Anticancer agents against malaria: time to revisit? Alexis Nzila 1,2 , John Okombo 1 , Ruy Perez Becker 3 , Roma Chilengi 1,2 , Trudie Lang 1,2 , and Tim Niehues 3 Alexis Nzila: [email protected] 1 Kenya Medical Research Institute (KEMRI)/Wellcome Trust Collaborative Research Programme, PO Box 230, 80108, Kilifi, Kenya 2 University of Oxford, Nuffield Department of Medicine, John Radcliffe Hospital, Oxford, UK 3 Helios Klinikum Krefeld Academic Hospital, Lutherplatz 40, 47805 Krefeld, Germany Abstract The emergence of artemisinin resistance could adversely impact the current strategy for malaria treatment; thus, new drugs are urgently needed. A possible approach to developing new antimalarials is to find new uses for old drugs. Some anticancer agents such as methotrexate and trimetrexate are active against malaria. However, they are commonly perceived to be toxic and thus not suitable for malaria treatment. In this opinion article, we examine how the toxicity of anticancer agents is just a matter of dose or ‘only dose makes the poison’, as coined in Paracelsus’ law. Thus, the opportunity exists to discover new antimalarials using the anticancer pharmacopoeia. The need for new antimalarial drugs The malaria parasite, the cause of one of the most significant infectious diseases, is characterised by an intrinsic ability to select for resistance against drugs. To slow down the pace of resistance selection, it has been recommended that antimalarials be used in combinations of at least two drugs with different modes of action, and the current recommendation is to use artemesinin-based combinations [1]. However, recent reports indicate that resistance to artemesinin is emerging in Southeast Asia, and there are concerns that the therapeutic life span of artemisinin combinations might be compromised [2]. The choices for combination therapy could be reduced and, as a result, drug resistance would remain a limitation to sustaining effective malaria treatment. One approach for discovering new antimalarials is to re-use drugs developed for the treatment of other diseases. The literature is replete with examples of new uses for old drugs. For example, among the few drugs that are available in the treatment of malaria, quinine is used to treat muscle cramps [3], and chloroquine (CQ) (preferably hydroxyl-chloroquine) is used for the management of rheumatoid arthritis and systemic lupus erythematosus [4]. Artemisinin is being investigated for the treatment of schistosomiasis and cancer [5,6]. However, thus far, there is no drug that has been re-used for acute malarial treatment, in spite of the richness of the human pharmacopoeia. Only drugs, such as the antibiotic doxycycline, azithromycin and erythromycin, have been used, or are currently being investigated, for prophylaxis against malaria [7,8]. Dapsone, a drug used to treat leprosy and dermatitis herpetiformis [9], was combined with chlorproguanil (Lapdap ® ) [10]; however, this combination has been phased out because of dapsone toxicity. Antimalarial potential of methotrexate and trimetrexate, in vitro There is evidence that anticancer compounds methotrexate (MTX) and trimetrexate (TMX) are active against both pyrimethamine (PM)-sensitive and PM-resistant laboratory strains and Sponsored document from Trends in Parasitology Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129. Sponsored Document Sponsored Document Sponsored Document

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Anticancer agents against malaria: time to revisit?
Alexis Nzila1,2, John Okombo1, Ruy Perez Becker3, Roma Chilengi1,2, Trudie Lang1,2, andTim Niehues3Alexis Nzila: [email protected] Medical Research Institute (KEMRI)/Wellcome Trust Collaborative Research Programme,PO Box 230, 80108, Kilifi, Kenya2University of Oxford, Nuffield Department of Medicine, John Radcliffe Hospital, Oxford, UK3Helios Klinikum Krefeld Academic Hospital, Lutherplatz 40, 47805 Krefeld, Germany
AbstractThe emergence of artemisinin resistance could adversely impact the current strategy for malariatreatment; thus, new drugs are urgently needed. A possible approach to developing new antimalarialsis to find new uses for old drugs. Some anticancer agents such as methotrexate and trimetrexate areactive against malaria. However, they are commonly perceived to be toxic and thus not suitable formalaria treatment. In this opinion article, we examine how the toxicity of anticancer agents is just amatter of dose or ‘only dose makes the poison’, as coined in Paracelsus’ law. Thus, the opportunityexists to discover new antimalarials using the anticancer pharmacopoeia.
The need for new antimalarial drugsThe malaria parasite, the cause of one of the most significant infectious diseases, ischaracterised by an intrinsic ability to select for resistance against drugs. To slow down thepace of resistance selection, it has been recommended that antimalarials be used incombinations of at least two drugs with different modes of action, and the currentrecommendation is to use artemesinin-based combinations [1]. However, recent reportsindicate that resistance to artemesinin is emerging in Southeast Asia, and there are concernsthat the therapeutic life span of artemisinin combinations might be compromised [2]. Thechoices for combination therapy could be reduced and, as a result, drug resistance would remaina limitation to sustaining effective malaria treatment.
One approach for discovering new antimalarials is to re-use drugs developed for the treatmentof other diseases. The literature is replete with examples of new uses for old drugs. For example,among the few drugs that are available in the treatment of malaria, quinine is used to treatmuscle cramps [3], and chloroquine (CQ) (preferably hydroxyl-chloroquine) is used for themanagement of rheumatoid arthritis and systemic lupus erythematosus [4]. Artemisinin isbeing investigated for the treatment of schistosomiasis and cancer [5,6]. However, thus far,there is no drug that has been re-used for acute malarial treatment, in spite of the richness ofthe human pharmacopoeia. Only drugs, such as the antibiotic doxycycline, azithromycin anderythromycin, have been used, or are currently being investigated, for prophylaxis againstmalaria [7,8]. Dapsone, a drug used to treat leprosy and dermatitis herpetiformis [9], wascombined with chlorproguanil (Lapdap®) [10]; however, this combination has been phasedout because of dapsone toxicity.
Antimalarial potential of methotrexate and trimetrexate, in vitroThere is evidence that anticancer compounds methotrexate (MTX) and trimetrexate (TMX)are active against both pyrimethamine (PM)-sensitive and PM-resistant laboratory strains and
Sponsored document fromTrends in Parasitology
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent

field isolates of Plasmodium falciparum, including those carrying the Ileu164-Leu dhfr codon(dihydrofolate reductase, DHFR), with IC50 < 85 nM (inhibitor concentration that kills 50%of parasitaemia) [11], and IC90/99 values for these folate antagonist agents fall between 150and 350 nM; thus, if such a concentration can be achieved in vivo with an acceptable toxicityprofile, these compounds could potentially be used as antimalarials. However, anticanceragents in general, and MTX in particular, are perceived to be toxic and therefore not suitablefor malaria treatment. Yet the literature is replete with examples of new uses of anticanceragents in the treatment of non-neoplastic diseases (Table 1).
Anticancer toxicity and Paracelsus’ lawThe inhibition of pathways or enzymes in tumour cells also affects normal human cells. Someanticancer agents are directed against cancer-specific targets [e.g. Imatinib mesylate(Gleevec™) targets cancer-specific tyrosine kinases] and thus can be used at doses with arelatively better toxicity profile than most drugs [12]. However, most anticancer agents areused at doses that also lead to the inhibition of growth of normal cells, in addition to blockingtumourous cells. Most specifically affected are cells that multiply actively, such as bonemarrow cells, e.g. leukocytes, cells of the gastrointestinal mucosa and hair follicle cells,explaining why bone marrow suppression, mucositis and alopecia (hair loss) are among themost common side effects of anticancer compounds. However, according to Paracelsus’ law,for any drug (including anticancers), there is always a dose range at which a drug is safe.
Paracelsus’ law states ‘Sola dosis facit venenum (only dose makes the poison)’, meaning thatall substances are poisons and there are none which are not. The right dose differentiates apoison from a remedy; this principle is also known as the ‘dose–response effect’ [13,14] (Figure1). Thus, a molecule becomes a drug if the dose required to treat a complication ispharmacologically active with minimal toxicity.
The example of CQ is noteworthy. CQ is used at 10 mg kg–1 as a starting dose on days 1 and2, and at 5 mg kg–1 on day 3 [15]. At this dose regimen, CQ has an acceptable safety profile.However, a dose of 20 mg kg–1 is considered toxic [15], and fatal cases have been reportedfrom doses as low as 30 mg kg–1, only three times higher than the therapeutic dose [16,17].This indicates that a slight dose increase shifts the effect of CQ from the second range(acceptable safety profile with a pharmacological effect) to the third range (life-threateningtoxicity) (Figure 1). Thus, CQ has a very low safety margin, and yet it has been used widely(at the correct dose) and is considered to be one of the safest antimalarial agents available.
Different uses of MTX in humansMTX is another interesting example. MTX is used at high dose, up to 5000–12,000 mg persquare meter (m2) per week (130–300 mg kg–1) for several weeks for the treatment of cancer,and this dose can yield serum concentrations of >1000 μM, i.e. within the range ofconcentrations that is associated with MTX life-threatening toxicity [18]. By contrast, a 1000-fold lower dose of MTX (LD–MTX) [0.1–0.4 mg kg–1 (7.5–30 mg per adult)] is used onceweekly in the treatment of rheumatoid arthritis (RA), juvenile idiopathic arthritis in children(including infants <1 year old) and psoriasis [19,20].
The most common side effect of LD–MTX is mucositis (oral ulcers) and gastrointestinal (GI)tract disturbances, particularly nausea. Commonly, these adverse effects are observed afterseveral weeks and somewhat higher doses of MTX (more than 15 mg in adults) [19]. Indeed,in the treatment of adult RA, MTX starts at a dose of 7.5 mg once weekly. After a few weeks,this dose is increased by 2.5 mg per week (the timing of the increase could vary, depending onhow the patient is responding to the treatment), to reach a final dose of 25–35 mg per week.The toxicity of MTX is observed when it is used at doses >7.5 mg per week and several weeks
Nzila et al. Page 2
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent

after the first drug administration. The toxicity of MTX is a result of inhibition of the DHFRenzyme in normal cells. To prevent toxicity, a folate derivative is administered several hoursafter the dose of MTX. The addition of folate derivatives increases either dihydrofolate ortetrahydrofolate (DHFR substrate and product, respectively). In either case, the action of MTXagainst DHFR will be minimal because of the high folate content, leading to normal synthesisof pyrimidine and the restoration of cell growth. In juvenile arthritis, MTX is used at a higherdose, and there is still debate over the benefit of the addition of a folate derivative becausemucositis and GI tract disturbances are rare: this drug is better tolerated by children than byadults [19], probably as a result of high cell multiplication processes in children. Overall, it isclear that the toxicity of LD–MTX result from chronic use of doses >7.5 mg per week, as hasbeen clearly demonstrated with the use of MTX in the treatment of multiple sclerosis [21].
MTX is also being evaluated in the treatment of various disease conditions includinginflammatory bowel disease [22], urticaria [23], ankylosing spondylitis [24], idiopathichypertrophic cranial pachymeningitis [25], chronic cholestatic disorder [26], Wegener'sgranulomatosis [27], primary biliary cirrhosis [28], systemic lupus erythematosus [29] andinflammatory eye disease [30], haemophagocytic lymphohistiocytosis (HLH), a disease thataffects younger children, including infants (<12 months of age) [31].
Worldwide, it is estimated that 0.5–1 million adults and 50,000–100,000 children receive LD–MTX weekly for the treatment of RA and juvenile idiopathic arthritis, respectively. The drugis now being used in the African population, and its safety profile is similar to that reported inthe Western world [32].
Proof of concept of MTX as an antimalarial in humansThe antimalarial potential of MTX has been established for almost 40 years. Two relativelysmall clinical trials, involving seven patients, have demonstrated that doses as low as 2.5 mgper day for 3–5 days were effectively treating malaria infection in humans (Plasmodiumfalciparum and/or Plasmodium vivax) [33,34]. However, MTX has not come into widespreaduse because of concerns over toxicity [35,36]. At the time of the clinical trials (in the 1970 s),no information was available on the safety of LD–MTX. Indeed, LD–MTX started to be usedfor the treatment of arthritis from the 1980 s; before then, MTX was used only at high doses,associated with toxicity in cancer treatment. We now have 30 years experience on the safetyof LD–MTX in adults and in children, and thus this drug could now be re-evaluated as apotential antimalarial. Unlike its use in immune diseases, MTX would not be used on a chronicbasis against malaria; it would be used for 3–5 days only and thus the risk of toxicity wouldbe even lower.
The in vivo efficacy of LD–MTX is also supported by pharmacokinetics. Indeed, a daily doseof 5 mg in adults (0.035–0.1 mg kg–1) could yield serum MTX concentrations between 250and 500 nM [37,38], concentrations which exceed the IC90/99 concentrations required to killthe parasite in vitro [11]. Taken together, the information warrants further investigation of thisdrug as an antimalarial.
Potential of trimetrexate (TMX) as an antimalarialTMX is used primarily for the treatment of solid tumours [39]. Evidence is also available thatTMX has good activity against P. falciparum, and the addition of the folate derivative 5-methyltetrahydrofolate (5-Me-THF) does not reduce TMX activity [40]. Thus, this form of folatecould be used as an adjuvant, in combination with TMX, to increase its safety margin. 5-Me-THF would protect the host against drug toxicity and it would not negate the antimalarialactivity of TMX. The same rationale has been used in the combination TMX + folinic acid(FNA) for the treatment of Pneumocystis jiroveci infection (an opportunistic infection
Nzila et al. Page 3
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent

commonly found in association with HIV infection). TMX is active against P. jiroveci and thismicroorganism does not salvage folate derivatives because of lack of folate receptors andtransporters; thus, the addition of folate derivatives does not negate TMX activity. As a result,the combination of TMX + FNA is as active as TMX alone [41]. In cancer, a TMX dose of100 mg per day is used for several weeks and this is associated with toxicity. In P. jiroveciinfection, the same dose is used (100 mg per day for 28 days) but the addition of 200 mg ofFNA completely reverses the toxicity of the drug, making the combination TMX + FNA welltolerated. As a result, TMX has become useful against P. jiroveci infection [42].
For the treatment of malaria, a much lower dose of TMX would be required. Indeed, 100 mgof TMX yields plasma concentrations of ∼5000 nM [43] and this is ∼100 times higher thanthe IC50 value (i.e. <50 nM) of TMX against P. falciparum. Thus, it is reasonable to proposethat doses <10–20 mg of TMX would be effective to treat malaria. Such a low dose could besafe and the addition of 5-CH3-THF could even improve the therapeutic index of the drug.Although the potential exists for TMX to become an antimalarial, more work is needed toestablish whether this concept could be translated in vivo.
Other anticancer drugs in the treatment of malaria and other non-neoplasticdiseases
We have also shown that the folate antagonist pemetrexate is active against P. falciparum invitro, with activity in the nanomolar range (IC50 <50 nM) (A. Nzila et al. unpublished). Becausethe anticancers MTX, aminopterin and pemetrexate, all inhibitors of DHFR enzymes, are activeagainst P. falciparum, other anticancer inhibitors of DHFR, such as edatrexate, pralatrexate,and piritrexim might also be active against P. falciparum. Several other anticancer agents havealso been shown to have activity against P. falciparum malaria. For example, the inhibitors ofthe microtubulin assembly tubulozole, vinblastine, docetaxel, paclitaxel and dolastatin [44–48], the DNA crosslinking agent cisplatin [49] and the proteasome inhibitor Bortezomib [50]are effective antimalarials in vitro. If these drugs were active in vivo at low and tolerable doses,they could potentially become antimalarials. This is all the more possible in view of the factthat many anticancer drugs have already been used in the treatment of non-neoplastic diseasesat low dose (Table 1).
Concluding remarksThe burgeoning problem of antimalarial resistance highlights the need to have a strong pipelineof new drugs to treat malaria. Development of new drugs takes time and is associated withsignificant costs, explaining (at least partly) the paucity of available antimalarials, particularlyas this is not a profitable market and thus there is little incentive for the pharmaceutical industryto develop new treatments. The re-use of existing drugs is an attractive strategy to discovernew antimalarials as the development costs would be lower and the time to licensure shorter.Moreover, in the case of anticancer agents, the in-human toxicity profile would have alreadybeen well documented at much higher doses than could ever be achieved for any other productprimarily developed for malaria. Many antineoplastic drugs, including MTX, were shown tobe effective against the malaria parasite almost 40 years ago; however, their perceived toxicityhas prevented their development as antimalarials. It is now known, and as stated by Paracelsus(more than 400 years ago), that it is ‘the dose that makes a drug’; this principle has beenexploited in the use of such drugs at lower doses in several non-neoplastic diseases. It issurprising that the antimalarial potential of MTX has not been fully investigated. Exploitationof the available pharmacopoeia could be a cost-effective and efficient channel to develop muchneeded alternative treatments for challenging diseases such as malaria.
Nzila et al. Page 4
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent

References1. NostenF.WhiteN.J.Artemisinin-based combination treatment of falciparum malariaAm. J. Trop. Med.
Hyg.772007181192181654912. DondorpA.M.Artemisinin resistance in Plasmodium falciparum malariaN. Engl. J. Med.
3612009455467196412023. MillerT.M.LayzerR.B.Muscle crampsMuscle Nerve322005431442159026914. SibiliaJ.PasqualiJ.L.Systemic lupus erythematosus: news and therapeutic perspectivesPresse Med.
372008444459182420455. EfferthT.Willmar Schwabe Award 2006: Antiplasmodial and antitumor activity of artemisinin – from
bench to bedsidePlanta Med.732007299309173541636. UtzingerJ.Artemisinins for schistosomiasis and beyondCurr. Opin. Investig. Drugs820071051167. ChicoR.M.Azithromycin-chloroquine and the intermittent preventive treatment of malaria in
pregnancyMalar. J.72008255190872678. Croft, A.M. (2005) Malaria: prevention in travellers. Clin. Evid. 954–9729. WolfR.DapsoneDermatol. Online J8200221216521210. NzilaA.The past, present and future of antifolates in the treatment of Plasmodium falciparum
infectionJ. Antimicrob. Chemother.572006104310541661706611. KiaraS.M.In vitro activity of antifolate and polymorphism in dihydrofolate reductase of Plasmodium
falciparum isolates from the Kenyan coast: emergence of parasites with Ile-164-LeumutationAntimicrob. Agents Chemother.5320093793379819528269
12. PytelD.Tyrosine kinase blockers: new hope for successful cancer therapyAnticancer Agents Med.Chem.92009667619149483
13. LangmanL.J.KapurB.M.Toxicology: then and nowClin. Biochem.3920064985101673025414. RozmanK.K.DoullJ.Paracelsus, Haber and ArndtToxicology16020011911961124613915. TaylorW.R.WhiteN.J.Antimalarial drug toxicity: a reviewDrug Safety27200425611472008516. CannH.M.VerhulstH.L.Fatal acute chloroquine poisoning in
childrenPediatrics271961951021369044517. RiouB.Treatment of severe chloroquine poisoningN. Engl. J. Med.318198816333637918. ChabnerB.A.Antineoplastic agentsBruntonL.The Pharmacological Basis of Therapeutics9th
edn2006McGraw–Hill1315146519. NiehuesT.LankischP.Recommendations for the use of methotrexate in juvenile idiopathic
arthritisPaediatr. Drugs820063473561715464220. SwierkotJ.SzechinskiJ.Methotrexate in rheumatoid arthritisPharmacol. Rep.
5820064734921696379321. GrayO.M.A systematic review of oral methotrexate for multiple sclerosisMultiple
Sclerosis1220065075101690076622. DomenechE.Long-term methotrexate for Crohn's disease: safety and efficacy in clinical practiceJ.
Clin. Gastroenterol.4220083953991827789923. Montero MoraP.Autoimmune urticaria. Treatment with methotrexateRev. Alerg. Mex.
5120041671721579440524. ChenJ.Is methotrexate effective for the treatment of ankylosing spondylitis?Nat. Clin.
Practice3200749049125. BosmanT.Idiopathic hypertrophic cranial pachymeningitis treated by oral methotrexate: a case report
and review of literatureRheumatol. Int.2820087137181809497126. NovakK.SwainM.G.Role of methotrexate in the treatment of chronic cholestatic disordersClin. Liver
Dis.1220088196viii1824249827. SpecksU.Methotrexate for Wegener's granulomatosis: what is the evidence?Arthritis Rheum.
522005223722421605254028. GongY.GluudC.Methotrexate for primary biliary cirrhosisCochrane Database Syst. Rev.
2005CD0043851603492929. WongJ.M.EsdaileJ.M.Methotrexate in systemic lupus erythematosusLupus14200510110515751813
Nzila et al. Page 5
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent

30. StawellR.Methotrexate in inflammatory eye diseaseOcul. Immunol. Inflamm.11200379821453302631. HenterJ.I.HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic
lymphohistiocytosisPediatr. Blood Cancer4820071241311693736032. TahiriL.Therapeutic maintenance level of methotrexate in rheumatoid
arthritisSante1620061671721728439233. SheehyT.W.DempseyH.Methotrexate therapy for Plasmodium vivax malariaJ. Am. Med. Assoc.
214197010911434. WildbolzA.Methotrexate in the therapy of malariaTher. Umsch.301973218222456962935. FeroneR.Methotrexate therapy for P. vivax malariaJ. Am. Med. Assoc.215197111736. LaingA.B.Methotrexate in malariaTrans. R. Soc. Trop. Med. Hyg.661972518519504638937. ChladekJ.Low-dose methotrexate pharmacokinetics and pharmacodynamics in the therapy of severe
psoriasisBasic Clin. Pharmacol. Toxicol.9620052472481573322438. ChladekJ.Pharmacokinetics and pharmacodynamics of low-dose methotrexate in the treatment of
psoriasisBr. J. Clin. Pharmacol.5420021471561220763439. HallerD.G.Trimetrexate: experience with solid tumorsSemin. Oncol.241997S18-71-S18-7640. NduatiE.Effect of folate derivatives on the activity of antifolate drugs used against malaria and
cancerParasitol. Res.1022008122712341825977641. WalzerP.D.Activities of antifolate, antiviral, and other drugs in an immunosuppressed rat model of
Pneumocystis carinii pneumoniaAntimicrob. Agents Chemother.36199219351942141688442. FultonB.Trimetrexate. A review of its pharmacodynamic and pharmacokinetic properties and
therapeutic potential in the treatment of Pneumocystis cariniipneumoniaDrugs4919955635767789290
43. MarshallJ.L.DeLapR.J.Clinical pharmacokinetics and pharmacology of trimetrexateClin.Pharmacokinet.2619941902008194282
44. FennellB.J.Effects of the antimitotic natural product dolastatin 10, and related peptides, on the humanmalarial parasite Plasmodium falciparumJ. Antimicrob. Chemother.51200383384112654761
45. KokaS.Influence of paclitaxel on parasitemia and survival of Plasmodium berghei infected miceCellPhysiol. Biochem.23200919119819255513
46. SchrevelJ.Interactions between docetaxel (Taxotere) and Plasmodium falciparum-infectederythrocytesProc. Natl. Acad. Sci. U.S.A.911994847284767915841
47. SinouV.In vitro and in vivo inhibition of erythrocytic development of malarial parasites bydocetaxelAntimicrob. Agents Chemother.4019963583618834880
48. UsangaE.A.Mitotic inhibitors arrest the growth of Plasmodium falciparumFEBS Lett.209198623273542560
49. NairL.BhasinV.K.Cure with cisplatin (II) or murine malaria infection and in vitro inhibition of achloroquine-resistant Plasmodium falciparum isolateJpn. J. Med. Sci. Biol.4719942412527739147
50. KreidenweissA.Comprehensive study of proteasome inhibitors against Plasmodium falciparumlaboratory strains and field isolates from GabonMalar. J.7200818718816382
51. NanniniC.Effects of cyclophosphamide on pulmonary function in patients with scleroderma andinterstitial lung disease: a systematic review and meta-analysis of randomized controlled trials andobservational prospective cohort studiesArthritis Res. Ther.102008R12418937831
52. PrefontaineE.Azathioprine or 6-mercaptopurine for maintenance of remission in Crohn'sdiseaseCochrane Database Syst. Rev.2009CD00006719160175
53. WuJ.J.Thalidomide: dermatological indications, mechanisms of action and side-effectsBr. J.Dermatol.153200525427316086735
54. MateosJ.Vincristine is an effective therapeutic approach for transplantation-associated thromboticmicroangiopathyBone Marrow Transplant.37200633733816341229
55. BalasegaramM.Effectiveness of melarsoprol and eflornithine as first-line regimens for gambiensesleeping sickness in nine Medecins Sans Frontieres programmesTrans. R. Soc. Trop. Med. Hyg.103200928029018947846
56. HickmanJ.G.Human dermal safety studies with eflornithine HCl 13.9% cream (Vaniqa), a noveltreatment for excessive facial hairCurr. Med. Res. Opin.16200123524411268707
Nzila et al. Page 6
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent

57. BermanJ.J.Treatment of leishmaniasis with miltefosine: 2008 statusExpert Opin. Drug Metab.Toxicol.420081209121618721114
AcknowledgmentsWe thank the director of the Kenya Medical Research Institute for permission to publish this manuscript. This studywas supported by Pfizer-Royal Society Award, UK, the EU Commission under Framework 6 as part of the AntiMalIntegrated Project 018834, and the European and Developing Countries Clinical Trials Partnership (EDCPT) to A.N.We also thank Andrea Groth for reading and editing the manuscript.
Nzila et al. Page 7
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent
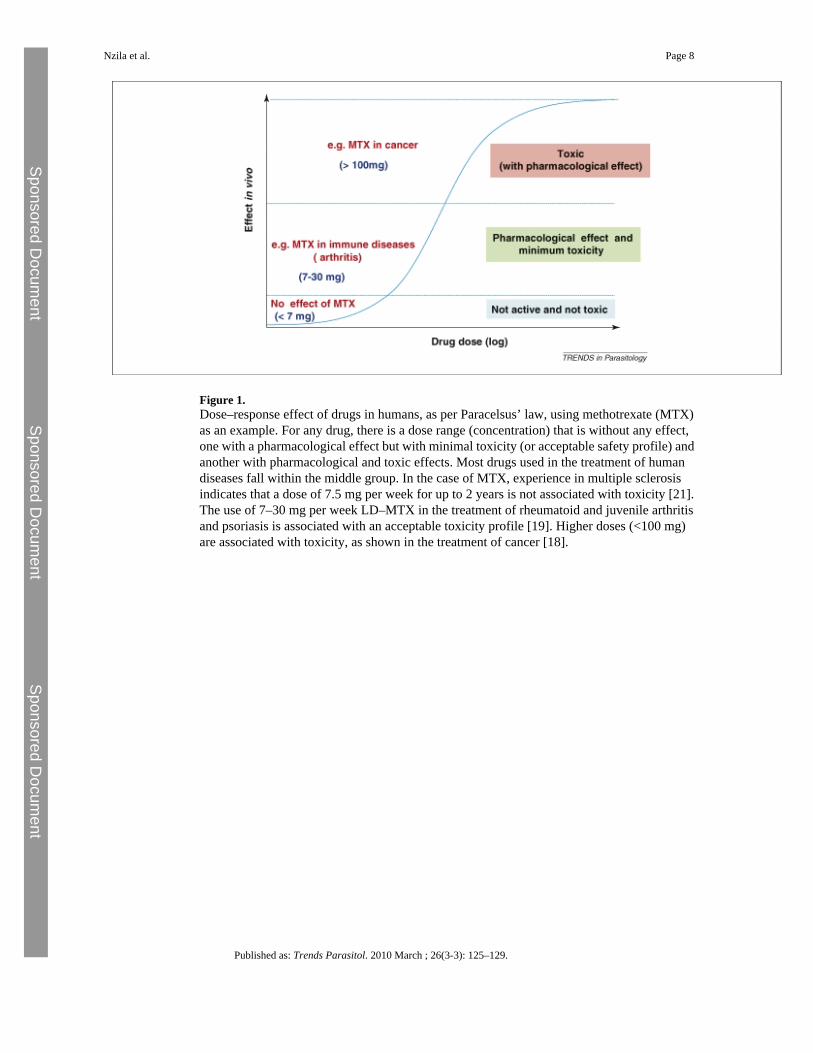
Figure 1.Dose–response effect of drugs in humans, as per Paracelsus’ law, using methotrexate (MTX)as an example. For any drug, there is a dose range (concentration) that is without any effect,one with a pharmacological effect but with minimal toxicity (or acceptable safety profile) andanother with pharmacological and toxic effects. Most drugs used in the treatment of humandiseases fall within the middle group. In the case of MTX, experience in multiple sclerosisindicates that a dose of 7.5 mg per week for up to 2 years is not associated with toxicity [21].The use of 7–30 mg per week LD–MTX in the treatment of rheumatoid and juvenile arthritisand psoriasis is associated with an acceptable toxicity profile [19]. Higher doses (<100 mg)are associated with toxicity, as shown in the treatment of cancer [18].
Nzila et al. Page 8
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent

Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent
Nzila et al. Page 9
Tabl
e 1
Sele
cted
onc
olog
ic d
rugs
use
d in
the
treat
men
t of n
on-n
eopl
astic
dis
ease
sa
Dru
gM
echa
nism
of a
ctio
nN
eopl
asm
(onc
olog
y)N
on-n
eopl
asm
Ref
s
Dos
e ra
ngeb
and
indi
catio
nsT
oxic
ity p
rofil
ecD
ose
rang
eb a
nd in
dica
tions
Tox
icity
pro
filec
MT
X20
0–20
00 m
g pe
r do
seG
rade
37.
5–35
mg
per
wee
kG
rade
0–1
[19,
20,2
2]Fo
late
ant
imet
abol
ite, i
nhib
its D
NA
synt
hesi
sA
LL, b
reas
t can
cer,
head
and
nec
kca
ncer
, NH
L, lu
ng ca
ncer
, ost
eosa
rcom
a,Tr
opho
blas
tic n
eopl
asm
Neu
rolo
gica
l, ga
stro
inte
stin
al a
ndde
rmat
olog
ical
sym
ptom
s. Pu
lmon
ary,
bon
em
arro
w, r
enal
and
hep
atic
toxi
city
Cro
hn's
dise
ase,
rheu
mat
oid
arth
ritis
, JIA
, pso
riasi
s,ps
oria
tic a
rthrit
isG
astro
inte
stin
al sy
mpt
oms,
trans
ient
elev
atio
n of
live
r enz
ymes
, liv
erdy
sfun
ctio
n
Cyc
loph
osph
amid
eD
NA
alk
ylat
ing
agen
t, in
hibi
ts D
NA
synt
hesi
s25
0–13
00 m
g pe
r do
seG
rade
375
–250
mg
per
dose
Gra
de 0
–1
[51]
ALL
, bre
ast c
ance
r, B
urki
tt ly
mph
oma,
HD
, NH
L, M
M, o
varia
n ca
ncer
,re
tinob
last
oma
Gas
troin
test
inal
, der
mat
olog
ical
and
cata
rrha
l sym
ptom
s. B
one
mar
row
, ren
al,
hepa
tic, p
ulm
onar
y to
xici
ty. A
cute
haem
orrh
agic
cys
titis
, inf
ertil
ity
Beh
cet's
synd
rom
e, id
iopa
thic
pul
mon
ary
fibro
sis,
ITP,
JIA
, lup
us n
ephr
itis,
NS,
SLE
, tra
nspl
ant r
ejec
tion
prop
hyla
xis,
mul
tiple
scle
rosi
s, W
egen
er's
gran
ulom
atos
is
Fatig
ue, g
astro
inte
stin
al, c
atar
rhal
and
derm
atol
ogic
al sy
mpt
oms
6-M
erca
pto-
puri
nePu
rine
anta
goni
st, i
nhib
itor o
f DN
A a
ndR
NA
synt
hesi
s15
0–35
0 m
g pe
r do
seG
rade
310
0–15
0 m
g pe
r do
seG
rade
1–2
[52]
ALL
, AM
L, C
ML
Gas
troin
test
inal
and
der
mat
olog
ical
sym
ptom
s. B
one
mar
row
, hep
atic
and
rena
lto
xici
ty
Cro
hn's
dise
ase,
ulc
erat
ive
colit
isG
astro
inte
stin
al sy
mpt
oms,
bone
mar
row
supp
ress
ion,
ele
vatio
n of
live
ren
zym
es
Tha
lidom
ided
Mec
hani
sm o
f act
ion
is n
ot c
ompl
etel
yun
ders
tood
. Sel
ectiv
ely
redu
ces l
evel
s of
TNF
200–
1200
mg
per
dose
Gra
de 2
–325
–100
mg
per
dose
Gra
de 0
–1
[53]
Kap
osi's
sarc
oma,
MM
, mal
igna
ntgl
iom
a, m
yelo
dysp
last
ic sy
ndro
me,
rena
lce
ll ca
ncer
Neu
rolo
gica
l and
der
mat
olog
ical
sym
ptom
s.B
one
mar
row
supp
ress
ion,
incr
ease
d ris
k of
thro
mbo
sis
Beh
cet's
synd
rom
e, C
rohn
's di
seas
e, S
LE, D
LEN
euro
logi
cal s
ympt
oms
Vin
cris
tine
Vin
ca a
lkal
oid:
inhi
bito
r of m
icro
tubu
lefo
rmat
ion,
stop
ping
cel
l div
isio
n2–
3.5
mg
per
wee
kG
rade
32
mg
per
mon
thG
rade
0–1
[54]
ALL
, HD
, mal
igna
nt g
liom
a,ne
urob
last
oma,
NH
Lrh
abdo
myo
sarc
oma,
Wilm
s’ tu
mou
r
Neu
rolo
gica
l, ga
stro
inte
stin
al a
ndde
rmat
olog
ical
sym
ptom
s. B
one
mar
row
supp
ress
ion,
neu
roto
xici
ty
ITP,
TTP
Neu
rolo
gica
l sym
ptom
s
DFM
Oe
Inhi
bito
r of o
rnith
ine
deca
rbox
ylas
e
>10
g pe
r do
see
Gra
de 4
0.4–
0.8
g pe
r da
y fo
r a
year
Gra
de 0
–1
[55,
56]
Pros
tate
can
cer
Dia
rrho
ea, a
bdom
inal
pai
n, a
lope
cia
and
otot
oxic
ity
Che
mop
rote
ctio
n ag
ains
t pro
stat
e ca
ncer
, act
inic
ker
atos
is
>0.4
g p
er d
ay fo
r se
vera
l mon
ths
Gra
de 0
–1
As a
cre
am H
irsut
ism
(fac
ial h
air)
>5 g
per
day
for
15 d
ays
Gra
de 1
–2
Slee
ping
sick
ness
(try
pano
som
iasi
s)G
astro
inte
stin
al sy
mpt
oms
Milt
efos
inef
Inhi
bito
rs of
phos
phol
ipid
bios
ynth
esis
ofce
ll m
embr
ane
xx
1.5–
2.5
mg
per k
g fo
r 28
days
Lei
shm
ania
sis
Gra
de1–
2[5
7]
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.

Sponsored Docum
ent Sponsored D
ocument
Sponsored Docum
ent
Nzila et al. Page 10
Dru
gM
echa
nism
of a
ctio
nN
eopl
asm
(onc
olog
y)N
on-n
eopl
asm
Ref
s
Dos
e ra
ngeb
and
indi
catio
nsT
oxic
ity p
rofil
ecD
ose
rang
eb a
nd in
dica
tions
Tox
icity
pro
filec
Nau
sea,
vom
iting
, dia
rrho
ea
a Abb
revi
atio
ns: A
LL =
acut
e lym
phoc
ytic
leuk
aem
ia, A
ML
= ac
ute m
yelo
geno
us le
ukae
mia
, CLL
= ch
roni
c lym
phoc
ytic
leuk
aem
ia, C
ML
= ch
roni
c mye
loge
nous
leuk
aem
ia, D
LE =
dis
coid
lupu
s ery
them
atos
us, H
D =
Hod
gkin
's di
seas
e, IT
P =
idio
path
ic th
rom
bocy
tope
nic p
urpu
ra,
JIA
= ju
veni
le id
iopa
thic
arth
ritis
, MM
= m
ultip
le m
yelo
ma,
NH
L =
non-
Hod
gkin
's ly
mph
oma,
NS
= ne
phrit
ic sy
ndro
me,
SLE
= sy
stem
ic lu
pus e
ryth
emat
osus
, TTP
= th
rom
botic
thro
mbo
cyto
peni
c pu
rpur
a.
b Adu
lt do
ses.
c Gra
de 0
= n
o ad
vers
e re
actio
n, G
rade
1 =
mild
adv
erse
reac
tions
, Gra
de 2
= m
oder
ate
adve
rse
reac
tions
, Gra
de 3
= se
vere
adv
erse
reac
tions
, Gra
de 4
= li
fe-th
reat
enin
g to
xici
ty.
d Not
use
d in
pre
gnan
cy b
ecau
se o
f its
eff
ect a
gain
st fo
etal
gro
wth
tera
toge
nici
ty.
e DFM
O (D
ifluo
ro-m
ethy
l-orn
ithin
e) w
as u
sed
as a
n ex
perim
enta
l dru
g in
can
cer b
ut w
as a
band
oned
bec
ause
eff
icac
y co
uld
only
be
achi
eved
with
dos
es a
ssoc
iate
d w
ith se
rious
life
-thre
aten
ing
toxi
city
. How
ever
, the
dru
g w
as re
vive
d fo
r can
cer c
hem
opro
tect
ion,
hirs
utis
m a
ndtry
pano
som
iasi
s tre
atm
ent a
t low
dos
e.
f Milt
efos
ine
was
initi
ally
dis
cove
red
as a
n an
tineo
plas
tic d
rug;
how
ever
, few
stud
ies w
ere
carr
ied
out i
n hu
man
s to
treat
can
cer,
thus
det
aile
d in
form
atio
n on
its t
oxic
ity a
nd d
ose
rang
es a
s an
antin
eopl
astic
is n
ot a
vaila
ble.
Published as: Trends Parasitol. 2010 March ; 26(3-3): 125–129.
Related Documents











