Vaccine 33 (2015) 3739–3745 Contents lists available at ScienceDirect Vaccine j our na l ho me page: www.elsevier.com/locate/vaccine Assessing stability and assembly of the hepatitis B surface antigen into virus-like particles during down-stream processing Maria Zahid a,1 , Heinrich Lünsdorf b , Ursula Rinas a,b,∗ a Leibniz University of Hannover, Technical Chemistry, Life Science, Callinstr. 5, 30167 Hannover, Germany b Helmholtz Centre for Infection Research, Inhoffenstr. 7, 38124 Braunschweig, Germany a r t i c l e i n f o Article history: Received 9 March 2015 Received in revised form 21 May 2015 Accepted 23 May 2015 Available online 12 June 2015 Keywords: Hepatitis B surface antigen virus-like particles Purification Aerosil-380 Ion-exchange chromatography Size-exclusion chromatography Transmission electron microscopy Vaccine a b s t r a c t The hepatitis B surface antigen (HBsAg) is a recombinant protein-based vaccine being able to form virus- like particles (VLPs). HBsAg is mainly produced using yeast-based expression systems, however, recent results strongly suggest that VLPs are not formed within the yeast cells during the cultivation but are formed in a gradual manner during the following down-stream procedures. VLPs are also not detectable during the first down-stream steps including mechanical and EDTA/detergent-assisted cell destruction. Moreover, VLPs are not detectable in the cell lysate treated with polyethylene glycol and colloidal silica. The first VLP resembling structures appear after elution of HBsAg from colloidal silica to which it binds through hydrophobic interaction. These first VLP resembling structures are non-symmetrical as well as heterodisperse and exhibit a high tendency toward cluster formation presumably because of surface exposed hydrophobic patches. More symmetrical and monodisperse VLPs appear after the following ion-exchange and size-exclusion chromatography most likely as the result of buffer changes during these purification steps (toward more neutral pH and less salt). Final treatment of the VLPs with the denaturant KSCN at moderate concentrations with following KSCN removal by dialysis does not cause unfolding and VLP disassembly but results in a re- and fine-structuring of the VLP surface topology. © 2015 Elsevier Ltd. All rights reserved. 1. Introduction The hepatitis B vaccine was the first recombinant protein-based vaccine introduced into the market in 1986 [1]. The recombinant vaccine was initially produced using the yeast Saccharomyces cere- visiae, later on also other yeast derived expression systems were employed for commercial production. The hepatitis B vaccine is based on the major hepatitis B surface antigen (HBsAg), a protein being able to assemble into so-called virus-like particles (VLPs). In the beginning, HBsAg VLPs of ∼22 nm were purified from the plasma of asymptomatic hepatitis B virus carriers, but due to safety issues and limited supply were later replaced by VLPs derived through recombinant production [2]. HBsAg is a very hydrophobic protein with long stretches of connected hydrophobic amino acids. Successful production of HBsAg VLPs has been obtained using mammalian and yeast based ∗ Corresponding author at: Helmholtz Centre for Infection Research, Inhoffenstr. 7, 38124 Braunschweig, Germany, Tel.: +49 53161817014. E-mail address: [email protected] (U. Rinas). 1 Current address: Department of Biology, SBA School of Science and Engineering, University of Management Sciences, Lahore 54792, Pakistan. expression systems. Secretory production of HBsAg VLPs has been achieved with mammalian systems, however with non economic production titers [3]. Moreover, the very slow secretion kinetics of HBsAg VLPs in mammalian cells also indicated that the in vivo assembly of HBsAg monomers into VLPs is a slow process com- pared to other VLP forming proteins [4]. In yeast, secretion is very inefficient and high-level production has only been achieved as intracellular product [5–7]. The purification of recombinant HBsAg from yeast cultures is well documented [5,8–29], and sev- eral studies have shown that purified yeast-derived HBsAg appears in characteristic ∼22 nm VLPs [7,30–32]. These particles are highly immunogenic and capable of eliciting potent neutralizing antibod- ies as they mimic the conformation of native viruses but lack the viral genome and can be used as safe and cheap vaccine [1,30,33,34]. The HBsAg monomers form the major part of the HBsAg VLPs, repre- senting around 60–70% of the VLP mass, the remaining part consists of lipids [35,36]. Previously, we have reported high level production of HBsAg using Pichia pastoris as expression host [7]. Despite extensive search no evidence for intracellular VLPs was found, however, we provided evidence that HBsAg remains in the endoplasmic reticulum (ER) where it does not form VLPs but where a major fraction of the protein assembles into well-ordered multi-layered http://dx.doi.org/10.1016/j.vaccine.2015.05.066 0264-410X/© 2015 Elsevier Ltd. All rights reserved.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Ai
Ma
b
a
ARRAA
KHpPAISTV
1
vvvebbIpit
cH
7
U
h0
Vaccine 33 (2015) 3739–3745
Contents lists available at ScienceDirect
Vaccine
j our na l ho me page: www.elsev ier .com/ locate /vacc ine
ssessing stability and assembly of the hepatitis B surface antigennto virus-like particles during down-stream processing
aria Zahida,1, Heinrich Lünsdorfb, Ursula Rinasa,b,∗
Leibniz University of Hannover, Technical Chemistry, Life Science, Callinstr. 5, 30167 Hannover, GermanyHelmholtz Centre for Infection Research, Inhoffenstr. 7, 38124 Braunschweig, Germany
r t i c l e i n f o
rticle history:eceived 9 March 2015eceived in revised form 21 May 2015ccepted 23 May 2015vailable online 12 June 2015
eywords:epatitis B surface antigen virus-likearticlesurification
a b s t r a c t
The hepatitis B surface antigen (HBsAg) is a recombinant protein-based vaccine being able to form virus-like particles (VLPs). HBsAg is mainly produced using yeast-based expression systems, however, recentresults strongly suggest that VLPs are not formed within the yeast cells during the cultivation but areformed in a gradual manner during the following down-stream procedures. VLPs are also not detectableduring the first down-stream steps including mechanical and EDTA/detergent-assisted cell destruction.Moreover, VLPs are not detectable in the cell lysate treated with polyethylene glycol and colloidal silica.The first VLP resembling structures appear after elution of HBsAg from colloidal silica to which it bindsthrough hydrophobic interaction. These first VLP resembling structures are non-symmetrical as well asheterodisperse and exhibit a high tendency toward cluster formation presumably because of surface
erosil-380on-exchange chromatographyize-exclusion chromatographyransmission electron microscopyaccine
exposed hydrophobic patches. More symmetrical and monodisperse VLPs appear after the followingion-exchange and size-exclusion chromatography most likely as the result of buffer changes during thesepurification steps (toward more neutral pH and less salt). Final treatment of the VLPs with the denaturantKSCN at moderate concentrations with following KSCN removal by dialysis does not cause unfolding andVLP disassembly but results in a re- and fine-structuring of the VLP surface topology.
© 2015 Elsevier Ltd. All rights reserved.
. Introduction
The hepatitis B vaccine was the first recombinant protein-basedaccine introduced into the market in 1986 [1]. The recombinantaccine was initially produced using the yeast Saccharomyces cere-isiae, later on also other yeast derived expression systems weremployed for commercial production. The hepatitis B vaccine isased on the major hepatitis B surface antigen (HBsAg), a proteineing able to assemble into so-called virus-like particles (VLPs).
n the beginning, HBsAg VLPs of ∼22 nm were purified from thelasma of asymptomatic hepatitis B virus carriers, but due to safety
ssues and limited supply were later replaced by VLPs derivedhrough recombinant production [2].
HBsAg is a very hydrophobic protein with long stretches ofonnected hydrophobic amino acids. Successful production ofBsAg VLPs has been obtained using mammalian and yeast based
∗ Corresponding author at: Helmholtz Centre for Infection Research, Inhoffenstr., 38124 Braunschweig, Germany, Tel.: +49 53161817014.
E-mail address: [email protected] (U. Rinas).1 Current address: Department of Biology, SBA School of Science and Engineering,niversity of Management Sciences, Lahore 54792, Pakistan.
ttp://dx.doi.org/10.1016/j.vaccine.2015.05.066264-410X/© 2015 Elsevier Ltd. All rights reserved.
expression systems. Secretory production of HBsAg VLPs has beenachieved with mammalian systems, however with non economicproduction titers [3]. Moreover, the very slow secretion kineticsof HBsAg VLPs in mammalian cells also indicated that the in vivoassembly of HBsAg monomers into VLPs is a slow process com-pared to other VLP forming proteins [4]. In yeast, secretion isvery inefficient and high-level production has only been achievedas intracellular product [5–7]. The purification of recombinantHBsAg from yeast cultures is well documented [5,8–29], and sev-eral studies have shown that purified yeast-derived HBsAg appearsin characteristic ∼22 nm VLPs [7,30–32]. These particles are highlyimmunogenic and capable of eliciting potent neutralizing antibod-ies as they mimic the conformation of native viruses but lack theviral genome and can be used as safe and cheap vaccine [1,30,33,34].The HBsAg monomers form the major part of the HBsAg VLPs, repre-senting around 60–70% of the VLP mass, the remaining part consistsof lipids [35,36].
Previously, we have reported high level production of HBsAgusing Pichia pastoris as expression host [7]. Despite extensive
search no evidence for intracellular VLPs was found, however,we provided evidence that HBsAg remains in the endoplasmicreticulum (ER) where it does not form VLPs but where a majorfraction of the protein assembles into well-ordered multi-layered
3 ine 33
llHsca[
Vwpbc
2
2
ocT
2
db
2s
pppMC
2
Gfiofoa
2
wpfp3ww1pic1afit
740 M. Zahid et al. / Vacc
amellar structures [28]. The layering order of HBsAg in these lamel-ar structures strongly suggested the presence of well-orderedBsAg subunits [28], which should be solubilizable without getting
tructurally disordered to reassemble into VLPs under appropriateonditions. Purification of Pichia-derived HBsAg using the protocollso employed in this study was finally leading to well-defined VLPs7,28,37] with excellent immunogenic properties [37].
In this contribution we show that the assembly of HBsAg intoLPs occurs gradually during downstream processing. Moreover,e also analyzed the resistance of HBsAg VLPs toward buffer com-onents employed during down-stream processing in order toetter understand the criteria determining the stability and theonditions allowing the assembly of HBsAg into VLPs.
. Materials and methods
.1. Strain and culture conditions
The construction of the P. pastoris strain GS115 carrying 8 copiesf the HBsAg gene under the control of the AOX1 promoter andonditions used for HBsAg production were described before [7,38].he host strain GS115 was from Invitrogen (Carlsbad, CA).
.2. Purification of recombinant HBsAg
The purification of HBsAg was carried out essentially asescribed before [28,37] with minor modifications as outlinedelow.
.2.1. Step 1: Cell lysis and EDTA/detergent mediatedolubilization of HBsAg
A cell pellet corresponding to 100 g wet cell mass was resus-ended in 1 L ice cold lysis buffer (25 mM sodium phosphate buffer,H 8.0, 5 mM EDTA, 0.6% (v/v) Tween-20). This cell suspension wasassed through a microfluidizer (M110L, Microfluidics, Newton,A, USA) for 12–14 times at a pressure of 12,000 psi and ∼4 ◦C.
ell lysis was confirmed by light microscopy.
.2.2. Step 2: Polyethylene glycol (PEG) precipitationPolyethylene glycol 6000 (PEG6000, Sigma-Aldrich Chemie
mbH, Germany) was added slowly to 1 L of the cell lysate to anal concentration of 5% (w/v) followed by addition of 5 M NaCl tobtain a final concentration of 0.5 M NaCl. The mixture was stirredor 2 h and precipitation allowed for additional 12–16 h at 4 ◦C with-ut stirring. Finally, the suspension was clarified by centrifugationt 3345 × g for 25 min. All steps were carried out at 4 ◦C.
.2.3. Step 3: Aerosil-380 adsorptionAerosil-380 (Evonik, Hanau, Germany) was gently mixed twice
ith Aerosil equilibration buffer (25 mM sodium phosphate buffer,H 7.2, 0.5 M NaCl) and re-collected by centrifugation at 3345 × gor 15 min and 4 ◦C. The clarified supernatant obtained after PEGrecipitation (∼800 mL) was added to the pre-equilibrated Aerosil-80 pellet (0.13 g of dry Aerosil-380 pre-equilibrated per g of initialet cell mass) and the pH adjusted to pH 7.2. This suspensionas stirred for 4 h at 4 ◦C and centrifuged at 4 ◦C and 3345 × g for
5 min. The pellet was washed twice with 25 mM sodium phos-hate buffer (pH 7.2), centrifuged as above, finally resuspended
n 800 mL Aerosil elution buffer (50 mM sodium carbonate-bi-arbonate buffer, pH 10.8, 1.2 M urea) and kept at 37 ◦C for
2 h with stirring. This suspension was then centrifuged at 25 ◦Cnd 8665 × g for 150 min. The solution was clarified by vacuum-ltration (0.2 �m) and stored for 25 h at 4 ◦C before proceeding tohe next step.(2015) 3739–3745
2.2.4. Step 4: Ion-exchange chromatographyThe clarified Aerosil-380 eluate was further processed by anion-
exchange chromatography. Prior to chromatography, the pH wasadjusted to pH 8.0 using phosphoric acid. DEAE Sepharose FFresin (20 mL, GE Healthcare), washed with 1 M NaOH and pre-equilibrated with 25 mM sodium carbonate-bi-carbonate buffer,pH 8.0, was used to capture HBsAg from the Aerosil-380 eluate andsubsequently transferred to a chromatography column. This self-packed column was washed with washing buffer (50 mM Tris–HCl,pH 8.0, conductivity ∼5 mS/cm) until the absorbance at 280 nmin the eluate returned to baseline. The bound HBsAg was elutedusing a salt step (50 mM Tris–HCl, pH 8.0, 0.5 M NaCl, conductiv-ity ∼55 mS/cm). The protein containing fractions (absorbance at280 nm) were analyzed by SDS–PAGE. Fractions containing HBsAgwere pooled and concentrated by ultrafiltration (Vivaspin mem-brane, 10,000 MWCO, Sartorius Stedium Biotech GmbH, Germany).
2.2.5. Step 5: Size-exclusion chromatographyThe pooled and concentrated HBsAg-containing fractions
obtained after ion-exchange chromatography were loaded onto agel filtration column (Sephacryl S-300, 26/60, GE Healthcare) pre-equilibrated in phosphate buffered saline (PBS), pH 7.2 (137 mMNaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.76 mM KH2PO4, pH 7.2). Elu-tion was carried out at 1 mL/min using PBS, pH 7.2 and proteincontaining fractions were monitored at 280 nm.
2.2.6. Step 6: Potassium thiocyanate (KSCN) treatment anddialysis of the final VLP stock
The HBsAg positive fractions were pooled and treated with KSCNto a final concentration of 1.2 M. This mixture was incubated inan orbital shaker (Certomat® BS-1, B. Braun Biotech International,Germany) at 100 rpm and 37 ◦C for ∼5 h. The KSCN treated HBsAgwas extensively dialyzed against PBS (pH 7.2) using a celluloseacetate membrane of 14 kDa cut off (Visking, Carl-Roth, Karlsruhe).Finally, this purified HBsAg stock was filter sterilized and stored at4 ◦C.
2.3. Analytical methods for HBsAg determination
The concentration of HBsAg was determined using a quantita-tive Sandwich ELISA (Hepanostika micro ELISA, bioMerieux, France)following the manufacturer’s instructions. This ELISA was origi-nally developed for analyzing HBsAg in human sera and most likelydetects preferentially the immunogenic (“bioactive”) versions ofHBsAg (e.g. VLPs and rod-shaped structures). The clarified sam-ples were diluted appropriately in PBS (pH 7.2) and analyzed intriplicates. Concentrations of HBsAg in pure HBsAg solutions weredetermined spectrophotometrically at 280 nm [Abs 0.1% (=1 g/l)3.168 cm−1, http://web.expasy.org/protparam/].
2.4. Other protein analytical methods
Total protein concentrations were determined using the bicin-choninic acid (BCA) method [39]. SDS–PAGE analysis of proteinsamples was done essentially as described before [7] using 12%polyacrylamide gels. Gels were stained using colloidal CoomassieBrilliant Blue G-250 according to the “Blue silver” protocol [40].
2.5. Transmission electron microscopy
Electron microscopy of HBsAg VLPs was carried out essentiallyas described previously [7,28]. Briefly, samples collected during
the downstream procedures were analyzed using the energy-filtered transmission electron microscope Libra 120PLUS (Zeiss,Oberkochen, Germany). The samples were diluted accordingly(0.2–0.5 mg/mL protein), adsorbed for 1 min to a glow-discharged
ine 33
C2vwGoaa
2
HbdwHa(awcta
2
PfipP5(pcpcis
pbqp
ranUiatscarm2aw
3
V
M. Zahid et al. / Vacc
-Formvar coated Cu-grids (300 mesh) and negatively stained with% (w/v) uranylacetate, pH 4.5. The zero-loss images, under the sur-eyance of realtime FFT (iTEM software, OSIS, Münster, Germany),ere taken with a 2048 × 2048 CCD camera (Tröndle, Moorenweis,ermany) using a slit-width of 15 eV and an objective aperturef 60 �m. Purified HBsAg VLPs were diluted with PBS (pH 7.2) toppropriate concentration (0.2 mg/mL) and images were recordeds described above.
.6. Image processing and semi-quantitative VLP assessment
TEM images were analyzed via ImageJ (National Institutes ofealth, Bethsda, MD, USA) to measure VLP diameters and num-ers. For the semi-quantitative assessment of VLP formation duringownstream processing only symmetrical and clearly defined VLPsith a diameter of 22 ± 2 nm were considered in the differentBsAg containing fractions, also containing host cell contaminantsnd none VLP HBsAg (e.g. HBsAg agglomerates). In each sampleduplicates) the number of HBsAg VLPs was determined in a definedrea of the TEM image and the total amount of HBsAg in the sampleas determined by SDS–PAGE. VLP counts are then given as relative
ounts and determined by dividing the number of VLPs through theotal amount of HBsAg. Purified HBsAg VLPs after KSCN treatmentsnd dialysis against PBS, pH 7.2 were set as 100%.
.7. Stability testing of HBsAg VLPs
Purified preformed HBsAg VLPs (0.8 mg/mL) stored at 4 ◦C inBS (pH 7.2) were diluted into buffers of various composition to anal concentration of 0.2 mg/mL: (A): lysis buffer (25 mM sodiumhosphate buffer, pH 8.0, 5 mM EDTA, 0.6% (v/v) Tween-20), (B):EG precipitation buffer (25 mM sodium phosphate buffer, pH 8.0,
mM EDTA, 0.6% (v/v) Tween-20, 5% (w/v) PEG6000, 0.5 M NaCl),C): Aerosil equilibration buffer (25 mM sodium phosphate buffer,H 7.2, 0.5 M NaCl), and (D): Aerosil elution buffer (50 mM sodiumarbonate–bi-carbonate buffer, pH 10.8, 1.2 M urea). Dilution ofurified preformed HBsAg VLPs into PBS (pH 7.2) to a final con-entration of 0.2 mg/mL was used as control. All samples werencubated at 4 ◦C for at least 24 h up to a maximum of 7 days andubsequently processed for electron microscopy.
Moreover, purified preformed HBsAg VLPs (0.125 mg/mL in PBS,H 7.2) were extensively dialyzed against anion-exchange elutionuffer (50 mM Tris–HCl, pH 8.0, 0.5 M NaCl) at 4 ◦C and subse-uently subjected to electron microscopy using the non-dialyzedurified preformed HBsAg VLPs in PBS (pH 7.2) as control.
Structural rearrangements of purified preformed HBsAg VLPs inesponse to treatment with chaotropic reagents (GdnSCN, GdnHCl,nd KSCN) were monitored via fluorescence spectroscopy (Lumi-escence spectrometer Perkin Elmer LS50B, PerkinElmer Ltd.,nited Kingdom). Stock solutions of pure HBsAg VLPs (0.8 mg/mL
.e., 30 �M) were first mixed with a 1 mM bis-ANS stock solution in 1:10 ratio to ensure complete saturation of the dye binding sites inhe HBsAg (monomer) and the mixture left at 25 ◦C for 10 min. Sub-equently, the HBsAg.bis-ANS solution was mixed with appropriateoncentrations of the denaturant (final concentration 2 �M HBsAgnd 20 �M bis-ANS) and kept at 25 ◦C [41]. Preceding experimentsevealed that the fluorescence in this denaturant/HBsAg.bis-ANSixture did not change within an incubation period of 3 min to
4 h. Thus, all measurements were done after 3 min incubationnd the fluorescence spectra obtained at 25 ◦C using an excitationavelength of 400 nm.
. Results and discussion
Generation and purification of P. pastoris derived HBsAgLPs encompass multiple steps outlined below (Fig. 1). The first
(2015) 3739–3745 3741
step includes cell breakage and the release of HBsAg from theendoplasmic reticulum (ER) where it is found assembled intodefined multi-layered lamellar structures [28]. This is a criticalstep as it includes the mechanical destruction of cells and cellcompartments and the EDTA/detergent-assisted solubilization ofmembranes and membranous structures including the releaseand solubilization of HBsAg from the ER embedded lamellas. Thesubsequent steps include removal of the majority of host cellcontaminants by precipitation (PEG6000), hydrophobic adsorptionof HBsAg to colloidal silica (Aerosil-380) and final purification andmaturation of HBsAg using different chromatographic steps andKSCN treatment, respectively. The presence or absence of HBsAgVLPs during these different consecutive steps of purification wereprobed by electron microscopy in order to determine the crucialfactors allowing the assembly of HBsAg into VLPs during down-stream processing. Moreover, the effects of buffer componentsemployed during HBsAg purification were analyzed regardingtheir impact on the integrity of preformed HBsAg VLPs. Finally,the concentration-dependent impact of various structure breakingchaotropic compounds, including KSCN, on the coherence of HBsAgVLPs was assessed to better understand the effect of KSCN on thematuration of HBsAg VLPs.
3.1. Monitoring the assembly of HBsAg VLPs during thepurification process
Cell lysis was carried out by high pressure homogenization usinga buffer containing a detergent and a chelating compound. This stepcombines cell lysis with the solubilization of HBsAg from the multi-layered lamellar deposits found in the ER. A survey of this crudecell lysate using electron microscopy did not reveal any structuresresembling VLPs (Fig. 1A). Addition of PEG to this crude cell lysateresulted in partial precipitation of host cell proteins while keep-ing the HBsAg in solution. An analysis of the supernatant obtainedafter centrifugation of the PEG precipitate as well as the PEG super-natant/colloidal silica suspension by electron microscopy also didnot show any VLP resembling structures (Fig. 1B and C). The firstVLP-like structures appeared after elution of proteins bound tocolloidal silica (Fig. 1D). Binding of HBsAg to colloidal silica (Aerosil-380) in clarified PEG extracts at neutral pH through hydrophobicadsorption represents an important step during purification as theelution of bound proteins from colloidal silica already leads to aHBsAg preparation with a purity of 60–70% [37]. An electron micro-scopic investigation of this eluate revealed that the size of theseirregular VLP-like structures were in the range of HBsAg VLPs butclearly neither uniform in size nor in shape (Fig. 1D). Moreover, aclustering of these large irregular HBsAg VLP-like structures intoagglomerates also point at best to very immature VLPs. The pHadjustment of the colloidal silica eluate (pH 10.8) to pH 8.0 prior toanion-exchange chromatography already modified the morphol-ogy of these large irregular VLP-like structures and, moreover,increased the particle size in the particulate background presum-ably consisting of smaller size HBsAg assemblies (“mini-VLPs”)(Fig. 1E). Thus, the pH shift from alkaline to more neutral pH alreadyhad a significant effect on the morphology of the HBsAg assemblies(large irregular VLP-like structures and “mini-VLPs”). The size dis-tribution of the VLP-like structures further improved in the eluatefraction obtained after ion-exchange chromatography, however,these particles still revealed a high tendency to form clusters andstill exhibited an irregular morphology (Fig. 1F). Clearly defined andless disperse VLPs were detectable in the eluate fraction collectedafter size-exclusion chromatography (Fig. 1G) and monodispersity
further improved after KSCN treatment followed by dialysis againstPBS (Fig. 1H). Moreover, the KSCN treatment also resulted in VLPswith a more fine-structured surface as has been previously reportedusing atomic force microscopy [41]. The appearance of well-defined
3742 M. Zahid et al. / Vaccine 33 (2015) 3739–3745
Fig. 1. Monitoring for the appearance of HBsAg VLPs during the purification procedure. Sections encircled in red and green correspond to purification steps with noneo micros atogrs t and
r this a
Vsbfo2rstKim
3d
t
r very immature and to steps with clearly detectable VLPs, respectably. Electronuspension (pH 7.2), (D) Aerosil eluate (pH 10.8), (E) load for anion-exchange chromize-exclusion chromatography (pH 7.2), and (H) HBsAg VLPs after KSCN treatmeneferences to color in this figure legend, the reader is referred to the web version of
LPs during the down-stream process is also illustrated using aemi-quantitative approach (Fig. 2). For this purpose, the num-ers of VLPs were determined in the different HBsAg containingractions obtained during downstream-processing consideringnly symmetrical and clearly defined VLPs with a diameter of2 ± 2 nm. These semi-quantitative data corroborate that symmet-ical and clearly defined VLPs of uniform size appeared first inignificant numbers after size-exclusion chromatography and fur-her increased in numbers after KSCN treatment. Thus, the finalSCN treatment after size inclusion chromatography did not only
mprove the fine-structure of the VLP surface but also improvedonodispersity, e.g. increased the numbers of VLPs of uniform size.
.2. Stability of HBsAg VLPs in buffers and solutions employed
uring the purification processThe first VLP-like elements were observed after elution of pro-eins bound to colloidal silica, namely Aerosil-380, resulting in
graphs of (A) crude cell lysate (pH 8.0), (B) PEG supernatant (pH 8.0), (C) Aerosilaphy (pH 8.0), (F) load for size-exclusion chromatography (pH 8.0), (G) eluate afterdialysis against PBS (pH 7.2). Bars correspond to 100 nm. (For interpretation of therticle.)
a HBsAg preparation of ∼60–70% purity. However, these VLP-like elements were neither uniform in size nor in structure andexhibited a high tendency to form clusters (Fig. 1D). Thus, weanalyzed the impact of the Aerosil elution buffer and the buffersemployed in the preceding purification steps on the integrity ofpurified preformed HBsAg VLPs. Purified preformed HBsAg VLPswere incubated in EDTA/detergent containing lysis buffer as well asin PEG precipitation buffer, and in Aerosil equilibration and elutionbuffer, and the VLP morphology was subsequently analyzed by elec-tron microscopy. As control purified preformed HBsAg VLPs werediluted to the same amount/concentration into PBS (pH 7.2) andalso assessed by electron microscopy. The results of these studiesclearly revealed that the buffers employed during the first purifica-tion steps did not cause disassembly of preformed HBsAg VLPs but
led to clumping and deformation of the VLPs (Fig. 3). In particular,incubation of preformed HBsAg VLPs in lysis buffer as well as in PEGprecipitation buffer caused severe clumping of VLPs but also led to apronounced distortion of the VLP symmetry (Fig. 3). Thus, the buffer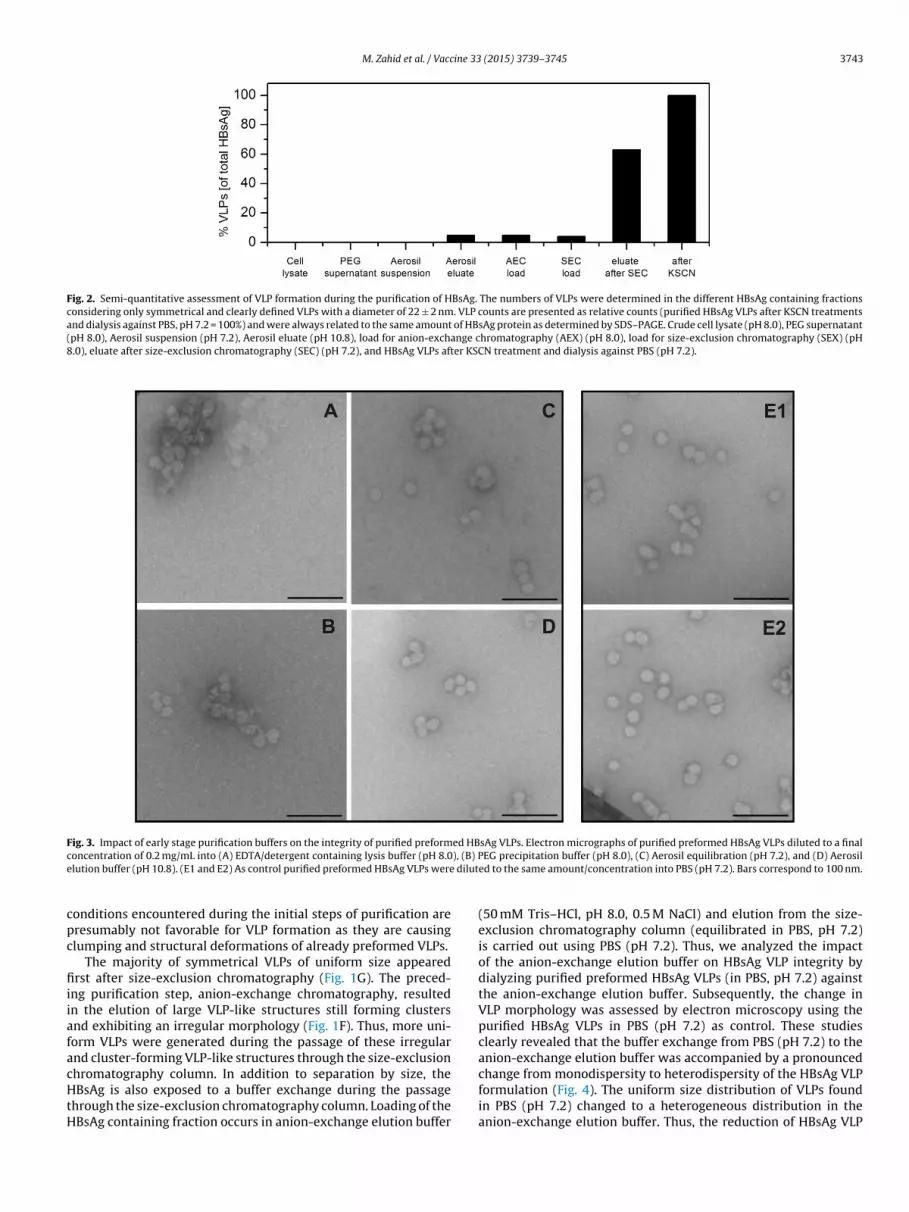
M. Zahid et al. / Vaccine 33 (2015) 3739–3745 3743
Fig. 2. Semi-quantitative assessment of VLP formation during the purification of HBsAg. The numbers of VLPs were determined in the different HBsAg containing fractionsconsidering only symmetrical and clearly defined VLPs with a diameter of 22 ± 2 nm. VLP counts are presented as relative counts (purified HBsAg VLPs after KSCN treatmentsand dialysis against PBS, pH 7.2 = 100%) and were always related to the same amount of HBsAg protein as determined by SDS–PAGE. Crude cell lysate (pH 8.0), PEG supernatant(pH 8.0), Aerosil suspension (pH 7.2), Aerosil eluate (pH 10.8), load for anion-exchange chromatography (AEX) (pH 8.0), load for size-exclusion chromatography (SEX) (pH8.0), eluate after size-exclusion chromatography (SEC) (pH 7.2), and HBsAg VLPs after KSCN treatment and dialysis against PBS (pH 7.2).
Fig. 3. Impact of early stage purification buffers on the integrity of purified preformed HBsAg VLPs. Electron micrographs of purified preformed HBsAg VLPs diluted to a finalc ), (B) Pe dilut
cpc
fiiiafacHtH
oncentration of 0.2 mg/mL into (A) EDTA/detergent containing lysis buffer (pH 8.0lution buffer (pH 10.8). (E1 and E2) As control purified preformed HBsAg VLPs were
onditions encountered during the initial steps of purification areresumably not favorable for VLP formation as they are causinglumping and structural deformations of already preformed VLPs.
The majority of symmetrical VLPs of uniform size appearedrst after size-exclusion chromatography (Fig. 1G). The preced-
ng purification step, anion-exchange chromatography, resultedn the elution of large VLP-like structures still forming clustersnd exhibiting an irregular morphology (Fig. 1F). Thus, more uni-orm VLPs were generated during the passage of these irregularnd cluster-forming VLP-like structures through the size-exclusion
hromatography column. In addition to separation by size, theBsAg is also exposed to a buffer exchange during the passagehrough the size-exclusion chromatography column. Loading of theBsAg containing fraction occurs in anion-exchange elution buffer
EG precipitation buffer (pH 8.0), (C) Aerosil equilibration (pH 7.2), and (D) Aerosiled to the same amount/concentration into PBS (pH 7.2). Bars correspond to 100 nm.
(50 mM Tris–HCl, pH 8.0, 0.5 M NaCl) and elution from the size-exclusion chromatography column (equilibrated in PBS, pH 7.2)is carried out using PBS (pH 7.2). Thus, we analyzed the impactof the anion-exchange elution buffer on HBsAg VLP integrity bydialyzing purified preformed HBsAg VLPs (in PBS, pH 7.2) againstthe anion-exchange elution buffer. Subsequently, the change inVLP morphology was assessed by electron microscopy using thepurified HBsAg VLPs in PBS (pH 7.2) as control. These studiesclearly revealed that the buffer exchange from PBS (pH 7.2) to theanion-exchange elution buffer was accompanied by a pronounced
change from monodispersity to heterodispersity of the HBsAg VLPformulation (Fig. 4). The uniform size distribution of VLPs foundin PBS (pH 7.2) changed to a heterogeneous distribution in theanion-exchange elution buffer. Thus, the reduction of HBsAg VLP
3744 M. Zahid et al. / Vaccine 33 (2015) 3739–3745
Fig. 4. Impact of late stage purification buffer on the integrity of purified preformed HBsAg VLPs. Effect of buffer exchange from size-exclusion chromatography elution buffer(PBS, pH 7.2) to anion-exchange chromatography elution buffer (50 mM Tris–HCl, pH 8.0, 0.5 M NaCl). (A) Size frequency distribution and (B and C) electron micrographs ofHBsAg VLPs (� in A and B) before (in PBS, pH 7.2) and (� in A and C) after dialysis against anion-exchange chromatography elution buffer. Bars correspond to 100 nm.
F ncents
hdecHe
3c
tVcboiisAecpwdHr
ig. 5. Structural rearrangements and unfolding of HBsAg VLPs through increasing copectroscopy.
eterodispersity after size-exclusion chromatography is mainlyue to the buffer exchange but not to the passage through the size-xclusion chromatography column. However, the size-exclusionhromatography step removed remaining host cell proteins as theBsAg VLPs elute with the void volume and host cell contaminantslute afterwards [37].
.3. Stability of HBsAg VLPs toward structure-breakinghaotropic compounds
There is widespread usage of KSCN during the final steps ofhe purification of HBsAg to improve the maturation of HBsAgLPs [8,10,14,20,21,28]. KSCN is a structure-breaking chaotropicompound though less harsh than GdnSCN and GdnHCl [42]. Toetter understand the mechanism of KSCN mediated maturationf HBsAg VLPs during the final purification step the denaturantnduced unfolding of HBsAg VLPs was studied in the presence ofncreasing concentrations of GdnSCN, GdnHCl, and KSCN. Unfoldingtudies were carried out in the absence and presence of bis-NS, a hydrophobic fluorescent dye which preferentially binds toxposed hydrophobic patches of protein folding intermediates, so-alled “molten globules” and not to native or completely unfoldedroteins [43,44]. Incubation of purified preformed HBsAg VLPs
ith increasing denaturant concentrations revealed that unfol-ing, respectively, the formation of a “molten globular state” of theBsAg VLPs apparent through the increased bis-ANS fluorescenceequired denaturant concentrations of 0.5 M and 1.2 M of GdnSCN
rations of (A) GdnSCN, (B) GdnHCl, and (C) KSCN monitored via bis-ANS fluorescence
and GdnHCl, respectively (Fig. 5). As treatment of purified matureHBsAg VLPs with KSCN up to 5 M KSCN did not lead to a detectable“molten globular state” of the HBsAg VLPs (Fig. 5) and KSCN is a lessharsh denaturant compared to GdnSCN and GdnHCl it is concludedthat KSCN, in particular at the concentrations employed during thefinal step of HBsAg VLP maturation, does not cause disassembly ofHBsAg VLPs nor does it cause a significant transfer of hydrophobicpatches from the VLP interior toward the VLP surface. The resultsstrongly suggest that the KSCN most likely causes a restructuringof surface exposed stretches leading to a more uniform exterior ofthe HBsAg VLPs.
4. Conclusions
Our previous studies strongly suggested that the HBsAg VLPsare not formed within the yeast cells during the cultivation butmust be formed after cell breakage during the following down-stream procedures [28]. Our studies now show that VLP formationoccurs in a gradual manner during down-stream processing withthe first VLP resembling structures appearing after elution of boundHBsAg from colloidal silica. These VLP-like structures were stillnon-symmetrical and heterodisperse and exhibited a high ten-dency toward cluster formation presumably because of surface
exposed hydrophobic patches. More uniform and monodisperseVLPs appeared after the following chromatographic purificationprocedures most likely the result of buffer changes during thesepurification steps (toward more neutral pH and less salt). Final
ine 33
tcus
C
A
AE
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
M. Zahid et al. / Vacc
reatment of the VLPs with the denaturant KSCN at moderate con-entrations with following KSCN removal by dialysis did not causenfolding and VLP disassembly but resulted in a re- and fine-tructuring of the VLP surface topology.
onflict of interest statement
None.
cknowledgements
Maria Zahid wishes to express her gratitude to the Deutscherkademischer Austauschdienst (DAAD) of Germany and the Higherducation Commission (HEC) of Pakistan for her fellowship.
eferences
[1] Roldao A, Mellado MC, Castilho LR, Carrondo MJ, Alves PM. Virus-like particlesin vaccine development. Expert Rev Vaccines 2010;9(Oct):1149–76.
[2] Frost LJ, Reich MR. Hepatitis B vaccine: access to vaccines. In: Creative com-mons attribution-noncommercial-share Alike 3.0 United States License; 2008.p. 67–90. 〈http://www.accessbook.org/downloads/chapter 4 AccessBook.pdf〉(Chapter 4, Access: How do good health technologies get to poor people in poorcountries).
[3] Stephenne J. Recombinant versus plasma-derived hepatitis B vaccines: issues ofsafety, immunogenicity and cost-effectiveness. Vaccine 1988;6(Aug):299–303.
[4] Patzer EJ, Nakamura GR, Yaffe A. Intracellular transport and secretion of hep-atitis B surface antigen in mammalian cells. J Virol 1984;51(Aug):346–53.
[5] Kuroda S, Miyazaki T, Otaka S, Fujisawa Y. Saccharomyces cerevisiae can releasehepatitis B virus surface antigen (HBsAg) particles into the medium by itssecretory apparatus. Appl Microbiol Biotechnol 1993;40(Nov):333–40.
[6] Kim EJ, Park YK, Lim HK, Park YC, Seo JH. Expression of hepatitis B surface anti-gen S domain in recombinant Saccharomyces cerevisiae using GAL1 promoter. JBiotechnol 2009;20(May (141)):155–9.
[7] Gurramkonda C, Adnan A, Gäbel T, et al. Simple high-cell density fed-batchtechnique for high-level recombinant protein production with Pichia pastoris:application to intracellular production of Hepatitis B surface antigen. MicrobCell Fact 2009;8:13.
[8] Wampler DE, Lehman ED, Boger J, McAleer WJ, Scolnick EM. Multiple chemicalforms of hepatitis B surface antigen produced in yeast. Proc Natl Acad Sci USA1985;82(Oct):6830–4.
[9] Uemura Y, Ohmura T, Ohmizu A, et al. Process for the purification of HBsAg. USPatent 4,694,074, 1987.
10] Friedman A, Lehman ED, McAleer WJ, Schaefer TF, Scolnick EM, Wampler DE.Immunogenic HBsAg derived from transformed yeast. US Patent 4,707,542,1987.
11] van Wijnendaele F, Simonet G. Method for the isolation and purification ofHepatitis B surface antigen using polysorbate. US Patent 4,649,192, 1987.
12] van Wijnendaele F, Gilles D, Simonet G. Process for the extraction and purifi-cation of proteins from culture media producing them. US Patent 4,683,294,1987.
13] van Wijnendaele F, Gilles D, Simonet G. Process for the extraction and purifi-cation of proteins from culture media producing them. US Patent 4,857,317,1989.
14] Yamazaki S. Process for purifying recombinant hepatitis antigens. US Patent5,001,915, 1991.
15] Nobuya O, Kyosuke M, Fukusaburo J, Hiroshi M. Lyophilized hepatiits B vaccine.EP 0 156 242 B1, 1992.
16] Hsieh JH, Shih SC, Chi WK, Chu YD, Lin AN. Isolation of Hepatitis B surfaceantigen from transformed yeast cells. US Patent 5,462,863, 1995.
17] Kee GS, Jin J, Balasundaram B, Bracewell DG, Pujar NS, Titchener-Hooker NJ.Exploiting the intracellular compartmentalization characteristics of the S. cere-visiae host cell for enhancing primary purification of lipid-envelope virus-likeparticles. Biotechnol Prog 2010;26(Jan):26–33.
18] Pointek M, Weniger M. Method for obtaining recombinant HBsAg. US Patent6,428,984 B1, 2002.
19] Huang Y, Bi J, Zhang Y, et al. A highly efficient integrated chromatographicprocedure for the purification of recombinant hepatitis B surface antigen fromHansenula polymorpha. Protein Expr Purif 2007;56(Dec):301–10.
[
[
(2015) 3739–3745 3745
20] Agraz A, Quinones Y, Exposito N, et al. Adsorption-desorption of recombi-nant hepatitis B surface antigen (r-HBsAg) from P. pastoris on a diatomaceousearth matrix: optimization of parameters for purification. Biotechnol Bioeng1993;42(Nov):1238–44.
21] Venkata RK, Satyanarayana PK, Venkat SK. A process of purifying the hepatitisB surface antigen from the extracts of Pichia pastoris. Indian Patent IN180249,1998.
22] Venkata RK, Satyanarayana PK, Venkat SK, Venkata SA.;1; Process of purifyingthe hepatitis B surface antigen from the extracts of Pichia pastoris. Indian PatentIN180250, 1998.
23] Hardy E, Martinez E, Diago D, Diaz R, Gonzalez D, Herrera L. Large-scale produc-tion of recombinant hepatitis B surface antigen from Pichia pastoris. J Biotechnol2000;77(Feb):157–67.
24] Bardiya N. Expression in and purification of Hepatitis B surface anti-gen (S-protein) from methylotrophic yeast Pichia pastoris. Anaerobe2006;12:194–203.
25] Ottone S, Nguyen X, Bazin J, Berard C, Jimenez S, Letourneur O. Expres-sion of hepatitis B surface antigen major subtypes in Pichia pastorisand purification for in vitro diagnosis. Protein Expr Purif 2007;56(Dec):177–88.
26] Srinivas VK, Krishnamurthy E. A novel process for the preparation and purifi-cation of recombinant proteins. Indian Patent IN202961, 2007.
27] Liu R, Lin Q, Sun Y, et al. Expression, purification, and characterization of hep-atitis B virus surface antigens (HBsAg) in yeast Pichia pastoris. Appl BiochemBiotechnol 2009;158(Aug):432–44.
28] Lünsdorf H, Gurramkonda C, Adnan A, Khanna N, Rinas U. Virus-like particleproduction with yeast: ultrastructural and immunocytochemical insights intoPichia pastoris producing high levels of the Hepatitis B surface antigen. MicrobCell Fact 2011;10:48.
29] Patil A, Khanna N. Novel membrane extraction procedure for the purifi-cation of hepatitis B surface antigen from Pichia pastoris. J Chromatogr B2012;898(Jun):7–14.
30] Valenzuela P, Medina A, Rutter WJ, Ammerer G, Hall BD. Synthesis andassembly of hepatitis B virus surface antigen particles in yeast. Nature1982;298(Jul):347–50.
31] McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hille-man MR. Human hepatitis B vaccine from recombinant yeast. Nature1984;307(Jan):178–80.
32] Cregg JM, Tschopp JF, Stillman C, et al. High-level expression and efficientassembly of hepatitis B surface antigen in the methylotrophic yeast, Pichiapastoris. Biotechnology (NY) 1987;5:479–85.
33] Grgacic EV, Anderson DA. Virus-like particles: passport to immune recognition.Methods 2006;40(Sep):60–5.
34] Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geom-etry, kinetics and molecular patterns. Nat Rev Immunol 2010;10(Nov):787–96.
35] Gavilanes F, Gonzalez-Ros JM, Peterson DL. Structure of hepatitis B surface anti-gen. Characterization of the lipid components and their association with theviral proteins. J Biol Chem 1982;257(Jul):7770–7.
36] Diminsky D, Schirmbeck R, Reimann J, Barenholz Y. Comparison between hep-atitis B surface antigen (HBsAg) particles derived from mammalian cells (CHO)and yeast cells (Hansenula polymorpha): composition, structure and immuno-genicity. Vaccine 1997;15(Apr):637–47.
37] Gurramkonda C, Zahid M, Nemani SK, et al. Purification of hepatitis B surfaceantigen virus-like particles from recombinant Pichia pastoris and in vivo analysisof their immunogenic properties. J Chromatogr B 2013;940:104–11.
38] Vassileva A, Chugh DA, Swaminathan S, Khanna N. Effect of copy number onthe expression levels of hepatitis B surface antigen in the methylotrophic yeastPichia pastoris. Protein Expr Purif 2001;21(Feb):71–80.
39] Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicin-choninic acid. Anal Biochem 1985;150(Oct):76–85.
40] Candiano G, Bruschi M, Musante L, et al. Blue silver: a very sensitivecolloidal Coomassie G-250 staining for proteome analysis. Electrophoresis2004;25(May):1327–33.
41] Zhao Q, Wang Y, Freed D, et al. Maturation of recombinant hepatitis B virussurface antigen particles. Hum Vaccin 2006;2(Jul):174–80.
42] Ropson IJ, Gordon JI, Frieden C. Folding of a predominantly �-structureprotein: rat intestinal fatty acid binding protein. Biochemistry 1990;29:9591–9.
43] Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas’ AF, Gilman-shin RI. Study of the molten globule intermediate state in protein folding by ahydrophobic fluorescent probe. Biopolymers 1991;31(Jan):119–28.
44] Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for proteincharacterization. Pharm Res 2008;25(Jul):1487–99.
Related Documents











