Assembly and Annotation of a 22Gb Conifer Genome, Loblolly Pine Jill Wegrzyn Pieter de Jong, Chuck Langley, Dorrie Main, Keithanne Mockaitis, Steven Salzberg, Kristian Stevens, Nick Wheeler, Jim Yorke, Aleksey Zimin, David Neale Univ. of Calfornia, Davis; Children’s Hospital of Oakland Research Institute; Indiana Univ.; Washington State Univ.; Univ. of Maryland; and Johns Hopkins Univ.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Assembly and Annotation of a 22Gb Conifer Genome, Loblolly Pine
Jill Wegrzyn
Pieter de Jong, Chuck Langley, Dorrie Main, Keithanne Mockaitis, Steven Salzberg, Kristian Stevens, Nick Wheeler, Jim Yorke, Aleksey Zimin, David Neale
Univ. of Calfornia, Davis; Children’s Hospital of Oakland Research Institute; Indiana Univ.; Washington State Univ.; Univ. of Maryland; and Johns Hopkins Univ.

PineRefSeq
GoalTo provide the benefits of conifer reference genome
sequences to the research, management and policy communities.
Specific Objectives– Provide a high-quality reference genome sequence of loblolly
pine looking toward sugar pine and then Douglas-fir.– Provide a complete transcriptome resource for gene
discovery, reference building, and aids to genome assembly– Provide annotation, data integration, and data distribution
through Dendrome and TreeGenes databases.

The Large, Complex Conifer Genomes Present a Challenge
• Challenges– The estimated 22 Gigabase loblolly pine genome is 8 times larger than
the human genome– Conifer genomes generally possess large gene families (duplicated and
divergent copies of a gene), and abundant pseudo-genes.– The vast majority of the genome appears to be repetitive DNA
• Approaches to Resolving Challenges– Complementary sequencing strategies that seek to reduce complexity
through use of actual or functional haploid genomes and reduced size of individual assemblies.
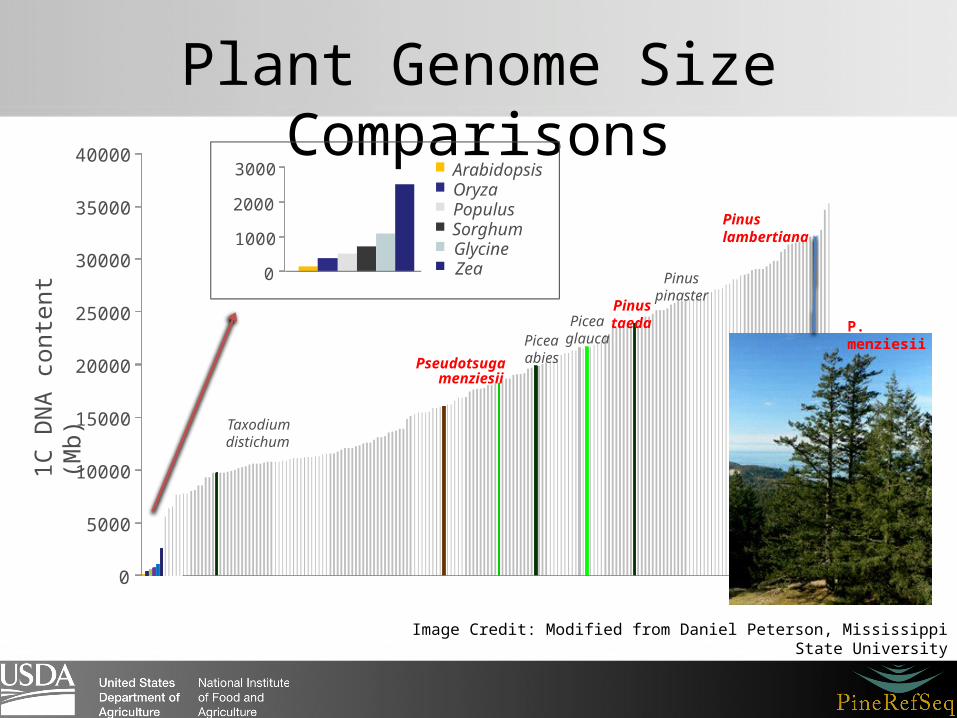
Plant Genome Size Comparisons
Image Credit: Modified from Daniel Peterson, Mississippi State University
0
5000
10000
15000
20000
25000
30000
35000
40000
0
1000
2000
3000 ArabidopsisOryzaPopulusSorghumGlycineZea
Pseudotsugamenziesii
Taxodiumdistichum
Piceaabies
Piceaglauca
Pinustaeda
Pinuspinaster
1C D
NA
con
tent
(M
b)
Pinus lambertiana
P. menziesii

Existing and Planned Angiosperm Tree Genomes
Species Genome Size1
In Progress With Draft Assemblies
Populus trichocarpa Black cottonwood 423 Mbp
Populus nigra Black poplar 480 Mbp
Eucalyptus grandis Rose gum 691 Mbp
Eucalyptus globulus Blue gum 530 Mbp
Eucalyptus camaldulensis River red gum 624 Mbp
Corymbia citriodora Lemon-scented gum 370 Mbp
Betula nana Dwarf birch 450 Mbp
Fraxinus excelsior European ash 900 Mbp
Malus domestica Apple 881 Mbp
Prunus persica Peach 227 Mbp
Citrus sinensis Sweet orange 319 Mbp
Azadirachta indica Neem 363 Mbp
In Progress Or Planned
Castanea mollissima Chinese Chestnut 800 Mbp
Quercus robur Pedunculate Oak 740 Mbp
Populus spp and ecotypes Various various
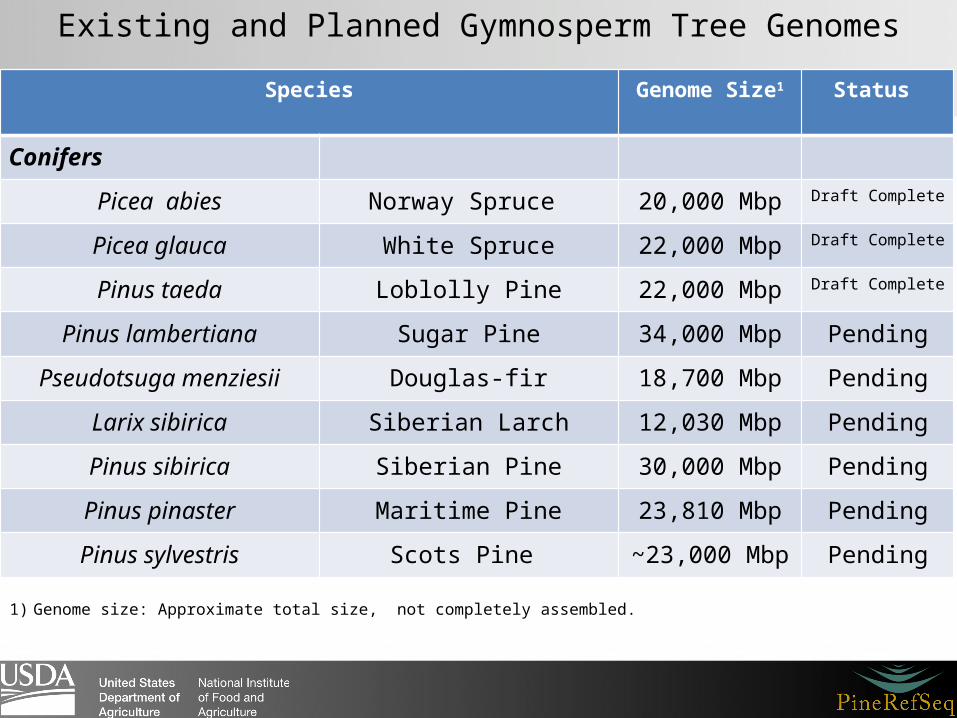
Existing and Planned Gymnosperm Tree Genomes
Species Genome Size1 Status
Conifers
Picea abies Norway Spruce 20,000 Mbp Draft Complete
Picea glauca White Spruce 22,000 Mbp Draft Complete
Pinus taeda Loblolly Pine 22,000 Mbp Draft Complete
Pinus lambertiana Sugar Pine 34,000 Mbp Pending
Pseudotsuga menziesii Douglas-fir 18,700 Mbp Pending
Larix sibirica Siberian Larch 12,030 Mbp Pending
Pinus sibirica Siberian Pine 30,000 Mbp Pending
Pinus pinaster Maritime Pine 23,810 Mbp Pending
Pinus sylvestris Scots Pine ~23,000 Mbp Pending
1) Genome size: Approximate total size, not completely assembled.

Elements of the Conifer Genome Sequencing Project

Acquiring the DNA
HaploidHaploid megagametophyte tissue1NShotgun sequenced
DiploidDiploid needle tissue2N40 Kb cloned fosmids, pooledand sequenced
Figure Credit: Nicholas Wheeler, University of California, Davis

Sequencing Strategy
65X 12X

Technology for De Novo Sequencing of the Conifer Genomes
Parallel and Complementary ApproachesWhole Genome
Shotgun Sequencing (Haploid/Diploid)
Illumina GAIIx
IlluminaHiSeq
Sequencing of Pooled Fosmid
Clones (Diploid)1
IlluminaGAIIx
IlluminaHiSeq
Max Output: 95 GigabasesMax. paired end reads - 640 million
Max Output: 300 GigabasesMax. paired end reads - 3 billion
1 Effectively haploid

Sequencing Strategy
Today

Megagametophyte Whole Genome Shotgun (M-WGS)
• Not enough haploid DNA in a megagametophyte to implement a complete list of WGS ingredients.
• Compromise: Obtain DNA for longer insert linking libraries (> 1kbp) from diploid needle tissue.
• Prepare only short insert Illumina libraries from megagametophye tissue.

Each DNA sample is then run on an Agilent Bioanalyzer to determine a preliminary estimate of insert size and coefficient of variation.
If within spec, selected DNA samples are converted into Illumina libraries
M-WGS Short Insert LibrariesPreliminary QC and Size Selection

• Libraries are subsequently QCed on the Illumina MiSeq
M-WGS Short Insert LibrariesLibrary QC and Titration

A k-mer Genome Size Estimate
How deep to sequence the libraries?Experimentally – hybridizationComputationally (WGS) – choose substring of the reads of length k
P. taeda genome size ≅ total k-mers in genome
total k-mers in P. taeda genome ≅
total k-mers in P. taeda reads expected number of times a
genomically unique k-mer is observed in the reads

k-mer Genome Size Estimates
Loblolly pine Pinus taeda:31-mers total: 3.736 x 1011
Expected k-mer depth: 18.11Estimated genome size: 20.63 GB
High Copy 31-mers 1.09% of distinct 31-mers33% of all 31-mers
24-mers total: : 4.092 x 1011
Expected k-mer depth: 19.79Estimated genome size: 20.68 GB
Sugar pine Pinus lambertiana:31-mers total: 2.776 x 1011
Expected k-mer depth: 8.12Estimated genome size: 34.19 GB
High Copy 31-mers 0.35% of distinct 31-mers33% of all 31-mers
24-mers total: 3.031 x 1011
Expected k-mer depth: 8.89Estimated genome size: 33.98 GB

truly large genomes

P. taeda Version 0.9 Library Statistics
• Haploid short insert libraries – 10 short insert libraries 200 - 640bp– 1.4Tbp GA2x, HiSeq, MiSeq sequence– 65 fold coverage
• Diploid jumping libraries– 47 jumping libraries 1300 – 5500bp– 280Gbp GA2x sequence– 12 fold coverage
• 13 Fosmid DiTag Libraries
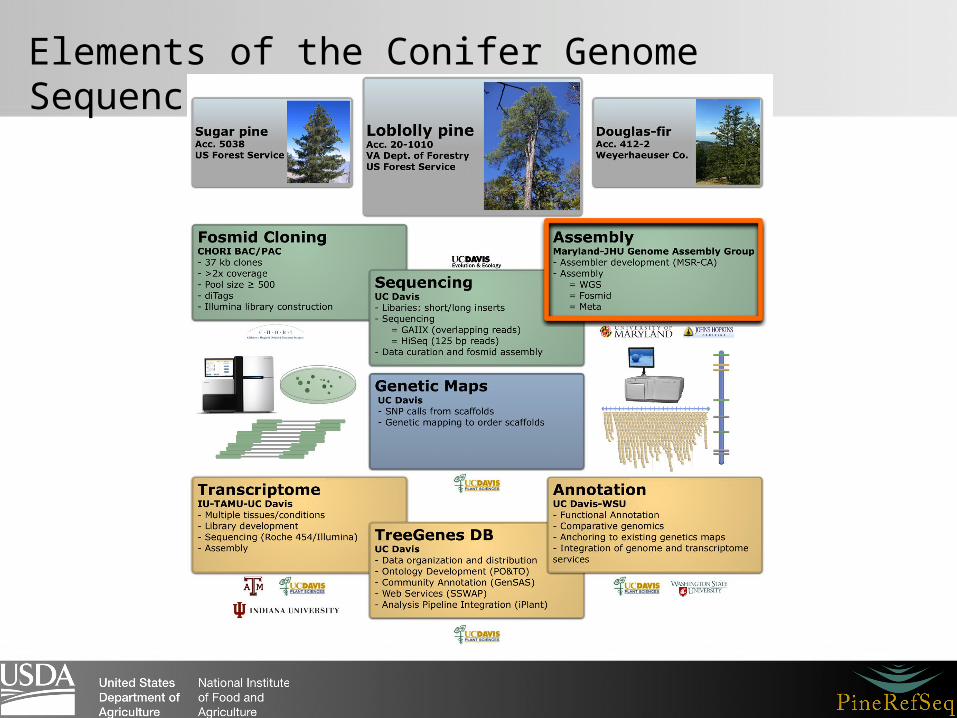
Elements of the Conifer Genome Sequencing Project

• 65X coverage in paired ends from a single seed
• 1/3 in GAIIx, 160-bp overlapping pairs• 2/3 in HiSeq, 100-bp pairs
• 1.7 billion reads from “jumping” libraries from pine needles, diploid DNA

Collect jumping reads from same haplotype
1.7 billion jumping reads (4 Kbp)
93 million Di-Tag reads (36 Kbp)
Keep only pairs where both reads
match haploid DNA
Filter: both reads had to be covered by 52-mers from megagametophyte data

How to get all these reads into a single assembly run?
16 billion paired reads

Recent Assemblers for Illumina Data
• MSR-CA (Aleksey Zimin, UMD)
– Based on Celera assembler– 454, Illumina, and Sanger reads
• Allpaths-LG• SOAPdenovo• Velvet• ABySS• Contrail• SGA

Two Classes of Assembly Algorithms
• Overlap-Layout-Consensus (OLC)– Used by most assemblers for previous generation (Sanger)
sequencing– Celera Assembler, PCAP, Phusion, Arachne, etc
• De Bruijn Graph– Used by most assemblers for Illumina data– SOAPdenovo, Allpaths-LG, Velvet, Abyss, etc
• We use a combined approach that combines the benefits of both OLC and the De Bruijn Graph in our MSR-CA assembler

Combine Benefits of OLC and De Bruijn Graph
• Benefits of OLC– Can deal with variable
length reads and reads from different sequencing platforms
– Overlaps can be long and thus more reliable
– Overlaps do not have to be exact
– Can resolve repeats of up to read size
• Drawbacks of OLC– Computationally intensive,
number of overlaps grows quickly with the number of reads and coverage
• Benefits of DeBruijn Graph– Computationally faster
• Drawbacks of DeBruijn Graph– Errors in the reads create
spurious branches in the graph requiring error correction
• Max. size of k-mer is limited by the shortest read size
• All overlaps in the graph are exact overlaps of k-1 bases
• Repeats of longer than k bases cannot be resolved– Without space consuming side
information
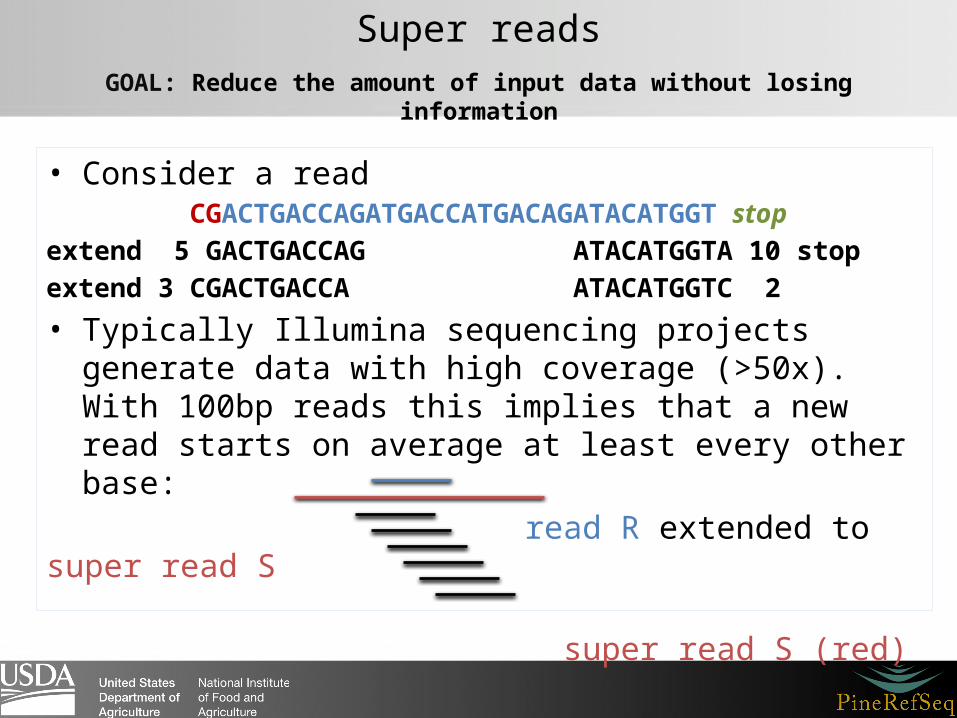
• Consider a read CGACTGACCAGATGACCATGACAGATACATGGT stopextend 5 GACTGACCAG ATACATGGTA 10 stopextend 3 CGACTGACCA ATACATGGTC 2
• Typically Illumina sequencing projects generate data with high coverage (>50x). With 100bp reads this implies that a new read starts on average at least every other base:
read R extended to super read S super read S (red) the other reads extend to the S as well
Super readsGOAL: Reduce the amount of input data without losing information

Super-Reads Compress the Data
16 billion paired reads
150 million super-reads
• 100-fold compression• 50% of sequence is in super reads
> 500 bp• Super-read total: 52 Gbp

MaSuRCA assembler performance
• 64-core computer with 1 Terabyte of RAM• Time/memory to assemble:
• QuORUM error correction: 10 days / 800 GB• Super-reads construction plus filtering: 11 days /
400 GB• Contig and scaffold construction: 60+ days / 450 Gb
• uses CABOG assembler
• Gap filling with super-reads: 8 days / 300 Gb

MSR-CA Output
Contigs: contiguous sequences that do not appear to be repetitive (may contain internal repeats). These end up in scaffolds.
Scaffolds: ordered and oriented collections of contigs, built using mate pair data. A scaffold can consist of just one contig (a "single-contig" scaffold).
Degenerate contigs: contigs that appeared to be repeats according to the coverage statistics. Only placed in scaffolds when linked to contigs via mate pairs. Most of them will end up being placed in more than one location, but many will not appear in any scaffold.

P. taeda WGS V0.6 (June 2012)• Approximately 35X coverage
– 7 billion reads (50 million jumping library reads)– Compressed to 377 million Super-reads
• Total Sequence: 18,321,727,393 bp
• Total contig sequence: 14,606,783,345 bp
• N50 1,199bp (9.16 Gbp is contained in contigs of 1199 bp or longer)
• Total scaffold sequence (with imputed gaps): 18,428,460,141bp
• N50 1,230bp (9.21 Gbp is contained in scaffolds of 1230 bp or longer)
• Degenerate contig sequence 3.8Gb

P. taeda WGS V0.8 (January 2013)
• Approximately 65X coverage – 16 billion reads (1.7 billion jumping library reads)– Compressed to 150 million Super-reads
• Total Sequence: 22,518,572,092 bp
• N50 Contig: 7,083bp
• N50 Scaffold: 15,885 bp

P. taeda WGS V0.9 (March 2013)• Total Sequence: 20.1 Gbp
• Total contig sequence: 2.3 Gbp
• N50 8,200bp (11.6 million)
• Total scaffold sequence (with imputed gaps): 17.8 Gbp
• N50 30,700bp (4.8 million)

Ongoing Efforts• Improve MSR-CA scaffolding
• Transcriptome + WGS assembly
• Fosmid pool sequencing and assembly
• GBS to anchor and orient scaffolds
• Sugar pine genome: 35 Gigabases!

Elements of the Conifer Genome Sequencing Project

Sequencing StrategyMolecular approach to complexity reduction
End of summer
2013

Fosmid Pooling:Genome partitioning for reduced assembly complexity
• The immense and complex diploid pine genome can be economically and efficiently partitioned into smaller, functionally haploid, pieces using pools of fosmid clones.
• Fosmids in a pool should have a combined insert size far less than a haploid genome size; to ensure haploid genome representation.
• The sequence data obtained from a single fosmid pool may be up to 80 X deep.
• The sequence data obtained from a pool must be screened for vector and E. coli contamination
• Ideally: larger clones (BACs) are more desirable, more likely to span repeats

Fosmid Sequence Components
• Haploid fosmids with vector tagged ends• Primary coverage from short insert libraries • Additional coverage from long insert libraries
from equi-molar pool of pools.• Fosmid end sequences (diTags) link ends of the assembly
and count fosmids in a pool

Fosmid PoolsDetermining the Best Assembler for the Job
quartilesAssembler Stat Count Q1 Q2 Q3 N50 Sum
Allpaths-LGscf 987 2499 7781 30271 26298 14 x 106 ctg 1524 2355 6031 12509 10324 14 x 106
scf30K+ 248 33595 35682 38361 30114 9 x 106
MSR-CAscf 2162 506 1375 9224 14753 15 x 106 ctg 3519 503 1339 5000 6826 14 x 106
scf30K+ 136 32603 35087 38119 30147 5 x 106
SOAPscf 3251 123 185 495 33389 15 x 106 ctg 23873 76 175 348 1515 15 x 106
scf30K+ 322 33907 35766 38683 33389 12 x 106
Assembly results for a relatively large pool of approximately 600 P. taeda fosmids

Use Cases for Fosmid Pools
• Assembler Evaluation
• Repeat Library Construction
• SNP Identification

Genomic SequencePinus taeda BACs and Fosmids
Pinus taeda BACs Pinus taeda Fosmids
Total number of sequences 103 90,973
Average sequence length 115,130 2,918
Median sequence length 118,782 475
N50 sequence length (bp) 127,167 16,204
Shortest sequence length 1,392 201
Longest sequence length 235,088 75,791
Total length (bp) 11,858,447 265,511,345
GC % 37.98% 38.09%
A : C : T : G% 31.27 : 18.79 : 31.32 : 18.62 30.94:19.07:30.97:19.03
Combined sequence resource represents roughly 1% of the estimated 22 GB genome

Similarity and De Novo RepeatIdentification
Tandem Repeat Finder (TRF)
Homology (Censor against RepBase)Summary of Repbase v17.07• Number of entries: 28,155• Number of species represented: 715• Number of repeat families: 280
• Angiosperm entries: 131• Gymnosperm entries (conifer):15
De Novo (REPET/TEannot)• Self-alignment (all vs all) with BLAST to find HSPs is followed by clustering with Grouper, Recon, and Piler • 3 sets of clusters are aligned with a MSA (MAP) to derive a consensus sequence• Structural search runs simultaneously (LTR Harvest) to detect highly diverged LTRs• Final Blastclust to cluster potential sequences

Tandem RepeatsComparison across sequenced angiosperms and other gymnosperms
(partial)
Dinucleotide
Trinucle
otide
Tetra
nucleotide
Pentan
ucleotide
Hexan
ucleotide
Heptan
ucleotide
Octanucle
otideTo
tal0.00
10.00
20.00
30.00
40.00
50.00
60.00
70.00
Pinus taeda Taxus maireiPicea glaucaCucumis sativusPopulus trichocarpaArabidopsis thalianaVitis vinifera
Num
ber o
f Micr
osat
ellit
e lo
ci/M
bp
Pinus taeda (BAC + Fosmid) Picea glauca (BAC) Taxus mairei (Fosmid)
Micro Mini Sat Micro Mini Sat Micro Mini Sat
Most frequent period 2 21 123 2 27 122 2 24 230
Cumulative length 126,254 216,194 154,835 876 843 389 1,411 587 1,598
Num. of loci 64,740 10,508 1,258 11 10 1 32 19 2
Most frequent period
(%)
0.05% 0.08% 0.06% 0.33% 0.32% 0.15% 0.09% 0.04% 0.10%
Total cumulative length
(bp)
241,822 4,323,361 2,650,740 925 6,603 1,864 3,024 15,875 5,871
Total (%) 0.09% 1.56% 0.96% 0.35% 2.49% 0.70% 0.19% 0.98% 0.36%
Total tandem content: 2.6%3.31% of BACs2.59% of fosmids

Homology Search ResultsCensor (BLAST-style) comparisons against Repbase
Cucumis sativus
Taxus mairei
Arabidopsis thaliana
Pinus taeda
Picea glauca
Populus trichocarpa
Vitis vinifera
0.00% 10.00% 20.00% 30.00% 40.00% 50.00% 60.00% 70.00% 80.00%
Class I: LTR: CopiaClass I: LTR: GypsyClass II: DNA TransposonClass I: Non-LTR: LINE
Percent of sequence sets
Partial and Full-length Interspersed Alignments (compared across species)
Full-length Alignments Only
Pinus taeda (BACs) by homology
Pinus taeda (fosmids) by homology
Pinus taeda (BACs) by de novo
Pinus taeda (fosmids) by de novo
0.00% 1.00% 2.00% 3.00% 4.00% 5.00% 6.00% 7.00% 8.00%
Full Length Sequences80-80-80 Rule (Wicker et al. 2007)
• 80 bp in length• 80% identity• 80% coverage

Summary of Combined Homology and De Novo Approach
• 88% repetitive (partial and full-length)• 29% repetitive (full-length only defined by 80-80-80)
– 87% of the full-length content is characterized as LTR retrotransposons
• Repeats are highly diverged– Only 23% identified by homology for full and partial elements– Repbase contains just 15 (+5) gymnosperm elements– 6,270 novel families discovered with no homology
• 5,155 are single copy
• High copy elements are either Gypsy or Copia LTRs• Nested repeats common in LTR retrotransposons

Novel Repeat ElementsDiverged LTRs are annotated as 6,270 novel families
Top 400 elements only cover 12% of the combined sequence sets
Repeat family Full-Length Copies Length (bp) Percent of Sequence Set
TPE1 159 1,077,598 0.39%
PtPiedmont (93122) 133 969,109 0.35%
IFG7 162 956,018 0.34%
PtOuachita (B4244) 47 576,871 0.21%
Corky 78 469,286 0.17%
PtCumberland (B4704) 67 431,492 0.16%
PtBastrop (82005) 38 378,631 0.14%
PtOzark (100900) 32 378,020 0.14%
PtAppalachian (212735)
67 367,653 0.13%
PtPineywoods (B6735) 68 322,632 0.12%
PtAngelina (217426) 24 309,248 0.11%
Gymny 24 291,479 0.11%
PtConagree (B3341) 50 285,850 0.10%
PtTalladega (215311) 33 274,826 0.10%
Total 982 7,088,713 2.56%

Novel Repeat Elements
MSA with annotations of the novel Copia LTR -PtPineywoods
MSA with annotations of the novel Gypsy LTR - PtAppalachian

Elements of the Conifer Genome Sequencing Project

Loblolly transcriptome from 30 unique RNA collections
Carol Loopstra (RNA) and Keithanne Mockaitis (sequencing)

Progressive Transcript Profiling
Build a useful transcriptome reference early in project:
generate long reads for ease of assembly, scaffolding of existing shorter data
integrate community data into assemblies
Vegetative Organsvegetative budscandlesstemsneedlesroots
Early Stress Signaling Responsescoldheatelevated UVcompression
Reproductive Developmentmegastrobilimicrostrobili
Early Developmentseedsyoung seedlings

Transcriptome Assembly
• Considerable variation in de novo transcriptome assemblies– Used a compare and compete methodology to
select the final transcripts– Two Trinity versions and Velvet/Oasis (6 different
k-mer sizes)– First analysis: Basic clustering methods with 454
and other protein evidence to determine optimal full-length proteins

Coding transcripts, clustered outputs by assembler
Transcript Class Trinity 2012.10.05
Trinity 2013.02.25
Velvet 1.2.08Oases 0.2.08
complete CDS 58,707 115,353 395,370
complete CDS, UTR poor 8,023 10,033 39,833
complete CDS, UTR very short/absent 1,076 1,393 7,298
total complete protein (non-unique) 67,806 126,779 442,501
partial protein coding 196,252 404,722 2,041,836
total 264,058 531,501 2,484,337
Protein coding loci, estimated from transcript evidence alone (32 sets):
87,602 unique complete
64,610 mapped to the WGS assembly
preliminary results, Keithanne Mockaitis

Improving Transcriptome Assembly• Improved transcript grouping with exon-aware clustering methods
Transcript Class Total (Improved Clustering) Mapped to genome v0.9 Unmapped
Primary, complete CDS 87,241 83,271 3970
Alternate, complete CDS 642,175 642,175 4092
Partial CDS 69,044 61,114 7930
Alternative partial CDS 617,248 607,490 9758
• Duplicates/Paralogs• Pseudogenes• Too much compression of Unigene set?
Mapping Occurrences Complete transcripts mapped
1 9840
2 3574
3 2542
4 2624
>/=5 64690

Examining Gene FamiliesMyB Transcription Factors
Homeobox Transcription Factors

Improving the Assembly with Transcriptome
• Map WGS (v0.9) against the transcripts with nucmer
• Iteratively compute alignments and merge scaffolds.
• 12,000+ scaffolds merged during first pass• . . . V1.0

Elements of the Conifer Genome Sequencing Project

Source: Jiao et al., Ancestral polyploidy in seed plants and angiosperms, Nature, Vol. 473, May 5, 2011

Mapping Full-Length Orthologous ProteinsAlignments: exonerate protein2genome, heuristic, 70% query coverage, 70% similarity
~220,000 query proteins total
• Physcomitrella patens: 2,761 out of 25,506 (10.8%)• Selaginella moellendorffii: 2,025 out of 16,821 (12.0%)• ‘Basal’ angiosperm:
– Amborella trichopoda: 4,076 out of 25,347 (16.1%)• Angiosperms:
– Arabidopsis thaliana: 4,777 out of 27,986 (17.1%)– Populus trichocarpa: 4,023 out of 18,588 (21.6%)– Sorghum bicolor: 3,368 out of 24,122 (18.1%)– Vitis vinifera: 3,833 out of 18,441 (20.8%)– Glycine max: 9,970 out of 52,178 (19.1%)
• Gymnosperms:• Picea: 6,696/11,065 (60.5%)
– The majority of these are Picea sitchensis (Ralph et al., 2008)• Pinus: 345/426 (81.0%)

Mapping Proteins~220K full-length proteins and CEGMA analysis
• BLAT/Exonerate with ~220K proteins– Requiring 70% similarity and 70% query coverage,
45,101 proteins aligned to 11,897 unique scaffolds/contigs
• CEGMA– Examines conserved eukaryotic core genes (KOGS)– 240 full-length and 197 partial proteins (of 458)– 113 full-length proteins of the 248 in the highly
conserved category

Training MAKERPinus taeda resources:ADEPT2 Project ClustersExon Capture (Neves et al. 2013)PineRefSeq Transcriptome454 Transcriptome (Lorenz et al. 2012)
Pinus Resources:TreeGenes UniGenesWhitebark pine (RNASeq)Sugar pine transcriptome (454 + RNASeq)Limber pine transcriptome (RNASeq)Lodgepole pine (454) (Parchman et al. 2010)Longleaf pine (454) (Lorenz et al. 2012)
Picea Resources:TreeGenes UniGenesSitka spruce (Sanger/454) (Ralph et al. 2008)Norway spruce (454) (Chen et al. 2013)Congenie transcriptome (Nysterdt et al. 2013)Norway spruce (454) (Lorenz et al. 2013)White spruce (454) (Rigault et al. 2011)
. . . Just finished at iPlant (TACC)Running on 8,000 cores…

WebApollo on TreeGenesIntrons
• Exon conservation highlighted• Supporting EST evidence• Intron 2: Size 131,138• Intron 3: Size 179,620

Gene family conservation
An Example of: jcf7180063228536 4.97Mbp scaffold
WebApollo on TreeGenes

Conifer specific proteins
An Example of: jcf7180063228536
WebApollo on TreeGenes

Elements of the Conifer Genome Sequencing Project

Dendrome ProjectTreeGenes Database to Distribute Transcriptome and Genome

GENome Sequence Annotation Server (GenSAS) Community Annotation

GENome Sequence Annotation Server (GenSAS) Opening Screen
After the selected programs run, clicking on the green icon will bring users to a map interface
List of available programs to run with optional paramaterization
Sequence choices from database or uploaded file

GENome Sequence Annotation Server (GenSAS) Map Interface
Click on features to see more info
Quick Navigation Shortcuts
Click to jump positions
Task Name
Export Layer and Save

The Maryland Genome Assembly Group featuring co-PD Steven Salzberg and Daniela Puiu (Johns Hopkins U) and co-PD Jim Yorke and
Aleksey Zimin (U of Maryland)
PD David Neale (r), co-PD Jill Wegrzyn (c), and (l to r) John Liechty, Ben Figueroa, Patrick McGuire, Pedro J. Martinez-
Garcia, Hans Vasquez-GrossUC Davis
Co-PD Chuck Langley (r) and (l to r) Marc Crepeau, Kristian Stevens, and
Charis Cardeno UC Davis
(l to r) Co-PD Pieter de Jong, Ann Holtz-Morris, Maxim Koriabine,
Boudewijn ten HallersCHORI BAC/PAC
Co-PD Carol Loopstra and Jeff Puryear TAMU
Co-PD Keithanne Mockaitis and Zach Smith Indiana U
Co-PD Dorrie MainWSU
DP
SS AZJY
Related Documents











