
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript


Editorial
Announcement of the first impact factor for The World Journal of Biological PsychiatryHans-Jurgen Moller, Rainer Rupprecht ................................................................................................. 130
Reviews
Peripheral thyroid dysfunction in depressionKonstantinos N. Fountoulakis, Sotiris Kantartzis, Melina Siamouli, Panagiotis Panagiotidis,
Stergios Kaprinis, Apostolos Iacovides, George Kaprinis .................................................................. 131
Melatonin in mood disordersVenkataramanujan Srinivasan, Marcel Smits, Warren Spence, Alan D. Lowe, Leonid Kayumov,
Seithikurippu R. Pandi-Perumal, Barbara Parry, Daniel P. Cardinali ............................................... 138
Original Investigations
Striatal dopamine transporter availability and DAT-1 gene in adults with ADHD: No higherDAT availability in patients with homozygosity for the 10-repeat allele
Johanna Krause, Stefan H. Dresel, Klaus-Henning Krause, Christian La Fougere,Peter Zill, Manfred Ackenheil ............................................................................................................ 152
Association study of the glycogen synthase kinase-3b gene polymorphism with prophylacticlithium response in bipolar patients
Aleksandra Szczepankiewicz, Janusz K. Rybakowski, Aleksandra Suwalska,Maria Skibinska, Anna Leszczynska-Rodziewicz, Monika Dmitrzak-Weglarz,Piotr M. Czerski, Joanna Hauser ....................................................................................................... 158
The influence of concomitant neuroleptic medication on safety, tolerability and clinicaleffectiveness of electroconvulsive therapy
Caroline Nothdurfter, Daniela Eser, Cornelius Schule, Peter Zwanzger, Alain Marcuse,Ines Noack, Hans-Jurgen Moller, Rainer Rupprecht, Thomas C. Baghai .......................................... 162
Viewpoint
Disasters and mental health: New challenges for the psychiatric professionJuan J. Lopez-Ibor Jr ............................................................................................................................. 171
Case Reports
Sulbutiamine, an ‘innocent’ over the counter drug, interferes with therapeutic outcome ofbipolar disorder
Athanasios Douzenis, Ioannis Michopoulos, Lefteris Lykouras ........................................................... 183
Autism and Williams syndrome: A case reportSabri Herguner, Nahit Motavalli Mukaddes ......................................................................................... 186
Instructions to Authors ....................................................................................................... 191
The World Journal of Biological PsychiatryVolume 7, No 3, 2006
Contents

EDITORIAL
Announcement of the first impact factor for The World Journal of
Biological Psychiatry
Over the last two decades the impact factor has
developed into the ‘gold currency’ in the world of
scientific publications, and every scientist, including
scientifically active psychiatrists, considers himself
rich and happy when he possesses as much of this
‘gold currency’ as possible. Although this is only a
virtual currency that cannot be used to buy any-
thing, it has become increasingly relevant, some-
times even decisive, for both an individual’s
academic career and the rating of institutions. It is
also seen as an indicator of a journal’s quality and,
together with other factors, determines among other
things how attractive a journal is to authors.
Three years after acceptance of The World Journal
of Biological Psychiatry for indexing by Thomson ISI
we are thrilled to announce that our first impact
factor has put us in the upper league of psychiatric
journals. The World Journal of Biological Psychiatry
has achieved an impact factor of 2.800 for 2005, and
is ranked 31st of 94 journals in this field (Source:
2005 JCR Science Edition). To put this into
perspective, Schizophrenia Bulletin is ranked 30th
with an impact factor of 2.871, and Acta Psychiatrica
Scandinavica 28th with an impact factor of 2.968, to
name just two examples.
Our thanks go out to all those who have supported
The World Journal of Biological Psychiatry over the
past 6 years, without whom this great success would
never have been possible. Those to be thanked
include all authors, reviewers, associate editors and
editorial board members, members of WFSBP
Guideline Task Forces who invested time and effort
to prepare global treatment guidelines for various
psychiatric indications, the staff at Taylor & Francis
whose knowledge of and expertise in the world of
scientific publishing have advanced the Journal even
further since commencement of our cooperation at
the beginning of last year, the staff at the WFSBP
Global Headquarters, and last but not least the staff
at the Editorial Office.
Without wanting to dampen the excitement, some
problematic consequences associated with the per-
haps too great importance of the impact factor
should be mentioned here. An impact factor can
be manipulated by various means. For example, it is
known that among colleagues who are particularly
experienced with respect to the ‘impact currency’,
impact circles have already existed for a longer time
which push up the impact factor of all involved
through mutual citations. Anyone who does not join
such an impact circle can optimise his impact factor
by regularly citing his own articles. Also journals that
are worried about their impact factor have developed
regulating systems to improve it, for example by
motivating authors to cite earlier publications from
the journal whenever possible. A particular absurdity
is the fact that papers with bold hypotheses and
perhaps first relevant results achieve an especially
high impact factor, even when shortly afterwards
they are shown to be false. This absurdity even
happens with publications reporting falsified results.
Together with the newly introduced online manu-
script submission and administration system, Manu-
script Central, the impact factor can be expected to
cause an increase in the number and quality of
papers submitted to The World Journal of Biological
Psychiatry. Despite the elation about a relatively
good impact factor, it should not become decisive
for the future manuscript policy of the Journal of the
WFSBP, but the original idea behind the foundation
of the Journal should not be forgotten: a truly global
psychiatric journal that brings the whole world of
biological psychiatry to the whole world.
Hans-Jurgen Moller Rainer Rupprecht
Chief Editor Assistant Chief Editor
The World Journal of Biological Psychiatry, 2006; 7(3): 130
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970600889521

REVIEW
Peripheral thyroid dysfunction in depression
KONSTANTINOS N. FOUNTOULAKIS, SOTIRIS KANTARTZIS, MELINA SIAMOULI,
PANAGIOTIS PANAGIOTIDIS, STERGIOS KAPRINIS, APOSTOLOS IACOVIDES &
GEORGE KAPRINIS
Laboratory of Psychophysiology, 3rd Department of Psychiatry, Aristotle University of Thessaloniki, University Hospital
AHEPA, Thessaloniki, Greece
AbstractThe involvement of the thyroid gland and thyroid hormones is generally believed to be important in the aetiopathogenesisof major depression. Major support comes from studies in which alterations in components of the hypothalamic�pituitary�thyroid (HPT) axis have been documented in patients with primary depression. However, screening thyroid tests areoften routine and add little to the diagnostic evaluation. Overt thyroid disease is rare among depressed inpatients. Thefinding that depression often co-exists with autoimmune subclinical thyroiditis suggests that depression may causealterations in the immune system, or that in fact it could be an autoimmune disorder itself. The outcome of treatment andthe course of depression may be related to thyroid status as well. Augmentation of antidepressant therapy with the co-administration of thyroid hormones (mainly T3) is a well-documented treatment option for refractory depressed patients.Review of the literature suggests that there are no conclusive data on the role of thyroid function in depression. It is clearthat depression is not characterised by an overt thyroid dysfunction, but it is also clear that a subgroup of depressed patientsmay manifest subtle thyroid abnormalities, or an activation of an autoimmune process. There is a strong possibility that thepresence of a subtle thyroid dysfunction is a negative prognostic factor for depression and may demand specific therapeuticintervention.
Key words: Depression, thyroid function, psychoneuroendocrinology
Introduction
There is a general and widespread belief among
psychiatrists that depression is characterised by
subtle neuroendocrinological disorders. The invol-
vement of the thyroid gland and thyroid hormones is
believed to be important.
The thyroid hormones (Reed and Pangaro 1995),
L�3,5,3?,5?-tetraiodothyronine (T4) and L�3,5,3?-triiofothyronine (T3) are synthesised by the follicu-
lar epithelial cells of the thyroid gland. This synthesis
requires the availability of iodine and is increased by
thyroid-stimulating hormone (TSH) from the ante-
rior pituitary gland. Some T4 is converted to T3
before release. These steps are under the influence of
TSH or other proteins that bind to the TSH
receptor. However, most T3 (80�85%) is derived
from extrathyroidal conversion of T4 in peripheral
tissue, mostly in the liver and kidney. The balance
between production and degradation is mediated by,
among other things, nutrition, non-thyroidal illness,
exercise, pregnancy and medications. Less than 1%
of the total circulating amount of each hormone is
free in the plasma (free T3-FT3, free T4-FT4).
Thyroid hormone regulation is directed through the
hypothalamic �pituitary� thyroid� peripheral tissue
axis. The system extends higher to neuroendocrine
modulation at the hypothalamus and lower to
peripheral thyroid hormone metabolism. This sys-
tem has autocrine (enzyme autoregulation), para-
crine (somatostatin, TRH) and hemocrine
autoregulation that is also influenced by environ-
mental factors (energy balance, circadian variation,
illness).
Depression itself is not a homogeneous disorder.
It is traditionally classified into two opposite poles
(Roth 1959; Van Praag et al. 1965; Overall et al.
1966; Fountoulakis et al. 1999), today named
‘melancholic’ (APA 1994) or ‘somatic’ syndrome
(WHO 1993) versus ‘atypical’ features (that is
Correspondence: K.N. Fountoulakis, MD PhD, 1st Parodos, Ampelonon Street 55535, Pournari Pylaia, Thessaloniki, Greece. Tel: �/30
2310 994622. Fax: �/30 2310 266570. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(3): 131�137
(Received 23 November 2004; accepted 9 November 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500474739

‘reverse neurovegetative symptoms’, increased appe-
tite, weight gain, increased sleep and interpersonal
rejection sensitivity (Sargant 1960; Dally and Rohde
1961; Liebowitz et al. 1988)).
Of the various hypothalamic�pituitary-end organ
axes, the thyroid and adrenal systems are those most
often implicated in affective disorders. Patients
with primary thyroid disease have high rates of
depression, and patients with Addison’s disease or
Cushing’s syndrome have relatively high rates of
affective and anxiety symptoms. However, the major
support for the involvement of endocrine axes in the
pathophysiology of mood disorders comes from
studies in which alterations in components of the
hypothalamic�pituitary�thyroid (HPT) (Legros
et al. 1985; Staner et al. 1992; Rao et al. 1996)
and the hypothalamic�pituitary�adrenal (HPA)
axes (Mendlewicz et al. 1984; Kocsis et al. 1985;
Evans and Golden 1987; Nelson and Davis 1997)
have been documented in patients with primary
depression.
Thyroid dysfunction and depression
It has been argued that depression might be char-
acterised by a ‘low-thyroid function syndrome’
(Legros et al. 1985; Staner et al. 1992; Rao et al.
1996). Hypothyroidism is associated with anxiety
(Iacovides et al. 2000) or refractory depression,
suggesting that this characterises one biological
subtype of refractory depression. However, screen-
ing thyroid tests are often routine for depressed
inpatients, and data suggest that thyroid screening
may add little to the diagnostic evaluation. Overt
thyroid disease is rare among depressed inpatients
(Ordas and Labbate 1995), and the role of thyroid
hormones in the pathophysiology of affective dis-
orders remains to be clarified (Joffe and Sokolov
1994).
According to Musselman and Nemeroff (1996),
concerning the HPT axis, depressed patients have
been reported to have:
a. alterations in thyroid-stimulating hormone re-
sponse to thyrotropin-releasing hormone
(TRH);
b. an abnormally high rate of antithyroid antibo-
dies; and
c. elevated cerebrospinal fluid (CSF) TRH con-
centrations.
Moreover, tri-iodothyronine has been shown to
augment the efficacy of various antidepressants,
although opposite reports exist. All of the HPA axis
alterations in depression studied thus far are state-
dependent, whereas the HPT axis alterations may be
partially trait and partially state markers.
There are several papers suggesting that the
thyroid function of depressed patients is within the
normal range, hypothyroidism and hyperthyroidism
are extremely uncommon and that the presence of
subtle thyroid function abnormalities does not have
an impact on treatment outcome (Joffe 1987; Harris
et al. 1989; Fava et al. 1995; Joffe et al. 1996;
Haggerty et al. 1997; Pop et al. 1998). However, on
the contrary, there are even more papers supporting
the idea of a subclinical thyroid dysfunction, espe-
cially in melancholic or refractory patients, possibly
of an autoimmune origin (Banki et al. 1985;
Kjellman et al. 1985; Nemeroff et al. 1985; Gewirtz
et al. 1988; Marchesi et al. 1988; Nemeroff 1989;
Rao et al. 1989; Rupprecht et al. 1989; Howland
1993; Bunevicius et al. 1994; Maes et al. 1994a; Rao
et al. 1996) suggesting that subclinical hypothyroid-
ism may lower the threshold for the occurrence of
depression (Haggerty et al. 1993), or generally to
any mental disorder (O’Donnell et al. 1988; Stein
and Uhde 1989; Haggerty et al. 1990). Also, it has
been suggested that patients with bipolar disorder
are particularly sensitive to variations in thyroid
function within the normal range (Cole et al. 2002).
So, most patients with depression, although gen-
erally viewed as chemically euthyroid, may have
alterations in their thyroid function (Joffe et al.
1992; Custro et al. 1994; De Mendonca Lima et
al. 1996) including slight elevation of the serum FT4
(especially in melancholic patients) (Maes et al.
1993), blunted TSH response to thyrotropin-releas-
ing hormone (TRH) stimulation (Loosen 1985),
and loss of the nocturnal TSH rise, and this may
reflect brain hypothyroidism in the context of
systemic euthyroidism (Bauer et al. 1990; Jackson
1998; Sullivan et al. 1999).
Thyroid dysfunction, however, may constitute an
expression of a coordinated neuroendocrine-im-
mune response to nonthyroidal illness (Maes et al.
1994b), and this is in accord with the finding that
depressive symptoms are associated with positive
thyroid antibody status in the postpartum period
(Harris et al. 1992). The general idea is that while
patients with symptomless autoimmune thyroiditis
are clinically euthyroid, what might be symptomless
for the endocrinologist might be a syndrome pre-
senting with psychiatric symptoms to the psychiatrist
(Gold et al. 1982).
High (but within the normal range) serum TSH
(beyond the upper 25th percentile) is reported to be
positively associated with recurrent depression the
presence of somatic disease condition and the
number of suicide attempts (Berlin et al. 1999).
132 K. N. Fountoulakis et al.
Some data suggest that the 5-HT reduced activity
is more pronounced in those patients without HPT
axis abnormality. In this frame, HPT dysregulation
may be regarded as a compensatory mechanism for
diminished central 5-HTactivity (Duval et al. 1999),
which is a suggestion similar to another one pro-
posed concerning the hypothalamus�pituitary�adrenal axis (Fountoulakis et al. in press). These
just reflect the fact that the true relationship of
peripheral indices to brain function is always an open
question (Frye et al. 1999).
The availability of thyroid testing led to a bulk of
research on thyroid dysfunction in non-thyroidal
illness. Data suggest that, within a given patient’s
status, change of thyroid function is determined by
the severity and duration of illness as well as the
presence of mitigating influences that are associated
with the specific underlying disorders. These thyroid
disturbances in the frame of non-thyroidal illness
constitute a diagnostic problem which needs focused
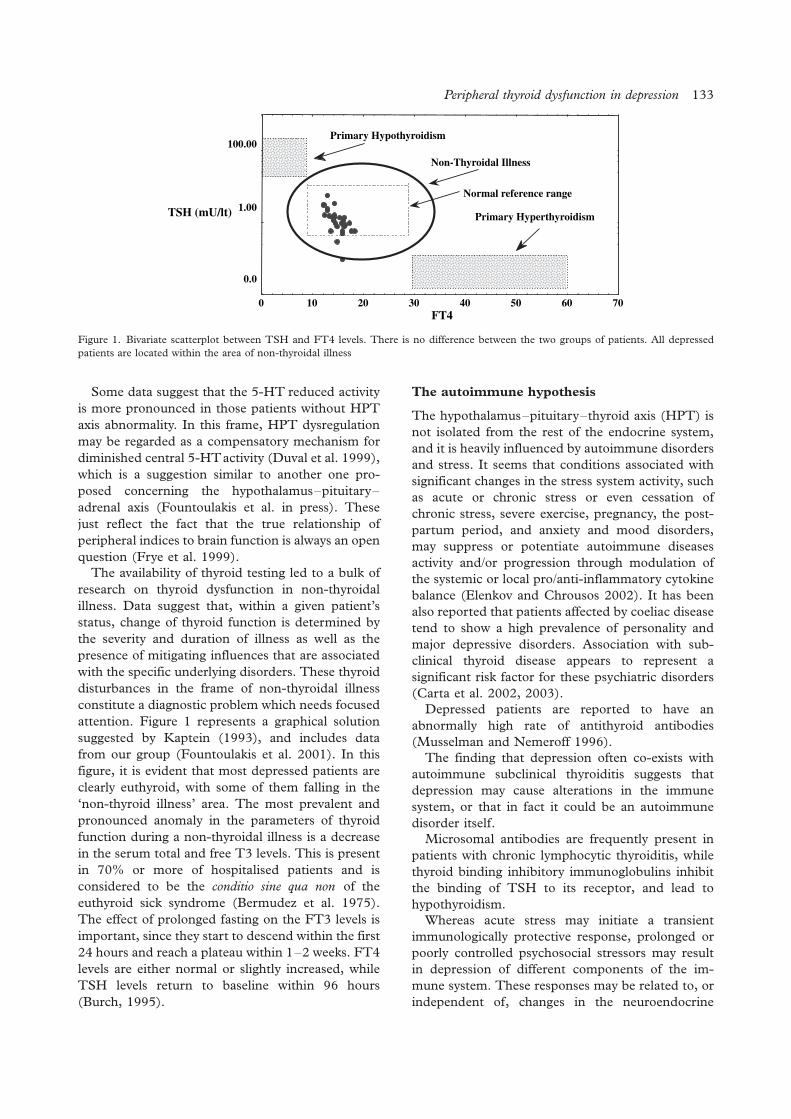
attention. Figure 1 represents a graphical solution
suggested by Kaptein (1993), and includes data
from our group (Fountoulakis et al. 2001). In this
figure, it is evident that most depressed patients are
clearly euthyroid, with some of them falling in the
‘non-thyroid illness’ area. The most prevalent and
pronounced anomaly in the parameters of thyroid
function during a non-thyroidal illness is a decrease
in the serum total and free T3 levels. This is present
in 70% or more of hospitalised patients and is
considered to be the conditio sine qua non of the
euthyroid sick syndrome (Bermudez et al. 1975).
The effect of prolonged fasting on the FT3 levels is
important, since they start to descend within the first
24 hours and reach a plateau within 1�2 weeks. FT4
levels are either normal or slightly increased, while
TSH levels return to baseline within 96 hours
(Burch, 1995).
The autoimmune hypothesis
The hypothalamus�pituitary�thyroid axis (HPT) is
not isolated from the rest of the endocrine system,
and it is heavily influenced by autoimmune disorders
and stress. It seems that conditions associated with
significant changes in the stress system activity, such
as acute or chronic stress or even cessation of
chronic stress, severe exercise, pregnancy, the post-
partum period, and anxiety and mood disorders,
may suppress or potentiate autoimmune diseases
activity and/or progression through modulation of
the systemic or local pro/anti-inflammatory cytokine
balance (Elenkov and Chrousos 2002). It has been
also reported that patients affected by coeliac disease
tend to show a high prevalence of personality and
major depressive disorders. Association with sub-
clinical thyroid disease appears to represent a
significant risk factor for these psychiatric disorders
(Carta et al. 2002, 2003).
Depressed patients are reported to have an
abnormally high rate of antithyroid antibodies
(Musselman and Nemeroff 1996).
The finding that depression often co-exists with
autoimmune subclinical thyroiditis suggests that
depression may cause alterations in the immune
system, or that in fact it could be an autoimmune
disorder itself.
Microsomal antibodies are frequently present in
patients with chronic lymphocytic thyroiditis, while
thyroid binding inhibitory immunoglobulins inhibit
the binding of TSH to its receptor, and lead to
hypothyroidism.
Whereas acute stress may initiate a transient
immunologically protective response, prolonged or
poorly controlled psychosocial stressors may result
in depression of different components of the im-
mune system. These responses may be related to, or
independent of, changes in the neuroendocrine
FT4
TSH (mU/lt)
0 10 20 30 40 50 60 70
Primary Hypothyroidism
Primary Hyperthyroidism
Non-Thyroidal Illness
100.00
1.00
0.0
Normal reference range
Figure 1. Bivariate scatterplot between TSH and FT4 levels. There is no difference between the two groups of patients. All depressed
patients are located within the area of non-thyroidal illness
Peripheral thyroid dysfunction in depression 133

system. As the rather prolific literature in this infant
area of psychoneuroimmunology reveals, there are
many complex levels of interaction that require
further investigation (Schindler, 1985), concerning
a close relationship between delayed hypersensitivity
to neural tissue antigens and immunopsychiatric
diseases, and they may imply that cell-mediated
immune mechanisms may be involved in the patho-
genesis of certain mental disorders (Jankovic 1985).
It is also believed (although not well documented)
that depression is accompanied by various direct and
indirect indicators of a moderate activation of the
inflammatory response system. Increased produc-
tion of proinflammatory cytokines, such as inter-
leukin-1, interleukin-6 and interferon (IFN-g), may
play a crucial role in the immune and acute phase
response in depression (van West and Maes 1999).
However, the research in the area of psychoim-
munology is very delicate and one should be very
careful in interpreting results. Apart from problems
arising from the limitations in laboratory techniques
themselves, depressed patients may suffer from
secondary alterations of immune function, while
controls may be ‘super-normal’.
Gestation and the postpartum period
Although some authors report that the presence of
abnormal thyroid function tests is not related to a
distinct clinical picture (Kent et al. 1999), others
suggest that serum T4 levels may be lower in
seasonally affected patients (Sher et al. 1999), and
an elevated level of peroxidase antibodies may be
related to depression in perimenopausal women
(Pop et al. 1998). However, again, the literature is
split and the results are inconclusive (Kuijpens et al.
2001; Lucas et al. 2001; Harris et al. 2002). The
most robust relationship between thyroid dysfunc-
tion and depression concerns gestation and the
postpartum period. Thyroid antibody-positive wo-
men are prone to hypothyroidism, which is often
preceded by transient hyperthyroidism after delivery
(Harris et al. 1992; Harris 1999). Also, lower range
total and free thyroxine concentrations during late
pregnancy may be related to postpartum depressive
symptoms (Pedersen 1999).
Response to treatment and long-term outcome
It was mentioned above that successful therapy
alleviates thyroid dysfunction, if present. There are
not many studies on this issue. One study reported a
significant reduction of 11.2% in thyroxine during
treatment with 20 mg paroxetine in 25 severely
depressed patients (Konig et al. 2000). It has also
been reported that 4 weeks of sertraline treatment
leads to increased plasma cortisol levels, while a 24-
week treatment leads to increased plasma T3 levels
in depressed patients (Sagud et al. 2002). On the
contrary, other authors report no effect on thyroid
function (Schule et al. 2005) Another study
(blinded, placebo-controlled) reported that repeated
transcranial magnetic stimulation over the prefrontal
cortex administered to healthy individuals, produces
acute elevations of mood and serum TSH (Szuba et
al. 2001). The same results are reported concerning
sleep deprivation. However, it is suggested that sleep
deprivation responders compensate by secreting
more TSH with normal bioactivity while, on the
contrary, non-responders compensate by secreting
TSH with increased bioactivity (Orth et al. 2001).
Responders were also reported to have lower T3
uptake levels than non-responders in both prospec-
tive and retrospective studies (David et al. 2000).
The outcome of treatment and the course of
depression seem to relate to thyroid status as well.
Time to recurrence is reported to be inversely
related to T3 levels but not to T4 levels (Joffe and
Marriott 2000). But again, opposite reports exist
(Joffe 1999).
There is an open question concerning the role of
subtle thyroid dysfunction in the long-term outcome
of depression. There are scarce reports that cognitive
deficits caused by hypothyroidism persist after
patients return to euthyroid status, with concentra-
tion, recall and short-term memory appearing to be
most severely affected (Leentjens and Kappers
1995). Several risk factors have been proposed for
Alzheimer’s disease (AD), among them depression
(Broe et al. 1990; Kokmen et al. 1991), and prior
thyroid disease (Heyman et al. 1984).
Augmentation of antidepressant therapy with the
co-administration of thyroid hormones (mainly T3)
is a well-documented treatment option for refractory
depressed patients, and although no clear biochem-
ical or clinical predictors of preferential response to
T3 have been found, its effect may be related to
thyroid function even within the normal range.
Surprisingly, only minimal side-effects have been
reported in the literature (Nemeroff 1996; Joffe
1997, 1998; Post et al. 1997; Cadieux, 1998;
Sussman and Joffe 1998; Thase et al. 1998; Shelton
1999; Dording 2000; Fava 2000; Joffe and Sokolov
2000; Marangell 2000; Bauer 2002; Bauer et al.
2002; Agid and Lerer 2003; Altshuler et al. 2003;
Pridmore and Turnier-Shea 2004).
Recently, a study of our group suggested that
depressed patients who responded well to treatment
might have lower FT4 and TSH as well as TBII,
but higher FT3 levels in comparison to poor
responders. That study suggested that when
the function 20.86�1.52*[FT4]�0.98*[TSH]�/
134 K. N. Fountoulakis et al.

0.74*[FT3]�0.07*[TBII] takes values above zero,
the patient is likely to be a good responder (with
89.47% chance of correct prediction) (Fountoulakis
et al. 2004).
However, this function is yet to be validated, and
has been reported in only one study. It needs further
research and validation, and currently is not recom-
mended for clinical use.
Conclusion
Thus, a review of the literature suggests that there
are no conclusive data on the role of thyroid function
in depression. It is clear that depression is not
characterised by an overt thyroid dysfunction. It is
also clear that a subgroup of depressed patients may
manifest subtle thyroid abnormalities, or an activa-
tion of an autoimmune process; however, the cause
of this phenomenon and its implications are unclear.
There is a strong possibility that the presence of a
subtle thyroid dysfunction is a negative prognostic
factor for depression and may demand specific
therapeutic intervention.
Statement of interest
The authors have no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
Agid O, Lerer B. 2003. Algorithm-based treatment of major
depression in an outpatient clinic: Clinical correlates of
response to a specific serotonin reuptake inhibitor and to
triiodothyronine augmentation. Int J Neuropsychopharmacol
6:41�49.
Altshuler LL, Frye MA, Gitlin MJ. 2003. Acceleration and
augmentation strategies for treating bipolar depression. Biol
Psychiatry 53:691�700.
Banki C, Vojnik M, Arato M, Papp Z, Kovacs Z. 1985.
Dexamethasone suppression and multiple hormonal responses
(TSH, prolactin and growth hormone) to TRH in some
psychiatric disorders. Eur Arch Psychiatry Neurol Sci
235:32�37.
Bauer M, Baur H, Berghofer A, Strohle A, Hellweg R, Muller-
Oerlinghausen B, Baumgartner A. 2002. Effects of supraphy-
siological thyroxine administration in healthy controls and
patients with depressive disorders. J Affect Disord 68:285�94.
Bauer M, Whybrow P, Winokur A. 1990. Rapid cycling bipolar
affective disorder I: Association with grade I hypothyroidism.
Arch Gen Psychiatry 47:427�432.
Bauer M. 2002. Thyroid hormone augmentation with levothyr-
oxine in bipolar depression. Bipolar Disord 4(Suppl 1):109�110.
Berlin I, Payan C, Corruble E, Puech A. 1999. Serum thyroid-
stimulating-hormone concentration as an index of severity of
major depression. Int J Neuropsychopharmcol 2:105�110.
Bermudez F, Surks M, Oppenheimer J. 1975. High incidence of
decreased serum triiodothyroine concentration in patients with
non-thyroidal disease. J Clin Endocrinol Metab 41:27�31.
Broe G, Henderson A, Creasey H, McCusker E, Korten A, Jorm
A, Longley W, Anthony J. 1990. A case control study of
Alzheimer’s disease in Austria. Neurology 40:1698�1707.
Bunevicius R, Kazanavicius G, Telksnys A. 1994. Thyrotropin
response to TRH stimulation in depressed patients with
autoimmune thyroiditis. Biol Psychiatry 36:543�547.
Burch H. 1995. Abnormal thyroid function test results in
euthyroid persons. In: Becker K, editor. Principles and practice
of endocrinology and metabolism. Philadelphia, PA: JB Lip-
pincott Company. pp 323�332.
Cadieux RJ. 1998. Practical management of treatment-resistant
depression. Am Fam Phys 58:2059�2062.
Carta M, Hardoy M, Boi M, Mariotti S, Carpiniello B, Usai P.
2002. Association between panic disorder, major depressive
disorder and celiac disease: A possible role of thyroid auto-
immunity. J Psychosom Res 53:789�793.
Carta M, Hardoy M, Usai P, Carpiniello B, Angst J. 2003.
Recurrent brief depression in celiac disease. J Psychosom Res
55:573�574.
Cole D, Thase M, Mallinger A, Soares J, Luther J, Kupfer D,
Frank E. 2002. Slower treatment response in bipolar depres-
sion predicted by lower pretreatment thyroid function. Am J
Psychiatry 159:116�121.
Custro N, Scafidi V, LoBaido R, Nastri L, Abbate G, Cuffaro M,
Gallo S, Vienna G, Notarbartolo A. 1994. Subclinical hy-
pothyroidism resulting from autoimmune thyroiditis in female
patients with endogenous depression. J Endocrinol Invest
17:641�646.
Dally P, Rohde P. 1961. Comparison of antidepressant drugs in
depressive illnesses. Lancet i:18�20.
David M, Owen J, Abraham G, Delva N, Southmayd S,
Wooltorton E, Lawson J. 2000. Thyroid function and response
to 48-hour sleep deprivation in treatment-resistant depressed
patients. Biol Psychiatry 48:323�326.
De Mendonca Lima C, Vandel S, Bonin B, Bertschy G, Bizouard
P. 1996. Thyroid function in depressed patients. Encephale
22:85�94.
Dording CM. 2000. Antidepressant augmentation and combina-
tions. Psychiatr Clin North Am 23:743�55.
Duval F, Mokrani M, Bailey P, Correa H, Diep T, Crocq M,
Macher J. 1999. Thyroid axis activity and serotonin function in
major depressive episode. Psychoneuroendocrinology 24:695�712.
Elenkov I, Chrousos G. 2002. Stress hormones, proinflammatory
and antiinflammatory cytokines, and autoimmunity. Ann NY
Acad Sci 966:290�303.
Evans D, Golden R. 1987. The dexamethasone suppression test:
A review. In: Loosen P, editor. Handbook of clinical psycho-
neuroendocrinology. New York: John Wiley and Sons. pp 313�335.
Fava M. 2000. New approaches to the treatment of refractory
depression. J Clin Psychiatry 61(Suppl 1):26�32.
Fava M, Labbate L, Abraham M, Rosenbaum J. 1995. Hypothyr-
oidism and hyperthyroidism in major depression revisited. J
Clin Psychiatry 56:186�192.
Fountoulakis K, Fotiou F, Iacovides A, Karamouzis M, Goulas A,
Demetriadou A, Kaprinis G. 2001 Relationship between
pupillometric findings, DST and melancholia. Acta Psychiatr
Belg 101:202�235.
Fountoulakis K, Iacovides A, Grammaticos P, Kaprinis G, Bech P.
2004. Thyroid function and clinical subtypes of major depres-
sion: An exploratory study. BMC Psychiatry 4:6.
Fountoulakis K, Iacovides A, Nimatoudis I, Kaprinis G, Ierodia-
konou C. 1999. Comparison of the diagnosis of melancholic
and atypical features according to DSM-IV and somatic
syndrome according to ICD-10 in patients suffering from
major depression. Eur Psychiatry 14:426�434.
Peripheral thyroid dysfunction in depression 135

Frye M, Dunn R, Gary K, Kimbrell T, Callahan A, Luckenbaugh
D, Cora-Locatelli G, Vanderham E, Winokur A, Post R. 1999.
Lack of correlation between cerebrospinal fluid thyrotropin-
releasing hormone (TRH) and TRH-stimulated thyroid-stimu-
lating hormone in patients with depression. Biol Psychiatry
45:1049�1052.
Gewirtz G, Malaspina D, Hatterer J, Feureisen S, Klein D,
Gorman J. 1988. Occult thyroid dysfunction in patients with
refractory depression. Am J Psychiatry 145:1012�1014.
Gold M, Pottash A, Extein I. 1982. Symptomless autoimmune
thyroiditis in depression. Psychiatry Res 6:261�269.
Haggerty J, Evans D, Golden R, Pedersen C, Simon J, Nemeroff
C. 1990. The presence of antithyroid antibodies in patients
with affective and nonaffective psychiatric disorders. Biol
Psychiatry 27:51�60.
Haggerty J, Stern R, Mason G, Beckwith J, Morey C, Prange A.
1993. Subclinical hypothyroidism: A modifiable risk factor for
depression? Am J Psychiatry 150:508�510.
Haggerty J, Silva S, Marquardt M, Mason G, Chang H, Evans D,
Golden R, Pedersen C. 1997. Prevalence of antithyroid
antibodies in mood disorders. Depression Anxiety 5:91�96.
Harris B. 1999. Postpartum depression and thyroid antibody
status. Thyroid 9:699�703.
Harris B, Fung H, Johns S, Kologlu M, Bhatti R, McGregor A,
Richards C, Hall R. 1989. Transient post-partum thyroid
dysfunction and postnatal depression. J Affect Disord
17:243�249.
Harris B, Othman S, Davies J, Weppner G, Richards C, New-
combe R, Lazarus J, Parkes A, Hall R, Phillips D. 1992.
Association between postpartum thyroid dysfunction and
thyroid antibodies and depression. Br Med J (Clin Res Edn)
305:152�156.
Harris B, Oretti R, Lazarus J, Parkes A, John R, Richards C,
Newcombe R, Hall R. 2002. Randomised trial of thyroxine to
prevent postnatal depression in thyroid-antibody-positive wo-
men. Br J Psychiatry 180:327�330.
Heyman A, Wilkinson W, Stafford J. 1984. Alzheimer’s disease: A
study of epidemiological aspects. Ann Neurol 15:335�341.
Howland R. 1993. Thyroid dysfunction in refractory depression:
implications for pathophysiology and treatment. J Clin Psy-
chiatry 54:47�54.
Iacovides A, Fountoulakis K, Grammaticos P, Ierodiakonou C.
2000. Difference in symptom profile between generalized
anxiety disorder and anxiety secondary to hyperthyroidism.
Int J Psychiatry Med 30:71�81.
Jackson I. 1998. The thyroid axis and depression Thyroid 8:951�956.
Jankovic BD. 1985. Neural tissue hypersensitivity in psychiatric
disorders with immunologic features. J Immunol 135:853�857s.
Joffe R. 1987. Antithyroid antibodies in major depression. Acta
Psychiatr Scand 76:598�599.
Joffe R. 1999. Peripheral thyroid hormone levels in treatment
resistant depression. Biol Psychiatry 45:1053�1055.
Joffe R, Marriott M. 2000. Thyroid hormone levels and recur-
rence of major depression. Am J Psychiatry 157:1689�1691.
Joffe R, Bagby R, Levitt A. 1992. The thyroid and melancholia.
Psychiatry Res 42:73�80.
Joffe R, Segal Z, Singer W. 1996. Change in thyroid hormone
levels following response to cognitive therapy for major
depression. Am J Psychiatry 153:411�413.
Joffe RT. 1997. Refractory depression: treatment strategies, with
particular reference to the thyroid axis. J Psychiatry Neurosci
22:327�331.
Joffe RT. 1998. The use of thyroid supplements to augment
antidepressant medication. J Clin Psychiatry 59(Suppl 5):26�29; Discussion 30�31.
Joffe RT, Sokolov ST. 1994. Thyroid hormones, the brain, and
affective disorders. Crit Rev Neurobiol 8:45�63.
Joffe RT, Sokolov ST. 2000. Thyroid hormone treatment of
primary unipolar depression: A review. Int J Neuropsychophar-
macol 3:143�147.
Kaptein E. 1993. Clinical application of free thyroxine determina-
tions. Clin Lab Med 13:653�672.
Kent G, Stuckey B, Allen J, Lambert T, Gee V. 1999. Postpartum
thyroid dysfunction: Clinical assessment and relationship to
psychiatric affective morbidity. Clin Endocrinol 51:429�438.
Kjellman B, Ljunggren J, Beck-Friis J, Wetterberg L. 1985. Effect
of TRH on TSH and prolactin levels in affective disorders.
Psychiatry Res 14:353�363.
Kocsis J, Davis J, Katz M, Koslow S, Stokes P, Casper R,
Redmond D. 1985. Depressive behavior and hyperactive
adrenocortical function. Am J Psychiatry 142:1291�1298.
Kokmen E, Beard R, Chandra V, Offord K, Schoenberg B,
Ballard D. 1991. Clinical risk factors for Alzheimer’s disease: A
population-based case-control study. Neurology 41:1393�1397.
Konig F, Hauger B, vonHippel C, Wolfersdorf M, Kaschka W.
2000. Effect of paroxetine on thyroid hormone levels in severely
depressed patients. Neuropsychobiology 42:135�138.
Kuijpens J, Vader H, Drexhage H, Wiersinga W, van Son M, Pop
V. 2001. Thyroid peroxidase antibodies during gestation are a
marker for subsequent depression postpartum. Eur J Endocri-
nol 145:579�584.
Leentjens AF, Kappers EJ. 1995. Persistent cognitive defects after
corrected hypothyroidism. Psychopathology 28:235�237.
Legros S, Mendlewicz J, Wybran J. 1985. Immunoglobulins,
autoantibodies and other serum protein fractions in psychiatric
disorders. Eur Arch Psychiatry Neurol Sci 265:9�11.
Liebowitz M, Quitkin F, Stewart J, McGrath P, Harrison W,
Markowitz J, Rabkin J, Tricamo E, Goetz D, Klein D. 1988.
Antidepressant specificity in atypical depression. Arch Gen
Psychiatry 45:129�137.
Loosen P. 1985. The TRH-induced TSH response in psychiatric
patients: A possible neuroendocrine marker. Psychoneuroen-
docrinology 10:237�260.
Lucas A, Pizarro E, Granada M, Salinas I, Sanmarti A. 2001.
Postpartum thyroid dysfunction and postpartum depression:
are they two linked disorders? Clin Endocrinol 55:809�814.
Maes M, Meltzer H, Cosyns P, Suy E, Schotte C. 1993. An
evaluation of basal hypothalamic�pituitary�thyroid axis func-
tion in depression: Results of a large-scaled and controlled
study. Psychoneuroendocrinology 18:607�620.
Maes M, D’Hondt P, Blockx P, Cosyns P. 1994a. A further
investigation of basal HPTaxis function in unipolar depression:
Effects of diagnosis, hospitalization, and dexamethasone ad-
ministration. Psychiatry Res 51:185�201.
Maes M, Scharp? S, Cosyns P, Meltzer H. 1994b. Relationships
between basal hypothalamic-pituitary-thyroid-axis activity and
plasma haptoglobin levels in depression. J Psychiatr Res
28:123�134.
Marangell LB. 2000. Augmentation of standard depression
therapy. Clin Ther 22(Suppl A):A25�38; Discussion A39�41.
Marchesi C, Chiodera P, DeRisio C, Dassz L, Govi A, DeFerri A,
Piagneri B, Minelli R, Bianconi L, Gnudi A. 1988. Dopami-
nergic control of TSH secretion in endogenous depression.
Psychiatry Res 25:277�282.
Mendlewicz J, Hubain P, Koumakis C. 1984. Further investiga-
tion of the dexamethasone suppression test in affective illness:
Relationship to clinical diagnosis and therapeutic response.
Neuropsychobiology 12:23�26.
Musselman DL, Nemeroff CB. 1996. Depression and endocrine
disorders: focus on the thyroid and adrenal system. Br J
Psychiatry Suppl 123�128.
136 K. N. Fountoulakis et al.

Nelson C, Davis J. 1997. DST studies in psychotic depression: A
meta-analysis. Am J Psychiatry 154:1497�1503.
Nemeroff C. 1989. Clinical significance of psychoneuroendocri-
nology in psychiatry: Focus on the thyroid and adrenal. J Clin
Psychiatry 50(Suppl):13�20.
Nemeroff C, Simon J, Haggerty J, Evans D. 1985. Antithyroid
antibodies in depressed patients. Am J Psychiatry 142:840�843.
Nemeroff CB. 1996. Augmentation strategies in patients with
refractory depression. Depress Anxiety 4:169�181.
O’Donnell M, Silove D, Wakefield D. 1988. Current perspectives
on immunology and psychiatry. Aust NZ J Psychiatry 22:366�382.
Ordas DM, Labbate LA. 1995. Routine screening of thyroid
function in patients hospitalized for major depression or
dysthymia? Ann Clin Psychiatry 7:161�165.
Orth D, Shelton R, Nicholson W, Beck-Peccoz P, Tomarken A,
Persani L, Loosen P. 2001. Serum thyrotropin concentrations
and bioactivity during sleep deprivation in depression. Arch
Gen Psychiatry 58:77�83.
Overall J, Hollister L, Johnson M. 1966. Nosology of depression
and differential response to drugs. J Am Med Assoc 195:946�950.
Pedersen C. 1999. Postpartum mood and anxiety disorders: A
guide for the nonpsychiatric clinician with an aside on thyroid
associations with postpartum mood. Thyroid 9:691�697.
Pop V, Maartens L, Leusink G, vanSon M, Knottnerus A, Ward
A, Metcalfe R, Weetman A. 1998. Are autoimmune thyroid
dysfunction and depression related? J Clin Endocrinol Metab
83:3194�3197.
Post RM, Leverich GS, Denicoff KD, Frye MA, Kimbrell TA,
Dunn R. 1997. Alternative approaches to refractory depression
in bipolar illness. Depress Anxiety 5:175�89.
Pridmore S, Turnier-Shea Y. 2004. Medication options in the
treatment of treatment-resistant depression. Aust NZ J Psy-
chiatry 38:219�225.
Rao M, Vartzopoulos D, Fels K. 1989. Thyroid function in
anxious and depressed patients. Pharmacopsychiatry 22:66�70.
Rao M, Ruhrmann S, Retey B, Liappis N, Fuger J, Kraemer M,
Kasper S, Møller H. 1996. Low plasma thyroid indices of
depressed patients are attenuated by antidepressant drugs and
influence treatment outcome. Pharmacopsychiatry 29:180�186.
Reed L, Pangaro L. 1995. Physiology of the thyroid gland I:
Synthesis and release, iodine metabolism and binding and
transport. In: Becker K, editor. Principles and practice of
endocrinology and metabolism. Philadelphia, PA: JB Lippin-
cott Company. pp 285�291.
Roth M. 1959. The phenomenology of depressive states. Can
Psychiatr Assoc J 4(Suppl):32�52.
Rupprecht R, Rupprecht C, Rupprecht M, Noder M, Mahlstedt J.
1989. Triiodothyronine, thyroxine, and TSH response to
dexamethasone in depressed patients and normal controls.
Biol Psychiatry 25:22�32.
Sagud M, Pivac N, Muck-Seler D, Jakovljevic M, Mihaljevic-Peles
A, Korsic M. 2002. Effects of sertraline treatment on plasma
cortisol, prolactin and thyroid hormones in female depressed
patients. Neuropsychobiology 45:139�43.
Sargant W. 1960. Some newer drugs in the treatment of
depression and their relation to other somatic treatments.
Psychosomatics 1:14�17.
Schindler BA. 1985. Stress, affective disorders, and immune
function. Med Clin North Am 69:585�597.
Schule C, Baghai T. C, Alajbegovic L, Schwarz M, Zwanzger P,
Eser D, Schaaf L, Moller H. J, Rupprecht R. 2005. The
influence of 4-week treatment with sertraline on the combined
T3/TRH test in depressed patients. Eur Arch Psychiatry Clin
Neurosci in press.
Shelton RC. 1999. Treatment options for refractory depression. J
Clin Psychiatry 60(Suppl 4):57�61; Discussion 62�63.
Sher L, Rosenthal N, Wehr T. 1999. Free thyroxine and thyroid-
stimulating hormone levels in patients with seasonal affective
disorder and matched controls. J Affect Disord 56:195�199.
Staner L, De La Fuente JM, Kerkhofs M, Linkowski P,
Mendlewicz J. 1992. Biological and clinical features of recur-
rent brief depression: A comparison with major depressed and
healthy subjects. J Affect Disord 26:241�245.
Stein M, Uhde T. 1989. Autoimmune thyroiditis and panic
disorder. Am J Psychiatry 146:259�260.
Sullivan G, Hatterer J, Herbert J. 1999. Low levels of transthyretin
in the CSF of depressed patients. Am J Psychiatry 156:710�715.
Sussman N, Joffe RT. 1998. Introduction: Augmentation of
antidepressant medication. J Clin Psychiatry 59(Suppl 5):3�4; Discussion 70�73.
Szuba M, O’Reardon J, Rai A, Snyder-Kastenberg J, Amsterdam
J, Gettes D, Wassermann E, Evans D. 2001. Acute mood and
thyroid stimulating hormone effects of transcranial magnetic
stimulation in major depression. Biol Psychiatry 50:22�27.
Thase ME, Howland RH, Friedman ES. 1998. Treating anti-
depressant nonresponders with augmentation strategies: An
overview J Clin Psychiatry 59(Suppl 5):5�12; Discussion 13�15.
Van Praag H, Uleman A, Spitz J. 1965. The vital syndrome
interview. Psychiatr Neurol Neurochir 68:329�349.
vanWest D, Maes M. 1999. Activation of the inflammatory
response system: A new look at the etiopathogenesis of major
depression. Neuroendocrinol Lett 20:11�17.
Peripheral thyroid dysfunction in depression 137

REVIEW
Melatonin in mood disorders
VENKATARAMANUJAN SRINIVASAN1, MARCEL SMITS2, WARREN SPENCE3,
ALAN D. LOWE3, LEONID KAYUMOV3, SEITHIKURIPPU R. PANDI-PERUMAL3,4,
BARBARA PARRY5 & DANIEL P. CARDINALI6
1Department of Physiology, School of Medical Sciences, University Sains Malaysia, Kubang Kerian, Kota Bharu, Kelantan,
Malaysia, 2Gelderse Vallei Hospital, Department of Neurology and Sleep Disorders, Ede, The Netherlands, 3Sleep and
Neuropsychiatry Institute (SNI), Scarborough, ON, Canada, 4Comprehensive Center for Sleep Medicine, Division of
Pulmonary, Critical Care, and Sleep Medicine, Mount Sinai School of Medicine, New York, NY, USA, 5Department of
Psychiatry, University of California, San Diego, La Jolla, CA, USA, and 6Departamento de Fisiologıa, Facultad de
Medicina, University of Buenos Aires, Buenos Aires, Argentina
AbstractThe cyclic nature of depressive illness, the diurnal variations in its symptomatology and the existence of disturbed sleep�wake and core body temperature rhythms, all suggest that dysfunction of the circadian time keeping system may underlie thepathophysiology of depression. As a rhythm-regulating factor, the study of melatonin in various depressive illnesses hasgained attention. Melatonin can be both a ‘state marker’ and a ‘trait marker’ of mood disorders. Measurement of melatonineither in saliva or plasma, or of its main metabolite 6-sulfatoxymelatonin in urine, have documented significant alterations inmelatonin secretion in depressive patients during the acute phase of illness. Not only the levels but also the timing ofmelatonin secretion is altered in bipolar affective disorder and in patients with seasonal affective disorder (SAD). A phasedelay of melatonin secretion takes place in SAD, as well as changes in the onset, duration and offset of melatonin secretion.Bright light treatment, that suppresses melatonin production, is effective in treating bipolar affective disorder and SAD,winter type. This review discusses the role of melatonin in the pathophysiology of bipolar disorder and SAD.
Key words: Melatonin, mood disorders, depression, bipolar affective disorder, seasonal affective disorder
Introduction
Mood disorders comprise a group of psychiatric
disorders in which pathological mood and related
psychomotor disturbances dominate the clinical
picture. The cluster of signs and symptoms is
sustained over a period of weeks to months and
tends to recur often in a period or in a cyclical
fashion (Kahn 1999). The most common mood
disorders are major depressive disorder, bipolar
affective disorder, mania and seasonal affective
disorder (SAD), winter type. Cycles of recurrence
are manifested in these disorders interspersed with
periods of euthymia. Since mood disorders are cyclic
in nature, disturbances in circadian rhythms have
been often implicated as one of the major precipitat-
ing factors for these disorders. However, it is not
clear whether this relationship is causal or is only an
epiphenomenon of the disease.
Periodic episodes of depression and mania are
usually linked to disorders of the time-keeping
system. Epidemiological studies reveal that insomnia
is a prominent comorbidity of depression (Riemann
et al. 2001). Sleep loss is a major risk factor for
occurrence of mania in patients with bipolar disorder
(Wehr 1991). Many studies place emphasis on the
importance of stable sleep�wake rhythms and
proper sleep hygiene for preventing relapses in
bipolar disorders (Frank et al. 1997).
A large number of studies undertaken in recent
years have clearly shown that the pineal hormone
melatonin (N-acetyl-5-methoxytryptamine) is in-
volved not only in the regulation of sleep and
sleep�wake rhythms but of many circadian functions
(Pandi-Perumal et al. 2005). In circadian rhythm
disorders, the disturbances in melatonin rhythm
and amplitude have become prominent features
Correspondence: Dr D.P. Cardinali, Departamento de Fisiologıa, Facultad de Medicina, UBA, Paraguay 2155, 1121 Buenos Aires,
Argentina. Tel/Fax: �/54 11 59509611. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(3): 138�151
(Received 5 October 2005; accepted 3 January 2006)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970600571822

(Srinivasan 1997; Srinivasan et al. 2006). Both the
amplitude and rhythm of melatonin secretion are
altered in patients suffering from both major depres-
sive disorder as well as in patients suffering from
bipolar affective disorder (Tuunainen et al. 2002;
Wetterberg 1999). The onset, offset and duration of
melatonin secretion has been found altered in major
depressive disorders and in bipolar affective disorder
patients.
This review discusses the role of melatonin in
mood disorders with emphasis in SAD. The changes
of melatonin secretion in major depressive disorder
and the response of melatonin to treatment with
antidepressants will be also analysed. The hypothesis
as to whether or not internal desynchronization and
changes in melatonin rhythmicity participates in the
genesis of bipolar affective disorder will be discussed.
Lastly, the activity of melatonin as an antidepressant
in delayed sleep phase syndrome (DSPS) with com-
orbid depression will be analysed in some detail.
Generally, the studies discussed were selected based
on their relevance and of the quality of their
scientific evidence.
Melatonin: Biosynthesis and physiological
effects
Melatonin is the major product secreted from the
pineal gland of all animals and in man. Extrapineal
synthesis of melatonin occurs in places like the
retina, the gastrointestinal tract, bone marrow and
lymphocytes; however, circulating melatonin only
derives from the pineal gland (Cardinali and Pevet
1998; Claustrat et al. 2005). Tryptophan serves
as the precursor for melatonin biosynthesis. It is
hydroxylated at C5 position and then decarboxy-
lated to form serotonin. Serotonin is N-acetylated
by the enzyme serotonin N-acetyltransferase, the
rate-limiting hormone to form N-acetylserotonin.
Serotonin N-acetyltransferase activity increases
30�70-fold at night. N-Acetylserotonin is finally
O-methylated by the enzyme hydroxyindole O-
methyltransferase (HIOMT) to form melatonin.
Melatonin production occurs at night in all species
irrespective of whether they are nocturnal or diurnal.
The rhythm of melatonin secretion is endogenous
and is driven by the suprachiasmatic nucleus of the
hypothalamus (SCN), the so-called ‘biological
clock’. The rhythm is synchronized to a 24-h day/
night cycle by light acting through the retinohy-
pothalamic pathway in animals and human beings
(Cardinali and Pevet 1998; Claustrat et al. 2005).
Exposure of animals to light at night rapidly
depresses pineal melatonin synthesis. Based on
denervation or nerve stimulation studies, a simple
model of pineal regulation was envisioned, compris-
ing two premises: (1) the neural route for environ-
mental lighting control of melatonin secretion is the
neuronal circuit ‘retina�retinohypothalamic tract�SCN�periventricular hypothalamus�intermediolat-
eral column of the thoracic chord gray�superior
cervical ganglion (SCG)�internal carotid nerves�pineal gland’, (2) norepinephrine released from
sympathetic terminals at night activates postsynaptic
b-adrenoceptors coupled to the adenylate cyclase�cAMP system, therefore increasing melatonin synth-
esis and release. However, the presence of functional
a-adrenoceptors as well as the characterization of
central peptidergic pinealopetal pathways point to a
complexity of mechanisms regulating melatonin
biosynthesis (Cardinali and Pevet 1998; Claustrat
et al. 2005).
Once formed, melatonin diffuses out into the
capillary blood and the cerebrospinal fluid (CSF)
(Arendt 2000; Tricoire et al. 2002). The delicate
connective tissue capsule of the pineal gland does
not prevent diffusion of melatonin into CSF. Mela-
tonin arrives early in the third ventricle CSF as
compared with the lateral ventricles. As melatonin
passes through all biological membranes with ease,
brain tissue has higher melatonin levels than other
tissues in the body (Reiter and Tan 2002). Indeed,
CSF melatonin levels have been found to be 5 to 10
times higher than those of melatonin in blood
(Tricoire et al. 2002).
Human plasma melatonin rhythm is remarkably
constant within the same individual and occurs with
invariant regularity from day to day and week to
week. However, melatonin production exhibits con-
siderable inter-individual differences (Macchi and
Bruce 2004), with more than 10-fold variability in
nocturnal plasma melatonin concentrations among
individuals (Zeitzer et al. 1999). The finding that,
compared to the general population, the variability
of rhythmicity in melatonin production is reduced in
siblings suggests that it may have a genetic basis
(Griefahn et al. 2003). Melatonin concentrations in
body fluids over a 24-h period have been found to be
useful for investigating the ‘free running rhythm
failure’ encountered in certain subtypes of depres-
sive patients, a fact which has been interpreted by
some (Wetterberg 1999) to support either a phase
advance or phase delay hypotheses of mood dis-
orders.
Melatonin is involved in the control of various
physiological functions like seasonal reproduction
(Reiter 1980), sleep regulation (Monti et al. 1999;
Wurtman and Zhdanova 1995), immune function
(Esquifino et al. 2004; Guerrero and Reiter 2002),
inhibition of tumor growth (Blask et al. 2002), blood
pressure regulation (Doolen et al. 1998; Scheer et al.
2004), retinal physiology (Dubocovich et al. 1999)
Melatonin in mood disorders 139

and control of circadian rhythms (Dawson and
Armstrong 1996; Kunz 2004), control of human
mood and behavior (Srinivasan 1997) and free
radical scavenging (Reiter et al. 2005). Melatonin
participates in many of the respective mechanisms
by acting through G-protein coupled membrane
receptors like MT1 and MT2 (Dubocovich et al.
2000; Reppert et al. 1994, 1995) and nuclear
receptors like RZR/ROR (Wiesenberg et al. 1995).
Within the G-protein coupled receptor family of
proteins, melatonin acts through a number of signal
transduction mechanisms that ultimately result in
specific physiological responses (Witt-Enderby et al.
2003). In addition to receptors in the proper sense,
melatonin acts through another binding site, origin-
ally thought to represent another membrane-bound
receptor (MT3), but later confirmed to be the
enzyme quinone reductase-2 (Nosjean et al. 2000).
Melatonin also binds directly to calmodulin and
cytoskeletal proteins (Benitez-King et al. 1996).
Melatonin in major depressive disorder
For a diagnosis of a major depressive disorder at
least five of the following symptoms must have been
present continuously for more than 2 weeks: (1)
depressed mood, (2) markedly diminished interest in
work, (3) significant weight loss, (4) insomnia or
hypersomnia, (5) psychomotor agitation, (6) fatigue
or loss of energy, (7) feelings of worthlessness,
(8) diminished ability to concentrate, (9) recurrent
thoughts of death (Kahn 1999). The nature and
extent of disruption of melatonin secretion in major
depressive disorder has been under intense investi-
gation during the last few decades, ever since
Wetterberg and co-workers (1979) formulated the
‘low melatonin syndrome’ hypothesis, i.e., the con-
cept that low melatonin secretion can be a biological
marker for susceptibility to depressive disorders.
Numerous studies have substantiated a deficiency
of melatonin secretion in depressives (Brown et al.
1985a,b; Claustrat et al. 1984; Miles and Philbrick
1988; Nair et al. 1984; Paparrigopoulos et al. 2001;
Sack and Lewy 1988; Venkoba rao et al. 1983;
Zeiten et al. 1987). In some studies clinical symp-
toms such as suicidal ideas correlated with the
decrease in melatonin levels (Venkoba Rao et al.
1983). It has been suggested that the low melatonin
levels seen in depressives are due to low norepi-
nephrine and serotonin levels in the brain (Arendt
1989).
Measurement of melatonin in the body fluids such
as plasma or saliva, or of its metabolite 6-sulfatox-
ymelatonin in the urine, is a reliable index of
noradrenergic activity. In a study undertaken in
patients with major depressive disorder, Paparrigo-
poulos et al. (2001) reported that the administration
of clonidine, a partial a2-adrenergic agonist, reduced
melatonin levels in depressives but not in healthy
controls. The authors concluded that depressive
symptoms could be due to a supersensitivity of a2-
adrenoceptors that in turn would result in reduced
norepinephrine release from sympathetic nerve
terminals and reduced melatonin secretion from
the pineal gland.
Additional evidence suggests a more complex
model of melatonin dysfunctionality than that of a
deficiency of melatonin production. For instance,
the number of studies reporting low melatonin levels
in depressives is at least equaled by those document-
ing increases in melatonin production (Crasson et al.
2004; Rubin et al. 1992; Sekula et al. 1997; Shafii
et al. 1997; Stewart and Halbreich 1989; Szymanska
et al. 2001; Thompson et al. 1988).. Rubin et al.
(1992) noted that in both men and women who were
diagnosed as having major depressive disorder,
nocturnal melatonin secretion increased significantly
above the average seen in normal subjects. The
authors could not find any relationship between
melatonin levels and depressive symptomatology nor
any particular type of depression. In addition to high
nocturnal melatonin levels, a late nocturnal peak
time of melatonin secretion was noted in patients
(Rubin et al. 1992). Higher nocturnal serum mela-
tonin levels were found in female depressives (Sekula
et al. 1997). In depressive patients classified into two
categories, those with Hamilton depression scores of
20�29 points and those with scores of 30�40 points,
high diurnal serum melatonin levels were observed
in parallel with higher Hamilton scores (i.e. with a
greater intensity of depressive symptoms) (Szy-
manska et al. 2001). With respect to nocturnal
melatonin levels, both groups of depressive patients
exhibited significantly higher melatonin levels when
compared to healthy controls of the same age group.
Treatment of patients with clorimipramine for a
period of 8 weeks significantly reduced mean mela-
tonin levels, although even after complete remission
those levels continued to be high as compared to
controls (Szymanska et al. 2001). The authors
attributed the elevated melatonin levels encountered
in their depressive patients to some kind of bio-
chemical defect in the retina or to a disrupted
homeostasis between the SCN and the pineal gland.
Crasson et al. (2004) also reported a significant
elevation of daytime urinary 6-sulfatoxymelatonin
secretions among depressives. In a study on 382
postmenopausal women, a positive family history
of depression was associated with longer duration
of 6-sulfatoxymelatonin excretion (Tuunainen et al.
2002). This study suggested a familial vulnerability
140 V. Srinivasan et al.

in endogenous melatonin signal in subjects prone to
depression.
The discordant results on melatonin levels in
depressive patients may well reflect subcategories
of illness. Inasmuch as a drop or rise in melatonin
levels is paralleled by comparable alterations in
serotonin levels, it may be relevant here to allude
briefly to the extensive research on the two bio-
chemical subtypes of endogenous depression: those
with low dopamine levels and those with low
dopamine plus low serotonin; the second group is
the most severe, the ‘impulsive depressives’, and at
the greatest risk for suicide. According to Wetterberg
(1999), patients with low melatonin syndrome are
different clinically and biochemically from patients
with normal or high melatonin secretion. Proper
identification of these subgroups of patients is
essential for designing a specific pharmacotherapy
to correct the underlying abnormality. In any event,
the disturbance of melatonin secretion in patients
with major depression supports the suspicion of
photoperiodic abnormality in depression.
A study performed in 459 postmenopausal women
revealed that those patients experiencing lower
levels of illumination had more depression and more
complaints of sleep disturbance (Kripke et al. 2004).
It was further noted that Hispanic African women
suffered more depression due to low levels of illumi-
nation than Native American women, thus revealing
ethnic differences in depression related to restrictions
in light exposure (Kripke et al. 2004). In a study of
pregnant women with major depressive disorder
according to Diagnostic and Statistical Manual of
Mental Disorders IV (DSM�IV) (American Psychia-
tric Association 1994), bright light treatment advan-
ced the melatonin rhythm and improved symptoma-
tology, thus supporting the applicability of bright light
therapy to treat antepartum depression (Epperson
et al. 2004).
Melatonin phase position in major depressive
disorder
In addition to changes in the amplitude of diurnal or
nocturnal melatonin secretion, a number of studies
have analysed melatonin rhythm in depressives. In
patients with major depressive disorder there is a
phase advance of melatonin rhythm (Beck-Friis et al.
1985a; Branchey et al. 1982; Claustrat et al. 1984;
Nair et al. 1984; Wehr et al. 1985). Beck Friis et al.
(1985a) studied the number of nocturnal melatonin
peaks that occurred in their sample before and after
01:00 h. The depressive patients with an abnormal
dexamethasone suppression test response had a
trend toward significantly earlier melatonin peaks
than those with a normal response to dexamethasone
or controls.
Indeed, a phase shift of melatonin secretion is a
prominent feature of major depressive disorder.
Rubin et al. (1992) noted a trend towards a later
peak (phase delay) in nocturnal melatonin secretion.
In a study on 14 depressive patients, Crasson and
co-workers (2004) found a delay of 77 min in the
serum melatonin peak when compared to normal
controls of the same age and at the same month of
sampling. As far as the onset in nocturnal melatonin
production, Rubin et al. (1992) reported that it
began at 21:00 h, and reached a maximum level at
around 03:00�05:00 h in depressives. This was in
contrast to that seen in normal subjects in which the
nocturnal melatonin onset began at around 23:00 h.
Thus, a trend towards earlier onset of melatonin
secretion was noted in depressed patients as com-
pared to normal healthy controls.
Sekula et al. (1997) found a significant correlation
between a delay in the offset of serum melatonin and
delayed acrophase with lifetime major depressive
disorder. A significant delay in the onset of urinary
6-sulfatoxymelatonin excretion was reported in post-
menopausal women with major depressive sympto-
matology (Tuunainen et al. 2002). The duration of
melatonin secretion was also longer than that
observed in healthy controls. Collectively, the results
suggest that it is the melatonin offset that is delayed
in major depressives. Offset melatonin time is signi-
ficant in determining the duration of melatonin
secretion.
Melatonin response to treatment with
antidepressants
The response of melatonin secretion to antidepres-
sant treatment has been studied. Chronic treatment
with desmethylimipramine for a period of 3 weeks
increased the amplitude of melatonin secretion
(Thompson et al. 1985). In another study, treatment
with imipramine for 2 weeks increased melatonin
excretion (Venkoba Rao et al. 1983). Sack and Lewy
(1986) reported a sustained increase in urinary 6-
sulfatoxymelatonin levels in depressed patients after
treatment with desipramine for a period of 3 weeks.
Golden et al. (1988) reported a significant increase
in 6-sulfatoxymelatonin excretion in depressives
following treatment with either imipramine or the
monoamine oxidase inhibitor bupropion. Experi-
mental studies in rats have also revealed that the
administration of imipramine increased pineal mel-
atonin content significantly (Srinivasan 1989).
A deficiency of norepinephrine at the synaptic
cleft of postganglionic sympathetic nerve fibres origi-
nating in the SCG resulted in a decrease of
Melatonin in mood disorders 141

melatonin production by the pineal gland as demon-
strated by the studies of Paparrigopoulos et al.
(2001). Taking the pineal neuroendocrine junction
as an end point is useful for considering the role of
catecholamines in the regulation of mood, and in
particular, how melatonin can be involved in this
process.
Several studies have supported the hypothesis that
melatonin secretion is an index of norepinephrine
activity in depressed patients. The evidence obtained
is consistent with Schildkraut’s original ‘catechola-
mine hypothesis of depression’ (Schildkraut 1965)
and can be considered an extension or corollary of it.
The clinical evidence indicating that antidepressant
drug treatment changes melatonin secretion in major
depressive disorder patients points to the possibility
that the pineal gland may play a role in the aetiology
of mood disorders. Higher serum melatonin levels
were found in patients suffering from major depres-
sive disorder which decreased after pharmacological
treatment (Varma et al. 2002). In another study in
patients diagnosed of major depressive disorder who
failed to respond to pharmacological therapy, elec-
troconvulsive therapy brought about a significant
decrease in depression symptomatology and urinary
6-sulfatoxymelatonin excretion (Krahn et al. 2000).
A close association between depressive symptoms
and delayed offset of 6-sulfatoxymelatonin excretion
occurred, indicating that the timing of melatonin
secretion may be important in depression (Tuunai-
nen et al. 2002).
Internal desynchronization and bipolar
affective disorder
Bipolar affective disorders are characterized by the
occurrence of mania or hypomania either preceded
or followed by episodes of depression. Bipolar I is
characterized by episodes of mania and depression,
while in bipolar II disorder, hypomania is preceded
or followed by major depression (Kahn 1999).
According to DSM-IV criteria for mania, the follow-
ing symptoms should be present for at least 1 week:
(1) inflated self esteem, (2) decreased need for sleep,
(3) more talkative, (4) flight of ideas, (5) distract-
ibility, (6) excessive involvement (Kahn 1999).
Several clinical features of bipolar disorder suggest
that disturbances in the timing or phase position of
circadian rhythms may play a role in precipitating
the disorders. Patients with bipolar disorder often
exhibit an infradian sleep/wake rhythm, i.e. the
patients forego sleep in a complete night between
two nights of normal sleep (Wehr et al. 1982).
Studies of sleep in recurrent depressive and
bipolar affective disorders have been useful for
theoretical considerations about the aetiology and
pathophysiology of the disease. Bunney and his co-
workers (1970) first noted that bipolar depressive
patients exhibited marked reduction in sleep during
the night before they switched from depression.
These observations were subsequently confirmed
(e.g., Sitaram et al. 1978). Manic-depressive pati-
ents at the beginning of a manic episode exhibited
one or more 48-h rest�activity cycles, i.e. the pati-
ents spent one complete sleepless night in between
two nights of normal sleep (Wehr and Goodwin
1979).
Similar responses have been observed in healthy
human subjects after a few weeks of exposure to
constant environmental conditions. The period of
rest/activity cycle lengthened to 45 h, whereas their
temperature and rapid eye movement (REM) sleep
rhythms remained synchronized to 25 h, resulting in
internal desynchronization (Wever 1986). The simi-
larities between bipolar patients and healthy human
subjects in an internal desynchronization situation
suggest that bipolar disorders imply internal desyn-
chronization. Kripke et al. (1978) hypothesized that
mania and depression are the result of a beat pheno-
menon generated when two rhythms go in and out of
phase. The hypothesis that internal desynchroniza-
tion causes a change in mood has gained support
from sleep deprivation studies also. Not only do
anxious patients exhibit significant sleep distur-
bance, but conversely sleep deprivation produces
elevations in anxiety symptoms (Bourdet and Gold-
enberg 1994). Indeed, depression has a significant
anxiety component, e.g., eight questions on the
Hamilton Anxiety Scale are shared in common
with the Hamilton Depression Scale.
The disturbance of circadian rhythms seen in
bipolar disorders can be due to disturbance in the
function of the SCN�pineal�melatonin link. In-
deed, melatonin has a regulating effect on the SCN
causing entrainment of circadian rhythms to a
natural 24-h cycle via MT2 receptors.
Melatonin amplitude and rhythm in bipolar
affective disorder
As a rhythm-regulating factor and a hormone
involved in the physiological regulation of the
sleep�wake rhythm, melatonin has drawn the atten-
tion of investigators studying bipolar affective dis-
orders. In view of its central role as an internal
synchronizer (Zeitgeber), melatonin fits more appro-
priately with bipolar illness than with any other type
of psychiatric disorder.
In a study on melatonin levels in unipolar and
bipolar depressive patients, Beck-Friis et al. (1985b)
noted significantly lower melatonin levels in euthy-
mic bipolar patients. Souetre et al. (1989) reported
142 V. Srinivasan et al.

reduced amplitude of melatonin secretion in 11
bipolar patients during the depressive phase that
came back to normal on remission. In a longitudinal
study of a single manic-depressive patient, an
increase in melatonin secretion was noted during a
manic phase was noted that was twice that of
melatonin levels during the euthymia and depressed
phases (Kennedy et al. 1989).
Based on melatonin level studies in bipolar
patients, Lewy et al. (1985) concluded that the
amplitude of melatonin secretion is state-dependent
rather than reflecting brain noradrenergic activity.
Kennedy and co-workers (1996) studied nine bipo-
lar patients during manic, depressed and euthymic
states. Serum melatonin levels were lower during
euthymic, depressed and manic phases, when com-
pared with healthy normal controls. The authors
concluded that the decreased melatonin production
is a trait marker and not a state marker of bipolar
disorders (Kennedy et al. 1996).
Numerous studies have demonstrated that the
phase of melatonin secretion varies systematically
with mood changes in bipolar affective disorder.
Lewy et al. (1979) were the first to report changes in
phase position of melatonin levels in bipolar pa-
tients. In a study of four manic patients, they found a
phase advance of melatonin levels when compared to
normal subjects. In a longitudinal study of a single
manic patient, Kennedy et al. (1989) noted a phase
advance of the nocturnal melatonin peak during the
manic phase which preceded that of the euthymic or
depressed phases by at least 1 h (i.e. the melatonin
peaks occurred at 03:00 h in the manic phase, at
04:00 h during the euthymic phase and at 05:00 h in
the depressed phase).
Several studies have demonstrated that bipolar
patients can be treated according to the chronobio-
logical principles, such as exposure to bright light or
administration of melatonin, which are used for
other phase disordered patients who do not have
depressive symptomatology (Leibenluft et al. 1997).
Bright light shifted melatonin levels, with morning
light advancing the melatonin rhythm and evening
light delaying melatonin rhythm (Minors et al.
1991). The use of morning bright light caused
bipolar patients in a hypomaniac phase to cycle
more dramatically. Liebenluft et al. (1997) observed
that the administration of 5�10 mg of melatonin in
the evening (or midday administration of bright
light) stabilized the phase of endogenous melatonin
rhythm. In another study, abnormalities of melato-
nin secretion were reported in bipolar I patients with
delayed peak melatonin time and baseline melatonin
levels lower than 60 pg/ml (Nurnberger et al. 2000).
To what extent the changes in melatonin observed
were related to the pharmacotherapy per se or to the
improvement in the depressive symptoms remains to
be defined.
Light suppression of nocturnal melatonin
secretion in bipolar affective disorder
The natural light�dark cycle plays an important role
in regulating circadian rhythms. Retinal stimulation
by light sends a tonic stimulatory signal to the SCN.
In turn, the SCN inhibits the firing of neurons in the
paraventricular nucleus (PVN) to regulate melatonin
secretion by the pineal gland. During nighttime
hours, when the SCN is not stimulated by light,
the neurons of the PVN are released from SCN
inhibition and pineal melatonin secretion is stimu-
lated. Signals initiating this process are transmitted
through the multisynaptic pathway involving the
median forebrain bundle, the intermediolateral col-
umn of the upper thoracic spinal cord and the SGG
(Moore 1996).
Lewy et al. (1980) were the first to demonstrate
that light of an intensity higher than 500 lux
suppresses melatonin secretion in human beings.
Subsequently, the same authors showed that sup-
pression of melatonin secretion by 500-lux light was
greater in manic-depressive patients (Lewy et al.
1981). In this study they found that exposure to
500 lux caused a 50% suppression of melatonin
levels in manic-depressive patients but had no effect
in controls. Therefore, an increased responsiveness
to light in manic-depressives could explain the
circadian rhythm abnormalities seen (such as phase
advances in temperature or REM sleep rhythms). In
another study of 11 euthymic bipolar patients,
exposure to 500 lux of light at 02:00 and 04:00 h
caused suppression of melatonin by 61.5% as
compared to age- and sex-matched control subjects,
in which exposure to light caused only 28% suppres-
sion of melatonin secretion (Lewy et al. 1985). The
authors concluded that an increased sensitivity to
light might be a trait marker for bipolar affective
disorder. The fact that some normal subjects also
exhibited increased sensitivity to light pointed to the
possibility that those normal individuals could be at
risk for developing a mood disorder.
In a study carried out in 29 euthymic bipolar
patients, 24 patients with unipolar depression and 50
non-psychiatrically ill control subjects, Nurnberger
et al. (2000) found a 29.8, 32.2 and 34.6% of light-
induced melatonin suppression in bipolar, unipolar
and control groups, respectively. Although no evi-
dence could be obtained for increased suppression
of melatonin secretion in bipolar patients, when
bipolar patients were sub-divided, patients with
bipolar I disorder showed a trend towards increased
dark-adjusted melatonin suppression (62.7%) as
Melatonin in mood disorders 143

compared to matched controls (40.0% suppression).
Taken together these findings, they support the
hypothesis that excessive light-induced suppression
of melatonin secretion may be a risk factor for
development of a mood disorder. What remains
to be established is what mechanism or factor(s)
mediates this light-induced suppression response.
Three meta-analyses published by Cochrane data-
bases support the efficacy of light therapy in mood
disorders (Forbes et al. 2004; Montgomery and
Dennis 2002; Tuunainen et al. 2004).
Melatonin in SAD
SAD, winter type, is characterized by recurrent
episodes of depression during winter months and
euthymia or hypomania in spring or summer. First
documented by Rosenthal et al. (1984), the major
symptoms of this disorder were noted to be
hypersomnia, hyperphagia, carbohydrate craving or
weight gain. Since some SAD patients suffer from
reduced appetite, weight loss and insomnia, at least
two clinical types of SAD (corresponding to the
atypical and melancholic feature specifier according
to DSM-IV) can be distinguished.
Patients suffering from SAD also exhibit delayed
circadian rhythms, and according to Lewy and co-
workers (1987, 1988) it is the phase delay of the
circadian pacemaker relative to the timing of the
sleep�wake cycle that underlies the pathogenesis of
the disease. While Lewy’s phase delay hypothesis of
winter depression has been supported by a number
of studies (Avery et al. 1997; Dahl et al. 1993; Lewy
et al. 1998; Sack et al. 1990), other investigations
(Eastman et al. 1993; Thompson et al. 1997; Wirz-
Justice et al. 1996) have failed to confirm it. To
understand the pathophysiology of SAD, Wehr et al.
(2001) studied the duration of melatonin secretion
in 57 patients and 62 healthy controls during
summer and winter seasons and under constant
dim light conditions. The levels of melatonin were
measured in plasma samples obtained every 30 min
for 24 h during each season. The results of the study
revealed that duration of active melatonin secretion
in summer tended to be shorter in patients (8.49/
1.4 h) than in healthy volunteers (8.99/1.2 h). This
difference was statistically different in the case of
men (8.59/1.4 h in SAD patients, 9.39/1.2 h in
healthy controls). There were no significant differ-
ences between patients and healthy controls in the
duration of melatonin secretion during winter (Wehr
et al. 2001).
Seasonal changes in the duration of melatonin
secretion can be manifested by changes in the time
of onset or offset of melatonin secretion. In SAD
patients the most prominent marker of seasonal
variance was the change in the time of melatonin
offset (Wehr et al. 2001). This study was the first to
document that patients with winter depression gen-
erate a biological signal of change in season in a
manner similar to that of photoperiodic mammals
(Wehr et al. 2001). By what mechanism patients
with SAD are able to produce changes in the
duration of melatonin secretion remains to be
defined.
One possibility is that the retina or neural circuits
that mediate responses to seasonal changes in day
length are only slightly affected by artificial light in
patients with SAD but respond well to the higher
luminance of sunlight. Some are of the opinion that
photosensitivity in SAD patients may be secondary
to a pineal dysfunction (Pacchierotti et al. 2001).
Nearly 90% of the SAD patients meet the criteria
of the DSM-IV for bipolar disorder II (American
Psychiatric Association 1994; Faedda et al. 1993;
Wehr et al. 1987). Patients with SAD have delayed
offset of melatonin secretion by 2 h (Terman et al.
1987). The importance of the circadian rhythm in
the causing SAD has gained much support from
bright light treatment studies. Exposure to bright
light in the morning has been shown to correct phase
position and acts as an effective antidepressant
(Lewy et al. 1998). As SAD patients appear to
have abnormally phase-delayed rhythms, application
of bright light for 1 or 2 h immediately upon
awakening is recommended.
By using plasma melatonin onset as the circadian
phase marker, Terman et al. (2001) looked for the
effects of morning and evening bright light on
melatonin phase-shift responses and clinical re-
sponses in SAD patients. Morning and evening
bright light exposure (10 000 lux, 30 min) produced
significant phase shifts in melatonin rhythm. A
significant correlation between the magnitude of
phase advances of melatonin secretion to morning
light and improvement in depression ratings was
detected (Terman et al. 2001). The direction and
magnitude of phase shifts depended upon the inter-
val between the dim light melatonin onset (DLMO)
and the time of light administration. According to
Terman et al. (2001), it is the DLMO to time of light
administration interval that provides a unifying
metric for circadian time. Consequently, they have
recommended that the best treatment for winter
depression was exposure to light having an intensity
of 10 000 lux for 30 min. For maximum effective-
ness, the initiation of the light exposure period must
be scheduled in circadian time, that is, about 8.5 h
after the DLMO. Significant correlations between
the magnitude of the phase advance after morning
light and improvement in depression ratings
144 V. Srinivasan et al.

supported the validity of this therapeutic strategy
(Terman et al. 2001).
The therapeutic effect of bright light treatment has
been demonstrated in several studies. Association of
winter type of SAD with reduced day length and
therapeutic response to bright light suggest that
either photoperiodic time measurement, or a delayed
circadian phase, or both, play a role in the aetiology
of the disease (Putilov et al. 2005). Changes in the
profiles of melatonin secretion in SAD could be
linked to the beneficial effects of bright light and the
remission during spring and summer. The delayed
phase and long duration of melatonin secretion in
SAD patients may be responsible for the symptoms
of hypersomnia and late awakening seen in this
disorder (Putilov et al. 2005). An abnormal melato-
nin secretion during daytime could account for the
SAD symptoms as well.
The association between season and mood
changes has also been studied in normal people.
Murray et al. (2003) tested Lewy’s phase shift
hypothesis of SAD in a normal community sample
of 244 adults in Melbourne. Seasonality of mood
was measured by using self-report analysis for a
3-year period. They also used a morningness�eveningness questionnaire to assess the self-report
estimates of circadian phase. A positive association
was found between lowered mood in winter and
winter phase delay among a random community
sample. This study provided additional support for
Lewy’s hypothesis in a normal population and
supported the role of seasonal influences in mood
disorders (Murray et al. 2003).
The efficacy of melatonin treatment to treat SAD
has been explored by Wirz-Justice et al. (1990).
Melatonin, at the same doses used to treat circadian
rhythm-related sleep disturbances, had no effect on
the depressive symptoms in SAD patients, whether
given early (07:00 h) or late (23:00 h) for a week.
Other placebo-controlled randomized studies on
melatonin/melatonin analog efficacy in the treatment
of mood disorders are listed in Table I.
Melatonin as an antidepressant in delayed
DSPS with comorbid depression
Weitzman et al. (1981) were the first to report the
presence of chronobiological abnormalities in half of
a sample of depressed patients, a finding which was
later supported by other investigators who found a
high prevalence of depression in adolescents with
DSPS (Ferber 1985). Several studies also supported
the view that a close association exists between
disorders of sleep onset and mood (Regestein and
Monk 1995; Regestein and Pavlova 1995; Thorpy et
al. 1988). In depressed states, circadian rhythms are
thought to be abnormally phase advanced with
respect to the sleep�wake cycle (Gwirtsman et al.
1989; Linkowski and Hubain 1995; MacLean et al.
1983; Shiromani et al. 1991). Since REM sleep is
predominantly controlled by circadian processes
(Borbely 1982), the apparent phase advance in
circadian rhythms provides a plausible explanation
for the short REM sleep latency and temporal
redistribution of REM sleep frequently observed in
depression (Coble et al. 1981; Hamilton and Sha-
piro 1990; Kupfer et al. 1984a,b; Reynolds and
Kupler 1988). Advancing sleep in some depressed
patients has been shown to have an antidepressant
effects (Wehr et al. 1979). There is evidence that
sleep deprivation in the second half of the night (but
not the first half) can be transiently helpful for
ameliorating depressive symptoms in many cases
Table I. Placebo-controlled randomised studies on melatonin/melatonin analogue efficacy in the treatment of mood disorders.
Melatonin/
melatonin
analogue
Dose Type of mood
disorder
Effect Reference
Melatonin 150�1150 mg/day Major depressive
disorder
increased dysphoria (Carman et al. 1976)
Melatonin 5 mg/day SAD loss of sleep & weight (Wirz-Justice et al. 1990)
Melatonin 10 mg/day Bipolar affective
disorder
No effect (Leibenluft et al. 1997)
Melatonin 5�10 mg/day,
slow release
Major depressive
disorder
Improved sleep, no effect on symptoms of
depression
(Dolberg et al. 1998)
Melatonin 5�10 mg/day Resistant depression No antidepressant effect but 20% decrease in
Hamilton scores, 36% decrease of insomnia
(Dalton et al. 2000)
Agomelatine 1�25 mg/day Major depressive
disorder
Significant antidepressant effect, even in
severely affected patients with sufficient
severity. Reduced anxiety associated with
depression
(Loo et al. 2002)
Melatonin 5 mg/day DSPS with comorbid
depression
Reduced fatigue, increased alertness, reduced
sleep disorder
Kayumov et al.
unpublished results
Melatonin in mood disorders 145

(Berger et al. 1997; Riemann et al. 1996; Schilgen
and Tolle 1980).
According to the internal coincidence theory of
depression, an internal phase angle disturbance
exists between sleep and other circadian rhythms
(Wehr and Wirz-Justice 1982). Thus, sleep induces
or exacerbates depression when sleep coincides with
the circadian phase that is sensitive to the effects of
sleep. In DSPS patients with comorbid depres-
sion, an internal phase angle disturbance may exist
because sleep is not as delayed as other circadian
rhythms, or conversely, because the circadian rhy-
thms are not as delayed as the sleep�wake cycle.
Late sleeping itself does not exacerbate nor precipi-
tate depression (Globus 1969; Surridge-David et al.
1987; Wehr et al. 1979), probably because sleep is
not as phase shifted as other circadian rhythms.
Theoretically, manipulations correcting the phase
angle disturbance between sleep�wake cycle and
circadian rhythms should result in an amelioration
of both DSPS and comorbid depression (Nagtegaal
et al. 2000).
It has been shown that exogenous melatonin
affects the phase of underlying biological rhythms
as well as the phase of the sleep�wake oscillator
(Arendt and Skene 2005; Cardinali et al. 2006).
Based on this fact, one of us assessed, in a rando-
mized, double-blind placebo-controlled study, the
effects of exogenous melatonin in eight patients
with established diagnosis of DSPS and comorbid
depression (Kayumov et al. unpublished results).
The diagnosis of depression was based on pre-study
clinical interviews and high scores on the CES-D
and Hamilton Depression scales (Radloff and Rae
1979). During melatonin treatment, the depression
scale scores decreased and increased again when the
subjects were placed on placebo. The patients with
depressive features treated with melatonin had sig-
nificantly more total sleep time than on placebo.
REM sleep latencies were within the normal range.
Interestingly, while on melatonin treatment the
patients showed a normal distribution of REM sleep
and an increase in REM sleep duration. The REM
density tended to be greater after melatonin treat-
ment (Kayumov et al., unpublished results).
An altered rhythm of melatonin secretion was
reported in DSPS patients (Shibui et al. 1999). A
relationship may exist between the magnitude of the
DSPS symptoms and the severity of comorbid
depressive features and abnormalities in the circa-
dian pattern of circulating melatonin as judged by
the excretion of major metabolite 6-sulfatoxymela-
tonin (Kayumov et al. unpublished results). The
patients with marked comorbid depression and
extreme symptoms of DSPS showed an abnormal
circadian pattern of melatonin secretion on placebo
treatment, with a peak during the diurnal period
(between 08.00 and 15.00 h). Melatonin adminis-
tration at a dose of 5 mg/day to DSPS patients with
comorbid depression normalized the circadian pat-
tern of 6-sulfatoxymelatonin excretion, with the
usual nocturnal rise and rapid decline of 6-sulfatox-
ymelatonin during daytime hours. This explains why
the patients did not report any hangover effects after
melatonin as judged by the subjective assessment of
the circadian pattern of sleepiness, fatigue, and
alertness. It is reasonable to conclude that a phase
advance in melatonin output and possibly in other
circadian rhythms along with a sleep�wake cycle
produced by exogenous melatonin resulted in ame-
lioration of symptoms of depression. This specula-
tion is supported by the findings of Nagtegaal et al.
(2000) showing that melatonin treatment alleviates
feelings of depression in DSPS patients as assessed
by the SF-36 mental health sub-scale. These find-
ings contradict the surprisingly widespread opinion
that melatonin may cause or exacerbate depression
(Chase and Gidal 1997; Cupp 1997). It must be
stressed that in DSPS patients with comorbid
depression, melatonin administration improved cir-
cadian profile scores of sleepiness, fatigue, and
alertness, thus having antidepressant properties.
Antidepressants with a more rapid response are
highly desirable for treating major depressive dis-
order. Agomelatine, a MT1/MT2 melatonin agonist
and selective antagonist of 5-HT2C receptors, has
been demonstrated to be active in several animal
models of depression. In a double-blind, rando-
mized multicentre multinational placebo-controlled
study, including 711 patients (238 males; 473
females, mean age 42.3 years) suffering from major
depressive disorder agomelatine (25 mg) was sig-
nificantly more effective ( 61.5%) than placebo
(46.3%) in the treatment of major depression (Loo
et al. 2002).
Conclusions
For quite some time, there has been a continuing
search for ‘biological markers’ of affective disorders
such as recurrent depressive and bipolar affective
disorders. A number of neurotransmitter substances
including norepinephrine, serotonin, acetylcholine
and dopamine, as well as hormones such as thyrox-
ine and cortisol, have all been implicated, but none
has provided a single model explaining the basis of
mood disorders. The occurrence of disturbed diur-
nal rhythms, such as delayed sleep onset, early
morning awakening, or body temperature variations
that are seen in depressives, suggest that dysfunction
of the circadian apparatus perhaps underlies the
pathogenesis of depression. An internal desynchro-
146 V. Srinivasan et al.

nization hypothesis has been advocated to explain
the periodic episodes of rapid cycling depression and
mania seen in bipolar disorders.
Indeed, health can be defined as the individual’s
ability to optimize internal rhythms so that they
synchronize with those of external light�dark cycles.
Both the pineal gland and its hormone melatonin are
involved in this adaptation. Measurement of mela-
tonin levels or that of its metabolites in body fluids
has shown that, in major depressive disorder, these
levels are altered during the active phase of illness.
Treatments with antidepressants have been shown
either to increase or to decrease melatonin levels in
depressive patients and after clinical remission,
although some studies could not find evidence to
support that hypothesis. Alteration in the phase
position of melatonin secretion has been documen-
ted in a number of studies of major depressive
disorder patients. Studies of melatonin levels in
bipolar affective disorders has demonstrated de-
creased levels during the depressive phase and
increased secretion during the manic phase.
Melatonin has been suggested as both a ‘state
marker’ as well as ‘trait marker’ in mood disorders,
although convincing evidence for this effect awaits
future studies. Phase advance in melatonin secretion
has also been reported in bipolar patients. Use of
bright light treatment is effective in restoring the
correct phase position of melatonin rhythm in
bipolar patients. Use of light brighter than 500 lux
suppresses nocturnal melatonin secretion in human
beings. This suppression is greater in bipolar
patients as compared to normal individuals. Such
supersensitivity to light has also been documented in
some normal subjects, perhaps indicating a propen-
sity to develop bipolar affective disorder. Like
recurrent depressive and bipolar affective disorders,
SAD, winter type, is also considered a mood
disorder. In SAD patients, a phase delay in circadian
rhythms has been documented and the use of bright
light in the morning has been found effective in
correcting this condition, including the melatonin
production cycle. Patients with winter depression
have been shown to exhibit altered onset duration
and offset of melatonin secretion as compared to
healthy subjects.
A change in the onset, duration and offset of
melatonin secretion was seen in patients during
summer and winter seasons. Patients with SAD
generate a biological signal in accordance with the
change in season. The use of bright light for treating
patients with SAD at the correct circadian time
rather than in clock time has been found beneficial
in correcting the underlying abnormalities seen.
Bright light or the judicious administration of
melatonin has been found beneficial in restoring
the disordered photoperiodic mechanism, involving
retina, SCN and pineal gland, disruptions of which
are all associated with, and may possibly be causally
involved in mood disorders, particularly SAD.
Melatonin regulates the rhythm of many func-
tions, and alterations in its secretory pattern have
been found in a number of psychiatric disorders.
Besides SAD, these include bipolar disorder and
unipolar depression, bulimia, anorexia, schizophre-
nia, panic disorder and obsessive compulsive dis-
order (Pacchierotti et al. 2001). At present, it is not
clear whether these changes are causal to or simply a
marker for other neurochemical alterations. Further,
it is not known if melatonin is equally involved in the
development of the pathophysiology of each of these
disorders.
Acknowledgements/Statement of interest
One of authors (VS) would like to acknowledge
Puan Rosnida Said, Department of Physiology,
School of Medical Sciences, University Sains
Malaysia, Malaysia for her secretarial assistance.
The authors have no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
American Psychiatric Association. 1994. Diagnostic and Statis-
tical Manual of Mental Disorders. 4th edn. DSM-IV. Washing-
ton, DC: American Psychiatric Press.
Arendt J. 1989. Melatonin: A new probe in psychiatric investiga-
tion? Br J Psychiatry 155:585�590.
Arendt J. 2000. Melatonin, circadian rhythms, and sleep. New
Engl J Med 343:1114�1116.
Arendt J, Skene DJ. 2005. Melatonin as a chronobiotic. Sleep
Med Rev 9:25�39.
Avery DH, Dahl K, Savage MV, Brengelmann GL, Larsen LH,
Kenny MA, Eder DN, Vitiello MV, Prinz PN. 1997. Circadian
temperature and cortisol rhythms during a constant routine are
phase-delayed in hypersomnic winter depression. Biol Psychia-
try 41:1109�1123.
Beck-Friis J, Kjellman BF, Aperia B, Unden F, von Rosen D,
Ljunggren JG, Wetterberg L. 1985a. Serum melatonin in
relation to clinical variables in patients with major depressive
disorder and a hypothesis of a low melatonin syndrome. Acta
Psychiatr Scand 71:319�330.
Beck-Friis J, Ljunggren JG, Thoren M, von Rosen D, Kjellman
BF, Wetterberg L. 1985b. Melatonin, cortisol and ACTH in
patients with major depressive disorder and healthy humans
with special reference to the outcome of the dexamethasone
suppression test. Psychoneuroendocrinology 10:173�186.
Benitez-King G, Rios A, Martinez A, Anton-Tay F. 1996. In vitro
inhibition of Ca2�/calmodulin-dependent kinase II activity by
melatonin. Biochim Biophys Acta 1290:191�196.
Berger M, Vollmann J, Hohagen F, Konig A, Lohner H,
Voderholzer U, Riemann D. 1997. Sleep deprivation combined
with consecutive sleep phase advance as a fast-acting therapy in
depression: an open pilot trial in medicated and unmedicated
patients. Am J Psychiatry 154:870�872.
Melatonin in mood disorders 147

Blask DE, Sauer LA, Dauchy RT. 2002. Melatonin as a
chronobiotic/anticancer agent: cellular, biochemical, and mo-
lecular mechanisms of action and their implications for
circadian-based cancer therapy. Curr Top Med Chem 2:113�132.
Borbely AA. 1982. A two process model of sleep regulation. Hum
Neurobiol 1:195�204.
Bourdet C, Goldenberg F. 1994. Insomnia in anxiety: Sleep EEG
changes. J Psychosom Res 38(Suppl 1):93�104.
Branchey L, Weinberg U, Branchey M, Linkowski P, Mendlewicz
J. 1982. Simultaneous study of 24-hour patterns of melatonin
and cortisol secretion in depressed patients. Neuropsychobiol-
ogy 8:225�232.
Brown R, Kocsis JH, Caroff S, Amsterdam J, Winokur A, Stokes
PE, Frazer A. 1985a. Differences in nocturnal melatonin
secretion between melancholic depressed patients and control
subjects. Am J Psychiatry 142:811�816.
Brown RP, Caroff S, Kocsis JH, Amsterdam J, Winokur A, Stokes
P, Frazer A. 1985b. Nocturnal serum melatonin in major
depressive disorder before and after desmethylimipramine
treatment. Psychopharmacol Bull 21:579�581.
Bunney WE Jr, Murphy DL, Goodwin FK, Borge GF. 1970. The
switch process from depression to mania: Relationship to drugs
which alter brain amines. Lancet 1:1022�1027.
Cardinali DP, Pevet P. 1998. Basic aspects of melatonin action.
Sleep Med Rev 2:175�190.
Cardinali DP, Furio AM, Reyes MP, Brusco LI. 2006. The use of
chronobiotics in the resynchronization of the sleep/wake cycle.
Cancer Causes Control in press.
Carman JS, Post RM, Buswell R, Goodwin FK. 1976. Negative
effects of melatonin on depression. Am J Psychiatry 133:1181�1186.
Chase JE, Gidal BE. 1997. Melatonin: Therapeutic use in sleep
disorders. Ann Pharmacother 31:1218�1226.
Claustrat B, Chazot G, Brun J, Jordan D, Sassolas G. 1984. A
chronobiological study of melatonin and cortisol secretion in
depressed subjects: plasma melatonin, a biochemical marker in
major depression. Biol Psychiatry 19:1215�1228.
Claustrat B, Brun J, Chazot G. 2005. The basic physiology and
pathophysiology of melatonin. Sleep Med Rev 9:11�24.
Coble PA, Kupfer DJ, Shaw DH. 1981. Distribution of REM
latency in depression. Biol Psychiatry 16:453�466.
Crasson M, Kjiri S, Colin A, Kjiri K, L’Hermite-Baleriaux M,
Ansseau M, Legros JJ. 2004. Serum melatonin and urinary 6-
sulfatoxymelatonin in major depression. Psychoneuroendocri-
nology 29:1�12.
Cupp MJ. 1997. Melatonin. Am Fam Physician 56:1421�1425,
1428.
Dahl K, Avery DH, Lewy AJ, Savage MV, Brengelmann GL,
Larsen LH, Vitiello MV, Prinz PN. 1993. Dim light melatonin
onset and circadian temperature during a constant routine in
hypersomnic winter depression. Acta Psychiatr Scand 88:60�66.
Dalton EJ, Rotondi D, Levitan RD, Kennedy SH, Brown GM.
2000. Use of slow-release melatonin in treatment-resistant
depression. J Psychiatry Neurosci 25:48�52.
Dawson D, Armstrong SM. 1996. Chronobiotics*Drugs that
shift rhythms. Pharmacol Ther 69:15�36.
Dolberg OT, Hirschmann S, Grunhaus L. 1998. Melatonin for
the treatment of sleep disturbances in major depressive
disorder. Am J Psychiatry 155:1119�1121.
Doolen S, Krause DN, Dubocovich ML, Duckles SP. 1998.
Melatonin mediates two distinct responses in vascular smooth
muscle. Eur J Pharmacol 345:67�69.
Dubocovich ML, Masana MI, Benloucif S. 1999. Molecular
pharmacology and function of melatonin receptor subtypes.
Adv Exp Med Biol 460:181�190.
Dubocovich ML, Cardinali DP, Delagrange P, Krause DN,
Strosberg D, Sugden D, Yocca FD. 2000. Melatonin receptors.
In: IUPHAR, editor. The IUPHAR compendium of receptor
characterization and classification. 2nd ed. London: IUPHAR
Media. pp 271�277.
Eastman CI, Gallo LC, Lahmeyer HW, Fogg LF. 1993. The
circadian rhythm of temperature during light treatment for
winter depression. Biol Psychiatry 34:210�220.
Epperson CN, Terman M, Terman JS, Hanusa BH, Oren DA,
Peindl KS, Wisner KL. 2004. Randomized clinical trial of
bright light therapy for antepartum depression: Preliminary
findings. J Clin Psychiatry 65:421�425.
Esquifino AI, Pandi-Perumal SR, Cardinali DP. 2004. Circadian
organization of the immune response: A role for melatonin.
Clin Appl Immunol Rev 4:423�433.
Faedda GL, Tondo L, Teicher MH, Baldessarini RJ, Gelbard HA,
Floris GF. 1993. Seasonal mood disorders. Patterns of seasonal
recurrence in mania and depression. Arch Gen Psychiatry
50:17�23.
Ferber R. 1985. Sleep, sleeplessness, and sleep disruptions in
infants and young children. Ann Clin Res 17:227�234.
Forbes D, Morgan DG, Bangma J, Peacock S, Pelletier N,
Adamson J. 2004. Light therapy for managing sleep, behaviour,
and mood disturbances in dementia. Cochrane Database Syst
Rev CD003946.
Frank E, Hlastala S, Ritenour A, Houck P, Tu XM, Monk TH,
Mallinger AG, Kupfer DJ. 1997. Inducing lifestyle regularity in
recovering bipolar disorder patients: Results from the main-
tenance therapies in bipolar disorder protocol. Biol Psychiatry
41:1165�1173.
Globus GG. 1969. A syndrome associated with sleeping late.
Psychosom Med 31:528�535.
Golden RN, Markey SP, Risby ED, Rudorfer MV, Cowdry RW,
Potter WZ. 1988. Antidepressants reduce whole-body norepi-
nephrine turnover while enhancing 6-hydroxymelatonin out-
put. Arch Gen Psychiatry 45:150�154.
Griefahn B, Brode P, Remer T, Blaszkewicz M. 2003. Excretion of
6-hydroxymelatonin sulfate (6-OHMS) in siblings during
childhood and adolescence. Neuroendocrinology 78:241�243.
Guerrero JM, Reiter RJ. 2002. Melatonin-immune system rela-
tionships. Curr Top Med Chem 2:167�179.
Gwirtsman HE, Halaris AE, Wolf AW, DeMet E, Piletz JE, Marler
M. 1989. Apparent phase advance in diurnal MHPG rhythm in
depression. Am J Psychiatry 146:1427�1433.
Hamilton M, Shapiro CM. 1990. Depression. In: Peck DF,
Shapiro CM, editors. Measuring human problems. A practical
guide. Chichester: John Wiley and Sons Ltd. pp 44�72.
Kahn D. 1999. Mood disorders. In: Cutler JG, Marcus ER,
editors. Textbook of psychiatry. Philadelphia, PA: WB Saun-
ders Company. pp 33�63.
Kennedy SH, Kutcher SP, Ralevski E, Brown GM. 1996.
Nocturnal melatonin and 24-hour 6-sulphatoxymelatonin le-
vels in various phases of bipolar affective disorder. Psychiatry
Res 63:219�222.
Kennedy SH, Tighe S, McVey G, Brown GM. 1989. Melatonin
and cortisol ‘switches’ during mania, depression, and euthymia
in a drug-free bipolar patient. J Nerv Ment Dis 177:300�303.
Krahn LE, Gleber E, Rummans TA, Pileggi TS, Lucas DL, Li H.
2000. The effects of electroconvulsive therapy on melatonin. J
ECT 16:391�398.
Kripke DF, Mullaney DJ, Atkinson M, Wolf S. 1978. Circadian
rhythm disorders in manic-depressives. Biol Psychiatry
13:335�351.
Kripke DF, Jean-Louis G, Elliott JA, Klauber MR, Rex KM,
Tuunainen A, Langer RD. 2004. Ethnicity, sleep, mood, and
illumination in postmenopausal women. BMC Psychiatry 4:8.
148 V. Srinivasan et al.

Kunz D. 2004. Chronobiotic protocol and circadian sleep
propensity index: New tools for clinical routine and research
on melatonin and sleep. Pharmacopsychiatry 37:139�146.
Kupfer DJ, Ulrich RF, Coble PA, Jarrett DB, Grochocinski V,
Doman J, Matthews G, Borbely AA. 1984a. Application of
automated REM and slow wave sleep analysis: I. Normal and
depressed subjects. Psychiatry Res 13:325�334.
Kupfer DJ, Ulrich RF, Coble PA, Jarrett DB, Grochocinski V,
Doman J, Matthews G, Borbely AA. 1984b. Application of
automated REM and slow wave sleep analysis: II. Testing the
assumptions of the two-process model of sleep regulation in
normal and depressed subjects. Psychiatry Res 13:335�343.
Leibenluft E, Feldman-Naim S, Turner EH, Wehr TA, Rosenthal
NE. 1997. Effects of exogenous melatonin administration and
withdrawal in five patients with rapid-cycling bipolar disorder. J
Clin Psychiatry 58:383�388.
Lewy AJ, Wehr T, Gold PW, Goodwin FK. 1979. Plasma
melatonin in manic depressive illness. In: Usdin E, Kopin IJ,
Barchas JD, editors. Catecholamines: Basic and clinical fron-
tiers. Vol II. Oxford: Pergamon Press. pp 1173�1175.
Lewy AJ, Wehr TA, Goodwin FK, Newsome DA, Markey SP.
1980. Light suppresses melatonin secretion in humans. Science
210:1267�1269.
Lewy AJ, Wehr TA, Goodwin FK, Newsome DA, Rosenthal NE.
1981. Manic-depressive patients may be supersensitive to light.
Lancet i:383�384.
Lewy AJ, Nurnberger JIJ, Wehr TA, Pack D, Becker LE, Powell
RL, Newsome DA. 1985. Supersensitivity to light: Possible
trait marker for manic-depressive illness. Am J Psychiatry
142:725�727.
Lewy AJ, Sack RL, Singer CM, White DM. 1987. The phase shift
hypothesis for bright light’s therapeutic mechanism of action:
Theoretical considerations and experimental evidence. Psycho-
pharmacol Bull 23:349�353.
Lewy AJ, Sack RL, Singer CM, White DM, Hoban TM. 1988.
Winter depression and the phase-shift hypothesis for bright
light’s therapeutic effects: History, theory, and experimental
evidence. J Biol Rhythms 3:121�134.
Lewy AJ, Bauer VK, Cutler NL, Sack RL, Ahmed S, Thomas
KH, Blood ML, Jackson JM. 1998. Morning vs evening light
treatment of patients with winter depression. Arch Gen
Psychiatry 55:890�896.
Linkowski P, Hubain PP. 1995. Sleep disorders and neuroendo-
crine regulation in depression]. Encephale 21(Spec No 7):25�28.
Loo H, Hale A, D’haenen H. 2002. Determination of the dose of
agomelatine, a melatoninergic agonist and selective 5-HT2C
antagonist, in the treatment of major depressive disorder: A
placebo-controlled dose range study. Int Clin Psychopharmacol
17:239�247.
Macchi MM, Bruce JN. 2004. Human pineal physiology and
functional significance of melatonin. Front Neuroendocrinol
25:177�195.
MacLean AW, Cairns J, Knowles JB. 1983. REM latency and
depression: computer simulations based on the results of phase
delay of sleep in normal subjects. Psychiatry Res 9:69�79.
Miles A, Philbrick DR. 1988. Melatonin and psychiatry. Biol
Psychiatry 23:405�425.
Minors DS, Waterhouse JM, Wirz-Justice A. 1991. A human
phase-response curve to light. Neurosci Lett 133:36�40.
Montgomery P, Dennis J. 2002. Bright light therapy for sleep
problems in adults aged 60�/. Cochrane Database Syst Rev
CD003403.
Monti JM, Alvarino F, Cardinali D, Savio I, Pintos A. 1999.
Polysomnographic study of the effect of melatonin on sleep in
elderly patients with chronic primary insomnia. Arch Gerontol
Geriatr 28:85�98.
Moore RY. 1996. Neural control of the pineal gland. Behav Brain
Res 73:125�130.
Murray G, Allen NB, Trinder J. 2003. Seasonality and circadian
phase delay: Prospective evidence that winter lowering of mood
is associated with a shift towards eveningness. J Affect Disord
76:15�22.
Nagtegaal JE, Laurant MW, Kerkhof GA, Smits MG, van der
Meer YG, Coenen AM. 2000. Effects of melatonin on the
quality of life in patients with delayed sleep phase syndrome. J
Psychosom Res 48:45�50.
Nair NP, Hariharasubramanian N, Pilapil C. 1984. Circadian
rhythm of plasma melatonin in endogenous depression. Prog
Neuropsychopharmacol Biol Psychiatry 8:715�718.
Nosjean O, Ferro M, Coge F, Beauverger P, Henlin JM, Lefoulon
F, Fauchere JL, Delagrange P, Canet E, Boutin JA. 2000.
Identification of the melatonin binding site MT3 as the
quinone reductase 2. J Biol Chem 275:31311�31317.
Nurnberger JI Jr, Adkins S, Lahiri DK, Mayeda A, Hu K, Lewy A,
Miller A, Bowman ES, Miller MJ, Rau L, Smiley C, Davis-
Singh D. 2000. Melatonin suppression by light in euthymic
bipolar and unipolar patients. Arch Gen Psychiatry 57:572�579.
Pacchierotti C, Iapichino S, Bossini L, Pieraccini F, Castrogio-
vanni P. 2001. Melatonin in psychiatric disorders: A review on
the melatonin involvement in psychiatry. Front Neuroendocri-
nol 22:18�32.
Pandi-Perumal SR, Zisapel N, Srinivasan V, Cardinali DP. 2005.
Melatonin and sleep in aging population. Exp Gerontol
40:911�925.
Paparrigopoulos T, Psarros C, Bergiannaki J, Varsou E, Dafni U,
Stefanis C. 2001. Melatonin response to clonidine administra-
tion in depression: indication of presynaptic alpha2-adrenocep-
tor dysfunction. J Affect Disord 65:307�313.
Putilov AA, Russkikh GS, Pandi-Perumal SR. 2005. Melatonin in
winter depression. In: Pandi-Perumal SR, Cardinali DP,
editors. Melatonin: Biological basis of its function in health
and disease. Georgetown, TX: Landes Bioscience. pp 257�267.
Radloff LS, Rae DS. 1979. Susceptibility and precipitating factors
in depression: sex differences and similarities. J Abnorm
Psychol 88:174�181.
Regestein QR, Monk TH. 1995. Delayed sleep phase syndrome:
A review of its clinical aspects. Am J Psychiatry 152:602�608.
Regestein QR, Pavlova M. 1995. Treatment of delayed sleep
phase syndrome. Gen Hosp Psychiatry 17:335�345.
Reiter RJ. 1980. The pineal and its hormones in the control of
reproduction in mammals. Endocr Rev 1:109�131.
Reiter RJ, Tan DX. 2002. Role of CSF in the transport of
melatonin. J Pineal Res 32:279.
Reiter RJ, Tan DX, Leon J, Kilic U, Kilic E. 2005. When
melatonin gets on your nerves: Its beneficial actions in
experimental models of stroke. Exp Biol Med (Maywood)
230:104�117.
Reppert SM, Weaver DR, Ebisawa T. 1994. Cloning and
characterization of a mammalian melatonin receptor that
mediates reproductive and circadian responses. Neuron
13:1177�1185.
Reppert SM, Godson C, Mahle CD, Weaver DR, Slaugenhaupt
SA, Gusella JF. 1995. Molecular characterization of a second
melatonin receptor expressed in human retina and brain: The
Mel1b melatonin receptor. Proc Natl Acad Sci USA 92:8734�8738.
Reynolds CF, Kupler D. 1988. Sleep in depression. In: Williams
RZ, Karacan I, Moore CA, editors. Sleep disorders, diagnosis
and treatment. New York: John Wiley. pp 147�164.
Riemann D, Hohagen F, Konig A, Schwarz B, Gomille J,
Voderholzer U, Berger M. 1996. Advanced vs. normal sleep
Melatonin in mood disorders 149

timing: effects on depressed mood after response to sleep
deprivation in patients with a major depressive disorder. J
Affect Disord 37:121�128.
Riemann D, Berger M, Voderholzer U. 2001. Sleep and
depression�Results from psychobiological studies: An over-
view. Biol Psychol 57:67�103.
Rosenthal NE, Sack DA, Gillin JC, Lewy AJ, Goodwin FK,
Davenport Y, Mueller PS, Newsome DA, Wehr TA. 1984.
Seasonal affective disorder. A description of the syndrome and
preliminary findings with light therapy. Arch Gen Psychiatry
41:72�80.
Rubin RT, Heist EK, McGeoy SS, Hanada K, Lesser IM. 1992.
Neuroendocrine aspects of primary endogenous depression.
XI. Serum melatonin measures in patients and matched control
subjects. Arch Gen Psychiatry 49:558�567.
Sack RL, Lewy AJ. 1986. Desmethylimipramine treatment
increases melatonin production in humans. Biol Psychiatry
21:406�410.
Sack RL, Lewy AJ. 1988. Melatonin and major affective disorders.
In: Miles A, Philbrich D, Thompson C, editors. Melatonin:
Clinical perspectives. New York: Oxford Medical Publications.
pp 205�227.
Sack RL, Lewy AJ, White DM, Singer CM, Fireman MJ,
Vandiver R. 1990. Morning vs evening light treatment for
winter depression. Evidence that the therapeutic effects of light
are mediated by circadian phase shifts. Arch Gen Psychiatry
47:343�351.
Scheer FA, Van Montfrans GA, Van Someren EJ, Mairuhu G,
Buijs RM. 2004. Daily nighttime melatonin reduces blood
pressure in male patients with essential hypertension. Hyper-
tension 43:192�197.
Schildkraut JJ. 1965. The catecholamine hypothesis of affective
disorders: A review of supporting evidence. Am J Psychiatry
122:509�522.
Schilgen B, Tolle R. 1980. Partial sleep deprivation as therapy for
depression. Arch Gen Psychiatry 37:267�271.
Sekula LK, Lucke JF, Heist EK, Czambel RK, Rubin RT. 1997.
Neuroendocrine aspects of primary endogenous depression.
XV: Mathematical modeling of nocturnal melatonin secretion
in major depressives and normal controls. Psychiatry Res
69:143�153.
Shafii M, MacMillan DR, Key MP, Kaufman N, Nahinsky ID.
1997. Case study: Melatonin in severe obesity. J Am Acad
Child Adolesc Psychiatry 36:412�416.
Shibui K, Uchiyama M, Okawa M. 1999. Melatonin rhythms in
delayed sleep phase syndrome. J Biol Rhythms 14:72�76.
Shiromani PJ, Klemfuss H, Lucero S, Overstreet DH. 1991.
Diurnal rhythm of core body temperature is phase advanced in
a rodent model of depression. Biol Psychiatry 29:923�930.
Sitaram N, Gillin JC, Bunney WE Jr.. 1978. The switch process in
manic-depressive illness. Circadian variation in time of switch
and sleep and manic ratings before and after switch. Acta
Psychiatr Scand 58:267�278.
Souetre E, Salvati E, Belugou JL, Pringuey D, Candito M, Krebs
B, Ardisson JL, Darcourt G. 1989. Circadian rhythms in
depression and recovery: Evidence for blunted amplitude as
the main chronobiological abnormality. Psychiatry Res
28:263�278.
Srinivasan V. 1989. Psychoactive drugs, pineal gland and affective
disorders. Prog Neuropsychopharmacol Biol Psychiatry
13:653�664.
Srinivasan V. 1997. Melatonin, biological rhythm disorders and
phototherapy. Indian J Physiol Pharmacol 41:309�328.
Srinivasan V, Smits G, Kayumov L, Pandi-Perumal SR, Cardinali
DP, Thorpy MJ. 2006. Melatonin in circadian rhythm sleep
disorders. In: Cardinali DP, Pandi-Perumal SR, editors.
Neuroendocrine correlates of sleep-wakefulness. New York:
Springer. pp 269�294.
Stewart JW, Halbreich U. 1989. Plasma melatonin levels in
depressed patients before and after treatment with antidepres-
sant medication. Biol Psychiatry 25:33�38.
Surridge-David M, MacLean A, Coulter ME, Knowles JB. 1987.
Mood change following an acute delay of sleep. Psychiatry Res
22:149�158.
Szymanska A, Rabe-Jablonska J, Karasek M. 2001. Diurnal
profile of melatonin concentrations in patients with major
depression: Relationship to the clinical manifestation and
antidepressant treatment. Neuroendocrinol Lett 22:192�198.
Terman JS, Terman M, Lo ES, Cooper TB. 2001. Circadian time
of morning light administration and therapeutic response in
winter depression. Arch Gen Psychiatry 58:69�75.
Terman M, Quitkin FM, Terman JS, Stewart JW, McGrath PJ.
1987. The timing of phototherapy: effects on clinical response
and the melatonin cycle. Psychopharmacol Bull 23:354�357.
Thompson C, Childs PA, Martin NJ, Rodin I, Smythe PJ. 1997.
Effects of morning phototherapy on circadian markers in
seasonal affective disorder. Br J Psychiatry 170:431�435.
Thompson C, Franey C, Arendt J, Checkley SA. 1988. A
comparison of melatonin secretion in depressed patients and
normal subjects. Br J Psychiatry 152:260�265.
Thompson C, Mezey G, Corn T, Franey C, English J, Arendt J,
Checkley SA. 1985. The effect of desipramine upon melatonin
and cortisol secretion in depressed and normal subjects. Br J
Psychiatry 147:389�393.
Thorpy MJ, Korman E, Spielman AJ, Glovinsky PB. 1988.
Delayed sleep phase syndrome in adolescents. J Adolesc Health
Care 9:22�27.
Tricoire H, Locatelli A, Chemineau P, Malpaux B. 2002.
Melatonin enters the cerebrospinal fluid through the pineal
recess. Endocrinology 143:84�90.
Tuunainen A, Kripke DF, Elliott JA, Assmus JD, Rex KM,
Klauber MR, Langer RD. 2002. Depression and endogenous
melatonin in postmenopausal women. J Affect Disord 69:149�158.
Tuunainen A, Kripke DF, Endo T. 2004. Light therapy for non-
seasonal depression. Cochrane Database Syst Rev CD004050.
Varma A, Kaul RK, Varma P, Kalra V, Malhotra V. 2002. The
effect of antidepressants on serum melatonin levels in endo-
genous depression. J Assoc Phys India 50:1262�1265.
Venkoba Rao A, Parvathi Devi S, Srinivasan V. 1983. Urinary
melatonin in depression. Indian J Psychiatry 25:167�172.
Wehr TA. 1991. Sleep-loss as a possible mediator of diverse causes
of mania. Br J Psychiatry 159:576�578.
Wehr T, Goodwin FK. 1979. Tricyclics modulate frequency of
mood cycles. Chronobiologia 6:377�385.
Wehr TA, Wirz-Justice A. 1982. Circadian rhythm mechanisms in
affective illness and in antidepressant drug action. Pharmacop-
sychiatria 15:31�39.
Wehr TA, Wirz-Justice A, Goodwin FK, Duncan W, Gillin JC.
1979. Phase advance of the circadian sleep-wake cycle as an
antidepressant. Science 206:710�713.
Wehr TA, Goodwin FK, Wirz-Justice A, Breitmaier J, Craig C.
1982. 48-hour sleep-wake cycles in manic-depressive illness:
Naturalistic observations and sleep deprivation experiments.
Arch Gen Psychiatry 39:559�565.
Wehr TA, Sack DA, Duncan WC, Mendelson WB, Rosenthal NE,
Gillin JC, Goodwin FK. 1985. Sleep and circadian rhythms in
affective patients isolated from external time cues. Psychiatry
Res 15:327�339.
Wehr TA, Sack DA, Rosenthal NE. 1987. Seasonal affective
disorder with summer depression and winter hypomania. Am J
Psychiatry 144:1602�1603.
150 V. Srinivasan et al.

Wehr TA, Duncan WC Jr, Sher L, Aeschbach D, Schwartz PJ,
Turner EH, Postolache TT, Rosenthal NE. 2001. A circadian
signal of change of season in patients with seasonal affective
disorder. Arch Gen Psychiatry 58:1108�1114.
Weitzman ED, Czeisler CA, Coleman RM, Spielman AJ, Zimmer-
man JC, Dement W, Richardson G, Pollak CP. 1981. Delayed
sleep phase syndrome. A chronobiological disorder with sleep-
onset insomnia. Arch Gen Psychiatry 38:737�746.
Wetterberg L. 1979. Clinical importance of melatonin. Prog Brain
Res 52:539�547.
Wetterberg L. 1999. Melatonin and clinical application. Repro-
duction Nutrition Development 39:367�382.
Wever RA. 1986. Characteristics of circadian rhythms in human
functions. J Neural Transm Suppl 21:323�373.
Wiesenberg I, Missbach M, Kahlen JP, Schrader M, Carlberg C.
1995. Transcriptional activation of the nuclear receptor RZR
alpha by the pineal gland hormone melatonin and identification
of CGP 52608 as a synthetic ligand. Nucleic Acids Res
23:327�333.
Wirz-Justice A, Graw P, Krauchi K, Gisin B, Arendt J, Aldhous
M, Poldinger W. 1990. Morning or night-time melatonin is
ineffective in seasonal affective disorder. J Psychiatr Res
24:129�137.
Wirz-Justice A, Graw P, Krauchi K, Sarrafzadeh A, English J,
Arendt J, Sand L. 1996. Natural’ light treatment of seasonal
affective disorder. J Affect Disord 37:109�120.
Witt-Enderby PA, Bennett J, Jarzynka MJ, Firestine S, Melan
MA. 2003. Melatonin receptors and their regulation: Biochem-
ical and structural mechanisms. Life Sci 72:2183�2198.
Wurtman RJ, Zhdanova I. 1995. Improvement of sleep quality by
melatonin. Lancet 346:1491.
Zeiten M, Potkin S, Urbanchek M. 1987. Melatonin in depres-
sion. Psychiatr Ann 17:676�681.
Zeitzer JM, Daniels JE, Duffy JF, Klerman EB, Shanahan TL,
Dijk DJ, Czeisler CA. 1999. Do plasma melatonin concentra-
tions decline with age? Am J Med 107:432�436.
Melatonin in mood disorders 151

ORIGINAL INVESTIGATION
Striatal dopamine transporter availability and DAT-1 gene in adultswith ADHD: No higher DAT availability in patients with homozygosityfor the 10-repeat allele
JOHANNA KRAUSE1, STEFAN H. DRESEL2, KLAUS-HENNING KRAUSE3,
CHRISTIAN LA FOUGERE3, PETER ZILL4 & MANFRED ACKENHEIL4
1Outpatient Clinic for Psychiatry and Psychotherapy, Ottobrunn, Germany 2Department of Nuclear Medicine,3Friedrich-Baur-Institute, and 4Department of Neurochemistry, Ludwig-Maximilians-University, Munich, Germany
AbstractIn 29 adults with attention deficit hyperactivity disorder (ADHD) striatal dopamine transporter (DAT) availability wasassessed by [99mTc]TRODAT-1 SPECT and correlated with 3? VNTR polymorphism of the DAT gene on chromosome5p15.3. Seventeen patients showed homozygosity for the 10-repeat allele, two homozygosity of the 9 allele and 10 wereheterozygous (9�10). No statistically significant difference in DAT availability was found between patients with 10�10carriers (DAT 1.289/ 0.34) and with at least one 9 allele (DAT 1.319/0.27); when smokers were excluded, DAT availabilitywas 1.389/0.28 in the 10�10 carriers (n�/12) and 1.429/0.19 in the 9�10 and 9�9 carriers (n�/7). In conclusion, nohigher striatal DAT was found in patients with homozygosity of the 10 allele of the DAT gene in this study. These resultsdiffer from a study in 11 Korean children with ADHD, in which 10�10 carriers showed higher DATavailability in [123I]IPTSPECT. Discrepancies may be explained by differences in patient’s age, ethnical differences, different imaging techniques orthe limited number of patients included in both studies.
Key words: Attention deficit hyperactivity disorder (ADHD), brain imaging techniques, dopamine transporter (DAT),
TRODAT-1 SPECT, DAT gene
Introduction
Neuroimaging studies with MRI, PET and SPECT
suggest involvement of striatal structures in ADHD
(Bush et al. 2005; Castellanos and Tannock 2002;
Krause et al. 2003; Spencer et al. 2005). Most of the
DAT-SPECT imaging studies showed a higher
availability of DAT binding sites in adult patients
with ADHD (Cheon et al. 2003; Dougherty et al.
1999; Dresel et al. 2000, Krause et al. 2000, Madras
et al. 2005). From molecular genetic studies an
involvement of a polymorphism of the DAT1 gene in
ADHD was described (e.g., a higher rate of homo-
zygosity of the 10-repeat allele in the 3? untranslated
region of exon 15 of the DAT gene on chromosome
5p15.3) (Chen et al. 2003; Cook et al. 1995;
Cornish et al. 2005; Faraone et al. 2005; Gill et al.
1997; Waldman et al. 1998). It has been postulated,
that individuals with the 10 repeat allele may have
increased DAT availability (Kirley et al. 2002). In
accordance with this assumption, Cheon et al.
(2005) found higher DAT availability in the seven
of 11 ADHD children with 10�10 genotype. To our
best knowledge this is the first study, which investi-
gated the influence of 3? VNTR polymorphism on
striatal DAT availability in adult patients with
ADHD.
Patients and methods
Striatal DAT binding was measured in 29 adult
patients (19 males, 10 females, age 19�54 years,
mean9/SD 37.69/10.0) with ADHD, using
[99mTc]TRODAT-1 SPECT. The patients were
diagnosed by a board-certified psychiatrist (J.K.).
Diagnosis of ADHD was made according to DSM-
IV criteria (American Psychiatric Association 1994)
by structured interviews. No patient has ever been
treated with stimulants. Exclusion criteria were a
known history of alcohol or drug abuse or past or
Correspondence: Johanna Krause, MD, Schillerstr. 11a, D-85521 Ottobrunn, Germany. Tel: �/49 89 6012471. Fax: �/49 89 60019387.
E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(3): 152�157
(Received 1 September 2005; accepted 8 December 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500518444
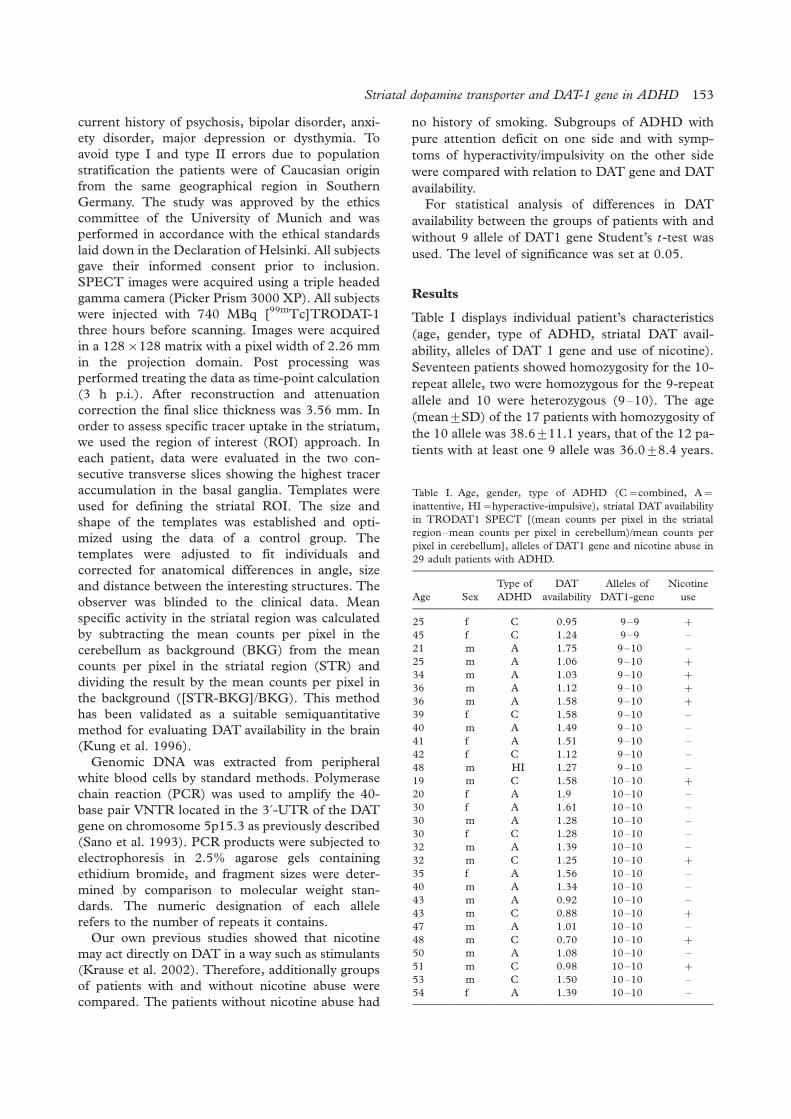
current history of psychosis, bipolar disorder, anxi-
ety disorder, major depression or dysthymia. To
avoid type I and type II errors due to population
stratification the patients were of Caucasian origin
from the same geographical region in Southern
Germany. The study was approved by the ethics
committee of the University of Munich and was
performed in accordance with the ethical standards
laid down in the Declaration of Helsinki. All subjects
gave their informed consent prior to inclusion.
SPECT images were acquired using a triple headed
gamma camera (Picker Prism 3000 XP). All subjects
were injected with 740 MBq [99mTc]TRODAT-1
three hours before scanning. Images were acquired
in a 128�/128 matrix with a pixel width of 2.26 mm
in the projection domain. Post processing was
performed treating the data as time-point calculation
(3 h p.i.). After reconstruction and attenuation
correction the final slice thickness was 3.56 mm. In
order to assess specific tracer uptake in the striatum,
we used the region of interest (ROI) approach. In
each patient, data were evaluated in the two con-
secutive transverse slices showing the highest tracer
accumulation in the basal ganglia. Templates were
used for defining the striatal ROI. The size and
shape of the templates was established and opti-
mized using the data of a control group. The
templates were adjusted to fit individuals and
corrected for anatomical differences in angle, size
and distance between the interesting structures. The
observer was blinded to the clinical data. Mean
specific activity in the striatal region was calculated
by subtracting the mean counts per pixel in the
cerebellum as background (BKG) from the mean
counts per pixel in the striatal region (STR) and
dividing the result by the mean counts per pixel in
the background ([STR-BKG]/BKG). This method
has been validated as a suitable semiquantitative
method for evaluating DAT availability in the brain
(Kung et al. 1996).
Genomic DNA was extracted from peripheral
white blood cells by standard methods. Polymerase
chain reaction (PCR) was used to amplify the 40-
base pair VNTR located in the 3?-UTR of the DAT
gene on chromosome 5p15.3 as previously described
(Sano et al. 1993). PCR products were subjected to
electrophoresis in 2.5% agarose gels containing
ethidium bromide, and fragment sizes were deter-
mined by comparison to molecular weight stan-
dards. The numeric designation of each allele
refers to the number of repeats it contains.
Our own previous studies showed that nicotine
may act directly on DAT in a way such as stimulants
(Krause et al. 2002). Therefore, additionally groups
of patients with and without nicotine abuse were
compared. The patients without nicotine abuse had
no history of smoking. Subgroups of ADHD with
pure attention deficit on one side and with symp-
toms of hyperactivity/impulsivity on the other side
were compared with relation to DAT gene and DAT
availability.
For statistical analysis of differences in DAT
availability between the groups of patients with and
without 9 allele of DAT1 gene Student’s t-test was
used. The level of significance was set at 0.05.
Results
Table I displays individual patient’s characteristics
(age, gender, type of ADHD, striatal DAT avail-
ability, alleles of DAT 1 gene and use of nicotine).
Seventeen patients showed homozygosity for the 10-
repeat allele, two were homozygous for the 9-repeat
allele and 10 were heterozygous (9�10). The age
(mean9/SD) of the 17 patients with homozygosity of
the 10 allele was 38.69/11.1 years, that of the 12 pa-
tients with at least one 9 allele was 36.09/8.4 years.
Table I. Age, gender, type of ADHD (C�/combined, A�/
inattentive, HI�/hyperactive-impulsive), striatal DAT availability
in TRODAT1 SPECT [(mean counts per pixel in the striatal
region�mean counts per pixel in cerebellum)/mean counts per
pixel in cerebellum], alleles of DAT1 gene and nicotine abuse in
29 adult patients with ADHD.
Age Sex
Type of
ADHD
DAT
availability
Alleles of
DAT1-gene
Nicotine
use
25 f C 0.95 9�9 �/
45 f C 1.24 9�9 �21 m A 1.75 9�10 �25 m A 1.06 9�10 �/
34 m A 1.03 9�10 �/
36 m A 1.12 9�10 �/
36 m A 1.58 9�10 �/
39 f C 1.58 9�10 �40 m A 1.49 9�10 �41 f A 1.51 9�10 �42 f C 1.12 9�10 �48 m HI 1.27 9�10 �19 m C 1.58 10�10 �/
20 f A 1.9 10�10 �30 f A 1.61 10�10 �30 m A 1.28 10�10 �30 f C 1.28 10�10 �32 m A 1.39 10�10 �32 m C 1.25 10�10 �/
35 f A 1.56 10�10 �40 m A 1.34 10�10 �43 m A 0.92 10�10 �43 m C 0.88 10�10 �/
47 m A 1.01 10�10 �48 m C 0.70 10�10 �/
50 m A 1.08 10�10 �51 m C 0.98 10�10 �/
53 m C 1.50 10�10 �54 f A 1.39 10�10 �
Striatal dopamine transporter and DAT-1 gene in ADHD 153
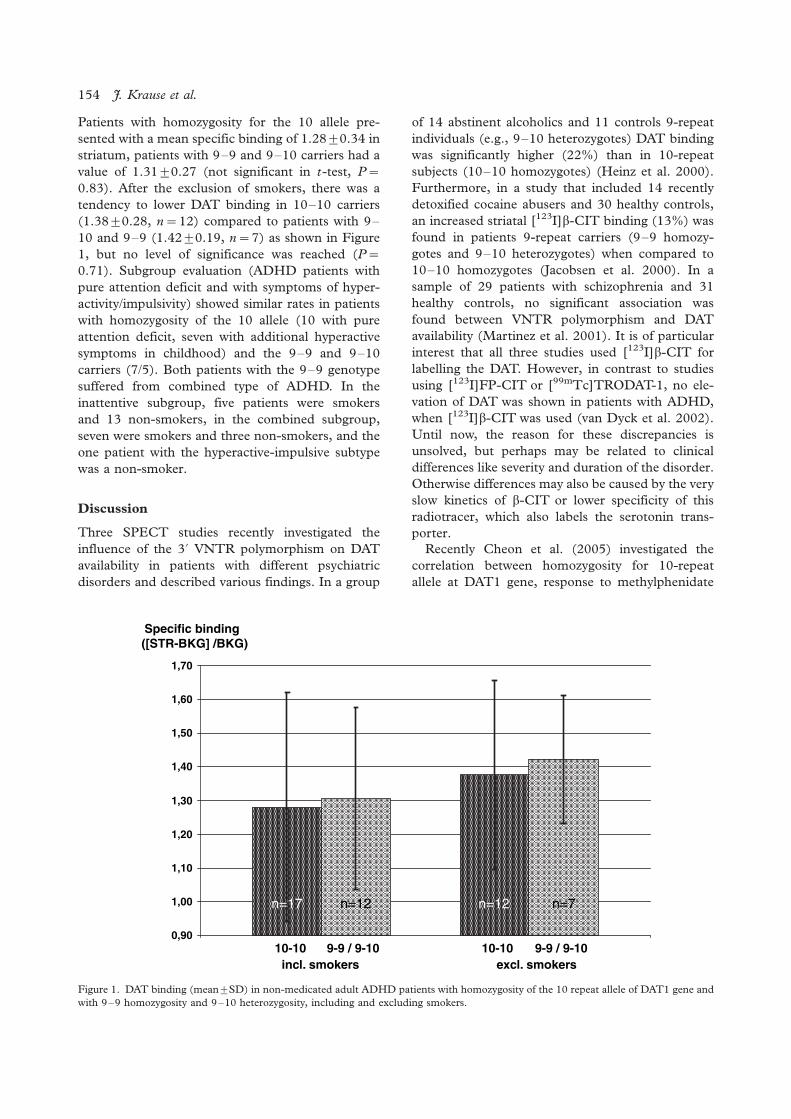
Patients with homozygosity for the 10 allele pre-
sented with a mean specific binding of 1.289/0.34 in
striatum, patients with 9�9 and 9�10 carriers had a
value of 1.319/0.27 (not significant in t-test, P�/
0.83). After the exclusion of smokers, there was a
tendency to lower DAT binding in 10�10 carriers
(1.389/0.28, n�/ 12) compared to patients with 9�10 and 9�9 (1.429/0.19, n�/ 7) as shown in Figure
1, but no level of significance was reached (P�/
0.71). Subgroup evaluation (ADHD patients with
pure attention deficit and with symptoms of hyper-
activity/impulsivity) showed similar rates in patients
with homozygosity of the 10 allele (10 with pure
attention deficit, seven with additional hyperactive
symptoms in childhood) and the 9�9 and 9�10
carriers (7/5). Both patients with the 9�9 genotype
suffered from combined type of ADHD. In the
inattentive subgroup, five patients were smokers
and 13 non-smokers, in the combined subgroup,
seven were smokers and three non-smokers, and the
one patient with the hyperactive-impulsive subtype
was a non-smoker.
Discussion
Three SPECT studies recently investigated the
influence of the 3? VNTR polymorphism on DAT
availability in patients with different psychiatric
disorders and described various findings. In a group
of 14 abstinent alcoholics and 11 controls 9-repeat
individuals (e.g., 9�10 heterozygotes) DAT binding
was significantly higher (22%) than in 10-repeat
subjects (10�10 homozygotes) (Heinz et al. 2000).
Furthermore, in a study that included 14 recently
detoxified cocaine abusers and 30 healthy controls,
an increased striatal [123I]b-CIT binding (13%) was
found in patients 9-repeat carriers (9�9 homozy-
gotes and 9�10 heterozygotes) when compared to
10�10 homozygotes (Jacobsen et al. 2000). In a
sample of 29 patients with schizophrenia and 31
healthy controls, no significant association was
found between VNTR polymorphism and DAT
availability (Martinez et al. 2001). It is of particular
interest that all three studies used [123I]b-CIT for
labelling the DAT. However, in contrast to studies
using [123I]FP-CIT or [99mTc]TRODAT-1, no ele-
vation of DAT was shown in patients with ADHD,
when [123I]b-CIT was used (van Dyck et al. 2002).
Until now, the reason for these discrepancies is
unsolved, but perhaps may be related to clinical
differences like severity and duration of the disorder.
Otherwise differences may also be caused by the very
slow kinetics of b-CIT or lower specificity of this
radiotracer, which also labels the serotonin trans-
porter.
Recently Cheon et al. (2005) investigated the
correlation between homozygosity for 10-repeat
allele at DAT1 gene, response to methylphenidate
0,90
1,00
1,10
1,20
1,30
1,40
1,50
1,60
1,70
Specific binding([STR-BKG] /BKG)
incl. smokers excl. smokers
n=17 n=12 n=12 n=7
10-10 9-9 / 9-10 10-10 9-9 / 9-10
Figure 1. DAT binding (mean9/SD) in non-medicated adult ADHD patients with homozygosity of the 10 repeat allele of DAT1 gene and
with 9�9 homozygosity and 9�10 heterozygosity, including and excluding smokers.
154 J. Krause et al.

and DAT availability measured with [123I]IPT
SPECT in 11 korean children with ADHD. They
found that the seven children with 10�10 homo-
zygosity had higher DAT binding ratio than the
others. With regard to treatment with methylphe-
nidate, the response was better in the four children
without homozygosity, who all responded well to
methylphenidate, whereas only two of the seven
children with homozygosity were responders. The
finding of better response to methylphenidate in
patients without homozygosity is in accordance with
the results of Winsberg and Comings (1999), who
found in their sample of 30 African�American
children a generally very low response rate of
53%, as well as of Roman et al. (2002), who
investigated 50 Brazilian youths with ADHD. In
accordance we identified three 10�10 carriers with
reduced DAT availability, who all were non-respon-
ders to methylphenidate (unpublished results). In a
sample of 18 non-smoking adults with ADHD we
found that response to methylphenidate generally
seems to be depending on DAT availability: patients
with low DAT availability showed no response
(Krause et al. 2005). In contrast to the finding of
Winsberg and Comings (1999) and Roman et al.
(2002) a study, which investigated Irish children
with ADHD, reported a significant better response
to methylphenidate in the 53 children with 10�10
homozygosity as compared to 42 children with at
least one 9-repeat DAT1 allele (Kirley et al. 2003).
An interesting result concerning the effect of
another stimulant, d-amphetamine, on self-report
measures (euphoria, anxiety, feel drug) found Lott
et al. (2005) in 96 healthy volunteers with a double-
blind crossover design: in this study the effects of
amphetamine were indistinguishable from placebo
in all eight subjects with 9�9 genotype, whereas the
9�10 and 10�10 subjects showed the expected
effects of amphetamine. Accordingly, in a double-
blind, placebo-controlled, crossover study with
forced weekly titrations of three dose conditions of
methylphenidate in 47 children with ADHD Stein
et al. (2005) documented, that the six children with
9�9 homozygosity responded poorly to methylphe-
nidate in comparison to the children with one or
two copies of the 10-repeat allele. Additional
studies with higher number of patients are war-
ranted to confirm these preliminary results, show-
ing an atypical stimulant response in people with
9�9 genotype. In our study only two patients
showed a 9�9 genotype; one had a low DAT
availability, but was a heavy smoker, so that the
low DAT could be caused by nicotine abuse
(Krause et al. 2003), the other presented with
slightly elevated DAT availability. It is of further
interest, that Ujike et al. (2003) found, that 9
repeat allele of the DAT gene seems to be a strong
risk factor for prolonged metamphetamine psy-
choses, confirming earlier results of Gelernter
et al. (1994), who saw links between the 9 allele
and cocaine-induced paranoia.
Concerning the subgroups of ADHD and their
relation to DAT1 gene we could not confirm the
results of Waldman et al. (1998), who found
differences between the purely inattentive subtype
and the hyperactive-impulsive or combined sub-
types, the former showing lower frequency of the
10 allele of the DAT 1 gene than the latter. However,
the number of patients in this study is too small to
allow a final conclusion. Our results are in accor-
dance with the study of Stein et al. (2005), who
found no significant differences between patients
with 9�10 alleles (68% with combined type, 32%
with inattentive type) and 10�10 alleles (63% with
combined type, 37% with inattentive type); but from
six patients with 9�9 genotype 5 (83%) had the
combined and only 1 the inattentive type. In our
study both patients with the 9�9 genotype showed
the combined type.
In conclusion, the impact of the abnormalities of
VNTR polymorphism of DAT1 gene seen in
ADHD on the striatal DAT is far from clear. Our
first results definitely show no higher striatal DAT
availability in the ADHD patients with homozygos-
ity of the 10 allele compared with those having the 9
allele. It should be mentioned that the heteroge-
neous results of our study compared with some
published reports might be due to ethnical differ-
ences in our patient sample or to the limited sample
size. Furthermore, differences possibly could result
from the use of different imaging techniques; our
study was the only one, in which [99mTc]TRODAT-
1 was used as radiopharmaceutical, whereas in the
other studies [123I]IPT or [123I]b-CIT were used for
labelling the DAT. In the three studies with [123I]b-
CIT (Heinz et al. 2000; Jacobsen et al. 2000;
Martinez et al. 2001) no patients with ADHD
were investigated, and the only other study, which
dealed with ADHD patients, using [123I]IPT
(Cheon et al. 2005), included even much fewer
patients than our study. Although our patients were
Caucasians from the same geographical region of
Southern Germany we cannot fully exclude popula-
tion stratification effects. The results of our study
are preliminary and it appears to be important to
investigate relations between the different subtypes
of ADHD, DAT availability and genetics in higher
number of patients. In this context sequence analy-
sis of the tandem repeat region in the 3?-UTR of the
DAT gene in patients with ADHD would be of
interest.
Striatal dopamine transporter and DAT-1 gene in ADHD 155

Statement of interest
The authors have no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
American Psychiatric Association. 1994. Diagnostic and Statis-
tical Manual of Mental Disorders. 4th ed. Washington, DC:
American Psychiatric Association.
Bush G, Valera EM, Seidman LJ. 2005. Functional neuroimaging
of attention-deficit/hyperactivity disorder: A review and sug-
gested future directions. Biol Psychiatry 57:1273�1284.
Castellanos FX, Tannock R. 2002. Neuroscience of attention-
deficit/hyperactivity disorder: the search for endophenotypes.
Nat Rev Neurosci 3:617�628.
Chen CK, Chen SL, Mill J, Huang YS, Lin SK, Curran S, Purcell
S, Sham P, Asherson P. 2003. The dopamine transporter gene
is associated with attention deficit hyperactivity disorder in a
Taiwanese sample. Mol Psychiatry 8:393�396.
Cheon KA, Ryu YH, Kim JW, Cho DY. 2005. The homozygosity
for 10-repeat allele at dopamine transporter gene and dopa-
mine transporter density in Korean children with attention
deficit hyperactivity disorder: Relating to treatment response to
methylphenidate. Eur Neuropsychopharmacol 15:95�101.
Cheon K-A, Ryu YH, Kim Y-K, Namkoong K, Kim C-H, Lee JD.
2003. Dopamine transporter availability in the basal ganglia
assessed with [123I]IPT SPET in children with attention deficit
hyperactivity disorder. Eur J Nucl Med 30:306�311.
Cook EH Jr, Stein MA, Krasowski MD, Cox NJ, Olkon DM,
Kieffer JE, Leventhal BL. 1995. Association of attention-deficit
disorder and the dopamine transporter gene. Am J Hum Genet
56:993�998.
Cornish KM, Manly T, Savage R, Swanson J, Morisano D, Butler
N, Grant C, Cross G, Bentley L, Hollis CP. 2005. Association
of the dopamine transporter (DAT1) 10/10-repeat genotype
with ADHD symptoms and response inhibition in a general
population sample. Mol Psychiatry 10:686�698.
Dougherty DD, Bonab AA, Spencer TJ, Rauch SL, Madras BK,
Fischman AJ. 1999. Dopamine transporter availability in
patients with attention deficit hyperactivity disorder. Lancet
354:2132�2133.
Dresel S, Krause J, Krause KH, la Fougere C, Brinkbaumer K,
Kung HF, Hahn K, Tatsch K. 2000. Attention deficit
hyperactivity disorder: binding of [99mTc]TRODAT-1 to the
dopamine transporter before and after methylphenidate treat-
ment. Eur J Nucl Med 27:1518�1524.
Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ,
Holmgren MA, Sklar P. 2005. Molecular genetics of attention-
deficit/hyperactivity disorder. Biol Psychiatry 57:1313�1323.
Gelernter J, Kranzler HR, Satel SL, Rao PA. 1994. Genetic
association between dopamine transporter protein alleles
and cocaine-induced paranoia. Neuropsychopharmacology 11:
195�200.
Gill M, Daly G, Heron S, Hawi Z, Fitzgerald M. 1997.
Confirmation of association between attention deficit hyper-
activity disorder and a dopamine transporter polymorphism.
Mol Psychiatry 2:311�313.
Heinz A, Goldman D, Jones DW, Palmour R, Hommer D, Gorey
JG, Lee KS, Linnoila M, Weinberger DR. 2000. Genotype
influences in vivo dopamine transporter availability in human
striatum. Neuropsychopharmacology 22:133�139.
Jacobsen LK, Staley JK, Zoghbi SS, Seibyl JP, Kosten TR, Innis
RB, Gelernter J. 2000. Prediction of dopamine transporter
binding availability by genotype: A preliminary report. Am J
Psychiatry 157:1700�1703.
Kirley A, Hawi Z, Daly G, McCarron M, Mullins C, Millar N,
Waldman I, Fitzgerald M, Gill M. 2002. Dopaminergic system
genes in ADHD: Toward a biological hypothesis. Neuropsy-
chopharmacology 27:607�619.
Kirley A, Lowe N, Hawi Z, Mullins C, Daly G, Waldman I,
McCarron M, O’Donnell D, Fitzgerald M, Gill M. 2003.
Association of the 480 bp DAT1 allele with methylphenidate
response in a sample of Irish children with ADHD. Am J Med
Genet B Neuropsychiatr Genet 121:50�54.
Krause KH, Dresel SH, Krause J, Kung HF, Tatsch K. 2000.
Increased striatal dopamine transporter in adult patients with
attention deficit hyperactivity disorder: Effects of methylphe-
nidate as measured by single photon emission computed
tomography. Neurosci Lett 285:107�110.
Krause KH, Dresel SH, Krause J, Kung HF, Tatsch K, Ackenheil
M. 2002. Stimulant-like action of nicotine on striatal dopamine
transporter in the brain of adults with attention deficit
hyperactivity disorder. Int J Neuropsychopharmacol 5:111�113.
Krause KH, Dresel SH, Krause J, la Fougere C, Ackenheil M.
2003. The dopamine transporter and neuroimaging in atten-
tion deficit hyperactivity disorder. Neurosci Biobehav Rev
27:605�613.
Krause J, la Fougere C, Krause KH, Ackenheil M, Dresel SH.
2005. Influence of striatal dopamine transporter availability on
the response to methylphenidate in adult patients with ADHD.
Eur Arch Psychiatry Clin Neurosci 255: 428�431.
Kung HF, Kim HJ, Kung MP, Meegalla SK, Plossl K, Lee HK.
1996. Imaging of dopamine transporters in humans with
technetium-99m TRODAT-1. Eur J Nucl Med 23:1527�1530.
Lott DC, Kim SJ, Cook EH Jr, de Wit H. 2005. Dopamine
transporter gene associated with diminished subjective re-
sponse to amphetamine. Neuropsychopharmacology 30:602�609.
Madras BK, Miller GM, Fischman AJ. 2005. The dopamine
transporter and attention-deficit/hyperactivity disorder. Biol
Psychiatry 57:1397�1409.
Martinez D, Gelernter J, Abi-Dargham A, van Dyck CH, Kegeles
L, Innis RB, Laruelle M. 2001. The variable number of tandem
repeats polymorphism of the dopamine transporter gene is not
associated with significant change in dopamine transporter
phenotype in humans. Neuropsychopharmacology 24:553�560.
Roman T, Szobot C, Martins S, Biederman J, Rohde LA, Hutz
MH. 2002. Dopamine transporter gene and response to
methylphenidate in attention-deficit/hyperactivity disorder.
Pharmacogenetics 12:497�499.
Sano A, Kondoh K, Hakimoto Y, Kondo I. 1993. A 40-nucleotide
repeat polymorphism in the human dopamine transporter gene.
Hum Genet 91:405�406.
Spencer TJ, Biederman J, Madras BK, Faraone SV, Dougherty
DD, Bonab AA, Fischman AJ. 2005. In vivo neuroreceptor
imaging in attention-deficit/hyperactivity disorder: A focus on
the dopamine transporter. Biol Psychiatry 57:1293�1300.
Stein MA, Waldman ID, Sarampote CS, Seymour KE, Robb AS,
Conlon C, Kim SJ, Cook EH. 2005. Dopamine transporter
genotype and methylphenidate dose response in children with
ADHD. Neuropsychopharmacology 30:1374�1382.
Ujike H, Harano M, Inada T, Yamada M, Komiyama T, Sekine Y,
Sora I, Iyo M, Katsu T, Nomura A, Nakata K, Ozaki N. 2003.
Nine- or fewer repeat alleles in VNTR polymorphism of the
dopamine transporter gene is a strong risk factor for prolonged
methamphetamine psychosis. Pharmacogenomics J 3:242�247.
van Dyck CH, Quinlan DM, Cretella LM, Staley JK, Malison RT,
Baldwin RM, Seibyl JP, Innis RB. 2002. Unaltered dopamine
156 J. Krause et al.

transporter availability in adult attention deficit hyperactivity
disorder. Am J Psychiatry 159:309�312.
Waldman ID, Rowe DC, Abramowitz A, Kozel ST, Mohr JH,
Sherman SL, Cleveland HH, Sanders ML, Gard JM, Stever C.
1998. Association and linkage of the dopamine transporter
gene and attention-deficit hyperactivity disorder in children:
Heterogeneity owing to diagnostic subtype and severity. Am J
Hum Genet 63:1767�1776.
Winsberg BG, Comings DE. 1999. Association of the dopamine
transporter gene (DAT1) with poor methylphenidate response.
J Am Acad Child Adolesc Psychiatry 38:1474�1477.
Striatal dopamine transporter and DAT-1 gene in ADHD 157

ORIGINAL INVESTIGATION
Association study of the glycogen synthase kinase-3b genepolymorphism with prophylactic lithium response in bipolar patients
ALEKSANDRA SZCZEPANKIEWICZ1, JANUSZ K. RYBAKOWSKI2, ALEKSANDRA
SUWALSKA2, MARIA SKIBINSKA1, ANNA LESZCZYNSKA-RODZIEWICZ2,
MONIKA DMITRZAK-WEGLARZ1, PIOTR M. CZERSKI1 & JOANNA HAUSER1,2
1Laboratory of Psychiatric Genetics, Department of Psychiatry, Poznan University of Medical Sciences, Poznan, Poland, and2Department of Adult Psychiatry, Poznan University of Medical Sciences, Poznan, Poland
AbstractA relationship between response to lithium prophylaxis and T-50C polymorphism of glycogen synthase kinase-3b (GSK-3b) gene was investigated in 89 bipolar patients (41 male and 48 female) who have been taking lithium for at least 5 years.The patients were delineated as excellent responders, partial responders and non-responders to lithium. The resultsobtained suggest that this polymorphism may not be related to the degree of prophylactic lithium response.
Key words: Association, bipolar disorder, glycogen synthase kinase-3b gene, lithium response
Introduction
The enzyme, glycogen synthase kinase-3b (GSK-
3b), is essential player in a number of intracellular
signaling pathways and regulates several transcrip-
tion factors and cytoskeletal elements. GSK-3b is
pro-apoptotic factor and plays a significant role in
neuroprotection processes. The pathogenic impor-
tance of GSK-3b has been recently implicated in
such illnesses as bipolar mood disorder and Alzhei-
mer’s disease (Gould et al. 2004b).
The gene for GSK-3b was mapped to 3q21.1
region and several polymorphisms of this gene have
been described. The �50 T/C polymorphism is
localized in an untranscribed region of the promoter
that is not putative transcriptional binding site (Russ
et al. 2001). However, it is located in an effective
promoter region (nucleotide �171 to �/29) of the
gene encoding GSK-3b and decreased expression of
the gene was found after deletion of this promoter
region (Lau et al. 1999).
Lithium is one of the most commonly used drugs
in the prophylaxis and treatment of bipolar disorder.
The mechanisms of mood-stabilizing effect of
lithium incorporate its effect on the processes of
intracellular signalling and neuronal plasticity. Many
possible targets of mood-normalizing action of
lithium have been delineated such as phosphatidyli-
nositol system, brain-derived neurotrophic factor
(BDNF) and also GSK-3b (Manji and Zarate
2002). In our recent study, an association was
demonstrated between lithium prophylactic re-
sponse and BDNF gene polymorphisms (Ryba-
kowski et al. 2005).
Lithium exerts inhibitory effect on the GSK-3
activity at concentrations relevant for bipolar dis-
order treatment, thus promoting impaired cellular
resilience and synaptic plasticity in this illness
(Gould et al. 2004a; Li et al. 2002). In addition,
lithium also affects circadian clocks which distur-
bances may play pathogenic role in bipolar disorder
(Padiath et al. 2004). GSK3-b is the mammalian
orthologue of an enzyme which regulates molecular
clock in Drosophila (Martinek et al. 2001) and it was
observed that long-term lithium administration, via
reduction of GSK-3 activity, lengthens the circadian
clock (Iwahana et al. 2004).
Recently, Benedetti et al. (2005) reported that the
efficacy of long-term lithium prophylaxis is influ-
enced by the T-50C polymorphism in GSK-3b gene.
In their sample, a better efficacy of lithium was
observed in the carriers of mutant C allele of this
Correspondence: Aleksandra Szczepankiewicz, Laboratory of Psychiatric Genetics, Poznan University of Medical Sciences, ul. Szpitalna
27/33, 60-572 Poznan, Poland. Tel: �/48 61 8491311. Fax: �/48 61 8480-392. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(3): 158�161
(Received 20 July 2005; accepted 22 December 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970600554711

polymorphism. In the present study we make an
attempt to verify their findings in thoroughly char-
acterized group of bipolar patients receiving long-
term lithium prophylaxis.
Experimental procedures
Patients
The study was performed on 89 patients with
bipolar disorder (n�/82 of bipolar I and n�/7 of
bipolar II) according to DSM-IV and ICD-10
criteria using SCID, recruited from Wielkopolska
region, attending the Outpatient Lithium Clinic at
the Department of Psychiatry, Poznan University of
Medical Sciences. All patients involved in the study
(41 males with a mean age of 49 years, SD�/12; 48
females with a mean age of 47 years, SD�/15) have
been treated with lithium carbonate for at least 5
years (5�27 years, mean 15 years). Serum concen-
tration of lithium has been maintained in the range
between 0.5�0.8 mmol/l.
The course of illness was assessed retrospectively,
based on the analysis of medical outpatient charts,
inpatient records and semi-structured reviews. The
efficacy of lithium treatment was assessed according
to the following criteria: excellent lithium responders
(ER) had no affective episodes on lithium; partial
lithium responders (PR) showed 50% reduction in
the episode index (number of episodes per year to
pre-lithium period); lithium non-responders (NR)
showedB/50% reduction, no change or worsening in
the episode index.
All patients gave written consent to the study.
The study was approved by the Local Bioethics
Committee.
Genotyping
The DNA was extracted from 10 ml of EDTA
anticoagulated whole blood using the salting out
method (Miller et al. 1988). A 344-basepair frag-
ment of the GSK-3b gene was amplified by PCR
reaction with set of primers described by Russ et al.
(2001) using PTC-200 (MJ Research) thermal
cycler. PCR product (4.0 ml) was then digested
with AluI restriction endonuclease (MBI Fermen-
tas). The uncut PCR product size was 344 bp. After
RFLP analysis, the following alleles were observed:
uncut C allele (127 bp), for Tallele the bands of 220
and 124 bp. Restriction analysis for uncut C allele
was repeated to confirm the results.
Statistical analysis
The Pearson’s chi-square (x2)-test and Fisher’s exact
test were applied to test differences in the genotypic
and allelic (respectively) distribution between groups
of lithium responders. Additionally, stepwise logistic
regression analysis was performed including the
GSK-3b polymorphism and lithium response as
covariates. Calculations were performed using the
computer programme SPSS version 11.5. For small
numbers of patients in cells (see Table I, below 5) we
performed Fisher�Freeman�Halton test with Stat-
Xact-4 v 4.0.1 programme.
Results
Genotype distribution of analysed population was in
concordance with Hardy�Weinberg equilibrium
(P�/0.18). Among this group, 23 patients (25.8%)
were classified as excellent responders (ER), 47
(52.8%) as partial responders (PR) and 19
(21.3%) as non-responders (NR) to lithium prophy-
laxis. Clinical data of these patients are presented in
Table I.
No significant differences in genotype distribu-
tions and allele frequencies between GSK-3b T-50C
polymorphism and the degree of lithium response
was found (P�/0.646 and P�/0.675, respectively).
Data are shown in Table II. Analysis by gender did
not reveal any significant differences in genotype and
allele frequencies, either (P�/0.788 for males, P�/
0.649 for females).
Due to very small number in the case of
homozygous patients, we performed the Fisher�Freeman�Halton test considering small amount in
some cells in Table I (below 5). However, no
Table I. Clinical characteristics of the bipolar patients on lithium prophylaxis.
Total (n�/89) ER (n�/23) PR (n�/47) NR (n�/19)
Age, years [mean9/SD] 54.89/12.3 57.89/14.2 53.29/12.2 54.19/8.4
Gender [M:F] 38:51 11:12 16:31 10:9
Family history of psychiatric illness, N (%) 41 (46.6%) 11 (40.7%) 24 (53.3%) 6 (37.5%)
Age at onset, years [mean9/SD] 31.59/10.7 33.09/11.6 31.19/10.9 30.19/8.5
Duration of illness before lithium, years [mean9/SD] 7.49/7.4 9.79/9.6 5.69/5.9 8.79/6.0
Duration of lithium treatment, years [mean9/SD] 14.69/7.3 14.09/7.1 15.39/7.9 13.89/5.8
Affective episodes before lithium, N [mean�/SD] 6.29/4.1 7.09/3.6 6.09/4.5 5.89/3.9
Affective episodes on lithium, N [mean�/SD] 3.39/3.9 0 3.59/2.7 8.29/4.8
Association study of GSK-3b T-50C polymorphism and lithium response 159
statistically significant differences were found be-
tween the analysed groups (P�/0.6977).
Discussion
The main finding of our study is lack of significant
association between T-50C polymorphism in the
GSK-3b gene and the degree of prophylactic re-
sponse to lithium carbonate in carefully character-
ized group of bipolar patients. Thus, the results
obtained in our analysis are not consistent with the
previous study performed by Benedetti et al. (2005).
In our study, in comparison to that of Benedetti’s
group, a nearly identical number of bipolar patients
was involved (89 vs. 88, respectively). On the other
hand, the duration of lithium prophylaxis in our
study was much longer (at least 5�27 years on
lithium) what enabled more precise assessment of
quality of lithium prophylactic effect. Our division of
bipolar patients on lithium into three groups (ex-
cellent, partial and non-responders) makes it possi-
ble to analyse an association between GSK-3b and
prophylactic lithium response in the groups of
bipolar patients with distinct clinical characteristics.
Excellent lithium responders are characterised by
episodic course of disease and low rates of comor-
bidity (MacQueen et al. 2005). On the other hand,
family studies showed that lithium non-responders
had 10 times higher prevalence of schizophrenia in
first-degree relatives compared to lithium responders
(Grof et al. 1994). Therefore, our analysis may be
complementary to that of Benedetti et al. (2005) and
extend it.
A closer inspection of Table II reveals that the
ratio of ER/NR in carriers of C allele (patients with
T/C�/C/C genotypes) was higher than ER/NR ratio
in patients with T/T genotype (1.5 and 0.9, respec-
tively). Therefore, this may be a slight hint to
suppose that ER may be more likely to have C allele
than NR. Although it may resemble a relationship
found by Italian investigators, this is not supported
by any statistical significance for neither genotypes
nor alleles.
In conclusion, the role of GSK-3b in the patho-
genesis of bipolar illness and in the prophylactic
action of lithium remains to be further elucidated.
Some association has been found between GSK-3bgene polymorphism with some features of illness
such as later onset or antidepressant response to
total sleep deprivation (Benedetti et al. 2004a,b). On
the other hand, studies of GSK-3b in postmortem
brains of bipolar patients were negative (Beasley et
al. 2002; Lesort et al. 1999). Although the study of
Benedetti et al. (2005) suggested an association
between GSK-3b gene polymorphism and lithium
prophylactic effect, we were not able to confirm their
results with our population of patients.
Acknowledgements/Statement of interest
This study was supported by the Polish Committee
of Scientific research (KBN), grants no. 2P05B
01 226 and 2P05B 00 226. Dr P.M.C. is the
recipient of a 2004 Annual Stipend for Young
Scientists from the Foundation for Polish Science
(FNP).
References
Beasley C. 2002. An investigation of the Wnt-signalling pathway
in the prefrontal cortex in schizophrenia, bipolar disorder and
major depressive disorder. Schizophr Res 58:63�67.
Benedetti F. 2004a. A single nucleotide polymorphism in glycogen
synthase kinase 3-beta promoter gene influences onset of illness
in patients affected by bipolar disorder. Neurosci Lett 355:37�340.
Benedetti F. 2004b. A glycogen synthase kinase 3-beta promoter
gene single nucleotide polymorphism is associated with age at
onset and response to total sleep deprivation in bipolar
depression. Neurosci Lett 368:123�126.
Benedetti F. 2005. Long-term response to lithium salts in bipolar
illness is influenced by the glycogen synthase kinase 3-beta-50
T/C SNP. Neurosci Lett 376:51�55.
Gould TD. 2004a. In vivo evidence in the brain for lithium
inhibition of glycogen synthase kinase-3. Neuropsychopharma-
cology 29:32�38.
Gould TD. 2004b. Glycogen synthase kinase-3: A target for novel
bipolar disorder treatments. J Clin Psychiatry 65:10�21.
Grof P. 1994. Lithium response and genetics of affective
disorders. J Affect Disord 32:85�95.
Table II. Comparison of genotypes and alleles of T-50C GSK-3b polymorphism in bipolar patients stratified by response to lithium
prophylaxis.
Lithium response Genotypes Alleles
T/T T/C C/C T C
ER 8 (34.8%) 14 (60.9%) 1 (4.3%) 30 (65.2%) 16 (34.8%)
PR 16 (34.0%) 25 (53.2%) 6 (12.8%) 57 (60.6%) 37 (39.4%)
NR 9 (47.4%) 8 (42.1%) 2 (10.5%) 26 (68.4%) 12 (31.6%)
Difference, ER vs. PR vs. NR � x2 �/2.475, df�/4, P�/0.649 for genotypes, P�/0.675 for alleles.
ER (excellent lithium responders): no affective episodes on lithium; PR (partial lithium responders): 50% reduction in the episode index,
defined as a number of episodes per year compared to pre-lithium period; NR (non-responders): B/50% reduction, no change or worsening
in the episode index, defined as a number of episodes per year compared to pre-lithium period.
160 A. Szczepankiewicz et al.

Iwahana E. 2004. Effect of lithium on the circadian rhythms of
locomotor activity and glycogen synthase kinase-3 protein
expression in the mouse suprachiasmatic nuclei. Eur J Neurosci
19:2281�2287.
Lau KF. 1999. Molecular cloning and characterization of the
human glycogen synthase kinase-3beta promoter. Genomics
60:121�128.
Lesort M. 1999. Glycogen synthase kinase-3beta, beta-catenin,
and tau in postmortem bipolar brain. J Neural Transm
106:1217�1222.
Li X. 2002. Glycogen synthase kinase-3beta, mood stabilizers,
and neuroprotection. Bipolar Disord 4:137�144.
MacQueen GM. 2005. The phenotypes of bipolar disorder:
Relevance for genetic investigations. Mol Psychiatry 10:811�826.
Manji HK, Zarate CA. 2002. Molecular and cellular mechanisms
underlying mood stabilization in bipolar disorder: Implications
for the development of improved therapeutics. Mol Psychiatry
7(Suppl 1):S1�7.
Martinek S, Inonog S, Manoukian AS, Young MW. 2001. A role
for the segment polarity gene shaggy/GSK-3 in the Drosophila
circadian clock. Cell 105:769�779.
Miller SA. 1988. A simple salting out procedure for extracting
DNA from human nucleated cells. Nucleic Acids Res 16:1215.
Padiath QS. 2004. Glycogen synthase kinase 3beta as a likely
target for the action of lithium on circadian clocks. Chronobiol
Int 21:43�55.
Russ C. 2001. Identification of sequence variants and analysis of
the role of the glycogen synthase kinase 3 beta gene and
promoter in late onset Alzheimer’s disease. Mol Psychiatry
6:320�324.
Rybakowski JK. 2005. Prophylactic lithium response and poly-
morphism of the brain-derived neurotrophic factor gene.
Pharmacopsychiatry 38:166�170.
Association study of GSK-3b T-50C polymorphism and lithium response 161

ORIGINAL INVESTIGATION
The influence of concomitant neuroleptic medication on safety,tolerability and clinical effectiveness of electroconvulsive therapy
CAROLINE NOTHDURFTER, DANIELA ESER, CORNELIUS SCHULE, PETER
ZWANZGER, ALAIN MARCUSE, INES NOACK, HANS-JURGEN MOLLER,
RAINER RUPPRECHT & THOMAS C. BAGHAI
Department of Psychiatry and Psychotherapy, Ludwig-Maximilians-University, Munich, Germany
AbstractBackground: Electroconvulsive therapy (ECT) is still considered to be the most efficacious treatment option in majordepressive disorder and treatment-resistant schizophrenia. Unfortunately, in some cases patients do not respond sufficientlyto conventional unilateral ECT, or even to bilateral or high dose ECT. In these cases, concomitant pharmacotherapy can bea useful augmentation strategy to improve clinical effectiveness. Interestingly, there is not much data about ECT andconcomitant neuroleptic medication. Method: We evaluated 5482 treatments in 455 patients in our retrospective study tosee whether there might be differences between combination therapies (ECT and concomitant neuroleptic medication) andECT monotherapy. We focused on clinical effectiveness and tolerability; furthermore we investigated treatment modalitiesand ictal neurophysiological parameters that might influence the treatment. Results: A total of 18.2% of all treatments weredone with no psychotropic medication, 2.8% with a neuroleptic monotherapy. Seizure duration according to EEGderivations turned out to be significantly longer in patients treated with neuroleptics of lower antipsychotic potency, whereasseizure duration in EMG was shorter in treatments done with atypical substances. Postictal suppression was highest intreatments done with atypical neuroleptics, whereas the same group was lowest regarding convulsion energy and convulsionconcordance indices. The best therapeutic effectiveness was seen in treatments done with atypical substances. Adverseeffects were not influenced significantly by concomitant neuroleptic medication. Conclusion: Our study suggests that theremight be a clinical benefit by combining ECT treatment with neuroleptic medication; especially atypical substances seem toenhance improvement. The tolerability of ECT treatment was not influenced by concomitant neuroleptic medication.
Key words: Electroconvulsive therapy, major depression, schizophrenia, neuroleptics, clinical effectiveness
Introduction
ECT is still considered to be a very efficacious and
well-tolerated treatment option in different psychia-
tric diseases, such us major depressive disorder or
schizophrenia, especially in cases of pharmacother-
apy resistance (Tharyan et al. 2002; ECT Review
Group 2003). In major depressive disorder, there is
evidence that ECT is even the most efficacious
therapeutic regimen (ECT Review Group 2003).
As far as schizophrenic disorders are concerned,
clozapine is still considered to be the standard
medication in patients who do not respond to other
neuroleptics (Fink 1990), but with this medication
there still remain 40% of patients who do not show a
significant improvement of their symptoms (Meltzer
et al. 1989). Taken together, one-fifth of all schizo-
phrenic patients do not respond sufficiently to
pharmacotherapeutic treatment. In the case of
therapy resistance, atypical neuroleptics, especially
clozapine, have to be considered as a treatment
option (Schafer et al. 2004). This shows the
necessity of alternative therapeutic strategies. Pub-
lished data support the idea that neuroleptics in
combination with ECT might be more efficacious
than single neuroleptic medication. Kales et al.
(1999) found that combining ECT and clozapine
seems to be a safe and effective alternative treatment
option in some patients who suffer from refractory
schizophrenia. Tang and Ungvari (2002, 2003)
showed that ECT might be a worthful augmentation
strategy in schizophrenic patients who do not
sufficiently respond to a neuroleptic medication,
whereas there are hints that such a positive effect
Correspondence: Thomas C. Baghai MD, Department of Psychiatry and Psychotherapy, Ludwig-Maximilians-University,
Nussbaumstrasse 7, D-80336 Munich, Germany. Tel: �/49 89 7095 2717. Fax: �/49 89 7095 2715. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(3): 162�170
(Received 4 May 2005; accepted 29 September 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500395280

of this combination might be limited over time.
Suzuki et al. (2004) showed that the relapse rate of
patients suffering from catatonic schizophrenia after
response to acute ECT is high after 6 months,
although neuroleptic pharmacotherapy was contin-
ued. These findings and the fact that ECT still is one
of the best tolerated and most efficacious therapies
raised the question of the influence of concomitant
neuroleptic medication on safety, tolerability and
clinical effectiveness of electroconvulsive therapy.
Clinical effectiveness of ECT and treatment history
The clinical effectiveness of ECT depends on
different treatment parameters, such as stimulation
energy and stimulation modus. In general, unilateral
ECT is a very widespread method of treatment.
Unfortunately, not all patients respond to unilateral
ECT, so that in some cases bilateral stimulation is
necessary. Using this method, lower stimulation
energy is needed, e.g., according to the half-age
method (Abrams 2002). Another possibility of
intensifying the therapy is the use of high-dose
ECT. Using higher stimulation energy in a unilateral
treatment mode can be even as efficacious as
bitemporal or bifrontal ECT (Sackeim et al. 2000).
Augmentation strategies in ECT
Furthermore, additional psychopharmacotherapy
can be used for the augmentation of an ECT
treatment series. Concerning antidepressant conco-
mitant medication, study results indicate that there
is a benefit of combining ECT with tricyclic anti-
depressants (Lauritzen et al. 1996). In the case of
concomitant antipsychotic treatment, published data
are very rare. There is consensus that combining
ECT and clozapine can be a useful augmentation
strategy to improve clinical response in schizophre-
nic patients (Fink 1990; Kupchik et al. 2000), but if
there is a clinical benefit in prescribing neuroleptics
during ECT, this treatment still has to be investi-
gated in more detail.
Study plan
In our retrospective investigation, we addressed the
following questions: Is the combination of ECT and
neuroleptics beneficial for patients with regard to
ictal electrophysiological parameters that might be of
prospective significance for clinical effectiveness?
Are safety or tolerability of ECT treatment altered
if combined with neuroleptic medication in compar-
ison to an ECT monotherapy? Does concomitant
neuroleptic medication influence the incidence of
cognitive or cardiovascular side effects? Are there
differences between different classes of neuroleptics
with regard to ictal electrophysiological parameters
and clinical effectiveness?
Materials and methods
Patients
A total of 5482 ECT treatments in 455 patients,
treated from 1995 to 2003, were analysed in our
retrospective study. We recorded 518 treatment
series; in 63 cases one patient received more than
one treatment series during our period of observa-
tion. A total of 1075 treatments (19.6%) were done
with a concomitant psychopharmacological mono-
therapy (14.4% of treatments were done with no
concomitant medication at all), of which we ana-
lysed 452 treatments (42%) that were done with a
neuroleptic monotherapy. Table I gives patient
characteristics, diagnostic groups according ICD-
10 (World Health Organization 1992), demographic
data and ECT treatment parameters. The patients
were subdivided into two major diagnostic groups:
patients with schizophrenia or schizoaffective
Table I. Demographic data and ECT treatment parameters according to the major diagnostic groups (ICD-10) of ECT treatments. Most
of the treatments were done with patients who suffer from major depressive disorder; as expected, they were older than patients who suffer
from schizophrenia. Depressed patients were more often unilaterally stimulated with lower energy, whereas schizophrenic patients were
mainly bilaterally treated with higher energy.
Diagnoses (ICD-10) x2-Test, ANOVA
F2 F3 F ; x2; P
n (%) 170 (38.5%) 267 (60.5%)
Sex (M/F) 40.9/59.1% 41.5/58.5% 0.492
Age (mean9/SD) 47.49/14.1 54.89/13.8 B/0.0001
ECT treatment modalities
No. of treatments (mean9/SD, n ) 11.59/5.3 10.69/2.8 0.61
Duration (mean9/SD, days) 75.49/95.7 31.79/10.5 B/0.0001
Unilateral (%)/bilateral (%) 24.5/75.5% 85.8/14.2% B/0.0001
Charge (mean9/SD, mC) 3349/193 2629/110 B/0.0001
F2�/schizophrenia (ICD-10); F3�/unipolar and bipolar major depression (ICD-10).
Electroconvulsive therapy and concomitant neuroleptic medication 163

disorder (F2), patients with unipolar or bipolar
major depression (F3). Four treatments were carried
out in patients with rare diagnoses (such as Gilles-
de-la-Tourette syndrome) which were not explicitly
mentioned in the results. The patients gave their
written informed consent for ECT treatment and
anaesthesia separately.
ECT treatments (Weiner et al. 1988)
The Thymatron DGxTM device was used from 1995
to June 2000 for ECT treatments; we then used the
Thymatron System-IVTM from July 2000. Both
devices deliver stimuli in the form of a constant
current (900 mA) bi-directional pulse wave. The
voltage was B/450 V, impulse width was 0.5�1 ms
(mean9/standard deviation (SD): 0.559/0.13 ms),
frequency was 20�70 Hz (52.99/16.8 Hz), length of
stimulus train was 0.14�8 s (5.29/2.0 s).
Stimulus intensity was chosen according to the
modified age method in the case of unilateral
stimulation and the half-age method in the case of
bilateral stimulation (Abrams 2002). The lower
charge limit during the first stimulation was
30% (151 mC), the upper charge limit was 60%
(302 mC). In a very few cases (B/5%) dose titration
was necessary before starting the initial treatment
session. In these cases, unilaterally treated patients
received a first stimulus 2.5�5-fold over threshold,
bilaterally treated patients a stimulus 5% over
threshold. In the case of EEG B/30 s, EMGB/25 s
and postictal suppression index B/80%, we re-
stimulated in 10% dosage elevation steps. The
maximal charge was 504 mC during 1995�2001
and 1008 mC since January 2002. The mean
postictal suppression index was 83.89/21.3%, and
the mean stimulation energy was 57.29/29.9%
(288.39/150.7 mC).
In the case of unilateral stimulation, the treatment
electrodes were placed according to the d’Elia
method as follows (d’Elia 1970; d’Elia and Raotma
1975): one electrode was placed temporally 1 cm
above the centre-point of a line between the outer
eyelid angle and the external acoustic meatus;
the other electrode was placed 12�13 cm distant,
2�3 cm lateral to the vertex. In therapy-resistant
cases, or if former inefficient unilateral ECT treat-
ments were reported, we used bitemporal stimula-
tion. In these cases, electrodes were placed on both
sides according the description above for the tem-
poral electrode. During 1995�1999, electrode pla-
cement was not recorded during clinical routine
treatments, so that these data are lacking in 46.5%
of the analysed treatments. From 2000, 30.3%
unilateral and 23.2% bitemporal treatments were
recorded.
Seizure monitoring included one-lead EEG and
EMG monitoring up to June 2002 and two-lead
EEG, EMG and ECG monitoring from July 2002.
The position of the one-lead EEG electrodes was left
frontopolar and over the ipsilateral mastoid (FP1�A1
according the international 10�20 system). The
two-lead EEG electrodes were positioned left and
right frontopolar (FP1�A1 and FP2�A2) and over
the ipsilateral mastoid. EMG conduction was placed
5�10 cm over the flexor carpi ulnaris muscle. The
mean seizure duration was 33.49/18.3 s (EEG) and
20.39/12.8 s (EMG).
Anaesthesia
Thiopental was used in 71.9% of treat-
ments (mean dosage9/SD: 3609/88 mg), propofol
was used in 15.0% (1759/64 mg), methoh-
exital was used in 4.4% (1359/31 mg) and
etomidate was used in 2.2% (359/5 mg). The
choice of anaesthetic agent was made according to
the clinical need to achieve adequate seizure
control. Patients with higher seizure threshold
more often received methohexital than other
anaesthetics. For muscle relaxation, succinylcholine
or pyridostigmine, together with atracurium, for
precurarisation were used.
Psychotropic medication
There were 787 treatments done without any
psychiatric medication; 1075 treatments were done
with concomitant non-benzodiazepine hypnotics.
The analysis of all electrophysiological and clinical
parameters revealed no clinically relevant or statis-
tically significant differences between these two
groups.
As far as treatment with neuroleptics as a
monotherapy is concerned, 36.7% of the treat-
ments were done with concomitant medication in
the form of atypical substances, 17.2% with high
potency classical neuroleptics, and 8.2% with
medium and 36.7% with low potency neuroleptics
(Benkert et al. 2003). In the case of atypical
substances, clozapine (41%) and olanzapine
(18%) were given in most cases; antipsychotics
of high neuroleptic potency were predomi-
nantly haloperidol (57%) and benperidol (16%).
Perazine (38%) was the most commonly pre-
scribed medium potency neuroleptic; prothipendyl
(46%) and promethazine (36%) were mainly given
in the case of low potency neuroleptic medication.
The distribution of concomitant neuroleptic med-
ication to the different diagnostic groups is shown
in Table II.
164 C. Nothdurfter et al.

Clinical assessments
We recorded ECT treatments and clinical data from
the ECT treatment documentation and ECT device
printouts in a relational database (Microsoft Access
2000) and from patient records. The severity of
disease, according to the Clinical Global Impression
scale (CGI, Item 1; National Institute of Mental
Health 1976), and diagnosis (ICD-10) were as-
sessed. CGI scores were recorded after each electro-
convulsive treatment, and additionally every week in
the patient records. Furthermore we registered
information about therapeutic response (CGI, Item
3.1) and adverse effects with special regard to
cognitive impairment (CGI, Item 3.2) systematically
after each treatment (serious adverse events were
recorded in free form). We also recorded clinical,
technical and electrophysiological ECT (stimulation
parameters, EEG and EMG including the length of
convulsions, postictal suppression, seizure energy
and seizure concordance). All prescribed medication
(psychiatric medication, substances used for anaes-
thesia, and others, including dosages) were also
recorded.
Electrophysiological measures
To estimate the success of ECT treatment, the
minimal duration of generalised convulsions mea-
sured using EEG and EMG is necessary, but not
sufficient. Measurements of at least 25 s duration in
the EEG and 20 s in the EMG are required (Coffey
et al. 1995). In most cases, computer-recorded EEG
and EMG estimation of the seizure duration is of
sufficient quality (Swartz et al. 1994). On the other
hand, there is evidence that there is no significant
correlation between the clinical therapeutic effec-
tiveness of ECT and the duration of convulsions
(Abrams 1972, 2002; Nobler et al. 1993). In the
case of unilateral ECT, a higher stimulation energy
may result in shorter duration of convulsions (Frey
et al. 2001a), although the clinical effectiveness is
higher (Sackeim et al. 2000). For these reasons,
other measures can be utilised to decide whether
restimulation may be recommended during an
individual treatment session. One of these is the
postictal suppression index (PSI). The PSI shows
how fast and complete the EEG amplitude flattens
directly after the convulsions. It is calculated from
the quotient of the mean EEG amplitude during a
3-s derivation 0.5 s after the end of convulsions and
the mean amplitude of a 3-s passage during the
convulsions. There is a high correlation between its
unit ‘‘% suppression’’ and the probability of ther-
apeutic efficacy (Suppes et al. 1996). If the PSI is
under 80%, restimulation should be taken into
consideration (Weiner et al. 1991: Nobler et al.
1993). As a measure of the intensity of the ictal
response after electrical stimulation (mV �/s), the
convulsion energy index (CEI) is used (Weiner
et al. 1991). It is calculated from the product of
the mean EEG amplitude and the duration of
convulsions. The convulsion concordance index
(CCI) is a measure for the intracerebral general-
isation of the convulsions (Swartz et al. 1986). It is
calculated in the following way:
100�EEG � EMG
EEH � EMG
(the duration of the convulsions is represented by
EEG and EMG). If the index is below 51%, the
patient should be re-stimulated. Senior psychiatrists
provided regular monitoring of EEG and EMG
quality and all treatment procedures.
Statistical analyses
SPSS for Windows (Release 12.0.1, SPSS Inc.,
Chicago, IL 60606, USA) was used for all statistical
analyses. For the comparison of mean differences in
demographic and clinical variables between treat-
ment group independent samples Student’s t-tests
and x2-tests were used. A one-way analysis of
variance (ANOVA procedure) was performed to
detect whether there were significant differences in
electrophysiological variables and clinical mean
scores between the treatment groups. In the case of
multiple testing, Bonferroni correction was used;
0.05 was set as the level of significance.
Results
Psychotropic medication was given in 81.8% of all
treatments, in 23.4% as a psychotropic monother-
apy. A total of 14.6% of the treatments done with
neuroleptics was as a monotherapy. Within the
group of mono-pharmacotherapeutic treatments
with neuroleptics, which was of special interest in
Table II. Description of the major diagnostic groups (ICD-10)
and concomitant neuroleptic medication. Most of the treatments
were done with low potency neuroleptics or atypical substances,
less than half the treatments were done with high potency or
medium potency neuroleptics.
Diagnoses (ICD-10)
Neuroleptic monotherapy (%) F2 F3 Mean
High potency 32.9% 67.1% 17.2%
Medium potency 44.7% 55.3% 8.2%
Low potency 8.2% 89.3% 38.6%
Atypical substances 78.7% 21.3% 38.6%
F2�/schizophrenia (ICD-10); F3�/unipolar and bipolar major
depression (ICD-10).
Electroconvulsive therapy and concomitant neuroleptic medication 165
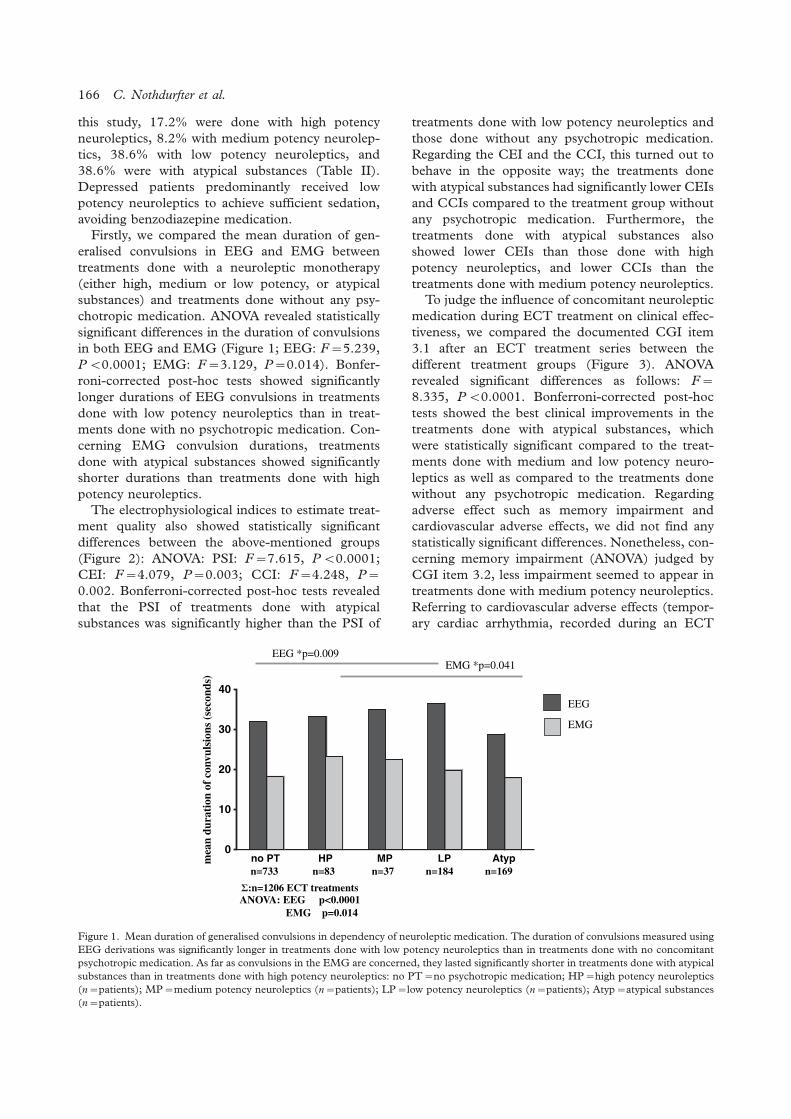
this study, 17.2% were done with high potency
neuroleptics, 8.2% with medium potency neurolep-
tics, 38.6% with low potency neuroleptics, and
38.6% were with atypical substances (Table II).
Depressed patients predominantly received low
potency neuroleptics to achieve sufficient sedation,
avoiding benzodiazepine medication.
Firstly, we compared the mean duration of gen-
eralised convulsions in EEG and EMG between
treatments done with a neuroleptic monotherapy
(either high, medium or low potency, or atypical
substances) and treatments done without any psy-
chotropic medication. ANOVA revealed statistically
significant differences in the duration of convulsions
in both EEG and EMG (Figure 1; EEG: F�/5.239,
P B/0.0001; EMG: F�/3.129, P�/0.014). Bonfer-
roni-corrected post-hoc tests showed significantly
longer durations of EEG convulsions in treatments
done with low potency neuroleptics than in treat-
ments done with no psychotropic medication. Con-
cerning EMG convulsion durations, treatments
done with atypical substances showed significantly
shorter durations than treatments done with high
potency neuroleptics.
The electrophysiological indices to estimate treat-
ment quality also showed statistically significant
differences between the above-mentioned groups
(Figure 2): ANOVA: PSI: F�/7.615, P B/0.0001;
CEI: F�/4.079, P�/0.003; CCI: F�/4.248, P�/
0.002. Bonferroni-corrected post-hoc tests revealed
that the PSI of treatments done with atypical
substances was significantly higher than the PSI of
treatments done with low potency neuroleptics and
those done without any psychotropic medication.
Regarding the CEI and the CCI, this turned out to
behave in the opposite way; the treatments done
with atypical substances had significantly lower CEIs
and CCIs compared to the treatment group without
any psychotropic medication. Furthermore, the
treatments done with atypical substances also
showed lower CEIs than those done with high
potency neuroleptics, and lower CCIs than the
treatments done with medium potency neuroleptics.
To judge the influence of concomitant neuroleptic
medication during ECT treatment on clinical effec-
tiveness, we compared the documented CGI item
3.1 after an ECT treatment series between the
different treatment groups (Figure 3). ANOVA
revealed significant differences as follows: F�/
8.335, P B/0.0001. Bonferroni-corrected post-hoc
tests showed the best clinical improvements in the
treatments done with atypical substances, which
were statistically significant compared to the treat-
ments done with medium and low potency neuro-
leptics as well as compared to the treatments done
without any psychotropic medication. Regarding
adverse effect such as memory impairment and
cardiovascular adverse effects, we did not find any
statistically significant differences. Nonetheless, con-
cerning memory impairment (ANOVA) judged by
CGI item 3.2, less impairment seemed to appear in
treatments done with medium potency neuroleptics.
Referring to cardiovascular adverse effects (tempor-
ary cardiac arrhythmia, recorded during an ECT
0
10
20
30
40
no PT HP MP LP Atypmea
n du
rati
on o
f co
nvul
sion
s (s
econ
ds)
n=733 n=83 n=37 n=184 n=169
:n=1206 ECT treatmentsANOVA: EEG p<0.0001 EMG p=0.014
EEG
EMG
EEG *p=0.009EMG *p=0.041
Figure 1. Mean duration of generalised convulsions in dependency of neuroleptic medication. The duration of convulsions measured using
EEG derivations was significantly longer in treatments done with low potency neuroleptics than in treatments done with no concomitant
psychotropic medication. As far as convulsions in the EMG are concerned, they lasted significantly shorter in treatments done with atypical
substances than in treatments done with high potency neuroleptics: no PT�/no psychotropic medication; HP�/high potency neuroleptics
(n�/patients); MP�/medium potency neuroleptics (n�/patients); LP�/low potency neuroleptics (n�/patients); Atyp�/atypical substances
(n�/patients).
166 C. Nothdurfter et al.
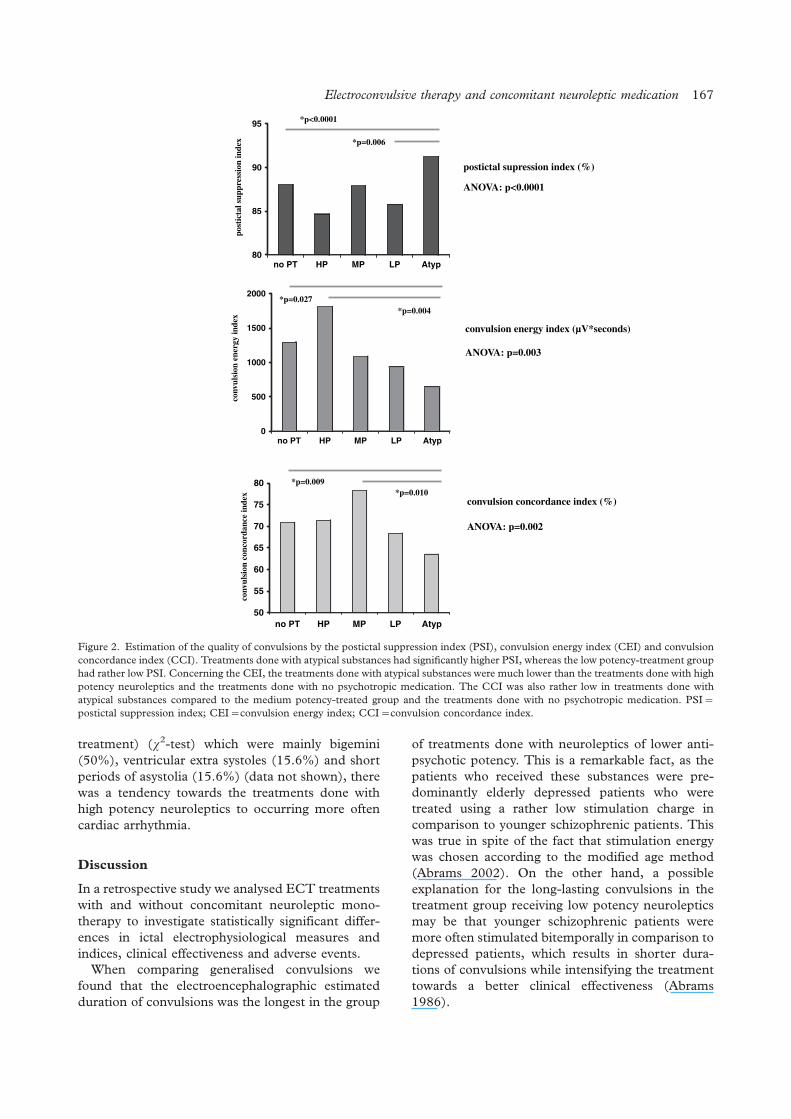
treatment) (x2-test) which were mainly bigemini
(50%), ventricular extra systoles (15.6%) and short
periods of asystolia (15.6%) (data not shown), there
was a tendency towards the treatments done with
high potency neuroleptics to occurring more often
cardiac arrhythmia.
Discussion
In a retrospective study we analysed ECT treatments
with and without concomitant neuroleptic mono-
therapy to investigate statistically significant differ-
ences in ictal electrophysiological measures and
indices, clinical effectiveness and adverse events.
When comparing generalised convulsions we
found that the electroencephalographic estimated
duration of convulsions was the longest in the group
of treatments done with neuroleptics of lower anti-
psychotic potency. This is a remarkable fact, as the
patients who received these substances were pre-
dominantly elderly depressed patients who were
treated using a rather low stimulation charge in
comparison to younger schizophrenic patients. This
was true in spite of the fact that stimulation energy
was chosen according to the modified age method
(Abrams 2002). On the other hand, a possible
explanation for the long-lasting convulsions in the
treatment group receiving low potency neuroleptics
may be that younger schizophrenic patients were
more often stimulated bitemporally in comparison to
depressed patients, which results in shorter dura-
tions of convulsions while intensifying the treatment
towards a better clinical effectiveness (Abrams
1986).
80
85
90
95
no PT HP MP LP Atyp
0
500
1000
1500
2000
no PT HP MP LP Atyp
50
55
60
65
70
75
80
no PT HP MP LP Atyp
post
icta
l sup
pres
sion
inde
x co
nvul
sion
ene
rgy
inde
x co
nvul
sion
con
cord
ance
inde
x
*p<0.0001
*p=0.006
postictal supression index (%)
ANOVA: p<0.0001
convulsion energy index (µV*seconds)
ANOVA: p=0.003
*p=0.027 *p=0.004
convulsion concordance index (%)
ANOVA: p=0.002
*p=0.009 *p=0.010
Figure 2. Estimation of the quality of convulsions by the postictal suppression index (PSI), convulsion energy index (CEI) and convulsion
concordance index (CCI). Treatments done with atypical substances had significantly higher PSI, whereas the low potency-treatment group
had rather low PSI. Concerning the CEI, the treatments done with atypical substances were much lower than the treatments done with high
potency neuroleptics and the treatments done with no psychotropic medication. The CCI was also rather low in treatments done with
atypical substances compared to the medium potency-treated group and the treatments done with no psychotropic medication. PSI�/
postictal suppression index; CEI�/convulsion energy index; CCI�/convulsion concordance index.
Electroconvulsive therapy and concomitant neuroleptic medication 167
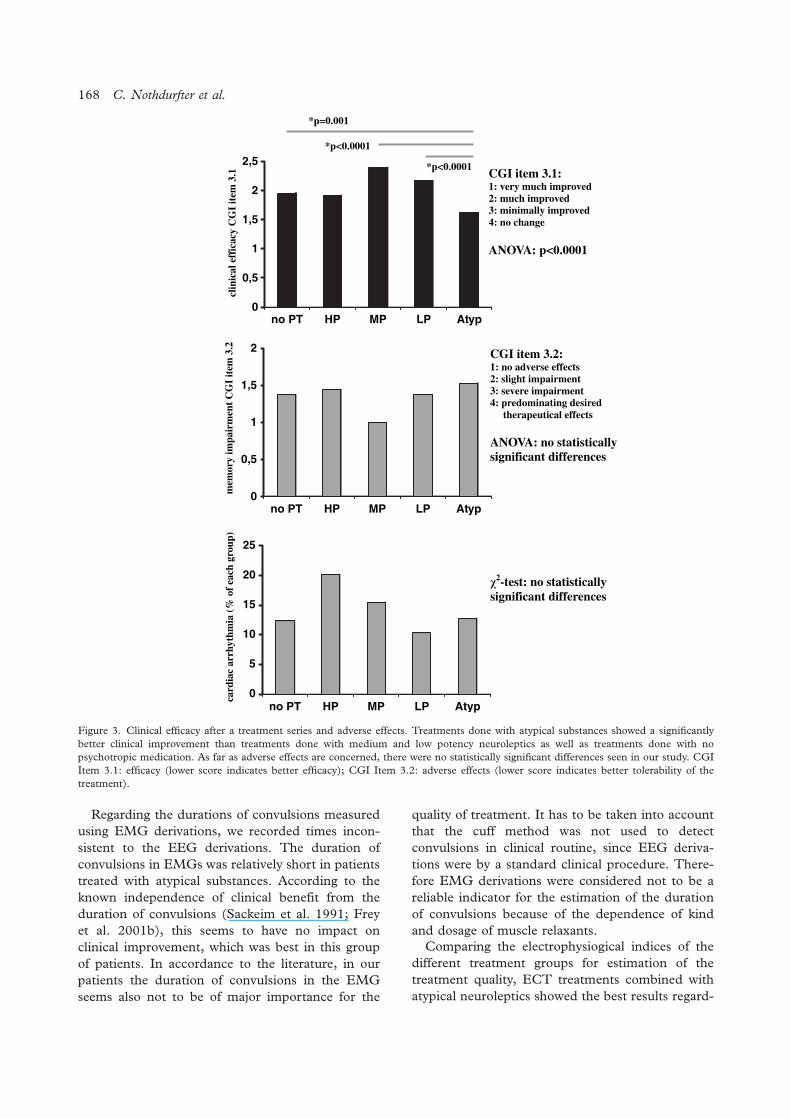
Regarding the durations of convulsions measured
using EMG derivations, we recorded times incon-
sistent to the EEG derivations. The duration of
convulsions in EMGs was relatively short in patients
treated with atypical substances. According to the
known independence of clinical benefit from the
duration of convulsions (Sackeim et al. 1991; Frey
et al. 2001b), this seems to have no impact on
clinical improvement, which was best in this group
of patients. In accordance to the literature, in our
patients the duration of convulsions in the EMG
seems also not to be of major importance for the
quality of treatment. It has to be taken into account
that the cuff method was not used to detect
convulsions in clinical routine, since EEG deriva-
tions were by a standard clinical procedure. There-
fore EMG derivations were considered not to be a
reliable indicator for the estimation of the duration
of convulsions because of the dependence of kind
and dosage of muscle relaxants.
Comparing the electrophysiogical indices of the
different treatment groups for estimation of the
treatment quality, ECT treatments combined with
atypical neuroleptics showed the best results regard-
0
0,5
1
1,5
2
2,5
no PT HP MP LP Atyp
0
0,5
1
1,5
2
no PT HP MP LP Atyp
0
5
10
15
20
25
no PT HP MP LP Atyp
clin
ical
eff
icac
y C
GI
item
3.1
CGI item 3.1: 1: very much improved 2: much improved 3: minimally improved 4: no change
ANOVA: p<0.0001
mem
ory
impa
irm
ent
CG
I it
em 3
.2
CGI item 3.2:1: no adverse effects 2: slight impairment3: severe impairment4: predominating desired therapeutical effects
ANOVA: no statisticallysignificant differences
card
iac
arrh
ythm
ia (
% o
f ea
ch g
roup
)
2-test: no statistically significant differences
*p=0.001
*p<0.0001
*p<0.0001
Figure 3. Clinical efficacy after a treatment series and adverse effects. Treatments done with atypical substances showed a significantly
better clinical improvement than treatments done with medium and low potency neuroleptics as well as treatments done with no
psychotropic medication. As far as adverse effects are concerned, there were no statistically significant differences seen in our study. CGI
Item 3.1: efficacy (lower score indicates better efficacy); CGI Item 3.2: adverse effects (lower score indicates better tolerability of the
treatment).
168 C. Nothdurfter et al.

ing postictal suppression (PSI), thus supporting the
hypothesis that this index seems to be a very reliable
parameter for judgment of therapy quality (Suppes
et al. 1996). This is further confirmed by the clinical
improvement judged by CGI item 3.1, which was
also best in the patients treated with atypical sub-
stances. On the other hand, the convulsion energy
index (CEI) was highest in patients treated with high
potency neuroleptics, whereas the convulsion con-
cordance index (CCI) was favourable in the group of
medium potency treated patients. The inhomogene-
ity of these electrophysiological indices shows that
they might have to be seen critically during a therapy
combining ECT and neuroleptics (especially CEI
and CCI). So far, there have been no published
studies investigating the influence of neuroleptic
medication on the electophysiological indices sys-
tematically.
As already mentioned, the clinical effectiveness
(according CGI item 3.1) turned out to be best in
patients treated with ECT and concomitant atypical
neuroleptics, which underlines the quality of ECT
treatments judged by the PSI. Treatments done with
concomitant medium and low potency substances
showed less convincing results, which might be
explained by the fact that these patients were more
severely ill and agitated, and thus needed more
sedative medication in form of low and medium
potency substances.
We also compared adverse effects (CGI item 3.2),
especially with regard to memory impairment
between the different groups of patients treated
with an ECT monotherapy or different neuroleptics
additionally to ECT treatment. Here we did not find
any statistically significant differences, which sup-
ports our assumption that the combination of ECT
and neuroleptic medication is not an additional risk
(O’Brien et al. 1993). It has to be mentioned that
there was no incidence of severe adverse effects at
all; mild cardiovascular side effects occurred only
temporarily during the ECT treatment and did not
need further intervention in most cases.
Due to the retrospective character of our investi-
gation and the inhomogeneous pharmacological
treatments using concomitant neuroleptics, our
analyses were oriented at treatment sessions rather
than to treatment courses of individual patients. In
clinical routine, treatment regimes very often change
with regard to ECT and pharmacotherapy treatment
modalities (data not shown). In this way the analysis
of singular treatment series in patients who are
treatment resistant and who have received more
treatments than other patients, may have more
influence on our analysis than patients showing a
better treatment response. An advantage of this kind
of analysis might be the fact that changes in ECT
and concomitant neuroleptic medication treatment
modalities within one ECT series cannot influence
the results significantly.
It is noteworthy that the different groups analysed
in our study differ not only in their treatment
modalities, but also concerning clinical and demo-
graphic data (Table I). We analysed predominantly
treatments of depressed patients (60.5%), who were,
as expected, significantly older (7.4 years) than
schizophrenic patients. Depressed patients very
often received low potency neuroleptics with sedat-
ing properties in order to avoid benzodiazepines
during ECT, whereas most of the treatment-resistant
schizophrenic patients received atypical neurolep-
tics. These differences can of course make the
interpretation of our results more difficult.
In summary, our results show an excellent toler-
ability of ECT treatment as a monotherapy as well as
in combination with neuroleptic medication. The
study gives further hints that especially atypical
substances are beneficial with regard to the clinical
effectiveness of ECT treatments. The retrospective
design of the study is of course of a certain
limitation. Our study provides data that demand
further controlled analyses using double-blind and
placebo-controlled conditions in order to prove
whether concomitant neuroleptic medication should
be recommended in ECT treatment to enhance
responder rates and shorten the time until clinical
improvement, and cause as few adverse effects as
possible. Furthermore it could be of interest to
further analyse the influence of individual neurolep-
tics on ECT treatment parameters, for there are
hints that there are differences, especially in the
group of atypical substances. It has been found that
clozapine and olanzapine are epileptogenic, whereas
quetiapine seems to reduce seizure activity (Amann
et al. 2003; Gazdag et al. 2004). In this way,
recommendations for the choice of concomitant
neuroleptic medication during an ECT treatment
might be made even easier.
Acknowledgements
The authors would like to thank Mrs M. Ertl, Mrs S.
Rauch and Mr K. Neuner for patient nursing, ECT
organisation and database management. Parts of this
study were done in the framework of the doctoral
thesis of Mrs Ines Noack which has been submitted
to the Faculty of Medicine, University of Munich.
Statement of interest
Each author certifies that he or she has no commer-
cial associations that might pose a conflict of interest
in connection with the submitted article.
Electroconvulsive therapy and concomitant neuroleptic medication 169

References
Abrams R. 1972. Recent clinical studies of ECT. Semin Psychia-
try 4:3�12.
Abrams R. 1986. A hypothesis to explain divergent findings
among studies comparing the efficacy of unilateral and bilateral
ECT in depression. Convuls Ther 2:253�257.
Abrams R. 2002. Stimulus titration and ECT dosing. J ECT
18:3�9.
Amann BL, Pogarell O, Mergl R, Juckel G, Grunze H, Mulert C,
Hegerl U. 2003. EEG abnormalities associated with antipsy-
chotics: a comparison of quetiapine, olanzapine, haloperidol
and healthy subjects. Hum Psychopharmacol 18:641�646.
Benkert O, Hippius H. 2003. Kompendium der Psychiatrsichen
Pharmakotherapie. Heidelberg: Springer.
Coffey CE, Lucke J, Weiner RD, Krystal AD, Aque M. 1995.
Seizure threshold in electroconvulsive therapy: I. Initial seizure
threshold. Biol Psychiatry 37:713�720.
d’Elia G. 1970. Unilateral electroconvulsive therapy. Acta Psy-
chiatr Scand Suppl 215:1�98.
d’Elia G, Raotma H. 1975. Is unilateral ECT less effective than
bilateral ECT? Br J Psychiatry 126:83�89.
ECT Review Group. 2003. Efficacy and safety of electroconvul-
sive therapy in depressive disorders: A systematic review and
meta-analysis. Lancet 361:799�808.
Fink M. 1990. Clozapine and electroconvulsive therapy. Arch
Gen Psychiatry 47:290�291.
Frey R, Heiden A, Scharfetter J, Schreinzer D, Blasbichler T,
Tauscher J, Felleiter P, Kasper S. 2001a. Inverse relation
between stimulus intensity and seizure duration: Implications
for ECT procedure. J ECT 17:102�108.
Frey R, Heiden A, Scharfetter J, Schreinzer D, Blasbichler T,
Tauscher J, Felleiter P, Kasper S. 2001b. Inverse relation
between stimulus intensity and seizure duration: Implications
for ECT procedure. J ECT 17:102�108.
Gazdag G, Barna I, Ivanyi Z, Tolna J. 2004. The impact of
neuroleptic medication on seizure threshold and duration in
electroconvulsive therapy. Ideggyogy Sz 57:385�390.
Kales HC, DeQuardo JR, Tandon R. 1999. Combined electro-
convulsive therapy and clozapine in treatment-resistant schizo-
phrenia. Prog Neuropsychopharmacol Biol Psychiatry 23:547�556.
Kupchik M, Spivak B, Mester R, Reznik I, Gonen N, Weizman A,
Kotler M. 2000. Combined electroconvulsive-clozapine ther-
apy. Clin Neuropharmacol 23:14�16.
Lauritzen L, Odgaard K, Clemmesen L, Lunde M, Ohrstrom J,
Black C, Bech P. 1996. Relapse prevention by means of
paroxetine in ECT-treated patients with major depression: A
comparison with imipramine and placebo in medium-term
continuation therapy. Acta Psychiatr Scand 94:241�251.
Meltzer HY, Bastani B, Kwon KY, Ramirez LF, Burnett S, Sharpe
J. 1989. A prospective study of clozapine in treatment-resistant
schizophrenic patients. I. Preliminary report. Psychopharma-
cology (Berlin) (Suppl) 99:S68�72.
National Institute of Mental Health. 1976. 028 CGI. Clinical
global impressions. In: Guy W, Bonato RR, editors. Manual for
the EDCEU Assessment Battery, 2nd rev. Chevy Chase, MD:
National Institute of Mental Health. p 12-1�12-6.
Nobler MS, Sackeim HA, Solomou M, Luber B, Devanand DP,
Prudic J. 1993. EEG manifestations during ECT: Effects of
electrode placement and stimulus intensity. Biol Psychiatry
34:321�330.
O’Brien PD, Berrios GE. 1993. Concurrent psychotropic medica-
tion has no negative influence on the outcome of electrocon-
vulsive therapy. Encephale 19:347�349.
Sackeim HA, Devanand DP, Prudic J. 1991. Stimulus intensity,
seizure threshold, and seizure duration: impact on the efficacy
and safety of electroconvulsive therapy. Psychiatr Clin North
Am 14:803�843.
Sackeim HA, Prudic J, Devanand DP, Nobler MS, Lisanby SH,
Peyser S, Fitzsimons L, Moody BJ, Clark J. 2000. A prospec-
tive, randomized, double-blind comparison of bilateral and
right unilateral electroconvulsive therapy at different stimulus
intensities. Arch Gen Psychiatry 57:425�434.
Schafer I, Lambert M, Naber D. 2004. Atypical antipsychotics in
therapy refractory schizophrenia. Nervenarzt 75:79�91.
Suppes T, Webb A, Carmody T, Gordon E, Gutierrez-Esteinou R,
Hudson JI, Pope HG Jr. 1996. Is postictal electrical silence a
predictor of response to electroconvulsive therapy? J Affect
Disord 41:55�58.
Suzuki K, Awata S, Matsuoka H. 2004. One-year outcome after
response to ECT in middle-aged and elderly patients with
intractable catatonic schizophrenia. J ECT 20:99�106.
Swartz CM, Abrams R, Rasmussen K, Pavel J, Zorumski CF,
Srinivasaraghavan J. 1994. Computer automated versus vi-
sually determined electroencephalographic and electromyo-
graphic seizure duration. Convuls Ther 10:165�170.
Swartz CM, Larson G. 1986. Generalization of the effects of
unilateral and bilateral ECT. Am J Psychiatry 143:1040�1041.
Tang WK, Ungvari GS. 2002. Efficacy of electroconvulsive
therapy combined with antipsychotic medication in treat-
ment-resistant schizophrenia: A prospective, open trial. J
ECT 18:90�94.
Tang WK, Ungvari GS. 2003. Efficacy of electroconvulsive
therapy in treatment-resistant schizophrenia: A prospective
open trial. Prog Neuropsychopharmacol Biol Psychiatry
27:373�379.
Tharyan P, Adams CE. 2002. Electroconvulsive therapy for
schizophrenia. Cochrane Database Syst Rev CD000076.
Weiner RD, Coffey CE, Krystal AD. 1991. The monitoring and
management of electrically induced seizures. Psychiatr Clin
North Am 14:845�869.
Weiner RD, Weaver LA Jr, Sackeim HA. 1988. Reporting of
technical parameters in ECT publications: Recommendations
for authors. Convuls Ther 4:88�91.
World Health Organization. 1992. The ICD-10 Classification of
Mental and Behavioural Disorders: Clinical descriptions and
diagnostic guidelines. Geneva: World Health Organization.
170 C. Nothdurfter et al.

VIEWPOINT
Disasters and mental health: New challenges for the psychiatricprofession
JUAN J. LOPEZ-IBOR JR
Department of Psychiatry and Medical Psychology, Complutense University, Institute for Psychiatry and Mental Health,
San Carlos Clinical Hospital, Madrid, Spain
Articles published in the Viewpoint section of this Journal may not meet the strict editorial and scientific standards that are appliedto major articles in The World Journal of Biological Psychiatry. In addition, the viewpoints expressed in these articles do notnecessarily represent those of the Editors or the Editorial Board.
AbstractA disaster is the consequence of an extraordinary event that destroys goods, kills people, produces physical or psychologicalharm but, above all, which overcomes the adaptive possibilities of the social group. Disasters have strong politicalbackground and consequences. They shake the life of a community and raise questions about safety, social organization andthe meaning of life. Disasters confront psychiatrists with challenges far beyond regular clinical activities or researchstrategies. During early interventions after a disaster, psychiatrists often have to work out of their usual clinical premises, incontact with unfamiliar professionals (i.e. rescue personnel) and with individuals who should not be considered as ‘cases’,and therefore without keeping regular clinical records. In the latter stages they have to confront many factors which tend tocause the clinical consequences of those affected and who developed a psychiatric condition to be chronic. Reactions tostress occur in stages, each one characterised by a specific psychological mechanism. Symptoms include flashbacks,difficulties in remembering, avoidance of stimuli, blunting of responses, high arousal level and obsessive ruminations. Thestrong biological and psychosocial factors which are unchained after a disaster should be recognised and chanelled. Theexperience of psychiatry with the bio-psycho-social model can help to understand what disasters are, how some negativeaspects of them could be prevented, and how their consequences, both clinical as well as social, can be reduced.
Key words: Disaster, catastrophe, stress, posttraumatic stress disorder
Introduction
The psychological reactions to and psychopatholo-
gical consequences of disasters have not received,
until recent times, all the attention they deserve.
Only in 1948 were they recognized in the Interna-
tional Classification of Diseases (ICD-6; WHO
1948), and they did not appear until 1980 in the
Diagnostic and Statistical Manual of the American
Psychiatric Association (DSM-III; APA 1980).
Furthermore the descriptions and criteria are very
unsatisfactory in both classification systems which,
on the other hand, are very different from each other
(WHO 1992; APA 1996; Lopez-Ibor Jr 2002).
There are several reasons for this situation. The
first one is to consider that it is up to human nature
to be able to face up to all calamities, independently
from the fact that occasionally some individuals
could yield, due to a lack of personal strength. The
other one is to accept that the personality is stable
and does not change throughout lifetime, even after
experiencing extreme situations. The concentration
camps of Nazi Germany changed these views:
tolerance has limits, there are situations under which
any individual would succumb. Experiences in
communist concentration camps showed how the
combination of physical deprivation, isolation and
psychological humiliation shatter the most solid
defences. The study of concentration camp survivors
led Venzlaff (1958), and later von Baeyer et al.
(1964), to describe for the first time persistent
transformations of the personality.
Correspondence: J. J. Lopez-Ibor Jr, World Psychiatric Association, Nueva Zelanda, 28035 Madrid, Spain. Tel: �/34 91 330 3572.
Fax: �/34 91 316 2749. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(3): 171�182
(Received 23 August 2005; accepted 21 October 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500428735

A third reason for the relative little interest in this
subject, is that it has mainly been studied by military
psychiatrists, who are usually confronted with many
difficulties when trying to publish and share their
experiences with other colleagues, as they may well
be on opposing sides in a war or confrontation. This is
changing, and there is a greater collaboration between
the civil and the military worlds, and this allows the
transference of the great experience accumulated
during military conflicts to civil disasters.
What is a disaster
Research in disasters is very difficult, because it is
hard to integrate it into the rescue tasks which
have priority and because it usually recalls negative
reactions from the perspective of the victims. In-
vestigation is therefore opportunistic and after-the-
event (Kasl et al. 1981). Furthermore, the involve-
ment of psychiatrists in the rescue and care of
victims is usually late and often because of secondary
reasons, for instance, in the case of court litigation.
This also reduces the opportunities for research
(Lopez-Ibor et al. 1985). On the other hand,
reactions to the most different disasters are extra-
ordinarily similar. This allows to accumulate and
share experiences, create groups of experts and
prepare plans and intervention groups.
It is almost impossible to find an acceptable
definition by a majority of what a disaster is. Korver
(1987) collected, many years ago, forty different
definitions of disaster; many more that have ap-
peared since can be added. Some authors (Freud
1969; Winnik 1969; Furst et al. 1971; Crocq 1992,
1993; Benyacar 2002) consider that the notion of
what a traumatic event is, is inherent to the complex-
ity of human existence and therefore inaccessible to
a consensus. However, it is necessary to define what
a disaster is in order to face up to the risks derived
from them and their consequences. Quarantelli
(1998) states that if the experts do not reach an
agreement as to whether a disaster is a physical event
or a social construct, the field will have serious
intellectual problems and that the worries about
what a disaster is should not mean that we become
involved in a futile academic exercise. On the
contrary, agreement consists of delving into what
are the important and significant characteristics of
the phenomenon, of the conditions that lead to it
and to its consequences. But further definition is
needed in order to understand the phenomenon,
because any concrete disaster unchains the question
on its sense. Therefore, the conceptualisation of
what a disaster is, is necessary in order to cope and
to understand it.
Most dictionaries define disasters and cata-
strophes in similar ways, as events in which there is
a lot of harm and destruction. The word catastrophe
has a larger and deeper semantic scope than a
disaster.
Human losses, number of injured persons, mate-
rial and economic losses and the harm produced to
the environment are often considered in order to
define a disaster. For some the number of 25
deceased has to be exceeded (Dombrowsky 1998),
for others this figure has to be larger, more than 100
deceased and more than 100 injured or losses worth
more than $US 1 million (Sheehan and Hewitt
1969), and for others the limit is still higher: an event
leading to 500 deaths or $US 10 million in damages
(Tobin and Montz 1997). According to Wright
(1997), experience shows that when an event affects
more than 120 persons, except for cases of war, non-
routine interventions and the coordination between
different organisations are needed, something which
is already indicating other important characteristics
of a disaster.
To define a disaster based on the magnitude of the
damage caused has many inconveniences. First, it
may be difficult to evaluate the damages in a first
stage, or to assign them later on. Second, they are of
no use for comparative studies in different countries
or social situations and they are affected by inflation
(Dynes 1998). Third, disasters are very different.
Earthquakes of the same intensity are only a fright in
California nowadays but would have been a cata-
strophe before 1989, and would be a catastrophe in
many developing countries at present. There may
even exist disasters with zero harm. The best
example for this was the broadcasting in 1935 by
Orson Welles of The War of the Worlds (Holmsten and
Lubertozzi 2001). More than one million persons
showed intense panic reactions because of what they
believed to be a Martian invasion.
Disasters are often considered as elements of the
physical environment harmful to human beings and
caused by forces foreign to them (Burton and Kates
1964; Burton et al. 1993). It has also been assumed
that disasters are the consequence of not very
frequent and non-routine events. Disasters are
normally unforeseen and catch the population and
administration affected off-guard. However, there
are disasters which repeat themselves, for example in
areas affected by flooding, and others which are
persistent, as in many forms of terrorism. In these
cases, a culture of adaptation and resignation to
disasters develops.
Disasters are normally considered as ‘chance’
events and are therefore unavoidable. In the past
they were ascribed to a divine punishment, and even
nowadays it is not strange to read that an event
172 J. J. Lopez-Ibor Jr

‘reached Biblical proportions’, or that nature’s
powers have been unchained as they were when
God had to punish evilness of human beings with the
Flood. In fact, the etymology of disaster, from Latin
desastrum (des ‘lack, loss’, astrum ‘heavenly body,
star’), indicates bad luck or fortune. The word
started to be used in a generalised way in France,
from where it extended to other countries as a
consequence of the secularisation brought about by
the Enlightenment.
An important characteristic of disasters is their
centrality (Green, 1982). A disaster is peripheral
when the persons affected were in the place by
chance (for example, in a plane crash) and when the
victims come back to their intact social surrounding
where they can rely on the support of relatives.
Catastrophes are disasters of a great magnitude, of a
great centrality. A total breakdown of everyday
functioning takes place in them, with the disappear-
ance of normal social functioning, loss of immediate
leaderships, insufficient health and emergency sys-
tems, in such a way that survivors do not know
where to go to receive help.
Human-made disasters are normally distinguished
from those consequences of natural disasters.
Among the former, some are not intentional, that
is to say they are a consequence of human errors. In
this case, the responsibility is considered to be
institutional, and compensations from insurance
companies are granted. Possible calculated risks
should be guaranteed based on their probability.
There are also disasters which are a consequence
of a clear intention, e.g., as conventional war would
be. In these cases, individuals are able to start up
more or less legitimate or efficient coping or defence
mechanisms to confront the aggression.
In other occurrences, violence is due to terrorist
attacks, assaults by rapists or similar events. This is
an anonymous violence the goal of which is to cause
harm to anybody, something that prevents victims
from developing any kind of defence. This kind of
violence may affect any person, in any place of the
world, at any time.
However, it is not possible to accept that there are
purely natural disasters, since the human hand is
always present. This is Steinberg’s thesis (2000); he
has studied a large series of disasters in the USA. It
has to be taken into account that the degree of
development of a community is a determinant fact.
Between 1960 and 1987, 41 out of the 109 worst
natural disasters took place in developing countries,
in which 758,850 persons died, while the remaining
59% of disasters took place in developed countries,
in which 11,441 persons died (Benz, 1989). In
another series studied between 1990 and 1998,
94% of the 568 greater disasters occurred in poor
countries causing 97% of deaths related to them. It
is curious enough that these proportions are similar
to the ones caused by famine, HIV or by refugees
(Easterly 2001).
Definitions of disasters based on the idea of an
exceptional agent are not fully satisfying. In fact,
when reviewing them other elements already appear
which are related to social conditions. The flooding
of an uninhabitated non-cultivated plain with no
ecological value is not a disaster, human involvement
is needed.
Therefore the impact of a danger on a social group
is related to the mechanisms and adaptive capability
the community has developed to face up to the effects
of potential destructive events. If they are efficient,
we can speak of an emergency, not of a disaster.
Disasters have been defined from this perspective as
external attacks that break social systems (Burton
and Kates 1964), which exert a disruptive effect on
the social structure (Benyacar 2002). The social,
political and economic environment is as determi-
nant as the natural environment, it is what makes
dangers turn into disasters (Blaikie et al. 1994), and
the disruption may create more difficulties than its
physical consequences (Quarantelli 1988).
The United Nations Coordinating Committee for
Disasters (UNDRUCO 1984) stipulates that a
disaster, seen from a sociological point of view, is
an event located in time and space, producing
conditions under which the continuity of the struc-
tures and of the social processes turns problematic,
and The American College of Emergency Medicine
(1976) points out that a disaster is a massive and
speedy disproportion between hostile elements of
any kind and the available survival resources needed
in order to re-balance the situation in the shortest
period of time possible (Dynes et al. 1987). This is
similar to that appearing in a definition by the WHO
(1991).
Crocq et al. (1987) point out the importance of
the loss of social organisation after a disaster. For
them the most constant characteristic is the altera-
tion of social systems that secure the harmonious
functioning of a society (information systems, circu-
lation of persons and goods, energy production and
consumption, food and water distribution, health
care, public order and security keeping, as well as
everything related to corpses and funerary ceremo-
nies in cemeteries).
In summary, disasters are events as a consequence
of a danger that affect a social group and which
produce such material and human losses that the
resources of the community becomes overrun and,
therefore, the usual social mechanisms for coping
with emergencies are insufficient.
Disasters and mental health 173

As a consequence, it has to be pointed out that a
disaster is something exceptional, not only because
of its magnitude: it is not enough to mobilise more
material and staff, these events go beyond the
jurisdiction of organisations and institutions, unfa-
miliar tasks have to be carried out, changes in the
organisations of the institutions in charge of respond-
ing to the disaster are enforced, new organisations
appear and persons and institutions which normally
do not respond to emergencies, are mobilised.
Several things are needed in order to produce a
catastrophe: an extraordinary event able to destroy
material goods, to cause the death of persons or to
produce injuries and suffering (Cohen 1999), or an
event after which a community lacks any adequate
social guidelines to react (Anderson 1968). This
leads to the need for intervention and of external
support, to a personal sensation of helplessness, to
feeling threatened, to tensions between social sys-
tems and individuals (Schulberg 1974), as well as to
a deterioration of the links that unite the population
and of the prevalence of the sense of pertaining to
the community (Erikson et al. 1976).
Disasters not only affect social functioning, they
are also the consequence of a certain social vulner-
ability up to then hardly perceived. They reveal
previous failures. Vulnerability decreases with the
degree of development of civilizations, which in
essence precisely aims to protect human beings
from the negative consequences of their behaviour
and from the forces unchained by nature (Gilbert
1998).
This social vulnerability is present even in the
clinical reactions to disasters. Among the risk factors
for post-traumatic stress disorder, more often iden-
tified in the USA, are the following: female sex, to be
Hispanic (Ruef et al. 2000), personal and familiar
history of psychiatric disorders, experiences with
previous traumas, especially during childhood, social
instability, low intelligence, neurotic traits, low self-
esteem, negative beliefs of oneself and the world and
an external locus of control (van Zelst et al. 2003).
Curiously enough there is a preventing factor, and
that is political activism.
Disasters are political events. Several authors have
dealt with this important aspect of a disaster. If
politics is an allocation of values, the link between
politics and disasters is determined by the allocation
of values on the authorities regarding security in the
period previous to the event, the survival possibilities
during the emergency stage and the opportunities to
survive during recovery and reconstruction (Olson
2000).
A disaster is also a political opportunity to develop
innovative initiatives, essential to diminish the present
and future consequences of the danger. A thorough
statistical study (Drury et al. 1998) on the relation-
ship between the severity of a disaster and political
instability showed that the repercussion is restrained
by the repression exercised by an authoritarian regime
and by a high level of development, but not because
of inequality of income (this last fact against the
hypothesis of the authors).
There is also a political use of disasters analysed
by Edelman (1977). Governments usually behave in
different ways when confronted with a problem and
with a crisis. In the case of a problem, they try to
cause a systematic deflation of the attention on the
inequality of the goods and services offered to the
population. On the contrary, in the case of a crisis,
they try a systematic inflation of threats, allowing
them to legitimate and demand an increase of
authority. When crises repeat themselves, authori-
tarianism increases; this is not restrained by the
presence of problems which are being dampened. In
this way, governments maintain their liturgies on an
increase of authoritarian power.
The political management of a crisis is based on a
political and organisational symbology and opens up
new opportunities for those responsible for new
initiatives and for other players who will achieve
visibility and prominence. In this way, the directive
elite exploits the resource of symbology in order to
influence the collective conceptualisation of the
situation and to enhance the chosen actions. Con-
crete strategies are started like, for example, the
framing of the crisis in a determined context, the
ritualisation maintained by collective action and the
masking of possible alternative conceptualisations
(Edelman 1977). It has to be pointed out that
Edelman’s remarks can be applied to any kind of
crisis, for example, in the management of corpora-
tions.
Disasters are a great opportunity to appoint
scapegoats, efforts to burden the guilt on a person
or a group are constant. But scapegoating is not a
means of finding and assigning responsibility. It is a
means of avoiding finding and assigning true re-
sponsibility. Whenever one finds the scapegoat
mentality at work, responsibility has been abrogated,
not shouldered (Allinson, 1993). Therefore, the
thesis of something accidental or unavoidable cannot
be accepted, because in the long run it turns into a
prophecy that fulfils itself and prevents us from
concentrating on the real causes.
Reactions to disasters
Stages in the reactions to disasters
One aspect of stress has not been considered in
depth in ICD-10 (WHO 1992) or in DSM-IV (APA
174 J. J. Lopez-Ibor Jr

1996), namely that reactions to stress develop in
phases. There are various descriptions of the phases
of a disaster, including a different number of them,
generally between three and seven. However, it has
to be pointed out that all of them are characterised
by a great homogeneity of the factors associated to
each one of them, and that they cannot be con-
sidered as rigid periods in time since their display has
a great diversity in different disasters. That is to say,
the important thing is to know that the mechanisms
and the psychological and psychopathological con-
sequences vary along time and that therefore they
have to be anticipated, although in each moment
various given reactions may appear with different
cadences.
Glass (1959) has described them, adding for the
first time the dominant psychological mechanism in
each one of them. His description is based on the
bombings on the civil population during Second
World War, but it may apply to all disasters.
The first phase, pre-impact period, is previous
to the event itself. Denial mechanisms are dominant
in this stage: the bomb will not fall here , Hitler will not
invade Poland , the mountain of accumulated slag will
not fall , the dam will resist , and so on. This phase is
important since it facilitates the disaster and mainly
because the reactions to it include not only the event
itself, but also the consequences which made it
possible.
The second phase is the one of alarm period. In
it an inefficient hyperactivity takes place. It is a phase
of panic, of heroic and altruistic behaviour, but is not
always efficient.
The third phase described by Glass is the one of
the impact period, this refers to the explosion of
the bomb. During this stage there is no psychological
reaction due to its shortness. This stage is not
present in other types of disasters and may well be
dismissed.
Following comes a phase of recoil period, in
which the mechanism put into action before the start
begins to make its effect. Combat exhaustion,
hypoactivity, apathy and disappointment appear. In
military psychiatry it is known as combat exhaustion
or the ‘syndrome of the old sergeant’.
The fifth and last phase described by Glass is the
post-impact period. In this phase feelings of rage
and hostility may appear directed towards those
possibly responsible, but also against the society in
which the disaster took place and against its leaders.
All disasters leave their print in the political life of a
country.
However, there is another phase to which Glass
did not pay attention, maybe because he wrote about
the phases very soon after the events during the
Second World War. It is a phase of reconciliation
(Lopez-Ibor et al. 1986, 1987) in which the social
group again comes to terms with itself, buries its
dead and its ghosts, gives a new meaning to the lives
of its individuals, the ones who died and the ones
who survived, be they injured or not. This stage is
not always reached, or it is not reached in a radical
enough way. The monuments for those fallen in war,
or to battles, try to be this.
Levels in the reactions to disasters
Reactions to disasters develop in three levels: biolo-
gical, psychological and social. A challenge for
research is to find out insomuch how they develop
in parallel and how each level influences the others.
Biological level . Stress and stressing agent are the
same. Stress is the reaction that appears when the
individual is threatened by an environmental factor
or factors, which are the stressing agents or stressors
(physical, chemical, psychological or social), that
disturb or threaten to disturb a state of internal
balance (homeostasis). They are a group of unspe-
cific responses that are set off before having been
able to identify the specific threat. Their purpose is
to prepare the individual for action: for fight or
flight.
In a first stage, an activation of the hypothalamus�hypophysis�suprarenal axis, with a rapid and brief
CRF-ACTH glucocorticoid secretion increase, takes
place. Later, a sustained response characterises
chronic stress: a more general activation of the
central nervous system takes place, specially of the
neurons of the paraventricular nucleus and an
increase of CRF and VP giving way to a a-adrenergic
and 5-HT1a, 5-HT, nicotinic, cholinergic, interleu-
kin 1, angiotensin II, TRF, neuropeptide Y and
vasopressin stimulation (Dıaz-Marsa et al., 2000).
The consequence of all this is an increase in the
activity of several neurotransmitters, among others
dopamine in the limbic system, in the cortex, in the
hypophysis and in the cerebellum, and hormones
like epinephrine, with an increase of blood pressure,
pulse rate, fat catabolism and the metabolism of
carbohydrates. Also, corticoids and various physio-
logical responses increase, insulin and growth hor-
mone secretions and also immunological responses
decrease, causing among other things, stress ulcers
(Yehuda 1997).
Hormonal secretion has a characteristic temporal
pattern. Immediately after the impact of the stres-
sing factor, in a few minutes GH secretion falls and
secretion of b-endorphins, ACTH and LH show a
significant increase, this is also the case for TSH but
to a lesser degree. This last recedes in minutes, but
LH takes some hours to decrease. LH secretion
Disasters and mental health 175

recovers its basal level after 4�5 hours, although
afterwards it may remain under basal levels for
longer. ACTH levels are maintained over basal levels
for 12 hours or more, with b-endorphins for up to
24 hours.
In recent years, research has been centred on the
biological processes unleashed after exposure to
disruptive situations (Bremner et al. 1995; Yehuda
2000). Bremner et al. have studied in a systematic
way the possible brain injuries as a consequence of
stress and McEwen (1999) studied the dysfunctions
and plasticity of the hippocampus (Bremner 1999;
McEwen 1999; Gould et al. 1999). Research on the
functional specificity of the amygdala and its trophic
mechanisms in disruptive situations that produce a
dysfunction of emotional memory has to be added
(Roozendal et al. 1997; Post et al. 1998; Cahil and
McGaugh 1998).
Neuroimaging studies have provided some impor-
tant data. First, a reduction of the volume of the
hippocampus in persons who have been exposed to
stressful situations, for example US army veterans
(Hedges et al. 2003) or women with a history of
sexual abuse during childhood (Bremner et al.
2003). In a PET the blood flow is generally
increased, but specially in the cerebellum and in
the precentral, superior temporal and right fusiform
circumvolutions. The cerebellosum and extra-stria-
tal flow positively correlate with depression and post-
traumatic scales (Bonne et al. 2003).
Psychological level. Bakan (1968) has underlined the
strong parallelism between Freud’s ideas (Freud
1926) and the concept of reaction to stress described
by Selye. It is curious that Freud’s death instinct
came from the observation of dreams of traumatic
events, which could not be explained by the libido
and the desiderative character of dreams. He came
to the notion of a non-libidinous, autodestructive
principle: death instinct, thanatos. The individual
psychological response of human beings to external
aggressions or threats (real or imagined) is anxiety
that, at the same time, unchains various defence or
coping mechanisms. In some circumstances, defence
mechanisms anticipate themselves in order to face
up to threats still not identified that may result in
harm for the individual’s organism and, in the end,
may even entail a risk for survival. The notion of
autodestructive mechanisms in individuals was men-
tioned by Freud in his description of the death
instinct or thanatos and is present in the concept
of adaptation illnesses.
In this context it’s worth remembering Sartre’s
(1939) concept on emotion: its a substitute beha-
viour in which the affected person, in front of an
irrational unbearable senseless situation, assumes a
metaphoric relationship with the world allowing
him/her to continue living. For example, sadness is
the possibility to survive the loss of a beloved person,
shutting oneself in on oneself, becoming isolated
from a world lacking any sense after the loss of the
beloved person. Moreover, all emotions have their
vegetative correlate. They are the serious face of
emotion.
Emotion ends when the moment and the possibi-
lity is given to elaborate the trauma and to give it a
significance, that is to say, when it is rationalised
(Lopez-Ibor et al. 1999). Rationalisation implies a
verbalisation.
A series of particular behaviours maintains the
validity of the trauma, like the avoidance of those
things that may remind of it, suppression of thoughts
and remembrances, ruminations, a behavioural pat-
tern of avoidance the individual considers as ‘safe’,
dissociation mechanisms and alcohol and drug
abuse (Ehlers and Clark 2000).
Many psychological theories, not incompatible
among them, have been postulated. For Foa
(1986, 1997), there is an association of banal stimuli
with fear. For Charney et al. (1993), there are two
different conditionings of fear, one related to envir-
onmental stimuli that cause discomfort and another
one which is an operating and instrumental deter-
minant that gives way to avoidance. For Brewin et al.
(1999), trauma has a double representation in
memory, one is accessible to verbalisation and the
other one to the situation.
In any case, after a traumatic event, a transforma-
tion of the vision of the world, of oneself and of the
future, takes place. Memory becomes dissociated
from its context (Ehlers and Clark 2000), requiring a
re-adaptation process to reality consisting of a re-
elaboration of the trauma (Horowitz 1993). On the
other hand, new beliefs appear and other false and
old ones are surmounted (Janoff-Bulman 1992) like,
for example, ‘the world is a safe place’ or ‘the worst
always happens to me’ or ‘it never rains but it pours’.
In any case, it occurs as the appearance of a new
emotional conscience which, as any emotional state,
is always accompanied by a vegetative correlate.
Social level . This bio-psychological model has to be
expanded to become bio-psycho-social, including
reactions to collective stresses, like, for example, in
disasters and catastrophes. Here, the overwhelming
external threat has a much lesser role for individual
vulnerability than for neurotic disorders. What
happens in the initial stages has consequences on
the more advanced ones. The impact of an event for
which an individual is not prepared (no protection or
immunity is available, or negation has prevented
preparation for action) unchains an exaggerated
176 J. J. Lopez-Ibor Jr

response, smoothing the way for consecutive re-
sponses: strain and chronic states.
In the aetiopathology of these manifestations,
group factors and the psychopathology of the masses
have to be taken into account. The nature and
characteristics of group membership and factors like
lack of security, isolation, intra-group conflicts, role
distribution during immediate reactions and the
elaboration of sequelae are more important. Also,
imitation and identification mechanisms have an
influence.
Among the factors of macrosocial predisposition
to be pointed out are the low cohesion of the group,
which functions as a mass (a mass is a totally
unstructured group lacking any cohesion), and the
lack of working experience are common. Therefore
those groups recently created or re-created are very
vulnerable to panic. The qualitative�quantitative
failing of the organisational frame is another factor
to be taken into account. There are groups with very
vulnerable precarious levels characterised by ex-
treme nationalistic ideas and collective defence
values, and there are there are intrinsic fragile groups
like children, frustrated individuals, and women, in
general. Benyacar (2002) has also pointed out
personal vulnerability with changes in recent biolo-
gical events such as marriage, divorce, death of
partner, home moving or changes in job conditions.
Integrative model . The reaction stages and levels
subsequent to a disaster are integrated and sum-
marised in Table I (Lopez-Ibor et al., 1985).
Clinical manifestations of reactions to stress
In order to better understand what a disaster is, it is
necessary to briefly think about the characteristics of
the clinical manifestations of the reactions which
may evidence the following:
1. Affective and emotional symptoms, like anxiety,
phobias, depression, irritability, apathy and
withdrawal. All of them unspecific symptoms
appearing in many other disorders. [Indican una
mala respuesta ante el estres .]
ICD-10 follows the model Kretschmer
(1948) proposed for hysteria, in which two
very primitive defence mechanisms are present,
the tempest of movements, for example hyster-
ical movements, which is what chickens do
when they flutter around when feeling threa-
tened in the farmyard. The second defence
mechanism is the dead reflex, present in
hysterical paralysis, which is a mimicry of
threatened animals as when an ostrich buries
its head in the sand. Lopez Ibor Sr. (1950)
extended this description to all neurotic phe-
nomena in general and called them ‘sobresalto ’
and ‘sobrecogimiento ’ (lit. startle and terror
reactions).
2. Physiological disorders, among them to be
pointed out, a constant vegetative hyperactiva-
tion evoked by the memory of the trauma or by
other traumas.
3. Sleep disorders and dreaming, specially insom-
nia and untimely awakening, mainly caused by
daydreaming and nightmares of the traumatic
event.
4. Memory disorders, consisting of two apparently
different things. On the one hand, a voluntary
evocation of the memories of the trauma is not
possible, and when it is possible to do so the
memories are confused, sketchy, unorganised
and scarcely elaborated. On the other hand,
memories burst in at untimely moments during
consciousness, generally like flash-backs, with
very lively and intrusive images, sometimes
unchained by clues that bring the event to the
memory. In other words, the memory is dis-
sociated from its context (Ehlers and Clark
2002).
It has to be taken into account that many
theories on the function of sleep and day-
dreaming coincide in importance of including
Table I. The reaction stages and levels in front of a disaster.
Biological level general
adaptation syndrome
Psychological level post-traumatic
stress disorder Social level disasters
1. Pretrauma Negative or
insufficient defenses
(congenital or acquired)
Lack of immunological
barriers
Non-adaptative defence
mechanisms, pathological life
styles, poor self-identification
Poor planification,
denial, lack of identity
and social stability
2. Acute response HHA Axis Ergotrophic
Trofotrophic
Alert period (anxiety, fright).
Withdrawal period (anguish, fright)
Alert period (hyperactivity)
Withdrawal period (apathy)
3. Chronic maladaptation Adaptative illnesses
(immunological)
Post-impact period (rage,
resentment). Illness as a form
and way of survival)
Institutionalisation
4. Recovery New immunological
mechanisms
To live with the losses Sense
for existence is recovered
Reconciliation
Disasters and mental health 177

the stimuli previous to the sleep. Sleep is an
active process of memory consolidation, a
cognitive process for the re-elaboration of
experiences. The main function of REM sleep
is to forget everything unnecessary, and there-
fore daydreaming becomes the organised con-
science and interprets the information the
individual has available (Crick and Mitchinson
1983). Therefore, a non-elaborated trauma
erupts time and again during daydreaming
and also during wakeful consciousness because
it has not been elaborated, that is to say, it has
added the forgotten heritage from our past.
This interpretation coincides with some psy-
chological theories that refer to a double
representation of the trauma in the memory,
one accessible to verbalisation and another one
to the situation (Brewin 1999). Others impinge
on the two conditionings of fear, one a con-
ditionant of environmental stimuli that un-
chains discomfort, and another an operative
or instrumental one, responsible for avoidance
(Charney 1993).
5. Behaviours that reinforce and maintain the
trauma in the conscience, generally with beha-
viours of avoidance which prevent from over-
coming the object of phobia related to the
trauma. The avoidance to remember, the
suppression of the thought and memories, the
grumbling, ‘safe’ behaviours, dissociation and
alcohol or drug consumption do not help to
overcome the trauma, but give place to an
increase in the frequency of the symptoms since
the suppression creates an increase of intrusive
thoughts and prevents any positive changes in
behaviour.
6. Repercussions on the basic beliefs of oneself
and the world. The idle effort to find a sense to
this experience and the fact of worrying once
and again about the trauma accompanied by
feelings of guilt (because of having survived the
beloved ones) and of shame. This factor leads,
on the one hand, to seek external comfort and,
on the other hand, to reject it due to the shame
produced by the inability to cope with the
consequences, giving way to feelings like rage
and hostility towards the community, consider-
ing them even responsible for the catastrophe
and its consequences.
Do disasters have a meaning?
Up to now, I have been using the words disaster and
catastrophe as synonymous, although pointing out
some differences. One of them is the magnitude of
the impact on society. In fact, in many languages a
catastrophe is a disaster of great proportions.
The word catastrophe comes from the Greek
katastrvf/h (Katastrophe), from kata (kata) ‘under-
neath’ �stofoin (strephein) ‘to turn around’ and
etymologically refers to the movement of the chorus
on stage. Its primitive significance is the outcome,
especially when it is a very dramatic one, of a poem or
a theatre play.
A disaster is not bad luck, it is an empirical
falsification of human action, the proof of the
incorrectness of human beings’ conception of nature
and culture (Dombrowsky 1998). It is also a state of
lack of certainty, of inability to spot real or supposed
dangers, especially when it is something impreg-
nated in mental schemes to be able to understand
the dominant reality in a community (Gilbert 1958).
A disaster not only affects structures and social
functioning, also many mental schemes break down.
All of a sudden death anxiety and the loss of sense of
invulnerability becomes obvious (Lifton 1969).
Frankel (1962), who survived a Nazi concentration
camp, and Brull (1969) and others, have pointed out
that, after such an experience, sense of the context of
existential experience disappears. The vision of the
world, of oneself, of the future, changes. Therefore,
during the phase of overcoming the trauma a process
of re-adaptation to reality, a re-elaboration of the
trauma (Horowitz, 1993), the establishment of new
beliefs, and the overcoming of other old and false
beliefs, like the ‘world is a safe place’ and those
negative ones established after a disaster like ‘all the
worst always happens to me’, ‘it never rains but it
pours’, and so on (Janoff-Bulman 1992), is needed.
Victims or damaged?
The worst thing that can happen is the victimisation
of those affected and here psychiatry can play an
important role (Benyacar 2002).
The victim is a person who remains trapped by
the situation, petrified in that position, and passes
from being an individual to become an object of the
social, losing in this way his/her subjectivity. It is a
biblical concept, inherent to the expiatory needs of
society. Since the moment of the disruptive event,
society’s needs for restoration intermingle with the
needs of the individual. The demand that the outer
world repairs the harm he/she has suffered arises.
The damnified is the person who has suffered a
damage, prone to be repaired or irreparable in its
whole or partly. The concept ‘damnified’ connotes
psychic mobility, as well as the preserving of the
individual’s subjectivity. Therefore, mental health
services have to assist all those affected not as victims
but as damnified.
178 J. J. Lopez-Ibor Jr

Compensations in disasters
Reactions to disasters and their definition have been
always been marked by compensation. Literature on
compensation neurosis is old (Kinzie et al. 1996). In
fact, definitions that emphasise the presence of a
stressing agent of great magnitude which would
affect almost any persons, as DSM-III (APA 1980)
did, turn even witnesses into victims. Since a disaster
destroys social frames and is a consequence of it, it is
natural that any individual turns to society to ask
that the harm suffered be repaired. This is why there
is a tendency for victims to maximise ‘secondary
benefits’, perpetuating the psychic harm in order to
receive compensation, be it economic, affective or of
any other kind. This is reinforced by the fact that
psychic harm affects persons who functioned within
parameters of normality before the disaster and if
afterwards they cannot do so it is because an external
factor, of a social nature, has caused them harm.
According to Cohen (1999), most persons and their
relatives affected by a disaster have functioned in an
adequate way before the tragic event, but their
ability to solve problems becomes limited because
of the threat inherent to the situation.
Compensations in disasters are indispensable and
have to include psychic harms. However, the reper-
cussion on the mental health of the damnified must
also be evaluated, if he/she turns into a victim the
risk exists to make out of the disaster and the
compensation his/her new life style. It is also true
that mental health professionals are there to avoid
iatrogenia and should help the victims to overcome
this situation so as to avoid the situation where the
disability becomes chronic. It is also true that society
can impose its limits subsequent to any possible
victimisation abuses.
Mental health professionals should participate in
the judging of indemnifications and in the decision
to include victims in reintegration programmes to
their everyday activities (Benyacar 2002).
Therapeutic interventions
From all what has been exposed before, it can be
said that an intervention in disorders secondary to
severe stress has to be multifaceted, embracing all
social, psychological and biological aspects.
Strategy
A mental health programme has to start with the
following (Benyakar et al. 1994, 2002; Cohen 1999):
� Evaluation of the needs.
� Definition of intervention objectives.
� Take into consideration other possible options.
� Design of a program.
� To put into practice and carry out a care-giving
project in mental health.
Intervention has to be quick, as immediate as
possible, following the strategy of preventing the
consolidation of hard-to-treat clinical conditions,
which could even lead to an irreversible transforma-
tion of the personality.
Intervention has to be integrated (Collazo 1985;
Lebigot 1998), following the model of liaison
psychiatry (Soria et al. 1983a,b). It has to be carried
out as near as possible to the site where the events
took place (Cohen and Ahearn 1989) and include
individual and collective reactions (Crocq et al.
1987; Andreoli 2000).
One of the characteristics of this kind of situation
is the inability of the victim to ask for help, and
therefore an in-person intervention is needed in
which the mental health professional tries to directly
contact the victims in order to avoid the develop-
ment of psychic disorders that could be unchained
by lack of possibility or ability to cope with the event
in an adequate way.
Benyacar (2002) recommends the three basic
elements proposed during the First World War
(Salmon 1919): immediacy, proximity and expec-
tancy.
Immediacy refers to the fact that the victims
should be assisted immediately after the event.
Proximity refers to the fact that the victim should
be assisted as near as possible to the place where the
event took place. The principle of expectancy main-
tains that the professional and the colleagues of the
victim have to keep expectancy and express the will
that the victim goes back to his/her usual activities
developed before the event, as soon as possible.
Mental health professionals have to be integrated
into the disaster intervention staff in all its different
levels, the immediate ones, as well as subsequent
ones (Collazo 1983, 1985; Lebigot 1999).
Care should be provided to the population as a
whole and not only those who have suffered physical
harm. It should include close relatives directly
involved or those not so close, passers-by and
witnesses, emergency staff, and also mental health
professionals and others, including social leaders. It
has to be taken into account that for each physically
affected victim there will be three persons who will
suffer consequences in their mental health (Benya-
kar 1997), and for each death or severely injured
person there will be up to 400 psychically affected.
Extremely dramatic news, rumours, information
‘wars’ and collective panics have to be avoided, and
it has to be tried to provide adequate information.
Disasters and mental health 179

Panic situations have occurred without the slightest
trace of harm or threat.
Training, preparedness and anticipation are es-
sential factors in order to prevent a disaster and its
consequences.
Psychological interventions
In this kind of intervention, the professional acts as a
mediator so that the victim is able to articulate in his/
her psyche all what has happened using his/her own
idiosyncratic psychic abilities.
Cohern and Ahearn (1989) point out that an
important goal for mental health intervention is the
adequate use of techniques for restoring the ability of
the victims to solve the stressful situation in which
they are in, and to help them to reorganise their world
through social interaction. A second goal is contin-
uous and active collaboration with other groups or
organisations that offer help, support and attention
to victims in particular and to the community in
general.
These authors add that clear therapeutic inter-
vention guidelines have to be taken into account.
Among them:
1. Risk factors:
� the patient’s mature or immature person-
ality;
� stress related to social behavioural func-
tions or expectancies, according to what the
victims themselves judge, and those of
others living with them;
� persisting environmental stress in the phy-
sical as well as in the social surroundings;
� crisis the victims may have experienced
before or after the disaster.
2. Social environment: the social environment in
which the victims are resettled is an important
variable that affects decisions about the type of
psychological intervention. The specific inter-
vention type for each environment has to be
clarified.
3. Medical and clinical resources. The diagnosis
of disorders that put life in a considerable risk,
require the intervention of medical staff and
have to be part of the evaluation process.
Essential intervention aspects are verbalisation, in-
terviewing and social support strategies. Group
therapies are also essential, as well as the incorpora-
tion of the affected persons in rescue activities, as
long as this is feasible.
The formulation of a differential diagnosis is
important, also the identification of severe cases
and those which cannot be treated on site and have
to be evacuated to rearguard positions or even to
psychiatric units.
‘Psychiatrisation’ of cases should be avoided, as
well as the problem of associating the stigma of
mental illnesses to the existing harm.
Individual psychological resources have to be
favoured and mobilised, trying to avoid victimisation
and an indiscriminate and exaggerated compensa-
tion system. An iatrogenic type compensation neu-
rosis should be avoided as much as possible.
Pharmacological treatments
Psychosocial meassures have to be accompanied by
pharmacological interventions (van der Kolk 1987;
Kandel 1999; Friedman 2002; Morgan et al. 2003)
aimed at diminishing non-adaptative phyisological
responses. Taking into account their role in the
treatment of anxiety, selective inhibitors of serotonin
reuptake are a good option.
Statement of interest
The author has no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
APA (American Psychiatric Association). 1980. Diagnostic and
statistical manual of mental disorders. 3rd ed. DSM III.
Washington, DC: APA. (Traduccion espanola. 1985. Manual
Diagnostico y Estadıstico de los Trastornos Mentales. 3a ed.
DSM-III. Barcelona: Masson.)
APA (American Psychiatric Association). 2000. Diagnostic and
statistical manual of mental disorders. 4th ed. Text revision,
DSM-IV-TR. Washington, DC: APA. (Traduccion espanola.
2002. Manual Diagnostico y Estadıstico de los Trastornos
Mentales. 4a ed. evision del texto. DSM-IV-TR. Barcelona:
Masson.)
Allinson RE. 1993. Global disasters: Inquiries into management
ethics. Prentice Hall: New York.
Anderson JW. 1968. Cultural adaptation to threatened disaster.
Hum Organizations 27:298�307.
Andreoli A. 2000. De la maladie traumatique aux interfaces du
traumatisme. Esquisse d’un modele de politique de traitement
et de soins. Rev Francophone Stress Trauma 1(1):33�44.
Baeyer W von, Hafner H, Kisker KP. 1964. Psychiatrie der
Verfolgten. Psychopathologische und gutachtliche Erfahrungen
an Opfern der Nationalsozialistischen verfolgung und vergle-
ichbarer Extrembelastungen. Berlin, Gottingen, Heidelberg:
Springer.
Bakan D. 1968. Disease, pain and sacrifice. Chicago, IL:
University of Chicago Press.
Benyakar M. 2002. Salud Mental en Situaciones de Desastres:
Nuevos Desafıos. Rev Neurol Neurocirug Psiquiatr Mejico
35(1):3�25.
Benz G. 1989. List of major natural disasters. 1960�1987.
Earthquake Volcanoes 20:226�228.
Blaikie P, Cannon T, Davis I, Wisner B. 1994. At risk: Natural
hazards, people’s vulnerability, and disasters. London and New
York: Routledge.
180 J. J. Lopez-Ibor Jr

Bonne O, Gilboa A, Louzoun Y, Brandes D, Yona I, Lester H,
Barkai G, Freedman N, Chisin R, Shalev AY. 2003. Resting
regional cerebral perfusion in recent posttraumatic stress
disorder. Biol Psychiatry 54(10):1077�1086.
Bremner JD, Vythilingam M, Vermetten E, Southwick SM,
McGlashan T, Nazeer A, Khan S, Vaccarino LV, Soufer R,
Garg PK, Ng CK, Staib LH, Duncan JS, Charney DS. 2003.
MRI and PET study of deficits in hippocampal structure and
function in women with childhood sexual abuse and posttrau-
matic stress disorder. Am J Psychiatry 160(5):924�932.
Bremner JD. 1999. Does stress damage the brain? Biol Psychiatry
45(7):797�805.
Bremner JD, Krystal JH, Southwick SM, Charney DS. 1995.
Functional neuroanatomical correlates of the effects of stress
on memory. J Trauma Stress 8(4):527�553.
Brewin CR, Andrews B, Rose S, Kirk M. 1999. Acute stress
disorder and posttraumatic stress disorder in victims of violent
crime. Am J Psychiatry 156(3):360�366.
Brull F. 1969. The trauma-theoretical considerations. Isr Am
Psychiatr Relat Discip 7(1):96�108.
Burton I, Kates RW. 1964. The perception of natural hazards in
resource management. Nat Resources J 3:412�441.
Burton I, Kates R, White G. 1993. The environment as hazard.
2nd ed. Guilford Press: New York.
Cahill L, McGaugh JL. 1998. Mechanisms of emotional arousal
and lasting declarative memory. Trends Neurosci 21(7):294�299.
Charney DS, Deutch AY, Krystal JH, Southwick SM, Davis M.
1993. Psychological mechanisms of posttraumatic stress dis-
order. Arch Gen Psychiatry 50(4):295�305.
Cohen R. 1999. Salud mental para vıctimas de desastres. Manual
para trabajadores. Organizacion Panamericana de la Salud,
Washington, DC: EUA.
Cohen RE, Ahearn FL Jr. 1989. Manual de la Atencion de Salud
Mental para Vıctimas de Desastres. Mexico: Harla.
Collazo C. 1985. Psychiatric casualties in Malvinas war: A
provisional report. In: Pichot P, Berner P, Wolf R, Thau K,
editors. Psychiatry: The state of the art Vol. 6. New York:
Plenum Press. pp 400�503.
Crick F, Mitchison G. 1983. The function of dream sleep. Nature
304(5922):111�114.
Crocq L. 1992. Panorama des sequelles des traumatisms psychi-
ques. Nervoses traumatiques, etats de stress post-traumatique
et autres sequelles. Psychol Med 24(5):427�432.
Crocq L. 1993. Le trauma et ses mythes. Psychol Med
25(10):992�999.
Crocq L, Doutheau C, Salham M. 1987. Les reactions emotion-
nelles dans les catastrophes. Encyclopedie Medico-Chirurgi-
cale. Paris: Editions Techniques.
Diaz-Marsa M, Molina R, Lozano MC, Carrasco JL. 2000. Bases
biologicas del trastorno por estres postraumatico. Actas Espa-
nolas Psiquiatrıa 28(6):379�384.
Dombrowsky Wolf R. 1998. Again and again � Is a disaster what
we call a disaster. In: Quarantelli EL, editor. What is a
‘disaster’. Chapter 3. London and New York: Routledge.
Drury AC, Olson RS. 1998. Disasters and political unrest: An
empirical investigation. J Contingencies Crisis Management
6:153�161.
Dynes RR, De Marchi B, Pelanda C. 1987. Sociology of disasters.
Milano: Franco Angelli.
Dynes RR. 1998. Coming to terms with community disaster
(Chapter 11). In: Quarantelli EL, editor. What is a disaster?.
London and New York: Routledge. pp 109�126.
Easterly W. 2001. The elusive quest for growth, economists’
adventures and misadventures in the tropics. Cambridge, MA:
MIT Press.
Edelman M. 1977. Political language: Words that succeed and
policies that fail. Boca Raton, FL: Academic Press.
Ehlers A, Clark DM. 2000. A cognitive model of posttraumatic
stress disorder. Behav Res Ther 38(4):319�345.
Erikson P, Drabek TE, Key WH, Crowe JL. 1976. Families in
disaster. Mass Emergencies 1:206�213.
Foa EB, Kozak MJ. 1986. Emotional processing of fear: Exposure
to corrective information. Psychol Bull 198 99(1):20�35.
Foa FB. 1997. Psychological processes related to recovery from a
trauma and an effective treatment for PTSD. Ann NY Acad Sci
21;821:410�424.
Frankel V. 1962. Man’s search of meaning. Boston, MA: Beacon
Press.
Freud, A. 1969. Foreword to Basic Psychoanalytic Concepts on
the theory of Dreams, Vol. II. Nagera H, editor. The
Hampstead Clinic Psychoanalitic Library, London, Maresfield
Reprints.
Freud S. 1922. Beyond the pleasure principle. The International
Psychoanalytical Library, No. 4. CJM Hubback (authorized
translation). 2nd edn. London: Hogarth Press.
Friedman MJ. 2002. Future pharmacotherapy for post-traumatic
stress disorder: Prevention and treatment. Psychiatr Clin North
Am 25(2):427�441.
Furst SS. 1978. The stimulus barrier and the pathogenicity of
trauma. Int J Psychoanal 59(2�3):345�352.
Gilbert JE. 1958. Human behaviour under conditions of disaster.
Med Serv J Can 14(5):318�324.
Glass AJ. 1959. Psychological aspects of disasters. J Am Med
Assoc 171:188�191.
Gould E, Tanapat P. 1999. Stress and hippocampal neurogenesis.
Biol Psychiatry 46(11):1472�1479.
Green BL. 1982. Assessing levels of psychological impairment
following disaster. J Nerv Ment Dis 170:544�552.
Hedges DW, Allen S, Tate DF, Thatcher GW, Miller MJ, Rice SA,
Cleavinger HB, Sood S, Bigler ED. 2003. Reduced hippocam-
pal volume in alcohol and substance naive Vietnam combat
veterans with posttraumatic stress disorder. Cogn Behav
Neurol 16(4):219�224.
Holmsten BY, Lubertozzi A. 2001. The complete war of the
worlds. Naperville, IL: Sourcebooks.
Horowitz MJ. 1993. Stress-response syndromes: A review of
posttraumatic stress and adjustment disorders. In: Wilson JP,
Raphael B, editors. International Handbook of Traumatic
Stress Syndromes. New York: Plenum Press.
Janoff-Bulman R. 1992. Shattered assumptions: Toward a new
psychology of trauma. New York: Free Press.
Kandel ER. 1999. Biology and the future of psychoanalysis: A
new intellectual framework for psychiatry revisited Am J
Psychiatry 156(4):505�524.
Kasl SV, Chisholm RF, Eskanazi B. 1981. The impact of the
accident at the Three Mile Island on the behavior and well-
being of nuclear workers. Part I: Perceptions and evaluations,
behavioral responses and work-related attitudes and feelings.
Part II: Job tension, psychophysiological symptoms and indices
of distress. Am J Psychiatry 71:472�495.
Kinzie JD, Goetz RR. 1996. A century of controversy surrounding
posttraumatic stress stress-spectrum syndromes: The impact
on DSM-III and DSM-IV. J Trauma Stress 9(2):159�179.
Korver AJH. 1987. What is disaster? Prehospital Disaster Med
2:152�153.
Kretschmer E. 1948. Hysterie, Reflex und Instinkt. Stuttgart:
Georg Thieme.
Lebigot F. 1998. The advantages of inmediate and postimmediate
care following psychic trauma. Presented at the 5th World
Congress of the International Association for Emergency
Psychiatry, Brussels, 15�17 October.
Disasters and mental health 181

Lifton RJ. 1979. The broken connection. New York: Simon &
Schuster.
Lopez Ibor JJ. 1950. La angustia vital. Madrid: Paz Montalvo.
Lopez-Ibor Alino JJ. 1986. Social reinsertation after catastrophes.
The toxic oil syndrome experience. Eur J Psychiatry 1(1):12�19.
Lopez-Ibor JJ. 2002. The classification of stress related disorders.
Psychopathology 35:107�111.
Lopez-Ibor Alino JJ, Jimenez Arriero MA. 1987. Psychosocial
rehabilitation in disasters: The experience of the Spanish Toxic
Oil Syndrome. Int Disability Studies 9:78�80.
Lopez-Ibor Alino JJ Jr, Soria J, Canas F, Rodrıguez-Gamazo M.
1985. Psychopathological aspects of the toxic oil syndrome
catastrophe. Br J Psychiatry 147:352�365.
Lopez-Ibor JJ Jr, Ortiz Alonso T, Lopez-Ibor MI. 1999. Lecciones
de Psicologia Medica. Barcelona: Masson, SA.
Lopez-Ibor JJ Jr, Saiz-Ruiz J, Moral L, Moreno I, Vinas R. 1991.
Neuroendocrine serotonergic challenges in clinical research. In:
Sandler M, Coppen A, Harnett S, editors. 5-Hydroxytrypta-
mine in psychiatry. Oxford: Oxford University Press.
McEwen BS. 1999. Stress and hippocampal plasticity. Annu Rev
Neurosci 22:105�122.
Morgan CA 3rd, Krystal JH, Southwick SM. 2003. Toward early
pharmacological posttraumatic stress intervention. Biol Psy-
chiatry 53(9):834�843.
Olson RS. 2000. Toward a politics of disaster: Losses, values,
agendas, and blame. Int J Mass Emergencies Disasters 18
(August):265�287.
Post RM, Weiss SR, Li H, Smith MA, Zhang LX, Xing G, Osuch
EA, McCann UD. 1998. Neural plasticity and emotional
memory. Dev Psychopathol 10(4):829�855.
Quarantelli EL. 1988. Community and organizational prepara-
tions for and responses to acute chemical emergencies and
disasters in the United States: Research findings and their
wider applicability. In: Gow HBF, Kay RW, editors. Emergency
planning for industrial hazards. London: Elsevier. pp 251�273.
Quarantelli EL, editor. 1998. What is a disaster? Perspectives on
the question. London and New York: Routledge.
Roozendaal B, Koolhaas JM, Bohus B. 1997. The role of the
central amygdala in stress and adaption. Acta Physiol Scand
Suppl 640:51�54.
Ruef AM, Litz BT, Schlenger WE. 2000. Hispanic ethnicity and
risk for combat-related posttraumatic stress disorder. Cultur
Divers Ethnic Minor Psychol 6(3):235�251.
Salmon TW. 1919. The war neuroses and their lessons. NY J Med
59:993�994.
Sartre JP. 1939. Esquisse d’une theorie des emotions. Parıs.
Hermann.
Schuffel W. 1993. The mining disaster of Borken, the implemen-
tation of a 3-year support programme and the help through
EuroActDIS. J R Soc Med 86(11):625�627.
Schulberg HC. 1974. Disaster, crisis theory and intervention
strategies. Omega 5:77�87.
Selye H. 1956. The stress of life. New York: McGraw-Hill.
Selye H. 1980. The stress concept today. In: Kutash IL, Schle-
singer LB, editors. Handbook of stress and anxiety. San
Francisco, CA: Jossey-Bass.
Sheehan L and Hewitt K. 1969. Pilot survey of global natural
disasters of the past twenty years. Natural Hazard Research.
University of Toronto.
Soria J, Canas F, Lopez-Ibor Alino JJ, Santo-Domingo J, Lopez-
Ibor JM, Acosta E. 1983a. Liaison psychiatry in massive
intoxication with toxic oil. Actas Luso-Espanolas Neurol
Psiquiatr Ci Afines 11(1):14�15.
Soria J, Canas F, Lopez-Ibor Alino JJ, Santo-Domingo J, Lopez-
Ibor JM, Acosta E. 1983b. Liaison psychiatry in toxic oil
syndrome. In: Lopez-Ibor JJ Jr, Saiz J, Lopez-Ibor JM, editors.
General hospital psychiatry. A challenge for the future of
psychiatry. Proceedings of the International Congress on
General Hospital Psychiatry. Amsterdam: Excerpta Medica. p
86�94.
Steinberg T. 2000. The acts of God: The unnatural history of
disasters in America. New york: Oxford University Press.
Tobin GA, Montz BE. 1997. Natural hazards: Explanation and
integration. New York and London: Guilford Press.
United Nations. 1992. Department of Humanitarian Affairs.
Internationally agreed glossary of basic terms related to disaster
management (DNA/93/36). Geneva, Switzerland: UN.
UNDRCO (United Nations Disaster Relief Coordinator Office).
1984. Disaster prevention and mitigation. Volume II. Prepa-
redness aspects. New York: United Nations.
van der Kolk BA. 1987. The drug treatment of post-traumatic
stress disorder. J Affect Disord 13(2):203�213.
van Zelst WH, de Beurs E, Beekman AT, Deeg DJ, van Dyck R.
2003. Prevalence and risk factors of posttraumatic stress
disorder in older adults. Psychother Psychosom 72(6):333�342.
Venzlaff U. 1958. Die psychoreaktiver Storungen nach entscha-
dingungspflichten Ereignissen. Berlin: Springer.
WHO. 1948. World Health Organization International Classifica-
tion of Diseases. 6th ed. (ICD-6). Geneva: WHO.
WHO. 1991. Psychosocial consequences of disasters-prevention
and management. Geneva: WHO.
WHO. 1992. The ICD-10 International Classification of Mental
and Behavioral Disorders. Clinical descriptions and diagnostic
guidelines. Geneva: WHO. [Traduccion espanola. 1992. Cla-
sificacion Internacional de las Enfermedades. Capıtulo V (F):
Trastornos Mentales y del Comportamiento. Madrid: Medi-
tor.]
Winnik HZ. 1969. Second thoughts about psychic trauma. Israel
Annals Psychiatry Relat Disciplines 7(1):82�95.
Wright SB. 1997. Northridge earthquake: property tax relief.
Disaster Legislation. Washington: The White House Publica-
tions.
Yehuda R. 1997. Sensitization of the hypothalamic-pituitary-
adrenal axis in posttraumatic stress disorder. In: Yehuda R,
McFarlane AC, editors. Psychobioloy of posttraumatic stress
disorder. New York: New York Academy of Sciences. pp 57�75.
Yehuda R. 2000. Biology of posttraumatic stress disorder. J Clin
Psychiatry 61(Suppl 7):14�21.
182 J. J. Lopez-Ibor Jr

CASE REPORT
Sulbutiamine, an ‘innocent’ over the counter drug, interferes withtherapeutic outcome of bipolar disorder
ATHANASIOS DOUZENIS, IOANNIS MICHOPOULOS & LEFTERIS LYKOURAS
Department of General Hospital Psychiatry, Athens University Medical School, ‘Attikon’ Hospital, Athens, Greece
AbstractA case of a patient with bipolar disorder with a history of hospitalizations and addiction to sulbutiamine is presented.Sulbutiamine is a precursor of thiamine that crosses the blood�brain barrier and is widely available without prescriptionin most countries or over the internet. Because of this patient’s need to consume ever increasing quantities of sulbutiamine,his psychiatric care was severely compromised through him defaulting appointments and frequent changes of psychiatrists.This paper reviews the current scientific knowledge about sulbutiamine, and some of the information and claims availableon the web about its use and potential. It is argued that doctors need to be aware of the potential misuse of medicationavailable over the counter or on the internet and its potential harmful influence.
Key words: Sulbutiamine, bipolar disorder, addiction
Introduction
Healthy adult men and healthy adult non-pregnant,
non-lactating women consuming a usual, varied
diet do not need vitamin supplements. Vitamins in
therapeutic amounts may be indicated for the
treatment of deficiency states, for pathological con-
ditions in which absorption and utilization of vita-
mins are reduced or requirements increased, and for
certain non-nutritional disease processes. The deci-
sion to employ vitamin preparations in therapeutic
amounts clearly rests with the physician (Council on
Scientific Affairs 1987).
ICD-10 has a special category for abuse of non-
dependence-producing substances, such as vitamins.
Although the medication may have been medically
prescribed or recommended in the first instance,
prolonged, unnecessary, and often excessive dosage
develops, which is facilitated by the availability of the
substances without medical prescription. Attempts
to discourage or forbid the use of the substance are
often met with resistance. Although it is usually clear
that the patient has a strong motivation to take the
substance, no dependence or withdrawal symptoms
develop as in the case of the psychoactive substances
specified in mental and behavioural disorders due
to psychoactive substance use (World Health Orga-
nization 1993).
Patients with psychiatric disorders are more likely
to use over the counter drugs than those with other
diseases (Mamtani and Cimino 2002). Substance
abuse is a major comorbidity in bipolar patients
(Strakowski and DelBello 2000; Cassidy et al.
2001). In fact, as many as 50% of individuals with
bipolar disorder have been found to have a lifetime
history of substance abuse or dependence (Sonne
and Brady 1999). Although rates decrease in older
age groups, substance abuse is still present at
clinically important rates in the elderly. Bipolar
patients with comorbid substance abuse may have
a more severe course (Cassidy et al. 2001).
The case report, presented below, shows the
relationship between the abuse of sulbutiamine, an
over the counter ‘psychoactive’ substance, and the
outcome of bipolar disorder.
Case report
Mr Z is a 42-year-old single man who works in
the local municipal services and lives with his
parents. At the age of 18 he suffered a manic episode
with agitation, aggressive behaviour and paranoid
Correspondence: Lefteris Lykouras, MD, PhD, FICPM, Professor in Psychiatry, Department of General Hospital Psychiatry, Athens
University Medical School, ‘Attikon’ Hospital, 1 Rimini St. 124 62, Athens, Greece. Tel: �/30 210 5832426. Fax: �/30 210 5326453.
E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(3): 183�185
(Received 14 July 2005; accepted 24 November 2005)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970500492616

ideation that required admission to a psychiatric
hospital. He subsequently suffered a depressive
episode that led to a serious suicide attempt.
Following this he had three further manic episodes
that required involuntary hospitalization. Apart from
the depressive and manic episodes his life was
organized, he had a steady job and managed to
have additional income from playing the stock
market. His latest admission was due to a manic
episode and he was discharged in January 2004
normothymic on two mood stabilizers (lithium and
carbamazepine), as well as a combination of two
different antipsychotics in high doses (haloperidol
20 mg and olanzapine 20 mg OD), benzodiazepines
(diazepam 40 mg and temazepam 10 mg OD) and
antiparkinsonics (biperiden 4 mg OD). Following
this discharge he has been followed-up by the
outpatient psychiatric department of our hospital.
Mr Z was first seen in March 2004 and has kept
regular contacts since. On assessment he presented
as a physically healthy man with mild pressure of
speech and flight of ideas, expressing grandiose ideas
that he did not act upon. During this session he also
mentioned that he had started receiving sulbutia-
mine (Arcalion) by a private prescription 4 months
before his latest admission. He was given this
medication when he complained of ‘lack of energy’
and ‘slowing down’. He found the medication help-
ful and on his own increased the dose. Before his last
admission he was taking ‘much more’ than he was
prescribed when assessed. Mr Z was adamant that
this prescription should be continued, although
sulbutiamine was not included in the Greek national
formulary. He was advised that according to medical
opinion there was no reason for him to receive
sulbutiamine and was also informed that the dose
would be gradually reduced and finally stopped.
Subsequent appointments revealed that the main
reason for attending his outpatient appointments
was to have prescriptions of sulbutiamine that he
consumed in vast quantities (more than 2 g/day). It
also emerged that one of the reasons he decided to
approach our services was the fact that his previous
psychiatric carers were unwilling to prescribe sulbu-
tiamine. It transpired that he requested similar
prescriptions from other clinics as well. When
confronted about his behaviour he became evasive
and agitated. He claimed that he used ‘Arcalion’
because it gave him ‘a high’, it raised his body
temperature and gave him a ‘warm feeling’. He also
felt stronger and consumed the medication before
swimming in the sea as it increased his stamina but
also made him withstand the cold water for a long
period that he could not tolerate otherwise (he is a
winter swimmer). It is worth noting that Mr Z never
overused the benzodiazepines prescribed and was
also willing to reduce the dose of diazepam he
was receiving so that he could continue the same
amount of sulbutiamine ‘while taking fewer pills
on the whole’.
Attempts to reduce the dose of sulbutiamine failed
and the patient managed to acquire prescriptions
from other doctors. This confrontation was the
main reason for repeated missing appointments,
unwillingness to perform regular blood tests and
non-compliance with the medication. On March
2005, after having missed several appointments, he
was urgently referred to our department. His clinical
picture was characterized by grandiose ideas, irrit-
ability, restlessness, pressure of speech, offensiveness
and inappropriate familiarity (he was hypomanic).
He was not taking his psychotropic medication
regularly, and insisted that medication was not
helpful. He kept saying: ‘the only drug that makes
me feel good is ‘‘Arcalion’’’. Reinstitution of his
previous medication and regular follow up led to
normothymia within 1 month.
Whenever sulbutiamine was reduced, he denied
experiencing somatic withdrawal symptoms, but
maintained that without sulbutiamine he felt less
energy, drowsiness and ‘unlike his normal self ’.
Eventually, he agreed to a gradual reduction and
now he receives 600 mg of sulbutiamine (initial dose
2 g) and 10 mg of diazepam (initial dose 40 mg)
without a change in his mental state and activities,
although he is complaining of feeling ‘less energetic’.
Discussion
Sulbutiamine is a hydrophobic molecule that easily
crosses the blood�brain barrier and gives rise to
thiamine and thiamine phosphate esters in the
brain (Van Reeth 1999). It does not have psycho-
stimulant properties or antidepressive effect, but it
can hasten the resorption of psycho-behavioural
inhibition occurring during major depressive epi-
sodes and thereby facilitate the rehabilitation of
patients in their social, professional and family life
functioning (Loo et al. 2000). Sulbutiamine may be
a useful adjunct to specific anti-infective treatment
(Shah 2003).
Information on the effects of sulbutiamine in
human brain is lacking. There is only one study
in which it was shown that acute sulbutia-
mine injection led to a significant decrease of the
dopamine levels in the prefrontal cortex and
3,4-dihydroxyphenylacetic acid levels (DOPAC) in
both the prefrontal and the cingular cortex in
rats, although homovallinic acid (HVA) concentra-
tion did not differ from the controls in these two
areas. Regarding glutaminergic transmission, acute
administration of sulbutiamine induced no change
184 A. Douzenis et al.

of density of N-methyl-D-aspartate (NMDA) and a-
amino-3-hydro-5-methyl-4-isoxazole propionic acid
(AMPA) receptors in cingular cortex, but signifi-
cantly decreased in the kainaite binding sites
(Trovero et al. 2000). The authors postulate that
sulbutiamine might exert a modulatory effect on
glutaminergic and dopaminergic transmission within
the prefrontal cortex. This could play a role in the
psychoactive effect of sulbutiamine. In this context,
it can be argued that it might also affect the
mechanism of action of the antipsychotic medi-
cation. It is also noted that searching in the litera-
ture did not reveal any report of pharmacokinetic
interaction between sulbutiamine and psychotropic
medication.
Thiamine deficiency in both man and animals is
known to produce memory dysfunction and cogni-
tive disorders which have been related to an impair-
ment of cholinergic activity. Sulbutiamine is
considered to improve memory formation in mice
and this behavioural effect could be mediated by
an increase in hippocampal cholinergic activity
(Micheau et al. 1985). In rhesus monkeys, sulbutia-
mine showed effect upon the mechanisms regulating
waking and light sleep, facilitating a state of wakeful-
ness (Balzamo and Vuillon-Cacciuttolo 1982).
There are no reports of any impairment caused by
overuse of sulbutiamine or other vitamins of the B
complex group except pyridoxine. It is reported that
one patient developed a severe sensory and a mild
motor neuropathy due to massive and prolonged
ingestion of pyridoxine (B6) (10 g daily for 5 years)
(Morra 1993).
A Google search on the internet showed almost
14,000 sites in which sulbutiamine (Arcalion) is
discussed. Some of them say that ‘(Arcalion) speeds
up the reflexes and reaction times in clinical tests,
promotes wakefulness and alertness’. In other sites
sulbutiamine is being sold among sexual merchan-
dise. Though sulbutiamine has no specific therapeu-
tic indication or clinical use, and it does not appear
to have an addictive profile, it seems to be used a
great deal by individuals. The same happens with
most vitamin preparations (Council on Scientific
Affairs 1987). It is well known that bipolar disorder
is associated with substance abuse and addiction
(Sonne and Brady 1999; Strakowski and DelBello
2000; Cassidy et al. 2001). Our patient fulfilled the
DSM-IV criteria for abuse and addiction of this
substance. His addiction to sulbutiamine interfered
with his treatment for bipolar disorder and the
therapeutic relationship with his doctors. Further-
more, it should be noted that the patient increased,
on his own, the sulbutiamine dosage by more than
2 g/day. This was temporally associated with the
emergence of the manic episode. In this regard the
large dose of sulbutiamine may have contributed to
the manic relapse. The widespread use of sulbutia-
mine can lead to the assumption that individuals
with mental illness can use it as well without their
doctors’ knowing. Bearing in mind the case pre-
sented, it might be clinically relevant to enquire of
psychiatric patients of young age about the use of
sulbutiamine.
Statement of interest
The authors have no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
Balzamo E, Vuillon-Cacciuttolo G. 1982. Facilitation of a state of
wakefulness by semi-chronic treatment with sulbutiamine
(Arcalion) in Macaca mulatta . Rev Electroencephalogr Neuro-
physiol Clin 12:373�378.
Cassidy F, Ahearn EP, Carroll BJ. 2001. Substance abuse in
bipolar disorder. Bipolar Disord 3:181�188.
Council on Scientific Affairs. 1987. Vitamin preparations as
dietary supplements and as therapeutic agents. J Am Med
Assoc ;257 10(14):1929�1936.
Loo H, Poirier MF, Ollat H, Elatki S. 2000. Effects of
sulbutiamine (Arcalion 200) on psycho-behavioral inhibition
in major depressive episodes. Encephale 26:70�75.
Mamtani R, Cimino A. 2002. A primer of complementary and
alternative medicine and its relevance in the treatment of
mental health problems. Psychiatr Q 73:367�381.
Micheau J, Durkin TP, Destrade C, Rolland Y, Jaffard R. 1985.
Chronic administration of sulbutiamine improves long term
memory formation in mice: Possible cholinergic mediation.
Pharmacol Biochem Behav 23:195�198.
Morra M, Philipszoon HD, D’Andrea G, Cananzi AR, L’Erario
R, Milone FF. 1993. Sensory and motor neuropathy caused by
excessive ingestion of vitamin B6: a case report. Funct Neurol
8:429�432.
Shah SN; Sulbutiamine Study Group. 2003. Adjuvant role of
vitamin B analogue (sulbutiamine) with anti-infective treat-
ment in infection associated asthenia. J Assoc Phys India
51:891�895.
Sonne SC, Brady KT. 1999. Substance abuse and bipolar
comorbidity. Psychiatr Clin North Am 22:609�627, ix.
Strakowski SM, DelBello MP. 2000. The co-occurrence of bipolar
and substance use disorders. Clin Psychol Rev 20:191�206.
Trovero F, Gobbi M, Weil-Fuggaza J, Besson MJ, Brochet D,
Pirot S. 2000. Evidence for a modulatory effect of sulbutiamine
on glutamatergic and dopaminergic cortical transmissions in
the rat brain. Neurosci Lett ;292 29(1):49�53.
Van Reeth O. 1999. Pharmacologic and therapeutic features of
sulbutiamine. Drugs Today (Barc) 35:187�192.
World Health Organization. 1993. The ICD-10. In: Classification
of Mental and Behavioural Disorders: Diagnostic Criteria for
Research. Geneva: World Health Organization.
Sulbutiamine interferes with therapeutic outcome of bipolar disorder 185

CASE REPORT
Autism and Williams syndrome: A case report
SABRI HERGUNER & NAHIT MOTAVALLI MUKADDES
Department of Child and Adolescent Psychiatry, Istanbul Faculty of Medicine, Istanbul University, Istanbul, Turkey
AbstractWilliams syndrome (WS) is a neurodevelopmental disorder caused by a deletion in the 7q11.23 region which includes atleast 17 genes. The presence of autistic features in WS is a controversial issue. While some authors describe WS as theopposite phenotype of autism, recent studies indicate that both share many common characteristics. We report a 12-year-old boy diagnosed as autistic disorder and WS with hemizygosity at the elastin locus and a karyotype of46,XY,del(7)(q11.21q11.23). Molecular genetic studies have shown that deletion at the elastin gene may account for thecardiovascular abnormalities seen in WS, but autistic features are likely caused by other genes flanking elastin.
Key words: Autistic disorder, Williams Syndrome, 7q11.23
Introduction
Autism is a severe neuropsychiatric disorder char-
acterized by impairments in reciprocal social inter-
action and communication, restricted and stereo-
typed patterns of interests and behaviours. Although
there are many studies on autistic disorder (AD) in
genetic syndromes, concurrence of AD and Williams
syndrome (WS) is reported to be very rare (Gillberg
and Coleman 2000).
WS is a rare neurodevelopmental disorder caused
by the hemizygous deletion of 7q11.23 and asso-
ciated with an ‘elfin face’ appearance, cardiovascular
abnormalities (particularly supravalvular aortic ste-
nosis), transient infantile hypercalcaemia, and
growth and developmental retardation (Pober and
Dykens 1996). Individuals with WS generally have
a unique neurological and behavioural profile.
They have poor spatial cognition, but have notably
intact language and musical abilities, and relatively
strong face-processing ability. Affected individuals
are highly sensitive to certain classes of sounds
and have mild mental retardation, with an average
IQ of 60. In addition, approximately 70% of WS
individuals suffer from attention deficit and hyper-
activity disorder (ADHD) and there is a high
incidence of anxiety and simple phobias (Bellugi
et al. 1999).
The presence of autistic features in WS is a
controversial issue. Children with WS are described
as outgoing, overfriendly, communicative, and hy-
persocial with good empathic skills. The excessively
social behaviour repertoire represents an important
component of its phenotype that distinguishes WS
from other developmental disorders (Jones et al.
2000). On the grounds of these features, it is
believed that WS is the opposite phenotype of
autism (Peterson and Panksepp 2004). On the other
hand, a recent study that compared subjects with
WS and other developmental disorders found that
WS group had more pragmatic language impair-
ments, poor social relationships and restricted inter-
ests. The researchers concluded that WS would
seem to share many characteristics of autistic dis-
order (Laws and Bishop 2004).
To our knowledge, there are only three case
reports about the concurrence of WS and AD
(Gillberg and Rasmussen 1994; Gosch and Pankau
1994; Reiss et al. 1985) without any specific genetic
information. Reiss et al. (1985) described two cases
with 46 XY karyotypes and one of the cases showed
pericentric inversion of chromosome 9 which had no
clinical significance. Gillberg and Rasmussen (1994)
reported four cases and examined them for chromo-
some 15 abnormalities but failed to demonstrate any
Correspondence: Dr Sabri Herguner, Istanbul Tip Fakultesi, Cocuk-Ergen Psikiyatrisi Anabilim Dali, 34393 � Capa, Istanbul, Turkey.
Tel: +90 533 7428150. Fax: +90 212 2349208. E-mail: [email protected]
The World Journal of Biological Psychiatry, 2006; 7(3): 186�188
(Received 21 September 2005; accepted 11 January 2006)
ISSN 1562-2975 print/ISSN 1814-1412 online # 2006 Taylor & Francis
DOI: 10.1080/15622970600584221

finding. The present case describes a child with WS
and AD who had 7q11.23 deletion.
Case report
EK, a 12-year-old boy, referred to our clinic due to
parental concerns regarding his impairment in
language development, social�emotional reciprocity,
and repetitive and self-mutilative behaviours. His
language was severely delayed, he had only 10 single
words and his speech was echolalic. He started using
single words at the age of 5 years and never used
two-word phrases. He had limited non-verbal com-
munication with poor eye contact and his response
to instructions was inconsistent. He had no pretend
play; his plays were mostly repetitive (like shaking
toys) and he had an unusual preoccupation with
collecting buttons, watching spinning wheels and
washing machines. He was not able to join his peers
or interact with them. He had stereotypic behaviours
such as peripheral gazing, rocking his head, and his
body, and self-mutilative behaviours including head-
banging and finger-biting.
EK was born at term after a normal pregnancy by
an unremarkable delivery. His motor development
was severely delayed; he sat at 12 months, first
walked at 40 months of age, and became continent
when he was 6 years old. He was referred to a
child neurology and genetic department at the age of
6 years for his psychomotor retardation. He was
diagnosed as WS due to his physical features; wide
mouth with thick lips, long philtrum, prominent
cheeks, periorbital fullness, epicanthal folds, long
neck, hypoplastic nails, hoarse voice, short stature
and microcephaly. His genetic analysis by fluores-
cence in situ hybridization (FISH) using a bio-
tin-labelled probe revealed hemizygosity at the
elastin locus with a karyotype of 46,XY,del(7)
(q11.21q11.23). In his family history, his aunt and
uncle had motor developmental delay. There was no
evidence of autistic spectrum disorders in his family
history.
His weight and height were below the 3rd percen-
tile, and his head circumference was at the 3rd
percentile. Blood chemistry analysis yielded normal
values. His cardiological examination and echocar-
diography revealed tricuspid insufficiency. His
neurological and ophthalmological examinations,
abdominal ultrasonography, electrocardiography,
cranial tomography and electroencephalography
were normal.
His total score in the Childhood Autism Rating
Scale (CARS) (Schopler et al. 1980) was 44,
indicating severe autism and total score on the
Autism Behavior Checklist (ABC) (Krug et al.
1980) was 111 points. He was diagnosed as AD
according to DSM-IV criteria (APA 1994).
Discussion
We present a case that was diagnosed as WS at 6
years and as AD at 12 years. This is the first case
with autistic disorder and WS with 7q11.23 deletion.
Deletions that span a number of genes and cause a
constellation of symptoms are called contiguous
gene syndromes. WS is one such disorder, caused
by the hemizygous deletion of a 1.5-Mb interval
encompassing at least 17 genes at 7q11.23 (Osborne
1999). In over 95% of the cases, the elastin gene
which encodes the major component of skin, blood
vessels, and lung tissues, is deleted. The deletion at
the elastin locus may account for the cardiac
abnormalities, most commonly supravalvular aortic
stenosis, but none of the other features seen in WS.
Supravalvular aortic stenosis also can occur as an
isolated trait with 7q11.23 deletion (Curran et al.
1993). Because it is expressed in high concentrations
in the brain, deletion in the LIM kinase 1 (LIMK1)
gene may be responsible for the impaired visuo-
spatial constructive cognition in this disorder (Don-
nai and Karmiloff 2000). In addition to ELN and
LIMK1, several genes have been described, but
whether their haploinsufficiency contributes to the
WS phenotype is not yet known (Wu et al. 1998).
But it had been postulated that other genes in this
region including CYLN2, RFC2, and STX1A could
account for the remaining features of WS such as
mental retardation, growth deficiency, infantile hy-
percalcaemia, and neurological alterations (Meng et
al. 1998). Taken together, autistic features may also
be caused by other genes flanking ELN.
Even though several full genome scans have failed
to identify susceptibility loci with certainty, numer-
ous genetic studies showed the presence of chromo-
some 7 abnormalities, mainly in 7q22.33, in autistic
spectrum disorders (Muhle et al. 2004). However,
no study has found 7q11.23 as a susceptible region.
Although it seems premature to mention clearly that
deleted genes in 7q11.23 are attributable for the
autism phenotype, increasing reports on autistic
features in WS raise the question of the probable
contribution of genes in 7q11.23 in the genetic
aetiology of AD.
Another important point in this case is the delayed
diagnosis of autistic disorder. Although he was
referred to the neurology clinic for his psychomotor
developmental retardation and had undergone neu-
rological and genetic investigations at age of 6 years,
his autistic features were not evaluated carefully by
the medical professionals until his referral to the
child psychiatry clinic at the age of 12 years. There
Autism and Williams Syndrome 187

may be several reasons for the delay in the diagnosis
of AD. Firstly, when a medical doctor makes a
diagnosis of a medical disorder in a child with
autistic disorder, it is quite likely that the medical
diagnosis will take precedence and the child will not
be referred for examination of the autistic symptoms
(Kielinen et al. 2004). Secondly, it could be related
to the lack of information and awareness on autism
in our society. Our clinical experience shows that
children with a medical condition and AD refer to
the child psychiatry clinics in older ages because
their behavioural symptoms are usually attributed to
their physical problems. It seems that both paedia-
tricians and parents mostly focus on the medical
problems than the behavioural�social development
of these children. To sum up, the case described here
highlights the need for consideration of the presence
of autism in sufferers of other medical conditions.
Since early diagnosis and intervention of autism
significantly improve patients’ long-term outcome,
multidisciplinary and comprehensive evaluation of
children with developmental delays appears vital.
Statement of interest
The authors have no conflict of interest with any
commercial or other associations in connection with
the submitted article.
References
Bellugi U, Lichtenberger L, Mills D, Galaburda A, Korenberg JR.
1999. Bridging cognition, the brain and the molecular genetics:
Evidence from Williams syndrome. Trends Neurosci 22:197�207.
Curran ME, Atkinson DL, Ewart AK, Morris CA, Leppert MF,
Keating MT. 1993. The elastin gene is disrupted by a
translocation associated with supravalvular aortic stenosis.
Cell 73:159�168.
American Psychiatric Association. 1994. Diagnostic and statistical
manual of mental disorders. 4th ed. Washington, DC: Amer-
ican Psychiatric Association Press.
Donnai D, Karmiloff-Smith A. 2000. Williams syndrome; From
genotype through to the cognitive phenotype. Am J Med Genet
97:164�171.
Gillberg C, Coleman M. 2000. The biology of the autistic
syndromes. 3rd ed. London: MacKeith Press (distributed by
Cambridge University Press).
Gillberg C, Rasmussen P. 1994. Brief report: Four case histories
and a literature review of Williams syndrome and autistic
behavior. J Autism Dev Disord 24:381�393.
Gosch A, Pankau R. 1994. Autistic behavior in two children with
Williams-Beuren syndrome. Am J Med Genet 53:83�84.
Jones W, Bellugi U, Lai Z, Chiles M, Reilly J, Lincoln A, Adolphs
R. 2000. Hypersociability in Williams syndrome. J Cogn
Neurosci Suppl 1:30�46.
Kielinen M, Rantala H, Timonen E, Linna SL, Moilanen I. 2004.
Associated medical disorders and disabilities in children with
autistic disorder: a population-based study. Autism 8:49�60.
Krug DA, Arick J, Almond P. 1980. Behavior checklist for
identifying severely handicapped individuals with high levels
of autistic behavior. J Child Psychol Psychiatry 21:221�229.
Laws G, Bishop D. 2004. Pragmatic language impairment and
social deficits in Williams syndrome: A comparison with
Down’s syndrome and specific language impairment. Int J
Lang Commun Disord 39:45�64.
Meng X, Lu X, Li Z, Green ED, Massa H, Trask BJ, Morris CA,
Keating MT. 1998. Complete physical map of the common
deletion region in Williams syndrome and identification and
characterization of three novel gene. Hum Genet 103:590�599.
Muhle R, Trentacoste SV, Rapin I. 2004. The genetics of autism.
Pediatrics 113:472�486.
Osborne LR. 1999. Williams-Beuren syndrome: unraveling the
mysteries of a microdeletion disorder. Mol Genet Metab 67:1�10.
Peterson BS, Panksepp J. 2004. Biological basis of childhood
neuropsychiatric disorders. In: Panksepp J, editor. Textbook of
biological psychiatry. New York: Wiley-Liss. pp 393�436.
Pober BR, Dykens EM. 1996. Williams syndrome: An overview of
medical, clinical and behavioral features. Child Adolesc Psy-
chiatr Clin North Am 5:929�943.
Reiss AL, Feinstein C, Rosenbaum KN, Borengasser-Caruso MA.
1985. Autism associated with Williams syndrome. Pediatrics
106:247�249.
Schopler E, Reichler RJ, DeVellis RF, Daly K. 1980. Toward
objective classification of childhood autism: Childhood autism
rating scale (CARS). J Autism Dev Disord 10:91�103.
Wu YQ, Sutton VR, Nickerson E, Lupski JR, Potocki L,
Korenberg JR, Greenberg F, Tassabehji M, Shaffer LG.
1998. Delineation of the common critical region in Williams
syndrome and clinical correlation of growth, heart defects,
ethnicity, and parental origin. Am J Med Genet 78:82�89.
188 S. Herguner & N. M. Mukaddes



The World Journal of Biological Psychiatry
Instructions to Authors
The aim of The World Journal of Biological Psychiatry is to increase theworldwide communication of knowledge in clinical and basic research onbiological psychiatry. The composition of The World Journal of BiologicalPsychiatry , with its diverse categories that allow communication of a greatvariety of information, ensures that it is of interest to a wide range ofreaders. It offers the opportunity to educate (through critical reviewpapers), to publish original work and observations (original papers andcase reports), and to express personal opinions (Viewpoints/Letters to theEditor). The World Journal of Biological Psychiatry is thus an extremelyimportant medium in the field of biological psychiatry all over the world.
Manuscripts must be written in standard and grammatical English andshould present new results as well as be of scientific value. Contributionswill be considered for the following categories:
/� Editorial/� Reviews/Mini-reviews/� Original investigations/Summaries of original research/� Brief reports/� Viewpoints/� Case reports/Case series/� Letters to the editors/� Book reviews/� Invitations/Announcements
The World Journal of Biological Psychiatry’s Manuscript Central, web-based manuscript submission and handling system, is now available.Please submit all manuscripts online via the Journal’s ManuscriptCentral site that is accessible via the Journal’s home page:www.tandf.no/wjbp
Click on ‘‘online submission’’ which directs you to Manuscript Central’slog in page. Here either create an account or enter an existing account tolog in to your ‘‘author centre’’ to upload manuscripts.
If you have difficulties in submitting your manuscript electronically, contactThe World Journal of Biological Psychiatry’s Editorial Office.
The receipt of the manuscript will be acknowledged by an e-mail whichincludes a manuscript ID number. Each manuscript will be assigned to atleast two reviewers. The manuscript ID number should be quoted in allcorrespondence with the Chief Editor and Editorial Office.
Submit manuscript text as a Word file and Figures as separate TIFF or EPSformat files. Use Times New Roman 12 point and double spacingthroughout the manuscript. Use a clear system of headings to divide upand clarify the text, with not more than three grades of headings. Thedesired position of figures and tables should be indicated in the manuscript.Submission of a manuscript implies: that the work described has not beenpublished before (except in the form of an abstract or as part of a publishedlecture, review or thesis); that it is not under consideration for publicationanywhere else; that its publication has been approved by all co-authors, ifany, as well as by responsible authorities � tacitly or explicitly � at theinstitute where the work has been carried out.
Manuscripts submitted for publication must contain a statement to theeffect that all human studies have been approved by the appropriate ethicscommittee and have therefore been performed in accordance with theethical standards laid down in the 1964 Declaration of Helsinki. It shouldalso be stated clearly in the text that all persons gave their informed consentprior to their inclusion in the study. Details that might disclose the identityof the subjects under study should be omitted.
Reports of animal experiments must state that the Principles of laboratoryanimal care (NIH publication No. 86-23, revised 1985) were followed, aswell as specific national laws (e.g., the current version of the German Lawon the Protection of Animals) where applicable.
The editors reserve the right to reject manuscripts that do not comply withthe above-mentioned requirements. The author will be held responsible forfalse statements or for failure to fulfill the above-mentioned requirements.
Manuscript specifications
1. The manuscript text file shall contain a title page. On the title page thefollowing shall be stated: The title of the article, which should beconcise but informative. A short title not exceeding 40 letters andspaces. The full names, affiliations and addresses of all the contribu-tors, and e-mail address, telephone number and fax number for thecorresponding author. The title page should also include details of thenumber of words, figures, and tables.
2. Each article must include a short Abstract of the essential results (notexceeding 200 words).
3. Key words. Up to five key words should be inserted below the abstract.Use terms from the Medical Subject Headings list from Index Medicus.
4. References. All references cited in the text are to be listed (double-spaced) at the end of the paper in alphabetical order under the name ofthe first author. If there are more than six authors, list the first sixauthors and use ‘et al.’ Articles ‘in press’ may be included, but muststate the journal that has accepted them. Personal communicationsshould be avoided. Journal titles should be abbreviated according toIndex Medicus.
Citations in the text should be by author and date in parentheses. Ifthere are two authors, both should be named (e.g., Agar and Douglas1955); if an article with more than two authors is cited, only the firstauthor’s name plus ‘et al.’ need be given (e.g., Komor et al. 1979); ifthere is more than one reference by the same author or team of authorsin the same year, then a, b, c, etc., should be added to the year, both inthe text and the list of references. When styling references, thefollowing examples should be followed:
Brain WR. 1958. The physiological basis of consciousness. Acritical review. Brain 81:426 �455.
Kuhlenbeck H. 1954. The human diencephalon. A summary ofdevelopment, structure, function and pathology. Basel: Karger.
Teuber HL. 1964. The riddle of frontal lobe function in man. In:Warren T, Akert CH, editors. The frontal and granular cortex andbehavior. New York: McGraw-Hill. pp 252�271.
5. Tables should be typed on separate sheets and numbered consecutivelywith Arabic numbers. All tables should include a brief descriptivecaption.
6. Figures. All illustrations, whether photographs, graphs or diagrams,should be numbered consecutively with Arabic numerals and sub-mitted on separate sheets. Each illustration should be referred to in thetext as ‘Figure 1’, ‘Figures 1�4’, etc. The figures should have a shorttitle followed by a concise description. All abbreviations and symbolsappearing in the figure must be explained in the legend. Remarks suchas ‘for explanation (or details) see text’ should be avoided. Each figureshould have a legend containing sufficient information to render thefigure intelligible without reference to the text. If a figure has beenpublished previously, acknowledge the original source and submitwritten permission from the copyright holder to reproduce it. Ifphotographs of persons are used, either the subjects must not beidentifiable or their written permission to use the photographs mustaccompany the manuscript. Figures can be sent electronically asseparate files in EPS or TIFF format.
7. Copyright. It is a condition of publication that authors vest copyrightin their articles, including abstracts, in Taylor & Francis GroupLtd. This enables us to ensure full copyright protection and todisseminate the article, and the journal, to the widest possiblereadership in print and electronic formats as appropriate. Authorsmay, of course, use the material elsewhere after publication providingthat prior permission is obtained from Taylor & Francis GroupLtd. Authors are themselves responsible for obtaining permission toreproduce copyright material from other sources. To view the ‘Copy-right Transfer Frequently Asked Questions’ please visit http://www.tandf.co.uk/joumals/copyright.asp
8. Proofs and offprints. Proofs will be sent as a PDF file to thecorresponding author’s e-mail address unless otherwise requested.The corrected proof must be returned to the publisher within 3 days.Authors will be liable to pay for excessive alterations in the proof.Offprints can be ordered by filling out the form accompanying theproofs. The corresponding authors will be supplied with a free copy ofthe relevant issue.
9. For further information about the journal, including links to the onlinesample copy and contents pages, visit the journal homepage:www.tandf.no/wjbp or contact the Editorial Office at the followingaddress:
Jacqueline Klesing, Editorial Assistant, Department of Psychiatry,Ludwig-Maximilians-University, Nussbaumstrasse 7, 80336 Munich,Germany. Tel: +49 89 5160 5531 Fax: +49 89 5160 5530. E-mail:[email protected]



Related Documents











