H. van der Goot (Editor) Trends in Drug Research III © 2002 Elsevier Science B.V. All rights reserved 181 Agonist channeling of a2-adrenoceptor function Karl E.O. Akerman, Johnny NSsman, Tomas Holmqvist and Jyrki P. Kukkonen Department of Physiology, Division of Cell Physiology, Uppsala University, BMC, Box 572, SE-75123 Uppsala, Sweden Introduction The a2-adrenergic receptors (a2-ARs) control the function of different organs via central and peripheral effects (Table 1). Ligands for these receptors have several therapeutic applications includmg anaesthesia and treatment of hypertension and glaucoma. Potential new indications may include obesity and psychiatric disorders. The central effects are thought to mainly be a result of presynaptic mhibition of noradrenaline release. However, a2-ARs are localised at postsynaptic sites as well. The mechanisms involved in the peripheral actions of a2-AR ligands are less well understood. Their effects can be both inhibitory and stimulatory depending on the organ in question [1]. Table 1. Physiological effects of ai adrenoceptor activation Central effects • Reduction in plasma adrenaline and nor- adrenaline • Inhibition of neuro- transmitter release • Sedation • Food intake • Hypotermia • Antinociception Peripheral effects Inhibitory • Insulin secretion • Vasopressin secretion • Tyroxine secretion • Saliva secretion • Gut movement (ileus) • Lipolysis Stimulatory • Vascular tonus (e.g. coronary & pulmonary venal tonus) • TSH secretion • Growth hormone secretion p Glycogenolysis • Platelet aggregation • Water absorption (gut) The variety of responses elicited by these receptors indicates that they may possess the ability to activate multiple signal transduction pathways [2]. The response elicited by the receptors depends on the tissue where these receptors are expressed as well as the subtype of receptor [3-10]. The classical cellular response observed upon activation of a2-ARs is an inhibition of sthnulated cAMP production [11]. However, changes in cAMP alone do not explain the physiological actions of a2-ARs. Three subtypes of a2- ARs, a2A, 0C2B and a2c, have been identified by cloning and pharmacological tools.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

H. van der Goot (Editor) Trends in Drug Research III © 2002 Elsevier Science B.V. All rights reserved 181
Agonist channeling of a2-adrenoceptor function
Karl E.O. Akerman, Johnny NSsman, Tomas Holmqvist and Jyrki P. Kukkonen Department of Physiology, Division of Cell Physiology, Uppsala University, BMC, Box 572, SE-75123 Uppsala, Sweden
Introduction The a2-adrenergic receptors (a2-ARs) control the function of different organs via
central and peripheral effects (Table 1). Ligands for these receptors have several therapeutic applications includmg anaesthesia and treatment of hypertension and glaucoma. Potential new indications may include obesity and psychiatric disorders. The central effects are thought to mainly be a result of presynaptic mhibition of noradrenaline release. However, a2-ARs are localised at postsynaptic sites as well. The mechanisms involved in the peripheral actions of a2-AR ligands are less well understood. Their effects can be both inhibitory and stimulatory depending on the organ in question [1].
Table 1. Physiological effects of ai adrenoceptor activation
Central effects
• Reduction in plasma adrenaline and noradrenaline
• Inhibition of neurotransmitter release
• Sedation • Food intake • Hypotermia • Antinociception
Peripheral effects Inhibitory • Insulin secretion • Vasopressin secretion • Tyroxine secretion • Saliva secretion • Gut movement (ileus) • Lipolysis
Stimulatory • Vascular tonus (e.g.
coronary & pulmonary venal tonus)
• TSH secretion • Growth hormone
secretion p Glycogenolysis • Platelet aggregation • Water absorption (gut)
The variety of responses elicited by these receptors indicates that they may possess the ability to activate multiple signal transduction pathways [2]. The response elicited by the receptors depends on the tissue where these receptors are expressed as well as the subtype of receptor [3-10]. The classical cellular response observed upon activation of a2-ARs is an inhibition of sthnulated cAMP production [11]. However, changes in cAMP alone do not explain the physiological actions of a2-ARs. Three subtypes of a2-ARs, a2A, 0C2B and a2c, have been identified by cloning and pharmacological tools.

182
Many of the systemic eflfects of a2-AR ligands are due to actions on presynaptic receptors, which inhibit transmitter release from in neuronal cells via inhibition cf voltage-gated N- or P/Q-type Ca ^ channels [12-14]. Like the inhibition of cAMP production this response is sensitive to pertussis toxin and therefore likely to be mediated by Gi/o-type G proteins.
Stimulation of cAMP accumulation Activation of a2-AR has also been shown to cause an increase rather than a decrease
of cAMP production both through recombinantly expressed [5,9,10,15] and endogenous a2-ARs [16-18]. In certain cells this stimulatory response is seen only at high agonist concentrations or if Gi-type G proteins are inactivated by pertussis toxin-treatment [6,10]. In many cell types the primary response seems to be stimulation of cAMP production and in many cases particularly the a2B-AR subtype has been implicated in the stimulatory coupling [10,19,20]. With all the subtypes, there are some agonists (e.g. clonidine, oxymetazoline, UK 14,304), though different for each subtype, that prefer coupling to an inhibitory response and have only a very weak stimulatory action [20-22]. The phenomenon where certam agonists can selectively activate specific signal pathways has been termed agonist trafficking (of receptor signals) [23]. The basis for this is that different active receptor conformations will preferentially activate specific G proteins.
Adenylyl cyclase (AC) can be stimulated directly through GSOL and directly or indirectly by other messengers [24] (Table 2). In many studies the stimulation cf cAMP production by a2-ARs and other Gj-coupled receptors has thus been interpreted as Gs coupling. However, many other possibilities exist such as effects via G protein p y subunits (Gjiy)? Ga ^ and protein kinase C (PKC). The effect of receptor activation would be dependent on the transduction mechanisms it utilises in the cells in question and on the respective AC expressed in the cells studied. In PC-12 cells, for instance, chelation of intracellular Ca ^ with BAPTA reduces the a2-AR-stimulated cAMP accumulation [5].
Table 2. Activation of adenylyl cyclase isoforms by different effectors Isoform activator Inhibitor Type l Type III, VIII Type II, IV, VII Type V, VI Type IX
Gsa, Ca'* G^a, Ca " G,a, PKC, GpY Gsa Gsa
GpY, Gitt Gia Gia (?) Gitt, Ca ^ Gia
To test whether py subunits would be involved in the stimulation of cAMP accumulation via the human a2B-AR in Sf9 cells the effects of Gpy and P-ARK (which functions as a Gpy scavenger) on forskolin-stimulated cAMP accumulation was tested (Fig. 1). While p-ARK enhanced forskolin-stimulated cAMP accumulation the GPy coexpression considerably reduced cAMP accumulation. This means that the adenylyl cyclase of Sf9 cells is inhibited by Py rather than stimulated. The a2B-AR-stimulated AMP accumulation in Sf9 cells can thus not be due to Py. The basal and forskolin-

183
stimulated cAMP accumulation are unaffected by stimulation of protein kinase C while a considerable stimulation of coexpressed type II adenylyl cyclase can be observed (manuscript in preparation). Ca "*" enhances the forskolin-stimulated cAMP accumulation [25]. However, stimulation of cAMP accumulation is seen also in cells depleted of intracellular Ca "*" [10]. Thus, the typical second-messenger- or CPy-niediated effects on adenylyl cyclase can be excluded in the case of the a2B-AR.
3-1
^2.5! 0
3 2 1
0
0 .5^
n J
p-ARK
control
n
—
r ^ n j = .
basal forsk basal forsk basal forsk
Fig. 1. Effect of P-ARK and Gpy expression on basal and forskolin-stimulated (forsk) cAMP accumulation in Sf9 cells. The methods used are essentially those described in references [10,22,25]. Cells were infected with recombinant baculovirus harboring the genes for either P-ARK (gift from Dr. A. DeBlasi, Consorzio Negri Sud, Santa Maria Imbaro, Chieti, Italy) or pi and Y2 (gifts from Dr. T. Haga University of Tokyo, Tokyo, Japan) subunits in the same recombinant virus. The infection time was 26 hours.
Activated ai-ARs have been shown to coprecipitate Gs proteins [6]. Mutagenesis studies on a2A-AR and a2B-AR suggest that the stimulatory response can be specifically modified by structural changes in the second intracellular loop [22] and in the N-terminal or C-terminal portion of the third intracellular loop [26]. Certain basic residues in the C-terminal portion are important for the stimulatory coupling [27]. The approximate positions of these domains in the intracellular loops of rhodopsin [28] are illustrated in Fig. 2. These findings, together with the fact that certain agonists can preferentially couple to inhibition of adenylyl cyclase, suggest that the inhibition and stimulation of adenylyl cyclase via a2-ARs require different structural determinants or are induced by different conformations of the receptor.

184
Fig. 2. The approximate position of domains important for stimulatory coupling super imposed on the intracellular surface of rhodopsin deduced from its crystal structure . The outline of the C-terminus second (i2) and third (i3) loops as well as the approximate positions of helices III, IV, V and VI are marked. Note that the i3 loop of a2-ARs is much longer than the corresponding loop in rhodopsin. The areas where stimulation of cAMP accumulation by a2-ARs are affected by mutagenesis are dotted black.
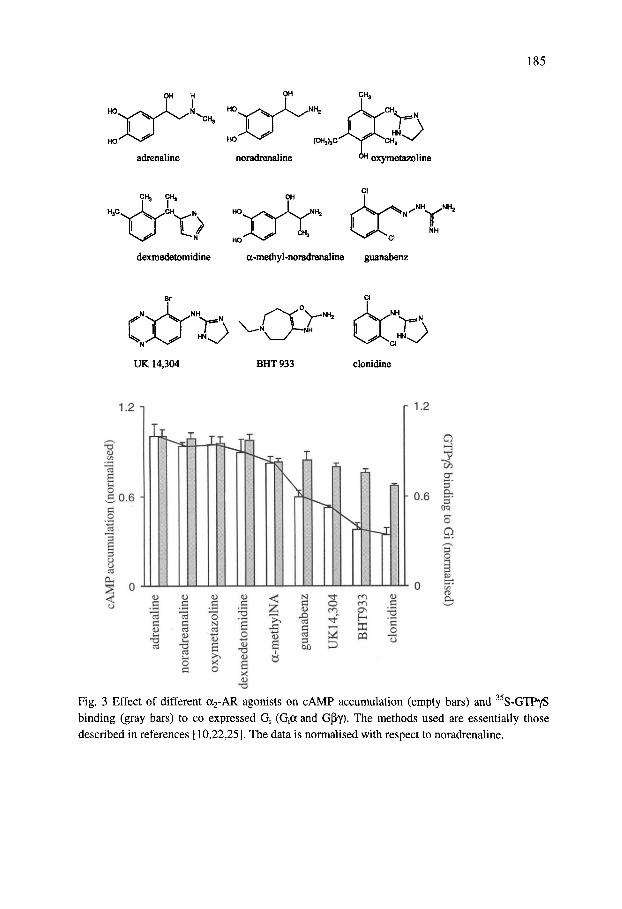
The ability of different a2-AR agonist to enhance cAMP accumulation as compared to their ability to activate coexpressed Gi, determined by ^^S-GTPyS binding, is shown in Fig. 3. Many of the ligands, like UK14,304 and clonidine, showed a weaker ability to stimulate cAMP accumulation while they were almost as effective as noradrenaline in coupling to Gi. With Gi coexpression these ligands inhibited rather than stimulated cAMP accumulation (Fig. 4). A small inhibiton was seen even with those ligands, which showed a high degree of stimulation when the Gi expression levels were further increased with longer incubation times. These results demonstrate that Gi can override the stimulatory coupling. They also indicate that the ability of the different ligands to couple to Gi is similar but the coupling to Gs is ligand-dependent. Thus when the receptor is activated by these ligands its ability to couple to stimulation of cAMP production is weaker than the coupling to inhibition. This would suggest that the Gs "affinity" of the receptor conformation induced by these ligands is lower. The results also suggest that Gi coexpression considerably attenuates the stimulatory coupling.
185
OH H Y
adrenaline
HO ^ (CH^C^
noradrenaline ^ oxymetazoline
CH3 CH3
HjC ^^xL ^CH N HO
dexmedetomidine a-methyl-noradrenaline guanabenz
t> "CP" NH2
UK 14,304 BHT933 clonidine
r 1.2
7^ •«
I O
O N
B
o
£ o
S X
T3
s
N C 0) ^ C
to/)
^ 0 m ^ T - H
^ ;D
m en G^ H UJ «
O
3 5'
crq
o
o
Fig. 3 Effect of different a2-AR agonists on cAMP accumulation (empty bars) and S-GTPyS binding (gray bars) to co expressed Q (Qa and Gpy). The methods used are essentially those described in references [10,22,25]. The data is normalised with respect to noradrenaline.

186
o c3
O
<D O CO o
0.3
0.1 H
^ ;T -0.1 H O a
-0.3
u s
I o
.s o N
s s o
*| o
T3
S X <u 'T::
2
S 1
d
n A T
N C 0)
-^ o
en en ON
(D C
"2 S
73
Fig. 4. Effect of Gi coexpression at 26 (light grey) and 38 hours of infection (dark grey) on agonist-mediated cAMP accumulation (a2B adrenoceptors expressed in Sf9 cells). Otherwise conditions are the same as in Fig. 3.
The data suggest that the two G proteins Gi and G^ interact with the same receptor. A model where two G proteins Ga and Gb competes for the same site of the receptor was developed (Fig. 4). This would not necessary mean that they compete for the same site but that coupling to one G protein excludes the coupling to another. The agonist activation of the receptor was modelled according to the equation:
r^l ^ [agonist][Rl^,_ ^ ^"^ [agonist] + EC 50-R
187
i[R]aci = concentration of activated receptors; [R]act-max = maximum concentration of receptors that can be activated; EC50-R = the concentration of agonist producing the half-maximal receptor activation). The efficacy could be regulated by changing the [R]act-max in an analogous fashion as in [25] and EC50-R. This activated receptor acts as an activator for two G proteins, Ga and Gb. The activation of Gb can be described according to the equation
[GbL=-[ ]„JGJ„
[^L+EC 50-G. 1 + G
50 y
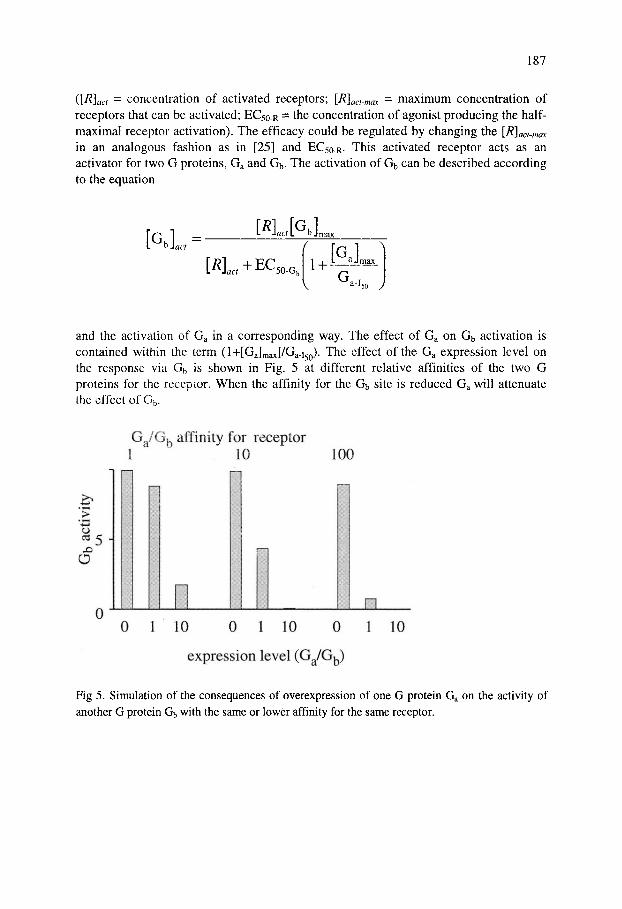
and the activation of Ga in a corresponding way. The effect of Ga on Gb activation is contained within the term (l+[Ga]max]/Ga-i5Q). The effect of the Ga expression level on the response via Gb is shown in Fig. 5 at different relative affinities of the two G proteins for the receptor. When the affinity for the Gb site is reduced Ga will attenuate the effect of G5.
0
Gj /G , affinity for receptor 1 10 100
_•. 0 1 10 0 1 10 0
expression level (G,JG^)
10
Fig 5. Simulation of the consequences of overexpression of one G protein G on the activity of another G protein Gb with the same or lower affinity for the same receptor.

188
Thus if the conformations induced by ligands such as UK 14,304 and clonidine have a lower "affinity" for Gs (=Gb) then Gi (=Ga) overexpression would lead to a considerable attenuation of the Gs response.
Coupling to Ca mobilisation
In many cells a2-ARs have been shown to couple to production of inositol-1,4,5-trisphosphate [4,29,30] and Ca "" mobilisation [3,30-35]. In Sf9 cells only the a2B receptor seems to be able to cause any significant Ca " mobilisation [30]. The agonist profile for this response is similar to that of stimulation of cAMP accumulation, e.g. UK 14,304 and clonidine are very weak agonists with respect to Ca "*" mobilisation. Fig. 6 shows that Gi coexpression considerably attenuates the Ca " mobilisation via a2B but Gs has little or no effect. Coexpression with Py subunits alone caused a small reduction in the noradrenaline-induced Ca^^ elevation.
[Ca - li /nM 400 n
300
200
100
0
. Q ^
1
^5^ O^ O'v < ^
Fig. 6 Effect of Gs, Gi and py coexpresion on 100 nM noradrenaline-stimulated Ca " mobilisation in cells expressing the a2B receptor. The methods used are essentially those described in references [10,22,25,]. Ca ^ measurement were performed using fura-2 as described in [30].
This result is in line with the effect of Gi coexpression on the stimulatory cAMP response and suggest that the Ca " mobilisation occurs through coupling to another G-protein with lower "affinity" for the receptor than Gi.
Ca " mobilisation via a2-AR activation has previously been observed in many other cells such as CHO or HEK293 cells expressing recombinant receptors. The response

189
seen in these cells is usually pertussis toxin sensitive and hence mediated by Gi/o proteins unlike the response in Sf9 cells. The properties of pertussis toxin sensitive Ca " mobilization are listed in table 3.
Table 3. Pertussis toxin sensitive Ca ^ mobilisation
• Is seen with recombinant Gi coupled receptors expressed in CHO, HEK, COS-7 etc as well as with native receptors in HEL erythroleukemia cells
• All a2 receptor subtypes are about equally effective • Sensitive to inhibitors of PLCP • Associated with an increase in IP3 • Sensitive to cotransfection with (Jy scavengers like (J-ARK
The pathway for activating Ca " mobilization in CHO cells is complex since it requires cooperating responses via other endogenously activated receptors such as purinergic P2Y receptors [35]. Thus a possible mechanism for this type of coupling is activation of PLCp by a cooperative action of Gq like proteins and py subunits from
Gi/o-
It is uncertain whether Ca "*" mobilisation in Sf9 cells and the pertussis toxin-sensitive Ca^^ mobilisation represent the same phenomenon since in Sf9 cells Gi inhibited rather than promoted the response. Interestingly the agonist profile for Ca^^ mobilisation via the a2B receptor is very similar to that seen in CHO cells both with respect when the maximal response and EC50 values are compared (Fig. 7).
In HEL human erythroleukemia cells, which have been used as models for platelets, there is a very robust Ca"* response to ai-AR activation via the endogenous a2A. In these cells synthetic compounds like imidazolines show a very similar ability to activate Gj coupling and Ca "*" mobilisation but the catecholamines are much more prone to induce Ca " mobilisation [36].

190
0) v> c
CO
O
(0
o c 2 CO
150
100
50
A. Maximum response
A catecholamine • imidazolines
best fit througii (0,0)
D-Medetomidine I Oxymetazoline
0 50 100
CHO (nomialised Ca^ response)
CO
1000 1 A catecholamine # imidazolines
best fit
100
10
Oxymetazoline
Noradr
2.4:1
10 100
CHO (nM)
Clonidine
1000
Fig. 7 Comparison of the ability of catecholamine and imidazoline ligands to induce Ca mobilisation via the a2B receptors in Sf9 and CHO cells. Methods are described in references [30, 35].

191
Conclusions a2, adrenoceptors, in particular a2B, can couple to several signal pathways. The
mutagenesis studies indicate that Gs and possibly Gq -type proteins couple to another site or conformation of the receptor than Gj. The ligand-specific coupling suggest that the conformation induced by these ligands is different as compared to native ligands. The UK 14,304-induced receptor conformation couples equally well Gi as the noradrenaline-induced conformation but less efficiently to Gs. The model with a different apparent affinity for the interaction of the secondary G protein for the receptor fits well with the experimental data. The outcome at the effector level will thus depend on the "affinity" of the G protein in question for the receptor as well as the G protein expression level. The more rigid synthetic ligands are thus more prone to promote coupling to the GJGo pathway. The more flexible natural ligands allow coupling to several pathways. Even more rigid ligands could be useful as probes for testing conformation-signal coupling. Coupling to different pathways could well explain the variety of effects of a2-AR agonists seen in different tissues
Acknowledgements r The original working this presentation was funded by The Medical Research Council of Sweden (MFR K98-14X-12205-02B), The Cancer Research Fund of Sweden, The Claes Groschinsky Foundation and The Ake Wiberg Foundation, the Academy of Finland, TEKES and The Sigrid Juselius Foundation.
References [I] R.R. Ruffolo, A.J. Nichols, J.M. Stadel, and J.P. Hieble, Annu. Rev. Pharmacol.
Toxicol. 33 (1993) 243. [2] G. Milligan, Trends Pharmacol. Sci. 14 (1993) 239. [3] M.C. Michel, L.F. Brass, A.Williams, G.M. Bokoch, V.J. LaMorte, and H.J.
Motulsky, J. Biol. Chem. 264 (1989) 4986. [4] S. Cotecchia, B.K. Kobilka, K.W. Daniel, R.D. Nolan, E.Y. Lapetina, M.G.
Caron, R.J. Lefkowitz, and J.W. Regan, J. Biol. Chem. 265 (1990) 63. [5] E. Duzic and S.M. Lanier, J. Biol. Chem. 267 (1992) 24045. [6] M.G. Eason, H. Kurose, B.D. Holt, J.R. Raymond, and S.B. Liggett, J. Biol.
Chem. 267 (1992) 15795. [7] E.E. MacNulty, S.J. McClue, I.C. Carr, T. Jess, M.J. Wakelam, and G. Milligan,
J. Biol. Chem. 267 (1992) 2149. [8] T.K. Aburto, C. Lajoie, and K.G. Morgan, Circ. Res. 72 (1993) 778. [9] C.C. Jansson, A. Marjamaki, K. Luomala, J.M. Savola, M. Scheinin, and K.E.O.
Akerman, Eur. J. Pharmacol. 266 (1994) 165. [10] C.C.Jansson, M. Karp, C. Oker-Blom, J. Nasman, J.M. Savola, and K.E.O.
Akerman, Eur. J. Pharmacol. 290 (1995.) 75. [II] J.W. Lomasney, S. Cotecchia, R.J. Lefkowitz, and M.G. Caron, Biochim.

192
Biophys. Acta 1095 (1991) 127. [12] M. Gollasch, J. Hescheler, K. Spicher, F.J. Klinz, G. Schultz, and W.Rosenthal,
Am. J. Physiol. 260 (1991) C1282. [13] S.Y. Song, K., Saito, K. Noguchi, and S. Konishi, Pflugers Arch. 418 (1991)592. [14] A. Surprenant, D.A. Horstman, H. Akbarali, and L.E. Limbird, Science 257
(1992)977. [15] S.B. Jones, S.P. Halenda,. and D.B. Bylund, Mol. Pharmacol. 39 (1991) 239. [16] S. Ullrich, and C.B. Wollheim,. J. Biol. Chem. 259 (1984) 4111. [17] S.B. Jones, M.L. Toews, J.T. Turner, and D.B. Bylund,. Proc. Natl. Acad. Sci.
USA 84 (1987) 1294. [18] S. Mhaouty, J. Cohen-Tannoudji, R. Bouet-Alard, I. Limon-Boulez, J. Maltier, J.
P. and C. Legrand, J. Biol. Chem. 270 (1995) 11012. [19] D.J. Pepperl, and J.W. Regan, Mol. Pharmacol. 44 (1993) 802. [20] K. Pohjanoksa, C.C. Jansson, K. Luomala A. Marjamaki, J.M. Savola and M.
Scheinin Eur. J. Pharmacol. 335 (1997) 53. [21] M.G. Eason, M.T. Jacinto, and S.B. Liggett, IVlol. Pharmacol. 45 (1994) 696. [22] J. Nasman, C.C. Jansson, and K.E.O. Akerman, J. Biol. Chem. 272 (1997) 9703. [23] T. Kenakin, Trends Pharmacol. Sci. 16 (1995) 232. [24] R.K. Sunahara, C.W. Dessauer, and A.G. Gilman, Annu. Rev. Pharmacol.
Toxicol. 36 (1996) 46. [25] J. Nasman, J.P. Kukkonen and K.E.O. Akerman, Insect Biochem. Mol. Biol.
(2001) In press [26] M.G. Eason, and S.B. Liggett, J. Biol. Chem. 271 (1996) 12826. [27] S.M. Wade, W.K. Lina, K.-L. Lan, D.A. Chung, M. Nanamori and R.M. Neubig.
Mol.Pharm58 (1999) 1005. [28] K. Palczewski, T. Kumasaka, T. Hori, C.A. Behnke, H. Motoshima, B.A. Fox, I.
Le-Trong, D.-C. Teller, T. Okada, R.-E. Stenkamp, M. Yamamoto, M. Miyano Science. 289 (2000) 739.
[29] M.O.K. Enkvist,H. Hamalainen, C.C. Jansson, J.P. Kukkonen, R. Hautala, M.J. Courtney,, and K.E.O. Akerman, J. Neurochem. 66 (1996) 2394.
[30] C.I. Holmberg, J.P. Kukkonen, A. Bischoff, J. Nasman, M.J. Courtney, M.C. Michel, and K.E.O. Akerman, Eur. J. Pharmacol. 363 (1998) 65.
[31] A.K. Salm, and K.D. McCarthy, Glia 3 (1990) 529. [32] W. Erdbrugger, P. Vischer, H.J. Bauch,. and M.C. Michel, J Cardiovasc
Pharmacol 22 (1993) 97. [33] G.W. Dorn, K.J.. Oswald, T.S. McCluskey, D.G. Kuhel, and S.B. Liggett,
Biochemistry 36 (1997) 6415. [34] J.P. Kukkonen, A. Renvaktar, R. Shariatmadari, and K.E.O. Akerman, J.
Pharmacol. Exp. Ther. 287, (1998) 667. [35] K.E.O. Akerman, J. Nasman, P.E. Lund, R. Shariatmadari, and J.P Kukkonen,
FEBS Lett. 430 (1998) 209. [36] J.P. Kukkonen, C.C. Jansson, and K.E.O. Akerman , Br. J. Pharmacol. 132
(2001)1477.
Related Documents











