Journal of Colloid and Interface Science 315 (2007) 47–53 www.elsevier.com/locate/jcis Adsorption of arsenic(III) and arsenic(V) from groundwater using natural siderite as the adsorbent Huaming Guo a,∗ , Doris Stüben b , Zsolt Berner b a School of Water Resources and Environment, China University of Geosciences, Beijing 100083, People’s Republic of China b Institute for Mineralogy and Geochemistry, University Karlsruhe (TH), Karlsruhe 76131, Germany Received 4 April 2007; accepted 19 June 2007 Available online 26 July 2007 Abstract Batch and column tests were performed utilizing natural siderite to remove As(V) and As(III) from water. One hundred milligrams of siderite was reacted at room temperature for up to 8 days with 50 mL of 1000 μg/L As(V) or As(III) in 0.01 M NaCl. Arsenic concentration decreased exponentially with time, and pseudoequilibrium was attained in 3 days. The estimated adsorption capacities were 520 and 1040 μg/g for As(V) and As(III), respectively. Column studies show that effluent As was below 1.0 μg/L after a throughput of 26,000 pore volumes of 500 μg/L As water, corresponding to about 2000 μg/g of As load in the filter. Results of scanning electron microscopy (SEM) and transmission electron microscopy (TEM) reveal that high As retention capacity of the filter arose from coprecipitation of Fe oxides with As and subsequently adsorption of As on the fresh Fe oxides/hydroxides. Arsenic adsorption in the filter from As-spiked tap water was relatively lower than that from artificial As solution because high HCO − 3 concentration restrained siderite dissolution and thus suppressed production of the fresh Fe oxides on the siderite grains. The TCLP (toxicity characteristic leaching procedure) results suggest that these spent adsorbents were inert and could be landfilled. © 2007 Elsevier Inc. All rights reserved. Keywords: Arsenate; Arsenite; Drinking water; Removal; Retention 1. Introduction Groundwater enriched with As species such as arsenate (As(V)), arsenite (As(III)), and organic arsenic has become one of the most serious problems in water environment, espe- cially in the southeast of Asia, including West Bengal, India, Bangladesh, and China [1,2]. It is particularly worse when the groundwater is utilized as drinking water [3,4]. Although organic As species can be presented as a result of in situ biomethylation, inorganic As as As(III) and As(V) are generally considered to be the dominant species in natural water. The oxidation state of As depends primarily on pH and redox conditions, with As(V) being the most stable form under aerobic conditions as the pH-dependent deprotonated oxyan- ions of arsenic acid (H 2 AsO − 4 and HAsO 2− 4 ) and As(III) the chemically dominant forms in reducing environment as a neu- * Corresponding author. Fax: +86 10 8232 1081. E-mail address: [email protected] (H. Guo). tral species (i.e., pK a1 = 9.2) at natural pH. Therefore, the As(III) is more difficult to remove from water at neutral pH by means of adsorption and coprecipitation due to the lack of elec- trostatic attraction [5,6]. However, most As-enriched ground- water is generally dominated by As(III), up to 96% of total As [2,7]. Furthermore, As(III) is about 60 times more toxic than As(V) [8,9]. Thus, the removal of As(III) from drinking water has received significant attention, as well as As(V) [10–14], and it is of major concern to many water utilities and governmental agencies. Among a variety of adsorbents for As removal, natural geo- materials are promising because they are relatively cheap, read- ily available in different particle sizes, and therefore may easily be used as column fillings in small-scale treatment plants. Ar- senic adsorption onto natural zeolite and volcanic stone [15], natural iron ores [16], oxisol [17], red mud [18], and fer- ruginous manganese ore [19] has been examined intensively. Contrastingly, few studies on As removal by natural siderite have been reported [20,21], where the combination of natural hematite and natural siderite has been utilized for As reten- 0021-9797/$ – see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.jcis.2007.06.035

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Journal of Colloid and Interface Science 315 (2007) 47–53www.elsevier.com/locate/jcis
Adsorption of arsenic(III) and arsenic(V) from groundwater using naturalsiderite as the adsorbent
Huaming Guo a,∗, Doris Stüben b, Zsolt Berner b
a School of Water Resources and Environment, China University of Geosciences, Beijing 100083, People’s Republic of Chinab Institute for Mineralogy and Geochemistry, University Karlsruhe (TH), Karlsruhe 76131, Germany
Received 4 April 2007; accepted 19 June 2007
Available online 26 July 2007
Abstract
Batch and column tests were performed utilizing natural siderite to remove As(V) and As(III) from water. One hundred milligrams of sideritewas reacted at room temperature for up to 8 days with 50 mL of 1000 µg/L As(V) or As(III) in 0.01 M NaCl. Arsenic concentration decreasedexponentially with time, and pseudoequilibrium was attained in 3 days. The estimated adsorption capacities were 520 and 1040 µg/g for As(V)and As(III), respectively. Column studies show that effluent As was below 1.0 µg/L after a throughput of 26,000 pore volumes of 500 µg/LAs water, corresponding to about 2000 µg/g of As load in the filter. Results of scanning electron microscopy (SEM) and transmission electronmicroscopy (TEM) reveal that high As retention capacity of the filter arose from coprecipitation of Fe oxides with As and subsequently adsorptionof As on the fresh Fe oxides/hydroxides. Arsenic adsorption in the filter from As-spiked tap water was relatively lower than that from artificial Assolution because high HCO−
3 concentration restrained siderite dissolution and thus suppressed production of the fresh Fe oxides on the sideritegrains. The TCLP (toxicity characteristic leaching procedure) results suggest that these spent adsorbents were inert and could be landfilled.© 2007 Elsevier Inc. All rights reserved.
Keywords: Arsenate; Arsenite; Drinking water; Removal; Retention
1. Introduction
Groundwater enriched with As species such as arsenate(As(V)), arsenite (As(III)), and organic arsenic has becomeone of the most serious problems in water environment, espe-cially in the southeast of Asia, including West Bengal, India,Bangladesh, and China [1,2]. It is particularly worse when thegroundwater is utilized as drinking water [3,4].
Although organic As species can be presented as a resultof in situ biomethylation, inorganic As as As(III) and As(V)are generally considered to be the dominant species in naturalwater. The oxidation state of As depends primarily on pH andredox conditions, with As(V) being the most stable form underaerobic conditions as the pH-dependent deprotonated oxyan-ions of arsenic acid (H2AsO−
4 and HAsO2−4 ) and As(III) the
chemically dominant forms in reducing environment as a neu-
* Corresponding author. Fax: +86 10 8232 1081.E-mail address: [email protected] (H. Guo).
0021-9797/$ – see front matter © 2007 Elsevier Inc. All rights reserved.doi:10.1016/j.jcis.2007.06.035
tral species (i.e., pKa1 = 9.2) at natural pH. Therefore, theAs(III) is more difficult to remove from water at neutral pH bymeans of adsorption and coprecipitation due to the lack of elec-trostatic attraction [5,6]. However, most As-enriched ground-water is generally dominated by As(III), up to 96% of total As[2,7]. Furthermore, As(III) is about 60 times more toxic thanAs(V) [8,9]. Thus, the removal of As(III) from drinking waterhas received significant attention, as well as As(V) [10–14], andit is of major concern to many water utilities and governmentalagencies.
Among a variety of adsorbents for As removal, natural geo-materials are promising because they are relatively cheap, read-ily available in different particle sizes, and therefore may easilybe used as column fillings in small-scale treatment plants. Ar-senic adsorption onto natural zeolite and volcanic stone [15],natural iron ores [16], oxisol [17], red mud [18], and fer-ruginous manganese ore [19] has been examined intensively.Contrastingly, few studies on As removal by natural sideritehave been reported [20,21], where the combination of naturalhematite and natural siderite has been utilized for As reten-

48 H. Guo et al. / Journal of Colloid and Interface Science 315 (2007) 47–53
tion. In comparison with natural hematite, the natural sideritegenerally had a higher adsorption capacity for As species inbatch tests, and preferentially adsorbed inorganic As in a rel-atively wider pH range [20]. Nevertheless, information of Asadsorption on natural siderite is very limited and more testingis necessary. There is a need to study the kinetics and isothermsof the siderite in adsorbing As from water, and to evaluate theeffectiveness of a siderite-packed column for As removal inachieving the permissible concentration in drinking water. Theobjectives of this study were to (1) evaluate the effectivenessof natural siderite in removing As(V) and As(III) from water;(2) examine the feasibility of the siderite as filter fillings for Asremoval from As-contaminated water; (3) evaluate the potentialutilization of the siderite as permeable reactive barrier media inremediation of As contamination in groundwater.
2. Materials and methods
2.1. Materials
Natural siderite, reported as SIO4 by Guo et al. [20], wasused in this study. Detailed chemical and mineralogical com-ponents of the sample have been provided earlier in Guo et al.[20]. Prior to experiments, the mineral sample was ground andsieved into five main particle size fractions, including <0.04,0.04–0.08, 0.08–0.10, 0.10–0.25, and 0.25–0.50 mm.
All reagents used were of analytical grade. Stock solutions(100 mg/L As) were prepared from sodium arsenite (AsNaO2;>99.0%, Fluka Chemical) for As(III) and sodium arsenate(Na2HAsO4·7H2O; >98.5%, Fluka Chemical) for As(V). In or-der to maintain a relatively constant ionic strength, all artificialAs solutions contained 0.01 M NaCl as background electrolyte.Tap water spiked with 100 µg/L As(V) and 100 µg/L As(III)was also used as inflow water to examine the effect of multi-components on the adsorption performance of siderite-packedfilters. Chemical composition of the tap water is shown in Ta-ble 1, indicating it was of Ca–HCO3 type.
All glassware and sample bottles were washed with a de-tergent solution, rinsed with tap water, soaked with 1.0% sub-boiled HNO3 for at least 12 h, and finally rinsed with Milli-Qwater three times.
2.2. Adsorption experiments
Batch experiments were conducted to obtain rate and equi-librium data by reacting 50 mL of solution containing a definedAs concentration with 0.1 g of sample material of known grainsize range at neutral pH at room temperature of 20 ± 2 ◦C in
100 mL light polyethylene bottles. Most experiments were, un-less otherwise stated, performed with a sample of a grain sizerange of 0.10–0.25 mm. After the required reaction time, thesuspension liquid was decanted and filtered through a 0.45 µmcellulose acetate filter. The filtered solution was analyzed forAs species. To study the effect of contact time on As uptake,experiments were carried out with initial As(V) or As(III) con-centrations of 1000 µg/L. Isotherm studies were conductedwith initial As(V) or As(III) concentrations between 250 and2000 µg/L, and a contact time of 72 h. To investigate the effectof the grain size range on As adsorption, batch tests were car-ried out with grain size ranges of <0.04, 0.04–0.08, 0.08–0.10,0.10–0.25, and 0.25–0.50 mm, and initial As(V) or As(III) con-centrations of 2000 µg/L.
Column experiments were conducted with the siderite of0.10–0.25 mm particle size fraction (SO), or with siderite-coated sand (SCS) prepared by homogeneously mixing 0.04–0.08 mm siderite with 1.0% HNO3-rinsed 0.10–0.25 mm quartzsand at a ratio of 1:5 in weight. The detailed procedure ofSCS preparation was described in Guo et al. [21]. Plexiglascolumns with an inner diameter of 30 mm and a height of150 mm, as fixed-bed upflow reactor, were used in the columnstudy, which yields a working volume of about 100 mL. Feed-water containing 250 µg/L As(V) and 250 µg/L As(III) waspumped through a SO-packed filter (SO-C1) with a peristalticpump (Model 205S, Watson Marlow Company) at a flow rate2.15 mL/min. In order to investigate the effect of multicompo-nent on As removal, another SO-packed column (SO-C2) waspenetrated by As-spiked tap water containing 100 µg/L As(III)and 100 µg/L As(V). A SCS-packed column (SCS-C) was usedto filter 500 µg/L As(V) solution at a flow rate of 1.48 mL/minto examine the capability of siderite-coated sand in removingAs from water. Effluent solutions from the filters were collectedat regular intervals and analyzed for residual As species.
2.3. Analytical methods
A flow injection hydride generation system (FIAS 200,Perkin-Elmer) coupled with atomic absorption spectrometry(AAS 4001, Perkin-Elmer) was used for As speciation analy-ses. With regard to different As species, hydride generation wascarried out in different acid media in the presence of reductant.After filling the sample loop the sample plug was pushed outby the acidic carrier solution and completely mixed with the se-lected acid in the mixing coil before reduction took place byentraining the potassium tetrahydroborate. The detailed analy-sis procedure was described by Rüde and Puchelt [22]. Thedetection limits of total As, As(III), and As(V) were 1, 0.2, and0.3 µg/L, respectively. Major anions were determined by ion
Table 1Chemical composition of tap water used in the column experiment (major ions in mg/L, trace elements in µg/L, and EC in µS/cm)
pH ECa HCO−3 Cl− NO−
3 SO2−4 HPO2−
4 K+ Na+ Ca2+ Mg2+ Cr Mn7.31 683 333 21.0 4.94 76.2 <0.01 1.83 10.7 113 12.5 0.13 4.35
Fe Co Ni Cu Zn As Rb Sr Mo Cd Sb Ba Pb8.40 0.09 1.50 58.3 1437 0.35 0.77 357 0.46 1.95 0.37 74.2 2.73
a EC means electrical conductivity.

H. Guo et al. / Journal of Colloid and Interface Science 315 (2007) 47–53 49
chromatography (IC, DX-100, DIONEX), major cations (i.e.,K+, Ca2+, Na+, and Mg2+) and Mn and Fe by flame atomic ab-sorption spectrometry (Model 1100B, Perkin-Elmer), and traceelements by high-resolution inductively coupled plasma massspectrometry (HR-ICP-MS, Axiom, VG Elemental).
Electrical conductivity (EC) and pH were monitored by aportable WTW EC meter (Model LF330, coupled with an ECprobe of TetraCon 325) and pH meter (Model pH330, coupledwith a pH probe of SenTix 43-1), respectively. The pH meterwas calibrated prior to use.
Morphological analysis of the pristine and spent SO grainswas performed by scanning electron microscopy (SEM) us-ing a LEO 1530 Gemini microscope (at 10 kV) with energy-dispersive X-ray analyses. For transmission electron micro-scopy (TEM), the spent SO grains were dispersed in glue andembedded in a hole at the center of a 3 mm metallic disk witha thickness of 500 µm. Each disk was mechanically grindedto a thickness between 100 and 150 µm. The grinding wasfollowed by a dimpling process until the specimen thicknessreached 10–50 µm in the thinnest regions. The final thinningprocedure was performed by Ar+ ion bombardment with ionenergy of 6 keV. The ion-thinning sections were observed witha Zeiss EM 912 TEM equipped with an X-ray spectrometer forelemental analysis. Energy dispersive spectroscopy (EDS) wasperformed at 120 kV with a beam current of 1 nA for ∼100 s.
2.4. The toxicity characteristic leaching procedure (TCLP)test
The spent adsorbents generated in the column filters weretested with TCLP to determine the spent filling materials as in-ert or hazardous in terms of leachability of adsorbed As [23]. Inthe TCLP, the spent adsorbent was treated with acetate buffer(5.7 mL of glacial CH3COOH added to 500 mL of Milli-Q wa-ter, plus 64.3 mL of 1 N NaOH and diluted to 1 L, pH 4.93),with a liquid/solid ratio of 20. The extraction was achieved byagitating the specimens for 18 h in a shaker, after which theliquid phase was separated off using 0.45 µm cellulose acetatefilter and analyzed for total As by HR-ICP-MS.
3. Results and discussion
3.1. Effect of contact time
The effect of contact time (0.5–194 h) on As adsorption inAs(V) batches and As(III) batches is shown in Fig. 1. The re-sults clearly demonstrate that adsorption efficiencies increasedrapidly with an increase in contact time up to 24 h, and max-imum removal capacities of 386 and 458 µg/g were achievedwith a contact time of 194 h in the As(V) batch and the As(III)batch, respectively. Furthermore, the amount of adsorbed Asfrom As(V) solution was a little greater than that from As(III)solution at a contact time between 0.5 and 2.5 h, while less thanthat from As(III) solution afterward. However, with the reactioncondition of adsorbent dosage of 10 g/L and contact time of24 h, the siderite with grain size fraction of 0.25–0.50 mm waspreviously found to remove As from As(V) solution (57.4 µg/g)
Fig. 1. Effect of contact time on As adsorption from As(V) solution and As(III)solution by the siderite, with reaction conditions: ionic strength = 0.01 M NaCl,adsorbent dosage = 2 g/L, initial As concentration = 1000 µg/L, grain sizefraction = 0.10–0.25 mm.
much more than from As(III) solution (22.1 µg/g) at neutralpH [20]. This difference suggests that the reaction conditionsand the grain size fraction influenced As adsorption on thesiderite.
The kinetics data also indicate that the As removal fromAs(V) solutions and As(III) solutions mainly occurred within72 h and there was no significant change in residual As con-centrations after this time up to 194 h, which means that apseudoequilibrium of As adsorption was roughly attained af-ter 72 h. The pseudoequilibrium time of 72 h was greater thanthat for As(V) adsorption on natural hematite (0.25–0.50 mm)reported as 24 h [20].
3.2. Effect of initial As concentration
The As loadings on the adsorbents (Qe, µg/g) were cal-culated from the pseudoequilibrium As concentrations (Ce,µg/L) using mass balance. The adsorption isotherm data (Qevs Ce) were fitted to Langmuir and Freundlich isotherm models[Eqs. (1) and (2), respectively] [24,25]. Nonlinear regressionwas performed with Statistica for Windows using the Quasi-Newton method. The Langmuir isotherm model assumes amonolayer surface coverage limiting the adsorption due to thesurface saturation, while the Freundlich isotherm model is anempirical model allowing for multilayer adsorption [26].
(1)Qe = Q0bCe
1 + bCe,
(2)Qe = KFCne ,
where Ce is the pseudoequilibrium concentration in the solution(µg/L), Qe is the amount adsorbed on the adsorbent at pseu-doequilibrium (µg/g), Q0 is the maximum adsorption capacity(µg/g), b is a constant related to the adsorption energy (L/µg),KF is the Freundlich constant denoting the adsorption capac-ity of the adsorbent [(µg/g)(L/µg)n], and n is the adsorptionintensity parameter. Values of 0.1 < n < 1 show favorable ad-sorption of As onto adsorbents [27].

50 H. Guo et al. / Journal of Colloid and Interface Science 315 (2007) 47–53
All the adsorption data obtained were fitted to both mod-els as shown in Fig. 2. However, with the adsorption of bothAs(V) and As(III), calculated correlation coefficients for theFreundlich isotherm model were a little higher compared tothose for the Langmuir (Table 2). Therefore, the Freundlichisotherm yielded a better fit to the experimental data with re-gard to the siderite with a grain size range of 0.10–0.25 mm. Incontrast, the As(V) adsorption on the same siderite with a grainsize range of 0.25–0.50 mm followed Langmuir isotherm moreclosely in comparison with the Freundlich isotherm [20]. Be-cause the Freundlich isotherm model [Eq. (2)] is an empiricalmodel allowing for multilayer adsorption, it could be speculatedthat the multilayer adsorption would be involved in the processof As removal by the fine grains (0.10–0.25 mm), whereas themonolayer adsorption by the coarse grains (0.25–0.50 mm), dueto the greater surface area of the fine grains in comparison withthe coarse ones [20].
3.3. Effect of grain size
The results demonstrate that the As removal from bothAs(III) solution and As(V) solution increased with the decreasein grain size fractions (see Fig. S1 in supporting information).The siderite with grain size fractions between 0.04–0.08 and
Fig. 2. Langmuir and Freundlich plots for As adsorption from As(V) so-lution and As(III) solution on the siderite, with reaction conditions: ionicstrength = 0.01 M NaCl; adsorbent dosage = 2 g/L; initial concentrations =250–2000 µg/L; contact time = 72 h; grain size fraction = 0.10–0.25 mm.
Table 2Correlation coefficients and isotherm parameters of both Langmuir and Fre-undlich models (reaction conditions: ionic strength = 0.01 M NaCl; adsorbentdosage = 2 g/L; initial concentrations = 250–2000 µg/L; contact time = 72 h;grain size fraction = 0.10–0.25 mm)
Langmuir model Freundlich model
Q0
(µg/g)b r2
(%)KF(µg/g)(L/µg)n
n r2
(%)
As(V) 516 0.0066 92.5 64.9 0.281 99.7As(III) 1040 0.0019 98.2 13.1 0.585 99.3
0.25–0.50 mm removed As from As(III) solution more thanfrom As(V) solution, while with the grain size fraction of<0.04 mm slightly less than from As(V) solution.
It was also found that Mn was released into solution duringadsorption reaction, the concentration of which in the As(III)batch was greater than that in the As(V) batch with regard toeach grain size fraction investigated (see Fig. S2 in support-ing information). The higher Mn concentration possibly meansmore Mn compounds had been reduced in the As(III) batch,in comparison with the As(V) batch. It has been known thatMn compounds oxidize As(III) readily [28,29]. The processesof As(III) removal by the siderite were not fully understood,but generally believed that the higher efficiency of As removalfrom As(III) solution possibly arose from the additional ad-sorption sites on the fresh surface from which Mn compoundswere introduced into the solution. From the results of the ki-netic study (Fig. 1), it is deduced that the enhanced adsorptionmay have taken effect when the adsorption proceeded for 2.5 h,after which the amount of As removed from As(III) solutionexceeded that from As(V) solution.
3.4. Column studies
Column experiments were conducted to evaluate the feasi-bility of the siderite as filter filling in removing As from con-taminated water. Total As in the SO-C1 effluent was below1.0 µg/L after a throughput of 26,000 pore volumes (PV: thevolume of water required to replace water in a certain volumeof saturated porous media; it is about 30 mL in the studied col-umn) of As solution containing 250 µg/L As(III) and 250 µg/LAs(V) (Fig. 3A). With a column packed with 0.25–0.50 mmsiderite in the lower half and 0.25–0.50 mm hematite in the up-per half, effluent As was close to 1.00 µg/L after a throughputof 7200 PV of the same As solution (data not shown). It is be-lieved that adsorption sites were still available for As retentionin the filter, and more As water was able to be remediated beforethe European drinking water standard (10 µg/L As) was ex-ceeded. A mass balance calculation indicates that total As loadin the filter was about 2000 µg/g before 1 µg/L of As break-through was observed, which was higher than the maximumAs(III) adsorption of 1040 µg/g calculated from the Langmuirisotherm (Table 2). Bang et al. also found that the As load onthe TiO2 in the filter was higher than that predicted by the batchadsorption isotherm [30]. They suggested that it should haveresulted from the long contact time of As with the adsorbentin the filter. In our study, the higher As load of the filter likelyattributed to the freshly formed Fe hydroxides in the columnduring its operation. A detailed explanation is provided later.The natural siderite as a filling material had a higher adsorp-tion capacity, in comparison with other natural geomaterials(Table 3).
In order to examine the effect of multicomponents on col-umn performance, SO-C2 was used to treat As-spiked tap wa-ter. Breakthrough curve of the filter is shown in Fig. 3B. Incomparison with artificial As solution, removal efficiency fromAs-spiked tap water was relatively lower, showing that the pres-ence of competing anions (i.e., NO−, SO2−, and HCO−) in
3 4 3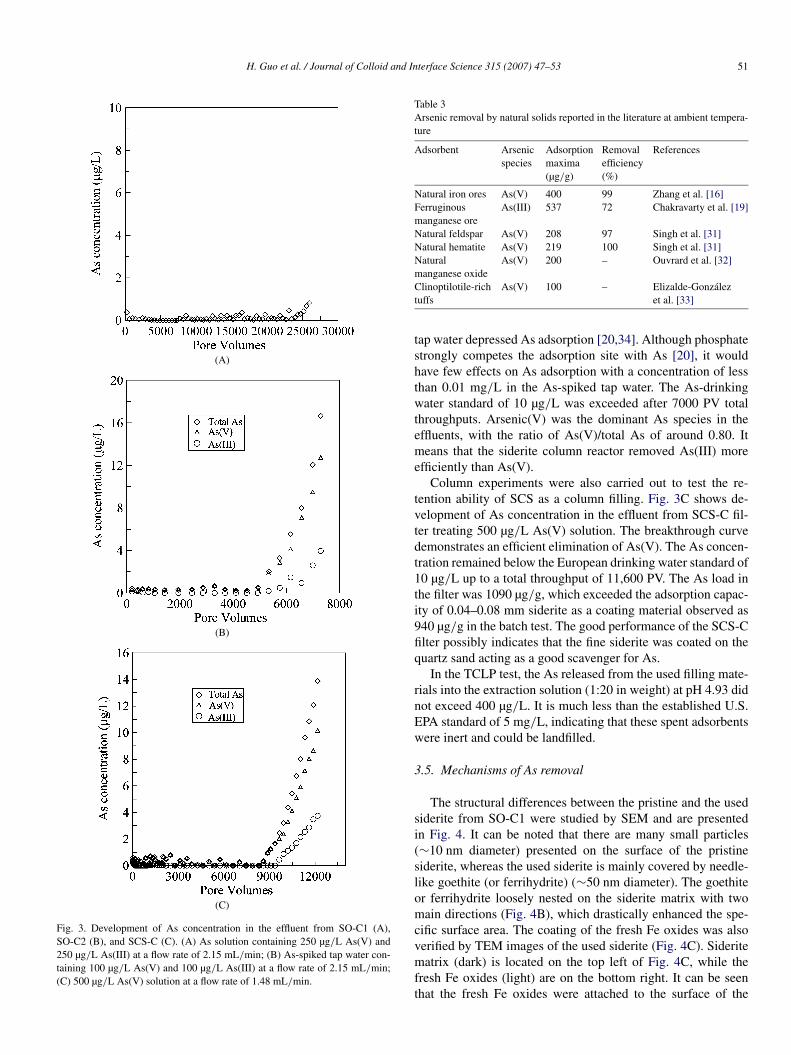
H. Guo et al. / Journal of Colloid and Interface Science 315 (2007) 47–53 51
(A)
(B)
(C)
Fig. 3. Development of As concentration in the effluent from SO-C1 (A),SO-C2 (B), and SCS-C (C). (A) As solution containing 250 µg/L As(V) and250 µg/L As(III) at a flow rate of 2.15 mL/min; (B) As-spiked tap water con-taining 100 µg/L As(V) and 100 µg/L As(III) at a flow rate of 2.15 mL/min;(C) 500 µg/L As(V) solution at a flow rate of 1.48 mL/min.
Table 3Arsenic removal by natural solids reported in the literature at ambient tempera-ture
Adsorbent Arsenicspecies
Adsorptionmaxima(µg/g)
Removalefficiency(%)
References
Natural iron ores As(V) 400 99 Zhang et al. [16]Ferruginousmanganese ore
As(III) 537 72 Chakravarty et al. [19]
Natural feldspar As(V) 208 97 Singh et al. [31]Natural hematite As(V) 219 100 Singh et al. [31]Naturalmanganese oxide
As(V) 200 – Ouvrard et al. [32]
Clinoptilotile-richtuffs
As(V) 100 – Elizalde-Gonzálezet al. [33]
tap water depressed As adsorption [20,34]. Although phosphatestrongly competes the adsorption site with As [20], it wouldhave few effects on As adsorption with a concentration of lessthan 0.01 mg/L in the As-spiked tap water. The As-drinkingwater standard of 10 µg/L was exceeded after 7000 PV totalthroughputs. Arsenic(V) was the dominant As species in theeffluents, with the ratio of As(V)/total As of around 0.80. Itmeans that the siderite column reactor removed As(III) moreefficiently than As(V).
Column experiments were also carried out to test the re-tention ability of SCS as a column filling. Fig. 3C shows de-velopment of As concentration in the effluent from SCS-C fil-ter treating 500 µg/L As(V) solution. The breakthrough curvedemonstrates an efficient elimination of As(V). The As concen-tration remained below the European drinking water standard of10 µg/L up to a total throughput of 11,600 PV. The As load inthe filter was 1090 µg/g, which exceeded the adsorption capac-ity of 0.04–0.08 mm siderite as a coating material observed as940 µg/g in the batch test. The good performance of the SCS-Cfilter possibly indicates that the fine siderite was coated on thequartz sand acting as a good scavenger for As.
In the TCLP test, the As released from the used filling mate-rials into the extraction solution (1:20 in weight) at pH 4.93 didnot exceed 400 µg/L. It is much less than the established U.S.EPA standard of 5 mg/L, indicating that these spent adsorbentswere inert and could be landfilled.
3.5. Mechanisms of As removal
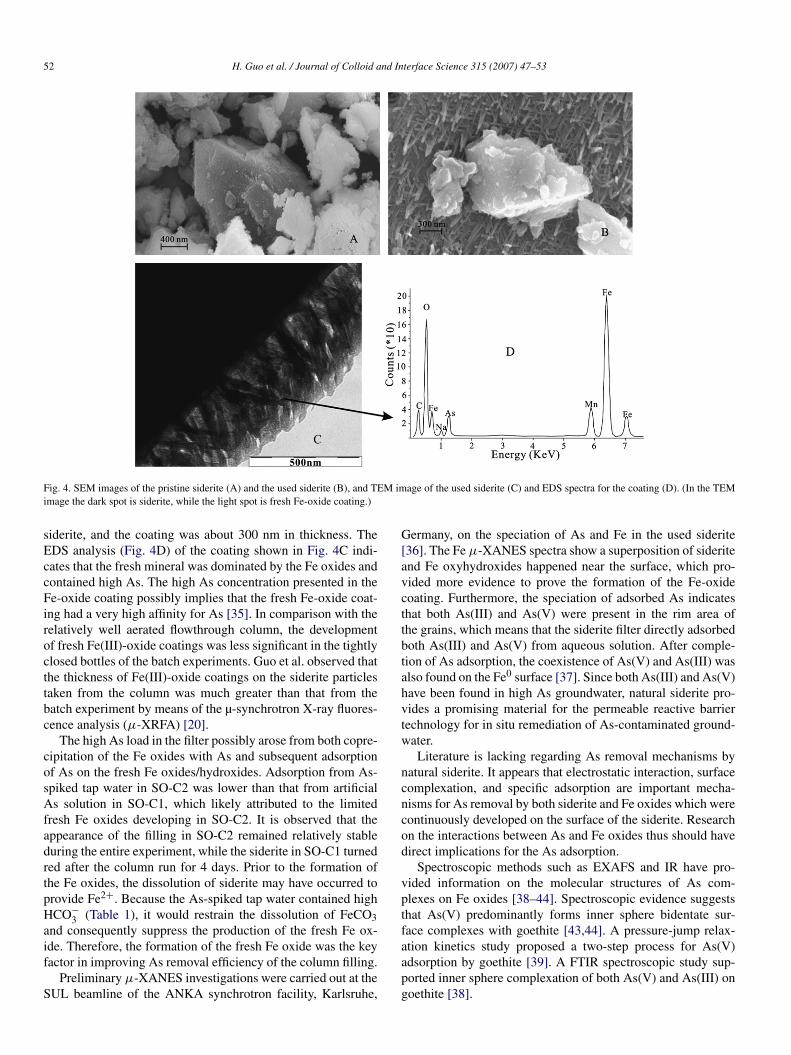
The structural differences between the pristine and the usedsiderite from SO-C1 were studied by SEM and are presentedin Fig. 4. It can be noted that there are many small particles(∼10 nm diameter) presented on the surface of the pristinesiderite, whereas the used siderite is mainly covered by needle-like goethite (or ferrihydrite) (∼50 nm diameter). The goethiteor ferrihydrite loosely nested on the siderite matrix with twomain directions (Fig. 4B), which drastically enhanced the spe-cific surface area. The coating of the fresh Fe oxides was alsoverified by TEM images of the used siderite (Fig. 4C). Sideritematrix (dark) is located on the top left of Fig. 4C, while thefresh Fe oxides (light) are on the bottom right. It can be seenthat the fresh Fe oxides were attached to the surface of the
52 H. Guo et al. / Journal of Colloid and Interface Science 315 (2007) 47–53
Fig. 4. SEM images of the pristine siderite (A) and the used siderite (B), and TEM image of the used siderite (C) and EDS spectra for the coating (D). (In the TEMimage the dark spot is siderite, while the light spot is fresh Fe-oxide coating.)
siderite, and the coating was about 300 nm in thickness. TheEDS analysis (Fig. 4D) of the coating shown in Fig. 4C indi-cates that the fresh mineral was dominated by the Fe oxides andcontained high As. The high As concentration presented in theFe-oxide coating possibly implies that the fresh Fe-oxide coat-ing had a very high affinity for As [35]. In comparison with therelatively well aerated flowthrough column, the developmentof fresh Fe(III)-oxide coatings was less significant in the tightlyclosed bottles of the batch experiments. Guo et al. observed thatthe thickness of Fe(III)-oxide coatings on the siderite particlestaken from the column was much greater than that from thebatch experiment by means of the µ-synchrotron X-ray fluores-cence analysis (μ-XRFA) [20].
The high As load in the filter possibly arose from both copre-cipitation of the Fe oxides with As and subsequent adsorptionof As on the fresh Fe oxides/hydroxides. Adsorption from As-spiked tap water in SO-C2 was lower than that from artificialAs solution in SO-C1, which likely attributed to the limitedfresh Fe oxides developing in SO-C2. It is observed that theappearance of the filling in SO-C2 remained relatively stableduring the entire experiment, while the siderite in SO-C1 turnedred after the column run for 4 days. Prior to the formation ofthe Fe oxides, the dissolution of siderite may have occurred toprovide Fe2+. Because the As-spiked tap water contained highHCO−
3 (Table 1), it would restrain the dissolution of FeCO3and consequently suppress the production of the fresh Fe ox-ide. Therefore, the formation of the fresh Fe oxide was the keyfactor in improving As removal efficiency of the column filling.
Preliminary μ-XANES investigations were carried out at theSUL beamline of the ANKA synchrotron facility, Karlsruhe,
Germany, on the speciation of As and Fe in the used siderite[36]. The Fe μ-XANES spectra show a superposition of sideriteand Fe oxyhydroxides happened near the surface, which pro-vided more evidence to prove the formation of the Fe-oxidecoating. Furthermore, the speciation of adsorbed As indicatesthat both As(III) and As(V) were present in the rim area ofthe grains, which means that the siderite filter directly adsorbedboth As(III) and As(V) from aqueous solution. After comple-tion of As adsorption, the coexistence of As(V) and As(III) wasalso found on the Fe0 surface [37]. Since both As(III) and As(V)have been found in high As groundwater, natural siderite pro-vides a promising material for the permeable reactive barriertechnology for in situ remediation of As-contaminated ground-water.
Literature is lacking regarding As removal mechanisms bynatural siderite. It appears that electrostatic interaction, surfacecomplexation, and specific adsorption are important mecha-nisms for As removal by both siderite and Fe oxides which werecontinuously developed on the surface of the siderite. Researchon the interactions between As and Fe oxides thus should havedirect implications for the As adsorption.
Spectroscopic methods such as EXAFS and IR have pro-vided information on the molecular structures of As com-plexes on Fe oxides [38–44]. Spectroscopic evidence suggeststhat As(V) predominantly forms inner sphere bidentate sur-face complexes with goethite [43,44]. A pressure-jump relax-ation kinetics study proposed a two-step process for As(V)adsorption by goethite [39]. A FTIR spectroscopic study sup-ported inner sphere complexation of both As(V) and As(III) ongoethite [38].

H. Guo et al. / Journal of Colloid and Interface Science 315 (2007) 47–53 53
4. Conclusions
This study confirms that the natural siderite is effective inremoving both As(V) and As(III) from solution. Arsenic ad-sorption increased exponentially with contact time in the batchtest, and generally reached pseudoequilibrium at a contact timeof 3 days. Arsenic removal from As(III) solution was muchhigher than from As(V) solution. Since As-enriched ground-water is generally dominated by As(III) [2,7,45,46], the sideritecan be utilized to efficiently remove As from groundwater with-out preoxidation of As(III) to As(V). Column studies show thatboth the siderite filter and the sand-siderite filter had a high Asretention capacity, with As loads of 2000 and 1090 µg/g, re-spectively. The high efficiency for As removal was attributed tothe adsorption of As on the pristine siderite and afterward onthe fresh Fe-oxide coatings.
The availability of the natural siderite at a low cost makes it apromising material for in situ remediation of As-contaminatedgroundwater resources. The positive effects of residence timemay be advantageous to potential permeable reactive barriers(PRB) composed of the natural siderite to remediate As in thefield. More studies are needed along this path to optimize theremediation efforts. Factors influencing the removal efficiencyshould be among the areas of future research. These factorsinclude temperature, dissolved organic constituents, redox con-dition, and microorganisms in groundwater.
Acknowledgments
H.M.G. is grateful to the Alexander von Humboldt Founda-tion, Germany, for providing a Research Fellowship to carry outthis research. Funding for this research has also been providedby the Natural Science Foundation of China (No. 40572145).The authors express their appreciation for analyses support ofour colleagues M. Fotouhi (TEM), V. Zibat (SEM), C. Moess-ner (HR-ICP-MS), T. Neumann (FI-AAS), and G. Preuss (GF-AAS).
Supporting information
Supporting information data for this article may be found inthe online version at DOI: 10.1016/j.jcis.2007.06.035.
References
[1] P.L. Smedley, D.G. Kinniburgh, Appl. Geochem. 17 (2002) 517.[2] H.M. Guo, Y.X. Wang, G.M. Shpeizer, S. Yan, J. Environ. Sci. Health
A 38 (2003) 2565.[3] National Research Council, Arsenic in Drinking Water: 2001 Update, Na-
tional Academy Press, Washington, DC, 2001.[4] J.O. Nriagu, in: W.T. Frankenberger (Ed.), Environmental Chemistry of
Arsenic, Dekker, New York, 2002, p. 1.[5] E.O. Kartinen, J.M. Martin, Desalination 103 (1995) 79.[6] M. Bissen, F.H. Frimmel, Acta Hydrochim. Hydrobiol. 31 (2003) 97.
[7] F. Wagner, Z. Berner, D. Stüben, in: J. Bundschuh, P. Bhattacharya, D.Chandrasekharam (Eds.), Natural Arsenic in Groundwater: Occurrences,Remediation and Management, Taylor & Francis, Balkema, 2005, p. 3.
[8] J.F. Ferguson, J. Gavis, Water Res. 6 (1972) 1259.[9] N.E. Korte, Q. Fernando, Crit. Rev. Environ. Control 21 (1991) 1.
[10] J.A. Wilkie, J.G. Hering, Colloids Surf. A 107 (1996) 97.[11] J.S. Ahn, C.M. Chon, H.S. Moon, K.W. Kim, Water Res. 37 (2003) 2478.[12] C.M. Su, R.W. Puls, Environ. Sci. Technol. 37 (2003) 2582.[13] Y.H. Kim, C.M. Kin, I.H. Choi, S. Rengaraj, J.H. Yi, Environ. Sci. Tech-
nol. 38 (2004) 924.[14] L.C. Roberts, S.J. Hug, T. Ruettimann, M.M. Billah, A.W. Khan, M.T.
Rahman, Environ. Sci. Technol. 38 (2004) 307.[15] M.P. Elizalde-González, J. Mattusch, W.D. Einicke, R. Wennrich, Chem.
Eng. J. 81 (2001) 187.[16] W. Zhang, P. Singh, E. Paling, S. Delides, Miner. Eng. 17 (2004) 517.[17] A.C.Q. Ladeira, V.S.T. Ciminelli, Water Res. 38 (2004) 2087.[18] H. Genç-Fuhrman, J.C. Tjell, D. McConchie, O. Schuiling, J. Colloid In-
terface Sci. 264 (2003) 327.[19] S. Chakravarty, V. Dureja, G. Bhattacharyya, S. Maity, S. Bhattacharjee,
Water Res. 36 (2002) 625.[20] H.M. Guo, D. Stüben, Z. Berner, Appl. Geochem. 22 (2007) 1039.[21] H.M. Guo, D. Stüben, Z. Berner, Sci. Total Environ. 377 (2007) 142.[22] T.R. Rüde, H. Puchelt, Fresenius J. Anal. Chem. 350 (1994) 44.[23] US EPA, Toxicity Characteristics Leaching Procedure, US Environmental
Protection Agency, Fed. Reg. 1999, p. 11798.[24] H. Freundlich, Z. Phys. Chem. 57 (1906) 385.[25] I. Langmuir, J. Am. Chem. Soc. 40 (1918) 1361.[26] S. Paikaray, S. Banerjee, S. Mukherji, Curr. Sci. India 88 (2005) 1580.[27] C. Namasivayam, K. Ranganathan, Ind. Eng. Chem. Res. 34 (1995) 869.[28] D.W. Oscarson, P.M. Huang, W.K. Liaw, U.T. Hammer, Soil Sci. Soc. Am.
J. 46 (1983) 644.[29] B. Daus, J. Mattusch, A. Paschke, R. Wennrich, H. Weiss, Talanta 51
(2000) 1087.[30] S. Bang, M. Patel, L. Lippincott, X. Meng, Chemosphere 60 (2005) 389.[31] D.B. Singh, G. Prasad, D.C. Rupainwar, Colloids Surf. 111 (1996) 49.[32] S. Ouvrard, M.O. Simonnot, M. Sardin, Water Sci. Technol. 1 (2001) 167.[33] M.P. Elizalde-González, J. Mattusch, R. Wennrich, P. Morgenstern, Mi-
croporous Mesoporous Mater. 46 (2001) 277.[34] H. Genç-Fuhrman, H. Bregnhøj, D. McConchie, Water Res. 39 (2005)
2944.[35] J.A. Munoz, A. Gonzalo, M. Valiente, Environ. Sci. Technol. 36 (2002)
3405.[36] J. Göttlicher, R. Steininger, J. Majzlan, U. Kramar, H.M. Guo, N. Zöger,
Annual Report, ANKA Angstroemquelle Karlsruhe, Germany, 2006,p. 172.
[37] C.M. Su, R.W. Puls, Environ. Sci. Technol. 35 (2001) 1487.[38] X. Sun, H.E. Doner, Soil Sci. 161 (1996) 865.[39] P.R. Grossl, M. Eick, D.L. Sparks, S. Goldberg, C.C. Ainsworth, Environ.
Sci. Technol. 31 (1997) 321.[40] X. Sun, H.E. Doner, Soil Sci. 163 (1998) 278.[41] S.C.B. Myneni, S.J. Traina, G.A. Waychunas, T.J. Logan, Geochim. Cos-
mochim. Acta 62 (1998) 3285.[42] M.L. Farquhar, J.M. Charnock, F.R. Livens, D.J. Vaughan, Environ. Sci.
Technol. 36 (2002) 1757.[43] B.A. Manning, M.L. Hunt, C. Amrhein, J.R. Yarmoff, Environ. Sci. Tech-
nol. 36 (2002) 5455.[44] G. Ona-Nguema, G. Morin, F. Juillot, G. Calas, J. Brown, Environ. Sci.
Technol. 39 (2005) 9147.[45] P. Huntsman-Mapila, T. Mapila, M. Letshwenyo, P. Wolski, C. Hemond,
Appl. Geochem. 21 (2006) 1376.[46] M. Berg, C. Stengel, P.T.K. Trang, P.H. Viet, M.L. Sampson, M. Leng, S.
Samreth, D. Fredericks, Sci. Total Environ. 372 (2007) 413.
Related Documents











