"An Eulerian path approach to global multiple alignment for DNA sequences” by Y. Zhang and M. Waterman * “An Eulerian path approach to local multiple alignment for DNA sequences” by Y. Zhang and M. Waterman ** Presented by Jaehee Jung Mar 4 2005 CPSC 689-604 *Journal of Computational Biology 10-6, pp. 803-819 (2003). ** Proc. National Academy of Science of USA 102-5, pp. 1285-1290 (2005).

"An Eulerian path approach to global multiple alignment for DNA sequences” by Y. Zhang and M. Waterman * “An Eulerian path approach to local multiple alignment.
Dec 21, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

"An Eulerian path approach to global multiple alignment for DNA sequences”
by Y. Zhang and M. Waterman *
“An Eulerian path approach to local multiple alignment for DNA sequences” by Y. Zhang and M. Waterman **
Presented by Jaehee Jung
Mar 4 2005
CPSC 689-604
*Journal of Computational Biology 10-6, pp. 803-819 (2003). ** Proc. National Academy of Science of USA 102-5, pp. 1285-1290 (2005).

2
Outline• Motivation
– Hamiltonian & Eulerian path– Superpath problem
• Global Alignment– Global Alignment Algorithm – Probability Analysis– Complexity– Discussion
• Local Alignment– Local Alignment Algorithm– Significance Estimation– Complexity– Discussion
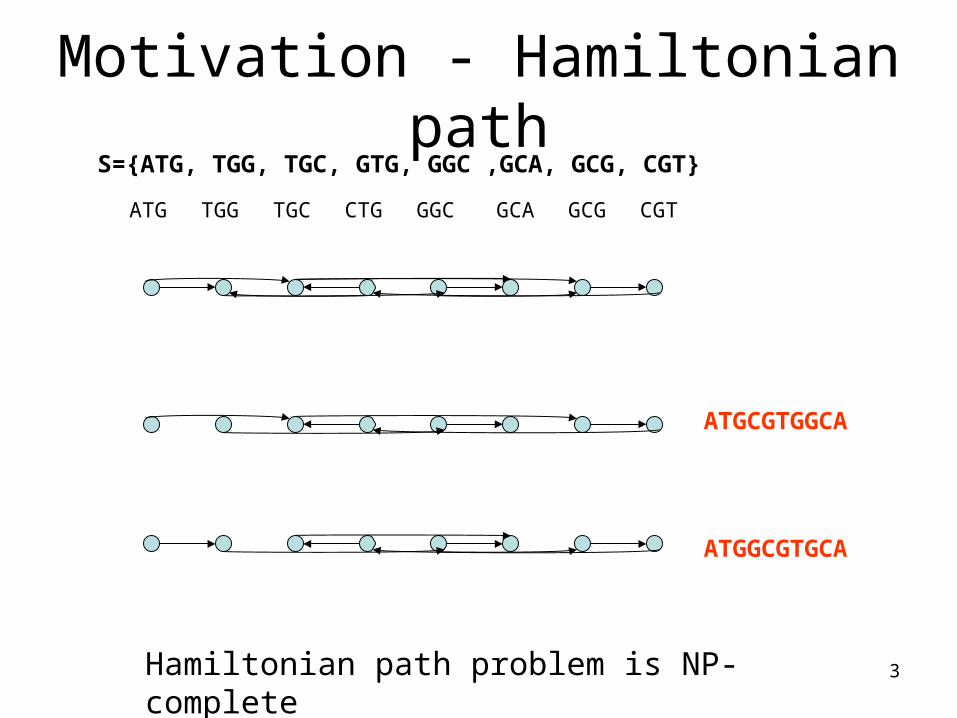
3
Motivation - Hamiltonian pathS={ATG, TGG, TGC, GTG, GGC ,GCA, GCG, CGT}
ATG TGG CTG GGC GCA GCG CGTTGC
ATGCGTGGCA
ATGGCGTGCA
Hamiltonian path problem is NP- complete

4
Motivation - Eulerian pathS={ATG, TGG, TGC, GTG, GGC ,GCA, GCG, CGT}
Vertices correspond to (l-1) tuples
Edges correspond to l-tuples from the spectrum
AT
GT CG
GC CA
GG
TG
AT
GT CG
GC CA
GG
TG
AT
GT CG
GC CA
GG
TG
ATGGCGTGCA ATGCGTGGCA
Eulerian path – visiting all edges correspond to sequence reconstruction

5
Global multiple alignment
• Global multiple alignment – Entire sequence are align into one configuration– Time and memory cost
• L : sequence length• N : number of sequences
• Multiple sequence alignment– Many heuristic algorithm
• Progressive alignment strategies – Aligning the closet pair of sequences
– Aligning the next close pair of sequences
» Ex: MULTAL, CLUSTALW, T-COFFEE
)( NL

6
Global multiple alignment
– Many heuristic algorithm (cont’d)• Iterative refinement strategies
– Local alignment to construct multiple alignment based on segment –segment comparison
– Refine the initial alignment iteratively by local alignment» Ex: DIALIGN
– Iteratively dividing the sequence into two groups and the realignment
» Ex: PRRP– Stochastic iterative strategies
» Ex: HMMT, SAM• ISSUE
– Robust under certain condition – Local optimal problem (iterative problem)=> Efficient time and memory space

7
Motivation
EULER[1][2] EulerAlign[3]Fragment assembly in DNA sequencing using Eulerian superpath approach
Global multiple DNA sequence alignment problem using Eulerian Paths
Easy to solve Eulerian path problem in Bruijn graph
Similar to Star method
Contribution: discard the traditional “overlap-layout-consensus”
“error-free” data by an error-correction procedure
Assume all input sequences are derived from a common ancestral sequence
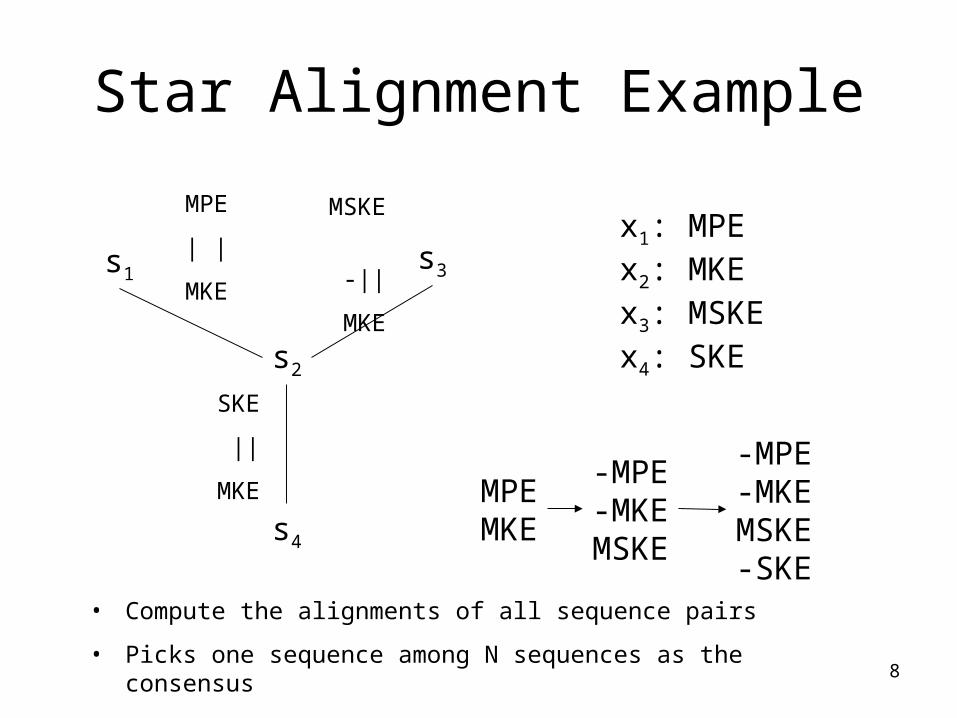
8
Star Alignment Example
s2
s1s3
s4
x1: MPEx2: MKEx3: MSKEx4: SKE
MPE
| |
MKE
MSKE
-||
MKE
SKE
||
MKE MPEMKE
-MPE-MKEMSKE
-MPE-MKEMSKE-SKE
• Compute the alignments of all sequence pairs
• Picks one sequence among N sequences as the consensus

9
Motivation - Eulerian Superpath
• Superpath Problem – EULER [2]– Given an Eulerian graph and a collection of
paths in this graph, find an Eulerian Path in this graph that contains all these paths as subpath
– Solve
• Transform graph G, system of path P -> G1 and P1
• Make a series of equivalent transformation
• (G , P) -> (G1 , P1) -> (G2 , P2) …. ->(Gk , Pk)

10
Motivation - Eulerian Superpath
• Equivalent transformation – X,Y detachment
vin Vmid vout
x y
P ->xP ->x P y->
P x,y
P y->
vin
Vmid
vout
z
P ->x
P x,y

11
Motivation - Eulerian Superpath
P
Px,y1
Px,y2
• Equivalent transformation– X,Y detachment
• P consistent with Px,y1 but inconsistent with Px,y2
• P is resolvable

12
Motivation - Eulerian Superpath
P
Px,y1
Px,y2
• Equivalent transformation – X,Y detachment
• P inconsistent with both Px,y1 and Px,y2
• Has no solution (did not encounter in *NM project)
*NM project: “difficult-to assemble” and “repeat-rich” bacterial genomes

13
Motivation - Eulerian Superpath
P
Px,y2
Px,y1
• Equivalent transformation– X,Y detachment
• P consistent with both Px,y1 and Px,y2
• Difficult situation – Analyze until all resolvable edges are analyzed

14
Motivation - Eulerian Superpath
• Equivalent transformation – X-cut
•P->x and Px-> without affecting the graph G
vin
vin
vin vin
vin
vin
vin
vin
vin vin
vin
vin
P x->P ->x
xx
y3y4 y2
y1
P ->xP x->
y3y4 y2
y1
vin
vin
vin vin
vin
vin
vin
vin
vin vin
vin
vin
P x->P ->x
xx
y3y4 y2
y1
P ->xP x->
y3y4 y2
y1

15
Eulerian global alignment -the algorithm
1. Construct a directed de Bruijn graph2. Transform the de Bruijn graph to DAG3. Extract a consensus path form the DAG
according to the edges4. Do fast pairwise alignment between the
consensus path and each input sequence
5. Construct the final multiple alignment according to the pairwise alignment

16
(1) – (2) – (3) – (4) – (5) Construct a directed de Bruijn graph
CCTTAG: CCTTA CTTAG:
CCTT CTTA CTTA TTAG+ +
CCTT CTTA CTTA TTAG
CCTT CTTA TTAG
Merge Vertices “CTTA
Construction of the de Bruijn graph for CCTTAG and k=5

17
de Bruijn Graph Construction
•Assume that there are no sequencing errors.•Construct the de Bruijn graph, taking all (k – 1)-mers appearing in the set of fragments as vertices.
TCACA ACAA GTCA•These errors have to be corrected before construction of the de Bruijn graph
read ACGGCTAT other reads CTAACTGC CTGCTA AACTGCT correction T
k = 3GT TC CA AC AA

18
(1) – (2) – (3) – (4) – (5) Construct a directed de Bruijn graph
1
2
3
4
5
6
8
9
0
7
0
4
1
2
3
5
6
7
8
9
8
910 9
9
9
9
8
9
8
9
An example of the initial de Bruijn graph
multiplicity

19
(1)– (2) – (3) – (4) – (5)Transformation the de Bruijn graph to DAG
• Transformation the de Bruijn graph to DAG – Tangle
• a vertex that has more than one incomings or outgoings edges
• Created by random matches, repeats, mutation DNA sequences
• Result cycle
– Goal : delete tangle, because of many cycles
vi

20
(1)– (2) – (3) – (4) – (5)Transformation the de Bruijn graph to DAG
• Claim
– E->Vi : left edge for vertex vi to be an edge that points to
vi
– If a vertex vi has two or more left edge{En->Vi
}n=1,2,3.. that
are contained in the same sequence path, there must exist a cycle in a graph
• Proof
– vi will visited when visiting E1->Vi
and vi wil visited will
when visiting E2->Vi
vi

21
(1) – (2) – (3) – (4) – (5)Transformation the de Bruijn graph to DAG
• Rule of transformation
– Sequence information in Evi-> partitioned two superedges E1
->vi->, E2->vi->
– Multiplicity for superedge E1->vi->, E2
->vi-> compute
vi vj
v´i
vj
vi
A tangle at vi is eliminated by making a copy vi’ of vertex vi and separating
E1->Vi
E2->Vi
EVi->
E1->Vi->
E2->Vi->

22
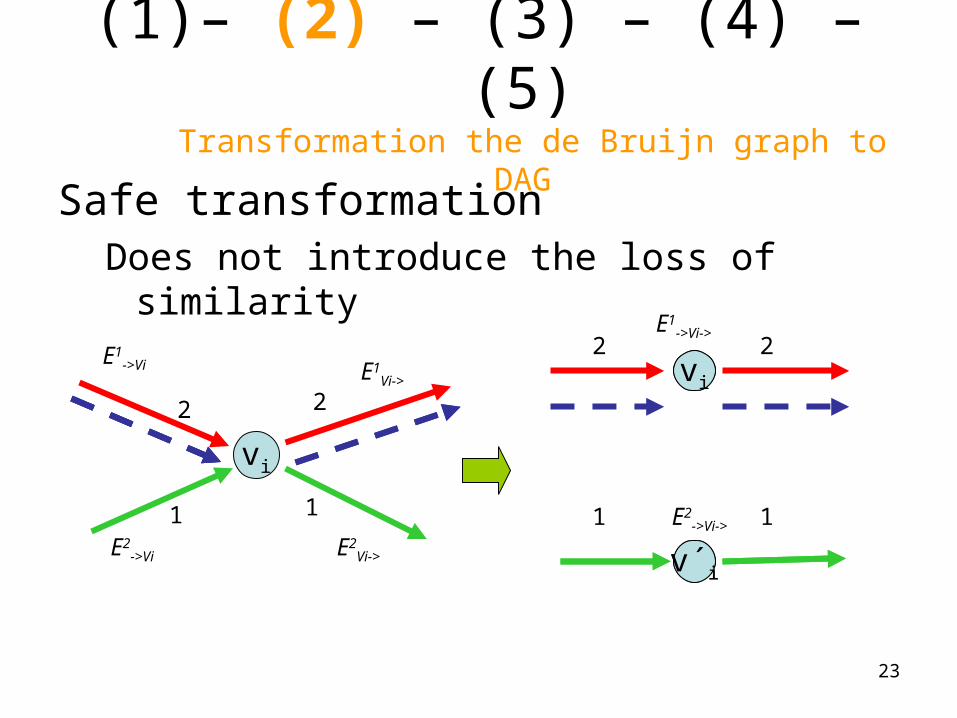
(1) – (2) – (3) – (4) – (5) Transformation the de Bruijn graph to DAG
• Rule of transformation
vi
v´i
vi
A tangle at vi is eliminated by making a copy vi ’ of vertex vi
E1->Vi
E2->Vi
E1Vi->
E1->Vi->
E2->Vi->
E2Vi->
23
(1) – (2) – (3) – (4) – (5) Transformation the de Bruijn graph to DAG
Safe transformationDoes not introduce the loss of similarity
vi
E1->Vi
E2->Vi
E1Vi->
E2Vi->
2
1
v´i
vi
v´i
vi
E1->Vi->
E2->Vi->
2
1
2
1
2
1

24
(1) – (2) – (3) – (4) – (5) Transformation the de Bruijn graph to DAG
Unsafe transformationIntroduce the loss of similarity
vi
2
1
1
2
E1->Vi
E2->Vi
E1Vi->
E2Vi-> v´i
vi
2
1
1
11
E1->Vi->
E2->Vi->
25
(1) – (2) – (3) – (4) – (5) Transformation the de Bruijn graph to DAG
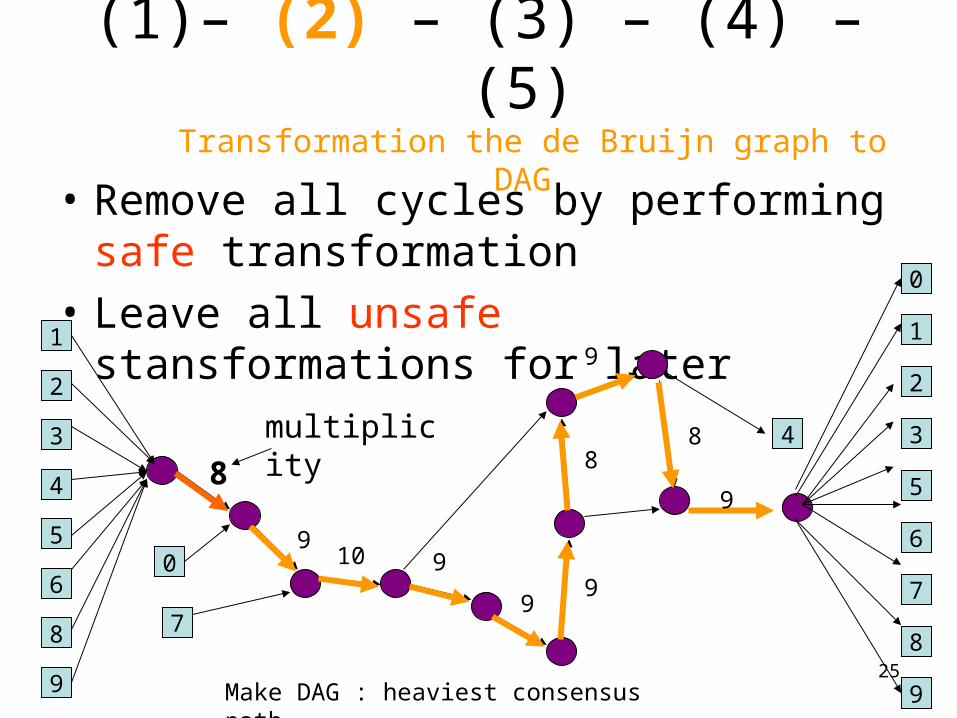
• Remove all cycles by performing safe transformation
• Leave all unsafe stansformations for later1
2
3
4
5
6
8
9
0
7
0
4
1
2
3
5
6
7
8
9
8
910 9
99
8
9
8
9
multiplicity
Make DAG : heaviest consensus path

26
(1) – (2) – (3) – (4) – (5) Extract a consensus path from DAG
• Greedy Algorithm– To find a heaviest path within linear time – Not optimal but satisfactory– Weight for each edge
• Proportional to its multiplicity and length

27
(1) – (2) – (3) – (4) – (5)Fast pairwise alignment
• Banded pairwise alignment algorithm– The positional shifts between two candidate
letters in two sequences are bonded by a constant
• Align the consensus sequence with each input sequence

28
(1) – (2) – (3) – (4) – (5) Construct the final multiple alignment
• Combine the alignment to construct the final multiple alignment

29
Probability Analysis
• Assume: all input sequence are derived from a common ancestral sequence S0
– N -> identical S0
– N: number of sequence– L : average sequence length– k :size k-tuple– :mutation rate
• No mutation : N sequence exactly same S0
multiplicity for each edge N
• With mutation : weight edge in S0
))(( 0seW

30
Probability Analysis
• Large Deviation Theorem (L.D.T) for binomial estimate
• If ,then consensus path exist and be accurate
•
)}(min{ 0seW )}(max{)}(min{ 00 seWseW
asNeNr
NXP NH ,)1(2
1
1
1~)(
)}(min{)1()}(max{ 00 seWkLNseW

31
Computational complexity
• Construction and transformation of the graph–
• Find the heaviest path –
• Banded pairwise alignment–
)(NLO
)(NLO
)|(| NLO

32
Discussion
• Choice of k-tuple size– The larger k, the fewer multiplicity for edge
• For Larger N
– The smaller k, the k is not unique in the sequence• For small N : get high multiplicity
– Estimate k using L.D.T
• Graph transformation may lose information– unsafe transformation, lose of similarity information
• Arbitrary scoring function

33
Local multiple alignment
• Difficulty– Locations, sizes, structures ,number of conserved
regions
• Local multiple alignment– PIMA, MACW,DIALIGN
• Subproblem of local alignment – Motif finding
• Gibbs motif sampler • Ex: MEME • Limitation
– size of data , the length of motif

34
Local multiple alignment
• Another Specific Problem of local alignment– Entire Genome Sequence– Large size sequence comparsion
• Local Alignment– Using pairwise sequence comparison
• Not accurate, error accumulate , ruin final result
– Comparing each sequence with a DB• Find only conserved regions

35
Local Alignment Algorithm
1. Construct de Bruijn graph by overlapping k-tuple
2. Cut “thin” edge by estimating the statistical significance of each edge with a Poisson heuristic
3. Resolve cycles in graph4. Extract a heaviest path as the consensus5. Construct and output a multiple alignment from
pairwise alignment6. Declump de Bruijn graph and return to step 5
to find other patterns
36
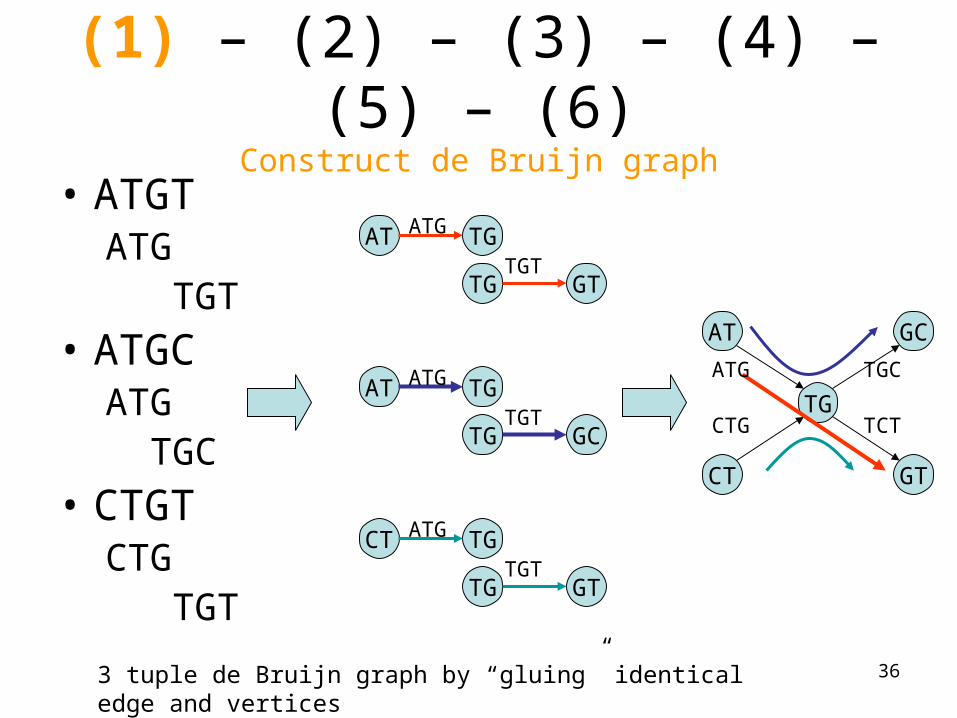
(1) – (2) – (3) – (4) – (5) – (6)Construct de Bruijn graph
• ATGTATG TGT
• ATGCATG TGC
• CTGTCTG TGT
AT TG
TG GT
ATG
TGT
AT TG
TG GC
ATG
TGT
CT TG
TG GT
ATG
TGT
AT
CT
TG
GT
GC
3 tuple de Bruijn graph by “gluing” identical edge and vertices
TCT
TGC
CTG
ATG
37
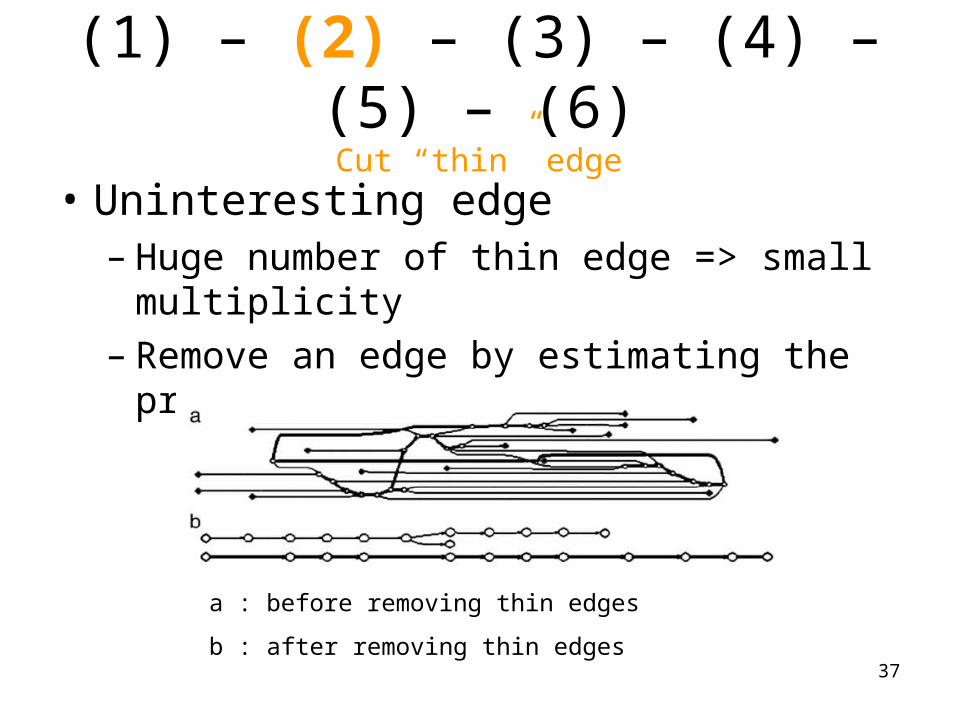
(1) – (2) – (3) – (4) – (5) – (6)Cut “thin” edge
• Uninteresting edge– Huge number of thin edge => small multiplicity– Remove an edge by estimating the probability
a : before removing thin edges
b : after removing thin edges

38
(1) – (2) – (3) – (4) – (5) – (6)Resolve cycles in graph
• Tandem repeat– Repeat present as a cycle in the graph
• Ambiguous to determine how many time a cycle
– Solve the superpath solution

39
(1) – (2) – (3) – (4) – (5) – (6) Extract a heaviest path as the consensus
• Heaviest path – Shortest path algorithm with negative edge
• Using topological sort
– Cost linear time (acyclic graph)

40
(1) – (2) – (3) – (4) – (5) – (6) Construct and output a multiple alignment
• Find the consensus– Banded version of local pairwise alignment– Declumping algorithm to find segments similar
to the consensus • Optimal alignment has p > p0
• P0 : assume the Poisson distribution
1)1)(max( 0pTP i

41
(1) – (2) – (3) – (4) – (5) – (6) Construct and output a multiple alignment
• Declumping algorithm
AT
AT
ATC ㅡㅡ AA T T CGC
ATCT T AA ㅡㅡ CGC
ATC A A T T ㅡㅡ CGC
ATC ㅡㅡ T T A A CGC

42
(1) – (2) – (3) – (4) – (5) – (6) Declumping graph
• Remove information of previously output local alignments
• Allows additional patterns • Ex: XYZ PYQ
– Do not remove the edge of Y– Reduce its multiplicity
• Repeat– Finding consensus – consensus alignment –
decumpling graph• Until no significant local alignment are left

43
Significance Estimation
• Estimate the P value of local multiple alignment– Remove thin edge formed by random matches– Rank multiple outputs by statistical significance
• Estimate minimum multiplicity of mutations free edge– Local alignment is complicated than in the global case
• Position and the orders of conserved regions in each sequences

44
Poisson clumping heuristic
• Pairwise alignment
– H is the optimal clump score
– p(2) is the probability that two letters are identical
– L1,L2 are the adjusted lengths of two sequences
– L1,L2 p(2)x is an approximation to the expected
number of clumps with score
• Multiple alignment
xpLL
exHP )2(211)(
)(1
)(),( nxn
i i pLn
Nbyxnh
n
in
xi pL
n
NehHp
1)())((1)(
,

45
Computation Efficiency
• k : tuple size
l : pattern length found in each iterations
N : number of sequences
L : average sequence length
• Time– Graph construction and transformation– Pairwise alignment with declumping
• Space
)(kNLO)(NLlO
)( 2lkNLO
The size of alignment matrix

46
Discussion
• Tuple size(10~20)
• How to detect true pattern other than concatenation different pattern
• Current version focus on DNA not protein sequence

47
Assignment #5
• When we using the de Bruijn graph in Eulerain graph, we just adopt in DNA because its characters are consist of four nucleotide like A,C,G,T. Give me an efficient algorithm to get the multiple sequence alignment for adopting protein (it is 20 characters) using the graph.– Hint: Not use de Bruijn graph and Eulerian
graph, Graph structure is embedded in the dynamic programming algorithm)
If you have question, Contact me [email protected]

48
Reference • [1] “A new algorithm for DNA sequence assembly”
by Idury, R., and Waterman,. Journal of Computational Biology. 2, 291–306. (1993)
• [2] “An Eulerian path approach to DNA fragment assembly”. by Pevzner, P.A., Tang, H., and Waterman,Proc. National Academy of Science of USA, PP9748–9753 (1998)
• [3] "An Eulerian path approach to global multiple alignment for DNA sequences" by Y. Zhang and M. Waterman, Journal of Computational Biology 10-6, pp. 803-819 (2003).
• [4] "An Eulerian path approach to local multiple alignment for DNA sequences" by Y. Zhang and M. Waterman, Proc. National Academy of Science of USA 102-5, pp. 1285-1290 (2005).
Related Documents











