Eur. J. Biochem. 214,549-561 (1993) 0 FEBS 1993 Aminotransferases : demonstration of homology and division into evolutionary subgroups Perdeep K. MEHTA, Terence I. HALE and Philipp CHRISTEN Biochemisches Institut der Universitat Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland (Received January 29March 10, 1993) - EJB 93 0145/3 A total of 150 amino acid sequences of vitamin €3,-dependent enzymes are known to date, the largest contingent being furnished by the aminotransferases with 51 sequences of 14 different en- zymes. All aminotransferase sequences were aligned by using algorithms for sequence comparison, hydropathy patterns and secondary structure predictions. The aminotransferases could be divided into four subgroups on the basis of their mutual structural relatedness. Subgroup I comprises aspar- tate, alanine, tyrosine, histidinol-phosphate, and phenylalanine aminotransferases ; subgroup I1 ace- tylornithine, ornithine, o-amino acid, 4-aminobutyrate and diaminopelargonate aminotransferases ; subgroup 111 D-alanine and branched-chain amino acid aminotransferases, and subgroup IV serine and phosphoserine aminotransferases. (N-1) Profile analysis, a more stringent application of profile analysis [Gribskov, M., McLachlan, A. D. and Eisenberg, D. (1987) Proc. Nut1 Acud. Sci. USA 84, 4355-43581, established the homology among the enzymes of each subgroup as well as among all subgroups except subgroup 111. However, similarity of active-site segments and the hydropathy patterns around invariant residues suggest that subgroup 111, though most distantly related, might also be homologous with the other aminotransferases. On the basis of the comprehensive alignment, a new numbering of amino acid residues applicable to aminotransferases (AT) in general is pro- posed. In the multiply aligned sequences, only four out of a total of about 400 amino acid residues proved invariant in all 51 sequences, i.e. Gly(314AT)197, Asp/Glu(340AT)222,Lys(385AT)258 and Arg(562AT)386, the number not in parentheses corresponding to the structure of porcine cytosolic aspartate aminotransferase. Apparently, the aminotransferases constitute a group of homologous proteins which diverged into subgroups and, with some exceptions, into substrate-specific individual enzymes already in the universal ancestor cell. The aminotransferases catalyze the reversible transfer of amino groups from amino acids to 0x0 acids, one of the numerous transformations of amino acids that are performed by vitamin-B,-dependent enzymes. Among the various groups of pyridoxal-5’-phosphate-dependent enzymes, the aminotransferases excel by the highest number of reported amino acid sequences, i.e. one third of the total of about 150 sequences of pyridoxal-5’-phosphate-dependent enzymes known to date. The homology of aspartate, tyrosine and his- tidinol-phosphate aminotransferases has already been re- ported. On alignment of 16 amino acid sequences of these enzymes, 12 amino acid residues, mainly ligands of the coen- zyme or the substrate, proved invariant out of a total of about 400 residues (Mehta et al., 1989). Since the publication of that report, the number of known aminotransferase sequences has doubled. In a continuation of our studies on the evolutionary rela- tionships among vitamin B,-dependent enzymes, all known sequences of aminotransferases were aligned on the basis of sequence similarity, hydropathy patterns and predicted sec- ondary structures. The aligned sequences could be divided into four subgroups. The intra- and inter-subgroup homology of the aminotransferases was quantitatively assessed. Correspondence to P. Christen, Biochemisches Institut der Uni- versitiit Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzer- land Abbreviations. AspAT, aspartate aminotransferase (EC 2.6.1 .l); AlaAT, alanine aminotransferase (EC 2.6.1.2); STAT, tyrosine ami- notransferase (EC 2.6.1 3; HisPAT, histidinol-phosphate amino- transferase (EC 2.6.1.9) ; AcomAT, acetylomithine aminotransferase (EC 2.6.1.11); OmAT, ornithine aminotransferase (EC 2.6.1.13); o- AaAT, o-amino acid aminotransferase (EC 2.6.1.18); GabaAT, 4- aminobutyrate aminotransferase (EC 2.6.1.19); D-A~~AT, D-alanine aminotransferase(EC 2.6.1.21); BcaaAT, branched-chain amino acid aminotransferase (EC 2.6.1.42) ; SerAT, serine aminotransferase (EC 2.6.1.51) ; PSerAT, phosphoserine aminotransferase (EC 2.6.1 S2) ; PheAT, phenylalanine aminotransferase(EC 2.6.1.58) ; DapaAT, dia- minopelargonate aminotransferase (EC 2.6.1.62). DATABASE AND METHODOLOGY Databases The list of aminotransferases with known amino acid se- quence (Table S l ) was compiled by screening the NBRF- PIR (Release 29), Swiss-Prot (Release 18) and EMBL (Re- lease 28) databases as well as the current literature. The amino acid sequence derived from cDNA assigned to valine aminotransferase (Liu et al., 1987) does not seem to be re- lated with the other aminotransferases and has been excluded from the present study.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Eur. J. Biochem. 214,549-561 (1993) 0 FEBS 1993
Aminotransferases : demonstration of homology and division into evolutionary subgroups Perdeep K. MEHTA, Terence I. HALE and Philipp CHRISTEN Biochemisches Institut der Universitat Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
(Received January 29March 10, 1993) - EJB 93 0145/3
A total of 150 amino acid sequences of vitamin €3,-dependent enzymes are known to date, the largest contingent being furnished by the aminotransferases with 51 sequences of 14 different en- zymes. All aminotransferase sequences were aligned by using algorithms for sequence comparison, hydropathy patterns and secondary structure predictions. The aminotransferases could be divided into four subgroups on the basis of their mutual structural relatedness. Subgroup I comprises aspar- tate, alanine, tyrosine, histidinol-phosphate, and phenylalanine aminotransferases ; subgroup I1 ace- tylornithine, ornithine, o-amino acid, 4-aminobutyrate and diaminopelargonate aminotransferases ; subgroup 111 D-alanine and branched-chain amino acid aminotransferases, and subgroup IV serine and phosphoserine aminotransferases. (N-1) Profile analysis, a more stringent application of profile analysis [Gribskov, M., McLachlan, A. D. and Eisenberg, D. (1987) Proc. Nut1 Acud. Sci. USA 84, 4355-43581, established the homology among the enzymes of each subgroup as well as among all subgroups except subgroup 111. However, similarity of active-site segments and the hydropathy patterns around invariant residues suggest that subgroup 111, though most distantly related, might also be homologous with the other aminotransferases. On the basis of the comprehensive alignment, a new numbering of amino acid residues applicable to aminotransferases (AT) in general is pro- posed. In the multiply aligned sequences, only four out of a total of about 400 amino acid residues proved invariant in all 51 sequences, i.e. Gly(314AT)197, Asp/Glu(340AT)222, Lys(385AT)258 and Arg(562AT)386, the number not in parentheses corresponding to the structure of porcine cytosolic aspartate aminotransferase. Apparently, the aminotransferases constitute a group of homologous proteins which diverged into subgroups and, with some exceptions, into substrate-specific individual enzymes already in the universal ancestor cell.
The aminotransferases catalyze the reversible transfer of amino groups from amino acids to 0x0 acids, one of the numerous transformations of amino acids that are performed by vitamin-B,-dependent enzymes. Among the various groups of pyridoxal-5’-phosphate-dependent enzymes, the aminotransferases excel by the highest number of reported amino acid sequences, i.e. one third of the total of about 150 sequences of pyridoxal-5’-phosphate-dependent enzymes known to date. The homology of aspartate, tyrosine and his- tidinol-phosphate aminotransferases has already been re- ported. On alignment of 16 amino acid sequences of these enzymes, 12 amino acid residues, mainly ligands of the coen-
zyme or the substrate, proved invariant out of a total of about 400 residues (Mehta et al., 1989). Since the publication of that report, the number of known aminotransferase sequences has doubled.
In a continuation of our studies on the evolutionary rela- tionships among vitamin B,-dependent enzymes, all known sequences of aminotransferases were aligned on the basis of sequence similarity, hydropathy patterns and predicted sec- ondary structures. The aligned sequences could be divided into four subgroups. The intra- and inter-subgroup homology of the aminotransferases was quantitatively assessed.
Correspondence to P. Christen, Biochemisches Institut der Uni- versitiit Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzer- land
Abbreviations. AspAT, aspartate aminotransferase (EC 2.6.1 .l); AlaAT, alanine aminotransferase (EC 2.6.1.2); STAT, tyrosine ami- notransferase (EC 2.6.1 3; HisPAT, histidinol-phosphate amino- transferase (EC 2.6.1.9) ; AcomAT, acetylomithine aminotransferase (EC 2.6.1.11); OmAT, ornithine aminotransferase (EC 2.6.1.13); o- AaAT, o-amino acid aminotransferase (EC 2.6.1.18); GabaAT, 4- aminobutyrate aminotransferase (EC 2.6.1.19); D-A~~AT, D-alanine aminotransferase (EC 2.6.1.21); BcaaAT, branched-chain amino acid aminotransferase (EC 2.6.1.42) ; SerAT, serine aminotransferase (EC 2.6.1.51) ; PSerAT, phosphoserine aminotransferase (EC 2.6.1 S2) ; PheAT, phenylalanine aminotransferase (EC 2.6.1.58) ; DapaAT, dia- minopelargonate aminotransferase (EC 2.6.1.62).
DATABASE AND METHODOLOGY
Databases
The list of aminotransferases with known amino acid se- quence (Table S l ) was compiled by screening the NBRF- PIR (Release 29), Swiss-Prot (Release 18) and EMBL (Re- lease 28) databases as well as the current literature. The amino acid sequence derived from cDNA assigned to valine aminotransferase (Liu et al., 1987) does not seem to be re- lated with the other aminotransferases and has been excluded from the present study.

550
Sequence comparison and alignment Standard alignment algorithms, e.g. the Needleman-
Wunsch algorithm (Needleman and Wunsch, 1970), and more sensitive sequence comparison methods (BESTFIT, PLOTSIMILARITY, PROFTLEGAP, etc.) served to align the amino acid sequences with a multiple sequence editor de- scribed previously (Mehta et al., 1989). For final optimization by hand, hydropathy plots (Kyte-Doolittle algorithm ; Kyte and Doolittle, 1982) and secondary structure predictions (Chou-Fasman algorithm; Chou and Fasman, 1974), as in- cluded in the UWGCG sequence analysis software package, version 7.0 (Devereux et al., 1984) were used.
Profile analysis Profile analysis (Gribskov et al., 1987) is a database-
searching program based on a position-specific scoring table, the profile, which is constructed from a set of aligned homol- ogous amino acid sequences, the so-called probe sequences, and a comparison table derived from the Dayhoff mutation data matrix (Dayhoff and Schwartz, 1978). The profile is thus specific for a certain group of related proteins and attri- butes position-specific score values to any of the 20 amino acids and position-specific penalties to a deletion or insertion at all positions along the sequence to be tested for relatedness with the probe sequences. The overall score value of the target sequence corresponds to the algebraic sum of the score values attributed to all its residues and gaps and is normal- ized for length of the polypeptide chain. The 2 score corre- sponds to the difference between the score of the target se- quence and the mean score of unrelated sequences expressed in terms of the standard deviation. The mean Z score for sequences unrelated to the profile thus is 0.0 and the standard deviation is 1 .O (for illustration, see Figs 2 and 3). A 2 score value is considered meaningful if it is higher than three stan- dard deviations. A score value higher than five standard devi- ations indicates a definitive relationship between the target sequence and the probe sequences (Gribskov et al., 1988).
Each enzyme used for the construction of the profile was attributed one vote regardless of the number of sequences available. This vote was subdivided among the sequences, the vote weight assigned to an individual sequence being inversely proportional to its degree of identity with the other sequences of the same enzyme. This procedure avoided an overrepresentation of closely related sequences. For instance, in the case of TyrAT the following vote weights were as- signed: TyrAT rat 0.25, human 0.25, Escherichia coli 0.50.
RESULTS Comparison and alignment of sequences
Analysis of 51 amino acid sequences of altogether 14 different aminotransferases with standard comparison and alignment programs failed to detect homology among the dif- ferent aminotransferases except in a few cases. Comparison of the sequences with BESTFIT and the combined programs COMPARE and DOTPLOT, which reveal similar segments in two sequences on a two-dimensional matrix display, indi- cated for most enzymes no more than a few short conserved stretches. In general, meaningful alignment can be obtained with standard programs only if sequence identity exceeds 30%. The majority of the aminotransferases, however, dis- play lower degrees of mutual overall sequence identity (for
degrees of identity and similarity of the aligned sequences, see Table S2). Therefore, we optimized the alignments man- ually using hydropathy plots for primary guidance and sec- ondary structure predictions as supportive evidence. Profile analysis served as a criterion for the quality of the alignment. Increased Z scores, particularly for distantly related se- quences that had been incorporated into the probe, indicated their improved alignment, i.e. optimization of the profile (Gribskov et al., 1988).
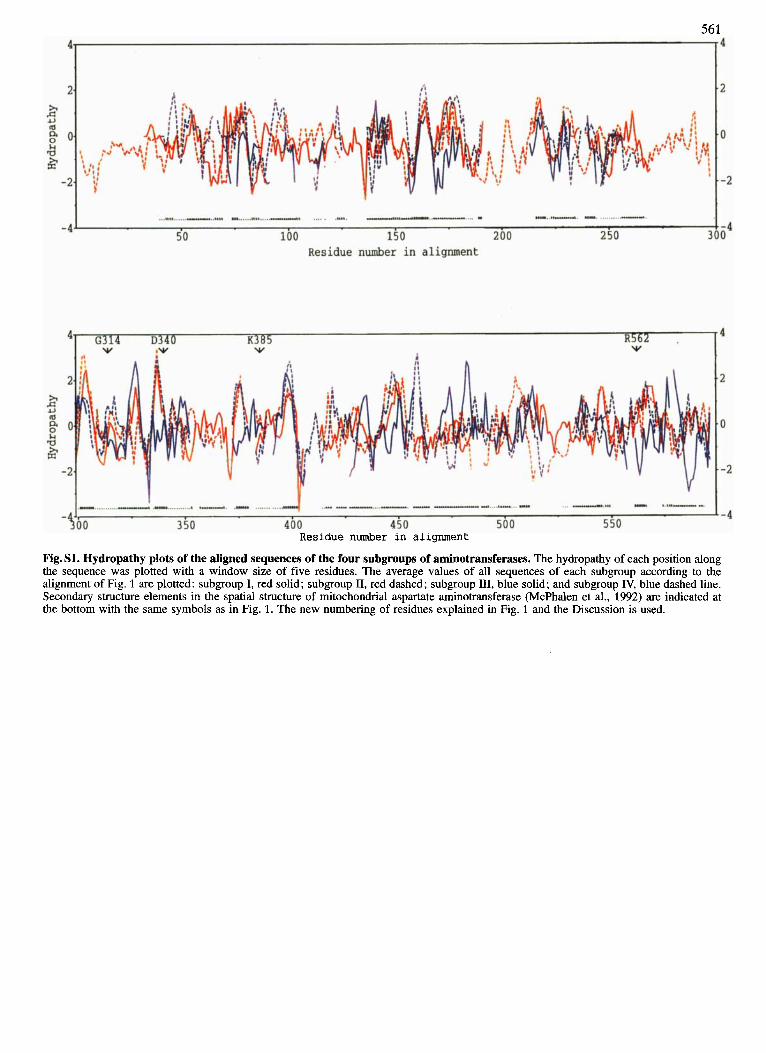
Iterative comparison and superpositioning of hydropathy plots and optimization of the alignment by introduction of gaps in variable regions led to the comprehensive alignment of all aminotransferase sequences. The average hydropathy plots of the four subgroups are shown in Fig. S1, the corre- sponding alignment of 32 selected sequences is presented in Fig. 1. In the alignment of all sequences, several invariant hydrophobic and hydrophilic stretches become obvious. In particular, conserved hydrophobic regions correspond to the seven-stranded #?-pleated sheet as defined in the refined three-dimensional structure of aspartate aminotransferase (McPhalen et al., 1992). Only four residues are conserved at corresponding positions in all sequences. In the case of Gly197, D-alanine aminotransferase is an exception in con- taining Ala at this position.
Subgroups of aminotransferases The 14 aminotransferases can be divided into four sub-
groups (Table 1) on the basis of the degree of their mutual similarities in primary structure (Table S2). For every en- zyme, the average degree of sequence identity with the other members of its own subgroup is higher than the average identity with any other subgroup.
Test of intra-subgroup homology of aminotransferases The alignment within each subgroup was quantitatively
objectified by (N-1) profile analysis, a modified application of the profile analysis of Gribskov et al. (1987). Details of this procedure are given under Database and Methodology. The term (N-l), where N is the number of enzymes, is used to indicate that all sequences of a given subgroup with the exception of the sequence(s) of one member enzyme were used to construct the profile. In any (N-1) profile, no infor- mation on the omitted target sequence(s), such as residue exchangeability and gaps, was incorporated into the align- ment of the probe sequences. The profile used for the analy- sis shown as an example in Fig. 2 was constructed from the aligned sequences of aspartate, alanine, tyrosine and phenyl- alanine aminotransferases, while none of the sequences of histidinol-phosphate aminotransferase were included into the probe. This (N-1) profile of four of the five members of sub- group I attributes higher scores to all histidinol-phosphate aminotransferase sequences than to any other of the 20000 proteins in the database with the exception of the probe se- quences themselves (Fig. 2). All score values of histidinol- phosphate aminotransferase sequences are clearly higher than the significance limit of 5 (see Database and Methodology) and thus indicate that omitted histidinol-phosphate amino- transferase is uniquely related to the enzymes used for the construction of the profile. Similar results were observed with the other four (N-1) profiles constructed within sub- group I by omitting the sequence(s) of every member enzyme in turn. The same procedure confirmed for each of the other subgroups that its members are more closely related among

551
Table 1. Subgroups of aminotransferases. The aminotransferases included are those with known amino acid sequences.
Sub- Enzyme ECno. Main substrates Structure of amino acid substrates group
amino acid 0x0 acid
I
I1
I11
IV
AspAT AlaAT TyrAT HisPAT PheAT
AcornAT
O d T w-AaAT GaBaAT DapaAT
D-AI~AT BcaaAT
SerAT PSerAT
2.6.1.1 2.6.1.2 2.6.1.5 2.6.1.9 2.6.1.58
2.6.1.11
2.6.1.13 2.6.1.18 2.6.1.19 2.6.1.62
2.6.1.21 2.6.1.42
2.6.1.51 2.6.1.52
L-aspartate L-alanine L-tyrosine L-histidinol-P L-phenylalanine
N-acet yl-L- ornithine L-omithine /3-alanine 4-aminobutyrate 7,8-diamino- pelargonate
L-serine
serine 3-phospho-L-
2-oxoglutarate 2-oxoglutarate 2-oxoglutarate 2-oxoglutarate pyruvate
2-oxoglutarate
2-oxoglutarate pyruvate 2-oxoglutarate S-adenosyl-4- methylthio-2- oxobutanoate
2-oxoglutarate 2-oxoglutarate
pyruvate 2-oxoglutarate
Coo' C G e I H-C-Nw HsH*-C-H
P H I
yc' CH 'CH,
coo' coo' I I
hN'-C-H H,W-C-H
CH20H dH,O-Pq*
co(*
P :.
themselves than with any member of any other subgroup (Ta- ble 2).
Test of inter-subgroup homology of aminotransferases Again, (N-1) profile analysis served to validate the inter-
subgroup homology and thus the significance of the align- ment among the subgroups. For the example shown in Fig. 3, a profile was generated from all aligned sequences of sub- groups I, I11 and IV while all sequences of subgroup 11 were omitted. This (N-1) profile clearly pulled all sequences of all five members of the omitted subgroup I1 out of the rest of the database. Analogous (N-1) profile analyses were con- ducted for subgroups I, 111 and IV. In all cases, with the exception of the sequences of subgroup 111, at least one of the sequences of the omitted subgroup scored higher than any protein not dependent on vitamin B6 in the database (Table 3) and exhibited a 2 score higher than 3 which is indicative of distant relationship (see Database and Methodology).
The relationship among subgroups I, I1 and IV is sup- ported by the following findings. The comprehensive sub- group I profile selected human serine aminotransferase of subgroup IV with a Z score of 3.3, the comprehensive sub-
group I1 profile selected aspartate aminotransferase of Sulfo- lobus solfataricus of subgroup I with a Z score of 4.3 and the (subgroup 11 minus 4-aminobutyrate aminotransferase) profile selected aspartate aminotransferase of S. solfataricus as well as histidinol-P aminotransferase of S. coelicolor, both of subgroup I, with 2 scores of 5.1 and 4.4, respectively.
Though subgroup 111, which comprises branched-chain amino acid and D-alanine aminotransferases, does not meet the criterion of (N-1) profile analysis, other evidence sug- gests that this subgroup might also be related with the other aminotransferases (see Discussion).
DISCUSSION
Alignment of sequences
The homology of all aminotransferases is not prima facie evident and is also not recognizable by standard algorithms for sequence comparison. However, the present study making use of hydropathy patterns, secondary structure predictions and profile analyses allowed the alignment of all aminotrans- ferase sequences and their division into four subgroups. For
Fig. 1. Aligned amino acid sequences of the four subgroups of aminotransferases. The abbreviations of the sources of the enzymes are explained in Table S1. Sequences which are more than 80% identical with one of those presented were excluded. At the top of each block a newly proposed numbering of residues applicable to aminotransferases in general is indicated (see Discussion). The numbering of residues that corresponds to the sequence of pig cytosolic aspartate aminotransferase (Ovchimikov et al., 1973) is given at the bottom. Secondary structure elements in the spatial structure of mitochondria1 aspartate aminotransferase (McPhalen et al., 1992) are indicated at the bottom of each block: a, a-helix; B, b-strand; t, 8-turn. Gaps, represented by dots, were introduced to optimize the alignment (see Database and Methodology). Residues are classified (Liithy et al., 1991) and colored as follows: hydrophobic aromatic, F, W, Y, dark brown; hydrophobic not aromatic, A, I, L, M, V, light brown; ambivalent (polar but uncharged or hydrophobic but small), C, G, P, S, T, yellow; hydrophilic basic, H, K, R, blue; and hydrophilic acidic and their amides, D, E, N, Q, red. The four invariant residues are marked by filled circles. In many aminotransferases the pyridoxal-5'-phosphate-binding Lys(385AT) has been identified by chemical analysis.

552
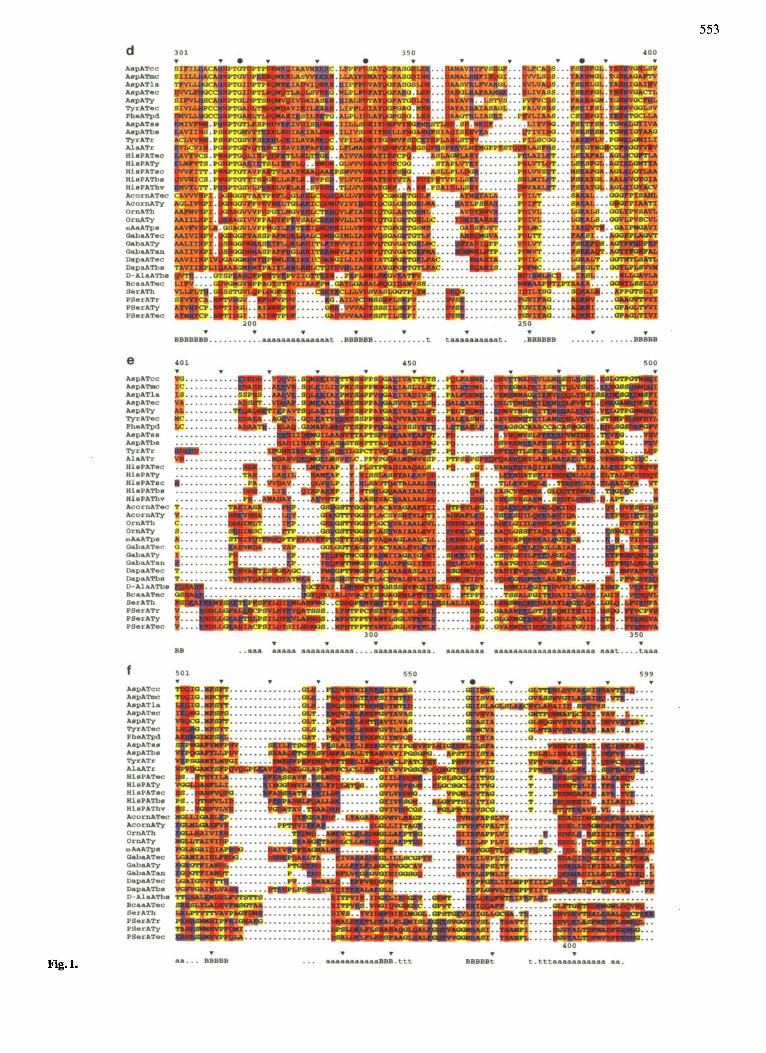
553
Fig. 1.
554
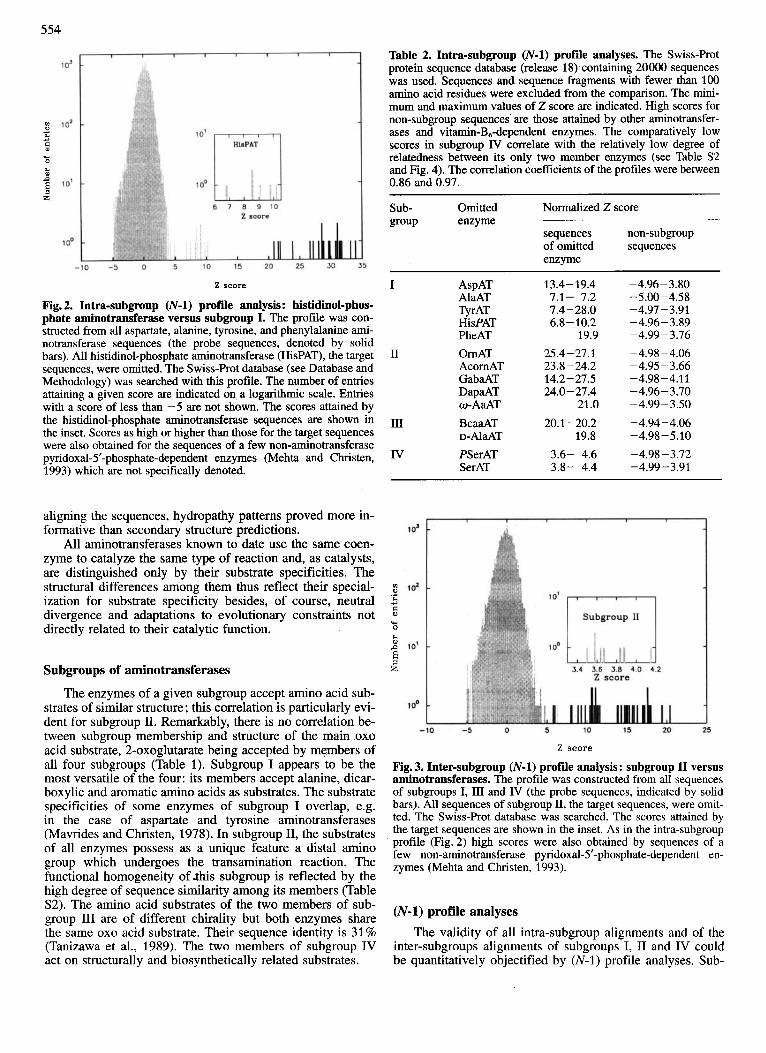
Table 2. Intra-subgroup (N-1) profile analyses. The Swiss-Rot protein sequence database (release 18) containing 20000 sequences was used. Sequences and sequence fragments with fewer than 100 amino acid residues were excluded from the comparison. The mini- mum and maximum values of 2 score are indicated. High scores for non-subgroup sequences are those attained by other aminotransfer- ases and vitamin-B,-dependent enzymes. The comparatively low scores in subgroup IV correlate with the relatively low degree of relatedness between its only two member enzymes (see Table S2 and Fig. 4). The correlation coefficients of the profiles were between 0.86 and 0.97.
Sub- Omitted Normalized Z score group enzyme
sequences non-subgroup of omitted sequences enzyme
2 score
Fig. 2. Intra-subgroup (N-1) profile analysis : histidinol-phos- phate aminotransferase versus subgroup I. The profile was con- structed from all aspartate, alanine, tyrosine, and phenylalanine ami- notransferase sequences (the probe sequences, denoted by solid bars). All histidinol-phosphate aminotransferase (HisPAT), the target sequences, were omitted. The Swiss-Rot database (see Database and Methodology) was searched with this profile. The number of entries attaining a given score are indicated on a logarithmic scale. Entries with a score of less than -5 are not shown. The scores attained by the histidinol-phosphate aminotransferase sequences are shown in the inset. Scores as high or higher than those for the target sequences were also obtained for the sequences of a few non-aminotransferase pyridoxal-5’-phosphate-dependent enzymes (Mehta and Christen, 1993) which are not specifically denoted.
I AspAT AlaAT TyrAT HisPAT PheAT
I1 OrnAT AcornAT GabaAT DapaAT
I11 BcaaAT D-A~~AT
IV PSerAT SerAT
o-AaAT
13.4- 19.4 7.1- 7.2 7.4-28.0 6.8- 10.2
19.9 25.4- 27.1 23.8-24.2 14.2-27.5 24.0- 21.4
21.0 20.1 -20.2
19.8 3.6- 4.6 3.8- 4.4
- 4.96 - 3.80 - 5 .OO -4.58 -4.97 - 3.91 -4.96-3.89 -4.99-3.76 -4.98-4.06 -4.95 - 3.66 -4.98 -4.1 1 -4.96-3.70 -4.99-3.50 -4.94-4.06 -4.98-5.10 -4.98-3.72 -4.99-3.91
aligning the sequences, hydropathy patterns proved more in- formative than secondary structure predictions.
All aminotransferases known to date use the same coen- zyme to catalyze the same type of reaction and, as catalysts, are distinguished only by their substrate specificities. The structural differences among them thus reflect their special- ization for substrate specificity besides, of course, neutral divergence and adaptations to evolutionary constraints not directly related to their catalytic function.
Subgroups of aminotransferases
The enzymes of a given subgroup accept amino acid sub- strates of similar structure ; this correlation is particularly evi- dent for subgroup II. Remarkably, there is no correlation be- tween subgroup membership and structure of the main 0x0 acid substrate, 2-oxoglutarate being accepted by members of all four subgroups (Table 1). Subgroup I appears to be the most versatile of the four: its members accept alanine, dicar- boxylic and aromatic amino acids as substrates. The substrate specificities of some enzymes of subgroup I overlap, e.g. in the case of aspartate and tyrosine aminotransferases (Mavrides and Christen, 1978). In subgroup 11, the substrates of all enzymes possess as a unique feature a distal amino group which undergoes the transamination reaction. The functional homogeneity of this subgroup is reflected by the high degree of sequence similarity among its members (Table S2). The amino acid substrates of the two members of sub- group I11 are of different chirality but both enzymes share the same 0x0 acid substrate. Their sequence identity is 31% (Tanizawa et al., 1989). The two members of subgroup IV act on structurally and biosynthetically related substrates.
Z score
Fig. 3. Inter-subgroup (N-1) profile analysis: subgroup I1 versus aminotransferases. The profile was constructed from all sequences of subgroups I, I11 and IV (the probe sequences, indicated by solid bars). All sequences of subgroup 11, the target sequences, were omit- ted. The Swiss-Prot database was searched. The scores attained by the target sequences are shown in the inset. As in the intra-subgroup profile (Fig. 2) high scores were also obtained by sequences of a few non-aminotransferase pyridoxal-5’-phosphate-dependent en- zymes (Mehta and Christen, 1993).
(N-1) profile analyses The validity of all intra-subgroup alignments and of the
inter-subgroups alignments of subgroups I, I1 and IV could be quantitatively objectified by (N-1) profile analyses. Sub-

555
Table 3. Inter-subgroup (N-1) profile analyses. The same database as in Table 2 was used. The minimum and maximum values of 2 score are indicated. High scores for non-aminotransferase sequences are those attained by other vitamin-B,-dependent enzymes. Note that in a11 subgroups except III at least one sequence achieved a higher score than any sequence of proteins not dependent on vitamin B,. The correlation coefficients of the profiles were between 0.74 and 0.85.
Omitted Normalized 2 score subgroup
sequences sequences of non- of omitted vitamin-B,-dependent subgroup proteins
Subgroup I 2.14-4.00 -4.92-2.95 Subgroup I1 3.50-4.15 -4.97-3.40 Subgroup 111 0.02-0.85 -4.99-3.89 Subgroup IV 2.93-3.50 -4.93 -3.06
Table 4. Invariant residues in aminotransferases. The residues are numbered according both to the numbering of cytosolic pig aspartate aminotransferase (Ovchinnikov et al., 1973) and to the new number- ing of the multiply aligned aminotransferases (Fig. 1 and Discus- sion). Their specific roles are indicated as they have been described on the basis of the high-resolution spatial structure of aspartate ami- notransferase (Kirsch et al., 1984).
Residue Specific role in aspartate aminotransferase
G197(314AT) participates in turn 194-197 located at domain interface
D/E222(340AT) salt bridge/H-bond to N1 of pyridoxal 5‘-phosphate
K258(385AT) Schiff base with pyridoxal5’-phosphate R386(562AT) salt bridge/€€-bond with a-carboxylate
group of substrate
group I11 proved an exception, receiving only insignificant score values in the inter-subgroup profile analysis. Its se- quences proved also most difficult to align with the other aminotransferases. Only three sequences are available to date for this subgroup. In the case of distantly related proteins, alignment is the more difficult the fewer sequences are avail- able. In the alignment proposed for subgroup I11 (Fig. l ) , the homology to other aminotransferases is not clearly evident except in a short segment around the pyridoxal-5‘-phosphate- binding lysine residue between branched-chain amino acid aminotransferase and E. coli acetylornithine aminotransfer- ase. The polypeptide chains of both members of subgroup I11 are the shortest of all aminotransferases, being 25% shorter than the average length of approximately 400 amino acid residues of the other aminotransferases. The alignment of branched-chain amino acid aminotransferase indicates that the small domain as, defined in aspartate aminotransferase (residues 15-47 and 326-410; Picot et al., 1991), is either absent or of radically different structure. The only link of 1)- alanine aminotransferase is with branched-chain amino acid aminotransferase, there is no apparent relationship with any other aminotransferase. The four invariant residues (Table 4) are also shared by subgroup 111. Its hydropathy profile is similar to those of the other aminotransferases within several p-strand regions (Figs S1 and 1). However, the determination of the three-dimensional structures of the enzymes or the application of novel algorithms for sequence analyses are re-
quired to settle conclusively the question of homology of subgroup I11 with the other aminotransferases.
If the three-dimensional structures of w-amino acid ami- notransferase (Watanabe et al., 1989, 1991), which on the basis of sequence similarity with omithine aminotransferase belongs to subgroup 11, and of phosphoserine aminotransfer- ase (for preliminary data on the three-dimensional structure, see Stark et al., 1991), which is a member of subgroup IV, are compared with the three-dimensional structure of aspartate aminotransferase of subgroup I, the folding patterns of the large domains of the three enzymes are found to be quite similar. This is consistent with the conclusion reached on the basis of sequence comparison that subgroups I, I1 and IV are homologous.
Invariant residues In the following discussion, a newly introduced number-
ing of amino acid residues applicable to aminotransferases (AT) in general is used (Fig. 1). The number of an amino acid residue is given in parentheses and is followed by the letters AT, e.g. Lys(385AT) denotes the pyridoxal-5’-phos- phate-binding lysine residue in the general numbering sys- tem. This residue corresponds to Lys258 in porcine cytosolic aspartate aminotransferase (Ovchinnikov et al., 1973). A par- ticular residue can thus be denoted by both its sequence- specific number and its AT number, e.g. Lys258(385AT) for porcine cytosolic aspartate aminotransferase.
In the alignment of 51 amino acid sequences of all four subgroups only four residues remain invariant (Table 4), in comparison to 11 invariant residues in subgroup I (5 en- zymes, 31 sequences), 19 invariant residues in subgroup I1 (5 enzymes, 11 sequences), 82 invariant residues in subgroup I11 (2 enzymes, 3 sequences) and 25 invariant residues in subgroup IV (2 enzymes, 6 sequences). With the exception of the four generally invariant residues, the residues invariant in subgroup I are not conserved in any of the other sub- groups. The number of known sequences in the other sub- groups, however, is too small to identify unambigously sub- group-specific invariant residues. Conclusive information on the structural or functional role of invariant residues, with the exception of pyridoxal-5’-phosphate-binding Lys(385AT), is as yet solely available for aspartate aminotransferase, one of the five members of subgroup I and the only enzyme out of a total of 14 aminotransferases with both spatial and primary structures known. The three-dimensional structures of other enzymes, i.e. w-amino acid (Watanabe et al., 1989) and pho- sphoserine aminotransferase (Stark et al., 1991), have only been reported in incomplete form; their primary structure and the assignment of residues to features of the spatial structure, respectively, have not been published as yet.
In spite of the addition of two new enzymes, i.e. alanine and phenylalanine aminotransferases, and 14 more se- quences, the first alignment of 16 sequences of three en- zymes of subgroup I, where 12 amino acid residues were reported invariant (Mehta et al., 1989), has remained valid and has retained all but one invariant residue, i.e. Ala(445AT) which had no defined role. The average identity of aspartate aminotransferases in pair-wise alignment de- creased from 49% to 41% on inclusion of aspartate amino- transferase of the archaebacterium Sulfolobus solfaturicus. Aspartate aminotransferase of S. solfuturicus is more closely related with the enzyme of the eubacterial thermophilic Bu- cillus species (33% identity) than to any other eubacterial or eucaryotic aspartate aminotransferase (average identity
556
_I -1 I I I I I
-1 I I
I I I I I I I l l I I I
_I I I I 1 I i i I I I . .
I I I -1 I ~, I
I I I -1 I I I 1-1 I I 1 I I I I I I I I I I I I I I -1 I I I I I I I I
_I I I I 1 I 1 I 1 I I I I I 1 I I I I
I 1 I
I I -1 I I I I I 11 I I 1 I I I I I I I I I I I I I I I I I I I I I I I I I I I 1 1 I I I I I I I l l I- I I I I I I I I I I I I I IV I I I- I I I I- I I I I I I I I 111
AspATcc
ArpATmc
A.pATy
AspATla
ArpATec
TyrATec
PheATpd
AspATss
A.pATb8
TyrATr
AIaATr
HlsPATec
HlrPATy
HIsPATsc
HIsPATbs
HlrPAThv
Acorn ATec
AcomATy
QabaATec
OrnATh
OrnATy
DapaATec
DapaATb.
o-APATPs
QabaATy
GabaATan
SerATh
PSerATr
PSwATye
PSerATec
D-AlaATbs
BcaaATec
I-I-I-I-I-I-I-I- 8 0 . 0 71 .0 6 4 . 0 56.0 48
.I-I-I- .o 40 .0
- 24
. I - I - I - .o 16.0
.I-I-I-I 8 . 0 0 . 0
Frodon of dlr8lmllar amlno add W8ldW8
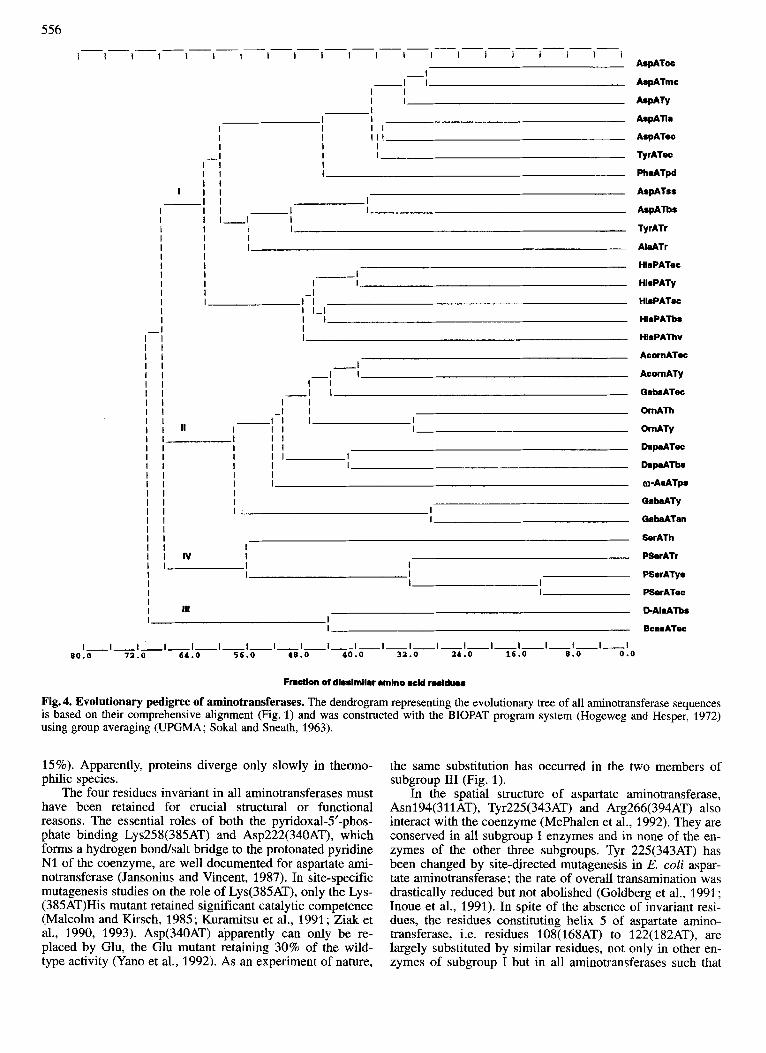
Fig. 4. Evolutionary pedigree of aminotransferases. The dendrogram representing the evolutionary tree of all aminotransferase sequences is based on their comprehensive alignment (Fig. 1) and was constructed with the BIOPAT program system (Hogeweg and Hesper, 1972) using group averaging (UPGMA; Sokal and Sneath, 1963).
15%). Apparently, proteins diverge only slowly in thermo- philic species.
The four residues invariant in all aminotransferases must have been retained for crucial structural or functional reasons. The essential roles of both the pyridoxal-5’-phos- phate binding Lys258(385AT) and Asp222(340AT), which forms a hydrogen bondsalt bridge to the protonated pyridine N1 of the coenzyme, are well documented for aspartate ami- notransferase (Jansonius and Vincent, 1987). In site-specific mutagenesis studies on the role of Lys(385AT), only the Lys- (385AT)His mutant retained significant catalytic competence (Malcolm and Kirsch, 1985; Kuramitsu et al., 1991; Ziak et al., 1990, 1993). Asp(340AT) apparently can only be re- placed by Glu, the Glu mutant retaining 30% of the wild- type activity (Yano et al., 1992). As an experiment of nature,
the same substitution has occurred in the two members of subgroup I11 (Fig. 1).
In the spatial structure of aspartate aminotransferase, Asn194(311AT), Tyr225(343AT) and Arg266(394AT) also interact with the coenzyme (McPhalen et al., 1992). They are conserved in all subgroup I enzymes and in none of the en- zymes of the other three subgroups. Tyr 225(343AT) has been changed by site-directed mutagenesis in E. coli aspar- tate aminotransferase ; the rate of overall transamination was drastically reduced but not abolished (Goldberg et al., 1991 ; Inoue et al., 1991). In spite of the absence of invariant resi- dues, the residues constituting helix 5 of aspartate amino- transferase, i.e. residues 108(168AT) to 122(182AT), are largely substituted by similar residues, not only in other en- zymes of subgroup I but in all aminotransferases such that

557
the respective segments of the hydropathy profiles are super- imposable (Fig. Sl) .
The importance of invariant Gly(314AT) and Arg- (562AT) had not been fully recognized previously. In aspar- tate aminotransferase, Gly197(314AT) lies at the interface of the coenzyme-binding and the small domain. This residue might not be substitutable in aminotransferases acting on L- amino acids because residues of another type might interfere with the folding of the polypeptide chain or with essential syncatalytic shifts in the open -closed conformational equi- librium of the enzyme (Picot et al., 1991). In D-alanine ami- notransferase, which is the most distantly related member of the aminotransferase family, Ala appears to be tolerated in this position. Arg386(562AT) binds the a-carboxylate group of the substrate (Kirsch et al., 1984). The substrates of all aminotransferases possess an a-anionic group, i.e. carboxyl- ate or phosphate in the case of histidinol-phosphate amino- transferase. Recently, Arg386(562AT) of aspartate amino- transferase has been replaced by a Lys, His, Tyr or Phe resi- due (Inoue et al., 1989; Danishefsky et al., 1991). All mu- tants showed drastically reduced enzymic activity, even a Lys residue proved an inadequate substitute for Arg386(562AT). Pyridoxal-5’-phosphate-dependent enzymes catalyze, among others, reactions that involve cleavage of any of the three non-C-N bonds at Ca of the coenzyme-substrate adduct. An important contribution to reaction specificity is thought to be provided by orienting the bond to be cleaved orthogonal to the plane of the coenzyme-substrate n system, thus facilita- ting its labilization by overlap of the 0 and R orbitals (Duna- than, 1966). In aspartate aminotransferase, the Ca-H bond has indeed been found in this orientation (Kirsch et al., 1984; McPhalen et al., 1992). Arg(562AT), by binding the a-car- boxylate group of the substrate, determines to a large extent the orientation of the substrate moiety relative to the plane of the coenzyme-substrate imine. The complete conservation of Arg(562AT) among all aligned sequences of aminotrans- ferases might relate to this particular mechanistic feature. It is to be noted that the enzymes of subgroup 11, which act on the o-amino groups of their substrates, also accept a-amino acids as substrates (Table 1).
Evolutionary pedigree In spite of the low degree of similarity of the amino acid
sequences of the various aminotransferases, the present study shows unequivocally that they all, with the possible excep- tion of subgroup 111, originate from a single universal ances- tor protein. The large differences in their sequences point to very early gene duplications and subsequent specialization for substrate specificity. The evolutionary pedigree (Fig. 4) drawn up for all sequences of all species clearly shows the clustering into the four subgroups with subgroup III being most distant from the other three. The homology of the two archaebacterial sequences, i.e. aspartate aminotransferase of S. solfataricus and histidinol-phosphate aminotransferase of Haloferax volcanii, with the respective enzymes of both pro- caryotes and eucaryotes indicates that the subgroups diverged already in the universal ancestor cell before the emergence of the three biological kingdoms. In general, the evolutionary branching within the subgroups corresponds with the special- ization of the enzymes for substrate specificity. There are two exceptions. First, in subgroup I, the aspartate amino- transferases of archaebacterial S. solfataricus and procaryotic Bacillus species are more closely related to tyrosine amino- transferase of rat than to the aspartate aminotransferases of
other procaryotic and of eucaryotic species (Cubellis et al., 1989). Similarly in subgroup II,4-aminobutyrate aminotrans- ferase of E. coli is closer to all other members of subgroup I1 than to the 4-aminobutyrate aminotransferases of yeast and Aspergillus nidulans. Apparently, in both subgroup I and I1 the specialization for substrate specificity occurred, in part, after the divergence of the three biological kingdoms. In fact, the substrate specificities of aspartate aminotransferase and tyrosine aminotransferase overlap to some extent (Mavrides and Christen, 1978).
We are grateful to Drs Peter Rice, Julie Thompson and Guido Dorigo for their contributions in composing the colored figures. We are also indebted to Dr. Jaap Heringa and Mr. Frederick W. Alexan- der for their assistance and to Prof. J. N. Jansonius and Dr. P. Argos for helpful discussions. This work was supported in part by the Swiss National Science Foundation (grant 3 1.27975-0.89).
REFERENCES Andr6, B. & Jauniaux, J. C. (1990) Nucleic Acids Res. 18, 3049. Barra, D., Martini, P., Montarani, G., Doonan, S. & Bossa, F. (1979)
Bartsch, K., von Johnn-Marteville, A., & Schulz, A. (1990) J. Bucte-
Bousquet-Lemercier, B., Pol, S., PavC-Preux, M., Hanoune, J. &
Carlomagno, M. S., Chiariotti, L., Alifano, P., Nappo, A. G. &
Chou, P. Y. & Fasman, G. D. (1974) Biochemistry 13,222-245. Conover, R. K. & Doolittle, W. F. (1990) J. Bacteriol. 172, 3244-
3249. Cronin, V. B., Maras, B., Barra, D. K. & Doonan, S . (1991) Bio-
chem. J. 277, 335-340. Cubellis, M. V., Rozzo, C., Nitti, G., Amone, M. I., Marino, G. &
Sannia, G. (1989) Eul: J. Biochem. 186, 375-381. Danishefsky, A. T., Onnufer, J. J., Petsko, G. A. & Ringe, D. (1991)
Biochemistry 30, 1980- 1985. Dayhoff, M. 0. & Schwartz, R. M. (1978) in Atlas of protein se-
quence and structure (Dayhoff, M. O., ed) vol. 5, suppl. 3, pp. 353 - 358, National Biomedical Research Foundation, Silver
FEBS Lett. 108, 103-106.
riol. 172, 7035-7062.
Barouki, R. (1990) Biochemistry 29,5293-5299.
Bruni, C. B. (1988) J. Mol. Biol. 203, 585-606.
Spring MD. Degols, G. (1987) Eul: J. Biochem. 169, 193-200. Devereux, J., Haeberli, P. & Smithies, 0. (1984) Nucleic Acids Res.
12, 387-395. Doonan, S., Martini, F., Angelaccia, S., Pascarella, S., Barra, D. &
Dunathan, H. C. (1966) Proc. Natl Acad. Sci. USA 55, 712-716. Duncan, K. & Coggins, J. R. (1986) Biochem. J. 234,49-57. Feild, M. J., Nguyen, D. C. & Armstrong, F. B. (1989) Biochemistry
Fotheringham, I. G., Dacey, S. A,, Taylor, P. P., Smith, T. J., Hunter, M. G., Finlay, M. E., Primrose, S. B., Parker, D. M. & Edwards, R. M. (1986) Biochem. J. 234, 593-604.
Gloeckler, R., Ohsawa, I., Speck, D., Ledoux, C., Bernard, S., Zin- sius, M., Villeval, D., Kison, T., Karnogawa, K. & Lemoine, Y. (1990) Gene 87, 63-70.
Goldberg, J. M., Swanson, R. V., Goodman, H. S. & Kirsch, J. F. (1991) Biochemistry 30, 305-312.
Graf-Hausner, U., Wilson, K. J. & Christen, P, (1983) J. Biol. Chem. 258, 8813-8826.
Grange, T., Guenet, C., Dietrich, J. B., Chasserot, S., Fromont, M., Befort, N., Jami, J., Beck, G. & Pictet, R. (1985) J. Mol. Biol.
Gribskov, M., McLachlan, A. D. & Eisenberg, D. (1987) Proc. Nut1
Gribskov, M., Homyak, M., Edenfield, J. & Eisenberg, D. (1988)
Gribskov, M., Luthy, R. & Eisenberg, D. (1990) Methods Enzymol.
Griffin, H. G. (1990) Nucleic Acids Res. 18, 4260.
Bossa, F. (1986) J. Mol. Evol. 23, 328-335.
28, 5306-5310.
184, 347-350.
Acad. Sci. USA 84,4355-4358.
Comput. Appl. Biol. Sci. 4, 61 -66.
183, 146-159.

558
Grisolia, V., Carlomagno, M. S., Nappo, A. G. & Bruni, C. B. (1985) J. Bacteriol. 164, 1317-1323.
Hayashi, H., Horio, Y., Tanaka, T., Taketoshi, M. & Wada, H. (1987) in Biochemistry of vitamin Bs (Korpela, T. & Christen, P., eds) pp. 39-42, Birkhhser, Basel.
Heirnberg, H., Boyen, A., Crabeel, M. & Glansdorff, N. (1990) Gene 90,69-78.
Henner, D. J., Band, L., Flaggs, G. & Chen, E. (1986) Gene 49, 147- 152.
Hogeweg, P. & Hesper, B. (1972) Biopat, program system for bio- logical pattern analysis, Bioinfonnatica, Utrecht.
Huynh, Q. K., Sakakibara, R., Watanabe, T. & Wada, H. (1981) J. Biochem. (Tokyo) 90, 863-875.
Inana, G., Totsuka, S., Redmond, M., Dougherty, T., Nagle, J., Shi- ono, T., Ohura, T., Kominami, E. & Katunuma, N. (1986) Proc. Natl Acad. Sci. USA 83, 1203-1207.
Inoue, Y., Kuramitsu, S., Inoue, K., Kagamiyama, H., Hiromi, K., Tanase, S. & Morino, Y. (1989) J. Biol. Chem. 264,9673-9681.
Inoue, K., Kuramitsu, S., Okamoto, A., Hirotsu, K., Higuchi, T., Morino, Y. & Kagamiyama, H. (1991) J. Biochem. (Tokyo) 190, 570-576.
Ishiguro M., Suzuki, M., Takio, K., Matsuzawa, T. & Titani, K. (1991a) Biochemistry 30, 6048-6053.
Ishiguro, M., Takio, K., Suzuki, M., Oyama, R., Matsuzawa, T. & Titani, K. (1991b) Biochemistry 30, 10451-10457.
Jansonius, J. N. & Vincent, M. G. (1987) in Biological macromole- cules and assemblies, active sites of enzymes (Jurnak, F. A. & McPherson, A., eds) vol. 3, pp. 187-285, Wiley, New York.
Kagamiyama, H., Sakakibara, R., Tanase, S., Morino, Y. & Wada, H. (1980) J. Biol. Chem. 255, 6153-6159.
Kirsch, J. F., Eichele, G., Ford, G. C., Vincent, M. G., Jansonius, J. N., Gehring, H. & Christen, P. (1984) J. Mol. Biol. 174, 497- 525.
Kuramitsu, S., Ogawa, T., Ogawa, H. & Kagamiyama, H. (1985a) J. Biochem. (Tokyo) 97, 993-999.
Kuramitsu, S., Okuno, S., Ogawa, T., Ogawa, H. & Kagamiyama, H. (1985b) J. Biochem. (Tokyo) 97, 1259-1262.
Kuramitsu, S., Yano, T., Inoue, K., Hiromi, K., Tanase, S., Morino, Y. & Kagamiyama, H. (1991) in Enzymes dependent on pyri- dona1 phosphate and other carbonyl compounds as cofactors (Fukui, T., Kagamiyama, H., Soda, K. & Wada, H., eds) pp. 179-181, Pergamon Press, Tokyo.
Kyte, J. & Doolittle, R. F. (1982) J. Mol. Biol. 157, 105-132. Limauro, D., Avitabile, A., Capellano, C., Puglio, A. M. & Bruni,
Liu, L., Wahlen, W., Das, A. & Berg, C. M. (1987) Nucleic Acids
Luthy, R., McLachlan, A. D. & Eisenberg, D. (1991) Proteins 10,
Malcolm, B. A. & Kirsch, J. F. (1985) Biochem. Biophys. Rex Com- rnun. 132, 915-921.
Martini, F., Angelaccio, S., Bma, D., Pascarella, S., Maras, B., Doo- nan, S. & Bossa, F. (1985) Biochim. Biophys. Acta 832, 46-51.
Mavrides, C. & Christen, P. (1978) Biochem. Biophys. Res. Com- rnun. 85, 769-773.
McPhalen, C. A., Vincent, M. G. & Jansonius, J. N. (1992) J. Mol. Biol. 225, 495-517.
Mehta, P. K. & Christen, P. (1993) Eur: J. Biochem. 211, 373-376. Mehta, P. K., Hale, T. I. & Christen, P. (1989) Eul: J. Biochem. 186,
Misrahi, M., Atger, M. & Milgrom, E. (1987) Biochemistry 26,
C. B. (1990) Gene 90, 31-41.
Res. 15, 9461-9469.
229- 239.
249- 253.
3975 - 3982.
Mueckler, M. M. & Pitot, H. C. (1985) J. Biol. Chem. 260, 12993- 12997.
Needleman, S. B. & Wunsch, C. D. (1970) J. Mol. Biol. 48, 443- 453.
Nishiwaki, K., Hayashi, N., hie, S., Chung, D.-H., Harashima, S. & Oshima, Y. (1987) Mol. Gen. Genet. 208, 159-167.
Obaru, K., Nomiyama, H., Shimada, K., Nagashima, F. & Morino, Y. (1986) J. Biol. Chem. 261, 16976-16983.
Oda, T., Miyajima, H., Suzuki, Y. & Ichiyama, A. (1987) Eur: J. Biochem. 168,537-542.
O’Gaora, P., Maskell, D., Coleman, D., Cafferkey, M. & Daugam, G. (1989) Gene 84, 23-30.
Otsuka, A. J., Buoncristiani, M. R., Howard, P. K., Flamm, J., John- son, C., Yamamoto, R., Uchida, K., Cook, C., Ruppert, J. & Matsuzaki, J. (1988) J. Biol. Chem. 263, 19577-19585.
Ovchinnikov, Yu. A., Egorov, C. A., Aldanova, N. A., Feigina, M. Yu., Lipkin, V. M., Abdulaev, N. G., Grishin, E. V., Kiselev, A. P., Modyanov, N. N., Braunstein, A. E., Polyanovsky, 0. L. & Nosikov, V. V. (1973) FEBS Lett. 29, 31-34.
Picot, D., Sandmeier, E., Thaller, C., Vincent, M. G., Christen, P. & Jansonius, J. N. (1991) Eur. J. Biochem. 196, 329-341.
Rettenmeier, R., Natt, E., Zentgraf, H. & Scherer, G. (1990) Nucleic Acids Res. 18, 3853-3861.
Richardson, I. B., Hurley, S . K. & Hynes, M. J. (1989) Mol. Gen. Genet. 217, 118-125.
Shlyapnikov, S. V., Myasnikov, A. N., Severin, E. S., Myagkova, M. A., Torchinsky, Y. M. & Braunstein, A. E. (1979) FEBS Lett.
Sokal, R. R. and Sneath, H. A. (1963) Principles of numerical taxon- omy, W. H. Freeman, San Francisco.
Stark, W., Kallen, J., Markovic-Housley, Z., Fol. B., Kania, M. & Jansonius, J. N. (1991) in Enzymes dependent on pyridoxalphos- phate and other carbonyl compounds as cofactors (Fukui, T., Kagamiyama, H., Soda, K. & Wada, H., eds) pp. 111-115, Per- gamon Press, Tokyo.
Sung, M.-H., Tanizawa, K., Tanaka, H., Kuramitsu, S., Kagamiyama H., Hirotsu, K., Okamoto, A., Higuchi, T. & Soda, K. (1991) J. Bid. Chem. 266, 2567-2572.
Takada, Y., Kaneko, N., Esumi, H., Purdue, P. E. & Danpure, C. J. (1990) Biochem. J. 268, 517-520.
Takagi, T., Taniguchi, T., Yamamoto, Y. & Shibatani, T. (1991) Bio- technol. Appl. Biochem. 13, 112-119.
Tanizawa, K., Asano, S., Masu, Y., Kuramitsu, S . , Kagamiyama, H., Tanaka, H. & Soda, K. (1989) J. Biol. Chem. 264,2450-2454.
Watanabe, N., Sakabe, K., Sakabe, N., Higashi, T., Sasaki, K., Aib- ara, S., Morita, Y., Yonaha, K., Toyama, S. & Fukutani, H. (1989) J. Biochem. (Tokyo) 150, 1-3.
Watanabe, N., Yonaha, K., Sakabe, K., Sakabe, N., Shigeo, A. & Morita, Y. (1991) in Enzymes dependent on pyridoxal phosphate and other carbonyl compounds as cofactors (Fukui, T., Kagami- yama, H., Soda, K. & Wada, H., eds) pp. 121-124, Pergamon Press, Tokyo.
Yano, T., Kuramitsu, S., Tanase, S., Morino, Y. & Kagamiyama, H. (1992) Biochemistry 31, 5878-5887.
Ziak, M., Jaussi, R., Gehring, H. & Christen, P. (1990) Eur: J. Bio- chem. 187, 329-333.
Ziak, M., Jager, J., Malashkevich, V. H., Gehring, H., Jaussi, R., Jansonius, J. N. & Christen, P. (1993) Eur. J. Biochem. 211, 475 -484.
106, 385-388.
Note added in proo$ The amino acid sequence of o-amino acid aminotransferase has in the meantime been published [Youaha, K., Nishie, M. & Aibara, S . (1992) J. Biol. Chem. 267, 12506-125101, The differences to the sequence shown in the multiple alignment of Fig. 1 have no consequences on the alignment or the conservation of residues.
559
Supplementary material to:
Aminotransferases: demonstration of homology and division into evolutionary subgroups
Perdeep K. MEHTA, Terence I. HALE and Philipp CHRISTEN
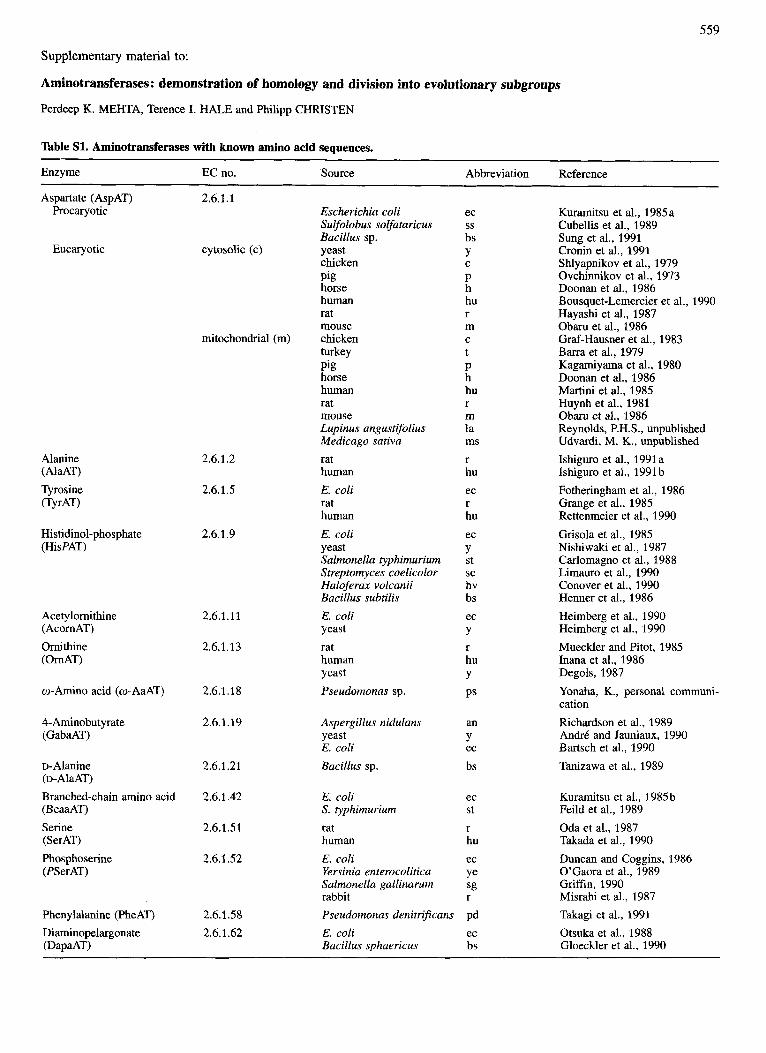
Table S1. Aminotransferases with known amino acid sequences.
Enzyme EC no. Source Abbreviation
Aspartate (AspAT) Procaryotic
Eucaryotic
Alanine (AlaAT) Tyrosine iTyrAT)
Histidinol-phosphate (HisPAT)
Acetylornithine (Acorn AT) Ornithine i 0 d T )
o-Amino acid (w-AaAT)
4-Aminobutyrate (GabaAT)
D- Alanine (D-A~~AT) Branched-chain amino acid (BcaaAT) Serine (SerAT) Phosphoserine (PSerAT)
Phenylalanine (PheAT) Diaminopelargonate (DapaAT)
2.6.1.1
cytosolic (c)
mitochondrial (m)
2.6.1.2
2.6.1.5
2.6.1.9
2.6.1.11
2.6.1.13
2.6.1.18
2.6.1.19
2.6.1.21
2.6.1.42
2.6.1.51
2.6.1.52
2.6.1.58 2.6.1.62
Escherichia coli Sulfolobus solfataricus Bacillus sp. yeast chicken Pig horse human rat mouse chicken turkey Pig horse human rat mouse Lupinus angustifolius Medicago sativa rat human E. coli rat human E. coli yeast Salmonella typhimurium Streptomyces coelicolor Haloferax volcanii Bacillus subtilis E. coli yeast rat human yeast Pseudomonas sp.
ec
bs Y
P h hu r m
t P h hu r m la ms r hu ec r hu ec Y st sc hv bs ec Y r hu Y PS
ss
C
C
Aspergillus nidulans an yeast Y E. coli ec Bacillus sp. bs
E. coli S. typhimurium rat human E. coli Yersinia enterocolitica Salmonella gallinarum rabbit Pseudomonas denitrificans E. coli Bacillus sphaericus
ec St r hu ec Ye sg r
Pd ec bs
Reference
Kuramitsu et al., 1985a Cubellis et al., 1989 Sung et al., 1991 Cronin et al., 1991 Shlyapnikov et al., 1979 Ovchinnikov et al., 1973 Doonan et al., 1986 Bousquet-Lemercier et al., 1990 Hayashi et al., 1987 Obaru et al., 1986 Graf-Hausner et al., 1983 Barra et al., 1979 Kagamiyama et al., 1980 Doonan et al., 1986 Martini et al., 1985 Huynh et al., 1981 Obaru et al., 1986 Reynolds, P.H.S., unpublished Udvardi, M. K., unpublished Ishiguro et al., 1991 a Ishiguro et al., 1991 b Fotheringham et al., 1986 Grange et al., 1985 Rettenmeier et al., 1990 Grisola et al., 1985 Nishiwaki et al., 1987 Carlomagno et al., 1988 Limauro et al., 1990 Conover et al., 1990 Henner et al., 1986 Heimberg et al., 1990 Heimberg et al., 1990 Mueckler and Pitot, 1985 Inana et al., 1986 Degols, 1987 Yonaha, K., personal communi- cation Richardson et al., 1989 An&& and Jauniaux, 1990 Bartsch et al., 1990 Tanizawa et al., 1989
Kuramitsu et al., 1985b Feild et al., 1989 Oda et al., 1987 Takada et al., 1990 Duncan and Coggins, 1986 O’Gaora et al., 1989 Griffin, 1990 Misrahi et al., 1987 Takagi et al., 1991 Otsuka et al., 1988 Gloeckler et al., 1990

560
Table S2. Percentage identity i.e. invariant residues (bold) and similarity i.e. invariant residues plus conservative substitutions @lain text) among aligned aminotransferases. The values given are based on the comprehensive alignment of all aminotransferases in Fig. 1. They, thus, are lower than the values obtained by pair-wise comparisons of the sequences. The classification of similar amino acid residues as proposed by Liithy et al. (1991) was applied; hydrophobic aromatic (F, Y, W); hydrophobic not aromatic (A, I, L, M, V); ambivalent (hydrophobic but small) or polar but uncharged (C, G, P, S , T); hydrophilic basic (H, K, R); hydrophilic acidic and their amides (D, E, N, Q). The abbreviations of the enzymes are explained in Table S1.
AspATcc AspATmc AspAlla AspATec AsPATY TyrATec PheATpd AspATss AspATbs TyrATr AlaATr HPATec HPATy HPATsc HPATbs HPATbv
.. 48 45 41 47 41 33 18 16 14 17 17 13 12 16 15 11 11 9 8 11 10 10 11 9 9 6 7 9 7 8 7 71 .. 50 41 44 41 33 18 17 15 14 16 14 13 14 18 8 11 9 9 11 8 8 10 10 8 8 9 9 8 8 7 62 68 .. 43 39 39 35 17 18 12 14 17 I5 13 14 15 9 9 6 8 13 7 9 8 11 9 6 7 11 9 10 9 61 62 65 .. 39 43 44 21 17 14 18 16 14 16 16 15 11 12 9 10 13 9 9 8 11 9 9 7 11 10 10 10 68 68 61 62 .. 37 31 21 16 15 16 15 13 14 14 14 11 11 11 9 13 10 9 9 10 7 7 9 7 7 9 8 62 66 63 65 63 .. 37 18 18 15 17 16 16 15 15 17 13 9 8 8 12 11 10 10 13 11 6 10 11 10 8 7 54 56 56 62 54 56 .. 18 18 14 I8 15 14 14 13 13 11 9 9 9 13 9 9 9 10 10 8 10 11 8 11 11 46 44 44 48 46 44 40 .. 35 24 19 19 15 19 15 16 12 10 11 13 12 13 10 8 10 11 10 8 5 8 8 '8 40 40 41 41 42 41 43 62 .. 25 22 18 16 17 18 I8 12 10 9 9 10 10 9 7 9 12 8 8 7 8 8 8 40 39 39 40 39 40 41 51 52 .. 19 19 16 16 14 16 12 13 9 10 10 12 11 10 11 10 9 11 8 10 8 9 37 37 37 39 38 38 40 42 48 45 .. 16 15 14 14 15 10 8 8 9 10 11 9 9 10 8 8 12 8 8 9 9 38 39 38 41 41 42 38 45 45 41 42 .. 36 33 29 31 12 12 13 12 12 11 12 11 13 13 10 14 8 12 10 9 33 37 36 37 36 39 34 40 40 40 39 61 .. 29 29 25 11 11 11 9 10 10 10 10 9 12 9 9 8 8 8 9 35 36 35 38 39 40 41 42 42 43 39 58 52 .. 34 30 12 11 10 11 12 10 12 12 10 11 10 11 11 11 11 9 37 36 37 41 37 38 36 38 41 35 37 55 53 56 .. 32 12 12 11 10 10 13 11 10 9 11 11 10 9 9 9 9 36 36 36 39 36 38 36 43 45 39 3R 54 52 52 54 .. 12 10 11 11 11 9 9 11 13 10 9 13 11 10 9 8
AcoATec 32 30 31 33 31 33 31 31 35 33 30 33 32 35 32 34 .. 40 34 30 26 37 23 22 29 27 7 10 12 10 10 9 AcoATy 33 31 30 32 29 33 31 31 32 35 30 33 33 37 33 34 61 .. 32 30 20 30 24 24 25 26 10 10 9 8 10 10 OmATh 30 28 29 29 30 29 30 32 34 31 31 37 33 39 37 37 57 52 .. 55 28 30 22 23 24 24 6 10 11 9 10 12 UmATy 31 31 30 33 31 33 35 35 36 32 30 38 35 38 37 33 55 48 69 .. 28 28 26 25 25 23 8 8 10 7 8 10 o-aaATps 31 32 33 34 32 33 34 31 32 33 33 36 35 37 33 34 51 43 49 52 .. 24 17 17 26 25 8 9 12 12 10 9 GabATec 33 31 29 34 33 35 33 34 35 33 34 31 35 37 37 37 61 53 55 53 46 .. 23 24 28 28 7 8 9 8 11 9 GabATy 29 28 25 28 28 32 29 30 28 30 25 32 30 32 29 28 45 44 43 44 39 45 ._ 58 19 17 7 8 9 8 10 11 GabATan 30 29 27 29 28 33 29 28 29 29 26 32 30 33 31 29 45 44 43 44 39 46 71 .. 21 17 9 8 10 8 10 10 DapATec 31 32 32 33 32 34 33 30 31 33 31 38 32 35 33 38 52 47 49 51 49 51 41 43 .. 38 7 9 12 8 10 9 DapA'h 31 32 29 29 30 32 30 31 34 30 30 37 35 32 32 35 52 46 48 47 48 50 40 39 59 .. 9 8 8 9 10 10
W T b s 29 29 21 29 30 28 30 32 34 33 30 31 32 32 33 31 25 29 28 32 27 28 26 29 27 27 .. 30 8 7 8 5 BcaATec 30 30 21 30 32 32 31 26 32 32 30 31 28 31 31 28 27 25 32 34 25 28 27 30 27 27 56 .. 12 10 9 9
SerATh 30 29 31 33 31 32 34 26 32 32 33 34 30 38 32 34 34 31 32 31 35 33 31 31 38 30 27 32 .. 16 17 15 PSATr 21 31 31 30 29 30 31 29 30 28 27 34 31 33 29 29 32 31 30 31 37 30 28 29 35 34 28 32 45 .. 47 49 PSATye 26 30 30 32 32 30 31 28 30 27 28 32 30 33 30 26 34 34 32 31 33 31 29 30 35 31 28 33 44 67 .. 75 PSATec 25 29 29 33 31 30 30 29 31 28 29 34 32 32 29 27 34 34 35 33 34 33 30 30 36 33 28 33 42 69 87 ..
... LLLI ...... I-....... "a ...... u ....... Y-. ..... .*.I_ ..-.- I .... " U-..l*u*. .y_ ..........
-41 50 100 150 200 250 3bo4 Residue number i n alignment
R562 14 41 G314 D340
.U ........... ............ * .YL. _Y ............ ... ..-. - -.--. 4 .... L ..".... - ... ..........._. t*t Y ... ...""".."I.
1-4 - 4 0 0 350 400 450 500 550
Residue number i n alignment
Fig. S1. Hydropathy plots of the aligned sequences of the four subgroups of aminotransferases. The hydropathy of each position along the sequence was plotted with a window size of five residues. The average values of all sequences of each subgroup according to the alignment of Fig. 1 are plotted: subgroup I, red solid; subgroup 11, red dashed; subgroup 111, blue solid; and subgroup IV, blue dashed line. Secondary structure elements in the spatial structure of mitochondrial aspartate aminotransferase (McPhalen et al., 1992) are indicated at the bottom with the same symbols as in Fig. 1. The new numbering of residues explained in Fig. 1 and the Discussion is used.
Related Documents











