Short communication Alternating Hemiplegia and Epilepsia Partialis Continua: A new phenotype for a novel compound TBC1D24 mutation Francesca Ragona a, 1 , Barbara Castellotti b, 1 , Barbara Salis a,c , Stefania Magri b , Jacopo C. DiFrancesco d,e , Nardo Nardocci a , Silvana Franceschetti e , Cinzia Gellera b , Tiziana Granata a, * a Department of Pediatric Neuroscience, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy b Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy c University of Sassari, Sassari, Italy d San Gerardo Hospital, University of Milano-Bicocca, Monza, Italy e Clinical Neurophysiology and Epilepsy Center, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy A R T I C L E I N F O Article history: Received 5 December 2016 Received in revised form 27 February 2017 Accepted 2 March 2017 Keywords: TBC1D24 gene Epilepsia Partialis Continua Alternating Hemiplegia Epilepsy Next generation sequencing Plegic attack A B S T R A C T Mutations in the TBC1D24 gene (MIM 613577) cause familial infantile myoclonic epilepsy (FIME; 605021) and early infantile epileptic encephalopathy-16 (EIEE16; 615338), both inherited with an autosomal recessive trait. The TBC1D24 gene encodes a member of the TBC family domain proteins, involved in cell signaling and oxidative stress resistance. We studied, by a Next Generation Sequencing (NGS) target re- sequencing gene approach, the DNA of a 5 year-old girl, affected by recurrent attacks of Alternating Hemiplegia (AH) and by recurrent episodes of Epilepsia Partialis Continua (EPC). The NGS study showed the presence of two different heterozygous, probably pathogenic variants in the TBC1D24 gene, inherited in trans from her parents: the c.116C>T (p.Ala39Val) and the c.457G>A (p.Glu153Lys). This study describes for the first time the association between TBC1D24 variants and AH expanding the phenotypic spectrum of TBC1D24-related diseases and suggesting that TBC1D24 molecular analysis should be considered in the diagnostic work up of AH patients. An additional peculiar feature is the association of AH and EPC. © 2017 British Epilepsy Association. Published by Elsevier Ltd. All rights reserved. 1. Introduction Alternating Hemiplegia (AH) is a rare childhood syndrome characterized by non-epileptic paroxysmal symptoms and stable neurological deficits, associated with ATP1A3 mutations in up to 85% of patients [1]. Epilepsia Partialis Continua (EPC) is a rare variant of epilepsy characterized by continuous focal jerkings of a body part over a period of hours or days, which may persist during sleep. In children, EPC is mostly associated with Rasmussen- encephalitis, mitochondriopathy and brain malformations [2]. Although half of AH patients experience epileptic seizures, no case of EPC has been reported [3]. We report the identification of two variants in TBC1D24 gene in a patient with AH and EPC. 2. Case report A 5 year-old girl presents, since the age of 4 months, paroxysmal symptoms of two types: (1) daily, prolonged episodes of unresponsiveness and hypoto- nia, variably associated with abnormal eye movements, nystag- mus, sialorrhea, and hemi or tetraplegia, which resolved with sleep. (2) bouts of rhythmic clonic jerks of a body part, lasting from minutes to hours, persisting during sleep; the jerks involved different body parts during the distinct attacks. Serial video-EEG-polygraph recordings clarified that the episodes of unresponsiveness were not associated with ictal EEG changes and that the focal jerks were consistent with EPC (Fig. 1). The diagnostic work up which included MRI, evoked potentials, CSF for glycorrhachia and neurotransmitters, karyotyping and CGH array, metabolic screening; molecular analyses of SCL2A1 and POLG1 genes were unrevealing. The age of onset and the semiology of the non-epileptic attacks fulfilled the diagnostic criteria for AH [1], therefore, despite the * Corresponding author at: Department of Pediatric Neuroscience, Fondazione IRCCS Istituto Neurologico Carlo Besta, Via Giovanni Celoria 11, 20133 Milan, Italy. E-mail address: [email protected] (T. Granata). 1 These authors equally contributed to the work. http://dx.doi.org/10.1016/j.seizure.2017.03.003 1059-1311/© 2017 British Epilepsy Association. Published by Elsevier Ltd. All rights reserved. Seizure 47 (2017) 71–73 Contents lists available at ScienceDirect Seizure journal homepage: www.elsevier.com/locate/yseiz

Alternating Hemiplegia and Epilepsia Partialis Continua: A new phenotype for a novel compound TBC1D24 mutation
Dec 10, 2022
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript
Alternating Hemiplegia and Epilepsia Partialis Continua: A new phenotype for a novel compound TBC1D24 mutationShort communication
Alternating Hemiplegia and Epilepsia Partialis Continua: A new phenotype for a novel compound TBC1D24 mutation
Francesca Ragonaa,1, Barbara Castellottib,1, Barbara Salisa,c, Stefania Magrib, Jacopo C. DiFrancescod,e, Nardo Nardoccia, Silvana Franceschettie, Cinzia Gellerab, Tiziana Granataa,* aDepartment of Pediatric Neuroscience, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy bUnit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy cUniversity of Sassari, Sassari, Italy d San Gerardo Hospital, University of Milano-Bicocca, Monza, Italy eClinical Neurophysiology and Epilepsy Center, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
A R T I C L E I N F O
Article history: Received 5 December 2016 Received in revised form 27 February 2017 Accepted 2 March 2017
Keywords: TBC1D24 gene Epilepsia Partialis Continua Alternating Hemiplegia Epilepsy Next generation sequencing Plegic attack
A B S T R A C T
Mutations in the TBC1D24 gene (MIM 613577) cause familial infantile myoclonic epilepsy (FIME; 605021) and early infantile epileptic encephalopathy-16 (EIEE16; 615338), both inherited with an autosomal recessive trait. The TBC1D24 gene encodes a member of the TBC family domain proteins, involved in cell signaling and oxidative stress resistance. We studied, by a Next Generation Sequencing (NGS) target re- sequencing gene approach, the DNA of a 5 year-old girl, affected by recurrent attacks of Alternating Hemiplegia (AH) and by recurrent episodes of Epilepsia Partialis Continua (EPC). The NGS study showed the presence of two different heterozygous, probably pathogenic variants in the TBC1D24 gene, inherited in trans from her parents: the c.116C>T (p.Ala39Val) and the c.457G>A (p.Glu153Lys). This study describes for the first time the association between TBC1D24 variants and AH expanding the phenotypic spectrum of TBC1D24-related diseases and suggesting that TBC1D24 molecular analysis should be considered in the diagnostic work up of AH patients. An additional peculiar feature is the association of AH and EPC.
© 2017 British Epilepsy Association. Published by Elsevier Ltd. All rights reserved.
Contents lists available at ScienceDirect
Seizure
1. Introduction
Alternating Hemiplegia (AH) is a rare childhood syndrome characterized by non-epileptic paroxysmal symptoms and stable neurological deficits, associated with ATP1A3 mutations in up to 85% of patients [1]. Epilepsia Partialis Continua (EPC) is a rare variant of epilepsy characterized by continuous focal jerkings of a body part over a period of hours or days, which may persist during sleep. In children, EPC is mostly associated with Rasmussen- encephalitis, mitochondriopathy and brain malformations [2]. Although half of AH patients experience epileptic seizures, no case of EPC has been reported [3]. We report the identification of two variants in TBC1D24 gene in a patient with AH and EPC.
* Corresponding author at: Department of Pediatric Neuroscience, Fondazione IRCCS Istituto Neurologico Carlo Besta, Via Giovanni Celoria 11, 20133 Milan, Italy.
E-mail address: [email protected] (T. Granata). 1 These authors equally contributed to the work.
http://dx.doi.org/10.1016/j.seizure.2017.03.003 1059-1311/© 2017 British Epilepsy Association. Published by Elsevier Ltd. All rights res
2. Case report
A 5 year-old girl presents, since the age of 4 months, paroxysmal symptoms of two types:
(1) daily, prolonged episodes of unresponsiveness and hypoto- nia, variably associated with abnormal eye movements, nystag- mus, sialorrhea, and hemi or tetraplegia, which resolved with sleep. (2) bouts of rhythmic clonic jerks of a body part, lasting from minutes to hours, persisting during sleep; the jerks involved different body parts during the distinct attacks.
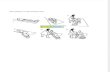
Serial video-EEG-polygraph recordings clarified that the episodes of unresponsiveness were not associated with ictal EEG changes and that the focal jerks were consistent with EPC (Fig. 1).
The diagnostic work up which included MRI, evoked potentials, CSF for glycorrhachia and neurotransmitters, karyotyping and CGH array, metabolic screening; molecular analyses of SCL2A1 and POLG1 genes were unrevealing.
The age of onset and the semiology of the non-epileptic attacks fulfilled the diagnostic criteria for AH [1], therefore, despite the
erved.
72 F. Ragona et al. / Seizure 47 (2017) 71–73
unusual association with EPC, the infant was given Flunarizine, which reduced the frequency of the attacks. Analysis for ATP1A3, performed after its identification as AH causative gene, failed to identify mutations and deletions. The clinical evolution was characterized by developmental delay, already evident at the onset, with general developmental quotient of 46 at the age of 4, trunk hypotonia and limb rigidity with dystonic postures. Episodes of EPC recurred many times a year, lasted from hours to days, and persisted during sleep (Fig. 1a–d). The jerks were not associated with ictal activity on the surface EEG; the jerk-locked back averaging (JLBA) showed that a positive transient over the contralateral central region preceded the jerks by 28 msec, consistent with cortical origin (Fig. 1e).
The attacks, usually associated with eye deviation, unilateral or bilateral nystagmus, tachypnea, and autonomic disorders, recurred many times a week (Supplementary Video S1), mostly triggered by baths, emotions, fatigue or fever, and always ceased with sleep. Topiramate, barbiturate, acetazolamide and benzodiazepines were ineffective.
The EEG follow-up showed the impoverishment of the background activity, and interictal bilateral asynchronous epileptic transients over the fronto-central regions. Given the worsening course and the lack of a definite diagnosis, enzymatic and genetic analyses for mitochondrial and lysosomal disorders were per- formed but were unrevealing.
Finally, the patient entered a Next Generation Sequencing (NGS) analysis through a gene panel of the 92 genes related to epileptic encephalopathies (see Supplementary material for details about methods of molecular study).
NGS analysis in the proband’s DNA revealed the presence of two variants of probably pathogenic significance in the TBC1D24 gene, inherited in trans from her parents: the c.116C > T (p.Ala39Val) and the c.457G > A (p.Glu153Lys).
The c.116C > T (p.Ala39Val) variant is novel and prediction analysis resulting in a probably damaging effect on the mutated protein; the c.457G > A (p.Glu153Lys) variant is already described (rs376712059) and prediction analysis resulting a probably damaging and deleterious effect on the mutated protein (see Supplementary material for details about results methods of molecular study).
The complete NGS data analysis did not reveal the presence of other variants of possible pathological significance or the presence of Variant of Unknown Significance (VUS).
3. Discussion
TBC1D24 encodes a protein containing a TBC domain involved in cell signaling and oxidative stress resistance [4]. Allelic variants in the TBC1D24 gene have been reported in different diseases with multisystem involvement and in a broad variety of epilepsies, from
F. Ragona et al. / Seizure 47 (2017) 71–73 73
benign myoclonic epilepsy, to early-onset epileptic encephalopa- thy with severe developmental delay and early death. The broad spectrum can be explained by the effect of TBC1D24 mutations on synthesis, stability and activity of the protein, and by the effect of TBC1D24-associated proteins and pathways [4].
This is the first report of TBC1D24 variants in a patient who fulfilled the clinical diagnostic criteria for Alternating Hemiplegia [1]. In the large series reported by Balestrini et al., in fact, most patients had, besides seizures, stable – predominantly dystonia, or ataxia – or paroxysmal symptoms. These latter included prolonged post-ictal hemiparesis and focal or axial dystonic episodes, but in no case a constellation of symptoms consistent with AH was reported [4]. A further peculiarity of our patient was the presence of EPC, which has never been described in AH. Epilepsy in AH actually occurs in about 50% of patients: seizures are usually focal, not frequent, and well controlled by AEDs, status epilepticus is uncommon [3]. By contrast, in our patient the epilepsy was severe, with frequent, and long- lasting episodes of EPC, which involved different parts of the body during distinct episodes. This multifocal localization, which is uncommon in EPC [2] probably reflects the diffuse brain dysfunction resulting from TBC1D24 mutation. Although we cannot exclude the presence of other novel genomic variants of possible pathological significance that may contribute to the complex phenotype described in our patient, our hypothesis is that the defective neurotransmission, determined by TBC1D24 mutation might have a role in determining both the seizures and the non-epileptic symptoms. Functional studies on cell models will hopefully clarify the mechanism by which the defective neuro- transmission determined by gene mutation may cause multisys- tem and neurological symptoms.
4. Conclusions
Our finding expands the phenotypic spectrum of TBC1D24- related diseases and suggests to consider TBC1D24 molecular
analysis in the diagnostic work-up of AH patients, especially in those negative for ATP1A3 mutations.
Conflict of interest
Authors have no competing interests. The research did not receive any specific grant from funding agencies in the public, commercial, or not-for profit sectors.
Acknowledgement
The authors thank the parents of patient for their collaboration.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j. seizure.2017.03.003.
References
[1] Panagiotakaki E, De Grandis E, Stagnaro M, Heinzen EL, Fons C, Sisodiya S, et al. Italian IBAHC Consortium; French AHC Consortium; International AHC Consortium. Clinical profile of patients with ATP1A3 mutations in alternating hemiplegia of childhood-a study of 155 patients. Orphanet J Rare Dis 2015;10:123–35.
[2] Kravljanac R, Djuric M, Jovic N, Djordjevic M, Zamurovic D, Pekmezovic T. Etiology, clinical features and outcome of epilepsia partialis continua in cohort of 51 children. Epilepsy Res 2013;104(March (1–2)):112–7, doi:http://dx.doi. org/10.1016/j.eplepsyres.2012.09.003.
[3] Panagiotakaki E, Gobbi G, Neville B, Ebinger F, Campistol J, Nevšímalová S, et al. Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain 2010;133:3598–610, doi: http://dx.doi.org/10.1093/brain/awq295.
[4] Balestrini S, Milh M, Castiglioni C, Lüthy K, Finelli MJ, Verstreken P, et al. TBC1D24 genotype-phenotype correlation epilepsies and other neurologic features. Neurology 2016;87(July (1)):77–85, doi:http://dx.doi.org/10.1212/ WNL.0000000000002807.
1 Introduction
Alternating Hemiplegia and Epilepsia Partialis Continua: A new phenotype for a novel compound TBC1D24 mutation
Francesca Ragonaa,1, Barbara Castellottib,1, Barbara Salisa,c, Stefania Magrib, Jacopo C. DiFrancescod,e, Nardo Nardoccia, Silvana Franceschettie, Cinzia Gellerab, Tiziana Granataa,* aDepartment of Pediatric Neuroscience, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy bUnit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy cUniversity of Sassari, Sassari, Italy d San Gerardo Hospital, University of Milano-Bicocca, Monza, Italy eClinical Neurophysiology and Epilepsy Center, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
A R T I C L E I N F O
Article history: Received 5 December 2016 Received in revised form 27 February 2017 Accepted 2 March 2017
Keywords: TBC1D24 gene Epilepsia Partialis Continua Alternating Hemiplegia Epilepsy Next generation sequencing Plegic attack
A B S T R A C T
Mutations in the TBC1D24 gene (MIM 613577) cause familial infantile myoclonic epilepsy (FIME; 605021) and early infantile epileptic encephalopathy-16 (EIEE16; 615338), both inherited with an autosomal recessive trait. The TBC1D24 gene encodes a member of the TBC family domain proteins, involved in cell signaling and oxidative stress resistance. We studied, by a Next Generation Sequencing (NGS) target re- sequencing gene approach, the DNA of a 5 year-old girl, affected by recurrent attacks of Alternating Hemiplegia (AH) and by recurrent episodes of Epilepsia Partialis Continua (EPC). The NGS study showed the presence of two different heterozygous, probably pathogenic variants in the TBC1D24 gene, inherited in trans from her parents: the c.116C>T (p.Ala39Val) and the c.457G>A (p.Glu153Lys). This study describes for the first time the association between TBC1D24 variants and AH expanding the phenotypic spectrum of TBC1D24-related diseases and suggesting that TBC1D24 molecular analysis should be considered in the diagnostic work up of AH patients. An additional peculiar feature is the association of AH and EPC.
© 2017 British Epilepsy Association. Published by Elsevier Ltd. All rights reserved.
Contents lists available at ScienceDirect
Seizure
1. Introduction
Alternating Hemiplegia (AH) is a rare childhood syndrome characterized by non-epileptic paroxysmal symptoms and stable neurological deficits, associated with ATP1A3 mutations in up to 85% of patients [1]. Epilepsia Partialis Continua (EPC) is a rare variant of epilepsy characterized by continuous focal jerkings of a body part over a period of hours or days, which may persist during sleep. In children, EPC is mostly associated with Rasmussen- encephalitis, mitochondriopathy and brain malformations [2]. Although half of AH patients experience epileptic seizures, no case of EPC has been reported [3]. We report the identification of two variants in TBC1D24 gene in a patient with AH and EPC.
* Corresponding author at: Department of Pediatric Neuroscience, Fondazione IRCCS Istituto Neurologico Carlo Besta, Via Giovanni Celoria 11, 20133 Milan, Italy.
E-mail address: [email protected] (T. Granata). 1 These authors equally contributed to the work.
http://dx.doi.org/10.1016/j.seizure.2017.03.003 1059-1311/© 2017 British Epilepsy Association. Published by Elsevier Ltd. All rights res
2. Case report
A 5 year-old girl presents, since the age of 4 months, paroxysmal symptoms of two types:
(1) daily, prolonged episodes of unresponsiveness and hypoto- nia, variably associated with abnormal eye movements, nystag- mus, sialorrhea, and hemi or tetraplegia, which resolved with sleep. (2) bouts of rhythmic clonic jerks of a body part, lasting from minutes to hours, persisting during sleep; the jerks involved different body parts during the distinct attacks.
Serial video-EEG-polygraph recordings clarified that the episodes of unresponsiveness were not associated with ictal EEG changes and that the focal jerks were consistent with EPC (Fig. 1).
The diagnostic work up which included MRI, evoked potentials, CSF for glycorrhachia and neurotransmitters, karyotyping and CGH array, metabolic screening; molecular analyses of SCL2A1 and POLG1 genes were unrevealing.
The age of onset and the semiology of the non-epileptic attacks fulfilled the diagnostic criteria for AH [1], therefore, despite the
erved.
72 F. Ragona et al. / Seizure 47 (2017) 71–73
unusual association with EPC, the infant was given Flunarizine, which reduced the frequency of the attacks. Analysis for ATP1A3, performed after its identification as AH causative gene, failed to identify mutations and deletions. The clinical evolution was characterized by developmental delay, already evident at the onset, with general developmental quotient of 46 at the age of 4, trunk hypotonia and limb rigidity with dystonic postures. Episodes of EPC recurred many times a year, lasted from hours to days, and persisted during sleep (Fig. 1a–d). The jerks were not associated with ictal activity on the surface EEG; the jerk-locked back averaging (JLBA) showed that a positive transient over the contralateral central region preceded the jerks by 28 msec, consistent with cortical origin (Fig. 1e).
The attacks, usually associated with eye deviation, unilateral or bilateral nystagmus, tachypnea, and autonomic disorders, recurred many times a week (Supplementary Video S1), mostly triggered by baths, emotions, fatigue or fever, and always ceased with sleep. Topiramate, barbiturate, acetazolamide and benzodiazepines were ineffective.
The EEG follow-up showed the impoverishment of the background activity, and interictal bilateral asynchronous epileptic transients over the fronto-central regions. Given the worsening course and the lack of a definite diagnosis, enzymatic and genetic analyses for mitochondrial and lysosomal disorders were per- formed but were unrevealing.
Finally, the patient entered a Next Generation Sequencing (NGS) analysis through a gene panel of the 92 genes related to epileptic encephalopathies (see Supplementary material for details about methods of molecular study).
NGS analysis in the proband’s DNA revealed the presence of two variants of probably pathogenic significance in the TBC1D24 gene, inherited in trans from her parents: the c.116C > T (p.Ala39Val) and the c.457G > A (p.Glu153Lys).
The c.116C > T (p.Ala39Val) variant is novel and prediction analysis resulting in a probably damaging effect on the mutated protein; the c.457G > A (p.Glu153Lys) variant is already described (rs376712059) and prediction analysis resulting a probably damaging and deleterious effect on the mutated protein (see Supplementary material for details about results methods of molecular study).
The complete NGS data analysis did not reveal the presence of other variants of possible pathological significance or the presence of Variant of Unknown Significance (VUS).
3. Discussion
TBC1D24 encodes a protein containing a TBC domain involved in cell signaling and oxidative stress resistance [4]. Allelic variants in the TBC1D24 gene have been reported in different diseases with multisystem involvement and in a broad variety of epilepsies, from
F. Ragona et al. / Seizure 47 (2017) 71–73 73
benign myoclonic epilepsy, to early-onset epileptic encephalopa- thy with severe developmental delay and early death. The broad spectrum can be explained by the effect of TBC1D24 mutations on synthesis, stability and activity of the protein, and by the effect of TBC1D24-associated proteins and pathways [4].
This is the first report of TBC1D24 variants in a patient who fulfilled the clinical diagnostic criteria for Alternating Hemiplegia [1]. In the large series reported by Balestrini et al., in fact, most patients had, besides seizures, stable – predominantly dystonia, or ataxia – or paroxysmal symptoms. These latter included prolonged post-ictal hemiparesis and focal or axial dystonic episodes, but in no case a constellation of symptoms consistent with AH was reported [4]. A further peculiarity of our patient was the presence of EPC, which has never been described in AH. Epilepsy in AH actually occurs in about 50% of patients: seizures are usually focal, not frequent, and well controlled by AEDs, status epilepticus is uncommon [3]. By contrast, in our patient the epilepsy was severe, with frequent, and long- lasting episodes of EPC, which involved different parts of the body during distinct episodes. This multifocal localization, which is uncommon in EPC [2] probably reflects the diffuse brain dysfunction resulting from TBC1D24 mutation. Although we cannot exclude the presence of other novel genomic variants of possible pathological significance that may contribute to the complex phenotype described in our patient, our hypothesis is that the defective neurotransmission, determined by TBC1D24 mutation might have a role in determining both the seizures and the non-epileptic symptoms. Functional studies on cell models will hopefully clarify the mechanism by which the defective neuro- transmission determined by gene mutation may cause multisys- tem and neurological symptoms.
4. Conclusions
Our finding expands the phenotypic spectrum of TBC1D24- related diseases and suggests to consider TBC1D24 molecular
analysis in the diagnostic work-up of AH patients, especially in those negative for ATP1A3 mutations.
Conflict of interest
Authors have no competing interests. The research did not receive any specific grant from funding agencies in the public, commercial, or not-for profit sectors.
Acknowledgement
The authors thank the parents of patient for their collaboration.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j. seizure.2017.03.003.
References
[1] Panagiotakaki E, De Grandis E, Stagnaro M, Heinzen EL, Fons C, Sisodiya S, et al. Italian IBAHC Consortium; French AHC Consortium; International AHC Consortium. Clinical profile of patients with ATP1A3 mutations in alternating hemiplegia of childhood-a study of 155 patients. Orphanet J Rare Dis 2015;10:123–35.
[2] Kravljanac R, Djuric M, Jovic N, Djordjevic M, Zamurovic D, Pekmezovic T. Etiology, clinical features and outcome of epilepsia partialis continua in cohort of 51 children. Epilepsy Res 2013;104(March (1–2)):112–7, doi:http://dx.doi. org/10.1016/j.eplepsyres.2012.09.003.
[3] Panagiotakaki E, Gobbi G, Neville B, Ebinger F, Campistol J, Nevšímalová S, et al. Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain 2010;133:3598–610, doi: http://dx.doi.org/10.1093/brain/awq295.
[4] Balestrini S, Milh M, Castiglioni C, Lüthy K, Finelli MJ, Verstreken P, et al. TBC1D24 genotype-phenotype correlation epilepsies and other neurologic features. Neurology 2016;87(July (1)):77–85, doi:http://dx.doi.org/10.1212/ WNL.0000000000002807.
1 Introduction
Related Documents