Anthocyanidin synthase from Gerbera hybrida catalyzes the conversion of (+)-catechin to cyanidin and a novel procyanidin Frank Wellmann a,1 , Markus Griesser b , Wilfried Schwab b , Stefan Martens a , Wolfgang Eisenreich c , Ulrich Matern a, * , Richard Lukac ˇin a,2 a Institut fu ¨ r Pharmazeutische Biologie, Philipps-Universita ¨ t Marburg, Deutschhausstrasse 17 A, D-35037 Marburg, Germany b Fachgebiet Biomolekulare Lebensmitteltechnologie, Technische Universita ¨t Mu ¨ nchen, Lise-Meitner-Str. 34, D-85354 Freising, Germany c Lehrstuhl fu ¨ r Organische Chemie und Biochemie, Technische Universita ¨t Mu ¨ nchen, Lichtenberg-Str. 4, D-85747 Garching, Germany Received 12 January 2006; revised 7 February 2006; accepted 8 February 2006 Available online 17 February 2006 Edited by Ulf-Ingo Flu ¨gge Abstract Anthocyanidins were proposed to derive from (+)- naringenin via (2R,3R)-dihydroflavonol(s) and (2R,3S,4S)-leuco- cyanidin(s) which are eventually oxidized by anthocyanidin synthase (ANS). Recently, the role of ANS has been put into question, because the recombinant enzyme from Arabidopsis exhibited primarily flavonol synthase (FLS) activity with negligi- ble ANS activity. This and other studies led to the proposal that ANS as well as FLS may select for dihydroflavonoid substrates carrying a ‘‘b-face’’ C-3 hydroxyl group and initially form the 3- geminal diol by ‘‘a-face’’ hydroxylation. Assays with recombi- nant ANS from Gerbera hybrida fully supported the proposal and were extended to catechin and epicatechin isomers as poten- tial substrates to delineate the enzyme specificity. Gerbera ANS converted (+)-catechin to two major and one minor product, whereas ent()-catechin (2S,3R-trans-catechin), ()-epicate- chin, ent(+)-epicatechin (2S,3S-cis-epicatechin) and ()-gallo- catechin were not accepted. The K m value for (+)-catechin was determined at 175 lM, and the products were identified by LC–MS n and NMR as the 4,4-dimer of oxidized (+)-catechin (93%), cyanidin (7%) and quercetin (trace). When these incuba- tions were repeated in the presence of UDP-glucose:flavonoid 3- O-glucosyltransferase from Fragaria · ananassa (FaGT1), the product ratio shifted to cyanidin 3-O-glucoside (60%), cyanidin (14%) and dimeric oxidized (+)-catechin (26%) at an overall equivalent rate of conversion. The data appear to identify (+)- catechin as another substrate of ANS in vivo and shed new light on the mechanism of its catalysis. Moreover, the enzymatic dimerization of catechin monomers is reported for the first time suggesting a role for ANS beyond the oxidation of leucocyani- dins. Ó 2006 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved. Keywords: Flavonoid biosynthesis; Oligomeric proanthocyanidins; (+)-Catechin; 2-Oxoglutarate-dependent dioxygenase; Anthocyanidin synthase 1. Introduction Flavonoids comprise a class of abundant secondary metab- olites which contribute in many ways to the growth and subsistence of plants [1,2]. Prominent examples are the antho- cyanin pigments and the related oligomeric proanthocyani- dins which are under investigation for their medicinal potential [3]. The biosynthesis of flavonoids has been studied extensively over the last decades [4]. However, while the early steps have been unequivocally unraveled the reactions leading to anthocyanidins and, in particular, to proanthocyanidins have remained under debate. All flavonoids derive from the flavanone (2S)-naringenin, which may be oxidized to the cor- responding flavone by the action of flavone synthase (FNS) or hydroxylated in 3b-configuration to the (2R,3R)-dihydrofl- avonol [5,6]. Substitution reactions may proceed at any stage. Reduction of the dihydroflavonol by dihydroflavonol 4-reduc- tase (DFR, Fig. 1) [7] leads to (2R,3S,4S)-leucoanthocyanidin which was considered as the immediate precursor of anthocy- anidin [8] initially based on supplemention experiments with acyanic flowers of genetically defined lines of Matthiola incana [9]. A branch pathway was proposed to convert cis-leucocy- anidin to (2R,3S)-trans-flavan-3-ol ((+)-catechin) [10] as the likely start unit to oligomeric proanthocyanidins (Fig. 1). The condensation to proanthocyanidins (PA) or condensed tannins (CT) was assumed to require flavan-3,4-diols (leuco- cyanidins) as extension units [10,11], but more recently (2R,3R)-cis-flavan-3-ols (()-epicatechins) (Fig. 1) derived from anthocyanidins by the action of anthocyanidin reductase (ANR) [12,13] have been suggested as the more likely precur- sors [11–14]. Nevertheless, the oligomerization reaction has not yet been accomplished in vitro and the precise mechanism remains to be established [15]. Although the stereoconfigura- tion of flavonoids at C-2 appears to be set at the level of (2S)-naringenin and resulting necessarily in (+)-catechin and ()-epicatechin (Fig. 1), the diastereomers ent()-catechin ((2S,3R)-trans-catechin) and ent(+)-epicatechin ((2S,3S)-cis- epicatechin) have also been reported as natural plant products (cf. [15]). Furthermore, in some plants a bypass may exist avoiding the 3b-hydroxylation of (2S)-flavanones and leading to (2R,4R)-flavan-4-ols (phlobaphenes) [16] and 3-deoxyanth- ocyanidins. ANS belongs to the 2-oxoglutarate iron-dependent oxygen- ases and was cloned first from Perilla frutescens [17]. Four recombinant ANSs were used subsequently with commercial Abbreviations: FaGT1, Fragaria · ananassa cv. Elsanta glucosyltrans- ferase * Corresponding author. Fax: +49 6421 282 6678. E-mail address: matern@staff.uni-marburg.de (U. Matern). 1 Present address: Werthenstein Chemie AG, CH-6105 Schachen, Switzerland. 2 Present address: Chromsystems Instruments & Chemicals GmbH, Heimburg-Str. 3, D-81243 Mu ¨ nchen, Germany. 0014-5793/$32.00 Ó 2006 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved. doi:10.1016/j.febslet.2006.02.004 FEBS Letters 580 (2006) 1642–1648

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

FEBS Letters 580 (2006) 1642–1648
Anthocyanidin synthase from Gerbera hybrida catalyzes the conversionof (+)-catechin to cyanidin and a novel procyanidin
Frank Wellmanna,1, Markus Griesserb, Wilfried Schwabb, Stefan Martensa,Wolfgang Eisenreichc, Ulrich Materna,*, Richard Lukacina,2
a Institut fur Pharmazeutische Biologie, Philipps-Universitat Marburg, Deutschhausstrasse 17 A, D-35037 Marburg, Germanyb Fachgebiet Biomolekulare Lebensmitteltechnologie, Technische Universitat Munchen, Lise-Meitner-Str. 34, D-85354 Freising, Germanyc Lehrstuhl fur Organische Chemie und Biochemie, Technische Universitat Munchen, Lichtenberg-Str. 4, D-85747 Garching, Germany
Received 12 January 2006; revised 7 February 2006; accepted 8 February 2006
Available online 17 February 2006
Edited by Ulf-Ingo Flugge
Abstract Anthocyanidins were proposed to derive from (+)-naringenin via (2R,3R)-dihydroflavonol(s) and (2R,3S,4S)-leuco-cyanidin(s) which are eventually oxidized by anthocyanidinsynthase (ANS). Recently, the role of ANS has been put intoquestion, because the recombinant enzyme from Arabidopsisexhibited primarily flavonol synthase (FLS) activity with negligi-ble ANS activity. This and other studies led to the proposal thatANS as well as FLS may select for dihydroflavonoid substratescarrying a ‘‘b-face’’ C-3 hydroxyl group and initially form the 3-geminal diol by ‘‘a-face’’ hydroxylation. Assays with recombi-nant ANS from Gerbera hybrida fully supported the proposaland were extended to catechin and epicatechin isomers as poten-tial substrates to delineate the enzyme specificity. Gerbera ANSconverted (+)-catechin to two major and one minor product,whereas ent(�)-catechin (2S,3R-trans-catechin), (�)-epicate-chin, ent(+)-epicatechin (2S,3S-cis-epicatechin) and (�)-gallo-catechin were not accepted. The Km value for (+)-catechin wasdetermined at 175 lM, and the products were identified byLC–MSn and NMR as the 4,4-dimer of oxidized (+)-catechin(93%), cyanidin (7%) and quercetin (trace). When these incuba-tions were repeated in the presence of UDP-glucose:flavonoid 3-O-glucosyltransferase from Fragaria · ananassa (FaGT1), theproduct ratio shifted to cyanidin 3-O-glucoside (60%), cyanidin(14%) and dimeric oxidized (+)-catechin (26%) at an overallequivalent rate of conversion. The data appear to identify (+)-catechin as another substrate of ANS in vivo and shed new lighton the mechanism of its catalysis. Moreover, the enzymaticdimerization of catechin monomers is reported for the first timesuggesting a role for ANS beyond the oxidation of leucocyani-dins.� 2006 Federation of European Biochemical Societies. Publishedby Elsevier B.V. All rights reserved.
Keywords: Flavonoid biosynthesis; Oligomericproanthocyanidins; (+)-Catechin; 2-Oxoglutarate-dependentdioxygenase; Anthocyanidin synthase
Abbreviations: FaGT1, Fragaria · ananassa cv. Elsanta glucosyltrans-ferase
*Corresponding author. Fax: +49 6421 282 6678.E-mail address: [email protected] (U. Matern).
1 Present address: Werthenstein Chemie AG, CH-6105 Schachen,Switzerland.2 Present address: Chromsystems Instruments & Chemicals GmbH,Heimburg-Str. 3, D-81243 Munchen, Germany.
0014-5793/$32.00 � 2006 Federation of European Biochemical Societies. Pu
doi:10.1016/j.febslet.2006.02.004
1. Introduction
Flavonoids comprise a class of abundant secondary metab-
olites which contribute in many ways to the growth and
subsistence of plants [1,2]. Prominent examples are the antho-
cyanin pigments and the related oligomeric proanthocyani-
dins which are under investigation for their medicinal
potential [3]. The biosynthesis of flavonoids has been studied
extensively over the last decades [4]. However, while the early
steps have been unequivocally unraveled the reactions leading
to anthocyanidins and, in particular, to proanthocyanidins
have remained under debate. All flavonoids derive from the
flavanone (2S)-naringenin, which may be oxidized to the cor-
responding flavone by the action of flavone synthase (FNS) or
hydroxylated in 3b-configuration to the (2R,3R)-dihydrofl-
avonol [5,6]. Substitution reactions may proceed at any stage.
Reduction of the dihydroflavonol by dihydroflavonol 4-reduc-
tase (DFR, Fig. 1) [7] leads to (2R,3S,4S)-leucoanthocyanidin
which was considered as the immediate precursor of anthocy-
anidin [8] initially based on supplemention experiments with
acyanic flowers of genetically defined lines of Matthiola incana
[9]. A branch pathway was proposed to convert cis-leucocy-
anidin to (2R,3S)-trans-flavan-3-ol ((+)-catechin) [10] as the
likely start unit to oligomeric proanthocyanidins (Fig. 1).
The condensation to proanthocyanidins (PA) or condensed
tannins (CT) was assumed to require flavan-3,4-diols (leuco-
cyanidins) as extension units [10,11], but more recently
(2R,3R)-cis-flavan-3-ols ((�)-epicatechins) (Fig. 1) derived
from anthocyanidins by the action of anthocyanidin reductase
(ANR) [12,13] have been suggested as the more likely precur-
sors [11–14]. Nevertheless, the oligomerization reaction has
not yet been accomplished in vitro and the precise mechanism
remains to be established [15]. Although the stereoconfigura-
tion of flavonoids at C-2 appears to be set at the level of
(2S)-naringenin and resulting necessarily in (+)-catechin and
(�)-epicatechin (Fig. 1), the diastereomers ent(�)-catechin
((2S,3R)-trans-catechin) and ent(+)-epicatechin ((2S,3S)-cis-
epicatechin) have also been reported as natural plant products
(cf. [15]). Furthermore, in some plants a bypass may exist
avoiding the 3b-hydroxylation of (2S)-flavanones and leading
to (2R,4R)-flavan-4-ols (phlobaphenes) [16] and 3-deoxyanth-
ocyanidins.
ANS belongs to the 2-oxoglutarate iron-dependent oxygen-
ases and was cloned first from Perilla frutescens [17]. Four
recombinant ANSs were used subsequently with commercial
blished by Elsevier B.V. All rights reserved.

FHT (2S)-flavanones (2R/3R)-dihydroflavonols
OOH
OH OH
OH
OH
R1
R2
OOH
OH
OH
OH
R1
R2
LAR
DFR
O+
OH
OH
OH
OH
R1
R2
OOH
OH
OH
OH
R1
R2
ANR
?
(2R,3S)-flavan-3-ols (+)-catechin
(2R,3S,4S)-leucocyanidins
?
proanthocyanidin ANS
(2R,3R)-flavan-3-ols (-)-epicatechin
anthocyanidins
FGT
anthocyanins
Fig. 1. Role of ANS in the biosynthesis of anthocyanins and proanthocyanidins. Broken arrows designate reactions which have not been confirmedexperimentally (R1, R2 = H or OH). ANS, anthocyanidin synthase; ANR, anthocyanidin reductase; DFR, dihydroflavonol 4-reductase; FHT,flavanone 3b-hydroxylase; FGT, flavonoid 3-O-glucosyltransferase; LAR, leucoanthocyanidin reductase.
F. Wellmann et al. / FEBS Letters 580 (2006) 1642–1648 1643
leucoanthocyanidin substrates and in combination with UDP-
glucose:flavonoid 3-O-glucosyltransferase to investigate the
reaction mechanism [8]. A mechanism was postulated that pro-
ceeds from leucoanthocyanidin via 2-flaven-3,4-diol (pseudo-
base) followed by isomerization of the 2,3-double bond to the
3,4-position concomitant with a shift of the hydroxyl group
from C-4 to C-2 and removal of the C-2 hydroxyl anion under
acidic conditions to yield anthocyanidin (flavylium ion) [8].
However, the synthesis of pure leucoanthocyanidin enantiomers
is rather difficult, and the instability of flavan-3,4-diols in aque-
ous solution as well as the substrate specificity of ANS observed
in vitro cast some doubt on the role of leucoanthocyanidin as a
natural precursor of anthocyanidin [18]. In a series of studies on
recombinant ANS from Arabidopsis thaliana evidence was
presented for the initial oxidationof the substrate atC-3 [19]. Fur-
thermore, the ANS formed predominantly quercetin and cis-
and trans-dihydroquercetin (DHQ) with cyanidin being aminor
product only [19,20], and the product pattern from (2R,3S,4S)-
cis-leucocyanidin vs. that from (2R,3S,4R)-trans-leucocyanidin
implied that cis-DHQ, trans-DHQ and cyanidin resulted mostly
from the unnatural (2R,3S,4R)-trans-leucocyanidin [20]. More-
over, co-crystallization of the ANS with Fe2+, 2-oxoglutarate
and racemic trans-DHQ or enantiomerically pure (2R,3R)-
DHQ as a substrate analogue in the absence or presence of
molecular oxygen supported the stereoselective C-3 hydroxyl-
ation of the substrate and surprisingly revealed two molecules
of the substrate analogue in the active site with (2R,3R)-trans-
DHQ closest to the iron atom, whereas either enantiomer was
bound at the other location [21]. Clearly, additional data are
required to define the substrate specificity of ANS.
Recombinant ANS from Gerbera hybrida was used to deter-
mine the activity with various flavan-3-ol substrates. The selec-
tive conversion of (+)-catechin to anthocyanidin and a dimeric
flavan-3-one sheds new light on the mode of action of ANS,
because neither the mechanism proposed via isomerisation of
3-flaven-2,3-diol concomitant with the C-4/C-2 shift of the hy-
droxyl group [8] nor the lack of configurational requirement at
C-2 apply in this instance. Furthermore, the dimerization reac-
tion might be considered as a precedent for proanthocyanidin
formation.
2. Materials and methods
2.1. ChemicalsBiochemicals of analytic grade were purchased from Roth
(Karlsruhe, Germany). Reference samples of (+)-catechin, (�)-catechin,(+)-epicatechin, (�)-epicatechin, (�)-gallocatechin, cyanidin chlorideand quercetin were from Roth (Karlsruhe, Germany) or Sigma(Deisenhofen, Germany). LiChroprep RP18 (40–63 lm) was obtainedfrom Merck (Darmstadt, Germany). The solutions of flavonoids werefreshly prepared in methanol when used for enzyme incubations.

1644 F. Wellmann et al. / FEBS Letters 580 (2006) 1642–1648
2.2. Cloning and expression of ANS from Gerbera hybridaRecombinant ANS was expressed in yeast [22]. The growing of yeast
transformants and protein isolation were performed as previously de-scribed [23].
2.3. Cloning and expression of FaGT1 from Fragaria · ananassa cv.
ElsantaThe construction of the cDNA library from strawberry fruit and
the identification of the UDP-glucose:flavonoid 3-O-glucosyltransfer-ase (FaGT1) sequence has been described [24]. The open readingframe of FaGT1 (GenBank Accession number AAU09442) whichcodes for a protein of 466 amino acids was cloned into the pET-29a (+) expression vector (Novagen, Schwalbach, Germany) usingthe EcoRI and the XhoI restriction sites. Protein expression was car-ried out with the Escherichia coli strain BL21 (DE3) pLysS (Nova-gen) which was grown at 37 �C until an A600 of approximately 0.5was reached. Induction of protein expression was initiated by addingisopropyl-1-thio-b-DD-galactopyranoside (IPTG) to a final concentra-tion of 1 mM, followed by further growth at 16 �C for 8 h. Proteinpurification was carried out using the Talon� metal affinity resin(BD Biosciences Clontech, Palo Alto, USA) following the manufac-turers instructions with minor modifications. In brief, cells were har-vested by centrifugation and were disrupted with chilled mortar,pestle and glass beads (Sigma, Deisenhofen, Germany). All bufferswere modified to contain 10% glycerol, 10 mM b-mercaptoethanoland 500 mM sodium chloride. The batch/gravity-flow column purifi-cation was applied and the recombinant protein was eluted using abuffer containing 150 mM imidazole. Protein concentrations weredetermined using the Bradford microprotein assay [25]. Negative con-trols were carried out with E. coli BL21 (DE3) pLysS cells harbour-ing an empty expression vector.
2.4. Enzyme assaysActive ANS from filtered (PD10 column, Pharmacia, Freiburg, Ger-
many) crude extract from yeast cells harbouring the ANS cDNA wasincubated at 37 �C for 15 min. The standard reaction mixture (100 ll)contained 100 lM (+)-catechin, 100 lM 2-oxoglutarate, 50 lM ammo-nium iron(II) sulfate, 2.5 mM sodium ascorbate, 2 mg/ml bovine cata-lase and 50 ll protein (0.8 lg/ll) in 200 mM potassium phosphatebuffer pH 6.0. The incubations were carried out in open vials undergentle shaking and the reaction was terminated by the addition of15 ll saturated aqueous EDTA solution. After centrifugation (5 min,10000 · g) the supernatant was directly subjected to LC–MSn analysisas described below.
2.5. Liquid chromatography – mass spectrometry (LC-MSn)The system used for the analysis of the enzyme assay products was a
Bruker Daltronics esquire 3000plus ion trap mass spectrometer (BrukerDaltronics, Bremen, Germany) connected with an Agilent 1100 HPLCsystem (Agilent Technologies, Waldbronn, Germany) equipped with aquaternary pump and a variable wavelength detector. Componentswere separated on a Eurospher 100 C18 column, particle size 5 lm,10 cm · 2 mm (Grom, Rottenburg, Germany) which was held at25 �C. Solvent A was water (Merck, Darmstadt, Germany) acidifiedwith 0.05% formic acid (Roth, Karlsruhe, Germany) and solvent Bwas acetonitrile (Merck, Darmstadt, Germany). Products were sepa-rated using a linear gradient from 100% A to 100% B in 30 min witha flow rate of 0.2 ml/min. The detection wavelength was either 280or 520 nm, as indicated in the text. The electrospray ionization voltageof the capillary was set to �4000 V and the end plate to �500 V. Nitro-gen was used as dry gas at a temperature of 330 �C and a flow rate of9 l/min. The full scan mass spectra were measured in a scan range from50 to 800 m/z with a scan resolution of 13000 m/z/s. Tandem massspectrometry was carried out using helium as collision gas(3.56 · 10�6 mbar) with the collision voltage set at 1 V. All spectrawere acquired in the positive ionization mode. Data analysis was per-formed using the DataAnalysis 3.1 software (Bruker Daltronics).
2.6. Kinetic propertiesRanges of flavonoid substrate concentrations of between 16 and
100 lM were used for Km determination. The apparent Michaelis con-stant for ANS was calculated from Lineweaver–Burk plot. The proteinamounts were quantified according to Bradford [25].
2.7. Concerted reaction of ANS and FaGT1The ANS standard tests were supplemented by addition of 50 ll par-
tially purified FaGT1 (�0.2 mg protein/ml) and 10 ll of 125 mMUDP-glucose to a final volume of 200 ll and incubated at 37 �C for60 min. The flavonoids were analyzed directly by LC–MSn as describedabove.
2.8. Isolation of the dimerFor preparative purposes numerous single incubations of a total
amount of 10 mg (+)-catechin and approximately 25 mg ANS proteinwere carried out under the above mentioned conditions. Successively,the flavonoids were isolated by repeated extraction with ethyl acetate(twice 75 ll), the organic fractions pooled and finally dried under vac-uum. After resolving the residue in 2 ml of 60% aqueous methanol thedimer was separated from the reaction products by preparative RP-18column chromatography (15 cm · 3 cm). Solvent A was water (Merck,Darmstadt, Germany) acidified with 0.05% formic acid (Roth,Karlsruhe, Germany) and solvent B was acetonitrile (Merck, Darms-tadt, Germany). Products were eluted using a stepwise gradient from100 ml of 100% A to 100 ml of 50% B, whereas during each step theconcentration of B was increased by 5%. A flow rate of 2 ml/minwas applied and 20-ml fractions were collected. Fractions were ana-lyzed by LC–MSn. Fractions 21 and 22 containing the dimer werepooled, concentrated to dryness, dissolved in d6-acetone and analyzedby NMR spectroscopy.
2.9. NMR spectroscopyNMR spectra of the catechin derived dimer were recorded at 25 �C
using an AVANCE 500 spectrometer (Bruker Instruments, Karlsruhe,Germany) at transmitter frequencies of 500.1 and 125.6 MHz for 1Hand 13C, respectively. Samples were dissolved in d6-acetone (1.5 mgin 0.5 mL). Two-dimensional COSY, NOESY, HMQC, and HMBCexperiments were performed according to standard Bruker software(XWINNMR). The mixing time was 1 s in the NOESY experiment.1H and 13C NMR chemical shifts were predicted by Specinfo. The sig-nal assignments are based on proton–proton (COSY, NOESY) andproton–carbon correlation experiments (HMQC, HMBC).
3. Results and discussion
3.1. ANS assays with catechins and epicatechins
Activity assays carried out recently with recombinant ANS
from A. thaliana and employing either (2R,3S,4S)-cis-leucocy-
anidin or (2R,3S,4R)-trans-leucocyanidin as a substrate sur-
prisingly revealed quercetin or (2R,3S)-cis-dihydroquercetin
as the primary product rather than the anticipated product
cyanidin which accounted for less than 5% only [20]. These
incubations require particular experimental care, because in
aqueous solution leucocyanidins epimerize easily and cyani-
din is degraded rapidly [8]. Analogous incubations were car-
ried out subsequently with ANS from G. hybrida expressed in
yeast cells [22] and using dihydrokaempferol as a substrate in
combination with dihydroflavonol reductase from G. hybrida,
which basically confirmed the low rate of pelargonidin forma-
tion (Martens et al., unpublished). Accordingly, a more de-
tailed examination of the physiological role of ANS appears
to be necessary. In a first approach, we used the partially
purified recombinant ANS from G. hybrida for activity assays
with enantiomerically pure (+)-catechin, (�)-catechin, and
(+)-epicatechin, respectively, each over a range of concentra-
tions from 16 to 100 lM. The assay conditions were adapted
from previous work [22,26], and crude extracts from yeast
cells harbouring the empty pYES2 expression vector were
routinely used as negative controls. The incubations were
subjected directly to LC–MSn analysis, thus avoiding the
acidification of the reaction mixture prior to the separation
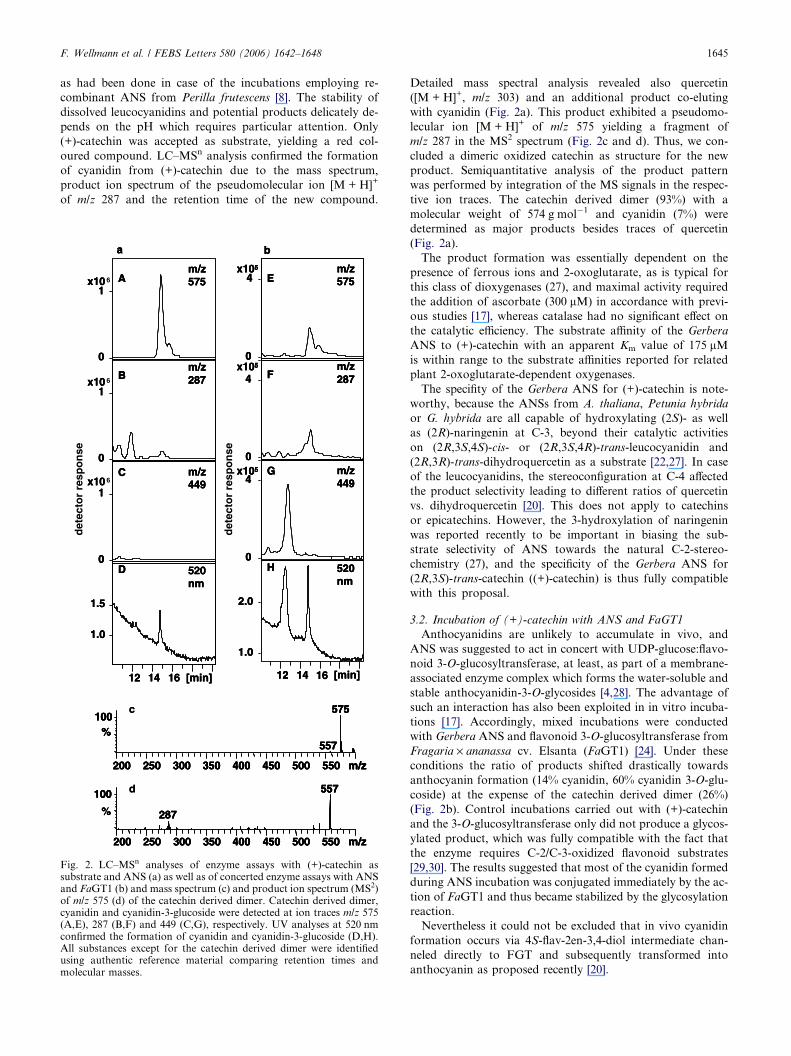
F. Wellmann et al. / FEBS Letters 580 (2006) 1642–1648 1645
as had been done in case of the incubations employing re-
combinant ANS from Perilla frutescens [8]. The stability of
dissolved leucocyanidins and potential products delicately de-
pends on the pH which requires particular attention. Only
(+)-catechin was accepted as substrate, yielding a red col-
oured compound. LC–MSn analysis confirmed the formation
of cyanidin from (+)-catechin due to the mass spectrum,
product ion spectrum of the pseudomolecular ion [M + H]+
of m/z 287 and the retention time of the new compound.
0
1
0
1
0
1
1.0
1.5
12 14 16 [min]
x10 6
x10 6
x10 6
A
B
C
D
m/z 575
m/z 287
m/z 449
520 nm
0
4
0
4
0
4
1.0
2.0
12 14 16 [min]
E
F
G
H
m/z 449
m/z 287
m/z 575
520 nm
x105
x105
x105
det
ecto
r re
spo
nse
det
ecto
r re
spo
nse
a b
287
557
200 250 300 350 400 450 500 550 m/z
100
557
575
200 250 300 350 400 450 500 550 m/z
100%
%
c
d
0
1
0
1
0
1
1.0
1.5
12 14 16 [min]
x10
x10
x10
A
B
C
D
m/z 575
m/z 287
m/z 449
520 nm
0
4
0
4
0
4
1.0
2.0
12 14 16 [min]
E
F
G
H
m/z 449
m/z 287
m/z 575
520 nm
x105
x105
x105
a b
0
1
0
1
0
1
1.0
1.5
12 14 16 [min]
x10
x10
x10
A
B
C
D
m/z 575
m/z 287
m/z 449
520 nm
0
4
0
4
0
4
1.0
2.0
12 14 16 [min]
E
F
G
H
m/z 449
m/z 287
m/z 575
520 nm
x105
x105
x105
a b
287
557
200 250 300 350 400 450 500 550 m/z
100
557
575
200 250 300 350 400 450 500 550 m/z
100%
% 287
557
200 250 300 350 400 450 500 550 m/z
100
287
557
200 250 300 350 400 450 500 550 m/z
100
557
575
200 250 300 350 400 450 500 550 m/z
100%
557
575
200 250 300 350 400 450 500 550 m/z
100%
%
c
d
Fig. 2. LC–MSn analyses of enzyme assays with (+)-catechin assubstrate and ANS (a) as well as of concerted enzyme assays with ANSand FaGT1 (b) and mass spectrum (c) and product ion spectrum (MS2)of m/z 575 (d) of the catechin derived dimer. Catechin derived dimer,cyanidin and cyanidin-3-glucoside were detected at ion traces m/z 575(A,E), 287 (B,F) and 449 (C,G), respectively. UV analyses at 520 nmconfirmed the formation of cyanidin and cyanidin-3-glucoside (D,H).All substances except for the catechin derived dimer were identifiedusing authentic reference material comparing retention times andmolecular masses.
Detailed mass spectral analysis revealed also quercetin
([M + H]+, m/z 303) and an additional product co-eluting
with cyanidin (Fig. 2a). This product exhibited a pseudomo-
lecular ion [M + H]+ of m/z 575 yielding a fragment of
m/z 287 in the MS2 spectrum (Fig. 2c and d). Thus, we con-
cluded a dimeric oxidized catechin as structure for the new
product. Semiquantitative analysis of the product pattern
was performed by integration of the MS signals in the respec-
tive ion traces. The catechin derived dimer (93%) with a
molecular weight of 574 g mol�1 and cyanidin (7%) were
determined as major products besides traces of quercetin
(Fig. 2a).
The product formation was essentially dependent on the
presence of ferrous ions and 2-oxoglutarate, as is typical for
this class of dioxygenases (27), and maximal activity required
the addition of ascorbate (300 lM) in accordance with previ-
ous studies [17], whereas catalase had no significant effect on
the catalytic efficiency. The substrate affinity of the Gerbera
ANS to (+)-catechin with an apparent Km value of 175 lMis within range to the substrate affinities reported for related
plant 2-oxoglutarate-dependent oxygenases.
The specifity of the Gerbera ANS for (+)-catechin is note-
worthy, because the ANSs from A. thaliana, Petunia hybrida
or G. hybrida are all capable of hydroxylating (2S)- as well
as (2R)-naringenin at C-3, beyond their catalytic activities
on (2R,3S,4S)-cis- or (2R,3S,4R)-trans-leucocyanidin and
(2R,3R)-trans-dihydroquercetin as a substrate [22,27]. In case
of the leucocyanidins, the stereoconfiguration at C-4 affected
the product selectivity leading to different ratios of quercetin
vs. dihydroquercetin [20]. This does not apply to catechins
or epicatechins. However, the 3-hydroxylation of naringenin
was reported recently to be important in biasing the sub-
strate selectivity of ANS towards the natural C-2-stereo-
chemistry (27), and the specificity of the Gerbera ANS for
(2R,3S)-trans-catechin ((+)-catechin) is thus fully compatible
with this proposal.
3.2. Incubation of (+)-catechin with ANS and FaGT1
Anthocyanidins are unlikely to accumulate in vivo, and
ANS was suggested to act in concert with UDP-glucose:flavo-
noid 3-O-glucosyltransferase, at least, as part of a membrane-
associated enzyme complex which forms the water-soluble and
stable anthocyanidin-3-O-glycosides [4,28]. The advantage of
such an interaction has also been exploited in in vitro incuba-
tions [17]. Accordingly, mixed incubations were conducted
with Gerbera ANS and flavonoid 3-O-glucosyltransferase from
Fragaria · ananassa cv. Elsanta (FaGT1) [24]. Under these
conditions the ratio of products shifted drastically towards
anthocyanin formation (14% cyanidin, 60% cyanidin 3-O-glu-
coside) at the expense of the catechin derived dimer (26%)
(Fig. 2b). Control incubations carried out with (+)-catechin
and the 3-O-glucosyltransferase only did not produce a glycos-
ylated product, which was fully compatible with the fact that
the enzyme requires C-2/C-3-oxidized flavonoid substrates
[29,30]. The results suggested that most of the cyanidin formed
during ANS incubation was conjugated immediately by the ac-
tion of FaGT1 and thus became stabilized by the glycosylation
reaction.
Nevertheless it could not be excluded that in vivo cyanidin
formation occurs via 4S-flav-2en-3,4-diol intermediate chan-
neled directly to FGT and subsequently transformed into
anthocyanin as proposed recently [20].

1646 F. Wellmann et al. / FEBS Letters 580 (2006) 1642–1648
3.3. Structure of the catechin-derived dimer
About 1.5 mg of the pure catechin-derived dimer was col-
lected from numerous incubations of (+)-catechin with recom-
binant ANS followed by separation on a preparative RP18
column. The formation of the dimer required (+)-catechin as
the substrate as well as an active ANS and was always accom-
panied by the formation of cyanidin. In contrast to cyanidin
the dimeric product was stable at room temperature. Its struc-
ture was analysed by NMR spectroscopy (Table 1).
In light of the molecular weight of the dimer, it was surpris-
ing that the 1H NMR spectrum displayed only a limited num-
ber of signals. In the downfield region of the 1H NMR
spectrum, a set of well-resolved signals were observed resem-
bling the full signal pattern of (+)-catechin. This indicates that:
(i) the structure of the dimer is closely related to the structure
of the catechin substrate; (ii) the molecule is characterized by
an inherent symmetry giving rise to isochronous signals of cor-
responding atoms in the symmetrical subunits of the molecule;
(iii) the two monomers are not connected via carbon atoms of
the aromatic rings bearing hydrogen atoms in the original cat-
echin substrate (e.g., C-8 which is the typical nucleophilic cen-
ter for condensations with catechin).
In the upfield region of the spectrum, a spin system compris-
ing hydrogen atoms at C-2–C-4 of the catechin substrate was
not observed for the dimer. On the other hand, two singlets
at 5.21 and 3.16 ppm were observed with intensities displaying
one hydrogen atom for each site per symmetrical subunit of
the molecule. A two-dimensional COSY experiment afforded
three pairs of correlated hydrogen atoms displaying the spin
systems of the aromatic rings in the catechin motif (i.e., H-
6–H-8, H-14–H-13, and H-14–H-10). Additionally, the signal
at 5.21 was correlated with the H-10 and H-14 signals and
was therefore clearly recognized as H-2. The singlet signal at
3.16 ppm gave weak correlation peaks with the H-6 and H-8
signals. Tentatively, this signal was therefore assigned as H-4.
Information about 13C NMR chemical shifts could be
gleaned from two-dimensional HMQC and HMBC experi-
ments revealing 24 pairs of connectivities between hydrogen
and carbon atoms via one bond (HMQC experiment) or via
multiple bonds (HMBC). This connectivity pattern established
the structure of the molecule as the C-4–C-4 dimer of catechin.
Table 1NMR data of the C-4–C-4 dimer derived from (+)-catechin
Position Chemical shifts (ppm) C
d1H d13C JH
Observed Predicted Observed Predicted
10 6.94(d, 1H) 6.49 114.7 117.9 2.14 6.81(dd, 1H) 6.58 119.4 123.3 8.13 6.75(d, 1H) 6.49 114.8 117.2 8.6 5.93(d, 1H) 5.71 95.2 95.3 2.8 5.81(d, 1H) 5.75 95.7 94.0 2.2 5.21(s, 1H) 5.84 82.3 95.34 3.16(s, 1H) 4.24 24.1 31.14a 94.6 99.79 130.5 128.311 144.6 144.612 145.2 142.98a 154.7 165.67 153.6 158.15 160.53 207.1
The configuration of the molecule cannot be derived unequiv-
ocally on the basis of the present data. However, the apparent
absence of a NOESY crosspeak between H-2 and H-4 could
indicate trans configuration of the respective atoms.
3.4. Mechanistic implications
Several studies have been conducted recently on flavonoid-
related 2-oxoglutarate dependent dioxygenases aiming primar-
ily at the mechanism of C-ring oxidation [6,19,22,27,31]. These
enzymes were commonly proposed to catalyze the hydroxyl-
ation of carbon-3 configurated either to the b-face (FHT/
FNS I) or to the a-face (ANS/FLS) of the flavonoid-ring plane
[27]. The initial hydroxylation suggested for the ANS reaction
is likely to occur at carbon-3, although oxidations at either C-2
or C-4 have not been fully excluded. In case of (+)-catechin,
only the a-face hydroxylation at C-3 or a concerted oxidation
at C-3/C-2 are plausible, because a hydroxylation at C-4 would
generate leucocyanidins as a source of different products [20]
(Fig. 3, I). It is thus conceivable to assume at the first stage
of the oxidation flav-3-en-3-ol and flav-2-en-3-ol with flavan-
3-one in tautomeric equilibrium, which spontaneously oxidize
further to anthocyanidin (cyanidin from (+)-catechin) in the
presence of oxygen [32] (Fig. 3, II). Alternatively, a second cy-
cle of ANS-catalyzed hydroxylation at carbon-3 might be pos-
tulated (Fig. 3, III), in analogy to the mechanistic proposal
suggested for the ANS-catalyzed formation of quercetin from
leucocyanidins [27].
The symmetric cyanidin derived dimer formed by ANS
in vitro has, to our knowledge, no precedent in the literature.
It was obviously produced by the ANS activity, most likely
from the flav-2-en-3-ol intermediate by abstraction of hydro-
gen from C-4 leaving a radical that stabilizes by spontaneous
dimerization (Fig. 3, IV). Natural procyanidins are highly di-
verse in size and composition, and the monomers are usually
lined up by C-4/C-8 or C-4/C-6 linkages [33]. So far only the
NADPH-dependent anthocyanidin (ANR) and leucoanthocy-
anidin (LAR) reductases have been characterized, which pro-
duce leucoanthocyanidin and catechin, respectively, the
precursors of procyanidins [12,33] (Fig. 1). Biomimetic models
have demonstrated a non-enzymatic condensation reaction,
suggesting the extension of a (+)-catechin start unit by
oupling constant (Hz) Correlation pattern
H COSY NOESY HMBC
2 14 2 25, 2.2 13, 10 2, 13 10, 25 14 14 142 82 6 2(w), 6(w)
10, 14 14, 10 10, 146(w), 8(w) 2
4(s), 2, 8(w)13, 21314, 102, 6(w)8(w)

OOH
OH
OH
OH
OH
I
C3-oxidation
OOH
OH
OH
OH
OH
OH
HOOH
OH
O
OH
OH
OOH
OH
OH
OH
OH
H Fe(III)
O
H
O+
OH
OH
OH
OH
OH
OOH
OH
OH
OH
OH
H H Fe(IV)
O
O+
OH
OH
OSugar
OH
OH
VII
III
(+)-catechin
flavan-3-one
flav-2-en-3-ol
FGT
FGT
C-4–C-4-catechin derived dimer
FGT
C4-hydrogen-abstraction
H2O- elimination
no enzyme
radical abreaction
OOH
OH
OH
OH
OH
H H
IVO OH
OH
OH
OH
OH
H H
Fe(IV)
O
O+
OH
OH
OH
OH
OH
H H
O Fe(II)
cyanidin cyanidin 3-glucoside
OOH
OH
O
OH
OH
O OH
OH
O
OH
OH
2
34
56
7 8 910
4a
8a
11
1213
14
V
II
C3-oxidation
VI·
Fig. 3. Oxidation of (+)-catechin catalyzed by ANS. Flav-2-en-3-ol intermediates might be further oxidized to cyanidin and catechin derived dimer,respectively, or may serve as substrate for other reactions in flavonoid or proanthocyanidin biosynthetic pathways.
F. Wellmann et al. / FEBS Letters 580 (2006) 1642–1648 1647
(2R,3S)/(2R,3R)-quinone methide derived via flav-3-en-ol from
leucoanthocyanidin. Flav-3-en-3-ols have more generally been
considered as precursors in flavonoid- and proanthocyanidin
biosynthesis [15,34] and were postulated to arise also during
the reverse NADPH/NADH-dependent reduction of anthocy-
anidins to 2,3-cis-flavan-3-ols catalysed by ANR [12]. Irrespec-
tive of chemical model reactions there is strong evidence for an
enzyme condensing catechins and leucocyanidins [35], how-
ever, the mechanism of condensation is poorly understood.
The dimerization of (+)-catechin by ANS might be considered
as a lead to proanthocyanidin condensation, in particular since
the ANS crystallized from A. thaliana has been shown recently
to accommodate two flavonoids in the active site pocket [21].
The catalytic activity documented in this report for ANS
clearly differs from the mode of action proposed recently for
the polymerisation of flavonoids in Arabidopsis seed coat testa,
which relies on a laccase-type enzyme oxidizing epicatechin to
the corresponding o-quinone [36]. This proposal followed the
assumption of polyphenol oxidases converting flavan-3-ols to
procyanidins [37].
References
[1] Harborne, J.B. and Williams, C.A. (2000) Advances in flavonoidresearch since 1992. Phytochemistry 55, 481–504.
[2] Cooper-Driver, G.A. (2001) Contributions of Jeffrey Harborneand co-workers to the study of anthocyanins. Phytochemistry 56,229–236.
[3] Kong, J.-M., Chia, L.-S., Goh, A.-K., Chia, T.-F. and Brouillard,R. (2003) Analysis and biological activities of anthocyanins.Phytochemistry 64, 923–933.
[4] Forkmann, G. and Heller, W. (1999) Biosynthesis of flavonoidsin: Comprehensive Natural Products Chemistry (Sankawa, U.,Ed.), Polyketides and Other Secondary Metabolites IncludingFatty Acids and their Derivatives, Vol. 1, pp. 713–748, PergamonPress, Oxford.

1648 F. Wellmann et al. / FEBS Letters 580 (2006) 1642–1648
[5] Lukacin, R. and Britsch, L. (1997) Identification of strictlyconserved histidine and arginine residues as part of the active sitein Petunia hybrida flavanone 3b-hydroxylase. Eur. J. Biochem.249, 748–757.
[6] Wellmann, F., Matern, U. and Lukacin, R. (2004) Significance ofC-terminal sequence elements for Petunia flavanone 3b-hydrox-ylase activity. FEBS Lett. 561, 149–154.
[7] Martens, S., Teeri, T. and Forkmann, G. (2002) Heterologousexpression of dihydroflavonol 4-reductases from various plants.FEBS Lett. 531, 453–458.
[8] Nakajima, J., Tanaka, Y., Yamazaki, M. and Saito, K. (2001)Reaction mechanism from leucoanthocyanidin to anthocyanidin3-glucoside, a key reaction for coloring in anthocyanin biosyn-thesis. J. Biol. Chem. 276, 25797–25803.
[9] Heller, W., Britsch, L., Forkmann, G. and Grisebach, H. (1985)Leucoanthocyanidins as intermediates in anthocyanidin biosyn-thesis in flowers of Matthiola incana R. BR.. Planta 163, 191–196.
[10] Stafford, H.A. (1990) Flavonoid Metabolism, CRC Press, Inc.,New York.
[11] Kitamura, S., Shikazono, N. and Tanaka, A. (2004) TRANS-PARENT TESTA 19 is involved in the accumulation of bothanthocyanins and proanthocyanidins in Arabidopsis. Plant J. 37,104–114.
[12] Xie, D.-Y., Sharma, S.B., Paiva, N.L., Ferreira, D. and Dixon,R.A. (2003) Role of anthocyanidin reductase, encoded by /BANYULS/ in plant flavonoid biosynthesis. Science 299, 396–399.
[13] Xie, D.-Y., Sharma, S.B. and Dixon, R.A. (2004) Anthocyanidinreductases from Medicago truncatula and Arabidopsis thaliana.Arch. Biochem. Biophys. 422, 91–102.
[14] Xie, D.-Y., Jackson, L.A., Cooper, J.D., Ferreira, D. and Paiva,N.L. (2004) Molecular and biochemical analysis of two cDNAclones encoding dihydroflavonol-4-reductase from Medicagotruncatula. Plant Physiol. 134, 979–994.
[15] Marles, M.A.S., Ray, H. and Gruber, M.Y. (2003) New perspec-tives on proanthocyanidin biochemistry and molecular regulation.Phytochemistry 64, 367–383.
[16] Chopra, S., Gevens, A., Svabek, C., Wood, K.V., Peterson, T.and Nicholson, R.L. (2002) Excision of the Candystripe1 trans-poson from a hyper-mutable Y1-cs allele shows that thesorghumY1 gene controls the biosynthesis of both 3-deoxyanth-ocyanidin phytoalexins and phlobaphene pigments. Physiol. Mol.Plant Pathol. 60, 321–330.
[17] Saito, K., Kobayashi, M., Gong, Z., Tanaka, Y. and Yamazaki,M. (1999) Direct evidence for anthocyanidin synthase as a 2-oxoglutarate-dependent oxygenase: molecular cloning and func-tional expression of cDNA from a red forma of Perilla frutescens.Plant J. 17, 181–189.
[18] Turnbull, J.J., Sobey, W.J., Aplin, R.T., Hassan, A., Schofield,C.J., Firmin, J.L. and Prescott, A.G. (2000) Are anthocyanidinsthe immediate products of anthocyanidin synthase? Chem.Commun., 2473–2474.
[19] Welford, R.W.D., Turnbull, J.J., Claridge, T.D.W., Schofield,C.J. and Prescott, A.G. (2001) Evidence for oxidation at C-3 ofthe flavonoid C-ring during anthocyanin biosynthesis. Chem.Commun., 1828–1829.
[20] Turnbull, J.J., Nagle, M.J., Seibel, J.F., Welford, R.W.D., Grant,G.H. and Schofield, C.J. (2003) The C-4 stereochemistry ofleucocyanidin substrates for anthocyanidin synthase affects prod-uct selectivity. Bioorg. Med. Chem. Lett. 13, 3853–3857.
[21] Wilmouth, R.C., Turnbull, J.J., Welford, R.W.D., Clifton, I.J.,Prescott, A.G. and Schofield, C.J. (2002) Structure and mecha-nism of anthocyanidin synthase from Arabidopsis thaliana.Structure 10, 93–103.
[22] Martens, S., Forkmann, G., Britsch, L., Wellmann, F., Matern,U. and Lukacin, R. (2003) Divergent evolution of flavonoid 2-oxoglutarate-dependent dioxygenases in parsley. FEBS Lett. 544,93–98.
[23] Urban, P., Mignotte, C., Kazmaier, M., Delorme, F. andPompon, D. (1997) Cloning, yeast expression, and characteriza-tion of the coupling of two distantly related Arabidopsis thalianaNADPH-cytochrome P450 reductases with P450 CYP73A5. J.Biol. Chem. 272, 19176–19186.
[24] Aharoni, A. and O’Conell, A.P. (2002) Gene expression analysisof strawberry achene and receptacle maturation using DNAmicroarrays. J. Exp. Bot. 53, 2073–2087.
[25] Bradford, M.M. (1976) A rapid and sensitive method for thequantification of microgram quantities of protein utilizing theprinciple of protein dye binding. Anal. Biochem. 72, 248–254.
[26] Schroder, G., Wehinger, E., Lukacin, R., Wellmann, F., Seefel-der, W., Schwab, W. and Schroder, J. (2004) Flavonoid methyl-ation: a novel 4 0-O-methyltransferase from Catharanthus roseus,and evidence that partially methylated flavanones are substratesof four different flavonoid dioxygenases. Phytochemistry 65,1085–1094.
[27] Turnbull, J.J., Nakajima, J., Welford, R.W.D., Yamazaki, M.,Saito, K. and Schofield, C.J. (2004) Mechanistic studies on three2-oxoglutarate-dependent oxygenases of flavonoid biosynthesis:anthocyanidin synthase, flavonol synthase, and flavanone 3b-hydroxylase. J. Biol. Chem. 279, 1206–1216.
[28] Winkel-Shirley, B. (2001) Flavonoid biosynthesis. A colorfulmodel for genetics, biochemistry, cell biology, and biotechnology.Plant Physiol. 126, 485–493.
[29] Yamazaki, M., Yamagishi, E., Gong, Z., Fukuchi-Mizutani, M.,Fukui, Y., Tanaka, Y., Kusumi, T., Yamaguchi, M. and Saito, K.(2002) Two flavonoid glucosyltransferases from Petunia hybrida:molecular cloning, biochemical properties and developmentallyregulated expression. Plant Mol. Biol. 48, 401–411.
[30] Fukuchi-Mizutani, M., Okuhara, H., Fukui, Y., Nakao, M.,Katsumoto, Y., Keiko, Y.-S., Kasumi, T., Hase, T. and Tanaka,Y. (2003) Biochemical and molecular characterization of a novelUDP-glucose:anthocyanin 3 0-O-glucosyltransferase, a key enzymefor blue anthocyanin biosynthesis, from gentian. Plant Physiol.132, 1652–1663.
[31] Lukacin, R., Wellmann, F., Britsch, L., Martens, S. and Matern,U. (2003) Flavonol synthase from Citrus unshiu is a bifunctionaldioxygenase. Phytochemistry 62, 287–292.
[32] Coetzee, J., Malan, E. and Ferreira, D. (2000) Synthesis andreactions of flav-3-en-3-ols. Tetrahedron 56, 1819–1824.
[33] Tanner, G.J., Francki, K.T., Abrahams, S., Watson, J.M.,Larkin, P.J. and Ashton, A.R. (2003) Proanthocyanidin biosyn-thesis in plants: purification of legume leucoanthocyanidinreductase and molecular cloning of its cDNA. J. Biol. Chem.278, 31647–31656.
[34] Stafford, H.A. (1983) Enzymic regulation of procyanidin biosyn-thesis; lack of a flav-3-en-3-ol intermediate. Phytochemistry 22,2643–2646.
[35] Abrahams, S., Tanner, G.J., Larkin, P.J. and Ashton, A.R. (2002)Identification and biochemical characterization of mutants in theproanthocyanidin pathway in Arabidopsis. Plant Physiol. 130,561–576.
[36] Pourcel, L., Routaboul, J.-M., Kerhoas, L., Caboche, M.,Lepiniec, L. and Debeaujon, I. (2005) TRANSPARENT TES-TA10 encodes a laccase-like enzyme involved in oxidativepolymerisation of flavonoids in Arabidopsis seed coat. Plant Cell17, 2966–2980.
[37] Dixon, R.A., Xie, D.Y. and Sharma, S.B. (2005) Procyanidins – afinal frontier in flavonoid research? New Phytol. 165, 9–28.
Related Documents








![pharmacokinetics of [14C] procyanidin B2 in male ratsdmd.aspetjournals.org/content/dmd/early/2009/11/12/dmd.109.030304... · wine. The average dietary ... bioavailability, pharmacokinetics](https://static.cupdf.com/doc/110x72/5b16eeb47f8b9a0b548b4fda/pharmacokinetics-of-14c-procyanidin-b2-in-male-wine-the-average-dietary-.jpg)


