JOURNAL OF VIROLOGY, Apr. 2007, p. 3608–3617 Vol. 81, No. 7 0022-538X/07/$08.000 doi:10.1128/JVI.02277-06 Copyright © 2007, American Society for Microbiology. All Rights Reserved. Assembly of Hepatitis Delta Virus: Particle Characterization, Including the Ability To Infect Primary Human Hepatocytes Severin Gudima, 1 Yiping He, 1 Anja Meier, 1 Jinhong Chang, 1 Rongji Chen, 1 Michal Jarnik, 1 Emmanuelle Nicolas, 1 Volker Bruss, 2 and John Taylor 1 * Fox Chase Cancer Center, Philadelphia, Pennsylvania, 1 and Department of Virology, University of Göttingen, Göttingen, Germany 2 Received 17 October 2006/Accepted 8 January 2007 Efficient assembly of hepatitis delta virus (HDV) was achieved by cotransfection of Huh7 cells with two plasmids: one to provide expression of the large, middle, and small envelope proteins of hepatitis B virus (HBV), the natural helper of HDV, and another to initiate replication of the HDV RNA genome. HDV released into the media was assayed for HDV RNA and HBV envelope proteins and characterized by rate-zonal sedimentation, immunoaffinity purification, electron microscopy, and the ability to infect primary human hepatocytes. Among the novel findings were that (i) immunostaining for delta antigen 6 days after infection with 300 genome equivalents (GE) per cell showed only 1% of cells as infected, but this was increased to 16% when 5% polyethylene glycol was present during infection; (ii) uninfected cells did not differ from infected cells in terms of albumin accumulation or the presence of E-cadherin at cell junctions; and (iii) sensitive quanti- tative real-time PCR assays detected HDV replication even when the multiplicity of infection was 0.2 GE/cell. In the future, this HDV assembly and infection system can be further developed to better understand the mechanisms shared by HBV and HDV for attachment and entry into host cells. Hepatitis B virus (HBV) is the natural helper virus of hep- atitis delta virus (HDV). In an infection of the liver, some hepatocytes can be infected by both viruses so that HBV rep- lication can provide the envelope (HBsAg) proteins for the assembly, release, and spread of virions containing the HDV RNA genome (6). Consider first the HBV assembly process as diverted by HDV. The HBV and the other mammalian hepadnaviruses encode three HBsAg proteins. They have a common C termi- nus and are known by their sizes as large, middle and small (L, M, and S). Relative to S, the M protein has a unique N- terminal domain referred to as preS2. Similarly, relative to M, L has a domain called preS1. As reviewed elsewhere, L, M, and S undergo a complex series of specific posttranslational mod- ifications and intermolecular associations, ultimately leading to the release of particles (15, 17). If the genome of HBV is replicating in such a cell, there can be formation of the viral nucleocapsids, some of which can be enveloped by HBsAg, leading to the release of infectious particles. Similarly, if the HDV genome is present, then in the additional presence of the delta (Ag) proteins, there can also be assembly and release of infectious HDV (33). While M is necessary for neither assembly nor infectivity (15, 35), the stoichiometry of L to S is a critical variable in these assembly processes, whether for HBV or HDV (33). The L protein is essential for both the assembly and the infectivity of HBV and the infectivity of HDV (34), and yet in the absence of S, L is unable even to be released from the cell (39). For HDV but not for HBV, the S protein is sufficient for assembly, but the particles are noninfectious (34). Superimposed on this complex pathway of infectious-particle assembly is a gross inefficiency. Typically, a 1,000- to 1,000,000- fold excess of empty particles that do not contain an HBV ge- nome is produced (15). From electron microscopy, the empty particles are seen to be either roughly spherical particles of 25-nm diameters or filaments of about 22-nm diameters but of variable lengths. The infectious HBV particle is about 42 nm in diameter (12, 13). The HDV particles are considered to be somewhat smaller, about 36 nm (30, 34). In an experimental in vitro situation, HDV assembly can be achieved by cotransfecting cells with two plasmids, one to pro- vide HBsAg and another to initiate the replication of the HDV RNA genome (31). Such particles were subsequently demon- strated to be infectious in an experimental animal (29). Largely from the work of Sureau and colleagues, it has become clear that HDV particles share some features with HBV in terms of assembly and especially in terms of the ability to attach to and enter host cells (23, 33). Others have also begun to use this approach to obtain virus particles with HDV genomes and various forms of hepadnavirus envelope proteins (1, 2, 14). The present study was undertaken to provide a detailed analysis of the requirements of HDV in vitro assembly and infectivity. This analysis included the following variables. (i) As mentioned above, HBV S is sufficient to achieve efficient as- sembly of HDV, but such particles are noninfectious, and it is only when L is also present that the particles are infectious (34). Therefore, we undertook to explore how varying the amount of L in the envelope would influence the abilities of the virus particles to initiate infection. (ii) Since HBsAg pro- teins are expressed only transiently, we assayed whether at later times after transfection there would be inefficient particle assembly. (iii) The replication of the HDV RNA strictly de- pends upon the presence of the HDV-encoded small delta protein (Ag-S) (24). During HDV replication, there occurs site-specific editing by adenosine deaminase acting on RNA * Corresponding author. Mailing address: Fox Chase Cancer Cen- ter, 333 Cottman Avenue, Philadelphia, PA 19111-2497. Phone: (215) 728-2436. Fax: (215) 728-3105. E-mail: [email protected]. Published ahead of print on 17 January 2007. 3608 on April 9, 2016 by guest http://jvi.asm.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF VIROLOGY, Apr. 2007, p. 3608–3617 Vol. 81, No. 70022-538X/07/$08.00�0 doi:10.1128/JVI.02277-06Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Assembly of Hepatitis Delta Virus: Particle Characterization, Includingthe Ability To Infect Primary Human Hepatocytes�
Severin Gudima,1 Yiping He,1 Anja Meier,1 Jinhong Chang,1 Rongji Chen,1 Michal Jarnik,1Emmanuelle Nicolas,1 Volker Bruss,2 and John Taylor1*
Fox Chase Cancer Center, Philadelphia, Pennsylvania,1 and Department of Virology, University of Göttingen, Göttingen, Germany2
Received 17 October 2006/Accepted 8 January 2007
Efficient assembly of hepatitis delta virus (HDV) was achieved by cotransfection of Huh7 cells with twoplasmids: one to provide expression of the large, middle, and small envelope proteins of hepatitis B virus(HBV), the natural helper of HDV, and another to initiate replication of the HDV RNA genome. HDV releasedinto the media was assayed for HDV RNA and HBV envelope proteins and characterized by rate-zonalsedimentation, immunoaffinity purification, electron microscopy, and the ability to infect primary humanhepatocytes. Among the novel findings were that (i) immunostaining for delta antigen 6 days after infectionwith 300 genome equivalents (GE) per cell showed only 1% of cells as infected, but this was increased to 16%when 5% polyethylene glycol was present during infection; (ii) uninfected cells did not differ from infected cellsin terms of albumin accumulation or the presence of E-cadherin at cell junctions; and (iii) sensitive quanti-tative real-time PCR assays detected HDV replication even when the multiplicity of infection was 0.2 GE/cell.In the future, this HDV assembly and infection system can be further developed to better understand themechanisms shared by HBV and HDV for attachment and entry into host cells.
Hepatitis B virus (HBV) is the natural helper virus of hep-atitis delta virus (HDV). In an infection of the liver, somehepatocytes can be infected by both viruses so that HBV rep-lication can provide the envelope (HBsAg) proteins for theassembly, release, and spread of virions containing the HDVRNA genome (6).
Consider first the HBV assembly process as diverted byHDV. The HBV and the other mammalian hepadnavirusesencode three HBsAg proteins. They have a common C termi-nus and are known by their sizes as large, middle and small (L,M, and S). Relative to S, the M protein has a unique N-terminal domain referred to as preS2. Similarly, relative to M,L has a domain called preS1. As reviewed elsewhere, L, M, andS undergo a complex series of specific posttranslational mod-ifications and intermolecular associations, ultimately leading tothe release of particles (15, 17). If the genome of HBV isreplicating in such a cell, there can be formation of the viralnucleocapsids, some of which can be enveloped by HBsAg,leading to the release of infectious particles. Similarly, if theHDV genome is present, then in the additional presence of thedelta (�Ag) proteins, there can also be assembly and release ofinfectious HDV (33).
While M is necessary for neither assembly nor infectivity (15,35), the stoichiometry of L to S is a critical variable in theseassembly processes, whether for HBV or HDV (33). The Lprotein is essential for both the assembly and the infectivity ofHBV and the infectivity of HDV (34), and yet in the absenceof S, L is unable even to be released from the cell (39). ForHDV but not for HBV, the S protein is sufficient for assembly,but the particles are noninfectious (34).
Superimposed on this complex pathway of infectious-particleassembly is a gross inefficiency. Typically, a 1,000- to 1,000,000-fold excess of empty particles that do not contain an HBV ge-nome is produced (15). From electron microscopy, the emptyparticles are seen to be either roughly spherical particles of25-nm diameters or filaments of about 22-nm diameters but ofvariable lengths. The infectious HBV particle is about 42 nm indiameter (12, 13). The HDV particles are considered to besomewhat smaller, about 36 nm (30, 34).
In an experimental in vitro situation, HDV assembly can beachieved by cotransfecting cells with two plasmids, one to pro-vide HBsAg and another to initiate the replication of the HDVRNA genome (31). Such particles were subsequently demon-strated to be infectious in an experimental animal (29). Largelyfrom the work of Sureau and colleagues, it has become clearthat HDV particles share some features with HBV in terms ofassembly and especially in terms of the ability to attach to andenter host cells (23, 33). Others have also begun to use thisapproach to obtain virus particles with HDV genomes andvarious forms of hepadnavirus envelope proteins (1, 2, 14).
The present study was undertaken to provide a detailedanalysis of the requirements of HDV in vitro assembly andinfectivity. This analysis included the following variables. (i) Asmentioned above, HBV S is sufficient to achieve efficient as-sembly of HDV, but such particles are noninfectious, and it isonly when L is also present that the particles are infectious(34). Therefore, we undertook to explore how varying theamount of L in the envelope would influence the abilities ofthe virus particles to initiate infection. (ii) Since HBsAg pro-teins are expressed only transiently, we assayed whether atlater times after transfection there would be inefficient particleassembly. (iii) The replication of the HDV RNA strictly de-pends upon the presence of the HDV-encoded small deltaprotein (�Ag-S) (24). During HDV replication, there occurssite-specific editing by adenosine deaminase acting on RNA
* Corresponding author. Mailing address: Fox Chase Cancer Cen-ter, 333 Cottman Avenue, Philadelphia, PA 19111-2497. Phone: (215)728-2436. Fax: (215) 728-3105. E-mail: [email protected].
� Published ahead of print on 17 January 2007.
3608
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

(7), allowing the production of an altered HDV mRNA. ThismRNA leads to the translation of the large delta protein (�Ag-L), which does not support HDV replication but is essential forthe envelopment by HBV-encoded proteins L, M, and S (9).The fraction of genomes that have undergone this necessaryediting increases steadily with time after replication has beeninitiated (38). However, while some specific editing has tooccur in order that �Ag-L might be produced, the edited HDVRNAs are no longer able to initiate HDV replication by infec-tion, since they cannot provide the essential �Ag-S. Thus, RNAediting is both a positive and a negative factor in obtaininginfectious HDV. In addition to this site-specific editing, we andothers have shown that additional nucleotide changes occur,many of which can be accounted for as editing at other sites (8,10, 20, 29). Therefore, we undertook to determine whether allthese changes would progressively decrease the infectivities ofthe released particles.
This study addresses these and other variables affecting theassembly of infectious HDV. The findings are important, sincebalancing of variables is not unique to in vitro assembly, and itcan be readily imagined that similar problems arise during viralassembly in the liver, whether co- or superinfected with HDV.In addition, an understanding of in vitro HDV assembly willallow us to modify the components of the HDV envelope andclarify the process by which HDV and HBV are able to attachto and infect susceptible cells.
MATERIALS AND METHODS
Antibodies. For immunoblots and immunostaining, �Ag was detected using arabbit polyclonal antibody (31) and HBsAg by a commercial rabbit polyclonalantibody (Fitzgerald Industries). The latter was confirmed by immunoblot anal-ysis to be largely specific for HBV S by a comparison with an antibody raisedagainst purified S (a gift from Camille Sureau). For immunoaffinity chromatog-raphy, we used S-26, a mouse monoclonal to HBV preS2 epitope QDPRVR,corresponding to positions 132 to 137 on genotype A, serotype adw2 (a gift fromVadim Bichko) (32). For immunostaining, we also used fluorescein isothiocya-nate-conjugated goat polyclonal antibody to human albumin (Antibodies Incor-porated), a mouse monoclonal to human E-cadherin (EMD Biosciences), andfluorescently labeled secondary antibodies (Jackson Immunoresearch Laborato-ries). For immunoblots, secondary antibodies labeled with infrared dyes wereobtained from LI-COR.
Plasmids. The plasmids used in this study have been described previously:pSVL(D3) initiates HDV genome replication (11). pSVBX24H and pSVB45Hexpress the HBV genotype A, serotype adw2 small envelope and all threeenvelope proteins, respectively (5). pCMV(LM�S�) expresses only the L protein(a gift from T. C. Benedict Yen).
Transfection. Virus assembly was performed with the human liver cell lineHuh7 (28). All transfections with plasmid DNA combinations were performedusing Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instruc-tions. A typical combination for the transfection of a 10-cm dish of Huh7 cellswas 2.5 �g pSVL(D3) and 10 �g of an HBV envelope-expressing plasmid.
Primary human hepatocytes and infection. Primary human hepatocytes,plated as confluent monolayers on rat tail collagen, were obtained commercially(Admet, Cambrex, and CellzDirect), typically in a 48-well configuration with200,000 hepatocytes per well. They were maintained in Hepatostim mediumsupplemented with 0.01 �g/ml epidermal growth factor, receptor grade, bothfrom BD Biosciences. Most infections were performed in the presence of 5%polyethylene glycol 8000 (PEG) (Sigma), with the media being changed after 6 h.
Virus. HDV virus-like particles were harvested from the media containingtransfected Huh7 cells and immediately clarified by centrifugation (10 krpm, 30min, 4°C in the HB-4 rotor of a Sorvall RC-5B). In some cases, this medium wasstored at �80°C. Particles present in clarified media were precipitated by theaddition of PEG to 10%, with stirring for 2 h at 4°C, followed by centrifugation(10 krpm, 30 min, 4°C). The pellet was resuspended in prechilled TAN buffer (50mM Tris-HCl [pH 8.0]–100 mM NaCl), using 1/100 of the original volume, afterwhich aliquots were stored at �80°C.
HBV was produced using the stable cell line HepAD38 (25), kindly providedby Christoph Seeger. The particles released between days 4 and 7 after inductionby tetracycline removal were concentrated as described above. Woodchuck hep-atitis virus (WHV) was obtained from sera of chronically infected woodchucks,kindly provided by William Mason.
Immunoaffinity purification of virus-like particles. The preS2-specific mousemonoclonal antibody S-26 was first complexed for 16 h at 4°C with proteinA-agarose beads (Sigma). This was then incubated for 16 h at 4°C with PEG-concentrated virus and then washed four times with cold NT2 buffer (50 mMTris-HCl [pH 7.5]–150 mM NaCl–1 mM MgCl2). The virus was competitivelyeluted by adding to the buffer 400 �g/ml of the peptide LQDPRVRG. Thiselution step was repeated four times. For samples to be examined by electronmicroscopy, the elutions were performed with only 10 �l. Throughout the puri-fication, aliquots of 2 �l were removed for RNA extraction and the monitoringof recoveries by quantitative real-time PCR (qPCR).
Immunoblots. Protein samples were resuspended in Laemmli buffer contain-ing 5% dithiothreitol and heated for 10 min at 95°C prior to electrophoresis onprecast 12% polyacrylamide gels (Duramide; Cambrex). As size markers, weused Rainbow Markers (Amersham) and MagicMark (Invitrogen). After elec-trotransfer to nitrocellulose membranes, proteins were detected with specificantibodies followed by secondary antibodies conjugated with infrared fluorescentdyes (LI-COR). Detection and quantitation were performed with an Odysseyapparatus (LI-COR). Unlike enzyme-based chemiluminescence, these dye-basedassays give a linear response over a �4,000-fold range (LI-COR and data notshown).
ELISA. HBV surface antigen (HBsAg) was detected using an enzyme-linkedimmunosorbent assay (ELISA) kit, ET-1-MAK-2 Plus, according to the manu-facturer’s instructions (DiaSorin).
RNA extraction, Northern analyses, and real-time PCR. All extractions usedTRI reagent (Molecular Research Center) according to the manufacturer’s in-structions. RNA concentrations were measured using an ND-1000 spectropho-tometer (Nanodrop).
Samples for Northern analyses were initially glyoxalated prior to electrophore-sis on gels of 1.5% agarose. Electrotransfer and hybridization with 32P-labeledRNA probes were as previously described (24). Radiation was detected andquantitated with a bio-imager (Fujifilm BAS-2500) and ImageQuant software,respectively.
Prior to qPCR assays, the RNA samples were subjected to digestion with RQ1RNase-free DNase (Sigma) and then reextracted with TRI reagent. This wasdone to remove HDV-specific plasmid DNA sequences carried over from theoriginal transfection of Huh7 cells. For HDV qPCR assays, we used the followingprimers, with their locations indicated using the HDV genome positions reportedby Kuo et al. (24): forward primer, 312-GGACCCCTTCAGCGAACA-329; andreverse primer, 393-CCTAGCATCTCCTCCTATCGCTAT-360. The TaqManprobe was 332-AGGCGCTTCGAGCGGTAGGAGTAAGA-357. The assayswere normalized relative to a series of 10-fold dilutions of a genomic HDV RNAstandard that had been transcribed in vitro and then gel purified. We deduce that1 pg HDV RNA standard is equal to 1 million molecules. For cell RNA samples,we assume 25 pg RNA per one cell (20) and thus deduce the number of HDVgenome equivalents (GE) per average cell. The qPCR assays for HBV and WHVfollow the reports of others (16, 27).
Immunostaining. Immunostaining was generally as described previously (21),with the following minor modifications. Cells on collagen in 48-well plates werefixed with 4% paraformaldehyde for 15 to 30 min at room temperature, washedtwice, and then permeabilized with N-octyl-glucopyranoside (EMD Biosciences).For detection of delta antigens, rabbit polyclonal antibody (1:1,000 dilution) wasused. E-cadherin and human albumin were analyzed with various dilutions of theabove-mentioned commercial antibodies. DNA was stained with 1 �g/ml DAPI(4�,6�-diamidino-2-phenylindole) (Sigma). Prepared samples were analyzed us-ing an inverted Nikon TE2000-U microscope with 40� or 20� objectives andspecific filter blocks, equipped with a Cascade 650 monochrome camera (Pho-tometrics), and utilizing MetaVue software (Universal Imaging). Images werefurther processed using Canvas 9.0 and Photoshop 7.0 software.
Electron microscopy. For electron microscopy studies, HDV was harvestedfrom transfected cells between days 7 and 10, concentrated using PEG, and thenpurified by immunoaffinity chromatography as described above. Negative stain-ing for transmission electron microscopy was performed as described previously(22). In brief, the viral suspension was adsorbed on a freshly glow-dischargedcollodion/carbon-coated electron microscopical grid for 4 min. The grid wasbriefly washed with phosphate-buffered saline (PBS) and the attached particlesfixed with 2% glutaraldehyde in PBS for 2 min. After three PBS and six double-distilled-water washes, the grid was stained with 2% aqueous solution of uranylacetate and air dried. The specimens were examined using a FEI Tecnai 12
VOL. 81, 2007 HDV ASSEMBLY AND INFECTIVITY 3609
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

electron microscope under 80-kV acceleration. The images were recorded on anAMT 2kx2k digital camera.
Rate-zonal sedimentation. Several sources of virus were used for Fig. 3. ForFig. 3A and B, the HDV was from media harvested between days 6 to 9 and 6 to12, respectively, after transfection with pSVL(D3) and pSVB45H. For Fig. 3C,the harvest was between days 7 and 10 after transfection with pSVL(D3) andpSVBX24H. For Fig. 3D, the harvest was between days 0 and 6 after transfectionwith pSVB45H. The PEG-concentrated virus was sonicated and treated in thepresence of 5 mM magnesium acetate-5 mM Tris-HCl (pH 8.0) with 0.1 mg/mlDNase I for 1 h at 37°C, after which EDTA was added to a 5 mM finalconcentration. The samples were dialyzed against STE (150 mM NaCl–10 mMTris-HCl [pH 7.5]–1 mM EDTA) to remove residual PEG and then clarified bybrief centrifugation prior to being layered onto a 10 to 30% (wt/wt) sucrosegradient in STE buffer. Centrifugation was performed as indicated in the text,using a Beckman SW41 rotor at 4°C. Fractions were collected from above andthen monitored for refractive index. Aliquots were extracted with TRI reagentfor RNA used in a subsequent qPCR assay for HDV RNA. Other aliquots werefirst digested with Pronase and sodium dodecyl sulfate and then extracted withTRI reagent to obtain the DNA used in qPCR assays for HBV and WHV DNA.
RESULTS
Time course of assembly. In order to achieve assembly ofHDV particles, Huh7 cells were transfected with two plasmids.One expressed HBsAg. The other expressed a trimer of HDVgenomic RNA and was sufficient to initiate the replication ofthe HDV genome, producing �Ag-S and �Ag-L. Figure 1A toC shows the presence of intracellular HBsAg, �Ag, and HDV
genomic RNA, respectively, at various times after transfection.Figure 1A shows the detection by immunoblot of two forms
of L and of S, consistent with the effects of N-linked glycosy-lation within the S domain (33). The M species were notdetected relative to L and S. The accumulation of L and Sdecreased with time, consistent with this being a transienttransfection. From quantitation, as indicated, the percentageof L relative to that of L plus S remained constant with time.
The immunoblot in Fig. 1B shows the intracellular accumu-lation up to day 15 for �Ag-S and �Ag-L. Note that the amountof �Ag-L (essential for assembly of particles) was initially un-detectable and then progressively increased relative to that of�Ag-S (essential for the replication of the HDV genome).
The qPCR data in Fig. 1C show that most of the accumu-lation of HDV genomic RNA was achieved by about 6 daysafter transfection.
Using the media from the same transfected cultures, weassayed for the presence of particles containing the HBsAgand HDV RNA genomic RNA. These data, shown in Fig. 2Aand B, refer to particles released in a series of 3-day harvests.Figure 2A shows that the release of HBsAg was highest in theharvest taken at 3 days and then progressively decreased. Fig-ure 2B shows that the release of HDV RNA containing parti-cles was undetectable at 3 days, increased at 6, 9, and 12 days,and then decreased by day 15. It should also be noted that thepercentage of L relative to that of L plus S for particles was lessthan the intracellular values (Fig. 1A) and that it decreasedwith time of harvest.
A plausible interpretation of these data is as follows. At 3days, the largest amount of the HBsAg protein was translatedand released into the media. However, insignificant amounts ofHDV GE were assembled at this time. At 6 days, there was nofurther accumulation of HBsAg, but the amount of releasedGE increased and was maximal at day 12. This is consistent
FIG. 1. Intracellular accumulation of HBsAg and HDV proteinsand RNA as a function of time after transfection of Huh7 cells. Asdescribed in the text, the transfection was performed with one plasmidto express the HBsAg and a second to initiate the replication of HDV.Panel A shows the accumulation of HBsAg, as detected by immuno-blot analysis. Panel B shows a similar analysis for �Ag. Panel C showsan analysis by qPCR to deduce the number of HDV RNA genomes(GE) per average cell. The immunoblot data in panels A and B wereevaluated to deduce the indicated relative amounts of the viral pro-teins. MW, molecular mass.
FIG. 2. Release of HBsAg and HDV RNA as a function of timeafter transfection of Huh7 cells. Transfection was performed as de-scribed for Fig. 1. Panel A shows an immunoblot analysis of the HBsAgreleased during 3-day intervals at the times indicated. Panel B showsquantitation by qPCR of HDV GE/ml of medium. MW, molecularmass.
3610 GUDIMA ET AL. J. VIROL.
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from
with an increase in the amount of edited HDV RNA, as evi-denced by the production of more �Ag-L, a component essen-tial for the assembly of HDV RNA-containing particles.
From the data in Fig. 1 and 2, we deduce that in the periodof days 9 to 12, when HDV release was maximal, 1,200 GEwere released per average cell. (This corresponds to the re-lease of one RNA-containing particle per cell per 4 min.) Inaddition, since the average cell contained 12,000 GE, we de-duce that 10% of the accumulated HDV genomic RNA wasassembled and released in 3 days.
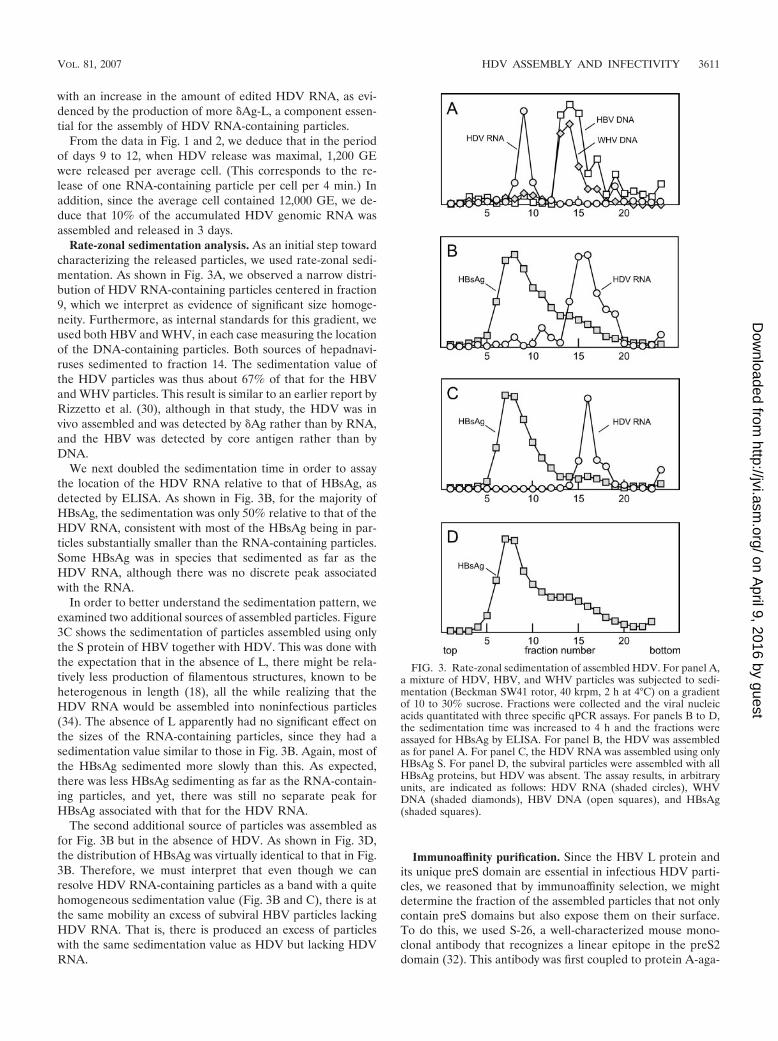
Rate-zonal sedimentation analysis. As an initial step towardcharacterizing the released particles, we used rate-zonal sedi-mentation. As shown in Fig. 3A, we observed a narrow distri-bution of HDV RNA-containing particles centered in fraction9, which we interpret as evidence of significant size homoge-neity. Furthermore, as internal standards for this gradient, weused both HBV and WHV, in each case measuring the locationof the DNA-containing particles. Both sources of hepadnavi-ruses sedimented to fraction 14. The sedimentation value ofthe HDV particles was thus about 67% of that for the HBVand WHV particles. This result is similar to an earlier report byRizzetto et al. (30), although in that study, the HDV was invivo assembled and was detected by �Ag rather than by RNA,and the HBV was detected by core antigen rather than byDNA.
We next doubled the sedimentation time in order to assaythe location of the HDV RNA relative to that of HBsAg, asdetected by ELISA. As shown in Fig. 3B, for the majority ofHBsAg, the sedimentation was only 50% relative to that of theHDV RNA, consistent with most of the HBsAg being in par-ticles substantially smaller than the RNA-containing particles.Some HBsAg was in species that sedimented as far as theHDV RNA, although there was no discrete peak associatedwith the RNA.
In order to better understand the sedimentation pattern, weexamined two additional sources of assembled particles. Figure3C shows the sedimentation of particles assembled using onlythe S protein of HBV together with HDV. This was done withthe expectation that in the absence of L, there might be rela-tively less production of filamentous structures, known to beheterogenous in length (18), all the while realizing that theHDV RNA would be assembled into noninfectious particles(34). The absence of L apparently had no significant effect onthe sizes of the RNA-containing particles, since they had asedimentation value similar to those in Fig. 3B. Again, most ofthe HBsAg sedimented more slowly than this. As expected,there was less HBsAg sedimenting as far as the RNA-contain-ing particles, and yet, there was still no separate peak forHBsAg associated with that for the HDV RNA.
The second additional source of particles was assembled asfor Fig. 3B but in the absence of HDV. As shown in Fig. 3D,the distribution of HBsAg was virtually identical to that in Fig.3B. Therefore, we must interpret that even though we canresolve HDV RNA-containing particles as a band with a quitehomogeneous sedimentation value (Fig. 3B and C), there is atthe same mobility an excess of subviral HBV particles lackingHDV RNA. That is, there is produced an excess of particleswith the same sedimentation value as HDV but lacking HDVRNA.
Immunoaffinity purification. Since the HBV L protein andits unique preS domain are essential in infectious HDV parti-cles, we reasoned that by immunoaffinity selection, we mightdetermine the fraction of the assembled particles that not onlycontain preS domains but also expose them on their surface.To do this, we used S-26, a well-characterized mouse mono-clonal antibody that recognizes a linear epitope in the preS2domain (32). This antibody was first coupled to protein A-aga-
FIG. 3. Rate-zonal sedimentation of assembled HDV. For panel A,a mixture of HDV, HBV, and WHV particles was subjected to sedi-mentation (Beckman SW41 rotor, 40 krpm, 2 h at 4°C) on a gradientof 10 to 30% sucrose. Fractions were collected and the viral nucleicacids quantitated with three specific qPCR assays. For panels B to D,the sedimentation time was increased to 4 h and the fractions wereassayed for HBsAg by ELISA. For panel B, the HDV was assembledas for panel A. For panel C, the HDV RNA was assembled using onlyHBsAg S. For panel D, the subviral particles were assembled with allHBsAg proteins, but HDV was absent. The assay results, in arbitraryunits, are indicated as follows: HDV RNA (shaded circles), WHVDNA (shaded diamonds), HBV DNA (open squares), and HBsAg(shaded squares).
VOL. 81, 2007 HDV ASSEMBLY AND INFECTIVITY 3611
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

rose and then incubated with virus coated with HBV L, M, andS protein (LMS virus). Bound virus was specifically eluted withthe peptide corresponding to the known epitope. A quantita-tion of the binding and elution of the HDV RNA-containingparticles was made by qPCR. As a negative control, a parallelstudy was performed with HDV RNA-containing particlescoated only with S (S virus).
From the results summarized in Table 1, we found that 77%(100 � 23) of the LMS-coated RNA-containing particles wasbound. Furthermore, 50% was subsequently eluted from theS-26 antibody, in contrast to 0.1% of the S particles. We thusdeduce that the binding and elution achieved a 500-fold puri-fication of LMS relative to that of S particles. We also concludethat most of the RNA-containing LMS virus contains at leastsome preS2 exposed on the surface. However, such studiescannot distinguish whether all or just some of the preS2 isexposed. This qualification is relevant in that others haveclaimed that only a fraction of the preS1 and preS2 domains ofHBV particles have been translocated to the outside of thevirion (26).
Electron microscopy evaluation. The above-described im-munoaffinity procedure was performed at a preparative level,to provide sufficient purified and concentrated virus to allowexamination by electron microscopy, with the results as pre-sented in Fig. 4A. Three main particle forms were detected:filamentous structures of relatively constant diameters butvariable lengths, along with roughly spherical particles that fellinto two main size classes (Fig. 4B). A quantitation of theparticle measurements is given in Table 2.
In a previous study of in vivo-assembled HDV (30), theHDV RNA-containing particles were roughly spherical parti-cles of 36-nm diameters. In Table 2, it can be seen that 29% ofour in vitro-assembled particles fit this description, with amean diameter of 36 � 4 nm. In a separate purification andelectron microscope study, only 14% of particles fit this de-scription (data not shown). Nevertheless, as considered furtherin Discussion, there remain important caveats.
Ability to infect primary human hepatocytes. We then askedwhether the particles assembled using L, M, and S were able toinitiate infection of primary human hepatocytes. Infection wasat a multiplicity of 300 GE/cell. After 6 days, total cell RNAwas extracted and assayed for HDV genomic RNA both byNorthern analyses and by qPCR. From the results summarizedin Table 3, it can be seen that genomic RNA was detected ininfected hepatocytes and the amount was more than that in theinitial inoculum.
Following the experience of others (1), we also tested infec-tion in the presence of PEG. As shown, 5% PEG provided amajor increase (typically 10- to 30-fold) in the amount of HDVRNA accumulated.
FIG. 4. Electron microscope examination of affinity-purified parti-cles. These were purified as described for Table 1. Panel A shows anexample of the eluted particles as examined by electron microscopy,with magnification as indicated. Many such fields were examined, theparticles quantitated, and their dimensions measured. Table 2 providesa summary of the quantitation. Panel B shows a histogram of thesedimensions for only the spherical particles.
TABLE 1. Immuno-affinity purification of HDVRNA-containing particlesa
Treatment group
Result (%) for indicatedsample group
LMS S
Unbound 23 99.4Eluted with peptide 50 0.1Remainder 27 0.5
a Samples of LMS or S particles were applied to protein A-Sepharose beads towhich S-26 antibody had been bound. The particles that then bound to the beadswere subjected to four consecutive elutions, using buffer containing peptide. Theremainder was released by treating beads with TRI reagent. All samples wereassayed for HDV RNA by qPCR. The data are expressed as percentages relativeto the total recovered RNA.
TABLE 2. Dimensions of particles purified byimmunoaffinity chromatography
Particle groupa Dimension(s) � SDb (nm) No. ofparticles
% oftotal
Filament (21.0 � 3.2) � (77 � 36) 119 26Small sphere (34 nm) 27.6 � 3.4 200 44Large sphere (�34 nm) 35.9 � 3.6 133 29
a Assembled HDV particles were purified by immunoaffinity chromatographyand then examined by electron microscopy, as shown in Fig. 4A.
b Dimensions were obtained for individual particles. A particle with an axialratio of �1.5 was defined as a filament. The remaining particles gave a bimodaldistribution (Fig. 4B) of small and large spheres.
TABLE 3. HDV replication following infection of primary humanhepatocytes with LMS- and S-coated particles
Infection groupaYield (GE/cell)b for indicated assay
Northern qPCR
LMS 490 990LMS plus PEG 9,420 36,200S 54 25S plus PEG 80 96
a Primary human hepatocytes were infected at a multiplicity of 300 GE (asassayed by qPCR) per cell with HDV particles assembled with either an LMS oran S envelope, as indicated. Infections were with or without 5% PEG, again asindicated.
b At 6 days after infection, total RNA was extracted and assayed for HDVgenomic RNA, using either Northern analyses or qPCR, as indicated. Usingknown amounts of an HDV RNA standard, it was possible to deduce the numberof HDV genome equivalents present per average cell.
3612 GUDIMA ET AL. J. VIROL.
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

As a negative control for these studies, we used S virus. Suchparticles, when applied at the same multiplicity of 300 GE/cell,were unable to give detectable replication, with or withoutPEG.
It should be noted that quantitation by Northern analysesgave results lower than those obtained by qPCR. We speculatethat this is because the Northern blot assays for full-lengthHDV RNAs, while the qPCR also detects species that are lessthan full-length. The qPCR was much more sensitive and coulddetect HDV RNAs even after cells were exposed to S particles.However, the amounts detected were typically 500 times lessthan those obtained with LMS particles. Also, the amountsdetected were typically 5 times less than the amounts of virusto which the cells were initially exposed. As others have sug-gested, this detected RNA may thus be a residual of the inoc-ulum (1).
We also used immunostaining to characterize the HDV in-
fection. Figures 5A and B show detection of �Ag in cells at 6days after infection in the absence and presence, respectively,of 5% PEG. We observed that PEG increased the fraction ofpositive cells from 1 to 16%. Also shown in Fig. 5A and B isstaining for albumin, considered a marker for mature, differ-entiated hepatocytes. Virtually all cells, as detected by DAPIstaining of chromosomal DNA, also stained for albumin. It isthus not obvious why, with a multiplicity of 300 GE/cell andwith the enhancement afforded by PEG, there was still infec-tion of only 16% of cells.
Immunostaining was also used to examine the infected cells athigher magnifications. In Fig. 5C and D, we detected not only �Agbut also intercellular-junction protein E-cadherin. It can be seenthat there are intercellular junctions in both infected and nonin-fected cells. Thus, neither the presence of intercellular junctionsnor the expression of albumin provided us with insights as to whyonly a fraction of the hepatocytes were infected.
FIG. 5. Immunocytochemistry of primary human hepatocytes at 6 days after infection with assembled HDV. In panels A and B, the infectionswere initiated at a multiplicity of 300 GE/cell, in the absence or presence of 5% PEG. The images detect �Ag (red), albumin (green), and DAPIstaining (blue). For panels C and D, the infection was initiated as for panel B, and the images were collected at higher magnifications, with �Ag(green), E-cadherin (red), and DAPI staining (blue). Images A and B were taken with a 20� objective, while C and D were taken with a 40�objective. All images were collected with a charge-coupled-device camera and superimposed using MetaVue software.
VOL. 81, 2007 HDV ASSEMBLY AND INFECTIVITY 3613
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from
Two patterns of �Ag staining were observed. The majoritywas of a general pattern of staining including both nucleus andcytoplasm, with some concentration within regions of the cy-toplasm (Fig. 5C). Rarely did we detect a distribution that waspredominantly nuclear (Fig. 5D).
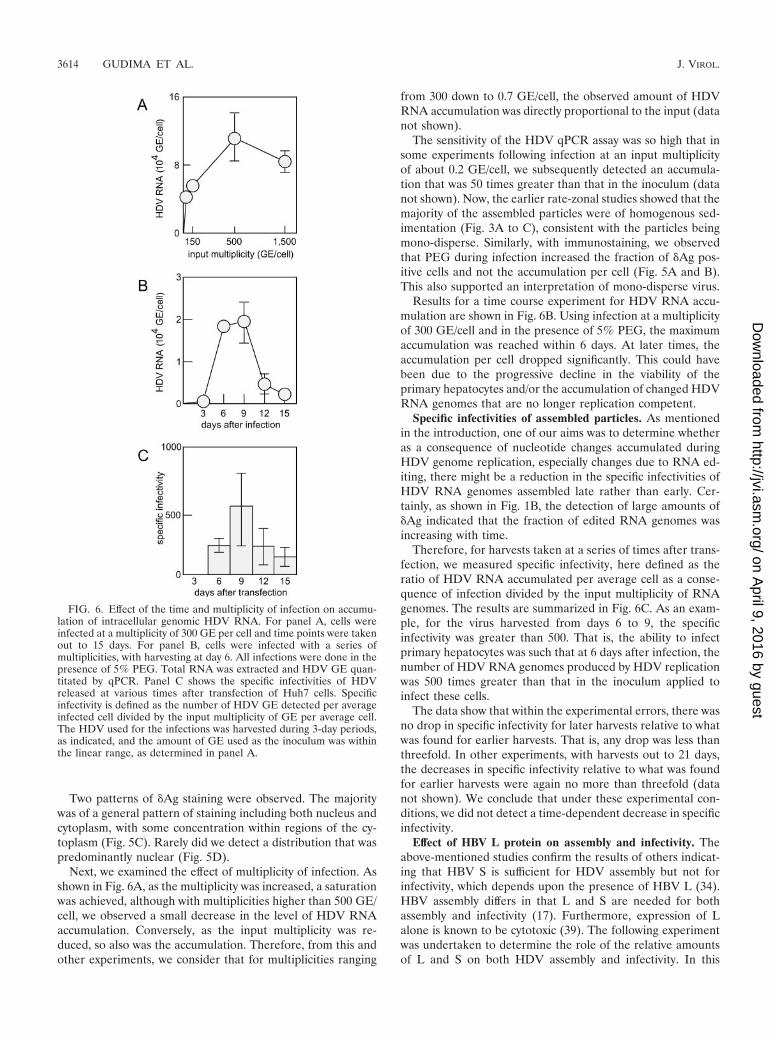
Next, we examined the effect of multiplicity of infection. Asshown in Fig. 6A, as the multiplicity was increased, a saturationwas achieved, although with multiplicities higher than 500 GE/cell, we observed a small decrease in the level of HDV RNAaccumulation. Conversely, as the input multiplicity was re-duced, so also was the accumulation. Therefore, from this andother experiments, we consider that for multiplicities ranging
from 300 down to 0.7 GE/cell, the observed amount of HDVRNA accumulation was directly proportional to the input (datanot shown).
The sensitivity of the HDV qPCR assay was so high that insome experiments following infection at an input multiplicityof about 0.2 GE/cell, we subsequently detected an accumula-tion that was 50 times greater than that in the inoculum (datanot shown). Now, the earlier rate-zonal studies showed that themajority of the assembled particles were of homogenous sed-imentation (Fig. 3A to C), consistent with the particles beingmono-disperse. Similarly, with immunostaining, we observedthat PEG during infection increased the fraction of �Ag pos-itive cells and not the accumulation per cell (Fig. 5A and B).This also supported an interpretation of mono-disperse virus.
Results for a time course experiment for HDV RNA accu-mulation are shown in Fig. 6B. Using infection at a multiplicityof 300 GE/cell and in the presence of 5% PEG, the maximumaccumulation was reached within 6 days. At later times, theaccumulation per cell dropped significantly. This could havebeen due to the progressive decline in the viability of theprimary hepatocytes and/or the accumulation of changed HDVRNA genomes that are no longer replication competent.
Specific infectivities of assembled particles. As mentionedin the introduction, one of our aims was to determine whetheras a consequence of nucleotide changes accumulated duringHDV genome replication, especially changes due to RNA ed-iting, there might be a reduction in the specific infectivities ofHDV RNA genomes assembled late rather than early. Cer-tainly, as shown in Fig. 1B, the detection of large amounts of�Ag indicated that the fraction of edited RNA genomes wasincreasing with time.
Therefore, for harvests taken at a series of times after trans-fection, we measured specific infectivity, here defined as theratio of HDV RNA accumulated per average cell as a conse-quence of infection divided by the input multiplicity of RNAgenomes. The results are summarized in Fig. 6C. As an exam-ple, for the virus harvested from days 6 to 9, the specificinfectivity was greater than 500. That is, the ability to infectprimary hepatocytes was such that at 6 days after infection, thenumber of HDV RNA genomes produced by HDV replicationwas 500 times greater than that in the inoculum applied toinfect these cells.
The data show that within the experimental errors, there wasno drop in specific infectivity for later harvests relative to whatwas found for earlier harvests. That is, any drop was less thanthreefold. In other experiments, with harvests out to 21 days,the decreases in specific infectivity relative to what was foundfor earlier harvests were again no more than threefold (datanot shown). We conclude that under these experimental con-ditions, we did not detect a time-dependent decrease in specificinfectivity.
Effect of HBV L protein on assembly and infectivity. Theabove-mentioned studies confirm the results of others indicat-ing that HBV S is sufficient for HDV assembly but not forinfectivity, which depends upon the presence of HBV L (34).HBV assembly differs in that L and S are needed for bothassembly and infectivity (17). Furthermore, expression of Lalone is known to be cytotoxic (39). The following experimentwas undertaken to determine the role of the relative amountsof L and S on both HDV assembly and infectivity. In this
FIG. 6. Effect of the time and multiplicity of infection on accumu-lation of intracellular genomic HDV RNA. For panel A, cells wereinfected at a multiplicity of 300 GE per cell and time points were takenout to 15 days. For panel B, cells were infected with a series ofmultiplicities, with harvesting at day 6. All infections were done in thepresence of 5% PEG. Total RNA was extracted and HDV GE quan-titated by qPCR. Panel C shows the specific infectivities of HDVreleased at various times after transfection of Huh7 cells. Specificinfectivity is defined as the number of HDV GE detected per averageinfected cell divided by the input multiplicity of GE per average cell.The HDV used for the infections was harvested during 3-day periods,as indicated, and the amount of GE used as the inoculum was withinthe linear range, as determined in panel A.
3614 GUDIMA ET AL. J. VIROL.
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

variation of the assembly procedure, we used separate plas-mids to express L and S. A series of different ratios of thesewere cotransfected along with the plasmid to initiate HDVreplication. The media were harvested from days 6 to 9, atwhich time the cells were also extracted. The media were thenassayed for HDV RNA titer and tested for the ability to initi-ate infection in primary human hepatocytes. Thus, we wereable to deduce the specific infectivity (as in Fig. 6C) for eachsample.
The results are summarized in Table 4. Note that for eachtransfection, we determined by immunoblot analysis the per-centage of L relative to that of L plus S in the transfected cells.The values ranged from 0 to 100%. Consider first the assemblyefficiency. As the percentage of L was increased from zero,there was a threefold reduction in the assembly efficiency.Further increases in the percentage of L progressively led tomore substantial reductions in assembly. At 100% L, no as-sembly could be detected, even though there was no obviouscell toxicity.
For the first four samples shown in Table 4, the assembly wassufficient to allow a determination by immunoblot analysis ofthe percentage of L relative to that of L plus S in the releasedparticles. Note that increases from 0 to 57% were observed asthe percentage of L in the cell was increased from 0 to 72%.
Aliquots of the media were also tested for their abilities toinfect primary human hepatocytes. Such data are shown inTable 4, but what are also shown, and are more informative,are the specific infectivities (as in Fig. 6C). For the particleswith 0% L, the specific infectivity was much less than 1, con-sistent with the particles being noninfectious. However, for allthe other samples, as long as there was enough virus to detect,there was the ability to subsequently detect a genuine specificinfectivity.
In summary, increasing the percentage of L in the trans-fected cells caused significant inhibition of particle release, butfor those particles released, the specific infectivity was stillsignificant. As long as some particles were released and theycontained at least 7% L, they were infectious. Increasing the
percentage of L gave no more than a 2.5-fold increase in thespecific infectivity.
DISCUSSION
We have described here the successful assembly of HDVRNA-containing particles from transfected Huh7 cells, alongwith evidence for their infectivity in susceptible primary humanhepatocytes. While some aspects of our study confirm previousstudies (1, 14, 34–37), we also provide significant extensions,along with novel findings, as discussed below.
(i) A detailed characterization of the assembly process wasmade. In the transfected cells, the S protein was most abun-dant, with somewhat lower levels of L and no detectable levelof M (Fig. 1A). In contrast, the particles released into themedium had a 2-fold-lower relative amount of L (Fig. 2A).This was true at all harvest times, either early, at 0 to 3 days,when HDV RNA was not assembled, or at later times, 6 to 15days, when the HDV was assembled. To specifically test thehypothesis that this reduction in the amount of assembled Lwas consistent with L protein interfering with the assemblymechanism, we considered assembly under conditions wherewe could control the L/S ratio within cells. We thus observedthat as the amount of L relative to that of S was increased, theability to achieve assembly of RNA-containing particles wasprogressively reduced to background levels (Table 4).
(ii) We showed that the released particles could be affinitypurified using a monoclonal antibody to the preS2 domain. Ina preparative analysis, 77% of the HDV RNA-containing par-ticles bound (Table 1). For a separate analytical analysis, thebinding was �90% (data not shown). Thus, the majority of thevirus particles contained sufficient molecules of preS regionexposed on the surface to interact with the antibody. Both theunbound virus and that which was eluted were infectious, butthe specific infectivity of the unbound virus was 6.5 times lessthan that of the eluted virus (data not shown). We considerthat many possible factors could contribute to the lower spe-cific infectivity of the 10% of unbound virus.
(iii) The ability to carry out preparative affinity purificationallowed us to examine the HDV particles by electron micros-copy. We observed an abundance of the small spheres andfilaments that are typically associated with natural HBV infec-tions and considered to be empty particles (15). In addition, wewere also able to detect larger spheres with a mean diameter of36 nm. In an earlier study using serum from an infected chim-panzee, Rizzetto and coworkers detected a similar particle thathad the same sedimentation value as particles containing thedelta antigen (3, 4, 30). However, they noted that for someanimals infected with HBV but not HDV, they could detectsimilar particles (30).
(iv) Like Rizzetto and coworkers (3, 4, 30), we also usedrate-zonal sedimentation and were able to detect a specificsedimentation behavior for HDV (Fig. 3A). Our study has anadvantage in that we could assay the RNA genome and not just�Ag. Also, as an internal control, we were able to assay bothHBV and WHV, thus demonstrating that they both have sig-nificantly larger sedimentation values (Fig. 3A). We also as-sayed for HBsAg particles and found that these were heterog-enous with the majority, having a sedimentation value less thanthat of HDV (Fig. 3B). A minor fraction had a higher sedi-
TABLE 4. Requirement of HBV L in HDV assemblyand infectivity
% La
in cells% La inparticles
Assemblyb
(107 GE/ml)Infectivityb
(GE/avg cell)Specific
infectivityb
0 0 46 8.6 0.0740 7 15 1050 2859 18 15 1350 3572 57 3.1 530 6780 NDc 1.4 210 6280 ND 0.73 95 5381 ND 0.37 51 5786 ND 0.16 30 7591 ND 0.07 3.0 1597 ND 0.05 2.2 21100 ND 0.00001 0.045 ND
a To achieve these varied percentages of L/L plus S, Huh7 cells were trans-fected with different combinations of plasmids that express only L or only S.However, the indicated values were deduced from an immunoblot, using anantibody specific for the HBV S domain.
b These values were determined as described in the legends to Fig. 2 and 6.c ND, not determined.
VOL. 81, 2007 HDV ASSEMBLY AND INFECTIVITY 3615
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

mentation value, overlapping with that of the HDV RNA-containing particles. And when particles were assembled usingHBsAg but in the absence of HDV, we again detected particlesin this region (Fig. 3D). Thus, like Bonino and coworkers for invivo-assembled HDV (3), we conclude that in vitro-assembledHDV particles may have a discrete sedimentation value butthat HBsAg particles of the same size that do not contain HDVRNA can be produced. Thus, the 36-nm particles detected byelectron microscopy can be a mix of particles with and withoutthe HDV RNA. For many other viruses, including HBV, thefull and empty particles can be separated via differences indensity; however, for HDV, where the genome is singlestranded and significantly smaller than that of any other animalvirus, there is not a sufficient density difference.
(v) As found for certain other studies (1, 14), we observedthat HDV assembled in vitro could infect susceptible cells andthat the extent of infection as assayed by the detection of HDVRNA could be significantly enhanced by the presence of 4 to5% PEG (Table 3). However, we also used immunostaining todetect �Ag and were able to show that PEG increased thefraction of hepatocytes infected from 1 to 16% (Fig. 5A andB). Thus, PEG increased the accumulation of progeny HDVRNA largely by increasing the fraction of cells infected ratherthan increasing the yield per infected cell.
(vi) These results raise the question of why, even with 5%PEG and a multiplicity of 300 GE/cell, we observed only 16%infection (Fig. 5B). That is, to what extent was the limitedinfection a consequence of the virus versus a consequence ofthe susceptibility of the cells? We found that the majority ofprimary hepatocytes, both infected and uninfected, seemedhomogeneous, as judged by immunostaining for liver proteinalbumin (Fig. 5A and B) or by assaying for the cell-cell junctionprotein E-cadherin (Fig. 5A to D).
The alternative to faulting the hepatocytes for the limitedextent of infection was to consider the infectivity of assembledvirus. (i) We tested the specific infectivities of virus harvestedduring 3-day periods at a series of times after transfection ofHuh7 cells. No significant differences (greater than threefold)were detected (Fig. 6C). (ii) To evaluate the effect of shorterharvest times on virus release and virus infectivity, we reducedthe harvest time to 18 h and even 6 h. We observed that theamount of GE accumulated per unit time was unchanged andthat the specific infectivities for these harvests were not sig-nificantly different (data not shown). Such data support theinterpretation that virus released into medium was not sig-nificantly inactivated as the consequence of a 3-day harvestperiod. (iii) For most of our studies, virus was assembled invitro from transfected Huh7 cells, and yet virus assembled invitro from COS7, a line of monkey kidney cells that had acomparable specific infectivity (data not shown). (iv) We alsotested virus assembled in vivo from HDV replicating in WHV-infected woodchucks. Several independent sources were testedand found to infect both primary human hepatocytes andprimary woodchuck hepatocytes (kindly provided by WilliamMason), and yet the specific infectivities were not in excess ofthose obtained for the in vitro-assembled HDV (data notshown). It may be relevant to note certain data from Glebe andcoworkers (19). They used HBV from infected patients toinfect tupaia primary hepatocytes and observed, as we did, that
with even a multiplicity of 10,000 particles per cell, only 20% ofcells could be infected.
In summary, we show clearly that not all hepatocytes couldbe infected, but we have not been able to determine to whatextent this was due to limitations of the virus and/or of thecultured hepatocytes.
(vii) It is clear from this study and earlier studies (34) that invitro-assembled HDV particles are infectious only if the Lprotein of HBV is also present, and yet it has been recognizedthat the minimum amount required to confer infectivity isunknown (33). In unique experiments, we reduced the fractionof available L relative to that of L plus S and measured boththe particle release and the corresponding specific infectivity.Reducing this from �57% to 7% increased the particle releasebut did not decrease the specific infectivity (Table 4). Recentstructural studies indicate that an infectious HBV particle con-tains about 240 molecules of L plus S (13, 18), while a 25-nmempty particle contains 48 (18). The value for the HDV par-ticle must be somewhere in between, so 7% L corresponds tosomewhere between 3 and 17 molecules per average particle.Thus, we assert that this number, even though ascribed peraverage particle, is sufficient for maximal specific infectivity ofHDV on susceptible cells. As experimental evidence for this,we reduced the fraction of L a further threefold, and thespecific infectivity decreased to a value not distinguishablefrom that of noninfectious S-only particles (data not shown).
We further predict that at least 17 molecules of L per aver-age particle are needed for maximal HBV infectivity. Thisdemonstrates a unique advantage for our system since a com-parable experiment for HBV would be very difficult to achievesince regions within preS1 are needed not only for infectivitybut also for nucleocapsid assembly.
In summary, the studies described here provide novel infor-mation about the HDV particles that can be assembled invitro, including their abilities to infect susceptible cells. Weconsider this experimental system to have many future appli-cations. Some will be linked to HDV, for purposes such as tobetter determine the requirements for assembly of genomicrather than antigenomic RNA. However, important contribu-tions will come from exploiting the unique advantage of thisHDV system for addressing unsolved questions relating to theattachment and entry of both HDV and HBV.
ACKNOWLEDGMENTS
J.M.T. was supported by grants AI-058269 and CA-06927 from theNIH and by an appropriation from the Commonwealth of Pennsylvania.
Glenn Rall, Richard Katz, and William Mason gave valuable com-ments on the manuscript. Thanks go to Vadim Bichko, Irina Shchaveleva,T. C. Benedict Yen, Camille Sureau, Chi Tarn, Donald Ganem, andHans Netter for essential materials and/or encouragement. The fluo-rescence imaging studies were carried out in the Fox Chase ImagingFacility. The electron microscopy was aided by the Fox Chase ElectronMicroscopy Facility. The qPCR was performed in the Biochemistryand Biotechnology Facility.
REFERENCES
1. Barrera, A., B. Guerra, H. Lee, and R. E. Lanford. 2004. Analysis of hostrange phenotypes of primate hepadnaviruses by in vitro infections of hepa-titis D virus pseudotypes. J. Virol. 78:5233–5243.
2. Barrera, A., B. Guerra, L. Notvall, and R. E. Lanford. 2005. Mapping of thehepatitis B virus pre-S1 domain involved in receptor recognition. J. Virol.79:9786–9798.
3. Bonino, F., K. H. Heermann, M. Rizzetto, and W. H. Gerlich. 1986. Hepatitis
3616 GUDIMA ET AL. J. VIROL.
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from

delta virus: protein composition of delta antigen and its hepatitis B virus-derived envelope. J. Virol. 58:945–950.
4. Bonino, F., W. Hoyer, J. W.-K. Shih, M. Rizzetto, R. H. Purcell, and J. L.Gerin. 1984. Delta hepatitis agent: structural and antigenic properties of thedelta associated-particles. Infect. Immun. 43:1000–1005.
5. Bruss, V., and D. Ganem. 1991. The role of envelope proteins in hepatitis Bvirus assembly. Proc. Natl. Acad. Sci. USA 88:1059–1063.
6. Casey, J. L. (ed.). 2006. Hepatitis delta virus, vol. 307. Springer, Berlin,Germany.
7. Casey, J. L., and J. L. Gerin. 1995. Hepatitis D virus RNA editing: specificmodification of adenosine in the antigenomic RNA. J. Virol. 69:7593–7600.
8. Casey, J. L., B. C. Tennant, and J. L. Gerin. 2006. Genetic changes inhepatitis delta virus from acutely and chronically infected woodchucks. J. Vi-rol. 80:6469–6477.
9. Chang, F. L., P. J. Chen, S. J. Tu, M. N. Chiu, C. J. Wang, and D. S. Chen.1991. The large form of hepatitis � antigen is crucial for the assembly ofhepatitis � virus. Proc. Natl. Acad. Sci. USA 88:8490–8494.
10. Chang, J., S. O. Gudima, and J. M. Taylor. 2005. Evolution of hepatitis deltavirus RNA genome following long-term replication in cell culture. J. Virol.79:13310–13316.
11. Chao, M., S.-Y. Hsieh, and J. Taylor. 1990. Role of two forms of the hepatitisdelta virus antigen: evidence for a mechanism of self-limiting genome rep-lication. J. Virol. 64:5066–5069.
12. Dane, D. S., C. H. Cameron, and M. Briggs. 1970. Virus-like particles inserum of patients with Australia antigen-associated hepatitis. Lancet i:695–698.
13. Dryden, K. A., S. F. Wieland, C. Whitten-Bauer, J. L. Gerin, F. V. Chisari,and M. Yeager. 2006. Native hepatitis B virions and capsids visualized byelectron cryomicroscopy. Mol. Cell 22:843–850.
14. Engelke, M., K. Mills, S. Seitz, P. Simon, P. Gripon, M. Schnolzer, and S.Urban. 2006. Characterization of a hepatitis B and hepatitis delta virusreceptor binding site. Hepatology 43:750–760.
15. Ganem, D., and R. J. Schneider. 2001. Hepadnaviridae: the viruses and theirreplication, p. 2923–2969. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A.Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4thed. Lippincott Williams and Wilkins, Philadelphia, PA.
16. Garcia-Navarro, R., B. Blanco-Urgoiti, P. Berraondo, R. Sanchez de la Rosa,A. Vales, S. Hervas-Stubbs, J. J. Lasarte, F. Borras, J. Ruiz, and J. Prieto.2001. Protection against woodchuck hepatitis virus (WHV) infection by genegun coimmunization with WHV core and interleukin-12. J. Virol. 75:9068–9076.
17. Gerlich, W. H., and M. Kann. 2005. Hepatitis B, p. 1226–1268. In B. W. J.Mahy and V. ter Meulen (ed.), Topley and Wilson’s microbiology and mi-crobial infections, vol. 2. ASM Press, Washington, DC.
18. Gilbert, R. J., L. Beales, D. Blond, M. N. Simon, B. Y. Lin, F. V. Chisari, D. I.Stuart, and D. J. Rowlands. 2005. Hepatitis B small surface antigen particlesare octahedral. Proc. Natl. Acad. Sci. USA 102:14783–14788.
19. Glebe, D., M. Aliakbari, P. Krass, E. V. Knoop, K. P. Valerius, and W. H.Gerlich. 2003. Pre-S1 antigen-dependent infection of Tupaia hepatocytecultures with human hepatitis B virus. J. Virol. 77:9511–9521.
20. Gudima, S. O., J. Chang, G. Moraleda, A. Azvolinsky, and J. Taylor. 2002.Parameters of human hepatitis delta virus replication: the quantity, quality,and intracellular distribution of viral proteins and RNA. J. Virol. 76:3709–3719.
21. Gudima, S. O., J. Chang, and J. M. Taylor. 2005. Reconstitution in cultured
cells of replicating HDV RNA from pairs of less than full-length RNAs.RNA 11:90–98.
22. Hayat, M. A., and S. E. Miller. 1990. Negative staining. McGraw-Hill, NewYork, NY.
23. Jaoude, G. A., and C. Sureau. 2005. Role of the antigenic loop of thehepatitis B virus envelope proteins in infectivity of hepatitis delta virus.J. Virol. 79:10460–10466.
24. Kuo, M. Y.-P., M. Chao, and J. Taylor. 1989. Initiation of replication of thehuman hepatitis delta virus genome from cloned DNA: role of delta antigen.J. Virol. 63:1945–1950.
25. Ladner, S. K., M. J. Otto, C. S. Barker, K. Zaifert, G. H. Wang, J. T. Guo,C. Seeger, and R. W. King. 1997. Inducible expression of human hepatitis Bvirus (HBV) in stably transfected hepatoblastoma cells: a novel system forscreening potential inhibitors of HBV replication. Antimicrob. Agents Che-mother. 41:1715–1720.
26. Lambert, C., S. Mann, and R. Prange. 2004. Assessment of determinantsaffecting the dual topology of hepadnaviral large envelope proteins. J. Gen.Virol. 85:1221–1225.
27. Loeb, K. R., K. R. Jerome, J. Goddard, M. Huang, A. Cent, and L. Corey.2000. High-throughput quantitative analysis of hepatitis B virus DNA inserum using the TaqMan fluorogenic detection system. Hepatology 32:626–629.
28. Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982.Growth of human hepatoma cell lines with differentiated functions in chem-ically defined medium. Cancer Res. 42:3858–3863.
29. Netter, H. J., T.-T. Wu, M. Bockol, A. Cywinski, W.-S. Ryu, B. C. Tennant,and J. M. Taylor. 1995. Nucleotide sequence stability of the genome ofhepatitis delta virus. J. Virol. 69:1687–1692.
30. Rizzetto, M., B. Hoyer, M. G. Canese, J. W. K. Shih, R. H. Purcell, and J. L.Gerin. 1980. � Agent: association of � antigen with hepatitis B surfaceantigen and RNA in serum of �-infected chimpanzees. Proc. Natl. Acad. Sci.USA 77:6124–6128.
31. Ryu, W.-S., M. Bayer, and J. Taylor. 1992. Assembly of hepatitis delta virusparticles. J. Virol. 66:2310–2315.
32. Sominskaya, I., V. Bichko, P. Pushko, A. Dreimane, D. Snikere, and P.Pumpens. 1992. Tetrapeptide QDPR is a minimal immunodominant epitopewithin the preS2 domain of hepatitis B virus. Immunol. Lett. 33:169–172.
33. Sureau, C. 2006. The role of the HBV envelope proteins in the HDVreplication cycle. Curr. Top. Microbiol. Immunol. 307:113–131.
34. Sureau, C., B. Guerra, and R. E. Lanford. 1993. Role of the large hepatitisB virus envelope protein in infectivity of the hepatitis delta virion. J. Virol.67:366–372.
35. Sureau, C., B. Guerra, and H. Lee. 1994. The middle hepatitis B virusenvelope protein is not necessary for infectivity of hepatitis delta virus.J. Virol. 68:4063–4066.
36. Sureau, C., J. R. Jacob, J. W. Eichberg, and R. E. Lanford. 1991. Tissueculture system for infection with human hepatitis delta virus. J. Virol. 65:3443–3450.
37. Sureau, C., A. M. Moriarty, G. B. Thornton, and R. E. Lanford. 1992.Production of infectious hepatitis delta virus in vitro and neutralization withantibodies directed against hepatitis B virus pre-S antigens. J. Virol. 66:1241–1245.
38. Wu, T.-T., V. V. Bichko, W.-S. Ryu, S. M. Lemon, and J. M. Taylor. 1995.Hepatitis delta virus mutant: effect on RNA editing. J. Virol. 69:7226–7231.
39. Xu, Z., V. Bruss, and T. S. Yen. 1997. Formation of intracellular particles byhepatitis B virus large surface protein. J. Virol. 71:5487–5494.
VOL. 81, 2007 HDV ASSEMBLY AND INFECTIVITY 3617
on April 9, 2016 by guest
http://jvi.asm.org/
Dow
nloaded from
Related Documents











