ORIGINAL ARTICLE Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway CG Concannon 1 , BF Koehler 1,2 , Claus Reimertz 2 , BM Murphy 1 , C Bonner 1 , N Thurow 2 , MW Ward 1 , A Villunger 3 , A Strasser 4 , D Ko¨gel 2,5 and JHM Prehn 1,5 1 Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin, Ireland; 2 Experimental Neurosurgery, Centre for Neurology and Neurosurgery, Johann Wolfgang Goethe University Clinics, Theodor-Stern-Kai 7, Frankfurt/Main, Germany; 3 Division of Experimental Pathophysiology and Immunology, Biocenter, Innsbruck Medical University, Innsbruck, Austria and 4 The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia The proteasome has emerged as a novel target for antineoplastic treatment of hematological malignancies and solid tumors, including those of the central nervous system. To identify cell death pathways activated in response to inhibition of the proteasome system in cancer cells, we treated human SH-SY5Y neuroblastoma cells with the selective proteasome inhibitor (PI) epoxomicin (Epoxo). Prolonged exposure to Epoxo was associated with increased levels of poly-ubiquitinylated proteins and p53, release of cytochrome c from the mitochondria, and activation of caspases. Analysis of global gene expression using high-density oligonucleotide microarrays revealed that Epoxo triggered transcriptional activation of the two Bcl-2-homology domain-3-only (BH3-only) genes p53 upregulated modulator of apoptosis (PUMA) and Bim. Subsequent studies in PUMA- and Bim-deficient cells indicated that Epoxo-induced caspase activation and apoptosis was predominantly PUMA-dependent. Further characterization of the transcriptional response to Epoxo in HCT116 human colon cancer cells demonstrated that PUMA induction was p53-dependent; with deficiency in either p53 or PUMA significantly protected HCT116 cells against Epoxo-induced apoptosis. Our data suggest that p53 activation and the transcriptional induction of its target gene PUMA play an important role in the sensitivity of cancer cells to apoptosis induced by proteasome inhibition, and imply that antineoplastic therapies with PIs might be especially useful in cancers with functional p53. Oncogene (2007) 26, 1681–1692. doi:10.1038/sj.onc.1209974; published online 18 September 2006 Keywords: proteasome; apoptosis; epoxomicin; Bcl-2 family; BH3-only protein; p53 Introduction The correct functioning of the ubiquitin-proteasome pathway is essential for the degradation of the majority of intracellular proteins. Several key regulatory proteins involved in cell proliferation and differentiation are regulated by proteasome-mediated proteolysis resulting in the activation or inhibition of specific cell signaling pathways (Adams, 2004a). The proteasome is also central to the regulation of cell death and apoptosis. The proapoptotic Bcl-2 family proteins Bim, Bik and Bid, which regulate the release of proapoptotic factors such as cytochrome c and Smac from mitochondria, are known substrates for targeted ubiquitination and degradation (Breitschopf et al., 2000; Marshansky et al., 2001; Ley et al., 2003). Indeed, caspases, the key proteases activated during apoptosis, are also regulated by the proteasome. An endogenous caspase inhibitor family of proteins, the inhibitor of apoptosis (IAPs), not only inhibit active caspases but also target them for destruction by acting as ubiquitin ligases and promoting polyubiquitination of activate caspases (Huang et al., 2000; Suzuki et al., 2001). In recent years modulation of proteasomal function with specific inhibitors has evolved as a novel target for the treatment of cancers such as multiple myeloma (Chauhan et al., 2005) as well as lung, colon and prostate cancer (Aghajanian et al., 2002; Papandreou et al., 2004; Chauhan et al., 2005). Proteasome inhibitors (PIs) have been demonstrated to overcome chemoresistance of tumor cells by enhancing chemo- sensitivity and even acting in synergy with other agents to induce apoptotic cell death of tumor cells (Adams, 2002; Park and Lenz, 2004). Indeed, clinical efficiency of Bortezomib (PS-341/Velcade), a reversible PI has recently been observed in multiple myeloma patients (Richardson et al., 2003). Inhibitors with a broader specificity and irreversible binding have likewise been identified and are currently being tested in preclinical trials (Melino, 2005). One such inhibitor is epoxomicin (Epoxo), a naturally found a 0 , b 0 epoxyketone peptide, which is a highly selective and irreversible inhibitor of the chymotryptic-, tryptic- and post-glutamyl peptidyl Received 29 March 2006; revised 27 July 2006; accepted 1 August 2006; published online 18 September 2006 Correspondence: Professor JHM Prehn, Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, 123 St Stephen’s Green, Dublin 2, Ireland. E-mail: [email protected] 5 These authors share equal senior authorship. Oncogene (2007) 26, 1681–1692 & 2007 Nature Publishing Group All rights reserved 0950-9232/07 $30.00 www.nature.com/onc

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ORIGINAL ARTICLE
Apoptosis induced by proteasome inhibition in cancer cells: predominantrole of the p53/PUMA pathway
CG Concannon1, BF Koehler1,2, Claus Reimertz2, BM Murphy1, C Bonner1, N Thurow2,MW Ward1, A Villunger3, A Strasser4, D Kogel2,5 and JHM Prehn1,5
1Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin, Ireland; 2Experimental Neurosurgery,Centre for Neurology and Neurosurgery, Johann Wolfgang Goethe University Clinics, Theodor-Stern-Kai 7, Frankfurt/Main,Germany; 3Division of Experimental Pathophysiology and Immunology, Biocenter, Innsbruck Medical University, Innsbruck, Austriaand 4The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia
The proteasome has emerged as a novel target forantineoplastic treatment of hematological malignanciesand solid tumors, including those of the central nervoussystem. To identify cell death pathways activated inresponse to inhibition of the proteasome system in cancercells, we treated human SH-SY5Y neuroblastoma cellswith the selective proteasome inhibitor (PI) epoxomicin(Epoxo). Prolonged exposure to Epoxo was associatedwith increased levels of poly-ubiquitinylated proteins andp53, release of cytochrome c from the mitochondria, andactivation of caspases. Analysis of global gene expressionusing high-density oligonucleotide microarrays revealedthat Epoxo triggered transcriptional activation of the twoBcl-2-homology domain-3-only (BH3-only) genes p53upregulated modulator of apoptosis (PUMA) and Bim.Subsequent studies in PUMA- and Bim-deficient cellsindicated that Epoxo-induced caspase activation andapoptosis was predominantly PUMA-dependent. Furthercharacterization of the transcriptional response to Epoxoin HCT116 human colon cancer cells demonstrated thatPUMA induction was p53-dependent; with deficiency ineither p53 or PUMA significantly protected HCT116cells against Epoxo-induced apoptosis. Our data suggestthat p53 activation and the transcriptional induction ofits target gene PUMA play an important role inthe sensitivity of cancer cells to apoptosis induced byproteasome inhibition, and imply that antineoplastictherapies with PIs might be especially useful in cancerswith functional p53.Oncogene (2007) 26, 1681–1692. doi:10.1038/sj.onc.1209974;published online 18 September 2006
Keywords: proteasome; apoptosis; epoxomicin; Bcl-2family; BH3-only protein; p53
Introduction
The correct functioning of the ubiquitin-proteasomepathway is essential for the degradation of the majorityof intracellular proteins. Several key regulatory proteinsinvolved in cell proliferation and differentiation areregulated by proteasome-mediated proteolysis resultingin the activation or inhibition of specific cell signalingpathways (Adams, 2004a). The proteasome is alsocentral to the regulation of cell death and apoptosis.The proapoptotic Bcl-2 family proteins Bim, Bik andBid, which regulate the release of proapoptotic factorssuch as cytochrome c and Smac from mitochondria, areknown substrates for targeted ubiquitination anddegradation (Breitschopf et al., 2000; Marshanskyet al., 2001; Ley et al., 2003). Indeed, caspases, the keyproteases activated during apoptosis, are also regulatedby the proteasome. An endogenous caspase inhibitorfamily of proteins, the inhibitor of apoptosis (IAPs), notonly inhibit active caspases but also target them fordestruction by acting as ubiquitin ligases and promotingpolyubiquitination of activate caspases (Huang et al.,2000; Suzuki et al., 2001).
In recent years modulation of proteasomal functionwith specific inhibitors has evolved as a novel target forthe treatment of cancers such as multiple myeloma(Chauhan et al., 2005) as well as lung, colon andprostate cancer (Aghajanian et al., 2002; Papandreouet al., 2004; Chauhan et al., 2005). Proteasomeinhibitors (PIs) have been demonstrated to overcomechemoresistance of tumor cells by enhancing chemo-sensitivity and even acting in synergy with other agentsto induce apoptotic cell death of tumor cells (Adams,2002; Park and Lenz, 2004). Indeed, clinical efficiencyof Bortezomib (PS-341/Velcade), a reversible PI hasrecently been observed in multiple myeloma patients(Richardson et al., 2003). Inhibitors with a broaderspecificity and irreversible binding have likewise beenidentified and are currently being tested in preclinicaltrials (Melino, 2005). One such inhibitor is epoxomicin(Epoxo), a naturally found a0, b0 epoxyketone peptide,which is a highly selective and irreversible inhibitor ofthe chymotryptic-, tryptic- and post-glutamyl peptidyl
Received 29 March 2006; revised 27 July 2006; accepted 1 August 2006;published online 18 September 2006
Correspondence: Professor JHM Prehn, Department of Physiologyand Medical Physics, Royal College of Surgeons in Ireland, 123St Stephen’s Green, Dublin 2, Ireland.E-mail: [email protected] authors share equal senior authorship.
Oncogene (2007) 26, 1681–1692& 2007 Nature Publishing Group All rights reserved 0950-9232/07 $30.00
www.nature.com/onc
hydrolytic-like activities of the proteasome (Meng et al.,1999), and was first identified and isolated from a speciesof actinomycetes on the basis of its antitumor activity(Hanada et al., 1992). Unlike several widely used PIs,Epoxo is highly specific for the proteasome withoutaffecting the activity of other non-proteasomal pro-teases including calpain, trypsin and cathepsin B (Menget al., 1999).
In addition to the post-translational modificationof proapoptotic proteins, inhibition of proteasomalfunction may also regulate several transcription-depen-dent processes. Indeed, the activity of several transcrip-tion factors, including p53 and nuclear factor kappa B(NF-kB) (Chowdary et al., 1994; Palombella et al.,1994), are known to be modulated by proteasomeactivity. This study was undertaken to analyse theunderlying transcriptional mechanisms leading toapoptosis triggered by inhibition of the proteasomesystem in cancer cells. Our data demonstratesthat although the two Bcl-2-homology domain 3-only(BH3-only) genes p53 upregulated modulator of apoptosis(PUMA) and Bim are transcriptionally activated byproteasome inhibition, p53-dependent activation ofPUMA plays a predominant role in this type of celldeath.
Results
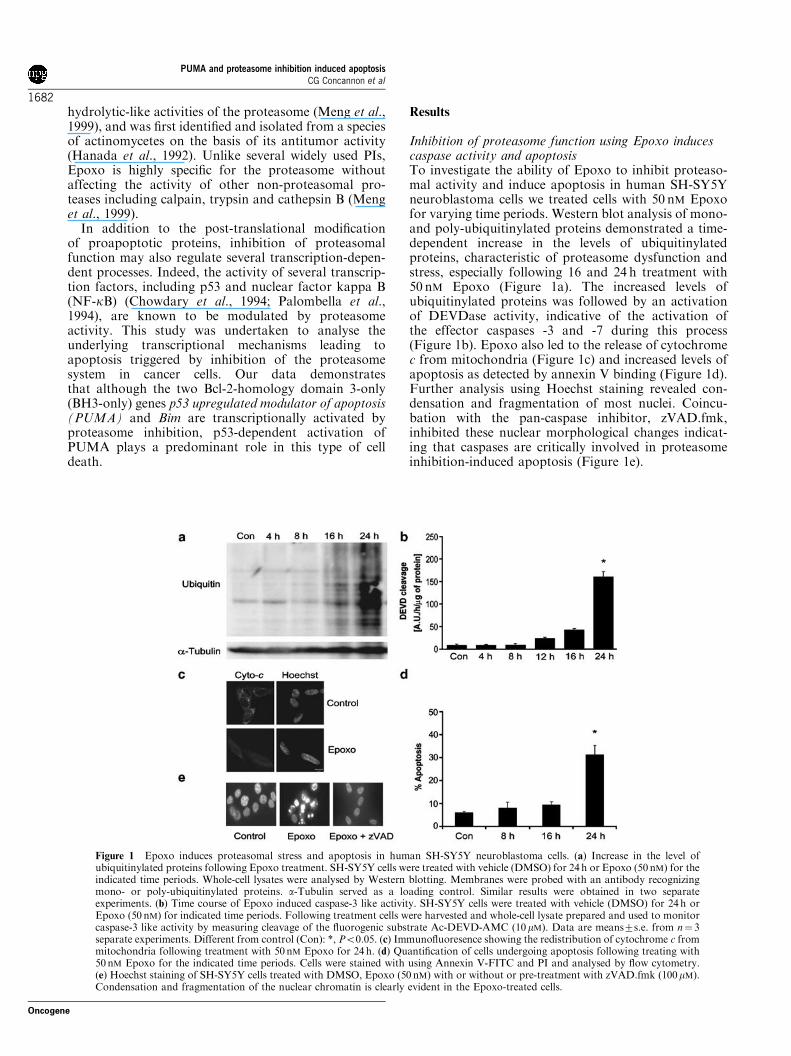
Inhibition of proteasome function using Epoxo inducescaspase activity and apoptosisTo investigate the ability of Epoxo to inhibit proteaso-mal activity and induce apoptosis in human SH-SY5Yneuroblastoma cells we treated cells with 50 nM Epoxofor varying time periods. Western blot analysis of mono-and poly-ubiquitinylated proteins demonstrated a time-dependent increase in the levels of ubiquitinylatedproteins, characteristic of proteasome dysfunction andstress, especially following 16 and 24 h treatment with50 nM Epoxo (Figure 1a). The increased levels ofubiquitinylated proteins was followed by an activationof DEVDase activity, indicative of the activation ofthe effector caspases -3 and -7 during this process(Figure 1b). Epoxo also led to the release of cytochromec from mitochondria (Figure 1c) and increased levels ofapoptosis as detected by annexin V binding (Figure 1d).Further analysis using Hoechst staining revealed con-densation and fragmentation of most nuclei. Coincu-bation with the pan-caspase inhibitor, zVAD.fmk,inhibited these nuclear morphological changes indicat-ing that caspases are critically involved in proteasomeinhibition-induced apoptosis (Figure 1e).
Figure 1 Epoxo induces proteasomal stress and apoptosis in human SH-SY5Y neuroblastoma cells. (a) Increase in the level ofubiquitinylated proteins following Epoxo treatment. SH-SY5Y cells were treated with vehicle (DMSO) for 24h or Epoxo (50nM) for theindicated time periods. Whole-cell lysates were analysed by Western blotting. Membranes were probed with an antibody recognizingmono- or poly-ubiquitinylated proteins. a-Tubulin served as a loading control. Similar results were obtained in two separateexperiments. (b) Time course of Epoxo induced caspase-3 like activity. SH-SY5Y cells were treated with vehicle (DMSO) for 24h orEpoxo (50 nM) for indicated time periods. Following treatment cells were harvested and whole-cell lysate prepared and used to monitorcaspase-3 like activity by measuring cleavage of the fluorogenic substrate Ac-DEVD-AMC (10mM). Data are means7s.e. from n! 3separate experiments. Different from control (Con): *, Po0.05. (c) Immunofluoresence showing the redistribution of cytochrome c frommitochondria following treatment with 50nM Epoxo for 24h. (d) Quantification of cells undergoing apoptosis following treating with50nM Epoxo for the indicated time periods. Cells were stained with using Annexin V-FITC and PI and analysed by flow cytometry.(e) Hoechst staining of SH-SY5Y cells treated with DMSO, Epoxo (50 nM) with or without or pre-treatment with zVAD.fmk (100mM).Condensation and fragmentation of the nuclear chromatin is clearly evident in the Epoxo-treated cells.
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1682
Oncogene
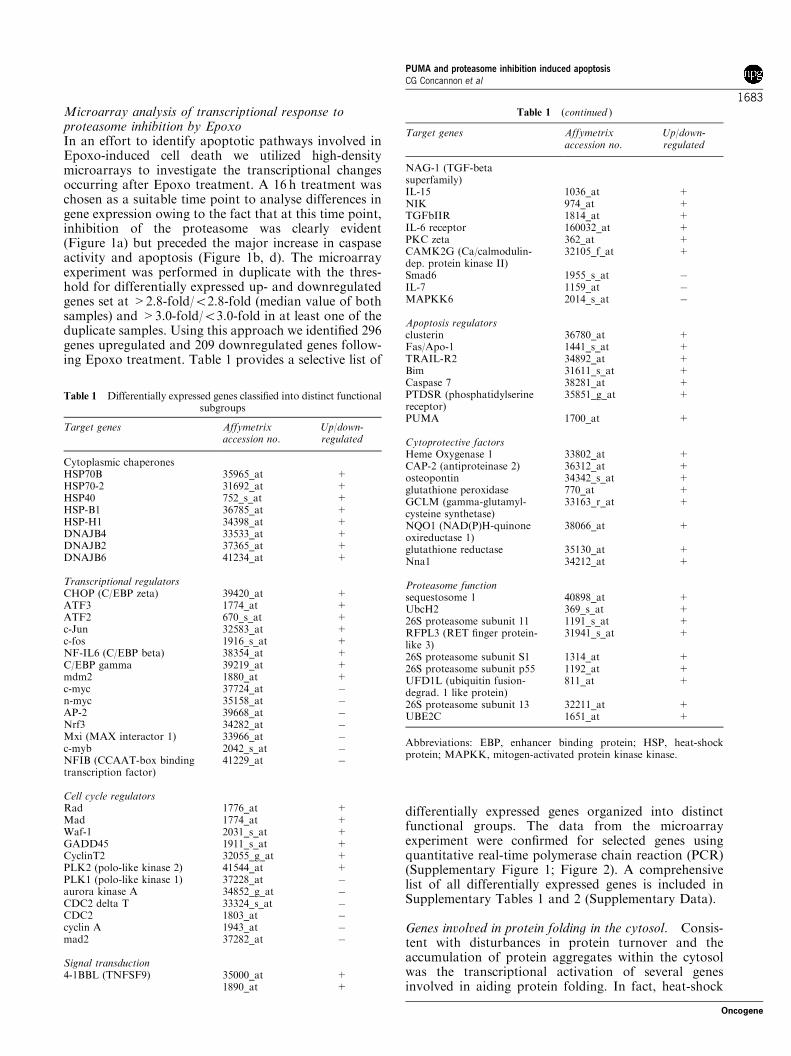
Microarray analysis of transcriptional response toproteasome inhibition by EpoxoIn an effort to identify apoptotic pathways involved inEpoxo-induced cell death we utilized high-densitymicroarrays to investigate the transcriptional changesoccurring after Epoxo treatment. A 16 h treatment waschosen as a suitable time point to analyse differences ingene expression owing to the fact that at this time point,inhibition of the proteasome was clearly evident(Figure 1a) but preceded the major increase in caspaseactivity and apoptosis (Figure 1b, d). The microarrayexperiment was performed in duplicate with the thres-hold for differentially expressed up- and downregulatedgenes set at >2.8-fold/o2.8-fold (median value of bothsamples) and >3.0-fold/o3.0-fold in at least one of theduplicate samples. Using this approach we identified 296genes upregulated and 209 downregulated genes follow-ing Epoxo treatment. Table 1 provides a selective list of
differentially expressed genes organized into distinctfunctional groups. The data from the microarrayexperiment were confirmed for selected genes usingquantitative real-time polymerase chain reaction (PCR)(Supplementary Figure 1; Figure 2). A comprehensivelist of all differentially expressed genes is included inSupplementary Tables 1 and 2 (Supplementary Data).
Genes involved in protein folding in the cytosol. Consis-tent with disturbances in protein turnover and theaccumulation of protein aggregates within the cytosolwas the transcriptional activation of several genesinvolved in aiding protein folding. In fact, heat-shock
Table 1 Differentially expressed genes classified into distinct functionalsubgroups
Target genes Affymetrixaccession no.
Up/down-regulated
Cytoplasmic chaperonesHSP70B 35965_at +HSP70-2 31692_at +HSP40 752_s_at +HSP-B1 36785_at +HSP-H1 34398_at +DNAJB4 33533_at +DNAJB2 37365_at +DNAJB6 41234_at +
Transcriptional regulatorsCHOP (C/EBP zeta) 39420_at +ATF3 1774_at +ATF2 670_s_at +c-Jun 32583_at +c-fos 1916_s_at +NF-IL6 (C/EBP beta) 38354_at +C/EBP gamma 39219_at +mdm2 1880_at +c-myc 37724_at "n-myc 35158_at "AP-2 39668_at "Nrf3 34282_at "Mxi (MAX interactor 1) 33966_at "c-myb 2042_s_at "NFIB (CCAAT-box bindingtranscription factor)
41229_at "
Cell cycle regulatorsRad 1776_at +Mad 1774_at +Waf-1 2031_s_at +GADD45 1911_s_at +CyclinT2 32055_g_at +PLK2 (polo-like kinase 2) 41544_at +PLK1 (polo-like kinase 1) 37228_at "aurora kinase A 34852_g_at "CDC2 delta T 33324_s_at "CDC2 1803_at "cyclin A 1943_at "mad2 37282_at "
Signal transduction4-1BBL (TNFSF9) 35000_at +
1890_at +
Table 1 (continued )
Target genes Affymetrixaccession no.
Up/down-regulated
NAG-1 (TGF-betasuperfamily)IL-15 1036_at +NIK 974_at +TGFbIIR 1814_at +IL-6 receptor 160032_at +PKC zeta 362_at +CAMK2G (Ca/calmodulin-dep. protein kinase II)
32105_f_at +
Smad6 1955_s_at "IL-7 1159_at "MAPKK6 2014_s_at "
Apoptosis regulatorsclusterin 36780_at +Fas/Apo-1 1441_s_at +TRAIL-R2 34892_at +Bim 31611_s_at +Caspase 7 38281_at +PTDSR (phosphatidylserinereceptor)
35851_g_at +
PUMA 1700_at +
Cytoprotective factorsHeme Oxygenase 1 33802_at +CAP-2 (antiproteinase 2) 36312_at +osteopontin 34342_s_at +glutathione peroxidase 770_at +GCLM (gamma-glutamyl-cysteine synthetase)
33163_r_at +
NQO1 (NAD(P)H-quinoneoxireductase 1)
38066_at +
glutathione reductase 35130_at +Nna1 34212_at +
Proteasome functionsequestosome 1 40898_at +UbcH2 369_s_at +26S proteasome subunit 11 1191_s_at +RFPL3 (RET finger protein-like 3)
31941_s_at +
26S proteasome subunit S1 1314_at +26S proteasome subunit p55 1192_at +UFD1L (ubiquitin fusion-degrad. 1 like protein)
811_at +
26S proteasome subunit 13 32211_at +UBE2C 1651_at +
Abbreviations: EBP, enhancer binding protein; HSP, heat-shockprotein; MAPKK, mitogen-activated protein kinase kinase.
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1683
Oncogene
protein (Hsp)70B, a potent molecular chaperone, wasidentified as the highest induced gene (>700-fold frommicroarray data). Other members of this group inducedinclude Hsp40, Hsp-B1, as well as several DNAJBchaperones. Significantly, all the chaperones identifiedare involved in protein folding within the cytosol andare not associated with protein folding within theendoplasmic reticulum (ER). This differs from thetranscriptional response to ER stress, which we havepreviously profiled in the identical cellular system(Reimertz et al., 2003). Here, proteasome inhibitiondoes appear to induce immunoglobulin heavy chain-binding protein/78 kDa glucose-regulated protein ex-pression although the impairment of ER-associatedprotein degradation (ERAD) may result in the accumu-lation of misfolded proteins within the ER and thesubsequent induction of ER stress response.
Genes involved in proteasomal function. The microarraydata identified differential expression of several genesinvolved in the function of the proteasome. Theseincluded genes encoding the 26S proteasome subunits11, 13, S1 and p55. Indeed, previous studies havedemonstrated transcriptional induction of proteasomalsubunits following proteasomal inhibition (Meinerset al., 2003).
Transcriptional regulators. Proteasomal stress resultedin increased expression of several members of the C/enhancer binding protein (EBP) family of transcriptionfactors including C/EBP-homologous protein (CHOP),C/EBP beta and C/EBP gamma. Similarly, two mem-bers of the ATF transcription factor family, ATF-2and ATF-3, were also induced. In addition, the micro-array analysis also revealed a transcriptional activationof both c-Jun and c-fos. The most prominently
downregulated genes after proteasome inhibition weremembers of the c-Myc family of transcription factorswhich control expression of genes involved in cellgrowth and metabolism (Dang, 1999).
Cell cycle regulation. Several genes involved in thecontrol of the cell cycle were found to be differentiallyexpressed. These included cyclin T2 and the p53 targetgenes Waf1 and Mdm2, suggesting that Epoxo treat-ment is associated with induction of p53-dependentcell signaling and induction of cell cycle arrest. Giventhat p53 expression levels and signaling is regulatedby ubiquitination by Mdm2, the identification ofp53-dependent targets is consistent with inhibition ofproteasomal function.
Antioxidant defence. Consistent with previously pub-lished data that suggest proteasomal stress is associatedwith increases in the production of reactive oxygenspecies (Kikuchi et al., 2003), is the transcriptionalactivation of several genes involved in the defenceagainst oxidative stress. These include genes involved inthe regeneration of the antioxidant protein glutathione(glutathione reductase) as well as the antioxidant genes,heme oxygenase-1 (HO-1) and glutathione peroxidase.
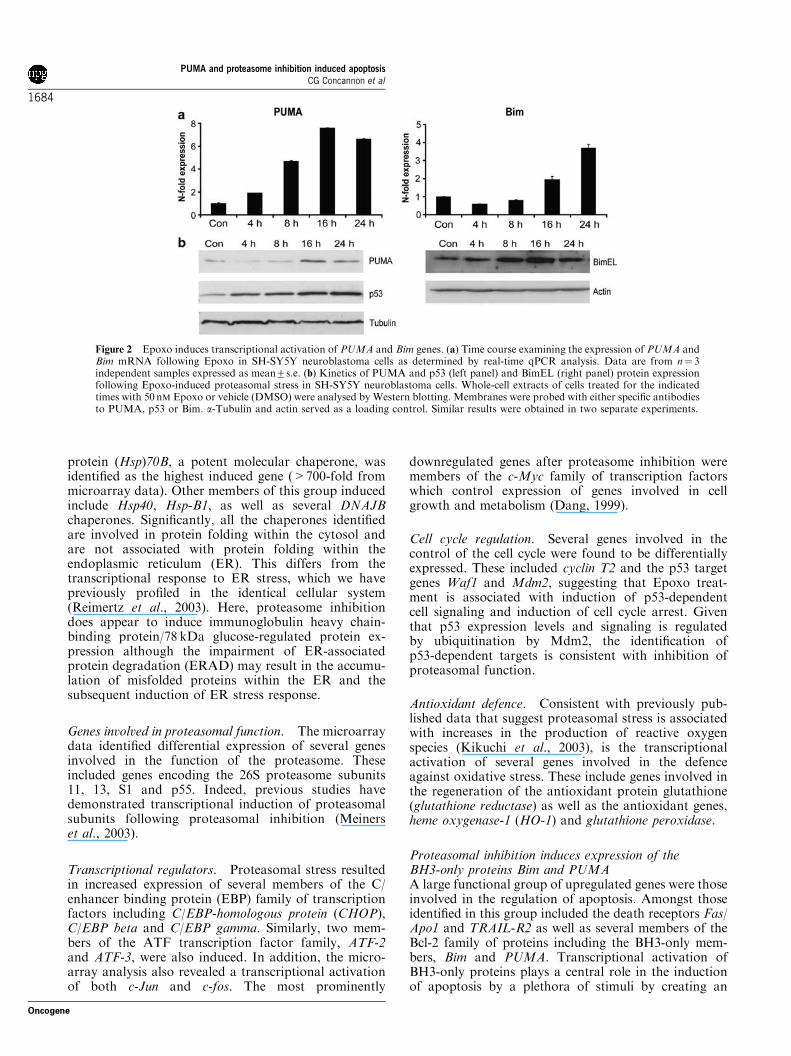
Proteasomal inhibition induces expression of theBH3-only proteins Bim and PUMAA large functional group of upregulated genes were thoseinvolved in the regulation of apoptosis. Amongst thoseidentified in this group included the death receptors Fas/Apo1 and TRAIL-R2 as well as several members of theBcl-2 family of proteins including the BH3-only mem-bers, Bim and PUMA. Transcriptional activation ofBH3-only proteins plays a central role in the inductionof apoptosis by a plethora of stimuli by creating an
Figure 2 Epoxo induces transcriptional activation of PUMA and Bim genes. (a) Time course examining the expression of PUMA andBim mRNA following Epoxo in SH-SY5Y neuroblastoma cells as determined by real-time qPCR analysis. Data are from n! 3independent samples expressed as mean7s.e. (b) Kinetics of PUMA and p53 (left panel) and BimEL (right panel) protein expressionfollowing Epoxo-induced proteasomal stress in SH-SY5Y neuroblastoma cells. Whole-cell extracts of cells treated for the indicatedtimes with 50 nM Epoxo or vehicle (DMSO) were analysed by Western blotting. Membranes were probed with either specific antibodiesto PUMA, p53 or Bim. a-Tubulin and actin served as a loading control. Similar results were obtained in two separate experiments.
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1684
Oncogene
imbalance between proapoptotic and antiapoptotic Bcl-2proteins resulting in the release of cytochrome c andapoptosis (Villunger et al., 2003a; Willis and Adams,2005). Because of the evidence of cytochrome c release inour model and in previous studies employing PIs(Wagenknecht et al., 2000; Mitsiades et al., 2002), wefurther characterized the transcriptional induction ofBim and PUMA following Epoxo treatment. Real-timequantitative PCR (qPCR) demonstrated an early tran-scriptional induction of PUMA mRNA, which wasevident following 8 h treatment and peaked at 16 h(Figure 2a, left panel) in SH-SY5Y neuroblastoma cells.In contrast, the induction of Bim mRNA showed a moredelayed kinetics with a steady increase over the entiretime period of 24 h (Figure 2a, right panel). This wasreflected on a protein level with increased expression ofPUMA evident following 16 h treatment with Epoxo,correlating with a maximal increase in the expression ofp53 at 16 and 24 h (Figure 2b, left panel). Westernblotting also revealed an increased expression of the Bimprotein following inhibition of the proteasome(Figure 2b, right panel), before the increase in mRNAexpression. This apparent discrepancy is likely caused bya decreased turnover of the BimEL protein afterinhibition of the proteasome (Figure 3b, right panel),as BimEL has previously been shown to be targeted fordegradation by the ubiquitin-proteasome pathway (Leyet al., 2003). Based on these observations, we concludethat enhanced Bim expression is triggered by two distinctmechanisms after proteasome inhibition: post-transla-tional protein stabilization and a delayed induction ofBim transcription.
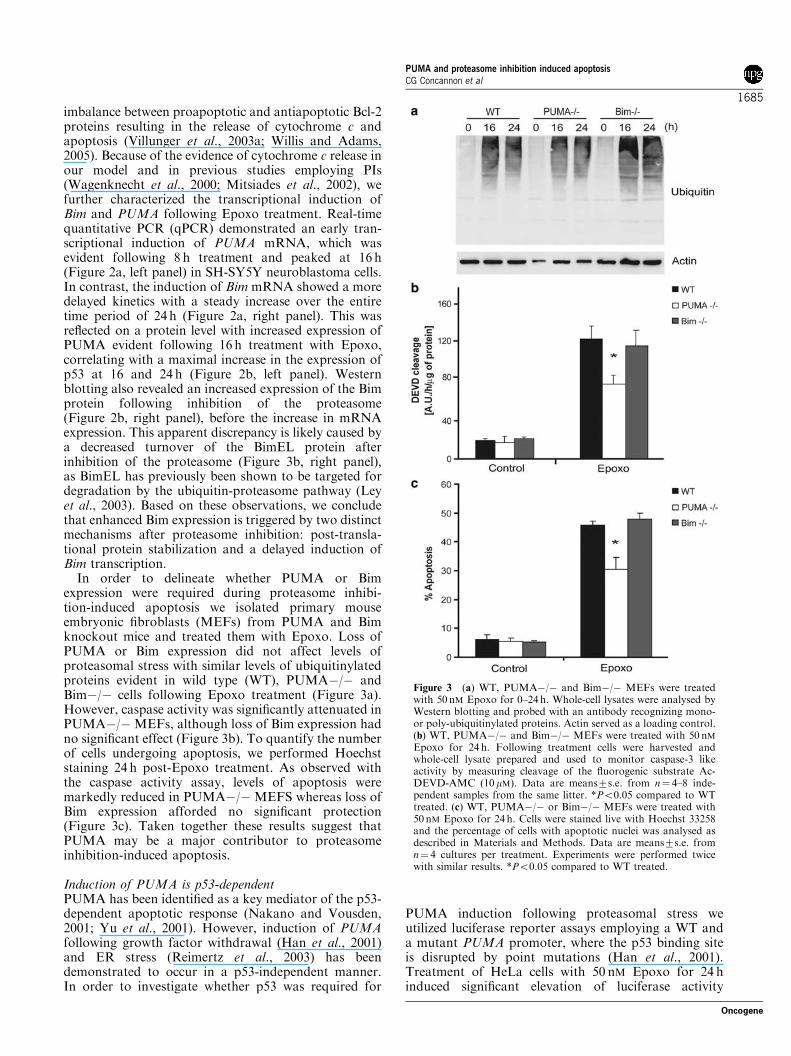
In order to delineate whether PUMA or Bimexpression were required during proteasome inhibi-tion-induced apoptosis we isolated primary mouseembryonic fibroblasts (MEFs) from PUMA and Bimknockout mice and treated them with Epoxo. Loss ofPUMA or Bim expression did not affect levels ofproteasomal stress with similar levels of ubiquitinylatedproteins evident in wild type (WT), PUMA"/" andBim"/" cells following Epoxo treatment (Figure 3a).However, caspase activity was significantly attenuated inPUMA"/" MEFs, although loss of Bim expression hadno significant effect (Figure 3b). To quantify the numberof cells undergoing apoptosis, we performed Hoechststaining 24 h post-Epoxo treatment. As observed withthe caspase activity assay, levels of apoptosis weremarkedly reduced in PUMA"/" MEFS whereas loss ofBim expression afforded no significant protection(Figure 3c). Taken together these results suggest thatPUMA may be a major contributor to proteasomeinhibition-induced apoptosis.
Induction of PUMA is p53-dependentPUMA has been identified as a key mediator of the p53-dependent apoptotic response (Nakano and Vousden,2001; Yu et al., 2001). However, induction of PUMAfollowing growth factor withdrawal (Han et al., 2001)and ER stress (Reimertz et al., 2003) has beendemonstrated to occur in a p53-independent manner.In order to investigate whether p53 was required for
PUMA induction following proteasomal stress weutilized luciferase reporter assays employing a WT anda mutant PUMA promoter, where the p53 binding siteis disrupted by point mutations (Han et al., 2001).Treatment of HeLa cells with 50 nM Epoxo for 24 hinduced significant elevation of luciferase activity
Figure 3 (a) WT, PUMA"/" and Bim"/" MEFs were treatedwith 50 nM Epoxo for 0–24h. Whole-cell lysates were analysed byWestern blotting and probed with an antibody recognizing mono-or poly-ubiquitinylated proteins. Actin served as a loading control.(b) WT, PUMA"/" and Bim"/" MEFs were treated with 50 nMEpoxo for 24 h. Following treatment cells were harvested andwhole-cell lysate prepared and used to monitor caspase-3 likeactivity by measuring cleavage of the fluorogenic substrate Ac-DEVD-AMC (10 mM). Data are means7s.e. from n! 4–8 inde-pendent samples from the same litter. *Po0.05 compared to WTtreated. (c) WT, PUMA"/" or Bim"/" MEFs were treated with50 nM Epoxo for 24 h. Cells were stained live with Hoechst 33258and the percentage of cells with apoptotic nuclei was analysed asdescribed in Materials and Methods. Data are means7s.e. fromn! 4 cultures per treatment. Experiments were performed twicewith similar results. *Po0.05 compared to WT treated.
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1685
Oncogene
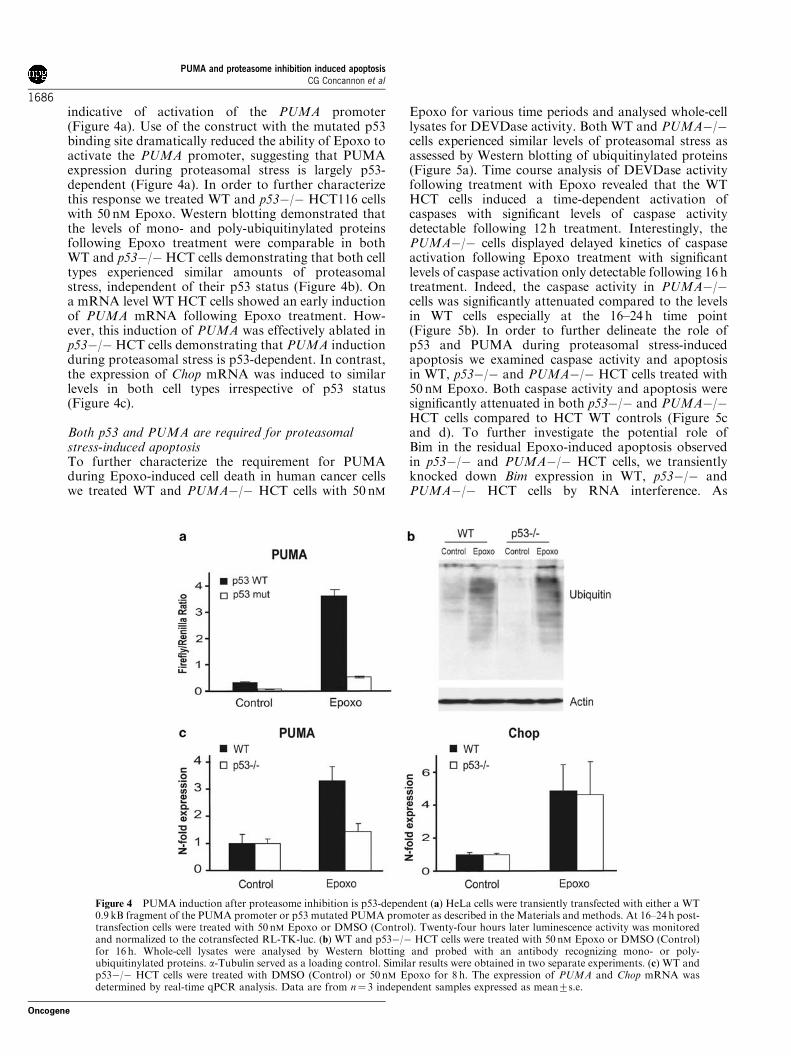
indicative of activation of the PUMA promoter(Figure 4a). Use of the construct with the mutated p53binding site dramatically reduced the ability of Epoxo toactivate the PUMA promoter, suggesting that PUMAexpression during proteasomal stress is largely p53-dependent (Figure 4a). In order to further characterizethis response we treated WT and p53"/" HCT116 cellswith 50 nM Epoxo. Western blotting demonstrated thatthe levels of mono- and poly-ubiquitinylated proteinsfollowing Epoxo treatment were comparable in bothWT and p53"/" HCT cells demonstrating that both celltypes experienced similar amounts of proteasomalstress, independent of their p53 status (Figure 4b). Ona mRNA level WT HCT cells showed an early inductionof PUMA mRNA following Epoxo treatment. How-ever, this induction of PUMA was effectively ablated inp53"/"HCT cells demonstrating that PUMA inductionduring proteasomal stress is p53-dependent. In contrast,the expression of Chop mRNA was induced to similarlevels in both cell types irrespective of p53 status(Figure 4c).
Both p53 and PUMA are required for proteasomalstress-induced apoptosisTo further characterize the requirement for PUMAduring Epoxo-induced cell death in human cancer cellswe treated WT and PUMA"/" HCT cells with 50 nM
Epoxo for various time periods and analysed whole-celllysates for DEVDase activity. Both WT and PUMA"/"cells experienced similar levels of proteasomal stress asassessed by Western blotting of ubiquitinylated proteins(Figure 5a). Time course analysis of DEVDase activityfollowing treatment with Epoxo revealed that the WTHCT cells induced a time-dependent activation ofcaspases with significant levels of caspase activitydetectable following 12 h treatment. Interestingly, thePUMA"/" cells displayed delayed kinetics of caspaseactivation following Epoxo treatment with significantlevels of caspase activation only detectable following 16htreatment. Indeed, the caspase activity in PUMA"/"cells was significantly attenuated compared to the levelsin WT cells especially at the 16–24 h time point(Figure 5b). In order to further delineate the role ofp53 and PUMA during proteasomal stress-inducedapoptosis we examined caspase activity and apoptosisin WT, p53"/" and PUMA"/" HCT cells treated with50 nM Epoxo. Both caspase activity and apoptosis weresignificantly attenuated in both p53"/" and PUMA"/"HCT cells compared to HCT WT controls (Figure 5cand d). To further investigate the potential role ofBim in the residual Epoxo-induced apoptosis observedin p53"/" and PUMA"/" HCT cells, we transientlyknocked down Bim expression in WT, p53"/" andPUMA"/" HCT cells by RNA interference. As
Figure 4 PUMA induction after proteasome inhibition is p53-dependent (a) HeLa cells were transiently transfected with either a WT0.9 kB fragment of the PUMA promoter or p53 mutated PUMA promoter as described in the Materials and methods. At 16–24 h post-transfection cells were treated with 50 nM Epoxo or DMSO (Control). Twenty-four hours later luminescence activity was monitoredand normalized to the cotransfected RL-TK-luc. (b) WT and p53"/" HCT cells were treated with 50 nM Epoxo or DMSO (Control)for 16 h. Whole-cell lysates were analysed by Western blotting and probed with an antibody recognizing mono- or poly-ubiquitinylated proteins. a-Tubulin served as a loading control. Similar results were obtained in two separate experiments. (c) WT andp53"/" HCT cells were treated with DMSO (Control) or 50 nM Epoxo for 8 h. The expression of PUMA and Chop mRNA wasdetermined by real-time qPCR analysis. Data are from n! 3 independent samples expressed as mean7s.e.
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1686
Oncogene
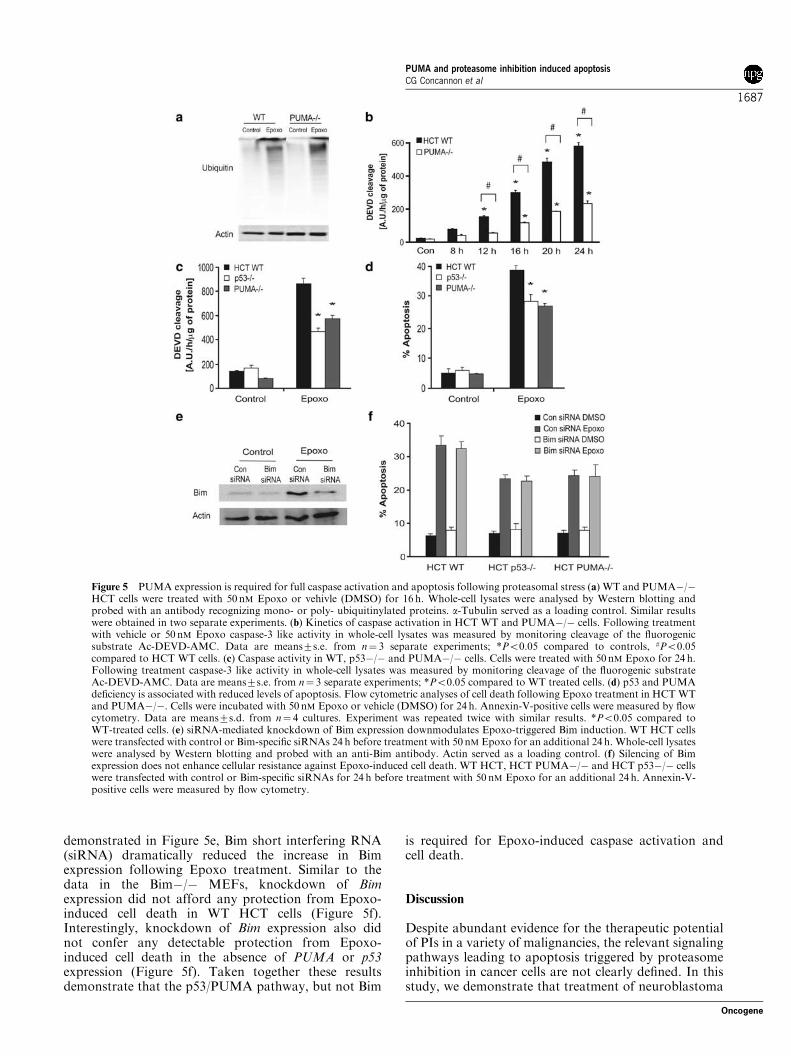
demonstrated in Figure 5e, Bim short interfering RNA(siRNA) dramatically reduced the increase in Bimexpression following Epoxo treatment. Similar to thedata in the Bim"/" MEFs, knockdown of Bimexpression did not afford any protection from Epoxo-induced cell death in WT HCT cells (Figure 5f).Interestingly, knockdown of Bim expression also didnot confer any detectable protection from Epoxo-induced cell death in the absence of PUMA or p53expression (Figure 5f). Taken together these resultsdemonstrate that the p53/PUMA pathway, but not Bim
is required for Epoxo-induced caspase activation andcell death.
Discussion
Despite abundant evidence for the therapeutic potentialof PIs in a variety of malignancies, the relevant signalingpathways leading to apoptosis triggered by proteasomeinhibition in cancer cells are not clearly defined. In thisstudy, we demonstrate that treatment of neuroblastoma
Figure 5 PUMA expression is required for full caspase activation and apoptosis following proteasomal stress (a) WT and PUMA"/"HCT cells were treated with 50 nM Epoxo or vehivle (DMSO) for 16 h. Whole-cell lysates were analysed by Western blotting andprobed with an antibody recognizing mono- or poly- ubiquitinylated proteins. a-Tubulin served as a loading control. Similar resultswere obtained in two separate experiments. (b) Kinetics of caspase activation in HCT WT and PUMA"/" cells. Following treatmentwith vehicle or 50 nM Epoxo caspase-3 like activity in whole-cell lysates was measured by monitoring cleavage of the fluorogenicsubstrate Ac-DEVD-AMC. Data are means7s.e. from n! 3 separate experiments; *Po0.05 compared to controls, #Po0.05compared to HCT WT cells. (c) Caspase activity in WT, p53"/" and PUMA"/" cells. Cells were treated with 50 nM Epoxo for 24 h.Following treatment caspase-3 like activity in whole-cell lysates was measured by monitoring cleavage of the fluorogenic substrateAc-DEVD-AMC. Data are means7s.e. from n! 3 separate experiments; *Po0.05 compared to WT treated cells. (d) p53 and PUMAdeficiency is associated with reduced levels of apoptosis. Flow cytometric analyses of cell death following Epoxo treatment in HCTWTand PUMA"/". Cells were incubated with 50 nM Epoxo or vehicle (DMSO) for 24 h. Annexin-V-positive cells were measured by flowcytometry. Data are means7s.d. from n! 4 cultures. Experiment was repeated twice with similar results. *Po0.05 compared toWT-treated cells. (e) siRNA-mediated knockdown of Bim expression downmodulates Epoxo-triggered Bim induction. WT HCT cellswere transfected with control or Bim-specific siRNAs 24 h before treatment with 50 nM Epoxo for an additional 24 h. Whole-cell lysateswere analysed by Western blotting and probed with an anti-Bim antibody. Actin served as a loading control. (f) Silencing of Bimexpression does not enhance cellular resistance against Epoxo-induced cell death. WT HCT, HCT PUMA"/" and HCT p53"/" cellswere transfected with control or Bim-specific siRNAs for 24 h before treatment with 50 nM Epoxo for an additional 24 h. Annexin-V-positive cells were measured by flow cytometry.
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1687
Oncogene

cells with the highly potent and selective PI Epoxoimpairs protein degradation and subsequently inducesactivation of caspases and apoptosis. In our modelprolonged proteasome inhibition was associated withthe concomitant induction of two members of theproapoptotic BH3-only family, PUMA and Bim. Inter-estingly, PUMA induction occurred in a p53-dependentmanner and cells deficient in PUMA expression weresignificantly protected from Epoxo-induced caspaseactivation, suggesting a requirement for PUMA duringproteasomal stress-induced apoptosis. Transcriptionalactivation and increased expression of BH3-only pro-teins is central in determining cell fate in response to avariety of stimuli, functioning as activation enablers andtransducing the cellular stress signal to other proapop-totic Bcl-2 family members such as Bax and Bak (forreview see Willis and Adams (2005)).
Owing to the broad number of substrates of theproteasome, a significant number of players have beenpreviously implicated in the proapoptotic activity of PIs,among them NF-kB, p53, Bcl-2 and caspases (for reviewsee Adams (2004b)). NF-kB is constitutively activated inmany cancer cells and is associated with cancerdevelopment and progression (Karin, 2006). NF-kBpotently inhibits apoptosis by activation of antiapopto-tic target genes such as Bcl-xL and IAPs) (Luo et al.,2005). As IkBa the endogenous inhibitor of NF-kB iscontinuously degraded via the proteasome pathway, PIsact as indirect inhibitors of NF-kB (Karin and Ben-Neriah, 2000). Indeed, inactivation of NF-kB has beensuggested to play a major role in the antitumor effect ofthe PI, Bortezomib (Velcade/PS-341), in multiplemyeloma (Hideshima et al., 2002) and melanoma cells(Amiri et al., 2004). Despite these observations, inhibi-tion of NF-kB is not required to sensitize hepatocellularcarcinoma cells, keratinocytes and lymphoma cells toapoptosis (Kurland and Meyn, 2001; Leverkus et al.,2003; Ganten et al., 2005). Our microarray analyses inSH-SY5Y neuroblastoma cells did not detect noticeableexpression changes in NF-kB-dependent cell deathregulators, such as Bcl-2, Bcl-xL, and the IAPs (datanot shown), and subsequent real-time qPCR analysis inHCT colon cancer cells also failed to detect anysignificant changes in the expression of these genes(Supplementary Figure 2). These data suggest thatinhibition of NF-kB might not play a major role inapoptosis induced by PIs in a significant number ofcancer types.
Similar controversy exists on the correlation betweenthe apoptosis-inducing efficiency of PIs and the p53status of various types of cancer cells, mainly owing tothe fact that this correlation seems to be cell type-dependent. Although PIs have been shown to inducep53-independent apoptosis in glioma cells (Wagen-knecht et al., 1999; Yin et al., 2005), the majority ofreports suggest that PIs induce a p53-dependent form ofapoptosis in most cancer cell types, such as leukemiacells (Shinohara et al., 1996; Masdehors et al., 2000), aswell as melanoma and myeloma cells (Qin et al., 2005).Consistent with previous studies we observed a promi-nent increase of p53 levels, which was associated with
transcriptional activation of the proapoptotic BH3-onlygene PUMA. Interestingly, ATF3, a recently identifiedpositive regulator of p53 stability (Yan et al., 2005), wasalso found to be transcriptionally induced and mayrepresent a positive feed back loop further amplifyingp53 activation during proteasomal stress. Of note, it hasbeen previously reported that PUMA accounts for mostof the p53-dependent apoptosis in the HCT116 coloncarcinoma cells employed in this study (Yu et al., 2001,2003). In our experiments, the extent of protectionagainst Epoxo-induced cell death was very similar inp53-deficient and PUMA-deficient HCT116 cells, sug-gesting that p53 mediates cell death largely throughPUMA induction, and that other transcriptional,proapoptotic targets of p53 such as Fas (Owen-Schaubet al., 1995) and Trail-R2 (Wu et al., 1997) may play aless pronounced role, although they may additionallysensitize cells to therapies involving death ligands.Interestingly, the most potently downregulated genefollowing proteasome inhibition was the oncogenictranscription factor, c-myc, which may have a rolein the antitumorigenic effect of PIs. Furthermore,c-myc has been shown to have an inhibitory effect onp53-dependent target gene expression and apoptosis(Ceballos et al., 2005).
Consistent with previous reports we found increasedexpression of Bim following inhibition of the protea-some (Nikrad et al., 2005), however, our data arguesagainst a significant role of Bim in our model.Interestingly, in agreement with our findings a recentstudy demonstrated increased Bim expression followingproteasome inhibition, although siRNA-mediatedknockdown of Bim afforded no protection (Ananet al., 2006). However, it remains plausible thatincreased expression of Bim in response to PIs may beof therapeutic benefit when used in combination withother agents such as TRAIL and paclitaxel (Nikradet al., 2005; Tan et al., 2005). In our model a lack ofPUMA expression was sufficient to offer significantprotection, suggesting that Bim induction is not able tocompensate for PUMA deficiency as has been reportedfor other BH3-only proteins in some models (Villungeret al., 2003a, b). However, although loss of Bimexpression afforded no protection in the cell linesemployed in this study it is still possible that PUMAand Bim have overlapping action during proteaso-mal-stress-induced apoptosis in certain cell types whichmay be best addressed by studying cells fromBim"/"PUMA"/" double knockout mice. Interest-ingly, several recent studies have also demonstrated aconcomitant induction of Bim and PUMA followingtreatment with the oxidative stress-inducing agents6-OHDA (Biswas et al., 2005) and arsenite (Wonget al., 2005). In both scenarios the inhibition of PUMAexpression was found to be sufficient to dramaticallyreduce the extent of apoptosis, whereas loss of Bimexpression incurred no advantage with equivalent levelsof cell death seen in WT and Bim"/" cells (Biswas et al.,2005; Wong et al., 2005). Hence, scenarios exist whereboth PUMA and Bim are induced, but PUMA plays amore prominent role for the execution of cell death.
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1688
Oncogene

Curiously, these functional studies argue against arecent study suggesting that a Bim BH3 peptide maybe a more potent inducer of cytochrome c release andapoptosis than its PUMA counterpart (Kuwana et al.,2005), suggesting that potency of BH3-only proteinsmay also be dependent on functional interactionswith other molecules within the dynamic environmentof a cell.
We have previously shown that cell death triggered byER stress also requires the transcriptional upregulationof PUMA (Reimertz et al., 2003). Defects in protea-some-mediated protein degradation trigger similarcellular stress responses as observed during ER stress,such as attenuation of protein synthesis and induction oftranscription factors of the ATF and C/EBP families(Reimertz et al., 2003) suggesting PUMA expression is ageneral response to mediate cell death during prolongeddefects in protein quality control and protein degrada-tion. Our data emphasizes the fundamental importanceof the p53/PUMA pathway in determining the level ofsensitivity of cancer cells to PIs.
Materials and methods
MaterialsCaspase substrate acetyl-DEVD-7-amido-4-methylcoumarin(Ac-DEVD-AMC) and inhibitor z-Val-Ala-Asp (O-methyl)-fluoromethyl ketone (zVAD.fmk) was from Bachem (StHelen’s, UK). Epoxo and Hoechst 33258 were purchasedfrom Sigma-Aldrich (Tallaght, Dublin, Ireland). All otherchemicals came in analytical grade purity from Sigma-Aldrich(Tallaght, Dublin, Ireland).
Cell lines and cultureHuman SH-SY5Y neuroblastoma cells, HeLa, HCT116 WT,HCT116 p53"/" and HCT116 PUMA"/" cells were grown inRosewell Park Memorial Institute medium 1640 mediumsupplemented with 10% heat-inactivated fetal calf serum,2mM glutamine, 100U/ml penicillin, and 100mg/ml strepto-mycin, in a humidified atmosphere of 5% CO2 in air at 371C.Primary MEFs were isolated fromWT, PUMA"/" or Bim"/"in a C57/BL/6 background from E14.5 day embryos usingstandard methods and were maintained in Dulbecco’s mod-ified Eagle’s medium with 10% heat-inactivated fetal calfserum, 100U/ml penicillin, and 100mg/ml streptomycin,in a humidified atmosphere of 5% CO2 in air at 37oC. Forall experiments fibroblasts were used between passages 1–3.The generation of the PUMA"/" and Bim"/" mice havebeen described previously (Bouillet et al., 1999; Villunger et al.,2003a).
Microarray analysisSH-SY5Y neuroblastoma cells were treated with 50 nM Epoxoor vehicle (0.1% dimethylsulfoxide (DMSO)) for 16 h. TotalRNA of duplicate samples was extracted using the RNeasyMidi Kit (Qiagen, Hilden, Germany). Complementary RNAwas synthesized from 40mg of the total RNA by using theSuperscript Choice Kit (Invitrogen, Paisley, UK). The cRNAwas prepared and biotin-labeled by in vitro transcription (EnzoBiochemical, Farmingdale, NY, USA). Labeled cRNA wasfragmented and hybridized for 16 h at 451C to an HG-U95Av2array (Affymetrix, Santa Clara, CA, USA). Hybridization,washing and staining, as well as scanning of the gene chips in a
GeneArray Scanner (Agilent Technologies, Palo Alto, CA,USA) were performed as described in the Affymetrix GeneExpression Analysis Technical Manual. The AffymetrixMicroarray Suite 5.0 was used to analyse the relativeabundance of each gene from the average difference ofintensities. For each individual gene, the signal ratio betweenperfect match and mismatch probe cells was used to determineits ‘Absolute Call’, indicating whether the correspondingtranscript was present (P), absent (A), or marginal (M). Foranalysis of differential target gene expression, the ‘DifferenceCall Decision Matrix’ was employed. Individual transcriptlevels were ranked as either increased (I), marginally increased(MI), decreased (D), marginally decreased (MD) or notchanged (NC). To be considered as upregulated, the AbsoluteCall of individual transcripts had to be present, and theDifference Call had to be increased in both duplicate samples.To be considered as downregulated, the Absolute Call ofindividual transcripts had to be present in the control samples(DMSO), and the Difference Call had to be decreased in bothduplicate samples.
Real-time qPCRTotal RNA was extracted using the RNeasy mini Kit (Qiagen).First strand cDNA synthesis was performed using 2mg of totalRNA as template and Moloney murine leukemia virus reversetranscriptase (Invitrogen) primed with 50 pmol of randomhexamers. Quantitative real-time PCR was performedusing the LightCycler (Roche Diagnostics, Basel, Switzerland)and the QuantiTech SYBR Green PCR kit (Qiagen) asper manufacturer’s protocol. Specific primers for eachgene analysed were designed using Primer3 software(http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Senseand antisense primers were: ATCTCAGTGCAATGGCTTCCand CAATGCATTCTCCACACCAG for Bim; GACGACCTCAACGCACAGTA and CACCTAATTGGGCTCCATCT forPUMA; GCCGAGAGAAAACAGTCCAG and GCCGAGAGAAAACAGTCCAG for WAF1; CCCCAAGATCCTGAAACAGA and CCGTTGCTGGACTGGATTAT for cJun;GGCAAGGAGCTGAACAAGAG and GATAGTGGCGTTCCTCTGGA for Hsp70b; GGTCCTGTCTTCAGATGAAAATG and CTTGGTGCAGATTCACCATTC for Chop. Eachprimer pair was tested with a logarithmic dilution of a cDNAmixto generate a linear standard curve, which was used to calculatethe primer pair efficiency. The PCR reactions were performed in20ml volumes with following parameters: 951C for 15minfollowed by 40 cycles of 941C for 20 s, 591C for 20 s, 721C for20 s. The generation of specific PCR products was confirmed bymelting curve analysis and gel electrophoresis. The data wasanalysed using the Lightcycler Software 4.0 with all samplesnormalized to b-actin.
Sodium dodecylsulfate–polyacrylamide gel electrophoresis andWestern blottingPreparation of cell lysates and Western blotting was carriedout as described (Reimertz et al., 2001). The resulting blotswere probed with a mouse monoclonal antiubiquitin antibody(Affiniti, Victoria, Canada) diluted 1:1000, a mouse mono-clonal anti-PUMA/Bbc3 antibody (Han et al., 2001) diluted1:1000, a rabbit polyclonal anti-Bim antibody (AAP-330;StressGen, Victoria, Canada) diluted 1:1000, or a mousemonoclonal anti-a-tubulin antibody (clone DM 1A; Sigma,Dublin, Ireland), diluted 1:5000. Horseradish peroxidaseconjugated secondary antibodies (Pierce, Northumberland,UK) were detected using SuperSignal West Pico Chemi-luminescent Substrate (Pierce) and imaged using a FujiFilmLAS-3000 imaging system (Fuji, Sheffield, UK).
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1689
Oncogene

Determination of caspase-3-like protease activityDEVDase (Caspase-3-like) activity was determined fluoro-metrically using carbobenzoxy-Asp-Glu-Val-Asp-7-amino-4-methyl-coumarin (DEVD-AMC) as substrate. Cleavage ofDEVD-AMC to liberate free AMC was monitored bymeasuring fluorescence after 1 and 2 h intervals. Proteincontent was determined using the Pierce Coomassie PlusProtein assay reagent (Perbio, Northumberland, UK). Caspaseactivity is expressed as change in fluorescent units per hour andper microgram protein.
Transient transfections and luciferase reporter gene assaysCells were transiently transfected in 24-well plates usingMetafectene (Biontex) as per manufacturer’s instruction.Transfection complexes consisted of 190 ng of a plasmidcontaining 0.9 kb of the PUMA promoter (a kind gift from DrT Chittenden) along with 10 ng of a plasmid encoding for theRenilla luciferase (phRL-TK-luc; Promega, Southampton,UK) to normalize for transfection efficiency between experi-ments. At 16–24 h post-transfection cells were treated withEpoxo or vehicle for 24 h. Luciferase activity was mea-sured using the Dual-Glo Luciferase Assay System (Promega)and resultant luminescence monitored using a BertholdLuminometer.
Hoechst staining of nuclear chromatinCells cultured on 24-well plates were stained live with Hoechst33258 (Sigma) at a final concentration of 1 mg/ml. Afterincubation for 10min, nuclear morphology was observed usingan Eclipse TE 300 inverted microscope (Nikon, Dusseldorf,Germany) and a 20 dry objective. For each time point andtreatment, a total number of 300 cells were analysed forapoptotic morphology in three subfields of each culture. Allexperiments were performed at least twice with similar results.
Gene silencing with siRNAThe annealed siRNA duplexes were purchased from SigmaProligo (Paris, France). HCT116 cells were transfectedwith 100nM of the appropriate duplex using Metafectene(Biontex, Martinsried/Planegg, Germany) as per manufacturer’sinstructions. Sequences used were: Bim sense 50-CAAUUGU
CUACCUUCUCGG(dTdT)-30; Bim antisense 50-CCGAGAAGGUAGACAAUUG(dTdT)-30; control sense 50-UUCUCCGAACGUGUCACGU(dTdT)-30; and control antisense 50-ACGUGACACGUUCGGAGAA(dTdT)-30. Twenty-four hourspost-transfection cells were treated with 50 nM Epoxo orvehicle (DMSO) as control for 24 h.
Flow cytometryFollowing treatments cells were stained with annexin V/propidium iodide (BioVision, Mountain View, CA, USA) asper manufacturer’s instructions. Samples were analysedimmediately by flow cytometry. In all cases, a minimum of104 events per sample were acquired. Flow cytometric analyseswere performed on a CyFlow ML (Partec, Munster, Germany)followed by analysis using FloMax software.
StatisticsData are given as means7s.e. For statistical comparison,t-test or one-way analysis of variance followed by Tukeytest were employed using SPSS software (SPSS GmbHSoftware, Munich, Germany). P-values smaller than 0.05 wereconsidered to be statistically significant.
Abbreviations
PUMA, p53 upregulated modulator of apoptosis; GRP78,immunoglobulin heavy chain-binding protein/78 kDa glucose-regulated protein; DMSO, dimethylsulfoxide; CHOP, C/EBP-homologous protein; Epoxo, epoxomicin; WT, wild type.
Acknowledgements
The authors wish to thank Drs B Vogelstein, L Zhang, andJ Yu for p53 and PUMA-deficient cells and Drs P Bouilletand J Adams for gifts of knockout mice. This study wassupported by grants from the DFG (PR 338/9-3 and 9-4) toJHMP and DK and Science Foundation Ireland (03/RP/B344)to JHMP.
References
Adams J. (2002). Proteasome inhibitors as new anticancerdrugs. Curr Opin Oncol 14: 628–634.
Adams J. (2004a). The proteasome: a suitable antineoplastictarget. Nat Rev Cancer 4: 349.
Adams J. (2004b). The proteasome: a suitable antineoplastictarget. Nat Rev Cancer 4: 349–360.
Aghajanian C, Soignet S, Dizon DS, Pien CS, Adams J,Elliott PJ et al. (2002). A phase I trial of the novelproteasome inhibitor PS341 in advanced solid tumormalignancies. Clin Cancer Res 8: 2505–2511.
Amiri KI, Horton LW, LaFleur BJ, Sosman JA, Richmond A.(2004). Augmenting chemosensitivity of malignant melano-ma tumors via proteasome inhibition: implication forbortezomib (VELCADE, PS-341) as a therapeutic agentfor malignant melanoma. Cancer Res 64: 4912–4918.
Anan A, Baskin-Bey ES, Bronk SF, Werneburg NW,Shah VH, Gores GJ. (2006). Proteasome inhibition induceshepatic stellate cell apoptosis. Hepatology 43: 335–344.
Biswas SC, Ryu E, Park C, Malagelada C, Greene LA.(2005). Puma and p53 play required roles in death evoked ina cellular model of Parkinson disease. Neurochem Res 30:839–845.
Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW,Kontgen F et al. (1999). Proapoptotic Bcl-2 relativeBim required for certain apoptotic responses, leukocytehomeostasis, and to preclude autoimmunity. Science 286:1735–1738.
Breitschopf K, Zeiher AM, Dimmeler S. (2000). Ubiquitin-mediated degradation of the proapoptotic active form ofbid. A functional consequence on apoptosis induction. J BiolChem 275: 21648–21652.
Ceballos E, Munoz-Alonso MJ, Berwanger B, Acosta JC,Hernandez R, Krause M et al. (2005). Inhibitory effect ofc-Myc on p53-induced apoptosis in leukemia cells. Microarrayanalysis reveals defective induction of p53 target genes andupregulation of chaperone genes. Oncogene 24: 4559–4571.
Chauhan D, Hideshima T, Anderson KC. (2005). Proteasomeinhibition in multiple myeloma: therapeutic implication.Annu Rev Pharmacol Toxicol 45: 465–476.
Chowdary DR, Dermody JJ, Jha KK, Ozer HL. (1994).Accumulation of p53 in a mutant cell line defective in theubiquitin pathway. Mol Cell Biol 14: 1997–2003.
Dang CV. (1999). c-Myc target genes involved in cell growth,apoptosis, and metabolism. Mol Cell Biol 19: 1–11.
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1690
Oncogene

Ganten TM, Koschny R, Haas TL, Sykora J, Li-Weber M,Herzer K et al. (2005). Proteasome inhibition sensitizeshepatocellular carcinoma cells, but not human hepatocytes,to TRAIL. Hepatology 42: 588–597.
Han J-w, Flemington C, Houghton AB, Gu Z, Zambetti GP,Lutz RJ et al. (2001). Expression of bbc3, a pro-apoptoticBH3-only gene, is regulated by diverse cell death andsurvival signals. Proc Natl Acad Sci USA 98: 11318–11323.
Hanada M, Sugawara K, Kaneta K, Toda S, Nishiyama Y,Tomita K et al. (1992). Epoxomicin, a new antitumor agentof microbial origin. J Antibiotics, 1746–1752.
Hideshima T, Chauhan D, Richardson P, Mitsiades C,Mitsiades N, Hayashi T et al. (2002). NF-kappa B as atherapeutic target in multiple myeloma. J Biol Chem 277:16639–16647.
Huang H-k, Joazeiro CAP, Bonfoco E, Kamada S,Leverson JD, Hunter T. (2000). The inhibitor of apoptosis,cIAP2, functions as a ubiquitin-protein ligase and promotesin vitro monoubiquitination of caspases 3 and 7. J Biol Chem275: 26661–26664.
Karin M. (2006). Nuclear factor-[kappa]B in cancer develop-ment and progression. Nature 441: 431.
Karin M, Ben-Neriah Y. (2000). Phosphorylation meetsubiquitination: the control of NF-[kappa]B activity. AnnuRev Immunol 18: 621–663.
Kikuchi S, Shinpo K, Tsuji S, Takeuchi M, Yamagishi S,Makita Z et al. (2003). Effect of proteasome inhibitor oncultured mesencephalic dopaminergic neurons. Brain Res964: 228.
Kurland JF, Meyn RE. (2001). Protease inhibitors restoreradiation-induced apoptosis to Bcl-2-expressing lymphomacells. Int J Cancer 96: 327–333.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C,Sullivan BA, Green DR et al. (2005). BH3 domains ofBH3-only proteins differentially regulate Bax-mediatedmitochondrial membrane permeabilization both directlyand indirectly. Mol Cell 17: 525–535.
Leverkus M, Sprick MR, Wachter T, Mengling T, Baumann B,Serfling E et al. (2003). Proteasome inhibition results inTRAIL sensitization of primary keratinocytes by removingthe resistance-mediating block of effector caspase matura-tion. Mol Cell Biol 23: 777–790.
Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ.(2003). Activation of the ERK1/2 signaling pathwaypromotes phosphorylation and proteasome-dependent de-gradation of the BH3-only protein, Bim. J Biol Chem 278:18811–18816.
Luo JL, Kamata H, Karin M. (2005). IKK/NF-kappaBsignaling: balancing life and death – a new approach tocancer therapy. J Clin Invest 115: 2625–2632.
Marshansky V, Wang X, Bertrand R, Luo H, Duguid W,Chinnadurai G et al. (2001). Proteasomes modulate balanceamong proapoptotic and antiapoptotic Bcl-2 family mem-bers and compromise functioning of the electron transportchain in leukemic cells. J Immunol 166: 3130–3142.
Masdehors P, Merle-Beral H, Maloum K, Omura S,Magdelenat H, Delic J. (2000). Deregulation of the ubiquitinsystem and p53 proteolysis modify the apoptotic response inB-CLL lymphocytes. Blood 96: 269–274.
Meiners S, Heyken D, Weller A, Ludwig A, Stangl K,Kloetzel PM et al. (2003). Inhibition of proteasome activityinduces concerted expression of proteasome genes andde novo formation of Mammalian proteasomes. J Biol Chem278: 21517–21525.
Melino G. (2005). Discovery of the ubiquitin proteasomesystem and its involvement in apoptosis. 12: 1155.
Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM.(1999). Epoxomicin, a potent and selective proteasomeinhibitor, exhibits in vivo antiinflammatory activity. ProcNatl Acad Sci USA 96: 10403–10408.
Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D,Fanourakis G, Gu X et al. (2002). Molecular sequelaeof proteasome inhibition in human multiple myeloma cells.99: 14374–14379.
Nakano K, Vousden KH. (2001). PUMA, a novel proapopto-tic gene, is induced by p53. Mol Cell 7: 683–694.
Nikrad M, Johnson T, Puthalalath H, Coultas L, Adams J,Kraft AS. (2005). The proteasome inhibitor bortezomibsensitizes cells to killing by death receptor ligand TRAILvia BH3-only proteins Bik and Bim. Mol Cancer Ther 4:443–449.
Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS,Santee SM, Fujiwara T et al. (1995). Wild-type human p53and a temperature-sensitive mutant induce Fas/APO-1expression. Mol Cell Biol 15: 3032–3040.
Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. (1994).The ubiquitinproteasome pathway is required for processingthe NF-[kappa]B1 precursor protein and the activation ofNF-[kappa]B. Cell 78: 773.
Papandreou CN, Daliani DD, Nix D, Yang H, Madden T,Wang X et al. (2004). Phase I trial of the proteasomeinhibitor bortezomib in patients with advanced solid tumorswith observations in androgen-independent prostate cancer.J Clin Oncol 22: 2108–2121.
Park DJ, Lenz HJ. (2004). The role of proteasome inhibitors insolid tumors. Ann Med 36: 296–303.
Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK,Chaturvedi V et al. (2005). Proteasome inhibitors triggerNOXA-mediated apoptosis in melanoma and myeloma cells.Cancer Res 65: 6282–6293.
Reimertz C, Kogel D, Lankiewicz S, Poppe M, Prehn JH.(2001). Ca(2+)-induced inhibition of apoptosis in humanSH-SY5Y neuroblastoma cells: degradation of apoptoticprotease activating factor-1 (APAF-1). J Neurochem 78:1256–1266.
Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH.(2003). Gene expression during ER stress-induced apoptosisin neurons: induction of the BH3-only protein Bbc3/PUMAand activation of the mitochondrial apoptosis pathway. JCell Biol 162: 587–597.
Richardson PG, Barlogie B, Berenson J, Singhal S,Jagannath S, Irwin D et al. (2003). A phase 2 study ofbortezomib in relapsed, refractory myeloma. N Engl J Med348: 2609–2617.
Shinohara K, Tomioka M, Nakano H, Tone S, Ito H,Kawashima S. (1996). Apoptosis induction resulting fromproteasome inhibition. Biochem J 317(Part 2): 385–388.
Suzuki Y, Nakabayashi Y, Takahashi R. (2001). Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosisprotein promotes proteasomal degradation of caspase-3 andenhances its anti-apoptotic effect in Fas-induced cell death.Proc Natl Acad Sci USA 98: 8662–8667.
Tan T-T, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P et al. (2005). Key roles of BIM-drivenapoptosis in epithelial tumors and rational chemotherapy.Cancer Cell 7: 227.
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G,Ausserlechner MJ et al. (2003a). p53- and drug-inducedapoptotic responses mediated by BH3-only proteins Pumaand Noxa. Science 302: 1036–1038.
Villunger A, Scott C, Bouillet P, Strasser A. (2003b). Essentialrole for the BH3-only protein Bim but redundant roles for
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1691
Oncogene

Bax, Bcl-2, and Bcl-w in the control of granulocyte survival.Blood 101: 2393–2400.
Wagenknecht B, Hermisson M, Eitel K, Weller M. (1999).Proteasome inhibitors induce p53/p21-independent apopto-sis in human glioma cells. Cell Physiol Biochem 9: 117–125.
Wagenknecht B, Hermisson M, Groscurth P, Liston P,Krammer PH, Weller M. (2000). Proteasome inhibitor-induced apoptosis of glioma cells involves the processing ofmultiple caspases and cytochrome c release. J Neurochem 75:2288–2297.
Willis SN, Adams JM. (2005). Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol 17: 617.
Wong HK, Fricker M, Wyttenbach A, Villunger A, MichalakEM, Strasser A et al. (2005). Mutually exclusive subsetsof BH3-only proteins are activated by the p53 and c-JunN-terminal kinase/c-Jun signaling pathways during corticalneuron apoptosis induced by arsenite. Mol Cell Biol 25:8732–8747.
Wu GS, Burns TF, McDonald III ER, Jiang W, Meng R,Krantz ID et al. (1997). KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17:141–143.
Yan C, Lu D, Hai T, Boyd DD. (2005). Activatingtranscription factor 3, a stress sensor, activates p53 byblocking its ubiquitination. EMBO J 24: 2425–2435.
Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL et al.(2005). Proteasome inhibitor PS-341 causes cell growtharrest and apoptosis in human glioblastoma multiforme(GBM). Oncogene 24: 344–354.
Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L.(2003). PUMA mediates the apoptotic response top53 in colorectal cancer cells. Proc Natl Acad Sci USA100: 1931–1936.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. (2001).PUMA induces the rapid apoptosis of colorectal cancercells. Mol Cell 7: 673–682.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
PUMA and proteasome inhibition induced apoptosisCG Concannon et al
1692
Oncogene
Related Documents











