo la remediación, la Química Sostenible
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
8
intenta que no se produzcan daños en el medio ambiente por efecto de la
actividad química.
Los procesos industriales basados en procesos biotecnológicos suelen ser
altamente sostenibles. Así lo reconoce la OCDE en sus publicaciones como
la ya clásica “Biothecnology for clean industrial products & products:
towards industrial sustainability” de 1998, donde hace notar que la
biotecnología es una herramienta poderosa para alcanzar el desarrollo
industrial sostenible. Ello se debe a que muchos de sus desarrollos están
basados en:18
- El uso directo de materias primas obtenidas de fuentes sostenibles.
- La obtención de productos como los polímeros biodegradables, de un
alto valor añadido a partir de aceites vegetales, fibras, hidratos de
carbono, etc.
- La conversión de la biomasa en productos intermedios de síntesis tales
como la glucosa, etanol, etc.
- La potencial producción por semisíntesis de productos agroindustriales o
farmacéuticos.
Existe una estrecha relación entre las Biotransformaciones y la Química
Sostenible debido en concreto a los siguientes puntos fundamentales del
trabajo en Biotecnología:1,13-18
- Minimización de residuos y control de efluentes.
- Simplificación de los procesos de purificación del producto final.
- Obtención de buenos rendimientos.
- Reducción al mínimo de la formación de productos concomitantes.
- Trabajar con disolventes no contaminantes.
- Evitar o reducir al mínimo el riesgo de producción de fuegos,
explosiones o emisiones contaminantes.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
9
1.5. Biotransformaciones
La aplicación de biotransformaciones catalizadas por células enteras es un
campo emergente que abre un horizonte de nuevas posibilidades para la
aplicación industrial de los biocatalizadores.
En biotransformaciones el uso de enzimas libres como biocatalizadores es
actualmente una metodología sintética plenamente aceptada y validada
para preparar los compuestos homoquirales utilizados en farmacéuticos,
aditivos alimentarios, o la industria de agroquímicos.19
Además, la biocatálisis al emplear células enteras como biocatalizadores en
las aplicaciones comerciales se ha limitado en gran medida a casos
especiales en que el microorganismo contiene la enzima necesaria para
reacciones de una sola etapa o donde la célula es un productor natural de
una sustancia química como el etanol o un producto complejo natural, como
la tetraciclina.19
Esta utilidad reducida se ha asociado con la falta de herramientas
experimentales generales y eficaces para desarrollo de cepas microbianas
para aplicaciones industriales. Sin embargo, los enormes avances en
ingeniería metabólica, la genómica, la metabolómica, la proteómica, la
evolución dirigida, etc., en los últimos años han abierto la puerta a la
creación apropiada de microorganismos con fines industriales.19
Estos avances están a favor de la introducción de procesos con células
enteras llamándose entonces biotecnología blanca. Hoy en día, algunos
procesos industriales están utilizando células enteras como
biocatalizadores, por ejemplo: nicotinamida a partir de 3-cianopiridina
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
10
utilizando Rhodococcus rhodochrous, acrilamida a partir del acrilonitrilo
utilizando R. rhodochrous, entre otros.20
1.5.1. Diferencias entre Fermentación y Biotransformación
Las diferencias más resaltantes entre un proceso fermentativo y una
biotransformación se encuentran descritas de manera sucinta en la
siguiente tabla:
Tabla 1-1. Principales diferencias entre una Fermentación y Biotransformación
FERMENTACION BIOTRANSFORMACION
Reacción
Producto del metabolismo del microorganismo: Existen varios pasos catalíticos en la transformación del sustrato en producto.
Existe un solo paso catalítico en la transformación del sustrato en producto.
Procesos largos. Muchas reacciones. Muchas enzimas.
Procesos acotados catalíticos. Una o pocas reacciones. Una o pocas enzimas.
Sustrato Fuentes de carbono y nitrógeno. Complejo natural.
Producto
La estructura química de los productos no suele ser semejante a la de los sustratos. Productos solo naturales
La estructura química de los productos es semejante a la de los sustratos. Productos naturales y no naturales.
Tolerancia al
producto final
Concentraciones bajas. Concentraciones bajas y altas.
Aislamiento del
producto final
Complejo. Sencillo
Productos secundarios Generalmente muchos. Generalmente pocos.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
11
1.6. Ventajas de las Biotransformaciones
Entre las principales ventajas que presentan las Biotransformaciones
(Biotecnología) con respecto a los procesos químicos tradicionales
podemos mencionar: 1-19
a) Reduce el número de pasos de síntesis dada la gran selectividad de los
biocatalizadores.
b) Evita la producción de subproductos. Esto es importante en la síntesis
de productos quirales dada la gran estereoselectividad de los
biocatalizadores.
c) Necesitan condiciones suaves de proceso pues los catalizadores
trabajan a temperatura ambiente, presión atmosférica, en medio acuoso
o en disolventes poco contaminantes.
d) Utiliza catalizadores muy activos y lo más selectivo posible, siendo los
biocatalizadores poco contaminantes.
e) Desarrolla metodologías analíticas que permiten el seguimiento en
tiempo real del proceso y el control previo de la posible formación de
substancias peligrosas; por ejemplo los biosensores.
Para mejor ilustración, se tomará como ejemplo la reducción química de
manera regio-, esterero-, o quimioselectiva de una molécula polifuncional,
es decir una molécula con varios grupos funcionales sensibles a reaccionar.
Para eso, a partir de los esquemas 1-1 y 1-2, se hace la comparación de la
reducción química, a través de un proceso químico y otro a través de un
proceso biotecnológico. Como se muestra en la siguiente tabla:
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
12
Tabla 1-2. Cuadro comparativo de los Esquemas 1-1 y 1-2
Reducción Regio-, estereo- o quimioselectiva de una molécula polifuncional
Proceso Químico Proceso Biotecnológico
Aparte de la reducción, necesita de otros 5 pasos más.
Consta de 2 pasos
Necesidad de proteger los otros grupos funcionales
Alta selectividad, no necesitan protegerse los otros grupos funcionales
Catalizadores contaminantes o peligrosos Catalizadores Biológicos
Consumo de disolventes
Trabaja en medios no contaminantes
Disolventes a recuperar Trabaja en condiciones
ambientalmente compatibles Productos concomitantes
Esquema 1-1: Proceso Químico de Reducción Selectiva de un grupo Y en
presencia de otro X sensible a la reducción. Ejemplo X: CHO e Y: C=O.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
13
Esquema 1-2: Proceso Biotecnológico de Reducción Selectiva de un
grupo Y en presencia de otro X sensible a la reducción. Ejemplo X:
CHO e Y: C=O.
1.7. Reacciones de Reducción
Las enzimas empleadas en reacciones redox son clasificadas en tres
categorías: deshidrogenasas, oxigenasas y oxidasas.21-23. Entre ellos, las
alcohol deshidrogenasas – también llamadas reductasas de carbonilo – han
sido ampliamente utilizadas para la reducción de grupos carbonilo
(aldehídos, cetonas) y las enoatoreductasas (Ver Figura 1-1), empleadas
con frecuencia en la reducción de dobles enlaces carbono-carbono.
Desde que la reducción implica la transformación de un carbono con
hibridación planar sp2 en un átomo tetraédrico sp3, esto va de la mano con
la formación de centros estereogénicos. Por lo contrario, el proceso inverso
correspondiente (por ejemplo la oxidación alcohólica o de
deshidrogenación) conduce a la destrucción de un centro estereogénico,
que generalmente es de uso ilimitado. Las oxidasas, que son responsables
de la transferencia de electrones, han jugado un papel menor en la
biotransformación de compuestos orgánicos no naturales, pero son cada
vez más utilizadas recientemente.6
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
14
Figura 1-1. Reacciones de reducción catalizados por deshidrogenasas.
1.7.1. Regeneración de la Coenzima
La distinción más importante de las enzimas redox, es el requerimiento de
coenzimas redox, los cuales donan o aceptan equivalentes químicos para la
reducción (u oxidación). Para la mayoría de enzimas redox, la nicotinamida
adenina dinucleótido [NAD(H)] y su respectivo fosfato [NADP(H)] son
aproximadamente requeridos en un porcentaje de 80% y 10%
respectivamente. Las Quinona, Flavinas (FMN, FAD) y pirroloquinolina
quinona (PQQ) son encontrados con menor frecuencia.6
Los coenzimas nicotinamidas son parecidos a un “complejo híbrido natural”,
que tienen dos características en común, es decir, son relativamente
moléculas inestables y costosas si se utilizan en cantidades
estequiométricas. Además, éstos no pueden ser reemplazados por otros
sustitutos más económicos creados por el hombre. Como sólo el estado de
oxidación es el que cambia durante la reacción, y el resto de la estructura
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
15
del complejo queda intacta, éstos pueden ser regenerados in situ mediante
una segunda reacción concurrente redox, que les permita entrar
nuevamente al ciclo de la reacción principal.24
El proceso de reducción estereoselectiva de cetonas permite la creación de
alcoholes homoquirales de gran interés como intermedios de síntesis. Al ser
enzimas intracelulares, dependen de una coenzima como NAD(P)H, NADH,
etc. que se consumen en cantidades equimoleculares respecto al substrato
a reducir. Esto implica que la regeneración de la coenzima hace al proceso
económicamente inviable con enzimas libres sin la regeneración del
cofactor (el precio aproximado de un mol de NADH es de 3.000$ USA y el
de un mol de NADPH 25.000$ USA).25
Así como la coenzima es muy costosa en cantidades catalíticas, esto lleva a
buscar una drástica reducción en los costos. La eficiencia de un proceso de
reciclado del cofactor se mide por el número de ciclos que se puede lograr
antes de que una molécula coenzima sea finalmente destruida. Este
reciclado se puede medir como número total de moles de producto formado
por un mol de coenzima durante toda su vida. Como regla general, unos
pocos miles de ciclos (103-104) son suficientes para las reacciones redox a
escala de laboratorio, mientras que para fines técnicos, ésta medida es
altamente deseado en el orden de 105. El impedimento económico para las
reacciones a gran escala es el costo de la coenzima, el cual ha sido
reconocido así por muchos años y gran parte de los esfuerzos en
investigación sobre las alcohol deshidrogenasas se han empleado con el fin
de resolver el problema del reciclado de la coenzima.26-29
La regeneración de la coenzima no es un problema cuando células
microbianas enteras son usadas como biocatalizadores en reacciones
redox. Además, como fuentes no costosas de equivalentes redox, se
pueden emplear ciertos carbohidratos, ya que el microorganismo posee
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
16
todas las enzimas y coenzimas necesarias que se necesiten en su
metabolismo.27
Es por ello que se utilizan células enteras como biocatalizadores ya que
éstas poseen sistemas para la regeneración de la coenzima. El único
problema existente es que dentro de las células suele haber más de una
enzima que cataliza el proceso, obteniéndose en algunos casos bajos
excesos enantioméricos (%e.e.). La reacción se puede representar como se
muestra en la siguiente figura:27
Figura 1-2. Sistema de Reciclado del Cofactor
1.8. Enzimas aisladas frente a células enteras: Ventajas y Desventajas
El estado fisiológico de los biocatalizadores que se utilizan en
Biotransformaciones puede ser muy diverso. La decisión final de utilizar
enzimas aisladas y purificadas, o microorganismos completos (células
enteras) en forma libre depende de varios factores, como son: 2,6
- Tipo de reacción.
- Necesidad o no necesidad de reciclar las coenzimas.
- Escala a la que se quiere desarrollar la biotransformación (mg, g o Kg).
La mayoría de las Biotransformaciones llevadas a cabo por
microorganismos parten de moléculas orgánicas relativamente complejas, y
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
17
utilizan sólo una reacción bioquímica del potencial enzimático del
microorganismo para llegar al producto deseado.
En la siguiente tabla se observa una secuencia de ventajas y desventajas
de usar enzimas aisladas o enzimas enteras: 2,6
Tabla 1-3. Ventajas y Desventajas de usar Sistemas con Enzimas Aisladas vs.
Células Enteras.2,6
Biocatalizador Forma Ventajas Inconvenientes
Enzimas Aisladas
En general
Dispositivo de trabajo simple. Mayor productividad debido a la
elevada tolerancia a concentración de sustrato.
Necesario el reciclaje de la coenzima
Disuelta en agua
Elevadas actividades catalíticas.
Posibles reacciones colaterales. Los sustratos lipófilos son solubles.
Recuperación de la enzima.
Suspendidas en disolventes orgánicos
Fácil de realizar, los sustratos lipófilos son solubles.
Recuperación de la enzima Actividades reducidas
Biocatalizador Forma Ventajas Inconvenientes
Células Enteras
En general
No es necesario el reciclaje de la coenzima.
No se requiere de purificación de la enzima.
Equipamiento caro. Proceso tedioso debido a los elevados volúmenes.
Baja productividad debido a la menor tolerancia a la concentración, baja tolerancia a disolventes orgánicos, reacciones colaterales debidas al
metabolismo incontrolado.
Cultivo en crecimiento Elevadas actividades
Elevada cantidad de biomasa, mayor cantidad de subproductos, difícil
control del proceso
Células en Reposo
Trabajo más fácil. Menor cantidad de
subproductos al estar el metabolismo controlado.
Menores actividades
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
18
1.9. Reducción de Aldehídos y Cetonas usando Células Enteras
En vez del aislamiento de la deshidrogenasa, el cual requiere de una
sofisticada regeneración de la coenzima, se emplean mayormente células
microbianas. Ellas contienen múltiples deshidrogenasas, las cuales están
aptas de aceptar sustratos no naturales, además tienen todas las
coenzimas necesarias y las rutas metabólicas para su regeneración. Por lo
tanto la regeneración o reciclado de la coenzima puede ser omitida ya que
se realiza automáticamente gracias al metabolismo de la célula viva.
Además, fuentes económicamente cómodas de carbono tales como
sacarosa o glucosa pueden ser usados como sustratos auxiliares para
reacciones asimétricas de reducción. Por otro lado, todas las enzimas y
coenzimas están bien protegidas dentro de su entorno celular natural.6
Sin embargo, estas ventajas tienen que ser tomadas en cuenta junto con
algunos inconvenientes importantes:
- La productividad de las conversiones microbianas son generalmente
bajas, ya que la mayoría de los sustratos no naturales son tóxicos para
los organismos vivos, y por lo tanto sólo son toleradas a bajas
concentraciones (0.1-0.3% por volumen aprox.).
- La gran cantidad de biomasa presente en el medio de reacción causa
bajos rendimientos y la recuperación del producto se torna problemático,
sobre todo cuando el producto es almacenada dentro de las células y no
se elimina al medio de reacción. Debido a que solo una menor fracción
(normalmente 0,5-2%) del sustrato auxiliar se utiliza para la
regeneración de la coenzima, la mayor parte se metaboliza, formando
subproductos polares, que a menudo impiden la purificación del
producto. Por lo tanto, el control de la reacción se hace más dificultosa.6
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
19
- Finalmente, es más probable que diferentes cepas de un
microorganismo cuenten con diferentes especificidades, por lo que es
importante usar exactamente el mismo cultivo para obtener resultados
comparables con la literatura.30
La estereoselectividad puede variar en gran medida debido a las siguientes
razones: Por un lado, un sustrato puede ser reducido por una sola vía
oxidorreductasa para dos enantiómeros que tienen valores similares de
energía libre. En otras palabras, el reconocimiento estereogénico permite
un ajuste alternativo del substrato dentro de una sola enzima. Si dos
enzimas compiten para el mismo sustrato, la pureza óptica del producto
está determinada por las velocidades relativas de las reacciones
individuales.30
Las técnicas generales siguientes se pueden aplicar para mejorar la
selectividad de las reacciones de reducción microbiana:
- Modificación del sustrato, por ejemplo, la variación de los grupos
protectores que pueden ser eliminados después de la transformación.32-
34
- La variación de los parámetros metabólicos de inmovilización.35-37
- El uso de células de diferentes edades.38
- La variación de las condiciones de fermentación.39-41
- Selección de microorganismos para obtener las cepas con las
propiedades óptimas.42,43
- La inhibición selectiva de una de las enzimas en competencia.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
20
El buen nivel de la tecnología de las reducciones de compuestos
carbonílicos, ha permitido su implementación a escalas industriales, y como
se ve hacen uso de células enteras, y tienen el cuidado respectivo en la
regeneración de sus cofactores. Ejemplos representativos son mostrados
en la siguiente figura: 6
Figura 1-3. Bioreducción de Compuestos Carbonílicos de Escala Industrial
1.9.1. ADH Deshidrogenasas
Las Alcohol deshidrogenasas (ADH) son enzimas ubicuas involucradas en
muchos procesos fisiológicos. Algunas de estas contienen Fe (II) para la
activación de la enzima, como es el caso de la ADH de Zymomonas
mobilis.44Otros ADHs no presentan un catión en la enzima, por ejemplo, el
ADH de la Drosophila lebanoniensis.45
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
21
Sin embargo, la mayoría de ADHs tienen muchos iones Zn(II) cerca al sitio
activo. Este es el caso del ADH de la levadura de pan 46, el cual contiene 4
átomos de Zn (II). Estos cationes pueden ser sustituidos por Co (II) después
de un tratamiento con material quelante seguido de la inserción de cobalto.
Estas enzimas catalizan la oxidación/reducción reversible de
alcoholes/aldehídos o cetonas usando NAD+/NADH como el hidruro
aceptor/donor. 45,46 Sólo en pocos casos el NADP(H) ha sido utilizado como
coenzima, por ejemplo el ADH del Clostridium beijerinckii.47El mecanismo
de reacción fue descrito por Theorell y Chanwe en 1951.48
Figura 1-4.Mecanismo de reacción
La etapa que controla la velocidad es la transferencia de hidruro en el
complejo ternario [enzima + co-enzima + compuesto orgánico].En el sitio
activo, la presencia de Ser, Tyr y Lys o Arg es reconocido generalmente
como una maquinaria catalítica fundamental para la etapa de transferencia
de hidruro, ya que el caso del ADH-2 de ratas, usando difracción de rayos X
y modelado molecular ha sido probado por Svensson.49
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
22
1.10. Screening de Nuevos Biocatalizadores
La primera cuestión a decidir es la fuente de las enzimas que vamos a
utilizar en el screening, pudiendo optar entre varias alternativas:
1.10.1. Screening a partir de bancos de genes
El screening en busca de nuevas enzimas a partir de bancos de genes
puede llegar a ser muy satisfactorio. Mediante el establecimiento de bancos
de genes en un pequeño grupo de microorganismos hospedadores
(bacterias como E. coli, o levaduras), sólo se necesita implementar un
limitado número de métodos de propagación de los genes a los organismos
permitiendo una realización sencilla de las técnicas sistemáticas del
screening.
El ADN utilizado para clonar puede proceder de ADN preparado a partir de
cultivos de microorganismos o a partir de microorganismos salvajes. Se
estima que sólo un 1% de los microorganismos existentes en la naturaleza
han sido cultivados en laboratorio.
1.10.2. Screening a partir de enzimas comerciales
Sin duda la forma más rápida y fácil de encontrar un biocatalizador que se
ajuste a nuestras necesidades es localizada en las colecciones de enzimas
comerciales, lo que permite un acceso rápido al biocatalizador en
cantidades suficientes. Existen pocas bibliotecas de enzimas de carácter
comercial, entre las que debemos destacar: Amano, Sigma-Aldrich, Fluka,
Toyobo, Diversa, ThermoGen, Altus Biologics y Roche (anteriormente
Boehringer-Mannheim).
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
23
1.10.3. Screening a partir de colecciones de microorganismos
Desde los orígenes de vida en la Tierra, como primeros habitantes, los
microorganismos han tenido mucho tiempo para desarrollar nuevos genes y
funciones (Wackett y Herschberger, 2000). Los microorganismos se
reproducen mucho más rápidamente que los organismos macroscópicos lo
que favorece que se desarrollen con mayor rapidez nuevas subespecies, es
decir nueva biodiversidad.
La mayoría de enzimas de importancia industrial proceden de especies
microbianas de tipo I o II en la escala de bioseguridad (consideradas en
líneas generales seguras). Entre ellas se incluyen géneros bacterianos
como Bacillus, Pseudomonas, levaduras, especialmente Saccharomyces y
hongos de las clases Ascomycota y Zygomycota.
1.11. Metodología de Screening
Debido a que el número de enzimas y el tamaño de colecciones de
microorganismos van aumentando, los métodos de screening a gran escala
son cada vez más importantes. Estos métodos incluyen el screening a gran
escala automatizado, y los screening consecutivos estructurados de modo
jerárquico. En un screening jerárquico, se realizan varios ensayos que se
combinan en series consecutivas hasta alcanzar, paso a paso, el objetivo
del screening. En primer lugar el screening nos permite seleccionar un
grupo de microorganismos positivos frente a una biotransformación o a la
producción de un metabolito, que pasan al siguiente paso del estudio. Las
pruebas más lentas, caras o complicadas son realizadas al final para
reducir la duración y el número de ensayos. Este tipo de screening es
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
24
rápido, útil y muy efectivo. En este trabajo se realizó un screening jerárquico
de tres fases:
a) Screening primario.- Es rápido y simple. En este tipo de selección se
deben eliminar todos los candidatos negativos y seleccionar los
positivos potenciales. Elimina todos los candidatos que carecen
absolutamente de la actividad deseada, reduciendo en gran medida el
tamaño de la colección objetivo de estudio. Sin embargo, el uso de
sustratos análogos y no el sustrato específico de estudio puede eliminar
positivos potenciales de la reacción de interés.
b) Screening secundario o intermedio.- En este paso se emplean mayor
número de sustratos y sirve para seleccionar los mejores candidatos
que pueden ser considerados como potenciales de la actividad
enzimática en estudio. Este segundo ensayo se realiza con criterios
cuantitativos claros que nos permitan reducir la colección a un número
razonable según la escala del screening específico a realizar a
continuación.
c) Screening específico.- En este paso se ensaya el sustrato específico
para evaluar la actividad enzimática en estudio, así el número de
candidatos es reducido.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

I. INTRODUCCIÓN
25
1.12. Diplogelasinospora grovesii.
Diplogelasinospora grovesii es un nuevo hongo filamentoso, el cual muestra
una alta actividad y enantioselectividad en la reducción de algunas cetonas
hidrofóbicas acíclicas o policíclicas.
Estos son hongos filamentosos, que tienen como unidad estructural a las
hifas, que pueden ser simples o ramificadas y estar tabicadas o no. El
crecimiento se produce por alargamiento de las hifas, y da lugar a una
masa filamentosa conocida como micelio. Su reproducción puede ser
asexual o sexual. En la reproducción asexual, a partir del micelio vegetativo
se diferencian hifas aéreas especiales (los conidióforos) que dan lugar a
esporas denominadas conidios. En el ciclo sexual se produce la fusión de
estructuras especializadas, los anteridios y ascogonios, pero sin fusión de
sus núcleos. El crecimiento de los hongos filamentosos dentro de un medio
líquido puede producirse en forma dispersa o como pequeñas colonias
denominadas pellets. La formación de los pellets puede ser debida a la
coagulación de esporas e hifas, o bien al crecimiento de hifas a partir de
una sola espora. Pero, en cualquier caso, es el resultado de la interacción
entre factores de tipo estructural (composición y naturaleza de las cargas de
la pared celular) y ambiental (tipo de nutrientes, relación entre carbono y
nitrógeno del medio, presencia del material particular, concentración de
inóculo, etc.) lo que determina una u otra forma de crecimiento. Además, el
microorganismo se aisla durante un proceso de screening de cepas
microbianas con alta actividad reductasa.2
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

II. OBJETIVOS Y PLAN DE TRABAJO
26
II. OBJETIVOS Y PLAN DE TRABAJO
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

II. OBJETIVOS Y PLAN DE TRABAJO
27
II. OBJETIVOS Y PLAN DE TRABAJO
2.1. OBJETIVOS De manera tal que existan nuevos métodos de reducción de cetonas, en vez de los métodos comerciales que son incompatibles con el medio ambiente, se propone el uso de las alcohol-deshidrogenasas que poseen los hongos filamentosos como inherentes de su metabolismo, siendo estas reacciones de cinética batch con biocatalizadores de células enteras.
Para lo cual se planteó los siguientes objetivos que se desarrollaron extensamente en la siguiente tesis de pregrado:
2.1.1. Objetivo General
Determinar los principales parámetros cinéticos en la obtención del 1-Feniletanol en medio acuoso empleando Diplogelasinospora grovesii como biocatalizador.
2.1.2. Objetivos Específicos
- Aislar y caracterizar un microorganismo con actividad reductasa.
- Realizar la curva de crecimiento del biocatalizador, para encontrar el tiempo en el cual se encuentra en mejores condiciones para hacer la bioreducción.
- Desarrollar métodos validados de análisis instrumental (HPLC, UV-VIS) para monitorear la reacción de reducción.
- Desarrollar cinéticas de reacción para determinar los principales parámetros cinéticos de la reacción.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

II. OBJETIVOS Y PLAN DE TRABAJO
28
2.2. PLAN DE TRABAJO Para alcanzar todos los objetivos planteados de esta tesis de pregrado, se desarrolló el siguiente plan de trabajo:
2.2.1. Se realizó la bioreducción para obtener 1-feniletanol como reacción test.
2.2.2. Se realizó la cualificación de cada uno de los componentes del
equipo HPLC, comprobando su linealidad, repetitividad, precisión y exactitud.
2.2.3. Se validó el método de análisis de la pareja acetofenona/1-
feniletanol, determinando: columna a utilizar, composición de la fase móvil, flujo (ml / min), longitud de onda (nm), determinado las condiciones óptimas de separación.
2.2.4. Se determinó el límite de cuantificación (LQ) que será muy importante a la hora de seguir la reacción de reducción por HPLC ya que nos indica la mínima conversión detectable.
2.2.5. Se realizó la curva de crecimiento del hongo filamentoso, para lo cual se desarrolló un protocolo de trabajo, desde el cuidado de almacenamiento del microorganismo, crecimiento en medio sólido y en medio líquido.
2.2.6. Se realizó las curvas de cinética de reacción a la reacción de reducción. Y se analizó y calculó los parámetros cinéticos que intervienen en la reacción.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
29
III. PARTE EXPERIMENTAL
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
30
III. PARTE EXPERIMENTAL
3.1. MATERIALES
3.1.1. Materiales de Vidrio.
Matraces Erlenmeyer de 100 ml y 250 ml. Marcas: Pirex, Kyntel y LBY Boro 3.3
Fiolas de 25 ml, 50 ml, 100 ml y 250 ml. Marcas: Kyntel y Fortuna
Vasos de Precipitación graduados de 50 ml, 100 ml, 250 ml y 400
ml. Marcas: Pirex, Kyntel y Duran.
Viales encapsulables para cromatografía líquida de 2ml.Marca: Agilent. Color: Ambar
Pipetas de 10 ml y 25 ml. Marcas: Pirex y Qualicolor.
Frascos para medio de cultivo de 250 ml, 500 ml y 1000
ml.Marca: Boeco
Tubos de ensayo de 16*100 mm – MARCA: PYREX Varillas de agitación. Lunas de reloj. Embudos de vidrio de vástago corto de 70 mm de diámetro.
3.1.2. Material de Plástico.
Tubos tipo Falcon de 14 ml.Marca: Fisherbrand
Placas Petri estériles. Marca: Miniplast-ein-shener
Probetas graduadas de 25 ml, 50 ml, 100 ml, 250 ml y 500 ml
Espátulas.
Puntas para micropipetas azules y amarillas.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
31
Rackspara viales.
Criotubos.
Porta puntas de micropipeta.
Sacabocados.
3.1.3. Material Auxiliar.
Guantes de látex.
Gafas de seguridad.
Porta objetos.
Asas de siembra metálicas.
Cinta Parafilm.
Filtros para viales de PTFE 0.2 µm.
Cinta para esterilización con vapor.
Tapones. Papel Aluminio.Algodón. Jeringas desechables de 1 ml. Marca: Qualimaxx Mascarillas. Bolsas para eliminación de puntas de pipeta contaminadas.
Bolsas para eliminación de residuos.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
32
3.1.4. Instrumental de Laboratorio.
Balanza de precisión. Marca: Sartorious AG Germany CPA 224S
Incubadora de agitación orbital de temperatura y RPM controlados.
Incubadora tipo estufa (JSGI – 150 T)
Autoclave JSR. Marca: JSAC-40.
Espectrofotómetro UV–VIS. Marca: HEWLETT 8452A PACKARD DIODE ARRAY
Cromatógrafo líquido de alta resolución. Marca: Agilent 1100 Vortex. Marca: Mixer.
Estufa. Marca: SP
Juego de micropipetas de volumen variable.
Lab Mate+(100-1000 µL) Pipet 4u (20 - 200µL) DragonMed(20 - 200 µL) Microlit(20 - 200 µL) Pipette pump de 10 ml.
Centrifuga.Marca: EBA 20.
Columna Cromatográfica mediterránea C-18 de fase reversa.
Refrigeradoras.Marcas: INRESA Y DAEVOO.
pH-metro portable. Marca: PH-009 (II) ATC.
Termómetro de -10°C hasta 400 °C.
Mechero bunsen.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
33
3.2. METODOS
3.2.1. Procedimiento para el Screening global
La colección de microorganismos utilizada como punto de partida fue
propuesta por el Dr. José Sinisterra Gago (UCM) y Dr. Alberto Quezada
(UNT), las cuales sonde diferentes grupos taxonómicos (bacterias,
levaduras, y ascomicetos), y de las cuales se seleccionó el microorganismo
apropiado como biocatalizador para la reacción de estudio.
3.2.1.1. Estado de la colección
Las cepas microbianas utilizadas en ésta tesis se encuentran almacenadas
a 5°C en solución de glicerol-agua al 30% v/v. Para ello se colocaron en
cada criotubo 1ml de solución glicerol-agua al 30%v/v y cubitos de medio
sólido que contienen microorganismo que ha crecido previamente en placas
Petri. Se utilizó glicerol por ser no tóxico para las células y por contar a nivel
molecular con espacios intersticiales amplios, generando una mejor
residencia del microorganismo, evitándose problemas de presionamiento
entre ellos.
3.2.1.2. Medios de Cultivo
La elección del medio de cultivo con sales minerales y un balance de
fuentes de carbono y nitrógeno adecuado, proporciona la posibilidad al
microorganismo de poner en marcha toda su maquinaria enzimática
responsable del éxito de nuestra biotransformación. Los microorganismos
crecieron en medios de cultivos recomendados por los papers publicados
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
34
del Servicio Industrial de Biotransformaciones del Parque Científico de
Madrid.2,19,25
De donde se extrajo la composición de cada medio, que fue utilizado para el
crecimiento de los microorganismos:
a. Medio de cultivo para Ascomicetos.
o Medio de cultivo HAGGS (Ajustar a pH = 6.5):
Glicina : 2 g/l
Harina de soja : 6 g/l
Almidón : 20 g/l
Solución elementos traza: 10 ml/l.
Solución Traza : 10 ml/l
Composición de la Solución de elementos traza:
FeSO4.7H2O : 1 g/l
MnSO4.4H2O : 1 g/l
CuCl2 : 0,025 g/l
CaCl2 : 0,10 g/l
H3BO3 : 0,056 g/l
ZnSO4.7H2O : 0,2 g/l
(NH4)6Mo7O24.4H2O : 0,019 g/l.
b. Medio de cultivo para Bacterias.
o Medio LB(Frenken y cols, 1992):
Triptona : 10 g/l
Extracto de levadura : 5 g/l
NaCl : 5 g/l
Tampón KH2PO4 pH= 6.5
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
35
c. Medio de cultivo para Levaduras
o Medio YM:
Extracto de levadura : 3 g/l
Extracto de malta : 3 g/l
Bactopeptona : 5 g/l
Bactodextrona(dextrosa): 10 g/l
3.2.1.3. Preparación del medio sólido de cultivo
Para la preparación de medio sólido, se añadió, a la composición descrita
anteriormente, agar tipo Americano 30 g/l. y se disuelve en agua purificada.
3.2.1.4. Esterilización de medios de cultivo.
Todos éstos medios deben se esterilizaron a 121°C y 1 atm de presión por
20 minutos.
3.2.1.5. Reacción test para el screening global catalizada por
células en condiciones de fermentación
Para evaluar la actividad reductasa de las cepas se seleccionó como
reacción test, la reducción de la acetofenona a 1-feniletanol; por ser una
reacción sencilla y de estudio, que nos permite evaluar de una manera
rápida y precisa la actividad de nuestras cepas y que puede ser analizada
por cromatografía líquida con un detector UV, dada la absorbancia de los
electrones pi deslocalizados de los anillos aromáticos frente a radiación
ultravioleta.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
36
La concentración inicial de sustrato fue de 10mM, que es lo suficientemente
baja como para permitir el crecimiento de los microorganismos y lo
suficientemente alta para permitir un grado de biotransformación del
sustrato suficiente para su extracción y posterior análisis cromatográfico.
Para llevar a cabo esta reacción se dejó crecer cada microorganismo en
erlenmeyers de 100ml con 20ml de medio de cultivo estéril, durante 48
horas para las bacterias y levaduras, y 72 horas para ascomicetos a 30°C y
200rpm. Luego se adicionó el sustrato hasta una concentración de 10mM.
Después de 72 horas de reacción a las mismas condiciones de agitación y
temperatura se tomó una muestra de 3ml, se centrifugó a 4000 rpm por
15min, se separó la biomasa del medio líquido, se tomó 1ml del medio
líquido, se filtró, y se analizó por cromatografía, para determinar en base a
la conversión de substrato, el mejor microorganismo que sea activo a la
reducción de cetonas.
3.2.2. Cualificación de los Equipos de Instrumentación Analítica.
En la siguiente tesis, se realizó una serie de cualificaciones y validaciones
de los equipos de instrumentación analítica más resaltantes que se usaron,
con la finalidad de tener mejor confiabilidad en precisión y en exactitud de
los datos obtenidos. Se cualificaron los equipos y se validaron sus métodos
de cualificación.
3.2.2.1. Cualificación de las Balanzas Analíticas.
Las mediciones de pesos para la siguiente investigación oscilan entre 0 a
100g., es por ello, que se realizó éste análisis en dos rangos de
cualificación:
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
37
- 1er. Rango de Cualificación: 0 a 10g.
- 2da. Rango de Cualificación: 0 a 100g.
Todas las mediciones se realizaron con agua destilada. Y para los
siguientes rangos, se tomaron los siguientes puntos intermedios:
- 1er. Rango de Cualificación: 0 a 10g.: 0g., 0.125g., 0.25g., 0.5g., 0.75g.,
1g. Con 4 repeticiones por cada medición realizada.
- 2da. Rango de Cualificación: 0 a 100g.: 0g., 25g., 50g., 75g., 100g. Con
4 repeticiones por cada medición realizada.
Con las rectas obtenidas de Peso Real vs. Peso Teórico, observamos la
variación de las pendientes al repetir cada cierto tiempo el mismo método
de cualificación, y así llevar un control de la exactitud y precisión de la
balanza analítica.
3.2.2.2. Cualificación del Espectrofotómetro UV.
Se preparó una solución madre de 0.528 mM de Benzoato de Sodio. Luego,
se empezó a medir las absorbancias de las diferentes concentraciones de
las diferentes diluciones, esto se realizó directamente en la celda de cuarzo
del espectrofotómetro con UV-Visible a tres longitudes de onda diferentes
(236nm, 254nm y 270nm). Estas diluciones se realizaron agregando de
manera cuantificada proporciones de Agua MiliQ (Agua Ultrapura), y se
obtuvo un espectro entre las concentraciones de 0 mM (Muestra Blanco)
hasta 0.528mM.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
38
Una vez tenido el espectro a cada longitud de onda, luego se pasó a
desarrollarse la prueba de linealidad y la prueba de repetitividad, del cual se
obtuvo los límites de detección y cuantificación para cada una de las
longitudes de onda.
Luego, éste método y los resultados obtenidos quedaron registrados, con el
propósito de ver la variación de los límites de detección y cuantificación
cada cierto periodo de tiempo, así se chequeó si los errores sistemáticos en
la medición pueden ser despreciables, los cuáles generaría la necesidad de
cambio de partes o todo el equipo: lámpara UV, etc.
3.2.2.3. Cualificación del HPLC
Antes de realizar cualquier análisis en el HPLC, primero se cualificó las
partes del HPLC, con el objetivo de asegurar la confiabilidad de cualquier
análisis al utilizar dicho equipo.
Se realizaron las cualificaciones de las siguientes partes:
a) Cualificación de la Bomba del HPLC.
Se comprobó el flujo real con el flujo teórico para cada uno de los 4 canales
del equipo cromatográfico. Se hizo pasar agua a través de cada canal y se
realizó la cualificación a los siguientes caudales nominales de: 0.5, 1.0, 1.5
y 2.0 mL/min.
Primero, se desconectó el conducto de desecho que se encuentra a la
salida del detector y se colocó un vaso para evitar caídas del eluyente
sobre el detector. Luego, se enumeró y se pesó 5 tubos eppendorf para el
flujo de 0,50 mL/min, y otra cantidad similar para los flujos de 1,0; 1,50 y
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
39
2,0 mL/min. Se activó el funcionamiento la bomba del equipo. Se estableció
el flujo de 0,500 mL/min para el canal “A” y se dejó pasar agua como fase
móvil durante el tiempo suficiente para que la presión se estabilice y que
por todo el sistema circule el solvente emitido por la bomba a través del
canal “A”.
De manera sincronizada se controló 2 min y se recogió el volumen de
eluyente que sale en ese tiempo. Además, se repetió ésta operación 5
veces (al mismo caudal), de tal manera que se obtuvieron las 5 alícuotas
del caudal. Se repitió los pasos anteriores empleando los flujos de 1,0; 1,50
y 2,0 mL/min.
Posteriormente, se pesó los tubos eppendorf que contienen la fase móvil
recogida(agua). Se determinó el volumen experimental recogido (usando la
densidad del eluyente a la temperatura del laboratorio) de las 05 lecturas
para cada uno de los flujos utilizado, luego se determinó el valor promedio
del volumen experimental recogido para cada caudal utilizado. De ésta
manera se repetió todos los pasos para el canal “B”, “C” y “D”.
b) Cualificación del Sistema de Inyección
Para tener mayor seguridad en los resultados tanto en reproducibilidad y
precisión, la cualificación del sistema de inyección se dividió en 2 partes:
Precisión del inyector y Magnitud del efecto memoria (efecto “carry over”).
- Precisión del inyector
Se realizó tal estudio con la columna Teknokroma Mediterranea Sea
C18 - 5 µm*15*0,46, con una fase movil: Metanol/agua(45/55 v/v), con
un flujo de 1ml/min, a una longitud de onda de 254nm, con una
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
40
temperatura de horno de la columna de 30°C, para un volumen de
inyección programado de 10ul.
Primero, se puso en funcionamiento el equipo. Luego, se seleccionó un
flujo de 1.00 mL/min y se dejó pasar fase móvil durante el tiempo
suficiente para que la presión se estabilice y para que circule el solvente
por todo el sistema de bombeo. Posteriormente, se inyectó 10 µl de la
disolución estándar. Por último, se repitió esta última operación 5 veces
con esas condiciones cromatográficas y se anotó el tiempo de retención
y el área para cada una de las inyecciones de la disolución estándar.
- Magnitud del efecto memoria (efecto “carry over”).
Este efecto de carry over es ocasionado por arrastre o retención del
analito analizado anteriormente. Por lo cual un mal lavado del sistema
de inyección después de un análisis, puede ocasionar falsos resultados
en los posteriores análisis cromatográficos.
Primero se programó a un flujo de 1ml/min la inyección de 10ul.
Posteriormente, se inyectó la disolución estándar y luego se inyecto una
muestra blanco compuesto de la misma fase movil. Se observó en el
cromatograma de este último si aparece señal en el tiempo de retención
de la disolución estándar.
Luego, se realizó la misma evaluación pero ésta vez antes de cada
inyección de la muestra blanco, se realizó un lavado programado del
sistema de inyección. Por último, se analizó si sigue presentándose
señal y se comparó el sistema de lavado.
c) Cualificación del Detector UV-Visble
Se llevó a cabo la evaluación de la linealidad de la detección a una
longitud de onda determinada en un rango de absorbancia. Esta
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
41
evaluación se realizó a 254nm, mediante una secuencia de tres
inyecciones de cinco disoluciones estándar de acetofenona (0.050,
0.150, 0.250, 0.350 y 0.500 mg/ml respectivamente en metanol/agua).
d) Cualificación de la Columna Cromatográfica
La columna cromatográfica puede presentar problemas de sangrado en
su fase estacionaria, por ello se necesita cualificar el equipo. Esta
evaluación se llevó a cabo con una secuencia de 3 inyecciones de una
disolución estándar que contuvo benceno (1 µl/ml), tolueno (1 µl/ml) y
naftaleno (1 µl/ml) en acetonitrilo. Éstos compuestos son similares pero
tienen distinto coeficiente de extensión molar a la longitud de onda de
254nm.
Se seleccionó un flujo de 0.700 ml/min y se dejó pasar la fase móvil
durante el tiempo suficiente para que la presión se estabilice. Se inyectó
20 µl de la disolución estándar. Luego, se repitió esta última operación 3
veces con esas condiciones cromatográficas. Por último, se anotó y
calculó el tiempo de retención, el área de pico, factor de capacidad (k´),
número de platos teóricos (N), número de platos teóricos por metro de
columna (N/m), altura de pico teórico reducido (h) y factor de asimetría
(As) del último pico (naftaleno) para cada una de las inyecciones.
- Número de platos teóricos (N)
Figura 3-1. Picos cromatográficos
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
42
N = tR2/ t2 ó N = 16 tR2/Wb
2 [3-1]
- Factor de capacidad (k’)
k' = ( tR - to ) / to [3-2]
Donde:
tR = tiempo de retención de la muestra
to = tiempo muerto de la columna
- Reducida del Plato Teórico (h o HETP)
h = m /N [3-3]
Donde m es la longitud de la columna y N el número de platos teóricos.
- Factor de Asimetría (As) (“tailing factor”)
As = a/b [3-4]
Se miden los valores de a y b como se indica en la figura. Si la columna
funciona bien este valor debe ser 1 o muy próximo al mismo.
Figura 3-2. Pico cromatográfico
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
43
3.2.3. Validación y Cualificación del Método de Análisis.
Se necesitó desarrollar una metodología particular de análisis para
determinar la relación de las concentraciones de Acetofenona y 1-
Feniletanol en el medio de reacción durante todo el proceso bioreducción.
Se emplearon los equipos cualificados previamente para la validación de un
buen método de análisis en el laboratorio de investigación de ésta tesis de
pregrado.
3.2.3.1. Determinación de la Longitud de Onda Óptima.
Se sabe que los analitos Acetofenona y 1-Feniletanol absorben radiación
UV, debido a la presencia de sus electrones pi deslocalizados en el anillo
aromático de cada uno de éstos. Además, se contó con un detector
ultravioleta en el HPLC. Es por ello, que primero, debido a ésta absorbancia
de cada uno de los analitos, se determinó la longitud de onda óptima en el
espectrofotómetro UV, a la cual se analizaron posteriormente en el detector
UV del HPLC.
Para determinar la longitud de onda óptima de análisis de la acetofenona y
el 1- Feniletanol, se encontró el Punto Isosbéstico entre los dos espectros
de absorbancia de cada uno de ellos.
En todo el rango del espectro ultravioleta, el Punto Isosbéstico fue la
longitud de onda, en donde la Acetofenona y el 1-Feniletanol tuvieron la
misma absorbancia, a la mínima concentración posible para cada uno de
ellos. Se encontraron diferentes cruces para ambos espectros de absorción
ya sea en zonas de cresta o valle.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
44
Para encontrar el correcto Punto Isosbéstico, se construyó los dos
espectros de absorbancia en un rango de longitudes de onda, apartir de
dos soluciones de 0.1mM de Acetofenona y otra de 0.1mM 1-Fenil Etanol,
con lo cual se observó una serie de valles y crestas para cada uno de los
espectros. Luego, se observó una serie de puntos isosbésticos al
superponer los dos espectros, éstas longitudes de onda a la cual cortan los
espectros, se anotaron para un siguiente estudio en la optimización de la
longitud de onda con cual trabajó el detector UV del HPLC para el análisis
de la relación de las diferentes concentraciones de Acetofenona y 1-
Feniletanol en el medio de reacción.
La búsqueda de la longitud de onda óptima para el análisis de la reacción,
se realizó con la propósito de obtener resultados de las concentraciones de
los analitos de manera exacta y precisa, obteniendo un método de análisis
que reporte límites de detección y cuantificación que aporten con mayor
exactitud y confianza en los análisis.
3.2.3.2. Validación del Método de Análisis Cromatográfico.
Para los análisis cromatográficos en HPLC, se seteó el detector ultravioleta
a la longitud de onda encontrada anteriormente. Para soluciones de 0.1mM
de concentración de Acetofenona y de 1-Feniletanol, se buscó la longitud
de onda óptima, flujo óptimo de fase reversa. Y, con todo ésto se encontró
un método validado para el análisis de éstos dos analitos.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
45
3.2.3.3. Obtención de las Curvas de Calibración para
Acetofenona y 1-Feniletanol.
Se utilizó el método cromatográfico validado para obtener las dos curvas de
calibración en los rangos de 0mM a 10mM para la Acetofenona y 1-Fenil
etanol, respectivamente. Para ello primero, se seteó todas las condiciones
validadas de análisis en el cromatógrafo HPLC, segundo se prepararon
diferentes concentraciones de los dos analitos en los viales de HPLC, cada
uno identificado con sus concentraciones respectivas, para luego ser
analizadas en el cromatógrafo.
Una vez terminado los análisis para cada concentración de los viales, se
reportaron las áreas, y con estos últimos se pasó a realizar las curvas de
calibración. Se halló una recta entre las áreas halladas vs. las
concentraciones de cada analito contenido en los viales, tanto para la
Acetofenona como el 1-Feniletanol. Las curvas de Calibrado entre Área vs
[Analito, mM], son:
- Para la acetofenona:
[3-5]
- Para el 1-Feniletanol:
[3-6]
Donde:
: Áreas halladas, que corresponden a cada vial.
: Pendiente de las curvas Área vs Concentración.
: Ordenadas de las curvas Área vs Concentración.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
46
3.2.3.4. Obtención de las Concentraciones de Acetofenona y 1-
Feniletanol durante la Reacción de Reducción
Se realizaron reacciones de 100mL en matraces de 250mL de capacidad, a
las concentraciones de 2.5mM, 5mM, 10mM y 20mM de Acetofenona. De
las cuales, se tomaron muestras del medio de reacción con micropipetas, a
cada periodo de tiempo expresado en horas. Se consideró el siguiente
orden en la toma de muestras:
a) Muestra Blanco: Muestra obtenida del medio de reacción, antes de
agregar la biomasa centrifugada de biocatalizador de los tubos falcons,
y antes de agregar la cantidad medida de Acetofenona. El contenido de
éste matraz es el siguiente: solución buffer de fosfatos y cantidad
medida de co-sustrato.
b) Muestra Cero: Muestra obtenida al inicio de la reacción, es decir a las
cero horas de reacción, inmediatamente después de agregar la biomasa
de catalizador. El contenido de éste matraz antes de tomar la muestra
es el siguiente: solución buffer de fosfatos, cosustrato (Glucosa),
sustrato (Acetofenona), biomasa de biocatalizador (células libres de
Diplogelasinospora grovesii).
c) Muestras durante el proceso de bioreducción: Por motivos de
obtener una buena gráfica de la cinética de reacción, y con ello una
velocidad inicial más exacta y precisa de la reacción, se tomó la mayor
cantidad de muestras que definan bien el inicio de la reacción, es decir
entre muestra y muestra tenían poco tiempo de separación. Es por ello,
que mayormente las tomas de muestras tuvieron la siguiente frecuencia:
0horas, 2horas, 4horas, 5 o 6 horas, 8 horas, 12 horas, 24 horas(1día),
48horas(2días), 72horas(3días), y así sucesivamente hasta obtener
unas asíntotas en la cinética de reducción al llegar a concentraciones
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
47
mínima de sustrato y una concentración máxima de producto en el
tiempo.
El protocolo para trasladar las muestras en viales para su posterior
análisis por cromatografía líquida en el HPLC, fue el siguiente:
Primero, se agitó moderadamente el matraz para homogenizar la
concentración en todo el medio de reacción antes de tomar la muestra.
Luego, se tomó la muestra con una micropipeta y se colocó en un tubo
de vidrio previamente identificado, luego se centrifugó la muestra y se
tomó con una micropipeta la cantidad exacta de muestra del líquido
centrifugado y se trasvasó en un vial de HPLC. Para reacciones que
poseen una concentración inicial de Acetofenona fuera de escala de la
curva de calibración, tal es el caso de 20Mm de Acetofenona, en el vial
HPLC se diluyó a la mitad su concentración con Metanol puro, para así
tener mejores resultados al usar la ecuación de la curva de calibración.
Posteriormente, se empleó una jeringa para succionar todo el contenido
del vial, luego se filtró la muestra usando un microfiltro, y posteriormente
se trasvasó éste contenido a otro vial limpio e identificado. Y finalmente
se prosiguió con los análisis cromatográficos de cada vial identificado.
Así se evitó el paso de sedimentos que podrían dañar los microductos
del HPLC, lo cual también se refleja en la calidad de los cromatogramas.
Para el cálculo de las concentraciones totales de cada uno de los
analitos en el medio de reacción (matraz), se recurrió a las siguientes
ecuaciones, las cuales necesitan como dato: las áreas encontradas de
los cronogramas, pendiente y ordenada de las regresiones.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
48
La concentración de Acetofenona en el medio de Reacción, se calcula
por medio de la siguiente ecuación:
[3-7]
La concentración de 1-Feniletanol en el medio de Reacción, se calcula
por medio de la siguiente ecuación:
[3-8]
3.2.4. Metodología Experimental para el proceso de biotransformación
con Diplogelasinospora grovesii
3.2.4.1. Asentamiento de la colección
El mejor microorganismo seleccionado para el proceso de reducción del 1-
feniletanol fue el ascomiceto Diplogelasinospora grovesii IMI 171018 y se
almacenó en criotubos en una solución de glicerol-agua al 30%v/v a -5°C.
La taxonomía de esta cepa es la siguiente:
Superreino: Eucariota.
Reino: Hongos.
División: Ascomicota.
Subdivisión: Euascomicotina.
Clase: Pirenomicetos.
Orden: Sordariales.
Familia: Sordariaceae.
Género: Diplogelasinospora.
Especie: grovesi.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
49
3.2.4.2. Crecimiento del ascomiceto Diplogesinospora grovesii
El proceso de replicación del ascomiceto Diplogesinospora grovesii se
realizó con el fin de purificar, aislar, reproducir y mantener al
microorganismo con su mejor actividad enzimática. Éste proceso de
replicación consistió en una secuencia de etapas que son las siguientes:
a. Crecimiento de ascomicetos en medio sólido
Primero, se esterilizó el material que se necesitó para el trabajo con
microorganismos, así se evitó problemas de contaminación y bajos
rendimientos en los ensayos. Luego, se realizó las mediciones en peso de
los componentes para el medio de cultivo sólido. Y por tratarse de medio
sólido se midió la cantidad de Agar correspondiente al volumen de medio
preparado.
Cada placa PETRI necesitó de 20 ml de medio de cultivo. Por otro lado, se
elaboró la siguiente tabla de pesos necesarios para diferentes volúmenes
de medio de cultivo, manteniendo las concentraciones indicadas para el
medio HAGGS:
Tabla 3-1. Proporciones en peso de insumos para Medio HAGGS a diferentes
volúmenes.
MEDIO DE CULTIVO HAGGS
(Medio de cultivo para hongos: H-79, H-97, H-141)
Componente 20 ml 50 ml 100 ml 250 ml 500 ml 1 litro
1 Glicina 0,04 g 0,1 g 0,2 g 0,5 g 1 g 2 g 2 Almidón 0,4 g 1 g 2 g 5 g 10 g 20 g 3 Harina de Soya 0,12 g 0,3 g 0,6 g 1,5 g 3 g 6 g 4 Solución traza 0,2 ml 0,5 ml 1 ml 2,5 ml 5 ml 10 ml 5 Agar 0,6 g 1,5 g 3 g 7,5 g 15 g 30 g
Medio sólido: 1,2,3,4 y 5 Medio líquido: 1,2,3 y 4
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
50
Todo el trabajo se realizó en presencia de una llama, con el fin de
aprovechar la atmósfera inerte que se genera por el calor liberado.
En un vaso de precipitación se diluyó el medio de cultivo sólido con agua
destilada hasta el volumen de medio cultivo a preparar, la mezcla se
homogenizó con agitación magnética, luego se trasvasó el medio de cultivó
a un frasco para medio de cultivo, y se autoclavó a 121 ºC y 1 atm por 20
min. Después del autoclavado, se llevó el frasco de medio de cultivo a la
zona de cultivo, donde con el mechero encendido y se trasvasó 20ml por
cada placa PETRI, luego se esperó que enfríe y solidifique.
Posteriormente, las placas con medio sólido se sometieron a radiación UV,
con la finalidad de obtener un medio libre de contaminación microbiana
indeseable.
En simultáneo a la esterilización ultravioleta del medio de cultivo sólido, se
tomó uno de los criotubos almacenados en frío como fuente del
microorganismo para la siembra. El criterio que se siguió para la selección
del criotubo apropiado fueron: la coloración de los microorganismos en los
criotubos (color verde vistoso) y la fecha de replicación más actualizada del
microorganismo. Posteriormente a la selección del mejor criotubo, se utilizó
un asa de cultivo esterilizada para extraer cubitos del microorganismo de
los criotubos y se sembró en la parte central de las placas PETRI que
contienen medio de sólido. Toda operación se realizó con el mayor cuidado
posible tanto en limpieza, desinfección y se utilizó el medio estéril generado
por los alrededores de la llama.
Finalmente, el crecimiento se llevó a cabo dentro de una incubadora a
condiciones de 1 atm y 30°C por el lapso de tiempo de 7 días. Estas
condiciones de crecimiento fueron las mismas para el crecimiento en medio
líquido y para las condiciones de la reacción estudiada. Se mantuvo
siempre éstas condiciones de operación invariables, así se evitó problemas
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
51
en el crecimiento del microorganismo como estrés enzimático, inhibición y
alteración de la actividad enzimática.
b. Crecimiento de ascomicetos en medio líquido.
Primero, se desarrolló el crecimiento de ascomicetos en medio sólido.
Pasado los 7 días de crecimiento en medio sólido, se retiraron las placas
PETRI de la incubadora y se seleccionó aquellas donde el microorganismo
creció radialmente de manera uniforme, limpia y vistosa. Las placas que no
tuvieron éste crecimiento se descartaron del proceso y se autoclavaron
antes de ser desechadas.
Segundo, se preparó medio de cultivo líquido siguiendo el mismo
procedimiento de preparación del medio HAGGS, se pesó los insumos para
el correspondiente volumen necesitado, a excepción del Agar. Se preparó el
medio líquido en un vaso de precipitación, se agregó agua destilada hasta
el volumen requerido y se agitó hasta tener una solución uniforme.
Tercero, se trasvasó 50ml del medio de cultivo líquido por cada matraz de
250ml. Se mantuvo siempre ésta proporción de 1 a 5 entre el medio de
cultivo líquido y la capacidad del matraz para abastecer al microorganismo
de suficiente oxígeno en su crecimiento. Luego, se tapó las bocas de los
matraces que contienen el medio líquido con tapones de jebe limpios, y
luego fueron envueltos con papel aluminio. En caso de falta de tapones de
jebe, se utilizó algodón para tapar la boca del matraz y luego se envolvió
con papel aluminio.
Cuarto, se autoclavó a 121 ºC por 20 min,y después de ello, se sometió a
radiación ultravioleta, para una esterilización total del medio líquido.
Quinto, se seleccionó una de las placas PETRI que contiene al
microorganismo crecido para su posterior crecimiento en medio líquido, y
las demás se aprovecharon para guardar el microorganismo en nuevos
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
52
criotubos con glicerol al 30% v/v, y así se obtuvo una nueva colección de
ascomicetos.
Sexto, en un lugar estéril, en presencia de llama y con los cuidados
respectivos, se sembró los microorganismos en el medio líquido. Para eso,
se extrajo de la zona más alejada diametralmente de la placa PETRI cubitos
de medio sólido con microorganismo, esta fue la zona que contiene los
ascomicetos más jóvenes y puros en comparación a la zona central que
fueron los ascomicetos más viejos. Y rápidamente, los cubitos con el
microorganismo crecido se introdujeron dentro del matraz con medio líquido
de cultivo estéril. Para ésta extracción se empleó un sacabocado
previamente esterilizado enel autoclave, junto con los matraces que
contenían el medio de cultivo líquido.
Finalmente, cada matraz se identificó con un sticker, y se pusieron a crecer
en la Incubadora de agitación orbital de temperatura y velocidad de
agitación controlado. El microorganismo creció a 30°C, 1 atm con agitación
orbital controlada de 200 rpm.
3.2.4.3. Replicación del ascomiceto Diplogesinospora grovesii
El proceso de replicación del ascomiceto Diplogesinospora grovesii se
realizó con el fin de purificar, aislar, reproducir y mantener al
microorganismo con su mejor actividad enzimática. Este proceso de
replicación consistió en una secuencia de crecimientos en medio sólido en
placas PETRI. Primero, se sembró el ascomiceto en medio sólido, en 2 a 4
placas PETRI, y se usó el procedimiento de crecimiento en medio sólido.
Luego de alcanzar el total crecimiento de ascomiceto en las placas PETRI
dentro de la incubadora, se seleccionó la placa que tuvo el mejor
crecimiento uniforme radial, mejor coloración y libre de contaminación o de
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
53
algún imperfecto que puedo ser observado. Posteriormente, se volvió a
sembrar en 2 a 4 placas con medio sólido, y como fuente del ascomiceto,
se usó la placa anteriormente seleccionada. Se cuidó que las condiciones
de crecimiento en la incubadora sean constantes. Por último, al finalizar el
crecimiento del ascomiceto en las nuevas placas PETRI, se obtuvo un
ascomiceto listo para los ensayos y pruebas correspondientes, o para un
nuevo banco de ascomicetos, que luego se guardaron en frío, en criotubos
con glicerol al 30%v/v.
3.2.4.4. Curva de Crecimiento del ascomiceto Diplogelasinospora
grovesii
Se determinó la curva de crecimiento con la intención de encontrar el
tiempo adecuado para aprovechar su máximo contenido enzimático en el
proceso de reducción de la acetofenona.
Para ello, se realizó todo el proceso de replicación y crecimiento del
microorganismo, es decir primero se cultivó en medio sólido y luego se
cultivó microorganismo en medio líquido.
De la incubadora con agitación orbital que se encontraba a 30°C, 1 atm y
200 rpm, se empezó a retirar 3 matraces por cada día para el análisis
correspondiente.(Contando como día cero, el día en que se realizó la
siembra y se puso los matraces a crecer en dicha incubadora)
El contenido de cada matraz extraído de la incubadora se trasvasó a un
tubo falcon, y luego se separó por medio de una centrífuga todo el líquido
sobrenadante. La biomasa se colocó en una luna de reloj, se medió su peso
inicial, y luego se puso a secar en una estufa hasta que el peso de la
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
54
biomasa se volvió constante. Se secó la biomasa alrededor de un lapso de
tiempo de 5 horas a 120°C. Y por último, se medió el peso de final.
De ésta manera se realizó la construcción de la curva de crecimiento del
microorganismo, a través de una gráfica que relacionó el peso de biomasa
vs tiempo de crecimiento del ascomiceto.
3.2.4.5. Preparación del Medio de Reacción
a. Preparación de la Solución Buffer
Para la preparación de la solución amortiguadora fue requisito que el pKa
del ácido débil deba tener un valor cercano al valor del pH requerido, así se
aseguró que las concentraciones del ácido y la base conjugada fueran
similares. (Raymond Chang, Química, Pág. 659).
Se tiene:
[3-9]
[3-10]
[3-11]
Partiendo de lo anterior, en ésta investigación se utilizó como medio de
reacción la solución buffer de fosfato. En éste caso, se utilizóel ácido débil
ya que posee un cercano al valor del pH requerido,
igual a 6.5.
Por ello, se escogió la siguiente ecuación para la preparación de la solución
buffer fosfato:
[3-12]
De acuerdo a la ecuación Henderson-Hasselbalch:
[3-13]
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
55
[3-14]
Se utilizó como fuente la base conjugada , sal ácidade di-Sodio
hidrogenofosfato dodecahidratado (Na2HPO4.12H2O / HNa2O4P.12H2O) de
Peso Molecular igual a 358,14 g/mol y como fuente del ácido, la sal ácida
de Sodio dihidrogenofosfato monohidratado (NaH2PO4.H2O / H2NaO4P.H2O)
con Peso Molecular igual a 137,99 g/mol.
Para un litro de solución buffer fosfato de pH=6.5, y con dato experimental
de
[3-15]
Así para éste sistema amortiguador de fosfatos se disolvió 2.9845 gramos
de Na2HPO4.12H2O y 5.8975 gramos NaH2PO4.H2O en suficiente cantidad
de agua miliQ y se llevó la disolución a un volumen de 1Litro.
b. Preparación de la Solución Reacción
Se sembró el ascomiceto en placas PETRI con medio sólido y luego se
colocaron en una incubadora a 30°C y a presión atmosférica para su
crecimiento por 7 días. Luego, se preparó medio de cultivo líquido en
matraces de 250 mL, y con un asa Henle se extrajo una cantidad de cepas
jóvenes de los ascomicetos de las placas PETRI, especialmente de los
extremos diametrales. Y luego, estos matraces se pusieron en agitación
orbital de 200rpm y a 30°C por 7 días.
En la preparación de la solución reacción, se utilizó acetofenona grado
HPLC, y como medio de reacción se utilizó el buffer de fosfato con pH=6.5
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
56
Además se utilizó un co-sustrato de glucosa al 0.5%(p/v) con el fin de
regenerar la co-enzima (cofactor) del microorganismo.
Se realizaron ensayos a 2.5mM, 10mM y 20mM de Acetofenona en
matraces de 250 mL, con el objetivo de analizar la influencia de la
concentración de sustrato en el comportamiento biocatalítico del ADH-
deshidrogenasa del microorganismo y se analizaron sus cinéticas de
reacción.
Después de agregar en el matraz de 250mL, la cantidad de sustrato y co-
sustrato, se tomó una muestra de 1mL del medio preparado en un vial de
HPLC, y después de un análisis cromatográfico, se obtuvo el dato de
muestra cero.
Posteriormente, teniendo la biomasa crecida del microorganismo en medio
líquido, se centrifugó en tubos falcon todo el contenido de los matraces de
medio líquido con ascomiceto crecido, con el fin de quedarnos con la
biomasa, libre de líquido sobrenadante y se realizó tres lavados de la
biomasa con buffer de fosfatos preparado de pH = 6.5.
Se midió el peso inicial de la biomasa de ascomicetos, y después, esta
biomasa se adicionó a los matraces con medio de reacción. Finalmente se
siguió la cinética de la reacción en agitación constante de 200 RPM en un
agitador orbital a temperatura controlada de 30°C.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
57
3.2.5. Estudio Cinético de Bioreducción
Cuando se llevan a cabo reacciones con biocatalizadores, no todas las
moléculas que reaccionan se encuentran disponibles para conversión
inmediata.
La reacción tiene lugar únicamente después que los reactantes han sido
transportados al lugar de reacción, por lo que los procesos de transferencia
de materia pueden ejercer una gran influencia sobre la velocidad global de
conversión. Debido a que la velocidad de reacción depende generalmente
de la concentración de sustrato, cuando las concentraciones del sistema
varían, el análisis cinético llega a ser muy complejo. Para biocatalizadores
debe conocerse la concentración de sustrato existente en cada punto del
interior de la pared celular con el fin de calcular la velocidad local de
conversión. En la mayoría de los casos éstas concentraciones no pueden
medirse aunque afortunadamente pueden calcularse utilizando la teoría de
la difusión-reacción.
Debido a que las concentraciones varían en el interior de los
biocatalizadores, las velocidades locales de reacción también varían
dependiendo de la posición. Cada célula responde a la concentración de
sustrato según su posición con una velocidad de reacción determinada por
los parámetros cinéticos del biocatalizador. Esta velocidad local de reacción
se denomina velocidad verdadera o velocidad intrínseca. Las velocidades
de reacción: locales y verdaderas, son difíciles de medir en biocatalizadores
sin alterar las condiciones de reacción. Sin embargo, es posible, medir la
velocidad global de reacción para el catalizador entero. En un sistema
cerrado, la velocidad global de conversión por reacción, hecho que en los
sistemas heterogéneos se denomina velocidad observada. Cuando los
niveles de sustrato cambian lentamente durante la reacción, los datos de
velocidad observada pueden obtenerse recogiendo muestras de la mezcla
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
58
de reacción a diferentes tiempos y analizando la variación de la
concentración de sustrato.
3.2.5.1. Velocidad inicial de reacción observada (Vo)
Las cinéticas de muchas reacciones biológicas son bien de orden cero, de
primer orden o una más compleja denominada cinética de Michaelis-
Menten. Para determinar la velocidad observada de cada biotransformación
se siguió el siguiente procedimiento: a erlenmeyers de 250mL se añadieron
100mL de tampón fosfato potásico pH= 6.5. Luego se añadió un peso
determinado de células libres, el co-sustrato (glucosa), y a continuación el
substrato a la concentración establecida (acetofenona), llevándose a cabo
la biotransformación a 30°C y 200 rpm. A diferentes tiempos de reacción se
sacaron muestras del medio de reacción en un tubo de ensayo, se agitó en
un Agitador Vortex por 10s, se centrifugó a 4500 rpm durante 2 minutos.
Del centrifugado se tomó 1mL en un vial; se añadió a éstas 0.5mL de
metanol, luego se filtró y luego la muestra filtrada se trasvasó a otro vial
para su posterior análisis cromatográfico de líquido en HPLC.
Para el análisis de las concentraciones del sustrato en el medio de
reacción, se empleó la curva de calibración del sustrato a diferentes
concentraciones dependiendo de las áreas obtenidas en el análisis
cromatográfico.
La concentración (milimolar) del producto formado al cabo de un tiempo (t)
de reacción se determinó por medio de la siguiente relación:
[3-16]
Donde:
= Concentración milimolar de producto al cabo de un tiempo t.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
59
Los datos así obtenidos se representaron en un plano XY, llevándose en
abcisas el tiempo de reacción(t) expresado en horas y en ordenadas la
concentración milimolar del producto al cabo de t horas de reacción.
Luego, dada la tendencia de los puntos graficados (y(t) aumenta
monótonamente desde y = 0 cuando t = 0 hasta una asíntota y = F a
valores elevados de t), con la ayuda del programa EXFIT del paquete
informático SIMFITV v5.5 ed.18, se realizó una regresión por mínimos
cuadrados ponderados, usando una seria de sumas exponenciales en
orden consecutivo (modelo c). Este programa ajusta y(t) a partir de una
serie de sumas como se indica a continuación:
a) A1.exp[-k1t] + A2.exp[-k2t] + ………… An.exp[-knt]
b) A1.exp[-k1t] + A2.exp[-k2t] + ………… An.exp[-knt] + C
c) B1.{ 1 - exp[-k1t] } + B2. {1 - exp[-k2t]} + … Bn. {1 - exp[-knt]}
d) B1.{ 1 - exp[-k1t] } + B2. {1 - exp[-k2t]} +… Bn. {1 - exp[-knt]} + C
El número(n) puede ser fijo o variable si se desean ajustar modelos en
secuencia de orden creciente. Los modelos (a y c) y (b y d) sólo varían en la
forma de los parámetros. A menudo sólo se requieren n = 1 (como en
nuestro caso) pero, si los datos son abundantes y de alta calidad tendría
sentido probar n = 2 ó n = 3. La función que mejor ajusta los datos
experimentales es de la forma.
y(t) =B . [ 1 – exp (-k.t)] [3-17]
La derivada de esta función es la expresión de la velocidad observada y
tiene la siguiente:
dy / dt = B . kexp (-k . t) [3-18]
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
60
La velocidad inicial quedará determinada cuando t tienda a cero, por lo que
la expresión anterior quedaría de la forma:
dy / dt = V0= B . k [3-19]
Donde:
B= Máxima concentración de producto alcanzada.
K = Constante aparente de velocidad.
La máxima conversión alcanzada se determinó por la relación:
Xmax(%)=(B/[S0]).100 [3-20]
El tiempo en que se alcanza la máxima conversión (Tmax), se determinó de
la siguiente manera. Dada la función cinética: y (t) =B. [1 – exp (-k.t)] su
valor asintótico en el límite es B. Si llamamos ɛ a la diferencia entre el valor
de B y el valor de la función por definición de límite se debe cumplir que
dado un: ɛ>0, debemos hallar un Tmax>0 tal que: t>Tmax entonces |y(t)-B|<ɛ.
Lo que indica que el valor absoluto de la diferencia entre la función y la
asíntota debe ser menor que ɛ (error). Resolviendo esta última expresión
tenemos: t > (1/k).ln(B/ɛ), con lo cual queda demostrado el límite para todo t
>Tmax. Por ejemplo, si queremos hallar con un error del 5% el tiempo de
máxima conversión de una reacción donde la máxima concentración de
producto alcanzado es 10Mm y el valor de k=0.28 h-1 tenemos: Tmax
=(1/0.28).ln(10/0.05)=19h.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
61
3.2.5.2. Orden de reacción (n)
Dadas las tendencias de las cinéticas antes mencionadas, el orden de
reacción se determinó experimentalmente de la siguiente manera. Se
supuso una cinética de pseudo-primer orden de la forma V0 = k.[S0]n. Se
realizó la reacción test con la cepa a dos concentraciones de sustrato
diferentes ([S1]=10Mm y [S1]=5Mm). Se determinó las velocidades iniciales
de ambas reacciones según:
V1= k.[S1]n [3-21]
V2= k.[S2]n [3-22]
Del cociente de ambas expresiones tenemos:
(V1/V2)=([S1]/[S2])n [3-23]
Tomando logaritmos y despejando el orden de reacción, n, tenemos:
n = [ln(V1/V2)]/[ln([S1]/[S2])] [3-24]
Si n=1, la suposición es correcta y la cinética de ésta reacción seria de
pseudo-primer orden.
3.2.5.3. Otras variables de cinética de reacción
a. Conversión: La conversión porcentual del sustrato, X (%) de cada
muestra al cabo de un tiempo (t), se calculó de la siguiente ecuación:
[3-25]
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

III. PARTE EXPERIMENTAL
62
b. Rendimiento: El rendimiento de la reacción de seudo-primer orden de
reacción se calcula con la siguiente ecuación:
[3-26]
c. Productividad: La productividad se encontró en base a la cantidad de
catalizador(biomasa), que se necesitó para obtener el producto deseado
en el tiempo que demandó la reacción, y se calcula de la siguiente
manera:
[3-27]
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
63
IV.RESULTADOS Y DISCUSIONES
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
64
IV. RESULTADOS Y DISCUSIONES
4.1. CUALIFICACIÓN DE LOS INSTRUMENTOS ANALÍTICOS
En esta tesis se cualificaron equipos e instrumentos utilizados para la
determinación de los parámetros cinéticos de la reacción de reducción de la
acetofenona; así como también se validaron los métodos de análisis, cuyos
procedimientos normalizados de trabajo (PNT) se encuentran en los
anexos. El propósito de cualificar un equipo y validar un método de análisis
es evitar y/o minimizar errores en las mediciones, que pueden afectar la
precisión y exactitud de los análisis. A modo de ejemplo se muestran las
cualificaciones de los principales equipos y sus métodos de análisis:
4.1.1. Cualificación de Balanza Analítica.
Tener una balanza analítica cualificada es importante porque ayuda a
confiar y obtener buenas mediciones de los pesos de las diferentes
sustancias utilizadas en los trabajos de investigación. En esta tesis se
cualificó la balanza Marca: Sartorious AG Germany CPA 224S, a fin de
conocer el estado del equipo y el grado de desviación de las mediciones
debido al uso, con la intención de tomar las medidas correctivas que nos
permitan tener resultados confiables.
La cualificación de la balanza analítica, se realizó en dos rangos de
medición. El primer rango comprendido entre 0 a 1g. útil para la preparación
de soluciones con bajas concentraciones (milimolar); y el segundo rango
comprendido entre 0 y 100g. para la preparación de medios de cultivo de
microorganismos.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
65
a. Primer Rango de Cualificación: 0 a 1g
La cualificación en este rango se realizó tomando los siguientes puntos: 0g.,
0.125g., 0.250g., 0.500g., 0.750g., 1.000g, con 4 repeticiones por cada una
de las mediciones realizadas. Las mediciones se realizaron midiendo
volúmenes de agua destilada (ρ = 1g/ml) en viales, empleando micropipetas
cualificadas. La regresión lineal se llevó a cabo empleando el método de
mínimos cuadrados del programa SIMFIT (Autor: W. G. Bardsley,
Universidad de Manchester, U. K.)
Figura 4-1. Variación del Peso: Rango 0 a 1g.
Cualificación de la Balanza Sartorius
y = 1.0086x - 0.0002
R2 = 1
0
0.2
0.4
0.6
0.8
1
1.2
0 0.2 0.4 0.6 0.8 1 1.2
Peso Teóricos (g)
Pe
sp
Re
ale
s (
g)
En la Figura 4-1, se observa que la regresión lineal tiene la siguiente
ecuación matemática:
En esta ecuación se puede observar que el valor de la pendiente es
aproximadamente uno (1), lo que nos indica que no existe variación
considerable entre los valores reales y los valores mostrados en la balanza
(teóricos), razón por la cual podemos inferir que la balanza Sartorious AG
Y = (1.0086 ± t.sx) X – (0.0002 ± t.sy)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
66
Germany CPA 224S se encuentra en buen estado y mostrando valores
fiables.
b. Segundo Rango de Cualificación: 0 a 100g
La cualificación en este rango se realizó tomando los siguientes puntos: 0g.,
25g., 50g., 75g., 100g., con 4 repeticiones por cada una de las mediciones
llevadas a cabo. Al igual que en el caso anterior la regresión lineal fue
llevada a cabo por el método de mínimos cuadrados con repeticiones,
utilizando el programa estadístico SIMFIT (Autor: W. G. Bardsley,
Universidad de Manchester, U. K.) como se muestra en la siguiente
figura:
Figura 4-2. Variación del Peso: Rango 0 a 100g
Cualificación de la Balanza Sartorius
y = 0.9929x - 0.0341
R2 = 1
0
20
40
60
80
100
120
0 20 40 60 80 100 120
Peso Teóricos (g)
Pe
sp
Re
ale
s (
g)
En la Figura 4-2, se observa la regresión lineal tiene la siguiente ecuación
matemática:
En esta ecuación se puede apreciar que la pendiente de la recta sufrió una
pequeña variación respecto a la pendiente del rango 0-1g; sin embargo,
esta variación no afecta la precisión de la balanza la cual puede
Y = (0.9929 ± t.sx) X – (0.0341 ± t.sy)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
67
considerarse que emite aún valores confiables; los que no podrían ser
considerados como tales cuando el valor de la pendiente fuera inferior a
0,97.
4.1.2. Cualificación de Espetrofotómetro Ultravioleta-Visible
Los analitos que intervienen en la determinación de los parámetros
cinéticos en esta tesis son: sustrato: acetofenona y producto: 1-feniletanol;
los cuales absorben en el rango ultravioleta (UV), debido a la
deslocalización de electrones que presentan ambos compuestos. En ese
sentido el equipo utilizado para determinar la longitud de onda a la cual se
produce la máxima absorción fue el espectrofotómetro UV-VIS Hewlett
Packard Mod 8452 A, Diode array; el cual se cualificó utilizando una
solución de benzoato de sodio como patrón validador del Espectrofotómetro
UV a tres diferentes longitudes de onda: 236nm, 254nm y 270nm. (Espectro
UV: 210nm a 310nm).
La cualificación y la validación del espectrofotómetro UV-Visible, nos ayuda
a determinar de manera confiable la longitud de onda del espectro UV (nm),
a la cual se analizará el sustrato y producto de la reacción, y con ello
programar el detector UV del cromatógrafo HPLC, y así finalmente, realizar
el seguimiento de la reacción y la cuantificación de los analítos.
A partir de una solución madre de benzoato de sodio 0.528 mM, se
determinó las absorbancias de diferentes diluciones con agua ultrapura
(miliQ). Estas diluciones se analizaron haciendo uso de la cubeta de
análisis (cuarzo). Las curvas de absorbancias (Lammbert-Beer) a 236nm,
254nm y 270nm fueron las siguientes:
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT
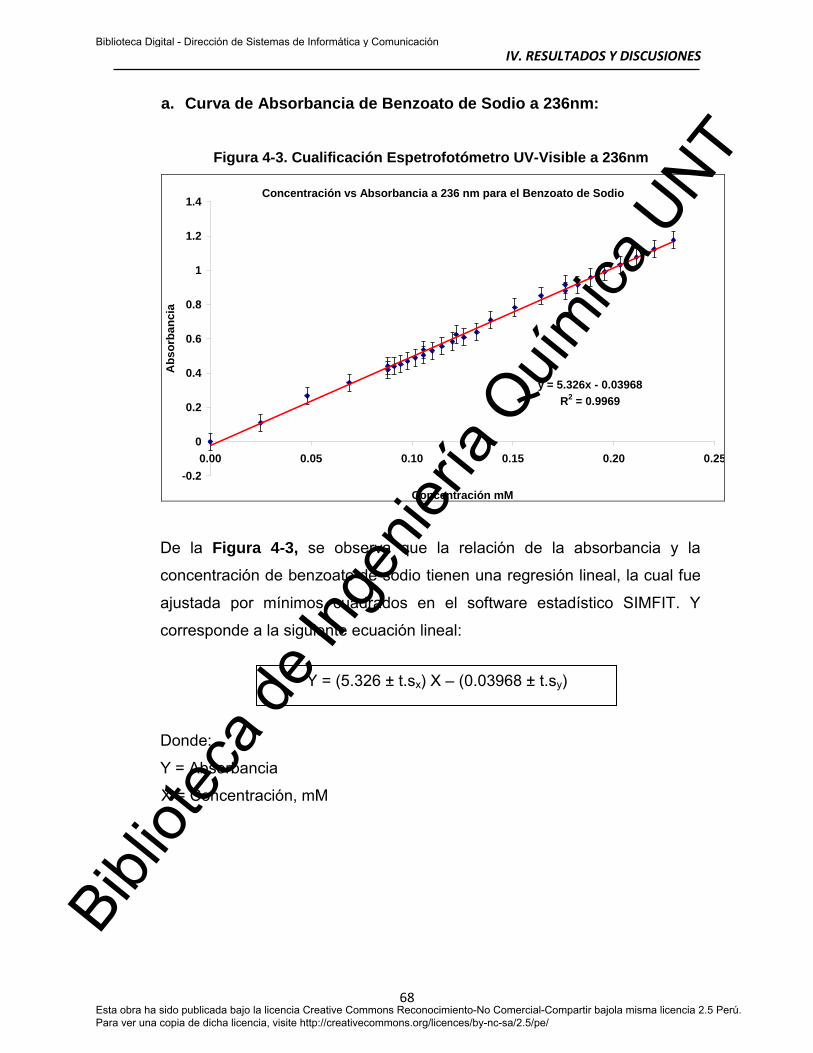
IV. RESULTADOS Y DISCUSIONES
68
a. Curva de Absorbancia de Benzoato de Sodio a 236nm:
Figura 4-3. Cualificación Espetrofotómetro UV-Visible a 236nm
Concentración vs Absorbancia a 236 nm para el Benzoato de Sodio
y = 5.326x - 0.03968
R2 = 0.9969
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0.00 0.05 0.10 0.15 0.20 0.25
Concentración mM
Ab
so
rban
cia
De la Figura 4-3, se observa que la relación de la absorbancia y la
concentración de benzoato de sodio tienen una regresión lineal, la cual fue
ajustada por mínimos cuadrados en el software estadístico SIMFIT. Y
corresponde a la siguiente ecuación lineal:
Donde:
Y = Absorbancia
X = Concentración, mM
Y = (5.326 ± t.sx) X – (0.03968 ± t.sy)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
69
b. Curva de Absorbancia de Benzoato de Sodio a 254nm:
Figura 4-4. Cualificación Espetrofotómetro UV-Visible a 254nm
Concentración vs Absorbancia a 254 nm para el Benzoato de Sodio
y = 0.8252x - 0.02265
R2 = 0.9975
-0.05
0.05
0.15
0.25
0.35
0.45
0.55
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Concentración mM
Ab
so
rban
cia
De la Figura 4-4, se observa que la relación de la absorbancia y la
concentración de benzoato de sodio tiene una regresión lineal, la cual fue
ajustada por mínimos cuadrados en el software estadístico SIMFIT. Y
corresponde a la siguiente ecuación lineal:
Donde:
Y = Absorbancia
X = Concentración, mM
Y = (0.8252 ± t.sx) X – (0.02265 ± t.sy)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
70
c. Curva de Absorbancia de Benzoato de Sodio a 270nm:
Figura 4-5. Cualificación Espetrofotómetro UV-Visible a 270nm
Concentración vs Absorbancia a 270 nm para el Benzoato de Sodio
y = 0.6307x - 0.0208
R2 = 0.9967
-0.05
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Concentración mM
Ab
so
rban
cia
De la Figura 4-5, la regresión lineal ajustada por mínimos cuadrados en el
software estadístico SIMFIT, corresponde a la siguiente ecuación lineal:
Donde:
Y = Absorbancia
X = Concentración, mM
Y = (0.6307 ± t.sx) X – (0.02080 ± t.sy)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
71
4.1.3. Cualificación de Cromatógrafo de Líquidos de Alta Performancia
4.1.3.1. Cualificación de la Bomba Cuaternaria
La cualificación de la bomba consiste en la comprobación de la exactitud y
precisión del caudal suministrado por la bomba a través de los 04 canales
del equipo cromatográfico. El procedimiento requiere utilizar la columna
estándar utilizada habitualmente en el equipo, (por ejemplo: C-8 o C-18) ya
que el sistema debe estudiar su funcionamiento como un todo durante un
análisis cromatográfico. La operación consiste en hacer pasar un solvente a
través de uno de los canales (en este caso H2O ultrapura) con caudales
nominales de 0,50; 1,0; 1,5 y 2,0 mL/min, con 5 repeticiones cada caudal,
y realizar la medición del solvente recogido a la salida del sistema.
a. Cualificación del Canal A
Figura 4-6. Cualificación del Canal A
Cualificación del Canal "A"
y = 0.9935x + 0.0101
R2 = 1
0
1
2
3
4
5
0 1 2 3 4
Flujo Experimental (mL/min)
Flu
jo T
eó
ric
o (
mL
/min
)
Desviación media (d) = Σdi/n=0.0174/6 = 0.0029
Desviación estándar (Sd) = [Σ(di – d)2/(n – 1)]1/2 = [6,49*10-4 /(6 – 1)]1/2
Sd = 0.0114
“t” de student calculada (tcal) = [d*(n)1/2]/Sd = [0.0029*(6)1/2]/0.0114
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
72
tcal = 0,623
De tablas el valor de “t” de student tabulada al 95% (ttab) y para n=6, se
tiene: T tab = 2,015
Como T cal<T tab, entonces se concluye que los valores obtenidos de
manera experimental son estadísticamente iguales a los valores teóricos
expresados por el instrumento.
b. Cualificación del Canal B
Figura 4-7. Cualificación del Canal B
Cualificación del Canal "B"
y = 0.9986x + 0.0059
R2 = 1
0
1
2
3
4
5
0 1 2 3 4
Flujo Experimental (mL/min)
Flu
jo T
eó
ric
o (
mL
/min
)
Desviación media (d) = Σdi /n = 0.0528/6 = 0.0088
Desviación estándar (Sd) = [Σ(di – d)2/(n – 1)]1/2 = [1,506*10-3/(6–1)]1/2
Sd = 0.0174
“t” de student calculada (tcal) = [d*(n)1/2]/Sd = [0.0088*(6)1/2]/0.0174
tcal = 1,2388
De tablas el valor de “t” de student tabulada al 95%(ttab) y para n=6, se
tiene: T tab = 2,015
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT
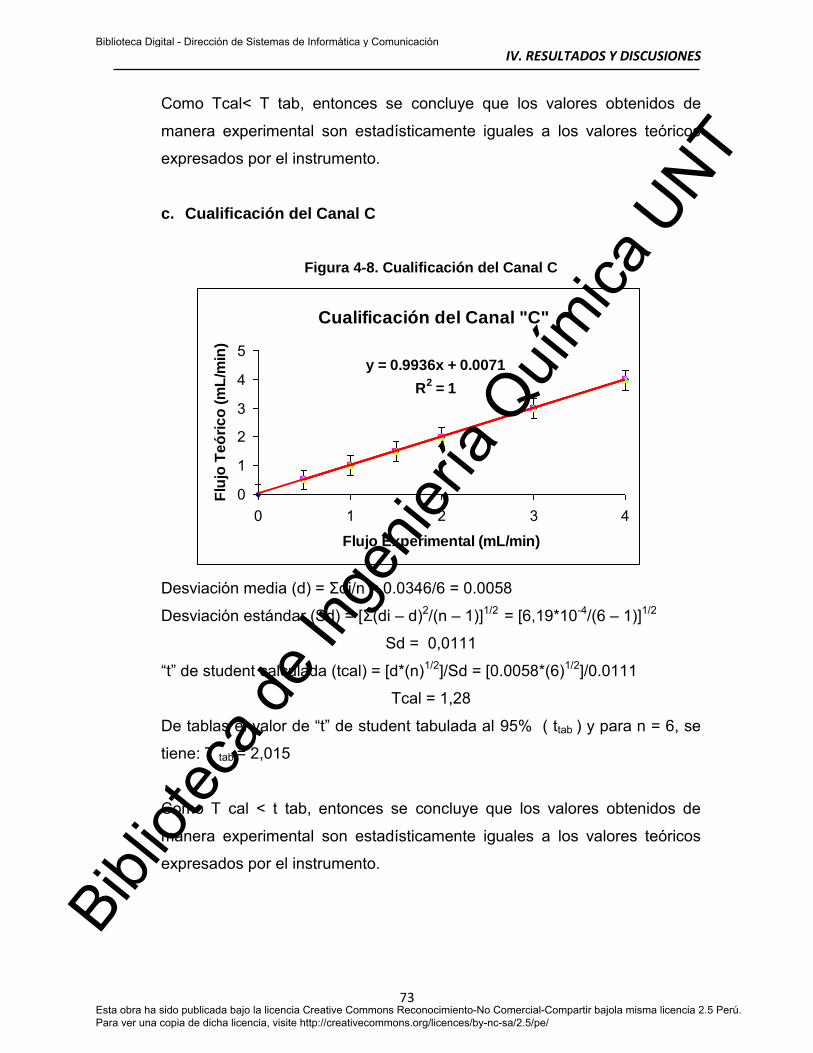
IV. RESULTADOS Y DISCUSIONES
73
Como Tcal< T tab, entonces se concluye que los valores obtenidos de
manera experimental son estadísticamente iguales a los valores teóricos
expresados por el instrumento.
c. Cualificación del Canal C
Figura 4-8. Cualificación del Canal C
Cualificación del Canal "C"
y = 0.9936x + 0.0071
R2 = 1
0
1
2
3
4
5
0 1 2 3 4
Flujo Experimental (mL/min)
Flu
jo T
eó
ric
o (
mL
/min
)
Desviación media (d) = Σdi/n = 0.0346/6 = 0.0058
Desviación estándar (Sd) = [Σ(di – d)2/(n – 1)]1/2 = [6,19*10-4/(6 – 1)]1/2
Sd = 0,0111
“t” de student calculada (tcal) = [d*(n)1/2]/Sd = [0.0058*(6)1/2]/0.0111
Tcal = 1,28
De tablas el valor de “t” de student tabulada al 95% ( ttab ) y para n = 6, se
tiene: T tab = 2,015
Como T cal < t tab, entonces se concluye que los valores obtenidos de
manera experimental son estadísticamente iguales a los valores teóricos
expresados por el instrumento.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
74
d. Cualificación del Canal D
Cualificación del Canal "D"
y = 0.9936x + 0.0071
R2 = 1
0
1
2
3
4
5
0 1 2 3 4
Flujo Experimental (mL/min)
Flu
jo T
eó
ric
o (
mL
/min
)
Figura 4-9. Cualificación del Canal D
Desviación media (d) = Σdi/n = 0.0220/6 = 0.0037
Desviación estándar (Sd) = [Σ(di – d)2 /(n – 1)]1/2 = [6.23*10-4 /(6 – 1) ]1/2
Sd = 0.0112
“t” de student calculada (tcal) = [d*(n)1/2]/Sd = [0.0037*(6)1/2]/0.0112
tcal = 0,809
De tablas el valor de “t” de student tabulada al 95%(ttab) y para n = 6, se
tiene: T tab = 2,015
Como Tcal < t tab, entonces se concluye que los valores obtenidos de
manera experimental son estadísticamente iguales a los valores teóricos
expresados por el instrumento.
4.1.3.2. Cualificación del Sistema de Inyección
La cualificación del sistema de inyección implica evaluar dos aspectos
fundamentales del sistema de inyección automático, que nos permite tener
la seguridad sobre los resultados obtenidos en cuanto a su reproducibilidad
y precisión: precisión del inyector y magnitud del efecto memoria (efecto
“carry over”).
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
75
a. Precisión del Inyector
Se llevó a cabo la evaluación de la precisión del sistema de inyección
manual mediante una secuencia de cinco inyecciones de una disolución
estándar de naftaleno (0.100 mg/ml en acetonitrilo) empleando las
siguientes condiciones cromatográficas:
Columna: Teknokroma Mediterranea Sea C18 - 5 µm*15*0,46
Fase móvil : Metanol/agua (45/55 v/v)
Flujo de la fase móvil : 1.00 ml/min.
Longitud de onda del detector : 254 nm.
Volumen de inyección : 10 µl.
Temperatura del horno de columna : 30 ºC.
Presión del equipo : 39 bar.
Tabla 4-1. Inyecciones de una disolución estándar de naftaleno (0.100 mg/ml en acetonitrilo)
De la Tabla 4-1, se observa que las desviaciones del tiempo de retención y
áreas son pequeñas, esto significa que el equipo reporta datos de mayor
precisión y exactitud.
Nº muestra tiempo retención (min) Área
1 4,693 2640,5
2 4,705 2538,8
3 4,703 2630,1
4 4,707 2612,3
5 4,707 2628,7
Promedio 4,703 2610,1
Desv. Estándar 0,0058 41,1055
Limite Superior (95%) 4,710 2661,13
Límite inferior (95%) 4,696 2559,07
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
76
b. Magnitud del efecto memoria (efecto “carry over”)
Este parámetro nos permite determinar la contaminación producida por la
inyección anterior, si no se limpia correctamente el “loop” del inyector
automático después de cada inyección.
Para determinar este parámetro se inyectó la disolución estándar y luego se
realizó una inyección en blanco (como blanco usamos la fase móvil) y se
observó si en el cromatograma aparece señal en el tiempo de retención de
compuesto de la disolución estándar (acetofenona).
A continuación se repitió el ensayo pero realizando la inyección con lavado,
y se verificó si hay algún cambio en la señal.
- Efecto Carry Over (Sin limpiar el sistema)
Tabla 4-2. Análisis cromatográfico de solución de acetofenona
Nº muestra Tiempo
retención (min)
Área (mV)
1 4,708 2460,8
2 4,712 2452,1
3 4,733 2523,8
Promedio 4,718 2478,9
Desv. Estándar 0,0134 39,1271
Limite Superior (95%) 4,7513 2576,105
Límite inferior (95%) 4,6847 2381,695
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
77
Tabla 4-3. Efecto carry over sin limpiar el sistema – Blanco: Fase Móvil
Nº blanco Tiempo
retención (min)
Área % Área
1 4,736 24,4 0,99
2 4,754 15,8 0,64
3 4,760 29,3 1,16
Promedio 4,750 23,167 0,93
Desv. Estándar 0,0125 6,8340 0,2634
Limite Superior (95%) 4,7811 40,145 1,7223
Límite Inferior (95%) 4,7190 6,1890 0,1422
De la Tabla 4-2 y 4-3, se observa que después de cada análisis
cromatográfico de la solución de acetofenona, queda pequeñas partículas
del analito en el sistema de inyección, las cuales se detectan en el
cromatograma al analizar una muestra blanco que contenga la misma fase
móvil. Este efecto de carry over, afecta la certeza de los análisis posteriores
si no se realiza un buen lavado interno del sistema de inyección.
- Efecto Carry Over (Con limpieza del sistema)
Tabla 4-4. Efecto carry over con lavado del sistema - Muestra: acetofenona
Nº muestra Tiempo
retención (min)
Área (mV)
1 4,741 2686,8
2 4,741 2688,5
3 4,743 2799,3
Promedio 4,742 2724,87
Desv. Estándar 0,0012 64,4668
Límite Superior(95%) 4,7451 2885,03
Límite Inferior(95%) 4,7382 2564,71
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
78
Tabla 4-5. Efecto Carry Over (utilizando opción inyección con lavado)
Nº blanco Tiempo
retención (min)
Área (mV) % Área
1 0 0 0
2 0 0 0
3 0 0 0
Promedio - - -
Desv. Estándar - - 0,0000
Limite Superior - - 0,0000
Límite Inferior - - 0,0000
De la Tabla 4-4 y 4-5, se observa que al realizar una lavado interno del
sistema de inyección después de cada análisis cromatográfico del analito,
se eliminan en su totalidad cualquier resto de partículas de analito que
puedan afectar los resultados de los siguientes análisis cromatográficos.
4.1.3.3. Cualificación del Detector UV-Visible
La cualificación del detector tiene como objetivo comprobar la linealidad de
la detección a una longitud de onda determinada (por ejemplo: 254 nm)
dentro de un rango de absorbancia que se considera normal (0.0 - 2.0 D.O).
Se llevó a cabo la evaluación del detector mediante una secuencia de tres
inyecciones de cinco disoluciones estándar de acetofenona (0.050, 0.150,
0.250, 0.350 y 0.500 mg/ml respectivamente en metanol/agua):
Para cada disolución estándar de acetofenona se calculó el valor promedio,
la desviación estándar y el error en la medida (superior e inferior) de las
áreas de los picos.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
79
Se representó los valores de concentración de acetofenona (mg/L) frente a
los valores de área de pico y se calculó la recta de regresión que pasa por
los puntos experimentales para comprobar la linealidad del detector.
Tabla 4-6. Cualificación del UV-Visible
nº disolución Standard
(mg/L) Acetofenona tR (min) Area (mV)
1 50 4,685 1073,0 1 50 4,688 1085,3 1 50 4,685 1113,4 2 150 4,741 3848,2 2 150 4,740 3730,7 2 150 4,745 3746,2 3 250 4,748 5426,4 3 250 4,747 5609,4 3 250 4,478 5529,7 4 350 4,676 9542,6 4 350 4,677 9316,3 4 350 4,678 9449,9 5 500 4,679 12619,1 5 500 4,680 12624,8 5 500 4,676 12831,2
Figura 4-10. Cualificación del UV-Visible
Cualificación de UV-Visible
y = 26.02x - 245.81
R2 = 1
-5002000450070009500
1200014500
0 100 200 300 400 500Concentración (mg/L)
Are
a (
mV
)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
80
De la Tabla 4-6 y de la Figura 4-10, se obtiene un coeficiente de
correlación con una buena linealidad entre la concentración estándar de
acetofenona y el área reportada por cromatografía.
4.1.3.4. Cualificación de la Columna
Las columnas envejecen con el tiempo por el sangrado de la fase
estacionaria, mal uso, suciedad, etc. Por ello este ensayo es vital y debería
de realizarse todos los días antes de empezar a trabajar.
La evaluación de la columna se llevó a cabo mediante una secuencia de
tres inyecciones de una disolución estándar que contenga una serie de
compuestos similares pero con distinto valor de coeficiente de extinción
molar a la longitud de onda elegida (v.g. 254 nm).
La disolución estándar preparada fue la siguiente: (1) benceno (1 µl/ml), (2)
tolueno (1 µl/ml) y (3) naftaleno (1 µl/ml) en acetonitrilo. Se inyectan 20 µl y
se debe asegurar que trabaja en condiciones óptimas de limpieza del
mismo.
Al no tener la hoja del fabricante preparamos la disolución estándar
indicada anteriormente y los resultados obtenidos los usaremos como
referencia del estado de la columna en evaluaciones posteriores.
Se emplearon las siguientes condiciones cromatográficas:
Teknokroma Mediterranea Sea C18 - 5 µm*15*0,46
Fase móvil : agua/acetonitrilo (35/65 v/v).
Flujo de la fase móvil : 0.700 ml/min.
Longitud de onda del detector : 254 nm.
Volumen de inyección : 20 µl.
Temperatura del horno de columna : 40 ºC.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
81
En la Tabla 4-7, se muestra el tiempo de retención, el área de pico, el factor
de capacidad (k´), el número de platos teóricos (N), el número de platos
teóricos por metro de columna (N/m), la altura de pico teórico reducido (h) y
el factor de asimetría (As) del último pico (naftaleno) para cada una de las
inyecciones.
Tabla 4-7. Cualificación de la columna
Los elevados valores de los platos teóricos y los valores cercanos al uno (1)
del factor de asimetría demuestran el buen funcionamiento de la columna
para nuestros análisis.
Pinchazo tR
(min) Área (mV) K´
Nº platos teóricos
Nºpl.t./m H As
1 6,244 10047,2 5,06 4240 282666,667 0,00000354 0,83 2 6,24 10086,3 5,06 4235 282333,333 0,00000354 0,83 3 6,244 9890,4 5,06 4334 288933,333 0,00000346 0,81
Promedio 6,243 10007,967 5,060 4269,667 284644,444 0,00000351 0,823 Desv.est. 0,002 103,676 0,000 55,770 3718,024 0,00000005 0,012 Lím. Sup. 6,250 10264,566 5,060 4408,219 293881,273 0,00000363 0,853 Lím. Inf. 6,236 9749,434 5,060 4131,115 275407,615 0,00000339 0,793
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
82
4.2. VALIDACIÓN DEL MÉTODO DE ANÁLISIS.
4.2.1. Longitud de Onda Óptima de análisis.
En esta tesis la cuantificación de los analitos se llevaron a cabo de manera
simultánea, es decir, se aprovechó la misma muestra para determinar la
concentración de acetofenona y 1-feniletanol. El inconveniente de este
procedimiento consiste en que ambos analitos absorben a diferentes
longitudes de onda, razón por la cual, se procedió a determinar el punto
isosbéstico de estos analitos (punto de máxima absorción de ambos
analitos juntos) a fin de resolver este inconveniente.
Los espectros UV-Visible de la acetofenona y del 1-Feniletanol a bajas
concentraciones (0.1 mM) se superpusieron para encontrar sus puntos
isobésticos con sus respectivas longitudes de onda. Éste estudio se
desarrolló con el propósito de encontrar una longitud de onda adecuada
para el análisis cromatográfico, ya que el Cromatógrafo de Líquidos de Alta
Performancia – HPLC tiene un detector UV. Por lo que se usó el
Espectrofotómetro UV-Visible para encontrar las longitudes de onda en las
cuales ambos espectros se cruzan. Las concentraciones de las soluciones
estándar de los analitos fueron las siguientes:
[Acetofenona]= 0,1008 mM
[1-Fenil Etanol]= 0.09742 mM
En todo el espectro de absorción de las soluciones estandar, se obtuvo los
siguientes cruces de ambos espectros al superponerse uno a otro:
- A 218 nm Absorbancia = 0.588 (Punto Isobéstico)
Tabla 4-8. Coeficientes de Extensión Molecular a 218nm
Coeficiente de Extensión Molecular L/cm.mol
Acetofenona 5833.33 1-Fenil etanol 6035.60
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
83
- A 270 nm Absorbancia = 0.266 Valle
De la Tabla 4-8, la longitud de onda adecuada es una longitud de onda en
la cual los picos cromatográficos detectados de la acetofenona y 1-
feniletanol reportan datos que reproducen las cantidades más exactas del
contenido de Acetofenona y 1-Feniletanol en las muestras tomadas en el
transcurso de las reacciones con células libres de ascomicetos. Por eso se
buscó una longitud de onda alrededor de 218nm que genere una
cromatografía con buena Resolución para la Acetofenona y el 1-Feniletanol.
A una longitud de onda igual a 217nm se obtiene el mejor resultado como
se muestra en la Tabla 4-9:
Tabla 4-9. Coeficientes de Extensión Molecular a 217nm
Coeficiente de Extensión Molecular L/cm.mol
Acetofenona 6789.7 1-Fenil etanol 4461.4
4.2.2. Validación de Método de Análisis Cromatográfico.
En ésta etapa se desarrollaron una serie de pruebas del punto isobéstico y
diferentes concentraciones de la fase móvil. Las mejores condiciones
cromatográficas encontradas para el análisis de la Acetofenona y del 1-
Feniletanol fueron las siguientes:
Columna: Teknokroma Mediterránea Sea C18 (µm * 15 * 0,46)
Fase móvil : Metanol/Agua (45/55).
Caudal fase móvil : 1,0mL/min.
Longitud de onda Detector : 217,4 nm.
Volumen de inyección : 10 µL.
Temperatura termostato : 30 ºC.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
84
4.2.3. Curvas de Calibración para Acetofenona y 1-Feniletanol.
a. Linealidad:
Una vez determinadas las condiciones adecuadas de análisis por HPLC, se
prosiguió a elaborar las curvas de calibrado; para esto se tuvieron que
preparar diferentes concentraciones de sustrato y producto entre valores de
0 a 10mM obteniendose los siguientes resultados como de muestran a
continuación.
Tabla 4-10. Curvas de Calibración para Sustrato y Producto en HPLC
Analito Curvas de Calibración Regresión
Acetofenona Area = 628.77 [mM] + 25.344 R² = 0.999 1-Feniletanol Area = 1065.2 [mM] + 254.08 R² = 0.999
En la Tabla 4-10, se muestran las ecuaciones correspondientes a las
curvas de calibración, con muy buena correlación lineal, que se utilizaron
para la determinación de las concentraciones de sustrato y producto en el
cromatógrafo HPLC.
b. Repetitividad:
El ensayo de repetitividad sirve para determinar las variaciones de error
sistemático, precisión y exactitud de las curvas de calibrado tanto para el
sustrato como para el producto. Es por ello que se tomó una muestra de
sustrato y producto (con tres repeticiones) y se calculó la desviación
estándar y su coeficiente de variación.
Tabla 4-11. Ensayo de Repetitividad para la Acetofenona (sustrato)
Standard Mm
Replica 1 Replica 2 Replica 3 Area Promedio
s %CV Tr Area Tr Area Tr Area
5.376 8.880 3394.670 8.881 3398.213 8.881 3400.086 3397.65633 2.75 0.08
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
85
Tabla 4-12. Ensayo de Repetitividad para la 1-Feniletanol (producto)
Standard Mm
Replica 1 Replica 2 Replica 3 Area Promedio
s %CV Tr Area Tr Area Tr Area 5.195 7.787 5903.147 7.787 5898.627 7.782 5891.656 5897.81006 5.79 0.10
En la Tabla 4-11 y en la Tabla 4-12, se observa que existen una buena
precisión de las curvas de calibrado ya que las mediciones obtenidas en el
ensayo de repetitividad poseen un coeficiente de variación menor al 2%.
c. Límites de Cuantificación y Detección:
El límite de detección se define como la mínima cantidad de analito en una
muestra que puede ser detectada pero no necesariamente cuantificada. El
límite de cuantificación es la mínima cantidad de analito en una muestra
que puede ser determinada cuantitativamente con una precisión y exactitud
adecuada. A continuación se muestran los límites de cuantificación y
detección obtenidos en esta tesis para la determinación de los parámetros
cinéticos de la reacción de reducción de la acetofenona.
Tabla 4-13. Límites de Cuantificación y Detección
Analitos Acetofenona 1-Feniletanol Límite de Cuantificación, mM. 0.79 0.60
Límite de Detección, Mm. 0.26 0.20
Los valores mostrados en la Tabla 4-13, son importantes para la
cuantificación de los analitos ya que evitan tener en cuanta en los análisis
falsas señales y/o picos equivocados que no permitirían tener un adecuado
seguimiento a la cinética de la reacción de reducción de la acetofenona.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
86
4.3. PROCESO DE BIOREDUCCIÓN CON Diplogelasinospora grovesii
4.3.1. Screening Taxonómico
Una vez constituida la colección de partida conformada por 9
microorganismos (3 bacterias, 3 levaduras y 3 hongos filamentosos) y
determinados los medios de cultivo adecuados para el crecimiento de los
microorganismos, se dio inicio al proceso de selección o screening
taxonómico. Tras consultar la bibliografía se seleccionó como reacción test,
para evaluar la actividad reductasa de cada uno de los microorganismos, la
reducción de la acetofenona; por ser una reacción sencilla que puede ser
analizada por HPLC para poder tener un criterio cuantitativo de las
actividades que presentan nuestros microorganismos. El objetivo de este
screening fue determinar qué microorganismos eran capaces de catalizar la
biotransformación de nuestro sustrato (acetofenona) sin que aparecieran
reacciones secundarias.
En la Tabla 4-14 se muestran los resultados del screening; con los
microorganismos de la colección de partida.
Tabla 4-14. Screening de microorganismos de la colección de partida.
Grupo Microorganismo Código Conversión (%)
Bacteria Rhodococcus rhodochrous DSMZ 11097 5
Bacteria Ralstonia eurotropha DSMZ 2839 8
Bacteria Rhodococcus erythropolis DSMZ 8424 10
Levadura Williopsis saturnus NCYC 2313 12
Levadura Williopsis californica CBS 2158 16
Levadura Pachysolen tannophilus NCYC 1597 13
Ascomiceto Phialemonium curvatum CBS 505.82 30
Ascomiceto Coniochaeta velutina CBS 981.68 50
Ascomiceto Diplogelasinospora grovesii IMI 171018 80
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
87
De la tabla anterior se puede deducir que de la colección estudiada, los
ascomicetos son el grupo taxonómico que presentan mayor actividad
reductasa mientras que las bacterias y levaduras son poco activos frente a
la reacción test propuesta.
En biotransformaciones catalizadas por células enteras la principal
limitación viene dada por el crecimiento no reproducible de la biomasa. Así,
para comprobar la reproducibilidad de los resultados obtenidos y evitar
seleccionar biocatalizadores con comportamientos poco reproducibles, se
llevó a cabo la reacción test por triplicado con los microorganismos que
presentan la mejor actividad reductasa.
Tabla 4-15. Reproducibilidad del screening.
Grupo Microorganismo Conversión (%)
s %CV 1º 2º 3º X
Ascomiceto Phialemonium curvatum 28 31 30 30 1,00 3
Ascomiceto Coniochaeta velutina 50 38 41 43 6,24 15
Ascomiceto Diplogelasinospora grovesii 82 80 81 81 1,00 1 X = conversión promedio; s = desviación estándar; %CV = coeficiente de variación.
De la Tabla 4-15, se deduce que los resultados obtenidos con el
ascomiceto Coniochaeta velutina son poco reproducibles al presentar datos
muy dispersos como lo demuestra el elevado valor del coeficiente de
variación (%CV=15). La reproducibilidad de los datos mejora con
Phialemonium curvatum, pero el sustrato presenta baja conversión
(X=30%); sin embargo, con Diplogelasinospora grovesii se obtiene la mejor
conversión de sustrato (X=81%) con el coeficiente de variación más bajo
(%CV=1%), razón por la cual se escogió a Diplogelasinospora grovesii
como el mejor candidato para realizar el estudio cinético de la reducción de
la acetofenona.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
88
con Diplogelasinospora grovesii se obtiene la mejor conversión de sustrato
(X=81%) con el coeficiente de variación más bajo (%CV=1%),
Diplogelasinospora grovesii IMI 171018 es un microorganismo que
muestra gran actividad como biocatalizador de células enteras para la
reducción de compuestos carbonílicos. Se trata de ascomiceto de
crecimiento muy rápido que desarrolla un color verde oscuro característico
aislado en Japón (Ver Figura 4-11). Existen referencias bibliográficas sobre
su capacidad para producir un compuesto de características
inmunosupresoras (Fujimoto y cols, 1998), pero nunca antes ha sido
descrito por sus características como biocatalizador.
Figura 4-11. D. grovesii en medio sólido HAGGS
4.3.2. Curva de Crecimiento
Es bien conocido que la facilidad de crecimiento de la cepa microbiana es
una de las características que llevan a seleccionar un microorganismo para
su posterior uso en biotransformaciones. Las células aisladas, cultivadas
en un volumen finito de medio de cultivo apropiado (reactor discontinuo),
van utilizando los nutrientes que tienen disponibles con la mayor eficacia y
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
89
rapidez que pueden, sintetizando sus propios componentes celulares y
dividiéndose en cuanto han podido duplicar su masa y su material genético.
Dado que en esta tesis intentamos llevar a cabo reacciones con células
libres, es importante la determinación del tiempo de crecimiento al cual se
tiene la mayor producción de enzima que interviene en el proceso; según la
bibliografía consultada, esto se logra cuando las células alcanzan la fase
estacionaria de crecimiento, esto se debe probablemente a que mientras
hay nutrientes en el medio el microorganismo crece incrementando la
biomasa y sólo al llegar a la fase estacionaria de crecimiento se inicia la
biotransformación (Cappaert y Larroche, 2004).
En la Tabla 4-16 se muestran los valores numéricos para la curva de
crecimiento del ascomiceto D. grovesii, expresados en peso seco.
Tabla 4-16. Peso Seco de D. grovesii para curva de crecimiento
Tiempo Peso Seco Promedio s %CV Días Gramos gramos
4 0,1860 0,1856 0,1837 0,19 0,0012 0,7% 5 0,2651 0,2639 0,2604 0,26 0,0024 0,9% 6 0,4264 0,4299 0,4242 0,43 0,0029 0,7% 7 0,4550 0,4579 0,4567 0,46 0,0015 0,3% 8 0,4548 0,4577 0,4566 0,46 0,0015 0,3% 9 0,2180 0,2171 0,2203 0,22 0,0017 0,8% 11 0,1809 0,1827 0,1847 0,18 0,0019 1,0% s = desviación estándar; %CV = coeficiente de variación;
Para observar mejor los resultados se construyó un gráfico de barras con
los tiempos de crecimiento del ascomiceto.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT
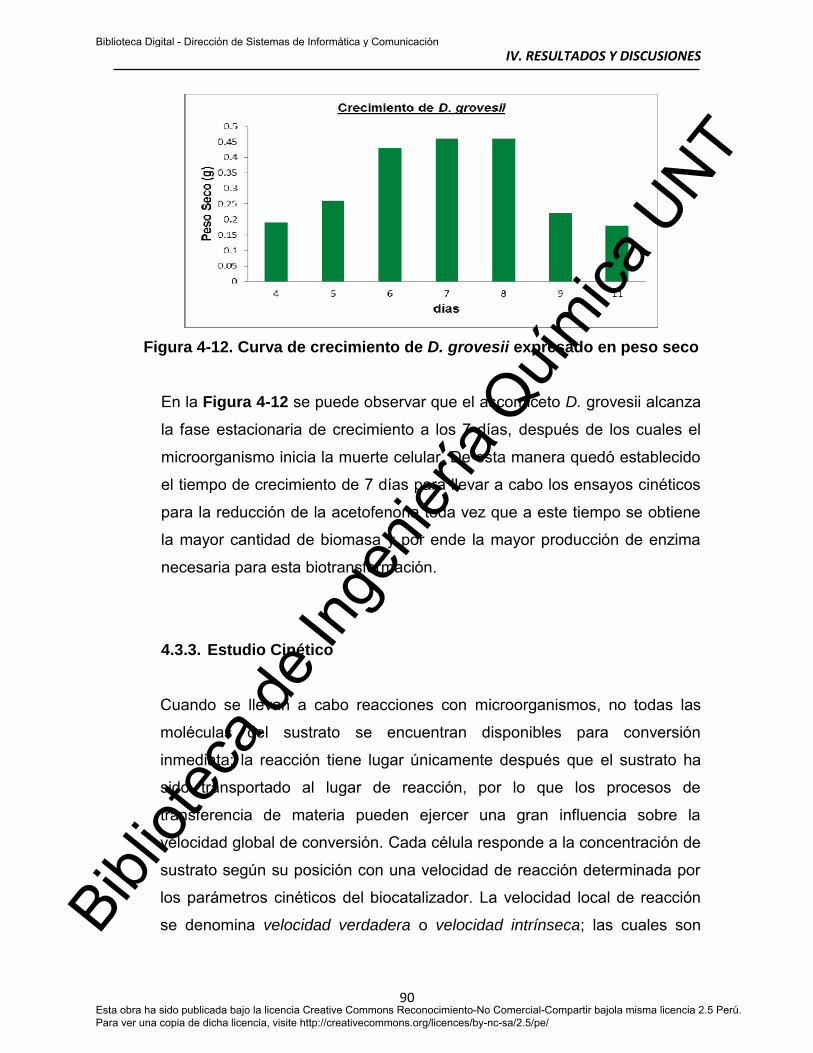
IV. RESULTADOS Y DISCUSIONES
90
Figura 4-12. Curva de crecimiento de D. grovesii expresado en peso seco
En la Figura 4-12 se puede observar que el ascomiceto D. grovesii alcanza
la fase estacionaria de crecimiento a los 7 días, después de los cuales el
microorganismo inicia la muerte celular. De esta manera quedó establecido
el tiempo de crecimiento de 7 días para llevar a cabo los ensayos cinéticos
para la reducción de la acetofenona toda vez que a este tiempo se obtiene
la mayor cantidad de biomasa y por ende la mayor producción de enzima
necesaria para esta biotransformación.
4.3.3. Estudio Cinético
Cuando se llevan a cabo reacciones con microorganismos, no todas las
moléculas del sustrato se encuentran disponibles para conversión
inmediata; la reacción tiene lugar únicamente después que el sustrato ha
sido transportado al lugar de reacción, por lo que los procesos de
transferencia de materia pueden ejercer una gran influencia sobre la
velocidad global de conversión. Cada célula responde a la concentración de
sustrato según su posición con una velocidad de reacción determinada por
los parámetros cinéticos del biocatalizador. La velocidad local de reacción
se denomina velocidad verdadera o velocidad intrínseca; las cuales son
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
91
difíciles de medir en microorganismos. Sin embargo, es posible, medir la
velocidad global de reacción para el catalizador entero. En un sistema
cerrado, la velocidad de desaparición de sustrato en el seno del líquido
debe ser igual a la velocidad global de conversión por reacción, hecho que
en los sistemas heterogéneos se denomina velocidad observada. Cuando
los niveles de sustrato cambian lentamente durante la reacción, los datos
de velocidad observada pueden obtenerse recogiendo muestras de la
mezcla de reacción a diferentes tiempos y analizando la variación de la
concentración de sustrato. A modo de ejemplo se muestra en la siguiente
tabla la disminución de la concentración inicial de sustrato acetofenona (7.2
mM).
Tabla 4-17. Resultados del análisis por HPLC del consumo de acetofenona.
Tiempo ACETOFENONA Área Promedio
s %CV [mM] vial
[mM] matraz H Area1 area2 area3
0 1537.28000 1536.99889 1537.29876 1537.19255 0.168 0.01% 2.40 7.21 2 1449.38973 1449.36789 1450.00123 1449.58628 0.360 0.02% 2.27 6.80 4 1371.74578 1372.10023 1371.74806 1371.86469 0.204 0.01% 2.14 6.42 8 1238.45886 1239.20012 1238.45788 1238.70562 0.428 0.03% 1.93 5.79 12 1132.85038 1132.87569 1132.85043 1132.85883 0.015 0.00% 1.76 5.28 24 928.21452 928.22568 928.23618 928.22546 0.011 0.00% 1.44 4.31 48 772.30403 772.38762 772.34721 772.34629 0.042 0.01% 1.19 3.56 72 731.39665 732.78765 732.73635 732.30688 0.789 0.11% 1.12 3.37 96 722.18183 722.18765 722.19876 722.18941 0.009 0.00% 1.11 3.32 144 718.89793 718.89876 718.78965 718.86211 0.063 0.01% 1.10 3.31 192 718.68278 718.68753 718.69876 718.68969 0.008 0.00% 1.10 3.31 240 718.66356 718.67543 718.68546 718.67482 0.011 0.00% 1.10 3.31 264 718.66345 718.67876 718.98744 718.77655 0.183 0.03% 1.10 3.31
Pendiente: 628.77; ordenada: 25.344; muestra: 1500 uL; dilución: 3.
En la Tabla 4-17 se puede observar que las áreas del sustrato en los
cromatogramas presentan poca dispersión como lo demuestran los bajos
valores de la desviación estándar y del coeficiente de variación, lo que da
solidez al valor promedio tomado para la determinación de la disminución
de la concentración de la acetofenona en función del tiempo de reacción.
Para observar mejor los resultados obtenidos, en la figura 4-03 se muestra
la cinética de consumo del sustrato acetofenona catalizada por D. grovesii.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
92
f ( t ) = [ A. exp (- K . t) ] + C
d f(t) / dt = A. exp (-K.t) . -K
Figura 4-13. Cinética de consumo de Acetofenona 7,2 mM, a 30 ºC y 200
rpm catalizada por D. grovesii (26 g de peso húmedo).
En la Figura 4-13 se puede observar que los datos experimentales tienen
una tendencia de decaimiento exponencial, razón por la cual todos los
ensayos de consumo de sustrato fueron ajustados con el programa EXFIT
del paquete estadístico SIMFIT v5.5 ed 18 a un modelo de decaimiento
exponencial de la forma:
Donde:
K = Constante aparente de velocidad de consumo de sustrato. (h-1)
C = Valor asintótico cuando el tiempo decrece indefinidamente. (mM)
A = Gradiente de concentración entre el estado inicial y el tiempo t. (mM)
La derivada de la función f(t) es la expresión de la velocidad observada de
consumo de sustrato y toma la siguiente forma:
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
93
d f(t) / dt = Vo = - A.K
La velocidad inicial del consumo de sustrato quedará determinada cuando t
tienda a cero; por lo que la expresión para calcular la velocidad inicial
observada de consumo de sustrato queda establecida como:
De igual manera cuando los niveles de producto cambian lentamente
durante la reacción, los datos de velocidad observada pueden obtenerse
recogiendo muestras de la mezcla de reacción a diferentes tiempos y
analizando la variación de la concentración de producto. A modo de ejemplo
se muestra en la siguiente tabla el incremento de la concentración de
producto 1-Feniletanol.
Tabla 4-18. Resultados del análisis por HPLC de la obtención del 1-Feniletanol.
Tiempo 1-FENILETANOL Area Promedio
s %CV [mM] vial
[mM] matraz H Area1 area2 area3
0 0.00000 0.00000 0.00000 0.00000 0.000 0.00% 0.00 0.00 2 308.4392 308.43786 308.65464 308.51057 0.125 0.04% 0.05 0.15 8 360.26123 360.26875 360.27893 360.26964 0.009 0.00% 0.10 0.30 12 409.66798 409.67875 409.66785 409.67153 0.006 0.00% 0.15 0.44 24 544.46389 544.46987 544.46785 544.46720 0.003 0.00% 0.27 0.82 48 762.44756 762.45674 762.48776 762.46402 0.021 0.00% 0.48 1.43 72 926.07894 926.87454 926.46373 926.47240 0.398 0.04% 0.63 1.89 96 1048.87654 1048.87567 1048.87645 1048.87622 0.000 0.00% 0.75 2.24 144 1210.56474 1210.67484 1210.98464 1210.74141 0.218 0.02% 0.90 2.69 192 1301.27375 1301.27746 1301.27565 1301.27562 0.002 0.00% 0.98 2.95 240 1352.53847 1352.54783 1352.53748 1352.54126 0.006 0.00% 1.03 3.09 264 1369.03127 1369.03284 1369.03245 1369.03219 0.001 0.00% 1.05 3.14 285 1369.02456 1369.02874 1369.02457 1369.02596 0.002 0.00% 1.05 3.14
Pendiente: 1065.2; ordenada: 254.08; muestra: 1500 uL; dilución: 3.
En la Tabla 4-18 se puede observar nuevamente una buena precisión en
los resultados obtenidos debido a los bajos valores de la desviación
estándar y del coeficiente de variación. Para observar mejor estos
resultados se muestra en forma gráfica la cinética de obtención de producto
1-Feniletanol catalizada por D. grovesii.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
94
y ( t ) = B . [ 1 - exp (- K . t) ]
d y(t) / dt = B. K. exp (-K.t)
Figura 4-14. Cinética de obtención del producto 1-feniletanol, a 30 ºC y
200 rpm catalizada por D. grovesii (26 g de peso húmedo).
En la Figura 4-14 se puede observar que los datos experimentales tienen
una tendencia de crecimiento exponencial, razón por la cual todos los
ensayos de obtención de producto fueron ajustados con el programa EXFIT
del paquete estadístico SIMFIT v5.5 ed 18 a un modelo de crecimiento
exponencial de la forma:
Donde:
K = Constante aparente de velocidad de aparición de producto. (h-1)
B = Máxima concentración de producto alcanzada. (mM)
La derivada de la función y(t) es la expresión de la velocidad observada de
aparición de producto y toma la siguiente forma:
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
95
d y(t) / dt = Vo = B.K
La velocidad inicial de aparición del producto quedará determinada cuando t
tienda a cero; por lo que la expresión para calcular la velocidad inicial
observada de aparición de producto queda establecida como:
4.3.4. Orden de Reacción
Se define como orden de reacción a la suma de los exponentes a los que
van elevados las concentraciones de substrato en la expresión cinética
global y que suele indicar el número de moléculas implicadas en el paso
limitante de la velocidad de reacción. En nuestro caso sería “n” en la
siguiente expresión: V = k [S]n. Este parámetro se determina o bien
mediante el ajuste de los datos cinéticos a las ecuaciones integradas de
velocidad o bien realizando experimentos con concentraciones múltiplos de
un valor. Se analizó la relación entre la velocidad inicial de reacción y la
concentración inicial de sustrato para llevar a cabo la reducción de la
acetofenona catalizada con células de D. grovesii en estado de no
crecimiento a 200 rpm, pH = 6.5 y a T=30ºC. El análisis de datos cinéticos
de la reacción test se realizó con el programa EXFIT del paquete SIMFIT
v5.5, y en todos los casos se ajustaron con buena correlación (R2) a un
modelo de decaimiento exponencial de la siguiente forma: f(t) = A . exp(-k.t)
+ C; lo que indicaría que la reacción sigue una aparente cinética de primer
orden. Dadas las tendencias antes mencionadas y conocidas las
velocidades iniciales de reacción a dos concentraciones de sustrato se
calculó el orden de reacción según la siguiente ecuación:
n = [ ln ( V1 / V2 ) ] / [ ln ( [ S1 ] / [ S2 ] ) ]
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
96
Los resultados de dos experimentos realizados para la determinar el orden
de reacción se muestran en la siguiente tabla.
Tabla 4-19: Orden de la Reacción de Reducción.
Exp. Cat Sustrato Vo Orden
G mM mM / h
1º 8.4749 2.90 0.03 1.2
9.5288 10.40 0.14
2º 26.0890 7.47 0.22 1.0
26.3571 18.70 0.55
En la Tabla 4-19 se puede observar que en cada uno de los dos
experimentos realizados el orden de la reacción de reducción de la
acetofenona catalizada por D. grovesii es prácticamente de orden uno
(pseudo primer orden), lo que justificaría el ajuste realizado de los datos
experimentales según el modelo de la ecuación de primer orden.
Asimismo se puede observar que la velocidad inicial de reacción en cada
uno de los experimentos realizados aumenta con el incremento de la
concentración de sustrato manteniendo cantidades similares de
biocatalizador, sin embargo, cabe resaltar que la disminución de la
velocidad inicial de reacción al incrementar la concentración inicial de
sustrato de 7.47 mM a 10.40 mM se debe al incremento de la cantidad de
biocatalizador empleada en la primera. Para poder evaluar el efecto del
biocatalizador en la cinética de la reacción de reducción de la acetofenona
se procedió a determinar las constantes específicas de la reacción, como se
indica en el siguiente a continuación.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
97
4.3.5. Parámetros Cinéticos
a. Parámetros Cinéticos del Sustrato Acetofenona:
En la siguiente tabla se muestran los principales parámetros cinéticos de la
reacción de reducción de la acetofenona catalizada por D. grovesii en
condiciones de no crecimiento. Los valores de las constantes A, K y C
fueron obtenidos con el programa EXFIT del paquete informático SIMFIT
v5.5 con buena correlación (R2=0.999).
Tabla 4-20: Parámetros cinéticos del Sustrato Acetofenona.
ACETOFENONA
Exp. Cat Sustrato A K C kesp Vo
G Mm Mm h-1 mM h-1 / gcat mM / h
1º 8.4749 2.9 2.018 0.01452 0.878 0.0050 0.03 9.5288 10.4 5.995 0.02291 4.386 0.0022 0.14
2º 26.0890 7.2 3.906 0.05678 3.308 0.0079 0.22 26.3571 18.7 7.423 0.07427 10.510 0.0040 0.55
En la Tabla 4-20 se puede observar que en cada uno de los dos
experimentos realizados la constante específica de velocidad kesp disminuye
ligeramente con el incremento de la concentración inicial de sustrato
manteniendo cantidades similares de biocatalizador, lo que puede deberse
a que el incremento de la concentración de sustrato origine toxicidad a la
célula causando inhibición de la enzima que cataliza el proceso de
reducción. Asimismo, podríamos afirmar que el valor de la constante
específica del consumo de acetofenona es del orden de 10-3 y que la
acetofenona inhibe al biocatalizador a concentraciones superiores a 7.2
mM.
b. Parámetros Cinéticos del Producto 1-Feniletanol:
En la siguiente tabla se muestran los principales parámetros cinéticos de la
obtención del 1-Feniletanol en la reacción catalizada por D. grovesii en
condiciones de no crecimiento. Los valores de las constantes B y K fueron
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
98
obtenidos con el programa EXFIT del paquete informático SIMFIT v5.5 con
buena correlación (R2=0.999).
Tabla 4-21. Parámetros cinéticos del Producto de Reacción 1-Feniletanol.
1-FENILETANOL
Exp. Cat Sustrato B K kesp Vo
G Mm Mm h-1 h-1 / gcat mM / h
1º 8.4749 2.9 1.323 0.0039 0.00047 0.005 9.5288 10.4 3.294 0.0039 0.00041 0.013
2º 26.0890 7.2 3.267 0.0120 0.00046 0.039 26.3571 18.7 1.464 0.0105 0.00040 0.015
En la Tabla 4-21 se puede observar que en el primer experimento realizado
para la obtención del 1-Feniletanol la velocidad inicial de reacción aumenta
con el incremento de la concentración de sustrato manteniendo cantidades
similares de biocatalizador, mientras que la constante específica de
velocidad no varía sustancialmente. En el segundo experimento se observa
que la velocidad inicial de la reacción disminuye a medida que incrementa
la concentración de sustrato, lo que podría deberse a que el producto de
reacción también inhiba la enzima que cataliza el proceso; mientras que la
constante específica de velocidad no varía y mantiene el mismo valor que el
primer experimento. Asimismo, podríamos afirmar que el valor de la
constante específica de la obtención del 1-Feniletanol es del orden de 10-4 y
que la enzima se inhibe a concentraciones de sustrato superiores a 7.2 mM
manteniendo casi constante la velocidad global de generación de 1-
Feniletanol.
De la comparación de las Tablas 4-18 y 4-19 podemos afirmar que la
velocidad específica de consumo del sustrato acetofenona es
aproximadamente diez veces mayor que la velocidad específica de
obtención del producto 1-feniletanol.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

IV. RESULTADOS Y DISCUSIONES
99
4.3.6. Productividad de la Biotransformación
En la siguiente tabla se muestran los resultados obtenidos de productividad
a fin de evaluar la actividad específica del biocatalizador D. grovesii en la
reacción de reducción de la acetofenona
Tabla 4-22. Productividad del 1-Feniletanol
Acetofenona 1-Feniletanol P x 10
4
Exp. Sustrato Cat X T Y T
mM G % H % H mM / gcat . h
1º 2.9 8.4749 70 255 46 827 1.9 10.4 9.5288 58 209 32 1068 3.2
2º 7.2 26.0890 54 77 45 347 3.6 18.7 26.3571 44 67 8 322 1.7
X=conversión; Y=Rendimiento; t=tiempo; P=Productividad.
En la Tabla 4-22 se puede observar que en cada uno de los experimentos
realizados la conversión y el rendimiento disminuyen al incrementar la
concentración de sustrato, lo que reafirma cierto grado de toxicidad para la
célula por parte del sustrato y producto de la reacción. Asimismo se puede
observar que a concentraciones bajas del sustrato acetofenona (2.9mM) y
altas (18.7mM) se obtienen bajas productividades por gramo de catalizador;
mientras que a concentraciones moderadas de sustrato acetofenona
(7.2mM y 10.4mM) las productividades mejoran sustancialmente.
La diferencia porcentual entre la conversión y el rendimiento de la reacción
hace suponer que la célula no sólo utiliza el sustrato acetofenona en la
reacción sino que consume parte de él para su aprovechamiento en el
incremento de la biomasa celular.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

V. CONCLUSIONES
100
V. CONCLUSIONES
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

V. CONCLUSIONES
101
V. CONCLUSIONES
De la labor realizada en esta tesis, sobre la determinación de parámetros
cinéticos en la obtención de 1-Feniletanol empleando Diplogelasinospora
grovesii como biocatalizador en medio acuoso; podemos extraer, a nuestro
juicio, las siguientes conclusiones:
5.1 La cualificación de los equipos e instrumentos de laboratorio y la validación
de los métodos de análisis disminuyen considerablemente los errores en las
mediciones, que pueden afectar la precisión y exactitud de los análisis para
la determinación de los parámetros cinéticos en la obtención del 1-
Feniletanol.
5.2 De la búsqueda jerarquizada (screening) a partir de la colección propuesta,
para la obtención del 1-Feniletanol por reducción biocatalítica de la
acetofenona, las bacterias y levaduras presentan escasa actividad
reductasa, mientras que los ascomicetos tienen la mayor actividad
reductasa; así mismo se encontró un nuevo microorganismo,
Diplogelasinospora grovesii, que es un ascomiceto que lleva a cabo la
reacción con crecimiento reproducible, buenos rendimientos sin obtención
de productos secundarios.
5.3 El sustrato, acetofenona, es biotransformado satisfactoriamente cuando se
añade en la fase estacionaria de su crecimiento (7 dias) lo que indica que
cuando las células dejan de reproducirse son metabólicamente más activas
para llevar a cabo la biosíntesis del 1-Feniletanol.
5.4 La cinética del proceso de obtención del 1-Feniletanol empleando
Diplogelasinospora grovesii como biocatalizador en medio acuoso es de
pseudo primer orden como lo demuestran los resultados de los parámetros
cinéticos obtenidos; asimismo, la enzima se inhibe a concentraciones de
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

V. CONCLUSIONES
102
sustrato (acetofenona) superiores a 7.2 mM manteniendo casi constante la
velocidad global de generación de 1-Feniletanol.
5.5 No todo el sustrato consumido por la célula (acetofenona) es utilizado en la
obtención del 1-Feniletanol debido a que parte de él es aprovechado para
incrementar la biomasa celular del ascomiceto.
5.6 El proceso de obtención del 1-feniletanol utilizado en esta tesis es
sostenible debido a que se lleva a cabo en medio acuoso, a temperatura
ambiente y presión atmosférica evitándose la utilización de disolventes
orgánicos contaminantes y catalizadores metálicos como el hidruro de litio y
aluminio y el borohidruro sódico que generan productos concomitantes en
la reacción.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

VI. BIBLIOGRAFÍA
104
VI.BIBLIOGRAFÍA
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

VI. BIBLIOGRAFÍA
105
VI. BIBLIOGRAFÍA
1. Domènech, Xavier. “Química Verde”. 2005. 1era Edición. Rubens Editorial.
2. Quezada, M.A. “Reacciones estereoselectivas de reducción de cetonas y
oxidación de alcoholes empleando células enteras inmovilizadas”. Tesis Doctoral. 2006. UCM. España.
3. Alcalde M., Ferrer M., Plou F.J. y Ballesteros A. “Environmental biocatalysis:
from remediation with enzymes to novel green process”. Trends in Biotechnol.,2006. 24, 281-287.
4. Sheldon R.A. y van Rantwijk F. “Biocatalysis for sustainable organic synthesis”. Aust. J. Chem. 2004.57, 281-289
5. Schmid A., Hollmann F., ByungPark J. y Bühler B. “The use of enzymes in
chemical industry in Europe”. Curr. Opin.Biotechnol.2002. 13, 359-366
6. Faber K: “Biotransformations in Organic Chemistry”. 2001. 4º Edición. Springer – Verlag.
7. Faber K, Kroutil W. “New Enzymes for biotransformations”. Cur. .OpinionChem. Biol. 2005. 9, 181-187.
8. Faber K. “Biotransformations in Organic Chemistry”. 5th Edition. Springer-Verlag.2004.
9. Parachin, N.S.; Carlquist, M.; Gorwa-Grauslund, M. F. “Bioreduction” In
Encyclopedia of Industrial Biotechnology: Bioprocess, Bioseparation, and Cell Technology., Flickinger, M.C., Ed. John Wiley and sons, Inc.: 2010.
10. Nayori, R. “Asymmetric Catalysis: Science and opportunities” (Nobel lecture). AngewChem. Int Edit2002, 41(12), 2008-2022.
11. Ma, J.-A.; Cahard, D. “Toward Perfect Catalytic Asymmetric Synthesis: Dual
Activation of the Electrophile and the Nucleophile. Angew. Chem. Int. Edit. 2004, 43(35), 4566-4583.
12. Burkhardt, E. R.; Matos, K. “Boron Reagents in Process Chemistry:
Excellent Tools for Selective Reductions” Chem Rev 2006, 106(7), 2617-2650.
13. Anastas, P.T.; Warner, J.C. “Green Chemistry: Theory and Practice”.
OxfordUniversityPress: Oxford, 1998.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

VI. BIBLIOGRAFÍA
106
14. Anastas, P.T. “Green Chemistry as applied to solvents” In. Clean Solvents – Alternative Media for Chemical reactions and Processing, Abraham, M.A.; Moens, L., Eds. 2002; Vol. 819, pp 1-9.
15. Tucker, J.L. “Green Chemistry: Cresting and Summit toward Sustainability”.
OrganicProcessResearch&Development, 2010, 14(2), 328-331.
16. Anastas, P.T. “Perspective on Green Chemistry: The most challenging
synthetic transformation. Tetrahedron2010, 66(5), 1026-1027.
17. Anastas, P.; Eghbali, N. “Green Chemistry: Principles and Practice”. Chem. Soc. Rev. 2010, 39(1), 301-312.
18. Anastas, P.T., Lankey R.L.: “Life cycle assessment and green chemistry: the
ying and yang of industrial ecologyGreen”. Green Chemistry 2000; 2 : 289-295.
19. J.V. Sinisterra. Biotransformations. Encyclopedia of Microbiology. (Moselio Schaechter, Editor), pp. 212-[251] Oxford: Elsevier
20. Arnold FH and Georgiou G (2003) Directed Enzyme Evolution: Screening and Selection Methods. Methods in Molecular Biology, vol. 230. USA: Humana Press.
21. May SW, Padgette SR (1983) Biotechnology 677
22. Hummel W, Kula MR (1989) Eur. J. Biochem. 184: 1
23. Willner I, Mandler D (1989) Enzyme Microb. Technol. 11: 467
24. Bj€orkling F, BouteljeJ,GatenbeckS,HultK,Norin T, Szmulik P (1985)
Tetrahedron 41: 1347.
25. Sánchez-Montero, J.M.; Sinisterra Gago, J.V. “Biocatálisis aplicada a la
Química Farmacéutica”. ANAL. REAL ACAD. NAC. FARM. 2007, 73 (4):
1199-1236.
26. Willner I, Mandler D (1989) Enzyme Microb. Technol. 11: 467
27. Chenault HK, Whitesides GM (1987) Appl. Biochem. Biotechnol. 14: 147
28. Wichmann R, Vasic-Racki D (2005) Adv. Biochem. Eng. Biotechnol. 92: 225
29. Van der Donk WA, Zhao HM (2003) Curr. OpinionBiotechnol. 14: 421
30. Chen CS, Zhou BN, Girdaukas G, Shieh WR, VanMiddlesworth F, Gopalan AS, Sih CJ (1984) Bioorg. Chem. 12: 98
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

VI. BIBLIOGRAFÍA
107
31. Shieh WR, Gopalan AS, Sih CJ (1985) J. Am. Chem. Soc. 107: 2993
32. Hoffmann RW, Ladner W, Helbig W (1984) Liebigs Ann. Chem. 1170
33. Nakamura K, Ushio K, Oka S, Ohno A (1984) Tetrahedron Lett. 25: 3979
34. Fuganti C, Grasselli P, Casati P, Carmeno M (1985) TetrahedronLett. 26: 101
35. Nakamura K, Kawai Y, Oka S, Ohno A (1989) Tetrahedron Lett. 30: 2245
36. Christen M, Crout DHG (1987) Bioreactors and Biotransformations, In: Moody GW, BakerPB (eds) Elsevier, London, p 213
37. Sakai T, Nakamura T, Fukuda K, Amano E, Utaka M, Takeda A (1986) Bull. Chem. Soc.Jpn. 59: 3185
38. Buisson D, Azerad R, Sanner C, Larcheveque M (1991) Tetrahedron Asymmetry 2: 987
39. Ushio K, Inoue K, Nakamura K, Oka S, Ohno A (1986) Tetrahedron Lett. 27: 2657
40. Kometani T, Kitatsuji E, Matsuno R (1991) Agric. Biol. Chem. 55: 867
41. Dahl AC, Madsen JO (1998) Tetrahedron Asymmetry 9: 4395
42. Buisson D, Azerad R (1986) Tetrahedron Lett. 27: 2631
43. Seebach D, Z€uger MF, Giovannini F, Sonnleitner B, Fiechter A (1984)
Angew. Chem. Int.Ed. 23: 151
44. Benach, J.; Atrian, S.; Gonzalez-Duarte, R.; Ladenstein, R. J. Mol. Biol. 1999, 289, 335–355.
45. Jornwall, H. Eur. J. Biochem. 1970, 16, 25–40.
46. Vanni, A.; Pessione, E.; Anfossi, L.; Baggiani, C.; Cavaleto, M.; Gulmini, M.; Giunta, C. J. Mol. Catal. B: Enzym. 2001, 9, 283–291
47. Korkhin, Y.; Kalb, A. J.; Peretz, M.; Bogin, O.; Burstein,Y.; Fromlow, F. J. Mol. Catal. 1998, 278, 967–981.
48. Theorell, H.; Chanwe, B. Acta Chem. Scand. 1951, 5,1127–1144.
49. Svensson, S.; Hoog, J. O.; Schneider, G. Y.; Sandalova, T.J. Mol. Biol. 2000, 302, 441–453.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

VII. ANEXOS
108
VII. ANEXOS
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: no procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 1 de 9
Realizado por: Grupo de Investigación
Fecha: 10 – 08 – 2010
Comprobado por: Fecha:
Aprobado por: Dr José Vicente Sinisterra Gago Fecha: 16 – 08 – 2010
Otros aprobados: Fecha:
Facultad de Ingeniería Química Departamento de Química
Grupo de Investigación en Biotransformaciones y Productos Naturales
F.I.Q.
Cualificación de la Balanza Sartorious AG Germany CPA 224S
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 2 de 9
INDICE
1. Objetivo 3
2. Definiciones 3
3. Desarrollo 3
3.1. Principios Generales 3
3.2. Materiales y Reactivos 3
3.3. Procedimiento operativo 4
3.4. Datos obtenidos 5
3.5. Cálculos y Resultados 7
4. Bibliografía 9
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 3 de 9
Cualificación de la balanza Sartorious AG Germany CPA 224S.
1. Objetivo
El objetivo del presente IT es cualificar la balanza Sartorious AG Germany CPA 224S.
2. Definiciones
Balanza Sartorious AG Germany CPA 224S
3. Desarrollo
4.1. Principios Generales
Es una balanza analítica con la que se cuenta en el laboratorio de
análisis Instrumental, la cual se utiliza en las prácticas de los alumnos de
la carrera de Ingeniería Química, y además en los análisis de
investigación de los diversos trabajos que se quiera realizar.
4.2. Materiales y Reactivos
- Balanza Sartorious AG Germany CPA 224S
- Viales de 3ml de capacidad
- Fiolas de 25ml, 50ml y 100ml
- Vaso de precipitación de 75 ml
- Papel blanco para pesar
- Agua destilada y purificada
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 4 de 9
4.3. Procedimiento operativo
1. Lavar y secar las fiolas y los viales.
2. Enchufar la balanza analítica digital.
3. Encender la balanza. Pulsar el botón del lado izquierdo de la
pantalla.
4. Nivelar la balanza con el indicador burbuja que se encuentra
en la parte posterior derecha de la balanza.
5. Tarar la balanza. Pulsar el botón “TARAR”, y mostrara un
formato numérico 0.0000 en la pantalla lo que significa que
esta apta para pesar.
6. Enumerar los viales a utilizar desde 0 hasta 4.
7. Pesar cada vial vacio por cuatro veces, y anotar pesos
correspondientes.
8. Agregar agua destilada a cada vial, en diferentes volúmenes
de : 0.125ml, 0.250 ml, 0.500ml, 0.75ml y 1 ml
9. Cada vial lleno con cierta cantidad de volumen de agua
destilada debe ser pesado por cuatro veces consecutivas,
luego anotar los pesos correspondientes.
10. Pesar las fiolas vacías de 25 ml , 50ml y 100 ml por cuatro
veces cada una.
11. Agregar agua destilada a las fiolas en sus volúmenes
indicados.
12. Cada fiola llena debe ser pesada cuatro veces consecutivas, y
anotar los pesos correspondientes.
13. Apagar la balanza. Pulse el mismo botón que se utilizo para
encender
14. Desenchufar la balanza
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 5 de 9
4.4. Datos obtenidos
- Rango de Cualificación de 0 a 1mL (Micropipetas)
Tabla N°1.
Tabla: Variación del peso respecto a un volumen definido.
Volumen (ml)
Peso vacío (g)
Peso lleno (g)
Peso del agua (g)
0,0000 0,0000 0,0000 0,0000 0,1250 2,5675 2,6925 0,1250 0,1250 2,5675 2,6924 0,1249 0,1250 2,5675 2,6925 0,1250 0,1250 2,5674 2,6924 0,1250 0,2500 2,5653 2,8186 0,2533 0,2500 2,5653 2,8186 0,2533 0,2500 2,5653 2,8185 0,2532 0,2500 2,5653 2,8185 0,2532 0,5000 2,5212 3,0244 0,5032 0,5000 2,5214 3,0245 0,5031 0,5000 2,5213 3,0246 0,5033 0,5000 2,5214 3,0246 0,5032 0,7500 2,5880 3,3450 0,7570 0,7500 2,5879 3,3451 0,7572 0,7500 2,5879 3,3451 0,7572 0,7500 2,5880 3,3451 0,7571 1,0000 2,6015 3,6095 1,0080 1,0000 2,6015 3,6095 1,0080 1,0000 2,6014 3,6095 1,0081 1,0000 2,6015 3,6095 1,0080
Sustancia empleada: Agua destilada
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 6 de 9
- Rango de Qualificación de 0 a 100mL (Fiolas)
Tabla N°2 Tabla: Variación del peso respecto a un volumen definido.
Volumen (ml)
Peso vacío
(g)
Peso lleno (g)
Peso del agua (g)
0 0,0000 0,0000 0,0000 25 32,6210 57,4679 24,8469 25 32,6210 57,4679 24,8469 25 32,6209 57,4680 24,8471 25 32,6209 57,4681 24,8472 50 41,9000 91,5981 49,6981 50 41,8999 91,5981 49,6982 50 41,8999 91,5981 49,6982 50 41,8998 91,5980 49,6982 75 51,1228 125,1712 74,0484 75 51,1228 125,1690 74,0462 75 51,1226 125,1675 74,0449 75 51,1224 125,1663 74,0439 100 58,6811 158,1665 99,4854 100 58,6808 158,1667 99,4859 100 58,6805 158,1666 99,4861 100 58,6806 158,1667 99,4861
Sustancia empleada: Agua destilada
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 7 de 9
4.5. Cálculos y Resultados
- Rango de Cualificación de 0 a 1mL (Micropipetas)
PAQUETE : SIMFIT PROGRAMA : LINFIT ACCIÓN : Regresión lineal AUTOR : W. G. Bardsley, Universidad de Manchester, U. K. Línea/Análisis polinomial nº 1 ============================ Resultados para ajuste con pesos (w = 1/s^2) Modelo = Recta ajustada por mínimos cuadrados Parámetro Valor Error Est. Límites confianza 95% p ordenada (c) -2.805E-04 3.465E-04 -1.006E-03 4.448E-04 0.4283 ** pendiente(m) 1.009E+00 5.582E-04 1.007E+00 1.010E+00 0.0000 (R-cuadrado = 1.0000, R = 1.0000, p = 0.0000)
Figura N°1
Y = (1.009 ± t.sx) X – (0.0002805 ± t.sy)
Cualificación de la Balanza Sartorius
y = 1.0086x - 0.0002
R2 = 1
0
0.2
0.4
0.6
0.8
1
1.2
0 0.2 0.4 0.6 0.8 1 1.2
Peso Teóricos (g)
Pe
sp
Re
ale
s (
g)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 8 de 9
- Rango de Qualificación de 0 a 100mL (Fiolas)
PAQUETE : SIMFIT PROGRAMA : LINFIT ACCIÓN : Regresión lineal AUTOR : W. G. Bardsley, Universidad de Manchester, U. K. Línea/Análisis polinomial nº 1 ============================ Resultados para ajuste con pesos (w = 1/s^2) Modelo = Recta ajustada por mínimos cuadrados Parámetro Valor Error Est. Límites confianza 95% p ordenada (c) -3.844E-02 1.872E-02 -7.834E-02 1.456E-03 0.0579 ** pendiente(m) 9.948E-01 3.767E-04 9.940E-01 9.956E-01 0.0000 (R-cuadrado = 0.9999, R = 1.0000, p = 0.0000)
Figura N°2
Y = (0.9948 ± t.sx) X – (0.03844 ± t.sy)
Cualificación de la Balanza Sartorius
y = 0.9929x - 0.0341
R2 = 1
0
20
40
60
80
100
120
0 20 40 60 80 100 120
Peso Teóricos (g)
Pe
sp
Re
ale
s (
g)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación de la Balanza Sartorious AG Germany CPA 224S.
Fecha de Emisión: 18 – 08 – 2010
Página: 9 de 9
4. Bibliografía
- Manual de Instrucciones de la Balanza Analítica: “HR Series
Instruction Manual”
- Certificado de la balanza
- Plan de Mantenimiento – Verificación – Calibración
- Certificado de calibración MS 90659, pesas de 10 mg -100 g. Clase
E2
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: no procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 1 de 16
Realizado por: Grupo de Investigación
Fecha: 10 – 08 – 2010
Comprobado por: Fecha:
Aprobado por: Dr José Vicente Sinisterra Gago Fecha: 16 – 08 – 2010
Otros aprobados: Fecha:
Facultad de Ingeniería Química Departamento de Química
Grupo de Investigación en Biotransformaciones y Productos Naturales
F.I.Q.
Cualificación del Equipo Espectrofotómetro UV- visible
Hewlett Packard Mod 8452 A
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 2 de 16
INDICE
1. Objetivo 3
2. Definiciones 3
3. Desarrollo 3
3.1. Principios Generales 3
3.2. Máquinas 3
3.3. Reactivos 4
3.4. Procedimiento Operativo 4
3.5. Datos obtenidos 7
3.6. Cálculos y Resultados 9
4. Bibliografía 16
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 3 de 16
Cualificación del Equipo Espectrofotómetro UV- visible Hewlett Packard
Mod 8452 A
1. Objetivo
El objetivo del presente PNT es cualificar el espectrofotómetro UV-
visible Hewlett Packard Mod 8452 A, Diode array spectrophotometer
2. Definiciones
Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A, Diode array
spectrophotometer
CPU - VTC
Pantalla Sansung SiMaster 3
Estabilizador Pre Line Voltaje Regulador
3. Desarrollo
3.1. Principios Generales
Es el único instrumento de UV que cuenta el laboratorio de análisis
Instrumental, el cual se utiliza en las practicas de los alumnos de la
carrera de Ingeniería Química, y además en los análisis de investigación
de los diversos trabajos que requiera utilizar técnicas
espectrofotométricas a baja longitud de onda.
3.2. Máquinas
- Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A, Diode
array spectrophotometer
- CPU- VTC
- Pantalla Sansung SiMaster
- Estabilizador Pre Line Voltaje Regulador
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 4 de 16
3.3. Reactivos
- Agua destilada y purificada
- Los reactivos han de ser calidad ultravioleta
- Todos los reactivos deben ser guardados en un armario con sus
respectivas
- Indicaciones de seguridad de líquidos inflamables.
- Las Cubetas UV son de cuarzo y se deben lavar después de cada
ensayo con agua destilada. Como disolvente de lavándose
recomiendan disolventes no grasos v.g. EtOH, MeOH o acetona.
- Las cubetas de plástico utilizadas en el visible son desechables y
solo se pueden lavar con agua bidestilada
3.4. Procedimiento operativo
1. Encender el estabilizador
2. Encender el UV. Botón atrás al lado de la toma de corriente. Se inicia el
Test del UV.
3. El aparto inicia el scaneo de las lámparas. Si el test fue correcto, “lamp”
aparecerá en verde.
4. Encender el PC.- aparece la página principal
5. Instrument on line1, aparece la petición de password. No tiene
password, se dá OK.
6. Ir a File. “Load method”. Se carga el método deseado o uno que se
utilizará, una vez modificado para hacer los análisis.
7. En la pantalla principal hay dos cuadros generales a la izquierda. En el
superior hay:
Task.- es la ventana de opción de trabajo: Fixed wavelengths, Spectrum
/Peaks, Ratio/equation y Quantification.
8. El cuadro de abajo se centra en como trabajar: Manual etc, y hacer las
muestras.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 5 de 16
9. Ir a “Instrument”
10. Lamps y seleccionar encender la lámpara “ON”
11. “Set up spectrometer”se pone el intervalo de las longitudes de onda a las
que vamos a trabajar, v.g.: 210 a 310nm (UV) o 340-800nm (visible).
Tambien poner el tiempo de integración, v.g. 0,5sec.
12. En UV inicia el encendido- aparece BUSY en su display y en PC “Lamp
swiching in progress (Abajo)
13. Hacer un espectro.-Seleccionar Método.
14. Ir a Tasks y después a “Spectrum/peaks”
15. Set up.-me permite elegir cuantos picos quiero evaluar vg: 3 y cuantos
valles evaluar vg:2
16. Longitud de onda. Tiene por defecto 180 - 800nm. Poner 210-310 si
queremos trabajar en el UV y 330-800nm si queremos hacer un espectro
en la zona visible. También puedo elegir poner absorbancia o
transmitancia
17. Poner la cubeta de UV en el espectrofotómetro con el medio
(transparente al UV). Ir al cuadro de abajo a izquierda y marcar Blank.
Sale el blanco con un barrido de toda la zona.
18. Se activa ahora “Sample”
19. Meter ahora la cubeta con la sustancia a analizar y hacer el espectro.
Sale el espectro en la pantalla marcándose los picos, la absorbancia y
los valles.
20. Medir a distintas longitudes de onda
Se prepara una solución de benzoato de sodio, que vamos a considerar
como padrón de referencia para el análisis.
21. Ir a Tasks y seleccionar Fixed wavelengths
22. Marcar el set up aparece un cuadro de dialogo y se selecciona las 3
longitudes de onda. vg: 236; 254 y 270nm
23. El date time hay que indicar “absorbancia” .
24. El display spectrum se pone la longitud de onda el de 200 y 210.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 6 de 16
25. Hacer el blanco, usando el disolvente. En este caso concreto se usa
agua bidestilada de calidad Milli-Q
26. Se añaden 2 ml en la cubeta, después con la micropipeta se agregan
100 microlitros y se da en el cuadro de abajo sample apareciendo las
absorbancias a las tres longitudes de onda seleccionadas. Repetir los
ensayos sucesivamente hasta completar el rango de concentraciones.
27. Ir midiendo los valores de absorbancia obtenidos a las distintas
concentraciones en cubeta.
28. Representar la variación de la absorbancia con la concentración y
calcular la linealidad
29. Repetir diez veces la medida de absorbancia de una muestra para
determinar la repetitividad
30. Apagar el espectrómetro UV
31. Cerrar el cuadro principal
32. Guardar el método. OK
33. Crear carpeta JVS1 y mandar guardar
34. Aparece menú principal Windows
35. Inicio, cerrar
36. Ir al botón de encendido (detrás al lado de la toma de corriente del UV) y
cerrar
37. Apagar el ordenador
38. Cerrar el regulador de voltaje
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 7 de 16
3.5. Datos obtenidos
Se tomaron los siguientes datos experimentales a partir de una muestra
benzoato de sodio de 0.528 mM, y se tomaron sus absorbancias a tres
longitudes de onda en el UV.
Cuadro N°1
Concentración de Benzoato de
Sodio (mM)
Absorbancia Absorbancia Absorbancia
PROCEDIMIENTO
236 nm 254 nm 270 nm
0.0000 0.0000 0.0000 0.0000
2mL Agua+ 0.1mL Muestra, agregamos continuamente
0.1mL muestra hasta volumen 3mL.
0.0250 0.1098 -0.0008 -0.0042 0.0480 0.2667 0.0231 0.0140 0.0689 0.3440 0.0351 0.0229 0.0880 0.4410 0.0502 0.0344 0.1056 0.5348 0.0655 0.0460 0.1219 0.6249 0.0797 0.0568 0.1389 0.7085 0.0928 0.0668 0.1509 0.7830 0.1051 0.0760 0.1639 0.8509 0.1177 0.0860 0.1760 0.9162 0.1318 0.0971 0.1320 0.6390 0.0831 0.0591
0.5mL Muestra + 1.5mL Agua,
agregamos continuamente 0.1mL Agua
hasta volumen 3mL
0.1257 0.6089 0.0786 0.0561 0.1200 0.5838 0.0777 0.0557 0.1148 0.5557 0.0706 0.0501 0.1100 0.5312 0.0667 0.0469 0.1056 0.5052 0.0625 0.0436 0.1015 0.4896 0.0613 0.0436 0.0978 0.4693 0.0570 0.0396 0.0943 0.4515 0.0553 0.0386 0.0910 0.4390 0.0562 0.0393 0.0880 0.4193 0.0502 0.0351 0.2640 0.2799 0.1157 0.0967
1mL Muestra +1mL Agua, agregamos continuamente
0.1mL Agua hasta volumen 3mL.
0.2514 1.2331 0.1780 0.1308 0.2400 1.1754 0.1684 0.1239 0.2296 1.1759 0.1677 0.1232 0.2200 1.1216 0.1596 0.1170 0.2112 1.0754 0.1527 0.1120 0.2031 1.0309 0.1462 0.1072 0.1956 0.9912 0.1401 0.1027 0.1886 0.9576 0.1398 0.1038 0.1821 0.9159 0.1287 0.0943 0.1760 0.8808 0.1235 0.0906
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 8 de 16
Concentración de Benzoato de
Sodio (mM)
Absorbancia Absorbancia Absorbancia
PROCEDIMIENTO
236 nm 254 nm 270 nm
0.5280 2.3675 0.4204 0.3188
2mL Muestra y 0.1mL Agua,
agregamos continuamente 0.1mL Agua
hasta volumen 3mL.
0.5029 2.3018 0.3996 0.3026 0.4800 2.2247 0.3768 0.2850 0.4591 2.1499 0.3597 0.2721 0.4400 2.0750 0.3440 0.2605 0.4224 2.0004 0.3294 0.2493 0.4062 1.9383 0.3167 0.2397 0.3911 1.8536 0.2996 0.2266 0.3771 1.8082 0.2918 0.2208 0.3641 1.7519 0.2804 0.2119 0.3520 1.6951 0.2713 0.2050 0.3960 1.8623 0.2929 0.2191
1.5mL Muestra + 0.5mL Agua,
agregamos continuamente 0.1mL Agua
hasta volumen 3mL.
0.3771 1.7823 0.2787 0.2084 0.3600 1.7086 0.2653 0.1987 0.3443 1.6369 0.2523 0.1886 0.3300 1.5711 0.2414 0.1806 0.3168 1.5097 0.2322 0.1740 0.3046 1.4557 0.2257 0.1692 0.2933 1.3992 0.2132 0.1594 0.2829 1.3494 0.2052 0.1533 0.2731 1.3009 0.1973 0.1473 0.2640 1.2577 0.1901 0.1421
Fuente: Módulo Experimental
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 9 de 16
3.6. Cálculos y Resultados
Longitud de onda 236 nm:
Concentración de Benzoato de
Sodio (mM)
Absorbancia Concentración
de Benzoato de
Sodio (mM)
Absorbancia Concentración
de Benzoato de
Sodio (mM)
Absorbancia
236 nm 236 nm 236 nm
0.0000 0.0000 0.1389 0.7085 0.3046 1.4557 0.0250 0.1098 0.1509 0.7830 0.3168 1.5097 0.0480 0.2667 0.1639 0.8509 0.3300 1.5711 0.0689 0.3440 0.1760 0.8808 0.3443 1.6369 0.0880 0.4410 0.1760 0.9162 0.3520 1.6951 0.0880 0.4193 0.1821 0.9159 0.3600 1.7086 0.0910 0.4390 0.1886 0.9576 0.3641 1.7519 0.0943 0.4515 0.1956 0.9912 0.3771 1.7823 0.0978 0.4693 0.2031 1.0309 0.3771 1.8082 0.1015 0.4896 0.2112 1.0754 0.3911 1.8536 0.1056 0.5052 0.2200 1.1216 0.3960 1.8623 0.1056 0.5348 0.2296 1.1759 0.4062 1.9383 0.1100 0.5312 0.2400 1.1754 0.4224 2.0004 0.1148 0.5557 0.2514 1.2331 0.4400 2.0750 0.1200 0.5838 0.2640 1.2577 0.4591 2.1499 0.1219 0.6249 0.2731 1.3009 0.4800 2.2247 0.1257 0.6089 0.2829 1.3494 0.5029 2.3018 0.1320 0.6390 0.2933 1.3992 0.5280 2.3675
LINEALIDAD: PAQUETE : SIMFIT PROGRAMA : LINFIT ACCIÓN : Regresión lineal AUTOR : W. G. Bardsley, Universidad de Manchester, U. K.
Línea/Análisis polinomial nº 1
============================ Para una longitud de onda 236 nm : Resultados para ajuste con pesos (w = 1/s^2) Modelo = Recta ajustada por mínimos cuadrados Parámetro Valor Error Est. Límites confianza 95% p ordenada (c) -3.968E-02 8.923E-03 -5.795E-02 -2.140E-02 0.0001 pendiente(m) 5.326E+00 7.745E-02 5.167E+00 5.484E+00 0.0000 (R-cuadrado = 0.9969, R = 0.9985, p = 0.0000)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 10 de 16
Figura N°1.
Concentración vs Absorbancia a 236 nm para el Benzoato de Sodio
y = 5.326x - 0.03968
R2 = 0.9969
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0.00 0.05 0.10 0.15 0.20 0.25
Concentración mM
Ab
so
rban
cia
Y = (5.326 ± t.sx) X – (0.03968 ± t.sy)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 11 de 16
Longitud de onda 254 nm:
Concentración de Benzoato de
Sodio (mM)
Absorbancia Concentración de Benzoato de
Sodio (mM)
Absorbancia Concentración de Benzoato de
Sodio (mM)
Absorbancia
254 nm 254 nm 254 nm
0.0000 0.0000 0.1389 0.0928 0.3046 0.2257 0.0250 -0.0008 0.1509 0.1051 0.3168 0.2322 0.0480 0.0231 0.1639 0.1177 0.3300 0.2414 0.0689 0.0351 0.1760 0.1318 0.3443 0.2523 0.0880 0.0502 0.1760 0.1235 0.3520 0.2713 0.0880 0.0502 0.1821 0.1287 0.3600 0.2653 0.0910 0.0562 0.1886 0.1398 0.3641 0.2804 0.0943 0.0553 0.1956 0.1401 0.3771 0.2787 0.0978 0.0570 0.2031 0.1462 0.3771 0.2918 0.1015 0.0613 0.2112 0.1527 0.3911 0.2996 0.1056 0.0625 0.2200 0.1596 0.3960 0.2929 0.1056 0.0655 0.2296 0.1677 0.4062 0.3167 0.1100 0.0667 0.2400 0.1684 0.4224 0.3294 0.1148 0.0706 0.2514 0.1780 0.4400 0.3440 0.1200 0.0777 0.2640 0.1901 0.4591 0.3597 0.1219 0.0797 0.2731 0.1973 0.4800 0.3768 0.1257 0.0786 0.2829 0.2052 0.5029 0.3996 0.1320 0.0831 0.2933 0.2132 0.5280 0.4204
LINEALIDAD:
PAQUETE : SIMFIT PROGRAMA : LINFIT ACCIÓN : Regresión lineal AUTOR : W. G. Bardsley, Universidad de Manchester, U. K.
Línea/Análisis polinomial nº 2
============================ Para una longitud de onda 254 nm : Resultados para ajuste con pesos (w = 1/s^2) Modelo = Recta ajustada por mínimos cuadrados Parámetro Valor Error Est. Límites confianza 95% p ordenada (c) -2.265E-02 9.959E-04 -2.465E-02 -2.065E-02 0.0000 pendiente(m) 8.252E-01 7.196E-03 8.107E-01 8.396E-01 0.0000 (R-cuadrado = 0.9975, R = 0.9987, p = 0.0000)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 12 de 16
Figura N°2
Concentración vs Absorbancia a 254 nm para el Benzoato de Sodio
y = 0.8252x - 0.02265
R2 = 0.9975
-0.05
0.05
0.15
0.25
0.35
0.45
0.55
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Concentración mM
Ab
so
rban
cia
Y = (0.8252 ± t.sx) X – (0.02265 ± t.sy)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT
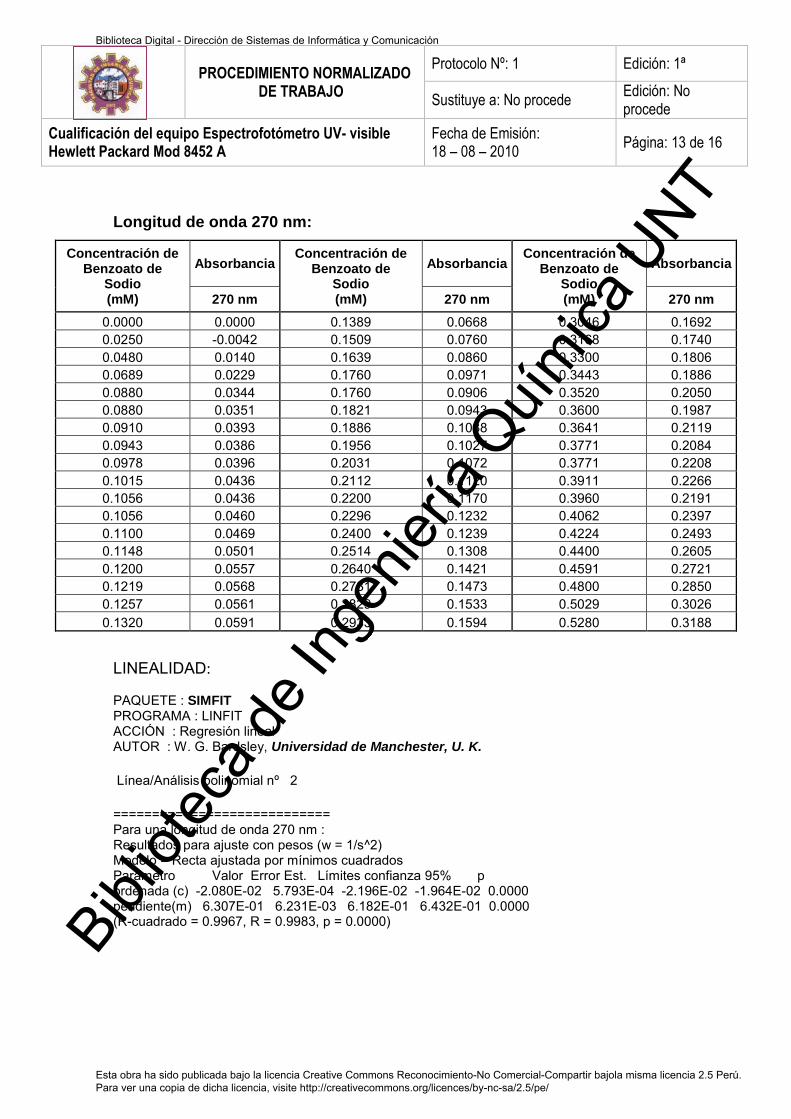
PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 13 de 16
Longitud de onda 270 nm:
Concentración de Benzoato de
Sodio (mM)
Absorbancia Concentración de
Benzoato de Sodio (mM)
Absorbancia Concentración de
Benzoato de Sodio (mM)
Absorbancia
270 nm 270 nm 270 nm
0.0000 0.0000 0.1389 0.0668 0.3046 0.1692 0.0250 -0.0042 0.1509 0.0760 0.3168 0.1740 0.0480 0.0140 0.1639 0.0860 0.3300 0.1806 0.0689 0.0229 0.1760 0.0971 0.3443 0.1886 0.0880 0.0344 0.1760 0.0906 0.3520 0.2050 0.0880 0.0351 0.1821 0.0943 0.3600 0.1987 0.0910 0.0393 0.1886 0.1038 0.3641 0.2119 0.0943 0.0386 0.1956 0.1027 0.3771 0.2084 0.0978 0.0396 0.2031 0.1072 0.3771 0.2208 0.1015 0.0436 0.2112 0.1120 0.3911 0.2266 0.1056 0.0436 0.2200 0.1170 0.3960 0.2191 0.1056 0.0460 0.2296 0.1232 0.4062 0.2397 0.1100 0.0469 0.2400 0.1239 0.4224 0.2493 0.1148 0.0501 0.2514 0.1308 0.4400 0.2605 0.1200 0.0557 0.2640 0.1421 0.4591 0.2721 0.1219 0.0568 0.2731 0.1473 0.4800 0.2850 0.1257 0.0561 0.2829 0.1533 0.5029 0.3026 0.1320 0.0591 0.2933 0.1594 0.5280 0.3188
LINEALIDAD: PAQUETE : SIMFIT PROGRAMA : LINFIT ACCIÓN : Regresión lineal AUTOR : W. G. Bardsley, Universidad de Manchester, U. K.
Línea/Análisis polinomial nº 2
============================ Para una longitud de onda 270 nm : Resultados para ajuste con pesos (w = 1/s^2) Modelo = Recta ajustada por mínimos cuadrados Parámetro Valor Error Est. Límites confianza 95% p ordenada (c) -2.080E-02 5.793E-04 -2.196E-02 -1.964E-02 0.0000 pendiente(m) 6.307E-01 6.231E-03 6.182E-01 6.432E-01 0.0000 (R-cuadrado = 0.9967, R = 0.9983, p = 0.0000)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 14 de 16
Figura N°3.
Concentración vs Absorbancia a 254 nm para el Benzoato de Sodio
y = 0.8252x - 0.02265
R2 = 0.9975
-0.05
0.05
0.15
0.25
0.35
0.45
0.55
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Concentración mM
Ab
so
rban
cia
Y = (0.8252 ± t.sx) X – (0.02265 ± t.sy)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 15 de 16
ENSAYO DE REPETITIVIDAD
Se realizaron diez ensayos de repetitividad a la misma concentración. Los
resultados obtenidos se muestran en la Tabla.
Concentración de Benzoato de Sodio (mM)
Absorbancia Absorbancia Absorbancia
236 nm 254 nm 270 nm
0.0780 0.2783 0.076492 0.065033 0.0780 0.27954 0.07225 0.065369 0.0780 0.27879 0.06263 0.064911 0.0780 0.27838 0.075562 0.064163 0.0780 0.28433 0.081802 0.070236 0.0780 0.2834 0.080505 0.06955 0.0780 0.28435 0.080994 0.070007 0.0780 0.28389 0.080582 0.069336 0.0780 0.28281 0.079391 0.068268 0.0780 0.28603 0.082489 0.071075 x(promedio) 0.28199 0.07727 0.06779 S(desviación standard) 0.00291 0.00606 0.00263 CV (%) 1.0 7.8 3.9
Longitud de Onda(nm) 236 254 270
Límeite de Cuantificación(LQ) 0.0055 0.0734 0.0417
Límeite de Detección (LD) 0.0016 0.0220 0.0125
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo Espectrofotómetro UV- visible Hewlett Packard Mod 8452 A
Fecha de Emisión: 18 – 08 – 2010
Página: 16 de 16
4. Bibliografía
- User’s Guide HP part nº 08452-90008 2rd ed. Agosto 1993.
- Using Your Software HP8452A Diode Array Spectrophotometer. HP
part nº 89531-90000 Abril 1989.
- General Scanning Software HP part nº 89532-90001 Enero 1992.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: no procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 1 de 35
Realizado por: Grupo de Investigación
Fecha: 10 – 08 – 2010
Comprobado por: Fecha:
Aprobado por: Dr José Vicente Sinisterra Gago Fecha: 16 – 08 – 2010
Otros aprobados: Fecha:
Facultad de Ingeniería Química Departamento de Química
Grupo de Investigación en Biotransformaciones y Productos Naturales
F.I.Q.
Cualificación del Equipo HPLC Agilent 1100 Series
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 2 de 35
INDICE
1. Objetivo 3
2. Responsabilidad de aplicación y alcance 3
3. Definiciones 3
4. Descripción del equipo 3
5. Desarrollo 4
5.1. Cualificación de los 04 canales de la
bomba cuaternaria 4
5.2. Cualificación del Sistema de Inyección 24
5.3. Cualificación del Detector UV-Vis
con arreglos de Diodos 29
5.4. Cualificación de la Columna Cromatográfica 31
6. Bibliografía 35
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 3 de 35
Cualificación del Equipo HPLC Agilent 1100 Series.
1. Objetivo
Conocer el protocolo a seguir para la cualificación del equipo HPLC
Agilent 1100 Series con inyector automático, de la Facultad de
Ingeniería Química - Departamento de Química.
2. Responsabilidad de aplicación y alcance
El contenido de este protocolo debe ser conocido por todo el personal
del laboratorio involucrado en la cualificación del equipo HPLC Agilent
1100 Series.
3. Definiciones
El presente documento permite conocer el procedimiento a seguir en la
cualificación de cada componente del equipo de Cromatografía Liquida
de Alta Resolución (HPLC), el cual incluye:
- Cualificación de los 04 canales de la bomba cuaternaria
- Cualificación del inyector automático
- Cualificación del termostato para la columna cromatográfica
- Cualificación de la Columna Cromatográfica.
- Cualificación del Detector UV-Vis con arreglos de Diodos.
4. Descripción del equipo
Equipo HPLC gilent 1100 Series con inyector automático, desgasificador,
sistema de bombas cuaternario, detector UV-visible.
Columna: Teknokroma Mediterranea Sea C18 - 15cm * 0,46 * 5 um. Nº
Serie NF 03706
Detector: VWD (Variable Wavelenght Detector)
Software: Agilent ChemStation.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 4 de 35
5. Desarrollo
5.1. Cualificación de la Bomba Cuaternaria
La Cualificación de la bomba, significa realizar la comprobación de la
exactitud y precisión del caudal suministrado por la bomba a través de
los 04 canales del equipo cromatográfico. El procedimiento requiere
utilizar la columna estándar utilizada habitualmente en el equipo, (por
ejemplo: C-8 o C-18) ya que el sistema se debe estudiar su
funcionamiento como un todo durante un análisis cromatográfico.
La operación consiste en hacer pasar un solvente a través de uno de los
canales (por ejemplo: H2O) con caudales nominales de 0,50; 1,0; 1,5 y
2,0 mL/min y realizar la medición del solvente recogido a la salida del
sistema.
a. Procedimiento
1. Desconectar el conducto de desecho que se encuentra a la salida del
detector y colocar un vaso para evitar caídas del eluyente sobre el
detector
2. Numerar y pesar 5 tubos eppendorf para el flujo de 0,50 mL/min, y
otra cantidad similar para los flujos de 1,0 ; 1,50 y 2,0 mLmin .
3. Activar el funcionamiento la bomba del equipo.
4. Establecer el flujo de 0,500 mL/min para el canal “A” y dejar pasar la
fase móvil durante el tiempo suficiente para que la presión se
estabilice y que por todo el sistema circule el solvente emitido por la
bomba a través del canal “A”.
5. De manera sincronizada controlar 2 min y recoger el volumen de
eluyente que sale en ese tiempo.
6. Repetir ésta operación 5 veces (al mismo caudal), de tal manera que
se obtengan las 5 alícuotas del caudal.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 5 de 35
7. Repetir los pasos (5) y (6), pero ahora empleando flujos de 1,0 ; 1,50
y 2,0 mL/min
8. Pesar los tubos eppendorf que contienen la fase móvil recogida.
9. Determinar el volumen experimental recogido (usando la densidad
del eluyente a la temperatura del laboratorio) de las 05 lecturas para
cada uno de los flujos utilizados.
10. Determinar el valor promedio del volumen experimental recogido
para cada caudal utilizado.
11. Repetir todos los pasos para el canal “B”, “C” y “D”.
NOTA.
Este paso es importante porque permite evaluar y predecir posibles
fallos de bombeo, ensuciamiento de los filtros, problemas de las fritas,
presencia de material particulado que impide un libre flujo de la fase
móvil, etc.
b. Presentación de los resultados
Calcular para cada caudal nominal el valor promedio, la desviación
estándar y el error de la medida (superior e inferior). Representar los
valores promedio frente a los valores nominales y calcular la recta de
regresión que pasa por los puntos experimentales.
NOTA:
Si el sistema esta correcto, entonces se obtendrá u a recta de pendiente
igual a 1,0 y una ordenada no significativa.
La Cualificación de la bomba, significa realizar la comprobación de la
exactitud y precisión del caudal suministrado por la bomba a través de
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 6 de 35
5.1.1. Cualificación del canal “A”
a. Datos Tomados:
Temperatura: 19 ºC
Densidad (19 ºC) = 0,99688 g/cm3
“t” Student al 99% = 3,747
Tiempo de recepción de agua en todas las pruebas = 2 minutos
Fase Móvil utilizada en todas las mediciones = Agua ultrapura miliQ
W tv = peso tubo vacío
Wtv + H2O = peso tubo vacío + agua recogida
Cuadro N°1.
Nª
Replica Flujo
(mL/min) Wtv (g)
Wtv + H2O (g)
W H2O (g)
Vol. Real (mL)
1 0,50 9.5909 10.5933 0.5012 0.5019
2 0,50 9.3325 10.3177 0.4926 0.4933
3 0,50 9.5894 10.574 0.4923 0.4930
4 0,50 9.395 10.3891 0.4971 0.4977
5 0,50 9.3444 10.3405 0.4981 0.4987
Prom 0.4969
Desv Std 0.0038
%C.V. 0.7627
Lim Max 0.5032
Lim Min 0.4910
Cuadro N°2
Nº Replica
Flujo (mL/min)
Wtv (g)
Wtv + H2O (g)
W H2O (g)
Vol. Real (mL)
1 1.0 9.6290 11.6483 1.0097 1.0110
2 1.0 9.5696 11.5709 1.0007 1.0020
3 1.0 9.3236 11.3265 1.0015 1.0028
4 1.0 9.6457 11.6516 1.0030 1.0043
5 1.0 9.6016 11.6091 1.0038 1.0051
Prom 1.0050
Desv Std 0.0036
%C.V. 0.3534
Limite Max 1.0110
Limite Min 0.9995
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 7 de 35
Cuadro N°3
Nº Replica
Flujo (mL/min)
Wtv (g)
Wtv + H2O (g)
W H2O (g)
Vol. Real (mL)
1 1.5 9.6523 12.6619 1.5048 1.5068
2 1.5 9.6199 12.6326 1.5064 1.5083
3 1.5 9.5719 12.5775 1.5028 1.5048
4 1.5 9.6392 12.6433 1.5021 1.5040
5 1.5 9.3157 12.3467 1.5155 1.5175
Prom 1.5083
Desv Std 0.0054
%C.V. 0.3594
Lim Max 1.5174
Lim Min 1.4999
Cuadro N°4
Nº Replica
Flujo (mL/min)
Wtv (g)
Wtv + H2O (g)
W H2O (g)
Vol. Real (ml)
1 2.0 9.6517 13.6185 1.9834 1.9860
2 2.0 9.6117 13.648 2.0182 2.0208
3 2.0 9.3242 13.3244 2.0001 2.0027
4 2.0 9.616 13.6202 2.0021 2.0047
5 2.0 9.6787 13.6888 2.0051 2.0077
Prom 2.0044
Desv Std 0.0125
%C.V. 0.6217
Lim Max 2.0253
Lim min 1.9850
Cuadro N°5
Nº Replica
Flujo (mL/min)
Wtv (g)
Wtv + H2O (g)
W H2O (g)
Vol. Real (ml)
1 3.0 9.6172 15.492 2.9374 2.9413
2 3.0 9.4789 15.4659 2.9935 2.9975
3 3.0 9.4795 15.4677 2.9941 2.9981
4 3.0 9.5439 15.5241 2.9901 2.9941
5 3.0 9.5457 15.5432 2.9988 3.0027
Prom 2.9867
Desv Std 0.0256
%C.V. 0.8566
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 8 de 35
Lim Max 3.0296
Lim min 2.9470
Cuadro N°6
Nº replica Flujo
(mL/min) Wtv (g)
Wtv + H2O (g)
W H2O (g)
Vol. Real (ml)
1 4,0 9.6091 17.5511 3.9710 3.9762
2 4,0 9.597 17.5221 3.9626 3.9678
3 4,0 9.3127 17.2742 3.9808 3.9860
4 4,0 9.6128 17.5759 3.9816 3.9868
5 4,0 9.4117 17.3807 3.9845 3.9898
Prom 3.9813
Desv Std 0.0091
%C.V. 0.2290
Lim Max 3.9966
Lim Min 3.9672
b. Resumen de Cualificación Canal “A” (Comparación por Pares de
Medidas)
Cuadro N°7
Nº Dato
Flujo Teórico (mL/min)
Flujo Exp (mL/min)
1 0.5 0,4969
2 1,0 1,0050
3 1,5 1,5083
4 2,0 2,0044
5 3.0 2,9867
6 4.0 3,9813
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 9 de 35
Figura N°1.
Cualificación del Canal "A"
y = 0.9935x + 0.0101
R2 = 1
0
1
2
3
4
5
0 1 2 3 4
Flujo Experimental (mL/min)
Flu
jo T
eó
ric
o (
mL
/min
)
Cuadro N°8.
Nº Flujo Teórico Flujo Exp Desv Individual (di - d)2
Dato (mL/min) (mL/min) (di)
1 0.5 0.4969 0.0031 0.000010
2 1.0 1.005 -0.0050 0.000025
3 1.5 1.5083 -0.0083 0.000069
4 2.0 2.0044 -0.0044 0.000019
5 3.0 2.9867 0.0133 0.000177
6 4.0 3.9813 0.0187 0.000350
Σ 0.0174 0.000649
Desviación media (d) = Σ di /n = 0.0174 / 6 = 0.0029
Desviación estándar (Sd) = [Σ (di – d)2/(n – 1)]1/2 = [6,49*10-4 /(6 – 1)]1/2 =
0.0114
“t” de student calculada (tcal) = [d * (n)1/2]/Sd = [0.0029* (6)1/2]/0.0114
= 0,623
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 10 de 35
De tablas el valor de “t” de student tabulada al 95% ( ttab ) y para n = 6,
se tiene
T tab = 2,015
Como Tcal < t tab , entonces se concluye que experimentalmente los
valores obtenidos son estadísticamente iguales a los valores teóricos
expresados por el instrumento.
5.1.2. Cualificación del canal “B”
a. Datos Tomados:
Temperatura: 19 ºC ;
Densidad (19 ºC) = 0,99688 g/cm3
“t” Student al 99% = 3,747
Tiempo de recepción de agua en todas las pruebas = 2 minutos
Fase Móvil utilizada en todas las mediciones = Agua ultrapura miliQ
W tv = peso tubo vacío
Wtv + H2O = peso tubo vacío + agua recogida
Cuadro N°9
No replica Flujo
(mL/min) Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 0.5 9.646 10.6091 0.9631 0.4822
2 0.5 9.3231 10.248 0.9249 0.4630
3 0.5 9.6152 10.5488 0.9336 0.4674
4 0.5 9.5746 10.5077 0.9331 0.4672
5 0.5 9.6003 10.5485 0.9482 0.4747
Prom 0.4709
Desv Std 0.0076
%C.V. 1.6087
Lim Max 0.4836
LimMin 0.4591
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 11 de 35
Cuadro N°10
No replica Flujo
(mL/min) Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 1.0 7.0082 8.9891 1.9809 0.9918
2 1.0 6.92 8.9217 2.0017 1.0022
3 1.0 7.1304 9.1282 1.9978 1.0002
4 1.0 7.0951 9.0948 1.9997 1.0012
5 1.0 7.0859 9.0842 1.9983 1.0005
Prom 0.9992
Desv Std 0.0042
%C.V. 0.4209
Lim Max 1.0062
Lim min 0.9926
Cuadro N°11
No replica Flujo
(mL/min) Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 1.5 6.8941 9.8923 2.9982 1.5011
2 1.5 6.8406 9.845 3.0044 1.5042
3 1.5 6.793 9.7952 3.0022 1.5031
4 1.5 6.9035 9.9212 3.0177 1.5108
5 1.5 6.7591 9.7691 3.01 1.5070
Prom 1.5052
Desv Std 0.0038
%C.V. 0.2519
Lim Max 1.5116
Lim min 1.4993
Cuadro N°12
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 2.0 6.8125 10.8129 4.0004 2.0028
2 2.0 6.8983 10.8955 3.9972 2.0012
3 2.0 6.9239 10.9176 3.9937 1.9995
4 2.0 6.9004 10.8943 3.9939 1.9996
5 2.0 6.9074 10.91 4.0026 2.0039
Prom 2.0014
Desv Std 0.0020
%C.V. 0.0984
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 12 de 35
Lim Max 2.0047
Lim min 1.9984
Cuadro N°13.
No replica Flujo
(mL/min) Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 3.0 6.8174 12.7925 5.9751 2.9915
2 3.0 7.1447 13.1623 6.0176 3.0128
3 3.0 7.0853 13.0444 5.9591 2.9835
4 3.0 7.0948 13.0875 5.9927 3.0003
5 3.0 6.8994 12.8665 5.9671 2.9875
Prom 2.9951
Desv Std 0.0117
%C.V. 0.3898
Lim Max 3.0147
Lim min 2.9770
Cuadro N°14.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 4.0 7.0703 14.9403 7.87 3.9402
2 4.0 7.1118 15.0611 7.9493 3.9799
3 4.0 7.0102 14.9629 7.9527 3.9816
4 4.0 7.0201 15.0146 7.9945 4.0025
5 4.0 6.9176 14.853 7.9354 3.9729
Prom 3.9754
Desv Std 0.0226
%C.V. 0.5681
Lim Max 4.0133
Lim min 3.9404
b. Resumen de Cualificación Canal “B” (Comparación por Pares de
Medidas)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT
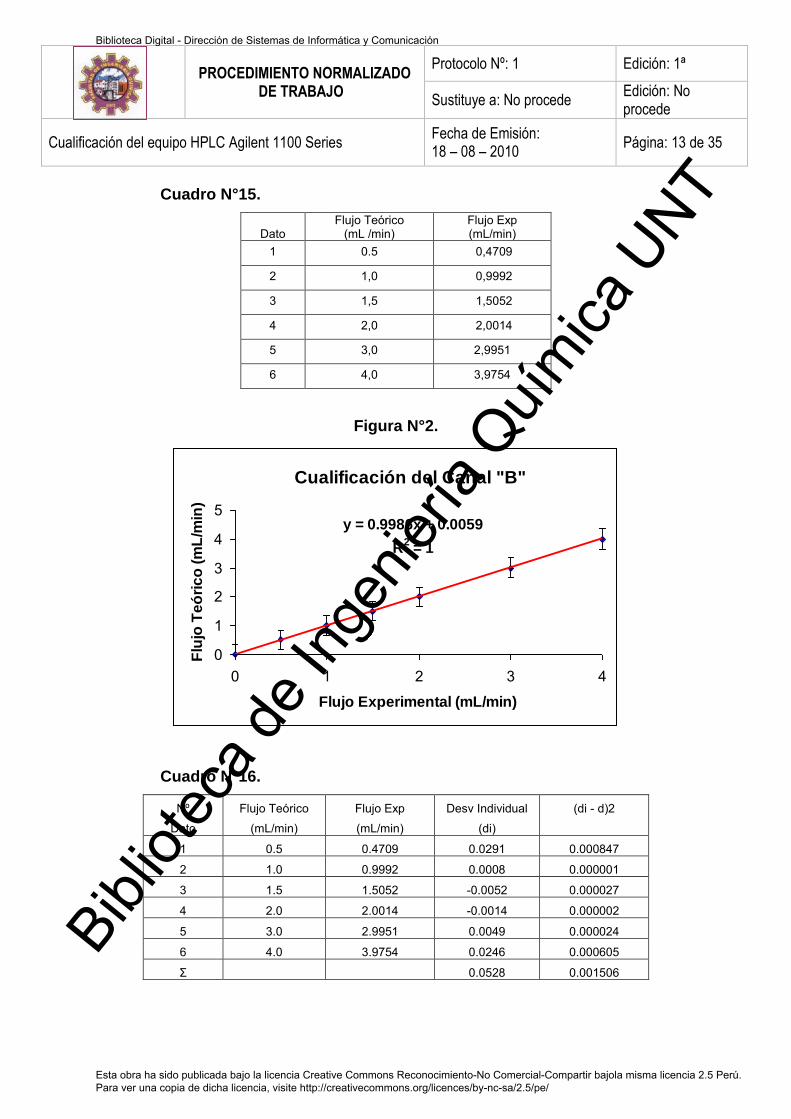
PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 13 de 35
Cuadro N°15.
Dato Flujo Teórico
(mL /min) Flujo Exp (mL/min)
1 0.5 0,4709
2 1,0 0,9992
3 1,5 1,5052
4 2,0 2,0014
5 3,0 2,9951
6 4,0 3,9754
Figura N°2.
Cualificación del Canal "B"
y = 0.9986x + 0.0059
R2 = 1
0
1
2
3
4
5
0 1 2 3 4
Flujo Experimental (mL/min)
Flu
jo T
eó
ric
o (
mL
/min
)
Cuadro N°16.
Nº Flujo Teórico Flujo Exp Desv Individual (di - d)2
Dato (mL/min) (mL/min) (di)
1 0.5 0.4709 0.0291 0.000847
2 1.0 0.9992 0.0008 0.000001
3 1.5 1.5052 -0.0052 0.000027
4 2.0 2.0014 -0.0014 0.000002
5 3.0 2.9951 0.0049 0.000024
6 4.0 3.9754 0.0246 0.000605
Σ 0.0528 0.001506
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 14 de 35
Desviación media (d) = Σ di /n = 0.0528 / 6 = 0.0088
Desviación estándar (Sd) = [Σ(di – d)2/(n – 1)]1/2 =[1,506*10-3/(6 – 1)]1/2
= 0.0174
“t” de student calculada ( tcal) =[d*(n)1/2]/Sd = [0.0088*(6)1/2]/0.0174
= 1,2388
De tablas el valor de “t” de student tabulada al 95% ( ttab ) y para n = 6,
se tiene
T tab = 2,015
Como Tcal < t tab , entonces se concluye que experimentalmente los
valores obtenidos son estadísticamente iguales a los valores teóricos
expresados por el instrumento.
5.1.3. Cualificación del canal “C”
a. Datos Tomados:
Temperatura: 19 ºC ;
Densidad (19 ºC) = 0,99688 g/cm3
“t” Student al 99% = 3,747
Tiempo de recepción de agua en todas las pruebas = 2 minutos
Fase Móvil utilizada en todas las mediciones = Agua ultrapura miliQ
W tv = peso tubo vacío
Wtv + H2O = peso tubo vacío + agua recogida
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 15 de 35
Cuadro N°17.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 0.5 7.0794 8.0811 1.0017 0.5015
2 0.5 7.1741 8.175 1.0009 0.5011
3 0.5 7.2181 8.2179 0.9998 0.5006
4 0.5 7.1144 8.1165 1.0021 0.5017
5 0.5 7.2225 8.2164 0.9939 0.4976
Prom 0.5005
Desv Std 0.0017
%C.V. 0.3350
Lim Max 0.5033
Lim min 0.4979
Cuadro N°18.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 1.0 7.5557 9.5556 1.9999 1.001271679
2 1.0 7.5194 9.5234 2.004 1.003324388
3 1.0 7.5342 9.5314 1.9972 0.999919894
4 1.0 7.4965 9.4875 1.991 0.9968
5 1.0 7.4856 9.4733 1.9877 0.9952
Prom 0.9993
Desv Std 0.0033
%C.V. 0.3309
Lim Max 1.0048
Lim min 0.9942
Cuadro N°19.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 1.5 7.5704 10.5648 2.9944 1.4992
2 1.5 7.5183 10.5054 2.9871 1.4955
3 1.5 7.5241 10.5266 3.0025 1.5032
4 1.5 7.5438 10.5371 2.9933 1.4986
5 1.5 7.5195 10.5128 2.9933 1.4986
Prom 1.4990
Desv Std 0.0028
%C.V. 0.1836
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 16 de 35
Lim Max 1.5037
Lim min 1.4948
Cuadro N°20.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 2.0 7.5023 11.5005 3.9982 2.0017
2 2.0 7.5305 11.5192 3.9887 1.9970
3 2.0 7.4749 11.4638 3.9889 1.9971
4 2.0 7.5455 11.5434 3.9979 2.0016
5 2.0 7.574 11.56 3.986 1.9956
Prom 1.9986
Desv Std 0.0029
%C.V. 0.1427
Lim Max 2.0034
Lim min 1.9942
Cuadro N°21.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 3.0 7.4709 13.4576 5.9867 2.9973
2 3.0 7.5563 13.5225 5.9662 2.9870
3 3.0 7.613 13.5917 5.9787 2.9933
4 3.0 7.518 13.4843 5.9663 2.9871
5 3.0 7.5291 13.5039 5.9748 2.9913
Prom 2.9912
Desv Std 0.0044
%C.V. 0.1456
Lim Max 2.9985
Lim Min 2.9845
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 17 de 35
Cuadro N°22.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 4.0 7.4654 15.4133 7.9479 3.9792
2 4.0 7.5011 15.4529 7.9518 3.9812
3 4.0 7.4843 15.4339 7.9496 3.9800
4 4.0 7.526 15.4686 7.9426 3.9765
5 4.0 7.5144 15.4384 7.924 3.9672
Prom 3.9768
Desv Std 0.0056
%C.V. 0.1416
Lim Max 3.9863
Lim min 3.9681
W tv = peso tubo vacío
Wtv + H2O = peso tubo vacío + agua recogida
b. Resumen de Cualificación Canal “C” (Comparación por Pares de
Medidas)
Cuadro N°23.
No Dato
Flujo Teórico (mL/min)
Flujo Exp (mL/min)
1 0,5 0,5005
2 1,0 0,9993
3 1,5 1,4990
4 2,0 1,9986
5 3,0 2,9912
6 4,0 3,9768
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 18 de 35
Figura 3.
Cualificación del Canal "C"
y = 0.9936x + 0.0071
R2 = 1
0
1
2
3
4
5
0 1 2 3 4
Flujo Experimental (mL/min)
Flu
jo T
eó
ric
o (
mL
/min
)
Cuadro N°24.
Nº Flujo Teórico Flujo Exp Desv Individual (di - d)2
Dato (mL/min) (mL/min) (di)
1 0.5 0.5005 -0.0005 0.000000
2 1.0 0.9993 0.0007 0.000000
3 1.5 1.499 0.0010 0.000001
4 2.0 1.9986 0.0014 0.000002
5 3.0 2.9912 0.0088 0.000077
6 4.0 3.9768 0.0232 0.000538
Σ 0.0346 0.000619
Desviación media (d) = Σ di /n = 0.0346/6 = 0.0058
Desviación estándar (Sd) = [Σ (di – d)2 / (n – 1)]1/2 = [6,19*10-4/(6 – 1) ]1/2
= 0,0111
“t” de student calculada ( tcal )= [d*(n)1/2]/Sd = [0.0058*(6)1/2]/0.0111
= 1,28
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 19 de 35
De tablas el valor de “t” de student tabulada al 95% (ttab) y para n = 6, se
tiene:
T tab = 2,015
Como Tcal < t tab , entonces se concluye que experimentalmente los
valores obtenidos son estadísticamente iguales a los valores teóricos
expresados por el instrumento.
5.1.4. Cualificación del canal “D”
a. Datos Tomados:
Temperatura: 19 ºC ;
Densidad (19 ºC) = 0,99688 g/cm3
“t” Student al 99% = 3,747
Tiempo de recepción de agua en todas las pruebas = 2 minutos
Fase Móvil utilizada en todas las mediciones = Agua ultrapura miliQ
W tv = peso tubo vacío
Wtv + H2O = peso tubo vacío + agua recogida
Cuadro N°25
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 0.5 9.6058 10.6054 0.9996 0.5005
2 0.5 9.4025 10.4227 1.0202 0.5108
3 0.5 9.3324 10.3594 1.027 0.5142
4 0.5 9.3525 10.3844 1.0319 0.5166
5 0.5 9.6567 10.6781 1.0214 0.5114
Prom 0.5107
Desv Std 0.0062
%C.V. 1.2094
Lim Max 0.5210
Lim min 0.5011
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 20 de 35
Cuadro N°26.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 1.0 9.3392 11.3496 2.0104 1.0065
2 1.0 9.582 11.5852 2.0032 1.0029
3 1.0 9.6158 11.5929 1.9771 0.9899
4 1.0 9.5867 11.5653 1.9786 0.9906
5 1.0 9.6916 11.6904 1.9988 1.0007
Prom 0.9981
Desv Std 0.0075
%C.V. 0.7519
Lim Max 1.0107
Lim min 0.9865
Cuadro N°27.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 1.5 9.5733 12.5795 3.0062 1.5051
2 1.5 9.5572 12.5569 2.9997 1.5018
3 1.5 9.582 12.5958 3.0138 1.5089
4 1.5 9.6133 12.6186 3.0053 1.5046
5 1.5 9.6095 12.6074 2.9979 1.5009
Prom 1.5043
Desv Std 0.0031
%C.V. 0.2082
Lim Max 1.5095
Lim min 1.4994
Cuadro N°28.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 2.0 9.3697 13.3615 3.9918 1.9985
2 2.0 9.3369 13.3157 3.9788 1.9920
3 2.0 9.6712 13.6595 3.9883 1.9968
4 2.0 9.6503 13.6252 3.9749 1.9901
5 2.0 9.6241 13.6063 3.9822 1.9937
Prom 1.9942
Desv Std 0.0034
%C.V. 0.1727
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 21 de 35
Lim Max 2.0000
Lim min 1.9889
Cuadro N°29.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 3.0 9.6417 15.6063 5.9646 2.9862
2 3.0 9.4181 15.3813 5.9632 2.9855
3 3.0 9.617 15.6021 5.9851 2.9965
4 3.0 9.3298 15.2875 5.9577 2.9828
5 3.0 9.6308 15.6092 5.9784 2.9932
Prom 2.9888
Desv Std 0.0057
%C.V. 0.1919
Lim Max 2.9985
Lim min 2.9799
Cuadro N°30.
No replica
Flujo (mL/min)
Wtv (g)
Wtv+H2O (g)
W H2O (g)
Vol. Real (ml)
1 4.0 9.4933 17.4421 7.9488 3.9797
2 4.0 9.6121 17.555 7.9429 3.9767
3 4.0 9.3297 17.3107 7.981 3.9958
4 4.0 9.6228 17.5707 7.9479 3.9792
5 4.0 9.6284 17.5738 7.9454 3.9780
Prom 3.9819
Desv Std 0.0079
%C.V. 0.1975
Lim Max 3.9950
Lim min 3.9696
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 22 de 35
b. Resumen de Cualificación Canal “D” (Comparación por Pares de
Medidas)
Cuadro N°31.
No Dato
Flujo Teórico (mL/min)
Flujo Exp (mL/min)
1 0,5 0.5107
2 1,0 0.9981
3 1,5 1.5043
4 2,0 1.9942
5 3,0 2.9888
6 4,0 3.9819
Figura N°4.
Cualificación del Canal "D"
y = 0.9936x + 0.0071
R2 = 1
0
1
2
3
4
5
0 1 2 3 4
Flujo Experimental (mL/min)
Flu
jo T
eó
ric
o (
mL
/min
)
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 23 de 35
Cuadro N°32.
Nº Flujo Teórico Flujo Exp Desv individual (di - d)2
Dato (mL/min) (mL/min) Di
1 0.5 0.5107 -0.0107 0.000114
2 1.0 0.9981 0.0019 0.000004
3 1.5 1.5043 -0.0043 0.000018
4 2.0 1.9942 0.0058 0.000034
5 3.0 2.9888 0.0112 0.000125
6 4.0 3.9819 0.0181 0.000328
Σ 0.0220 0.000623
Desviación media (d) = Σ di/n = 0.0220 / 6 = 0.0037
Desviación estándar (Sd) = [Σ(di – d)2/(n – 1)]1/2 = [6.23*10-4/(6 – 1)]1/2
= 0.0112
“t” de student calculada ( tcal ) = [d*(n)1/2]/Sd = [0.0037*(6)1/2]/0.0112
tcal = 0,809
De tablas el valor de “t” de student tabulada al 95% ( ttab ) y para n = 6,
se tiene
T tab = 2,015
Como Tcal < t tab , entonces se concluye que experimentalmente los
valores obtenidos son estadísticamente iguales a los valores teoricos
expresados por el instrumento.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 24 de 35
5.2. Cualificación del Sistema de Inyección
Implica evaluar dos aspectos fundamentales del sistema de inyección
automático, que nos permitirá tener la seguridad sobre los resultados
obtenidos en cuanto a su reproducibilidad y precisión:
A. Precisión del inyector.
B. Magnitud del efecto memoria (efecto “carry over”).
A. Precisión del inyector
Se llevará a cabo la evaluación de la precisión del sistema de inyección
manual mediante una secuencia de cinco inyecciones de una disolución
estándar de naftaleno (0.100 mg/ml en acetonitrilo) empleando las
siguientes condiciones cromatográficas:
Columna: Teknokroma Mediterranea Sea C18 - 5 µm*15*0,46
Fase móvil : Metanol/agua (45/55 v/v).
Flujo de la fase móvil : 1.00 ml/min.
Longitud de onda del detector : 254 nm.
Volumen de inyección : 10 µl.
Temperatura del horno de columna: 30 ºC.
Presión del equipo : 39 bar.
a. Procedimiento
1. Poner en funcionamiento el equipo.
2. Seleccionar un flujo de 1.00 mL/min y dejar pasar la fase móvil
durante el tiempo suficiente para que la presión se estabilice y que
por todo el sistema circule el solvente de la bomba.
3. Inyectar 10 µl de la disolución estándar.
4. Repetiremos esta última operación 5 veces con esas condiciones
cromatográficas.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 25 de 35
5. Anotar el tiempo de retención y el área para cada una de las
inyecciones de la disolución estándar.
b. Presentación de los resultados
Calcular el valor promedio, la desviación estándar y el error en la medida
(superior e inferior) de los tiempos de retención y las áreas de pico.
B. Magnitud del efecto memoria (efecto “carry over”)
Este parámetro nos permite determinar la contaminación producida por
la inyección anterior si no se limpia correctamente el “loop” del inyector
automático después de cada inyección.
Para determinar este parámetro se debe inyectar la disolución estándar
y luego se debe inyectar una inyección en blanco (como blanco usamos
la fase móvil) y se observa si en el cromatograma aparece señal en el
tiempo de retención de compuesto de la disolución estándar
(acetofenona).
A continuación se repetirá el ensayo pero realizando la inyección con
lavado, para verificar si hay algún cambio en la señal.
a. Procedimiento
1. Poner en funcionamiento el equipo.
2. Seleccionar un flujo de 1,00 ml/min y dejar pasar la fase móvil
durante el tiempo suficiente para que la presión se estabilice y que
por todo el sistema circule el solvente de la bomba.
3. Programar el instrumento para que inyecte secuencialmente 10 µl de
la disolución estándar y luego 10 µl del blanco.
4. Repetiremos los pasos c) y d) tres veces cada uno.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 26 de 35
5. Anotar el tiempo de retención y el área para cada una de las
inyecciones de la disolución estándar y del blanco.
6. Repetir el ensayo pero dejando usando la opción “inyección con
lavado”
7. Anotar el tiempo de retención y el área para cada una de las
inyecciones de la disolución estándar y del blanco.
b. Presentación de los resultados
Calcular el valor promedio, la desviación estándar y el error en la medida
(superior e inferior) de los tiempos de retención y de las áreas de pico.
Asimismo se calculará la relación de áreas (en %) entre el pico
encontrado del blanco y el pico de la disolución estándar.
5.2.1. Precisión del inyector:
Cuadro N°33.
Nº muestra tiempo retención (min) Área
1 4,693 2640,5
2 4,705 2538,8
3 4,703 2630,1
4 4,707 2612,3
5 4,707 2628,7
Promedio 4,703 2610,1
Desv. Estándar 0,0058 41,1055
Limite Superior (95%) 4,710 2661,13
Límite inferior (95%) 4,696 2559,07
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 27 de 35
5.2.2. Magnitud del efecto memoria (efecto “carry over”)
5.2.2.1. Efecto Carry Over (Sin limpiar el sistema)
Cuadro N°34.
Nº muestra tiempo retención (min)
Área (mV)
1 4,708 2460,8
2 4,712 2452,1
3 4,733 2523,8
Promedio 4,718 2478,9
Desv. Estándar 0,0134 39,1271
Limite Superior (95%) 4,7513 2576,105
Límite inferior (95%) 4,6847 2381,695
Cuadro N°35.
Nº blanco Tiempo retención (min) Área % Área
1 4,736 24,4 0,99
2 4,754 15,8 0,64
3 4,760 29,3 1,16
Promedio 4,750 23,167 0,93
Desv. Estándar 0,0125 6,8340 0,2634
Limite Superior (95%) 4,7811 40,145 1,7223
Límite Inferior (95%) 4,7190 6,1890 0,1422
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 28 de 35
5.2.2.2. Efecto Carry Over (utilizando opción inyección con
lavado)
Cuadro N°36.
Nº muestra Tiempo retención (min)
Área (mV)
1 4,741 2686,8
2 4,741 2688,5
3 4,743 2799,3
Promedio 4,742 2724,87
Desv. Estándar 0,0012 64,4668
Límite Superior(95%) 4,7451 2885,03
Límite Inferior(95%) 4,7382 2564,71
Cuadro N°37.
Nº blanco Tiempo retención (min) Área (mV) % Área
1 0 0 0
2 0 0 0
3 0 0 0
Promedio - - -
Desv. Estándar - - 0,0000
Limite Superior - - 0,0000
Límite Inferior - - 0,0000
Programando la opción con lavado desde la ventana del inyector
automático, se le deja realizar la serie de pinchazos correspondientes,
con lo cual conseguimos que el “loop” se limpie correctamente no
quedando residuos de muestra de la inyección anterior.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 29 de 35
5.3. Cualificación del Detector UV-Vis
La cualificación del detector tiene como objetivo comprobar la linealidad
de la detección a una longitud de onda determinada (por ejemplo: 254
nm) dentro de un rango de absorbancia que se considera normal (0.0 -
2.0).
Se llevará a cabo la evaluación del detector mediante una secuencia de
tres inyecciones de cinco disoluciones estándar de acetofenona (0.050,
0.150, 0.250, 0.350 y 0.500 mg/ml respectivamente en metanol/agua)
empleando las siguientes condiciones cromatográficas:
Columna: Teknokroma Mediterranea Sea C18 - 5 µm*15*0,46
Fase móvil: metanol/agua (45/55 v/v).
Flujo de la fase móvil: 1,00 ml/min.
Longitud de onda del detector: 254 nm.
Volumen de inyección: 10 µl.
Temperatura del horno de columna: 30 ºC.
Presión del equipo: 39 bar.
a. Procedimiento
1. Poner en funcionamiento el equipo.
2. Seleccionar un flujo de 1.00 ml/min y dejar pasar la fase móvil
durante el tiempo suficiente para que la presión se estabilice y que
por todo el sistema circule el solvente de la bomba.
3. Inyectar 10 µl de la disolución estándar nº1.
4. Repetiremos esta última operación 3 veces con esas condiciones
cromatográficas.
5. Repetir el ensayo con el resto de disoluciones estándar.
6. Anotar el tiempo de retención y el área para cada una de las
inyecciones de las 5 disoluciones estándar.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 30 de 35
b. Presentación de los resultados
Para cada disolución estándar de naftaleno calcular el valor promedio, la
desviación estándar y el error en la medida (superior e inferior) de las
áreas de los picos.
Representar los valores de concentración de acetofenona (mg/L) frente a
los valores de área de pico y calcular la recta de regresión que pasa por
los puntos experimentales para comprobar la linealidad del detector.
5.3.1. Cualificación del Detector
Cuadro N°38.
nº disolución standard
(mg/L) naftaleno
tR (min) Area (mV)
1 50 4,685 1073
1 50 4,688 1085,3
1 50 4,685 1113,4
2 150 4,741 3848,2
2 150 4,74 3730,7
2 150 4,745 3746,2
3 250 4,748 5426,4
3 250 4,747 5609,4
3 250 4,478 5529,7
4 350 4,676 9542,6
4 350 4,677 9316,3
4 350 4,678 9449,9
5 500 4,679 12619,1
5 500 4,68 12624,8
5 500 4,676 12831,2
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 31 de 35
Cuadro N°39.
Disol. 1 Disol. 2 Disol. 3 Disol. 4 Disol. 5
Promedio 1090,6 3775,0 5521,8 9436,3 12691,7
Desv. Estándar 20,7 63,8 91,8 113,8 120,8
Error superior 1152,7 3966,5 5797,1 9777,6 13054,2
Error inferior 1028,4 3583,5 5246,6 9095,0 12329,2
Figura N°5.
Cualificación de UV-Visible
y = 26.02x - 245.81
R2 = 1
-5002000450070009500
1200014500
0 100 200 300 400 500Concentración (mg/L)
Are
a (
mV
)
5.4. Cualificación de la Columna
Las columnas envejecen con el tiempo por el sangrado de la fase
estacionaria, mal uso, suciedad,etc. Por ello este ensayo es vital y debe
realizarse todos los días antes de empezar a trabajar.
Se llevará a cabo la evaluación de la columna mediante una secuencia
de tres inyecciones de una disolución estándar que contenga una serie
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 32 de 35
de compuestos similares pero con distinto valor de coeficiente de
extinción molar a la longitud de onda elegida (v.g. 254 nm). Debemos
estar seguros que las absorciones se encuentran en la zona lineal
determinada anteriormente.
La disolución estándar preparada es la siguiente: benceno (1 µl/ml),
tolueno (1 µl/ml) y naftaleno (1 µl/ml) en acetonitrilo. Se inyectan 20 µl y
se debe asegurar que trabaja en condiciones óptimas de limpieza del
mismo.
Al no tener la hoja del fabricante preparamos la disolución estándar
indicada anteriormente y los resultados obtenidos los usaremos como
referencia del estado de la columna en evaluaciones posteriores.
Empleando las siguientes condiciones cromatográficas:
Columna : Teknokroma Mediterranea Sea C18 - 5 µm*15*0,46
Fase móvil : agua/acetonitrilo (35/65 v/v).
Flujo de la fase móvil : 0.700 ml/min.
Longitud de onda del detector : 254 nm.
Volumen de inyección : 20 µl.
Temperatura del horno de columna : 40 ºC.
a. Procedimiento
1. Poner en funcionamiento el equipo.
2. Seleccionar un flujo de 0.700 ml/min y dejar pasar la fase móvil
durante el tiempo suficiente para que la presión se estabilice y que
por todo el sistema circule el solvente de la bomba.
3. Inyectar 20 µl de la disolución estándar.
4. Repetiremos esta última operación 3 veces con esas condiciones
cromatográficas.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 33 de 35
5. Anotar el tiempo de retención, el área de pico, factor de capacidad
(k´), número de platos teóricos (N), número de platos teóricos por
metro de columna (N/m), altura de pico teórico reducido (h) y factor
de asimetría (As) del último pico (naftaleno) para cada una de las
inyecciones.
b. Presentación de los resultados
- Factor de capacidad (k’)
k' = ( tR - to ) / to
Donde:
tR = tiempo de retención de la muestra
to = tiempo muerto de la columna
- Número de platos teóricos (N)
N = tR2/ t2 ó N = 16 tR2 / Wb
2
Una columna es tanto más eficaz cuanto más estrecho es el pico.
Como los picos se van ensanchando, para unas mismas
condiciones cromatográficas este análisis se hace con el último
pico.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 34 de 35
- Altura Reducida del Plato Teórico (h o HETP):
h = m /N
Donde m es la longitud de la columna y N el número de platos
teóricos
- Factor de Asimetría (As) (“tailing factor”):
As = a/b
Se miden los valores de a y b como se indica en la figura. Si la
columna funciona bien este valor debe ser 1 o muy próximo al
mismo.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT

PROCEDIMIENTO NORMALIZADO
DE TRABAJO
Protocolo Nº: 1 Edición: 1ª
Sustituye a: No procede Edición: No procede
Cualificación del equipo HPLC Agilent 1100 Series Fecha de Emisión: 18 – 08 – 2010
Página: 35 de 35
5.4.1. Cualificación de la columna
Teknokroma Mediterranea Sea C18
5 µm*15*0,46 - Equipo: Agilent 1100 Series
Cuadro N°40.
Pinchazo tR (min) Área (mV) K´ Nº platos teóricos
nºpl.t./m h As
1 6,244 10047,2 5,06 4240 282666,667 0,00000354 0,83
2 6,24 10086,3 5,06 4235 282333,333 0,00000354 0,83
3 6,244 9890,4 5,06 4334 288933,333 0,00000346 0,81
Promedio 6,243 10007,967 5,060 4269,667 284644,444 0,00000351 0,823
Desv.est. 0,002 103,676 0,000 55,770 3718,024 0,00000005 0,012
Lím. Sup. 6,250 10264,566 5,060 4408,219 293881,273 0,00000363 0,853
Lím. Inf. 6,236 9749,434 5,060 4131,115 275407,615 0,00000339 0,793
6. Bibliografía
- Agilent ChemStation – Understanding Your ChemStation. - Agilent 1100 Series – Qualification workbook.
- Protocolo Normalizado de Trabajo nº1 ed. 1ª: “Manual rápido de
manejo del HPLC Agilent 1100 Series y del software Agilent
ChemStation”.
Biblioteca Digital - Dirección de Sistemas de Informática y Comunicación
Esta obra ha sido publicada bajo la licencia Creative Commons Reconocimiento-No Comercial-Compartir bajola misma licencia 2.5 Perú. Para ver una copia de dicha licencia, visite http://creativecommons.org/licences/by-nc-sa/2.5/pe/
Bibliot
eca d
e Ing
enier
ía Quím
ica U
NT
Related Documents











