Analysis of the GAD1 promoter: Trans-acting factors and DNA methylation converge on the 5 0 untranslated region Ying Chen, Erbo Dong, Dennis R. Grayson * The Psychiatric Institute, Department of Psychiatry, College of Medicine, University of Illinois Chicago,1601 W. Taylor St., Chicago, IL 60612, USA article info Article history: Received 20 August 2010 Received in revised form 10 September 2010 Accepted 16 September 2010 Keywords: Schizophrenia Methylation Regulation Histone deacetylase inhibitors Interneurons abstract GAD67 corresponds to one of two enzymes that decarboxylates glutamate to produce g-aminobutyric acid, the main inhibitory neurotransmitter in the mammalian central nervous system, hence defining the cellular phenotype of a diverse set of inhibitory interneurons of the brain. Reduced cortical GAD67 mRNA levels have consistently been reported in schizophrenia and bipolar disorder with psychosis. The human gene encoding GAD67, GAD1 , is located on chromosome 2q31.1 and the transcriptional start site resides within a large CpG island that spans a region extending from upstream through the first exon. We have analyzed the GAD1 promoter using transient transfection analysis of upstream and downstream sequences in NT2 cells, a human neuroprogenitor cell line. Interestingly, results from these studies show that cis-acting regulatory elements are located downstream of the RNA start site and are in the region corresponding to the first exon. Trans-acting factors such as Pitx2 and the Dlx family of transcription factors are active in promoting downstream reporter expression even when all of the 5 0 flanking sequences are removed. However, those constructs that contain an internal deletion from þ66 to þ173 bp fail to support expression even when these factors are provided in trans. We have previously shown that the Class I histone deacetylase inhibitor MS-275 potently activates GAD1 mRNA expression in NT2 cells suggesting the possibility that the promoter is sensitive to drugs that induce chromatin remodeling. Using methyl DNA immuneprecipitation of MS-275-treated NT2 cells, we provide data showing that Class I HDAC inhibition mediated an increase in GAD1 expression and that this was accompanied by decreased GAD1 promoter methylation. Moreover, the reduced levels of GAD1 DNA methylation are highest in those regions proximal to the location of the in vitro defined cis-acting regulatory elements. Our data suggest that changes in promoter methylation associated with gene regulation are not random but overlap the locations of proximal cis-acting elements. This article is part of a Special Issue entitled ‘Trends in Neuropharmacology: In Memory of Erminio Costa’. Ó 2010 Elsevier Ltd. All rights reserved. 1. Introduction Nearly two decades ago it was shown that the enzyme responsible for decarboxylating glutamate to produce g-amino- butyric acid (GABA) was encoded by two related genes on different chromosomes in mammals (Bu and Tobin, 1994). The correspond- ing mRNAs (glutamate decarboxylase 1 (GAD1) and GAD2) when translated specify the major isoforms of the enzymes that define the neurotransmitter phenotype of the major class of inhibitory interneurons in the brain. These two isoforms differ in molecular size based on their migration on SDS polyacrylamide gels (GAD65, GAD67). GAD1 maps to chromosome 2q31.1 while GAD2 maps to chromosome 10p11.23. Both isoenzymes share structural similari- ties (w65% at the amino acid level) but their responsiveness to the pyridoxal 5 0 -phosphate cofactor varies as does their subcellular distribution (Bu and Tobin, 1994). GAD65 exists predominantly as an apo enzyme that is responsive to pyridoxal 5 0 -phosphate which makes the availability of the cofactor rate limiting in terms of enzyme activity. In contrast, GAD67 is nearly always saturated. GAD65 is localized predominantly in synapses and produces synaptic GABA, while GAD67 is present largely in cell bodies and Abbreviations: ChIP, chromatin immuneprecipitation; DNMT, DNA methyl- transferase; GABA, gamma-aminobutyric acid; GAD25, glutamate decarboxylase25- the truncated protein that arises from a splice variant of GAD67; GAD1, the locus corresponding to GAD67, glutamic acid decarboxylase67; G3PDH, glyceraldehyde 3- phosphate dehydrogenase; HDAC, histone deacetylase; meDIP, methyl DNA immuneprecipitation; MS-275 (SNDX-275), N-(2-aminophenyl)-4-[N-(pyridin-3-yl- methoxycarbonyl) aminomethyl] benzamide; MS-IN, the inactive stereoisomer of MS-275, N-(3-aminophenyl)-4-[N-(pyridin-3-yl-methoxy carbonyl) aminomethyl] benzamide; NT2, N-tera 2 neuronal progenitor cells; SNP, single nucleotide poly- morphism; TK, thymdine kinase; TSS, transcriptional start site; VPA, valproic acid. * Corresponding author. Tel.: þ1 312 413 4577. E-mail address: [email protected] (D.R. Grayson). Contents lists available at ScienceDirect Neuropharmacology journal homepage: www.elsevier.com/locate/neuropharm 0028-3908/$ e see front matter Ó 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.neuropharm.2010.09.017 Neuropharmacology 60 (2011) 1075e1087

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

lable at ScienceDirect
Neuropharmacology 60 (2011) 1075e1087
Contents lists avai
Neuropharmacology
journal homepage: www.elsevier .com/locate/neuropharm
Analysis of the GAD1 promoter: Trans-acting factors and DNA methylationconverge on the 50 untranslated region
Ying Chen, Erbo Dong, Dennis R. Grayson*
The Psychiatric Institute, Department of Psychiatry, College of Medicine, University of Illinois Chicago, 1601 W. Taylor St., Chicago, IL 60612, USA
a r t i c l e i n f o
Article history:Received 20 August 2010Received in revised form10 September 2010Accepted 16 September 2010
Keywords:SchizophreniaMethylationRegulationHistone deacetylase inhibitorsInterneurons
Abbreviations: ChIP, chromatin immuneprecipittransferase; GABA, gamma-aminobutyric acid; GAD25the truncated protein that arises from a splice variancorresponding to GAD67, glutamic acid decarboxylase6phosphate dehydrogenase; HDAC, histone deacetimmuneprecipitation; MS-275 (SNDX-275), N-(2-aminmethoxycarbonyl) aminomethyl] benzamide; MS-IN,MS-275, N-(3-aminophenyl)-4-[N-(pyridin-3-yl-methbenzamide; NT2, N-tera 2 neuronal progenitor cells;morphism; TK, thymdine kinase; TSS, transcriptional* Corresponding author. Tel.: þ1 312 413 4577.
E-mail address: [email protected] (D.R. Gra
0028-3908/$ e see front matter � 2010 Elsevier Ltd.doi:10.1016/j.neuropharm.2010.09.017
a b s t r a c t
GAD67 corresponds to one of two enzymes that decarboxylates glutamate to produce g-aminobutyricacid, the main inhibitory neurotransmitter in the mammalian central nervous system, hence defining thecellular phenotype of a diverse set of inhibitory interneurons of the brain. Reduced cortical GAD67 mRNAlevels have consistently been reported in schizophrenia and bipolar disorder with psychosis. The humangene encoding GAD67, GAD1, is located on chromosome 2q31.1 and the transcriptional start site resideswithin a large CpG island that spans a region extending from upstream through the first exon. We haveanalyzed the GAD1 promoter using transient transfection analysis of upstream and downstreamsequences in NT2 cells, a human neuroprogenitor cell line. Interestingly, results from these studies showthat cis-acting regulatory elements are located downstream of the RNA start site and are in the regioncorresponding to the first exon. Trans-acting factors such as Pitx2 and the Dlx family of transcriptionfactors are active in promoting downstream reporter expression even when all of the 50 flankingsequences are removed. However, those constructs that contain an internal deletion from þ66to þ173 bp fail to support expression even when these factors are provided in trans. We have previouslyshown that the Class I histone deacetylase inhibitor MS-275 potently activates GAD1 mRNA expression inNT2 cells suggesting the possibility that the promoter is sensitive to drugs that induce chromatinremodeling. Using methyl DNA immuneprecipitation of MS-275-treated NT2 cells, we provide datashowing that Class I HDAC inhibition mediated an increase in GAD1 expression and that this wasaccompanied by decreased GAD1 promoter methylation. Moreover, the reduced levels of GAD1 DNAmethylation are highest in those regions proximal to the location of the in vitro defined cis-actingregulatory elements. Our data suggest that changes in promoter methylation associated with generegulation are not random but overlap the locations of proximal cis-acting elements.
This article is part of a Special Issue entitled ‘Trends in Neuropharmacology: InMemory of Erminio Costa’.� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
Nearly two decades ago it was shown that the enzymeresponsible for decarboxylating glutamate to produce g-amino-butyric acid (GABA) was encoded by two related genes on different
ation; DNMT, DNA methyl-, glutamate decarboxylase25-t of GAD67; GAD1, the locus7; G3PDH, glyceraldehyde 3-ylase; meDIP, methyl DNAophenyl)-4-[N-(pyridin-3-yl-the inactive stereoisomer ofoxy carbonyl) aminomethyl]SNP, single nucleotide poly-start site; VPA, valproic acid.
yson).
All rights reserved.
chromosomes in mammals (Bu and Tobin, 1994). The correspond-ing mRNAs (glutamate decarboxylase 1 (GAD1) and GAD2) whentranslated specify the major isoforms of the enzymes that definethe neurotransmitter phenotype of the major class of inhibitoryinterneurons in the brain. These two isoforms differ in molecularsize based on their migration on SDS polyacrylamide gels (GAD65,GAD67). GAD1 maps to chromosome 2q31.1 while GAD2 maps tochromosome 10p11.23. Both isoenzymes share structural similari-ties (w65% at the amino acid level) but their responsiveness to thepyridoxal 50-phosphate cofactor varies as does their subcellulardistribution (Bu and Tobin, 1994). GAD65 exists predominantly asan apo enzyme that is responsive to pyridoxal 50-phosphate whichmakes the availability of the cofactor rate limiting in terms ofenzyme activity. In contrast, GAD67 is nearly always saturated.GAD65 is localized predominantly in synapses and producessynaptic GABA, while GAD67 is present largely in cell bodies and

Y. Chen et al. / Neuropharmacology 60 (2011) 1075e10871076
dendrites and produces GABA tonically. Reports have shown that ofthe two isoforms, GAD67 provides over 90% of basal GABA synthesisand is produced at limiting levels in the brain (Asada et al., 1997;Kash et al., 1997; Chattopadhyaya et al., 2007). A novel splicevariant of GAD67 that results in a truncated protein called GAD25has been described (Chessler and Lernmark, 2000). The shorterisoform lacks the cofactor binding site and is devoid of enzymaticactivity. The transcript specifies a protein that identically matchesthe first 213 amino acids of the human GAD67 protein. While thefunction of this truncated protein remains unclear, its expressionprofile is distinct from the full length GAD67 transcript. That is,GAD25 is produced in pancreatic islets, testis, adrenal cortex, andother endocrine tissues but not in neurons of the brain (Chesslerand Lernmark, 2000). This is particularly interesting as the 50
flanking sequences and 50 untranslated regions of these two tran-scripts are identical.
Numerous single nucleotide polymorphisms (SNPs) in regions ofGAD1 have been associated with schizophrenia (Straub et al., 2007;Marenco et al., 2010) and cleft lip with and without cleft palate(Kanno et al., 2004). Targetedmutations of themouseGad1 gene alsoconfirm the importance of GABA signaling to the development ofa normal palate (Condie et al., 1997). The differential expression ofboth GAD65 and GAD67 mRNAs has been examined in variouspsychiatric diseases including schizophrenia and bipolar disorderwith psychosis (Heckers et al., 2002). GAD1mRNA and protein levelsin schizophrenia have been extensively studied since the initialobservations some years ago (Akbarian et al., 1995; reviewed inGuidotti et al., 2005; Akbarian and Huang, 2006; Gonzalez-Burgoset al., 2010) and are reproducibly reduced in various corticalregions and in the hippocampus (Kim and Webster, 2010). Thereduction in GAD67 and reelin in schizophrenia post-mortem tissuewas reported as being one of themore reproduciblefindingswhen allbrain regions are considered together (Torrey et al., 2005). A recent insitu hybridization study showed that GAD1 mRNA down-regulationin schizophrenia is considerably more widespread with decreasesobserved in orbitofrontal cortex, anterior cingulate cortex, superiortemporal gyrus, striatum, and thalamus (Thompson et al., 2009).
Several studies characterizing the mouse Gad1 promoter haveappeared in the literature (Szabo et al., 1996; Yanagawa et al., 1997;Westmoreland et al., 2001). Like the human gene, the mousepromoter is embedded in a CpG island and contains putativebinding sites for factors that bind GC-rich elements like Sp1 andegr1 (Szabo et al., 1996). This group reported the presence ofmultiple promoters/RNA start sites based on primer extensionanalysis of adult mouse brain polyA enriched RNA (Szabo et al.,1996). A similar conclusion was drawn during an analysis of thehuman gene using both primer extension and S1 nuclease mapping(Bu and Tobin, 1994). More recently, it has been shown that there isa recognition site for the mouse orthologue of unc-30, a C. elegansgene required for the differentiation of so-called “type D neurons”that showa GABAergic phenotype (Westmoreland et al., 2001). Thisgroup showed that both the worm unc-30 and mouse bicoid-likehomeobox factor Pitx2 activate the Gad1 promoter in vitro and bindto sequences in the mouse promoter. Dlx homeobox transcriptionfactors have been linked to a specification of the GABAergicphenotype during development (Anderson et al., 1999). Of partic-ular interest is the finding that ectopic expression of the homeoboxgenes Dlx2 and Dlx5 (and not Dlx1) robustly induces the expressionof both Gad65 and Gad67 in embryonic forebrain slices (Stühmeret al., 2002a). Whether this induction is due to a direct interac-tion of these factors with cis-acting elements within or distal to thecorresponding promoters has not been established.
The human genes corresponding to GAD1 and GAD2 andencoding GAD67 and GAD65 were cloned from genomic librariesand characterized and mapped to different chromosomes some
years ago (Bu and Tobin, 1994). The coding region of GAD67 geneconsists of 17 exons spanning over 45 kb of genomic sequence.These authors also mapped the intron/exon boundaries of bothdecarboxylases and described an initial characterization of thehuman GAD1 promoter (Erlander and Tobin, 1992). S1 nucleaseprotection experiments were used to identify two transcriptionstart sites (TSS) and these investigators performed a sequenceanalysis of the upstream or 50 flanking regions. Examination of thetwo promoters (P1 and P2) suggests that they direct expressionthrough distinct mechanisms. P1 is GC rich, contains numerous GCboxes and lacks consensus TATA and CAAT boxes generally associ-ated with RNA polymerase II mediated transcription. In contrast, P2is less GC rich and contains a TATA box upstream of the corre-sponding start site thus sharing many features associated withtissue specific promoters (Erlander and Tobin, 1992). In general,these authors found that P1 is a stronger promoter in terms ofdirecting expression of a downstream reporter in C6 and Rat1 celllines. In spite of these findings and based on the current NCBIdatabase entry (NM_000817), these start sites map downstream(P2: þ53 bp and P1: þ193 bp, respectively) of the currently posi-tioned start of transcription (see Fig. 1). In the current study, wedesigned a series of constructs to evaluate the regulatory regions ofthe GAD1 promoter in the neural progenitor cell line, NT2. Weexamined the RNA start site in these cells, defined regions relevantto regulation, and also looked at the activation of the promoter inresponse to the HDAC inhibitor MS-275. Our findings provideevidence that the 50 flanking sequence contains the cis-actingelements important for directing expression and that methylationlevels of the promoter inversely correlate with promoter activity.
2. Methods
2.1. Cell culture and total RNA isolation
NT2 cells (Stratagene, La Jolla, CA, USA)weremaintained as previously described(Chen et al., 2002). RNA was isolated following ultracentrifugation through CsCl.
2.2. GAD1 promoters/reporter and deletion constructs
To generate the GAD67 promoter constructs, we designed a series of primers toPCR amplify regions upstream of the GAD1 promoter �1940 bp relative to the TSS.NT2 genomicDNAwasfirst amplified using primers GAD1 gþ417 andGAD1 g�2333.The product of this PCR reaction was used as a template in subsequent nestedamplifications. Primer sequences are listed in Table 1. PCR products were direc-tionally subcloned into the pGL3-Basic luciferase vector (Promega/Fisher). Also, an884-bp region of the GAD67 promoter (from�627 toþ64 bp relative to the TSS) wasamplified with the forward primer GAD1 SpeI-627 and reverse primer GAD1HindIIIþ257. This ampliconwas subcloned into the pCpGL luciferase reporter vector(Klug and Rehli, 2006). Deletion constructs were generated by deleting a middleportion of the amplicon using overlap extension PCR. Primers GAD1 (D1) and GAD1XhoI�627 or GAD1 XhoI�319 were used to produce one 50 fragment, while primersGAD1 (D2) and GADI HindIII þ257 were used to produce a second 30 fragment. Thenoverlap extension was performed with the two fragments and the outside primers(GAD1 XhoI �627 or GAD1 XhoI �319 and GADI HindIII þ257). Following PCR,amplicons were subcloned into the pGL3-basic construct.
2.3. Transfection/co-transfection assays and reporter measurements
The dual-luciferase reporter assay system (Promega/Fisher) was used tomeasure reporter activity (see Chen et al., 2002, 2007). NT2 cells were transfectedusing Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen).We routinely used 3e4 mg of each test DNA with 5 ng pRL-TK vector (Renilla lucif-erase) for each well of a 6-well plate. Media were changed 6 h after transfection. Celllysates were prepared 48 h after transfection and aliquots (5e20 ml) were used fordetermination of luciferase activity in a TD20/20 luminometer (Turner Design). Data(minimum of three transfections/construct) are expressed as the ratio of the signalobtained from the SV40 promoter/luciferase vector transfected in parallel (pGL3-Control vector; Promega/Fisher).
To test the effects of the Dlx, Pitx and Sp1 transcription factors on GAD1promoter activity, NT2 cells were co-transfected with 200 ng promoter construct ordeletion construct and 3 mg the specific expression vector. Cells were lysed after 48 hand reporter activity was measured. To test for effects of Dlx, Pitx and Sp1 on

Dot matrix
Mouse Gad1 Promoter
Hu
man
G
AD
1 P
ro
mo
ter
A Comparison of mouse (top) and human (bottom) regulatory regions.
Mouse -206 TCCTTTCTGGTCGCAAACCCGTGAGCTGGATTTATAATCGCCCTACAAAGCTCCAGAGGC ||| ||| || |||||| ||||||||||||||||||||||||||| ||||||||||||||
Human -11 TCC-TTCCGGGCGCAAAACCGTGAGCTGGATTTATAATCGCCCTATAAAGCTCCAGAGGC
Mouse -146 AGTCAGACACCTGCAAAGGAGCCCCAG-CGCTCCGCGGACGAGCTGCCCCCGCGAGCAAC ||||| |||||||| ||||||||| | |||||||| ||| |||||||||||||||||||
Human +49 GGTCAGGCACCTGCAGAGGAGCCCC-GCCGCTCCGCCGACTAGCTGCCCCCGCGAGCAAC
Mouse -87 GGCCTCGTGATTCCCCCCGCCGAGCGGGTCCCCGCCTCCCCACTCCGCCCCCGCCTCCCC|||||||||||| |||| ||||| | ||||||||||||||||||| |||||||||| |||
Human +108 GGCCTCGTGATTTCCCC-GCCGATCCGGTCCCCGCCTCCCCACTCTGCCCCCGCCTACCC
Mouse -27 CA-AGCCCAGCGGCCGCCTCTCCGGATCTCTCCCTTCTTCAGGCTCTCCCGTGCCGGACC| |||| || |||||||||||| ||||||| ||||| | ||| ||| ||||| | |
Human +167 CGGAGCCGTGCAGCCGCCTCTCCGAATCTCTCTCTTCTCCTGGCGCTCGCGTGC-G-AG-
Mouse +33 AGGGATCGT-GCAAGCA--AGGAAGCAGCCCTGG-GGTGACACCCAGCACG-T-ACTCCT||||| | | || || | |||||||||| ||| |||||| || ||| | | || |||
Human +224 AGGGAAC-TAGCGAGAACGAGGAAGCAGC--TGGAGGTGACGCCGGGCA-GATTACGCCT
Mouse +64 GTGACAGAGCCGAG.... (169 bp total in first exon)|| ||| ||||||
Human +280 GT--CAGGGCCGAG. .. (359 bp total in first exon)
B
Fig. 1. Comparison of the mouse and human GAD1 sequences proximal to the transcriptional start sites. A) Shows a Blast-n alignment of the mouse Gad1 and human GAD1sequences for comparing ‘somewhat similar’ sequences allowing for gaps. For the mouse sequence, we used NCBI entry AF354680, submitted based on the previous analysis of themouse promoter (Westmoreland et al., 2001). The human sequences were from our own study and extend from �783 through þ293 bp of cloned genomic DNA. The transcribedregions of each region are underlined based on their respective NCBI entries (Gad1: NM_00807 and GAD1: NM_000817). In addition, the 10 base human dec-1 sequence is boxed.This region is a 9/10 match with element 1 of the Drosophila DDC gene which was previously shown to be required for neuronal expression of the Drosophila dopa decarboxylasegene (Bray et al., 1988). This match falls to 8 out of 10 when this element is compared with the mouse sequence. B) The corresponding dot matrix representation of the alignmentwas linear with 83% identity over the 263 mouse/316 human overlap. There were 20/316 gaps within the region of identity. Places where gaps are prevalent are indicated by thevertical arrows. The dot matrix plot shows a linear representation of the paired alignment shown in A, with discontinuities reflected by breaks in the line. The highest degree ofsequence identity with these two stretches corresponds to near the 30 end of each input sequence. With respect to the human sequence, this represents nearly all of the 50
untranslated sequence. In contrast, the mouse region showing the greatest amount of sequence identity extends from w200 bp upstream of the start site through the first 70 bp of50 untranslated portion of exon 1. Because the previously identified major start site (P1) of human GAD1 mapped to within 1 bp of the mouse Gad1 sequence, we confirmed thecurrently positioned human start site (NCBI entry: NM_000817) using primers that extend up- and downstream of this region (see Fig. 2).
Y. Chen et al. / Neuropharmacology 60 (2011) 1075e1087 1077
endogenous GAD mRNA expression, 4 mg expression vector was used to transfectNT2 cells. RNA was isolated after 72 h.
2.4. Methylated DNA immunoprecipitation assay
Methylated DNA immuneprecipitation (MeDIP) was used to analyze the meth-ylation status of regions flanking GAD67 promoter and first exon. NT2 cells weretreated with either 5 mMMS-IN or 5 mMMS-275 for 48 h. Genomic DNAwas isolatedand sheared by sonication (Bioruptor UCD-200; Diagenode, Liege, Belgium) yielding200- to 800-bp fragments. DNA was diluted in TriseEDTA buffer (10 mM Tris and1mM EDTA, pH 8.0) and denatured for 10min in boiling water. An aliquot was savedas input (control) DNA. The denatured DNA was immunoprecipitated overnightusing a monoclonal antibody against 5-methylcytosine (Calbiochem, San Diego, CA)in MeDIP buffer (10mM sodium phosphate at pH 7.0, 140mMNaCl, and 0.05% TritonX-100). Protein A/G-Sepharose beads (Biovision, Mountain View, CA), prewashedwith phosphateebuffered saline/0.1% bovine serum albumin, were used to bindimmunoprecipitated complexes. After a 2-h incubation with the reaction mixture,beads were washed three times with MeDIP buffer, resuspended in 500 ml ofdigestion buffer (50 mM Tris, pH 8.0, 10 mM EDTA, and 0.5% SDS) containing
proteinase K (0.28 mg/ml), and incubated overnight with shaking at 55 �C. DNAwasrecovered by centrifugation after phenol/chloroform extraction and ethanolprecipitation. The resulting pellet was resuspended in 30 ml of nuclease-free water.
2.5. Real-time PCR analysis of MeDIP samples and endogenous GAD mRNA
To analyze the above samples for the amounts of DNA precipitated, real-timePCR reactions were carried out with 40 ng of input DNA and 1/15 of the immune-precipitated methylated DNA samples. The GAD1 and G3PDH primers are listed intable (MeDIP). Real-time PCR reactions were performed using am Mx3000P real-time PCR system and data were analyzed with MxPro software (Stratagene, La Jolla,CA). PCR amplifications (in duplicates) were carried out in a 20-ml reaction volume,using Maxima SYBR Green qPCR Master Mix (Fermentas). For all three promoterregions, PCR cycling conditions were as follows: initial denaturation at 95 �C for10 min, 40 cycles of denaturation (95 �C, 30 s), annealing (62 �C, 1 min), andextension (72 �C, 1 min), followed by dissociation at 95 �C for 1 min and 55 �C for30 s. The dissociation curves of the PCR products confirmed that only the genes ofinterest were amplified. G3PDH was used as an endogenous reference for this assay.

Table 1Complete list of primers used in this study.
PromoterConstruction
Oligonucleotide primer (50 / 30)
GAD1 HindIII þ257 AAGCTT-CACCTCCAGCTGCTTCCTCGTTCTGAD1 XhoI �1940 CTCGAG-GAAGCTGGACGGACTCAGCTTCAGGAD1 XhoI �1526 CTCGAG-CACTTCAAGCAGCGCAGAGCAGCTGAD1 XhoI �1281 CTCGAG-CCACAGCGAGATCCCTCTTGCTGAGAD1 XhoI �741 CTCGAG-GCTTCTCCCAACGCAGCGGTTCTTGAD1 XhoI �627 CTCGAG-CCTTCCGGGCACCTCTGTGCTCGGGAD1 XhoI �503 CTCGAG-GAGCGAGCGCATAGCAAAAGGGACGAD1 XhoI �319 CTCGAG-AGAGAGCCAACGAGGACACGACCCGAD1 XhoI �153 CTCGAG-GAAGAAGAAACGCTCAGAAAGGCAGAD1 XhoI þ1 CTCGAG-GCAAAACCGTGAGCTGGATTTATAGAD1 XhoI þ101 CTCGAG-GAGCAACGGCCTCGTGATTTCCCGAD1 XhoI þ193 CTCGAG-AATCTCTCTCTTCTCCTGGCGCTCGAD1 SpeI �627 ACTAGT-CCTTCCGGGCACCTCTGTGCTCGGGAD1 g þ417 CCTGGCTGAAAAGCCACGGTTGGGGAD1 g �2333 AGAAGGACGGGCGCTGTCCTTCGT
Deletion ConstructsGAD1(Alt-555) 50 CTCGAG-ACGTGATTAATCAGGAGGCCTTCTGAD1(Alt-469) 50 CTCGAG-TTTTCTCTGCCGGTGGCACTGGGAD1(Altþ25) 30 AAGCTT-TAAATCCAGCTCACGGTTTTGCGCGAD1 (internal D50) TCAGGCACCTGCAGA(þ65)d(þ172)
CGTGCAGCCGCCTCTGAD1 (internal D30) AGAGGCGGCTGCACG(þ172)d(þ65)
TCTGCAGGTGCCTGA
MeDIPGAD1 MeDIP-442 CTGTGGCCAGGTGTGGTACTTTGAD1 MeDIP-313 CTCTCTACGGAGCTGGGATTGTTGTAGAD1 MeDIP-214 GCGGTTTTGCCTCAGTGCTTTTGAD1 MeDIP-237 GAAAAGCACTGAGGCAAAACCGCCGAD1 MeDIP-68 CTTTGGAAATAGGCAGGCTGTCGGGAD1 MeDIPþ21 AAATCCAGCTCACGGTTTTGCGCGAD1 MeDIP-142 ACGCTCAGAAAGGCAGAATTCTTCGGAD1 MeDIP-43 GCGTGTTGAGTACGTTCTGGATTACTCGAD1 MeDIPþ121 GGAAATCACGAGGCCGTTGCTGAD1 MeDIPþ101 AGCAACGGCCTCGTGATTTCCGAD1 MeDIPþ255 CTCCAGCTGCTTCCTCGTTCT
G3PDH promoter50 �122 bp CGGCTACTAGCGGTTTTACG30 þ42 bp AGGAGGAGCAGAGAGCGAAGReal-time-PCRGAD67 cDNA: Forward 50-AGGCAATCCTCCAAGAACCT-30
Reverse 50-GGTGGAGCGATCAAATGTCT-30
G3PDH cDNA: Forward 50-CGAGATCCCTCCAAAATCAA-30
Reverse 50-TTCACACCCATGACGAACAT-30
All primers with an indicated restriction site were used for cloning into a reporter(generally pGL3 (Promega) or pCpGL (pCpGL is a luciferase reporter that is free ofCpG dinucleotides, Klug and Rehli, 2006). The restriction site is shown in bold andeach site was preceded by 3 random bps to facilitate enzyme digestion. Numberingrefers to the first base following the hyphen. For the internal deletions, numberingrefers to the position of the base (total deletion was from þ66 bp to þ171 bp).
Y. Chen et al. / Neuropharmacology 60 (2011) 1075e10871078
RT-PCR was used to measure the endogenous GAD mRNA levels. Reverse tran-scription of total RNA with random hexamers and M-MLV RT was performed aspreviously described (Chen et al., 2002). Following RT, 100 ng cDNA was used forreal-time analysis. The values obtained were then normalized against the signalobtained using the G3PDH primers. The real-time PCR analysis was performed threetimes per sample from a minimum of three different RNA isolations. The data wereanalyzed for significance using a one-way Quantitative ANOVA. For confirming the50 ends of the GAD1 primary transcript, we performed RT as above with 200 ng RNAand random hexamers. The specific primers used and the various controls areindicated in the legend to Fig. 2C.
2.6. MS-275 treatment and Southern blotting
Cytosines in CpG dinucleotides were methylated in vitro by incubating 10 mgpCpG-GAD1-627 promoter/reporter plasmid with 25 units of SssI DNA methylase(see Kundakovic et al., 2009). The extent of methylation was examined by digestingaliquots of the reaction with corresponding restriction enzymes (HpaII vs. MspI). Toexamine the capability of MS-275 to alter methylation patterns, in vitro methylatedconstructs were transiently transfected and after 24 h cells were incubated with
5 mM MS-275 or 5 mM MS-IN. For the dose-response experiments, NT-2 cells wereincubated with vehicle (DMSO) or the indicated concentrations of either MS-275 orMS-IN for 48 h.
For the Southern blot, samples were subjected to electrophoresis on 2.0%agarose gels and then transferred to Hybond-Nþ membrane (GE Healthcare). Blotswere probed with a 32P-labeled GAD1 50 flanking probe (�627 to þ257 bp)synthesized using a random primer labeling kit. Membranes were hybridized andwashed as recommended by the manufacturer. The demethylation assay measuresthe fraction of the GAD1 promoter that was demethylated using the HpaII restrictionenzyme, which cleaves unmethylated 50-CCGG-30 but does not cleave methylated50-CCGG-30 sequences. For autoradiography, blots were exposed to Storage PhosphorScreens (Molecular Dynamics) for from 2 to 4 days. Signals were visualized usinga STORM 840 Optical Scanner (Amersham) and quantified using imagequant soft-ware (Molecular Dynamics).
2.7. Statistical analyses
All experimental results are expressed as mean � SEM of three independentexperiments (a minimum of three separate measurements were obtained perexperiment). For most of the transfection experiments, data are expressed relative(%) to the data obtained from an SV40 containing construct transfected in parallel(pGL3 promoter vector; Promega/Fisher) after normalizing for transfection effi-ciency (Figs. 3A, B and 5A). Bonferroni t-test was used to assess the significance ofthe differences between groups (Figs. 3C and 4AeC). Analyses were conducted usingSigmaStat software (SysStat, Richmond, CA, USA). T-test was used to compare twogroups (Figs. 4D and 6).
3. Results
3.1. Analysis of the genomic sequences surrounding the GAD1RNA start site
We compared sequences associated with the published mousepromoter with the human promoter 50 flanking region and firstexon (Fig. 1). To compare the extent of sequence identity wegenerated a Blast (2seq) alignment of 625 bp of the mousepromoter (NCBI accession AF354680; Westmoreland et al., 2001)along with from �783 to þ193 bp of the human sequences. Theregion of strongest overlap coincided with the alignment shown inFig. 1A. In the region of overlap between the mouse (total test:625 bp) and human (total test: 1076 bp) species, there is w83%sequence identity (263/316 or 83%). The transcribed portions ofeach sequence are underlined. The boxed/bolded sequence in themouse promoter (�179 to�168 bp) corresponds to the 30 portion ofthe previously identified Pitx2/UNC-30 binding site (Westmorelandet al., 2001). The 50 portion of this split site is located upstream(�238 to �233 bp) and is a perfect match between species. Theboxed human sequence (þ129 to þ138 bp) corresponds to theDrosophila decamer element that was originally identified andshown to be relevant to transcription of the human promoter. Thisregion, which is partially conserved in the mouse (8 out of 10), wasshown to bind a protein present in rat brain, rat liver, and Rat1 cellnuclear extracts (Erlander and Tobin, 1992). Of particular interestwith respect to the disputed TSS sites is that the original P1promoter is positioned 1 bp before the TSS of the mouse primarytranscript. That is, position þ193 bp of the human DNA, whichrepresents P1, is 1 bp 50 to the start site mapped in mice. The dotmatrix curve displays the sequence alignment as a linear repre-sentation (Fig. 1B). The degree of sequence identity in this region isremarkably high andwhile there aremultiple gaps in the alignment(see vertical arrows), these gaps are small and generally from 1 to2 bp in length.
3.2. Analysis of the GAD1 CpG island and transcriptional start site
We have previously carried out experiments examining dele-tions of the RELN promoter using transient transfection of NT2 cells(Chen et al., 2002, 2007). At the time, we became aware that theRELN promoter was embedded in a large CpG island that flanks the
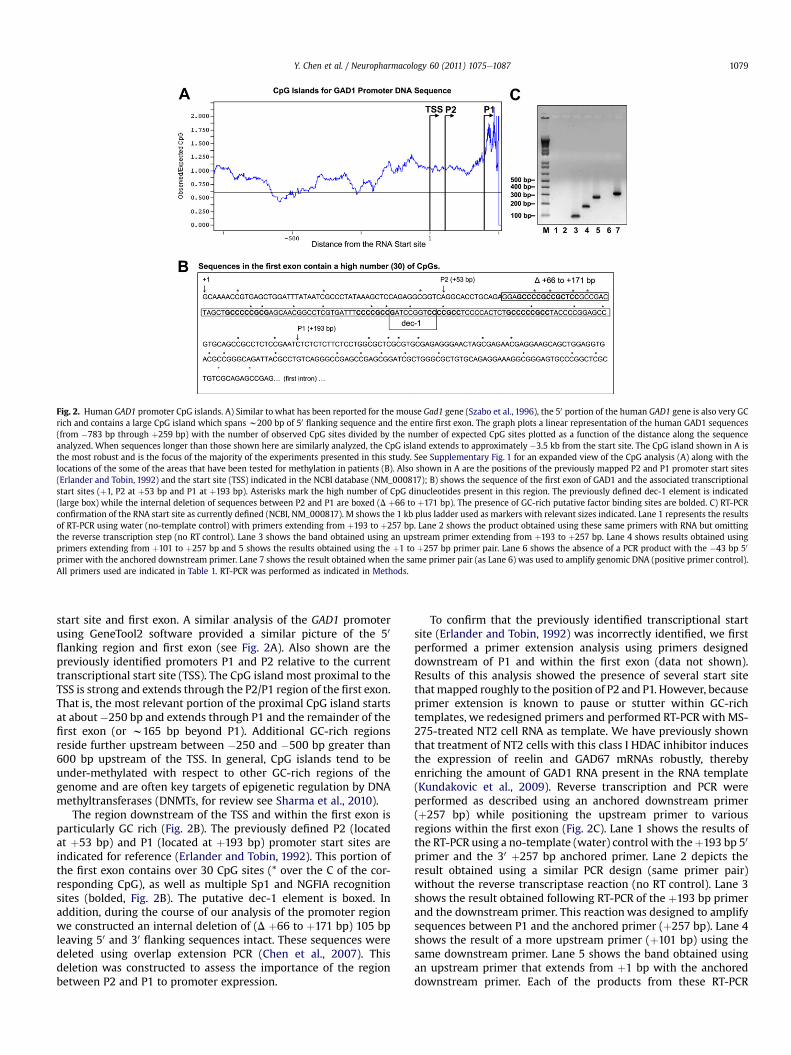
Fig. 2. Human GAD1 promoter CpG islands. A) Similar to what has been reported for the mouse Gad1 gene (Szabo et al., 1996), the 50 portion of the human GAD1 gene is also very GCrich and contains a large CpG island which spans w200 bp of 50 flanking sequence and the entire first exon. The graph plots a linear representation of the human GAD1 sequences(from �783 bp through þ259 bp) with the number of observed CpG sites divided by the number of expected CpG sites plotted as a function of the distance along the sequenceanalyzed. When sequences longer than those shown here are similarly analyzed, the CpG island extends to approximately �3.5 kb from the start site. The CpG island shown in A isthe most robust and is the focus of the majority of the experiments presented in this study. See Supplementary Fig. 1 for an expanded view of the CpG analysis (A) along with thelocations of the some of the areas that have been tested for methylation in patients (B). Also shown in A are the positions of the previously mapped P2 and P1 promoter start sites(Erlander and Tobin, 1992) and the start site (TSS) indicated in the NCBI database (NM_000817); B) shows the sequence of the first exon of GAD1 and the associated transcriptionalstart sites (þ1, P2 at þ53 bp and P1 at þ193 bp). Asterisks mark the high number of CpG dinucleotides present in this region. The previously defined dec-1 element is indicated(large box) while the internal deletion of sequences between P2 and P1 are boxed (D þ66 to þ171 bp). The presence of GC-rich putative factor binding sites are bolded. C) RT-PCRconfirmation of the RNA start site as currently defined (NCBI, NM_000817). M shows the 1 kb plus ladder used as markers with relevant sizes indicated. Lane 1 represents the resultsof RT-PCR using water (no-template control) with primers extending from þ193 to þ257 bp. Lane 2 shows the product obtained using these same primers with RNA but omittingthe reverse transcription step (no RT control). Lane 3 shows the band obtained using an upstream primer extending from þ193 to þ257 bp. Lane 4 shows results obtained usingprimers extending from þ101 to þ257 bp and 5 shows the results obtained using the þ1 to þ257 bp primer pair. Lane 6 shows the absence of a PCR product with the �43 bp 50
primer with the anchored downstream primer. Lane 7 shows the result obtained when the same primer pair (as Lane 6) was used to amplify genomic DNA (positive primer control).All primers used are indicated in Table 1. RT-PCR was performed as indicated in Methods.
Y. Chen et al. / Neuropharmacology 60 (2011) 1075e1087 1079
start site and first exon. A similar analysis of the GAD1 promoterusing GeneTool2 software provided a similar picture of the 50
flanking region and first exon (see Fig. 2A). Also shown are thepreviously identified promoters P1 and P2 relative to the currenttranscriptional start site (TSS). The CpG island most proximal to theTSS is strong and extends through the P2/P1 region of the first exon.That is, the most relevant portion of the proximal CpG island startsat about �250 bp and extends through P1 and the remainder of thefirst exon (or w165 bp beyond P1). Additional GC-rich regionsreside further upstream between �250 and �500 bp greater than600 bp upstream of the TSS. In general, CpG islands tend to beunder-methylated with respect to other GC-rich regions of thegenome and are often key targets of epigenetic regulation by DNAmethyltransferases (DNMTs, for review see Sharma et al., 2010).
The region downstream of the TSS and within the first exon isparticularly GC rich (Fig. 2B). The previously defined P2 (locatedat þ53 bp) and P1 (located at þ193 bp) promoter start sites areindicated for reference (Erlander and Tobin, 1992). This portion ofthe first exon contains over 30 CpG sites (* over the C of the cor-responding CpG), as well as multiple Sp1 and NGFIA recognitionsites (bolded, Fig. 2B). The putative dec-1 element is boxed. Inaddition, during the course of our analysis of the promoter regionwe constructed an internal deletion of (D þ66 to þ171 bp) 105 bpleaving 50 and 30 flanking sequences intact. These sequences weredeleted using overlap extension PCR (Chen et al., 2007). Thisdeletion was constructed to assess the importance of the regionbetween P2 and P1 to promoter expression.
To confirm that the previously identified transcriptional startsite (Erlander and Tobin, 1992) was incorrectly identified, we firstperformed a primer extension analysis using primers designeddownstream of P1 and within the first exon (data not shown).Results of this analysis showed the presence of several start sitethatmapped roughly to the position of P2 and P1. However, becauseprimer extension is known to pause or stutter within GC-richtemplates, we redesigned primers and performed RT-PCR with MS-275-treated NT2 cell RNA as template. We have previously shownthat treatment of NT2 cells with this class I HDAC inhibitor inducesthe expression of reelin and GAD67 mRNAs robustly, therebyenriching the amount of GAD1 RNA present in the RNA template(Kundakovic et al., 2009). Reverse transcription and PCR wereperformed as described using an anchored downstream primer(þ257 bp) while positioning the upstream primer to variousregions within the first exon (Fig. 2C). Lane 1 shows the results ofthe RT-PCR using a no-template (water) control with theþ193 bp 50
primer and the 30 þ257 bp anchored primer. Lane 2 depicts theresult obtained using a similar PCR design (same primer pair)without the reverse transcriptase reaction (no RT control). Lane 3shows the result obtained following RT-PCR of the þ193 bp primerand the downstream primer. This reaction was designed to amplifysequences between P1 and the anchored primer (þ257 bp). Lane 4shows the result of a more upstream primer (þ101 bp) using thesame downstream primer. Lane 5 shows the band obtained usingan upstream primer that extends from þ1 bp with the anchoreddownstream primer. Each of the products from these RT-PCR

retomorP 1DAG namuH
002 pb
SST )718000_MN(
*2P
1 noxE
*1P
pb 002-pb 006-pb 000,1-
SST ot evitaleR
Δ pb 913-
pb 52+ ot 964-
pb 52+ ot 555-
C
pb 726-
pb 305-
pb 913-
pb 351-
pb 1+
pb 101+
pb 391+
B
pb 147-
pb 0491-
pb 6251-
pb 1821-
***A
147- 1821- 6251- 0491- nleR
0
02
04
06
08
001
021
041
061
Pe
rc
en
t o
f S
V4
0 p
ro
mo
te
r
’A
0
02
04
06
08
001
021
041
061
391+ 101+ 1+ 351- 913- 305- 726- P3LGp
’B
Pe
rc
en
t o
f S
V4
0 p
ro
mo
te
r
’C
913- Δ 726- 913- Δ 52+/555- 52+/964- 726-
0
02
04
06
08
001
021
041
061
* * * *
Pe
rc
en
t o
f S
V4
0 p
ro
mo
te
r
Δ pb 726-
pb 271+ ot 56+
Fig. 3. Promoter/reporter constructs and transient transfection analysis of the human GAD1 regulatory sequences. A linear representation of the human GAD1 promoter is indicatedat the top showing the 50 flanking sequence, transcriptional start site (TSS, NM_000817), and the first exon (shaded gray box below the line). Also noted just below the line are threeregions that were examined for the presence of DNA methylation due, in part, to the CpG island shown in Fig. 2. These regions were analyzed by different investigators usingdifferent methodologies. The box centered at about �800 bp was examined by Mill et al. (2008). The open box located between �600 and �400 bp was analyzed by Kundakovicet al. (2009) while the third positioned closest to �200 bp was the focus of Huang and Akbarian (2007). The P2 and P1 start sites previously identified by Erlander and Tobin (1992)are indicated below the first exon. Promoter/reporter constructs shown in groups A and B represent deletions with the indicated 50 end and an anchored or common 30 end locatedat þ257 bp. Group C GAD1/reporter constructs correspond to either two internal deletions (sequences deleted are from 65 through þ171 bp) with different amounts of 50 flankingsequence (�627 and �319 bp). The remaining two group C constructs contain an upstream 30 anchor (þ25 bp) and either �469 or �555 bp of 50 flanking sequence. These constructswere transiently transfected into NT2 cells along with a constant amount of Renilla luciferase (for normalizing transfection efficiency) and reporter activity was analyzed in celllysates preparedw48 h post-transfection (Chen et al., 2007). A) Data from the longest constructs are shown and expressed as a percent of the SV40 promoter (Chen et al., 2002). Theactivity of the RELN promoter (�514 bp) is shown for comparative purposes (Chen et al., 2002). B) Data obtained from the indicated constructs are shown as in A’. The vector pGL3-Prepresents pGL3 luciferase activity driven by the SV40 promoter used to normalize these data. C) Results of the internal deletions and alternative 30 constructs are compared withthe corresponding parent constructs.
Y. Chen et al. / Neuropharmacology 60 (2011) 1075e10871080
reactions (Lanes 3e5) showed a single band of the size predictedbased on the distances between primer pairs. Lane 6 shows thatwhen a primer positioned at �43 bp relative to the current TSS isused with the downstream primer, no RT-PCR product is obtained.To confirm that this latter result was not an artifact of the primerpair, we used the same primers to amplify 30 ng of genomic DNA inparallel (Lane 7). These primers are indicated in Table 1 under theheading ‘Promoter Construction’. Each of these corresponds to thethose listed in the Table but were made without the additionalrestriction site. While these data do not precisely position the startsite, they support the concept that the previous datamay have beenan artifact associated with the methods used in the mappingprocedures (Erlander and Tobin, 1992).
3.3. In vitro dissection of the GAD1 promoter
Similar to our previous work with the reelin promoter (Chenet al., 2002, 2007), we generated a series of promoter constructscorresponding to different regions of the GAD1 50 flanking and firstexon and cloned these upstream of a luciferase reporter construct(Fig. 3A, B, C). The GAD1 flanking sequences and first exon arerepresented to scale at the top of Fig. 3. Open and hatched barsunder this line represent regions of the promoter that have beenpreviously examined for the presence of DNA methylation (Huanget al., 2007; Mill et al., 2008; Kundakovic et al., 2009) and alsoexon 1. While these researchers also looked at regions up- and
downstream of the region shown in Fig. 3, we have not includedthese other areas as they are outside of the map coordinates dis-played. For additional detail regarding regions previously examinedfor methylation, please see Supplementary Fig. 1b. The currentlydefined TSS (NM_000817) is indicated. The longest upstreamregions examined (�1940 bp,�1526 bp, and�1281 bp) fall into thegroup A vectors (Fig. 3A) that contain varying amounts of 50
flanking sequence anchored to a common 30 (þ257 bp) down-stream primer. These were cloned from NT2 genomic DNA usinga nested PCR with the genomic primers listed in Table 1 (50 GAD1g �2333 and 30 GAD1 g þ417 bp). The group B constructs (Fig. 3B)also contain different amounts of upstream sequences anchored tothe same exon 1 30 primer (þ257 bp). These include the �627 bp,�503 bp, �319 bp, �153 bp, þ1 bp, þ101 bp, and þ193 bp GAD1/luciferase promoter reporter constructs. These constructs weregenerated using PCR and a parent template and were designed toprovide a series of evenly distributed promoter fragments that notonly span the upstream region but extend into and up to the P1promoter start site (þ193 bp). The group C promoter/reportervectors contain an internal deletion (fromþ65 throughþ171 bp) oran alternative 30 anchored primer (Fig. 3C). We used overlapextension PCR with the primers indicated in Table 1 (see deletionconstruct primers) and either the�627 bp or the�319 bp upstreamandþ257 bp downstream as outer primer combinations. The groupC alternative constructs were generated using nested PCR withthe �627 template and either the �555 bp �469 bp upstream

Dlx1 Dlx2 Dlx5
noi
ss
er
px
E
dl
oF
0
2
4
6
8
10
12
******
*** ***
*** ***
Pitx2a Pitx2c cΔ39
noi
ss
er
px
E
dl
oF
0
4
8
12
16
20
***
**
GAD1-627
GAD1-319 Deletion
GAD1-627 Deletion
GAD1-319
GAD1-486/+25
GAD1-319
GAD1-319Deletion
GAD1-486/+25
0
0.5
1.0
1.5
2.0
2.5
3.0
-1940 -1526 -627 -319 +1 +101
noi
ss
er
px
E
dl
oF
**
Sp1 co-transfectionA
B
C
HP
D3
G/
AN
Rm
7
6D
AG
%
0
50
100
150
200
250
300
350
Con Egr1 Sp1 Pitx2a Pitx2cDlx2 Dlx2+5
** *
*
*** ***D
Fig. 4. Co-transfection assays to assess the trans-activating capabilities of selected transcription factors on regions of the GAD1 promoter. Based on previously published reportsregarding factors regulating the mouse Gad1 promoter (Szabo et al., 1996; Yanagawa et al., 1997; Westmoreland et al., 2001; Stühmer et al., 2002a), we performed a series of co-transfection experiments to assess the trans-activating abilities of these factors in directing expression of the human GAD1 promoter. A) Results obtained following co-transfection ofthemouse Dlx1, 2 and 5 expression vectors and 5 distinctGAD1 promoter/reporter constructs. These included the parent deletion constructs GAD1-319 and the corresponding internaldeletion (GAD1D-319); GAD1-627 and GAD1D-627 and then the 30 truncated version GAD1-486 toþ25 bp. In each case, the amount of GAD1/reporter was limiting and the amount ofexpression cassette was in 5-fold molar excess. After 72 h, cells were lysed and reporter activity was measured. In each case, the corresponding empty vector was co-transfected inparallel for comparison. Results (fold induction) are expressed relative to the parallel readings from the cells receiving the GAD1 construct plus empty expression vector. * indicatesstatistically significant difference (p< 0.001). B) Pitx2, a bicoid-like homeodomain transcription factor defective in Rieger syndrome, was previously shown to transactivate themouseGad1 promoter (Westmoreland et al., 2001). Here we compared the effects of two splice variants of Pitx2, Pitx2a, and Pitx2c on the activation of GAD1 in vitro. Pitx2 cD39 representsa C-terminal truncated version of Pitx2c in which the DNA binding activity is increased (relative to wild type) but the corresponding protein shows reduced trans-acting activity. Co-transfections were carried out as described in A. Results are normalized to the activity obtained by co-transfecting the GAD1 construct with the same expression vector lacking aninsert. C) In this case we used a variety of longer constructs to analyze the effects of Sp1 on promoter activity. As in A) and B), co-transfections were performed and analyzed bycomparing the GAD1 construct plus Sp1 vs. GAD1 with the empty vector. Co-transfections were carried out a minimum of 3 independent times. D) Effects of exogenous transcriptionfactors on the expression of the GAD67 endogenous mRNA. Expression vectors corresponding to Egr1, Sp1, Pitx2a, Pitx2c, Dlx2 and Dlx2 þ 5 were transfected into NT2 cells and RNAwas harvested as described in Methods. The levels of GAD67mRNA are expressed as a ratio to the signal obtained with G3PDH primers. T-test was used to compare numbers betweeneach group and control (Con). Data are expressed as mean � S.E.M.*, p < 0.05; **, p < 0.01; ***, p < 0.001 vs. control group (one-way ANOVA followed by Bonferroni test).
Y. Chen et al. / Neuropharmacology 60 (2011) 1075e1087 1081
primer with the þ25 bp downstream primer. This latter pair ofconstructs had all sequences downstream of þ25 bp deleted withupstream sequences relatively intact.
The above constructs were introduced into neural progenitorcells (NT2 cells) and analyzed for activity (Fig. 3A, B and C). Whilesubtle variations in the strength of each A group promoterconstruct were evident, these variations did not suggest the pres-ence of upstream elements within these regions. The same is alsotrue of the group B promoter constructs except once sequencesthrough the start site and past the P2 promoter are removed,significant loss of activity occurs. That is, not until all 50 flankingsequences and 192 bp of the first exon are removed is therea significant decrease in promoter activity. This is not due to theremoval of the GAD1 Kozak sequence because all GAD1 sequencespresent in these constructs are located upstream of the trans-lational start site that is present in exon 2. The translational startsite is at position þ423 relative to the mRNA sequence and exon 1contains a total of 359 bp. Moreover, there is a consensus Kozaksequence located in the pGL3 reporter vector. These data suggestthat all of the sequences responsible for regulating GAD1 expres-sion are located downstream of the RNA start site and within thefirst exon. To confirm these data, wemade the C group of constructs(Fig. 3C). We were primarily interested in the GC-rich region
between P2 and P1 that had previously been shown to be relevantto GAD1 promoter/reporter expression using transient transfectionanalysis of cell lines (Erlander and Tobin, 1992). A comparison ofeither �627 bp or �319 bp promoter constructs with the corre-sponding internal deletion (þ65 to þ171 bp) showed that removalof these 107 bp reduced activity to basal levels of expression(Fig. 3C). The same is true of the alternative promoter constructs inwhich the 50 flanking sequences and TSS were left intact butsequences downstream of þ25 bp were removed. This 107 bpregion of the 50 untranslated region contains two Sp1-likesequences and the putative Drosophila dec-1 element which sitsbetween these two sites.
3.4. Co-transfection of the GAD1 promoter/reporter constructswith candidate transcription factors
Based on previous analyses of the mouse Gad1 (Szabo et al.,1996; Yanagawa et al., 1997; Westmoreland et al., 2001) andhuman GAD1 promoters (Erlander and Tobin, 1992), we examinedthe action of several trans-acting factors on the activity of ourpromoter constructs and the behavior of the endogenous gene inNT2 cells. To do this, we co-transfected vectors that direct theexpression of specific factors along with our promoter and internal

Y. Chen et al. / Neuropharmacology 60 (2011) 1075e10871082
deletion constructs. The goal of these experiments was to testwhether these factors positively impacted expression of eitherdownstream reporter activity or the endogenous GAD1 mRNA inNT2 cells. We first focused on the Dlx (1, 2, and 5) family of tran-scription factors because of their importance to tangential migra-tion of cortical interneurons and because of experiments showingthat ectopic expression of Dlx homeobox genes induces theexpression of GAD65 and GAD67 (Stühmer et al., 2002a,b). We alsotested the ability of the bicoid transcription factor family Pitx2 toactivate the human GAD1 constructs based on previous workshowing that the mouse promoter was activated in trans by Pitx2,the murine homologue of UNC-30 (Westmoreland et al., 2001).
Fig. 4A shows results obtained when expression vectors corre-sponding to mouse Dlx 1, 2 and 5 were co-transfected along withthe indicated GAD1 constructs and reporter activity was measured72 h later. In each case, the corresponding reporter activity wascompared with the activity of the same vector co-transfected withan empty expression vector cassette. While the magnitude of thefold increase in expression varied between Dlx family members(i.e., Dlx1 vs. Dlx2 vs. Dlx5), results obtained with both the�627 bpand �319 parent constructs were comparable in each case(compare Dlx1 and GAD1-319 with Dlx1 and GAD1-627, etc.). Incontrast, the fold increase with contracts containing either theinternal deletion (�319 deletion or �627 deletion) or the down-stream deletion (�486/þ25) showed significantly reduced induc-tive responses to the exogenously introduced Dlx transcriptionfactors. The observation that all three Dlx transcription factorsco-activate GAD1 expression suggests that these transcriptionfactors subserve overlapping or redundant functions.
In addition, we tested the activities of two splice variants of thePitx2 primary transcript (Pitx2a and Pitx2c) and Pitx2 CD39. Pitx2gene mutations are responsible for the genetic disorder Riegersyndrome which is characterized by dental hypoplasia, craniofacialdysmorphism and other features associated with heart, limb andanterior pituitary development (Semina et al., 1996). The CD39truncated version of Pitx2c contains a C-terminal 39 amino aciddeletion that shows enhanced DNA binding activity (Amendt et al.,1999). As with the Dlx expression vectors, these constructs wereco-transfected along with the GAD1-319 bp parent template, theGAD1D-391 internal deletion, and the GAD1-486 bp/þ25 bp trun-cated promoter. Reporter activity derived from each is comparedwith a CMV empty vector cassette (Fig. 4B). Splice variant Pitx2aresulted in the most robust response while Pitx2c was e half asactive. Both of these were unable to boost expression whendownstream GAD1 promoter sequences are removed either byinternal deletion or by truncating the 30 portion (�486 bpto þ25 bp). The Pitx2 CD39 vector failed to significantly induce theexpression of any of the GAD1 promoter vectors over the CMVcontrol (empty) vector.
Finally, we examined the activity of an Sp1 expression vector intrans-activating a series of GAD1 promoter deletions using theco-transfection assays described above (Fig. 4C). In the upstreamand first exon spanning CpG islands, there are multiple Sp1/Sp3binding sites that are characteristic of GC-rich promoter regions.Unlike the more pronounced effect we had seen with the reelinpromoter (Chen et al., 2007), the absolute induction observed withSp1 was considerably weaker. However, we did see a positive resultwith the �1940 and �319 deletion constructs compared with theempty cassette (Fig. 4C). This suggests the presence of cis-actingelements recognized by Sp1 in these regions. However, this findingawaits confirmation with site-directed mutations before we canposition these sites with certainty. In addition to Sp1, we alsoperformed co-transfections with an Egr1 (NGF1A) expressionvector, as Egr1 also recognizes a GC-rich binding site. The Egr familyof transcription factors (1e4) are highly expressed in brain and
based on recognition site similarities, may have comparable func-tions (Poirier et al., 2008). There is evidence that the rat Gad1promoter responds to Egr1/NGFI-A in the context of maternal careand that methylation of this recognition site interferes with thisinteraction (Zhang and Meaney, 2010). However, we did not finda significant induction with CMV-Egr1 on any of our GAD1promoter vectors (data not shown). This suggests that if Egr1regulates GAD1, it does so outside the sequences examined.
Because of previous reports showing a positive effect of some ofthe Dlx proteins on GAD65 and GAD67 expression (Stühmer et al.,2002a), we examined the effect of these transcription factors andothers (Egr1, Sp1, Pitx2a and Pitx2c) on the levels of endogenousGAD67 mRNA in NT2 cells (Fig. 4D). More specifically, the previousstudy reported that ectopic expression of Dlx2 and Dlx5 inducedthe expression of GAD65 immunoreactivity and also inducedGAD67 mRNA. We attempted to replicate this finding in NT2 cellsby introducing the mouse constructs expressing Dlx2 and thenDlx2 þ Dlx5 by transient transfection. We harvested RNA 72 h laterand quantified the level of GAD67 mRNA using competitive RT-PCRwith an internal standard (Fig. 4D). Compared to the control (emptyexpression cassette), Dlx2 induced GAD67 mRNA approximatelytwo-fold while the double Dlx2 þ Dlx5 combination had a similareffect. While the extent of induction was not as robust as seenwithpromoter constructs, the effect was clearly evident. This was alsotrue with the other transcription factors tested: Egr1, Sp1, Pitx2a,and Pitx2c. These results may be low in terms of fold inductionbecause of the effects of local chromatin structure. The endogenousGAD1 mRNA expressed in untreated NT2 cells is low and increasesnearly 10-fold by the action of histone deacetylase inhibitors(Kundakovic et al., 2009).
While we did not study the expression of GAD25, the likelihoodis high that these two splice variants share many features commonto the 50 untranslated region and first exon. Because GAD25 isexpressed in testis and pancreatic islets but not in brain (Chesslerand Lernmark, 2000), it suggests that the regulation of this tran-script may arise from sequences located elsewhere in the gene.
3.5. Transient transfection of an in vitro methylated GAD1 promoterconstruct and the effect of MS-275
We have previously analyzed the effect of methylation on thereelin promoter in vitro (Kundakovic et al., 2009). These data showthat when an artificially methylated promoter is transfected intoNT2 cells and then treatedwithMS-275, the reporter activity comesback in a time- and dose-dependent manner. In contrast, if the cellsare not treated with the HDAC inhibitor, the activity remainslow (close to baseline) throughout the time course. Torepeat this experiment using the GAD1 promoter, we clonedthe �627/þ257 bp GAD1 construct into the pCpGL luciferasereporter vector which lacks CpG dinucleotides in the luciferasereporter and remaining plasmid backbone (Klug and Rehli, 2006).The GAD1 promoter construct was methylated in vitro with SssImethylase. HpaII digestion was used to confirm that the methyla-tion went to completion. The methylated promoter/reporter wastransfected into NT2 cells as previously described, along with smallamounts of (5 ng) of pRL-TK (Renilla luciferase) to normalize fortransfection efficiency (Kundakovic et al., 2009). Twenty-four hourafter transfection, MS-275 (5 mM)was added to the culture dish andleft for the next 48 h. In parallel, the inactive stereo enantiomerMS-IN was used to treat a second group of plates as a control tonormalize the fold induction. At the end of the treatment, cellswere harvested and lysed and reporter activity was measured. Asshown in Fig. 5A, methylated GAD1 promoter-mediated reporteractivity increased in a dose-dependent manner with increasingamounts of the Class I HDAC inhibitor MS-275. Similar results were

48 h MS-275 (μM)
noi
tc
ud
nI
re
tr
op
eR
dl
oF
0
10
20
30
40
50
0 0.25 1.0 2.5 5.0 7.5
A
B
GAD1 -627 bp pCpGL
-627 bp +254 bp
-622 bp -459 bp -241 bp
C
TSS
-5 bp +131/+167 bp
Hpa II/MspI sites
Fig. 5. Transfection of a methylated GAD1 promoter and treatment with the HDACinhibitor MS-275 reduces methylation. The GAD1 �627 to þ257 bp promoter wascloned into plasmid CpGL that lacks CpG dinucleotides in the vector backbone andluciferase coding regions. The construct was methylated in vitro with SssI methylaseand transfected into NT2 cells. After 24 h, the cells were treated with MS-275 (48 h) atwhich time cells were harvested and lysed for measuring luciferase activity. A) Showsreporter activity as a function of increasing amounts of MS-275. Data are expressedrelative to the activity obtained in parallel with MS-IN. B) To assess the effect of MS-275 on the methylation pattern of the transfected construct, we isolated DNA followingtransfection and MS-275 or MS-IN drug treatment (5 mM, 48 h). DNA was either notdigested or digested with HpaII (cuts only at unmethylated CCGG) or MspI (cuts ateither methylated or unmethylated CCGG). Groups I and II: SssI methylated GAD1promoter (�627) isolated after NT2 cell transfection treated with MS-275. Groups IIIand IV: SssI methylated GAD1 promoter fragment isolated after NT2 cell transfectiontreated with MS-275 IN (control). Group V: SssI methylated GAD67 promoter fragmentonly. Arrows indicate digest products or partially digested fragments. C) Line drawingshowing the location of HpaII (unmethylated) or MspI (either methylated or non-methylated) CCGG sites. The arrows position these sites relative to the cloning sitesand transcriptional start site. The anticipated fragments from an MspI digest would be�236 bp, 218 bp, 163 bp, 126 bp and 35 bp (from highest to lowest).
Y. Chen et al. / Neuropharmacology 60 (2011) 1075e1087 1083
previously obtained by treating 293 cells with VPA followingtransfection of amethylated CMV-GFP reporter (Detich et al., 2003).This group showed that VPA induces dose-dependent histoneacetylation, DNA demethylation and expression of the ectopicCMV-GFP construct and that the plasmid remained episomal.
Our previous experiments with the RELN promoter suggestedthe possibility that this increased activity may be due to an openingof the chromatin surrounding the episomal plasmid promoter andnot due to a direct effect of methylation on transcription(Kundakovic et al., 2009). To test this possibility, we also examinedthe methylation status of the transfected plasmid using methyla-tion-sensitive restriction enzymes and Southern blotting (Fig. 5B).Forty-eight hour after treating transfected cultures with MS-275,DNA was harvested, digested, and prepared for analysis. Following
transfer, blots were hybridized to the 32P-labeled GAD1 insert probeand exposed for various times to Phosphor Screens. Screens werescanned using a Molecular Dynamics Storm 860 scanner. Datashown in Fig. 5B show a representative blot obtained as described.Arrows to the left of the image indicate the presence of bandscorresponding to either fully or partially digested bands. Groups Iand II (Fig. 5B) are replications (different transfections) showing thepattern obtained following MS-275 treatment. The presence ofmultiple bands in the HpaII digested lanes strongly suggest thatNT2 cell treatment with the HDAC inhibitor facilitates demethyla-tion of the transfected construct. In contrast, groups III and IV showthe results obtained following treatment of the transfected cultureswith MS-IN. The corresponding digests are (lane 2) are blank. MspIdigestion on the other hand shows at least partial digestion of thetransfected DNA (Group III and IV, lanes 3). Group V shows thebands obtained using in vitro methylated DNA that was digestedwithout prior transfection. This represents the control group fordigest comparisons. For example, HpaII does not digest thistemplate while MspI does so almost completely. Fig. 5C showsa linear representation of the cloned �627 bp/þ257 bp GAD1promoter/pCpGL and the location of the HpaII/MspI sites withinthis sequence. Because of the relatively low level of resolution ofSouthern blotting and because of what appears to be partial digestswith some of the groups, we opted to examine the degree ofmethylation of the endogenous GAD1 promoter following treat-ment with MS-275 (Kundakovic et al., 2009).
3.6. Analysis of the methylation status of the endogenous GAD1promoter following treatment with either MS-IN or MS-275
We had previously shown that MS-275 induces expression ofthe reelin and GAD67 mRNAs in a time- and dose-dependentmanner. We had also compared regions within the reelin andGAD67 promoters for changes in methylation following treatmentof NT2 cells with MS-275 and MS-IN (Kundakovic et al., 2009).Using MeDIP assays, we had analyzed the region between �579 bpand �320 bp (see Fig. 3, top) to show a small but significantreduction in methylation in this region as a consequence of HDACinhibitor (MS-275 vs. MS-IN) or DNA methylation inhibitor (doxo-rubicin vs. vehicle) treatments. In the current study, we focus onadditional regions, particularly in light of our findings regarding thedownstream (within the 50 flanking sequences) regulatory regionsof the gene. For this purpose we designed an additional set of sixprimer pairs (see Table 1) for use in this assay. We hypothesize thatthe degree of methylation would be higher in those regions of thepromoter critical for regulation and anticipate that the amount ofmethylation would decrease when the promoter is more active.
A comparative analysis of the methylation of six overlappingregions of the GAD1 promoter are shown. In Fig. 6A, we providea linear representation of the promoter alongwith the start site andpreviously defined promote regions labeled. In addition, the loca-tion of each primer pair used in the analysis is indicated with the 50
and 30 endpoints of each primer listed (also see Table 1). NT2 cellswere treated for 48 h with either MS-275 (5 mM) or MS-IN (5 mM)and genomic DNA was harvested for analysis. Following sonicationof the genomic DNA into fragments of w300e500 bp in length,methylated sequences were immuneprecipitated with an antibodyagainst 5-methylcytosine (see Methods). Amounts of DNA presentin each region were measured using quantitative real-time PCRcomparing identical regions (primer pairs) from cells treated witheither MS-IN or MS-275 (Fig. 6B). While significant differenceswere observed in one upstream region (from�442 to�214 bp), thelargest differences occurred between regions overlapping theregulatory domains defined by the transient transfection experi-ments (�43 toþ121 bp andþ101 toþ255 bp).We also show results

Fig. 6. MS-275 treatment of NT2 cells reduces methylation of the GAD1 promoter. A)Shows a representation of the GAD1 promoter 50 flanking sequence and first exon. Thetranscriptional start site (þ1) is shown along with the 359 bp first exon (NM_000817).Contained within the first exon are the two previously defined (Erlander and Tobin,1992) promoters (P2 and P1). The sequences deleted for the analysis of the regula-tory region are indicated (þ66 to þ171 bp). Horizontal bars underneath show theamplicons that were examined following 5-methylcytosine immuneprecipitation.Primers used to define these amplicons are listed in Table 1. MeDIP and real-time PCRamplification were performed as indicated in Methods. B) Results of the meDIP anal-ysis of MS-IN and MS-275-treated NT2 cells. Cells were treated with either compound(5 mM) for 48 h. Genomic DNA was isolated, sonicated, and immuneprecipitated. Real-time PCR analysis was performed to evaluate the amounts of DNA precipitated cor-responding to each amplicon. Data are expressed as a percent of the input DNA foreach sample. Analysis of the G3PDH control region showed no differences in amountsof DNA precipitated by this antibody regardless of NT2 cell treatment. Data areexpressed this way rather than as a % of control because of the varying amounts ofmethylation that occur across the length of the promoter. *p < 0.05.
Y. Chen et al. / Neuropharmacology 60 (2011) 1075e10871084
obtained with the glyceraldehyde 3-phosphate dehydrogenase(G3PDH) regulatory control region and find no change in responseto the HDAC inhibitor treatment.
4. Discussion
In the current study we have focused on regulatory elementsassociated with the human GAD1 promoter. The mouse and humanpromoters show a high degree of sequence identity (Fig. 1). Overthe regions shown, as much as 83% of the sequences betweenspecies are identical. This includes sequences corresponding to thePitx2 binding site (Westmoreland et al., 2001) and particularly inthe 50 flanking region of the human gene. Interestingly, the start siteof the two mRNAs is displaced and differs by close to 195 bp. Thisdifference seems to suggest that either the mouse TSS is physicallylocated further downstream of the corresponding human start siteor that the humanTSS is closer towhere it was originally positioned(Erlander and Tobin, 1992). To rule out this latter possibility, weperformed RT-PCR experiments and show that NT2 cell RNA yieldsbands of the expected sizes based on the current position of thestart site (NCBI: NM_000817). Provided that this finding holds truein GABAergic neurons of the brain, it suggests that the bulk of theregulatory region of the promoter lies downstream of the RNA startsite. Deleting the entire 50 flanking sequence through þ1 does notresult in an appreciable decrease in expression. Indeed, transcrip-tion factors such as Dlx 1, 2 and 5, Pitx2a, and Pitx2c all fail to actwhen sequences from þ66 to þ171 bp are deleted from thepromoter constructs. While we have not precisely mapped thecorresponding recognition sites, we are able to conclude that thisregion of the promoter is important for regulation by these factors.It seems that Sp1 might interact at additional sites more distal fromthe deleted sequences but the effect is not nearly as robust. We
have also shown that each of the factors that activate the GAD1promoter constructs in trans also have a positive effect onexpression of the endogenous gene (Fig. 4). While the effects of theendogenously added transcription factors varied and were notnearly as strong as on the transfected templates, one has to keep inmind that endogenous GAD1 is embedded in chromatin that maynot be as readily accessible as the transfected promoter templates.In other words, the effect of these transcription factors on thenative GAD1 gene is likely restricted by the local chromatinconformation. Since the gene is transcriptionally active but at verylow levels, it may be that the introduction of additional transcrip-tion factors shifts the promoter to a more open state but the extentto which this is transcriptionally productive may be limited.
Both the 50 flanking sequences and first exons of themouseGad1and human GAD1 gene are GC rich and show characteristics ofa large CpG island. CpG islands tend to be under-methylatedcompared to the remainder of the genome and actively undergochanges in methylation status during cell specification (Illingworthand Bird, 2009). This is consistentwith ourfindings that bothmouseGad1 (Tremolizzo et al., 2002, 2005; Noh et al., 2005; Dong et al.,2005, 2007) and human GAD1 (Kundakovic et al., 2007, 2009) maybe regulated by changes in methylation. In fact, a recent study hasshown that methylation of the rat Gad1 promoter in the hippo-campus can be influenced bymaternal care following birth and thatthis increased methylation interferes with NGF1A activation of therat Gad1 promoter (Zhang et al., in press). In addition, this increasedmethylation and that of the glucocorticoid receptor promoter areaccompanied by higher levels of Dnmt1 mRNA. Increased Dnmt1mRNA expression has also been noted in GABAergic neurons of theschizophrenia cortex (Veldic et al., 2004, 2005).
We showhere that themethylation status of regions of theGAD1promoter that likely impact expression is reduced in response toMS-275 (Fig. 6). As one might anticipate an inverse correlationbetween methylation status and transcriptional activity, this resultis not surprising. However, we do not yet know whether thisrepresents active DNA demethylation as a consequence of chro-matin remodeling. Our previous studies show that MS-275 inducespromoter activation and that this is accompanied by the dissocia-tion of a large repressor complex from the reelin and GAD67regulatory regions using chromatin immuneprecipitation analyses(Kundakovic et al., 2009). We have extended this to include addi-tional MeDIP data in response to MS-275 that confirm and extendour previous findings. We also show preliminary data that areconsistent with DNA demethylation using an in vitro methylatedGAD67 promoter reporter construct (Fig. 5). The activation of theexogenous promoter following MS-275 treatment is clear but thepresence of some bands that may be due to partial digestionproducts confound the interpretation of these data. While NT2 cellsare a dividing cell line, the methylated plasmid vector is replicationdeficient following transient transfection. Others have shown thatthe histone deacetylase inhibitor VPA induces active DNA deme-thylation and expression of an ectopically methylated CMVpromoter in HEK-293 cells (Detich et al., 2003). There have beensome studies on the fate of exogenously introduced DNA once itenters a cell. Jeong and Stein (1994), using micrococcal nucleasedigestion of isolated nuclei to show that regularly arranged nucle-osome-like particles fomred on the transfected DNA but that thesizes of the ladders producedwere anomolous suggesting a possibleassociation with the nuclear matrix. In addition, they showed thatw10% of transfected DNA generated a typical nucleosome ladder.Buschhausen et al. (1987) showed that microinjected DNA adoptsa nucleosomal strucutre after a latency period lasting w8 h. Theseauthors conclude that the transcription of methylated DNA is notblocked until after these nucleosomal structures form and recruitmethyl DNA binding proteins. They suggest that the methyl moiety

Y. Chen et al. / Neuropharmacology 60 (2011) 1075e1087 1085
on the cytosine does not per se inhibit transcription until thetemplate acquires sufficient amounts of repressive chromatin thatprohibits both transcription factor and RNA polymerase access. Theuse of artificial templates to study chromatin modifications haslimitations and for this reason we sought to shift focus towards theeffects on the endogenous GAD1 gene.
Several groups have examined themethylation status of GAD1 inhuman post-mortem brain as a function of either psychiatric illnessand/or age at death (Siegmund et al., 2007; Huang and Akbarian,2007; Mill et al., 2008). For the most part, these studies focus onmultiple genes and promoters using either array-relatedapproaches, PCR based analyses, or candidate gene analysis andbisulphite sequencing. Siegmund et al. (2007) found that methyl-ation of the GAD1 gene fit into a pattern characteristic of otherneuronally expressed genes (GABRA2, HOXA1, NEUROD1, NEUROD2and others) that showed a sharp developmental rise in methylationwhich was accompanied by decreased levels of the correspondingmRNA. Additional studies that focused on patterns of methylationas a function of psychiatric diagnosis found no difference in GAD1genomic DNA and DNA methylation (Huang and Akbarian, 2007;Mill et al., 2008). In one of these studies, the investigators foundan 8-fold decrease in repressive chromatin-associated methylationcorresponding to GAD1 upstream sequences (Huang and Akbarian,2007). However, in each of the cases that have been examined thusfar, only one of these groups has looked at sequences within thefirst exon (Mill et al., 2008), where we find the bulk of the regu-latory elements associated with GAD1 expression are located.These authors found no significant differences in the methylationstatus in this region either. For a map of the human 50 flankingsequence and a summary of the promoter proximal regionsexamined thus far, see Supplemental Fig. 1. When examininga complex tissue such as cortex, it may be difficult to measuremethylation differences because of neuronal heterogeneity, whichwould tend to minimize signal differences. This is particularlyrelevant as methylation patterns are cell type-specific and will varyacross different neurons in the brain. Because our experimentswere performed in NT2 cells, we are not able to draw conclusionsregarding whether the differences that might exist in the humanbrain. However, we are confident that we have identified theappropriate region of the promoter where methylation is likely toimpact transcriptional activity.
According to the International HapMap Data Project Phase III,there are some 44 genotyped SNPs that span the 44.46 kb ofchromosome 2 that corresponds to the genomic sequence encodingthe GAD1 mRNA sequence (NCBI: NM_000817). Additional SNPshave been mapped further upstream and downstream of thisregion. The finding of association of some GAD1 promoter SNPssuggests that this gene is a candidate susceptibility gene for bipolaraffective disorder and schizophrenia (Lundorf et al., 2004). Morerecently, there has been evidence of distorted transmission of SNPsin two independent schizophrenia family-based samples (Straubet al., 2007). Of particular interest was the finding that one SNPin the 50 untranslated region that influenced cognition was asso-ciated with decreased GAD1 mRNA expression. This SNP(rs3749034) represents a G to A mutation at position þ275 relativeto the TSS. This position has been, based on in silico predictions,proposed as part of a transcription factor binding site for eitherAREB6 or MYOD1 (Straub et al., 2007). This site falls outside of thewindow (þ66 to þ171 bp) we have identified as important totranscriptional regulation of GAD1 although we cannot rule out thispossibility based on our analysis. Our meDIP data and preliminarybisulphite sequencing data indicate that the cytosinewhich directlyprecedes this SNP is methylated when the gene is transcriptionallyless active. It may be that this mutation somehow impacts localmethylation patterns and hence indirectly affects expression.
However, this conclusion is premature and will require experi-mental data prior to confirmation. Several other SNPs in the GAD1gene have been identified that positively modulate GABA levels inthe anterior cingulate cortex (Marenco et al., 2010). These SNPs arealso located outside of the range we have focused on withrs1978340 well upstream of the TSS while rs769390 is downstreamin intron 6.
Our previous studies have shown that the GAD1 promoterresponds much like the RELN promoter in terms of its response toepigenetic drugs (Kundakovic et al., 2009). Both reelin and GAD1mRNAs are down-regulated in prefrontal cortex of schizophreniapost-mortem tissue samples (Guidotti et al., 2000). These sameneurons overexpress the mRNA and protein corresponding to DNAmethyltransferase I (Veldic et al., 2004, 2005). Since both reelin andGAD1 are expressed in the same GABA neurons expressing DNMT1,we studied the behavior of these promoters in parallel to betterunderstand their response to factors that affect methylation. Forexample, both reelin and GAD67 mRNAs increase in parallel inresponse to treating NT2 cells with the DNMT inhibitor doxorubicin(Kundakovic et al., 2007). In addition, we have shown that theHDAC inhibitor MS-275 activates both reelin and GAD67 mRNAswith the same EC50 and in the same temporal context (Kundakovicet al., 2009). Moreover, treatment of mice with the methyl donormethionine inhibits reelin and GAD67mRNA expression in primarycortical neurons in vitro (Noh et al., 2005) and in vivo (Dong et al.,2005). In addition, we have shown that this effect can be reversedby co-administering the HDAC inhibitor VPA (Tremolizzo et al.,2005; Dong et al., 2007). Along these same lines, we haverecently described how HDAC inhibitors facilitate the action ofantipsychotic subtypes to induce chromatin remodeling by alteringthe methylation status of these two promoters (Guidotti et al.,2009). Clearly, unraveling the extent to which methylation,histone modifications, and chromatin remodeling interact to finetune levels of expression in different neurons of the brain isa complex problem. However, it should be recognized that regu-latory regions are experimentally defined and not generated insilico. The extent to which methylation maps overlap with regula-tory regions of promoters is not yet knownwith certainty. However,it would seem more appropriate to define these regions prior toundertaking an in depth study ofmethylation in a tissue as complexas the brain. While DNA methylation signatures in different brainregions may distinguish region-specific functional specializations(Ladd-Acosta et al., 2007), it will be necessary to look at individualcells to better understand the basis for how these differentialpatterns arise. As interindividual differences in DNA methylationpatterns can be extensive in the brain (Zhang et al., 2010), knowingwhere in the genome to look may prove to be as important as thecellular resolution used in obtaining samples for analysis.
Acknowledgements
We are grateful to following: Dr. John L. Rubenstein, Dept. ofPsychiatry at the University of California San Francisco, for thegenerous gift of the mouse Dlx1, Dlx2, Dlx5 and control expressioncassettes; Dr. Dona LeeWong, Harvard Medical School and Directorof the Laboratory of Molecular and Developmental Neurobiology,McLean Hospital for sending us the pCMV-Egr1 and control vector:Dr. Andrew Russo, Department of Physiology and Biophysics,University of Iowa for the gift of the Pitx2 vectors; and Dr. RobertTjian, Professor of Molecular and Cell Biology, University of Cal-ifornia Berkeley, for providing the Sp1 expression vector andcontrol. The authors would like to thank Dr. Alessandro Guidotti formany thoughtful discussions. We acknowledge the editorialexpertise of Roberta Baruch for help in proofing this manuscript.We would like to dedicate this manuscript to the memories of

Y. Chen et al. / Neuropharmacology 60 (2011) 1075e10871086
a friend and colleague Dr. Robert H. Costa and his father and ourmentor, Dr. Ermino Costa. Robert died on September 1, 2006 frompancreatic cancer. Dr. Erminio Costa passed away on November 28,2009 due to complications of multiple myeloma.
Appendix. Supplementary material
Supplementarymaterial associatedwith this paper can be found,in the online version, at doi:10.1016/j.neuropharm.2010.09.017.
References
Akbarian, S., Huang, H.-S., 2006. Molecular and cellular mechanisms of alteredGAD1/GAD67 expression in schizophrenia and related disorders. Brain Res. Rev.52, 293e304.
Akbarian, S., Kim, J.J., Potkin, S.G., Hagman, J.O., Tafazzoli, A., Bunney, W.E.,Jones, E.G., 1995. Gene expression for glutamic acid decarboxylase is reducedwithout loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen.Psychiatry 52, 258e266.
Amendt, B.A., Sutherland, L.B., Russo, A.F., 1999. Multifunctional role of the Pitx2homeodomain protein C-terminal tail. Mol. Cell. Biol. 19, 7001e7010.
Anderson, S.A., Eisenstat, D.D., Shi, L., Rubenstein, J.L.R., 1999. Differential origins ofneocortical projection and local circuit neurons: role of Dlx genes in neocorticalinterneuronogensis. Cereb. Cortex 9, 646e654.
Asada, H., Kawamura, Y., Maruyama, K., Kume, H., Ding, R., Ji, F.Y., Kanbara, N.,Kuzume, H., Sanbo, M., Yagi, T., Obata, K., 1997. Cleft palate and decreased braingamma-aminobutyric acid in mice lacking the 67- kDa isoform of glutamic aciddecarboxylase. Proc. Natl. Acad. Sci. U S A 94, 6496e6499.
Bray, S.J., Johnson, W.A., Hirsh, J., Heberlein, U., Tjian, R., 1988. A cis-acting elementand associated binding factor required for CNS expression of the Drosophilamelanogaster dopa decarboxylase gene. EMBO J., 177e188.
Bu, D.-F., Tobin, A.J., 1994. The exon-intron organization of the genes (GAD1 andGAD2) encoding two human glutamate decarboxylases (GAD67 and GAD65)suggests that they derive from a common ancestral GAD. Genomics 21, 222e228.
Buschhausen, G., Wittig, B., Graessmann, M., Graessmann, A., 1987. Chromatinstructure is required to block transcription of the methylated herpes simplexvirus thymidine kinase gene. Proc. Natl. Acad. Sci. U S A 84, 1177e1181.
Chattopadhyaya, B., Di Cristo, G., Wu, C.Z., Knott, G., Kuhlman, S., Fu, Y., Palmiter, R.D.,Huang, Z.J., 2007. GAD67-mediated GABA synthesis and signaling regulateinhibitory synaptic innervation in the visual cortex. Neuron 54, 889e903.
Chen, Y., Sharma, R.P., Costa, R.H., Costa, E., Grayson, D.R., 2002. On the epigeneticregulation of the human reelin promoter. Nucleic Acids Res. 30, 2930e2939.
Chen, Y., Kundakovic, M., Agis-Balboa, R.C., Pinna, G., Grayson, D.R., 2007. Induction ofthe reelinpromoterby retinoic acid ismediatedby Sp1. J. Neurochem.103, 65e655.
Chessler, S.D., Lernmark, A., 2000. Alternative splicing of GAD67 results in thesynthesis of a third form of glutamic-acid decarboxylase in human islets andother non-neural tissues. J. Biol. Chem. 275, 5188e5192.
Condie, B.G., Gain, G., Gottlieb, D.I., Capecchi, M.R., 1997. Cleft palate in mice witha targeted mutation in the g-aminobutyric acid-producing enzyme glutamicacid decarboxylase 67. Proc. Natl. Acad. Sci. U S A 94, 11451e11455.
Detich, N., Bovenzi, V., Szyf, M., 2003. Valproate induces replication-independentactive DNA demethylation. J. Biol. Chem. 278, 27586e27592.
Dong, E., Agis-Balboa, R.C., Simonini, M.V., Grayson, D.R., Costa, E., Guidotti, A.,2005. Reelin and glutamic acid decarboxylase67 promoter remodeling in anepigenetic methionine-induced mouse model of schizophrenia. Proc. Natl.Acad. Sci. U S A 102, 12578e12583.
Dong, E., Guidotti, A., Grayson, D.R., Costa, E., 2007. Demethylation of reelin andglutamic acid decarboxylase 67 brain promoters is induced by hyperacetylationof histones. Proc. Natl. Acad. Sci. U S A 104, 4676e4681.
Erlander, M.G., Tobin, A.J., 1992. A transcriptional regulatory element of the geneencoding the 67,000-Mr form of human glutamate decarboxylase is similar toa Drosophila regulatory element. J. Neurochem. 58, 2182e2190.
Gonzalez-Burgos, G., Hashimoto, T., Lewis, D.A., 2010. Alterations of cortical GABAneurons and network oscillations in schizophrenia. Curr. Psychiatry Rep. 12,335e344.
Guidotti, A., Auta, J., Davis, J.M., Di Giorgi-Gerevini, V., Dwivedi, Y., Grayson, D.R.,Impagnatiello, F., Pandey, G., Pesold, C., Sharma, R., Uzunov, D., Costa, E., 2000.Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression inschizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen.Psych. 57, 1061e1069.
Guidotti, A., Auta, J., Davis, J.M., Dong, E., Grayson, D.R., Veldic, M., Zhang, X.,Costa, E., 2005. GABAergic dysfunction in schizophrenia: a new treatment onthe horizon. Psychopharmacology 180, 191e205.
Guidotti, A., Dong, E., Kundakovic, M., Satta, R., Grayson, D.R., Costa, E., 2009.Characterization of the action of antipsychotic subtypes on valproate-inducedchromatin remodeling. Trends Pharmacol. Sci. 30, 55e60.
Heckers, S., Stone, D., Walsh, J., Schick, J., Koul, P., Benes, F.M., 2002. Differentialhippocampal expression of glutamic acid decarboxylase 65 and 67 messengerRNA in bipolar disorder and schizophrenia. Arch. Gen. Psychiatry 59,521e529.
Huang, H.-S., Akbarian, S., 2007. GAD1 mRNA expression and DNA methylation inprefrontal cortex of subjects with schizophrenia. PloS ONE 2, e809.
Huang, H.-S., Matevossian, A., Whittle, C., Kim, S.Y., Schumacher, A., Baker, S.O.,Akbarian, S., 2007. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters.J. Neurosci 27, 11254e11262.
Illingworth, R.S., Bird, A.P., 2009. CpG islands-a rough guide. FEBS Lett. 583,1713e1720.
Jeong, S.W., Stein, A., 1994. Micrococcal nuclease digestion of nculei reveals extendednucleosome ladders having anomolous DNA lengths for chromatin assembled onnon-replicating plasmids in transfected cells. Nuc. Acids Res. 22, 370e375.
Kanno, K., Suzuki, Y., Yamada, A., Aoki, Y., Kure, S., Matsubara, Y., 2004. Associationbetween nonsyndromic cleft lip with or without cleft palate and the glutamatedecarboxylase 67 gene in the Japanese population. Am. J.Med. Genet.127A,11e16.
Kash, S.F., Johnson, R.S., Tecott, L.H., Noebels, J.L., Mayfield, R.D., Hanahan, D.,Baekkeskov, S., 1997. Epilepsy in mice deficient in the 65-kDa isoform of glu-tamic acid decarboxylase. Proc. Natl. Acad. Sci. USA 94, 14060e14065.
Kim, S., Webster, M.J., 2010. The Stanley Neuropathology Consortium integrativedatabase: a novel, web-based tool for exploring neuropathological markers inpsychiatric disorders and the biological processes associated with abnormali-ties of those markers. Neuropsychopharmacology 35, 473e482.
Klug, M., Rehli, M., 2006. Functional analysis of promoter CpG methylation usinga CpG-free luciferase reporter vector. Epigenetics 1, 127e130.
Kundakovic, M., Chen, Y., Costa, E., Grayson, D.R., 2007. DNA methyltransferaseinhibitors coordinately induce expression of the human reelin and glutamicacid decarboxylase genes. Mol. Pharmacol. 71, 644e653.
Kundakovic, M., Chen, Y., Guidotti, A., Grayson, D.R., 2009. The reelin and GAD67promoters are activated by epigenetic drugs that facilitate the disruption oflocal repressor complexes. Mol. Pharmacol. 75, 342e354.
Ladd-Acosta, C., Pevsner, J., Sabunciyan, S., Yolken, R.H., Webster, M.J., Dinkins, T.,Callinan, P.A., Fan, J.-B., Potash, J.B., Feinberg, A.P., 2007. DNA methylationsignatures within the human brain. Am. J. Hum. Genet. 81, 1304e1315.
Lundorf,M.D., Buttenshon, H.N., Foldager, L., Blackwood, D.H.R.,Muir,W.J.,Murray, V.,Pelosi, A.J., Kruse, T.A., Ewald, H., Mors, O., 2004. Mutational screening andassociation study of glutamate decarboxylase I as a candidate susceptibility genefor bipolar affective disorder and schizophrenia. Am. J. Med. Genet.135B, 94e101.
Marenco, S., Savostyanova, A.A., van der Veen, J.W., Geramita, M., Stern, A.,Barnett, A.S., Kolachana, B., Radulescu, E., Zhang, F., Callicott, J.H., Straub, R.E.,Shen, J., Weinberger, D.R., 2010. Genetic modulation of GABA levels in the ante-rior cingulate cortex by GAD1 and COMT. Neuropsychopharmacol 35,1708e1717.
Mill, J., Tang, T., Kaminsky, Z., Khare, T., Yazdanpanah, S., Bouchard, L., Jia, P.,Assadzadeh, A., Flanagan, J., Schumacher, A., Wang, S.-C., Petronis, A., 2008.Epigenomic profiling reveals DNA-methylation changes associated with majorpsychosis. Am. J. Hum. Genet. 82, 696e711.
Noh, J.S., Sharma, R.P., Veldic, M., Salvacion, A.A., Jia, X., Chen, Y., Costa, E., Guidotti, A.,Grayson, D.R., 2005. DNA methyltransferase 1 regulates reelin mRNA expressionin primary mouse cortical cultures. Proc. Natl. Acad. Sci. U S A 102, 1749e1754.
Poirier, R., Cheval, H., Mailhes, C., Garel, S., Charnay, P., Davis, S., Laroche, S., 2008.Distinct functions of Egr gene family members in cognitive processes. Front.Neurosci. 2, 47e55.
Semina, E.V., Reiter, R., Leysens, N.J., Alward, L.M., Small, K.W., Datson, N.A., Siegel-Bartelt, J., Bierke-Nelson, D., Bitoun, P., Zabel, B.U., Carey, J.C., Murray, J.C., 1996.Cloning and characterization of a novel bicoid-related homeobox transcriptionfactor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 14, 392e399.
Sharma, R.P., Gavin, D.P., Grayson, D.R., 2010. CpG methylation in neurons: message,memory, or mask? Neuropsychopharmacol epub ahead of print.
Siegmund, K.D., Connor, C.M., Campan, M., Long, T.I., Weisenberger, D.J.,Biniszkiewicz, D., Jaenisch, R., Laird, P.W., Akbarian, S., 2007. DNA methylationin the human cerebral cortex is dynamically regulated throughout the life spanand involves differentiated neurons. PloS ONE 9, e895.
Straub, R.E., Lipska, B.K., Egan, M.F., Goldberg, T.E., Callicott, J.H., Mayhew, M.B.,Vakkalanka, R.K., Kolachana, B.S., Kleinman, J.E., Weinberger, D.R., 2007. Allelicvariation in GAD1 (GAD67) is associated with schizophrenia and influencescortical function and gene expression. Mol. Psychiatry 12, 854e869.
Stühmer, T., Anderson, S.A., Ekker, M., Rubenstein, J.L.R., 2002a. Ectopic expressionof the Dlx genes induces glutamic acid decarboxylase and Dlx expression.Development 129, 245e252.
Stühmer, T., Puelles, L., Ekker, M., Rubenstein, J.L.R., 2002b. Expression from a Dlx geneenhancer marks adult mouse cortical GABAergic neurons. Cereb. Cortex 12, 75e85.
Szabo, G., Katarova, Z., Kortvely, E., Greenspan, R.J., Urban, Z., 1996. Structure of themouse gene encoding the 67-kD form of glutamic acid decarboxylase. DNA CellBiol. 15, 1081e1091.
Thompson, M., Weickert, C.S., Wyatt, E., Webster, M.J., 2009. Decreased glutamicacid decarboxylase67 mRNA expression in multiple brain areas of patients withschizophrenia and mood disorders. J. Psych. Res. 43, 970e977.
Torrey, E.F., Barci, B.M., Webster, M.J., Bartko, J.J., Meader-Woodruff, J.H.,Knable, M.B., 2005. Neurochemical markers for schizophrenia, bipolar disorder,and major depression in post-mortem brains. Biol. Psychiatry 57, 252e260.
Tremolizzo, L., Carboni, G., Ruzicka, W.B., Mitchell, C.P., Sugaya, I., Tueting, P.,Sharma, R., Grayson, D.R., Costa, E., Guidotti, A., 2002. An epigenetic mousemodel for molecular and behavioral neuropathologies related to schizophreniavulnerability. Proc. Natl. Acad. Sci. U S A 99, 17095e17100.
Tremolizzo, L., Doueiri, M.S., Dong, E., Grayson, D.R., Pinna, G., Tueting, P., Rodri-guez-Menendez, V., Costa, E., Guidotti, A., 2005. Valproate corrects the

Y. Chen et al. / Neuropharmacology 60 (2011) 1075e1087 1087
schizophrenia-like epigenetic behavioral modifications induced by methioninein mice. Biol. Psychiatry 57, 500e509.
Veldic, M., Caruncho, H.M., Liu, W.S., Davis, J., Satta, R., Grayson, D.R., Guidotti, A.,Costa, E., 2004. DNA-Methyltransferase-1 mRNA is selectively overexpressed intelencephalic GABAergic interneurons of schizophrenia brains. Proc. Natl. Acad.Sci. U S A 101, 348e353.
Veldic, M., Guidotti, A., Maloku, E., Davis, J.M., Costa, E., 2005. In psychosis, corticalinterneurons overexpress DNA-methyltransferase I. Proc. Natl. Acad. Sci. U S A102, 2152e2157.
Westmoreland, J.J., McEwen, J., Moore, B.A., Jin, Y., Condie, B.G., 2001. Conservedfunction of Caenorhabditis elegans UNC-30 and mouse Pitx2 in controllingGABAergic neuron differentiation. J. Neurosci. 21, 6810e6819.
Yanagawa, Y., Kobayahi, T., Kamei, T., Ishii, K., Nishijima, M., Takaku, A., Kobayashi, T.,Tamura, S., 1997. Structure and alternative promoters of the mouse glutamicacid decarboxylase gene. Biochem. J. 326, 573e578.
Zhang, T.-Y., Meaney, M.J., 2010. Epigenetics and the environmental regulation ofthe genome and its function. Annu. Rev. Psychol. 61, 439e466.
Zhang, D., Cheng, L., Badner, J.A., Chen, C., Chen, Q., Luo, W., Craig, D.W.,Redman, M., Gershon, E.S., Liu, C., 2010. Genetic control of individualdifferences in gene-specific methylation in human brain. Am. J. Hum. Genet.86, 411e419.
Zhang, T.-Y., Hellstrom, I.C., Bagot, R.C., Wen, X., Diorio, J., Meaney, M.J., Maternalcare and DNA methylation of a glutamic acid decarboxylase 1 promoter in rathippocampus. J. Neurosci. Manuscript in press.
Related Documents











