Cite this: Lab Chip, 2013, 13, 1295 Aqueous two-phase microdroplets with reversible phase transitions3 Received 4th October 2012, Accepted 23rd January 2013 DOI: 10.1039/c3lc41122b www.rsc.org/loc Jonathan B. Boreyko, a Prachya Mruetusatorn, b Scott T. Retterer ac and C. Patrick Collier* a Aqueous two-phase systems contained within microdroplets enable a bottom-up approach to mimicking the dynamic microcompartmentation of biomaterial that naturally occurs within the cytoplasm of cells. Here, we demonstrate the generation of femtolitre aqueous two-phase droplets within a microfluidic oil channel. Gated pressure pulses were used to generate individual, stationary two-phase microdroplets with a well-defined time zero for carrying out controlled and sequential phase transformations over time. Reversible phase transitions between single-phase, two-phase, and core–shell microbead states were obtained via evaporation-induced dehydration and water rehydration. In contrast to other microfluidic aqueous two-phase droplets, which require continuous flows and high-frequency droplet formation, our system enables the controlled isolation and reversible transformation of a single microdroplet and is expected to be useful for future studies in dynamic microcompartmentation and affinity partitioning. 1 Introduction Aqueous solutions containing two different polymers can be immiscible when mixed together above critical concentrations. The resulting interface of aqueous two-phase systems (ATPS) exhibits a remarkably low surface tension (y1–100 mNm 21 ), 1,2 allowing for biomolecules to readily traverse between the phases without denaturation. For this reason, ATPS are of great interest for the partitioning and self-assembly of biomaterials by exploiting their affinity for one of the polymer phases. 3–5 Over the past decade, various microfluidic devices have utilized polymer solutions such as poly(ethylene glycol) (PEG) and dextran to generate aqueous two-phase flows capable of partitioning cells, 6–10 proteins, 11,12 and DNA. 13 Recently there has been much interest in containing ATPS within droplets to mimic the differences in local composition within the cytoplasm of cells, known as microcompartmenta- tion. 14 This has been motivated by evidence that aqueous phase-separation is an important mechanism for cellular microcompartmentation, 15,16 including the observation that P granules in germ cells are liquid droplets, and therefore some intracellular compartments are entirely aqueous in composi- tion. 17 An acoustically levitated aqueous two-phase droplet was used to demonstrate the affinity partitioning of biotinylated liposomes, 18 however the millimetric droplet size is incom- mensurate with cellular length scales. Synthetic cells have been developed by encapsulating ATPS inside of spherical lipid bilayer membranes. 19 In these systems, proteins could be preferentially sorted into the PEG or dextran-rich phase via affinity partitioning 20 or by tuning the pH. 21 This encapsula- tion method, while effective, results in a wide variety of vesicle sizes and morphologies, such as multilamellar vesicles and vesicles where each phase is additionally encapsulated by separate lipid bilayers. 20 The concentrations of the encapsu- lated polymers and molecules also tend to vary between vesicles, 22 and during osmotic deflation the lipid bilayer is wetted by both phases 23 and becomes unstable, resulting in vesicle budding 24–26 and fission. 27 Formation of ATPS inside of vesicles has not yet been demonstrated in a microfluidic device, although a recent work showed successful encapsulation of a single phase of dextran inside of monodisperse lipid membranes within a micro- channel. 28 Without resorting to lipid membranes 28 or levita- tion, 18 a double emulsion is required to contain an ATPS within a microdroplet. Obtaining even a single emulsion (e.g. a dextran drop inside a continuous PEG phase) within a microfluidic ATPS is not trivial, however, as the low surface tension makes it difficult to break up streams or jets into individual microdroplets. Currently, single emulsion (w/w) microfluidic ATPS have been achieved by electrically 29,30 or mechanically 31,32 perturbing a jet, or by fabricating rounded multi-leveled microchannels to induce flow instabilities. 33 Very recently, all-aqueous (w/w/w) double emulsions have also been obtained in a microchannel using electrical 34 and a Center for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN, 37831-6493, USA. E-mail: [email protected] b Department of Materials Science and Engineering, The University of Tennessee, Knoxville, TN, 37996, USA c Biological and Nanoscale Systems Group, Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN, 37831-6445, USA 3 Electronic supplementary information (ESI) available: Bright field movie sequences corresponding to Fig. 1–2 and 4–6. See DOI: 10.1039/c3lc41122b Lab on a Chip PAPER This journal is ß The Royal Society of Chemistry 2013 Lab Chip, 2013, 13, 1295–1301 | 1295

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Cite this: Lab Chip, 2013, 13, 1295
Aqueous two-phase microdroplets with reversiblephase transitions3
Received 4th October 2012,Accepted 23rd January 2013
DOI: 10.1039/c3lc41122b
www.rsc.org/loc
Jonathan B. Boreyko,a Prachya Mruetusatorn,b Scott T. Rettererac and C.Patrick Collier*a
Aqueous two-phase systems contained within microdroplets enable a bottom-up approach to mimicking
the dynamic microcompartmentation of biomaterial that naturally occurs within the cytoplasm of cells.
Here, we demonstrate the generation of femtolitre aqueous two-phase droplets within a microfluidic oil
channel. Gated pressure pulses were used to generate individual, stationary two-phase microdroplets with
a well-defined time zero for carrying out controlled and sequential phase transformations over time.
Reversible phase transitions between single-phase, two-phase, and core–shell microbead states were
obtained via evaporation-induced dehydration and water rehydration. In contrast to other microfluidic
aqueous two-phase droplets, which require continuous flows and high-frequency droplet formation, our
system enables the controlled isolation and reversible transformation of a single microdroplet and is
expected to be useful for future studies in dynamic microcompartmentation and affinity partitioning.
1 Introduction
Aqueous solutions containing two different polymers can beimmiscible when mixed together above critical concentrations.The resulting interface of aqueous two-phase systems (ATPS)exhibits a remarkably low surface tension (y1–100 mN m21),1,2
allowing for biomolecules to readily traverse between thephases without denaturation. For this reason, ATPS are ofgreat interest for the partitioning and self-assembly ofbiomaterials by exploiting their affinity for one of the polymerphases.3–5 Over the past decade, various microfluidic deviceshave utilized polymer solutions such as poly(ethylene glycol)(PEG) and dextran to generate aqueous two-phase flowscapable of partitioning cells,6–10 proteins,11,12 and DNA.13
Recently there has been much interest in containing ATPSwithin droplets to mimic the differences in local compositionwithin the cytoplasm of cells, known as microcompartmenta-tion.14 This has been motivated by evidence that aqueousphase-separation is an important mechanism for cellularmicrocompartmentation,15,16 including the observation that Pgranules in germ cells are liquid droplets, and therefore someintracellular compartments are entirely aqueous in composi-tion.17 An acoustically levitated aqueous two-phase droplet was
used to demonstrate the affinity partitioning of biotinylatedliposomes,18 however the millimetric droplet size is incom-mensurate with cellular length scales. Synthetic cells havebeen developed by encapsulating ATPS inside of sphericallipid bilayer membranes.19 In these systems, proteins could bepreferentially sorted into the PEG or dextran-rich phase viaaffinity partitioning20 or by tuning the pH.21 This encapsula-tion method, while effective, results in a wide variety of vesiclesizes and morphologies, such as multilamellar vesicles andvesicles where each phase is additionally encapsulated byseparate lipid bilayers.20 The concentrations of the encapsu-lated polymers and molecules also tend to vary betweenvesicles,22 and during osmotic deflation the lipid bilayer iswetted by both phases23 and becomes unstable, resulting invesicle budding24–26 and fission.27
Formation of ATPS inside of vesicles has not yet beendemonstrated in a microfluidic device, although a recent workshowed successful encapsulation of a single phase of dextraninside of monodisperse lipid membranes within a micro-channel.28 Without resorting to lipid membranes28 or levita-tion,18 a double emulsion is required to contain an ATPSwithin a microdroplet. Obtaining even a single emulsion (e.g. adextran drop inside a continuous PEG phase) within amicrofluidic ATPS is not trivial, however, as the low surfacetension makes it difficult to break up streams or jets intoindividual microdroplets. Currently, single emulsion (w/w)microfluidic ATPS have been achieved by electrically29,30 ormechanically31,32 perturbing a jet, or by fabricating roundedmulti-leveled microchannels to induce flow instabilities.33
Very recently, all-aqueous (w/w/w) double emulsions havealso been obtained in a microchannel using electrical34 and
aCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak
Ridge, TN, 37831-6493, USA. E-mail: [email protected] of Materials Science and Engineering, The University of Tennessee,
Knoxville, TN, 37996, USAcBiological and Nanoscale Systems Group, Biosciences Division, Oak Ridge National
Laboratory, Oak Ridge, TN, 37831-6445, USA
3 Electronic supplementary information (ESI) available: Bright field moviesequences corresponding to Fig. 1–2 and 4–6. See DOI: 10.1039/c3lc41122b
Lab on a Chip
PAPER
This journal is � The Royal Society of Chemistry 2013 Lab Chip, 2013, 13, 1295–1301 | 1295
mechanical32,35 pulsations. Interestingly, both PEG-in-dextran-in-PEG and dextran-in-PEG-in-dextran systems were possible,although these types of all-aqueous double emulsions areinherently unstable and break down to their phase separatedstates within seconds35 or minutes.34 A dextran-in-PEG-in-oil(w/w/o) ATPS, on the other hand, is both stable and conduciveto droplet pinch-off due to the large surface tension of thewater/oil interface. Previously, aqueous two-phase droplets inan oil microchannel have been obtained using aT-junction36,37 or flow-focusing cross junctions.38
Here, we report the generation of femtolitre-volumeaqueous two-phase droplets in an oil microchannel. Incontrast to other reports of aqueous two-phase microdroplets,which rely on continuous flows/jets and high-frequencydroplet formation,32,34–38 our device exploits the interfacialtension between the oil and aqueous phases to generateultrasmall two-phase droplets with a well-defined time zeroand without crossflow in the oil phase. This allows us tomonitor individual droplets for extended times, and to carryout programmed sequential phase transitions on the samedroplet. Due to the large surface area to volume ratio of themicrodroplets, we observe that single-phase droplets withinitially low polymer concentrations transition to two-phasedroplets during evaporation, and subsequently transition yetagain to core–shell microbeads. These phase transitions arefully reversible by rehydration via fusion with an additionaldroplet of pure water. The controlled generation and inter-conversion of single-phase and two-phase microdroplets isexpected to be of interest for dynamic microcompartmentationand affinity partitioning, and the reversible creation of core–shell microbeads may be of interest for the controlled deliveryof encapsulated biomaterials.
2 Experimental
The microfluidic devices were composed of poly(dimethylsi-loxane) (PDMS) bonded to PDMS-coated glass cover slips andwere fabricated using the same procedure detailed in previousreports.39,40 To allow for droplet fusion, two opposing aqueousside-channels (1 mm 6 1 mm, see Fig. 1b–c) connect to eitherside of a central oil channel of larger dimensions (14.6 mmwide 6 18 mm high). The generation and fusion of dropletswas obtained by applying pressure pulses to the side-channels;the abrupt change in channel height caused droplet formationto occur by virtue of shape-induced shear when the backingpressure exceeded the capillary pressure of the 1 mm 6 1 mmside-channels (124–131 kPa for pure water).39
PEG 35 kDa and dextran 100 kDa polymers (Sigma-Aldrich)were mixed with deionized water at varying concentrationsand injected into the side-channels. In some experiments,PEG-only and dextran-only solutions were injected in theirrespective side-channels, while in other experiments a solutioncontaining both PEG and dextran was in one side-channel toallow for pure deionized water in the other. Purified soybeanoil (Sigma-Aldrich) was always used as the continuous phase inthe central channel. The cross-flow in the oil channel wasusually set to zero by balancing the slight hydrostatic pressurewith a constant backing pressure; however, in select experi-ments a cross-flow was intentionally introduced by increasingthe backing pressure. Bright field images were acquired usingan inverted optical microscope (Eclipse TE 300, NikonInstruments) and a CCD camera (CoolSNAP HQ, RoperScientific). For all experiments, the temperature was held toa constant 25 uC in a microscope cage incubator (OkolabH201).
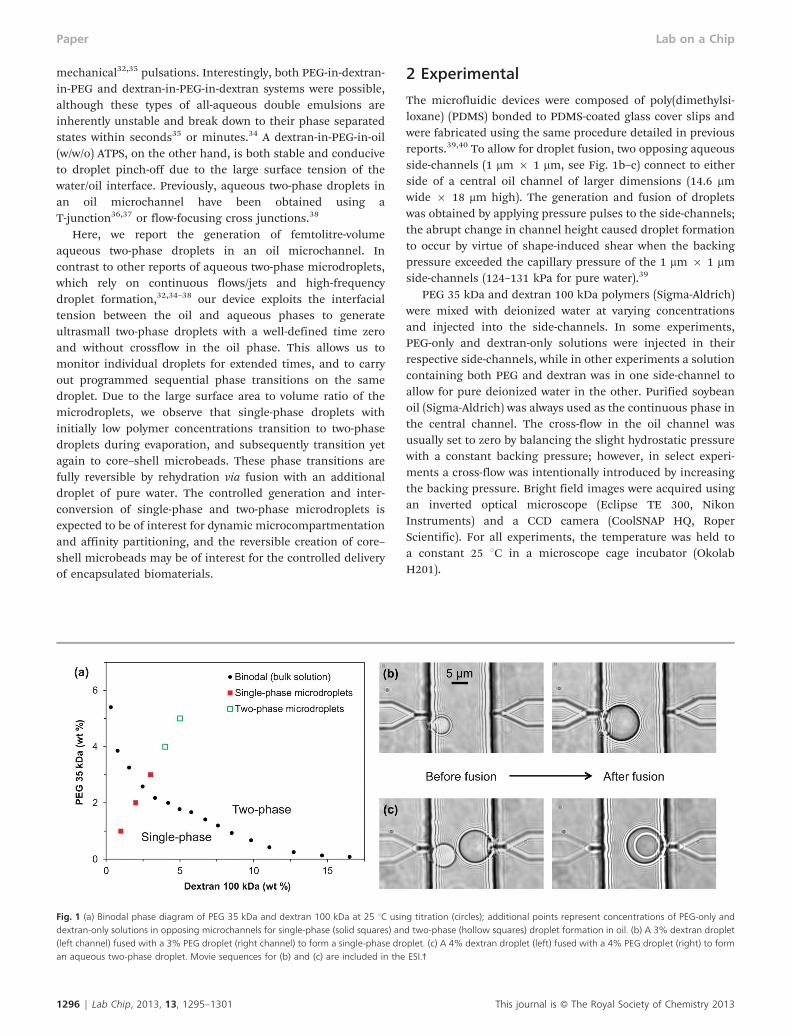
Fig. 1 (a) Binodal phase diagram of PEG 35 kDa and dextran 100 kDa at 25 uC using titration (circles); additional points represent concentrations of PEG-only anddextran-only solutions in opposing microchannels for single-phase (solid squares) and two-phase (hollow squares) droplet formation in oil. (b) A 3% dextran droplet(left channel) fused with a 3% PEG droplet (right channel) to form a single-phase droplet. (c) A 4% dextran droplet (left) fused with a 4% PEG droplet (right) to forman aqueous two-phase droplet. Movie sequences for (b) and (c) are included in the ESI.3
1296 | Lab Chip, 2013, 13, 1295–1301 This journal is � The Royal Society of Chemistry 2013
Paper Lab on a Chip

3 Results and discussion
3.1 Generation of single-phase and two-phase microdroplets
A bulk phase diagram for aqueous mixtures of PEG 35 kDa anddextran 100 kDa was constructed using cloud point titration2,3
(circular data points in Fig. 1a). Dextran solutions wereprepared in 10 mL test tubes with concentrations rangingfrom 0–20 wt%, and a stock solution of 10 or 20 wt% PEG wasadded dropwise until each solution became turbid uponshaking. The critical concentrations of PEG and dextrancompose the binodal line, above which phase separationoccurs and below which a single-phase mixture results.
Using the binodal as a guideline, both single-phase and two-phase droplets were obtained in the oil microchannel.Separate PEG and dextran solutions were inserted into theirrespective side-channels, and their concentrations were variedfrom 1–5 wt% to cover both sides of the binodal. A pressurepulse was initialized in both side-channels to actuate a PEGdrop and a dextran drop in the oil channel, and the resultingdroplet coalescence and phase behavior were observed andcharacterized. To minimize evaporation effects, the PDMSchip was saturated with deionized water by soaking it for atleast 24 h prior to experiments. Initial polymer concentrationsof 3% or smaller resulted in single-phase behavior uponcoalescence (Fig. 1b), while phase separation with a dextran-rich droplet inside of a PEG-rich droplet occurred forconcentrations of 4% and above (Fig. 1c). It was confirmedthat the inner drop was always the dextran-rich phase bycombining a PEG drop and a dextran drop of differingvolumes, in which case the volume of the inner phase alwayscorresponded to that of the initial dextran drop.
The initial concentrations of PEG and dextran in the side-channels will not be exactly the same as their final concentra-tions in a merged drop with larger water volume. This isparticularly true for non-partitioned single-phase droplets,where the polymer concentrations will become roughly halvedupon coalescence. This is why the 3% PEG and dextranconcentrations used in the side-channels were single-phaseupon droplet fusion, despite these initial concentrations beingslightly over the binodal (Fig. 1a). Regardless, the overall phasebehavior of fused droplets in the microchannel was wellapproximated by the bulk phase diagram, and for simplicity allpolymer concentrations will be referring to the well-controlledinitial concentrations injected into the inlet reservoirs of theside-channels.
The aqueous two-phase microdroplets generated here areunique in several respects. After coalescence, the diameter of adroplet typically ranged from 5 to 15 mm, depending on theamplitude and duration of the applied pressure pulses. Thisresults in aqueous two-phase droplet volumes of V y 100 fL,on the order of typical cellular volumes and one,38 three,36 andfive37 orders of magnitude smaller than previously reportedaqueous two-phase droplets in oil channels. The small dropletsize also promotes spontaneous mixing and phase separation,as the surface energy released upon coalescence becomesincreasingly significant at micrometric length scales andresults in dramatic droplet oscillations.41 The complete mixingof two microdroplets upon coalescence was previously
observed to occur in less than 1 ms,40 an order of magnitudefaster than the diffusive mixing time scale and in goodagreement with the near-instantaneous phase separationobserved here upon droplet fusion. This is in contrast toprevious reports, which typically required serpentine channelsto achieve complete mixing and phase separation of largeraqueous two-phase droplets.36,38 Finally, while other devicesrequired continuous flows to generate aqueous two-phasedroplets,32,34–38 the pressure pulses used here allow for thecontrolled formation of a single, stationary two-phase dropletwith a well-defined time zero. This enables a controlled studyof droplet behavior over extended times and can be used toobserve dynamic phase transitions.
3.2 Evaporation-induced phase transitions
With static single-phase and two-phase droplets reliablygenerated in a microchannel, it is of interest to investigatethe possibility of phase transitions occurring over time. Apassive technique for inducing a single-phase to two-phasetransition is evaporation, which causes polymer concentra-tions to gradually increase until they cross the binodal lineand phase separate.18,20,35 This approach is especially optimalfor the femtolitre-sized droplets used here, for example adroplet with a 6 mm diameter has a surface area to volumeratio of 1 mm21. In general, evaporation is enabled by the smallamounts of water molecules that are able to diffuse from themicrodroplets into the oil, where the maximum possibleconcentration of water depends on the type of oil used (#0.3volume% in soybean oil).42 The permeability of PDMS allowsfor the subsequent transport of the water vapor through thewalls of the device due to an osmotic pressure difference.43 Inthe future, the evaporation rate of the microdroplets could betuned or even reversed to condensation (swelling) by control-ling the osmotic pressure across the PDMS with an externalreservoir.44–46 Here, dry PDMS samples were utilized in placeof the water-soaked samples used in the previous section,which increased the droplet shrinkage rate from y10 mm2
min21 to y100 mm2 min21 at 25 uC by increasing the osmoticpressure difference. When microdroplets with the largesurface area to volume ratios used here evaporated at thisrate, dramatic evaporation-induced phase transitions occurredin a brief time span of only a few minutes (Fig. 2).
To ensure that an actuated droplet would initially be wellwithin the single-phase regime, an aqueous solution contain-ing low concentrations of 1% PEG and 1% dextran was used inone of the side-channels. Note that this simplified approachdiffers from the technique in the previous section of using aPEG solution in one side-channel and a dextran solution in theopposing side-channel (Fig. 1), which was necessary at higherpolymer concentrations to prevent phase separation fromoccurring within a side-channel before droplet actuation. Asingle-phase droplet was generated from the side-channel intothe static oil channel (Fig. 2a), and three distinct types ofphase transitions were observed over a five minute evaporationtime as polymer concentrations increased. First, multipledextran droplets began to nucleate within the droplet,indicating the beginnings of phase separation (Fig. 2b).Second, the dextran drops continued to grow in size andcoalesce together, culminating in complete phase separation
This journal is � The Royal Society of Chemistry 2013 Lab Chip, 2013, 13, 1295–1301 | 1297
Lab on a Chip Paper
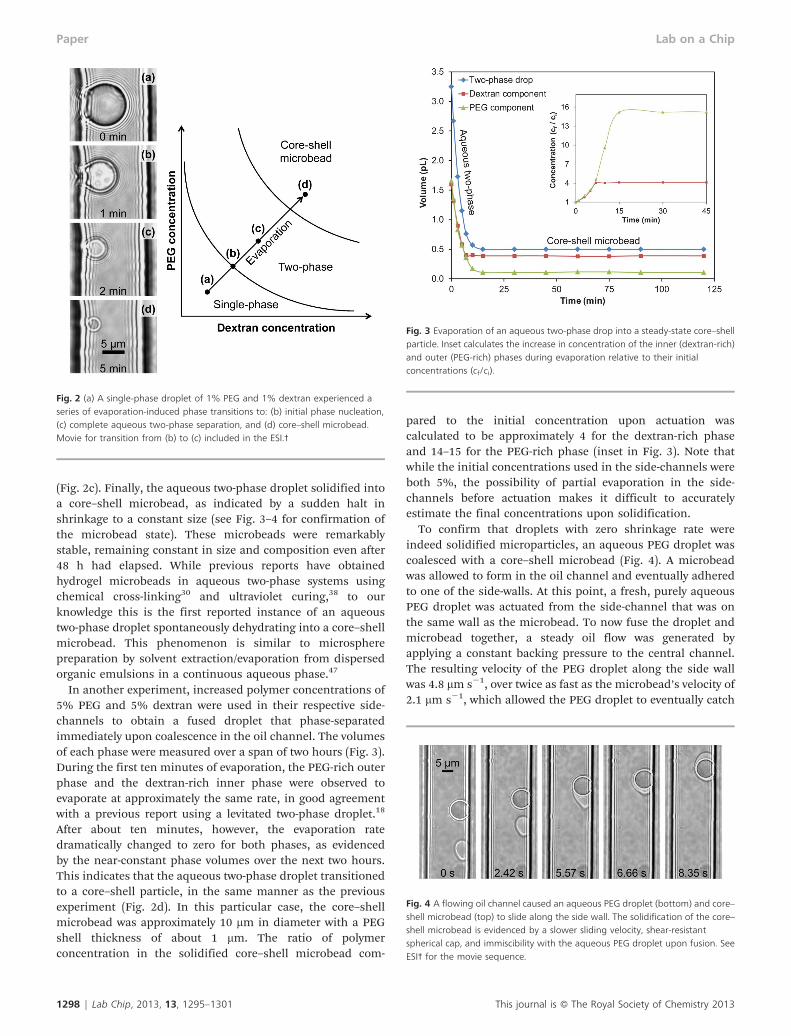
(Fig. 2c). Finally, the aqueous two-phase droplet solidified intoa core–shell microbead, as indicated by a sudden halt inshrinkage to a constant size (see Fig. 3–4 for confirmation ofthe microbead state). These microbeads were remarkablystable, remaining constant in size and composition even after48 h had elapsed. While previous reports have obtainedhydrogel microbeads in aqueous two-phase systems usingchemical cross-linking30 and ultraviolet curing,38 to ourknowledge this is the first reported instance of an aqueoustwo-phase droplet spontaneously dehydrating into a core–shellmicrobead. This phenomenon is similar to microspherepreparation by solvent extraction/evaporation from dispersedorganic emulsions in a continuous aqueous phase.47
In another experiment, increased polymer concentrations of5% PEG and 5% dextran were used in their respective side-channels to obtain a fused droplet that phase-separatedimmediately upon coalescence in the oil channel. The volumesof each phase were measured over a span of two hours (Fig. 3).During the first ten minutes of evaporation, the PEG-rich outerphase and the dextran-rich inner phase were observed toevaporate at approximately the same rate, in good agreementwith a previous report using a levitated two-phase droplet.18
After about ten minutes, however, the evaporation ratedramatically changed to zero for both phases, as evidencedby the near-constant phase volumes over the next two hours.This indicates that the aqueous two-phase droplet transitionedto a core–shell particle, in the same manner as the previousexperiment (Fig. 2d). In this particular case, the core–shellmicrobead was approximately 10 mm in diameter with a PEGshell thickness of about 1 mm. The ratio of polymerconcentration in the solidified core–shell microbead com-
pared to the initial concentration upon actuation wascalculated to be approximately 4 for the dextran-rich phaseand 14–15 for the PEG-rich phase (inset in Fig. 3). Note thatwhile the initial concentrations used in the side-channels wereboth 5%, the possibility of partial evaporation in the side-channels before actuation makes it difficult to accuratelyestimate the final concentrations upon solidification.
To confirm that droplets with zero shrinkage rate wereindeed solidified microparticles, an aqueous PEG droplet wascoalesced with a core–shell microbead (Fig. 4). A microbeadwas allowed to form in the oil channel and eventually adheredto one of the side-walls. At this point, a fresh, purely aqueousPEG droplet was actuated from the side-channel that was onthe same wall as the microbead. To now fuse the droplet andmicrobead together, a steady oil flow was generated byapplying a constant backing pressure to the central channel.The resulting velocity of the PEG droplet along the side wallwas 4.8 mm s21, over twice as fast as the microbead’s velocity of2.1 mm s21, which allowed the PEG droplet to eventually catch
Fig. 2 (a) A single-phase droplet of 1% PEG and 1% dextran experienced aseries of evaporation-induced phase transitions to: (b) initial phase nucleation,(c) complete aqueous two-phase separation, and (d) core–shell microbead.Movie for transition from (b) to (c) included in the ESI.3
Fig. 3 Evaporation of an aqueous two-phase drop into a steady-state core–shellparticle. Inset calculates the increase in concentration of the inner (dextran-rich)and outer (PEG-rich) phases during evaporation relative to their initialconcentrations (cf/ci).
Fig. 4 A flowing oil channel caused an aqueous PEG droplet (bottom) and core–shell microbead (top) to slide along the side wall. The solidification of the core–shell microbead is evidenced by a slower sliding velocity, shear-resistantspherical cap, and immiscibility with the aqueous PEG droplet upon fusion. SeeESI3 for the movie sequence.
1298 | Lab Chip, 2013, 13, 1295–1301 This journal is � The Royal Society of Chemistry 2013
Paper Lab on a Chip

up and fuse with the microbead. Furthermore, the shape of thePEG droplet noticeably sheared during oil flow, in starkcontrast to the microbead which remained spherical. Finally,upon coalescence the PEG droplet surrounded the core–shellmicrobead but the two were unable to mix together, a behaviornever observed for purely aqueous coalescence events. Takentogether, these observations strongly indicate that a transitionto a microbead state had indeed occurred. This is corroboratedby the additional observation that PEG and/or dextransolutions, when left static inside of the 1 mm 6 1 mm side-
channels for a long enough period of time, eventuallyevaporated into solidified clogs that could not be cleared evenwith strong backing pressures of several atmospheres.
3.3 Hydration-induced reversible phase transitions
Evaporation led to a series of phase transitions from single-phase to two-phase to core–shell particles, but some type ofhydration mechanism is required for reversible phase transi-tions in the opposite direction.20 A mixture containing both1% PEG and 1% dextran was placed in one of the side-channels, which allowed pure deionized water to be utilized inthe opposing side-channel for hydration experiments. As withthe previous section, dry PDMS samples were used tomaximize the evaporation rate and accelerate phase transi-tions to a solidified state.
A large droplet was actuated from the polymer side-channel,and was subsequently allowed to evaporate until phaseseparation occurred. Due to the large volume of the outerPEG phase, the inner dextran droplet was the first to transitioninto a particle during continued evaporation, at which pointadditional solution was injected to obtain two immiscibledextran particles inside of a PEG microbead after completeevaporation. A pure water droplet was then actuated from theother side-channel (t = 0 ms in Fig. 5), which upon coalescence(121 ms) immediately transitioned the core–shell particle intoan aqueous two-phase droplet (242 ms), as evidenced by thesudden miscibility and coalescence of the inner dextranphases. Finally, the aqueous two-phase droplet was allowedto evaporate back into a microbead (77 min), which now has asingle dextran core instead of two due to their fusion duringhydration. This dehydration and rehydration cycle wasperformed four times in succession on the same droplet(movies included in ESI3).
By decreasing the volume of a microbead and increasing thevolume of an actuated water droplet, a complete hydrationtransition back to a single-phase aqueous droplet was alsoobserved (Fig. 6). As with the previous hydration experiment,the phase transition occurred immediately after coalescence.Approximately one minute after hydration, the dextran phasebegan to nucleate within the droplet due to evaporation(similar to Fig. 2b), and after ten minutes transitioned backinto a microbead. Interestingly, in this particular case thedextran concentration and droplet volume were small enoughthat solidification into a microbead occurred before the phase
Fig. 5 A microbead with two dextran cores and a PEG shell (left) was fused witha pure water droplet (right) to hydrate back into an aqueous two-phase droplet.The two-phase droplet eventually transitioned back into a core–shell microbeadafter evaporation. Movie sequences included in the ESI.3
Fig. 6 A microbead engulfed in a large water droplet transitioned back into a single-phase aqueous droplet. Subsequent evaporation resulted in partial phaseseparation and a return to a solidified state. Movie sequences included in the ESI.3
This journal is � The Royal Society of Chemistry 2013 Lab Chip, 2013, 13, 1295–1301 | 1299
Lab on a Chip Paper

separation was fully completed, as indicated by the multipledextran cores present within the PEG shell.
These microbeads are fundamentally different from thehydrogel capsules48 and hydrogel microbeads30,38 that haverecently been obtained with microfluidic ATPS. Hydrogels areformed by some type of polymerization,49 such as chemicalcross-linking30 and/or UV curing,38 and remain stable evenafter prolonged immersion in water.30 The core–shell particlesobtained here, on the other hand, are simply the physicalsolidification of aqueous PEG and dextran solutions due toevaporation increasing the polymer concentration to a criticallevel. As a result, these microbeads may be immediatelytransformed back into an aqueous phase when hydrated withwater (Fig. 5–6). While the microbeads generated here wereisolated to study reversible phase transitions, applying acontinuous flow could generate a large number of microbeadsfor collection, provided that the channel is long enough toenable the evaporation-induced phase transition from theaqueous state.
4 Conclusion
We demonstrated the generation of aqueous two-phasemicrodroplets in an oil microchannel. In contrast to otheraqueous two-phase microdroplets, which require continuousflows and high-frequency droplet formation, our system allowsfor the controlled production of an isolated two-phasemicrodroplet with no cross flow and a well-defined time zero.The large surface area to volume ratio of the femtolitredroplets was exploited to passively obtain evaporation-inducedphase transitions from single-phase to two-phase to core–shellmicrobead states. Reversible transitions from a core–shellmicrobead back to an aqueous two-phase or single-phasedroplet were achieved by rehydration via fusion with anadditional water droplet.
The small femtolitre volume, static positioning, andreversible single-phase and two-phase states of our micro-droplets provides an ideal system for future studies in thedynamic microcompartmentation and affinity partitioning ofbiomaterials. Furthermore, the reversible transitions betweenaqueous two-phase and solidified core–shell states couldpotentially be used for the controlled delivery of biomaterials.
Acknowledgements
This research was conducted at the Center for NanophaseMaterials Sciences, which is sponsored at Oak Ridge NationalLaboratory by the Scientific User Facilities Division, Office ofBasic Energy Sciences, U. S. Department of Energy.
References
1 J. Ryden and P. A. Albertsson, J. Colloid Interface Sci., 1971,37, 219–222.
2 Y. Liu, R. Lipowsky and R. Dimova, Langmuir, 2012, 28,3831–3839.
3 P. A. Albertsson, Partitioning of Cell Particles andMacromolecules, Wiley, New York, 1986.
4 H. Walter, G. Johansson and D. E. Brooks, Anal. Biochem.,1991, 197, 1–18.
5 A. M. Azevedo, P. A. J. Rosa, I. F. Ferreira and M. R. Aires-Barros, Trends Biotechnol., 2009, 27, 240–247.
6 M. Yamada, V. Kasim, M. Nakashima, J. Edahiro andM. Seki, Biotechnol. Bioeng., 2004, 88, 489–494.
7 K. H. Nam, W. J. Chang, H. Hong, S. M. Lim, D. I. Kim andY. M. Koo, Biomed. Microdevices, 2005, 7, 189–195.
8 M. Tsukamoto, S. Taira, S. Yamamura, Y. Morita,N. Nagatani, Y. Takamura and E. Tamiya, Analyst, 2009,134, 1994–1998.
9 J. R. SooHoo and G. M. Walker, Biomed. Microdevices, 2009,11, 323–329.
10 J. P. Frampton, D. Lai, H. Sriram and S. Takayama, Biomed.Microdevices, 2011, 13, 1043–1051.
11 G. Munchow, S. Hardt, J. P. Kutter and K. S. Drese, LabChip, 2007, 7, 98–102.
12 R. J. Meagher, Y. K. Light and A. K. Singh, Lab Chip, 2008,8, 527–532.
13 T. Hahn and S. Hardt, Soft Matter, 2011, 7, 6320–6326.14 D. P. Jones, Microcompartmentation, CRC, Boca Raton, FL,
1988.15 H. Walter and D. E. Brooks, FEBS Lett., 1995, 361, 135–139.16 X. Ge, A. J. Conley, J. E. Brandle, R. Truant and C. D.
M. Filipe, J. Am. Chem. Soc., 2009, 131, 9094–9099.17 C. P. Brangwynne, C. R. Eckmann, D. S. Courson,
A. Rybarska, C. Hoege, J. Gharakhani, F. Julicher and A.A. Hyman, Science, 2009, 324, 1729–1732.
18 S. Santesson, I. B. R. Ramirez, P. Viberg, B. Jergil andS. Nilsson, Anal. Chem., 2004, 76, 303–308.
19 M. R. Helfrich, L. K. Mangeney-Slavin, M. S. Long, K.Y. Djoko and C. D. Keating, J. Am. Chem. Soc., 2002, 124,13374–13375.
20 M. S. Long, C. D. Jones, M. R. Helfrich, L. K. Mangeney-Slavin and C. D. Keating, Proc. Natl. Acad. Sci. U. S. A., 2005,102, 5920–5925.
21 L. M. Dominak, E. L. Gundermann and C. D. Keating,Langmuir, 2010, 26, 5697–5705.
22 L. M. Dominak and C. D. Keating, Langmuir, 2007, 23,7148–7154.
23 Y. Li, R. Lipowsky and R. Dimova, J. Am. Chem. Soc., 2008,130, 12252–12253.
24 M. S. Long, A. S. Cans and C. D. Keating, J. Am. Chem. Soc.,2008, 130, 756–762.
25 Y. Li, R. Lipowsky and R. Dimova, Proc. Natl. Acad. Sci. U. S.A., 2011, 108, 4731–4736.
26 Y. Li, H. Kusumaatmaja, R. Lipowsky and R. Dimova, J.Phys. Chem. B, 2012, 116, 1819–1823.
27 M. Andes-Koback and C. D. Keating, J. Am. Chem. Soc.,2011, 133, 9545–9555.
28 S. Matosevic and B. M. Paegel, J. Am. Chem. Soc., 2011, 133,2798–2800.
29 Y. S. Song, Y. H. Choi and D. H. Kim, J. Chromatogr., A,2007, 1162, 180–186.
30 I. Ziemecka, V. van Steijn, G. J. M. Koper, M. Rosso, A.M. Brizard, J. H. van Esch and M. T. Kreutzer, Lab Chip,2011, 11, 620–624.
1300 | Lab Chip, 2013, 13, 1295–1301 This journal is � The Royal Society of Chemistry 2013
Paper Lab on a Chip

31 H. C. Shum, J. Varnell and D. A. Weitz, Biomicrofluidics,2012, 6, 012808.
32 A. Sauret and H. C. Shum, Appl. Phys. Lett., 2012, 100,154106.
33 D. Lai, J. P. Frampton, H. Sriram and S. Takayama, LabChip, 2011, 11, 3551–3554.
34 I. Ziemecka, V. van Steijn, G. J. M. Koper and M.T. Kreutzer, Soft Matter, 2011, 7, 9878–9880.
35 Y. Song and H. C. Shum, Langmuir, 2012, 28, 12054–12059.36 K. Vijayakumar, S. Gulati, A. J. deMello and J. B. Edel,
Chem. Sci., 2010, 1, 447–452.37 S. H. S. Lee, P. Wang, S. K. Yap, T. A. Hatton and S. A. Khan,
Biomicrofluidics, 2012, 6, 022005.38 S. Ma, J. Thiele, X. Liu, Y. Bai, C. Abell and W. T. S. Huck,
Small, 2012, 8, 2356–2360.39 S. Y. Jung, S. T. Retterer and C. P. Collier, Lab Chip, 2010,
10, 2688–2694.40 S. Y. Jung, S. T. Retterer and C. P. Collier, Lab Chip, 2010,
10, 3373–3376.41 J. B. Boreyko and C. H. Chen, Phys. Rev. Lett., 2009, 103,
184501.
42 M. He, C. Sun and D. T. Chiu, Anal. Chem., 2004, 76,1222–1227.
43 T. C. Merkel, V. I. Bondar, K. Nagai, B. D. Freeman andI. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 2000, 38,415–434.
44 C. L. Hansen, S. Classen, J. M. Berger and S. R. Quake, J.Am. Chem. Soc., 2006, 128, 3142–3143.
45 J. U. Shim, G. Cristobal, D. R. Link, T. Thorsen, Y. Jia,K. Piattelli and S. Fraden, J. Am. Chem. Soc., 2007, 129,8825–8835.
46 J. U. Shim, S. N. Patil, J. T. Hodgkinson, S. D. Bowden, D.R. Spring, M. Welch, W. T. S. Huck, F. Hollfelder andC. Abell, Lab Chip, 2011, 11, 1132–1137.
47 S. Freitas, H. P. Merkle and B. Gander, J. Controlled Release,2005, 102, 313–332.
48 L. K. Fiddes, E. W. K. Young, E. Kumacheva and A.R. Wheeler, Lab Chip, 2007, 7, 863–867.
49 E. Tumarkin and E. Kumacheva, Chem. Soc. Rev., 2009, 38,2161–2168.
This journal is � The Royal Society of Chemistry 2013 Lab Chip, 2013, 13, 1295–1301 | 1301
Lab on a Chip Paper
Related Documents











