Reudelhuber J. Ferreira, Robson A. S. Santos, Thomas Walther, Rhian M. Touyz and Timothy L. Chantal Mercure, Alvaro Yogi, Glaucia E. Callera, Anna B. Aranha, Michael Bader, Anderson Angiotensin(1-7) Blunts Hypertensive Cardiac Remodeling by a Direct Effect on the Heart Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2008 American Heart Association, Inc. All rights reserved. is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Circulation Research doi: 10.1161/CIRCRESAHA.108.184911 2008;103:1319-1326; originally published online October 9, 2008; Circ Res. http://circres.ahajournals.org/content/103/11/1319 World Wide Web at: The online version of this article, along with updated information and services, is located on the http://circres.ahajournals.org/content/suppl/2008/10/10/CIRCRESAHA.108.184911.DC1.html Data Supplement (unedited) at: http://circres.ahajournals.org//subscriptions/ is online at: Circulation Research Information about subscribing to Subscriptions: http://www.lww.com/reprints Information about reprints can be found online at: Reprints: document. Permissions and Rights Question and Answer about this process is available in the located, click Request Permissions in the middle column of the Web page under Services. Further information Editorial Office. Once the online version of the published article for which permission is being requested is can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Circulation Research in Requests for permissions to reproduce figures, tables, or portions of articles originally published Permissions: by guest on March 15, 2014 http://circres.ahajournals.org/ Downloaded from by guest on March 15, 2014 http://circres.ahajournals.org/ Downloaded from by guest on March 15, 2014 http://circres.ahajournals.org/ Downloaded from by guest on March 15, 2014 http://circres.ahajournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ReudelhuberJ. Ferreira, Robson A. S. Santos, Thomas Walther, Rhian M. Touyz and Timothy L.
Chantal Mercure, Alvaro Yogi, Glaucia E. Callera, Anna B. Aranha, Michael Bader, AndersonAngiotensin(1-7) Blunts Hypertensive Cardiac Remodeling by a Direct Effect on the Heart
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2008 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.108.1849112008;103:1319-1326; originally published online October 9, 2008;Circ Res.
http://circres.ahajournals.org/content/103/11/1319World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2008/10/10/CIRCRESAHA.108.184911.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from

Angiotensin(1-7) Blunts Hypertensive Cardiac Remodelingby a Direct Effect on the Heart
Chantal Mercure, Alvaro Yogi, Glaucia E. Callera, Anna B. Aranha, Michael Bader,Anderson J. Ferreira, Robson A. S. Santos, Thomas Walther,
Rhian M. Touyz, Timothy L. Reudelhuber
Abstract—Angiotensin-converting enzyme 2 (ACE2) converts the vasopressor angiotensin II (Ang II) into angiotensin (1-7)[Ang(1-7)], a peptide reported to have vasodilatory and cardioprotective properties. Inactivation of the ACE2 gene in micehas been reported by one group to result in an accumulation of Ang II in the heart and an age-related defect in cardiaccontractility. A second study confirmed the role of ACE2 as an Ang II clearance enzyme but failed to reproduce thecontractility defects previously reported in ACE2-deficient mice. The reasons for these differences are unclear but couldinclude differences in the accumulation of Ang II or the deficiencies in Ang(1-7) in the mouse models used. As a result, theroles of ACE2, Ang II, and Ang(1-7) in the heart remain controversial. Using a novel strategy, we targeted the chronicoverproduction of either Ang II or Ang(1-7) in the heart of transgenic mice and tested their effect on age-related contractilityand on cardiac remodeling in response to a hypertensive challenge. We demonstrate that a chronic accumulation of Ang IIin the heart does not result in cardiac contractility defects, even in older (8-month-old) mice. Likewise, transgenic animals withan 8-fold increase in Ang(1-7) peptide in the heart exhibited no differences in resting blood pressure or cardiac contractilityas compared to age-matched controls, but they had significantly less ventricular hypertrophy and fibrosis than theirnontransgenic littermates in response to a hypertensive challenge. Analysis of downstream signaling cascades demonstratesthat cardiac Ang(1-7) selectively modulates some of the downstream signaling effectors of cardiac remodeling. These resultssuggest that Ang(1-7) can reduce hypertension-induced cardiac remodeling through a direct effect on the heart and raise thepossibility that pathologies associated with ACE2 inactivation are mediated in part by a decrease in production of Ang(1-7).(Circ Res. 2008;103:1319-1326.)
Key Words: angiotensin � hypertension � hypertrophy � remodeling
Angiotensin-converting enzyme 2 (ACE2), a homologueof the ACE enzyme1,2 expressed primarily in the vas-
cular endothelium of the heart, kidney, and forebrain,3 re-moves a single amino acid from the carboxy-terminus ofangiotensin II (Ang II) to generate angiotensin(1-7) [Ang(1-7)]. Inactivation of ACE2 in mice has been reported to resultin an age-dependent cardiac contractility defect associatedwith a decrease in systolic blood pressure in older (6-month-old) male mice, an upregulation of hypoxia-associated genesin the heart, and an increase in accumulation of Ang II incardiac tissue.4 These findings led the authors to suggest thatACE2 functions as a clearance enzyme for Ang II in the heartand that the accumulation of Ang II results in the observedcardiac defects over time. This possibility was supported bythe finding that the compound inactivation of both ACE2 andACE (thereby rendering the animals unable to synthesize AngII) corrects the cardiac contractility defect seen in the olderACE2 knockout male mice.4 However, ACE2 inactivationwould also be expected to reduce Ang(1-7) content in the
heart and compound inactivation of both ACE and ACE2could lead to restoration of Ang(1-7) levels by causing anaccumulation of Ang I. Thus, it is not clear whether thecontractility defects reported by Crackower et al4 result froman accumulation of cardiac Ang II or a deficit in Ang(1-7)production.
More recently, Gurley et al5 reported that inactivation ofACE2 resulted in a modest increase in basal blood pressure inthe C57BL/6 mouse strain that correlated with a deficiency inplasma Ang II clearance after bolus administration, confirm-ing that ACE2 can function as an Ang II clearance enzyme.Notably, there was no blood pressure effect of ACE2 inacti-vation in the 129/SvEv strain of mice, and no cardiaccontractility defects were detected in any of the ACE2knockout mice. The reason for the differences in the findingsby these 2 groups are not entirely clear but could be attributedto varying genetic backgrounds in the mice used or effects ofACE2 inactivation in noncardiac tissues or in other environ-mental or physiological variables. As a result, it is still
Original received January 21, 2008; revision received September 23, 2008; accepted October 1, 2008.From the Clinical Research Institute of Montreal (IRCM) (C.M., T.L.R.), Montreal, Canada; Kidney Research Center (A.Y., G.E.C., A.B.A., R.M.T.),
University of Ottawa, Ottawa, Canada; Max-Delbruck Center for Molecular Medicine (M.B.), Berlin-Buch, Germany; Federal University of Minas Gerais(A.J.F., R.A.S.S.), Belo Horizonte, Brazil; Centre of Biomedical Research (T.W.), Hull York Medical School, University of Hull, UK.
Correspondence to Timothy L. Reudelhuber, PhD, IRCM, 110 avenue des Pins Ouest, Montreal (QC) H2W 1R7, Canada. E-mail [email protected]© 2008 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.108.184911
1319 by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from

unclear whether cardiac dysfunction can result from a chronicincrease in Ang II or the loss of a protective effect ofAng(1-7) specifically in the heart.
In the current study, we have used a novel technique todirectly test for the role of the 2 angiotensin peptides on theheart. Our results show that Ang(1-7) can protect the heartfrom hypertension-induced remodeling and provide the firstevidence to our knowledge for a direct cardiac effect ofAng(1-7) in vivo.
Materials and MethodsAnimalsThe mice used in this study were housed in a 12/12 hour light/darkcycle with free access to normal mouse chow and water. All of theexperiments described herein were approved by the institutionalAnimal Ethics Committee and are in compliance with guidelinesissued by the Canadian Council on Animal Care.
Production of Transgenic MiceProduction of mice with chronic elevations of cardiac angiotensin II(Ang II) in the FVB/N line has been previously described.6 The miceused in this article correspond to the 1B founder line and have beenbackcrossed onto the C57BL/6 line for �8 generations. Generationof the mice expressing Ang(1-7) in the heart was performed byexpressing a fusion protein under the control of the mouse �-myosinheavy chain promoter (�-MHC, a gift from Dr. Jeffrey Robbins,University of Cincinnati). Transgenic mouse generation was per-formed using standard protocols. Line VII-7 was derived frominjection of inbred C57BL/6 embryos, whereas the VII-7b andVII-7c lines were derived from C57BL/6 X C3H F1 embryoinjections. All of the physiological characterization was performed ininbred mice (line VII-7) or in mice that had been backcrossed for �6generations (F6) onto the C57BL/6 (Harlan, Toronto, Canada) strain.Genomic integration of the transgene was determined by polymerasechain reaction analysis of DNA obtained from tails that underwentbiopsies.
Transgene Expression and Peptide ProductionTransgene expression was confirmed by extracting total tissue RNAby the guanidine isothiocyanate method and performing an RNaseprotection assay on 10 �g of total RNA for the transgene and histoneH4 (an internal control) as previously described.7 Production ofAng(1-7) in tissues of transgenic mice was measured by radioimmu-noassay by a modification of a previously published method.8
Briefly, animals were euthanized by CO2 inhalation and tissues wererapidly extracted and homogenized in 5 mL of 4 mol/L guanidiniumhydrochloride. The lysate was cleared by centrifugation (10 000g, 15min) and peptides were extracted using a High Flow Bond ElutLRC-C18 cartridge (Varian, Harbor City, Calif). Sequential washeswith 20 mL of 99.9% acetonitrile/0.1% HFBA, 20 mL of 0.1%HFBA, 3 mL of 0.1% HFBA containing 0.1% fat-free bovine serumalbumin, 10 mL of 10% acetonitrile/0.1% HFBA, and 3 mL of 0.1%HFBA were used to activate the columns. After sample application,the columns were washed with 20 mL of 0.1% HFBA and 3 mL of20% acetonitrile/0.1% HFBA. The adsorbed peptides were elutedwith 3 mL of 99.9% acetonitrile/0.1% HFBA into polypropylenetubes rinsed with 0.1% fat-free bovine serum albumin. After evap-oration, Ang(1-7) levels were measured by high-performance liquidchromatography-coupled radioimmunoassay as described byBotelho et al.9
Hypertensive ChallengeOsmotic minipumps (Alza Corp, Palo Alto, Calif) were implantedsubcutaneously in anesthetized (2% isoflurane) 12-week-old maletransgenic and control mice for the administration of Ang II (350ng/kg/min) or saline. Animals were allowed to recover for 1 week,after which blood pressure was measured for 9 days and the animals
were subjected to echocardiography. After physiological character-ization (19 days after minipump implantation), the mice wereeuthanized and the hearts were collected for histology and assess-ment of ventricular hypertrophy.
Physiological CharacterizationCardiac geometry and function were evaluated with a Sonos 5500(Philips) echo Doppler equipped with a 15- to 6-L 6 to 15-MHzultraband Intraoperative Linear Array probe. All studies were per-formed on lightly anesthetized (2% isoflurane, 5 min) mice main-tained at 37°C. M-mode–derived measures of left ventricle (LV)area were obtained, using the leading edge-to-leading-edge conven-tion adopted by the American Society of Echocardiography.10
Temporal changes between LV systolic diameter (LVD) and LVdiastolic diameter were used for the calculation of percent shorteningfraction, as follows [(LVD diameter�LVD)/LVD diameter]�100.Three heart beats were averaged for each measurement.
Systolic blood pressure was measured by computer-automatedtail-cuff plethysmography (model BP-2000; Visitech Systems, Apex,NC). Briefly, mice were trained to the apparatus for 9 continuousdays and measurements were recorded only for the last 4 days. Meanvalues for these last 4 days were calculated for each animal.
Assessment of Cardiac RemodelingVentricular hypertrophy was assessed by measuring the ratio ofcombined ventricular wet weight to total body weight (body weightdid not vary significantly between the animal groups studies). Fordetermination of cardiomyocyte hypertrophy and cardiac collagencontent, mice were anesthetized by intraperitoneal injection of 3 mgpentobarbital sodium (MTC Pharmaceuticals, Cambridge, Ontario).Blood was chased from major vessels by whole-body perfusion ofsaline solution through the heart, followed by in situ organ fixationusing 40 mL Bouin fixative solution (0.9% picric acid, 10%formaldehyde, and 5% glacial acetic acid). Organs were quicklyremoved and postfixed for 5 hours. Fixed tissue was stored in 70%ethanol at 4°C until analyzed.
To determine myocyte cross-sectional area, heart sections werestained with hematoxylin and eosin and examined with a ZeissAxiophot a magnification of 60�. To ensure that the sections wereperpendicular, only myocytes in which the nucleus was centrallylocated within the cell were used. The circumference of the myocyteswas traced using imaging software (Northern Eclipse 7.0; EM-PIXImaging Inc), and the myocyte cross-sectional area (�m2) wasdetermined by averaging the calculated area of 180 to 200 individualcardiomyocytes within the ventricular free wall over 5 to 6 sectionsper animal.
To quantify cardiac fibrosis, sections were stained with Sirius RedF3BA (0.5% in saturated aqueous picric acid; Sigma-Aldrich,Oakville, Ontario) for assessment of interstitial and perivascularcollagen content, as previously described.11 Quantification of fibro-sis was performed using an image analysis system (Northern Eclipse7.0; EM-PIX Imaging Inc). Seven fields were analyzed on 3independent sections per mouse to determine the collagen area/totalarea. A single investigator unaware of the experimental groupsperformed the analysis.
Modulation of Cardiac RNA ExpressionTotal RNA was isolated from mouse ventricles with TRIzol (Invitro-gen). Quantitative reverse transcriptase polymerase chain reactionwas performed on cDNA generated with the Omniscript RT Kit(QIAGEN, Inc) with the Quantitect SYBR Green polymerase chainreaction kit (QIAGEN Inc) in a MX3005 real-time polymerase chainreaction machine (Stratagene). The oligonucleotides were designedto have a melting temperature of 60°C and were used with an annealingtemperature of 58°C. The oligonucleotides used for quantitative poly-merase chain reaction were 5�-CCGATAGATCTGCCCTCTTG-3� (for-ward) and 5�-TCCAGGAGGGTATTCACCAC-3� (reverse) for atrial na-triuretic peptide; 5�-CAGCTCTTGAAGGACCAAGG-3� (forward) and5�-AGAGACCCAGGCAGAGTCAG-3� (reverse) for brain natriureticpeptide; 5�-TGGTGGGAATATTTGGAAAC-3� (forward) and 5�-
1320 Circulation Research November 21, 2008
by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from
GATGATGCAGGTGACTTTGG-3� (reverse) for the Ang II AT1 receptor(detects both AT1a and AT1b); 5�-GTCATTGACCTGGCACTTCC-3�(forward) and 5�-TGGCTAGGCTGATTACATGC-3� (reverse) for theAng II AT2 receptor; 5�-TGAGTGGCTGTCTTTTGACG-3� (forward)and 5�-CGCACACAGCAGTTCTTCTC-3� (reverse) for transforminggrowth factor beta (TGF beta 1); and 5�-TCTGGGCAAGGAGA-GATTTG-3� (forward) and 5�-CCGCCAAACTTCTTGGATTC-3� (re-verse) for 40S ribosomal protein S16. Expression of the correspondingRNA is shown relative to S16, arbitrarily set to 1.0 for control.
Western Blotting AnalysisMice were euthanized by CO2 inhalation and the hearts were rapidlyexcised and frozen in liquid nitrogen. Frozen hearts were homoge-nized in lysis buffer (50 mmol/L Tris/HCl, pH 7.4; 5 mmol/L EGTAand 2 mmol/L EDTA, 5% Triton 100, 0.1 mmol/L PMSF, 1 mmol/Lpepstatin A, 1 mmol/L leupeptin, and 1 mmol/L aprotinin). Thirtymicrograms of protein were separated by electrophoresis on a 10%polyacrylamide gel and probed for content of GAPDH, SHP-1, andSHP-2, as well as phosphorylated c-Src, p38MAPK, and ERK1/2 bystandard methodologies (see online supplement for expanded meth-ods at http://circres.ahajournals.org/cgi/content/full/CIRCRESAHA.108.184911/DC1).
Phosphatase AssayProtein tyrosine phosphatase assays for SHP-1 and SHP-2 wereperformed by a modification of previously published methods12 (seeonline supplement at http://circres.ahajournals.org/cgi/content/full/CIRCRESAHA.108.184911/DC1).
Statistical AnalysisUnpaired t test (Figure 1–3) and 1-way ANOVA with Bonferroni
multiple comparison post test (Figure 4–7) were performed usingGraphPad Prism version 3.0 for Windows (GraphPad Software, SanDiego, Calif).
ResponsibilityThe authors had full access to the data and take responsibility for itsintegrity. All authors have read and agree to the article as written.
Control
TG Ang II
A
B
LVDd LVDs
LVDd LVDs
Conrt ol
nA GT
g II0
10
20
30
40
50gninetrohs lanoitcarf %
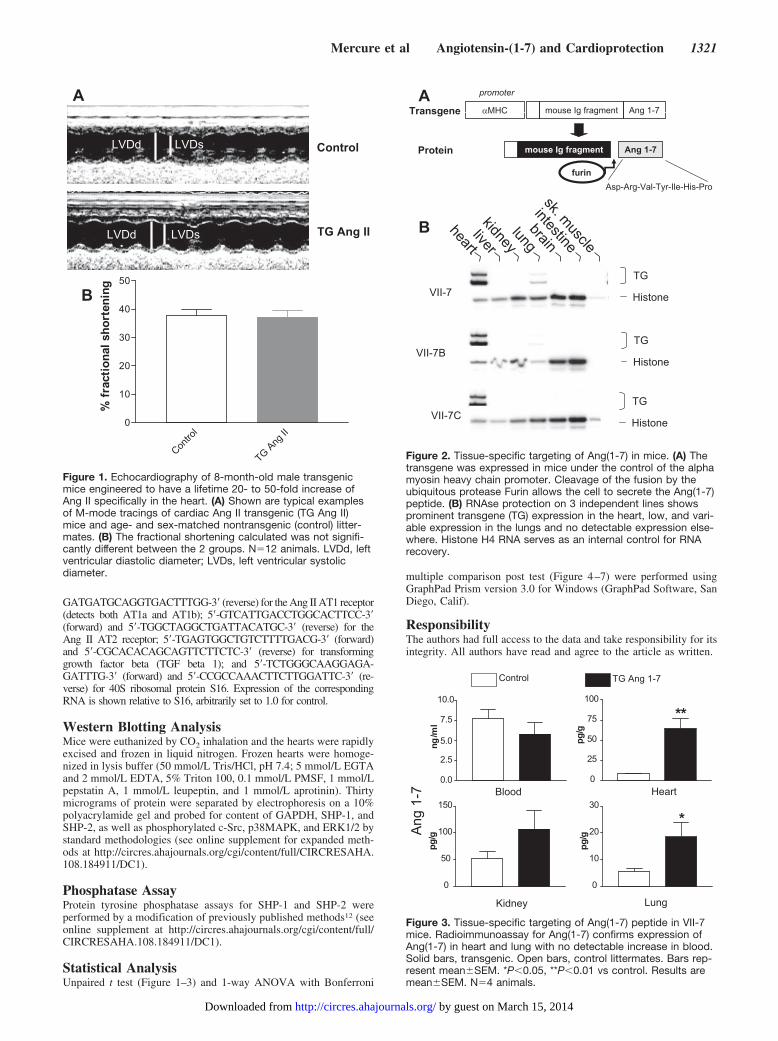
Figure 1. Echocardiography of 8-month-old male transgenicmice engineered to have a lifetime 20- to 50-fold increase ofAng II specifically in the heart. (A) Shown are typical examplesof M-mode tracings of cardiac Ang II transgenic (TG Ang II)mice and age- and sex-matched nontransgenic (control) litter-mates. (B) The fractional shortening calculated was not signifi-cantly different between the 2 groups. N�12 animals. LVDd, leftventricular diastolic diameter; LVDs, left ventricular systolicdiameter.
TG
Histone
TG
Histone
TG
Histone
VII-7
VII-7B
VII-7C
heartkidney
intestine
lungbrain
sk. m uscle
liver
furin
promoter
αMHC mouse Ig fragment Ang 1-7
mouse Ig fragment Ang 1-7Protein
TransgeneA
B
Asp-Arg-Val-Tyr-Ile-His-Pro
Figure 2. Tissue-specific targeting of Ang(1-7) in mice. (A) Thetransgene was expressed in mice under the control of the alphamyosin heavy chain promoter. Cleavage of the fusion by theubiquitous protease Furin allows the cell to secrete the Ang(1-7)peptide. (B) RNAse protection on 3 independent lines showsprominent transgene (TG) expression in the heart, low, and vari-able expression in the lungs and no detectable expression else-where. Histone H4 RNA serves as an internal control for RNArecovery.
0.0
2.5
5.0
7.5
10.0
lm/
gn
0
25
50
75
100
**
g/gp
0
50
100
150
g/gp
0
10
20
30
*
g/gp
Blood Heart
Kidney Lung
Control TG Ang 1-7
7-1 gnA
Figure 3. Tissue-specific targeting of Ang(1-7) peptide in VII-7mice. Radioimmunoassay for Ang(1-7) confirms expression ofAng(1-7) in heart and lung with no detectable increase in blood.Solid bars, transgenic. Open bars, control littermates. Bars rep-resent mean�SEM. *P�0.05, **P�0.01 vs control. Results aremean�SEM. N�4 animals.
Mercure et al Angiotensin-(1-7) and Cardioprotection 1321
by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from
ResultsCardiac Ang II and ContractilityTransgenic mice expressing an Ang II-generating fusionprotein in the heart exhibit a chronic 20- to 50-fold increasein cardiac Ang II.6 For the current study these mice werecrossed for 8 generations into the C57BL/6J background. Totest whether this local increase in cardiac Ang II would leadto age-dependent contractility defects, 8-month-old maletransgenic mice and control littermates were subjected toechocardiography. Results show no discernible difference inheart rate, blood pressure, chamber dimensions (not shown),or contractility (Figure 1) between transgenic and controlmice (fractional shortening 37.8�2.2% control, 37.2�2.4%Ang II transgenic). These results demonstrate that chronicincreases of Ang II in the heart are not sufficient to causecontractility defects, even in older mice.
Generation of Mice With Chronic Increases inCardiac Ang(1-7)To test whether cardiac Ang(1-7) could play a cardioprotec-tive role, we used a similar technology to generate transgenicmice expressing an Ang(1-7)-releasing fusion protein underthe control of the cardiac-specific �-MHC promoter (Figure2A). Three independent founder lines were obtained thatshow the expected expression of the transgene in the heart(Figure 2B) and a lower and variable level of expression ofthe transgene in the lung, as is commonly reported for thispromoter.13 We have not detected any underrepresentation orany increase in sudden death in any of the lines of miceoverexpressing Ang(1-7) in the heart over the past 9 gener-ations, although we were unable to generate offspring fromline VII-7B (data not shown). These results demonstrate that
overproduction of Ang(1-7) in the heart is not lethal. Usingline VII-7, we measured tissue-specific production of theAng(1-7) peptide in heart, lung, blood, and kidney (Figure 3).As expected, the expression of the transgene led to �8-foldincrease in Ang(1-7) in the heart. A 2- to 3-fold increase ofAng(1-7) was also detected in the lung of transgenic mice thatmay be attributable to the previously reported residual activ-ity of the promoter in pulmonary vessels branching from theheart,13 and there was a nonsignificant trend toward anincrease in the kidney although the transgene was notexpressed in this tissue (Figure 2B). Notably, there was nodetectable increase in the content of Ang(1-7) in the blood oftransgenic animals, thereby demonstrating that we weresuccessful in deriving a mouse model of cardiac overexpres-sion of Ang(1-7).
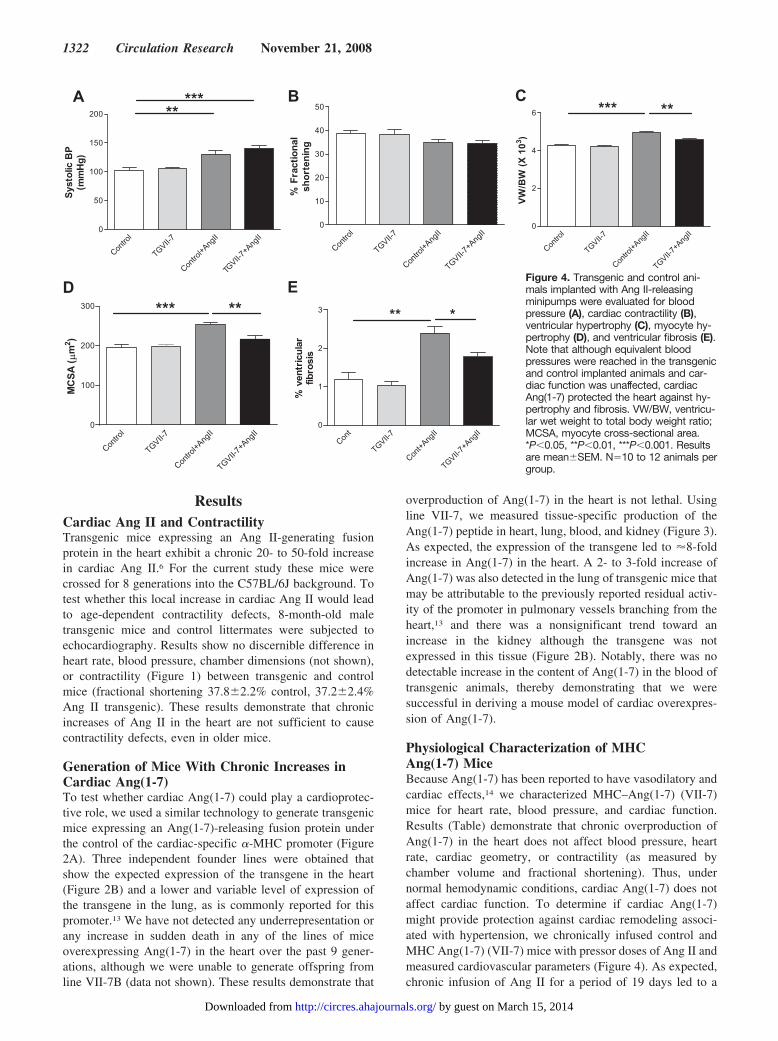
Physiological Characterization of MHCAng(1-7) MiceBecause Ang(1-7) has been reported to have vasodilatory andcardiac effects,14 we characterized MHC–Ang(1-7) (VII-7)mice for heart rate, blood pressure, and cardiac function.Results (Table) demonstrate that chronic overproduction ofAng(1-7) in the heart does not affect blood pressure, heartrate, cardiac geometry, or contractility (as measured bychamber volume and fractional shortening). Thus, undernormal hemodynamic conditions, cardiac Ang(1-7) does notaffect cardiac function. To determine if cardiac Ang(1-7)might provide protection against cardiac remodeling associ-ated with hypertension, we chronically infused control andMHC Ang(1-7) (VII-7) mice with pressor doses of Ang II andmeasured cardiovascular parameters (Figure 4). As expected,chronic infusion of Ang II for a period of 19 days led to a
C
E
A B
C
lortno7-IIVGT
Cno
rtlo
A+IgnI
GTV
A+7-II
IIgn
0
100
200
300 *** **
(AS
CM
µm2 )
lortnoCIVGTI 7-
oCn
lort+
IIgnA
GTIVI
+7-
IIgnA
0
50
100
150
200 *****
PB cilotsyS)g
Hm
m(
rtnoC
loVGT
-II 7
rtnoC
lo +AIIgn
IIgnA+7-IIVGT
0
2
4
6 *** **
01 X( W
B/W V
3 )
tnoC IVGTI 7-
oCtn
IIgnA+
GTIVI 7- +A
ngII
0
1
2
3 ** *ralucirtnev
%s is or bif
Cno
rt ol
TGVI -I 7 IIgnA+lortnoC TG-IIV7+
AngII
0
10
20
30
40
50
la noi tc arF %
gn inetroh sD
Figure 4. Transgenic and control ani-mals implanted with Ang II-releasingminipumps were evaluated for bloodpressure (A), cardiac contractility (B),ventricular hypertrophy (C), myocyte hy-pertrophy (D), and ventricular fibrosis (E).Note that although equivalent bloodpressures were reached in the transgenicand control implanted animals and car-diac function was unaffected, cardiacAng(1-7) protected the heart against hy-pertrophy and fibrosis. VW/BW, ventricu-lar wet weight to total body weight ratio;MCSA, myocyte cross-sectional area.*P�0.05, **P�0.01, ***P�0.001. Resultsare mean�SEM. N�10 to 12 animals pergroup.
1322 Circulation Research November 21, 2008
by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from
significant increase in systolic blood pressure that was equiv-alent in transgenic and control mice (Figure 4A). Likewise,both groups showed a trend toward a decrease in fractionalshortening that did not reach statistical significance (Figure4B). In contrast, although control mice responded to the AngII infusion with �20% increase in ventricular weight, trans-genic VII-7 mice had a significantly diminished hypertrophicresponse (Figure 4C). These same differences were reflected
in the size of cardiomyocytes (Figure 4D). Likewise, cardiacfibrosis increased by �2-fold in control mice infused for 19days with Ang II while the fibrotic response was approxi-mately halved in transgenic VII-7 mice (Figure 4E). Inconclusion, chronic production of Ang(1-7) in the heartblunts cardiac remodeling in response to Ang II-inducedhypertension.
Molecular Markers of Cardiac RemodelingIn an effort to better define the site of action of Ang(1-7) incardioprotection, the expression of markers and mediators of
lortnoC GTV
II -7
Cortno +l Ang
I I IIgnA+7-IIVGT
0
2
4
6
8 *** *noisserpxe P
NA
lortnoC GTVI -I 7
oCntr
lo +
IIgnA A+7-IIVGT
nIIg
0
1
2
3
4
5 *** **
noisserpxe PN
B
Conrt ol -IIVGT
7
oCn rt ol
nA+
IIg
TIVG
7-I
nA+
IIg0.0
0.5
1.0
1.5 *
FGT
βn ois serpxe 1 -
A
B
C
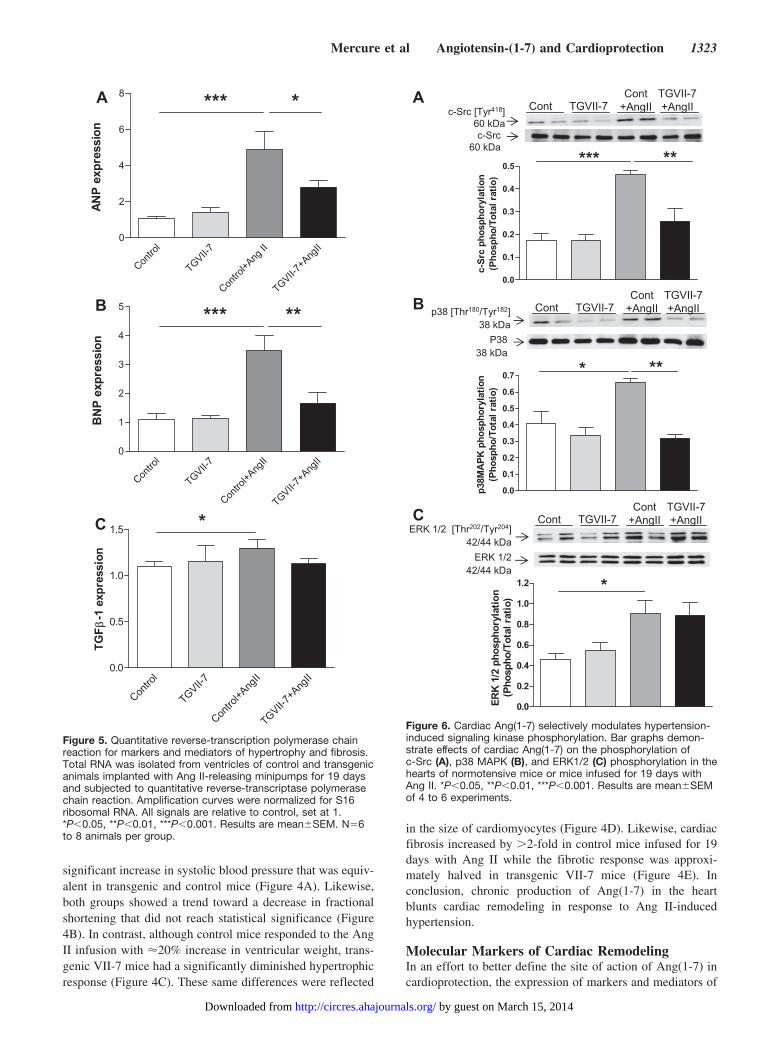
Figure 5. Quantitative reverse-transcription polymerase chainreaction for markers and mediators of hypertrophy and fibrosis.Total RNA was isolated from ventricles of control and transgenicanimals implanted with Ang II-releasing minipumps for 19 daysand subjected to quantitative reverse-transcriptase polymerasechain reaction. Amplification curves were normalized for S16ribosomal RNA. All signals are relative to control, set at 1.*P�0.05, **P�0.01, ***P�0.001. Results are mean�SEM. N�6to 8 animals per group.
0.0
0.1
0.2
0.3
0.4
0.5
noitalyrohpsohp crS-c)oitar latoT/ohpsohP (
*** **
c-Src [Tyr418]60 kDac-Src
60 kDa
A
B
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7noitalyrohpsohp KP
AM83p
)oitar lat oT/ ohpsohP(
* **
p38 [Thr180/Tyr182]38 kDa
P3838 kDa
0.0
0.2
0.4
0.6
0.8
1.0
1.2
noitalyrohpsohp 2/1 KRE
)oitar latoT/ ohpsohP(Cont TGVII-7
Cont+AngII
TGVII-7+AngII
*
ERK 1/2 [Thr202/Tyr204]42/44 kDa
ERK 1/242/44 kDa
C
Cont TGVII-7Cont
+AngIITGVII-7+AngII
Cont TGVII-7Cont
+AngIITGVII-7+AngII
Figure 6. Cardiac Ang(1-7) selectively modulates hypertension-induced signaling kinase phosphorylation. Bar graphs demon-strate effects of cardiac Ang(1-7) on the phosphorylation ofc-Src (A), p38 MAPK (B), and ERK1/2 (C) phosphorylation in thehearts of normotensive mice or mice infused for 19 days withAng II. *P�0.05, **P�0.01, ***P�0.001. Results are mean�SEMof 4 to 6 experiments.
Mercure et al Angiotensin-(1-7) and Cardioprotection 1323
by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from
LVH was measured in control and transgenic VII-7 mice withand without infusion of Ang II (Figure 5). As has beenpreviously shown, ventricular expression of atrial natriureticpeptide messenger RNA is significantly enhanced with LVH,being increased nearly 5-fold in control mice after 19 days ofAng II infusion (Figure 5A). In contrast, mice expressingAng(1-7) in the heart increase atrial natriuretic peptide geneexpression by �3-fold in response to the Ang II infusion.Likewise, brain natriuretic peptide expression is increased�4-fold by Ang II infusion in control animals, whereas it isinduced �2-fold in transgenic VII-7 (Figure 5B). Localexpression of Ang(1-7) also eliminated the slight increase inventricular transforming growth factor-�1 RNA seen with
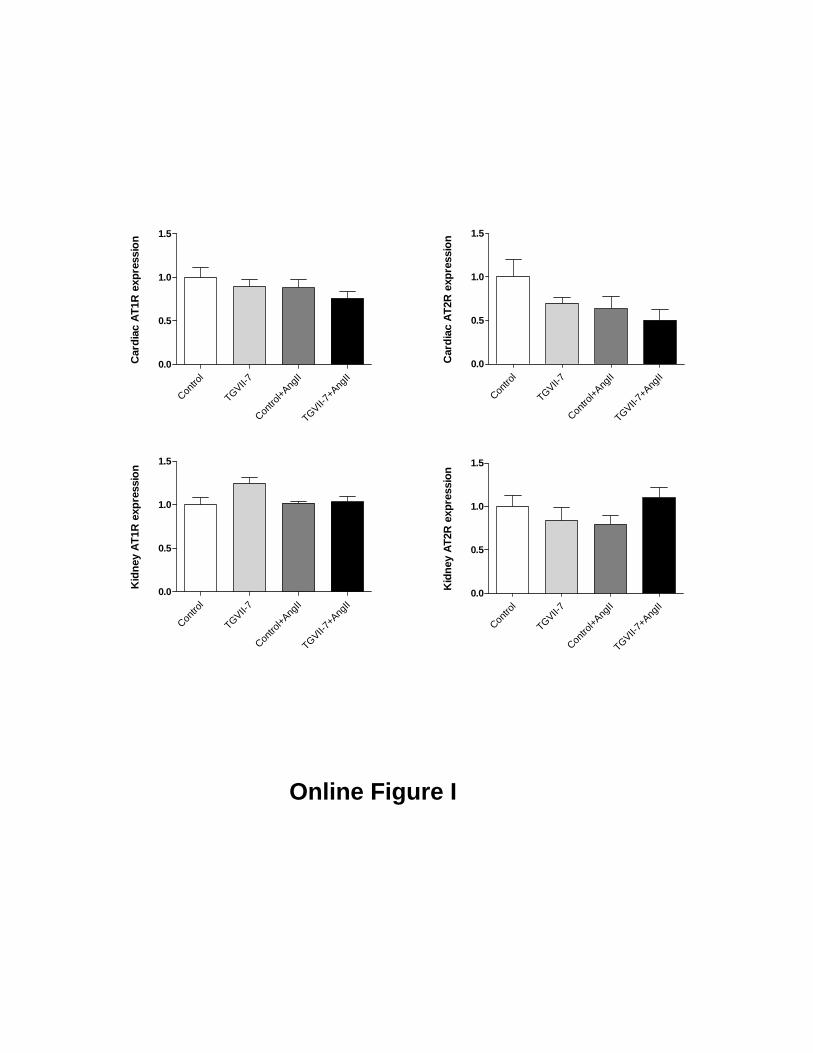
Ang II infusion (Figure 5C). In contrast, we found no effectof the transgene on expression of AT1 (AT1a and AT1b) orAT2 receptors in either the hearts or the kidneys of mice (seeonline supplement for Figure I at http://circres.ahajournals.org/cgi/content/full/CIRCRESAHA.108.184911/DC1).These results confirm that the effect of Ang(1-7) on cardiacremodeling is reflected by changes at the molecular level butthat they are not explained by an alteration in the expressionof angiotensin receptor levels.
Ang(1-7) Selectively Modulates Growth SignalingPathways in Ang II-Infused MiceTo investigate the mechanisms by which local Ang(1-7)reduces hypertension-induced cardiac remodeling, we as-sessed a number of intracellular signaling pathways known tobe associated with hypertrophic remodeling, including c-Src,p38, and ERK1/2 mitogen-activated protein kinase (MAPK)(Figure 6). Chronic production of Ang(1-7) in the hearts ofmice had no effect of the basal level of phosphorylation ofthese signaling kinases, as compared to control animals. Inaddition, the phosphorylation of all of these signaling mole-cules was increased in response to Ang II infusion in controlmice as has been previously reported.15–17 In contrast, miceexpressing Ang(1-7) in the heart showed a significant de-crease in phosphorylation of c-Src (Figure 6A) and p38kinase (Figure 6B) seen after Ang II infusion, whereas theincrease in ERK1/2 phosphorylation was unaffected by ex-pression of the transgene (Figure 6C). These results demon-strate a selective effect of Ang(1-7) on intracellular signalingpathways that mediate cardiac remodeling.
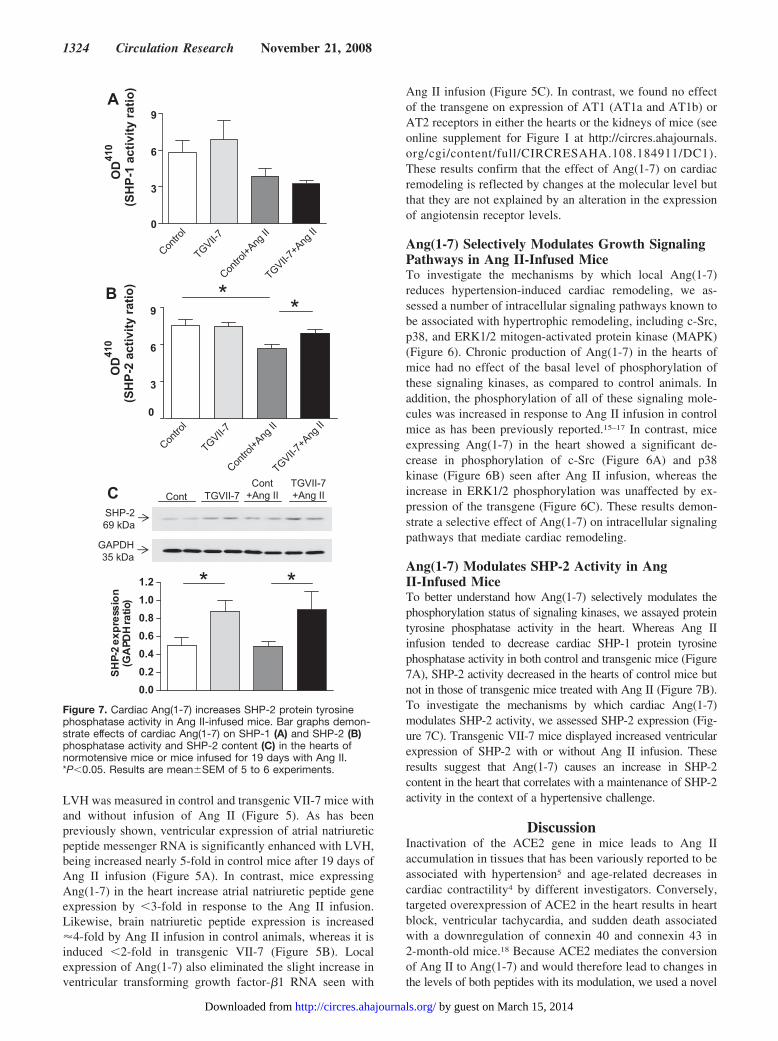
Ang(1-7) Modulates SHP-2 Activity in AngII-Infused MiceTo better understand how Ang(1-7) selectively modulates thephosphorylation status of signaling kinases, we assayed proteintyrosine phosphatase activity in the heart. Whereas Ang IIinfusion tended to decrease cardiac SHP-1 protein tyrosinephosphatase activity in both control and transgenic mice (Figure7A), SHP-2 activity decreased in the hearts of control mice butnot in those of transgenic mice treated with Ang II (Figure 7B).To investigate the mechanisms by which cardiac Ang(1-7)modulates SHP-2 activity, we assessed SHP-2 expression (Fig-ure 7C). Transgenic VII-7 mice displayed increased ventricularexpression of SHP-2 with or without Ang II infusion. Theseresults suggest that Ang(1-7) causes an increase in SHP-2content in the heart that correlates with a maintenance of SHP-2activity in the context of a hypertensive challenge.
DiscussionInactivation of the ACE2 gene in mice leads to Ang IIaccumulation in tissues that has been variously reported to beassociated with hypertension5 and age-related decreases incardiac contractility4 by different investigators. Conversely,targeted overexpression of ACE2 in the heart results in heartblock, ventricular tachycardia, and sudden death associatedwith a downregulation of connexin 40 and connexin 43 in2-month-old mice.18 Because ACE2 mediates the conversionof Ang II to Ang(1-7) and would therefore lead to changes inthe levels of both peptides with its modulation, we used a novel
Contro
l
TGVII-7
TGVII-7+A
ngII
Contro
l+Ang
II
* *
0
3
6
9
DO
014
)oitar ytivitca 2-PHS(
0.00.20.40.60.81.01.2
noisserpxe 2-PHS
)oitar HDP
AG(
Cont TGVII-7TGVII-7+Ang II
Cont+Ang II
SHP-269 kDa
GAPDH35 kDa
* *
B
A
Contro
l
TGVII-7
TGVII-7+A
ngII
Contro
l+Ang
II0
3
6
9D
O014
)oitar ytivitca 1-PHS(
C
Figure 7. Cardiac Ang(1-7) increases SHP-2 protein tyrosinephosphatase activity in Ang II-infused mice. Bar graphs demon-strate effects of cardiac Ang(1-7) on SHP-1 (A) and SHP-2 (B)phosphatase activity and SHP-2 content (C) in the hearts ofnormotensive mice or mice infused for 19 days with Ang II.*P�0.05. Results are mean�SEM of 5 to 6 experiments.
1324 Circulation Research November 21, 2008
by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from
approach to test for the effect of selective increases of each ofthese peptides individually in the hearts of transgenic mice.
Our results show that mice with a chronic and very large (20-to 50-fold) increase in Ang II exclusively in the heart did notexhibit the decreases in cardiac contractility previously reportedin mice deficient for ACE-2,4 even at advanced age, making itunlikely that this cardiac phenotype is attributed to an accumu-lation of Ang II in the heart. This possibility is further supportedby our finding that local production of Ang(1-7) in the heart canprovide protection against cardiac remodeling associated withhypertension. The possibility that Ang(1-7) is important for heartfunction is also supported by other gene manipulation studies inmice. The complete inactivation of the RAS by knocking outangiotensinogen also leads to impaired systolic function inmice,19 suggesting that the lack of some product of the RAS isresponsible. Furthermore, Santos et al20 demonstrated that micedefective for Mas, the putative Ang(1-7) receptor, exhibit de-creased fractional shortening. The decreased cardiac contractilityseen in this latter study was obvious in normotensive mice andwas attributed to increased overproduction of certain extracellu-lar matrix proteins in the hearts of Mas-deficient mice.20 Al-though overproduction of Ang(1-7) in the heart did not lead to anapparent decrease in collagen content in the normotensive micein our studies, our results do confirm a role for Ang(1-7) in thecontrol of cardiac collagen content in hypertension. It is alsonotable that the chronic overproduction of Ang(1-7) weachieved in our studies did not result in any detectable adverseeffects on the heart and did not decrease survival of ourtransgenic mice.
Whereas our current results support a direct effect ofAng(1-7) on the heart, the target cell is more difficult toidentify because the approach we used is likely to lead to ageneral increase in Ang(1-7) in the interstitial spaces aroundcardiomyocytes. It seems quite likely, however, that thecardiomyocyte is a primary target since the antihypertrophiceffect of local Ang(1-7) is mirrored by effects on cardiomyo-cyte size (Figure 4). Ang(1-7) selectively binds to cardiomyo-cytes in vitro and in vivo, and this binding disappears in Masreceptor-deficient mice.20,21 It is less clear how Ang(1-7) actsto reduce hypertensive cardiac fibrosis because there has beenno direct demonstration of Ang(1-7) receptors on fibroblasts.However, by using mice chimeric for Ang II AT1 receptordeficiency, Matsusaka et al22 elegantly demonstrated thatcardiac fibroblast proliferation in response to a hypertensivechallenge is orchestrated by the cardiomyocytes, raising thepossibility that Ang(1-7) interferes with the signaling mech-anism before it leaves the cardiomyocyte.
Interestingly, increasing cardiac Ang(1-7) led to a slight(1.5�), but significant increase in ventricular atrial natriureticpeptide gene expression in the normotensive animals (Figure5A). In addition to being a marker of hypertrophy, we havepreviously obtained evidence that ventricular atrial natriureticpeptide may also play a role in cardioprotection throughstimulation of intracellular cGMP accumulation in cardio-myocytes.23 Nevertheless, we found no increase in cardiaccGMP content in the hearts of mice producing Ang(1-7)locally (data not shown). Rather, it appears that Ang(1-7)selectively modulates MAPK signaling in the heart.
Cardiac hypertrophy can result in response to a number ofstimuli, including hormones, cytokines, and biomechanicalstress (stretch). All of these stimuli result in enhanced MAPKsignaling in cardiomyocytes and, through the activation ofkinase cascades, lead to the ultimate phosphorylation ofterminal kinases such as ERK, p38, and JNK kinases.17
These, in turn, regulate cardiomyocyte gene expressionthrough selective transcription factor phosphorylation. Bio-mechanical stress signals can also be transduced throughintegrin-mediated activation of focal adhesion kinase–Srckinase complex leading to the activation of small GTPasessuch as Ras and Rho.24 We tested whether Ang(1-7) mightprovide protection from cardiac remodeling by modulating�1 of these hypertrophic signaling pathways. Our findingsdemonstrate that Ang(1-7) modulates some (c-Src and p38MAPK) but not all (ERK1/2) growth signaling pathways inthe heart in response to hypertension. The inhibitory effect ofAng(1-7) on p38 MAPK activation might also explain theapparent decrease in hypertension-induced transforminggrowth factor �-1 expression seen in the transgenic mice(Figure 5C) because p38 is one of the known mediators ofboth transforming growth factor �-1 expression and activa-tion.25 The lack of effect on ERK1/2 signaling is surprisingbecause its phosphorylation is closely correlated with hyper-trophic remodeling. However, Purcel et al26 recently reportedthat mice with targeted inactivation of the ERK1/2 genesshowed no reduction in the development of pathologicalhypertrophy. This led the authors to suggest that ERK1/2 mayactually play a protective role in response to pathologicalstimuli. A local increase in Ang(1-7) also correlates with anincrease in the ventricular content of SHP-2 protein tyrosinephosphatase and an increase in its activity in the face of AngII infusion. This increase in phosphatase activity may explainthe reduced phosphorylation of c-src and p38 MAPK, and thiswill remain an interesting area for further study.
Table. Physiological Characterization of TG Mice
Mice Heart Rate, bpm Systolic BP, mm Hg %FS, mm LVDd, mm LVDs
Control 653�13 106.8�2.9 38.1�1.3 4.17�0.09 2.58�0.14
Ang(1-7) TG (line VII-7) 617�12 113.4�2.8 39.1�1.7 4.12�0.08 2.51�0.08
Ang-(1-7) TG (line VII-7C) 600�12* 95.3�2.9† 38.4�1.8 3.96�0.10 2.45�0.12
Values represent the mean�standard error. N�12 animals/group.%FS indicates percent fractional shortening; bpm, heart rate in beats per minute; LVDd, left ventricular diastolic diameter; LVDs,
left ventricular systolic diameter; TG, transgenic.*P�0.05 vs control.†P�0.01 vs control.
Mercure et al Angiotensin-(1-7) and Cardioprotection 1325
by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from

Although it is also possible that Ang(1-7) inhibits the Ang IIreceptor directly to reduce cardiac remodeling resulting fromAng II infusion, we found no evidence that chronic overproduc-tion of Ang(1-7) decreased AT1 receptor expression in the heartsor kidneys of our transgenic mice. It has also been suggested thatAng(1-7) can downregulate Ang II signaling by promotingheterodimerization between the Mas receptor and the Ang IIAT1 receptor.27 However, a specific reduction of Ang II signal-ing by Ang(1-7) is an unlikely explanation for our results forseveral reasons. First, local production of Ang II in the heartdoes not result in the cardiac remodeling we see in our AngII-infused mice,6 suggesting that it is not attributed to a directeffect of Ang II signaling in cardiomyocytes. Thus, a localproduction of Ang(1-7) in the heart could not reverse remodelingby blocking local Ang II signaling. Second, we had previouslydemonstrated that circulating Ang(1-7) blunts cardiac remodel-ing attributed to isoproterenol injection.8 Thus, Ang(1-7) mustact to block signaling of �1 type of hypertrophic stimulus,consistent with a role in the signaling pathways common to thesestimuli. Finally, Ang(1-7) did not block the stimulation ofERK1/2 phosphorylation, a classical downstream target of AngII receptor activation in tissue culture. Taken together, our resultssuggest that Ang(1-7) acts on cardiomyocytes to block signalingpathways that trigger cardiac remodeling in an acute hyperten-sive model. Whether the Ang(1-7) peptide can also providecardioprotection from chronic hypertensive heart disease will bea topic of future interest.
AcknowledgmentsThe authors acknowledge the expert technical assistance of AugustoMontezano and Yong Wang.
Sources of FundingThis work was supported by grants MOP 13569 and 44018 from theCanadian Institutes of Health Research to T.L.R. and R.M.T.,respectively.
DisclosuresNone.
References1. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N,
Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. Anovel angiotensin-converting enzyme-related carboxypeptidase (ACE2)converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–E9.
2. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ.A human homolog of angiotensin-converting enzyme. Cloning and func-tional expression as a captopril-insensitive carboxypeptidase. J BiolChem. 2000;275:33238–33243.
3. Gembardt F, Sterner-Kock A, Imboden H, Spalteholz M, Reibitz F,Schultheiss HP, Siems WE, Walther T. Organ-specific distribution ofACE2 mRNA and correlating peptidase activity in rodents. Peptides.2005;26:1270–1277.
4. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE,Oliveira-dos-Santos AJ, da CJ, Zhang L, Pei Y, Scholey J, Ferrario CM,Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. An-giotensin-converting enzyme 2 is an essential regulator of heart function.Nature. 2002;417:822–828.
5. Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, Haystead TA,Donoghue M, Breitbart RE, Acton SL, Rockman HA, Coffman TM.Altered blood pressure responses and normal cardiac phenotype inACE2-null mice. J Clin Invest. 2006;116:2218–2225.
6. van Kats JP, Methot D, Paradis P, Silversides DW, Reudelhuber TL. Useof a biological peptide pump to study chronic peptide hormone action in
transgenic mice. Direct and indirect effects of angiotensin II on the heart.J Biol Chem. 2001;276:44012–44017.
7. Lochard N, Silversides DW, van Kats JP, Mercure C, Reudelhuber TL.Brain-specific restoration of angiotensin II corrects renal defects seen inangiotensinogen-deficient mice. J Biol Chem. 2003;278:2184–2189.
8. Santos RA, Ferreira AJ, Nadu AP, Braga AN, de Almeida AP,Campagnole-Santos MJ, Baltatu O, Iliescu R, Reudelhuber TL, Bader M.Expression of an angiotensin-(1-7)-producing fusion protein producescardioprotective effects in rats. Physiol Genomics. 2004;17:292–299.
9. Botelho LM, Block CH, Khosla MC, Santos RA. Plasma angiotensin(1-7)immunoreactivity is increased by salt load, water deprivation, and hem-orrhage. Peptides. 1994;15:723–729.
10. Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R,Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I. Recom-mendations for quantitation of the left ventricle by two-dimensionalechocardiography. Am Society of Echocardiography Committee onStandards, Subcommittee on Quantitation of Two-Dimensional Echocar-diograms. J Am Soc Echocardiogr. 1989;2:358–367.
11. Touyz RM, Mercure C, He Y, Javeshghani D, Yao G, Callera GE, YogiA, Lochard N, Reudelhuber TL. Angiotensin II-dependent chronic hyper-tension and cardiac hypertrophy are unaffected by gp91phox-containingNADPH oxidase. Hypertension. 2005;45:530–537.
12. Lee K, Esselman WJ. Inhibition of PTPs by H(2)O(2) regulates the activationof distinct MAPK pathways. Free Radic Biol Med. 2002;33:1121–1132.
13. Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J.Tissue-specific regulation of the alpha-myosin heavy chain genepromoter in transgenic mice. J Biol Chem. 1991;266:24613–24620.
14. Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and func-tional roles of angiotensin-converting enzyme 2 and angiotensin-(1-7) inregulation of cardiovascular function. Am J Physiol Heart Circ Physiol.2005;289:H2281–H2290.
15. Sugden PH. Ras, Akt, and mechanotransduction in the cardiac myocyte.Circ Res. 2003;93:1179–1192.
16. Sugden PH, Clerk A. “Stress-responsive” mitogen-activated proteinkinases (c-Jun N-terminal kinases and p38 mitogen-activated proteinkinases) in the myocardium. Circ Res. 1998;83:345–352.
17. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intra-cellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600.
18. Donoghue M, Wakimoto H, Maguire CT, Acton S, Hales P, Stagliano N,Fairchild-Huntress V, Xu J, Lorenz JN, Kadambi V, Berul CI, BreitbartRE. Heart block, ventricular tachycardia, and sudden death in ACE2transgenic mice with downregulated connexins. J Mol Cell Cardiol.2003;35:1043–1053.
19. Walther T, Steendijk P, Westermann D, Hohmann C, Schulze K, Heringer-Walther S, Schultheiss HP, Tschope C. Angiotensin deficiency in mice leads todilated cardiomyopathy. Eur J Pharmacol. 2004;493:161–165.
20. Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, Paula RD, CruzJS, Ramos AS, Rosa KT, Irigoyen MC, Bader M, Alenina N, Kitten GT,Ferreira AJ. Impairment of in vitro and in vivo heart function in angioten-sin-(1-7) receptor MAS knockout mice. Hypertension. 2006;47:996–1002.
21. Tallant EA, Ferrario CM, Gallagher PE. Angiotensin-(1-7) inhibitsgrowth of cardiac myocytes through activation of the mas receptor. Am JPhysiol Heart Circ Physiol. 2005;289:H1560–H1566.
22. Matsusaka T, Katori H, Inagami T, Fogo A, Ichikawa I. Communicationbetween myocytes and fibroblasts in cardiac remodeling in angiotensinchimeric mice. J Clin Invest. 1999;103:1451–1458.
23. Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF.Expression of constitutively active guanylate cyclase in cardiomyocytesinhibits the hypertrophic effects of isoproterenol and aortic constrictionon mouse hearts. J Biol Chem. 2003;278:47694–47699.
24. Torsoni AS, Constancio SS, Nadruz W Jr, Hanks SK, Franchini KG.Focal adhesion kinase is activated and mediates the early hypertrophicresponse to stretch in cardiac myocytes. Circ Res. 2003;93:140–147.
25. Gordon KJ, Blobe GC. Role of transforming growth factor-beta super-family signaling pathways in human disease. Biochim Biophys Acta.2008;1782:197–228.
26. Purcell NH, Wilkins BJ, York A, Saba-El-Leil MK, Meloche S, RobbinsJ, Molkentin JD. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy invivo. Proc Natl Acad Sci U S A. 2007;104:14074–14079.
27. Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, Gembardt F, Kellett E, Martini L, Vanderheyden P,Schultheiss HP, Walther T. G-protein-coupled receptor Mas is a physiologicalantagonist of the angiotensin II type 1 receptor. Circulation. 2005;111:1806–1813.
1326 Circulation Research November 21, 2008
by guest on March 15, 2014http://circres.ahajournals.org/Downloaded from

Mercure et al. Angiotensin-(1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart EXPANDED METHODS: Western Blotting analysis Mice were euthanized by CO2 inhalation and the hearts were rapidly excised and frozen in liquid nitrogen. Frozen hearts were homogenized in lysis buffer [50 mmol/l Tris/HCl (pH 7.4), 5 mmol/l EGTA and 2 mmol/l EDTA, 5% Triton 100, 0.1 mmol/l PMSF, 1 mmol/l pepstatin A, 1 mmol/l leupeptin and 1 mmol/l aprotinin]. Thirty micrograms of protein were separated by electrophoresis on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. Nonspecific binding sites were blocked with 5% skim milk or 1% BSA in Tris-buffered saline solution with Tween for 1 hour at 24°C. Membranes were then incubated with primary antibodies (1:1000) overnight at 4°C. Antibodies were as follows: anti–c-Src (Tyr418) (Biosource International), anti-SHP-2 (Santa Cruz), anti anti–p38 MAPK Thr180/Tyr182), anti-ERK1/2 (Thr202/Tyr204) and anti–SHP-2 (Tyr 580) (the latter 3 from Cell Signaling Technology, Inc). Immunoblots for nonphosphoproteins (c-Src, p38 MAPK, ERK1/2, Akt) or GAPDH (Chemicon) were carried out in the same membranes used as a loading control. After incubation with secondary antibodies, signals were revealed by chemiluminescence, visualized by autoradiography, and quantified densitometrically. Results were normalized by the total protein or GAPDH. Phosphatase assay Hearts were homogenized in immunoprecipitation buffer without sodium orthovanadate (15 mM KCl, 10 mM HEPES, 2 mM MgCl2, 0.1% NP40, 1mM PMSF, 1μg/mL aprotinin, 1μg/mL leupeptin, 1μg/mL pepstatin A, pH 7.4). After centrifugation (5000 g, 4 oC, 5 min), supernatants containing 800 μg protein were then incubated with 2 μg of monoclonal SHP-1 or SHP-2 antibody (Santa Cruz) overnight at 4 oC on a rocking platform followed by immunoprecipitation with 30 μL GammaBind Plus Sepharose beads (GE Healthcare) for 3 additional hours. Immune complexes were washed 2 times in immunoprecipitation buffer and once in phosphatase assay buffer (25 mM Imidazole pH 7.2, 45 mM NaCl, 1 mM EDTA, 5 mm dithiothreitol, 1mg/mL BSA). Immune complexes were incubated in 100 μL phosphatase assay buffer with a final concentration of 20 mM p-nitrophenyl phosphate (P-NPP, Thermo Fisher Scientific) for 30 min at 37oC with shaking. Optical density of the supernatants was measured at 410 nm. After measurement of PTP activity, the proteins were recovered from the beads and subjected to SDS-PAGE gel for the western blotting analysis. The absorbance values obtained from PTP immune complexes were then normalized by the respective densitometry values of SHP-1 or SHP-2 imunoblots. FIGURE LEGEND: Online FIGURE I: Quantitative RT-PCR for angiotensin II AT1 and AT2 receptors (AT1R and AT2R, respectively). Total RNA was isolated from ventricles and kidneys of control and transgenic animals implanted with Ang II-releasing minipumps for 19 days and subjected to quantitative reverse-transcriptase polymerase chain reaction. Amplification curves were normalized for S16 ribosomal RNA. All signals are relative to Control, set at 1. Results are mean±SEM. N=6-8 animals per group.
michelle
Text Box
Supplement Material
Contro
l
TGVII-7
Contro
l+Ang
II
TGVII-7+A
ngII
0.0
0.5
1.0
1.5
Car
diac
AT1
R e
xpre
ssio
n
Contro
l
TGVII-7
Contro
l+Ang
II
TGVII-7+A
ngII
0.0
0.5
1.0
1.5
Car
diac
AT2
R e
xpre
ssio
n
Contro
l
TGVII-7
Contro
l+Ang
II
TGVII-7+A
ngII
0.0
0.5
1.0
1.5
Kid
ney
AT1R
exp
ress
ion
Contro
l
TGVII-7
Contro
l+Ang
II
TGVII-7+A
ngII
0.0
0.5
1.0
1.5K
idne
y AT
2R e
xpre
ssio
n
Online Figure I
Related Documents