Reactions of Monomeric Methyllithium with CO, CNMe and NCMe: Theoretical Study By Aislinn Fegan 10902953 Directed by: Dr Matthias Tacke I hereby declare that all the work presented in this thesis is my own, unless clearly indicated by citation. Student Signature: Submission Date: 1 For Examiners’ use only. Examiner’s initials:

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Reactions of Monomeric Methyllithium with CO, CNMe and
NCMe: Theoretical StudyBy
Aislinn Fegan
10902953
Directed by:
Dr Matthias Tacke
I hereby declare that all the work presented in this thesis is my own, unless clearly indicated by citation.
Student Signature:
Submission Date:
1
For Examiners’ use only.
Examiner’s initials:

Contents
Abstract…………………………………………………………………………………………….….3
Chapter 1: Introduction……………………………………………………………………………….4
1.1 Nature of C-Li bond………………………………………………………………………………5
1.2 Aggregation of organolithium compounds……………………………………………………..7
1.3 Solvent Choice: Liquid Xenon…………………………………………………………………..8
1.4 Reaction of tetrameric methyllithium with CO and CNMe……………………………………9
1.5 Reaction of hexameric methyllithium with CO and CNMe………………………………….10
Chapter 2: Computational Chemistry Methods…………………………………………………..12
2.1 Molecular Mechanics…………………………………………………………………………...14
2.2 Schrödinger Equation…………………………………………………………………………..16
2.3 Semi-Empirical Method…………..................................................................................17
2.4 Quantum Methods………………………………………………………………….…………..18
2.5 Ab initio Methods………….………………………………………………………………..…..19
2.6 Geometry Optimisation…………………………………………………………………………21
Chapter 3: Results and Discussion……………………………………………………………….22
3.1 Reaction of monomeric methyllithium with carbon monoxide……………………………...23
3.2 Reaction of monomeric methyllithium with methylisonitrile (CNMe)………………………29
3.3 Reaction of monomeric methyllithium with acetonitrile (NCMe)…………………………...34
Chapter 4: Conclusion……………………………………………………………………………..39
4.1 Conclusion………………………………………………………………………………………40
Acknowledgements…………………………………………………………………………………42
References…………………………………………………………………………………………..43
2

Abstract
In this computational project monomeric methyllithium was allowed to react with carbon
monoxide, methylisonitrile and acetonitrile (CO, CNMe and NCMe). The structures,
energies and characteristic IR frequencies of the intermediates and products of these
reactions were calculated using high level ab initio calculations.
These results for monomeric methyllithium were compared to the behaviour of tetrameric
and hexameric methyllithium reacting with the same species. The reaction sequence for
tetrameric and hexameric methyllithium are similar, forming the same key intermediates.
In the experiment, coordination of CO to methyllithium is first found forming a lithium
carbonyl species, at very low temperatures (-100°C). This is unexpected behaviour of
lithium since lithium is not a transition-metal, yet is displaying transition-metal like behaviour
by forming carbonyl complexes. This lithium carbonyl then rearranges via formal insertion
into the lithium-carbon bond to form a lithium acetyl species, at higher temperatures, which
is again expected for transition metal complexes only. This second species finally reacts
further, with further warming up, to produce a species with no C-O stretching frequencies
above 1500 cm-1. The results of calculations are compared with spectroscopic results which
show the existence of lithium carbonyl and lithium acetyl species (as well as their isonitrile
counterparts) at low temperature.
To date, there has been no study of monomeric methyllithium, so in this project this reaction
sequence was modelled using monomeric methyllithium so that we can study the behaviour
of lithium with these species in a simpler system.
3

Chapter 1
Introduction
4

Introduction
1.1 Nature of the C-Li bond
Organolithium compounds contain carbon-lithium bonds and constitute a very important
class of organometallic reagents. These reagents have been used in organic and
organometallic synthesis for decades but much is still unknown about the structure and
reactivity of these compounds. Theoretical studies have played an important role in the
development of our understanding of organolithium compounds (structure, bonding and
reactions).
The nature of the C-Li bond is still a dilemma for chemists due to its “dual nature”
(possessing both ionic and covalent character) 1,2 The “degree of covalency” of the carbon-
lithium bond varies with temperature, solvent, and structure of the organic component. This
“dual nature” of the C-Li bond is very important and explains why this bond behaves
differently in different compounds. The ionic nature of the monomeric MeLi increases on
solvation and tetrameric MeLi has more ionic C-Li bonding. The bonding is governed by
electrostatic interactions. The C-Li bond in methyllithium is a tight-ion pair with little covalent
bonding, but the covalent component cannot be neglected. 3,4,5 As shown in the diagram
below is 13C-6Li/7Li spin-spin coupling in methyllithium.
5
13C-6Li/7Li spin-spin coupling
Streitweiser, A. Williams, J.E Alexandratos, S. Mc Kelvey, J.M. J. Am. Chem. Soc. (1976), 98, 4778
Images taken from: www.scs.illinois.edu/denmark/presentations/2013/gm-2013-3-12.pdf
Figure 1 Figure 2
Figure 3
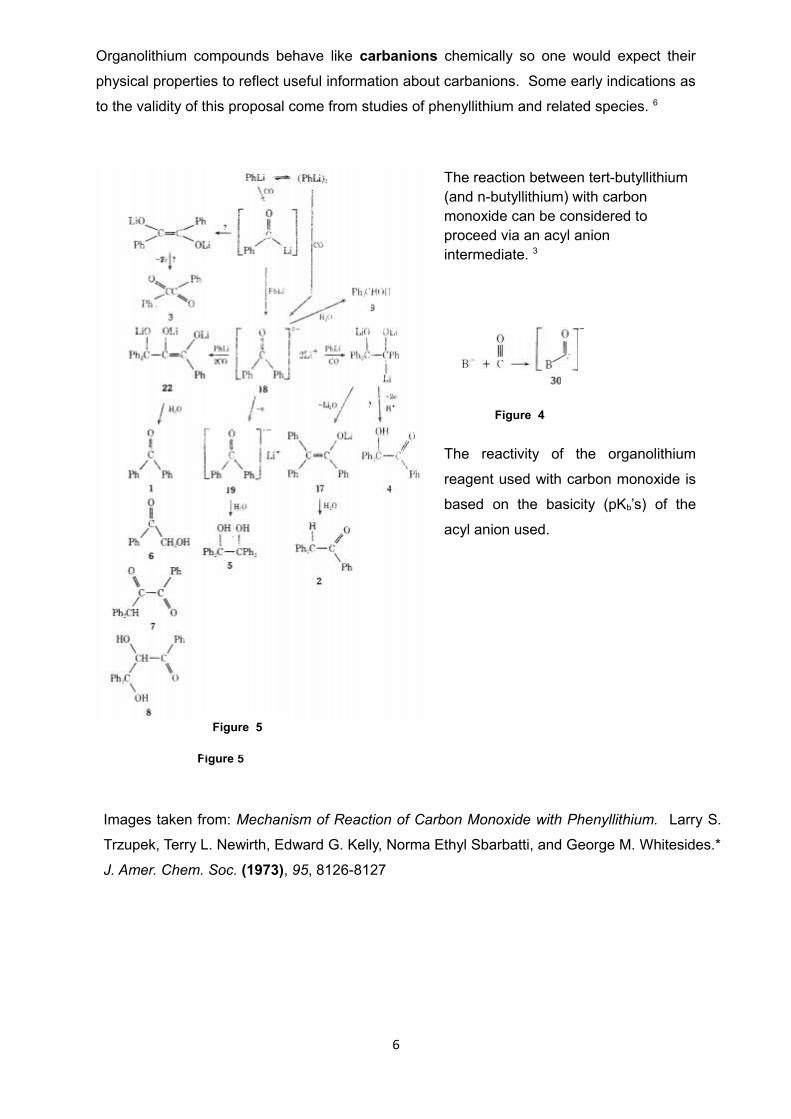
Organolithium compounds behave like carbanions chemically so one would expect their
physical properties to reflect useful information about carbanions. Some early indications as
to the validity of this proposal come from studies of phenyllithium and related species. 6
The reaction between tert-butyllithium (and n-butyllithium) with carbon monoxide can be considered to proceed via an acyl anion intermediate. 3
The reactivity of the organolithium
reagent used with carbon monoxide is
based on the basicity (pKb’s) of the
acyl anion used.
1.2 Aggregation of organolithium compounds
1.2 Aggregation of organolithium compounds
6
Images taken from: Mechanism of Reaction of Carbon Monoxide with Phenyllithium. Larry S.
Trzupek, Terry L. Newirth, Edward G. Kelly, Norma Ethyl Sbarbatti, and George M. Whitesides.*
J. Amer. Chem. Soc. (1973), 95, 8126-8127
Figure 5
Figure 4
Figure 5

1.2 Aggregation of organolithium compounds
Organolithium compounds form the largest single group of synthetically useful
organometallic compounds with ~2 new ones being announced every day. 1,2 So to fully
understand the chemistry behind these versatile reagents it is necessary to look at their
structures. Organolithium compounds exhibit an astonishing variety of structures from
variants of ion-pairs to covalent clusters such as cubic tetramers and octahedral hexamers.
Aggregation is a nearly ubiquitous characteristic of organolithium compounds. 7 This high
tendency for aggregation is due to the inherent strong dipole moments within the
compounds, and can be influenced by solvent choice and steric effects. In strongly
coordinating solvents, dimers or even monomers exist, but in weakly or non-coordinating
solvents, tetramers or hexamers dominate. THF/ether favours tetramer formation in
methyllithium. The hexameric form of methyllithium is favoured over the tetrameric form and
so was used as the model of choice in reaction. However, THF disaggregates hexameric
methyllithium and so a non-coordinating solvent (liquid Xenon) was used to allow hexamer to
exist.
Monomeric methyllithium was the model of choice because this compound hasn’t been
studied before, and it is of interest to see how lithium behaves in a simpler system.
7
Monomeric Methyllithium
HyperChem was used to draw
this molecule
Tetrameric Methyllithium
Image taken from:
en.wikipedia/org/wiki/methyllithiu
m (public domain)
Hexameric Methyllithium
Image taken from:
en.wikipedia/org/wiki/methyllithiu
m (public domain)
Figure 6
Figure 7
Figure 8

1.3 Solvent choice: Liquid Xenon (LXe)
Organolithium reagents such as tetrameric tert-butyl lithium and hexameric n-butyllithium are
able to interact with CO or isonitrile in a newly developed LXe cell constructed from one
piece of single-crystal silicon. 8,9
Liquid Xenon was the solvent of choice for a number of reasons. Liquid Xenon is a
weakly/non coordinating solvent which prevents disaggregation of organolithium
compounds, and allowing tetramers/hexamers to exist. Liquid Xenon is chemically inert and
has high polarisability meaning it is able to have significant interactions with the solute but
not chemically/structurally disrupt the reaction. Liquid Xenon is used as the reaction medium
because it suppresses electron-transfer reactions which are known to complicate the
reaction.10 IR spectroscopy is being used in these reactions for characterisation of
complexes formed so it is necessary to use an optically transparent solvent which won’t
appear as peaks on the IR spectra, so liquid Xenon is ideal. Also liquid Xenon allows for
measurements at temperatures between -112°C and -20°C, which is the range in which the
wanted intermediates exist. 11
Since liquid Xenon is a poor solvent it was appropriate to use the gas phase approach in the
calculations, 12 both in the reaction of hexameric methyllithium and monomeric methyllithium.
8

1.4 Reaction of tetrameric methyllithium with carbon monoxide: 9
In the first step of this reaction, if performed at sufficiently low temperatures (-100°C) carbon
monoxide is complexed with n-butyl lithium, to form a lithium carbonyl adduct. The carbon
monoxide molecule then inserts itself into the lithium-carbon bond in the second step at
higher temperatures (-30°C) to form a lithium acetyl intermediate. Further warming up to
-20°C results in decomposition of these intermediates and the proposed product would be a
lithiated oxycarbene, with a strong lithium-oxygen bond, the driving force of this reaction
being due to the oxophilicity of lithium. 12 This reaction was then modelled using calculations
using ab initio HF/6-31G**, for comparison with experimental data.
9
Figure 9
Figure 10
The addition of CO to (LiMe)4 releases -7.8
kcal/mol with the formation of the linear lithium
carbonyl structure. In contrast to the experiment,
the calculated compound doesn’t show any
backbonding to CO as indicated by a higher C-O
stretching frequency. The insertion of CO into the
lithium-carbon bond is now exothermic (-4.2
kcal/mol) and the resulting acetyl group
coordinates to the lithium in a µ3 fashion, which
helps to find an exothermic reaction pathway for
insertion. This lithium acetyl intermediate contains
a double-bonded CO group which appears at the
same time as the decomposition of lithium
carbonyl species at higher temperatures (-30°C).
This lithium acetyl shits the carbonyl stretching
frequency to 1635 cm-1 which then reacts further
resulting in a species with no C-O stretching
frequencies above 1500 cm-1
indicating a bond
order of less than 2.
Figure 9 and 10 taken from: Carbonyl and Benzene Complexes of Lithium: Transition-Metal-
Like Behaviour of Lithium in Organolithium Compounds. Matthias Tacke, Eur. J. Inorg. Chem.
(1998), 537-541
Figure 10

1.5 Reaction of hexameric methyllithium with CO 12
In this experiment, n-butyl lithium was allowed to react with CO and CNMe, and an infrared
study in liquid Xenon was used to study the complexes formed. To mimic this experiment,
calculations were made using hexameric methyllithium reacting with CO and CNMe.
Methyllithium was used instead of n-butyl lithium because it is a smaller molecule which
would require less computational expense and time, yet reacts to form the same complexes.
All energies noted in the calculations were calculated as the electronic energy corrected by a
zero point energy using B3LYP with the 6-31G(d,p) basis set. These Density Functional
Theory (DFT) results from calculations are compared with spectroscopic results from
experiment.
10
Firstly, CO inserts into C-Li bond of hexameric methyllithium to
form lithium carbonyl. The CO bond length reduces from
113.8 ppm(free carbonyl) to 113.4 ppm indicated an increased
bond order, supported by an increase in CO stretching
frequency from 2209 cm-1(free CO) to 2241 cm-1(complexed
CO). However, in experiment, the CO stretching decreased
from 2139 cm-1to 2047 cm-1(due to some backbonding in
lithium carbonyl).
The carbon monoxide molecule then inserts into the Li-C bond
of the lithium carbonyl to form lithium acetyl. This is an
exothermic reaction releasing 12.2 kcal/mol due to an
increased stability of lithium acetyl. The acetyl group resides
on a Li3 face and the oxygen bridges between 2 lithium atoms.
The CO bond length has now been elongated from 113.4 ppm
to 129.2 ppm indicating a decreased bond order (bond order of
2). The calculated CO stretching frequency decreased to 1424
cm-1. The experiment also showed a decrease in CO
stretching frequency to 1635 cm-1.
It is reasonable to assume that the lithium acetyl reacts further
and dimerizes to form a lithiated oxycarbene. The species
formed in experiment showed a bond order of less than 2,
allowing Li-O bonds to strengthen even further, the driving
force of this reaction being the oxophilicity of lithium. This is a
highly exothermic reaction in calculations with a complexation
enthalpy of -42.3 kcal/mol.
Figure 11: Taken from: Reactions of Methyllithium With CO and CNMe: Theoretical study. Matthias
Tacke, International Journal of Quantum Chemistry (2006) Vol106, 692-696

Reaction of Hexameric methyllithium with CNMe 12
The experimental reaction of t-butylisonitrile with n-butyl lithium was modelled in calculations
using DFT for the reaction sequence (with methyllithium and methylisonitrile). Below the
reaction sequence is being described:
11
Figure 12: Taken from “Reactions of Methyllithium With CO and CNMe: Theoretical study” Matthias Tacke, International Journal of Quantum Chemistry (2006) Vol106, 692-696
Firstly, methylisonitrile adds to hexameric methyllithium to
form a lithium methylisonitrile complex, CN bond length
reduces from 117.7 ppm (free CNMe) to 116.8 ppm
(complexed) showing an increase in bond order, supported
by an increase in CN stretching frequency from 2250 cm -1 to
2303 cm-1. However, in experiment the CN frequency
reduces from 2179 cm-1 to 2135 cm-1.
In the second reaction step, the isonitrile inserts into the Li-C
bond of the complex to form the structurally interesting
lithiated Schiff base molecule. The MeCNMe group
bridges over the 3 lithium atoms and the nitrogen bridges
the two lithium atoms (similar to oxygen). The CN bond
length is elongated from 116.8 ppm to 132.0 ppm which
indicates a CN double bond. The experimental results for
CN stretching frequency are in agreement with calculations,
with 1540 cm-1(calculated) and 1510 cm-1(experiment)
indicating a bond order of 2. This is a highly exothermic
reaction releasing 19.4 kcal/mol to produce the stable
lithiated Schiff base molecule. Due to the stability of the
lithiated Schiff base molecule, there is no further reaction
step in calculations.
Hexameric methyllithium forms complexes with CO and
CNMe with complexation enthalpies of 4.8 and 8.5 kcal/mol,
respectively. These values relate directly to the donor
capabilities of these ligands, where nitrogen is a better
donor ligand than oxygen, highlighted by the more stable
complex formed.

Chapter 2
Computational Chemistry Methods
12

Computational Chemistry Methods
There are 4 different flavours to computational chemistry: Molecular Mechanics, Semi
Empirical Methods, Ab initio, and Density Functional Theory.
These 4 methods differ in how they compute the geometries and energies of molecules.
Molecular mechanics is suitable for use in calculations for larger molecules, using empirical
parameters for calculations. Quantum mechanics methods (including ab initio and DFT) give
more accurate optimisation of smaller molecules, solving an exact approximate of the
Schrödinger equation. Semi-Empirical methods attempt to simplify difficult mathematical
calculations by combining with some empirical data from the lab. The computational time
and expense increases as you move up to higher level methods.
Molecular Mechanics
13
Figure 13: Taken from “Introduction to Molecular Modelling and Computational Chemistry”
http://www.chem.arizona.edu/~lichtend/C518_Fall2009/introtocompchem/IntroToCC.html

HyperChem is the molecular modelling program used in this project, which allowed us to
choose between four types of force fields using the molecular mechanics method, namely
MM+, AMBER, OPLS, and BIO+. Molecular Mechanics (MM+) was used in this project to
obtain reasonable input geometries for the calculations before using higher level methods.
Molecular mechanics is suitable for studying larger molecules e.g. proteins, using empirical
data and so requires less computational time and expense than the higher level methods.
In molecular mechanics, the nucleus and electrons are ignored and each atom is seen as a
single entity. A chemical bond is viewed as a spring connecting two spheres (atoms)
therefore by changing the tension of the spring we can adjust the bond strength and bond
energy of the molecules. The energy of a molecule varies with geometry because these
springs resist being stretched or bent away from their “natural” length or angle, but they also
resist being pushed too closely together (atom crowding). The main principle behind
molecular mechanics is to express the energy of a molecule as a function of this resistance
to bond stretching, bending, torsional energy, van der Waals energy, electrostatic energy and
cross terms. 14
E = Estr
+ Ebend
+ Etors
+ Evdw
+ Eel
+ Ecross
When numbers are input into the equation, this is called ‘parameterizing the forcefield’. A
forcefield can be parameterized by reference to experiment (empirical data) or by obtaining
the numbers from high level ab initio or DFT calculations, or even a combination. No set of
force field parameters are complete and are being updated on a regular basis.
Kstretch could be obtained experimentally, from Infrared spectra as the bond stretching
frequency depends on the force constant (kstretch). Leq could be obtained from X-ray
diffraction, electron diffraction or microwave spectroscopy. STO-35 calculations
(representations of wavefunctions) can be carried out in order to find these parameters. This
method also applies for parameterising the angle bending term. Ab initio calculations are
used to calculate the parameters the torsional and nonbonding interactions terms. Once all
parameters are obtained, the force field can be parameterised and calculations can be
carried out to calculate the total potential energy.
14

For small-medium sized molecules, as used in this project, the forcefield MM+ was used.
The Allinger group has been responsible for the “MM” series. MM calculations on small-
medium sized molecules are fast and can be quite accurate.
Molecular mechanics can also be used to calculate geometries and energies of very large
molecules. Two of the most widely used forcefields are CHARMM (Chemistry at HARvard
using Molecular Mechanics) and the computational package AMBER (Assisted Model
Building with Energy Refinement). These programmes perform an extremely important
aspect of designing pharmacologically active drugs by modelling biopolymers.
Molecular mechanics offers fast speeds for determining quite accurate potential energy and
geometries for large molecules, with no expensive hardware required. However, between
75-80% of known molecules do not have good parameters, but these parameters are being
updated annually – AMBER, MM series, OPLS-UA and OPLS-AA. The internal structure of
molecules cannot be studied using molecular mechanics method because electron
interactions are not considered. Also it is not possible to study reactions as bond breaking
cannot occur using this method. 14
15

Schrödinger Equation
The Schrödinger equation was proposed by the Austrian physicist Erwin Schrödinger in
1926. It is an important equation that is fundamental to quantum mechanics. 13,14
This equation integrates both optics and classical mechanics. The equations for the Law of
Conservation of Energy are used in terms in wavefunction. This is used to solve wave
functions of atomic particles (electrons, protons and atoms), for example ‘particle-in-a-box’
method. 15
There are two types of Schrödinger equations, time-dependent and time-independent. The
time-independent equation predicts that wavefunctions can form stationary states (orbitals)
which do not change over time. 13
The time-dependent equation describes the wavefunction
as a function of position and time. In chemistry we are more concerned with stationary
states so we focus on the time-independent Schrödinger equation.
Time-independent Schrödinger equation:
E = proportionality constant
Ψ = wavefunction
H = Hamiltonian
In this equation, the Hamiltonian operator equals the total energy operator, consisting of
kinetic energy and potential energy.
From classical mechanics, the Law of Conservation of Energy states:
Total Energy = Kinetic Energy + Potential Energy
So, the Schrödinger equation uses this fundamental principle in terms of its wavefunction.
Quantum Mechanics methods attempt to solve the Schrödinger equation in order to
calculate geometries and energies for molecules.
16
Figure 14: Taken from “The Schrödinger Equation”
http://chemwiki.ucdavis.edu/Physical_Chemistry/Quantum_
Mechanics/Quantum_Theory/Principle_of_Quantum_Mecha
nics/Schr%C3%B6dinger_Equation

Semi Empirical Method
Semi empirical method attempts to simplify difficult mathematical equations used in quantum
methods by combining with empirical data from experiments. Semi empirical method
attempts to approximately solve the Schrödinger equation. Semi empirical methods are
used for calculations of molecules with 5-100 atoms which allows for larger molecules to be
studied as compared to ab initio but with less computational time and expense.
The mathematics involved in semi empirical methods is restricted to valence electrons. For
missing electrons, parameterised datasets are added.
The 3 semi empirical methods are as follows:
AM1 – Predicts heat of formation
PM3 – Method used in this project. More powerful version of AM1. Very good method for
organic systems.
NDO – Neglect of Differential Overlap MINDO, INDO, ZINDO, SINDO
Electronic potential energy is calculated using computer techniques to solve the quantum
mechanical Schrödinger equation.
Potential Energy = sum of repulsions of nuclei and attractions arising from electrons
17
Figure 15: Semi-empirical methods - screenshot of options menu using
HyperChem software.

Quantum Methods
The quantum mechanics methods in HyperChem differ in how they approximate the
Schrödinger equation and how they compute potential energy. The ab initio and DFT
methods expand molecular orbitals into a linear combination of atomic orbitals (LCAO) and
do not immediately introduce any further approximates.
Ab initio Hartree-Fock calculations then approximate the form of the final wavefunction
determining the energy while DFT calculations approximate the relationship of the energy to
the electron density.
Quantum mechanics requires no information about location or geometry of bonds in a
molecular system. Parameters for elements are independent of chemical environment
(unlike molecular mechanics). Quantum mechanics can also describe bond breaking.
18

Ab initio Methods
The term ‘ab initio’ is Latin for ‘from the beginning’ meaning that all results come from
significant computational analysis of the Schrödinger’s equation. No empirical data derived
from experiment is included in these calculations which make this method one of the most
computationally expensive but also one of the most accurate methods.
Ab initio methods are not solvable directly, an iterative technique must be used – SCF (Self
Consistent Field method).
The primary deficiency:
E(Exact) = E(Hartree-Fock) + E(Correlation)
There are 3 categories of ab initio:
Hartree-Fock – determination of wavefunction and energy of quantum body in stationary
state
Møller-Plesset – calculation of Hartree-Fock wavefunction (electron in ground state) and
wavefunction of electrons in excited states
Configuration Interaction – electrons are in different configurations, similar to Møller-
Plesset where excited wavefunctions are mixed in.
Hartree-Fock Method
This method was used in this project for ab initio calculations. Hartree-Fock method is also
called the self-consistent field method – Hartree-Fock equation is an approximate solution to
the Schrödinger equation requiring the final field as computer from the charge distribution to
be ‘self-consistent’ with the assumed initial field. HyperChem ends the iterations when the
coefficients or computer energy no longer change, the solution is then ‘self-consistent’. 15,16
Energy is calculated from: columbic repulsion of nuclei, electron kinetic energy and electron-
nuclei attraction, columbic repulsion of electrons and other electron-electron interactions
EHF = Enuclear + Ecore + Ecoulomb + Eexchange
In Hartree-Fock calculations, the correlated electron-electron repulsion is not specifically
taken into account, only its average effect is included in the calculation. Many types of
19

calculations start with Hartree-Fock calculations and subsequently correct for electron-
electron repulsion = electronic correlation.
Hartree-Fock equations can neglect correlations due to many-body interactions, and this
effect is not negligible. The requirement for a computationally practicable scheme that
successfully incorporates the effects of both exchange and correlation leads us to consider
density functional theory.
Methods used in this project:
HyperChem 7.0 is the molecular modelling program used in this project. For each molecule
we used molecular mechanics (MM+) firstly to obtain reasonable input geometries before
moving to higher level methods. We then calculated the geometries and energies of the
molecules using semi empirical methods (PM3), before moving to ab initio Hartree-Fock.
For the ab inito calculations, the molecule was optimised on the small basis set firstly HF/3-
21G*, then the medium basis set HF/6-31G*, and finally the large basis set HF/6-31G**.
20

Geometry Optimisation
When studying the geometry of a molecule in computational chemistry we use the Cartesian
coordinates to look at bond angles, bond distances and dihedral angles. We use this
information to find the optimal molecular geometry. The objective of geometry optimization
is to find the point at which the energy is a minimum because this is where the molecule is
most stable and most likely to be found in nature.
The aim is to find a point at which the arrangement of atoms results in a net inter-atomic
force of close to zero, and the position on the potential energy surface is a stationary point.
Potential energy surfaces are characterized by distinct points; local maxima, global maxima,
local minima, global minima, saddle point (represents transition structure – optimal
geometry). 19
First derivative of the energy is where the gradient needs to be calculated, at a minima
gradient this equals zero. The first step to geometry optimisation is when a user specifies a
beginning geometry as Cartesian coordinates, then a basis set is specified, and the program
then computes the energy and the gradient at that point. The program continues computing
the energy and gradients, deciding if a stationary point (convergence) has been reached and
the geometry is varied based on the size of the gradient. New integrals are calculated, new
self-consistent field calculations are done and new energy and gradients are calculated.
These steps are repeated until the program reaches convergence i.e. finds a stationary
point. Once a stationary point has been reached, we need to detect whether this is the
geometry of the product or the transition state. To do this, we looked at the infrared spectra
for this geometry to detect any negative bond stretching frequencies which indicate this is
the transition state, and the molecule has not been fully optimised. 18
There are a number of different algorithms for performing optimizations which can also
calculate the second derivative of the energy with respect to the coordinates known as the
Hessian. The Hessian serves to specify the ‘curvature of the surface’ for that particular
geometry thus optimizing the determination of how to vary the geometry for the next step.
21

Chapter 3
Results and Discussion
22
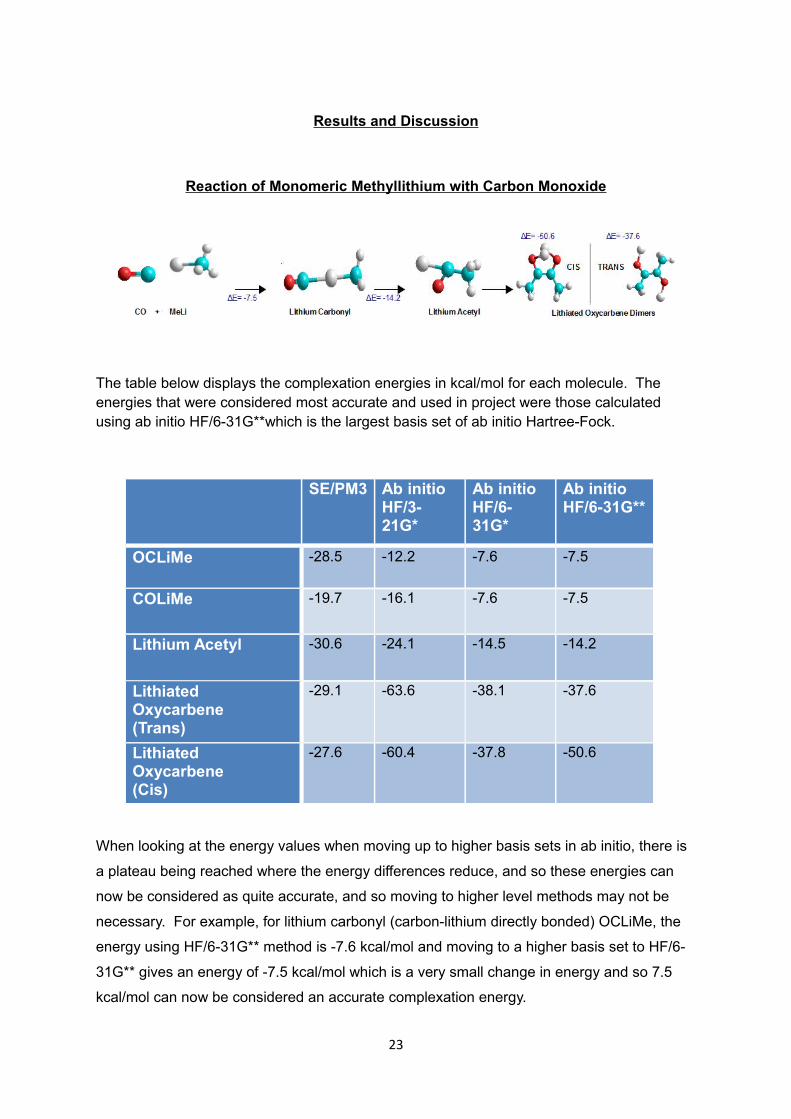
Results and Discussion
Reaction of Monomeric Methyllithium with Carbon Monoxide
The table below displays the complexation energies in kcal/mol for each molecule. The energies that were considered most accurate and used in project were those calculated using ab initio HF/6-31G**which is the largest basis set of ab initio Hartree-Fock.
SE/PM3 Ab initioHF/3-21G*
Ab initioHF/6-31G*
Ab initioHF/6-31G**
OCLiMe -28.5 -12.2 -7.6 -7.5
COLiMe -19.7 -16.1 -7.6 -7.5
Lithium Acetyl -30.6 -24.1 -14.5 -14.2
Lithiated Oxycarbene(Trans)
-29.1 -63.6 -38.1 -37.6
Lithiated Oxycarbene(Cis)
-27.6 -60.4 -37.8 -50.6
When looking at the energy values when moving up to higher basis sets in ab initio, there is
a plateau being reached where the energy differences reduce, and so these energies can
now be considered as quite accurate, and so moving to higher level methods may not be
necessary. For example, for lithium carbonyl (carbon-lithium directly bonded) OCLiMe, the
energy using HF/6-31G** method is -7.6 kcal/mol and moving to a higher basis set to HF/6-
31G** gives an energy of -7.5 kcal/mol which is a very small change in energy and so 7.5
kcal/mol can now be considered an accurate complexation energy.
23

Formation of Lithium Carbonyl Complex
Formed using ab initio HF/6-31G** method
A very similar trend is seen in the reaction between monomeric methyllithium with carbon
monoxide as compared to the same reaction using hexameric methyllithium with carbon
monoxide. To form lithium carbonyl 7.5 kcal/mol is released in an exothermic reaction where
carbon monoxide adds to the lithium-carbon bond (the same value whether oxygen or
carbon bonded). This is similar to the hexameric methyllithium reaction which released 4.8
kcal/mol, which is only a small energy release. The addition of CO to methyllithium forms
lithum carbonyl adduct via σ-bond between carbon and lithium.
24
+
2441 cm-1
ΔE = -7.5 kcal/mol
2497 cm-1

Formation of Lithium Acetyl Complex
Formed using ab initio HF/6-31G** method
The carbon monoxide then inserts into the lithium-carbon bond to form lithium acetyl
releasing more energy in an exothermic reaction – 14.2 kcal/mol released, this is due to
lithium acetyl being more stable due to the unusual structure of this molecule. The geometry
of lithium acetyl was unexpected and showed an attraction between lithium and oxygen.
The bond angle Li-C=O was 64.5°. There is an electrostatic interaction between the lithium
and oxygen in this structure, where there is subcoordination between lithium and oxygen.
The dashed yellow lines in the diagram below indicate ionic bonds and the carbon to oxygen
bond remains covalent confirmed by CO stretching frequency of 1690 cm -1. As we can see
the CO stretching frequency reduces from 2497 cm-1 to 1690 cm-1 indicating a decrease in
bond order, from triple bond to double bond.
25
2497 cm-1
1690 cm-1
ΔE = -14.2 kcal/mol

Dimerisation to form Lithiated Oxycarbene complexes
Formed using ab inito HF/6-31G**
The lithium acetyl then dimerizes to form lithiated oxycarbene complexes, with cis(left) and
trans (right) isomers. This is a highly exothermic reaction for both cis and trans isomers,
with the oxophilicity of lithium being the driving force, where now the oxygen and lithium are
now directly bonded. The cis isomer is significantly more stable than the trans isomer (-50.6
26
ΔE = -37.6 kcal/molΔE = -50.6 kcal/mol
1690 cm-1
1479 cm- 1501 cm-1
1479 cm-1

kcal/mol for the cis isomer compared to -37.6 kcal/mol for the trans isomer) so using
HyperChem the molecule was rotated about its plane to determine reasons why this
geometry is more stable. In the image shown below, we can see the geometry of lithiated
oxycarbene (cis) after being rotated on its side, the yellow dashed lines indicate an ionic
bond, an electrostatic interaction between the oxygens and the lithiums in this 4 membered
ring that has been formed. The bond angles in this 4 membered ring are 90.5° indicating a
symmetrical 4 membered ring, connected by ionic bonds, where there is subcoordination
between the oxygen and lithium. This is a highly stabilising effect and explains why the cis
isomer is much more stable than the trans isomer.
The following table displays CO bond lengths in each complex, and the corresponding CO
stretching frequency for each complex. As the CO bond order decreases, the corresponding
bond length increases.
ΔE (HF/6-31G**) kcal/mol
v(CO)
cm-1
CO Bond Length
(Angstroms)
Free CO N/A 2441 1.11
OCLiMe -7.5 2497 1.12
COLiMe -7.5 2393 1.14
Lithium Acetyl -14.2 1690 1.25
Lithiated Oxycarbene (Trans)
-37.6 1501 1.34
Lithiated Oxycarbene(Cis)
-50.6 1479 1.39
27

The CO stretching frequency is higher for lithium carbonyl (carbon-lithium bonded) than for
lithium carbonyl (oxygen bonded), meaning the CO triple bond in OCLiMe is stronger than
COLiMe. This could be due to oxygen being a greater electron donor than carbon, therefore
donating electrons to lithium in COLiMe and so weakening the CO bond, therefore reducing
the bond order of CO (characterized by a decrease in CO stretching frequency).
The CO stretching frequency then decreases to 1690 cm-1 in lithium acetyl indicating a bond
order of 2, and the CO bond lengthens to 1.25Å.
For lithiated oxycarbene dimers, there is no single low lying CO stretching frequency, but
instead various C-O modes. The CO stretching frequencies provided in the table above
indicate the antisymmetric C-O modes of vibration with the highest intensity in IR spectra.
These values of 1501 cm-1 and 1479 cm-1 indicate a bond order of less than 2 in lithiated
oxycarbene dimers
28

Reaction of monomeric methyllithium with methylisonitrile (CNMe)
When looking at the energy values for enthalpies of formation for each complex, one can
see a very small difference in energies between ab initio HF/6-31G* and HF/6-31G** and so
a plateau has been reached therefore these values can be considered quite accurate.
SE/PM3 Ab initioHF/3-21G*
Ab initioHF/6-31G*
Ab initioHF/6-31G**
MeNCLiMe -35.8 -23.2 -19.2 -19.2
Lithiated Schiff Base MoleculeC-Li bonded
-8.6 -7.6 -10.8 -10.3
Lithiated Schiff Base Dimer(Trans)
-14.1 -11.2 -7.4 -7.4
Lithiated Schiff Base Dimer(Cis)
-19.0 -11.4 -11.0 -11.1
The first reaction between methylisonitrile and methyllithium is a highly exothermic reaction
releasing 19.2 kcal/mol, which released more energy on complexation than the first adduct
formed by reaction of CO with methyllithium (-7.5kcal/mol). This relates to the donor
capabilities of these ligands, where methylisonitrile is a better donor ligand to methyllithium
than carbon monoxide. The subsequent reactions are still exothermic but not to as great of
an extent (-10.3 kcal/mol to form lithiated Schiff base molecule, and -7.4 kcal/mol and -11.1
kcal/mol to form lithiated Schiff base dimer.
29

Complex formed by Methylisonitrile and Methyllithium
Formed using ab initio HF/6-31G** method
Methylisonitrile adds directly to carbon-lithium bond in methyllithium to form lithium isonitrile
complex. This is a highly stable molecule given the complexation energy is -19.2 kcal/mol
and so is a favourable reaction. CN stretching frequency increases to 2497 cm -1 in
complexed CN indicating an increased bond order, possibly due to carbon being directly
bonded to lithium as opposed to nitrogen. This result indicates that the CN bond in the
complex has more triple bond character than free CNMe.
30
ΔE = -19.2 kcal/mol
2467 cm-1
2497 cm-1

Formation of Lithiated Schiff Base Complex
Formed using ab initio HF/6-31G** method
Methylisonitrile then inserts into the carbon-lithium bond of methyllithium isonitrile complex to
form the structurally interesting lithiated Schiff base complex. The CN bond stretching
frequency decreases from 2497 cm-1 to 1715 cm-1 indicating a decreased bond order to a
bond order of 2. The yellow dashed lines in the diagram represent the ionic bonds in this
structure, where there is subcoordination between lithium and nitrogen with a Li-C-N bond
angle of 66.2°.
31
ΔE = -10.3 kcal/mol
2497 cm-
1715 cm-1
2497 cm-1

Lithiated Schiff Base Dimerisation
Formed using ab initio HF/6-31G** method
Dimerisation of the lithiated Schiff base took place to form the –cis and –trans isomers of
lithiated Schiff base dimers. The cis isomer is more stable, with a complexation enthalpy of
-11.1 kcal/mol (compared to -7.4 kcal/mol for the trans isomer). Again by looking at the
structure in more detail and rotating the molecule using HyperChem we were able to see a
4-membered ring structure being formed.
32
ΔE (Cis) = -11.1 kcal/molΔE (Trans) = -7.4 kcal/mol
1715 cm-1
1480 cm-
1441 cm-11480 cm-1

This is an electrostatic interaction, highlighted by the yellow dashed lines in the diagram
below, with subcoordination between oxygens and lithiums in the structure. This is a
symmetrical 4 membered ring, with a bond angle of 90.5°. This ring structure is stabilising
for the cis isomer because it allows lithium to gain easier access to electrons within this ring.
ΔE(HF/6-31G**) (kcal/mol)
V(CN)
(cm-1
)
CN Bond Length(Angstroms)
MeNC N/A 2467 1.15
MeNCLiMe -19.2 2497 1.14
Lithiated Schiff BaseC-Li bonded
-10.3 1715 1.29
Lithiated Schiff BaseDimer (Trans)
-7.4 1480 1.43
Lithiated Schiff Base Dimer (Cis)
-11.1 1441 1.46
CN stretching frequency increases from 2467 cm-1 to 2497 cm-1 when complexed to form
methyllithium acetonitrile complex, indicating the CN bond has strengthened and has more
triple bond character, supported by the decrease in bond length for CN bond from 1.15 Å to
1.14 Å. The CN stretching frequency then decreases to 1715 cm-1 in lithiated Schiff base,
and the CN bond lengthens to 1.29 Å indicating a bond order of 2
33

Reaction of monomeric methyllithium with Acetonitrile (NCMe)
The table below displays the complexation energies for each molecule using semi-empirical
methods and 3 basis sets of ab initio. The energy values reach a plateau using ab initio
Hartree-Fock method, so the energies using ab initio HF/6-31G** were the values
considered to be the most accurate.
SE/PM3 Ab initioHF/3-21G* (kcal/mol)
Ab initioHF/6-31G* (kcal/mol)
Ab initioHF/6-31G** (kcal/mol)
MeCNLiMe -59.5 -26.8 -19.8 -19.9
Lithiated Schiff Base moleculeN-Li bonded
-29.7 -23.9 -24.9 -24.6
Lithiated Schiff base dimer(Trans)
-14.0 -12.0 -9.8 -10.2
Lithiated Schiff base dimer(Cis)
-31.4 -17.5 -13.2 -12.1
34

The formation of this methyllithium-acetonitrile adduct has a negative enthalpy of -19.9
kcal/mol. This is a largely favourable reaction due to the donor capabilities of nitrogen,
donating electrons to lithium, weakening the carbon to nitrogen bond. There is possible π-
backbonding 8 in this adduct as indicated by a decrease in CN stretching frequency. This
electron transfer strengthens the lithium-carbon bond and weakens the carbon-nitrogen
bond.
35
ΔE = -19.9 kcal/mol
2620 cm-1
2393 cm-1
Formation of Lithiated Schiff Base Complex
Formed using ab initio HF/6-31G** method
Complex formed by reaction of Acetonitrile and Methyllithium
Formed using ab initio HF/6-31G** method

This is a largely exothermic reaction releasing 24.6 kcal/mol. This reaction is favourable due
to nitrogen and lithium being directly bonded in this lithiated Schiff base molecule. The
acetonitrile inserts into the carbon to lithium bond in methyllithium to produce this short-lived
intermediate complex where nitrogen is directly bonded to lithium. CN stretching frequency
reduces from 2393cm-1 to 1720cm-1 indicating a decreased bond order, to a bond order of 2.
36
ΔE = -24.6 kcal/mol
2393 cm-1
1720 cm-1

Lithiated Schiff Base Dimerisation
Formed using ab initio HF/6-31G** method
37
1350 cm-
ΔE (Cis) = -12.1 kcal/molΔE (Trans) = -10.2 kcal/mol
1350 cm-1 1338 cm-1
The dimerization of lithiated Schiff base molecules is a favourable reaction, releasing 10.2
kcal/mol and 12.1 kcal for trans and cis isomers, respectively. Organolithium compounds
tend to form oligomeric molecules, hence negative complexation enthalpies. Both lithiated
Schiff base complexes (carbon and nitrogen bonded) dimerise to form the above isomers
of lithiated Schiff base dimers.

The cis isomer is more stable, with a complexation enthalpy lower than trans, indicating
higher stability, again due to the formation of a 4-membered ring, stabilizing lithium
molecules due to greater access to electron density.
The table below displays CN stretching frequencies in each complex and the corresponding
CN bond lengths. We can see that the CN stretching frequency reduces to form the first
complex MeCNLiMe which is in accordance with the experimental results for the reaction
between n-butyl lithium and t-butyl isonitrile where the CN stretching frequency reduced from
2179 cm-1 to 2135 cm-1.
ΔE (HF/6-31G**) kcal/mol
v (CN)
cm-1
CN Bond Length(Angstroms)
MeCN N/A 2632 1.14
MeCNLiMe -19.9 2393 1.13
Lithiated Schiff BaseN-Li bonded
-24.6 1720 1.24
Lithiated Schiff Base Dimer (Trans)
-10.2 1350 1.43
Lithiated Schiff Base Dimer (Cis)
-12.1 1338 1.46
38

Chapter 4
Conclusion
Conclusion
39

Monomeric methyllithium reacted with CO to form a main group lithium carbonyl adduct in
the first reaction step. This species then rearranged via formal insertion to form an ionic
species – lithium acetyl. There is subcoordination between lithium and oxygen in this lithium
acetyl intermediate, an intra-molecular electrostatic interaction. The lithium acetyl then
dimerizes to form lithiated oxycarbene dimers, with strong lithium-oxygen bonds, with a large
enthalpy of formation, which is the essential step to form room temperature stable products.
For further investigations, methylisonitrile and acetonitrile were used to react with monomeric
methyllithium. In the experiment t-butyl isonitrile was used to react with n-butyl
methyllithium, and followed a similar reaction sequence to CO since tBuNC is isoglobal to
CO. In the first reaction step, CNMe/NCMe reacts with monomeric methyllithium to form a
linear adduct where the triple bond is still intact, which was an exothermic reaction. The
CNMe/NCMe group then formally inserted into the carbon-lithium bond to form a lithiated
Schiff base molecule, which was the final product in experiments using tert-butyl lithium due
to the stability of this molecule at -20°C. However, in calculations, this lithiated Schiff base
molecule was dimerized to form lithiated Schiff base dimer molecules.
The reaction of methyllithium with CO offered promise for the use in synthesising aldehydes
from lithium acetyl reacting with water. This would have been very beneficial as aldehydes
are useful tools in organic synthesis. However, using the results of this project, the formation
of these ionic intermediates prevents the use of this reaction being used in the synthetic
preparation of aldehydes. These ionic intermediates are stable only for a very short period
of time at very low temperatures, before the intermediates oligomerize (typical behaviour of
organolithium compounds) so lithium acetyl is non-accessible to be used in the synthesis of
aldehydes.
Since 2004, the year of publication of “Reactions of Methyllithium with CO and CNMe:
Theoretical Study” (Matthias Tacke, Rosaria Leyden, Laurence P. Cuffe), there has been no
further work on the reactions of methyllithium with these ligands, and therefore no papers
have been published. Perhaps the spectroscopic work will stimulate further experiments and
theoretical calculations about the nature of chemical bonding in organolithium to quantify
differences between main group and d-element chemistry. 17 IR spectroscopy was used in
this project to monitor the characteristic IR frequencies for intermediates and products.
Another technique that is primarily used for structural analysis of organolithium compounds
is NMR spectroscopy. To date, there has been no study of monomeric methyllithium by
NMR spectroscopy so this is an opportunity for future work. 18
40

Acknowledgements
41

First and foremost, I would like to take this opportunity to say a huge thank you to Dr. Tacke
for all of his support and guidance throughout the course of this project. I would like to thank
him for his patience and understanding throughout the past year. His expertise and
experience in computational chemistry have proven invaluable to me, and have led to the
success of the completion of my project. I have really enjoyed learning under his
supervision.
I would also like to thank my colleague Laura Finnegan for her support and encouragement
throughout the project.
Figure Reference List
42

Figure 1 www.scs.illinois.edu/denmark/presentations/2013/gm-2013-3-12.pdf
Figure 2 www.scs.illinois.edu/denmark/presentations/2013/gm-2013-3-12.pdf
Figure 3 The Carbon to Lithium Bond Streitweiser, A.; Williams, J.E.; Alexandratos, S.; Mc
Kelvey, J.M. J. Am. Chem. Soc. (1976), 98, 4778
Figure 4 Mechanism of Reaction of Carbon Monoxide with Phenyllithium. Larry S. Trzupek,
Terry L. Newirth, Edward G. Kelly, Norma Ethyl Sbarbatti, and George M Whitesides.*
J. Amer. Chem. Soc. (1973), 95, 8126-8127
Figure 5 Mechanism of Reaction of Carbon Monoxide with Phenyllithium. Larry S. Trzupek,
Terry L. Newirth, Edward G. Kelly, Norma Ethyl Sbarbatti, and George M Whitesides.*
J. Amer. Chem. Soc. (1973), 95, 8126-8127
Figure 6 Molecule drawn using software HyperChem
Figure 7 en.wikipedia/org/wiki/methyllithium Image available for use by public domain by
user Benjah-bmm27
Figure 8 en.wikipedia/org/wiki/methyllithium Image available for use by public domain by
user Benjah-bmm27
Figure 9 Carbonyl and Benzene Complexes of Lithium: Transition-Metal-Like Behaviour of
Lithium in Organolithium Compounds. Matthias Tacke, Eur. J. Inorg. Chem. (1998),
537-541
Figure 10 Carbonyl and Benzene Complexes of Lithium: Transition-Metal-Like Behaviour of
Lithium in Organolithium Compounds. Matthias Tacke, Eur. J. Inorg. Chem. (1998),
537-541
Figure 11: Reactions of Methyllithium With CO and CNMe: Theoretical study. Matthias
Tacke, International Journal of Quantum Chemistry (2006) 106, 692-696
Figure 12: Reactions of Methyllithium With CO and CNMe: Theoretical study. Matthias
Tacke, International Journal of Quantum Chemistry (2006) 106, 692-696
43

Figure 13: Introduction to Molecular Modelling and Computational Chemistry. Retrieved
from
http://www.chem.arizona.edu/~lichtend/C518_Fall2009/introtocompchem/IntroToCC.html
Figure 14: The Schrödinger Equation. Retrieved from
http://chemwiki.ucdavis.edu/Physical_Chemistry/Quantum_Mechanics/Quantum_Theory/Pri
nciple_of_Quantum_Mechanics/Schr%C3%B6dinger_Equation
Figure 15: Screenshot from HyperChem software
44

References
1. “Theoretical studies in organolithium compounds”, Zvi Rappoport, Ilan Marek
(Editors) (2006) pg.2 The Chemistry of Organolithium Compounds
2. “Theoretical studies in organolithium compounds”, Zvi Rappoport, Ilan Marek
(Editors) (2006) pg.2 The Chemistry of Organolithium Compounds
3. The Carbon to Lithium Bond Streitweiser, A.; Williams, J.E.; Alexandratos, S.; Mc
Kelvey, J.M. J. Am. Chem. Soc. (1976), 98,4778
4. Streitwieser, A. J. Org. Chem. 2009, 74, 4433
5. D. Seyferth. RC. Hui, R.M Weinstein, W-L. Wang, Nova Acta Leopoldina (1985), 59,
335.
6. Mechanism of Reaction of Carbon Monoxide with Phenyllithium. Larry S. Trzupek,
Terry L. Newirth, Edward G. Kelly, Norma Ethyl Sbarbatti, and George M
Whitesides.* J. Amer. Chem. Soc. (1973), 95, 8126-8127
7. J. L. Wardell, in Comprehensive Organometallic Chemistry (Editors: G. Wilkinson, F.
G. A. Stone, E. W. Abel), Pergamon, Oxford, England, 1982; W. N. Setzer, P. von R.
Schleyer, Adv. Organomet. Chem. 1985, 24, 353; Lithium Chemistry
8. The Reaction of Rli Species with CO and Isonitriles; IR Spectroscopic Investigations
in liquid Xenon and Ab Initio Calculations of the Intermediates. M. Tacke. Chem. Ber.
(1995), 128, 1051-1053
9. A Novel Liquid Xenon IR Cell Constructed from a Silicon Single Crystal. M. Tacke,
P. Sparrer, R. Teuber, H-J Stadter, F. Schuster J. Mol. Struct. (1995), 349, 251-252
45

10. Carbonyl and Benzene Complexes of Lithium: Transition-Metal-Like Behaviour of
Lithium in Organolithium Compounds. Matthias Tacke, Eur. J. Inorg. Chem. (1998),
537-541
11. Reactions of Methyllithium With CO and CNMe: Theoretical study. Matthias Tacke,
International Journal of Quantum Chemistry (2006) 106, 692-696
12. “Introduction to Molecular Mechanics” C. David Sherill. School of Chemistry and
Biochemistry, Georgia Institute of Technology.
http://vergil.chemistry.gatech.edu/courses/chem6485/pdf/molmech-lecture.pdf
Lecture slide 6
13. The Schrödinger Equation. Retrieved from
http://chemwiki.ucdavis.edu/Physical_Chemistry/Quantum_Mechanics/Quantum_Th
eory/Principle_of_Quantum_Mechanics/Schr%C3%B6dinger_Equation
14. Molecular Quantum Mechanics P.W Atkins and R.S Friedman 3rd
Edition
15. Exploring Chemistry with Electronic Structure Methods. Second Edition. James B.
Foresman and AEleen Frisch.
16. Geometry Optimization taken from
-www.shodor/org/chemviz/optimization/teachers/background.html
17. Complexes of Decamethylsilicocene Cp2*
Si(CO) and Cp2*Si(N2) M. Tacke, Ch.
Klein, D.J Stufkens, A. Oskam, P. Jutzi, E.A Bunte Z. Anorg. Allg. Chem. (1993),
619, 865-868
18. Nuclear Magnetic Resonance G.A Webb. R. Soc. Chem. (1996), 166-
167Streitwieser, A. J. Org. Chem. 2009, 74, 4433
19. Geometry Optimisation HyperChem Practical Guide, pg 16. Can be viewed online
at: http://cheminfo.chemi.muni.cz/ktfch/janderka/Manuals/compchem.pdf
46

47