Lecture 1 Autism Spectrum Disorders (ASD) David Saffen, Ph.D. Professor/Principal Investigator Department of Cellular and Genetic Medicine Fudan University, Shanghai, China Email: [email protected]

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Lecture 1Autism Spectrum Disorders (ASD)
David Saffen, Ph.D.Professor/Principal Investigator
Department of Cellular and Genetic MedicineFudan University, Shanghai, China
Email: [email protected]

OutlineA. Introduction
- Brief history of autism spectrum disorders (ASD)- Core clinical features - Diagnosis- Comorbidities- Epidemiology and societal burden- Behavioral and drug therapies- Prognosis and management- Famous individuals with ASD
B. Causes of ASD: genetics, neurobiology, environment - Genetics- Neurobiology- Environmental risk factors
C. Developing new therapies to treat ASD- Mouse models- Novel therapeutic approaches
D. References, journal presentations, internet resources, additional slides

A. Introduction

Autism spectrum disorders (ASD) comprise a group of neurodevelopmental disorders characterized by:
1) social impairments, 2) language difficulties 3) repetitive and stereotyped behaviors.
The term "spectrum" refers to the wide range of symptoms and levels of impairment in affected individuals.
Diagnosis is usually made in early childhood
Some children are mildly impaired by their symptoms, but others are severely disabled.

Early behaviors often first noticed by parents
Children with ASD often:- do not “babble” as infants; are very slow to develop language; display
continuing language deficits (e.g. mutism, echolalia, poverty of speech)
- make little eye contact
- do not look at or listen to people in their environment and fail to respond when talked to
- do not readily share their enjoyment of toys or activities by pointingor showing things to others; show little interest in other children
- do not play with toys in a normal manner; engage in “stimming” or other obsessive or stereotypical behaviors
- respond unusually when others show anger, distress, or affection

Pioneer investigators who identified autism as a distinct neurodevelopmental disorder
Hans Asperger(1906-1980)
Austrian pediatricianUniversity Children’s
Hospital, Vienna
Leo Kanner(1896-1981)
American psychiatrist(child and adolescent)
Johns Hopkins University

Core clinical features
• Abnormal development and use of language
• Impairments in reciprocal social interactions
• Restricted interests
• Repetitive and ritualized behaviors
Note: ASD is a behaviorally defined disorder

Clinical course of ASD
• ASD diagnosis is usually made in early childhood: ages 2 – 5
• Parents often report onset of symptoms within the first 12 – 18 months
• Also observed: normal development through the first 18 – 24 months, followed by regression (25 –40% of cases)
• Although the spectrum and severity of symptoms vary, many individuals with ASD experience life-long deficits.
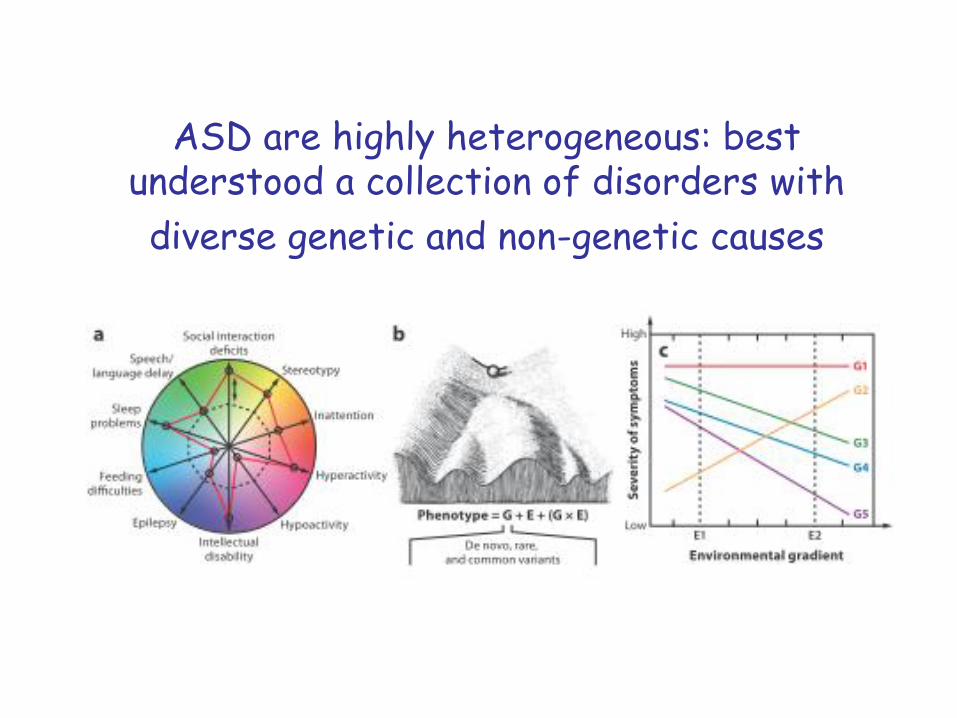
ASD are highly heterogeneous: best understood a collection of disorders with
diverse genetic and non-genetic causes

Hierarchical approach to understanding ASD
Ecker, Spooren & Murphy, 2013

Classification of autism spectrum disorders (ASD)
• DSM-IV-TRTM (Diagnostic and Statistical Manual-Fourth Edition; American Psychiatry Association)
- Autistic disorder (Classical autism)- Asperger’s disorder (Asperger’s syndrome)- Pervasive developmental disorder not
otherwise specified (PDD-NOS)- Rett’s disorder (Rett syndrome)- Childhood disintegrative disorder (CDD)

Diagnosis: Autism spectrum disorder
A. Social communication1) deficits in social-emotional reciprocity 2) deficits in nonverbal communicative
behaviors used for social interactions3) deficits in developing, maintaining and
understanding relationships
B. Restricted, repetitive behaviors1) stereotyped or repetitive movements, use of objects or speech2) insistence on sameness, inflexible adherence to routines, or
ritualized patterns of behavior 3) highly restricted, fixed interests that are abnormal in intensity
or focus4) hyper- or hypo-reactivity to sensory input or unusual interest in
sensory aspects of the environment
C. Symptoms must be present in the early developmental period
D. Symptoms cause clinically significant impairment in social, occupationalor other important areas of current functioning
DSM-5 (Diagnostic and Statistical Manual-Fifth Edition, 2013)

• ICD-10 (International Statistical Classification of Diseases and Related Health Problems, 10th Revision; World Health Organization)
(F84) Pervasive developmental disorders (PDDs):- Childhood autism- Atypical autism- Rett’s syndrome- Other childhood disintegrative disorder- Overactive disorder associated with mental
retardation and stereotyped movements- Asperger syndrome
Note: ICD-11 to be published in 2018
Chinese classification of Mental Disorders, Third Edition (CCMD-3), Chinese Society of Psychiatry (CSP), 2001

Epidemiology and societal burden
• Present in 0.7 – 1.1% of population (prevalence)(Possibly over-diagnosed in the US and underdiagnosed in China)
• Incidence:Classic autism: 1-2 per 1000Asperger syndrome: 0.6 per 1000Pervasive developmental disorder-not otherwise specified (PDD-NOS): 3.7 per 1000Rett’s syndrome: 0.1 per 1000 Childhood disintegrative disorder (CDD) 0.02 per 1000
• More common among males compared to females (4.7-to-1)
• Prognosis: many affected individuals are never able to live independently; Estimated annual cost (2012): $126B (US)
1 in 68 (ASD at age 8)
Boys: 1 in 42Girls: 1 in 189

ASD Comorbidities and secondary symptoms
ASD is typically associated with other psychiatric or medical conditions:- intellectual disability (40 – 60%)- epilepsy (30%)- attention-deficit/hyperactivity disorder (ADHD)- anxiety and aggression (anxiety + ADHD: 30-50%)- mood disorders- tics- motor control problems- sleep disorders- gastrointestinal problems- abnormal responses to sensory stimuli
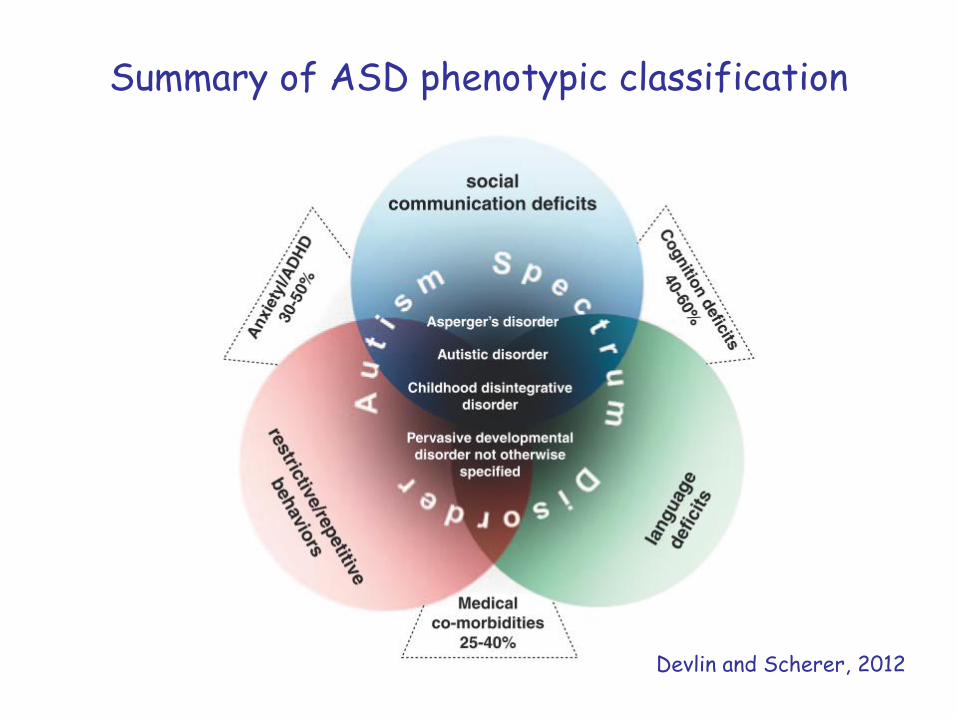
Summary of ASD phenotypic classification
Devlin and Scherer, 2012

Behavioral therapies
There is accumulating evidence that early, focused behavioral therapies are effective in reducing the core symptoms of ASD.
For children:- Early intensive behavioral intervention (EIBI)- Applied behavior analysis (ABA)- Structured teaching- Speech and language therapy
For adolescents and adults with ASD- Social skill therapy- Occupational therapy - Assisted living

Drug used to treat patients with ASDThere are currently no drugs available to treat the causes or core
symptoms of ASD. Rather, medications are used to treat comorbid symptoms, including:
- attention-deficit/hyperactivity (ADHD)(stimulants: e.g., methylphenidate)
- depression and anxiety(antidepressants: e.g., fluoxetine, sertraline)
- stereotypy irritability, temper tantrums, aggression, self-harming acts (antipsychotic drugs: e.g., respiridone, aripripazole)
Note: psychotropic drugs have serious side-effects in some patients:- antidepressants: suicidal ideation - antipsychotic drugs: obesity and type II diabetes

Famous individuals with ASD
Temple Grandin, PhDProfessor Animal Sciences
Tito Mukhopadhyay(with mother)
Tim Page, Music Critic, Pulitzer prize
winner
Daryl HannahActress
Vernon L Smith, PhD Noble Prize in
Economics 2002
Danny Beath,Photographer

Autistic savants
Daniel TammetBritish author
Stephen WiltshireBritish architectural artist
Matt Savage American jazz musician
Richard WawroScottish artist
Henrietta Seth F.Hungarian poet, writer, artist

B. Causes of ASD: Genetics, neurobiology, environment

Evidence for genetic contributions to ASD risk
• Concordance for monozygotic twins: 82-92%;Concordance for dizygotic twins: 1 – 20%
• Recurrence risk in families with one child with ASD: estimated 5 – 20%
• Estimated heritability (h2): 0.36 - 0.90
• Epigenetic, stochastic and environmental factor are also thought to contribute to ASD susceptibility
Note: taken together, these results completely refute the once widely held belief that autism is a rare disorder caused by “poor parenting”: Bruno Bettelheim,The empty fortress: infantile autism and the birth of the self, Free Press, N.Y., 1967.
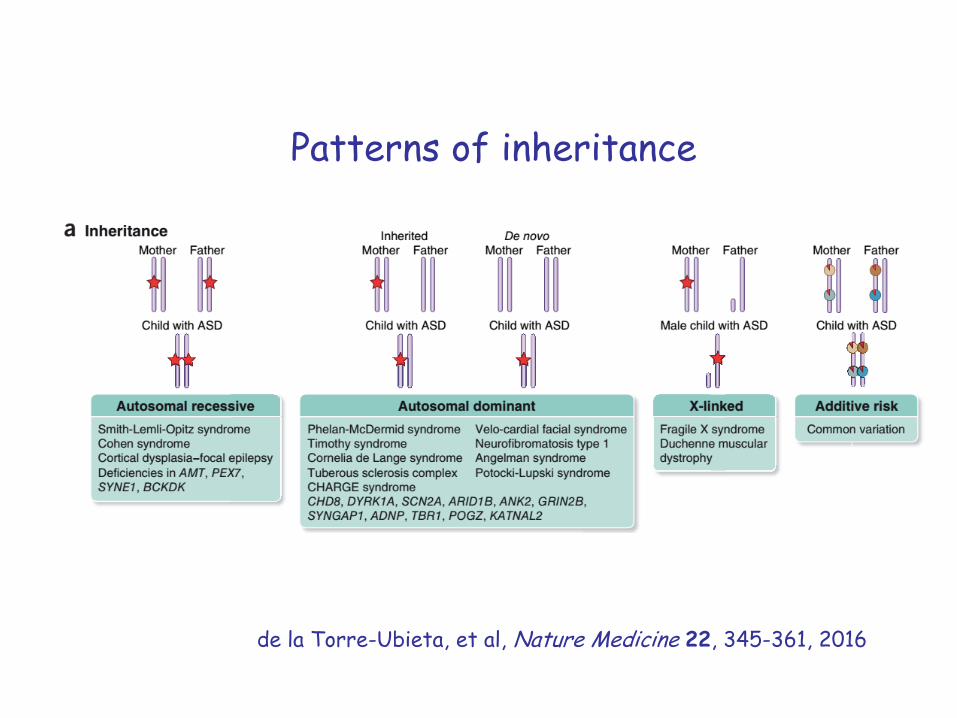
Patterns of inheritance
de la Torre-Ubieta, et al, Nature Medicine 22, 345-361, 2016
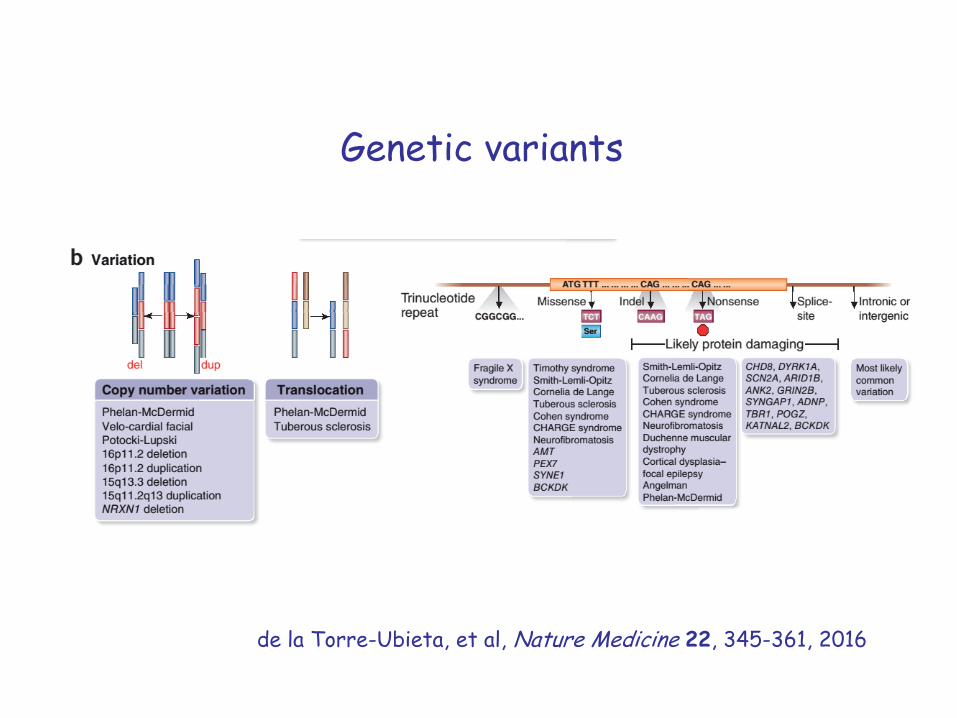
Genetic variants
de la Torre-Ubieta, et al, Nature Medicine 22, 345-361, 2016
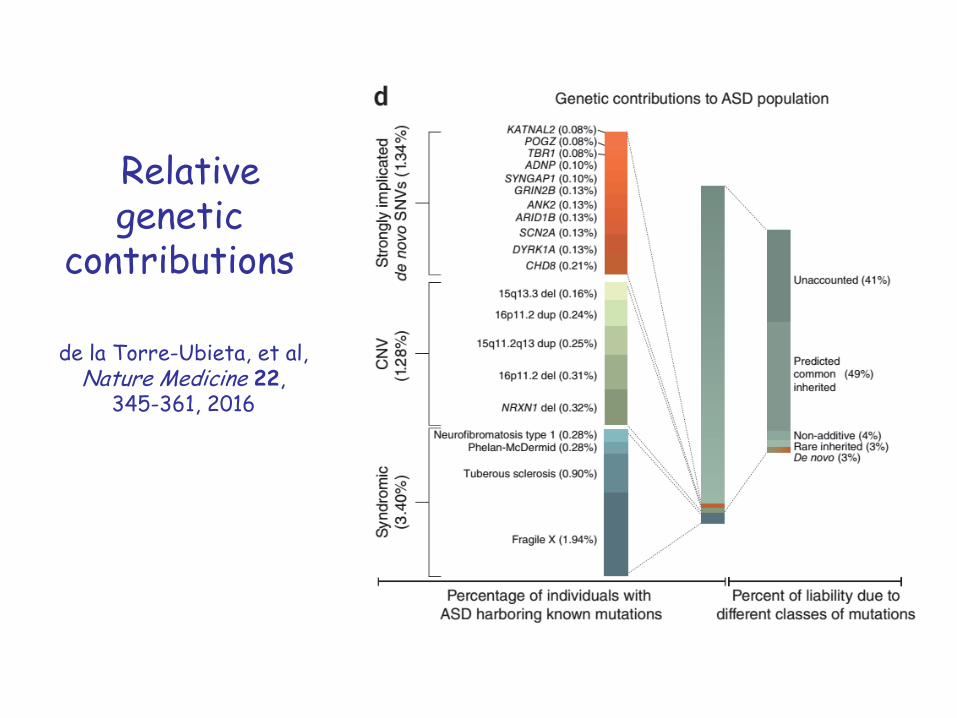
Relativegenetic
contributions
de la Torre-Ubieta, et al, Nature Medicine 22,
345-361, 2016
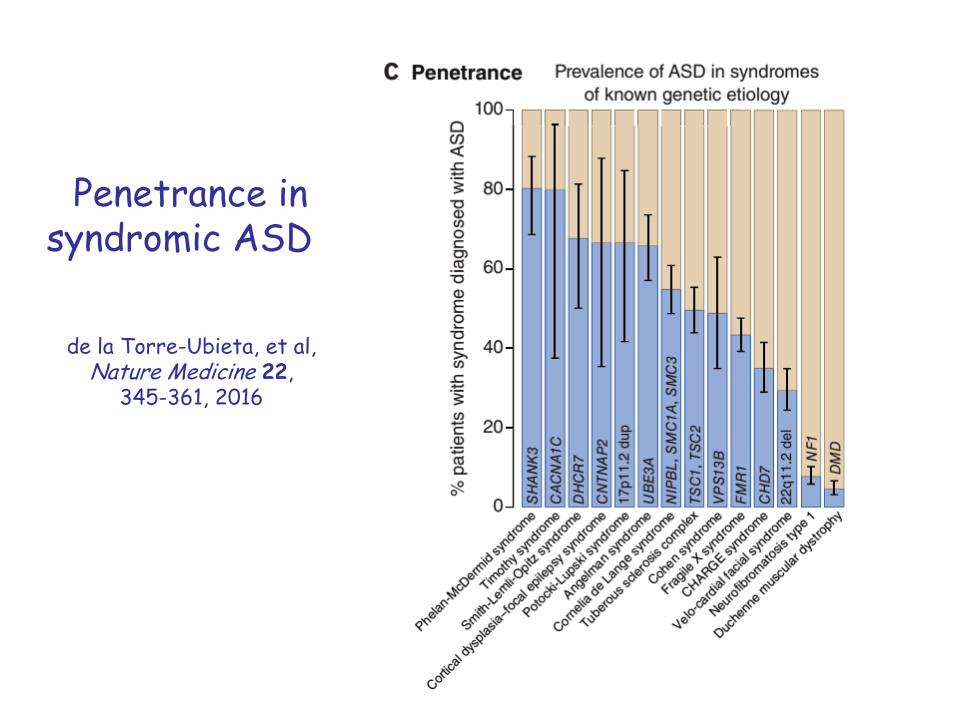
Penetrance in syndromic ASD
de la Torre-Ubieta, et al, Nature Medicine 22,
345-361, 2016

Molecular, cellular and brain-system level contributions
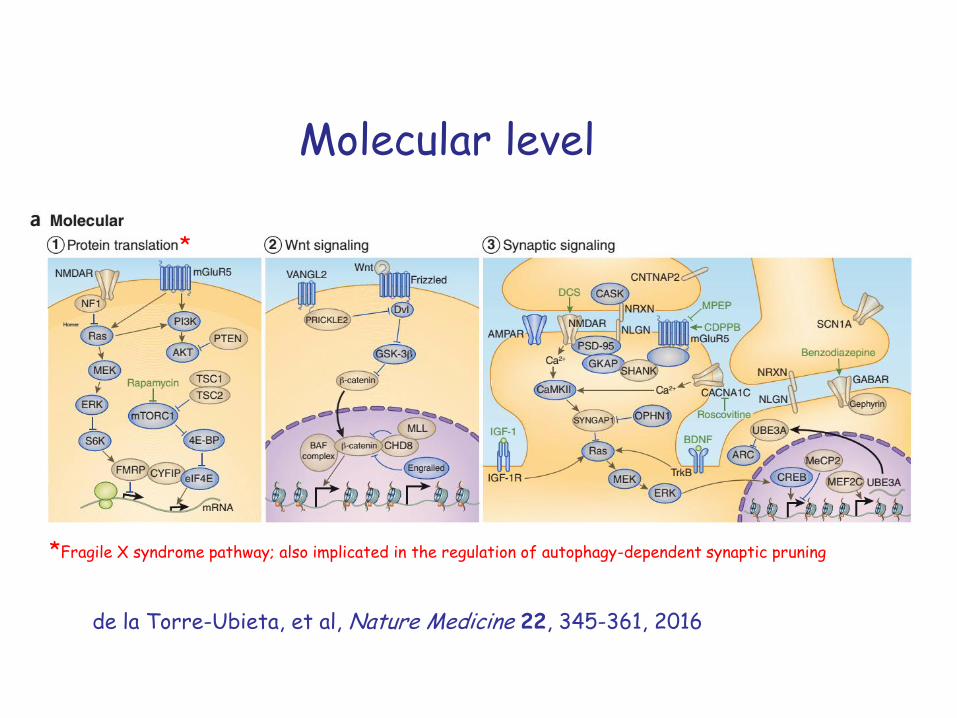
Molecular level
*
*Fragile X syndrome pathway; also implicated in the regulation of autophagy-dependent synaptic pruning
de la Torre-Ubieta, et al, Nature Medicine 22, 345-361, 2016
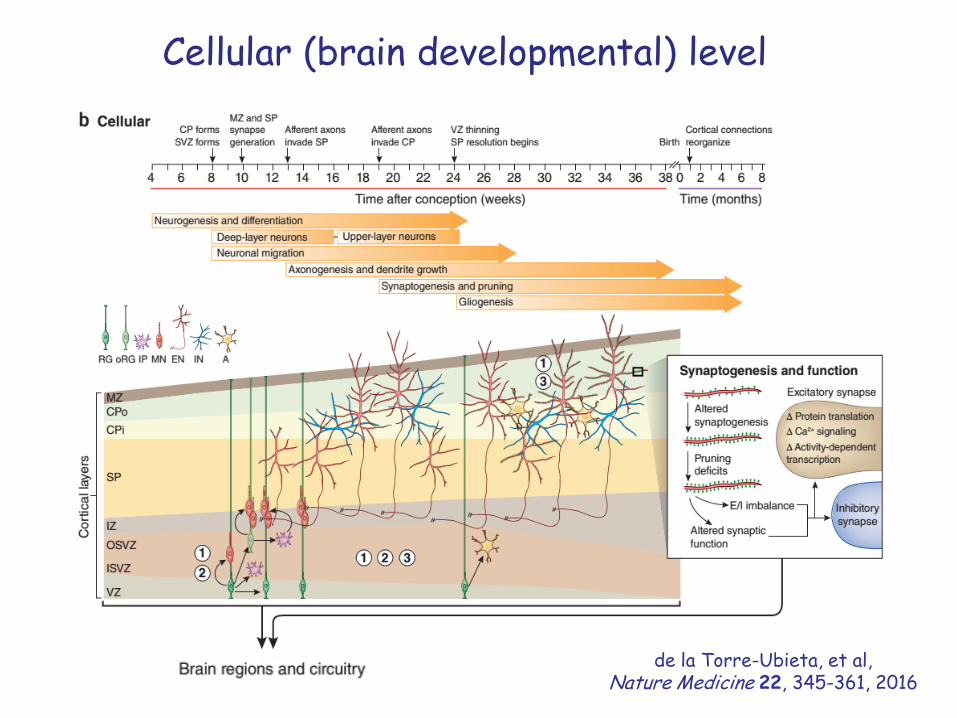
Cellular (brain developmental) level
de la Torre-Ubieta, et al, Nature Medicine 22, 345-361, 2016
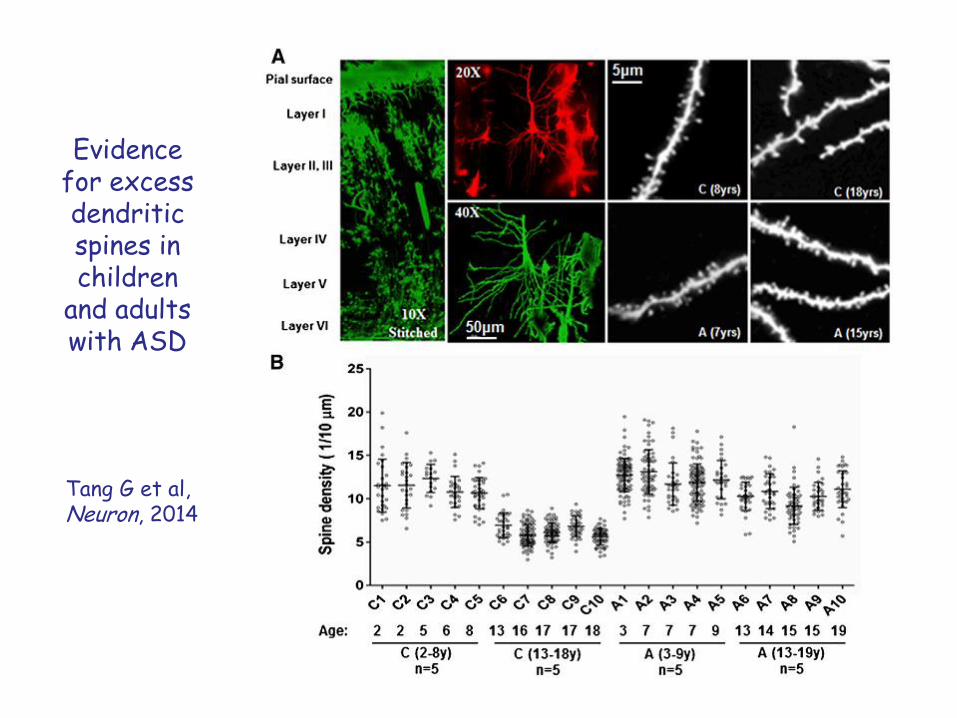
Evidence for excess dendritic spines in children
and adults with ASD
Tang G et al,Neuron, 2014
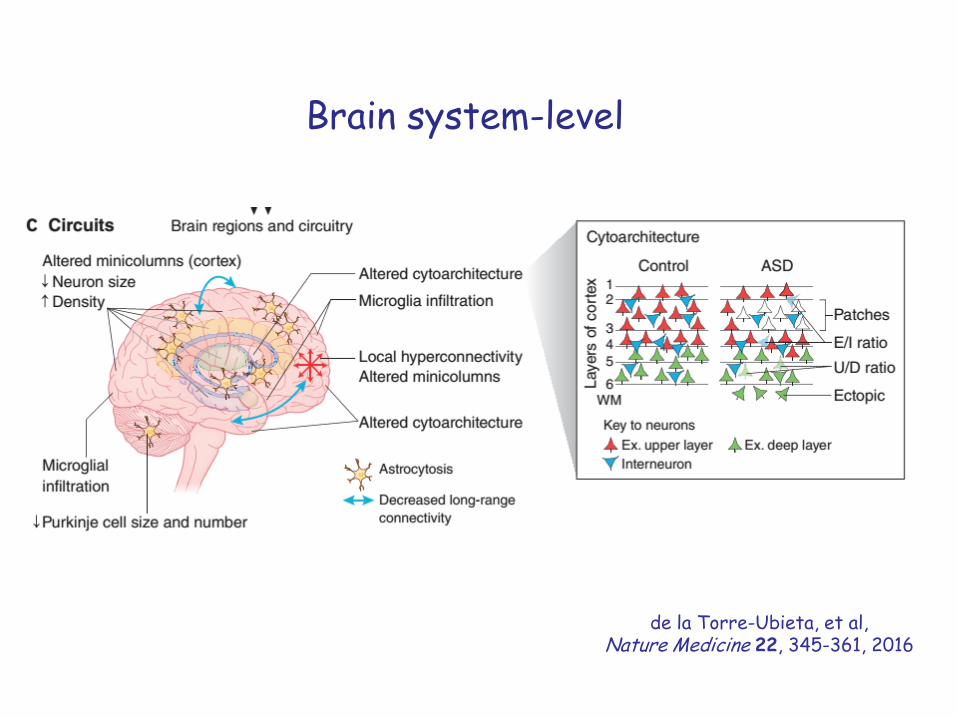
Brain system-level
de la Torre-Ubieta, et al, Nature Medicine 22, 345-361, 2016

Legend for slides 28, 29, 31
Legend for slide 30
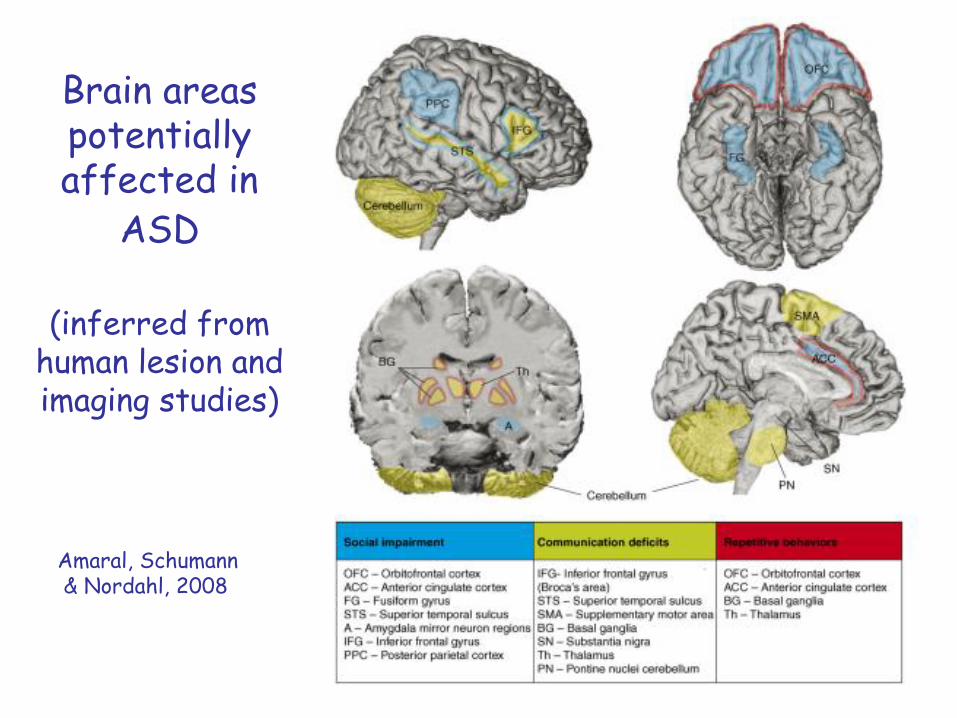
Brain areas potentially affected in
ASD
(inferred from human lesion and imaging studies)
Amaral, Schumann& Nordahl, 2008
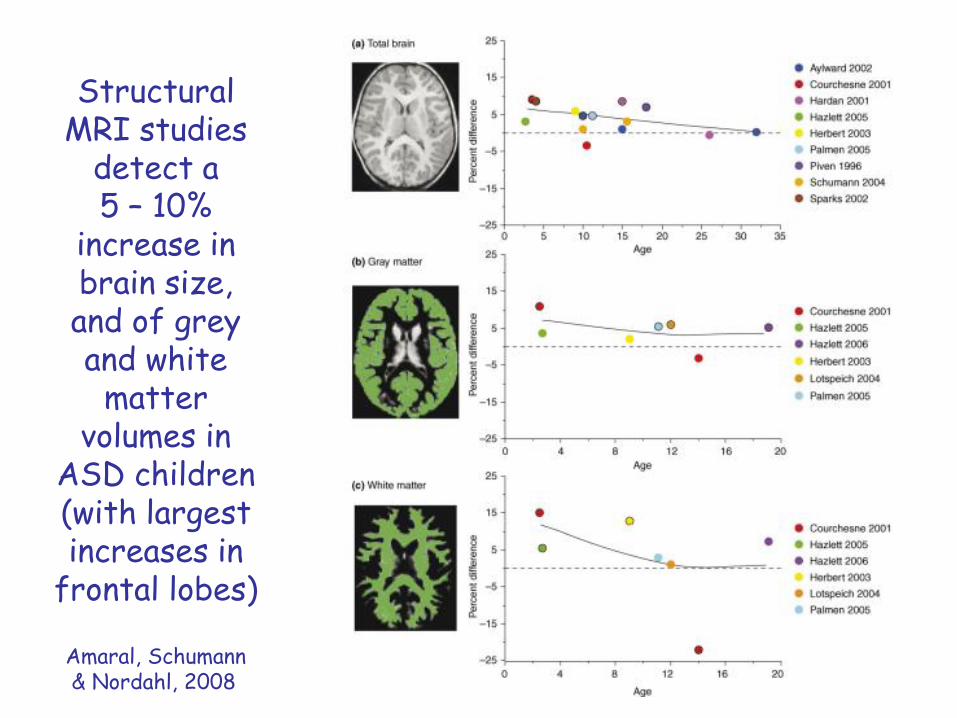
Structural MRI studies
detect a 5 – 10%
increase in brain size, and of grey and white
matter volumes in
ASD children(with largest increases in
frontal lobes)
Amaral, Schumann& Nordahl, 2008
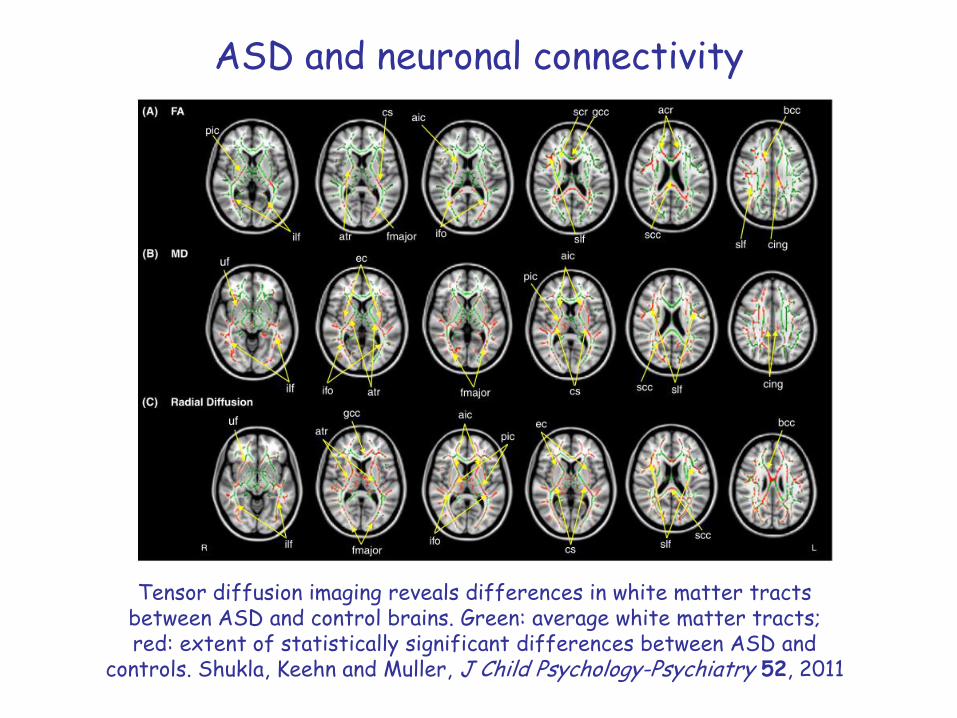
ASD and neuronal connectivity
Tensor diffusion imaging reveals differences in white matter tracts between ASD and control brains. Green: average white matter tracts; red: extent of statistically significant differences between ASD and
controls. Shukla, Keehn and Muller, J Child Psychology-Psychiatry 52, 2011

Environmental factors linked to increased ASD risk
• Pregnancy complications, including: early birth, need for induced labor, distress during delivery, low birth weight
• Maternal: obesity, diabetes, high blood pressure, low serum levels of thyroid hormone (thyroxine) or folic acid
• Prenatal exposure to: rubella (German measles) virus, certain drugs (e.g. thalidomide, valproic acid)
• Advanced paternal age (likely a genetic effect due to increased mutational burden of sperm)
• Note: there is no scientific evidence for links between vaccinations or the mercury-based vaccine preservative thimerosal and ASD

C. Developing new therapies to treat ASD
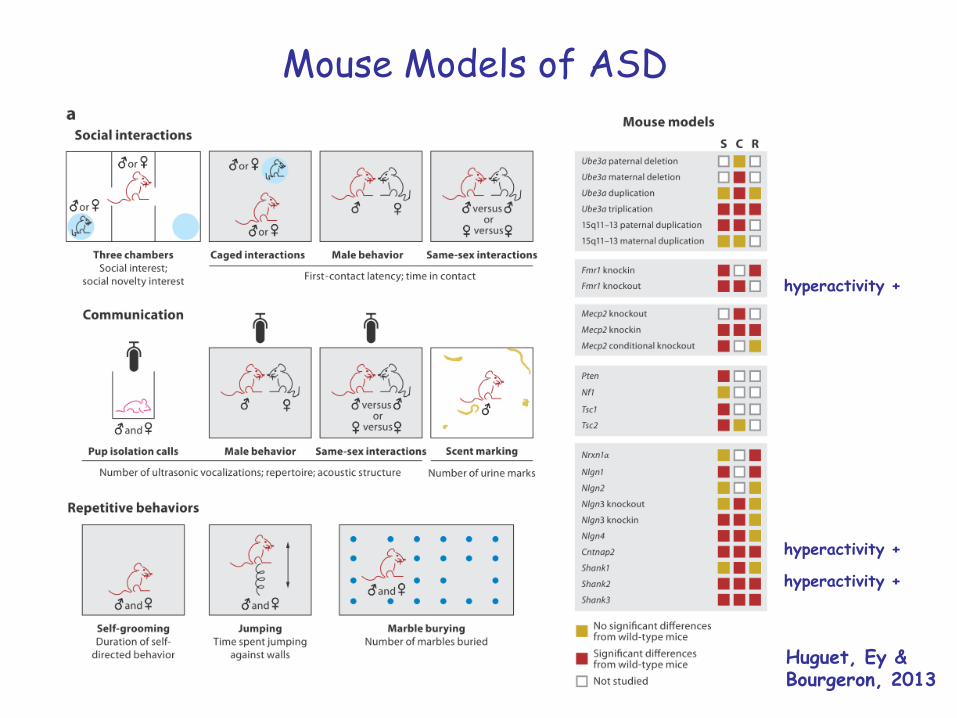
Mouse Models of ASD
Huguet, Ey &Bourgeron, 2013
hyperactivity +
hyperactivity +
hyperactivity +

Possible therapeutic approaches for ASD:Modulation of the fragile-x pathway
• Clinical trials to treat ASD patients with the mGluR5 antagonistAFQ056 are in progress. (Levenga J et al, Neurobiol. Dis. 42, 2011; Jacquemont S, et al, Sci. Transl. Med. 3: 64ra1, 2011)
• The PI3K inhibitor LY294002 reverses increased protein synthesis, AMPAR internalization, and spine density abnormalities in Fmr1 KO mice (Gross C, et al, J. Neuroscience 30, 10624-36, 2010).
• Rapamycin, which inhibits mTORC1 activity by binding the mTORC1 activating protein FKBP, reverses learning deficits and autism-like phenotypes in Tsc2-/+ mice, Ehninger D,
• eIF4E inhibitors of reverse autistic-like traits in E4-BP2 KO mice. (Gkogkas CG et al, Nature 493, 2013)
• GABAA synaptic transmission is impaired in Fmr1 KO mice, suggesting that GABAAreceptor agonists may be effective in treating FXS (Henderson C, et al, Science Translational Medicine 4, 152ra128, 2012). Clinical trials are in progress (Berry-Kravis EM, et al, Science Translational Medicine 4, 152ra127, 2012).

Recent experiments suggest that postnatal therapies can reverse
ASD-related deficits
Examples:
• Conditional reactivation of Mecp2 expression in immature and mature mice restores morphological defects in motor cortex. Guy J et al, Science 315, 2007
• Introduction of wild-type microglia into Mecp2 defective mice arrested development of the disorder. Derecki et al, Nature484, 2012
• Re-expression of Nlgn3 in juvenile KO mice, restored synaptic function and mGluR-dependent synaptic plasticity. Baudouin SJ, et al. Science 338, 2012

Summary
• Despite extensive heterogeneity in the clinical features and causes for ASD, recent research has identified possible common etiological roots for these disorders, including:
- Defective development and functioning of synapses- Abnormal excitation-inhibition imbalance- Abnormal brain connectivity
• Genetic analysis has identified specific genes and biological pathways that contribute to ASD
• Clinical trials of novel drugs are now in progress to ameliorate or reverse key ASD symptoms. The observation that ASD-related abnormalities can sometimes be reverse in post natal mice, provides hope that treatment of children or adults with ASD may also be effective.

D. References, ASD resources, andadditional lecture-related slides

References (1)• De la Torre-Ubieta, et al, Advancing the understanding of autism disease mechanisms
through genetics, Nature Medicine 22, 345- 361, 2016
• Geschwind DH, Gene hunting in autism spectrum disorder: on the path to precision medicine, Lancet, April 17, 2015
• Huguet G, Ey E and Bourgeron T, The genetic landscapes of autism spectrum disorders, Annual Reviews in Genomics and Human Genetics, July 23, 2013
• Devlin B and Scherer S, Genetic architecture in autism spectrum disorder,Current Opinion in Genetics & Development 22, 229-237, 2012
• Amaral DG, Schumann CM and CW Nordahl, Neuroanatomy of autism, Trends in Neurosciences 31, 137-145, 2008
• Nelson SB and Valakh V, Excitation/Inibitory balance and circuit homeostasis in autism spectrum disorder, Neuron 87, August, 2015
• Tang G, et al, Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits, Neuron, 2014
• Gilman SR, et al, Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses, Neuron 70, 898-907, 2011
• Iossifov I, et al, De novo gene disruptions in children on the autistic spectrum, Neuron 74, 285-299, 2012

References (2)
• Sanders S, et al, De novo mutations revealed by whole exome sequencing are strongly associated with autism, Nature 485, 237-241, 2013
• Klei L et al, Common genetic variants, acting additively, are a major source of risk for autism, Molecular Autism 3: 9, 2012
• Psychiatric Genomics Consortium, Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs, Nature Genetics, August 11, 2013
• Alley CS, Gillberg C and Wilson P, Neurobiological abnormalities in the first few years of life in individuals later diagnosed with Autistic Spectrum Disorder: A review of recent data, Behavioural Neurobiology, 1-21, 2013
• Ecker C, et al., Neuroimaging autism spectrum disorder: brain structure and function across the lifespan, Lancet, April 17, 2015
• Zoghbi HY and Bear MF, Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities, Cold Spring Harbor Perspectives in Biology 2012; 4:a009886
• Ecker C, Spooeren W & Murphy DGM, Translational approaches to the biology of autism: false dawn or a new era?” Molecular Psychiatry 18, 432-442, 2013
• Guy J et al, Reversal of neurological deficits in a mouse model of Rett syndrome, Science 315, 1143-1147, 2007
• Derecki et al, Wild-type microglia arrest pathology in a mouse model of Rett syndrome, Nature484, 105-109, 2012,

References (3)• Baudouin SJ, et al. Science 338, Shared synaptic pathophysiology in syndromic and
nonsyndromic rodent models of autism, Science 338, 128-132, 2012
• Santoro MR, Bray SM and Warren ST, Molecular mechanisms of fragile X syndrome: a twenty-year perspective, Annual Review Mechanisms of Disease 7, 219-245, 2012
• Wang H and Doering LC, Reversing autism by targeting downstream mTOR signaling, Frontiers in Cellular Neuroscience, March 13, 2013
• Levenga J et al, AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome, Neurobiological Disorders. 42, 311-317, 2011
• Jacquemont S, et al, Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 3: 64ra1, 2011
• Gross C, et al, J, Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome, Neuroscience 30, 10624-36, 2010
• Gkogkas et al, Autism-related deficits via dysregulated eIF4E-dependent translational control, Nature 493, 2013
• Berry-Kravis EM, et al, Effects of STX (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled phase II trial, Science Translational Medicine 4, 152ra127, 2012
• Uhlhass PJ and Singer W, Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks, Neuron 75, 963-980, 2012

Journal Presentation
Marchetto MC et al, Altered proliferation and networks in neural cells derived from idiopathic autistic individuals, Molecular Psychiatry, May 5, 2016, 1-16

Internet resources
• National Institute of Mental Health (NIMH)(http://www.nimh.nih.gov/health/topics/autism-spectrum-disorders-pervasive-developmental-disorders/index.shtml)
• Autism Science Foundation(http://www.autismsciencefoundation.org)
• Autism Speaks(www.autismspeaks.org/) and(http://www.autismspeaks.org/what-autism/video-glossary)
• National alliance on Mental Illness (NAMI) (http://www.nami.org)
• Simmons Foundation Autism Research Initiative (SFARI)(https://sfari.org/)
• AutismKB(http://autismkb.cbi.pku.edu.cn/)

Additional reading/resources
- Donavan J and Zucker C, Autism’s First Child, Atlantic Monthly, Oct 10, 2010
- Tammet D, Born on a Blue Day: Inside the Extraordinary Mind of an Autistic Savant, Simon and Schuster, 2006
• Panel Discussion on ASD (Eric Kandel, Uta Frish, Matthew Slate, Gerald Fishbach, Alison Singer) (http://www.charlierose.com/watch/60073097)
• Professor Uta Frish’s homepage (https://sites.google.com/site/utafrith/Home)
• Savant Syndrome Homehttps://www.wisconsinmedicalsociety.org/professional/savant-syndrome/
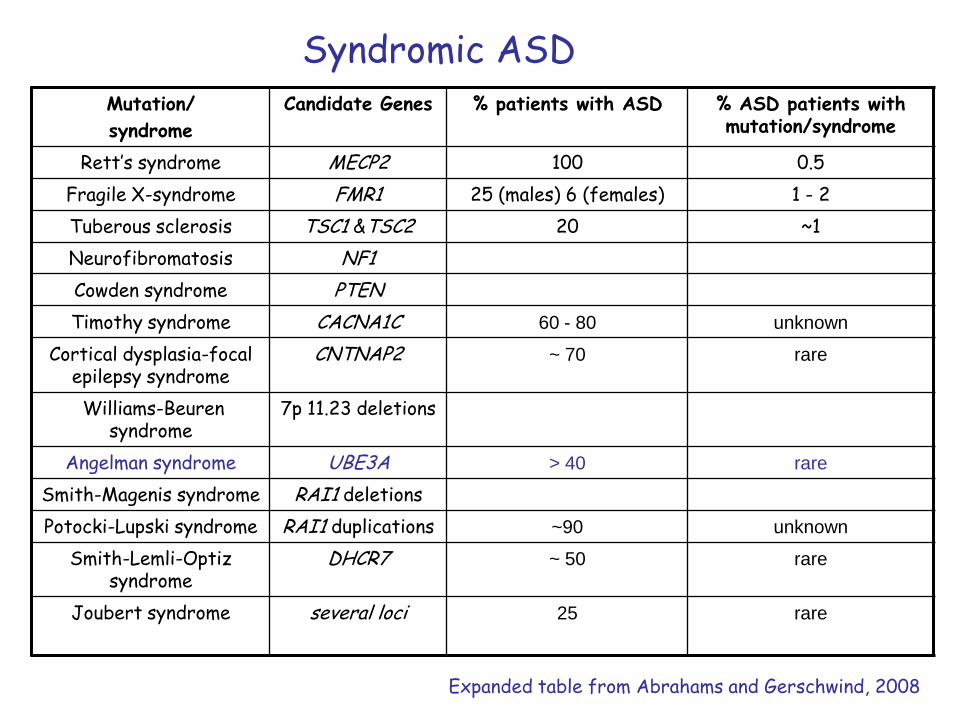
Syndromic ASDMutation/
syndrome
Candidate Genes % patients with ASD % ASD patients with mutation/syndrome
Rett’s syndrome MECP2 100 0.5
Fragile X-syndrome FMR1 25 (males) 6 (females) 1 - 2
Tuberous sclerosis TSC1 &TSC2 20 ~1
Neurofibromatosis NF1
Cowden syndrome PTEN
Timothy syndrome CACNA1C 60 - 80 unknown
Cortical dysplasia-focal epilepsy syndrome
CNTNAP2 ~ 70 rare
Williams-Beuren syndrome
7p 11.23 deletions
Angelman syndrome UBE3A > 40 rare
Smith-Magenis syndrome RAI1 deletions
Potocki-Lupski syndrome RAI1 duplications ~90 unknown
Smith-Lemli-Optiz syndrome
DHCR7 ~ 50 rare
Joubert syndrome several loci 25 rare
Expanded table from Abrahams and Gerschwind, 2008
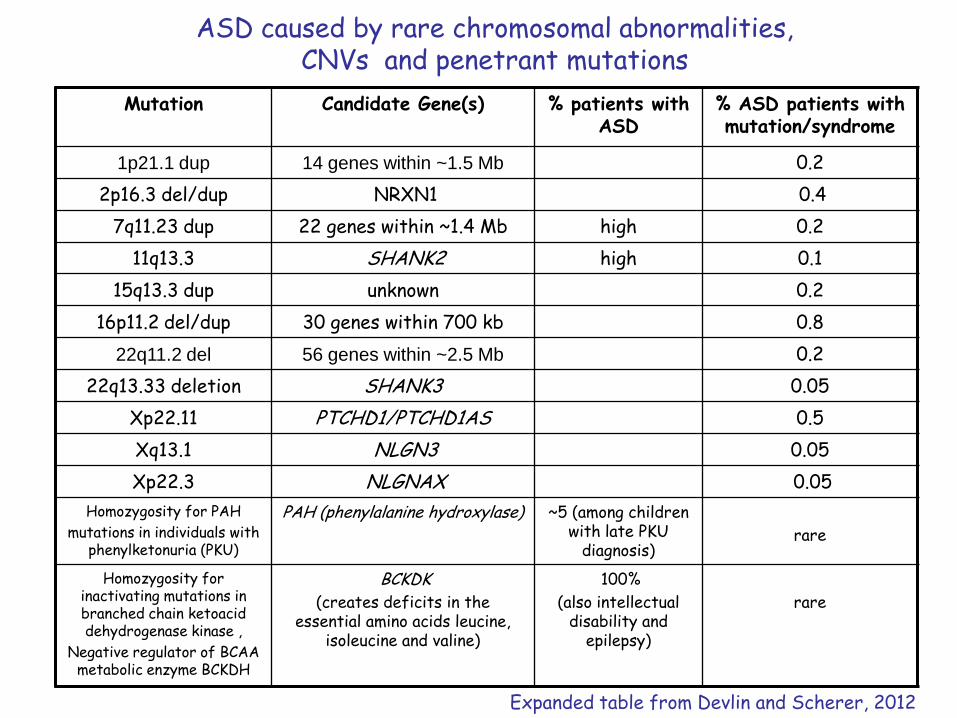
ASD caused by rare chromosomal abnormalities, CNVs and penetrant mutations
Mutation Candidate Gene(s) % patients with ASD
% ASD patients with mutation/syndrome
1p21.1 dup 14 genes within ~1.5 Mb 0.2
2p16.3 del/dup NRXN1 0.4
7q11.23 dup 22 genes within ~1.4 Mb high 0.2
11q13.3 SHANK2 high 0.1
15q13.3 dup unknown 0.2
16p11.2 del/dup 30 genes within 700 kb 0.8
22q11.2 del 56 genes within ~2.5 Mb 0.2
22q13.33 deletion SHANK3 0.05
Xp22.11 PTCHD1/PTCHD1AS 0.5
Xq13.1 NLGN3 0.05
Xp22.3 NLGNAX 0.05
Homozygosity for PAH
mutations in individuals with phenylketonuria (PKU)
PAH (phenylalanine hydroxylase) ~5 (among children with late PKU
diagnosis)rare
Homozygosity for inactivating mutations in branched chain ketoacid dehydrogenase kinase ,
Negative regulator of BCAA metabolic enzyme BCKDH
BCKDK
(creates deficits in the essential amino acids leucine,
isoleucine and valine)
100%
(also intellectual disability and
epilepsy)
rare
Expanded table from Devlin and Scherer, 2012

ASD associated with common genetic variants
• Large-scale genome-wide association studies suggest that hundreds to thousands of common genetic variants, each having only a small effect, are required to account for the observed heritability of ASD. Klei L et al, Molecular Autism 3, 2012.
• These estimates, however, are based on the assumption of strictly additive effects for liability variants.
• By contrast, gene-gene or gene-environment interactions or the clustering of liability variants within biological pathways that relevant to autism may increase the effect size of individual variants within specific ASD subpopulations. Melhem and Devlin, Genomic Medicine 2, 2010

Devlin and Scherer, 2012
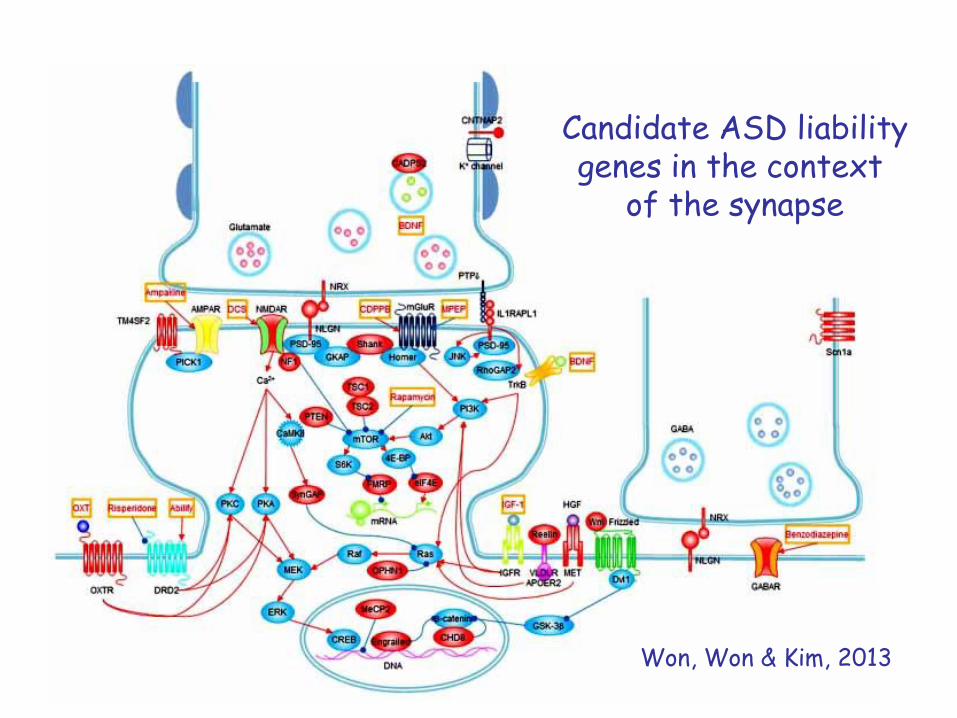
Candidate ASD liabilitygenes in the context
of the synapse
Won, Won & Kim, 2013
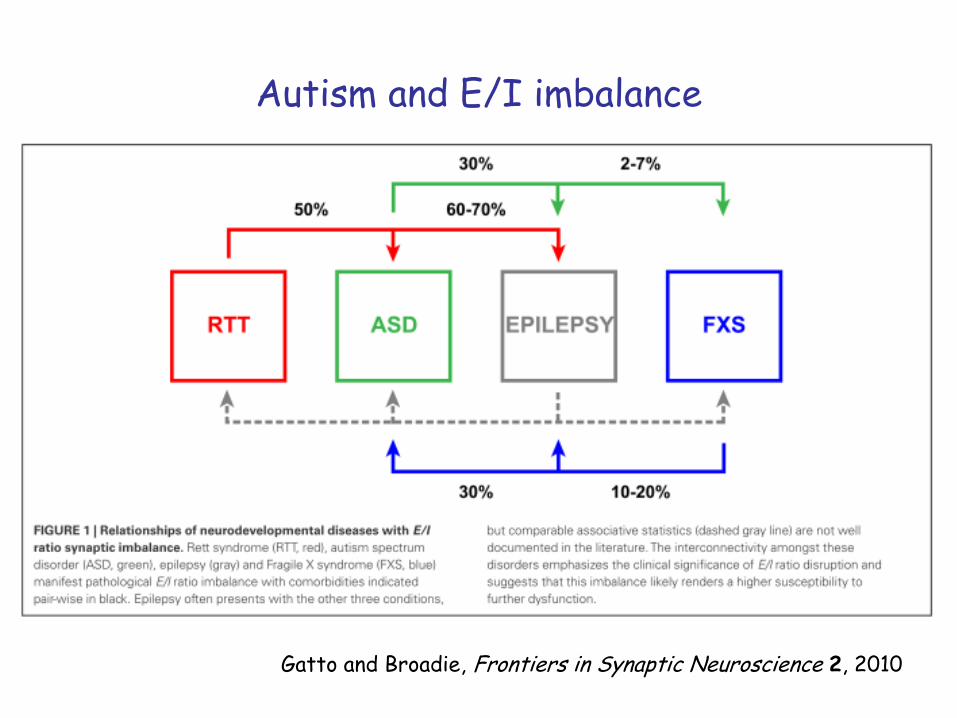
Autism and E/I imbalance
Gatto and Broadie, Frontiers in Synaptic Neuroscience 2, 2010

For example, many genes disrupted by de novononsense, splice site or frame-shift mutations in children with ASD encode proteins that function
within the “Fragile X protein (FMRP) pathway”
Iossifov I et al, Neuron 74, 2012
One of the most significant achievements of the genetic analysis of ASD has been the identification of specific
biological pathways that contribute to the disorder

Fragile X syndrome
• The most common cause of inherited mental disability and most common known cause of autism
• Severity of symptoms varies greatly among individuals, ranging from mild learning problems and normal IQ to severe mental retardation and autism
• Prevalence is ~ 1/4000 males and ~ 1/8000 females; males are generally more severely affected compared to females.
• The name “fragile X” derives from a microscopically visible “break” in the long arm of the metaphase X chromosome located just above the tip of the chromosome. This region is designated, Xq27.3
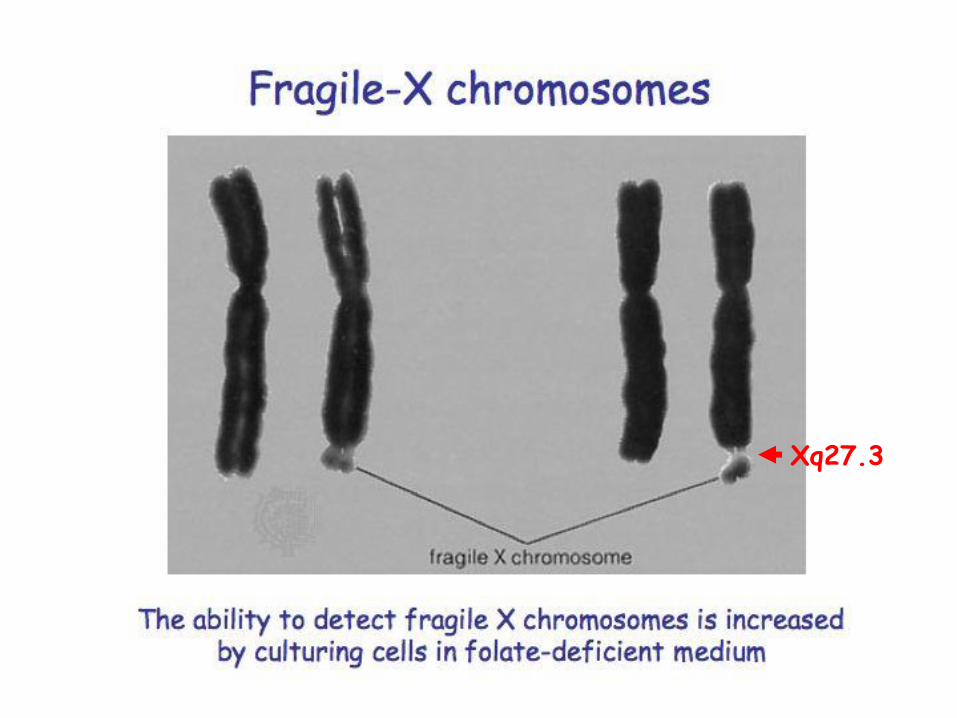
Xq27.3
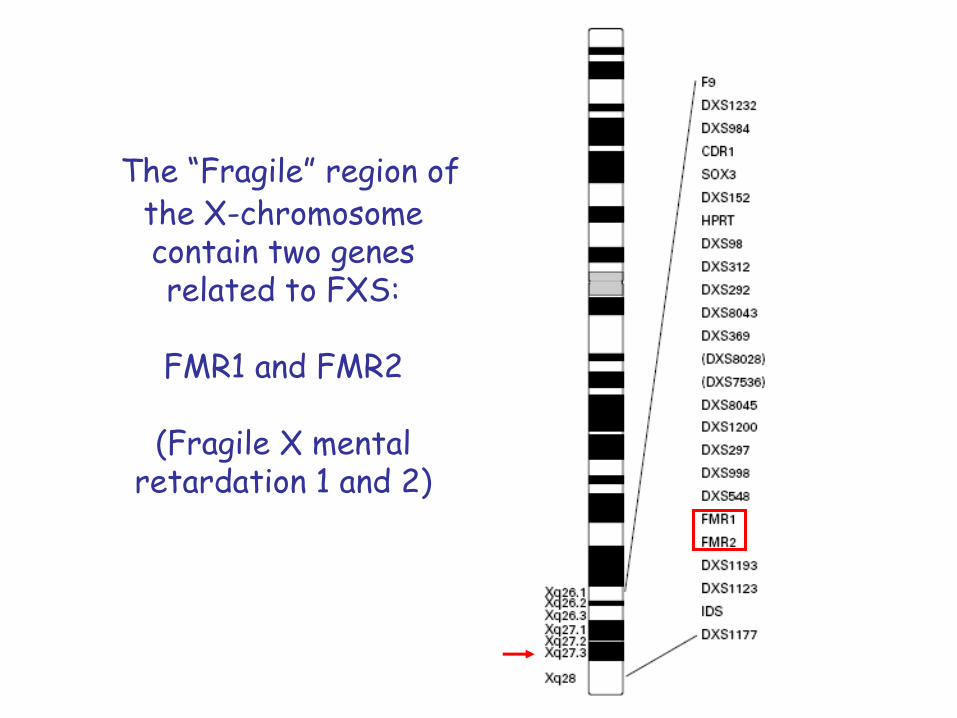
The “Fragile” region of the X-chromosomecontain two genes related to FXS:
FMR1 and FMR2
(Fragile X mental retardation 1 and 2)
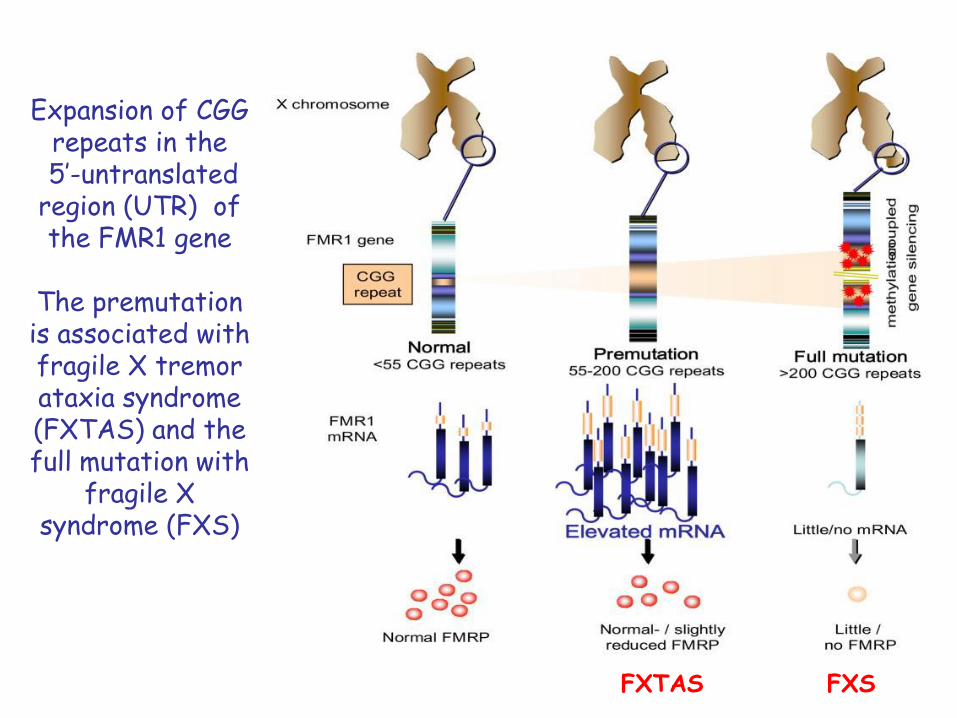
Expansion of CGG repeats in the5’-untranslated
region (UTR) of the FMR1 gene
The premutation is associated with fragile X tremor ataxia syndrome (FXTAS) and the full mutation with
fragile X syndrome (FXS)
FXTAS FXS
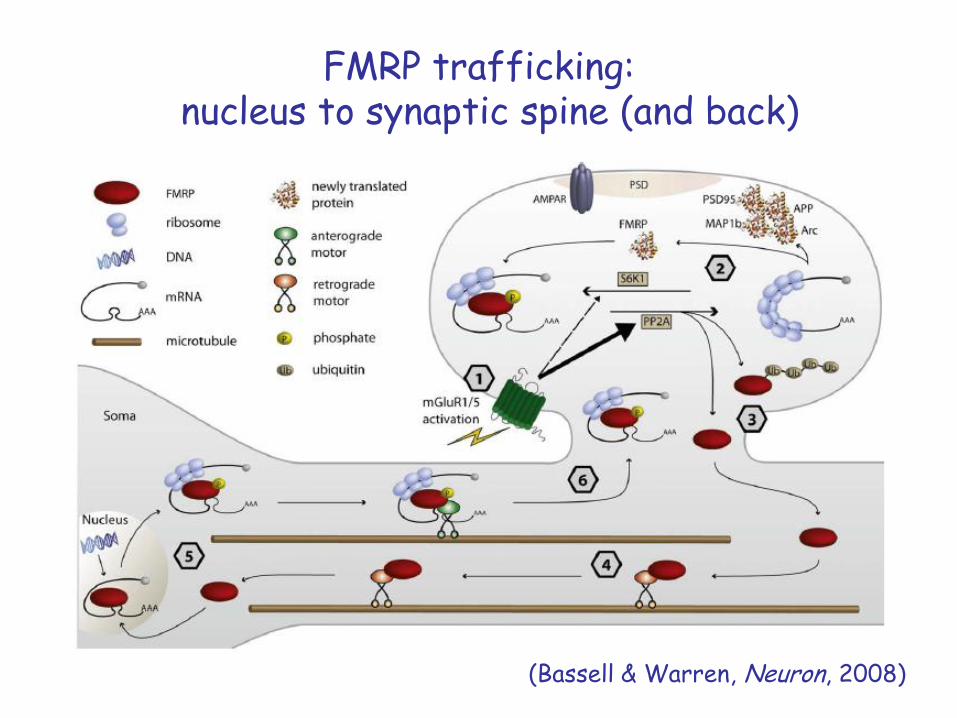
FMRP trafficking:nucleus to synaptic spine (and back)
(Bassell & Warren, Neuron, 2008)
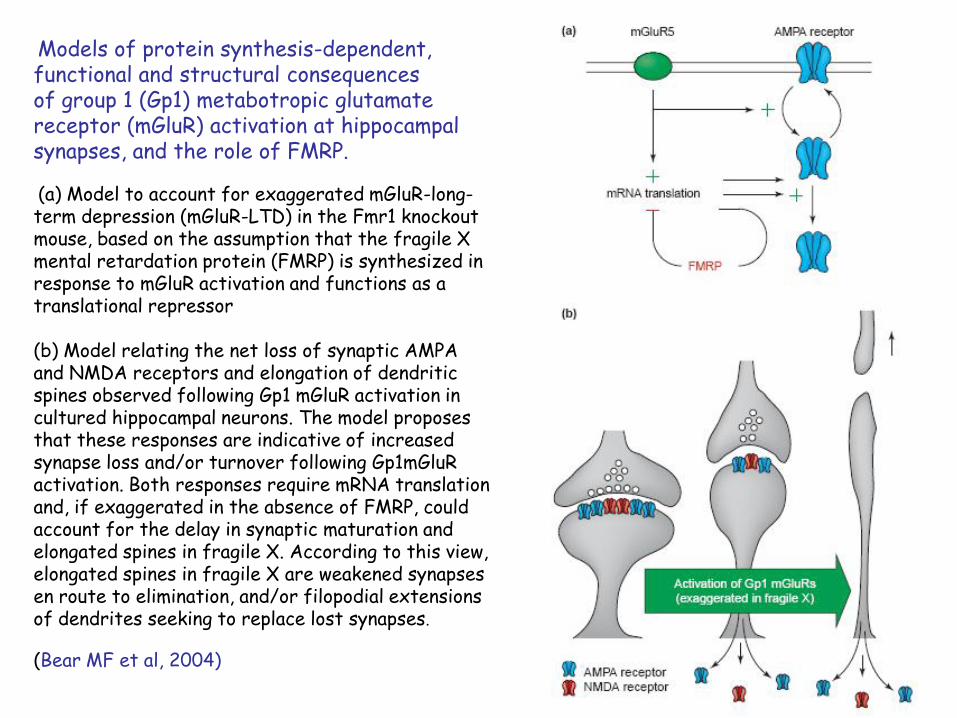
Models of protein synthesis-dependent, functional and structural consequencesof group 1 (Gp1) metabotropic glutamate receptor (mGluR) activation at hippocampal synapses, and the role of FMRP.
(a) Model to account for exaggerated mGluR-long-term depression (mGluR-LTD) in the Fmr1 knockout mouse, based on the assumption that the fragile X mental retardation protein (FMRP) is synthesized in response to mGluR activation and functions as a translational repressor
(b) Model relating the net loss of synaptic AMPA and NMDA receptors and elongation of dendritic spines observed following Gp1 mGluR activation in cultured hippocampal neurons. The model proposes that these responses are indicative of increased synapse loss and/or turnover following Gp1mGluR activation. Both responses require mRNA translation and, if exaggerated in the absence of FMRP, could account for the delay in synaptic maturation and elongated spines in fragile X. According to this view, elongated spines in fragile X are weakened synapses en route to elimination, and/or filopodial extensions of dendrites seeking to replace lost synapses.
(Bear MF et al, 2004)
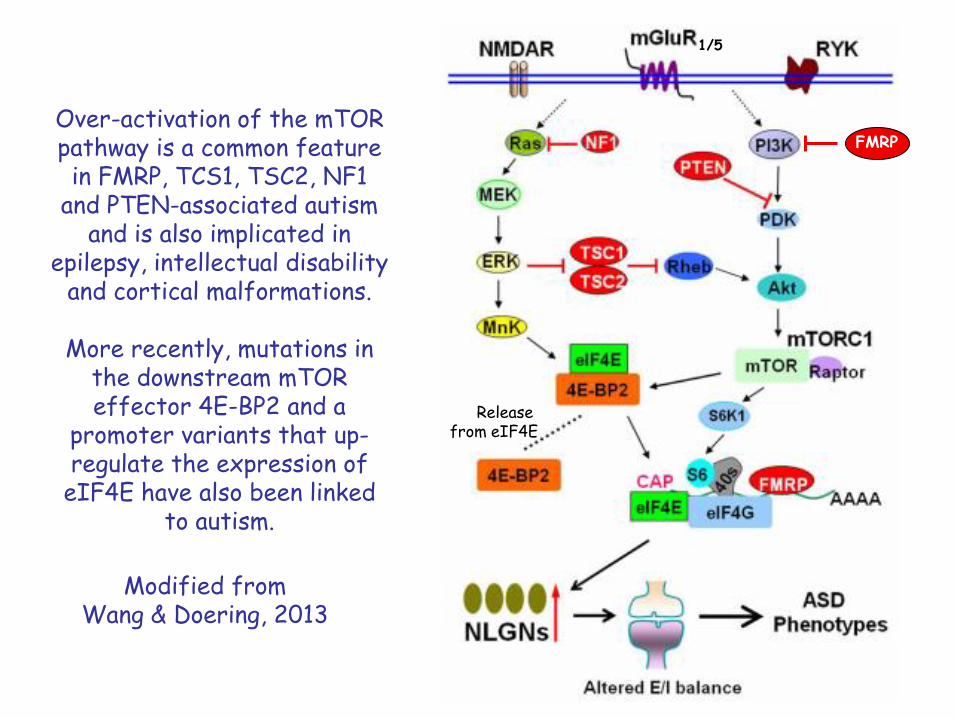
Over-activation of the mTOR pathway is a common feature in FMRP, TCS1, TSC2, NF1
and PTEN-associated autism and is also implicated in
epilepsy, intellectual disability and cortical malformations.
More recently, mutations in the downstream mTOR effector 4E-BP2 and a
promoter variants that up-regulate the expression of
eIF4E have also been linked to autism.
Modified fromWang & Doering, 2013
FMRP
1/5
Release from eIF4E
Related Documents











