COMMENTARY Assessing the Performance of Amorphous Solid Dispersions ANN NEWMAN, 1 GREGORY KNIPP, 2 GEORGE ZOGRAFI 3 1 Seventh Street Development Group, Lafayette, Indiana 47901 2 Department of Industrial and Physical Pharmacy, School of Pharmacy, Purdue University, West Lafayette, Indiana 47907 3 School of Pharmacy, University of Wisconsin—Madison, Madison, Wisconsin 53705 Received 24 August 2011; revised 21 November 2011; accepted 7 December 2011 Published online 27 December 2011 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/jps.23031 ABSTRACT: The characterization and performance of stable amorphous solid dispersion sys- tems were evaluated in 40 research papers reporting active pharmaceutical ingredient (API) dissolution and bioavailability from various systems containing polymers. The results from these studies were broadly placed into three categories: amorphous dispersions that improved bioavailability (∼82% of the cases), amorphous dispersions possessing lower bioavailability than the reference material (∼8% of the cases), and amorphous dispersions demonstrating similar bioavailabilities as the reference material (∼10% of the cases). A comparative anal- ysis of these studies revealed several in vitro and in vivo variables that could have influ- enced the results. The in vitro factors compared primarily centered on dissolution testing and equipment, content and amount of dissolution media, sink or nonsink conditions, agitation rates, media pH, dissolution characteristics of the polymer, and dispersion particle size. The in vivo factors included reference materials used for bioavailability comparisons, animal species utilized, fasting versus fed conditions, and regional differences in gastrointestinal (GI) con- tent and volume. On the basis of these considerations, a number of recommendations were made on issues ranging from the assessment of physical stability of API–polymer dispersions to in vivo GI physiological factors that require consideration in the performance evaluation of these systems. © 2011 Wiley Periodicals, Inc. and the American Pharmacists Association J Pharm Sci 101:1355–1377, 2012 Keywords: amorphous; bioavailability; dissolution; in vitro–in vivo correlation (IVIVC); polymers; solid dispersion; solid dosage form; absorption; stabilization INTRODUCTION In recent years, there has been an increased inter- est in the use of amorphous forms of active pharma- ceutical ingredients (APIs) in various formulations, especially when their crystalline forms are shown to exhibit very poor aqueous solubility. This often leads to inadequate rates of dissolution and oral bioavailability. 1,2 Amorphous forms tend to exhibit high levels of supersaturation in aqueous media rela- tive to the crystal, and thus higher apparent solubil- ity. These increases arise from the lack of a highly ordered crystal with lattice energies that must be overcome to attain adequate solubility of the crystal. Correspondence to: Ann Newman (Telephone: +765-650-4462; Fax: +765-742-1062; E-mail: [email protected]) Journal of Pharmaceutical Sciences, Vol. 101, 1355–1377 (2012) © 2011 Wiley Periodicals, Inc. and the American Pharmacists Association As amorphous forms are thermodynamically unsta- ble relative to the crystal, we would expect a spon- taneous tendency for the solid to revert back to the crystalline form. This commonly occurs during stor- age in the solid state at various relative humidities and temperatures, 3 and when the amorphous form encounters in vitro and in vivo dissolution media, 4 which in both cases negates the solubility advantage of the amorphous form. The challenge, therefore, is to inhibit crystallization over the time period of product storage and to maintain a sufficient level of supersat- uration upon oral administration without crystalliza- tion. Currently, a major strategy used to obtain good physical stability, as well as enhanced dissolution and oral bioavailability, is to use amorphous solid disper- sions. In these systems, the API is combined with a water-soluble polymer to produce a single-phase amorphous mixture of the API and the polymer. 5 JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 1355

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

COMMENTARY
Assessing the Performance of Amorphous Solid Dispersions
ANN NEWMAN,1 GREGORY KNIPP,2 GEORGE ZOGRAFI3
1Seventh Street Development Group, Lafayette, Indiana 47901
2Department of Industrial and Physical Pharmacy, School of Pharmacy, Purdue University, West Lafayette, Indiana 47907
3School of Pharmacy, University of Wisconsin—Madison, Madison, Wisconsin 53705
Received 24 August 2011; revised 21 November 2011; accepted 7 December 2011
Published online 27 December 2011 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/jps.23031
ABSTRACT: The characterization and performance of stable amorphous solid dispersion sys-tems were evaluated in 40 research papers reporting active pharmaceutical ingredient (API)dissolution and bioavailability from various systems containing polymers. The results fromthese studies were broadly placed into three categories: amorphous dispersions that improvedbioavailability (∼82% of the cases), amorphous dispersions possessing lower bioavailabilitythan the reference material (∼8% of the cases), and amorphous dispersions demonstratingsimilar bioavailabilities as the reference material (∼10% of the cases). A comparative anal-ysis of these studies revealed several in vitro and in vivo variables that could have influ-enced the results. The in vitro factors compared primarily centered on dissolution testing andequipment, content and amount of dissolution media, sink or nonsink conditions, agitationrates, media pH, dissolution characteristics of the polymer, and dispersion particle size. Thein vivo factors included reference materials used for bioavailability comparisons, animal speciesutilized, fasting versus fed conditions, and regional differences in gastrointestinal (GI) con-tent and volume. On the basis of these considerations, a number of recommendations weremade on issues ranging from the assessment of physical stability of API–polymer dispersionsto in vivo GI physiological factors that require consideration in the performance evaluationof these systems. © 2011 Wiley Periodicals, Inc. and the American Pharmacists AssociationJ Pharm Sci 101:1355–1377, 2012Keywords: amorphous; bioavailability; dissolution; in vitro–in vivo correlation (IVIVC);polymers; solid dispersion; solid dosage form; absorption; stabilization
INTRODUCTION
In recent years, there has been an increased inter-est in the use of amorphous forms of active pharma-ceutical ingredients (APIs) in various formulations,especially when their crystalline forms are shownto exhibit very poor aqueous solubility. This oftenleads to inadequate rates of dissolution and oralbioavailability.1,2 Amorphous forms tend to exhibithigh levels of supersaturation in aqueous media rela-tive to the crystal, and thus higher apparent solubil-ity. These increases arise from the lack of a highlyordered crystal with lattice energies that must beovercome to attain adequate solubility of the crystal.
Correspondence to: Ann Newman (Telephone: +765-650-4462;Fax: +765-742-1062; E-mail: [email protected])Journal of Pharmaceutical Sciences, Vol. 101, 1355–1377 (2012)© 2011 Wiley Periodicals, Inc. and the American Pharmacists Association
As amorphous forms are thermodynamically unsta-ble relative to the crystal, we would expect a spon-taneous tendency for the solid to revert back to thecrystalline form. This commonly occurs during stor-age in the solid state at various relative humiditiesand temperatures,3 and when the amorphous formencounters in vitro and in vivo dissolution media,4
which in both cases negates the solubility advantageof the amorphous form. The challenge, therefore, is toinhibit crystallization over the time period of productstorage and to maintain a sufficient level of supersat-uration upon oral administration without crystalliza-tion.
Currently, a major strategy used to obtain goodphysical stability, as well as enhanced dissolution andoral bioavailability, is to use amorphous solid disper-sions. In these systems, the API is combined witha water-soluble polymer to produce a single-phaseamorphous mixture of the API and the polymer.5
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 1355

1356 NEWMAN, KNIPP, AND ZOGRAFI
Such miscibility appears to be essential for main-taining long-term physical stability of the amorphousAPI, as well as appropriate levels of supersaturationupon dissolution. A review of the literature reveals,however, that the term solid dispersion is often usedmore generally to describe a variety of solid mixturesof API and excipients with the intent of improvingoral bioavailability, and can include mixtures of crys-talline API and polymers, solid complexes of API withcomplexing agents such as the cyclodextrins, and API“dissolved” in solid lipid-based excipients.2 The fo-cus for this paper will be restricted to amorphousAPI–polymer solid dispersions and the term amor-phous solid dispersion will be used to describe thesesystems.
The major polymers in pharmaceutical use forpreparing amorphous solid dispersions are either wa-ter soluble at all pH conditions or those exhibitingthe dissolution properties of enteric coating systemsunder more alkaline conditions. Pharmaceutical poly-mers used for amorphous solid dispersions includepoly(vinylpyrrolidone) (PVP), poly(vinylpyrrolidone/vinyl acetate copolymer), hydroxypropylmethyl cellu-lose (HPMC), hydroxypropylmethyl cellulose acetate/succinate (HPMCAS), hydroxypropylmethyl cellulosephthalate (HPMCP), and the acrylic acid-based en-teric Eudragit systems.
Amorphous dispersions typically are formed bycombining the API and polymer in a melt extruder,or by dissolving them in a solvent, followed by dryingsuch as in spray drying or lyophilization.6,7 The soliddispersion may contain surfactants, such as Tween80, Span 80, Vitamin E polyethylene glycol 1000 suc-cinate (D-"-tocopheryl polyethylene glycol 1000 suc-cinate), or Cremophor [Polyoxyl 35 castor oil, UnitedStates Pharmacopeia (USP)/National Formulary], tofacilitate processing and/or to facilitate dissolution.For example, surfactants appear to be particularlyuseful for facilitating the formation of amorphousdispersions during the melt extrusion process andfor promoting dissolution from dispersions containinghigh levels of hydrophobic API. The resulting disper-sion generally is considered miscible when only oneglass transition temperature, Tg, is observed usingdifferential scanning calorimetry,8,9 and when phaseseparation is not detectable by other analytical tech-niques such as X-ray diffraction9 and solid-state nu-clear magnetic resonance.10
In most pharmaceutical situations, these single-phase amorphous dispersions contain amounts of APIin excess of the equilibrium solubility of the API crys-tals in the polymer; in other words, an amount ofAPI that is supersaturated relative to the solubil-ity of the crystalline form. This supersaturation ismaintained by the apparent miscibility of the compo-nents in the amorphous state, but often with a ther-modynamic tendency for phase separation into two
amorphous phases and eventual API crystallization.True miscibility of amorphous components would rep-resent a thermodynamically stable one-phase systemas with the miscibility of two liquids. Recent studieshave reported values for the true equilibrium solu-bility of selected crystalline APIs in polymers to helpascertain the maximum API load in the dispersionthat would not have any thermodynamic tendencyto crystallize.11,12 These values are generally quitelow at temperatures near and below the Tg, which, ofcourse, limits the amount of API (i.e., the dose) thatcan be produced as a true thermodynamically stabledispersion. Hence, in the case of dosage levels gener-ally required, there is a need to use supersaturatedsingle-phase dispersions. It should be mentioned inthis regard that differences between processing condi-tions and handling and storage conditions may causea supersaturated one-phase system to undergo phaseseparation. For example, a one-phase system formedat high temperatures, as in a melt extruder, mightphase separate when cooled to lower temperatures.In such a case, the distribution of phase-separatedcomponents may or may not be homogeneous. In-deed, it is possible that even one-phase systems uponprocessing may exhibit clustering or other forms ofinhomogeneity.
In general, relatively small amounts of polymer invarious dispersions have been shown to significantlyinhibit crystallization both in the solid state beforeadministration and after introduction of the disper-sion into dissolution media and gastrointestinal (GI)tract fluids.4,13,14 The ability of polymers to inhibitcrystallization in stored samples can be linked to theTg of the polymer relative to that of the API andthe ability of the polymer to raise the overall Tg of thedispersion, which reduces API molecular mobility atnormally encountered storage temperatures and rel-ative humidities.3,8 Polymers capable of specificallyinteracting with the API in the dispersion, such asthrough hydrogen bonding, have been demonstratedto inhibit crystallization at very low concentrations.In these systems, the effects on Tg are not signifi-cant, and the hydrogen bonding provides stability bydirectly interfering with the nucleation and crystalgrowth processes, where stronger interaction ener-gies provide greater resistance to crystallization.13–15
The various factors that can influence the extent andrate of crystal nucleation and growth of amorphousAPI are quite complex, making it difficult to predictlong-term stability as a function of temperature andrelative humidity from studies conducted under ac-celerated conditions. However, it has been suggestedthat as a “rule of thumb” one can avoid crystallizationof amorphous API over long periods by storing sam-ples at temperatures that are in the range of 50 Kor greater below the Tg of the dispersion. Such stor-age temperatures below Tg appear to be where the
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1357
molecular mobility of the API has been reduced totimescales of a few years.15
Once the solid dispersion encounters in vitro orin vivo dissolution media, supersaturation in solutionmust be maintained over a period of time that willensure complete dissolution and potentially increasebioavailability.2,4,16 If the API and polymer dissolverapidly, supersaturation must be maintained by fac-tors, including the dissolved polymer, that can inhibitsolution-mediated nucleation and crystalline growthof the API. In this regard, the ability of some poly-mers in solution to prevent crystallization of a su-persaturated API solution has been demonstrated.17
It is also possible that the API–polymer combina-tion does not dissolve immediately, but rather re-mains intact as a more slowly dissolving structure,thus reducing the level of API supersaturation andtendency to crystallize. It is also very likely that insuch situations any surfactants present can furtherprevent crystallization by promoting the formation ofmicelles and other colloidal aggregates that interferewith solution-mediated crystallization. It has beenshown, for example, that since the chemical structureof HPMCAS appears to have the characteristics of anamphiphilic- or surfactant-like polymer, hydrophobicdrugs likely can interact with HPMCAS to form col-loidal structures in aqueous solution, and that thismechanism may be responsible for the excellent abil-ity of this polymer to maintain high levels of APIsupersaturation.16 Bile salt micelles and other lipidsfound in vivo in the GI tract may also function in thismanner to maintain high levels of supersaturation.In summary, it appears very likely that a very criti-cal part of obtaining long-term supersaturation uponin vivo release is the tendency for the API in the amor-phous state to remain associated as colloidal struc-tures with the polymer, with surfactants if present inthe dispersion, and with natural surfactant-like ma-terials found in the GI tract. Consequently, in vitrostudies should be conducted to assess the possibleimportance of this mechanism, including the use ofvarious simulated intestinal fluids.
The major challenge faced by scientists using amor-phous solid dispersions, therefore, is storage sta-bility and maintenance of postadministration su-persaturation. Preparation of acceptable dispersionsystems includes rationally choosing methods ofpreparation, the best polymer and polymer molecu-lar weight grade, the optimal API loading (or poly-mer concentration), and analytical and formulationmethodologies that can predict and alleviate any in-stabilities. Once this is determined, performance ofthe dispersion then needs to be evaluated.
This Commentary will discuss the issues associatedwith establishing and characterizing stable amor-phous solid dispersion systems that will ultimatelyincrease the ability of investigators to provide a clini-
cally reproducible, safe, and efficacious response uponadministration. In particular, the paper will addressa specific set of questions related to assessing clinicalperformance including:
(1) Is there sufficient experimental evidence to saythat amorphous dispersions always improveperformance, such as bioavailability?
(2) Has it been possible to demonstrate an in vitro–in vivo relationship (IVIVR) with amorphousdispersions through the use of appropriate dis-solution methodology?
(3) What in vivo issues should be considered whensetting up animal/human studies for proof ofconcept and potential clinical support withamorphous dispersions?
EXPERIMENTAL EVIDENCE FOR AMORPHOUSSOLID DISPERSION PERFORMANCE
Many of the papers on amorphous solid dispersionsstate that dispersions can help improve clinical per-formance (bioavailability, and in some cases bioactiv-ity) because of the increased solubility exhibited bythe amorphous drug. However, there is still a need todemonstrate the validity of this assumption in prac-tice. A reasonable way to approach such a question isto critically evaluate the numerous published studiesperformed to date, determine the number of studiesthat have demonstrated improved performance for anamorphous dispersion, and critically assess and dis-cuss key determining factors.
It is important to define the relationship betweenbioavailability and bioactivity for a pharmaceuticalsystem. Bioavailability in practice refers to the rateand extent of absorption into the blood/plasma froma dosage form and is related to pharmacokinetics(PK). Bioactivity is due to the API molecular struc-ture and the interactions that occur with a biolog-ical target or targets to give a pharmacological re-sponse. Generally, bioactivity changes arising froman amorphous solid dispersion are related to the min-imum effective and minimum toxic blood/plasma con-centrations that define the Therapeutic window.18
In practice, an amorphous dispersion may increasebioactivity through the subsequent increase in theexposure level (bioavailability). In a significant num-ber of cases, an increase in bioactivity observed for anamorphous dispersion may be the result of targetedbioavailability,19 where the concentration of the APIis increased primarily in the area of the therapeutictarget due to the increased exposure. Therefore, theincrease in exposure can influence the pharmacody-namics (PD) observations made but does not alter theAPI’s bioactivity.
To assess the question of improved bioavailabil-ity for amorphous solid dispersions, a reasonably
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1358 NEWMAN, KNIPP, AND ZOGRAFI
significant sample of research papers were collectedwhere bioavailability, or exposure, of amorphous soliddispersions was reported. An attempt was made toinclude as many studies as possible to get represen-tative data, although it is acknowledged that the re-sulting list cannot be considered comprehensive. Over80 papers were found that included the use of soliddispersions in animal or human studies. The reportswere reviewed to determine if sufficient characteriza-tion data were available to confirm that the drug wasamorphous. If the drug was amorphous, the excipi-ents in the studies were then evaluated and studieswith polymers as the main component were included.Ternary systems with a polymer and a surfactant orlipid were also included if the polymer was the maincomponent. Systems containing the API dispersedonly in surfactants and lipid formulation components,such as Gelucire R© and phospholipids, were not in-cluded because they were considered lipid-based sys-tems that rely on a different supersaturation mech-anism, for example, micelles and microemulsions.Cyclodextrin-based systems were also not includedin our analysis because complexation is the predom-inate mechanism used to produce a supersaturatedsystem.
On the basis of these criteria, a total of 40 stud-ies were surveyed, as summarized in, Table 1.20–69
For the first assessment, results from the biologicalstudies were broadly placed into three categories: (1)the amorphous dispersion improved the bioavailabil-ity, (2) the amorphous dispersion bioavailability waslower than that of a reference entity, and (3) the amor-phous dispersion demonstrated the same bioavailabil-ity as the reference entity. In some cases, the amor-phous dispersion was compared with more than onereference entity; if the amorphous dispersion showedbetter performance than one entity but not all, itwas placed in category 1 (improves performance). Onthe basis of these criteria, we can conclude that theuse of an amorphous dispersion produced improvedbioavailability in 82% of the cases (33 reports). Theamorphous dispersion exhibited poorer bioavailabil-ity in approximately 8% of the cases (three reports),and the same bioavailability was reported in nearly10% of the cases (four reports). These data suggestthat in the majority of reported cases, an amorphousdispersion did improve the performance of a poorlysoluble API. However, there is the possibility thatstudies showing negative results were not reportedand internal unreported results may be different.
The first assessment discussed above did not takeinto consideration the reference materials used forcomparison with the amorphous dispersion. A va-riety of reference entities were reported includingcrystalline material, physical mixtures of polymerand crystalline drug, simple capsule or tablet for-mulations, oral solutions or suspensions, and mar-
keted products (beads, capsules, tablets, and suspen-sions). The majority of studies compared the amor-phous solid dispersion with the crystalline material(49%). Marketed products (26%), solutions (7%), andother dosage forms (9%) were also used. Amorphousdispersions increased bioavailability approximatelytwo to 82 times over the respective crystalline ma-terials when an improvement was obtained and upto seven times when compared with marketed prod-ucts. The appropriate material used for comparisonobviously will vary depending on the stage of devel-opment, properties of the API, and available alterna-tives. For many poorly soluble early development can-didates, comparison of the amorphous solid dispersionto the crystalline material in a small bioavailabilitystudy can give valuable information on whether tomove a compound forward. For marketed products,an amorphous dispersion may be part of the life cyclemanagement of the drug by making a modified drugproduct with improved performance and a simpli-fied formulation. This can be accomplished by addingother benefits such as a reduction in the number ofdoses, which results in increased patient compliance.For these later stage studies, comparison with themarketed product is more applicable.
It is interesting to note that the formulations usedfor crystalline reference materials also varied. Neatcrystalline drug in a capsule was used in 62% of thestudies and 14% of the studies used a formulationcontaining excipients. Another common option was asuspension of the powder, which was used for both thecrystalline material and the amorphous solid disper-sions in 24% of the studies. None of the suspensionstudies reported the amount of drug dissolved in thesuspension vehicle, which may be significantly dif-ferent for crystalline and amorphous materials. Forinstance, varying amounts of drug in solution couldsignificantly impact the bioavailability or alter the po-tential nucleation and formation of crystalline mate-rial from an amorphous form. It is recommended thatif suspension formulations are used, the amount ofdrug dissolved should be determined. Several otherfactors during storage or use may also significantlyaffect the net bioavailability of a suspension includ-ing solution stability, buffer capacity, and interfacialphenomena occurring in the suspension, and shouldalso be investigated for amorphous solid dispersions.These factors related to the dosing of a suspension re-quire careful consideration when preparing a protocolfor an in vivo pharmacokinetic study.
IN VITRO–IN VIVO PREDICTABILITY
Overview
Although it is acknowledged that other types ofin vitro testing are necessary for development, suchas permeability testing with cell cultures, dissolution
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1359
Table 1. List of Amorphous Solid Dispersions Evaluated for In Vitro–In Vivo Performance
Study Drug Polymers Species Dissolution Media IVIVR Reference
1 Albendazole PVP Rabbit NA NA 20,212 Albendazole HPMC, HPMCP Rabbit Phosphate buffer (pH 6.8) Y 223 AMG 517 HPMCAS. HPMC Monkey Phosphate buffer (pH 6.8) Y 234 Compound 1 Gelucire, PEG/polysorbate 80 Dog 0.05 N HCl Y 245 Compound 1 PVP Dog pH 2 buffer (0.1 N HCl), pH
6.8 with 0.1% SLSY 25
6 Cyclosporine A Polyoxyethylene stearate (S40) Rat Water NA 267 ER-34122 HPMC Dog Phosphate buffer (pH 6.8) Y 278 Esomeprazole Zn PEG Human Phosphate buffer (pH 6.8) Y 289 Fenofibrate PVP Eudragit Dog 0.1 M HCl (0.36% SDS) NA 2910 Frusemide (furosemide) PVP Human SGF without enzymes, SIF Y 30–3211 Halofantrine PEG, gelucire, TPGS Dog 0.1 N HCl, water Y 3312 Hoffman-La Roche drug PVP, polaxamer Dog SGF without enzymes N 3413 Indomethacin HPC, HPMC, HEC Human Water (around pH 7) Y 3514 Indomethacin PEG, HPMCP Rabbit Phosphate buffer (pH 7.2),
NaClY 36
15 Indomethacin PEG Rabbit Phosphate buffer (pH 7.2),NaCl
N 37
16 Itraconazole HPMC Rat 0.1 N HCl Y 3817 Itraconazole CAP, PVAP Rat 0.1 N HCl, (pH 6.8) N 3918 Itraconazole HPMCP Dog NA NA 4019 Itraconazole PEG Human SGF without enzymes, SIF Y 4120 Itraconazole HPMC Rat SGF without enzymes NA 42,4321 Itraconazole Eudragit, PVP/VA Human SGF without enzymes N 44–4722 Ketoconazole PEG Rat SIF without enzymes Y 4823 KRN633 PVP Rat SGF Y 4924 Lonidamine PEG, PVP Rat Water Y 5025 MFB-1041 HPMC, HPMCP, CMEC Dog Phosphate buffer (pH 1.2 and
6.8)Y 51
26 Nifedipine PEG, phosphatidylcholine Rat Water NA 5227 Nifedipine PEG, PEG MME Rabbit SGF N 5328 Nifedipine PVP Dog Water Y 5429 Nifedipine PVP Dog Water Y 5530 Nifedipine PVP Dog Water with 0.1% HCO-60 Y 5631 Nimodipine HPMC, PVP/VA, Eudragit Dog Acetate buffer (pH 4.5) with
0.05% SDSY 57,58
32 Nitrendipine HPMCP, Eudragit, EC Dog Water (with 0.5% SDS) Y 5933 Pranlukast Gelatin Rat Phosphate buffer (pH 3, 5,
and 7)Y 60
34 R103757 HPMC Dog/human 0.1 N HCl N 6135 Ritonavir PEG Dog 0.1 N HCl Y 62,6336 Ritonavir and lopinavir Dopovidone, sorbitan monolaurate Human NA NA 6437 RP69698 PEG Dog Water NA 6538 Tacrolimus PEG, PVP, HPMC Dog pH 1.2 Y 6639 Tolbutamide PEG Rabbit pH 2 buffer N 67,6840 Tolbutamide PVP Dog Phosphate buffer (pH 6.8) Y 69
CAP, cellulose acetate phthalate; CMEC, carboxymethylethylcellulose; EC, ethyl cellulose; HEC, hydroxyethyl cellulose; HPC, hydroxypropyl cellulose;HPMC, hydroxypropyl methylcellulose; HPMCAS, hydroxypropyl methylcellulose acetate succinate; HPMCP, hydroxypropyl methylcellulose phthalate; PEG,polyethylene glycol; PEG MME, polyethylene glycol monomethyl ether; PVAP, polyvinyl acetate phthalate; PVP, polyvinyl pyrrolidone; PVP/VA, polyvinylpyrrolidone vinylacetate; TPGS, D-"-tocopheryl polyethylene glycol 1000 succinate; NA, not available; HCl, hydrochloric acid; SIF, simulated intestinal fluid;SGF, simulated gastric fluid; NaCl, sodium chloride; SLS, sodium lauryl sulfate; SDS, sodium dodecyl sulfate; HCO-60, surfactant.
testing will be the primary in vitro method discussedin this Commentary based on the large amount of lit-erature directly related to dissolution of amorphoussolid dispersions.
One reason for collecting in vitro dissolution datafor amorphous solid dispersions is to try to predictthe in vivo biological performance. There are twotypes of predictions for in vitro and in vivo dataknown as (IVIVRs) and in vitro–in vivo correlations(IVIVCs).70 IVIVR is a broad term that includes quali-
tative and semiquantitative associations between thedata. IVIVC, on the contrary, is a predictive math-ematical model that requires an evaluation of pre-dictability and degree of validation as outlined by theUS Food and Drug Administration (FDA).71,72 IVIVCcan also be related to the Biopharmaceutics Clas-sification System (BCS) classification of the drug.73
For Class II drugs, IVIVC can be expected if thein vitro dissolution rate is similar to the in vivo disso-lution rate. In early development, IVIVR is the likely
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1360 NEWMAN, KNIPP, AND ZOGRAFI
method used to compare dissolution data and earlyanimal studies. These studies, particularly for BCSClass II and IV compounds, can be extremely valuablein moving the drug development process forward witha minimum of in vivo work. For both Class I and ClassIII compounds, the achievement of an IVIVR/IVIVCis more related to physiological factors including gas-tric emptying rates and permeability, respectively. Inmany cases, dissolution methods are revisited afterbiological data are available and modified to providethe IVIVR/IVIVC.70
Of the 40 studies discussed above, 32 papers in-cluded details on dissolution studies and these datawere used to determine a qualitative IVIVR. In 78%of these cases, there was a relationship between thedissolution and bioavailability data; the other 22%did not show an IVIVR. Therefore, it appears thatit would be useful to more closely examine factorsthat are important in the dissolution and absorptionprocesses used in these types of studies. Factors in-volved in in vitro testing that might have influencedresults in the studies reviewed will be discussed first,followed by factors related to the in vivo conditions.
In Vitro Factors
To begin any analysis of dissolution data, it is im-portant to consider the intrinsic properties of thesolid API and the particles making up the formu-lation. As expected, particle size clearly has beenshown to affect dissolution results as shown with astudy with the API, nitrendipine.74 Different parti-cle sizes of the amorphous solid dispersions (200 nm,620 nm, 2.7:m, 4.1:m, 20.2:m) resulted in large dif-ferences in the dissolution rates in fasted state sim-ulated intestinal fluid (FaSSIF), which translated tosignificantly different bioavailability values (61.4%,51.5%, 29.4%, 26.7%, and 24.7%, respectively) in rats.In our survey, the role of particle size was reportedand evaluated in 26 studies. In these cases, the ma-terials were sieved before being used for dissolutionexperiments or animal studies, and a wide range ofparticle sizes were observed (7–10 to 850–1700:m),as shown in Table 2. Studies that did not show anIVIVR reported a variety of particle sizes, including90–150:m,53 <250:m,61 <355:m,42,43 <425:m,34
and 850–1700:m.69 Although these data mostly fallon the high end of the particle size range, there doesnot appear to be a direct relationship between parti-cle size and the lack of IVIVR. It is not clear if thedifferences in predictability arise from uneven sur-faces created during the dissolution of amorphousdispersions, dissolution media differences, physiolog-ical confounders, or other unknown factor(s). A sug-gestion for some amorphous solid dispersion sys-tems would include a more discriminatory study withdifferent particle size ranges versus dissolution or
Table 2. Number of Studies Utilizing Different Particle SizeRanges of Amorphous Solid Dispersions
Reported Particle Size (:m) No. of Studies
7–10 145–250 190–150 190–250 1<115 1<125 1<149 3149–250 2149–420 1<150 2150–250 1<177 1<180 1<250 2a
250–420 1280–900 1<300 1<355 1<425 1<500 1850–1700 1
Italics represents particle sizes where no IVIVR was obtained.aOnly one of the two studies using a particle size <250 showed no
IVIVR.
bioavailability to develop stronger relationships orcorrelations.
The role of different dissolution media and/or thedifferences in the physicochemical properties of thecompounds could also be contributing factors.75–77
When a dissolution method is first being developed,the purpose of the method needs to be established.In most cases, a quality control (QC) method to dif-ferentiate drug product properties is the ultimategoal. However, in early development, the goal maybe to predict the performance in a biological systemwhen animal data are not available. Although a sin-gle dissolution test method for IVIVR/IVIVC and QCpurposes would be ideal, this may not be possiblefor many systems. In many cases, IVIVR/IVIVC testmethods would not be practical for QC testing be-cause the extent of dissolution is too low or becausethe method is too complicated for routine testing. Arecent study highlighted the fact that refinement ofearly screening tools used to predict clinical outcomes,such as dissolution testing, would have a significantimpact on reducing later stage attrition.78 For thisreason, it is important that dissolution methods con-tinue to evolve to provide the necessary data for var-ious stages of development.
The first consideration is the choice of the typeof dissolution vessel to use, as described in theUSP under the general chapters of Dissolution andDrug Release.79 Apparatus 1 (basket method) andApparatus 2 (paddle method) are the most com-mon dissolution methods used. However, Apparatus 3
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1361
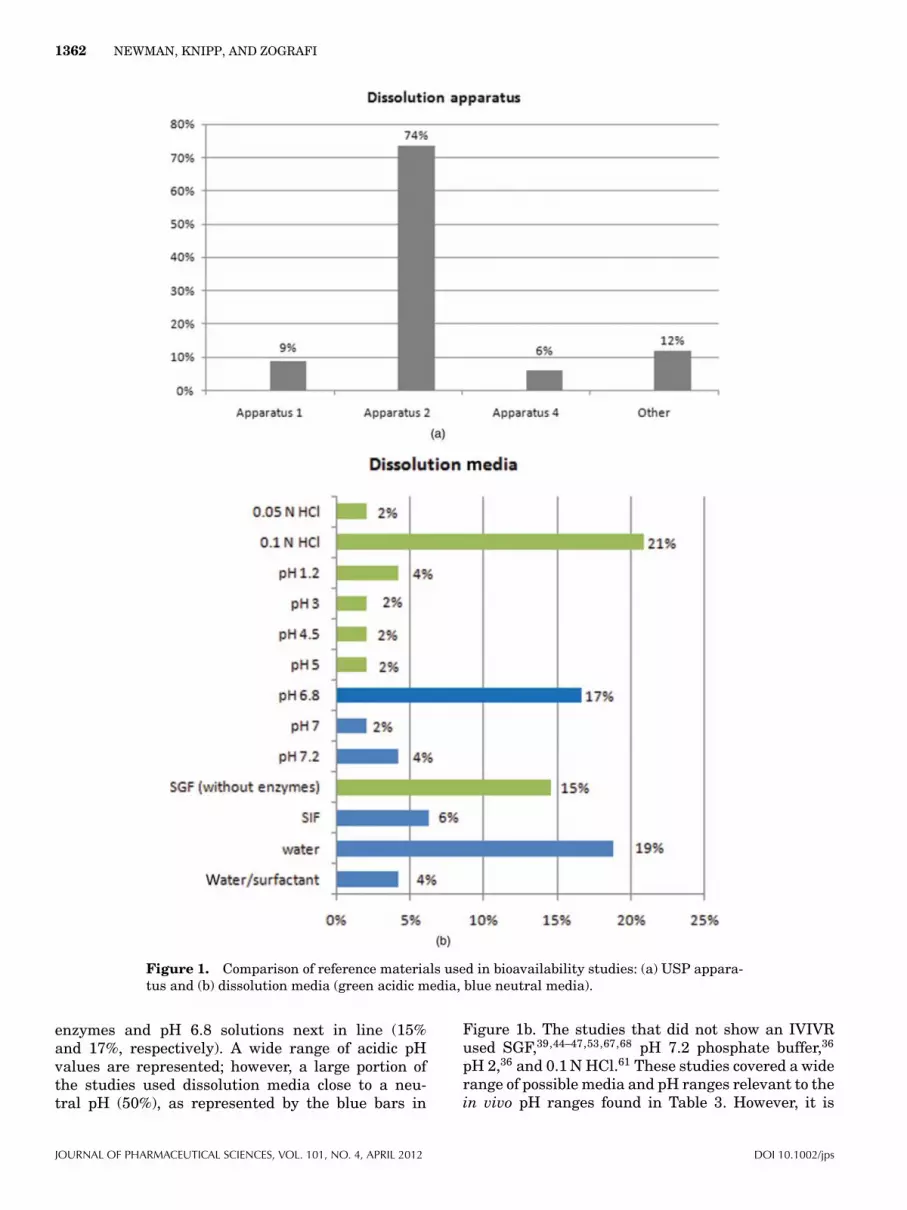
(reciprocating cylinder) and Apparatus 4 (flow-through cell) offer advantages for low solubilitydrugs by producing sink conditions and allowingcontrolled pH and volume changes of the dissolu-tion media throughout the test. It has also beenshown that Apparatus 3 can give similar resultscompared with Apparatus 2 if the right conditionsare used.80 Other two phase dissolution systemshave been reported that attempt to mimic the GItract (stomach and duodenum) to give a better cor-relation with bioavailability studies.81–83 In one ofthese systems,81 the crystallization of an amorphousform during dissolution was observed and the dis-solution results still compared favorably with dogbioavailability studies. In our survey, the majorityof studies used Apparatus 2 (71%) as shown in Fig-ure 1a. However, other dissolution systems wereused including a specific method developed for sup-pository dissolution36 and smaller laboratory scalemethods.69
When choosing dissolution media, it is generallyrecommended that dissolution experiments be per-formed under sink conditions. The USP defines sinkconditions as three times the volume of dissolutionmedia that is required to saturate a particular drugin that media,84 whereas the British Pharmacopeiastates that sink conditions normally occur in a vol-ume of dissolution medium that is at least five to10 times the saturation volume.85 Although it is rou-tine to assume sink conditions in such situations, it isrecommended that for poorly soluble drugs other fac-tors such as nonsink conditions and possible precipi-tation also be considered. In our survey, it is interest-ing to note that only 8% specified sink conditions, 6%specified nonsink conditions, and 86% did not spec-ify the conditions used. It is not known whether theconditions were not specified because sink conditionswere assumed or that it was simply not determined.
Media selection is one of the first method develop-ment parameters to consider and this is where thepurpose of the method needs to be established. A QCmethod that will look at batch to batch quality test-ing should be partly based on the available apparentsolubility data and the dose range of the drug prod-uct required to ensure that sink conditions are met. If
Table 3. Characteristics of Regional Environments alongthe Human Gastrointestinal Tract86
RegionLength
(m)Surface
area (m2) pHResidence
Time
Esophagus 0.3 0.02 6.8 >30 secStomach 0.2 0.2 1.8–2.5 1–5 hrDuodenum 0.3 0.02 5–6.5 >5 minJejunum 3 100 6.9 1–2 hrIleum 4 100 7.6 2–3 hrColon 1.5 3 5.5–7.8 15–48 hr
Table 4. Variation in the Regional Human of GastrointestinalFluid Volumes in the Fasted and Fed States87
Stomach[volume (mL)]a
Smallintestine
[volume (mL)]
Largeintestine
[volume (mL)]
FastedMinimum 13 45 1Maximum 72 319 44Median 47 83 8Mean (SD) 45 (18) 105 (72) 13 (12)
FedMinimum 534 20 2Maximum 859 156 97Median 701 39 18Mean (SD) 686 (9.3) 54 (41) 11(26)
aVolume of the stomach after the meal represents the filling volume(not only fluid).
the method is to be used to investigate biopharmaceu-tical properties, it is important that it closely simu-lates the environment in the GI tract rather than onlyproduce sink conditions. Therefore, the use of biorel-evant media is strongly recommended. When devel-oping dissolution methods for amorphous solid dis-persions in early development, the physiological pHrange of 1.2–7.2 should generally be targeted. The dis-solution medium composition, volume, and the exper-imental time should also be varied to gain additionalinformation relevant to biopharmaceutical conditions(Tables 3 and 4).
Another method for choosing the medium is to cal-culate the dose–solubility ratio.74 It is suggested thatthis ratio be used to help guide the selection of anoptimum formulation by considering the dose to beused and the solubility in the dissolution media. Adose solubility ratio greater than 250 indicates thatthe GI conditions are less than favorable for drug dis-solution. As an example, nitrendipine exhibits a dosesolubility ratio greater than 8000 based on a 40 mgdose and a solubility of 5 :g/mL in FASSIF.74 On thebasis of this ratio, it is probable that some of the solidwill not dissolve instantly in the GI tract and sinkconditions will not be achieved in vivo. The inabilityto reach sink conditions may also be affected by therelative differences in the fluid volumes that are notuniformly distributed throughout the GI tracts, par-ticularly when comparing different patients (Table 4).The different effects of absorption and dissolution ki-netics in physiological systems on sink conditions canalso play a role.
Various dissolution media were utilized in the re-ports on solid amorphous dispersions that were exam-ined for this Commentary. As shown in Figure 1b, 13different dissolution media were used, with pH val-ues ranging from acidic to neutral. The most commonmedia used were 0.1 N HCl solutions (21%) and wa-ter (18%) with simulated gastric fluid (SGF) without
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012
1362 NEWMAN, KNIPP, AND ZOGRAFI
Figure 1. Comparison of reference materials used in bioavailability studies: (a) USP appara-tus and (b) dissolution media (green acidic media, blue neutral media).
enzymes and pH 6.8 solutions next in line (15%and 17%, respectively). A wide range of acidic pHvalues are represented; however, a large portion ofthe studies used dissolution media close to a neu-tral pH (50%), as represented by the blue bars in
Figure 1b. The studies that did not show an IVIVRused SGF,39,44–47,53,67,68 pH 7.2 phosphate buffer,36
pH 2,36 and 0.1 N HCl.61 These studies covered a widerange of possible media and pH ranges relevant to thein vivo pH ranges found in Table 3. However, it is
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1363
Figure 2. Comparison of dissolution parameters: (a) media volume and (b) agitation rates.
interesting to note in Table 3 that there is a significantpH range for both the stomach (pH 1.8–2.5) and intes-tine (pH 5–7.6), which is not typically captured duringmost dissolution testing. It is recommended that drugdissolution also be evaluated as to the importance ofregional dependence, such as enteric-coated formula-tions that are largely targeted to the intestines. Withthese types of dosage forms, dissolution media and pHselection will be critical when developing an IVIVR/IVIVC and these factors need to be considered earlyin the predictive dissolution testing design stage.87–89
A comparison of the amount of dissolution mediaused in these studies is given in Figure 2a. Fifty-two percent of the studies used 900 mL and 32% used500 mL; these are typical volumes for many dissolu-tion studies. It was interesting that 91% of all dis-solution studies were performed in 500 mL of mediaor greater, whereas actual GI fluid volumes tend tobe considerably lower in most cases as shown in Ta-
ble 4. Smaller volumes were also used in the studies(25 mL of solution69) likely due to limited materialsbeing available, which is common in early develop-ment. For paddle and basket methods, the agitationcan also play an important role, and yet speeds rang-ing from 25 to 200 rpm were reported in our sur-vey, as shown in Figure 2b. The majority of studies(60%) were performed using 100 rpm with the nextmost common options being 50 rpm (18%) and 75 rpm(10%). Although stirring rate is an important factor indissolution testing, it is suggested that investigationsrelated to mixing and agitation rates encounteredin vivo also be conducted.90–96
In Vivo Factors
An important decision point for in vivo studies is theanimal model to be used, especially in early develop-ment studies. In our survey, five species were used inthe in vivo studies as shown in Figure 3. The most
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1364 NEWMAN, KNIPP, AND ZOGRAFI
Figure 3. Breakdown of species used in bioavailabilitystudies using amorphous solid dispersions.
common animal used for bioavailability assessmentwas the dog (41%). The rat was used in 24% ofthe studies, with rabbits, humans, and monkeys uti-lized to a lesser degree. One study used dogs toinitially test formulation concepts (tablet with crys-talline drug, film-coated beads, oral solution) and thenmoved to human studies to test specific solids.61 An-other study used dogs as their model but pretreatedthem with histamine “to reduce the gastric pH andcreate an environment that more closely mimics hu-man physiology.”62,63 Rabbits with low acidity (pH <
5) were used to study the effect of pH because gastricpH was reported to have a significant effect on the ab-sorption of albendazole.20,21 In our survey, the studiesthat did not show IVIVR used rats (one study), rab-bits (three studies), dogs (two studies), and humans(two studies).
Current drug product safety regulations mandatethat the PK and PD of new chemical entities are tobe tested in both rodent and nonrodent laboratoryanimals prior to human administration.97,98 The se-lection of the most appropriate nonrodent species is acomplex issue that should take into account severalfactors including: (1) physiological (e.g., metabolismand morphology) similarities between the referenceanimal and the corresponding human organ, (2) pre-dictive power of the process being modeled in the testanimal relative to the analogous process in humans,and (3) cost and availability of the experimental ani-mals. Manageability and behavior characteristics arealso factors, and can include growth rate, size at mat-uration, reproduction rate, and interaction with hu-mans in a laboratory environment.97 On the basis ofthese considerations, several different animal speciesare currently and extensively used as models to pre-dict human bioavailability of various drug candidatesincluding rodents, dogs, primates, and more recentlypigs and minipigs.97,98
In order to build a more simplistic view of re-gional GI diversity, the gastrointestinal transit anddrug absorption (GITA) model has been proposed.99
The GITA divides the GI tract into eight regions:
stomach, duodenum, upper jejunum, lower jejunum,upper ileum, lower ileum, cecum, and colon. The di-vision enables one to consider only the relevant fac-tors within these regions that need to be consideredwith respect to drug dissolution and absorption. Whenlooking at the GI tract in more detail, differences inregional fluid volumes are apparent, as summarizedin Table 4. In contrast to the majority of the dissolu-tion medium volumes (500–900 mL) used in the pa-pers being reviewed, Table 4 reveals that the fastedGI tract fluid volumes are approximately 100 mL orlower.98,87 The mean regional fluid volumes in thestomach and along the small and large intestines un-der the fasted state have been determined to be ap-proximately 45, 105, and 13 mL, respectively. In thefed state, the mean regional free fluid volumes in thestomach (meal and fluid combined), the small andlarge intestines were observed to be approximately686, 54, and 11 mL, respectively, which are signifi-cantly different than the fasted state.87,89 Flow ratesthrough the small intestine (jejunum) were deter-mined to be 0.6–1.2 or 2–4.2 mL/min in the fed andfasted states, respectively, suggesting that a differ-ence in hydrostatic pressure also existed in vivo.100,86
Taken together, this suggests that lower dissolutionvolumes may more accurately reflect the in vivo dis-solution process and it is recommended that smallervolumes be used when IVIVR is needed. Furthermore,churning and agitation will differ based on the fedand fasted states; therefore, a closer inspection ofthe dissolution apparatus speeds and a better assess-ment of the agitation on dissolution also needs to beconsidered.91–96
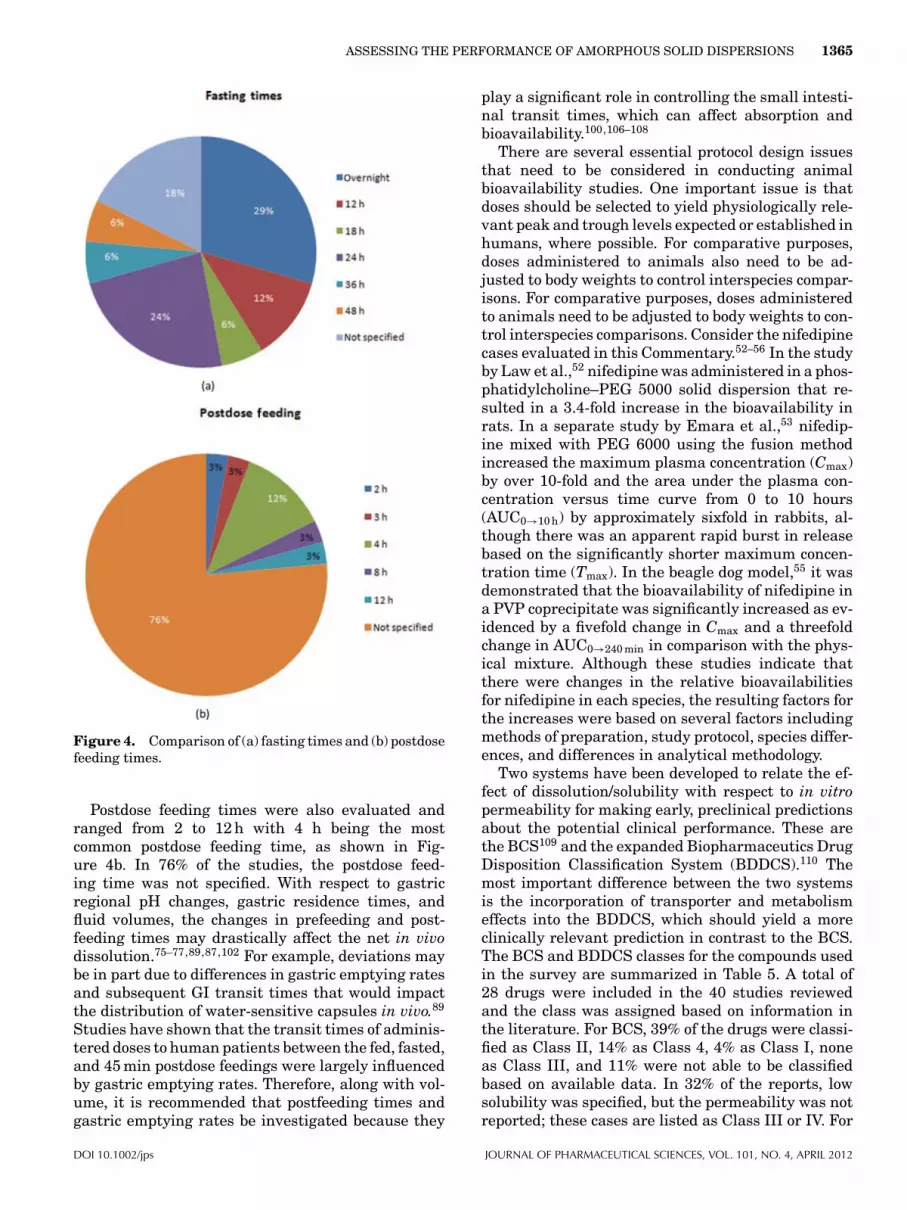
The presence of food will change the pH in the stom-ach; however, the changes are not uniform through-out the three distinct regions of the stomach (fun-dus, body, and antrum).101,102 It was revealed that thegastric body and fundal pHs raised to a pH 4.5 in atime-dependent manner postfeeding,101 whereas theantral pH remained at 2 throughout the time thatfood was present. These results may suggest thatthe stomach be considered as two separate regionsin the fed state when utilizing the GITA model or aderivative.99,103–105 On the basis of the volume differ-ences observed in the fed and fasted state, it is sug-gested that this factor be investigated in the in vivostudies. In our survey, 80% of the studies specifiedthat a fasted state was used for the bioavailabilitystudies. Free access to food was allowed in 7% of thecases, whereas the fed state was reported for 9% ofthe papers. The fasted versus fed state was not re-ported for 5% of the studies. For those studies usingthe fasted state, a variety of fasting times were re-ported ranging from overnight to 48 h, as shown inFigure 4a. Overnight fasting was the most common(29%), followed by 24 h (24%) and 12 h (12%). In 18%of the cases, the fasting time was not specified.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps
ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1365
Figure 4. Comparison of (a) fasting times and (b) postdosefeeding times.
Postdose feeding times were also evaluated andranged from 2 to 12 h with 4 h being the mostcommon postdose feeding time, as shown in Fig-ure 4b. In 76% of the studies, the postdose feed-ing time was not specified. With respect to gastricregional pH changes, gastric residence times, andfluid volumes, the changes in prefeeding and post-feeding times may drastically affect the net in vivodissolution.75–77,89,87,102 For example, deviations maybe in part due to differences in gastric emptying ratesand subsequent GI transit times that would impactthe distribution of water-sensitive capsules in vivo.89
Studies have shown that the transit times of adminis-tered doses to human patients between the fed, fasted,and 45 min postdose feedings were largely influencedby gastric emptying rates. Therefore, along with vol-ume, it is recommended that postfeeding times andgastric emptying rates be investigated because they
play a significant role in controlling the small intesti-nal transit times, which can affect absorption andbioavailability.100,106–108
There are several essential protocol design issuesthat need to be considered in conducting animalbioavailability studies. One important issue is thatdoses should be selected to yield physiologically rele-vant peak and trough levels expected or established inhumans, where possible. For comparative purposes,doses administered to animals also need to be ad-justed to body weights to control interspecies compar-isons. For comparative purposes, doses administeredto animals need to be adjusted to body weights to con-trol interspecies comparisons. Consider the nifedipinecases evaluated in this Commentary.52–56 In the studyby Law et al.,52 nifedipine was administered in a phos-phatidylcholine–PEG 5000 solid dispersion that re-sulted in a 3.4-fold increase in the bioavailability inrats. In a separate study by Emara et al.,53 nifedip-ine mixed with PEG 6000 using the fusion methodincreased the maximum plasma concentration (Cmax)by over 10-fold and the area under the plasma con-centration versus time curve from 0 to 10 hours(AUC0→10 h) by approximately sixfold in rabbits, al-though there was an apparent rapid burst in releasebased on the significantly shorter maximum concen-tration time (Tmax). In the beagle dog model,55 it wasdemonstrated that the bioavailability of nifedipine ina PVP coprecipitate was significantly increased as ev-idenced by a fivefold change in Cmax and a threefoldchange in AUC0→240 min in comparison with the phys-ical mixture. Although these studies indicate thatthere were changes in the relative bioavailabilitiesfor nifedipine in each species, the resulting factors forthe increases were based on several factors includingmethods of preparation, study protocol, species differ-ences, and differences in analytical methodology.
Two systems have been developed to relate the ef-fect of dissolution/solubility with respect to in vitropermeability for making early, preclinical predictionsabout the potential clinical performance. These arethe BCS109 and the expanded Biopharmaceutics DrugDisposition Classification System (BDDCS).110 Themost important difference between the two systemsis the incorporation of transporter and metabolismeffects into the BDDCS, which should yield a moreclinically relevant prediction in contrast to the BCS.The BCS and BDDCS classes for the compounds usedin the survey are summarized in Table 5. A total of28 drugs were included in the 40 studies reviewedand the class was assigned based on information inthe literature. For BCS, 39% of the drugs were classi-fied as Class II, 14% as Class 4, 4% as Class I, noneas Class III, and 11% were not able to be classifiedbased on available data. In 32% of the reports, lowsolubility was specified, but the permeability was notreported; these cases are listed as Class III or IV. For
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1366 NEWMAN, KNIPP, AND ZOGRAFI
Table 5. Summary of the Evaluated Drugs in BCS and BCDDSClasses
ClassPercent inBCS Class
Percent inBCDDS Class
Class I 4 0Class II 39 48Class III 0 4Class IV 14 4NA 11 44Class II or IVa 32 –
aReported to be poorly soluble but no permeability data available; usedonly for BCS classification.
BDDCS, a higher percentage (61%) was not classi-fied because of a lack of data. For those that wereclassified, 32% were found to be Class II, 4% werefound to be Class III, 4% were found to be Class IV,and none were found to be Class I. In our survey, thecompounds that did not show an IVIVR were mostlyBCS Class II (indomethacin,37 itraconazole,39,44–47
R103757,62,63 and tolbutamide69) and one was Class I(nifedipine53).
Another topic that is often not considered is therole of gender on polymer or API performance in anamorphous solid dispersion. For example, it has beendemonstrated that PEG 400 can affect the bioavail-ability of a BCS Class III compound, ranitidine, differ-ently in males compared with females.111 The effectsof gender on metabolism and transport are fairly wellestablished in most species. However, gender-baseddifferences in bioavailability enhancement from spe-cific polymers are often overlooked and should beinvestigated for many systems. This may be of partic-ular importance when employing biodegradable poly-mers that are subject to metabolism.112
PHARMACEUTICAL CONSIDERATIONS WHENDEVELOPING AMORPHOUS SOLIDDISPERSIONS
As our review of the literature shows, amorphous dis-persions are a viable choice for early and late pharma-ceutical development. However, it is important to rec-ognize that there are a number of factors that shouldbe considered when developing these materials intodrug products, as listed in Table 6. This final sec-tion will highlight issues associated with physical at-tributes, in vitro testing, and in vivo evaluation thatare believed to be important for testing and devel-oping amorphous solid dispersions, with particularemphasis on ensuring therapeutic performance.
Physical Attributes
The analysis of the available literature and the ap-pearance of recent commercial products reveal thesignificant practical advantages of using the amor-phous form of an API. Their ability to enhance dis-
solution and oral bioavailability of a poorly solubleAPI can be utilized during various stages of devel-opment, ranging from early safety assessment to themarketed product. From a physical perspective, threeareas must be examined: formulation and process-ing of dispersions, insuring physical stability duringprocessing and storage, and assuring that sufficientlevels of supersaturation are maintained after disso-lution in vivo.
The first level of concern is the choice of a polymer–API composition that can be conveniently processedas a solid dispersion and then formulated into eithercapsules or tablets. This begins with the choice of themethod of processing, be it melt extrusion or spraydrying.6,7 The choice of a method must be based onan understanding of the physical characteristics ofthe API, the polymer and the final dispersion. Partic-ularly in the case of melt extrusion, a moderate Tg,still greater than that of the API, is desirable, andsurfactants may be required as part of the dispersionto facilitate processing. During spray drying, relativesolubilities of components in suitable solvents will beimportant to ensure homogeneous one-phase systems.
The ability to form a one-phase “miscible” polymermixture needs to be evaluated early in the process.Both thermodynamic and kinetic factors need to beoptimized to prevent crystallization over the time pe-riod for handling and storage of the amorphous soliddispersion. In this regard, attention must be paid tothe chemistry of the polymer, its molecular weight,the extent to which the API and polymer are actuallymiscible, the Tg, and level of hygroscopicity.5 Whetheror not other excipients used to form the amorphoussolid dispersion, such as surfactants, have a signifi-cant effect must also be considered. It is recommendedthat the following physical properties be evaluated foramorphous dispersions: particle size, hygroscopicity,mechanical properties, wettability, chemical stability,and the nature of the mixing of the API and excipi-ents making up the dispersion. The possible impact onphysical stability of other excipients added to the dis-persion and formulation to facilitate manufacturingprocesses and therapeutic performance should alsobe evaluated.
The most difficult physical factor to control isthe ability of the dispersion to resist crystallizationonce it has encountered the dissolution media. Rapiddissolution of API in the supersaturated state cer-tainly would appear to be an asset in obtaining goodbioavailability. However, it may be equally importantfor the polymer to provide protection against solution-mediated crystallization by remaining in close contactwith the API.17 This may be accomplished by directinhibition in solution by the dissolved polymer, or bynot immediately releasing the entire dose of API inthe supersaturated state, yet still providing an ade-quate level of supersaturation. The challenge is not to
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1367
Table 6. Pharmaceutical Considerations for Developing Amorphous Solid Dispersions
Area Considerations Recommendations
Physical Choice of polymer Polymer characteristics (solubility, melting point, wettability,hygroscopicity, effect of pH, dissolution rate, etc.) and large scalemanufacturing process (spray drying and melt extrusion) need tobe considered early in the development of the amorphous soliddispersion.
Polymer–API ratio Optimize the amount of polymer required to provide the long-termphysical stability desired in the solid state and crystallizationinhibition in solution.
Miscibility Determine if a one-phase miscible system has been produced.Manufacturing processes Consider that the amorphous solid dispersion produced at elevated
temperatures may be miscible but may separate upon cooling toroom temperature or under other conditions.
Hygroscopicity Water uptake and the effect on Tg, physical stability, andcrystallization need to be evaluated.
Dissolution Dissolution apparatus andmedia conditions
Dissolution method development should focus on conditions thatmimic the GI tract (biorelevant media that match the deliveryroute, pH, stirring rate, and volume); these conditions may notbe applicable for routine dissolution testing to discriminatedifferences between lots.
Sink versus nonsink conditions Both sink and nonsink conditions should be investigated andcompared with data from the in vivo studies; for poorly solubledrugs, nonsink conditions and the possibility of crystallization inthe GI tract need to be evaluated.
Polymer controlleddissolution/wettability
Incorporate polymer properties into the dissolution methoddevelopment (gel formation of the polymer, floating ofparticle/dispersion particles, solid clinging topaddles/shafts/glassware).
Supersaturation in dissolutiontesting over hours
Test supersaturation over biologically relevant time frames inbiorelevant media to determine if the API will stay in solution orcrystallize out.
Biological Fasting- versus Fed-pH effects Food effects need to be evaluated; specifics on fasting times andpostdose feedings need to be detailed in reported studies.
Sink versus nonsink conditions Dose range-dependent sink conditions along the GI tract (pH,volumes, and residence times) need to be considered andinvestigated; these will be dependent on the solubility of the APIand the amorphous solid dispersion.
Local effect of polymers on pH pH effects of polymers, especially polymers used for entericcoatings, need to be evaluated throughout the entire GI tract.
Species differences Investigate the use of alternative animal models, such as pigs andminipigs, that are a better model for nonprimate studies.
Transporters and metabolism Determine if APIs are substrates for human transporters and/ormetabolizing isoforms and the affect this will have on absorption.
Gastrointestinal physiology For highly lipophilic molecules, lymphatic absorption and not onlyblood absorption need to be determined in early studies; useextended GITA model to predict regions of optimal absorption;use feedback from in silico models to help refine formulations.
have the polymer become the rate-limiting factor ina negative manner. It is suggested that these factorsbe tested and that the mechanisms be investigatedwhen using amorphous solid dispersions.
In Vitro Testing
Along with the routine parameters that need to be op-timized for dissolution testing, other factors need to beconsidered when developing methods for amorphoussolid dispersions. It is important that the methodsbe tailored to provide the desired information dur-ing development. For many studies involving amor-
phous solid dispersions, the goal will be to predictperformance; therefore, it is recommended that dis-solution parameters be chosen to mimic the biologicalsystems and provide an IVIVR. Method developmentwill include decisions on the appropriate apparatus,the media used to mimic biological conditions, andsink versus nonsink conditions. A number of studiesshow that the two-phase dissolution method employ-ing both gastric and intestinal fluids can successfullypredict the performance for oral solid forms.81–83 Itis also important to match the media with the pro-posed delivery route. In one study using supposito-ries, it is postulated that the lack of IVIVR was dueto the smaller amount of in vivo fluid available in the
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1368 NEWMAN, KNIPP, AND ZOGRAFI
animal model (rabbit) compared with that used in thedissolution test.37 Use of simulated colonic fluid113
during the dissolution testing may also have helpedthe IVIVR. Dissolution methods to better mimicin vivo factors such as mixing and agitation due tothe muscle layers have also been developed and needto be considered for routine use.91–96
The presence of a high amount of polymer in solidamorphous dispersions can also play an importantrole in designing the dissolution experiment. Manypolymers have very different properties comparedwith APIs and excipients and these need to be in-corporated into the method development. Physicalcharacteristics114 such as gel formation of the poly-mer, floating of polymer/dispersion particles on top ofthe dissolution media, and polymer/dispersion parti-cles clinging to paddle shafts or glassware, need to beinvestigated early in order to incorporate best prac-tices and develop an effective method.
As discussed above, the polymers can also have asecondary effect that will result in crystallization in-hibition and extended supersaturation. Understand-ing the extent of this supersaturation advantage canbe easily incorporated into the method developmentand it is recommended that time frames similar tothose observed in biological systems (as outlined inTable 3) be used for method development and thatpossible precipitation be evaluated. Dissolution timesof 30–60 min will be somewhat representative of gas-tric residence time but will provide no informationon the time spent in the rest of the GI tract. Pre-cipitation of the drug during an in vivo study maysignificantly affect performance, especially for poorlysoluble drugs.
When considering dissolution media for amorphousdispersions, it is important to consider not only thesolubility characteristics of the drug but to also con-sider the dissolution characteristics for the poly-mer, especially in the case of pH-sensitive entericpolymers,39,44–47 such as polymethacrylates (EudragitL and S) and HPMC derivatives (HPMCAS andHPMCP), which are insoluble in gastric fluid butdissolve rapidly in intestinal fluid. Using a gastricpH range around 2 to determine the dissolution ofdispersion made with an enteric polymer that dis-solves at neutral pH will be difficult to relate backto in vivo studies. An example to consider uses itra-conazole, a weak base, and dispersions made withHPMC and Eudragit E-100.44–47 Dissolution studiesperformed in SGF without enzymes showed a signifi-cantly faster dissolution rate for the Eudragit E-100amorphous solid dispersions versus the HPMC amor-phous solid dispersions because the Eudragit E-100is soluble in gastric fluid below pH 5. However, theEudragit E-100 amorphous solid dispersions demon-strated a lower human bioavailability compared withthe HPMC amorphous solid dispersion.
The use of surfactants is also a consideration. Theyare commonly used when trying to increase solubilityand achieve sink conditions. However, on the basis ofdesire to mimic GI conditions, it is our recommenda-tion that it is better to go with simulated media asdescribed previously.77
Sink versus nonsink conditions will continue to bean area of discussion for dissolution methods. Fordifferentiating lot-to-lot variations, sink conditionsare recommended by regulatory agencies. However,sink conditions in a dissolution bath are not relevantto physiological systems where competing processessuch as absorption and regional differences in GI vol-umes may act to influence the actual supersaturationsolubilities and dissolution rates. For comparison toin vivo data, it is suggested that different sink andnonsink conditions be investigated to achieve anIVIVR.
In Vivo Evaluation
Drug candidates often fail in Phase I clinical trialsbecause of poor oral bioavailability, whereas a lackof in vivo efficacy or unexpected toxicity tend to bethe predominant reasons for failures in Phase II andIII clinical testing.115 It is becoming more obviousthat important factors are not consistently predictedor observed based on current preclinical screeningstudies. This indicates that better early in vitro andin vivo models are required in preclinical develop-ment to characterize the physicochemical properties,bioavailability, and predict the developability of newchemical entities.
There is significant interest in the utility of amor-phous solid dispersions for early and late studies,yet our limited survey of the literature showed thatbioavailability improvement was not obtained in eightout of the 40 reports reviewed. As discussed previ-ously, crystallization in the GI tract and unoptimizeddissolution parameters could be playing a role. How-ever, another possible reason is that little is knownabout the effects of physiology upon the clinical per-formance of amorphous solid dispersions. This factprompts us to end our Commentary with suggestionsconcerning the in vivo considerations that need to beevaluated prior to clinical testing in order to reduceclinical attrition.
The driving force for developing amorphous disper-sion systems is the ability to increase the apparentsolubility and subsequent absorption, which leads toincreased bioavailability. One limitation of currentdissolution and solubility testing is that they oftendo not accurately mimic clinical (biorelevant) condi-tions found in the patient.88 Improvement of predic-tive IVIVR/IVIVC between dissolution testing and ob-served bioavailability will only be fully realized byincorporating the critical physiological factors that
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1369
Table 7. Representative Factors for Amorphous Solid Dispersion Formulations on Absorption/Bioavailability Due to Increased Solubility
Properties Factors Observations Effect on Bioavailability (BA)
Physicochemical Lipophilicity Relationship between increasing lipophilicityand absorption is parabolic.
High lipophilicity—API stays in membrane ordepots in fat with small increase in BA;moderate increases BA; lowlipophilictydepends on polarity, slightincrease in BA.
Stability Physical stability (solid state) is poor; compoundmay nucleate to a crystalline form.
Decrease BA
Chemical stability is generally poor in theformulation and/or in solution as a function ofpH; absorption of the API will depend onregional pHs.
Depends on the region of release. Can increaseBA if released in region with optimal stabilityor decrease where API is unstable.
Ionization Increased solubility of the charged APIdepending upon pH; weak bases would havesolubility problems at a higher pH, andconversely weak acids at a lower pH.
Decrease in BA by passive transcellular routewhen solubility is increased for the ionizeddrug. Amorphous form release needs to beregionally targeted in the GI tract.
Size There is an inverse reciprocal relationshipbetween molecular size and permeability.
In general, larger molecules have lower BA.
Solubility limited Dissolution and permeability are normally fast. When solubility controls the BA, the gut canbecome saturated with API and increasingdose will not effect BA. For an amorphousform where the compound can maintain thesupersaturated state, it would be possible toovercome the solubility-limited absorption.
Formulation Surface areaincrease (particlesize reduction)
Increases the solubility. Poorly soluble compounds will demonstrateincreased BA, dependent upon lipophilicity.
Excipients Lipophilic excipients May decrease dissolution and BA (e.g.lubricants), although lipid drug deliverysystems can increase BA.
Disintegrants can increase dissolution Generally increase BA, depending on the API’sphysicochemical properties.
Binders have the opposite effect of disintegrants. Generally can decrease BA by restrictingdissolution.
Surfactants can increase solubility andpermeability, can be used in dispersions.
Surfactants can lower surface tension, formmicelles and increase BA; can interact withMDR transporters to increase BA (see below).
Polymers can increase solubility, can also beused to generate dispersions or co-crystals
Can increase BA; however, API-polymerinteractions can also lower BA. Higherconcentrations of polymers can reduce releaserate and potentially lower BA.
Dissolution limited Tdiss is greater than the small intestineresidence time. Permeability and solubilitycan be fast.
BA can increase with increasing dose; excipientchanges may increase dissolution and BA.
Physiological Lumen contents The gastrointestinal fluids can changeregionally; pH varies along the GI tract; floraand components such as bile salts also vary;food can dramatically vary environment;cellular debris and mucus also present.
Depending on the region, lipophilic APIs may besequestered in lipid/bile micelles. This mayalter BA in an unpredictable manner. Uptakeby flora lowers BA. Differences in BA effectsare API dependent. Biorelevant dissolutionmedia is limited in modeling the luminalcontents.
Epithelium The complex mucus and glycocalyx layer is noteffectively modeled in traditional in vitro celllines.
The nature of the mucus and glycocalyx canlimit the BA of lipophilic solutes. It also canalter the pH at the membrane surface due tobuffering effects.
Lipid bilayer Contents of the lipid bilayer can varysignificantly based on diet. Parallel artificialmembrane permeability assays (PAMPA)may help assess the net effect on passivetranscellular permeation.
This effect is often overlooked. The lipid bilayeris composed of fatty acids incorporated intophospholipids. There is also a polarizeddistribution of phospholipids, with somephospholipids such as phosphatidylserineand phosphotidylethanolamine appearingonly on the inner leaflet of the bilayer. Chargeeffects can alter BA.
(Continued)
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1370 NEWMAN, KNIPP, AND ZOGRAFI
Table 7. Continued.
Properties Factors Observations Effect on Bioavailability (BA)
Efflux transporters Can result in multidrug resistance [e.g.,P-glycoprotein (Pgp), multidrugresistance-like transporters (MRPs)] andlimit permeability when present on theluminal facing membrane or increase itwhen on the basal facing membrane. Canact in concert depending on substrateaffinity and capacity. Function through aMichaelis–Menten saturable process, thusincreasing solubility; can increasepermeability. Excipients can also disruptthe function of some isoforms.
The main concern for BA is usually withluminal facing efflux pumps that can limitpump and absorbed substrate from thecytoplasm back into the lumen, thusreducing permeation of an API. Theprimary focus on ABC efflux pumps is onthe Pgp, BCRP, and MRP1 and 2 isoforms;however, approximately 50 ABCtransporters have been identified. Thereare also a number of SLC transporters thatwork to efflux drugs from the cytoplasminto the extracellular space, [e.g., organicanion transporter (OAT) isoforms].
Influx transporters Influx transporters can increase thepermeability of a variety of substrates,normally polar.
Increases the BA of more polar APIs. Thegeneral substrates for an influxtransporter seldom require amorphousdispersions.
Metabolism Normally result in the formation ofchemically modified metabolites that aremore polar than the API for excretion.There are two major forms of metabolism:Phase I that results in chemicalmodification of an API through severaldifferent pathways (e.g., deamidation oroxidation) and Phase II results in chemicalconjugates (e.g., glucuronidation)
Reduces BA by removing the API and makingit more polar for excretion. Amorphousdispersions can increase the absorption oflipophilic compounds by the passivetranscellular pathway and increase thepotential to saturation of metabolizingenzymes in the GI epithelium andpotentially the liver, thus increasing theBA.
Permeability limited The permeability across the epithelial barrierwill remain low regardless of the solubility.Dissolution can be fast, particularly fordispersions.
Amount of drug absorbed increases with anincreasing dose via the passivetranscellular pathway. Amorphousdispersions can increase the BA.
impact in vivo dissolution, including modifying solu-bility and dissolution methods to utilize more relevantmedia, and by optimizing permeability models thatare superior to the current cell lines that possessesinherent variability.88,116–119 In Table 7 we have sum-marized several of the rate-determining physiologicalfactors that influence the clinical performance of anamorphous dispersion that require greater consider-ation in preclinical testing.
Central to amorphous solid dispersion performancewill be the extent to which a variety of factors in-fluence both the extent of supersaturation and per-meability in the GI tract. The physiological diver-sity of GI fluids, particularly in fasted and fed states,presents a major issue when associated with defin-ing sink conditions. The physical considerations as-sociated with in vitro dissolution tend to be ap-paratus dependent and are not directly relevantto in vivo bioavailability controlling factors.120 Thefact that fluid volumes are nonhomogeneously dis-tributed throughout the GI tract, unlike a homoge-neous dissolution bath, is an issue considering thatthe Noyes–Whitney equation relies in part on theassumption that the GI fluid is uniform in com-position and continuously distributed under sinkconditions. Clearly, regional GI fluid compositionand volume changes may potentially impact solu-
bility and dissolution differently along the GI tractcompared with what is observed in a dissolutionbath.87,101,102,121,122
It is also important to note that since solubilitymay differ across media and as a function of time (Ta-ble 3), the apparent thermodynamic equilibrium sol-ubility must be appropriate for assessing dose range-dependent sink conditions along the GI tract. Whenconsidering poorly soluble BCS Class II and IV drugsfor amorphous dispersions, it is critical that the role offree physiological water volumes on regional absorp-tion is also considered.123 Simulating GI tract condi-tions are important from a biorelevancy and regula-tory point of view as well. According to the FDA,88
a biorelevant product is defined as follows: “A drugproduct designed, developed and manufactured ac-cording to Quality Target Product Profile with speci-fication (such as dissolution/release acceptance crite-ria) consistent with the desired in vivo performanceof the product.” More simply stated, biorelevancy isbased on the premise of “linking process, product, andpatient for patient benefit.”88 Biorelevancy is an im-portant consideration on several levels and needs topart of the drug development plan.
There are a number of regional differences in fluidcomposition along the GI tract leading to signifi-cant physiological diversity that testing methods are
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1371
trying to reproduce. Volumes in the stomach can bechanged significantly in the fasted versus fed states,which will alter the extent of dissolution. Tradition-ally, the focus has been placed on longer residencetimes and pH variations for drug release in the fedstate. However, it is recommended that other fac-tors be considered upon feeding, such as ionic species,bile salts, buffer capacity, and so on that can alterthe dissolution process or the dissolved state of thedrug.86,108 Fed and fasted states are also manageddifferently, as seen in our survey where a variety offasting and postfeeding times were reported (Figs. 4aand 4b). Taken together, food effects can have a pro-found effect on in vivo drug product performance asillustrated in Figure 5 and it is suggested that thisfactor be considered when using amorphous solid dis-persions in in vivo testing.
Animal studies are usually performed under con-trolled diets to reduce potential confounding factorsthat might arise, particularly drug–nutrient interac-tions. In a controlled clinical setting, a human trialmay also be managed under controlled conditions.However, this is certainly not the case for most hu-man patients, particularly when one considers di-etary diversity on a global scale. These differencesin diet can affect the average residence times in theGI tract and may drastically alter gastric empty-ing rates. Fluid volumes between humans and mostanimals also differ and prefeeding and postfeedingtimes will also have a potential effect on gastric emp-tying rates and bioavailability.87,101,108,120 Further-more, interindividual variations due to environmen-tal, genetic, and dietary factors are often observed inhumans.19 This is often not the case when using ho-mogeneous animal model strains for preclinical test-ing. Factors including dissolution media compositionand volume, measurement times that reflect actualresidence times in the region, rates of competing nu-cleation, and apparent solubility changes in each re-gion will impact the in vivo dissolution of amorphousdispersions and ultimately the product performance.
The complexity of physiological factors associatedwith the predictions of human bioavailability signif-icantly increase when species differences are consid-ered in the preclinical in vivo PK screening studies.Comprehensive reviews contrasting the GI character-istics across commonly used animal models in phar-maceutical screening can serve as a good referencefor species selection for oral formulations and com-pared with humans.124,125 Several common speciesthat are used to investigate and estimate humanbioavailability are mouse, rat, rabbits, dogs, andmonkeys (Fig. 3).98,126 Changes along the GI tractsof the rats and dogs appeared to vary more whencompared with human values, whereas the pig, an-other nonprimate species increasingly being used insafety pharmacology studies, was more similar to
Figure 5. Representative physiological rate-determiningfactors that can impact the extent of oral bioavailability ob-served for a drug: (a) general overview of the function ofdrug transporters and metabolizing enzymes present in theGI epithelial cells, (b) net permeation/absorption of drugsacross a cell barrier is the sum of the permeability coeffi-cients via each parallel pathway, and (c) a number of addi-tional physiological factors that can influence the amountof free drug available for absorption.
humans.91–97,124 Monkeys actually had higher vari-ability than any other species when compared with
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1372 NEWMAN, KNIPP, AND ZOGRAFI
Table 8. Transporter and Enzyme Interactions for Drugs in Survey
Drug BCS class BDDCS Potential Transporter Enzyme Interactions
Albendazole 2 2 NA CYP3A4, CYP2J2Cyclosporine A 2 2 P-gp CYP3A4, CYP3A5, CYP3A7Esomeprazole Zn 4 3 NA CYP2C9, CYP2C19, CYP3A4,Fenofibrate 2 2 P-gp, 1 URAT1 CYP2C9Frusemide (Furosemide) 4 4 URAT, hNPT4 NAHalofantrine NA NA NA CYP2D6, CYP3A4, CYP3A5Indomethacin 2 2 OAT 1 and 3, OCT 1 and
2, MRP isoformsCYP2C9, CYP2C19, (CYP2D6 and
CYP1A2 minor)Itraconazole 2 2 BCRP, P-gp CYP3A4Ketoconazole 2 2 BCRP, P-gp CYP2C19, CYP3A4Lonidamine 2 or 4 low solubility NA MCT1 NANifedipine 1 2 CYP3A4 NANimodipine 2 2 BCRP, hENT2 CYP3A4Nitrendipine 2 2 BCRP, hENT2 CYP3A4Pranlukast 2 or 4 low solubility NA NA CYP3A4Ritanovir 4 2 BCRP, P-gp, MRP-1 CYP2D6, CYP3A4, CYP3A5Lopinavir 4 2 P-gp, MRP-2 CYP3A4Tacrolimus 2 2 BCRP, P-gp CYP3A4, CYP3A5Tolbutamide 2 2 NA CYP2C9
NA, not available; hNPT4/SLC17A3, human sodium phosphate transporter 4; P-gp, P-glycoprotein; URAT1, urate transporter 1; uridine diphosphateUGP, glucuronosyltransferases (UGTs).
humans, with stomach pH ranges of 4.7–5.0 beingnormal.124 This is important when considering the ef-fects that can occur with weak acids and weak basesduring development. To illustrate this, several studieshave reported highly variable correlations betweenthe human bioavailability and bioavailability deter-mined in rodents, dogs, and monkeys for a number ofcompounds.126,127
The utilization of animal models to investigatebioavailability differences between amorphous dis-persions of an API with their reference materials iscritical to assess in vivo performance and potentiallyhuman clinical performance. It is recommended thatthe selection of a predictive animal model for test-ing the bioavailability of an amorphous dispersionshould be made on a case-by-case basis, where the an-ticipated physiological rate-determining factors willdictate the selection of the species that is most anal-ogous to humans for testing the specific compound.Furthermore, it is recommended that alternative ani-mal models be investigated that may be underutilizeddespite their potentially higher physiological similar-ities to humans. For example, pigs and minipigs havebeen reported as a better model for nonprimate safetypharmacology studies89,97,98,127 and should be consid-ered for future testing of amorphous solid dispersions.
Modeling studies may also be useful when evalu-ating the in vivo performance of amorphous solid dis-persions. The separation of different regions in theGITA model helps to account for regional changesand may enable better prediction of amorphous dis-persion release using regionally targeted formula-tions, especially when using enteric coating polymersin the dispersions.128,129 Moreover, the data obtained
from preclinical and regional in vivo absorption stud-ies may be incorporated into in silico population-based PK (PBPK) models including Simcyp103 orGastroPlus,130,131 where databases can be built andIVIVRs/IVIVCs can be evaluated in vitro, in vivo, orin silico using physiologically based PBPK models.It must be emphasized that in general the predic-tive capabilities of these programs increase with therelevant amount of in vivo data added. Thus, animalstudies are essential for improving in silico-based pre-dictive human PK and can aid in reducing clinicalattrition. There have been a number of other absorp-tion models that have been advanced for predictingabsorption.105 It is suggested that continuous feed-back from these programs be used to direct refine-ment of formulations that will ultimately improveupon clinical performance, particularly during laterstages of clinical testing and throughout life cyclemanagement. A number of other factors need to beconsidered for in vivo studies that involve processesafter the drug has dissolved. First is the mechanismof absorption (Fig. 5). For highly lipophilic molecules,it is suggested that lymphatic absorption and not onlyblood absorption be considered during early screeningstudies to improve lead candidate selection and re-duce clinical attrition.132,133 An extension of the GITAmodel can be used to predict regions of optimal ab-sorption (absorption windows) where complex physio-logical factors can be used along with physicochemicalproperties to tailor dosage form design aimed at in-creasing bioavailability.99,102–104,134 Second is the roleof transporters and drug metabolizing enzymes dur-ing the absorption process. Absorption may be im-pacted by the potential for drug–X interactions (X =
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1373
drug, food, endogenous substrate, and/or xenobiotic),as well as several other factors that need to be iden-tified. An example is the marketed product Kaletra,an amorphous dispersion of lopinavir and ritonavir.In the Kaletra formulation, ritonavir is used to in-hibit the CYP3A4 enzyme, which then increases thebioavailability of lopinavir.135,136 Table 8 reveals thatmany of the amorphous dispersion compounds re-viewed were also substrates for human transportersand/or metabolizing isoforms. The effects of thesevariations cannot be ignored and need to be consid-ered in utilizing the BCS model. Method standardiza-tion and placing a greater emphasis on the BDDCSmight be merited to obtain better IVIVRs or IVIVCs.Third is variation in permeability testing to classifydrugs in the BCS and BDDCS.137 It is recommendedthat careful consideration be given to the permeabil-ity/absorption model used to classify a compound be-cause cell lines and tissue sections may offer differ-ent confounding variables, which can lead to differentresults and confuse early in vivo studies.119 The re-cently proposed Developability Classification Systemoffers an opportunity to expand upon the BCS, andpotentially the BDDCS. This system incorporates im-portant factors that influence drug performance in-cluding the measurement of intestinal permeabilityand solubility in the context of formulation composi-tion and its characteristics such as particle size.138
In summary, there are a number of physiologicallyrelevant factors that need to be incorporated into thedesign of both in vitro and in vivo methods in orderfor a true IVIVR/IVIVC to be realized. In the amor-phous solid dispersion papers that were surveyed,there were several issues that required greater con-sideration and were not fully discussed. First andforemost, a greater understanding of clinically rele-vant conditions needs to be incorporated into in vitrotesting in order to enhance the predictability of de-veloped IVIVRs/IVIVCs. Dissolution media and vol-umes also need to be closely monitored with respectto the fed and fasted states, particularly in the stom-ach. The regional effects of metabolism and activetransport cannot be neglected as they impact the ob-served bioavailability in vivo. Cell models or perme-ability testing surrogates including tissue segmentsmust also be characterized to determine the extent oftheir relevancy to the human in vivo region targetedfor amorphous dispersion release. Furthermore, care-ful consideration of differences across the differentspecies used require a greater understanding, includ-ing their GI physiology, GI regional fluid volumes andpHs, mixing forces, luminal compositions, dietary ef-fects, roles of gender, and regional residence timescomparative to those observed in humans. Animalmodels can offer a good prediction of human responseprovided they are carefully selected based on the de-sired research outcomes that are being sought.
CONCLUSION
In this Commentary, we have attempted to considerthe important factors that affect the therapeutic per-formance of amorphous solid dispersions from anin vitro and in vivo perspective and to make recom-mendations where possible. We have used a limitednumber of publications as a basis for describing a widerange of factors that can influence the stability andperformance of dispersions. In particular, we wish toemphasize the importance of a greater understandingof the important physiological factors associated withthe GI tract that play an important role in optimiz-ing dissolution and bioavailability of API presented insolid dosage forms containing amorphous dispersions.
REFERENCES
1. Serrajuddin ATM. 1999. Solid dispersions of poorly water-soluble drugs: Early promises, subsequent problems, and re-cent breakthroughs. J Pharm Sci 88:1058–1066.
2. Brouwers J, Brewster ME, Augustijns P. 2009. Supersaturat-ing drug delivery systems: Answer to solubility-limited oralbioavailability. J Pharm Sci 98:2549–2572.
3. Bhugra C, Pikal MJ. 2008. Role of thermodynamic, molecu-lar and kinetic factors in crystallization from the amorphousstate. J Pharm Sci 97:1329–1349.
4. Alonzo DE, Zhang GGZ, Zhou D, Gao Y, Taylor LS. 2010. Un-derstanding behavior of amorphous pharmaceutical systemsduring dissolution. Pharm Res 27:608–618.
5. Padden BE, Mill JM, Robbins T, Zocharski PD, Prasad L,Spence JK, LaFountaine J. 2011. Amorphous solid disper-sions as enabling formulations for discovery and early devel-opment. Amer Pharm Rev 1:66–73.
6. Dobry DE, Settell DM, Baumann JM, Ray RJ, Graham LJ,Beyerinck RA. 2009. A model-based methodology for spray-drying process development. J Pharm Innov 4:133–142.
7. Breitenbach J. 2002. Melt extrusion: From process to drugdelivery technology. Eur J Pharm Biopharm 54:107–117.
8. Shamblin SL, Taylor LS, Zografi G. 1998. Mixing behavior ofcolyophilized binary systems. J Pharm Sci 87:694–701.
9. Newman A, Engers D, Bates S, Ivanisevic I, Kelly RC,Zografi G. 2008. Characterization of amorphous API–polymer mixtures using X-ray powder diffraction. J PharmSci 97:4840–4856.
10. Pham TN, Watson SA, Edwards AJ, Chavda M, Clawson JS,Strohmeier M, Vogt G. 2010. Analysis of amorphous solid dis-persions using 2D solid-state NMR and1HT1relaxation mea-surements. Mol Pharm 7:1667–1691.
11. Ta J, Sun Y, Zhang GGZ, Yu L. 2009. Solubility of smallmolecule crystals in polymers: D-Mannitol in PVP, in-domethacin in PVP/VA, and nifedipine in PVP/VA. PharmRes 26:855–864.
12. Sun Y, Tao Y, Zhang GGZ, Yu L. 2010. Solubilities of crys-talline drugs in polymers: An improved analytical methodand comparisons of solubilities of indomethacin and nifedip-ine in PVP, PVP/VA and PVAc. J Pharm Sci 99:4023–4031.
13. Matsumoto T, Zografi G. 1999. Physical properties ofsolid molecular dispersions of indomethacin with poly(vinylpyrrolidone) and poly(vinyl pyrrollidone–co–acetate) inrelation to indomethacin crystallization. Pharm Res16:1722–1728.
14. Taylor LS, Zografi G. 1997. Spectroscopic characterization ofinteractions between PVP and indomethacin in amorphousmolecular dispersions. Pharm Res 14:1691–1698.
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1374 NEWMAN, KNIPP, AND ZOGRAFI
15. Hancock BC, Shamblin S, Zografi G. 1995. Molecular mo-bility of amorphous pharmaceutical solids below their glasstransition temperature. Pharm Res 12:799–806.
16. Friesen DT, Shanker R, Crew M, Smithey DT, Curatolo WJ,Nightingale JAS. 2008. Hydroxypropyl methylcellulose ac-etate succinate-based spray-dried dispersions: An overview.Mol Pharm 5:1003–1019.
17. Curatolo W, Nightingale JA, Herbig SM. 2009. Utility of hy-droxypropylmethylcellulose acetate succinate (HPMCAS) forinitiation and maintenance of drug supersaturation in the GImilieu. Pharm Res 26:1419–1431.
18. Rowland M, Towser TN. 1995. Clinical pharmacokinetics:Concepts and applications. 3rd Ed. Philadephia, Pennsylva-nia: Lippincott Williams & Wilkins, pp 2–6, 57–60, 95, and99.
19. Elmquist WF. 2005. Targeted bioavailability: A fresh lookat pharmacokinetic and pharmacodynamic issues in drug de-livery. In Drug delivery: Principles and practices; Wang B,Siahaan T, Soltero R, Eds. Hoboken: John Wiley and Sons,Inc., pp 73–82.
20. Kalaiselvan R, Mohanta GP, Madhusadan S, Manna PK,Manavaian R. 2007. Enhancement of bioavailability and an-tihelmintic efficacy of albendazole by solid dispersions andcyclodextrin complexation techniques. Pharmazie 62:604–607.
21. Kalaiselvan R, Mohanta GP, Manavaian R. 2006. The inhi-bition of albendazole solid molecular dispersions. Pharmazie61:618–624.
22. Kohri N, Yamayoshi Y, Xin H, Iseki K, Sato N, Todo S,Miyazaki K. 1999. Improving the oral bioavailability of alben-dazole in rabbits by the solid dispersion technique. J PharmPharmacol 51:159–164.
23. Kennedy M, Hu J, Ga P, Li L, Ali-Reynolds A, Chai B, GuptaV, Ma C, Mahajan N, Akrami A, Surapaneni S. 2008. En-hanced bioavailability of a poorly soluble VR1 antagonsit us-ing an amorphous solid dispersion approach: A case study.Mol Pharm 5:981–993.
24. Joshi HN, Tejwani W, Jemal M, Bathala MS, Varia SA, Ser-ajuddin ATM. 2004. Bioavailability enhancement of a poorlywater soluble drug by solid dispersion in a polyetyleneglycol-polysorbate 80 mixture. Int J Pharm 269:251–258.
25. Lakshman JP, Cao Y, Kowalski J, Serajuddin ATM. 2008. Ap-plication of melt extrusion in the development of a physicallyand chemically stable high-energy amorphous solid disper-sion of a poorly water-soluble drug. Mol Pharm 5:994–1002.
26. Liu C, Wu J, Shi B, Zhang Y, Gao T, Pei Y. 2006. Enhancingthe bioavailability of cyclosporine A using solid dispersioncontaining polyethylene (40) stearate. Drug Dev Ind Pharm32:115–123.
27. Kushida I, Ichikawa M, Asakawa N. 2002. Improvement ofdissolution and oral absorption of WR-34122, a poorly wa-ter soluble dual 5-lipoxygenase/cyclooxygenase inhibitor withanti-inflammatory activity by preparing solid dispersion. JPharm Sci 91:258–266.
28. Xie Y, Xie P, Song X, Tang X, Song H. 2008. Preparation ofesomeprazole zinc solid dispersion and study on its pharma-cokinetics. Int J Pharm 360:53–57.
29. He H, Yang R, Tang X. 2010. In vitro and in vivo evaluation offenofibrate solid dispersion prepared by hot-melt extrusion.Drug Dev Ind Pharm 36:681–687.
30. Doherty C, York P. 1998. The in vitro pH–dissolution de-pendence and in vivo bioavailability of frusemide–PVP soliddispersions. J Pharm Pharmacol 41:73–78.
31. Doherty C, York P. Mechanisms of dissolution of frusemide/PVP solid dispersions. Int J Pharm 34:197–205.
32. Doherty C, York P. Accelerated stability of an X-ray amor-phous frusemide–polyvinylpyrrolidone solid dispersion. DrugDev Ind Pharm 15:1969–1987.
33. Khoo S-M, Porter CJH, Charman WN. 2000. The formulationof halofantrine as either non-solubilising PEG 6000 or solu-bilising lipid based solid dispersions: Physical stability andabsolute bioavailability assessment. Int J Pharm 205:65–78.
34. Chokshi RJ, Zia H, Sandhu HK, Shah NH, Malick WA. 2007.Improving the dissolution rate of poorly water soluble drugby solid dispersion and solid solution—Pros and cons. DrugDel 14:33–45.
35. Chowdary KPR, Babu KVVS. 1994. Dissolution, bioavail-ability and ulcerogenic studies on solid dispersions of in-domethacin in water soluble cellulose polymers. Drug DevInd Pharm 20:799–813.
36. Ohnishi N, Yokoyama T, Umeda T, Kiyohara Y, Kuroda T,Kita Y, Kuroda K. 1986. Preparation of sustained-releasesuppositories of indomethacin using a solid dispersion systemand evaluation of bioavailability in rabbits. Chem Pharm Bull34:2999–2304.
37. Ohnishi N, Kiyohara Y, Kita Y, Kuroda K, Yokoyama T. 1987.Effect of the molecular weight of polyethylene glycol on thebioavailability of indomethacin in sustained-release suppos-itories prepared with solid dispersions. Chem Pharm Bull35:3511–3515.
38. DiNunzio JC, Brough C, Miller DA, Williams RO, McGinityJW. 2010. Fusion processing of itraconazole solid dispersionsby KinetiSol dispersing: A comparative study to hot melt ex-trusion. J Pharm Sci 99:1239–1253.
39. DiNunzio JC, Miller DA, Yang W, McGinity JW, Williams RO.2008. Amorphous compositions using concentration enhanc-ing polymers for improved bioavailability of itraconazole. MolPharm 5:968–980.
40. Engers D, Teng J, Jimenez-Novoa J, Gent P, Hossack S,Campbell C, Thomson J, Ivanisevic I, Templeton A, ByrnS, Newman A. 2010. A solid-state approach to enable earlydevelopment compounds: Selection and animal bioavailabil-ity studies of an itraconazole amorphous solid dispersion. JPharm Sci 99:3901–3922.
41. Kapsi SG, Ayres JW. 2001. Processing factors in developmentof solid solution formulation of itraconazole for enhance-ment of drug dissolution and bioavailability. Int J Pharm229:193–203.
42. Lee SL, Nam K, Kim MS, Jun SW, Park J-S, Woo JS, HwangS-J. 2005. Preparation and characterization of solid disper-sions of itraconazole by using aerosol solvent extraction sys-tem for improvement in drug solubility and bioavailability.Arch Pharm Res 28:866–874.
43. Nam K-W, Lee S, Hwang S-J, Woo JS. 2002. Enhanc-ing water-solubility of poorly soluble drug, itraconazolewith water-soluble polymer using supercritical fluid process-ing(abstract #245). Proceedings of the Controlled Release So-ciety, 29thAnnual Meeting, July 20-25, Seoul, South Korea.
44. Six K, Daems T, de Hoon J, Van Hecken A, Depre M, BoucheM-P, Prinsen P, Verreck G, Peeters J, Brewster ME, Vanden Mooter G. 2005. Clinical study of solid dispersions ofitraconazole prepared by hot-stage extrusion. Eur J PharmSci 24:179–186.
45. Six K, Leuner C, Dressman J, Verreck G, Peeters J, Bla-ton N, Augustjins P, Kinget R. 2002. Thermal properties ofhot-stage extrudates of itraconazole and Eudragit E100.Phase separation and polymorphism. J Therm Anal Cal68:591–601.
46. Six K, Berghmans H, Leuner C, Dressman J, Van WerdeK, Mullens J, Benoist L, Thimon M, Meublat L, Verreck G,Peeters J, Brewster M, Van den Mooter G. 2003. Characteri-zation of solid dispersions of itraconazole and hydroxypropy-lmethylcellulose prepared by melt extrusion, Part II. PharmRes 20:1047–1054.
47. Six K, Verreck G, Peeter J, Brewster M, Van den Mooter G.2004. Increased physical stability and improved dissolution
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1375
properties of itraconazole, a class II drug, by solid dispersionsthat combine fast- and slow-dissolving polymers. J Pharm Sci93:124–131.
48. Heo M-Y, Piao S-S, Kim T-W, Cao Q-R, Kim A, Lee B-J.2005. Effect of solubilizing and microemulsifying excipientsin polyethylene glycol 6000 solid dispersions on enhanceddissolution and bioavailability of ketoconazole. Arch PharmRes 28:604–611.
49. Matsunaga N, Nakamura K, Yamamoto A, Taguchi E, Tsun-oda H, Takahashi K. 2006. Improvement by solid dispersionsof the bioavailability of KRN633, a selective inhibitor of vegfreceptor-2 tryosine kinase, and identification of its potentialtherapeutic window. Mol Cancer Ther 5:80–88.
50. Palmieri FP, Cantalamessa F, Martino PD, Nasuti X,Martelli S. 2002. Lonidamine solid dispersions: In vitro andin vivo evaluation. Drug Dev Ind Pharm 28:1241–1250.
51. Kai T, Akayama Y, Nomura S, Sata M. 1996. Oral absorptionimprovement of poorly soluble drug using solid dispersiontechnique. Chem Pharm Bull 44:569–571.
52. Law SL, Lo WY, Lin FM, Chang CH. 1992. Dissolutionand absorption of nifedipine in polyethylene glycol soliddispersion containing phosphatidylcholine. Int J Pharm84:161–166.
53. Emara LH, Badr RM, Abd Elbary A. 2002. Improving thedissolution and bioavailability of nifedipine using solid dis-persions and solubilizers. Drug Dev Ind Pharm 28:795–807.
54. Sugimoto I, Kuchiki A, Nakagawa H. 1981. Stability ofnifedipine–polyvinylpyrrollidone coprecipitate. Chem PharmBull 29:1715–1723.
55. Sugimoto I, Kuchiki A, Nakagawa H, Tohgo K, Kondo S,Iwane I, Takahashi K. 1980. Dissolution and absorption ofnifedipine from nifedipine–polyvinylpyrrollidone coprecipi-tate. Drug Dev Ind Pharm 6:137–160.
56. Uekama K, Uekama K, Ikegami K, Wang Z, Horiuchi Y,Hirayama F. 1992. Inhibitory effect of 2-hydroxypropyl-$-cyclodextrin on crystal growth of nifedipine during storage:Superior dissolution and oral bioavailability compared withpolyvinyl pyrrolidone K-30. J Pharm Pharmacol 44:73–78.
57. Zheng X, Yang R, Zhang Y, Wang Z, Tang X, Zheng L. 2007.Part II: Bioavailability in Beagle dogs of nimodipine soliddispersions prepared by hot-melt extrusion. Drug Dev IndPharm 33:783–789.
58. Zheng X, Yang R, Tang X, Zheng L. 2007. Part I: Characteri-zation of solid dispersions of nimodipine prepared by hot-meltextrusion. Drug Dev Ind Pharm 33:791–802.
59. Cui F, Yang M, Jiang Y, Cun D, Lin W, Fan Y, KawashimaY. 2003. Design of sustained-release nitrendipine microspe-heres having solid dispersion structure by quasi-emulsionsolvent diffusion method. J Control Rel 91:375–384.
60. Chono S, Takeda E, Seki T, Morimoto K. 2008. Enhance-ment of the dissolution rate and gastrointestinal absorptionof prankulast as a model poorly water-soluble drug by grind-ing with gelatin. Int J Pharm 347:71–78.
61. Verrek G, Vandecruys R, De Conde V, Baert L, Peeters J,Brewster M. 2004. The use of three different solid disper-sion formulations—melt extrusions, film-coated beads, and aglass thermoplastic system—to improve the bioavailability ofa novel microsomal triglyceride transfer protein inhibitor. JPharm Sci 93:1217–1228.
62. Law D, Schmitt EA, Marsh KC, Everitt EA, Wang W, FortJJ, Krill SL, Qiu Y. 2004. Ritanovir–PEG 8000 amorphoussolid dispersions: In vitro and in vivo evaluations. J PharmSci 93:563–570.
63. Law D, Krill SL, Schmitt EA, Fort JJ, Qiu Y, Wang W, PorterWR. 2001. Physicochemical considerations in the prepara-tion of amorphous ritanovir–poly(ethylene glycol) 8000 soliddispersions. J Pharm Sci 90:1015–1025.
64. Klein C E, Chiu Y-L, Awni W, Zhu T, Heuser RS, Doan T, Bre-itenbach J, Morris JB, Brun SC, Hanna GJ. 2007. The tabletformulation of lopinavir/ritanovir provides similar bioavail-ability to the soft–gelatin capsule formulation with less phar-macokinetic variability and diminished food effect. J AcquirImmune Defic Syndr 44:401–410.
65. Sheen P-C, Khetarpal VK, Cariola CM, Rowlings CE. 1995.Formulation studies of a poorly water-soluble drug, insolid dispersions to improve bioavailability. Int J Pharm118:221–227.
66. Yamashita K, Nakate T, Okimoto K, Ohike A, TkunagaY, Ibuki R, Higaki K, Kimura T. 2003. Establishment ofnew preparation method for solid dispersion formulation oftacrolimus. Int J Pharm 267:79–91.
67. Kedzierewicz F, Zinutti C, Hoffman M, Maindent P. 1993.Bioavailability study of tolbutamide g-cyclodextrin inclusioncompounds. Solid dispersions and bulk powder. Int J Pharm94:69–74.
68. Kedzierewicz F, Hoffman M, Maincent F. 1990. Comparisonof tolbutamide $-cyclodextrin inclusion compounds and soliddispersions. Int J Pharm 58:221–227.
69. Kimura K, Hirayama F, Arima H, Uekama K. 2000. Ef-fects of aging on crystallization, dissolution and absorptioncharacteristics of amorphous tolbutamide-2-hydroxypropyl-$-cyclodextrin complex. Chem Pharm Bull 48:646–650.
70. Brown CK, Chokshi HP, Nickerson B, Reed RA, Rohrs BR,Shah PA. 2004. Acceptable analytical practices for dissolutiontesting of poorly soluble compounds. Pharm Tech. Decemberissue: 56–64.
71. Food and Drug Administration. 2000.Guidance for industry:Waiver of in vivo bioavailability and bioequivalence studiesfor immediate release solid oral dosage forms based on a Bio-pharmaceutics Classification System. Rockville, Maryland.
72. Food and Drug Administration. 1997.Guidance for industry:Extended release oral dosage forms: Development, evalua-tion, and application of in vitro/in vivo correlations. Rockville,Maryland
73. Loebenberg R, Amidon GL. 2000. Modern bioavailability,bioequivalence and biopharmaceutics classification system.New scientific approaches to international regulatory stan-dards. Eur J Pharm Biopharm 5:3–12.
74. Xia D, Cui F, Piao H, Cun D, Piao H, Jiang Y, Ouyang M,Quan P. 2010. Effect of crystal size on the in vitro dissolu-tion and oral absorption of nitrendipine in rats. Pharm Res27:1965–1976.
75. Jantratid E, Janssen N, Reppas C, Dressman J. 2008. Dis-solution media simulating conditions in the proximal humangastrointestinal tract: An update. Pharm Res 25:1663–1676.
76. Mudie DM, Amidon GL, Amidon GE. 2010. Physiological pa-rameters for oral delivery and in-vitro testing. Mol Pharm7:1388–1405.
77. Bevernage J, Fortier T, Brouwers J, Tack J, Annaert P, Au-gustijns P. 2011. Excipient-mediated supersaturation stabi-lization in human intestinal fluids. Mol Pharm 8:564–570.
78. Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, MunosBH, Lindborg SR, Schacht AL. 2010. How to improve R & Dproductivity: The pharmaceutical industry’s grand challenge.Nat Rev Drug Discov 9:203–214.
79. General Chapters <711>, “Dissolution” and <724> “DrugRelease.” 2004.In The United States Pharmacopeia 27–National Formulary 22. United States PharmacopeialConvention, Inc., Rockville, Maryland, pp 2303–2304and2305–2312.
80. Yu LX, Wang JT, Hussain AS. 2002. Evaluation of USP appa-ratus 3 for dissolution testing of immediate- release products.AAPS PharmSci 4, 1–5.
81. Carino SR, Sperry DC, Hawley M. 2010. Relative bioavail-ability of three different solid forms of PNU-141659 as
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012

1376 NEWMAN, KNIPP, AND ZOGRAFI
determined with the artificial stomach-duodenum model. JPharm Sci 99:3923–3930.
82. Gao Y, Carr A, Spence JK, Wang WW, Turner TM, LipariJM, Miller JM. 2010. A pH–dilution method for estimation ofbiorelevant drug solubility along the gastrointestinal tract:Application to physiologically based pharmacokinetic model-ing. Mol Pharm 7:1516–1526.
83. Polster CS, Stassi F, Wu S-Y, Sperry DC. 2010. Use of ar-tificial stomach–duodenum model for investigation of dos-ing fluid effect on clinical trial variability. Mol Pharm7:1533–1538.
84. General Chapter <1092>. 2006. The dissolution procedure:Development and validation. In The United States 41Phar-macopeia 34–National Formulary 29. United States Pharma-copeial Convention, Inc., Rockville, Maryland.
85. Supplementary Chapter I E. 1998.Dissolution of solid oraldosage forms.” In British Pharmacopeia1998, Vol. II, ppA327–A331.
86. Fadda HM, Sousa T, Carlsson AS, Abrahamsson B, WilliamsJG, Kumar D, Basit AW. 2010. Drug solubility in luminalfluids from different regions of the small and large intestineof humans. Mol Pharm 7:1527–1532.
87. Fadda HM, McConnell EL, Short MD, Basit AW. 2009. Meal-induced acceleration of tablet transit through the humansmall intestine. Pharm Res 26:356–360.
88. Selen A. 2009. Biorelevant dissolution testing: Characteriz-ing the product for the patient. short course #3: developingbiorelevant dissolution test methods with an emphasis onQbD. Los Angeles, California: American Association of Phar-maceutical Scientists.
89. Schiller C, Frohlich CP, Giessmann T, Siegmund W, Mon-nikes H, Hosten N, Weitschies W. 2005. Intestinal fluidvolumes and transit of dosage forms as assessed by mag-netic resonance imaging. Aliment Pharmacol Ther 22:971–979.
90. Weitschies W, Wedemeyer RS, Kosch O, Fach K, Nagel S,Soderlind E. 2005. Impact of the intragastric location of ex-tended rlease tablets on food interactions. J Control Release108:375–85.
91. Minekus M, Marteau P, Havenaar R, Huis In T Veld J. 1995.A multicomparmental dynamic computer-controlled modelsimulating the stomach and small intestine. Altern Lab Ani-mals 23:197–209.
92. Minekus M, Smeets-Peeters M, Bernalier A, Marol-Bonnin S,Havenaar R, Marteau P, Alric M, Fonty G, Huis In T Veld JH.1995. A computer-controlled system to simulate conditionsof the large intestine with peristaltic mixing, water absorp-tion and absorption of fermentation products. Appl MicrobiolBiotechnol 53:108–114.
93. Garbacz G, Wedemeyer R-S, Nagel S, Giessmann T,Monnikes H, Wilson CG. 2008. Irregular absorption pro-files observed from diclofenac extended release tablets canbe predicted using a dissolution test apparatus that mimicsin vivo physical stresses. Eur J Pharm Biopharm 70:421–428.
94. D’Arcy DM, Corrigan OI, Healy AM. 2006. Evaluation of hy-drodynamics in the basket dissolution apparatus using com-putational fluid dynamics–dissolution rate implications. EurJ Pharm Sci 27:259–267.
95. Wei JD, Ho HO, Chen CH, Ke WT, Chen ET, Sheu MT. 2010.Characterisation of fenofibrate dissolution delivered by a self-microemulsifying drug-delivery system. J Pharm Pharmacol62:1685–1696.
96. Garbacz G, Golke B, Wedemeyer R-S, Axell M, Soderlind E,Abrahamsson B. 2009. Comparison of dissolution profiles ob-tained from nifedipine extended release once a day productsusing different dissolution test apparatuses. Eur J Pharm Sci38:147–155.
97. Webster J, Bollen P, Grimm H, Jennings M. 2010. Ethical im-plications of using the minipig in regulatory toxicology stud-ies. J Pharmacol Toxicol Methods 62:160–166.
98. Curtis MJ. 2010. The RETHINK project: Impact of toxi-city testing in the minipig as an alternative approach inregulatory toxicity testing. J Pharmacol Toxicol Methods62(3):157–162.
99. Kimura T, Higaki K. 2002. Gastrointestinal transit and drugabsorption. Biol Pharm Bull 25:149–164.
100. Collins PJ, Horowitz M, Cook DJ, Harding PE, ShearmanDJC. 1983. Gastric emptying in normal subjects—A repro-ducible technique using a single scintillation camera andcomputer system. Gut 24:1117–1125.
101. McLaughlan G, Fullarton GM, Crean GP, McColl KE. 1989.Comparison of gastric body and antral pH: A 24 hour ambu-latory study in healthy volunteers. Gut 30:573–578.
102. Washington N, Washington C, Wilson CG. 2001.Physiologicalpharmaceutics: Barriers to drug absorption, 2nd Ed. London,UK: Taylor and Francis.
103. Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, Tucker G. 2009. Population-based mechanis-tic prediction of oral drug absorption. AAPS J 11:225–237.
104. Rostami-Hodjegan A, Tucker G. 2007. Simulation and predic-tion of in vivo drug metabolism in human populations fromin vitro data. Nat Rev Drug Disc 6:140–148.
105. Huang W, Lee SL, Yu LX. 2009. Mechanistic approaches topredicting oral drug absorption. AAPS J 11:217–224.
106. Coupe AJ, Davis SS, Wilding IR. 1991. Variation in gastroin-testinal transit of pharmaceutical dosage forms in healthysubjects. Pharm Res 8:360–364.
107. Podczeck F, Mitchell CL, Newton JM, Evans D, Short MB.2007. The gastric emptying of food as measured by gamma-scintigraphy and electrical impedance tomography (EIT) andits influence on the gastric emptying of tablets of differentdimensions. J Pharm Pharmacol 59(11):1527–1536.
108. McConnell EL, Fadda HM, Basit AW. 2008. Gut instincts:Explorations in intestinal physiology and drug delivery. IntJ Pharm 364:213–226.
109. Yu LX, Amidon GL. 2002. Biopharmaceutics ClassificationSystem: The scientific basis for biowaiver extensions. PharmRes 19(7):921–925.
110. Wu CY, Benet LZ. 2005. Predicting drug disposition via ap-plication of BCS: Transport/absorption/elimination interplayand development of a biopharmaceutics drug disposition clas-sification system. Pharm Res 22:11–23.
111. Ashiru DAI, Patel R, Basit AW. 2008. Polyethylene glycol400 enhances the bioavailability of a BCS class III drug(ranitidine) in male subjects but not females. Pharm Res25:2327–2333.
112. Kabanov AV, Okano T. 2003. Challenges in polymer thera-peutics: State of the art and prospects of polymer drugs. AdvExp Med Biol 519:1–27.
113. Vertzoni M, Diakidou A, Chatzilias M, Soderlind E, Abra-hamsson B, Dressman JB, Reppas C. 2010. Biorelevant me-dia to simulate fluids in the ascending colon of humans andtheir usefulness in predicting intracolonic drug solubility.Pharm Res 27:2187–2196.
114. Puri V, Dantuluri K, Bansal AK. 2011. Investigation of atyp-ical dissolution behavior of an amorphous solid dispersion. JPharm Sci 100:2640–2648.
115. Rolan P, Danhof M, Stanskic D, Peck C. 2007. Current issuesrelating to drug safety especially with regard to the use ofbiomarkers: A meeting report and progress update. Eur JPharm Sci 30:107–112.
116. Jantratid E, Janssen N, Reppas C, Dressman J. 2008. Dis-solution media simulating conditions in the proximal humangastrointestinal tract: An update. Pharm Res 25:1663–1676.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012 DOI 10.1002/jps

ASSESSING THE PERFORMANCE OF AMORPHOUS SOLID DISPERSIONS 1377
117. Volpe DA. 2010. Application of method suitability for drugpermeability classification. AAPS J 12:670–678.
118. Volpe DA. 2008. Variability in Caco-2 and MDCK cell-basedintestinal permeability assays. J Pharm Sci 97:712–725.
119. Yamashita S, Tachiki H. 2009. Analysis of risk factors inhuman bioequivalence study that incur bioinequivalence oforal drug products. Mol Pharm 6:48–59.
120. Abrahamsson B, Lennernas H. 2005.Application of the Bio-pharmaceutics Classification System now and in the future.Drug bioavailability: Estimation of solubility, permeability,absorption and bioavailability. In Methods and principles inmedicinal chemistry; vanWaterbeemd H, Lennerhas H, Ar-tursson P, Eds. Vol. 18. Weinheim, Germany: Wiley-VCH.
121. Lennerhas H. 1998. Human intestinal permeability. J PharmSci 87:403–410.
122. Maury J, Nicoletti C, Guzzo-Chambraud L, Maroux S.1995. The filamentous brush border glycocalyx, a mucin-like marker of enterocyte hyper-polarization. Eur J Biochem228:323–331.
123. Sutton SC. 2009. Role of physiological intestinal water in oralabsorption. AAPS J 11:277–285.
124. Desso JM, Williams AL. 2008. Contrasting the gastrointesti-nal tracts of mammals: Factors that influence absorption.Ann Rep Med Chem 43:353–371.
125. Kararli TT. 1995. Comparison of the gastrointestinalanatomy, physiology, and biochemistry of humans and com-monly used laboratory animals. Biopharm Drug Dispos16:351–380.
126. Grass GM, Sinko PJ. 2002. Physiologically-based phar-macokinetic simulation modeling. Adv Drug Deliv Rev54:433–451.
127. Bode G, Clausing P, Gervais F, Loegsted J, Luft J, NoguesV, Sims J, and under the auspices of the Steering Group ofthe RETHINK Project. 2010. The utility of the Minipig as ananimal model in regulatory toxicology. J Pharmacol ToxicolMethods 62:196–220.
128. Kagan L, Hoffman A. 2008. Systems for region selective drugdelivery in the gastrointestinal tract: Biopharmaceutical con-siderations. Expert Opin Drug Deliv 5:681–692.
129. Johnson KC. 2003. Dissolution and absorption modeling:Model expansion to simulate the effects of precipitation, wa-ter absorption, longitudinally changing intestinal permeabil-ity, and controlled release on drug absorption. Drug Dev IndPharm 29:833–842.
130. Bolger MB, Lukacova V, Woltosz WS. 2009. Simulationsof the nonlinear dose dependence for substrates of influxand efflux transporters in the human intestine. AAPS J11:353–363.
131. Lukacova V, Woltosz WS, Bolger MB. 2009. Prediction ofmodified release pharmacokinetics and pharmacodynamicsfrom in vitro, immediate release, and intravenous data. AAPSJ 11:323–334.
132. Trevaskis NL, Charman WN, Porter CJ. 2008. Lipid-based delivery systems and intestinal lymphatic drug trans-port: A mechanistic update. Adv Drug Deliv Rev 60:702–716.
133. Porter CJ, Charman WN. 2001. Intestinal lymphatic drugtransport: An update. Adv Drug Deliv Rev 50:61–80.
134. Aoki Y, Morishita M, Asai K, Akikusa B, Hosoda S, TakayamaK. 2005. Region-dependent role of the mucous/glycocalyx lay-ers in insulin permeation across rat small intestinal mem-brane. Pharm Res 22:1854–1862.
135. van Waterschoot RA, ter Heine R, Wagenaar E, van derKruijssen CM, Rooswinkel RW, Huitema AD, Beijnen JH,Schinkel AH. 2010. Effects of cytochrome P450 3A (CYP3A)and the drug transporters P-glycoprotein (MDR1/ABCB1)and MRP2 (ABCC2) on the pharmacokinetics of lopinavir.Br J Pharmacol 160:1224–1233.
136. ter Heine R, Van Waterschoot RA, Keizer RJ, Beijnen JH,Schinkel AH, Huitema AD. 2011. An integrated pharmacoki-netic model for the influence of cyp3a4 expression on thein vivo disposition of lopinavir and its modulation by riton-avir. J Pharm Sci 100:2508–2515.
137. Benet LZ, Broccatelli F, Oprea TI. 2011. BDDCS applied toover 900 drugs. AAPS J 13:519–547.
138. Butler JM, Dressman JB. 2010. The developability classifi-cation system: Application of biopharmaceutics concepts toformulation development. J Pharm Sci 99:4940–4954.
DOI 10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 101, NO. 4, APRIL 2012
Related Documents











