Assignment of protein function in the postgenomic era Alan Saghatelian & Benjamin F Cravatt Genome sequencing projects have provided researchers with an unprecedented boon of molecular information that promises to revolutionize our understanding of life and lead to new treatments of its disorders. However, genome sequences alone offer only limited insights into the biochemical pathways that determine cell and tissue function. These complex metabolic and signaling networks are largely mediated by proteins. The vast number of uncharacterized proteins found in prokaryotic and eukaroyotic systems suggests that our knowledge of cellular biochemistry is far from complete. Here, we highlight a new breed of ‘postgenomic’ methods that aim to assign functions to proteins through the integrated application of chemical and biological techniques. One of the preeminent challenges facing twenty-first century scientists is the determination of the molecular, cellular and (patho)physiological functions for the numerous proteins encoded by eukaryotic and pro- karyotic genomes. It is clear that this problem cannot be addressed by the analysis of genome sequences alone. Indeed, a significant frac- tion of every genome sequenced to date, from bacterial to human, is composed of uncharacterized proteins. Even those proteins for which tentative or apparently robust assignments of activity can be made are susceptible to reinterpretation as our understanding of the molecular complexity of life increases. For example, it is now clear that proteins with highly related sequences can perform different functions in vivo, and, conversely, proteins may show similar activities while lacking dis- cernible sequence or structural similarity 1 . The additional realization that one gene can code for tens if not hundreds of different proteins as a result of post-transcriptional and post-translational processing and modification indicates that the number of proteins in need of characterization vastly exceeds the number of genes in the genome 2 . These various issues combine to present a formidable, but exciting, set of experimental problems for contemporary biological and chemical researchers, all of which distill down to predominantly a single ques- tion: how should we undertake the assignment of protein function in the postgenomic era? The biochemical properties of proteins are typically determined in vitro with purified material. Although this classical ‘test tube bio- chemistry’ approach has succeeded in explaining the activities of many proteins, it does suffer some shortcomings. First, proteins do not func- tion in isolation in vivo, but rather as parts of complex metabolic and signaling networks. Proteins are also regulated by post-translational mechanisms in vivo, including covalent modification and protein- protein interactions (Fig. 1). These dynamic events create a context dependency to the performance of proteins in living systems that can be difficult, if not impossible, to replicate in vitro. From a methodological perspective, in vitro studies are biased toward proteins that are more straightforward to express recombinantly and for which activity assays are available. As such, many challenging classes of proteins, including membrane-associated and uncharacterized proteins, are not effectively addressed by classical methods. Finally, the examination of individual proteins ‘one at a time’ is an uncomfortably slow process, especially when confronted with the thousands of proteins in current need of functional characterization. The limitations of classical biochemical methods have inspired the development of tools and technologies that can, in a direct and systematic way, characterize the activities of proteins in complex bio- logical settings. Among these ‘systems biology’ approaches, genetic techniques, such as targeted gene disruption (gene ‘knockouts’ 3 ) and RNA interference (RNAi) 4 , have proven particularly powerful, owing to their remarkable specificity and generality (Fig. 2). Nonetheless, genetic methods suffer from a lack of temporal control over protein activity and are not well suited for dissecting the functions of dif- ferent protein isoforms. The subject of this review is a distinct breed of systems biology methods that take advantage of chemistry, often in combination with genetic, protein biochemistry and analytical techniques, to tackle the problem of characterizing protein function on a global scale (Fig. 2). As will be seen, a common trait of these methods is that they are truly ‘postgenomic’ in the sense that they require complete genome sequences for optimal performance. Thus, although the limitations of pure reliance on genome sequences will emerge as a recurring theme, these comments should not be viewed as a negative critique of genome sequencing efforts. On the contrary, the chemical biology approaches described herein provide perhaps the most profound endorsement of the value of complete genome sequences, which are enabling us to make experimental inquiries never before deemed possible. Genomic methods for the assignment of protein function Prokaryotic organisms provide perhaps the richest and most diverse source of metabolic enzymes on the planet. These enzymes often The Skaggs Institute for Chemical Biology and Departments of Cell Biology and Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. Correspondence should be addressed to B.F.C. ([email protected]). 130 VOLUME 1 NUMBER 3 AUGUST 2005 NATURE CHEMICAL BIOLOGY REVIEW © 2005 Nature Publishing Group http://www.nature.com/naturechemicalbiology

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Assignment of protein function in the postgenomic eraAlan Saghatelian & Benjamin F Cravatt
Genome sequencing projects have provided researchers with an unprecedented boon of molecular information that promises to revolutionize our understanding of life and lead to new treatments of its disorders. However, genome sequences alone offer only limited insights into the biochemical pathways that determine cell and tissue function. These complex metabolic and signaling networks are largely mediated by proteins. The vast number of uncharacterized proteins found in prokaryotic and eukaroyotic systems suggests that our knowledge of cellular biochemistry is far from complete. Here, we highlight a new breed of ‘postgenomic’ methods that aim to assign functions to proteins through the integrated application of chemical and biological techniques.
One of the preeminent challenges facing twenty-first century scientists is the determination of the molecular, cellular and (patho)physiological functions for the numerous proteins encoded by eukaryotic and pro-karyotic genomes. It is clear that this problem cannot be addressed by the analysis of genome sequences alone. Indeed, a significant frac-tion of every genome sequenced to date, from bacterial to human, is composed of uncharacterized proteins. Even those proteins for which tentative or apparently robust assignments of activity can be made are susceptible to reinterpretation as our understanding of the molecular complexity of life increases. For example, it is now clear that proteins with highly related sequences can perform different functions in vivo, and, conversely, proteins may show similar activities while lacking dis-cernible sequence or structural similarity1. The additional realization that one gene can code for tens if not hundreds of different proteins as a result of post-transcriptional and post-translational processing and modification indicates that the number of proteins in need of characterization vastly exceeds the number of genes in the genome2. These various issues combine to present a formidable, but exciting, set of experimental problems for contemporary biological and chemical researchers, all of which distill down to predominantly a single ques-tion: how should we undertake the assignment of protein function in the postgenomic era?
The biochemical properties of proteins are typically determined in vitro with purified material. Although this classical ‘test tube bio-chemistry’ approach has succeeded in explaining the activities of many proteins, it does suffer some shortcomings. First, proteins do not func-tion in isolation in vivo, but rather as parts of complex metabolic and signaling networks. Proteins are also regulated by post-translational mechanisms in vivo, including covalent modification and protein-protein interactions (Fig. 1). These dynamic events create a context dependency to the performance of proteins in living systems that can be
difficult, if not impossible, to replicate in vitro. From a methodological perspective, in vitro studies are biased toward proteins that are more straightforward to express recombinantly and for which activity assays are available. As such, many challenging classes of proteins, including membrane-associated and uncharacterized proteins, are not effectively addressed by classical methods. Finally, the examination of individual proteins ‘one at a time’ is an uncomfortably slow process, especially when confronted with the thousands of proteins in current need of functional characterization.
The limitations of classical biochemical methods have inspired the development of tools and technologies that can, in a direct and systematic way, characterize the activities of proteins in complex bio-logical settings. Among these ‘systems biology’ approaches, genetic techniques, such as targeted gene disruption (gene ‘knockouts’3) and RNA interference (RNAi)4, have proven particularly powerful, owing to their remarkable specificity and generality (Fig. 2). Nonetheless, genetic methods suffer from a lack of temporal control over protein activity and are not well suited for dissecting the functions of dif-ferent protein isoforms. The subject of this review is a distinct breed of systems biology methods that take advantage of chemistry, often in combination with genetic, protein biochemistry and analytical techniques, to tackle the problem of characterizing protein function on a global scale (Fig. 2). As will be seen, a common trait of these methods is that they are truly ‘postgenomic’ in the sense that they require complete genome sequences for optimal performance. Thus, although the limitations of pure reliance on genome sequences will emerge as a recurring theme, these comments should not be viewed as a negative critique of genome sequencing efforts. On the contrary, the chemical biology approaches described herein provide perhaps the most profound endorsement of the value of complete genome sequences, which are enabling us to make experimental inquiries never before deemed possible.
Genomic methods for the assignment of protein functionProkaryotic organisms provide perhaps the richest and most diverse source of metabolic enzymes on the planet. These enzymes often
The Skaggs Institute for Chemical Biology and Departments of Cell Biology and Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. Correspondence should be addressed to B.F.C. ([email protected]).
130 VOLUME 1 NUMBER 3 AUGUST 2005 NATURE CHEMICAL BIOLOGY
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

function as large complexes to produce natural products with remark-able biological activities5. To date, more than 200 prokaryotic genomes have been fully sequenced, with many more projects under way (http://www.tigr.org/tigr-scripts/CMR2/CMRHomePage.spl). This rich source of molecular information has spawned the birth of a new discipline termed ‘genomic enzymology’ to signify a movement away from single-enzyme phenomenology to the study of these proteins in the context of entire biological systems1. Genomic enzymology aims to integrate find-ings from multiple fields of research, including comparative genomics, structural biology and enzyme kinetics, to determine the functions of uncharacterized enzymes. This multidisciplinary approach has revealed several cases where the sequence, structure and function of enzymes diverge in surprising ways.
Divergent functions from similar sequences. The (β/α)8-(TIM) bar-rel, which is the most prevalent fold found among enzymes, has pro-vided a fertile subject for postgenomic investigations6. Initiated with the discovery of two (β/α)8-barrel proteins, mandelate racemase and muconate lactonizing enzyme, that share conserved structures and active site functional groups, but catalyze different reactions7, this enzyme superfamily has since been shown to possess members that perform a wide range of mechanistically diverse chemical transforma-tions. Thus, (β/α)8-barrel enzymes provide a clear case where gene or protein sequence alone cannot be used to predict function.
Complete genome sequences have afforded enzymologists the first opportunity to systematically compare (β/α)8-barrel proteins from distinct organisms in efforts to characterize their unique functions. This general strategy of comparative genomics8 is particularly useful for prokaryotic organisms because their genes are often organized into operons, defined as a set of adjacent genes all under the transcrip-tional control of the same operator. Gerlt and colleagues have exploited ‘genomic context’ to predict activities for several uncharacterized (β/α)8-barrel enzymes. For example, the genes for YcjG and YkfB, two distantly related homologs of o-succinylbenzoate synthase (OSBS), were both found in the vicinity of genes involved in murein peptido-glycan metabolism9. Knowledge of this metabolic pathway, coupled with an understanding of the scope of potential reactions catalyzed by (β/α)8-barrel enzymes, led the researchers to propose that YcjG and YkfB are L-Ala-D/L-Glu epimerases (Fig. 3a). Subsequent biochemical analysis of these proteins confirmed that they have L-Ala-D/L-Glu epim-erase activity, representing the first examples of enzymes that catalyze this reaction. Notably, neither enzyme was able to catalyze the OSBS
reaction, underscoring the remarkable functional diversity that can exist among sequence-related proteins.
Structural insights into the plasticity of (β/α)8-barrel proteins have been gained by a comparison of orotidine 5′-monophosphate decar-boxylase (OMPDC) and 3-keto-L-gulonate 6-phosphate decarboxyl-ase (KGPDC), two homologous enzymes that catalyze mechanistically unrelated reactions (metal-independent and Mg2+-dependent decar-boxylations, respectively)10. An overlay of the active site structures of OMPDC and KGPDC revealed that conserved residues occupy nearly identical positions but serve distinct roles in catalysis. In OMPDC, these residues are involved in substrate binding, whereas in KGPDC they serve as ligands to coordinate a Mg2+ ion required for stabilization of an enediolate anion intermediate (this intermediate is not formed in OMPDC-catalyzed decarboxylations). The authors conclude that OMPDC and KGPDC should be designated as members of a supra-family to underscore that, even though these enzymes have significant sequence and structural similarity, they use distinct catalytic mecha-nisms (Fig. 3b). More generally, these discoveries offer an instructive warning to those eager to predict the functions of uncharacterized pro-teins solely on the basis of sequence or structural information.
Examples of homologous enzymes that have disparate activities can also be found outside the (β/α)8-barrel family of enzymes. Whitman and colleagues have shown that different members of the 4-oxalocrotonate tautomerase (4-OT) family can catalyze not only tautomerase and iso-merase, but also dehalogenase reactions11,12. Pohl and colleagues have used innovative mass spectrometry (MS)–based substrate screening methods to determine that an archaeal enzyme originally annotated on the basis of sequence analysis as a glucose-1-phosphate deoxythymi-dylyltransferase is actually a bifunctional acetyltransferase–N-acetyl-glucosamine-1-phosphate uridylyltransferase13. Notably, the MS-based technologies described in this study should prove of general value for the rapid and systematic biochemical characterization of new carbohydrate-related enzymes encoded by prokaryotic and eukaryotic genomes.
Similar functions from divergent sequences. Genomic enzymolo-gists have also uncovered interesting cases where enzymes that share no sequence or structural similarity perform similar functions. For example, Begley and colleagues used comparative genomic analyses to identify the first tryptophan-to-quinolinate pathway in bacteria14, thereby dispelling the generally held belief that this process is unique to eukaryotes. Before this discovery, prokaryotes were assumed to bio-synthesize quinolinate exclusively from aspartate and dihydroxyacetone
O P O—O
OH
O
OH
HOHO
AcHNO
Protein-proteininteractions
Endogenousinhibitors
PTMs
Lack of proteinbinding partners
Noinhibitors
No PTMs
In vivo In vitro
Figure 1 Comparison of the endogenous (in vivo) and exogenous (in vitro) environments typically experienced by proteins, highlighting several natural modes for their post-translational regulation.
Figure 2 Genome sequences have served as a foundation for the advancement of global experimental approaches to investigate the function of proteins.
Genome
sequences
GenomicsGene knockouts/RNAi
Genomic enzymology
Assignment of
protein function
Chemical profiling
Molecular engineering
ProteomicsTargeted
Untargeted
Metabolomics
NATURE CHEMICAL BIOLOGY VOLUME 1 NUMBER 3 AUGUST 2005 131
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

phosphate. However, screening of genome archives identified clusters of neighboring genes encoding homologs of key enzymes involved in the eukaryotic tryptophan-to-quinolinate pathway in multiple bacterial species. These bacterial enzymes were shown to produce quinolinate from tryptophan by both biochemical and genetic methods. The predic-tive power of genomic context was once again exploited to identify an unannotated gene with a metal-dependent hydrolase sequence as the N-formylkynurenine formamidase component of the bacterial trypto-phan-to-quinolinate pathway. These studies demonstrate that bacteria can produce quinolinate, the universal de novo precursor to the pyridine ring of NAD, through at least two distinct routes whose enzymatic con-stituents share no sequence, structural or mechanistic homology.
Dunaway-Mariano and colleagues have also provided an intriguing example of sequence-unrelated enzymes that perform similar reac-tions. By comparing the 4-chlorobenzoate (4-CBA) dehalogenation pathway operons from different bacteria, the authors readily identified conserved sequence homologs responsible for the 4-CBA-coenzyme A (4-CBA-CoA) ligase and 4-CBA-CoA dehalogenase activities in this pathway15. Notably, however, the 4-hydroxybenzoate-CoA thioesterase activity was found to be catalyzed by distinct sets of sequence-unrelated enzymes (Fig. 3c). This divergence in protein sequences among 4-CBA
pathways from different bacteria suggests that they did not, as originally suspected, evolve as a recent adaptation to exposure to industrially produced 4-CBA, leading the authors to speculate that natural sources of 4-CBA may exist.
Proteomic methods for the assignment of protein functionThe comparative genomics methods described above are extremely pow-erful for prokaryotic organisms, where genes of related function are often spatially linked in the genome. However, additional technologies are needed to facilitate the global characterization of proteins encoded by eukaryotic genomes. These technologies generally fit under the term proteomics16,17. The daunting size and diversity of eukaryotic proteomes has inspired efforts to profile specific classes of proteins on the basis of shared functional properties18. Within this realm of ‘targeted pro-teomics’, chemical strategies are emerging as a powerful approach to analyze protein function in complex biological systems19,20.
Profiling enzyme activities in proteomes. Conventional genomic and proteomic methods comparatively quantify the expression levels of transcripts21 and proteins22, respectively. From these abundance-based measurements, changes in protein function are inferred. However, many
+
ycjG
ycjI ycjY
ycjZ mppA
dppA dppB dppC dppD dppE ykfA ykfB ykfC ykfD
E. coli
B. subtilis
=YcjG/YkfB
L-Ala-D-Glu L-Ala-L-Glu
Substrate Intermediate Product
OMPCD
OMPDC "suprafamily"
KGPDC
HN
N
O
O CO2-
HN
N
O
OCO2
-
H HN
N
O
O H
CO2-
HO HO
H OHHO H
CH2OPO32-
H OHHO H
CH2OPO32-
O C O
H
O-Mg2+
HO CH2OH
OH OH
HO HCH2OPO3
2-
Substrate Product
Arthrobacter4-HBA-CoA
thioesterases
Pseudomonas
HOO
SCoAHO
O
OHHomologous
Nonhomologous
a
b c
Figure 3 Genomic enzymology. (a) The uncharacterized proteins YcjG and YkfB were determined to be L-Ala-D/L-Glu epimerases by integration of knowledge of genomic context (the murein peptidoglycan metabolic pathway) and the mechanistic versatility of their parent enzyme class (enolase superfamily)9. (b) OMPDC and KGPDC are homologous enzymes that catalyze mechanistically distinct reactions using different substrates10. OMPDC decarboxylates orotidine 5′-monophosphate in a metal-independent manner, whereas KGPDC catalyzes the metal (Mg2+)-dependent decarboxylation of 3-keto-L-gulonate 6-phosphate. These intriguing differences have led to the proposal that these enzymes be considered members of a suprafamily, to designate that they share significant sequence similarity but use distinct catalytic mechanisms. (c) The 4-hydroxybenzoate-CoA (HBA) thioesterase enzymes in the 4-chlorobenzoate dehalogenation pathways of Arthrobacter and Pseudomonas share no sequence homology but perform the same reaction15.
RG/BG TAG
— 1 2 3 4 5 6
m/z
Rel
. int
.
LC-MSCE-MS
SDS-PAGE
— 1 2 3 4 Inhibtor sensitivity
SDS-PAGE
a
b
Figure 4 Activity-based protein profiling (ABPP). (a) General strategy for ABPP. Proteomes are treated with chemical probes that label active enzymes but not enzymes inhibited by intra- or intermolecular regulators or those lacking complementary binding sites. RG, reactive group; BG, binding group; TAG, biotin and/or fluorophore. Probe-labeled proteomes can be analyzed through several platforms, including gel24,27–29 or capillary32 electrophoresis and LC-MS31,32. (b) Inhibitor discovery by ABPP. The potency and selectivity of inhibitors can be profiled in parallel by performing competitive ABPP reactions in whole proteomes29,57,58.
132 VOLUME 1 NUMBER 3 AUGUST 2005 NATURE CHEMICAL BIOLOGY
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

proteins, and in particular enzymes, are regulated by post-translational mechanisms in vivo23, meaning that their expression level may not cor-relate with their activity. To address this important issue, a chemical proteomic strategy has been introduced, referred to as activity-based protein profiling (ABPP)24–26, that uses active site–directed probes to read out the functional state of large numbers of enzymes directly in whole proteomes (Fig. 4a). ABPP probes selectively label active enzymes but not their inactive (for example, zymogen or inhibitor-bound) forms27, facilitating the characterization of changes in enzyme activity that occur in the absence of alterations in protein or transcript expres-sion25,28,29. Additionally, because ABPP probes label enzymes on the basis of shared catalytic properties rather than mere expression level, they provide data sets enriched in low-abundance proteins that can be read out in a range of formats, including gels28,29, microarrays30, liquid chromatography31, and capillary electrophoresis32. These features have enabled ABPP to be applied to a wide range of biological samples, permitting, for example, the identification of new enzyme activities elevated in aggressive cancer cells and tumors28,33,34, invasive malaria parasites35 and obese livers36.
To date, we and others have generated ABPP probes for more than
20 enzyme classes, including members of all major families of prote-ases (serine24, cysteine37–41, metallo42,43, aspartyl44, proteasomal45,46), phosphatases47,48, kinases49, glycosidases50 and glutathione S-transfer-ases51 and a range of oxidoreductases31,51–53 (Table 1). These probes have derived from a combination of two complementary approaches, referred to as directed and nondirected ABPP54. In directed ABPP, probes are designed to target a specific class of enzymes by the incorporation of known affinity labels and/or binding groups into their structures. Directed probes can achieve covalent modification of enzyme active sites by either electrophilic labeling of complementary nucleophilic residues24,37–41,45,46 or photo-cross-linking42–44. Because each directed probe typically targets a large set of mechanistically related enzymes, they have enabled the identification of unannotated members of enzyme superfamilies that could not be classified by sequence analysis55,56. In nondirected ABPP, combinatorial libraries of candidate probes are syn-thesized and screened against complex proteomes to identify specific protein labeling events. Nondirected ABPP probes have used diverse reactive groups, including sulfonate esters51,52 and α-chloroacetamides36 (Table 1), to target several mechanistically distinct enzyme classes. Detailed analyses have provided strong evidence that, in most cases,
Table 1 List of ABPP probes and their respective enzyme targets
Structure
Directed
Nondirected SO
nTag
O
OR
NH
ClO
O
R1
HN
O
NH
CONH2
R2
Tag
FP
EtO On
Tag
X = Halide or Acyloxy
NH
Tag
O
O
EtOO
R
O
HN
O
O
NH
O
O
OHNH
HOTag
NH
HN
NH
OHO
OR
OHN
NH
O
O
CF3
Tag
N N
HNN
NH
HNO
O
O
HN
O OHO
HN
O
NH
O
COOH
O
Tag
R
O
N3HO
HOF
F
NH
OH
O
NH
Tag
O
O
O
OH
MeO
O
O
OTag
O
Enzyme class
Serine hydrolases
Cysteine proteases
Cathepsins
Ubiquitin hydrolases
Metalloprotease
Metalloprotease
Gamma-secretase
Serine/threonine
phosphatases
Tyrosine
phosphatases
Glycosidases
Kinases
Oxidoreductases
Enoyl-CoA hydratases
Glutathione-S-transferases (GSTs)
Hydroxypyruvate reductase
GSTs
Electrophile/
Photo-cross-linker
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Photo-cross-linker
Photo-cross-linker
Photo-cross-linker
References
36
37, 38, 40
HNX
O
ONH
R
HO2C
Tag
NH
UbitquitinO
SO
O
Tag39
23, 24, 26
41
42
43
NH
OS
O
OTag Proteasome 45Electrophile
HN
PO
—O OHTag
Br
O
46
47
48
49
50–52
35
NATURE CHEMICAL BIOLOGY VOLUME 1 NUMBER 3 AUGUST 2005 133
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

nondirected ABPP probes selectively label the active sites of their enzyme targets31, indicating that they should provide an accurate assessment of the functional state of these proteins in proteomes.
Although promiscuous probes are of great value for profiling many enzyme activities in parallel, the assignment of protein function also relies on the development of specific reagents to perturb individual members of large enzyme classes. Toward this end, ABPP has proven effective as a primary screen for the discovery of both reversible57 and irreversible29,58 enzyme inhibitors (Fig. 4b). Inhibitor screening by ABPP has several advantages over conventional substrate assays. First, enzymes can be assayed in their native proteomic context without the need for recombinant expression or purification. Second, inhibitors can be developed for uncharacterized enzymes that lack known substrates57. Finally, because inhibitors are evaluated against many enzymes in para-llel, selective agents can be readily discriminated from nonspecific compounds that target many enzymes. Inhibitor screening by ABPP has culminated in the design of selective covalent probes for several proteases, which have been used to test the function of these enzymes in living systems and as in vivo imaging agents29,34.
One drawback of the original ABPP method is that probes were encumbered with a large reporter tag, which can perturb their cel-lular uptake and distribution. Tag-free versions of ABPP were recently introduced that exploit either the Huisgen’s azide-alkyne cycloaddition reaction (click chemistry)59 or the Staudinger ligation46 to attach the reporter group to activity-based probes after the covalent labeling of protein targets. These advanced ABPP approaches have been used to profile enzyme activities in vivo, resulting in the discovery of enzymes that are selectively labeled in living cells, but not in vitro60.
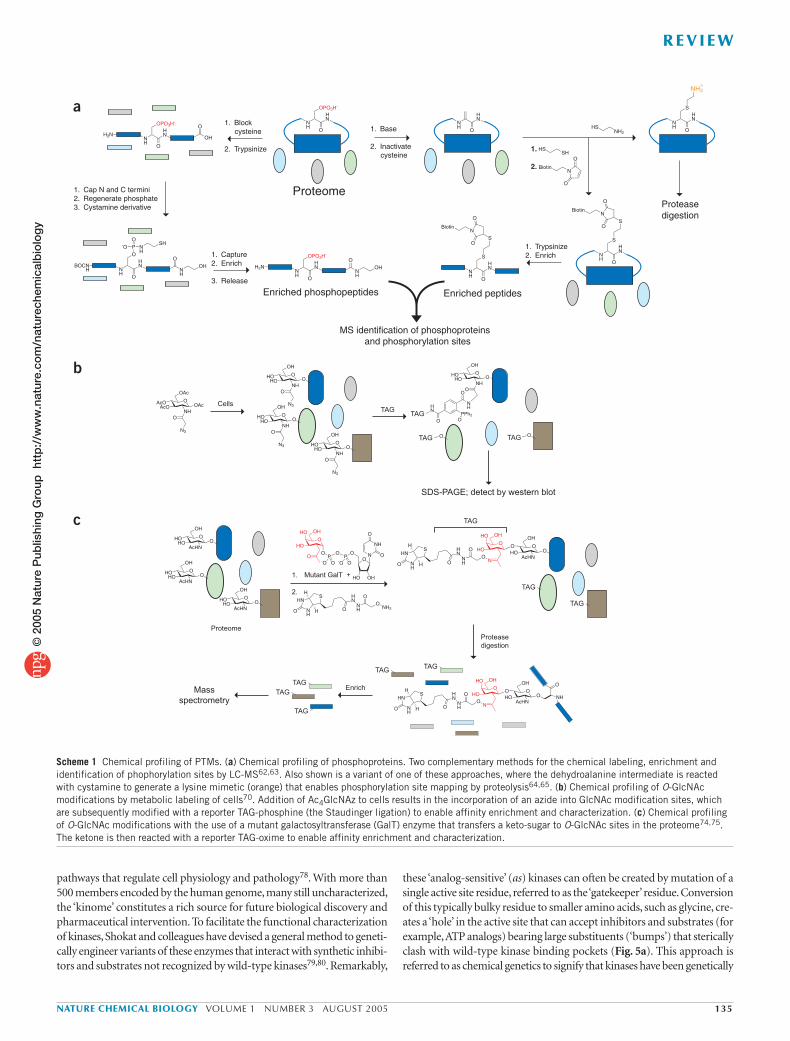
Profiling post-translational modifications in proteomes. Chemical strategies have also been developed to profile many types of post-trans-lational modifications (PTMs) in cells and tissues. Protein phosphoryla-tion events dynamically regulate the function of numerous intracellular proteins and cell signaling pathways61. Phosphorylation sites are dif-ficult to predict from sequence analysis alone; therefore, a major goal of proteomics is to comprehensively map this PTM in both space and time in cells and tissues. The Aebersold and Chait groups have described methods to chemically tag phosphorylated peptides in proteomes for their selective enrichment and characterization62,63. In the Aebersold approach, phosphopeptides are converted to phosphoramidates, which are then conjugated with cystamine and captured by reaction with iodoacetyl groups immobilized on a solid support62 (Scheme 1a, left). Chait and colleagues induced the base-catalyzed β-elimination of phosphoserine and phosphothreonine residues to dehydroalanine, which could then serve as a Michael acceptor for ethanedithiol and subsequently be modified with a maleamide biotin tag63 (Scheme 1a, right). Each of these approaches has distinct advantages and limita-tions, with the Aebersold method being applicable to all three types of phosphorylation (serine, threonione, tyrosine) but requiring more steps, whereas the Chait method is more efficient, but not applicable to phosphotyrosine and susceptible to cross-reaction with O-linked N-acetylglucosamine (O-GlcNAc)–modified peptides (see later discus-sion). Notably, a creative variant of this latter approach was recently introduced by the Hathaway and Shokat groups, who showed that dehy-droalanine peptides can be modified with cysteamine to create trypsin cleavage sites for phosphorylation mapping by selective proteolysis64,65 (Scheme 1a).
Another dynamic PTM that is gaining considerable interest among cell biologists is the O-GlcNAc modification of serine and threonine residues66. O-GlcNAc has been shown to modulate a range of cellular processes, including proteasomal degradation67 and gene silencing68.
Like phosphorylation, O-GlcNAc modifications are dynamic and cannot be easily predicted from the analysis of protein sequences; thus, technologies are needed for their direct identification in complex cell and tissue preparations. Hart and colleagues showed that O-GlcNAc modifications can be selectively eliminated over phosphorylation sites under mild basic conditions, thus making them susceptible to analy-sis through the Michael acceptor chemical tagging method described above69. More recently, the Bertozzi and Hsieh-Wilson groups have described alternative strategies for tagging and enriching O-GlcNAc–modified proteins from proteomes. Bertozzi and colleagues treated cells with N-azidoacetylglucosamine (GlcNAz), resulting in the in situ deliv-ery of a bio-orthogonal group (azide) to sites of O-GlcNAc modifica-tion in the proteome70 (Scheme 1b). GlcNAz-modified proteins could then be derivatized through the Staudinger ligation with a reporter-tagged triarylphosphine ester for purification and characterization. Notably, the Bertozzi group and others have exploited this metabolic labeling method to profile additional PTMs, including mucin-type O-linked glycosylation71 and lipid modifications such as farnesylation72,73.Hsieh-Wilson and colleagues have taken a different approach and engi-neered a β-1,4-galactosyltransferase to append a ketone-containing galactose selectively onto the C-4 hydroxyl of GlcNAc74 (Scheme 1c). These ketone-tagged proteins are then reacted with an aminooxy biotin reagent to allow their affinity purification by avidin chromatography75. One advantage of this strategy is that it can be applied to any proteomic sample, including those not readily amenable to metabolic engineering. For example, 25 O-GlcNAc–modified proteins were identified from rat forebrain, 23 of which were not previously known to be modified with this carbohydrate75. Finally, a chemical labeling approach involving oxidative cleavage and hydrazide reaction on solid phase has been devel-oped to profile N-linked glycoproteins76. The inclusion of an isotope-labeling step permitted comparative quantification of glycoproteins in different proteomic samples.
In summary, chemical tagging has emerged as a preferred strategy for the selective enrichment and characterization of a range of PTMs in proteomes. The molecular information garnered by these approaches extends far beyond that which can be gleaned from standard genom-ics or proteomics. It should also be noted that the success of chemical proteomic methods has been bolstered by tremendous advances in the sensitivity and accuracy of modern protein MS techniques22. We antici-pate that chemical proteomics will continue to play an integral part in the inventorying of PTMs and assessment of the dynamic manner in which these events regulate protein function in vivo.
Engineering specific enzyme-inhibitor pairs. Enzyme superfamilies present an intriguing paradox for postgenomic researchers. On the one hand, recognition from sequence, structural and/or biochemical analyses that a protein belongs to a particular enzyme class restricts its potential activities in vivo and therefore should, in principle, facilitate functional characterization. However, the daunting size of enzyme superfamilies complicates experimental efforts to assign unique physiological activities to individual members. For example, classical genetic approaches, such as targeted gene disruption, are susceptible to compensatory changes by other superfamily members that mask enzyme function. Likewise, inhibitors of enzymes from large classes often lack the requisite selectivity for in vivo pharmacological applica-tion77. Recently, chemical biology strategies have been introduced that integrate the complementary power of organic synthesis and protein engineering to create tools for investigating the function of individual members of enzyme superfamilies.
Protein kinases constitute the largest enzyme class in mammalian proteomes and are key elements of most, if not all, signal transduction
134 VOLUME 1 NUMBER 3 AUGUST 2005 NATURE CHEMICAL BIOLOGY
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y
pathways that regulate cell physiology and pathology78. With more than 500 members encoded by the human genome, many still uncharacterized, the ‘kinome’ constitutes a rich source for future biological discovery and pharmaceutical intervention. To facilitate the functional characterization of kinases, Shokat and colleagues have devised a general method to geneti-cally engineer variants of these enzymes that interact with synthetic inhibi-tors and substrates not recognized by wild-type kinases79,80. Remarkably,
these ‘analog-sensitive’ (as) kinases can often be created by mutation of a single active site residue, referred to as the ‘gatekeeper’ residue. Conversion of this typically bulky residue to smaller amino acids, such as glycine, cre-ates a ‘hole’ in the active site that can accept inhibitors and substrates (for example, ATP analogs) bearing large substituents (‘bumps’) that sterically clash with wild-type kinase binding pockets (Fig. 5a). This approach is referred to as chemical genetics to signify that kinases have been genetically
NH
O
HN
HSSH
N
O
O
NH
O
HN
S
SN
O
O
Biotin
NH
O
HN
S
SN
O
O
Biotin
1. Base
2. Inactivate cysteine
1.
2. Biotin
1. Trypsinize2. Enrich
Enriched peptides
H2N
O
OH
O
NH
OHNH
O
O
HN
P-OO
NH
SH
BOCNH
NH
OPO3H-
O
HN
Proteome
NH
OPO3H-
O
HN
1. Block cysteine
2. Trypsinize
1. Cap N and C termini2. Regenerate phosphate3. Cystamine derivative
1. Capture2. Enrich
3. Release
H2NNH
OPO3H-
O
HN
O
NH
OH
Enriched phosphopeptides
MS identification of phosphoproteins and phosphorylation sites
HSH2N
NH
O
HN
S
H3N+
Proteasedigestion
a
b
c
TAG
TAG
TAG
O
OH
HOHO
AcHNO
O
OH
HOHO
AcHNO
O
OH
HOHO
AcHNO
O
OH
HO
O
HO
OP
OP
O
-O O -O OO
HO OH
NH
N
O
O
HN
NH
O H
HN
HS
ONH
OO
NH2
1.
2.
O
OH
OHO
AcHNO
O
OH
HO
HO
HN
NH
O H
HN
HS
ONH
OO
N
TAG
TAG
TAG
Protease
digestion
O
OH
OHO
AcHNO
O
OH
HO
HO
HN
NH
O H
HN
HS
ONH
OO
N
NH
O
TAG
TAGMass
spectrometry
O
OAc
AcOAcO
NHOAc
N3
O
Cells
O
OH
HOHO
NHO
N3
O
O
OH
HOHO
NHO
N3
O
O
OH
HOHO
NHO
N3
O
O O
NH
O
PPh2
O
HN
O
OH
HOHO
NHO
O
O
SDS-PAGE; detect by western blot
Enrich
Proteome
Mutant GalT +
TAG
TAG
TAG
TAG
Scheme 1 Chemical profiling of PTMs. (a) Chemical profiling of phosphoproteins. Two complementary methods for the chemical labeling, enrichment and identification of phophorylation sites by LC-MS62,63. Also shown is a variant of one of these approaches, where the dehydroalanine intermediate is reacted with cystamine to generate a lysine mimetic (orange) that enables phosphorylation site mapping by proteolysis64,65. (b) Chemical profiling of O-GlcNAc modifications by metabolic labeling of cells70. Addition of Ac4GlcNAz to cells results in the incorporation of an azide into GlcNAc modification sites, which are subsequently modified with a reporter TAG-phosphine (the Staudinger ligation) to enable affinity enrichment and characterization. (c) Chemical profiling of O-GlcNAc modifications with the use of a mutant galactosyltransferase (GalT) enzyme that transfers a keto-sugar to O-GlcNAc sites in the proteome74,75. The ketone is then reacted with a reporter TAG-oxime to enable affinity enrichment and characterization.
NATURE CHEMICAL BIOLOGY VOLUME 1 NUMBER 3 AUGUST 2005 135
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

engineered for sensitivity to specific chemical reagents.Chemical genetics has been successfully implemented to test the roles
of many kinases in signaling events, both in culture and in vivo. For example, chemical inhibition of an as mutant of the budding yeast kinase Prk1p revealed a role for this protein in regulating actin patches involved in endocytosis81. An as mutant of CamKIIα was introduced by trans-genic methods selectively into the forebrains of mice, providing evidence that the activity of this kinase is required during the first postlearning week for memory consolidation82. Both of these studies highlight one of the primary advantages of chemical genetic methods, namely, tight temporal control over dynamic, kinase-regulated cellular and behav-ioral processes. An additional benefit was demonstrated by Witte and colleagues, who showed that chemical genetics can be used to infer the mechanism of action of kinase-directed inhibitors83. Specifically, selec-tive inhibition of an as variant of the oncogenic kinase BCR-ABL did not eliminate immature myeloid leukemic cells, which contrasts with the cytotoxic effects of the BCR-ABL–directed inhibitor imatinib mesylate (Gleevec) in these cells (Fig. 5b). These results suggest that the thera-peutic efficacy of imatinib is caused by the inhibition of multiple kinases in addition to BCR-ABL (for example, KIT, which is also expressed in immature myeloid leukemic cells). Finally, as variants can also be used in combination with ATP analogs to profile kinase substrate selectivity in proteomic extracts, as exemplified by the studies of Morgan and col-leagues on the cyclin-dependent kinase Cdk1 (ref. 84).
Given the impressive success of chemical genetic applications to kinases, an obvious next question is whether this approach can be generally extended to other enzyme superfamilies. Although studies that speak to this issue are still relatively scarce, initial results with
ATPases85, GTPases86,87, cyclophilins88 and phosphatases89 are encouraging. Indeed, an as variant of myosin 1c has revealed a specific role for this motor protein in controlling adapative responses in hair cells of the inner ear85. Moreover, recent advances reported by Shokat and colleagues to address kinase family members intolerant to original gatekeeper mutations offer methodological and biophysical guidelines that should assist future chemical genetics endeavors relating to this enzyme family and others90.
Engineering specific post-translational modifications in proteins. The historical view of a one-to-one relationship between gene and pro-tein has been replaced by the provocative realization that numerous protein products derive from a single gene2. These protein variants arise by multiple mechanisms, including alternative splicing, proteo-lytic processing and PTM. Accordingly, as described above, a major goal of proteomics is to inventory these epigenetic events to achieve a more comprehensive understanding of the diversity of protein species that exist in vivo. However, an equally if not more important problem
N
N NN
NH2
N
N NN
NH2
Otherkinases
KIT
BCR-ABL
BCR-ABLas
OtherKinases
KIT
Imatinibmesylate
Otherkinases
KIT
BCR-ABL Eliminates BCR-ABL+ KIT+expressing cells
NaPP1
Otherkinases
KIT
Insufficient to eliminate BCR-ABL+ KIT+
expressing cells
Wild type
as mutants
v-Srcc-Fync-AblCDK2CAMKIIv-Src-I338GcFyn-T339Gc-Abl-T315ACDK2-F80GCAMKII-F89G
10.60.61822
0.00150.00650.00700.0150.097
281.03.42924
0.00430.00320.12
0.00500.0080
IC50(µM) IC50(µM)
BCR-ABLas
a
b
Figure 5 Chemical genetic analysis of kinases. (a) Kinases are genetically engineered to show sensitivity to inhibitors that are not active against wild-type kinases80. These as mutant kinases have been used to investigate the mechanism of action of the cancer drug imatinib mesylate (b), showing that it produces maximal cytotoxic effects by inhibiting multiple kinases (BCR-ABL and KIT)83.
Protein expression
Synthetic PTMmodified peptide
NH
OHS N
H
HN
PHO O—O
O
Nonhydrolyzablephosphotyrosine
PTM
S NAcyl Shift
O PO
OO—
NO2
O PO
OO—
—hν Inhibition of
14-3-3 proteins
hν
Active
O PS—
HOO
OP
S
HO O
O
OH
Inactive
ATP(γ)S
PDK1kinase
PKA
InteinO
SNH2
O
SNH2
hν
Activecofilin
Inactivecofilin
S CO2—
NO2
SH
RRLYRSLP RRLYRSLP
Alkylate
Alkylate
a
b
c
d
Scheme 2 Introducing specific PTMs into proteins via chemical methods. (a) The EPL method91, where proteins bearing specific phosphorylation sites are semisynthesized for biophysical and cell biological studies. Inset shows a nonhydrolyzable analog of phosphotyrosine used to characterize the role of phosphorylation in SHP-2 (refs. 92,93). (b–d) Methods for the creation of caged phosphorylation sites that can be unveiled by light. (b) A caged phosphovariant of PKA generated with thiophosphorylation chemistry95. (c) Caged phosphopeptides that bind 14-3-3 proteins generated by solid-phase synthesis97,100. (d) A negative-regulatory phosphorylation site in cofilin is regulated by conversion to either cysteine (active) or a caged cysteine variant (inactive)101.
136 VOLUME 1 NUMBER 3 AUGUST 2005 NATURE CHEMICAL BIOLOGY
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

is to determine the unique functions performed by different protein isoforms. Indeed, the current (and rapidly expanding) catalog of PTMs far exceeds the number of cases where the physiological significance of these events has been deduced. The ability to create homogeneous preparations of proteins bearing specific PTMs is essential for their bio-chemical and cell biological characterization. Multiple chemical biology research programs have been initiated with this goal in mind.
Cole, Muir and colleagues have pioneered the development and application of expressed protein ligation (EPL) methods for the semi-synthesis of post-translationally modified proteins. These approaches exploit the versatile native chemical ligation reaction, in which two protein fragments, one containing a C-terminal thioester (generated through fusion to an intein protein) and the other containing an N-terminal cysteine, react and rearrange through an S-to-N acyl shift to form a native peptide bond91 (Scheme 2a). Significantly, protein substrates for EPL can derive from recombinant or synthetic sources, greatly expanding the range of chemical modifications that can be site-specifically incorporated into proteins. Cole and colleagues haveused EPL to generate homogeneous preparations of singly and doub-ly phosphorylated forms of the tyrosine phosphatase SHP-2 (refs. 92,93). In these studies, a nonhydrolyzable phosphotyrosine mimetic was incorporated into SHP-2, permitting dissection of the roles of spe-cific phosphorylation events in regulating the biochemical and cellular activity of this phosphatase. Muir and colleagues have exploited EPL to decipher the contribution of individual phosphorylation events to the oligomerization of Smad2 and its recognition as a substrate for transforming growth factor β kinase94.
Chemical approaches have also been devised for the creation of caged phosphopeptides or phosphoproteins, allowing exquisite spatial and temporal control over their function in vitro and in living cells. Bayley and colleagues have described the semi-synthesis of thiophosphory-lated variants of protein kinase A (PKA), which is accomplished by incubation of this protein with a kinase in the presence of ATP(γ)S95 (Scheme 2b). The thiophosphoryl group can then be selectively caged on the basis of its enhanced nucleophilicity. Thiophosphoryl groups
have the advantage of being resistant to phosphatase cleavage and thus show enhanced stability in living cells. One limitation of this approach, however, is that enzyme-catalyzed thiophosphorylation reactions do not yet constitute a generally applicable way to generate any phosphory-lated protein.
Imperiali and colleagues have pioneered a complementary approach that uses versatile 1-(2-nitrophenyl) ethyl–modified serine, threonine and tyrosine building blocks for the synthesis of caged phosphoproteins or phosphopeptides96,97 (Scheme 2c). These caged phosphoamino acids can be incorporated into proteins or peptides through several mecha-nisms, including direct synthesis97, in vitro translation using nonsense suppression methods98 and EPL99. Imperiali, Yaffe and colleagues have applied caged phosphopeptides to disrupt the function of the 14-3-3 proteins in living cells100. Notably, with the use of a consensus phospho-peptide motif that interacts with all 14-3-3 proteins (Scheme 2c), this chemical biology approach allowed the synchronous inactivation of an entire family of adaptor proteins, a task that would have been difficult to accomplish with traditional genetic methods.
Finally, Lawrence and colleagues have shown that the activity of the actin-severing protein cofilin can be controlled by mutagenesis of a serine phosphorylation site to either cysteine (active) or a caged cysteine variant modified with α-bromo-(2-nitrophenyl) acetic acid (inactive)101 (Scheme 2d). Photoactivation of the caged profilin in liv-ing cells revealed new roles for this protein in actin-based cytoskeletal dynamics. This approach may prove generally useful for studying the activity of proteins that are negatively regulated by phosphorylation.
It is clear from the examples provided here that chemical biology approaches offer powerful methods and tools to test the functional significance of specific PTMs. To date, most of these studies have focused on protein phosphorylation, and it will be exciting to witness as approaches such as EPL and caged amino acid derivatives are extended to the analysis of additional PTMs. Toward this end, the ability to site-specifically incorporate a range of amino acid derivatives into proteins remains a paramount objective. Additionally, many proteins, such as integral membrane proteins, are not easily produced in purified form
H2NO
OH O
OH
O O
MeO
GlcO
OGlcGlcOOH
HO O
OHOH
O
OH
O
OO
OMe
HO
HO
OMeOH
NH2
O
OH
O
OH
O
HOO
O
OHO
O
OH
O
HONH2
+ +
NH
SO3—
O
n
OH
O
n H2NOH
H2NSO3
—
NH
OHO
n
OH
O
n
+
+
NH2
RO
OH
NR OH N
R
HO
SNH2
CO2HN
R
HOSGlucose
N
R
— O3SOSGlucose
PAL1
mBCAT
FAAHKaempferol
ConiferylCoumarins
Leu Gluα-KG α-KIC
CYP +cysteine
SUR1 +glucose
PAPS +sulfotransferaseCYP
Primarysulfur
metabolism
NAE
NAT
FFA Ethanolamine
TaurineFFA
Phe
a
b
c
d
Scheme 3 Identification of endogenous substrates of enzymes by global metabolite profiling. (a,b) Targeted metabolite profiling methods have been used to explain (a) the role of PAL1 in the biosynthesis of phenylpropanoids (kaempferol, coniferyl, coumarins)105 and (b) a role for mBCAT in the generation of branched chain α-keto acids106. Deletion of mBCAT leads to molecular and physiological phenotypes in mice that are reminiscent of human maple syrup disease. (c) Discovery (untargeted) metabolite profiling identified a structurally new class of brain natural products, the NATs, that are regulated by FAAH in vivo107. (d) Untargeted metabolite profiling, in combination with transcriptional profiling, identified the plant sulfotransferases involved in GLS metabolism110.
NATURE CHEMICAL BIOLOGY VOLUME 1 NUMBER 3 AUGUST 2005 137
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

through either chemical synthesis or recombinant expression. Recent advances by the Schultz and Muir groups that extend technologies such as non-natural amino acid incorporation102,103 and EPL104 to in vivo settings should expand the spectrum of post-translationally modified proteins amenable to molecular and cellular analysis.
Metabolomic methods for the assignment of protein functionA protein’s function can be defined on many levels—molecular, cel-lular and physiological. Although these functions are all interrelated (for example, an enzyme performs chemistry to generate a product (molecular) that affects a cell (cellular), which in turn affects the higher organism (physiological)), the technologies required for their experi-mental investigation are distinct. For example, genetic approaches such as gene knockouts3 and RNAi4, as well as the proteomic methods delin-eated in the previous section, can be combined with cell- and organism-based phenotypic screens to characterize the cellular and physiological functions of proteins. However, the endogenous biochemical activities of proteins are less straightforward to address with these methods. For enzymes, in particular, the identification of natural substrates often remains an elusive goal. Recent advances in global metabolite profiling offer powerful new ways to address this problem.
Enzymes regulate biological processes through the conversion of spe-cific substrates to products. Therefore, the identification of an enzyme’s full complement of physiological substrates is of paramount importance. The systematic characterization of endogenous substrates of enzymes requires the advancement of new technologies that can broadly profile the molecular composition of cells and tissues. The field of metabolo-mics has emerged with this objective in mind. Metabolome researchers are confronted with several technical challenges that, in many ways, sur-pass those of genomics and proteomics. First, as opposed to transcripts and proteins, metabolites are not directly linked to the genetic code, but are instead the products of the concerted action of complex enzymatic networks. Similarly, metabolites are not linear polymers composed of a defined set of monomers, but rather a remarkably diverse collection of structures with differing chemical and physical properties. These dis-tinguishing features have inspired the development of both targeted and untargeted analytical methods for metabolite profiling that, when coupled with genomic and proteomic technologies, provide a powerful platform for forging direct connections among enzymes, substrates and the higher-order physiological processes they regulate.
Assignment of enzyme function by targeted metabolite profiling. Targeted metabolomic experiments have proven effective at characteriz-ing the physiological relationship between a defined set of enzymes and natural products. For example, by profiling the metabolic consequences of disrupting distinct phenylalanine ammonia lyase (PAL) enzymes in Arabidopsis thaliana, Rhode and colleagues succeeded in differentiating their unique biochemical functions105. PAL enzymes had long been recognized to participate in the biosynthesis of the phenylpropanoid class of natural products (for example, lignins, flavonoids, coumarins); however, which PAL enzymes performed these chemical transforma-tions in vivo has remained unclear. Targeted liquid chromatography-ultraviolet (LC-UV) spectroscopy revealed a primary role for PAL1 in the biosynthesis of phenylpropanoids, with a secondary role for PAL2 (Scheme 3a). The authors also performed complementary genomic studies on PAL gene mutants, leading to the identification of additional metabolic pathways altered in these organisms. These results provided the first global view of the extent to which individual PAL enzymes are integrated into the larger metabolic networks of plant cells.
Targeted metabolite profiling has also been used as a screen to ‘mole-cularly phenotype’ mutant organisms originating from forward genetic
screens. Wu and colleagues profiled plasma amino acid and acylcarnitine levels in ethylnitrosourea (ENU)-treated mice by direct-inject tandem MS analysis, resulting in the discovery of a mutant mouse line with highly elevated levels of branched-chain amino acids (BCAAs)106. These mice also showed signs of wasting (reduced body weight and motility). Similar phenotypes are observed in humans with defects in branched-chain α-keto acid dehydrogenase (BCKD), leading to maple syrup urine disease. Surprisingly, however, the mutant mice did not show reduced BCKD activity, but rather a loss in the function of the mitochondrial branched-chain aminotransferase (mBCAT) enzyme, which converts BCAAs to branched-chain α-keto acids (Scheme 3b), the natural sub-strates of BCKD. These results indicate that the disruption of one of multiple enzymes in the BCAA biosynthetic pathway leads to a common set of metabolic and physiological defects.
Assignment of enzyme function by untargeted metabolite profiling. The targeted profiling methods described above provide a robust and sensitive way to quantify known metabolites in complex biological samples. However, these approaches are not suited for the discovery of new metabolites and may therefore fail to report on the full spectrum of substrates used by a given enzyme in vivo. To address this problem, we have recently introduced a complementary liquid chromatography-mass spectrometry (LC-MS)–based strategy for ‘untargeted’ metabolo-mics referred to as discovery metabolite profiling (DMP)107. In contrast to targeted LC-MS, in which the levels of specific metabolites are mea-sured by selected ion monitoring (SIM) in conjunction with isotopic standards, DMP operates as a global, standard-free method, where the relative levels of numerous metabolites are quantified in parallel by the direct measurement of their mass ion intensities in a broad mass scanning mode (Fig. 6).
DMP was applied to characterize the tissue metabolomes of wild-type mice and mice lacking the enzyme fatty acid amide hydrolase [FAAH–/– mice], which degrades the N-acyl ethanolamine (NAE) class of signaling lipids in vivo (Scheme 3c), including the endocannabinoid anandamide108. Consistent with previous targeted LC-MS studies109, DMP accurately measured an approximately ten-fold increase in the central nervous system (CNS) levels of NAEs in FAAH–/– mice. The detection of low-abundance NAEs such as anandamide indicated that DMP maintained good sensitivity (estimated to be within one order of magnitude of targeted methods). Interestingly, DMP also detected a second class of CNS metabolites that were highly elevated in FAAH–/–
mice. These metabolites did not belong to any of the known classes of endogenous lipids, suggesting that they constituted new natural products. This premise was confirmed by subsequent analytical and synthetic chemistry efforts, which identified the uncharacterized class of FAAH-regulated lipids as conjugates of fatty acids and the amino acid taurine [N-acyl taurines (NATs)] (Scheme 3c). The function of the FAAH-NAT metabolic pathway in mammalian biology is under active investigation. Finally, other major classes of known lipids, including fatty acids, phospholipids, monoacylglycerols and ceramides, were not altered in FAAH–/– mice, supporting a specific role for this enzyme in the metabolism of endogenous amidated lipids. These results under-score the power of untargeted profiling methods such as DMP, which can provide a more comprehensive portrait of the endogenous sub-strates of enzymes and, through doing so, illuminate unanticipated connections between the proteome and metabolome.
Untargeted metabolite profiling has more recently been imple-mented, in combination with gene-expression profiling, to identify a class of sulfotransferases involved in glucosinolate (GLS) metabolism in plants110. The GLS metabolic pathway has been an active subject of research for many years, and most of its participating enzymes had been
138 VOLUME 1 NUMBER 3 AUGUST 2005 NATURE CHEMICAL BIOLOGY
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

previously characterized, with the exception of the sulfotransferases. Saito and colleagues monitored in an untargeted manner the time-dependent changes in the metabolome and transcriptome induced by sulfur deprivation using Fourier transform MS and DNA microarrays, respectively. A collection of GLS metabolites and known biosynthetic genes were found to show similar expression profiles. Interestingly, within this collection of coregulated genes, the authors also found three putative sulfotransferases. Recombinant expression and biochemical analysis demonstrated that these proteins can indeed convert desulfo-allyl GLS to sulfonated allyl GLS (Scheme 3d, red), confirming their identification as GLS sulfotransferases. These findings indicate that the integration of metabolomic and transcriptomic data can reveal func-tions for uncharacterized enzymes.
In summary, global metabolite profiling methods provide access to a valuable portion of biomolecular space that is largely inaccessible to genomic and proteomic methods. Significantly, much of the informa-tion provided by metabolomics, especially as relates to protein function assignment, may be difficult if not impossible to procure by classical biochemical methods. For example, the substrate selectivity of FAAH, as determined in vitro with purified preparations of enzyme, was largely unpredictive of the substrates used by this enzyme in vivo107. These findings reinforce the importance of characterizing enzymes within the context of the cells and tissues in which they are naturally expressed, where potentially competitive and/or interdependent metabolic path-ways are taken into account. Future efforts will probably focus on expanding the portion of the metabolome that is addressable by DMP methods. Lipids, which can be enriched by organic extraction, were an obvious first target. The extension of DMP to other classes of metabo-lites will require the advent of methods for their selective enrichment. Another important challenge will be to devise ways to accelerate the determination of metabolite structures, possibly through the creation of searchable databases that contain the chemical and physical features of all known metabolites (for example, their mass fragmentation pat-terns, solvent solubility profiles, susceptibility to chemical modifica-tion). Solutions to these provocative problems, which may very well come from the field of chemical biology, should greatly enhance our understanding of how enzymes and their substrates interact to form the higher-order metabolic and signaling networks that govern cell and organismal behavior.
Summary and Future DirectionsIn this review, we have attempted to achieve two major goals. First, we hope to have captured the remarkable power of integrated chemical and biological approaches (dubbed ‘chemical biology’) for the deter-mination of protein function. Whether it be the characterization of new metabolic pathways in bacteria, or the identification of enzyme activities and protein PTMs in mammalian proteomes, or the disco-very of physiological substrates of enzymes, chemical biology efforts are perhaps best defined by their motivation to unite methods and tools from historically separate disciplines. This integrated approach, where complementary techniques such as genetics, protein engineer-ing, organic synthesis and analytical chemistry are brought to bear on a single research problem, has allowed inquiries into the functional state of proteins in highly complex biological samples, providing insights into their molecular, cellular and physiological activities that extend far beyond the information accessible to any individual discipline.
Our second objective was to illuminate the essentiality of genome sequences to research endeavors that aim to describe protein function on a systematic and global scale. Indeed, many of the technologies described in this review are founded in classical experimental approaches, such as operon analysis, affinity labeling and site-directed mutagenesis. However, their conversion into global strategies for the characterization of protein function was only possible with the availability of complete genome sequences. For example, the sequencing of several hundred bacterial genomes has fostered the emergence of new fields of research, such as genomic enzymology, where biochemical, structural and comparative genomic methods converge to reveal unexpected functional relation-ships among members of enzyme super- and suprafamilies. Likewise, the molecular information procured by chemical proteomic investigations of protein activity and PTM state would be virtually uninterpretable without searchable databases of MS fragmentation patterns of all pro-teins encoded by a given genome. Finally, and perhaps most impor-tantly, genome sequences provide a context in which to evaluate the full complement of proteins from a given family to discern their unique
Targeted analysis
DMP (untargeted)
Mas
s io
n in
tens
ityRetention time
m/z 347.5
Deuteratedstandard
Endogenousmetabolite
Rel
ativ
e in
tens
ity
m/z340 360
Knockout
20
40
60
80
100
12
3
4
Mas
s io
n in
tens
ity
Retention time
m/z 200–1200
Wild type
20
40
60
80
100
Knockout
12
3
4
Mas
s io
n in
tens
ity
Retention time
m/z 200–120020
40
60
80
100
Mas
s io
n in
tens
ity
Retention time
m/z 347.5
Deuteratedstandard
Endogenousmetabolite
Rel
ativ
e in
tens
ity
m/z340 360
Wild type
20
40
60
80
100
RatioKO/WT
Global metabolite analysis
25
20
15
10
5
0
–5
–10
KO/WTratio
Retentiontime
m/z
90 80 70 60 50
40
30
20 200 400
600
800
1,000
a
b
Figure 6 Comparison of targeted and untargeted LC-MS methods for comparative metabolite analysis. (a) General scheme for targeted LC-MS analysis, in which metabolites are detected by SIM (shown for a metabolite with a mass of 347.5) and their levels are quantified by comparing mass signals to those of isotopically distinct internal standards. (b) General scheme for DMP107, an untargeted global LC-MS approach, in which metabolites are detected in the broad mass scanning mode (for example, 200–1200 mass units) and their levels quantified by measurement of direct mass ion intensities (that is, without the inclusion of internal standards). Enzyme-regulated metabolites are identified by comparison of mass ion intensity ratios between wild-type and knockout samples.
NATURE CHEMICAL BIOLOGY VOLUME 1 NUMBER 3 AUGUST 2005 139
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

and overlapping functions. In this regard, enzymes such as kinases and hydrolases, for which there are several hundred representatives in the human genome, stand as a humbling reminder of how much still remains to be learned about even the most ‘familiar’ classes of proteins.
In looking to the future, we recognize several challenges that con-front researchers interested in applying postgenomic methods to the functional characterization of proteins. First, as the reader has probably noticed, this review has placed a disproportionate emphasis on enzymes over other types of proteins. We believe that this bias is rooted in several factors. First, the function of enzymes, which generally equates with catalytic activity, is, at least on first pass, simpler to define than for many other proteins. Enzymes also possess well-defined active sites, which harbor conserved catalytic residues that can be exploited to cre-ate class-wide approaches for experimental analysis (for example, sites for comparative sequence alignment, affinity labeling or mutagenesis). Nonetheless, we anticipate that many of the principles established for the postgenomic examination of enzymes can be extended to other types of proteins, such as receptors and ion channels, which also share common molecular and structural features that should make them vulnerable to unified strategies for functional characterization.
A more general problem relates to the somewhat arbitrary definition of ‘protein function’ itself, which can vary depending on the mode of scientific investigation. Indeed, it is becoming clear that many proteins in both prokaryotic and eukaroytic organisms have been misannotated on the basis of a cursory inspection of their sequences111. Although increas-ingly sophisticated sequence analysis programs such as Shotgun112,113 should help to rectify these errors, experimental approaches still stand as the only satisfactory way to verify the actual activities performed by proteins. Several of the methods described in this review use molecular engineering to create specific variants of proteins for functional analysis. Although these approaches offer the compelling advantage of isolating one protein (or even one PTM state of a protein) for characterization in complex biological settings, they generally involve heterologous expres-sion systems and, thus, often do not address the function of proteins in their native environment. In contrast, chemical profiling methods can be applied to any biological system (even primary human biopsies) to assess the functional state of natively expressed proteins. However, these profiling technologies provide only an associative relationship between the activ-ity or PTM state of proteins and a particular (patho)physiological event. Follow-up studies are needed to deduce whether the identified proteins actually contribute to the higher-order processes under investigation. Given that molecular engineering and chemical profiling methods possess complementary strengths and weaknesses, we suggest that future chemi-cal biology efforts be aimed at fully developing these technologies such that a complete spectrum of experimental approaches is available for the functional characterization of all protein classes in the proteome. Before the achievement of this ultimate goal, we can be satisfied with the fact that chemistry, by providing unique reagents and methods for interrogating the function of proteins in complex biological systems, promises to have a prominent role in most, if not all, forms of postgenomic inquiry.
ACKNOWLEDGMENTSThe authors apologize to many wonderful colleagues whose work we were unable to cite because of space limitations. We gratefully acknowledge the support of the US National Institutes of Health (CA087660, DA017259, DA015197), the Helen L. Dorris Child and Adolescent Neuropsychiatric Disorder Institute, and the Skaggs Institute for Chemical Biology. A.S. is a Merck Fellow of the Life Sciences Research Foundation.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Published online at http://www.nature.com/naturechemicalbiology/
1. Gerlt, J.A. & Babbitt, P.C. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu. Rev. Biochem. 70, 209–246 (2001).
2. Walsh, C.T. Posttranslational Modification of Proteins. Expanding Nature’s Inventory (Roberts & Company, Greenwood Village, Colorado, USA, 2005).
3. Austin, C.P. et al. The knockout mouse project. Nat. Genet. 36, 921–924 (2004).4. Carpenter, A.E. & Sabatini, D.M. Systematic genome-wide screens of gene function.
Nat. Rev. Genet. 5, 11–22 (2004).5. Clardy, J. & Walsh, C. Lessons from natural molecules. Nature 432, 829–837
(2004).6. Gerlt, J.A. & Raushel, F.M. Evolution of function in (β/α)8-barrel enzymes. Curr. Opin.
Chem. Biol. 7, 252–264 (2003).7. Neidhart, D.J., Kenyon, G.L., Gerlt, J.A. & Petsko, G.A. Mandelate racemase and
muconate lactonizing enzyme are mechanistically distinct and structurally homolo-gous. Nature 347, 692–694 (1990).
8. Osterman, A. & Overbeek, R. Missing genes in metabolic pathways: a comparative genomics approach. Curr. Opin. Chem. Biol. 7, 238–251 (2003).
9. Schmidt, D.M., Hubbard, B.K. & Gerlt, J.A. Evolution of enzymatic activities in the enolase superfamily: functional assignment of unknown proteins in Bacillus subtilis and Escherichia coli as L-Ala-D/L-Glu epimerases. Biochemistry 40, 15707–15715 (2001).
10. Wise, E., Yew, W.S., Babbitt, P.C., Gerlt, J.A. & Rayment, I. Homologous (β/α)8-barrel enzymes that catalyze unrelated reactions: orotidine 5′-monophosphate decarboxyl-ase and 3-keto-L-gulonate 6-phosphate decarboxylase. Biochemistry 41, 3861–3869 (2002).
11. Wang, S.C., Person, M.D., Johnson, W.H., Jr & Whitman, C.P. Reactions of trans-3-chloroacrylic acid dehalogenase with acetylene substrates: consequences of and evidence for a hydration reaction. Biochemistry 42, 8762–8773 (2003).
12. de Jong, R.M., Brugman, W., Poelarends, G.J., Whitman, C.P. & Dijkstra, B.W. The X-ray structure of trans-3-chloroacrylic acid dehalogenase reveals a novel hydration mechanism in the tautomerase superfamily. J. Biol. Chem. 279, 11546–11552 (2004).
13. Mizanur, R.M., Jaipuri, F.A. & Pohl, N.L. One-step synthesis of labeled sugar nucleo-tides for protein O-GlcNAc modification studies by chemical function analysis of an archaeal protein. J. Am. Chem. Soc. 127, 836–837 (2005).
14. Kurnasov, O. et al. NAD biosynthesis: identification of the tryptophan to quinolinate pathway in bacteria. Chem. Biol. 10, 1195–1204 (2003).
15. Zhuang, Z., Gartemann, K.H., Eichenlaub, R. & Dunaway-Mariano, D. Characterization of the 4-hydroxybenzoyl-coenzyme A thioesterase from Arthrobacter sp. strain SU. Appl. Environ. Microbiol. 69, 2707–2711 (2003).
16. Patterson, S.D. & Aebersold, R. Proteomics: the first decade and beyond. Nat. Genet. 33, 311–323 (2003).
17. de Hoog, C.L. & Mann, M. Proteomics. Annu. Rev. Genomics Hum. Genet. 5, 267–293 (2004).
18. Corthals, G.L., Wasinger, V.C., Hochstrasser, D.F. & Sanchez, J.C. The dynamic range of protein expressioin: a challenge for proteomic research. Electrophoresis 21, 1104–1115 (2000).
19. Adam, G.C., Sorensen, E.J. & Cravatt, B.F. Chemical strategies for functional pro-teomics. Mol. Cell. Proteomics 1, 781–790 (2002).
20. Verhelst, S.H. & Bogyo, M. Chemical proteomics applied to target identification and drug discovery. Biotechniques 38, 175–177 (2005).
21. Brown, P.O. & Botstein, D. Exploring the new world of the genome with DNA microar-rays. Nat. Genet. 21, 33–37 (1999).
22. Aebersold, R. & Mann, M. Mass spectrometry-based proteomics. Nature 422, 198–207 (2003).
23. Kobe, B. & Kemp, B.E. Active site-directed protein regulation. Nature 402, 373–376 (1999).
24. Liu, Y., Patricelli, M.P. & Cravatt, B.F. Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. USA 96, 14694–14699 (1999).
25. Jessani, N. & Cravatt, B.F. The development and application of methods for activity-based protein profiling. Curr. Opin. Chem. Biol. 8, 54–59 (2004).
26. Berger, A.B., Vitorino, P.M. & Bogyo, M. Activity-based protein profiling: applications to biomarker discovery, in vivo imaging and drug discovery. Am. J. Pharmacogenomics 4, 371–381 (2004).
27. Kidd, D., Liu, Y. & Cravatt, B.F. Profiling serine hydrolase activities in complex proteomes. Biochemistry 40, 4005–4015 (2001).
28. Jessani, N., Liu, Y., Humphrey, M. & Cravatt, B.F. Enzyme activity profiles of the secreted and membrane proteome that depict cancer invasiveness. Proc. Natl. Acad. Sci. USA 99, 10335–10340 (2002).
29. Greenbaum, D. et al. Chemical approaches for functionally probing the proteome. Mol. Cell. Proteomics 1, 60–68 (2002).
30. Sieber, S.A., Mondala, T.S., Head, S.R. & Cravatt, B.F. Microarray platform for profil-ing enzyme activities in complex proteomes. J. Am. Chem. Soc. 126, 15640–15641 (2004).
31. Adam, G.C., Burbaum, J.J., Kozarich, J.W., Patricelli, M.P. & Cravatt, B.F. Mapping enzyme active sites in complex proteomes. J. Am. Chem. Soc. 126, 1363–1368 (2004).
32. Okerberg, E.S. et al. High-resolution functional proteomics by active-site peptide profiling. Proc. Natl. Acad. Sci. USA 102, 4996–5001 (2005).
33. Jessani, N. et al. Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo. Proc. Natl. Acad. Sci. USA 101, 13756–13761 (2004).
34. Joyce, J.A. et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 5, 443–453 (2004).
35. Greenbaum, D.C. et al. A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science 298, 2002–2006 (2002).
140 VOLUME 1 NUMBER 3 AUGUST 2005 NATURE CHEMICAL BIOLOGY
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

36. Barglow, K.T. & Cravatt, B.F. Discovering disease-associated enzymes by proteome reactivity profiling. Chem. Biol. 11, 1523–1531 (2004).
37. Greenbaum, D., Medzihradszky, K.F., Burlingame, A. & Bogyo, M. Epoxide electro-philes as activity-dependent cysteine protease profiling and discovery tools. Chem. Biol. 7, 569–581 (2000).
38. Thornberry, N.A. et al. Inactivation of interleukin-1 β converting enzyme by peptide (acyloxy)methyl ketones. Biochemistry 33, 3934–3940 (1994).
39. Faleiro, L., Kobayashi, R., Fearnhead, H. & Lazebnik, Y. Multiple species of CPP32 and Mch2 are the major active caspases present in apoptotic cells. EMBO J. 16, 2271–2281 (1997).
40. Borodovsky, A. et al. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem. Biol. 9, 1149–1159 (2002).
41. Kato, D. et al. Activity-based probes that targe diverse cysteine protease families. Nat. Chem. Biol. 1, 33–38 (2005).
42. Saghatelian, A., Jessani, N., Joseph, A., Humphrey, M. & Cravatt, B.F. Activity-based probes for the proteomic profiling of metalloproteases. Proc. Natl. Acad. Sci. USA 101, 10000–10005 (2004).
43. Chan, E.W., Chattopadhaya, S., Panicker, R.C., Huang, X. & Yao, S.Q. Developing photoactive affinity probes for proteomic profiling: hydroxamate-based probes for metalloproteases. J. Am. Chem. Soc. 126, 14435–14446 (2004).
44. Li, Y.M. et al. Photoactivated γ-secretase inhibitors directed to the active site cova-lently label presenilin 1. Nature 405, 689–694 (2000).
45. Groll, M., Nazif, T., Huber, R. & Bogyo, M. Probing structural determinants distal to the site of hydrolysis that control substrate specificity of the 20S proteasome. Chem. Biol. 9, 655–662 (2002).
46. Ovaa, H. et al. Chemistry in living cells: detection of active proteasomes by a two-step labeling strategy. Angew. Chem. Int. Edn Engl. 42, 3626–3629 (2003).
47. Kumar, S. et al. Activity-based probes for protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA 101, 7943–7948 (2004).
48. Shreder, K.R. et al. Design and synthesis of AX7574: a microcystin-derived, fluo-rescent probe for serine/threonine phosphatases. Bioconjug. Chem. 15, 790–798 (2004).
49. Liu, Y. et al. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem. Biol. 12, 99–107 (2005).
50. Vocadlo, D.J. & Bertozzi, C.R. A strategy for functional proteomic analysis of gly-cosidase activity from cell lysates. Angew. Chem. Int. Edn Engl. 43, 5338–5342 (2004).
51. Adam, G.C., Sorensen, E.J. & Cravatt, B.F. Proteomic profiling of mechanistically distinct enzyme classes using a common chemotype. Nat. Biotechnol. 20, 805–809 (2002).
52. Adam, G.C., Cravatt, B.F. & Sorensen, E.J. Profiling the specific reactivity of the proteome with non-directed activity-based probes. Chem. Biol. 8, 81–95 (2001).
53. Adam, G.C., Sorensen, E.J. & Cravatt, B.F. Trifunctional chemical probes for the con-solidated detection and identification of enzyme activities from complex proteomes. Mol. Cell. Proteomics 1, 828–835 (2002).
54. Speers, A.E. & Cravatt, B.F. Chemical strategies for activity-based proteomics. ChemBioChem 5, 41–47 (2004).
55. Jessani, N. et al. Class assignment of sequence-unrelated members of enzyme superfamilies by activity-based protein profiling. Angew. Chem. Int. Edn Engl. 44, 2400–2403 (2005).
56. Baxter, S.M. et al. Synergistic computational and experimental proteomics approaches for more accurate detection of active serine hydrolases in yeast. Mol. Cell. Proteomics 3, 209–225 (2004).
57. Leung, D., Hardouin, C., Boger, D.L. & Cravatt, B.F. Discovering potent and selec-tive reversible inhibitors of enzymes in complex proteomes. Nat. Biotechnol. 21, 687–691 (2003).
58. Greenbaum, D.C. et al. Small molecule affinity fingerprinting. A tool for enzyme family subclassification, target identification, and inhibitor design. Chem. Biol. 9, 1085–1094 (2002).
59. Speers, A.E., Adam, G.C. & Cravatt, B.F. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 125, 4686–4687 (2003).
60. Speers, A.E. & Cravatt, B.F. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 11, 535–546 (2004).
61. Johnson, S.A. & Hunter, T. Kinomics: methods for deciphering the kinome. Nat. Methods 2, 17–25 (2005).
62. Zhou, H., Watts, J.D. & Aebersold, R. A systematic approach to the analysis of protein phosphorylation. Nat. Biotechnol. 19, 375–378 (2001).
63. Oda, Y., Nagasu, T. & Chait, B.T. Enrichment analysis of phosphorylated proteins as a tool for probing the phosphoproteome. Nat. Biotechnol. 19, 379–382 (2001).
64. Rusnak, F., Zhou, J. & Hathaway, G.M. Identification of phosphorylated and glyco-sylated sites in peptides by chemically targeted proteolysis. J. Biomol. Tech. 13, 228–237 (2002).
65. Knight, Z.A. et al. Phosphospecific proteolysis for mapping sites of protein phos-phorylation. Nat. Biotechnol. 21, 1047–1054 (2003).
66. Zachara, N.E. & Hart, G.W. O-GlcNAc a sensor of cellular state: the role of nucleocy-toplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta 1673, 13–28 (2004).
67. Zhang, F. et al. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115, 715–725 (2003).
68. Yang, X., Zhang, F. & Kudlow, J.E. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repres-sion. Cell 110, 69–80 (2002).
69. Wells, L. et al. Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol. Cell. Proteomics 1, 791–804 (2002).
70. Vocadlo, D.J., Hang, H.C., Kim, E.J., Hanover, J.A. & Bertozzi, C.R. A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proc. Natl. Acad. Sci. USA 100, 9116–9121 (2003).
71. Hang, H.C., Yu, C., Kato, D.L. & Bertozzi, C.R. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. USA 100, 14846–14851 (2003).
72. Kho, Y. et al. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc. Natl. Acad. Sci. USA 101, 12479–12484 (2004).
73. Rose, M.W. et al. Enzymatic incorporation of orthogonally reactive prenylazide groups into peptides using geranylazide diphosphate via protein farnesyltransferase: implica-tions for selective protein labeling. Biopolymers 80, 164–171 (2005).
74. Khidekel, N. et al. A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications. J. Am. Chem. Soc. 125, 16162–16163 (2003).
75. Khidekel, N., Ficarro, S.B., Peters, E.C. & Hsieh-Wilson, L.C. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proc. Natl. Acad. Sci. USA 101, 13132–13137 (2004).
76. Zhang, H., Li, X.J., Martin, D.B. & Aebersold, R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 (2003).
77. Davies, S.P., Reddy, H., Caivano, M. & Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 (2000).
78. Manning, G., Whyte, D.B., Martinez, R., Hunter, T. & Sudarsanam, S. The protein kinase complement of the human genome. Science 298, 1912–1934 (2002).
79. Shah, K., Liu, Y., Deirmengian, C. & Shokat, K.M. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct sub-strates. Proc. Natl. Acad. Sci. USA 94, 3565–3570 (1997).
80. Bishop, A.C. et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 (2000).
81. Sekiya-Kawasaki, M. et al. Dynamic phosphoregulation of the cortical actin cyto-skeleton and endocytic machinery revealed by real-time chemical genetic analysis. J. Cell Biol. 162, 765–772 (2003).
82. Wang, H. et al. Inducible protein knockout reveals temporal requirement of CaMKII reactivation for memory consolidation in the brain. Proc. Natl. Acad. Sci. USA 100, 4287–4292 (2003).
83. Wong, S. et al. Sole BCR-ABL inhibition is insufficient to eliminate all myeloprolif-erative disorder cell populations. Proc. Natl. Acad. Sci. USA 101, 17456–17461 (2004).
84. Ubersax, J.A. et al. Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864 (2003).
85. Holt, J.R. et al. A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell 108, 371–381 (2002).
86. Weijland, A. & Parmeggiani, A. Toward a model for the interaction between elongation factor Tu and the ribosome. Science 259, 1311–1314 (1993).
87. Powers, T. & Walter, P. Reciprocal stimulation of GTP hydrolysis by two directly interacting GTPases. Science 269, 1422–1424 (1995).
88. Levitsky, K., Ciolli, C.J. & Belshaw, P.J. Selective inhibition of engineered receptors via proximity-accelerated alkylation. Org. Lett. 5, 693–696 (2003).
89. Hoffman, H.E., Blair, E.R., Johndrow, J.E. & Bishop, A.C. Allele-specific inhibitors of protein tyrosine phosphatases. J. Am. Chem. Soc. 127, 2824–2825 (2005).
90. Zhang, C. et al. A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nat. Methods 2, 435–441 (2005).
91. Muir, T.W. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 72, 249–289 (2003).
92. Lu, W., Gong, D., Bar-Sagi, D. & Cole, P.A. Site-specific incorporation of a phosphoty-rosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol. Cell 8, 759–769 (2001).
93. Lu, W., Shen, K. & Cole, P.A. Chemical dissection of the effects of tyrosine phos-phorylation of SHP-2. Biochemistry 42, 5461–5468 (2003).
94. Ottesen, J.J., Huse, M., Sekedat, M.D. & Muir, T.W. Semisynthesis of phosphovariants of Smad2 reveals a substrate preference of the activated T β RI kinase. Biochemistry 43, 5698–5706 (2004).
95. Zou, K., Cheley, S., Givens, R.S. & Bayley, H. Catalytic subunit of protein kinase A caged at the activating phosphothreonine. J. Am. Chem. Soc. 124, 8220–8229 (2002).
96. Rothman, D.M., Vazquez, M.E., Vogel, E.M. & Imperiali, B. General method for the synthesis of caged phosphopeptides: tools for the exploration of signal transduction pathways. Org. Lett. 4, 2865–2868 (2002).
97. Vazquez, M.E., Nitz, M., Stehn, J., Yaffe, M.B. & Imperiali, B. Fluorescent caged phosphoserine peptides as probes to investigate phosphorylation-dependent protein associations. J. Am. Chem. Soc. 125, 10150–10151 (2003).
98. Rothman, D.M. et al. Caged phosphoproteins. J. Am. Chem. Soc. 127, 846–847 (2005).
99. Hahn, M.E. & Muir, T.W. Photocontrol of Smad2, a multiphosphorylated cell-signaling protein, through caging of activating phosphoserines. Angew. Chem. Int. Edn Engl. 43, 5800–5803 (2004).
100. Nguyen, A., Rothman, D.M., Stehn, J., Imperiali, B. & Yaffe, M.B. Caged phospho-peptides reveal a temporal role for 14–3-3 in G1 arrest and S-phase checkpoint function. Nat. Biotechnol. 22, 993–1000 (2004).
NATURE CHEMICAL BIOLOGY VOLUME 1 NUMBER 3 AUGUST 2005 141
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y

101. Ghosh, M. et al. Cofilin promotes actin polymerization and defines the direction of cell motility. Science 304, 743–746 (2004).
102. Wang, L., Brock, A., Herberich, B. & Schultz, P.G. Expanding the genetic code of Escherichia coli. Science 292, 498–500 (2001).
103. Chin, J.W. et al. An expanded eukaryotic genetic code. Science 301, 964–967 (2003).
104. Giriat, I. & Muir, T.W. Protein semi-synthesis in living cells. J. Am. Chem. Soc. 125, 7180–7181 (2003).
105. Rohde, A. et al. Molecular phenotyping of the pal1 and pal2 mutants of Arabidopsis thaliana reveals far-reaching consequences on phenylpropanoid, amino acid, and carbohydrate metabolism. Plant Cell 16, 2749–2771 (2004).
106. Wu, J.Y. et al. ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J. Clin. Invest. 113, 434–440 (2004).
107. Saghatelian, A. et al. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry 43, 14332–14339 (2004).
108. Cravatt, B.F. et al. Molecular characterization of an enzyme that degrades neuro-modulatory fatty-acid amides. Nature 384, 83–87 (1996).
109. Cravatt, B.F. et al. Supersensitivity to anandamide and enhanced endogenous can-nabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 98, 9371–9376 (2001).
110. Hirai, M.Y. et al. Elucidation of gene-to-gene and metabolite-to-gene networks in Arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem., published online May 2005 (doi:10.1074/jbc.M502332200).
111. Babbitt, P.C. Definitions of enzyme function for the structural genomics era. Curr. Opin. Chem. Biol. 7, 230–237 (2003).
112. Pegg, S.C. & Babbitt, P.C. Shotgun: getting more from sequence similarity searches. Bioinformatics 15, 729–740 (1999).
113. Copley, S.D., Novak, W.R. & Babbitt, P.C. Divergence of function in the thioredoxin fold suprafamily: evidence for evolution of peroxiredoxins from a thioredoxin-like ancestor. Biochemistry 43, 13981–13995 (2004).
142 VOLUME 1 NUMBER 3 AUGUST 2005 NATURE CHEMICAL BIOLOGY
R E V I E W©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y
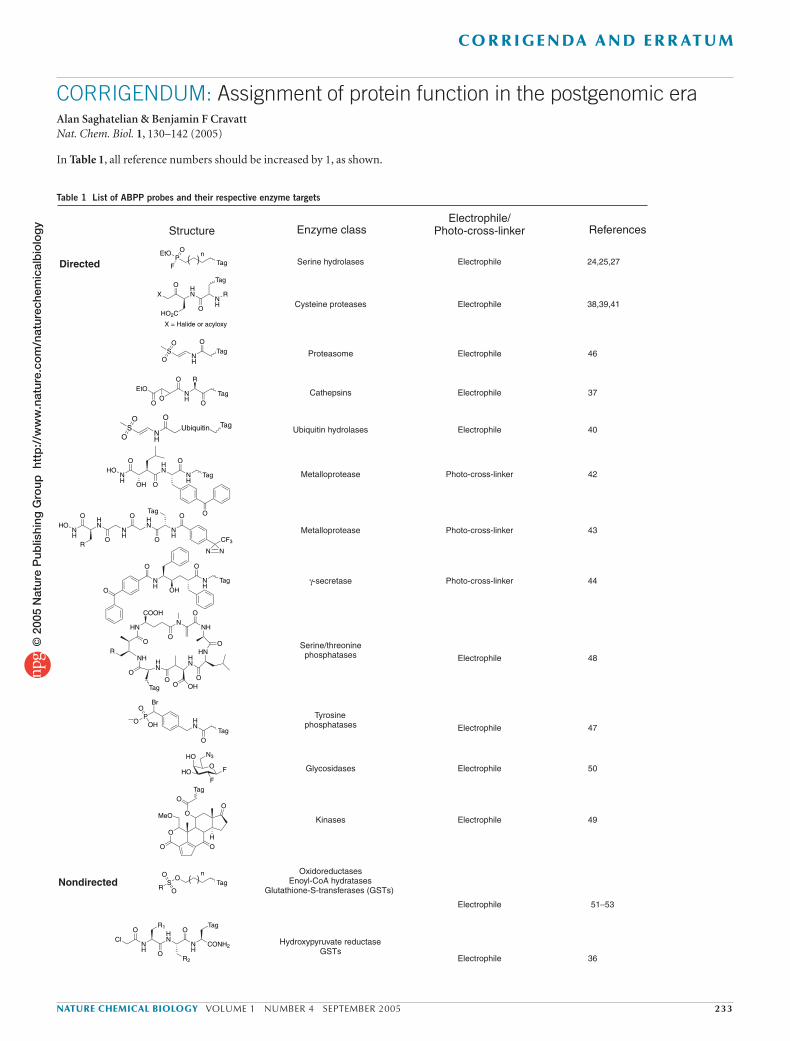
CORRIGENDUM: Assignment of protein function in the postgenomic eraAlan Saghatelian & Benjamin F CravattNat. Chem. Biol. 1, 130–142 (2005)
In Table 1, all reference numbers should be increased by 1, as shown.
Structure
Directed
Nondirected SO
nTag
O
OR
NH
ClO
O
R1
HN
O
NH
CONH2
R2
Tag
FP
EtO On
Tag
X = Halide or acyloxy
NH
Tag
O
O
EtOO
R
O
HN
O
O
NH
O
O
OHNH
HOTag
NH
HN
NH
OHO
OR
OHN
NH
O
O
CF3
Tag
N N
HNN
NH
HNO
O
O
HN
O OHO
HN
O
NH
O
COOH
O
Tag
R
O
N3HO
HOF
F
NH
OH
O
NH
Tag
O
O
O
OH
MeO
O
O
OTag
O
Enzyme class
Serine hydrolases
Cysteine proteases
Cathepsins
Ubiquitin hydrolases
Metalloprotease
Metalloprotease
γ-secretase
Serine/threoninephosphatases
Tyrosinephosphatases
Glycosidases
Kinases
OxidoreductasesEnoyl-CoA hydratases
Glutathione-S-transferases (GSTs)
Hydroxypyruvate reductaseGSTs
Electrophile/Photo-cross-linker
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Electrophile
Photo-cross-linker
Photo-cross-linker
Photo-cross-linker
References
37
38,39,41
HNX
O
ONH
R
HO2C
Tag
NH
UbiquitinO
SO
O
Tag40
24,25,27
42
43
44
NH
OS
O
OTag Proteasome 46Electrophile
HN
PO
—O OHTag
Br
O
47
48
49
50
51–53
36
Table 1 List of ABPP probes and their respective enzyme targets
NATURE CHEMICAL BIOLOGY VOLUME 1 NUMBER 4 SEPTEMBER 2005 233
CO R R I G E N DA A N D E R R AT U M©
200
5 N
atur
e P
ublis
hing
Gro
up h
ttp
://w
ww
.nat
ure.
com
/nat
urec
hem
ical
bio
log
y
Related Documents











