The Journal of Immunology Alternative Pathway Activation of Complement by Shiga Toxin Promotes Exuberant C3a Formation That Triggers Microvascular Thrombosis Marina Morigi,* ,1 Miriam Galbusera,* ,1 Sara Gastoldi,* Monica Locatelli,* Simona Buelli,* Anna Pezzotta,* Chiara Pagani,* Marina Noris,* Marco Gobbi, † Matteo Stravalaci, † Daniela Rottoli,* Francesco Tedesco, ‡ Giuseppe Remuzzi,* ,x and Carlamaria Zoja* Shiga toxin (Stx)-producing E.coli O157:H7 has become a global threat to public health; it is a primary cause of diarrhea- associated hemolytic uremic syndrome (HUS), a disorder of thrombocytopenia, microangiopathic hemolytic anemia, and acute renal failure with thrombi occluding renal microcirculation. In this study, we explored whether Stx triggers complement- dependent microvascular thrombosis in in vitro and in vivo experimental settings of HUS. Stx induced on human microvascular endothelial cell surface the expression of P-selectin, which bound and activated C3 via the alternative pathway, leading to thrombus formation under flow. In the search for mechanisms linking complement activation and thrombosis, we found that exuberant complement activation in response to Stx generated an increased amount of C3a that caused further endothelial P- selectin expression, thrombomodulin (TM) loss, and thrombus formation. In a murine model of HUS obtained by coinjection of Stx2 and LPS and characterized by thrombocytopenia and renal dysfunction, upregulation of glomerular endothelial P-selectin was associated with C3 and fibrin(ogen) deposits, platelet clumps, and reduced TM expression. Treatment with anti–P-selectin Ab limited glomerular C3 accumulation. Factor B-deficient mice after Stx2/LPS exhibited less thrombocytopenia and were protected against glomerular abnormalities and renal function impairment, indicating the involvement of complement activation via the alternative pathway in the glomerular thrombotic process in HUS mice. The functional role of C3a was documented by data showing that glomerular fibrin(ogen), platelet clumps, and TM loss were markedly decreased in HUS mice receiving C3aR antagonist. These results identify Stx-induced complement activation, via P-selectin, as a key mechanism of C3a-dependent microvascular thrombosis in diarrhea-associated HUS. The Journal of Immunology, 2011, 187: 172–180. S higa toxin (Stx)-producing Escherichia coli (STEC) O157: H7 is a foodborne or waterborne pathogen, responsible for worldwide spread of hemorrhagic colitis complicated by diarrhea-associated hemolytic uremic syndrome (D+HUS), a dis- order of thrombocytopenia, microangiopathic hemolytic anemia, and acute renal failure that mainly affects infants and small chil- dren (1–3). Death or end-stage renal disease occurs in ∼12% of patients with D+HUS, and 25% of survivors demonstrate long- term renal sequelae (4). Over the last two decades, E. coli O157: H7 has been the cause of multiple outbreaks, becoming a public health problem in both developed and developing countries (1, 5–7). Apart from supportive therapy, there are presently no speci- fic treatments for D+HUS (8), and strategies including STEC- component vaccines, Stx receptor mimics, and Abs against Stx are still under investigation (9). After STEC ingestion, Stx is transported in the circulation to the capillary bed of target organs, including the kidney (5, 6). Glomerular endothelium that ex- presses the receptor globotriaosyl ceramide (Gb3)/CD77 (10, 11) is the main target of the toxic effects of Stx1 and 2 (12), which activate a cascade of signals contributing to vascular dysfunction, leukocyte recruitment, and thrombus formation (13–19). We pre- viously reported that Stx promoted von Willebrand factor (VWF)- dependent thrombus growth on microvascular endothelium under flow via upregulation of P-selectin (17). In patients with D+HUS, elevated plasma levels of P-selectin were measured during the acute phase, which reflected increased P-selectin expression by activated endothelial cells and platelets (20). P-selectin overex- pressed on activated platelets and transfected Chinese hamster ovary cells had the capacity to activate the complement (C) system by acting as C3b binding protein, thereby generating the ana- phylatoxins C3a and C5a and the membrane attack complex (C5b- 9) (21). C3a and C5a, through binding to G-protein coupled re- ceptors, elicit inflammatory responses such as cytokine produc- tion, adhesive molecule expression, and increased vascular perme- ability, leading to endothelial cell activation (22–24). Whether C activation at renal endothelial level may contribute to microangiopathic lesions in D+HUS has never been addressed. *Mario Negri Institute for Pharmacological Research, Centro Anna Maria Astori, Science and Technology Park Kilometro Rosso, 24126 Bergamo, Italy; † Mario Negri Institute for Pharmacological Research, 20156 Milan, Italy; ‡ Department of Phys- iology and Pathology, University of Trieste, 34127 Trieste, Italy; and x Unita ` di Nefrologia e Dialisi, Azienda Ospedaliera Ospedali Riuniti di Bergamo, 24128 Bergamo, Italy 1 M.M. and M.G. contributed equally to this work. Received for publication February 16, 2011. Accepted for publication April 19, 2011. This work was supported in part by a Genzyme Renal Innovation Program grant. M.L. is the recipient of a fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare, Bergamo, Italy. Address correspondence to Carlamaria Zoja, Mario Negri Institute for Pharmacolog- ical Research, Centro Anna Maria Astori, Science and Technology Park Kilometro Rosso, Via Stezzano, 87, 24126 Bergamo, Italy. E-mail address: carlamaria.zoja@ marionegri.it The online version of this article contains supplemental material. Abbreviations used in this article: Bf 2/2 , factor B-deficient; BUN, blood urea nitrogen; C, complement; D+HUS, diarrhea-associated hemolytic uremic syndrome; HMEC, human microvascular endothelial cells; HS, human serum; KIU, kallikrein inhibitor unit; pM, picomolar; RU, resonance units; sCR-1, soluble C receptor-1; SPR, surface plasmon resonance; STEC, Shiga toxin-producing Escherichia coli; Stx, Shiga toxin; TM, thrombomodulin; t-PA, tissue-plasminogen activator; VWF, von Willebrand factor. Copyright Ó 2011 by The American Association of Immunologists, Inc. 0022-1767/11/$16.00 www.jimmunol.org/cgi/doi/10.4049/jimmunol.1100491

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The Journal of Immunology
Alternative Pathway Activation of Complement by ShigaToxin Promotes Exuberant C3a Formation That TriggersMicrovascular Thrombosis
Marina Morigi,*,1 Miriam Galbusera,*,1 Sara Gastoldi,* Monica Locatelli,* Simona Buelli,*
Anna Pezzotta,* Chiara Pagani,* Marina Noris,* Marco Gobbi,† Matteo Stravalaci,†
Daniela Rottoli,* Francesco Tedesco,‡ Giuseppe Remuzzi,*,x and Carlamaria Zoja*
Shiga toxin (Stx)-producing E.coli O157:H7 has become a global threat to public health; it is a primary cause of diarrhea-
associated hemolytic uremic syndrome (HUS), a disorder of thrombocytopenia, microangiopathic hemolytic anemia, and acute
renal failure with thrombi occluding renal microcirculation. In this study, we explored whether Stx triggers complement-
dependent microvascular thrombosis in in vitro and in vivo experimental settings of HUS. Stx induced on human microvascular
endothelial cell surface the expression of P-selectin, which bound and activated C3 via the alternative pathway, leading to
thrombus formation under flow. In the search for mechanisms linking complement activation and thrombosis, we found that
exuberant complement activation in response to Stx generated an increased amount of C3a that caused further endothelial P-
selectin expression, thrombomodulin (TM) loss, and thrombus formation. In a murine model of HUS obtained by coinjection of
Stx2 and LPS and characterized by thrombocytopenia and renal dysfunction, upregulation of glomerular endothelial P-selectin
was associated with C3 and fibrin(ogen) deposits, platelet clumps, and reduced TM expression. Treatment with anti–P-selectin Ab
limited glomerular C3 accumulation. Factor B-deficient mice after Stx2/LPS exhibited less thrombocytopenia and were protected
against glomerular abnormalities and renal function impairment, indicating the involvement of complement activation via the
alternative pathway in the glomerular thrombotic process in HUS mice. The functional role of C3a was documented by data
showing that glomerular fibrin(ogen), platelet clumps, and TM loss were markedly decreased in HUS mice receiving C3aR
antagonist. These results identify Stx-induced complement activation, via P-selectin, as a key mechanism of C3a-dependent
microvascular thrombosis in diarrhea-associated HUS. The Journal of Immunology, 2011, 187: 172–180.
Shiga toxin (Stx)-producing Escherichia coli (STEC) O157:H7 is a foodborne or waterborne pathogen, responsible forworldwide spread of hemorrhagic colitis complicated by
diarrhea-associated hemolytic uremic syndrome (D+HUS), a dis-order of thrombocytopenia, microangiopathic hemolytic anemia,and acute renal failure that mainly affects infants and small chil-dren (1–3). Death or end-stage renal disease occurs in ∼12% ofpatients with D+HUS, and 25% of survivors demonstrate long-
term renal sequelae (4). Over the last two decades, E. coli O157:
H7 has been the cause of multiple outbreaks, becoming a public
health problem in both developed and developing countries (1,5–7). Apart from supportive therapy, there are presently no speci-
fic treatments for D+HUS (8), and strategies including STEC-
component vaccines, Stx receptor mimics, and Abs against Stxare still under investigation (9). After STEC ingestion, Stx is
transported in the circulation to the capillary bed of target organs,
including the kidney (5, 6). Glomerular endothelium that ex-presses the receptor globotriaosyl ceramide (Gb3)/CD77 (10, 11)
is the main target of the toxic effects of Stx1 and 2 (12), whichactivate a cascade of signals contributing to vascular dysfunction,
leukocyte recruitment, and thrombus formation (13–19). We pre-
viously reported that Stx promoted von Willebrand factor (VWF)-dependent thrombus growth on microvascular endothelium under
flow via upregulation of P-selectin (17). In patients with D+HUS,
elevated plasma levels of P-selectin were measured during theacute phase, which reflected increased P-selectin expression by
activated endothelial cells and platelets (20). P-selectin overex-
pressed on activated platelets and transfected Chinese hamsterovary cells had the capacity to activate the complement (C) system
by acting as C3b binding protein, thereby generating the ana-
phylatoxins C3a and C5a and the membrane attack complex (C5b-9) (21). C3a and C5a, through binding to G-protein coupled re-
ceptors, elicit inflammatory responses such as cytokine produc-
tion, adhesive molecule expression, and increased vascular perme-ability, leading to endothelial cell activation (22–24).Whether C activation at renal endothelial level may contribute to
microangiopathic lesions in D+HUS has never been addressed.
*Mario Negri Institute for Pharmacological Research, Centro Anna Maria Astori,Science and Technology Park Kilometro Rosso, 24126 Bergamo, Italy; †MarioNegri Institute for Pharmacological Research, 20156 Milan, Italy; ‡Department of Phys-iology and Pathology, University of Trieste, 34127 Trieste, Italy; and xUnita di Nefrologiae Dialisi, Azienda Ospedaliera Ospedali Riuniti di Bergamo, 24128 Bergamo, Italy
1M.M. and M.G. contributed equally to this work.
Received for publication February 16, 2011. Accepted for publication April 19, 2011.
This work was supported in part by a Genzyme Renal Innovation Program grant.M.L. is the recipient of a fellowship from Fondazione Aiuti per la Ricerca sulleMalattie Rare, Bergamo, Italy.
Address correspondence to Carlamaria Zoja, Mario Negri Institute for Pharmacolog-ical Research, Centro Anna Maria Astori, Science and Technology Park KilometroRosso, Via Stezzano, 87, 24126 Bergamo, Italy. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: Bf2/2, factor B-deficient; BUN, blood urea nitrogen;C, complement; D+HUS, diarrhea-associated hemolytic uremic syndrome; HMEC,human microvascular endothelial cells; HS, human serum; KIU, kallikrein inhibitorunit; pM, picomolar; RU, resonance units; sCR-1, soluble C receptor-1; SPR, surfaceplasmon resonance; STEC, Shiga toxin-producing Escherichia coli; Stx, Shiga toxin;TM, thrombomodulin; t-PA, tissue-plasminogen activator; VWF, vonWillebrand factor.
Copyright� 2011 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1100491

There are anecdotes on reduced C3 and augmented C3b, C3c, andC3d serum levels in patients with active D+HUS (25–27). Highplasma levels of Bb and C5b-9 were recently measured in 17children with D+HUS, indicating C activation via alternativepathway during the onset of D+HUS (28). Another study reportedthat Stx2 activated C in the fluid phase in vitro (29). However, theuse of huge amounts of Stx2, together with the observation of noactive Stx in plasma of patients with D+HUS (30), raises concernsabout the pathophysiologic meaning of these results.In this study, we investigated in vitro whether Stx activated C via
the alternative pathway by promoting binding of C3 on micro-vascular endothelial P-selectin under flow conditions. The role of Cin favoring thrombus formation on endothelial cells in response toStx was evaluated. A novel mechanism by which C3a generated byStx-induced C activation impaired endothelial thromboresistancewas explored. To validate the in vitro findings, we assessed thecausal role of glomerular P-selectin on C3 deposition in a mousemodel of HUS obtained by coinjection of Stx2 and LPS. Theinvolvement of C activation via the alternative pathway in thedevelopment of microvascular thrombosis was studied applying theHUS model to factor B-deficient (Bf2/2) mice. Finally, the con-tribution of C3a in the thrombogenic process induced by Stx wasassessed by blocking C3a receptor in HUS mice.
Materials and MethodsEndothelial cell culture and incubation
The human microvascular endothelial cell line of dermal origin (i.e.,HMEC-1, from Dr. Edwin Ades and Francisco J. Candal [Centers forDisease Control and Prevention] and Dr. Thomas Lawley [Emory Uni-versity]) (31) was cultured as described (17). Purified Stx1 (provided byDr. Helge Karch and Dr. Martina Bielaszewska, Institute for Hygiene,University of Munster, Germany) (32) and commercially available Stx1and Stx2 (Toxin Technology) were used. Human serum (HS) from a poolof four healthy volunteers was used as a source of C. To remove plateletmicroparticles, serum was filtered with a 0.22-mm filter (Millipore) beforeeach experiment (33). Fifty percent of HS was chosen as the most ap-propriate nontoxic dose on the basis of preliminary experiments evaluatingC3 deposits.
To study the effect of Stxs on C3 accumulation and thrombus formation,HMEC-1 were treated with medium (control), purified Stx1, or commer-cially available Stx1 or Stx2 (50 picomolar [pM]) for 24 h in static con-ditions. Next, cells were perfused or not with 50% HS (3 min) followed ornot followed by whole blood in a parallel plate flow chamber at a constantflow rate of 1500 s21 (60 dynes/cm2) for 3 min (Supplemental Fig. 1).Soluble C receptor-1 (sCR-1; 100 mg/ml; CellDex) was incubated for20 min with HS before perfusion on HMEC-1. To evaluate whether Stx-induced C activation occurred via the alternative pathway, HS was pre-treated for 30 min with MgCl2-EGTA (5 mM; Sigma), an inhibitor of theclassical pathway of C activation.
P-selectin expression was examined in HMEC-1 incubated with purifiedStx1 (24 h, static condition). Costaining of P-selectin and C3 was performedon Stx-treated HMEC-1 perfused with HS. Functional blocking anti-P-selectin Ab (25 mg/ml; R&D Systems) or the irrelevant anti-CD44 Ab(25 mg/ml; Abcam), which binds to the endothelial cell surface, wereadded for 20 min to HMEC-1 at the end of Stx challenge and to serum(Supplemental Fig. 1). P-selectin was also blocked by PSGL-1 (20 mg/ml;R&D System), the soluble ligand of P-selectin, added to both Stx-treatedcells (20 min before perfusion) and serum. Involvement of C3a in throm-bus formation on Stx-treated HMEC-1 was investigated by adding theC3aR antagonist SB290157 (1 mM; Calbiochem) to the cells before per-fusion and to serum and blood (Supplemental Fig. 1). Endothelial ex-pression of thrombomodulin (TM) and P-selectin was evaluated afterincubation with purified C3a (1 mM, Calbiochem) in static condition. Theserine protease inhibitor aprotinin (200 kallikrein inhibitor unit [KIU]/ml;Sigma) was added to medium alone or to C3a. TM expression on HMEC-1was also investigated after 6 h exposure to tissue-plasminogen activator(t-PA; 10 mg/ml; Actyplase, Boehringer Ingeheim Pharma).
Immunofluorescence analysis of endothelial cells
HMEC-1 at the end of the perfusions were fixed in 3% paraformaldehydeand stained with the following Abs: FITC-conjugated anti-human C3c (11.3
mg/ml; Dako), anti-human C5b-9 (27.6 mg/ml; Calbiochem) followed byFITC-conjugated secondary Ab (13.6 mg/ml; Jackson ImmunoresearchLaboratories), anti-human P-selectin (20 mg/ml, R&D Systems) followedby Cy3-conjugated secondary Ab (13.6 mg/ml; Jackson ImmunoresearchLaboratories), and anti-human TM (1:50; R&D Systems) followed by Cy3-conjugated secondary Ab. Isotype-matched irrelevant Abs were used asa negative control. Coverslips were examined under confocal inverted lasermicroscopy (LSM 510 Meta; Zeiss, Jena, Germany). Fifteen fields persample were acquired, and the area of staining was quantified (pixel2/field)using Image J software (National Institutes of Health, Bethesda, MD). Forcolocalization studies, samples were imaged by confocal microscopy usinga z-scan (eight images acquired at 1.5-mm intervals by vertical projection).TM expression was measured by counting cells with moderate or strongintensity staining in 20 fields per sample. Data were expressed as a per-centage of fluorescent cells per total cells in each field.
Surface plasmon resonance analysis
Binding studies were performed using the ProteOn XPR36 Protein In-teraction Array system (Bio-Rad Laboratories), based on surface plasmonresonance (SPR) technology. Purified human C3b (Complement Technol-ogy) was covalently immobilized onto one flow channel surface of a ProteonGLC sensor chip (Bio-Rad), using amine coupling chemistry. The amountof C3b covalently immobilized onto the surface, expressed in resonanceunits (RU; 1 RU = 1 pg protein/mm2), was ∼7500 RU. A reference channel,with no ligand immobilized, was always prepared in parallel. Differentconcentrations, ranging from 21 to 310 nM, of recombinant human solubleP-selectin (R&D System) were then injected simultaneously over immo-bilized C3b (or over the reference surface in parallel) at a rate of 30 ml/minfor 2 min (association phase). The dissociation phase was evaluated in thefollowing 10 min. The running buffer, also used to dilute analytes, waseither phosphate-buffered saline pH 7.4, 0.005% Tween 20 (Bio-Rad) orTris HCl (10 mM, pH 7.4 containing 150 mM NaCl, 1.2 mM CaCl2 and 1.2mM MgCl2). All these assays were done at 25˚C.
Platelet adhesion assay under flow condition
Perfusion of heparinized whole blood (10 UI/ml) obtained from healthysubjects (prelabeled with the fluorescent dye mepacrine, 10 mM) wasperformed in a flow chamber (17). After 3 min of perfusion, the endothelialcell monolayer was fixed in acetone. Fifteen images per sample of plateletthrombi on endothelial cell surface were acquired by confocal invertedlaser microscope, and areas occupied by thrombi were evaluated by usingImage J software.
Experimental model of HUS
Male C57BL/6 mice were obtained from Charles River Italia. Bf2/2mice ofC57BL/6 genetic background (34) and C57BL/6 wild type mice were a giftfrom Dr. Marina Botto (Imperial College, London). For this study, Bf2/2
mice and control wild type littermates were obtained by heterozygote 3heterozygote matings at the Mario Negri Institute. Genotypes were de-termined by PCR (34). Animal care and treatment were performed inaccordance with institutional guidelines in compliance with national andinternational laws and policies. Animal studies were approved by the in-stitutional animal care and use committees of the Mario Negri Institute(Milan, Italy). HUS was induced in mice (26–28 g body weight) by i.p.injection of Stx2 (200 ng; Toxin Technology) plus LPS (75 mg, O111:B4,Sigma). Mice injected with saline served as controls. In parallel, somemice were injected with Stx2 or LPS alone. Stx2 was chosen because it isassociated with HUS clinical isolates more often than Stx1 (6) and becauseof the evidence that Stx2 injected in mice is more toxic and accumulates inthe kidney to a greater extent than Stx1 (35).
The dosing protocol was based on previous studies (16, 36). Mice weresacrificed at 3, 6, 24, or 48 h after injection. Blood platelet count and renalfunction measured by serum blood urea nitrogen (BUN; Reflotron test,Roche Diagnostics) were assessed. In separate experiments, C57BL/6 micewere given a 50-mg dose i.v. of a function-blocking anti-mouse P-selectinAb RB40.34 or an isotype-matched control Ab A110.1 (BD Pharmingen),1 h before Stx2/LPS. A second dose was administered 6 h later; mice weresacrificed 24 h after Stx2/LPS. The dose of RB40.34 was chosen on thebasis of previous studies (37). Other experiments included treatment ofmice with the C3aR antagonist SB290157 (15 mg/kg i.p.) or an equalvolume of vehicle (saline and dimethyl sulfoxide) four times per day (1 hbefore and 1, 4, and 8 h after Stx2/LPS injection).
Renal ultrastructural analysis
Fragments of kidney tissue were processed as described (16). Ultrathinsections were stained with uranyl acetate and lead citrate and were ex-
The Journal of Immunology 173
amined using a transmission electron microscope (Morgagni 268D; Philips,Brno, Czech Republic).
Immunofluorescence analysis of renal tissue
Periodate lysine paraformaldehyde (PLP)-fixed frozen sections (3 mm)were incubated with FITC-conjugated goat anti-rat fibrin(ogen) Ab (1:40;Nordic Immunology) and with rhodamine WGA lectin (Vector Labora-tories) dissolved in DAPI. Fibrin(ogen) deposition in glomerular capillarywalls was assessed by confocal microscopy and scored from 0 to 3(0, absent or trace; 1, weak granular, discontinuous staining; 2, stronglystaining; 3, occasional obliteration). P-selectin expression was evaluated infrozen sections incubated with anti-mouse P-selectin Ab (3 mg/ml; BDPharmingen) or with isotype-matched irrelevant Ab, followed by FITC-conjugated secondary Ab (1:150; Sigma-Aldrich). To determine the lo-cation of P-selectin in the glomerulus, adjacent sections were incubatedwith rabbit anti-VWF (1:100; Dako) followed by Cy3-conjugated anti-rabbit Abs (60 mg/ml; Jackson Immunoresearch Laboratories). Negativecontrols were obtained with secondary Ab alone. C3 deposits were eval-uated by staining with FITC-conjugated anti-mouse C3 Ab (10 mg/ml;Cappel). For C9 staining, rabbit anti-rat C9 (1:200; a gift from PaulMorgan, Cardiff, U.K.) and Cy3-conjugated secondary Abs (30 mg/ml;Jackson Immunoresearch Laboratories) were used. Isotype-matched irrel-evant Abs were used as negative controls. Glomerular C3 and C9 stainingwas scored from 0 to 3 (0, no staining or traces [,5%]; 1, stainingin,25% of the glomerular tuft; 2, staining affecting 26 to 50%; 3, stainingin .50%). TM was detected in PLP-fixed sections using rabbit anti-TMAb (1:25; Santa Cruz) followed by Cy3-conjugated goat anti-rabbit IgG(15mg/ml; Jackson Immunoresearch Laboratories). Glomerular TM ex-pression was estimated using Image J software.
Statistical analysis
Results are expressed as means 6 SEM. Data were analyzed by thenonparametric Mann–Whitney or Kruskal–Wallis tests, or by ANOVA asappropriate. The p values ,0.05 were considered statistically significant.
ResultsStx promotes endothelial C3 activation and deposition via thealternative pathway
To test whether C activation by Stxs involves C3 activation anddeposition, we evaluated C3 deposition on endothelial cell surface
in response to the toxin. HMEC-1 were incubated with purifiedStx1 (50 pM, 24 h) and then perfused in a parallel plate flowchamber (60 dynes/cm2, 3 min) with 50% HS as a source of C(Supplemental Fig. 1). Under these conditions, monolayer inte-grity was preserved (Supplemental Fig. 2A). A marked increase ofC3 deposits on Stx1-treated cells was observed with respect tounstimulated cells (Fig. 1A, 1B), indicating that endothelial cellsactivated by Stx fixed C. Similar results were obtained using non-purified, commercially available Stx1 and Stx2 (Supplemental Fig.2B). No C5b-9 deposits were detected on Stx-treated HMEC-1 afterthe short HS perfusion (not shown).When HS was incubated with the sCR-1—an inhibitor of
classical, alternative, and lectin pathways of C activation—endo-thelial C3 deposits induced by Stx1 were abolished, indicatingspecificity of the C3 signal (Fig. 1A, 1B). The role of the alter-native pathway of C activation was documented by the finding thatC3 deposition induced by Stx1 was not modified by inhibiting theclassical pathway with MgCl2-EGTA added to HS before cellperfusion (Fig. 1A, 1B).
C3 binds with high affinity to P-selectin expressed onStx-activated endothelial cells
Because we showed previously that Stx1 induced endothelial ex-pression of P-selectin (17), an adhesive protein that also acts asa C3b receptor on platelets and Chinese hamster ovary cells (21),experiments were performed to evaluate whether P-selectin hada role in C activation and C3 deposition on Stx-activated endo-thelial cells. First, we documented by immunofluorescence theincrease of P-selectin as a granular pattern on the endothelialapical side after exposure to purified Stx1 (Supplemental Fig. 3A).We then evaluated whether P-selectin could interact with C3 onHMEC-1 exposed to Stx and perfused with HS. By confocal mi-croscopy, the majority of C3 deposits colocalized with P-selectin(Fig. 1C). Such a colocalization was visible in a large number of
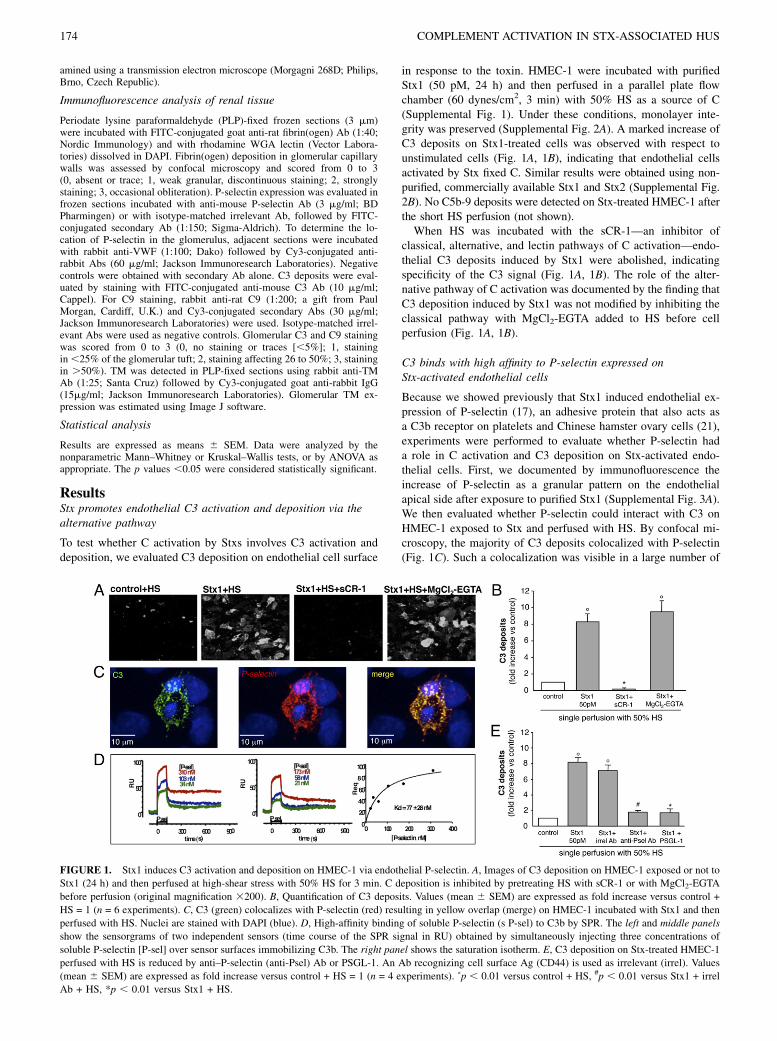
FIGURE 1. Stx1 induces C3 activation and deposition on HMEC-1 via endothelial P-selectin. A, Images of C3 deposition on HMEC-1 exposed or not to
Stx1 (24 h) and then perfused at high-shear stress with 50% HS for 3 min. C deposition is inhibited by pretreating HS with sCR-1 or with MgCl2-EGTA
before perfusion (original magnification 3200). B, Quantification of C3 deposits. Values (mean 6 SEM) are expressed as fold increase versus control +
HS = 1 (n = 6 experiments). C, C3 (green) colocalizes with P-selectin (red) resulting in yellow overlap (merge) on HMEC-1 incubated with Stx1 and then
perfused with HS. Nuclei are stained with DAPI (blue). D, High-affinity binding of soluble P-selectin (s P-sel) to C3b by SPR. The left and middle panels
show the sensorgrams of two independent sensors (time course of the SPR signal in RU) obtained by simultaneously injecting three concentrations of
soluble P-selectin [P-sel] over sensor surfaces immobilizing C3b. The right panel shows the saturation isotherm. E, C3 deposition on Stx-treated HMEC-1
perfused with HS is reduced by anti–P-selectin (anti-Psel) Ab or PSGL-1. An Ab recognizing cell surface Ag (CD44) is used as irrelevant (irrel). Values
(mean 6 SEM) are expressed as fold increase versus control + HS = 1 (n = 4 experiments). ˚p , 0.01 versus control + HS, #p , 0.01 versus Stx1 + irrel
Ab + HS, *p , 0.01 versus Stx1 + HS.
174 COMPLEMENT ACTIVATION IN STX-ASSOCIATED HUS

Stx-treated endothelial cells. Moreover, binding studies with SPRshowed a specific interaction between C3b and P-selectin. Fig. 1D(left and middle panels) shows the sensorgrams (time-course ofSPR signal) obtained when flowing different concentrations ofsoluble P-selectin over C3b immobilized on the sensor chip. Adose-dependent binding signal was detected. A clearly biphasicdissociation curve did not fit the simplest 1:1 interaction. Theinteraction is more complex, suggesting a conformational changein C3b molecules once bound to P-selectin, or the presence ofcooperativity among the multiple binding sites (SCR domains)present on P-selectin, as previously described for binding of C3bto CR1 (38, 39). However, the plot of the equilibrium responsescould be fitted by a simple saturation isotherm, allowing the es-timation of a Kd value of 77 6 28 nM (Fig. 1D, right panel).Next, we documented that P-selectin mediated C3 binding on
the surface of Stx-treated HMEC-1. Cell exposure to functionalblocking anti–P-selectin Ab, before and during HS perfusion, ledto a significant inhibition of C3 deposits in response to Stx1 (Fig.1E), whereas the addition of irrelevant Ab had no effect. More-over, a strong decrease of C3 accumulation was observed whenPSGL-1, the soluble ligand of P-selectin, was added to HMEC-1before and during HS perfusion (Fig. 1E).
Complement activation favors thrombus formation onStx-treated endothelial cells
To establish whether C activation and deposition on HMEC-1 couldbe instrumental to thrombus formation, Stx1-treated cells wereexposed to a double perfusion consisting of HS, followed by wholehuman blood prelabeled with the fluorescent dye mepacrine, or toa single perfusion with blood alone (Supplemental Fig. 1). WhenStx-1–treated cells were perfused with HS and blood, a more in-tense deposition of C3 and increased endothelial surface areacovered by thrombi were detected, as compared with unstimulatedcells (Table I). Notably, after double perfusion, C3 deposits werefollowed by thrombi detected on the endothelial surface to a sig-nificantly greater extent than on Stx-treated cells subjected tosingle blood perfusion (Table I). A representative image of orga-nized thrombi formed on the endothelial cell monolayer is shownin Supplemental Fig. 3B. Pretreatment of HS and blood with sCR-1 significantly (p , 0.01) limited platelet thrombi on HMEC-1exposed to the toxin, indicating a causal link between C3 de-position and thrombus formation (Fig. 2A, 2B).We next examined whether P-selectin blockade could limit C-
dependent thrombus formation on endothelial cells in responseto Stx. P-selectin inhibition in Stx1-treated cells exposed to doubleperfusion of HS and blood resulted in decreased C3 accumulation(Supplemental Fig. 3C), followed by a significant reduction inplatelet thrombi (Fig. 2A, 2B). Irrelevant Ab had no effect on
Stx-induced C3 deposition (Supplemental Fig. 3C) and thrombusformation (Fig. 2A, 2B). These data indicate that endothelial P-selectin upregulated by Stx is instrumental for C activation andC3-dependent thrombus formation on microvascular endothelium.
C3a generated by Stx-induced C activation impairs endothelialthromboresistance
To discover how C activation triggers thrombus formation onendothelial cells, we studied the effect of the anaphylatoxinC3a—a breakdown product generated during C activation andC3 deposition—on endothelial thromboresistance properties. Wefocused on TM, a glycoprotein highly expressed on endothelialcell surface, with cytoprotective and antithrombotic effects (40).TM was constitutively expressed on HMEC-1 (percentage offluorescent cells with moderate and strong intensity: 54.2 6 4.3and 6.7 6 1.1%). Treatment with Stx1 alone slightly reduced TMexpression, which further decreased after HS perfusion (Fig. 3A,Supplemental Fig. 3D). Addition of the C3aR antagonistSB290157 on Stx-treated HMEC-1 perfused with HS preventedTM loss (Fig. 3A, Supplemental Fig. 3D). Providing further evi-dence for a role of this anaphylatoxin, we found a marked re-duction of TM expression on the surface of HMEC-1 exposed toexogenous purified C3a (Fig. 3B). Because the cleavage of TM isthought to be dependent by serine protease activity, we examined t-PA, a serine protease present in endothelial Weibel-Palade bodiespossibly released upon C3a stimulation (41). Blockade of t-PAactivity by treatment of HMEC-1 with the serine protease in-hibitor aprotinin prevented TM shedding in response to exogenousC3a (Fig. 3B). Aprotinin alone as a control did not affect theconstitutive expression of TM (percentage of fluorescent cells withmoderate and strong intensity: aprotinin, 67.7 6 5.3 and 7.6 6 0.6versus control, 62.46 4.9 and 8.36 0.7). Consistently, the additionof t-PA to HMEC-1 significantly (p < 0.01) reduced TM expressionon the cell surface (percentage of fluorescent cells with moderateand strong intensity: t-PA, 51.36 2.1 and 1.76 0.3 versus control,62.2 6 1.2 and 6.6 6 0.9). The finding that C3a may favor exo-cytosis of the Weibel-Palade body’s content was also supported bythe increased apical staining of P-selectin on C3a-treated HMEC-1(Fig. 3C). The effects induced by C3a were critical for thrombusformation, as evidenced by the significant (p , 0.01) reduction of
Table I. Stx1 induces complement deposition and thrombus formationon HMEC-1 under flow condition
Control Stx1 (50 pM) Control Stx1 (50 pM)
Single Perfusion—
+ Blood Perfusion
Double Perfusion+50% HS
+ Blood Perfusion
C3 deposits 1.0 2.9 6 0.7a 2.5 6 0.8a 10.5 6 6.4a,b,c
Thrombi 1.0 7.5 6 3.4a 5.2 6 1.5a 29.0 6 18.8a,b,d
Area covered by C3 deposits or thrombi is expressed as fold increase over control(single perfusion) imposed as 1.0. Data are mean 6 SEM.
ap , 0.01 versus control plus blood perfusion.bp , 0.01 versus control plus HS plus blood perfusion.cp , 0.01 versus Stx1 plus blood perfusion.dp , 0.05 versus Stx1 plus blood perfusion.
FIGURE 2. C3 bound to P-selectin favors thrombus formation on en-
dothelial cells in response to Stx. A, Representative images of fluorescent
thrombi on control or Stx1-treated HMEC-1 perfused with HS and blood
pretreated or not with sCR-1 before perfusion. Stx-treated HMEC-1 are
also exposed to irrelevant (irrel) or anti-Psel Abs before double perfusion
(original magnification 3200). B, Quantification of fluorescent thrombi
(mean 6 SEM) expressed as pixel2 3 103 (n = 6 experiments). ˚p , 0.01
versus control + HS, *p , 0.01 versus Stx1 + HS, #p , 0.01 versus Stx1 +
irrel Ab + HS.
The Journal of Immunology 175
platelet thrombi on Stx-treated HMEC-1 perfused with HS andblood in the presence of the C3aR antagonist (Fig. 3D).
Glomerular C activation and deposition, via endothelialP-selectin, in a murine model of HUS
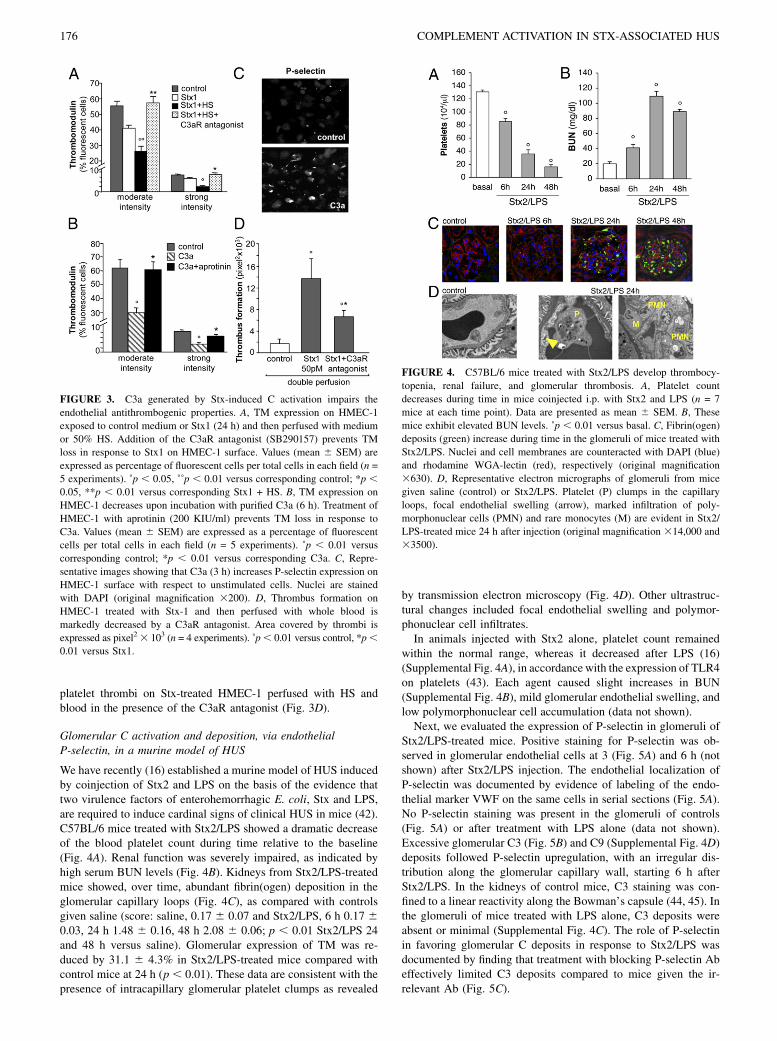
We have recently (16) established a murine model of HUS inducedby coinjection of Stx2 and LPS on the basis of the evidence thattwo virulence factors of enterohemorrhagic E. coli, Stx and LPS,are required to induce cardinal signs of clinical HUS in mice (42).C57BL/6 mice treated with Stx2/LPS showed a dramatic decreaseof the blood platelet count during time relative to the baseline(Fig. 4A). Renal function was severely impaired, as indicated byhigh serum BUN levels (Fig. 4B). Kidneys from Stx2/LPS-treatedmice showed, over time, abundant fibrin(ogen) deposition in theglomerular capillary loops (Fig. 4C), as compared with controlsgiven saline (score: saline, 0.17 6 0.07 and Stx2/LPS, 6 h 0.17 60.03, 24 h 1.48 6 0.16, 48 h 2.08 6 0.06; p , 0.01 Stx2/LPS 24and 48 h versus saline). Glomerular expression of TM was re-duced by 31.1 6 4.3% in Stx2/LPS-treated mice compared withcontrol mice at 24 h (p , 0.01). These data are consistent with thepresence of intracapillary glomerular platelet clumps as revealed
by transmission electron microscopy (Fig. 4D). Other ultrastruc-tural changes included focal endothelial swelling and polymor-phonuclear cell infiltrates.In animals injected with Stx2 alone, platelet count remained
within the normal range, whereas it decreased after LPS (16)(Supplemental Fig. 4A), in accordance with the expression of TLR4on platelets (43). Each agent caused slight increases in BUN(Supplemental Fig. 4B), mild glomerular endothelial swelling, andlow polymorphonuclear cell accumulation (data not shown).Next, we evaluated the expression of P-selectin in glomeruli of
Stx2/LPS-treated mice. Positive staining for P-selectin was ob-served in glomerular endothelial cells at 3 (Fig. 5A) and 6 h (notshown) after Stx2/LPS injection. The endothelial localization ofP-selectin was documented by evidence of labeling of the endo-thelial marker VWF on the same cells in serial sections (Fig. 5A).No P-selectin staining was present in the glomeruli of controls(Fig. 5A) or after treatment with LPS alone (data not shown).Excessive glomerular C3 (Fig. 5B) and C9 (Supplemental Fig. 4D)deposits followed P-selectin upregulation, with an irregular dis-tribution along the glomerular capillary wall, starting 6 h afterStx2/LPS. In the kidneys of control mice, C3 staining was con-fined to a linear reactivity along the Bowman’s capsule (44, 45). Inthe glomeruli of mice treated with LPS alone, C3 deposits wereabsent or minimal (Supplemental Fig. 4C). The role of P-selectinin favoring glomerular C deposits in response to Stx2/LPS wasdocumented by finding that treatment with blocking P-selectin Abeffectively limited C3 deposits compared to mice given the ir-relevant Ab (Fig. 5C).
FIGURE 3. C3a generated by Stx-induced C activation impairs the
endothelial antithrombogenic properties. A, TM expression on HMEC-1
exposed to control medium or Stx1 (24 h) and then perfused with medium
or 50% HS. Addition of the C3aR antagonist (SB290157) prevents TM
loss in response to Stx1 on HMEC-1 surface. Values (mean 6 SEM) are
expressed as percentage of fluorescent cells per total cells in each field (n =
5 experiments). ˚p , 0.05, ˚˚p , 0.01 versus corresponding control; *p ,0.05, **p , 0.01 versus corresponding Stx1 + HS. B, TM expression on
HMEC-1 decreases upon incubation with purified C3a (6 h). Treatment of
HMEC-1 with aprotinin (200 KIU/ml) prevents TM loss in response to
C3a. Values (mean 6 SEM) are expressed as a percentage of fluorescent
cells per total cells in each field (n = 5 experiments). ˚p , 0.01 versus
corresponding control; *p , 0.01 versus corresponding C3a. C, Repre-
sentative images showing that C3a (3 h) increases P-selectin expression on
HMEC-1 surface with respect to unstimulated cells. Nuclei are stained
with DAPI (original magnification 3200). D, Thrombus formation on
HMEC-1 treated with Stx-1 and then perfused with whole blood is
markedly decreased by a C3aR antagonist. Area covered by thrombi is
expressed as pixel2 3 103 (n = 4 experiments). ˚p, 0.01 versus control, *p,0.01 versus Stx1.
FIGURE 4. C57BL/6 mice treated with Stx2/LPS develop thrombocy-
topenia, renal failure, and glomerular thrombosis. A, Platelet count
decreases during time in mice coinjected i.p. with Stx2 and LPS (n = 7
mice at each time point). Data are presented as mean 6 SEM. B, These
mice exhibit elevated BUN levels. ˚p , 0.01 versus basal. C, Fibrin(ogen)
deposits (green) increase during time in the glomeruli of mice treated with
Stx2/LPS. Nuclei and cell membranes are counteracted with DAPI (blue)
and rhodamine WGA-lectin (red), respectively (original magnification
3630). D, Representative electron micrographs of glomeruli from mice
given saline (control) or Stx2/LPS. Platelet (P) clumps in the capillary
loops, focal endothelial swelling (arrow), marked infiltration of poly-
morphonuclear cells (PMN) and rare monocytes (M) are evident in Stx2/
LPS-treated mice 24 h after injection (original magnification 314,000 and
33500).
176 COMPLEMENT ACTIVATION IN STX-ASSOCIATED HUS

C3a derived by C activation, via the alternative pathway,contributes to a glomerular prothrombotic state in HUS mice
To establish the involvement of C activation in the microvascularthrombotic process, we used mice deficient for factor B, a zymogenthat carries the catalytic site of the alternative pathway C3 and C5convertases (46). After Stx2/LPS treatment, Bf2/2 mice exhibitedless thrombocytopenia and were protected against renal functionimpairment compared with wild type littermate mice (Fig. 6A,6B). The blockade of the alternative pathway of C activation alsoresulted in fewer fibrin(ogen) deposits (Fig. 6C; 48 h, score: sa-line, Bf2/2 0.19 6 0.04 versus wild type 0.22 6 0.06; Stx2/LPS,Bf2/2 0.70 6 0.23 versus wild type 2.15 6 0.09; p , 0.01). NoC3 deposits or platelet clumps were found in the glomeruli ofStx2/LPS-treated Bf2/2 mice. Moreover, while glomerular TMexpression was reduced by 42.8 6 7.3% at 48 h in wild type micegiven Stx2/LPS compared with saline-treated control mice (Fig.6D), no changes were observed in Bf2/2 mice regarding corre-sponding controls (Fig. 6D), thereby suggesting the causal role ofC activation in the loss of thromboresistance.The contribution of C3a in the thrombogenic process induced by
Stx was evaluated by blocking C3aR in HUS mice. Administrationof a C3aR antagonist to Stx2/LPS-treated C57BL/6 mice signifi-cantly (p , 0.05) limited glomerular fibrin(ogen) deposition andplatelet clumps (not shown) compared to mice receiving vehicle(Fig. 7A). In this study, we also assessed TM expression and found
that C3aR antagonist limited (p , 0.01) glomerular TM loss afterStx2/LPS (Fig 7B). A scheme of the mechanisms suggested by thepresent data is shown in Fig. 8.
DiscussionTo our knowledge, this study is the first to demonstrate by mul-tifaceted approaches that Stx triggers microvascular thrombosisvia the alternative pathway of C activation and indicates C3a asthe key factor in the development of the thrombotic processesin HUS. Previous findings of low serum C3 levels (25, 27) andaugmented breakdown C products (27, 28) suggested the involve-
FIGURE 5. Glomerular endothelial expression of P-selectin causes C3
deposits in C57BL/6 mice treated with Stx2/LPS. A, Staining of P-selectin
in glomeruli of mice injected with saline (control) or Stx2/LPS at 3 h.
VWF staining in an adjacent renal section of Stx2/LPS-treated mouse
reflects localization of P-selectin in glomerular endothelial cells (arrow).
Original magnification 3630. B, Glomerular C3 staining increases 6 h
after Stx2/LPS treatment with respect to saline (control) as assessed by
semiquantitative score. Data are mean 6 SEM (n = 7 mice for each time
point). Original magnification 3630. C, P-selectin blockade limits glo-
merular C3 deposits in C57BL/6 HUS mice. Mice are injected with the
anti-P-selectin (anti-Psel) or irrelevant (irrel) Abs and sacrificed after 24 h
(n = 5 mice for group). Control mice receive saline (n = 5). Data are mean6SEM. ˚p , 0.05, ˚˚p , 0.01 versus control, *p , 0.05 versus irrel Ab.
FIGURE 6. Activation of C, via the alternative pathway, favors C3a-
dependent glomerular prothrombotic state in mice with HUS. A, In Bf2/2
mice the fall in platelet count after Stx2/LPS treatment is significantly less
than in wild type littermates. B, Bf2/2 mice are protected from renal
function impairment, assessed as serum BUN, with respect to wild type
after Stx2/LPS (n = 6–10 mice per group). Data are mean 6 SEM. ˚p ,0.05, ˚˚p , 0.01 versus wild type. C, Few glomerular fibrin(ogen) deposits
in Bf2/2 mice given Stx2/LPS are present versus wild type mice exhibiting
abundant fibrin(ogen) staining (green). Nuclei are stained with DAPI
(blue), and cell membranes are stained with rhodamine WGA-lectin (red;
original magnification 3630). D, Representative images of TM expression
in glomeruli of wild type and Bf2/2 mice evaluated 48 h after saline or
Stx2/LPS injection. TM expression decreases in wild type but not in Bf2/2
mice after Stx2/LPS injection (original magnification 3630).
FIGURE 7. C3a contributes to the thrombogenic process in mice with
HUS. A, C3aR blockade limits fibrin(ogen) deposition in the glomeruli of
C57BL/6 HUS mice. Mice are treated with C3aR antagonist (SB290157)
or with vehicle (n = 4 mice per group), and sacrificed after 24 h. Control
mice receive saline (n = 4). Fibrin(ogen) deposits are assessed by semi-
quantitative score. Data are mean 6 SEM. B, Loss of glomerular TM
expression after Stx2/LPS injection is limited by treatment with a C3aR
antagonist. Data are mean 6 SEM of n = 4 mice per group. ˚˚p , 0.01
versus control, *p , 0.05, **p , 0.01 versus vehicle.
The Journal of Immunology 177

ment of C in D+HUS, although the functional relevance of theseabnormalities to the thrombotic process was not addressed. Ourdata provide evidence that Stx1 promotes C3 activation and de-position on microvascular endothelial cells via the alternativepathway, because no inhibition was found by blocking the clas-sical pathway. A relevant observation is that C3 deposition favorsthrombus growth under flow on the surface of Stx-treated HMEC-1, which is prevented by the C inhibitor sCR-1. This mechanismapplies also in vivo, because Bf2/2 mice with impaired C acti-vation via the alternative pathway were protected from renalprothrombotic processes when injected with Stx2/LPS comparedto wild-type mice that showed intraglomerular C3 and fibrin(ogen)deposition, TM loss, and platelet clumps associated with severethrombocytopenia.In the search for mechanisms underlying C activation elicited
by Stx and ultimately leading to thrombus formation, we haveidentified P-selectin as an endothelial binding site for C3. As shownpreviously (17) and documented in this study, upon challenge withStx the microvascular endothelium expresses P-selectin, an ad-hesive molecule implicated in VWF-dependent platelet deposi-tion (47). Rather than simply a marker of endothelial activation, P-selectin acts as a specific ligand for C3 through high-affinitybinding, based on the evidence that it colocalizes with the ma-jority of C3 deposits on Stx-treated HMEC-1. P-selectin is crucialin C-dependent thrombus formation because functional blockingof P-selectin leads to a marked reduction of C3 accumulation andlimits thrombus extension on Stx-treated endothelial cells. Theprocesses by which P-selectin favors C activation on endothelialcells are still poorly defined. It is possible that C3b bound to P-selectin may undergo conformational changes that either preventits inactivation by C regulatory molecules or favor C3b interactionwith factor B, thus leading to assembly of alternative pathway C3convertase.We have also provided a novel mechanism through which ac-
tivated C proteins foster thrombosis on Stx-activated endothelium.In our experimental setting, Stxs promoted C3 activation and de-position that resulted in C3a generation; therefore, we focused onthe endothelial effect of this anaphylatoxin. C3a powerfully in-creased P-selectin expression and reduced TM on the HMEC-1surface, indicating a major role for C3a as a driving factor in al-tering endothelial thromboresistance. The proof of this conceptrests on data showing that TM loss and thrombus growth on Stx-treated endothelium were markedly reduced by C3aR antagonist.
However, we cannot exclude a contribution of C5a in the pro-thrombotic effects triggered by Stx, considering that C5a shareswith C3a various inflammatory activities on endothelial cells(22–24). It is known that TM, a cofactor of thrombin in the activa-tion of protein C, regulates hemostasis and prevents local fibrinformation (40, 48, 49). Other properties of TM include anti-inflammatory and cytoprotective activities (40). It is thereforeconceivable that shedding of TM from the endothelial surface cancontribute to an increased risk of thrombosis associated with in-flammation. In this regard, increased levels of soluble TM havebeen detected in the plasma of patients with disseminated intra-vascular coagulation syndrome, pulmonary thromboembolism, andchronic renal disorders (50). Little is known about the mechanismsunderlying TM shedding and the proteases involved in its cleav-age. Serine proteases were reported to be implicated in thiol-induced proteolytic cleavage of TM in association with damageof endothelial cell membrane (51, 52). Based on the evidence thatt-PA is the serine protease stored in Weibel-Palade bodies of en-dothelial cells, we show that the inhibition of t-PA by aprotininmarkedly limited C3a-dependent TM loss on microvascular en-dothelial cells. This finding was also supported by the direct effectof exogenously added t-PA on TM shedding in HMEC-1. Ourfindings shed light on the nexus among endothelium, C, andthrombosis, considering that previous knowledge of the pro-thrombotic effect of C was restricted to studies on platelets (53).Proteins of the C system, including C3, C5, C6, C7, C8, and C9,significantly enhanced thrombin-induced aggregation and secre-tion in human platelets. Furthermore, the anaphylatoxin C3awas shown to directly induce aggregation of human platelets andrelease of serotonin (53). That the mechanisms of thrombosis maybe related to C activation has been also suggested by studies inpatients with familial or sporadic forms of atypical HUS or parox-ysmal nocturnal hemoglobinuria, both characterized by deficiencyin C-regulatory molecules, platelet hyperactivity, and thrombosis(33, 54).The mechanisms proposed in vitro have been validated in
a murine model of Stx-associated HUS, which closely mimicshuman D+HUS. The development of a mouse model able to re-produce the features of human HUS has been problematic, andstudies using E. coli O157:H7, purified Stx1 or Stx2 with orwithout LPS (16, 35, 36, 42, 55, 56), have given controversialresults. The role of E. coli LPS has been recognized in the mo-lecular pathogenesis of D+HUS, based on the evidence of serum
FIGURE 8. Stx alters endothelial thromboresistance by triggering exuberant C activation via C3a, leading to microvascular thrombosis. The proposed
scheme summarizes the sequence of events through which Stx binds to its specific endothelial receptor Gb3 and favors surface mobilization of P-selectin
and thrombus formation under flow by either interacting with platelets or by binding and/or activating C proteins. Excessive C3 activation (dependent on
increased C3 convertase assembly and activity) in response to Stx generates an increased amount of the anaphylatoxin C3a, which interacts with the specific
endothelial C3aR, thereby potentiating P-selectin expression and t-PA-dependent TM shedding with final thrombus formation.
178 COMPLEMENT ACTIVATION IN STX-ASSOCIATED HUS

Abs against O157 LPS in children with D+HUS (57). In a baboonmodel of HUS, LPS increased the toxicity of Stx (58). Consis-tently, both Stx2 and LPS were required to elicit a HUS-like re-sponse in mice (16, 36, 42). Our experiments document that inmice treated with Stx2/LPS that developed thrombocytopenia andrenal failure, enhanced P-selectin expression on glomerular endo-thelial cells preceded C3 deposits. Accumulation of C3 was as-sociated with fibrin(ogen) deposits, platelet clumps, and TM loss.P-selectin blockade limited glomerular C3 deposits, which arguesin favor of a causal relationship between endothelial P-selectin andC deposition. By means of Bf2/2 mice that were protected againstglomerular alterations and renal function impairment in responseto Stx2/LPS, we showed that C activation via the alternativepathway was instrumental to microvascular thrombosis in HUSmice. This study also highlights the critical role of C3a, derivedfrom glomerular C activation, in potentiating glomerular throm-botic processes. This finding is supported by data showing thattreatment with a C3aR antagonist markedly reduced fibrin(ogen)and platelet accumulation and limited the loss of TM in the glo-meruli of HUS mice.As suggested earlier (59), our results corroborate concepts that
may prove of general significance in the understanding of thecauses that trigger thrombosis and vascular occlusions. We pre-viously explored the heterogeneity of endothelial cells based onthe variable expression of specific molecules that determineda distinct response to stimuli (17) and demonstrated the link be-tween infections and thrombosis. In this study, we elaborated onthose concepts by documenting in a specific setting a phenomenonof interaction between C3 and P-selectin leading to the forma-tion of C3a, which appears to be instrumental in microvascularthrombosis, and may have wider implications for acute occlusivevascular accidents in a broader sense.In conclusion, as summarized in Fig. 8, we have demonstrated
by in vitro and in vivo experiments that Stx, by activating micro-vascular endothelial cells to express P-selectin, allows C3 deposi-tion through the alternative pathway, favoring thrombus formation.Moreover, we have identified C3a, generated by Stx-induced Cactivation, as the key factor responsible for further expression ofP-selectin, TM loss, and thrombus formation. These findings serveto suggest C inhibitors as possible therapeutic tools in the controlof D+HUS. In particular, C3aR antagonists, currently availablein preclinical studies (60) could represent a promising strategy.Currently, C inhibitors that have entered clinical studies were re-stricted to anti-C5 Abs that showed beneficial effects in paroxys-mal nocturnal hemoglobinuria (61), myocardial infarction (62),and atypical non–Stx-associated HUS (63, 64).
AcknowledgmentsWe thank Dr. A. Benigni for helpful suggestions and criticism, D. Corna and
C. Zanchi for technical assistance, Dr. E. Ades and Dr. F.J. Candal (Centers
for Disease Control and Prevention, Atlanta, GA) and Dr. T. Lawley (Emory
University) for developing developed HMEC-1, Dr. H. Karch and Dr.
M. Bielaszewska (Institute for Hygiene, University of Munster, Germany)
for providing purified Stx1, Dr. M. Botto (Imperial College, London, U.K.)
for the gift of the Bf2/2 mice, and CellDex (Needham, MA) for providing
sCR-1.
DisclosuresThe authors have no financial conflicts of interest.
References1. Tarr, P. I., C. A. Gordon, and W. L. Chandler. 2005. Shiga-toxin-producing
Escherichia coli and haemolytic uraemic syndrome. Lancet 365: 1073–1086.
2. Scheiring, J., S. P. Andreoli, and L. B. Zimmerhackl. 2008. Treatment andoutcome of Shiga-toxin-associated hemolytic uremic syndrome (HUS). Pediatr.Nephrol. 23: 1749–1760.
3. Noris, M., and G. Remuzzi. 2005. Hemolytic uremic syndrome. J. Am. Soc.Nephrol. 16: 1035–1050.
4. Garg, A. X., R. S. Suri, N. Barrowman, F. Rehman, D. Matsell, M. P. Rosas-Arellano, M. Salvadori, R. B. Haynes, and W. F. Clark. 2003. Long-term renalprognosis of diarrhea-associated hemolytic uremic syndrome: a systematic re-view, meta-analysis, and meta-regression. JAMA 290: 1360–1370.
5. Karmali, M. A. 2004. Infection by Shiga toxin-producing Escherichia coli: anoverview. Mol. Biotechnol. 26: 117–122.
6. Mark Taylor, C. 2008. Enterohaemorrhagic Escherichia coli and Shigella dys-enteriae type 1-induced haemolytic uraemic syndrome. Pediatr. Nephrol. 23:1425–1431.
7. Manning, S. D., A. S. Motiwala, A. C. Springman, W. Qi, D. W. Lacher,L. M. Ouellette, J. M. Mladonicky, P. Somsel, J. T. Rudrik, S. E. Dietrich, et al.2008. Variation in virulence among clades of Escherichia coli O157:H7 asso-ciated with disease outbreaks. Proc. Natl. Acad. Sci. USA 105: 4868–4873.
8. Michael, M., E. J. Elliott, J. C. Craig, G. Ridley, and E. M. Hodson. 2009.Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenicpurpura: a systematic review of randomized controlled trials. Am. J. Kidney Dis.53: 259–272.
9. Bitzan, M. 2009. Treatment options for HUS secondary to Escherichia coliO157:H7. Kidney Int. Suppl. 75: S62–S66.
10. Boyd, B., and C. Lingwood. 1989. Verotoxin receptor glycolipid in human renaltissue. Nephron 51: 207–210.
11. O’Loughlin, E. V., and R. M. Robins-Browne. 2001. Effect of Shiga toxin andShiga-like toxins on eukaryotic cells. Microbes Infect. 3: 493–507.
12. Obrig, T. G., C. B. Louise, C. A. Lingwood, B. Boyd, L. Barley-Maloney, andT. O. Daniel. 1993. Endothelial heterogeneity in Shiga toxin receptors andresponses. J. Biol. Chem. 268: 15484–15488.
13. Morigi, M., G. Micheletti, M. Figliuzzi, B. Imberti, M. A. Karmali, A. Remuzzi,G. Remuzzi, and C. Zoja. 1995. Verotoxin-1 promotes leukocyte adhesion tocultured endothelial cells under physiologic flow conditions. Blood 86: 4553–4558.
14. Forsyth, K. D., A. C. Simpson, M. M. Fitzpatrick, T. M. Barratt andR. J. Levinsky. 1989. Neutrophil-mediated endothelial injury in haemolyticuraemic syndrome. Lancet 2: 411–414.
15. Zoja, C., S. Angioletti, R. Donadelli, C. Zanchi, S. Tomasoni, E. Binda,B. Imberti, M. te Loo, L. Monnens, G. Remuzzi, and M. Morigi. 2002. Shigatoxin-2 triggers endothelial leukocyte adhesion and transmigration via NF-kappaB dependent up-regulation of IL-8 and MCP-1. Kidney Int. 62: 846–856.
16. Zanchi, C., C. Zoja, M. Morigi, F. Valsecchi, X. Y. Liu, D. Rottoli, M. Locatelli,S. Buelli, A. Pezzotta, P. Mapelli, et al. 2008. Fractalkine and CX3CR1 mediateleukocyte capture by endothelium in response to Shiga toxin. J. Immunol. 181:1460–1469.
17. Morigi, M., M. Galbusera, E. Binda, B. Imberti, S. Gastoldi, A. Remuzzi,C. Zoja, and G. Remuzzi. 2001. Verotoxin-1-induced up-regulation of adhesivemolecules renders microvascular endothelial cells thrombogenic at high shearstress. Blood 98: 1828–1835.
18. Guessous, F., M. Marcinkiewicz, R. Polanowska-Grabowska, S. Kongkhum,D. Heatherly, T. Obrig, and A. R. Gear. 2005. Shiga toxin 2 and lipopolysac-charide induce human microvascular endothelial cells to release chemokines andfactors that stimulate platelet function. Infect. Immun. 73: 8306–8316.
19. Zoja, C., S. Buelli, and M. Morigi. 2010. Shiga toxin-associated hemolyticuremic syndrome: pathophysiology of endothelial dysfunction. Pediatr. Nephrol.25: 2231–2240.
20. Kamitsuji, H., K. Nonami, N. Ishikawa, T. Murakami, A. Nakayama, Y. Umeki,and M. Nakajima. 1999. Plasma P-selectin in children with hemolytic uremicsyndrome caused by Escherichia coli O157:H7. J. Nara Med. Assoc. 50: 515–523.
21. Del Conde, I., M. A. Cruz, H. Zhang, J. A. Lopez, and V. Afshar-Kharghan.2005. Platelet activation leads to activation and propagation of the complementsystem. J. Exp. Med. 201: 871–879.
22. Monsinjon, T., P. Gasque, P. Chan, A. Ischenko, J. J. Brady, and M. C. Fontaine.2003. Regulation by complement C3a and C5a anaphylatoxins of cytokineproduction in human umbilical vein endothelial cells. FASEB J. 17: 1003–1014.
23. Schraufstatter, I. U., K. Trieu, L. Sikora, P. Sriramarao, and R. DiScipio. 2002.Complement c3a and c5a induce different signal transduction cascades in en-dothelial cells. J. Immunol. 169: 2102–2110.
24. Kohl, J. 2001. Anaphylatoxins and infectious and non-infectious inflammatorydiseases. Mol. Immunol. 38: 175–187.
25. Robson, W. L., A. K. Leung, G. H. Fick, and A. I. McKenna. 1992. Hypo-complementemia and leukocytosis in diarrhea-associated hemolytic uremicsyndrome. Nephron 62: 296–299.
26. Koster, F. T., V. Boonpucknavig, S. Sujaho, R. H. Gilman, and M. M. Rahaman.1984. Renal histopathology in the hemolytic-uremic syndrome following shig-ellosis. Clin. Nephrol. 21: 126–133.
27. Monnens, L., J. Molenaar, P. H. Lambert, W. Proesmans, and P. van Munster.1980. The complement system in hemolytic-uremic syndrome in childhood.Clin. Nephrol. 13: 168–171.
28. Thurman, J. M., R. Marians, W. Emlen, S. Wood, C. Smith, H. Akana,V. M. Holers, M. Lesser, M. Kline, C. Hoffman, et al. 2009. Alternative pathwayof complement in children with diarrhea-associated hemolytic uremic syndrome.Clin. J. Am. Soc. Nephrol. 4: 1920–1924.
29. Orth, D., A. B. Khan, A. Naim, K. Grif, J. Brockmeyer, H. Karch, M. Joannidis,S. J. Clark, A. J. Day, S. Fidanzi, et al. 2009. Shiga toxin activates complement
The Journal of Immunology 179

and binds factor H: evidence for an active role of complement in hemolyticuremic syndrome. J. Immunol. 182: 6394–6400.
30. Karmali, M. A., M. Petric, C. Lim, P. C. Fleming, G. S. Arbus, and H. Lior. 1985.The association between idiopathic hemolytic uremic syndrome and infection byverotoxin-producing Escherichia coli. J. Infect. Dis. 151: 775–782.
31. Ades, E. W., F. J. Candal, R. A. Swerlick, V. G. George, S. Summers,D. C. Bosse, and T. J. Lawley. 1992. HMEC-1: establishment of an immortalizedhuman microvascular endothelial cell line. J. Invest. Dermatol. 99: 683–690.
32. Meisen, I., A. W. Friedrich, H. Karch, U. Witting, J. Peter-Katalinic, andJ. Muthing. 2005. Application of combined high-performance thin-layer chro-matography immunostaining and nanoelectrospray ionization quadrupole time-of-flight tandem mass spectrometry to the structural characterization of high- andlow-affinity binding ligands of Shiga toxin 1. Rapid Commun. Mass Spectrom.19: 3659–3665.
33. Stahl, A. L., F. Vaziri-Sani, S. Heinen, A. C. Kristoffersson, K. H. Gydell,R. Raafat, A. Gutierrez, O. Beringer, P. F. Zipfel, and D. Karpman. 2008. FactorH dysfunction in patients with atypical hemolytic uremic syndrome contributesto complement deposition on platelets and their activation. Blood 111: 5307–5315.
34. Matsumoto, M., W. Fukuda, A. Circolo, J. Goellner, J. Strauss-Schoenberger,X. Wang, S. Fujita, T. Hidvegi, D. D. Chaplin, and H. R. Colten. 1997. Abro-gation of the alternative complement pathway by targeted deletion of murinefactor B. Proc. Natl. Acad. Sci. USA 94: 8720–8725.
35. Rutjes, N. W., B. A. Binnington, C. R. Smith, M. D. Maloney andC. A. Lingwood. 2002. Differential tissue targeting and pathogenesis of ver-otoxins 1 and 2 in the mouse animal model. Kidney Int. 62: 832–845.
36. Ikeda, M., S. Ito, and M. Honda. 2004. Hemolytic uremic syndrome induced bylipopolysaccharide and Shiga-like toxin. Pediatr. Nephrol. 19: 485–489.
37. Borges, E., R. Eytner, T. Moll, M. Steegmaier, M. A. Campbell, K. Ley,H. Mossmann, and D. Vestweber. 1997. The P-selectin glycoprotein ligand-1 isimportant for recruitment of neutrophils into inflamed mouse peritoneum. Blood90: 1934–1942.
38. Porteu, F., and L. Halbwachs-Mecarelli. 1988. Interaction of human monomericC3b with its receptor (complement receptor type 1, CR1) on neutrophils. Evi-dence for negative cooperativity. J. Biol. Chem. 263: 5091–5097.
39. Klickstein, L. B., T. J. Bartow, V. Miletic, L. D. Rabson, J. A. Smith, andD. T. Fearon. 1988. Identification of distinct C3b and C4b recognition sites in thehuman C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J. Exp. Med.168: 1699–1717.
40. Van de Wouwer, M., and E. M. Conway. 2004. Novel functions of thrombo-modulin in inflammation. Crit. Care Med. 32(5 Suppl): S254–S261.
41. Huber, D., E. M. Cramer, J. E. Kaufmann, P. Meda, J. M. Masse, E. K. Kruithof,and U. M. Vischer. 2002. Tissue-type plasminogen activator (t-PA) is stored inWeibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood99: 3637–3645.
42. Keepers, T. R., M. A. Psotka, L. K. Gross, and T. G. Obrig. 2006. A murinemodel of HUS: Shiga toxin with lipopolysaccharide mimics the renal damageand physiologic response of human disease. J. Am. Soc. Nephrol. 17: 3404–3414.
43. Aslam, R., E. R. Speck, M. Kim, A. R. Crow, K. W. Bang, F. P. Nestel, H. Ni,A. H. Lazarus, J. Freedman, and J. W. Semple. 2006. Platelet Toll-like receptorexpression modulates lipopolysaccharide-induced thrombocytopenia and tumornecrosis factor-alpha production in vivo. Blood 107: 637–641.
44. Turnberg, D., M. Lewis, J. Moss, Y. Xu, M. Botto, and H. T. Cook. 2006.Complement activation contributes to both glomerular and tubulointerstitialdamage in adriamycin nephropathy in mice. J. Immunol. 177: 4094–4102.
45. Cunningham, P. N., V. M. Holers, J. J. Alexander, J. M. Guthridge, M. C. Carroll,and R. J. Quigg. 2000. Complement is activated in kidney by endotoxin but doesnot cause the ensuing acute renal failure. Kidney Int. 58: 1580–1587.
46. Walport, M. J. 2001. Complement. First of two parts. N. Engl. J. Med. 344:1058–1066.
47. Frenette, P. S., R. C. Johnson, R. O. Hynes, and D. D. Wagner. 1995. Plateletsroll on stimulated endothelium in vivo: an interaction mediated by endothelialP-selectin. Proc. Natl. Acad. Sci. USA 92: 7450–7454.
48. Delvaeye, M., and E. M. Conway. 2009. Coagulation and innate immuneresponses: can we view them separately? Blood 114: 2367–2374.
49. Weiler-Guettler, H., P. D. Christie, D. L. Beeler, A. M. Healy, W. W. Hancock,H. Rayburn, J. M. Edelberg, and R. D. Rosenberg. 1998. A targeted point mu-tation in thrombomodulin generates viable mice with a prethrombotic state. J.Clin. Invest. 101: 1983–1991.
50. Takano, S., S. Kimura, S. Ohdama, and N. Aoki. 1990. Plasma thrombomodulinin health and diseases. Blood 76: 2024–2029.
51. Menschikowski, M., A. Hagelgans, G. Eisenhofer, O. Tiebel, and G. Siegert.2010. Reducing agents induce thrombomodulin shedding in human endothelialcells. Thromb. Res. 126: e88–e93.
52. Boehme, M. W., P. Galle, and W. Stremmel. 2002. Kinetics of thrombomodulinrelease and endothelial cell injury by neutrophil-derived proteases and oxygenradicals. Immunology 107: 340–349.
53. Polley, M. J., and R. Nachman. 1978. The human complement system inthrombin-mediated platelet function. J. Exp. Med. 147: 1713–1726.
54. Gralnick, H. R., M. Vail, L. P. McKeown, P. Merryman, O. Wilson, I. Chu, andJ. Kimball. 1995. Activated platelets in paroxysmal nocturnal haemoglobinuria.Br. J. Haematol. 91: 697–702.
55. Karpman, D., H. Connell, M. Svensson, F. Scheutz, P. Alm, and C. Svanborg.1997. The role of lipopolysaccharide and Shiga-like toxin in a mouse model ofEscherichia coli O157:H7 infection. J. Infect. Dis. 175: 611–620.
56. Paixao-Cavalcante, D., M. Botto, H. T. Cook, and M. C. Pickering. 2009. Shigatoxin-2 results in renal tubular injury but not thrombotic microangiopathy inheterozygous factor H-deficient mice. Clin. Exp. Immunol. 155: 339–347.
57. Bitzan, M., E. Moebius, K. Ludwig, D. E. Muller-Wiefel, J. Heesemann, andH. Karch. 1991. High incidence of serum antibodies to Escherichia coli O157lipopolysaccharide in children with hemolytic-uremic syndrome. J. Pediatr. 119:380–385.
58. Siegler, R. L., T. J. Pysher, R. Lou, V. L. Tesh, and F. B. Taylor Jr. 2001. Re-sponse to Shiga toxin-1, with and without lipopolysaccharide, in a primate modelof hemolytic uremic syndrome. Am. J. Nephrol. 21: 420–425.
59. Ruggeri, Z. M. 2001. Endothelial cells: they only look all alike. Blood 98: 1644.60. Bao, L., I. Osawe, M. Haas, and R. J. Quigg. 2005. Signaling through up-
regulated C3a receptor is key to the development of experimental lupus ne-phritis. J. Immunol. 175: 1947–1955.
61. Hillmen, P., C. Hall, J. C. Marsh, M. Elebute, M. P. Bombara, B. E. Petro,M. J. Cullen, S. J. Richards, S. A. Rollins, C. F. Mojcik, and R. P. Rother. 2004.Effect of eculizumab on hemolysis and transfusion requirements in patients withparoxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 350: 552–559.
62. Granger, C. B., K. W. Mahaffey, W. D. Weaver, P. Theroux, J. S. Hochman,T. G. Filloon, S. Rollins, T. G. Todaro, J. C. Nicolau, W. Ruzyllo,P. W. Armstrong; COMMA Investigators. 2003. Pexelizumab, an anti-C5 com-plement antibody, as adjunctive therapy to primary percutaneous coronaryintervention in acute myocardial infarction: the COMplement inhibition inMyocardial infarction treated with Angioplasty (COMMA) trial. Circulation108: 1184–1190.
63. Nurnberger, J., T. Philipp, O. Witzke, A. Opazo Saez, U. Vester, H. A. Baba,A. Kribben, L. B. Zimmerhackl, A. R. Janecke, M. Nagel, and M. Kirschfink.2009. Eculizumab for atypical hemolytic-uremic syndrome. N. Engl. J. Med.360: 542–544.
64. Gruppo, R. A., and R. P. Rother. 2009. Eculizumab for congenital atypicalhemolytic-uremic syndrome. N. Engl. J. Med. 360: 544–546.
180 COMPLEMENT ACTIVATION IN STX-ASSOCIATED HUS
Related Documents











