Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor- promoting mammary stromal myofibroblasts Yasushi Kojima a , Ahmet Acar a , Elinor Ng Eaton b , Kieran T. Mellody a , Christina Scheel b , Ittai Ben-Porath b , Tamer T. Onder b,c , Zhigang C. Wang d , Andrea L. Richardson e , Robert A. Weinberg b,c,f,1 , and Akira Orimo a,b,1 a Cancer Research-UK Stromal-Tumor Interaction Group, Paterson Institute for Cancer Research, The University of Manchester, Manchester M20 4BX, United Kingdom; b Whitehead Institute for Biomedical Research, Cambridge, MA 02142; c Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139; d Department of Cancer Biology, Dana-Farber Cancer Institute, Cambridge, MA 02139; e Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139; and f MIT Ludwig Center for Molecular Oncology, Cambridge, MA 02139 Contributed by Robert A. Weinberg, September 17, 2010 (sent for review July 22, 2010) Much interest is currently focused on the emerging role of tumor- stroma interactions essential for supporting tumor progression. Carcinoma-associated fibroblasts (CAFs), frequently present in the stroma of human breast carcinomas, include a large number of myofibroblasts, a hallmark of activated fibroblasts. These fibro- blasts have an ability to substantially promote tumorigenesis. However, the precise cellular origins of CAFs and the molecular mechanisms by which these cells evolve into tumor-promoting myofibroblasts remain unclear. Using a coimplantation breast tu- mor xenograft model, we show that resident human mammary fibroblasts progressively convert into CAF myofibroblasts during the course of tumor progression. These cells increasingly acquire two autocrine signaling loops, mediated by TGF-β and SDF-1 cyto- kines, which both act in autostimulatory and cross-communicating fashions. These autocrine-signaling loops initiate and maintain the differentiation of fibroblasts into myofibroblasts and the con- current tumor-promoting phenotype. Collectively, these findings indicate that the establishment of the self-sustaining TGF-β and SDF-1 autocrine signaling gives rise to tumor-promoting CAF myofibroblasts during tumor progression. This autocrine-signaling mechanism may prove to be an attractive therapeutic target to block the evolution of tumor-promoting CAFs. CXCR4 | Smad | tumor microenvironment | alpha-smooth muscle actin M yofibroblasts are often observed in the stroma of various human carcinomas that include those of the breast (1). The presence of these cells in large numbers is also associated with higher-grade malignancy and poor prognosis in patients (2–4). Myofibroblasts express α-smooth muscle actin (α-SMA) that dis- tinguishes these cells from fibroblasts and represents a hallmark of activated fibroblasts (5–10). The activated myofibroblast state of stromal fibroblasts also correlates with their ability to promote tu- mor growth (11–14). Although different types of mesenchymal cells and epithelial cells are proposed to be precursors of the myofibro- blasts present in tumors (15–20), their precise cellular origins and functional contributions to tumor growth still remain unclear. In recent years, the tumor-promoting roles of stromal fibroblasts and α-SMA-positive myofibroblasts, collectively termed carci- noma-associated fibroblasts (CAFs), have been studied (21). CAFs, when inoculated with carcinoma cells, have potently pro- moted the in vivo proliferation of carcinoma cells and tumor growth in mouse xenograft models (14, 21–25). We previously demon- strated that CAFs, prepared directly from invasive human mam- mary carcinomas, contain substantial numbers of myofibroblasts that secrete elevated levels of the proangiogenic chemokine, stro- mal cell-derived factor-1 (SDF-1, also called CXCL12) (14). SDF-1 signaling via its cognate receptor CXCR4, expressed on the surface of carcinoma cells, directly boosts the proliferation of these cells and can stimulate neoangiogenesis by recruiting circulating endo- thelial progenitor cells (EPCs) into the tumor stroma (14, 26). Previous research has shed little light on the molecular mech- anisms that mediate formation of the myofibroblastic state and the associated tumor-promoting capability of CAFs. The simi- larities between tumor-associated myofibroblasts and those pres- ent in sites of wound healing and chronic fibrosis have long been recognized (5, 27, 28). Though TGF-β-Smad autocrine signaling is apparently responsible for the activated state of myofibroblasts in fibrosis (29–31), it is not known if myofibroblastic CAFs also de- pend on TGF-β autocrine signaling and/or additional signaling to maintain their unique phenotypes. We therefore investigated the biochemical alteration(s) un- derlying the tumor-promoting myofibroblastic phenotype of CAFs and the cellular origins of these cells. Our findings show that the establishment of two autocrine signaling loops, mediated by TGF- β and SDF-1 cytokines, endows resident fibroblasts with the tu- mor-promoting myofibroblastic phenotype, thereby driving their differentiation into CAF myofibroblasts. Results Experimental Generation of CAFs from Human Mammary Fibroblasts. Myofibroblasts have been generated in vitro from fibroblasts through their transdifferentiation following exposure to TGF-β (32, 33). Moreover, the CAF populations in tumor-associated stroma are known to include both fibroblasts and myofibroblasts (14, 34). For these reasons, we speculated that preexisting nor- mal stromal fibroblasts could potentially convert into myofibro- blasts in vivo, specifically during the course of tumor progression. Such conversion has not previously been demonstrated. To test this hypothesis, primary normal human mammary fi- broblasts were isolated from reduction mammoplasty tissue and immortalized with hTERT, the catalytic subunit of the telomerase holoenzyme (35). Retroviral constructs encoding GFP and pu- romycin-resistance protein were also introduced into these fibro- blasts. The resulting fibroblasts were then mingled with MCF-7- ras human breast carcinoma cells expressing an activated ras oncogene (36), and the mixtures were injected s.c. into immu- nodeficient nude mice. As described in Fig. 1A, the tumor xenografts were resected at 42, 70, and 200 d after implantation and dissociated into single- cell suspensions. These cells were then cultured in vitro in the Author contributions: A.O. designed research; Y.K., A.A., E.N.E., K.T.M., C.S., and A.O. performed research; I.B.-P., T.T.O., Z.C.W., and A.L.R. contributed new reagents/analytic tools; Y.K., A.A., R.A.W., and A.O. analyzed data; and K.T.M., R.A.W., and A.O. wrote the paper. The authors declare no conflict of interest. Freely available online through the PNAS open access option. 1 To whom correspondence may be addressed. E-mail: [email protected] or aorimo@ picr.man.ac.uk. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1013805107/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1013805107 PNAS | November 16, 2010 | vol. 107 | no. 46 | 20009–20014 MEDICAL SCIENCES

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Autocrine TGF-β and stromal cell-derived factor-1(SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblastsYasushi Kojimaa, Ahmet Acara, Elinor Ng Eatonb, Kieran T. Mellodya, Christina Scheelb, Ittai Ben-Porathb,Tamer T. Onderb,c, Zhigang C. Wangd, Andrea L. Richardsone, Robert A. Weinbergb,c,f,1, and Akira Orimoa,b,1
aCancer Research-UK Stromal-Tumor Interaction Group, Paterson Institute for Cancer Research, The University of Manchester, Manchester M20 4BX, UnitedKingdom; bWhitehead Institute for Biomedical Research, Cambridge, MA 02142; cDepartment of Biology, Massachusetts Institute of Technology, Cambridge,MA 02139; dDepartment of Cancer Biology, Dana-Farber Cancer Institute, Cambridge, MA 02139; eDepartment of Pathology, Brigham and Women’s Hospital,Harvard Medical School, Cambridge, MA 02139; and fMIT Ludwig Center for Molecular Oncology, Cambridge, MA 02139
Contributed by Robert A. Weinberg, September 17, 2010 (sent for review July 22, 2010)
Much interest is currently focused on the emerging role of tumor-stroma interactions essential for supporting tumor progression.Carcinoma-associated fibroblasts (CAFs), frequently present in thestroma of human breast carcinomas, include a large number ofmyofibroblasts, a hallmark of activated fibroblasts. These fibro-blasts have an ability to substantially promote tumorigenesis.However, the precise cellular origins of CAFs and the molecularmechanisms by which these cells evolve into tumor-promotingmyofibroblasts remain unclear. Using a coimplantation breast tu-mor xenograft model, we show that resident human mammaryfibroblasts progressively convert into CAF myofibroblasts duringthe course of tumor progression. These cells increasingly acquiretwo autocrine signaling loops, mediated by TGF-β and SDF-1 cyto-kines, which both act in autostimulatory and cross-communicatingfashions. These autocrine-signaling loops initiate and maintainthe differentiation of fibroblasts into myofibroblasts and the con-current tumor-promoting phenotype. Collectively, these findingsindicate that the establishment of the self-sustaining TGF-β andSDF-1 autocrine signaling gives rise to tumor-promoting CAFmyofibroblasts during tumor progression. This autocrine-signalingmechanism may prove to be an attractive therapeutic target toblock the evolution of tumor-promoting CAFs.
CXCR4 | Smad | tumor microenvironment | alpha-smooth muscle actin
Myofibroblasts are often observed in the stroma of varioushuman carcinomas that include those of the breast (1). The
presence of these cells in large numbers is also associated withhigher-grade malignancy and poor prognosis in patients (2–4).Myofibroblasts express α-smooth muscle actin (α-SMA) that dis-tinguishes these cells from fibroblasts and represents a hallmark ofactivated fibroblasts (5–10). The activated myofibroblast state ofstromal fibroblasts also correlates with their ability to promote tu-mor growth (11–14). Although different types ofmesenchymal cellsand epithelial cells are proposed to be precursors of the myofibro-blasts present in tumors (15–20), their precise cellular origins andfunctional contributions to tumor growth still remain unclear.In recent years, the tumor-promoting roles of stromal fibroblasts
and α-SMA-positive myofibroblasts, collectively termed carci-noma-associated fibroblasts (CAFs), have been studied (21).CAFs, when inoculated with carcinoma cells, have potently pro-moted the in vivoproliferationof carcinomacells and tumorgrowthin mouse xenograft models (14, 21–25). We previously demon-strated that CAFs, prepared directly from invasive human mam-mary carcinomas, contain substantial numbers of myofibroblaststhat secrete elevated levels of the proangiogenic chemokine, stro-mal cell-derived factor-1 (SDF-1, also calledCXCL12) (14). SDF-1signaling via its cognate receptor CXCR4, expressed on the surfaceof carcinoma cells, directly boosts the proliferation of these cellsand can stimulate neoangiogenesis by recruiting circulating endo-thelial progenitor cells (EPCs) into the tumor stroma (14, 26).
Previous research has shed little light on the molecular mech-anisms that mediate formation of the myofibroblastic state andthe associated tumor-promoting capability of CAFs. The simi-larities between tumor-associated myofibroblasts and those pres-ent in sites of wound healing and chronic fibrosis have long beenrecognized (5, 27, 28). Though TGF-β-Smad autocrine signaling isapparently responsible for the activated state of myofibroblasts infibrosis (29–31), it is not known if myofibroblastic CAFs also de-pend on TGF-β autocrine signaling and/or additional signaling tomaintain their unique phenotypes.We therefore investigated the biochemical alteration(s) un-
derlying the tumor-promotingmyofibroblastic phenotype of CAFsand the cellular origins of these cells. Our findings show that theestablishment of two autocrine signaling loops, mediated by TGF-β and SDF-1 cytokines, endows resident fibroblasts with the tu-mor-promoting myofibroblastic phenotype, thereby driving theirdifferentiation into CAF myofibroblasts.
ResultsExperimental Generation of CAFs from Human Mammary Fibroblasts.Myofibroblasts have been generated in vitro from fibroblaststhrough their transdifferentiation following exposure to TGF-β(32, 33). Moreover, the CAF populations in tumor-associatedstroma are known to include both fibroblasts and myofibroblasts(14, 34). For these reasons, we speculated that preexisting nor-mal stromal fibroblasts could potentially convert into myofibro-blasts in vivo, specifically during the course of tumor progression.Such conversion has not previously been demonstrated.To test this hypothesis, primary normal human mammary fi-
broblasts were isolated from reduction mammoplasty tissue andimmortalized with hTERT, the catalytic subunit of the telomeraseholoenzyme (35). Retroviral constructs encoding GFP and pu-romycin-resistance protein were also introduced into these fibro-blasts. The resulting fibroblasts were then mingled with MCF-7-ras human breast carcinoma cells expressing an activated rasoncogene (36), and the mixtures were injected s.c. into immu-nodeficient nude mice.As described in Fig. 1A, the tumor xenografts were resected at
42, 70, and 200 d after implantation and dissociated into single-cell suspensions. These cells were then cultured in vitro in the
Author contributions: A.O. designed research; Y.K., A.A., E.N.E., K.T.M., C.S., and A.O.performed research; I.B.-P., T.T.O., Z.C.W., and A.L.R. contributed new reagents/analytictools; Y.K., A.A., R.A.W., and A.O. analyzed data; and K.T.M., R.A.W., and A.O. wrote thepaper.
The authors declare no conflict of interest.
Freely available online through the PNAS open access option.1To whom correspondence may be addressed. E-mail: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013805107/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1013805107 PNAS | November 16, 2010 | vol. 107 | no. 46 | 20009–20014
MED
ICALSC
IENCE
S

presence of puromycin for 5 d to eliminate any contaminatingcarcinoma cells and host stromal cells. The resulting puromycin-resistant cells were termed experimentally generated CAF1 (exp-CAF1) cells. These cells, resected 42 d after implantation, wereonce again mixed with MCF-7-ras cells and implanted s.c. intohost mice as before. The resulting second group of tumors, whichwere allowed to grow for an additional period of 200 d, wereonce again dissected, dissociated, and cultured in the presence ofpuromycin. The isolated puromycin-resistant cells were termedexperimentally generated CAF2 (exp-CAF2) cells (242 d old).As a control, normal GFP-labeled, puromycin-resistant, im-
mortalized human mammary stromal fibroblasts were injected
s.c. into nude mice as pure cultures without MCF-7-ras cells. Thefibroblasts that survived at the site of injection for the same periodas the exp-CAF-2 cells were isolated in the same way and termedcontrol fibroblast-2 cells (242 d old; Fig. S1A). The mesenchymalnature and human origin of exp-CAFs and the control fibroblastswere confirmed by immunofluorescence analysis (Fig. S1B).
Conversion of Resident Fibroblasts into Tumor-Promoting CAFMyofibroblasts Within the Tumor. To determine whether the ini-tially admixed normal human fibroblasts had converted into amyofibroblast-rich population during the course of tumor growth,we performed immunofluorescence using an anti-α-SMA anti-body.Weobserved that∼48%of the total cell population of 242-d-old exp-CAF2 cells stained positive for α-SMA, a far greaternumber than the ∼14% of myofibroblasts present in the 42-d-oldexp-CAF1 and the ∼2.5% present in the 242-d-old control fibro-blast-2 cell populations (Fig. 1B). The expression level of the ex-tracellular matrix glycoprotein tenascin-C, another marker ofmyofibroblasts (11, 37), was also dramatically elevated in exp-CAF2 cells relative to the control fibroblast-2 cells (Fig. 1B),providing additional evidence to support for the myofibroblasticstate of these cells.Western blot analysis further confirmed progressive up-regu-
lation of α-SMA expression in 42- (5.6-fold), 70- (27.2-fold),200- (29.1-fold) day-old exp-CAF1 cells, and exp-CAF2 cells(29.8-fold) relative to the control fibroblast-2 cells (Fig. 1C).Exp-CAF2 cells also retained their increased α-SMA expression,when propagated as pure populations for periods of 14 pop-ulation doublings (PDs) in vitro following their extraction fromtumors (Fig. 1D). Moreover, exp-CAF2 cell populations extrac-ted from four different tumor xenografts also showed similarincreased levels of α-SMA protein expression (Fig. S1C).We wished to determine whether the myofibroblast differen-
tiation was due to the transfer of the ras oncogene from theadmixed MCF-7-ras cells. We therefore checked for the pres-ence of oncogenic ras in the exp-CAFs by PCR and Western blotanalyses. No oncogenic ras gene and protein expression could bedetected in these cells (Fig. S1D). Collectively, these observa-tions indicate that myofibroblast differentiation is progressivelyincreased during tumor progression and, once established, thedifferentiated state of the resulting cell populations is stablymaintained in a cell-autonomous manner.To determine the ability of exp-CAFs to promote tumor growth,
we performed a tumor xenograft assay. In accordance with pre-vious observations (14), tumors from MCF-7-ras cells admixedwith 42-d-old exp-CAF1 or exp-CAF2 cells showed increasedgrowth kinetics and larger volumes by 1.4- or 2.2-fold, respectively,at 147 d after injection compared with tumors containing controlfibroblast-2 cells (Fig. 1E, i). Moreover, significant numbers ofGFP-positive exp-CAF2 and control fibroblast-2 cells were stillpresent in 150-d-old tumors, as determined by immunofluores-cence (Fig. S1E, e and f). Tumors containing 42-d-old exp-CAF1and exp-CAF2 cells also showed an increase inmicrovessel densityby 2.2- and 5.5-fold, respectively, compared with the control fi-broblast-containing tumors (Fig. 1E, ii and Fig. S1E). Taken to-gether, these various observations demonstrate that exp-CAFsclosely mimic the tumor-promoting behavior of CAFs extractedfrom actual human invasive breast carcinomas (14).
Role of TGF-β Autocrine Signaling Responsible for MyofibroblastDifferentiation in exp-CAFs. As TGF-β autocrine signaling regu-lates myofibrogenesis during fibrosis, we wondered if this was alsothe case for the myofibroblast differentiation observed in CAFs.We therefore measured TGF-β mRNA expression in these cellsby real-time PCR analysis. Levels of TGF-β1 and -β2 expressionprogressively increased in 42- (1.6- and 1.6-fold, respectively), 70-(2.0- and 2.2-fold), and 200- (2.4- and 2.4-fold) day-old exp-CAF1cells and exp-CAF2 cells (2.7- and 4.4-fold) compared with the
Fig. 1. exp-CAFs mimic the tumor-promoting behavior of CAFs preparedfrom breast cancer patients. (A) Isolation of exp-CAFs. Normal GFP-labeled,puromycin-resistant, immortalized human mammary fibroblasts were coin-jected with MCF-7-ras breast cancer cells s.c. into nude mice. Tumors weredissected at the indicated days and dissociated. The injected human fibro-blasts were isolated under puromycin selection in culture and were termedexp-CAFs. See result for details. (B) Immunofluorescence of exp-CAF2 cells andcontrol fibroblast-2 cells (control f.) to detect α-SMA (red) and tenascin-C (TN-C, red). Cell nuclei were stained with 4′,6-diamino-2-phenylindole (DAPI,blue). (Scale bar, 50 μm.) Error bars indicate SEM. (C andD)Western blotting offibroblasts using an anti-α-SMA antibody. The membranes were also probedby an anti-α-tubulin antibody. The ratios of the signal intensity of α-SMArelative to α-tubulin are indicated. PDs, population doublings. (E) MCF-7-rasbreast carcinoma cells were injected alone (n = 12) or coinjected with controlfibroblast-2 cells (control f.; n = 10), 42-d-old exp-CAF1 cells (n = 12), or 242-d-old exp-CAF2 cells (n = 10) s.c. into nudemice. Tumor volume (i) wasmeasuredat the indicated days and microvascular density (ii) was quantified in tumorsadmixedwith variousfibroblasts 150 d after injection. *P< 0.05. Error bars, SE.
20010 | www.pnas.org/cgi/doi/10.1073/pnas.1013805107 Kojima et al.

control fibroblast-2 cells (Fig. 2A, i). TGF-β3 expression, however,remained unchanged (Fig. S2A).We also observed increased TGF-βbioactivity in culture medium conditioned by 42-d-old exp-CAF1(2.7-fold) and exp-CAF2 (4.5-fold) cells compared with controlfibrobroblast-2 cells (Fig. 2A, ii).Given the increases in TGF-β expression and bioactivity ob-
served in exp-CAF myofibroblasts, we speculated that TGF-βligands may induce Smad signaling via their receptor in an auto-crine fashion, thereby contributing to the myofibroblastic stateof these cells. Indeed, immunofluorescence using an antibodyagainst Smad2/3 revealed intense nuclear staining in exp-CAF2cells (Fig. 2B, b) and in normal fibroblasts treated with TGF-β1(10 ng/mL) for 1 h (Fig. 2B, d). In contrast, the nuclear Smad2/3staining was rarely observed in control fibroblast-2 cells (Fig. 2B,a). Moreover, exp-CAF2 cells treated with SB431542 (10 μM),an inhibitor of the TGF-β type I receptor (TβRI) kinase, for 24 hsignificantly reduced the level of nuclear staining (Fig. 2B, c),indicating constitutive induction of Smad signaling via activationof the TβRI in these cells. Western blot analysis further confirmedprogressive elevation of phosphorylated Smad2 expression in 42-(2.9-fold), 70- (6.2-fold), and 200- (7.1-fold) day-old exp-CAF1cells and exp-CAF2 cells (14.3-fold) relative to the control fi-
broblast-2 cells (Fig. 2C). Taken together, these observationsindicate that exp-CAF-secreted TGF-β activates Smad2/3 sig-naling in these cells in an autocrine fashion, ostensibly throughthe TGF-β I/II receptor.To determine if TGF-β autocrine signaling contributes to
maintaining the myofibroblastic state and up-regulation of TGF-βsynthesis in exp-CAFs, we generated shRNAs to suppress signif-icantly either TGF-β type II receptor (TβRII) or Smad4 expres-sion (Fig. S2B). Inhibition of Smad signaling by treatment with theTβRII-shRNA, Smad4-shRNA, or SB431542 (10 μM) for 5 d at-tenuated expression levels of α-SMA (by 85%, 99%, or 85%, re-spectively), TGF-β1 (by 68%, 48–74%, or 65%), and TGF-β2 (by66%, 71%, or 75%) in exp-CAF2 cells, compared with the rele-vant controls (Fig. 2 D and E). Furthermore, activation of Smadsignaling by expressing a constitutively active TGF-β1 construct(38), or by adding recombinant TGF-β1, induced α-SMA andTGF-β expression in normal mammary fibroblasts (Fig. S2C),consistent with previous literature (39). Taken together, thesefindings suggest that activation of TβR-Smad signaling in exp-CAFs induces and maintains their myofibroblast differentiationand TGF-β synthesis, thereby generating a positive feedbackTGF-β signaling loop.
Role of Self-Stimulating SDF-1-CXCR4 Autocrine Signaling in MyofibroblastDifferentiation. Elevated expression levels of SDF-1 have pre-viously been observed in CAF myofibroblast populations in vitroand in vivo (14, 40–42). Consistently, real-time PCR analysisshowed progressive up-regulation of SDF-1 expression in the 42-(5.9-fold), 70- (10.3-fold), and 200- (38-fold) day-old exp-CAF1cells and exp-CAF2 cells (34-fold) relative to the control fibroblast-2 cells (Fig. 3A). An ELISA also showed increased levels of SDF-1released by 42-d-old exp-CAF1 (2.3-fold) and exp-CAF2 (20-fold)cells (Fig. S3A). Moreover, Western blot and immunofluores-cence analyses revealed a fivefold increase in expression of theSDF-1 cognate receptor, CXCR4, in exp-CAF2 cells (∼14 PDs)relative to the control fibroblasts (Fig. 3B), consistent with pre-vious literature (43). We therefore speculated that the releasedSDF-1 may act via CXCR4 upon these cells in an autocrinefashion, thus contributing to their myofibroblastic phenotype.We examined this possibility using two different shRNA con-
structs against SDF-1 or CXCR4. The SDF-1 and CXCR4 pro-tein expression levels were significantly inhibited by 72–76% andby 66–78%, respectively, in exp-CAF2 cells compared with theeffect of control GFP-shRNA (Fig. S3B and Fig. 3C). Immu-noblot and immunofluorescence analyses also demonstratedattenuated levels of α-SMA expression in exp-CAF2 cells expres-sing SDF-1-shRNA (by 67–77%), CXCR4-shRNA (by 62–75%),or treated with AMD3100, a CXCR4-specific inhibitor (44) for 6d (Fig. 3C and Fig. S3C). These data suggest that SDF-1-CXCR4autocrine signaling is required for maintaining the myofibro-blastic phenotype of exp-CAF2 cells.To determine whether triggering the SDF-1-CXCR4 signaling
induces the myofibroblastic phenotype, a retroviral construct en-coding CXCR4 cDNA (45) was introduced into human mam-mary fibroblasts (Fig. S3D). We observed that α-SMA expressionwas increased by ∼100-fold in these CXCR4-expressing cellswhen exposed to SDF-1 (200 ng/mL) for 72 h compared with thecontrol GFP-expressing fibroblasts (Fig. 3D). The ligand-inducedactivation of CXCR4 could also suffice to induce and continu-ously maintain elevated levels of SDF-1 expression in exp-CAFs(Fig. S3E). Taken together, these findings suggest that, like thedescribed TGF-β autocrine signaling, the SDF-1 autocrine sig-naling pathway also operates in a self-stimulating fashion andcontributes to the myofibroblastic differentiation in exp-CAFs.Importantly, MCF-7-ras tumors with admixed exp-CAF2 cells
expressing either CXCR4-shRNA exhibited a reduction in tumorvolume by 49% or 52%, respectively, and in neoangiogenesis by45% at 128 d after injection compared with tumors containing
Fig. 2. TGF-β autocrine signaling suffices to induce and maintain myofi-broblast differentiation in exp-CAFs. (A, i) TGF-β1 and TGF-β2 mRNA ex-pression in fibroblasts measured by real-time PCR. (ii) Active TGF-βconcentrations in the media conditioned by fibroblasts measured by a lu-ciferase assay. Error bars, SE. (B) Immunofluorescence for Smad2/3. Fibro-blasts were incubated with control DMSO (a and b), SB431542, an inhibitorof the TGF-β type I receptor kinase (c), or recombinant TGF-β1 (d ). Arrows(b, black; d, white) indicate nuclear Smad2/3 staining. (Scale bar, 100 μm.)(C ) Western blotting of fibroblasts using anti-pSmad2 and anti-β-actinantibodies. The ratios of the signal intensity of pSmad2 relative to β-actinare indicated. (D) Western blotting of fibroblasts treated with the in-dicated shRNAs or SB431542 (10 μM). The membrane stripped was probedusing different antibodies to detect α-SMA, pSmad2, Smad2/3, andα-tubulin. (E ) Real-time PCR analysis of the fibroblasts described above.Error bars, SE.
Kojima et al. PNAS | November 16, 2010 | vol. 107 | no. 46 | 20011
MED
ICALSC
IENCE
S
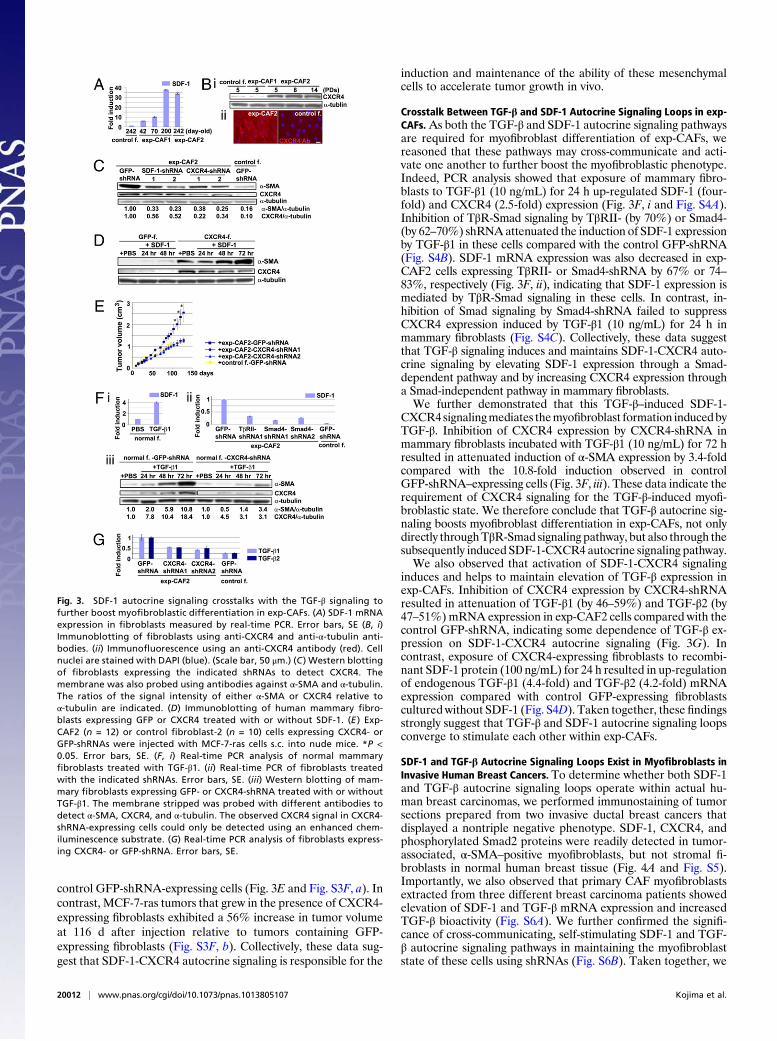
control GFP-shRNA-expressing cells (Fig. 3E and Fig. S3F, a). Incontrast, MCF-7-ras tumors that grew in the presence of CXCR4-expressing fibroblasts exhibited a 56% increase in tumor volumeat 116 d after injection relative to tumors containing GFP-expressing fibroblasts (Fig. S3F, b). Collectively, these data sug-gest that SDF-1-CXCR4 autocrine signaling is responsible for the
induction and maintenance of the ability of these mesenchymalcells to accelerate tumor growth in vivo.
Crosstalk Between TGF-β and SDF-1 Autocrine Signaling Loops in exp-CAFs. As both the TGF-β and SDF-1 autocrine signaling pathwaysare required for myofibroblast differentiation of exp-CAFs, wereasoned that these pathways may cross-communicate and acti-vate one another to further boost the myofibroblastic phenotype.Indeed, PCR analysis showed that exposure of mammary fibro-blasts to TGF-β1 (10 ng/mL) for 24 h up-regulated SDF-1 (four-fold) and CXCR4 (2.5-fold) expression (Fig. 3F, i and Fig. S4A).Inhibition of TβR-Smad signaling by TβRII- (by 70%) or Smad4-(by 62–70%) shRNAattenuated the induction of SDF-1 expressionby TGF-β1 in these cells compared with the control GFP-shRNA(Fig. S4B). SDF-1 mRNA expression was also decreased in exp-CAF2 cells expressing TβRII- or Smad4-shRNA by 67% or 74–83%, respectively (Fig. 3F, ii), indicating that SDF-1 expression ismediated by TβR-Smad signaling in these cells. In contrast, in-hibition of Smad signaling by Smad4-shRNA failed to suppressCXCR4 expression induced by TGF-β1 (10 ng/mL) for 24 h inmammary fibroblasts (Fig. S4C). Collectively, these data suggestthat TGF-β signaling induces and maintains SDF-1-CXCR4 auto-crine signaling by elevating SDF-1 expression through a Smad-dependent pathway and by increasing CXCR4 expression througha Smad-independent pathway in mammary fibroblasts.We further demonstrated that this TGF-β–induced SDF-1-
CXCR4signalingmediates themyofibroblast formation inducedbyTGF-β. Inhibition of CXCR4 expression by CXCR4-shRNA inmammary fibroblasts incubated with TGF-β1 (10 ng/mL) for 72 hresulted in attenuated induction of α-SMA expression by 3.4-foldcompared with the 10.8-fold induction observed in controlGFP-shRNA–expressing cells (Fig. 3F, iii). These data indicate therequirement of CXCR4 signaling for the TGF-β-induced myofi-broblastic state. We therefore conclude that TGF-β autocrine sig-naling boosts myofibroblast differentiation in exp-CAFs, not onlydirectly throughTβR-Smad signaling pathway, but also through thesubsequently induced SDF-1-CXCR4 autocrine signaling pathway.We also observed that activation of SDF-1-CXCR4 signaling
induces and helps to maintain elevation of TGF-β expression inexp-CAFs. Inhibition of CXCR4 expression by CXCR4-shRNAresulted in attenuation of TGF-β1 (by 46–59%) and TGF-β2 (by47–51%)mRNA expression in exp-CAF2 cells compared with thecontrol GFP-shRNA, indicating some dependence of TGF-β ex-pression on SDF-1-CXCR4 autocrine signaling (Fig. 3G). Incontrast, exposure of CXCR4-expressing fibroblasts to recombi-nant SDF-1 protein (100 ng/mL) for 24 h resulted in up-regulationof endogenous TGF-β1 (4.4-fold) and TGF-β2 (4.2-fold) mRNAexpression compared with control GFP-expressing fibroblastscultured without SDF-1 (Fig. S4D). Taken together, these findingsstrongly suggest that TGF-β and SDF-1 autocrine signaling loopsconverge to stimulate each other within exp-CAFs.
SDF-1 and TGF-β Autocrine Signaling Loops Exist in Myofibroblasts inInvasive Human Breast Cancers. To determine whether both SDF-1and TGF-β autocrine signaling loops operate within actual hu-man breast carcinomas, we performed immunostaining of tumorsections prepared from two invasive ductal breast cancers thatdisplayed a nontriple negative phenotype. SDF-1, CXCR4, andphosphorylated Smad2 proteins were readily detected in tumor-associated, α-SMA–positive myofibroblasts, but not stromal fi-broblasts in normal human breast tissue (Fig. 4A and Fig. S5).Importantly, we also observed that primary CAF myofibroblastsextracted from three different breast carcinoma patients showedelevation of SDF-1 and TGF-β mRNA expression and increasedTGF-β bioactivity (Fig. S6A). We further confirmed the signifi-cance of cross-communicating, self-stimulating SDF-1 and TGF-β autocrine signaling pathways in maintaining the myofibroblaststate of these cells using shRNAs (Fig. S6B). Taken together, we
Fig. 3. SDF-1 autocrine signaling crosstalks with the TGF-β signaling tofurther boost myofibroblastic differentiation in exp-CAFs. (A) SDF-1 mRNAexpression in fibroblasts measured by real-time PCR. Error bars, SE (B, i)Immunoblotting of fibroblasts using anti-CXCR4 and anti-α-tubulin anti-bodies. (ii) Immunofluorescence using an anti-CXCR4 antibody (red). Cellnuclei are stained with DAPI (blue). (Scale bar, 50 μm.) (C ) Western blottingof fibroblasts expressing the indicated shRNAs to detect CXCR4. Themembrane was also probed using antibodies against α-SMA and α-tubulin.The ratios of the signal intensity of either α-SMA or CXCR4 relative toα-tubulin are indicated. (D) Immunoblotting of human mammary fibro-blasts expressing GFP or CXCR4 treated with or without SDF-1. (E ) Exp-CAF2 (n = 12) or control fibroblast-2 (n = 10) cells expressing CXCR4- orGFP-shRNAs were injected with MCF-7-ras cells s.c. into nude mice. *P <0.05. Error bars, SE. (F, i) Real-time PCR analysis of normal mammaryfibroblasts treated with TGF-β1. (ii) Real-time PCR of fibroblasts treatedwith the indicated shRNAs. Error bars, SE. (iii) Western blotting of mam-mary fibroblasts expressing GFP- or CXCR4-shRNA treated with or withoutTGF-β1. The membrane stripped was probed with different antibodies todetect α-SMA, CXCR4, and α-tubulin. The observed CXCR4 signal in CXCR4-shRNA-expressing cells could only be detected using an enhanced chem-iluminescence substrate. (G) Real-time PCR analysis of fibroblasts express-ing CXCR4- or GFP-shRNA. Error bars, SE.
20012 | www.pnas.org/cgi/doi/10.1073/pnas.1013805107 Kojima et al.
conclude that the stromal myofibroblasts present within invasivehuman mammary carcinomas require both SDF-1 and TGF-βautocrine signaling loops in self-stimulating and cross-commu-nicating fashions to maintain myofibroblast differentiation.
DiscussionCAFs, myofibroblast-rich cell populations, extracted from hu-man carcinomas maintain an ability to promote tumorigenesis.These cells, passaged for 10 PDs in vitro without ongoing in-teraction with carcinoma cells, retained their ability to promotetumor growth when coinjected with carcinoma cells into immu-nodeficient mice (14, 46). However, the molecular mechanisms
underlying their tumor-promoting ability are poorly understood.Some have reported the importance of somatic genetic alter-ations in forming the tumor-promoting stroma, yet their existenceremains controversial (47–49). The cellular origins of tumor-associated myofibroblasts and the molecular processes that reg-ulate their myofibroblastic state also remain unclear.In the present experiments, we show that mammary fibroblasts
present within a tumor mass can be substantially converted intotumor-promoting CAFs, presumably through their myofibroblastdifferentiation. We cannot formally exclude the possibility thata small population of α-SMA-positive cells preexisting in normalmammary fibroblasts served as precursors of the tumor-associatedmyofibroblasts. This alternative origin is rendered less likely, how-ever, by the fact that such myofibroblastic conversion can be ef-ficiently induced in stromal fibroblasts exposed to media condi-tioned by carcinoma cells (50, 51).During the course of tumor progression, preexisting stromal
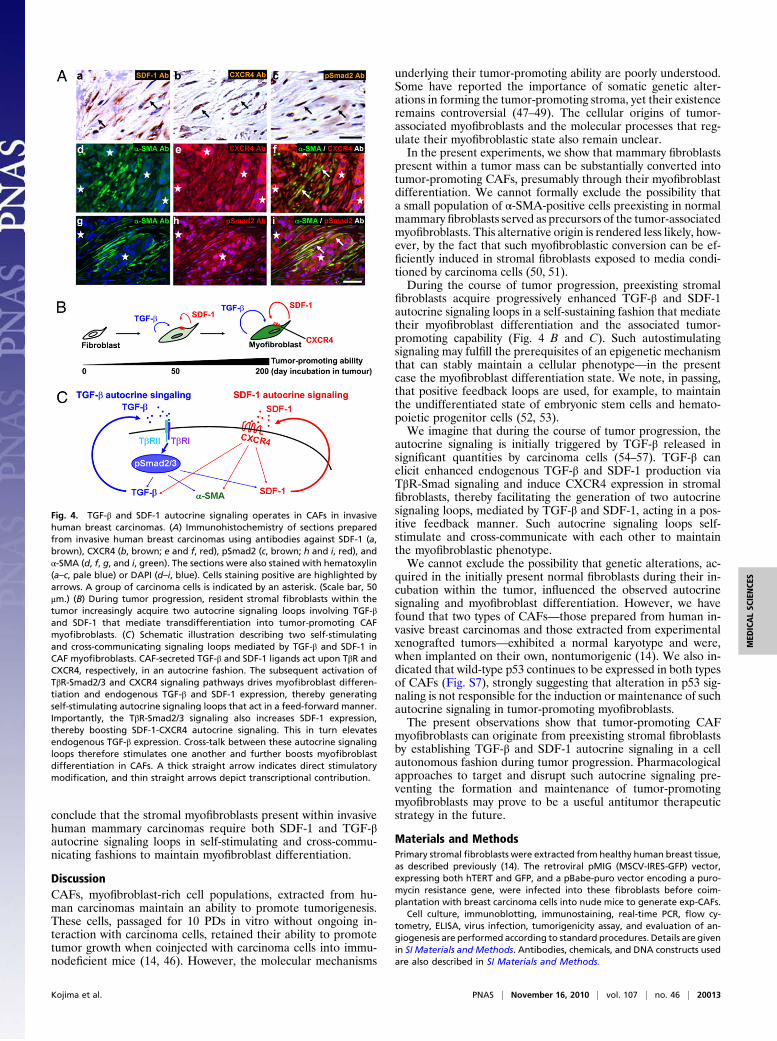
fibroblasts acquire progressively enhanced TGF-β and SDF-1autocrine signaling loops in a self-sustaining fashion that mediatetheir myofibroblast differentiation and the associated tumor-promoting capability (Fig. 4 B and C). Such autostimulatingsignaling may fulfill the prerequisites of an epigenetic mechanismthat can stably maintain a cellular phenotype—in the presentcase the myofibroblast differentiation state. We note, in passing,that positive feedback loops are used, for example, to maintainthe undifferentiated state of embryonic stem cells and hemato-poietic progenitor cells (52, 53).We imagine that during the course of tumor progression, the
autocrine signaling is initially triggered by TGF-β released insignificant quantities by carcinoma cells (54–57). TGF-β canelicit enhanced endogenous TGF-β and SDF-1 production viaTβR-Smad signaling and induce CXCR4 expression in stromalfibroblasts, thereby facilitating the generation of two autocrinesignaling loops, mediated by TGF-β and SDF-1, acting in a pos-itive feedback manner. Such autocrine signaling loops self-stimulate and cross-communicate with each other to maintainthe myofibroblastic phenotype.We cannot exclude the possibility that genetic alterations, ac-
quired in the initially present normal fibroblasts during their in-cubation within the tumor, influenced the observed autocrinesignaling and myofibroblast differentiation. However, we havefound that two types of CAFs—those prepared from human in-vasive breast carcinomas and those extracted from experimentalxenografted tumors—exhibited a normal karyotype and were,when implanted on their own, nontumorigenic (14). We also in-dicated that wild-type p53 continues to be expressed in both typesof CAFs (Fig. S7), strongly suggesting that alteration in p53 sig-naling is not responsible for the induction or maintenance of suchautocrine signaling in tumor-promoting myofibroblasts.The present observations show that tumor-promoting CAF
myofibroblasts can originate from preexisting stromal fibroblastsby establishing TGF-β and SDF-1 autocrine signaling in a cellautonomous fashion during tumor progression. Pharmacologicalapproaches to target and disrupt such autocrine signaling pre-venting the formation and maintenance of tumor-promotingmyofibroblasts may prove to be a useful antitumor therapeuticstrategy in the future.
Materials and MethodsPrimary stromal fibroblasts were extracted from healthy human breast tissue,as described previously (14). The retroviral pMIG (MSCV-IRES-GFP) vector,expressing both hTERT and GFP, and a pBabe-puro vector encoding a puro-mycin resistance gene, were infected into these fibroblasts before coim-plantation with breast carcinoma cells into nude mice to generate exp-CAFs.
Cell culture, immunoblotting, immunostaining, real-time PCR, flow cy-tometry, ELISA, virus infection, tumorigenicity assay, and evaluation of an-giogenesis are performed according to standard procedures. Details are givenin SI Materials and Methods. Antibodies, chemicals, and DNA constructs usedare also described in SI Materials and Methods.
Fig. 4. TGF-β and SDF-1 autocrine signaling operates in CAFs in invasivehuman breast carcinomas. (A) Immunohistochemistry of sections preparedfrom invasive human breast carcinomas using antibodies against SDF-1 (a,brown), CXCR4 (b, brown; e and f, red), pSmad2 (c, brown; h and i, red), andα-SMA (d, f, g, and i, green). The sections were also stained with hematoxylin(a–c, pale blue) or DAPI (d–i, blue). Cells staining positive are highlighted byarrows. A group of carcinoma cells is indicated by an asterisk. (Scale bar, 50μm.) (B) During tumor progression, resident stromal fibroblasts within thetumor increasingly acquire two autocrine signaling loops involving TGF-βand SDF-1 that mediate transdifferentiation into tumor-promoting CAFmyofibroblasts. (C) Schematic illustration describing two self-stimulatingand cross-communicating signaling loops mediated by TGF-β and SDF-1 inCAF myofibroblasts. CAF-secreted TGF-β and SDF-1 ligands act upon TβR andCXCR4, respectively, in an autocrine fashion. The subsequent activation ofTβR-Smad2/3 and CXCR4 signaling pathways drives myofibroblast differen-tiation and endogenous TGF-β and SDF-1 expression, thereby generatingself-stimulating autocrine signaling loops that act in a feed-forward manner.Importantly, the TβR-Smad2/3 signaling also increases SDF-1 expression,thereby boosting SDF-1-CXCR4 autocrine signaling. This in turn elevatesendogenous TGF-β expression. Cross-talk between these autocrine signalingloops therefore stimulates one another and further boosts myofibroblastdifferentiation in CAFs. A thick straight arrow indicates direct stimulatorymodification, and thin straight arrows depict transcriptional contribution.
Kojima et al. PNAS | November 16, 2010 | vol. 107 | no. 46 | 20013
MED
ICALSC
IENCE
S

ACKNOWLEDGMENTS. We thank Drs. Joseph Sodroski, Daniel Rifkin, LalageWakefield, and Luciano Zardi for reagents; Garry Ashton, Dr. Tsukasa Shibue,and Paul Chantry for technical assistance; Dr. David Sabatini for usefuldiscussion; Dr. Nic Jones for critical reading of this manuscript; and membersof R.A.W.’s and A.O.’s laboratories. This work was conducted using the corefacility at the Paterson Institute of Cancer Research and theW. M. Keck Foun-dation Biological Imaging Facility at the Whitehead Institute. This work was
supported by National Institutes of Health/National Cancer Institute GrantR21CA87081-02 (to R.A.W.), the German Academic Exchange Service (C.S.),National Institutes of Health Grants P01 CA080111 and R01 CA078461 (toR.A.W.), the Breast Cancer Research Foundation (R.A.W.), the MassachusettsInstitute of Technology/Ludwig Fund for Cancer Research (to R.A.W.), andCancer ResearchUKGrant C147/A6058 (to A.O.). R.A.W. is anAmerican CancerSociety Research Professor and a Daniel K. Ludwig Cancer Research Professor.
1. Sappino AP, Skalli O, Jackson B, Schürch W, Gabbiani G (1988) Smooth-muscledifferentiation in stromal cells of malignant and non-malignant breast tissues. Int JCancer 41:707–712.
2. Kellermann MG, et al. (2008) Mutual paracrine effects of oral squamous cellcarcinoma cells and normal oral fibroblasts: Induction of fibroblast to myofibroblasttransdifferentiation and modulation of tumor cell proliferation. Oral Oncol 44:509–517.
3. Cardone A, Tolino A, Zarcone R, Borruto Caracciolo G, Tartaglia E (1997) Prognosticvalue of desmoplastic reaction and lymphocytic infiltration in the management ofbreast cancer. Panminerva Med 39:174–177.
4. Maeshima AM, et al. (2002) Modified scar grade: A prognostic indicator in smallperipheral lung adenocarcinoma. Cancer 95:2546–2554.
5. Bissell MJ, Radisky D (2001) Putting tumours in context. Nat Rev Cancer 1:46–54.6. Serini G, Gabbiani G (1999) Mechanisms of myofibroblast activity and phenotypic
modulation. Exp Cell Res 250:273–283.7. Shimoda M, Mellody KT, Orimo A (2010) Carcinoma-associated fibroblasts are a rate-
limiting determinant for tumour progression. Semin Cell Dev Biol 21:19–25.8. Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401.9. Mueller MM, Fusenig NE (2004) Friends or foes—bipolar effects of the tumour stroma
in cancer. Nat Rev Cancer 4:839–849.10. Bhowmick NA, Neilson EG, Moses HL (2004) Stromal fibroblasts in cancer initiation
and progression. Nature 432:332–337.11. De Wever O, et al. (2004) Tenascin-C and SF/HGF produced by myofibroblasts in vitro
provide convergent pro-invasive signals to human colon cancer cells through RhoAand Rac. FASEB J 18:1016–1018.
12. Desmoulière A, Guyot C, Gabbiani G (2004) The stroma reaction myofibroblast: A keyplayer in the control of tumor cell behavior. Int J Dev Biol 48:509–517.
13. De Wever O, Demetter P, Mareel M, Bracke M (2008) Stromal myofibroblasts aredrivers of invasive cancer growth. Int J Cancer 123:2229–2238.
14. Orimo A, et al. (2005) Stromal fibroblasts present in invasive human breast carcinomaspromote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion.Cell 121:335–348.
15. Jeon ES, et al. (2008) Cancer-derived lysophosphatidic acid stimulates differentiationof human mesenchymal stem cells to myofibroblast-like cells. Stem Cells 26:789–797.
16. Mishra PJ, et al. (2008) Carcinoma-associated fibroblast-like differentiation of humanmesenchymal stem cells. Cancer Res 68:4331–4339.
17. Ishii G, et al. (2003) Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun 309:232–240.
18. Direkze NC, et al. (2004) Bone marrow contribution to tumor-associated myofibroblastsand fibroblasts. Cancer Res 64:8492–8495.
19. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R (2007) Discovery of endothelial tomesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res67:10123–10128.
20. Petersen OW, et al. (2003) Epithelial to mesenchymal transition in human breastcancer can provide a nonmalignant stroma. Am J Pathol 162:391–402.
21. Olumi AF, et al. (1999) Carcinoma-associated fibroblasts direct tumor progression ofinitiated human prostatic epithelium. Cancer Res 59:5002–5011.
22. Pietras K, Östman A (2010) Hallmarks of cancer: Interactions with the tumor stroma.Exp Cell Res 316:1324–1331.
23. Hu M, et al. (2008) Regulation of in situ to invasive breast carcinoma transition.Cancer Cell 13:394–406.
24. Yang G, et al. (2006) The chemokine growth-regulated oncogene 1 (Gro-1) links RASsignaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. ProcNatl Acad Sci USA 103:16472–16477.
25. Erez N, Truitt M, Olson P, Arron ST, Hanahan D (2010) Cancer-associated fibroblastsare activated in incipient neoplasia to orchestrate tumor-promoting inflammation inan NF-kappaB-dependent manner. Cancer Cell 17:135–147.
26. Balkwill F (2004) Cancer and the chemokine network. Nat Rev Cancer 4:540–550.27. Schäfer M, Werner S (2008) Cancer as an overhealing wound: An old hypothesis
revisited. Nat Rev Mol Cell Biol 9:628–638.28. Dvorak HF (1986) Tumors: Wounds that do not heal. Similarities between tumor
stroma generation and wound healing. N Engl J Med 315:1650–1659.29. Kim KK, et al. (2009) Epithelial cell alpha3beta1 integrin links beta-catenin and Smad
signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest119:213–224.
30. Varga J (2002) Scleroderma and Smads: Dysfunctional Smad family dynamicsculminating in fibrosis. Arthritis Rheum 46:1703–1713.
31. Mori Y, Chen SJ, Varga J (2003) Expression and regulation of intracellular SMADsignaling in scleroderma skin fibroblasts. Arthritis Rheum 48:1964–1978.
32. Rønnov-Jessen L, Petersen OW (1993) Induction of alpha-smooth muscle actin bytransforming growth factor-beta 1 in quiescent human breast gland fibroblasts.Implications for myofibroblast generation in breast neoplasia. Lab Invest 68:696–707.
33. Desmoulière A, Geinoz A, Gabbiani F, Gabbiani G (1993) Transforming growth factor-beta1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts andin quiescent and growing cultured fibroblasts. J Cell Biol 122:103–111.
34. Rønnov-Jessen L, Van Deurs B, Nielsen M, Petersen OW (1992) Identification, paracrinegeneration, and possible function of human breast carcinoma myofibroblasts in culture.In Vitro Cell Dev Biol 28A:273–283.
35. Vaziri H, Benchimol S (1998) Reconstitution of telomerase activity in normal humancells leads to elongation of telomeres and extended replicative life span. Curr Biol 8:279–282.
36. Kasid A, Lippman ME, Papageorge AG, Lowy DR, Gelmann EP (1985) Transfection ofv-rasH DNA into MCF-7 human breast cancer cells bypasses dependence on estrogenfor tumorigenicity. Science 228:725–728.
37. Mackie EJ, et al. (1987) Tenascin is a stromal marker for epithelial malignancy in themammary gland. Proc Natl Acad Sci USA 84:4621–4625.
38. Wakefield LM, Kondaiah P, Hollands RS, Winokur TS, Sporn MB (1991) Addition ofa C-terminal extension sequence to transforming growth factor-beta 1 interferes withbiosynthetic processing and abolishes biological activity. Growth Factors 5:243–253.
39. Barnard JA, Beauchamp RD, Coffey RJ, Moses HL (1989) Regulation of intestinalepithelial cell growth by transforming growth factor type beta. Proc Natl Acad SciUSA 86:1578–1582.
40. Tait LR, et al. (2007) Dynamic stromal-epithelial interactions during progression ofMCF10DCIS.com xenografts. Int J Cancer 120:2127–2134.
41. Ao M, et al. (2007) Cross-talk between paracrine-acting cytokine and chemokinepathways promotes malignancy in benign human prostatic epithelium. Cancer Res 67:4244–4253.
42. Allinen M, et al. (2004) Molecular characterization of the tumor microenvironment inbreast cancer. Cancer Cell 6:17–32.
43. Eck SM, Côté AL, Winkelman WD, Brinckerhoff CE (2009) CXCR4 and matrixmetalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and innormal mammary fibroblasts exposed to factors secreted by breast cancer cells. MolCancer Res 7:1033–1044.
44. De Clercq E (2003) The bicyclam AMD3100 story. Nat Rev Drug Discov 2:581–587.45. Babcock GJ, Farzan M, Sodroski J (2003) Ligand-independent dimerization of CXCR4,
a principal HIV-1 coreceptor. J Biol Chem 278:3378–3385.46. Orimo A, Weinberg RA (2006) Stromal fibroblasts in cancer: A novel tumor-promoting
cell type. Cell Cycle 5:1597–1601.47. Haviv I, Polyak K, Qiu W, Hu M, Campbell I (2009) Origin of carcinoma associated
fibroblasts. Cell Cycle 8:589–595.48. Weinberg RA (2008) Coevolution in the tumor microenvironment. Nat Genet 40:
494–495.49. Polyak K, Haviv I, Campbell IG (2009) Co-evolution of tumor cells and their
microenvironment. Trends Genet 25:30–38.50. Guo X, Oshima H, Kitmura T, Taketo MM, Oshima M (2008) Stromal fibroblasts
activated by tumor cells promote angiogenesis in mouse gastric cancer. J Biol Chem283:19864–19871.
51. Noma K, et al. (2008) The essential role of fibroblasts in esophageal squamous cellcarcinoma-induced angiogenesis. Gastroenterology 134:1981–1993.
52. Grass JA, et al. (2003) GATA-1-dependent transcriptional repression of GATA-2 viadisruption of positive autoregulation and domain-wide chromatin remodeling. ProcNatl Acad Sci USA 100:8811–8816.
53. Boyer LA, et al. (2005) Core transcriptional regulatory circuitry in human embryonicstem cells. Cell 122:947–956.
54. Jakowlew SB (2006) Transforming growth factor-beta in cancer and metastasis.Cancer Metastasis Rev 25:435–457.
55. Gold LI (1999) The role for transforming growth factor-beta (TGF-beta) in humancancer. Crit Rev Oncog 10:303–360.
56. Nørgaard P, Hougaard S, Poulsen HS, Spang-Thomsen M (1995) Transforming growthfactor beta and cancer. Cancer Treat Rev 21:367–403.
57. Gorsch SM, Memoli VA, Stukel TA, Gold LI, Arrick BA (1992) Immunohistochemicalstaining for transforming growth factor beta 1 associates with disease progression inhuman breast cancer. Cancer Res 52:6949–6952.
20014 | www.pnas.org/cgi/doi/10.1073/pnas.1013805107 Kojima et al.

Supporting InformationKojima et al. 10.1073/pnas.1013805107SI Materials and MethodsPlasmid Construction. Codon-optimized human CXCR4 cDNA (1)was kindly provided by Joseph G. Sodroski (Dana-Farber CancerInstitute, Harvard University, Boston) and cloned into the pBaberetroviral vector. A constitutively active form of swine TGF-β1cDNA vector, pPK9a (2), was kindly provided by Lalage M.Wakefield (National Cancer Institute, Bethesda, MD) and clonedinto the pBabe-neo retroviral vector. The shRNA oligonucleo-tides against GFP, CXCR4, SDF-1, p53, TβR-II, and Smad4genes were generated and cloned into lentivirus-derived pLKO-hygro- or pLKO-puro-vectors (3). Target sequences used arelisted in the table below.
Isolation of Human Mammary Fibroblasts and Tissue Culture. Normalhuman stromal fibroblasts were extracted from healthy breasttissue obtained from a reduction mammoplasty as describedpreviously (4) and cultured in DMEM supplemented with 10%calf serum (Valley Biomedical). The retroviral pMIG (MSCV-IRES-GFP) vector, expressing both hTERT and GFP, anda pBabe-puro vector encoding a puromycin resistance gene, wereinfected into these mammary fibroblasts to facilitate their im-mortalization. Mammary fibroblasts were also extracted fromtumor masses or noncancerous breast tissues obtained frombreast cancer patients, as described previously (4).
Subcutaneous Tumorigenicity Assays. MCF-7-ras human breastcarcinoma cells (1 × 106) and human mammary fibroblasts (3 ×106) were admixed and suspended in 400 μL of culture mediumwith 50% Matrigel (BD Biosciences). The mixture was injecteds.c. into immunodeficient nude mice. Tumorigenicity assay wasperformed as described previously (5).
Evaluation of Angiogenesis in MCF-7-ras Tumor Xenografts. Serialparaffin sections (taken at 2-mm intervals) were prepared fromtumor xenografts grown. A total of 30 sections, six independenttumors from each cohort, were immunostained using an anti-CD31 antibody, a marker of vascular endothelial cells. Micro-vessel density was assessed as previously described (6).
Retroviral and Lentiviral Infections. Retroviral and lentivirusinfections were performed as described previously (3, 5). Afterinfection, human mammary fibroblasts were cultured for 4–6 d inthe presence of the appropriate antibiotic for each plasmid:puromycin (1 μg/mL), neomycin (500 μg/mL), hygromycin (50μg/mL), or blasticidin (7.5 μg/mL).
Western Blot Analysis. Fibroblasts were seeded at 0.5 × 106 cellsper 6-cm dish and cultured for 48 h in DMEM supplementedwith 2% calf serum. A total of 10–30 μg of whole-cell lysate wasrun on NuPAGE 4–12% gradient gels (Invitrogen) and trans-ferred onto Hybond ECL membrane (GE Healthcare). Quanti-fication of band intensity was performed using Multi GaugeVersion 2.2 software (Fujifilm).
Real-Time RT-PCR Analysis. Total RNA was extracted using anRNeasy Plus Mini Kit (Qiagen) in accordance with the manu-
facturer’s protocol. SuperScript II reverse transcriptase (In-vitrogen) was used to synthesize cDNA. Real-time RT-PCRanalysis was performed as previously described (7). Data foreach sample were normalized relative to the expression level ofβ2-microglobulin gene. Primers used for RT-PCR analyses aredescribed in a table below.
Flow Cytometry. 1–5 × 105 human mammary fibroblasts wereanalyzed using FACSCalibur flow cytometry (Becton Dickinson)as previously described (4). The antibody used to detect CXCR4is listed in a table below. Nonviable cells were detected bystaining with 7-aminoactinomycin D (7-AAD; BD Biosciences).
Measurement of SDF-1 Protein Levels. Various human mammaryfibroblasts were cultured in DMEM with 2% serum for 48 h. Themedia conditioned by fibroblasts were collected and filteredthrough a 0.45-μm syringe filter. SDF-1 levels were measuredusing the Quantikine human SDF-1 immunoassay (R&D Sys-tems) in accordance with the manufacturer’s protocol.
Measurement of Levels of Biologically Active TGF-β. Levels of activeTGF-β were measured using mink lung epithelial cells (MLECs)expressing a PAI-1 promoter-driven luciferase reporter construct(8), a kind gift from Daniel B. Rifkin (New York UniversityMedical Center, New York). Cells (1.6 × 104) were seeded ontoa 96-well plate and incubated for 14 h in media conditioned byexp-CAFs or the control fibroblasts for 2 d. Luciferase activitywas measured using the Dual-Luciferase Reporter Assay System(Promega) in accordance with the manufacturer’s protocol.
Immunostaining of Human Breast Tissues and Cultured MammaryFibroblasts. Mammary fibroblasts were stained using variousantibodies as listed below. To quantify α-SMA-positive cells (%),the positive cell numbers relative to total cell numbers (>100counted cells) were evaluated in nine independent fields fromthree different wells of each fibroblast type under a fluorescencemicroscope. Paraffin sections prepared from human breast tis-sues were immunostained using either the Dako EnVision sys-tem (DAKO) or the avidin-biotin complex technique. Theantibodies used are listed in the table below.
Cell Cycle Analysis. Cell cycle analysis using propidium iodide wasperformed as described previously (9). Different fibroblastpopulations were exposed to 20 Gy of ionizing radiation (137Cs)and harvested 30 h after irradiation for analysis.
Statistical Analysis. Statistical analyses were performed usinga Student t test or ANOVA test, followed by a Dunnett’s mul-tiple comparison test using the SPSS version 13.0 software.Values of P < 0.05 were considered significant.
Antibodies and Chemicals. Primary antibodies used are listed inthe following table. AMD3100 octahydrochloride hydrate andSB431542 were purchased from Sigma-Aldrich. RecombinantSDF-1α and TGF-β1 proteins were obtained from R&D Systems.
Kojima et al. www.pnas.org/cgi/content/short/1013805107 1 of 9

Antibodies (clone name) Source Purpose
β-actin (8226) Abcam WBCD31 Santa Cruz Biotechnology IHCCollagen type1 1A Sigma IFPan-cytokeratin Sigma IFCXCR4 (12G5) R&D Systems IHCCXCR4-biotin (12G5) R&D Systems FCMCXCR4-PE (12G5) R&D Systems FCM, IFCXCR4 Leinco Technologies WBFibroblast surface protein(clone 1B10)
Abcam IF
Fibronectin BD Biosciences IFS100A4/FSP-1 (fibroblast-specificprotein-1)
Dako IF
SDF-1 (K15C) Gift from FernandoArenzana-Seisdedos*
IHC
α-SMA (1A4) Sigma IF, WB, IHCα-SMA-Cy3 (1A4) Sigma IFSmad2/3 BD Biosciences IF, WBPhosphorylated-Smad2 (138D4) Cell Signaling Technology WB, IHCSmad4 (B-8) Santa Cruz Biotechnology WBp53 Santa Cruz Biotechnology WBp21WAF1 Santa Cruz Biotechnology WBProlyl-4-hydroxylase (5B5) Sigma IFpan-RasArg-12 (Ab-1) Oncogene WBTGF-β RIIb R&D Systems WBTenascin C (BC-8) Gift from Luciano Zardi† IFα-tubulin (DM1A) Abcam WBVimentin (V9) Novocastra Laboratories IF
FCM, flow cytometry; IF, immunofluorescence; IHC, immunohistochemistry; WB, Western blotting.*Institut Pasteur, Paris.†Laboratory of Recombinant Therapeutic Proteins, CBA - Advanced Biotechnology Centre, Genoa, Italy.
shRNA target sequences
Sequence name Sequence Ref.
GFP-shRNA 5′-GCAAGCTGACCCTGAAGTTCA-3′ (4)CXCR4-shRNA1 5′-ATGGATTGGTCATCCTGGTCA-3′ (10)CXCR4-shRNA2 5′-ATCCTGGCCTTCATCAGTCTG-3′ (11)SDF-1-shRNA1 5′-TCCTCGTGCTGACCGCGCTCT-3′SDF-1-shRNA2 5′-AGCTATTCCTACTCTCTCCCC-3′TβR-II-shRNA1 5′-GAATGACGAGAACATAACACT-3′ (12)TβR-II-shRNA2 5′-GATTCAAGAGTATTCTCACTT-3′ (13)Smad4-shRNA1 5′-TCATCCTAGTAAATGTGTTAC-3′ (14)Smad4-shRNA2 5′-AGGTGTGCAGTTGGAATGTAA-3′ (15)p53-shRNA 5′-GACTCCAGTGGTAATCTAC-3′ (16)
PCR primers
Gene name Sense sequence Antisense sequence
α-SMA 5′-GTGTGTGACAATGGCTCTGG-3′ 5′-TGGTGATGATGCCATGTTCT-3′SDF-1 5′-CTAGTCAAGTGCGTCCACGA-3′ 5′-GGACACACCACAGCACAAAC-3′TGF-β1 5′-ACTGCAAGTGGACATCAACG-3′ 5′-TGCGGAAGTCAATGTACAGC-3′TGF-β2 5′-TTGACGTCTCAGCAATGGAG-3′ 5′-GGGTTCTGCAAACGAAAGAC-3′TGF-β3 5′- AAGTGGGTCCATGAACCTAA-3′ 5′- GCTACATTTACAAGACTTCAC-3′β2-microglobulin 5′-TGAGTGCTGTCTCCATGTTTGA-3′ 5′-TCTGCTCCCCACCTCTAAGTTG-3′v-H-ras 5′-GTGGTGGTGGGCGCTAGA-3′ 5′-TTCTTGACCTGTTGTGTCTAA-3′CXCR4 5′-AATCTTCCTGCCCACCATCT-3′ 5′-GACGCCAACATAGACCACCT-3′GAPDH 5′-ACCCAGAAGACTGTGGATGG-3′ 5′-TCTAGACGGCAGGTCAGGTC-3′
Kojima et al. www.pnas.org/cgi/content/short/1013805107 2 of 9

1. Babcock GJ, Farzan M, Sodroski J (2003) Ligand-independent dimerization of CXCR4,a principal HIV-1 coreceptor. J Biol Chem 278:3378–3385.
2. Wakefield LM, Kondaiah P, Hollands RS, Winokur TS, Sporn MB (1991) Addition ofa C-terminal extension sequence to transforming growth factor-beta 1 interferes withbiosynthetic processing and abolishes biological activity. Growth Factors 5:243–253.
3. Stewart SA, et al. (2003) Lentivirus-delivered stable gene silencing by RNAi in primarycells. RNA 9:493–501.
4. Orimo A, et al. (2005) Stromal fibroblasts present in invasive human breast carcinomaspromote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion.Cell 121:335–348.
5. Elenbaas B, et al. (2001) Human breast cancer cells generated by oncogenictransformation of primary mammary epithelial cells. Genes Dev 15:50–65.
6. Weidner N, Semple JP, Welch WR, Folkman J (1991) Tumor angiogenesis andmetastasis—correlation in invasive breast carcinoma. N Engl J Med 324:1–8.
7. Brisken C, et al. (2002) IGF-2 is a mediator of prolactin-induced morphogenesis in thebreast. Dev Cell 3:877–887.
8. Abe M, et al. (1994) An assay for transforming growth factor-beta using cellstransfected with a plasminogen activator inhibitor-1 promoter-luciferase construct.Anal Biochem 216:276–284.
9. Telford WG, King LE, Fraker PJ (1992) Comparative evaluation of several DNA bindingdyes in the detection of apoptosis-associated chromatin degradation by flowcytometry. Cytometry 13:137–143.
10. Li YM, et al. (2004) Upregulation of CXCR4 is essential for HER2-mediated tumormetastasis. Cancer Cell 6:459–469.
11. Lapteva N, Yang AG, Sanders DE, Strube RW, Chen SY (2005) CXCR4 knockdown bysmall interfering RNA abrogates breast tumor growth in vivo. Cancer Gene Ther 12:84–89.
12. Wesolowska A, et al. (2008) Microglia-derived TGF-beta as an important regulator ofglioblastoma invasion—an inhibition of TGF-beta-dependent effects by shRNAagainst human TGF-beta type II receptor. Oncogene 27:918–930.
13. Ogorelkova M, et al. (2006) Adenovirus-delivered antisense RNA and shRNA exhibitdifferent silencing efficiencies for the endogenous transforming growth factor-beta(TGF-beta) type II receptor. Oligonucleotides 16:2–14.
14. Sebestyén A, Hajdu M, Kis L, Barna G, Kopper L (2007) Smad4-independent, PP2A-dependent apoptotic effect of exogenous transforming growth factor beta 1 inlymphoma cells. Exp Cell Res 313:3167–3174.
15. He W, et al. (2006) Hematopoiesis controlled by distinct TIF1gamma and Smad4branches of the TGFbeta pathway. Cell 125:929–941.
16. Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of shortinterfering RNAs in mammalian cells. Science 296:550–553.
17. Levine AJ, Momand J, Finlay CA (1991) The p53 tumour suppressor gene. Nature 351:453–456.
18. el-Deiry WS, et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell75:817–825.
Kojima et al. www.pnas.org/cgi/content/short/1013805107 3 of 9

Fig. S1. (A) Isolation of control fibroblasts. As a control, normal GFP-labeled, puromycin-resistant, immortalized human mammary stromal fibroblasts wereinjected s.c. into nude mice as pure cultures without MCF-7-ras cells. Clusters of fibroblasts, which survived at the site of injection for 42 d after implantation,were dissected, dissociated, and cultured in puromycin-containing media. The resulting puromycin-resistant cells were termed control fibroblast-1 cells. Thesecells were once again injected alone s.c. into nude mice for an additional 200 d. The puromycin-resistant cells were similarly extracted and termed controlfibroblast-2 cells (242 d old). (B) The mesenchymal nature and human origin of the exp-CAFs and control fibroblasts. Immunofluorescence analysis of controlfibroblast-2 (control f.) and exp-CAF2 cells showed intense positive staining for mesenchymal markers, including human (but not mouse) vimentin, prolyl-4-hydroxylase, collagen 1A, fibronectin, S100A4/FSP-1, and fibroblast surface protein. In contrast, the epithelial marker pan-cytokeratin was not detected in thesecells. Collectively, these data indicate the human origin and mesenchymal nature of exp-CAF2 and control fibroblast-2 cells. GFP fluorescence of exp-CAF2 cellswas also shown. Cell nuclei are stained with DAPI (blue). (Scale bar, 50 μm.) (C) Stable expression of α-SMA in four independent exp-CAF2 cells. Western blotanalysis of fibroblasts using an anti-α-SMA antibody. The membrane stripped was reprobed by an anti-β-actin antibody. (D) No gene transfer of the oncogenicras into exp-CAFs in vivo. The set of PCR primers permitted only the specific amplification of DNA fragments containing two mutations present in the on-cogenic v-H-ras gene expressing in MCF-7-ras cells. Genomic- and RT-PCR analyses using these primers failed to detect the v-H-ras allele in the genomic DNA andcDNA of either control fibroblast-2 (control f.), 42-d-old exp-CAF1, or exp-CAF2 cells, whereas the v-H-ras allele was readily detected in the DNA of MCF-7-rascells. Primers to amplify glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were also used as an internal control. In addition, an anti-pan-Ras antibody thatrecognizes Arg12 mutant forms of the various Ras oncoproteins but not wild-type RasGly-12 proteins, readily detected the v-H-RasArg-12 protein expressed byMCF-7-ras cells. However, this antibody failed to detect such protein in either fibroblast population. The membrane treated with the anti-pan-Ras antibody wasalso probed by an anti-β-actin antibody. (E) Increased neoangiogenesis in the exp-CAF2-containing tumors. *P < 0.05. Error bars, SE. Staining of sectionsprepared from tumor xenografts admixed with exp-CAF2 (a), 42-d-old exp-CAF1 (b), or control fibroblast-2 cells (c) by Masson’s trichrome, staining collagen inblue and microvessels. Immunostaining of an exp-CAF2 cell-containing tumor section was performed using an anti-CD31 antibody (d, red). GFP fluorescence ofexp-CAF2 (e, green) and control fibroblast-2 cells (f, green) shown in the advanced tumors. Cell nuclei stained with DAPI (d–f, blue). (Scale bar, 100 μm.)
Kojima et al. www.pnas.org/cgi/content/short/1013805107 4 of 9
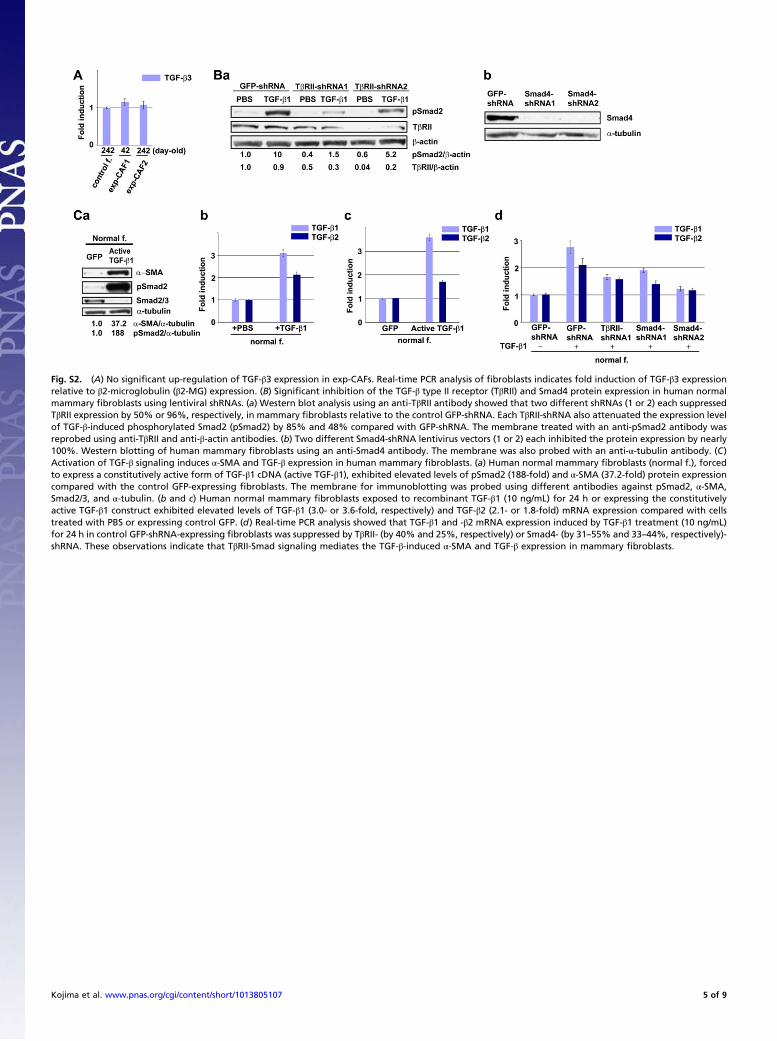
Fig. S2. (A) No significant up-regulation of TGF-β3 expression in exp-CAFs. Real-time PCR analysis of fibroblasts indicates fold induction of TGF-β3 expressionrelative to β2-microglobulin (β2-MG) expression. (B) Significant inhibition of the TGF-β type II receptor (TβRII) and Smad4 protein expression in human normalmammary fibroblasts using lentiviral shRNAs. (a) Western blot analysis using an anti-TβRII antibody showed that two different shRNAs (1 or 2) each suppressedTβRII expression by 50% or 96%, respectively, in mammary fibroblasts relative to the control GFP-shRNA. Each TβRII-shRNA also attenuated the expression levelof TGF-β-induced phosphorylated Smad2 (pSmad2) by 85% and 48% compared with GFP-shRNA. The membrane treated with an anti-pSmad2 antibody wasreprobed using anti-TβRII and anti-β-actin antibodies. (b) Two different Smad4-shRNA lentivirus vectors (1 or 2) each inhibited the protein expression by nearly100%. Western blotting of human mammary fibroblasts using an anti-Smad4 antibody. The membrane was also probed with an anti-α-tubulin antibody. (C)Activation of TGF-β signaling induces α-SMA and TGF-β expression in human mammary fibroblasts. (a) Human normal mammary fibroblasts (normal f.), forcedto express a constitutively active form of TGF-β1 cDNA (active TGF-β1), exhibited elevated levels of pSmad2 (188-fold) and α-SMA (37.2-fold) protein expressioncompared with the control GFP-expressing fibroblasts. The membrane for immunoblotting was probed using different antibodies against pSmad2, α-SMA,Smad2/3, and α-tubulin. (b and c) Human normal mammary fibroblasts exposed to recombinant TGF-β1 (10 ng/mL) for 24 h or expressing the constitutivelyactive TGF-β1 construct exhibited elevated levels of TGF-β1 (3.0- or 3.6-fold, respectively) and TGF-β2 (2.1- or 1.8-fold) mRNA expression compared with cellstreated with PBS or expressing control GFP. (d) Real-time PCR analysis showed that TGF-β1 and -β2 mRNA expression induced by TGF-β1 treatment (10 ng/mL)for 24 h in control GFP-shRNA-expressing fibroblasts was suppressed by TβRII- (by 40% and 25%, respectively) or Smad4- (by 31–55% and 33–44%, respectively)-shRNA. These observations indicate that TβRII-Smad signaling mediates the TGF-β-induced α-SMA and TGF-β expression in mammary fibroblasts.
Kojima et al. www.pnas.org/cgi/content/short/1013805107 5 of 9
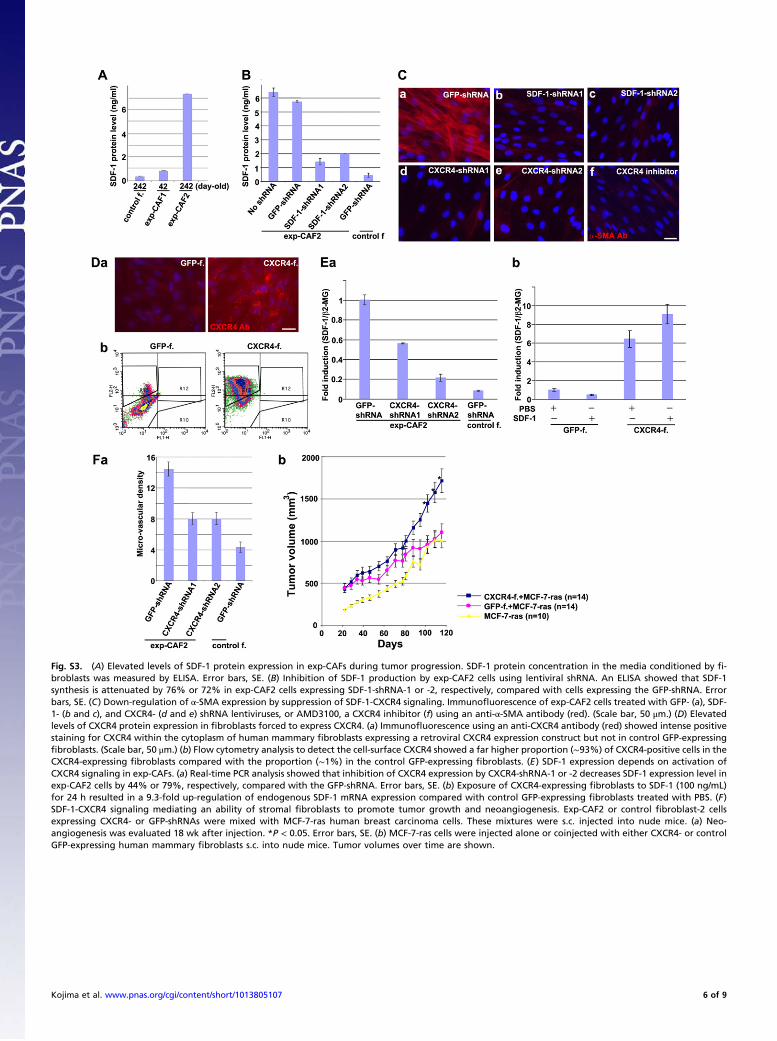
Fig. S3. (A) Elevated levels of SDF-1 protein expression in exp-CAFs during tumor progression. SDF-1 protein concentration in the media conditioned by fi-broblasts was measured by ELISA. Error bars, SE. (B) Inhibition of SDF-1 production by exp-CAF2 cells using lentiviral shRNA. An ELISA showed that SDF-1synthesis is attenuated by 76% or 72% in exp-CAF2 cells expressing SDF-1-shRNA-1 or -2, respectively, compared with cells expressing the GFP-shRNA. Errorbars, SE. (C) Down-regulation of α-SMA expression by suppression of SDF-1-CXCR4 signaling. Immunofluorescence of exp-CAF2 cells treated with GFP- (a), SDF-1- (b and c), and CXCR4- (d and e) shRNA lentiviruses, or AMD3100, a CXCR4 inhibitor (f) using an anti-α-SMA antibody (red). (Scale bar, 50 μm.) (D) Elevatedlevels of CXCR4 protein expression in fibroblasts forced to express CXCR4. (a) Immunofluorescence using an anti-CXCR4 antibody (red) showed intense positivestaining for CXCR4 within the cytoplasm of human mammary fibroblasts expressing a retroviral CXCR4 expression construct but not in control GFP-expressingfibroblasts. (Scale bar, 50 μm.) (b) Flow cytometry analysis to detect the cell-surface CXCR4 showed a far higher proportion (∼93%) of CXCR4-positive cells in theCXCR4-expressing fibroblasts compared with the proportion (∼1%) in the control GFP-expressing fibroblasts. (E) SDF-1 expression depends on activation ofCXCR4 signaling in exp-CAFs. (a) Real-time PCR analysis showed that inhibition of CXCR4 expression by CXCR4-shRNA-1 or -2 decreases SDF-1 expression level inexp-CAF2 cells by 44% or 79%, respectively, compared with the GFP-shRNA. Error bars, SE. (b) Exposure of CXCR4-expressing fibroblasts to SDF-1 (100 ng/mL)for 24 h resulted in a 9.3-fold up-regulation of endogenous SDF-1 mRNA expression compared with control GFP-expressing fibroblasts treated with PBS. (F)SDF-1-CXCR4 signaling mediating an ability of stromal fibroblasts to promote tumor growth and neoangiogenesis. Exp-CAF2 or control fibroblast-2 cellsexpressing CXCR4- or GFP-shRNAs were mixed with MCF-7-ras human breast carcinoma cells. These mixtures were s.c. injected into nude mice. (a) Neo-angiogenesis was evaluated 18 wk after injection. *P < 0.05. Error bars, SE. (b) MCF-7-ras cells were injected alone or coinjected with either CXCR4- or controlGFP-expressing human mammary fibroblasts s.c. into nude mice. Tumor volumes over time are shown.
Kojima et al. www.pnas.org/cgi/content/short/1013805107 6 of 9

Fig. S4. (A) Induction of CXCR4 expression by TGF-β1 in human mammary fibroblasts. RT-PCR analysis of TGF-β1-treated mammary fibroblasts detecting CXCR4and β2-microglobulin (β2-MG) expression. (B) SDF-1 expression induced by TGF-β is mediated through TβRII-Smad signaling. Real-time PCR analysis showed thatSDF-1 expression induced by TGF-β1 (10 ng/mL) for 24 h in normal mammary fibroblasts expressing GFP-shRNA, was attenuated in cells expressing TβRII- (by70%) or Smad4- (by 62–70%) shRNA. (C) TGF-β-induced CXCR4 expression is independent of the Smad signaling pathway. RT-PCR analysis of TGF-β1-treatedmammary fibroblasts detecting CXCR4 and β2-MG expression. Inhibition of Smad signaling by Smad4-shRNA-1 or -2 failed to suppress CXCR4 expression in-duced by treatment with TGF-β1 (10 ng/mL) for 24 h in human mammary fibroblasts compared with the effect of GFP-shRNA. (D) Induction of CXCR4 signalingelevates TGF-β expression in mammary fibroblasts. Real-time PCR analysis was performed using primers specific to TGF-β1 and 2. Either control GFP- or CXCR4-expressing human mammary fibroblasts were cultured in the presence or absence of SDF-1 protein (100 ng/mL). Exposure of CXCR4-expressing fibroblasts torecombinant SDF-1 protein (100 ng/mL) for 24 h resulted in up-regulation of endogenous TGF-β1 (4.4-fold) and TGF-β2 (4.2-fold) mRNA expression comparedwith control GFP-expressing fibroblasts cultured without SDF-1.
Fig. S5. α-SMA, CXCR4, phosphorylated Smad2, and SDF-1 proteins are not detected in stromal fibroblasts in normal breast tissue. Immunohistochemistry ofsections prepared from the normal human breast tissue using antibodies against α-SMA (A, green), CXCR4 (A, red), and pSmad2 (B, brown). Sections were alsostained with DAPI (A, blue) or hematoxylin (B, pale blue). Stromal fibroblast-like cells staining negative for α-SMA/CXCR4 (A) or pSmad2 (B) are highlighted byarrows. Arrowheads depict α-SMA-positive myoepithelial cells (A, green). Normal histology of mammary gland is indicated by asterisks. (Scale bar, 50 μm.) Wehave previously observed that stromal fibroblasts in the normal human breast tissue are negative for SDF-1 (4).
Kojima et al. www.pnas.org/cgi/content/short/1013805107 7 of 9

Fig. S6. (A) CAFs prepared from breast cancer patients show elevated levels of SDF-1 and TGF-β expression. Primary stromal mammary fibroblasts wereisolated from three different patients with invasive ductal breast cancer. CAFs (designated CAF1, CAF3, and CAF6) were extracted from the tumor mass, andcounterpart fibroblasts (counter.f.1, counter.f.3, and counter.f.6) were isolated from the noncancerous breast stroma of the same individual as a patient-specific control. The tumor-promoting ability of these CAFs has been confirmed in our previous work (4). Real-time PCR analysis and a luciferase assay showedincreased levels of SDF-1, TGF-β2, and active TGF-β expression in CAF1 (3.2-, 2.3-, and 1.6-fold, respectively), CAF3 (2.2-, 1.2-, and 0.8-fold), and CAF6 (4.4-, 2.7-,and 6.1-fold) cell populations in comparison with their control counterpart fibroblasts. Error bars, SE. (B) Autocrine signaling loops mediated by SDF-1 and TGF-β operate in the patient-derived CAFs in both self-stimulating and cross-communicating fashions. (a) Real-time PCR analysis showed that inhibition of SDF-1 orTGF-β signaling by SDF-1-, CXCR4-, TβRII-, and Smad4-shRNA in the patient-derived CAF1 cells suppressed the expression levels of SDF-1 (by 50–60%, 54–60%,88%, or 86–94%, respectively), TGF-β2 (50–67%, 61–69%, 48%, or 69–82%), and α-SMA (77–84%, 77–85%, 49%, or 73–85%,) compared with the effect of thecontrol GFP-shRNA. (b) α-SMA-positive cells (%) were measured by immunofluorescence analysis using an anti-α-SMA antibody. Inhibition of SDF-1 or TGF-βsignaling by SDF-1-, CXCR4-, TβRII-, or Smad4-shRNA in the patient-derived CAF1 cells decreased the proportion of α-SMA-positive cells (by 92–98%, 94–94%,38%, or 46–99%, respectively) compared with the control GFP-shRNA. Error bars, SE.
Kojima et al. www.pnas.org/cgi/content/short/1013805107 8 of 9
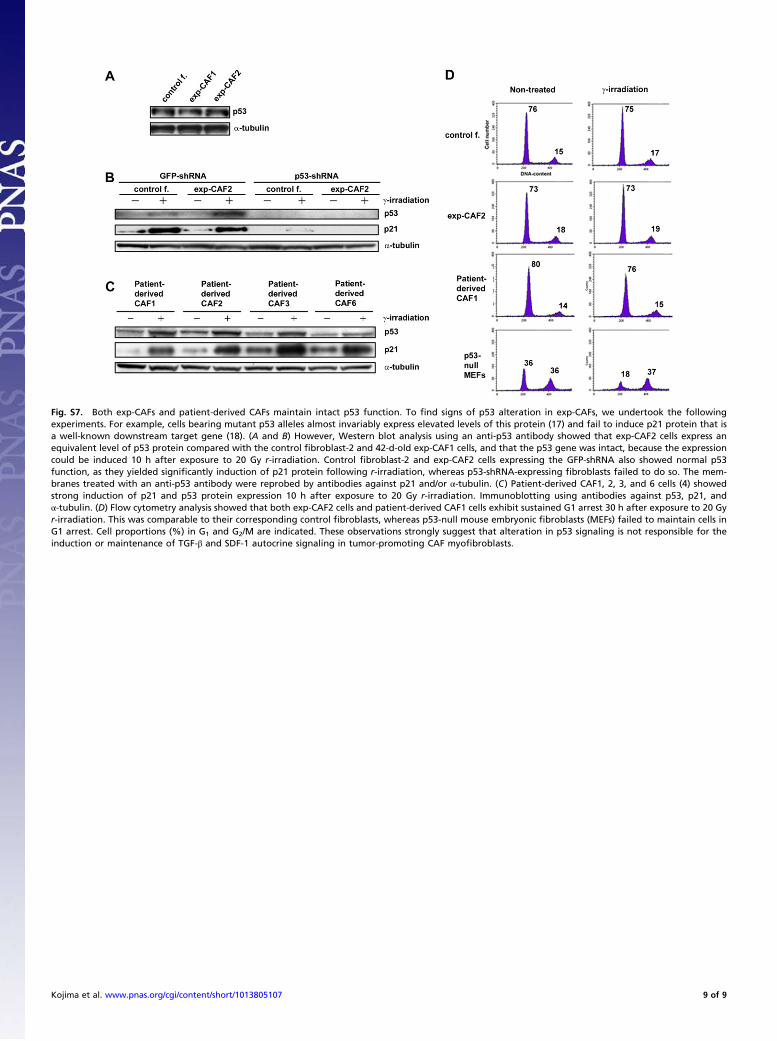
Fig. S7. Both exp-CAFs and patient-derived CAFs maintain intact p53 function. To find signs of p53 alteration in exp-CAFs, we undertook the followingexperiments. For example, cells bearing mutant p53 alleles almost invariably express elevated levels of this protein (17) and fail to induce p21 protein that isa well-known downstream target gene (18). (A and B) However, Western blot analysis using an anti-p53 antibody showed that exp-CAF2 cells express anequivalent level of p53 protein compared with the control fibroblast-2 and 42-d-old exp-CAF1 cells, and that the p53 gene was intact, because the expressioncould be induced 10 h after exposure to 20 Gy r-irradiation. Control fibroblast-2 and exp-CAF2 cells expressing the GFP-shRNA also showed normal p53function, as they yielded significantly induction of p21 protein following r-irradiation, whereas p53-shRNA-expressing fibroblasts failed to do so. The mem-branes treated with an anti-p53 antibody were reprobed by antibodies against p21 and/or α-tubulin. (C) Patient-derived CAF1, 2, 3, and 6 cells (4) showedstrong induction of p21 and p53 protein expression 10 h after exposure to 20 Gy r-irradiation. Immunoblotting using antibodies against p53, p21, andα-tubulin. (D) Flow cytometry analysis showed that both exp-CAF2 cells and patient-derived CAF1 cells exhibit sustained G1 arrest 30 h after exposure to 20 Gyr-irradiation. This was comparable to their corresponding control fibroblasts, whereas p53-null mouse embryonic fibroblasts (MEFs) failed to maintain cells inG1 arrest. Cell proportions (%) in G1 and G2/M are indicated. These observations strongly suggest that alteration in p53 signaling is not responsible for theinduction or maintenance of TGF-β and SDF-1 autocrine signaling in tumor-promoting CAF myofibroblasts.
Kojima et al. www.pnas.org/cgi/content/short/1013805107 9 of 9
Related Documents











