ABSTRACT Gamma Hydroxybutyrate (GHB): Mechanisms of Central Nervous System Toxicity Eric E. Lyng Mentor: Teodoro G. Bottiglieri, Ph.D. Gamma Hydroxybutyrate (GHB) is an endogenous metabolite of gamma- aminobutyric acid (GABA) and a putative neurotransmitter found in mammalian brain. The illicit use of GHB is a growing health care concern in the U.S. Low doses have euphoric and stimulatory effects while high doses act as a CNS depressant and can cause respiratory failure. In addition to fatalities and drug facilitated rape, in 2004 over 7000 GHB overdoses were reported in the U.S. Gamma Butyrolactone (GBL) and 1,4- Butanediol (1,4-BD), precursors of GHB, can cause similar effects after being converted to GHB in the body. While GHB is a Schedule I compound, recently it was given a Schedule III classification as a drug for treatment of cataplexy associated with narcolepsy. Individuals affected by the inherited disorder succinic semialdehyde dehydrogenase (SSADH) deficiency have significant elevation of GABA and GHB in body tissues, and a range of neurological complications. Currently there is no treatment option for a GHB overdose situation or for SSADH patients. GHB has been shown to inhibit striatal dopamine release leading to sedation and loss of locomotor activity in rodents. However, GHB’s mechanism of action is poorly

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ABSTRACT
Gamma Hydroxybutyrate (GHB): Mechanisms of Central Nervous System Toxicity
Eric E. Lyng
Mentor: Teodoro G. Bottiglieri, Ph.D.
Gamma Hydroxybutyrate (GHB) is an endogenous metabolite of gamma-
aminobutyric acid (GABA) and a putative neurotransmitter found in mammalian brain.
The illicit use of GHB is a growing health care concern in the U.S. Low doses have
euphoric and stimulatory effects while high doses act as a CNS depressant and can cause
respiratory failure. In addition to fatalities and drug facilitated rape, in 2004 over 7000
GHB overdoses were reported in the U.S. Gamma Butyrolactone (GBL) and 1,4-
Butanediol (1,4-BD), precursors of GHB, can cause similar effects after being converted
to GHB in the body. While GHB is a Schedule I compound, recently it was given a
Schedule III classification as a drug for treatment of cataplexy associated with
narcolepsy. Individuals affected by the inherited disorder succinic semialdehyde
dehydrogenase (SSADH) deficiency have significant elevation of GABA and GHB in
body tissues, and a range of neurological complications. Currently there is no treatment
option for a GHB overdose situation or for SSADH patients.
GHB has been shown to inhibit striatal dopamine release leading to sedation and
loss of locomotor activity in rodents. However, GHB’s mechanism of action is poorly

understood. This dissertation investigated acute and chronic effects of GBL, a precursor
to GHB, on locomotor function and monoamine neurotransmitter metabolism in mice.
Dose response studies were performed to characterize the effects of GBL. Compounds
aimed at increasing central dopaminergic activity or antagonism of GABAergic activity
were tested for their ability to antagonize the locomotor effects induced by GBL. In total,
eight different compounds were studied of which pergolide and nomifensine were
successful at antagonizing the loss of locomotor activity when administered either prior
to or after GBL. Chronic administration of GBL over the course of 14 days was
evaluated in mice using a rotarod system. These studies did not reveal any long term
detrimental effect of GBL on locomotor function.
This dissertation is the first report showing the ability of pergolide and
nomifensine to antagonize the loss of locomotor activity induced by GBL. These studies
provide insight into treatment options for GHB toxicity or overdose as well as for patients
with SSADH deficiency.


Copyright © 2006 by Eric E. Lyng
All rights reserved

TABLE OF CONTENTS
LIST OF FIGURES viii LIST OF TABLES x
LIST OF ABBREVIATIONS xii
ACKNOWLEDGMENTS xiv DEDICATION xv CHAPTER
1. Introduction 1
γ-Hydroxybutyric Acid 1
Metabolism of GHB 2
Localization in Tissues 7
GBL & 1,4-BD as Precursors to GHB 8
Medical Uses of GHB 9
GHB Toxicity and Drug Abuse 12
SSADH Deficiency: A Disorder of GABA and 16 GHB Metabolism
Neurotransmitter Function of GHB 20
GHB as a GABAA/B Agonist 22
GHB Receptors 24
GHB’s Role in Regulation of Dopaminergic Function 27
Aim of Dissertation 33
iii

2. Materials and Methods 34
Chemicals and Solvents 34
Chemicals 34
Solvents 34
Animals 35
Mice 35
Drug Injections 35
HPLC Methods 35
Monoamine Analysis 35
Brain Tissue Preparation 36
Behavioral Methods 39
TruScan® 39
Rotarod 41
Data and Statistics 42
3. Acute Effects of GBL on Locomotor Activity in Mice 44
Introduction 44
Materials & Methods 45
Results 46
Acute Study 1- 120 Minutes Post GBL 46
Summary of Behavioral Changes 51
Acute Study 2 – 30 Minutes Post GBL 53
Summary of Behavioral Changes 56
Discussion 56
iv

4. Acute Effects of GBL on Brain Neurotransmitter 64 Metabolism in Mice
Introduction 64
Materials & Methods 66
Results 66
Acute Study 1 - 120 minutes post GBL 66
Summary of Monoamine Metabolite Changes 68
Acute Study 2 - 30 minutes post GBL 68
Summary of Monoamine Metabolite Changes 70
Discussion 70
5. Pre-Treatment Effect of GABAA and GHB Receptor 76
Antagonists and Compounds Affecting Dopamine Metabolism on GBL Induced Loss of Locomotor Activity in Mice
Introduction 76
Pergolide 77
NCS-382 78
Pargyline 78
Nomifensine 79
Tolcapone 80
Apomorphine 80
Taurine 81
Bicuculline 82
Materials & Methods 82
Behavioral Results 83
v

Pergolide Treatment 83
NCS-382 Treatment 85
Pargyline Treatment 87
Nomifensine Treatment 89
Tolcapone Treatment 92
Apomorphine Treatment 92
Taurine Treatment 95
Bicuculline Treatment 95
Summary of Behavioral Changes 95
Monoamine Metabolites Results 98
Pergolide Treatment 98
Nomifensine Treatment 100
Summary of Monoamine Metabolite Changes 102
Discussion 102
6. Post-Treatment Effect of Dopamine Agonists on GBL Induced 114 Loss of Locomotor Activity in Mice
Introduction 114
Materials & Methods 115
Behavioral Results 116
GBL Treatment 116
Pergolide Treatment 118
Nomifensine Treatment 121
Summary of Behavioral Changes 124
vi

Monoamine Metabolite Results 124
GBL Treatment 124
Pergolide Treatment 126
Nomifensine Treatment 126
Summary of Monoamine Metabolite Changes 129
Discussion 131
7. Chronic Effect of GBL on Locomotor Activity 138
and Brain Monoamine Neurotransmitter Metabolism in Mice Introduction 138
Materials & Methods 141
Behavioral Results 143
Monoamine Metabolite Results 149
Discussion 149
8. General Conclusions 156
WORKS CITED 163
vii

LIST OF FIGURES
Figure Page
1. Structures of GABA and GHB 3 2. Metabolic pathway for GHB 4
3. Metabolism of γ-hydroxybutyric acid, 1,4-butanediol, 9
and γ-butyrolactone
4. Glutamate/GABA/Glutamine shuttle 19 5. HPLC chromatogram of monoamine metabolite standards 37 6. HPLC chromatogram of PCA extract from deproteinized 38
mouse half brain 7. Picture of the TruScan® system 40 8. The effect of GBL on movement episodes in mice over 130 minutes 47 9. The effect of GBL on movement time in mice over 130 minutes 49 10. The effect of GBL on distance moved in mice over 130 minutes 50 11. The effect of GBL on vertical plane entries in mice over 130 minutes 52 12. The effect of GBL on locomotor activity in mice over 40 minutes 54 13. Track plots of open field behavior in the TruScan® cage 55 14. The effects of GBL and pergolide on locomotor activity in mice 84
over 40 minutes 15. The effects of GBL and NCS-382 on locomotor activity in mice 86
over 40 minutes 16. The effects of GBL and pargyline on locomotor activity in mice 88
over 40 minutes 17. The effects of GBL and nomifensine on locomotor activity in mice 90 over 40 minutes
viii

18. The effects of GBL and tolcapone on locomotor activity in mice 93 over 40 minutes 19. The effects of GBL and apomorphine on locomotor activity in mice 94 over 40 minutes 20. The effects of GBL and taurine on locomotor activity in mice 96 over 40 minutes 21. The effects of GBL and bicuculline on locomotor activity in mice 97 over 40 minutes 22. The effect of GBL on locomotor activity in mice over 60 minutes 117 23. The effects of GBL and pergolide on locomotor activity in mice 119
over 60 minutes 24. The effects of GBL and nomifensine on locomotor activity in mice 122
over 60 minutes 25. The effects of chronic GHB administration on rotarod performance 144
in mice 26. Day one post injection responses to chronic GBL administration 145 27. Day seven post injection responses to chronic GBL administration 146 28. Day 14 post injection responses to chronic GBL administration 147
ix

LIST OF TABLES
Tables Page 1. Common street names of GHB 13 2. Dose response of GHB in both humans and rodents 14 3. The effect of GBL on dopamine metabolites and turnover in mice 67 over 130 minutes 4. The effect of GBL on serotonin metabolites and turnover in mice 67 over 130 minutes 5. The effect of GBL on dopamine metabolites and turnover in mice 69 over 40 minutes 6. The effect of GBL on serotonin metabolites and turnover in mice 69 over 40 minutes 7. Classification of compounds tested for GBL antagonism 77
8. The effects of GBL and pergolide on dopamine metabolites 99
and turnover in mice over 40 minutes 9. The effects of GBL and pergolide on serotonin metabolites 99
and turnover in mice over 40 minutes
10. The effects of GBL and nomifensine on dopamine metabolites 101 and turnover in mice over 40 minutes
11. The effects of GBL and nomifensine on serotonin metabolites 101
and turnover in mice over 40 minutes 12. Compounds tested for antagonism of GBL 103 13. The effect of GBL on dopamine metabolites and turnover in mice 125
over 60 minutes
14. The effect of GBL on serotonin metabolites and turnover in mice 125 over 60 minutes
x

15. The effects of GBL and pergolide on dopamine metabolites and 127 turnover in mice over 60 minutes
16. The effects of GBL and pergolide on serotonin metabolites and 127
turnover in mice over 60 minutes 17. The effects of GBL and nomifensine on dopamine metabolites and 128
turnover in mice over 60 minutes 18. The effects of GBL and nomifensine on serotonin metabolites and 130
turnover in mice over 60 minutes 19. The effects of chronic GBL administration on dopamine metabolites 150
and turnover in mice 20. The effects of chronic GBL administration on serotonin metabolites 150
and turnover in mice
xi

LIST OF ABBREVIATIONS
1,4-BD 1,4-Butanediol
3-OMD 3-O-Methyldopa
3-MT 3-Methoxytyramine
5-HIAA 5-Hydroxyindole-3-Acetic Acid
5-HT 5-Hydroxytryptamine Creatine Sulfate Complex
5-HTP 5-Hydroxy-L-Tryptophan
AADC Aromatic Amino Acid Decarboxylase
CNS Central Nervous System
COMT Catechol-O-Methyl Transferase
CSF Cerebral Spinal Fluid
DA Dopamine
DOPAC 3,4-Dihydroxyphenylacetic Acid
ED Extra Dimensional
EDTA Ethylenediaminetetraacetic acid
ER Emergency Room
GABA Gamma Aminobutyric Acid
GABA-T GABA Transaminase
GAD Glutamate Decarboxylase
GBL Gamma Butyrolactone
xii

GC/MS Gas Chromatography/Mass Spectrometry
GHB Gamma Hydroxybutyric Acid
GHB-DH GHB Dehydrogenase
HPLC High Performance Liquid Chromatography
HVA 4-Hydroxy-3-Methoxyphenylacetic
L-DOPA 3,4-Dihydroxy-L-Phenylalanine
MAO Monoamine Oxidase
NE Norepinephrine
NCS-382 6,7,8,9-tetrahydro-5-[H]benzocyclo- hepten-5-ol-4-ylideneacetic acid
NSD 1015 3-Hydroxybencilhydracine
OSS Sodium Octyl Sulfate
PCA Perchloric Acid
PD Parkinson’s Disease
SSA Succinic Semialdehyde
SSADH Succinic Semialdehyde Dehydrogense
SSR Succinic Semialdehyde Reductase
T-HCA trans-γ-hydrocrotonic acid TAU Taurine
TH Tyrosine Hydroxylase
VPE Vertical Plane Entries
WCST Wisconsin Card Sorting Task
xiii

ACKNOWLEDGMENTS
I would like to thank Dr. Teodoro Bottiglieri, Dr. Erland Arning, Matthew
Williams, and other members of the Baylor Institute of Metabolic Disease. I would also
like to thank Dr. Robert Kane and the Institute of Biomedical Studies for their support.
Finally, a thank you to Dr. Christopher Kearney, Dr. James Marcum, Dr. James
Matthews, and Dr. Jim Patton for serving on my dissertation committee.
xiv

To Laura, for all of your love, patience, and support.
I could not have done this without you.
xv

CHAPTER ONE
Introduction
γ-Hydroxybutyric Acid
γ-Hydroxybutyric acid (GHB) is a metabolite of the inhibitory neurotransmitter γ-
Aminobutyric Acid (GABA). The uses of GHB have included anesthesia, ethanol
withdrawal treatment, bodybuilding supplement, drug of abuse, drug facilitated rape, and
treatment for cataplexy associated with narcolepsy. GHB is an illegal substance in the
United States, being banned by the Food and Drug Administration (FDA) in 1990 (Okun
and others 2000) after several emergency room cases of respiratory depression, seizures,
and comas (Anonymous 2006). In March 2000, GHB was designated a Schedule I drug
under the Federal Controlled Substances Act of 1970 (Lloyd 2002). This means that
GHB has a high abuse potential, is not approved for medical use, and does not meet the
safety standards required for use in medical practice. On July 17, 2002, the FDA
approved Xyrem (Orphan Medical, Minnetonka, MN) (Anonymous 2002a), a drug with
an active ingredient of sodium oxybate or GHB, as a Schedule III Controlled Substance
to treat cataplexy attacks in patients with narcolepsy. Cataplexy is a condition
characterized by weak or paralyzed muscles. A Schedule III Controlled Substance has a
lower potential for abuse than Schedule I and II substances and is currently accepted for
medical treatment in the United States. Despite a Schedule III designation, there is
potential for a low to moderate physical dependence associated with Xyrem. Illicit use of
Xyrem is subject to Schedule I penalties. (Lloyd 2002).
1

2
Although GHB is no longer legally available to the general public, attempts to
obtain it from overseas as well as internet websites have been reported. Also available
illegally are the two precursors to GHB, γ-butyrolactone (GBL), and 1,4-butanediol (1,4-
BD). The abuse of GHB and its analogues is of growing concern in the United States.
According to the Drug Abuse Warning Network (DAWN), funded by the Substance
Abuse and Mental Health Services Administration (SAMHSA), hospital emergency room
visits involving GHB increased from 55 in 1994 to 4,969 in 2000 before declining to
3,340 in 2001. Among emergency department cases involving club drugs, GHB has been
cited more often than ecstasy each year since 2000 (Leinwand 2001). Understanding the
biochemical, physiological, and behavioral effects of GHB, both acute and chronic,
warrants attention and further investigation.
Metabolism of GHB GHB is a direct metabolite of GABA that differs only by the side chain on the γ
carbon (Roth and Giarman 1970; Wong and others 2003; de Fiebre and others 2004;
Waszkielewicz and Bojarski 2004). The structures of both GABA and GHB are shown
in Figure 1. GHB is an endogenous, short chain fatty acid found primarily in the
mammalian brain (Doherty and others 1978; Maitre 1997). One of the first experiments
concerning GHB synthesis involved injecting labeled [13C or 3H]-GABA into the lateral
ventricles of awake rats (Gold and Roth 1977). A maximum [3H]-GHB concentration in
brain tissue was observed within 20 minutes. In the brain, glutamate is the precursor to
GABA. Santaniello showed that radiolabeled glutamate lead to the formation of
radiolabeled GHB (Santaniello 1978), which verified that neuronally derived GABA

3
OHH2N
O
OHHO
OGABA GHB
Figure 1 Structures of GABA and GHB (Waszkielewicz and Bojarski 2004)
gave rise to GHB. GABA has two routes of metabolism (Figure 2): oxidation or
reduction (Matsuda and Hoshino 1977). It is not definitively known where synthesis of
GHB occurs, but previous work has indicated the mitochondria due to the localization of
GABA Transaminase (GABA-T) (Schousboe and others 1980). In the mitochondria,
GABA is converted to succinic semialdehyde (SSA) by GABA-T (Gold and Roth 1977).
SSA then undergoes oxidation via succinic semialdehyde dehydrogenase (SSADH)
resulting in succinate, which enters the Krebs cycle. Alternatively, in the cytosol, SSA is
reduced by succinic semialdehyde reductase (SSR), to GHB. Only 1-2% of GABA
metabolism occurs outside of the mitochondria resulting in GHB (Gold and Roth 1977).
Other studies report that GABA metabolism occurring in the reductive pathway that
yields GHB utilizes approximately 0.05% of GABA in vitro (Rumigny and others 1981)
and 0.16% of GABA in vivo (Gold and Roth 1977). Once GHB has been formed in the
cytosol, it is metabolized by GHB Dehydrogenase (GHB-DH) resulting in the formation
of SSA (Maitre 1997). SSA is further metabolized to succinate, which enters the Krebs
cycle or is converted back into GABA (Maitre 1997). Studies of SSADH deficient mice
have shown that accumulation of both GHB and GABA occurs in various tissues,
indicating conversion between the two compounds (Hogeman and others 2001).
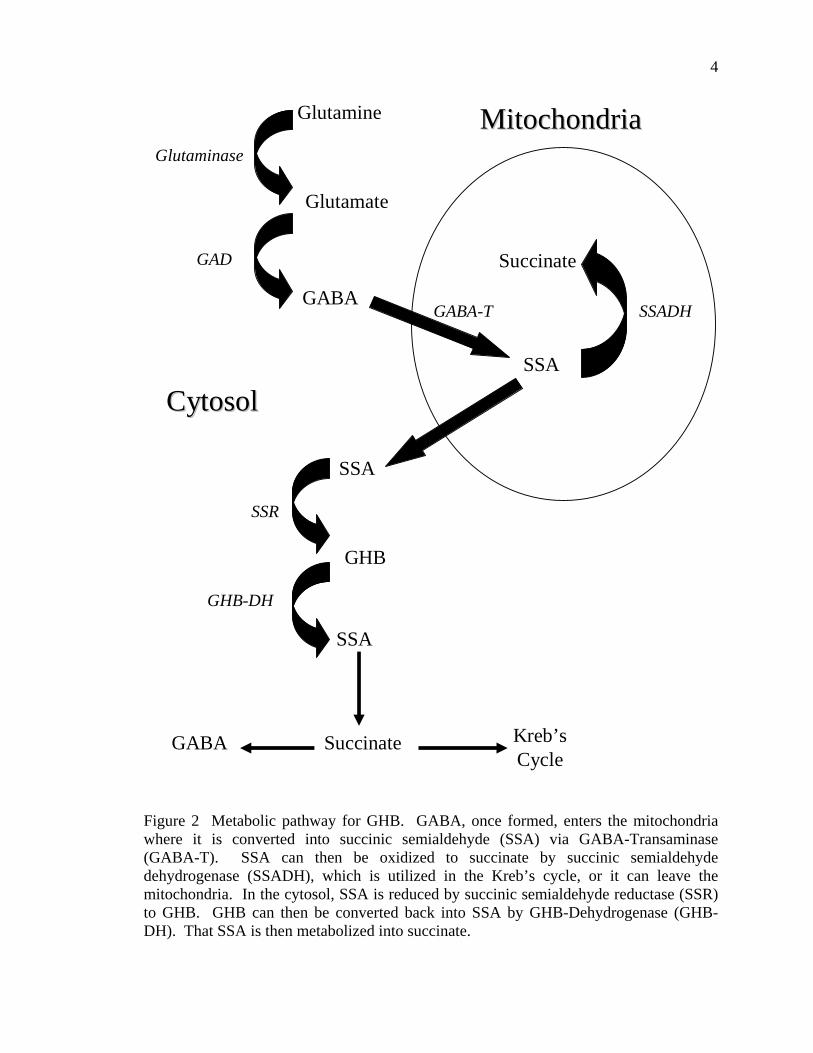
4
Glutamine
Glutamate
Glutaminase
GABAGABA-T
SSA
SSADH
Succinate
MitochondriaMitochondria
SSA
SSR
GHB
CytosolCytosol
GHB-DH
SSA
Succinate Kreb’sCycle
GABA
GAD
Figure 2 Metabolic pathway for GHB. GABA, once formed, enters the mitochondria where it is converted into succinic semialdehyde (SSA) via GABA-Transaminase (GABA-T). SSA can then be oxidized to succinate by succinic semialdehyde dehydrogenase (SSADH), which is utilized in the Kreb’s cycle, or it can leave the mitochondria. In the cytosol, SSA is reduced by succinic semialdehyde reductase (SSR) to GHB. GHB can then be converted back into SSA by GHB-Dehydrogenase (GHB-DH). That SSA is then metabolized into succinate.

5
The GHB synthesis pathway has been explored by inhibiting the enzymes located
in the pathway and monitoring the build up of related metabolites. In rats, GABA-T was
inhibited by γ-acetylenicGABA or aminooxyacetic acid (Eli and Cattabeni 1983). In this
study, rats showed decreased amounts of GHB indicating a block in synthesis. However,
Snead and others (Snead and others 1989) reported that when either of the compounds γ-
vinylGABA or aminooxyacetic acid were used to inhibit GABA-T, increased levels of
GHB were observed both in vitro and in vivo (Snead and other 1989). These contrasting
results imply that the presence of two forms of GABA-T may be present with each
playing a different role in the synthesis or degradation of GHB. It is traditionally thought
that GABA-T is an exclusively mitochondrial enzyme (Chan-Palay and others 1979).
However, it has been shown that the enzyme is located on post-synaptic membranes as
well as in the cytosol (Maitre 1997). The cytosolic presence of GABA-T would enable
conversion of GHB into GABA directly, and could give rise to a pool of GABA without
prior origins as glutamate. That GABA pool could possess different functional and
binding capabilities than glutamate derived GABA.
SSR plays a very important role in the synthesis of GHB. The location of SSR
has been elucidated by immunostaining experiments (Maitre and other 2000). When
neural tissue was immunostained for SSR, the cortex, hypothalamus, and hippocampus
were all positive, but glial cells were negative. SSR from rat brain was cloned in order to
investigate its role in GHB regulation via cDNA hybridization experiments
(Andriamampandry 1988). Those hybridization experiments showed no SSR mRNA
present in peripheral tissues such as kidney or liver. The highest GHB concentrations are
found in the cytosol and synaptosomal fraction (Snead 1987). SSR activity was also

6
found to be the highest in the cytosol and synaptosomal fraction (Rumigny and others
1981; Snead 1987). Furthermore, when SSR activity is inhibited, conversion of [3H]-
GABA into [3H]-GHB is reduced (Rumigny and others 1981). These observations imply
that the majority of GHB synthesis occurs in the cytosol and synaptosomal fraction.
Valproate is widely known to increase brain levels of GHB (Snead and others
1980). These increased levels have been observed by inhibition of a nonspecific
cytosolic aldehyde reductase in vivo that has been proposed in the metabolism of GHB to
GABA (Whittle and Turner 1978). This further implies that GHB’s effects might be due
to GHB being converted to a pool of GABA (Kaufman and others 1983; Kaufman and
Nelson 1987) that then acts in an inhibitory manner. However, valproate also
antagonizes some of the effects of GHB (Cash 1994) such as the EEG effects and ataxia
related behaviors. When examined in the rat brain, valproate was shown to be a
competitive inhibitor of SSADH (Cash 1994). Inhibition of SSADH would lead to
accumulation of SSA, which would be converted to GHB (Rumigny and others 1980,
1981). As SSADH deficiency is caused by decreased activity of SSADH, valproate has
been tested as a possible treatment and shown some promise (Rating 1984).
Several protective roles for GHB have been suggested based on the concentration
of GHB in peripheral tissues. Peripheral GHB has been found in the kidney at 10 times
neuronal levels and in the heart and muscle at five times neuronal levels (Nelson and
others 1981), often under stressful conditions. It is also known that under stressful
circumstances, serum concentrations of GHB increase (Mamelak 1989, 1997). One
theory for peripheral GHB is tissue protection during hypoxic episodes by decreasing
metabolic or energy consuming processes (Mamelak 1989, 1997). GHB has also been

7
shown to reduce cerebral glucose utilization, and alleviate intracranial pressure
(MacMillan 1980a; Haller and others 1990). When doses of approximately 5 mmol/kg of
either GHB or GBL were given, cerebral glucose metabolism was dramatically
decreased, implying a protective role in hypoxic situations (Godin 1968; Wolfson and
others 1977; MacMillan 1980b). Mamelak suggested that one of GHB’s roles might be
as an endogenous inhibitor of energy metabolism (Mamelak 1989), thus providing a
protective role for both neuronal and peripheral tissues. At subanesthetic doses, GBL
provided neurological protection against the effects of experimental ischaemia in
conscious rats (Lavyne 1983). High doses of GHB have also been shown to protect the
heart from secondary, damaging effects of cerebral ischaemia (Kolin 1991).
Localization in Tissues
The concentration of GHB in the mammalian brain is 1-4 μM (Doherty and others
1978; Snead and others 1989; Maitre 1997) and 0.3 mmol/g in the human brain
(Waszkielewicz and Bojarski 2004). More specifically, the highest concentrations of
GHB are found in the cytosol and then in the synaptosomal fraction (Snead 1987). Many
regions of the adult rat brain have GHB present in a heterogeneous distribution of
concentrations. In adult rats, GHB concentrations were 0.4μM in frontal cortex, 1.2μM
in the hippocampus, 1.8μM in the striatum and 4.6μM in the substantia nigra (Maitre
1997). The human and monkey have the highest concentrations of GHB in the brain with
a range of 11-25μM in the striatum (Maitre 1997). Interestingly, in humans, monkeys,
and rats, the highest levels of GHB were found not in adult brains, but in developing
brains (Maitre 1997). GHB can also be found in somatic and nerve cells as well as blood
and cerebral spinal fluid (CSF). In addition to its CNS localization, GHB is also

8
abundant in peripheral tissues. It has been identified in multiple organs with
concentrations of 12.4μM in the heart, 28.4μM in the kidney, 1.4μM in the liver, 10.2μM
in muscle, and 37μM in brown fat (Nelson and others 1981). As explanation of its
peripheral distribution, GHB is able to pass through the blood brain barrier freely,
although its role outside the CNS is not known (Laborit 1964, Waszkielewicz and
Bojarski 2004).
GBL & 1,4-BD as Precursors to GHB Numerous studies have indicated that GBL and 1,4-BD are prodrugs or precursors
of GHB, with GHB being the compound that exerts the pharmacological effect (Roth and
Giarman 1966; Roth and others 1966; Guidotti and Ballotti 1970; Snead 1982; Poldrugo
and Snead 1984; Snead and others 1989; Schneidereit 2000; Carai and others 2002;
Quang and others 2002a, 2002b). In vivo, GBL and 1,4-BD can both be converted to
GHB (Figure 3) (de Fiebre C and others 2004) and have been found in rat brains at levels
approximately 10% of endogenous GHB levels (Doherty and others 1975; Barker and
others 1985). 1,4-BD has been found in brain and an enzyme system for metabolism into
GHB (Figure 3) is present in both liver and brain (Barker and others 1985; Poldrugo and
Snead 1986). 1,4-BD is first converted to γ-hydroxybutyraldehyde by alcohol
dehydrogenase, which is then converted to GHB by aldehyde dehydrogenase (Carai and
others 2002). 1,4-BD is much more lipophillic than GHB, so when administered
peripherally, it passes through the blood brain barrier and renders its effects more quickly
(Waszkielewicz and Bojarski 2004). GBL has been shown to have a higher bio-

9
Oxidation
1,4-Butanediol
4-Hydroxybutanal
γ-Hydroxybutyric acid
Oxidation
OHHO
OHH
O
OHOH
O
O O
Dehydration
Hydrolysis
γ-Butyrolactone
Figure 3 Metabolism of γ-hydroxybutyric acid, 1,4-butanediol, and γ-butyrolactone (Waszkielewicz and Bojarski 2004) availability than GHB and is rapidly converted into GHB by a blood-born lactonase
(Roth and Giarman 1965; Fishbein and Bessman 1966; Roth and others 1966; Lettieri
and Fung 1978) with a half-life of less than one minute (Roth and Giarman 1966; Roth
and others 1966). While the conversion of GBL to GHB occurs in blood, GHB enters the
brain to exert its effects (Marcus and others 1976). This is supported by lack of lactonase
identified in brain cells (Fishbein and Bessman 1966). GBL also has greater lipid
solubility than GHB, so upon administration it might be absorbed into various tissues and
then slowly released for conversion to GHB. This could possibly extend the length of
time over which GBL exerts its effects (Lettieri and Fung 1978).
Medical Uses of GHB
Anesthetic. GHB was synthesized in 1960, and in 1964 Laborit proposed its use
as an anesthetic (Laborit 1964; Vickers 1969). Further studies revealed GHB to have
remarkable hypnotic action at high doses (0.5-2 g/kg i.p. in rats and 4-6 g i.v. in man),

10
which lead to use as an adjutant to anesthesia (Laborit 1973; Mamelak 1977; Hoes and
others 1980). As the dose of GHB increases, the effects progress from sedation to sleep
and then anesthesia (Laborit 1973). This progression has been observed in both animals
and humans. As a sedative, less GHB is needed to induce sleep than ethanol, but more
than in currently used hypnotic drugs (McCabe and others 1970). In humans, GHB doses
of 75-100 mg/kg result in a hypnotic state and rapid onset of sleep, while sedation doesn’t
occur in rodents until doses of 300 mg/kg or higher are used (Lapierre and others 1990;
Entholzner 1995). These authors also demonstrated that after administration, GHB is
quickly metabolized and cleared from the blood within eight hours.
Narcolepsy. More than 135,000 people in the United States are affected by
narcolepsy, which is characterized by an extremely strong tendency to fall asleep (NIH
2006). Observations of sleep quality and time spent in various stages of sleep have led to
the use of GHB as a treatment for narcolepsy as far back as the 1970s (Mamelak and
others 1986; Wong and others 2003). Taking GHB before sleep results in less time spent
in stage one sleep and more time in stages three and four (Lapierre and others 1990; Emri
and others 1996; Van Cauter and others 1997). More time spent in stages three and four
leads to more time spent in REM sleep (Entholzner and others 1995). One study using
GHB showed therapeutic effects with decreased cataplexy and an increase in the quality
of sleep in both control and narcoleptic patients (Scrima and others 1990). In a separate
clinical trial it was impossible to differentiate natural sleep from GHB induced sleep
using either behavioral or electroencephalgraphic data/observations (Mamelak and others
1977). Another trial showed few side effects and no EEG abnormalities other than those
seen in normal sleep (McCabe and others 1970). In contrast, GHB given to cats, rats,

11
rabbits, and monkeys lead to a reduced behavioral state that resembles nonconvulsive
epilepsy (Winters and Spooner 1965; Marcu and others 1967; Scotti De Carolis and
Massotti 1978; Benavides and others 1982; Snead 1992). One particular study followed
48 narcoleptic patients over the course of nine years (Maitre 1997). The patients took
2.25-3 grams of GHB each night. No tolerance or dependence developed, but narcoleptic
symptoms were reduced in most of the patients. One hypothesis from the study was that
GHB caused hyperdopaminergic activity, which altered acetylcholine release, which has
been suggested to play a role in narcolepsy (Maitre 1997).
One of the symptoms of narcolepsy is cataplexy which is a sudden loss of muscle
tone and control that can last anywhere from a few seconds to a few minutes (NIH 2006).
A cataplectic event is often accompanied by an emotion such as anger, excitement, or
amusement. An individual who experiences such cataplectic events may experience a
drooping of the jaw, buckling of the legs or may even collapse. GHB (Xyrem, Orphan
Medical, Minnetonka, MN) has recently been approved to treat narcolepsy with cataplexy
(Anonymous 2002a). The Xyrem International Study Group (Xyrem International Study
Group 2005) showed that cataplectic events were reduced by a median of 57%, 65%, and
85% in the 4.5, 6.0, and 9.0 g treatment groups respectively, compared to a median
decrease of 21% in the placebo group after eight weeks. Xyrem is a Schedule III,
federally controlled substance. It can be obtained only by a prescription and is provided
by only one central pharmacy (Anonymous 2002b). Primary side effects include nausea,
sleep difficulties, confusion, headache, vomiting, and enuresis. Xyrem is taken once
before bedtime and again between 2.5-4 hours later (Xyrem International Study Group
2005).

12
Dietary Supplement. In the 1980’s GHB was manufactured in the United States
and in the 1990’s it became a popular body building supplement when it was reported
that it increased growth hormone release in the body (Snead 1977; Van Cauter and others
1997). In 1990 GHB sales were banned by the FDA (Okun and others 2000). When
GHB was banned, sales of GBL and 1,4-BD rose as they were also found to act in a
similar way to GHB, by increasing the release of growth hormone (Freese and others
2002). In 1994 it became illegal to sell GBL and 1,4-BD (Waszkielewicz and Bojarski
2004), under the Dietary Supplement Health and Education Act (DSHEA). Currently all
three drugs are classified as Schedule I Compounds.
GHB Toxicity and Drug Abuse
In the late 1990’s GHB’s popularity had spread to the “night club” scene where it
became a drug of abuse, and more alarmingly, was reported in cases of drug-facilitated
rape (Raess and Tunnicliff 2002). Some of the common street names for GHB are shown
in Table 1. The sale of GHB was banned by the FDA in 1990 and classified as a
Schedule I drug in March 2000 (Smith and others 2002). Despite the ban of GHB, GBL
was not immediately banned, and was still available in health food stores as of January
2004 (Wong and others 2004). To date, GHB, GBL, and 1,4-BD are all classified as
Schedule I drugs by the FDA (Lloyd 2002). The exception to this is the recent approval
of GHB as a treatment for cataplexy associated with narcolepsy. Between August 1995
and September 1996, 69 acute poisonings and one death were attributed to GHB by
poison control centers in Texas and New York (Anonymous 1997). As of January 2004
there had been more than 7100 overdose reports or law enforcement encounters, 65
deaths, and 30 assaults due to GHB since 1990 (Shannon and Quang 2000). When GHB

13
Table 1 Common street names of GHB
Blue nitro G-riffick Oxy-sleep Cherry fX bombs Growth hormone booster Poor man's heroinCherry meth Insom-X Remforce Easy lay Invigorate Revivarant Everclear Lemon fX drops Salty water Firewater Liquid ecstasy Scoop Gamma G Liquid E Soap Georgia homeboy Liquid X Somatomax PM GHB Longevity Somsanit G.H. revitalizer Natural sleep-500 Vita-G Gib Nature's quaalude Water Goops Orange fX rush Wolfies Great hormones at bedtime Organic quaalude Zonked Grievous bodily harm Source: (O'Connell and others 2000)
is administered peripherally, it passes through the blood brain barrier to interact with
receptors that have specific distribution, ontogeny, and pharmacology. The receptors to
which GHB binds are present only in the brain and GHB exerts its effects on GABA,
dopamine, and opiate systems (Snead 1987; Schmidt-Mutter and others 1998). Acute
affects of GHB in humans include unconsciousness, hallucinations, ataxia, confusion, and
euphoria (Shannon and Quang 2000; Teter and Guthrie 2001). Chronic abuse of GHB
has lead to physical dependence as evidenced by withdrawal symptoms that accompany
discontinued use (Craig and others 2000; Catalano and others 2001; Nicholson and
Balster 2001). Observations in animals include changes in or loss of locomotor activity,
seizures, hyper/hypothermia, and loss of the righting ability (Davies 1978; Dudek and
Fanelli 1980; Kaufman and others 1990; Snead 1990; Cook and others 2002). However,
rodents can develop tolerance to these effects (Colombo and others 1995). GHB’s
adverse effects appear in a dose dependent fashion in both humans and rodents (Table 2)

14
(Colombo and others 1995). Some of these effects can be blocked by administration of
CGP 35348, a GABAB antagonist, implying a role for GHB at GABAB receptors (Xie
and Smart 1992; Enberg and Nissbrandt 1993; Williams and others 1995). Extremely
high doses (200-800 mg/kg) in rodents have shown a dose dependent loss of locomotor
activity and sedation (Nissbrandt and Engberg 1996; Martellota 1997; Schmidt-Mutter
and others 1999; Itzhak and Ali 2002). However, pretreatment with GABAB antagonists
can block those effects (Nissbrandt and Engberg 1996).
Table 2 Dose response of GHB in both humans and rodents
Dose or Concentration of GHB Physiological Effect Oral dose (humans) 10 mg/kg -1 Short-term amnesia 20-30 mg/kg -1 Sleep and drowsiness 50-60 mg/kg -1 Coma, cardiorespiratory depression, seizures Systemic dose (rodents) 10-50 mg/kg -1 Memory impairment 75-200 mg/kg -1 Absence seizures, spike-and-wave discharges on
EEG (threshold brain concentration for this effect is 240
uM) 200-300 mg/kg -1 Stupor, EEG slowing >300 mg/kg -1 Coma, EEG burst suppression; isoelectric period increases with dose 1.7 g/kg -1 LD50
Source: (Wong and others 2004)
In rats, chronic treatment with GHB lead to tolerance, withdrawal, and
conditioned place preference which is normally associated with the rewarding effects of
drugs of abuse (Martellotta and others 1997; Colombo and others 2002). Self-
administration of GHB, another characteristic of drug addiction, can be blocked by

15
administration of baclofen, another GABAB antagonist (Fattore and others 2001). Mice
subjected to chronic, high doses of GHB also showed what appeared to be a down
regulation of the GABAergic system (Gianutsos and Suzdak 1984).
Upon withdrawal of GHB, the same clinical symptoms appear as in ethanol
withdrawal (Craig and others 2000; Miotto and others 2001). Those symptoms include
tremors, anxiety, muscle cramps, severe agitation, disorientation, paranoia, auditory and
visual hallucinations, tachycardia, elevated blood pressure, diaphoresis, (Craig and others
2000), and insomnia, which usually lasted only three days (Nicholson and Balster 2001).
Ironically, some success has been achieved when using GHB as a treatment for ethanol
withdrawal (Berge and Carpanini 2000). GHB has been shown to reduce ethanol
withdrawal symptoms such as anxiety, restlessness, depression, nausea, tremors, and
sweating with a common side effect of dizziness (Gallimberti and others 1999). An
Italian study demonstrated that GHB was faster than diazepam at decreasing agitation,
anxiety, and depression scores in alcohol withdrawal (Addolorato and others 1999). A
separate ethanol withdrawal study showed that after six months of GHB administration
(50 mg/kg/day), 78% of patients reported as completely abstinent, with a reduced craving
for alcohol (Addolorado and others 1996). Of that 78%, 43 patients were still abstinent
six months later and 30 were still abstinent after one year.
It is difficult to measure GHB levels in the body by using traditional methods as
only 1% of circulating levels are excreted in the urine in the endogenous form (Hornfeldt
and others 2002). Gas chromatography/mass spectrometry (GC/MS) can measure
concentrations as low as 0.1 mg/l in plasma and 0.2 mg/l in urine, but the methodology
requires that GHB is first converted to GBL (Ferrara and others 1993). A separate

16
method that does not require the conversion to GBL can be utilized but is limited to
measuring samples in the 0.5-2.0 mg/l range (Louagie and others 1997; Couper and
Logan 2000).
As a drug of abuse, GHB is popular in nightclubs as a euphoric compound. It has
also been widely reported in cases of drug-facilitated rape, where it renders victims in an
unconscious state often with a certain level of amnesia afterwards. The amnesia is
induced by a hyperpolarization of neurons in the hippocampus, an important brain
structure for memory (Waszkielewicz and Bojarski 2004). In this process, GHB acts as a
GABAB agonist, which increases cGMP, causing hyperpolarization. Other symptoms of
GHB abuse are hypothermia and electroencephalographic changes (Snead 1978). During
ethanol induced liver failure or excessive alcohol consumption, the rate of GHB synthesis
and degradative pathways decrease (Maitre 1997; Zvosec and others 2001). This leads to
an increase in serum levels of GHB, resulting in increased toxicity.
Overdoses of GHB have resulted in seizures, cardiorespiratory depression, coma,
and death (Raess and Tunnicliff 2002). The mechanism of action for GHB is still not
completely understood and there is currently no antidote for GHB overdose (Shannon and
Quang 2000).
SSADH Deficiency: A Disorder of GABA and GHB Metabolism Succinic Semialdehyde Dehydrogenase (SSADH) deficiency is a rare autosomal
recessive disorder caused by a disruption in the metabolism of GABA (Wong and others
2003) that was first identified in 1981 (Jakobs and others 1981) and is characterized by a
significant lack of the SSADH enzyme (Gianutsos and Suzdak 1984; Rating and others
1984). Only 350 individuals worldwide have been identified with SSADH deficiency

17
and 37% of the 60 cases reviewed were the result of parental consanguinity (Pearl and
others 2003). Diagnosis most often occurs in childhood, but onset has been seen as late
as 25 years of age (Pearl and others 2003). Clinical symptoms include: ataxia, seizures,
psychomotor retardation, hypotonia, language delay, hyporeflexia, aggressiveness,
hyperkinesis, and oculomotor apraxia (Jakobs and others 1993; Gibson and others 1997).
Diagnostic markers used to identify this disorder are extremely high levels of GABA and
GHB, which are found in blood, urine, and cerebral spinal fluid (Cash 1994). It is not
known whether the disorder is a direct result of accumulated GHB in tissues, SSA in
tissues, or excess GABA that is formed from accumulated SSA. Another metabolite
present in SSADH screening is 4,5-dihydroxyhexanoic acid (DHHA). DHHA is not
normally detectable in mammalian tissues, but is found to accumulate in SSADH
patients. DHHA was first identified in the urine of SSADH patients in 1987 (Brown and
others 1987) and it was hypothesized to be formed from the condensation of succinic
semialdehyde with a two or three carbon intermediate such as acetyl-CoA, lactate, or
pyruvate (Shaw and Westerfeld 1968; Schoerken and Sprenger 1998). There is currently
no treatment for SSADH deficiency (Pearl and others 2003). Vigabatrin, an irreversible
GABA-T inhibitor used for its antiepileptic properties, has been the most popular choice
of treatment to be examined. As an inhibitor of GABA-T, vigabatrin should prevent the
formation of succinic semialdehyde, which in turn would prevent the formation of GHB,
resulting in increased GABA levels (Gibson and others 1989, 1995). However,
increasing GABA levels in a patient already suffering from a disorder characterized by

18
increased GABA levels may not be recommended. Furthermore, chronic use of
vigabatrin has been linked to vision difficulties and renal toxicity (Mtern and others
1996).
A mouse model for SSADH deficiency has been described (Gupta and others
2003). Deleting exon 7 of the mouse genome resulted in complete lack of SSADH
activity in both neural and peripheral tissues. These mice are born with the same
autosomal recessive pattern as human SSADH deficiency and exhibit symptoms of
ataxia, growth retardation, and seizures. Tonic clonic seizures develop between 16 and
22 days of life and lead to 100% mortality (Gupta and others 2003). In these mice,
GABA levels have been found at 20-60 times the endogenous amount and GHB levels
are elevated 2-3 fold (Hogema and others 2001; Gibson and others 2002). Another
characteristic marker is a significant decrease in glutamine levels found in both frontal
and parietal cortex, hippocampus, and cerebellum (Behar and others 1999; Sonnewald
and McKenna 2002; Gibson 2005). Despite decreased glutamine, glutamate levels
remain unaffected. This along with increased levels of GABA indicates a disruption of
the glutamine-glutamate “shuttle.” This shuttle (Figure 4) relies on glutamine to maintain
pools of neurotransmitters in the presynaptic neuron and the astrocytes. Treatment
studies have included CGP 35348 (Olpe and others 1990), a GABAB antagonist, and
NCS-382, a GHB antagonist, (Carai and others 2001). Both compounds significantly
increased the survival rate of SSADH knock out mice with NCS-382 extending life in
60% of the test population (Gupta and others 2003). In addition to pharmacotherapy, a
gene therapy targeting the liver has been developed (Chambliss and others 1995). This
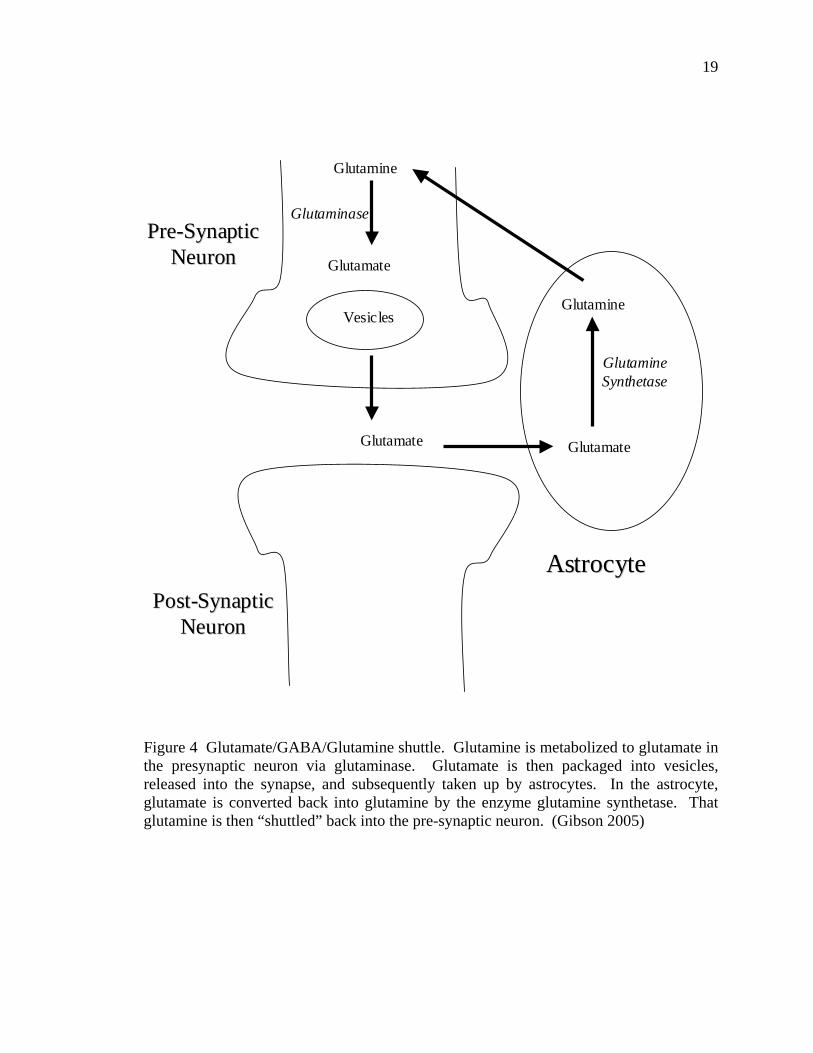
19
AstrocyteAstrocytePostPost--Synaptic Synaptic
NeuronNeuron
PrePre--Synaptic Synaptic NeuronNeuron
Glutamine
Glutamate
Glutaminase
Vesicles
Glutamate Glutamate
Glutamine
Glutamine Synthetase
Figure 4 Glutamate/GABA/Glutamine shuttle. Glutamine is metabolized to glutamate in the presynaptic neuron via glutaminase. Glutamate is then packaged into vesicles, released into the synapse, and subsequently taken up by astrocytes. In the astrocyte, glutamate is converted back into glutamine by the enzyme glutamine synthetase. That glutamine is then “shuttled” back into the pre-synaptic neuron. (Gibson 2005)

20
therapy was based on reports of SSADH activity in the liver being equal to or greater
than that in the brain. Recombinant viruses with the SSADH cDNA were injected into
the SSADH mouse intraperitoneally and lead to a 40% increase in life span (Gibson and
others 2002). Along with an increased life span, the gene therapy lead to decreased liver
concentrations of GHB and expression of active SSADH in the temporal lobe of the
brain.
Neurotransmitter Function of GHB
In order to be a classified as a neurotransmitter, a compound must be synthesized
in neural cells, have specific, high affinity binding sites, be released from neurons in a
polarizing or depolarizing manner, and a re-uptake mechanism must be present (Cash
1994). GHB satisfies these requirements but its role as a neurotransmitter remains
questionable as does its overall role both neuronally and peripherally. As previously
described, GHB is a metabolite of the inhibitory neurotransmitter GABA and its
synthesis and storage take place in brain cells (Galloway and others 1997). It is proposed
that GHB’s neurotransmitter functions are inhibitory in nature and are involved in
regulation of GABA and dopamine systems (Zvosec and others 2001). Traditionally,
neurotransmitters such as glutamate, GABA, and dopamine cannot exert their effects
when administered peripherally, as they cannot pass through the blood brain barrier.
However, GHB is able to pass through the blood brain barrier and exert its effects when
given peripherally (Cash 1994). This allows for the peripheral administration of GHB to
increase the brain concentration by 100 fold or more. Despite such increases, the brain
does not retain GHB.

21
As previously described, GHB is synthesized in either the mitochondria or cytosol
of neuronal cells. The primary localization of synthesis occurs in the cytosol and
synaptosomal fraction, as demonstrated by the localization of the highest concentrations
of SSR and GHB (Snead 1987). Synthesis in the synaptosomal fraction implies that
GHB can be released to exert an effect on other neurons. Studies with [3H]-GHB loaded
brain slices have shown that GHB is released via Ca2+ induced depolarization in a dose
dependent fashion (Maitre and others 1983; Muller and others 2002). In a follow up
experiment, by Maitre and others (Maitre and others 1983a) striatal slices were
depolarized by K+, which caused a release of approximately 50 fmol/min-1/mg-1 GHB wet
weight. When Ca2+ was removed from the medium, and the [3H]-GHB release decreased
by 50-60% (Maitre and others 1983a) Vertradine was tested in the same assay and found
to induce an even stronger release of [3H]-GHB measured at 70-80 fmol/min-1/mg-1 wet
weight. Using vertradine, different regions of rat brain slices were investigated further
for their [3H]-GHB releasing properties. Brain slices from the fronto-parietal cortex,
hippocampus, and striatum all showed strong release of GHB comparable to the
vertradine induced release of GHB. The cerebellum and pons-medulla, however, had
dramatically reduced [3H]-GHB release (Maitre and others 1983). Other studies have
shown that once released, both pre and postsynaptic transporters take up GHB. A Na+
dependent uptake system was identified as well as an active vesicular uptake system
(Muller and others 2002). It is hypothesized that the uptake system is the same vesicular
inhibitory amino acid transporter that is responsible for GABA and glycine. While this
uptake system is required for neurotransmitter classification, the Km value is
approximately 50 μM, which is 20 fold higher than endogenous amounts of GHB

22
(Benavides and others 1982). Furthermore, GABA competes with GHB at this transport
system. Labeled GHB accumulated in membrane vesicles, which was dependent on the
presence of at least one Na+ ion (Hechler and others 1985). In addition to being required,
the Na+, Cl-, and K+ ions were shown to stimulate this transport. In striatal slices from rat
brains, GHB uptake was also discovered to be Na+ and K+ dependent.
GHB as a GABAA/B Agonist There have been numerous studies regarding receptors to which GHB binds.
Possibilities include GABAA receptors, GABAB receptors, and GHB specific receptors.
There are some reports that GHB does not affect GABAA or GABAB receptors with any
regularity (Snead and Liu 1993; Feigenbaum and Howard 1996a; Mathivet and
Bernasconi 1997; Snead 2000). However, many other studies report that GHB does in
fact play a role at GABAA or GABAB receptors (Hosli and others 1983; Bourguignon and
others 1988; Zvosec and others 2001). However, the effect GHB has on GABAA
receptors is minimal (Kozhechkin 1980; Snead and Liu 1993) and as a consequence,
more studies have focused on the GABAB receptor. Additional studies have shown that
GHB has two of its own GHB specific receptors to which it binds (Maitre and others
1983c).
The GABAA receptor modulates fast inhibitory neurotransmission while the
GABAB receptor modulates slow inhibitory neurotransmission (Wong and others 2003).
GHB has been shown to play a weak role at the GABAA receptor (Snead and Nichols
1987; Snead and others 1992). When GHB does bind the GABAA receptor, it has an
agonist role at concentrations well above endogenous levels and the binding affinity is
extremely weak. GABAA agonists exacerbate GHB induced seizures, but GABAA

23
antagonists are unable to block those same seizures. Serra and others (Serra and others
1991) tested the effects that GHB might have on GABAA receptors by administering
GHB to animals before binding studies were performed in vitro. Neither GBL nor GHB
had an effect on the binding of GABAA agonists [3H]-muscimol, [3H]-flunitrazepam, or
[35S]-t-butylbicyclophosphorothionate. In contrast, GHB could, on occasion act as a
GABAA agonist in vivo, in tissue slices or in cell cultures (Snead and Liu 1993). The
results from these two studies indicate a lack of competitive binding between GHB and
GABA at GABAA receptors. That effect is thought to be a result of GHB’s role in
GABA release or the conversion of GABA into GHB.
Various binding studies have been utilized in the investigation of whether or not
the GHB receptor and the GABAB receptor are the same. When [3H]-GHB is bound,
GABAB antagonists are not able to displace it (Snead 1996; Kemmel and others 1998)
and GHB is only weakly able to displace the GABAB antagonist [3H] baclofen (Mathivet
and others 1997; Bernasconi and others 1999). In other studies using rat brain slices,
neither GHB nor its antagonist, NCS-382, competed for [3H]-GABA binding (Snead
1996). Supporting the theory of two different receptors, it has been reported that the
regional distribution (Snead 1994; Maitre 1997; Castelli and others 2000) and ontogeny
(Snead 1994; Snead 2000) for the GHB receptors and the GABAB receptor are different.
Studies in rats have shown that GHB administration causes a loss of locomotor
activity, which can be prevented by pretreatment with GABAB antagonists (Bottiglieri
and others 2001). However, if GABAB antagonists are given after GHB administration,
the loss of motor activity is not blocked (Bottiglieri and others 2001). This implies that
GHB, once bound to GABAB receptors, is not easily displaced. GABAB agonists can

24
also mimic the decrease in dopamine release in the striatum that results from GHB
administration. Furthermore, GABAB antagonists can antagonize that decrease in
dopamine release (DaPrada and Keller 1976; Kelly and Moore 1978; Broxterman 1981;
Waldmeier 1991; Engberg and Nissbrandt 1993; Nissbrandt and others 1994). Recent
studies have shown that the GABAB antagonists, CGP 35348 and 36742, when
administered before GHB can antagonize the reduction seen in striatal 3-MT levels and
loss of locomotor function. (Bottiglieri and others 2001). However, the levels of GHB
needed to displace GABA are 30-100 times greater than endogenous levels (Bernasconi
and others 1992; Ishige and others 1995; Ito 1995). Additionally, a GHB-GABAB
receptor complex has been suggested as another possibility (Cash 1994).
While the GHB has a weak affinity for GABAB receptors (Mathivet and others
1997; Bernasconi and others 1999) in the millimolar range (Lingenhoel and others 1999),
that affinity is much higher than the endogenous micromolar levels of GHB in the body
(Maitre 1997). Once the brain concentration of GHB increases by two to three fold,
GHB receptors are saturated and GABAB receptor mediated responses are present (Snead
2002; Kemmel and others 2003).
GHB Receptors According to several independent studies, it is believed that GHB has its own
specific receptors to which it binds, aside from GABA receptors (Bourguignon and others
1988; Snead 2000). GHB’s effects are induced primarily by binding to a GHB specific,
presynaptic, G-protein coupled receptor (Snead 2000). After binding to this receptor, the
downstream effects are dependent upon a decrease in adenyl cyclase activity
(Waszkielewicz and Bojarski 2004). Some studies in rat brain membranes showed that

25
GTP or its analogues could reduce the binding affinity of GHB, implying G-protein
linked receptors (Ratomponirina and others 1995). The distribution of the GHB high
affinity binding sites does not match the distribution of either GABAA or GABAB
binding sites (Maitre 1997). This again implies the presence of GHB specific receptors.
Radioactive labeling studies have been used extensively in the investigation of
GHB and the receptors to which it binds. Benavides and others (Benavides and others
1982) have described two classes of binding sites for GHB. One is a high affinity site
with a Kd of 30 to 90 nM and the other is a low affinity site with a Kd of 16 μM. The
binding characteristics of GHB are dependent on pH (Hechler and others 1990a). The
maximum binding occurs at pH 5.5, but is completely absent at pH 5.0 as well as pH 8.0.
Experiments are performed in a physiologically relevant range such as pH 6.0-7.5. In
this range there is very significant GHB binding. GHB binding is not, however, affected
by K+, Ca2+, Mn2+, or Cl-.
The studies regarding specific location of GHB binding sites in rat and human
brain have focused on the high affinity binding sites, as it was difficult to measure the
low affinity sites. Regional dissection of rat brain identified the areas with the greatest
concentration of GHB binding sites as the hippocampus, thalamus, olfactory tract,
striatum, and cortex (Maitre and others 2000). The liver, kidneys, muscle, and heart all
have high levels of GHB, however there are no high affinity binding sites in those tissues
(Nelson and others 1981; Snead and Liu 1984). Furthermore, in brain tissue cultures,
high affinity binding sites have been found only in neuronal cell lines (Hosli and Hosli
1983; Snead and Liu 1984; Maitre and others 2000). Other experiments in rat brain
identified the binding sites as located primarily in the synaptosomal fraction (Maitre and

26
others 1983c). Synaptosomal fractions from rat brain showed that the areas with the
highest amount of acetylcholine and acetylcholinesterase also had the highest density of
both high and low affinity GHB binding sites (Maitre and others 1983c). Those levels of
acetylcholine and acetylcholinesterase in the synaptosomal fraction were 10 times greater
than in nuclear, myelin, or mitochondrial fractions. This further indicates that GHB
binding sites are present only in vertebrate neurons and are concentrated in the
synaptosomal fraction.
Various compounds have been tested for their ability to displace [3H]-GHB from
its high affinity binding sites. The only compounds that were capable of displacing [3H]-
GHB were structural analogues (Benavides and others 1982a; Snead and Liu 1984).
GHB could not be displaced by GABA, baclofen, muscimol, isoguvacine, bicuculline,
dopamine, naloxone, or anti-epileptic drugs (ethosuccimide, trimethadione, and valproic
acid). It was also discovered that GBL, a previously mentioned precursor to GHB, was
not able to displace GHB. The first compound to show the capability to bind at a GHB
specific receptor was trans-γ-hydroxycrotonic acid (T-HCA) (Waszkielewicz and
Bojarski 2004). T-HCA is endogenous to the CNS, has a heterogeneous presence in the
brain nearly identical to that of GHB (Vayer and others 1988), and binds to the GHB
binding sites with four times the affinity of GHB itself (Hechler and others 1990b). In
contrast, Maitre and others (Maitre and others 2000) observed that T-HCA was present at
levels 5-10 times less then endogenous GHB, was able to displace GHB, and possessed
an affinity for the binding sites about 10 times greater than that of GHB.
NCS-382 was the first compound discovered to antagonize the GHB receptor
(Maitre and others 1990) although it had no effect on GABA binding (Maitre and others

27
2000). When administered, NCS-382 blocks nearly all neurophysiological and
neuropharmacological effects caused by administration of GHB (Hechler and others
1993).
In displacement studies, some of the most well known GABA receptor ligands
such as baclofen, picrotoxin, bicuculline, muscimol, and isoguvacine were not able to
displace GHB from its binding sites (Benavides and others 1982). GBL and 1,4-BD,
precursors to GHB, and structurally similar compounds were also unable to displace
GHB from its binding sites.
GHB’s Role in Regulation of Dopaminergic Function
Recent experimental observations and monoamine neurotransmitter analysis
studies suggest that administration of GHB has a profound effect on dopamine synthesis,
degradation, metabolism, regulation, modulation, release, and uptake.
The effect of GHB on dopamine synthesis has been studied by monitoring L-
Dopa (dopamine’s immediate metabolic precursor) levels and tyrosine hydroxylase (TH)
activity. TH is the rate-limiting enzyme (Nagatsu 1964) in dopamine synthesis, so if its
activity level increases, L-Dopa synthesis will increase. Increased L-Dopa will then lead
to a higher rate of dopamine synthesis. In one study performed in Sprague-Dawley
albino rats, the accumulation of L-dopa in brain tissue was determined after treatment
with GBL and 3-hydroxybencilhydracine (NSD 1015), an inhibitor of aromatic amino
acid decarboxylase (AADC) (Booth and others 1994). When AADC is inhibited, the rate
of accumulation of L-Dopa, the substrate for AADC, is a measure of TH activity. There
was a greater increase of L-dopa when NSD1015 and GBL were administered than there
was when NSD1015 and saline were administered. These findings indicated that GBL

28
results in an increase in TH activity. The authors proposed that GHB formed from GBL
acted to inhibit dopamine release, which activated presynaptic D3 receptors. Stimulation
of D3 receptors resulted in second messenger signaling that lead to increased TH activity.
That further reinforces the relationship between GHB and an increase in dopamine
synthesis.
The role GHB plays in dopamine release is thought to be that of an inhibitory
neurotransmitter/modulator (Feigenbaum and Howard 1996b; Madden and Johnson 1998;
Hedou and others 2000). Numerous studies have shown GHB to induce a significant,
reversible, 60 minute inhibition of impulse flow in mesolimbic and nigrostriatal
dopaminergic neurons (Aghajanian and Roth 1970; Roth and Suhr 1970; Roth and others
1973; Stock and others 1973; Walters and Roth 1974; Roth 1976). Other studies have
shown that newly synthesized dopamine is not released upon depolarization if GHB has
already been administered (Bustos and Roth 1972). This decrease in dopamine release
can also be seen when baclofen, a GABAB agonist, is given (Da Prada and Keller 1976)
and can be blocked when naloxone, a GABAB antagonist, is administered (Hechler and
others 1991, Feigenbaum and Howard 1996c). These observations show some
relationship between the GABAB receptor, GHB, and dopamine release. However, the
relationship remains poorly understood.
In vivo experiments using microdialysis have shown GHB to have a biphasic
effect on dopamine release (Hechler and others 1991). In vivo microdialysis experiments
showed a decrease in striatal dopamine release after the rodents were given 300 mg/kg
GHB or less. GHB doses over 300 mg/kg caused an increase in dopamine release
(Hechler and others 1991). Experiments with rats have shown brain dopamine levels to

29
double within one hour of GHB administration via i.v. injection (Gessa and others 1966).
One exception to the studies already mentioned is the striatum. No matter the dose of
GHB, striatal dopamine release decreases (Waszkielewicz and Bojarski 2004).
Furthermore, d-amphetamine, a compound that stimulates dopamine release, has been
used to prevent or reverse GHB induced increases of dopamine in the brain (Gessa and
others 1966; Gessa and others 1968; Aghajanian and Roth 1970; Roth and Suhr 1970;
Spano and others 1971; Hutchins and others 1972; Walters and Roth 1972; Kehr and
others 1972; Anden and others 1973; Roth and others 1973; Roth 1976). In addition to
reversing neurochemical changes, d-amphetamine has also shown the ability to
antagonize behavioral changes induced by GHB or GBL. Effects such as akinesia and
hypokinesia (Dudek and Fanelli 1980; Ellinwood and others 1983), catalepsy
(Feigenbaum and Howard 1996a), and sedation and loss of righting (Roth an Suhr 1970;
Dudek and Fanelli 1980) were all reversed by d-amphetamine.
As previously mentioned, in some cases, GHB has been shown to decrease
dopamine release. This leads to decreased dopamine in the synaptic cleft. The changes
observed in dopamine metabolism have also alluded to what role GHB may play in
dopaminergic regulation. Less dopamine in the synaptic cleft results in a decrease of
dopamine metabolites. Specifically, DOPAC and HVA levels should decrease after
administration of GHB and then increase upon the release of accumulated DA once GHB
has been metabolized. This was shown in several studies after high doses of GHB were
administered (Roffler-Tarlov 1971; Roth 1971; Roth and others 1980; Alter and others
1984). More recent studies indicate GHB can inhibit the release of neuronal dopamine.
Administration of GHB in rats leads to a marked reduction in the levels of striatal 3-

30
methoxy tyramine (3-MT) (Bottiglieri and others 2001). 3-MT is formed in the synapse
by postsynaptic catechol-O-methyl transferase (COMT) once dopamine has been
released. Decreased levels of 3-MT indicate inhibition of dopamine release. Further
studies showed that when pargyline was given both before and after GHB, striatal 3-MT
levels were normalized and loss of motor activity was blocked (Anderson and others
2002).
A discrepancy regarding the inhibition of dopamine release exists as some studies
have shown an increase in dopamine release. In a review of the literature, Fiegenbaum
and Howard (Feigenbaum and Howard 1996a) offer several explanations for the GHB
induced increases in dopamine release. In a study by Maitre and others (Maitre and
others1990) GHB reportedly stimulated dopamine release in vivo. In 1991, Hechler and
others (Hechler and others 1991) showed a GHB induced increase in dopamine release
both in vivo and in vitro. However, the calcium concentration in the dialysate was 3.4
mM in both of those studies. Previous work has shown that calcium concentrations
greater than 2.3 mM may alter the activity of centrally acting drugs or compounds
(Westerink and others 1988; Moghaddam and Bunney 1989; DeBoer and others 1990;
Timmerman and Westerink 1991). Thus, the high concentrations of calcium may have
artificially increased dopamine release. Other studies in cats have shown an increase in
dopamine release after GHB administration (Roth and others 1973; Mereu and others
1984). In that study, the cats were anesthetized with halothane. It has been demonstrated
that halothane causes an increase in the firing rate of nigral neurons (Roth and others
1973; Mereu and others 1984). This leads to the conclusion that the increase in dopamine
release was most likely caused by the halothane and not GHB. Furthermore, halothane

31
causes a more intense and longer lasting release of dopamine in the striatum of both cats
and rats (Nieoullon and others 1977; Savaki and others 1986; Spampinato and others
1986; Collin and others 1988; Osborne and others 1990; Stahle and others 1990).
Another anesthetic, chloral hydrate, was used during microdialysis experiments by
Nissbrandt and others (Nissbrandt and others 1994) that showed significant increases in
dopamine output. However, chloral hydrate has been shown to significantly alter striatal
release of dopamine in vivo (Lane and Blaha 1987; Zhang and others 1989; Hamilton and
others 1992). These examples suggest that the increases in dopamine release seen when
GHB is administered are likely due to components of microdialysis dialysates or
anesthetics that have their own profound effect on the regulation of dopamine release.
NCS-382 was first described 15 years ago (Maitre and others 1990) and was
identified as a compound that antagonized almost all neuropharmacological and
neurophysiological effects caused by GHB (Hechler and others 1993). NCS-382 also
antagonizes the sedative effects of GHB (Schmidt and others 1991). In vitro, NCS-382
competes with GHB at both high and low affinity binding sites and has also been shown
to prevent the increased dopamine release in the striatum caused by peripheral
administration of GHB (Maitre and others 1990). In vivo, NCS-382 antagonizes the
increase in dopamine accumulation and release as well as opioid like substance release
from rat striatum caused by GHB (Hechler and others 1991). As the dose of GHB is
increased, the decrease in dopamine release is followed quickly by an intracellular
accumulation of dopamine in the forebrain of rats (Gessa and others 1966; Gessa and
others 1968; Aghajanian and Roth 1970; Spano and others 1971; Pericic and Walters
1976; Lundborg and others 1980; Dyck and Kazakoff 1982).

32
Behaviorally, the decrease in dopamine release manifests as a decrease in
locomotor activity, depressed breathing, sedation, and temporary paralysis. Once the
GHB has been completely metabolized, a burst of dopamine release occurs (Cheramy
1977; Hechler and others 1991). This increases extracellular dopamine for a short period
of time. Upon this burst release of dopamine, the breathing quickly normalizes, paralysis
is abolished, and locomotor activity often increases to levels above those seen before
administration of GHB. When GHB or its analogues are administered in the 400-700
mg/kg range, release of DA in the striatum can reach as high as 600-1000% of basal
values (Hechler and others 1991). The GABAB antagonist NCS-382 can antagonize this
rapid, high level release.
High affinity GHB binding sites were prevalent in specific areas of the brain
(olfactory system, nucleus accumbens, caudate, putamen) that are all associated with
dopaminergic neurons (Snead and Liu 1984). Immunostaining experiments in the
striatum and substantia nigra of rats identified the colocalization of Tyrosine Hydroxylase
(TH), Glutamate Decarboxylase (GAD), and SSR in the same neurons (Maitre and others
2000). The presence of GAD and SSR in the same neurons suggests that GHB is
produced from glutamate in those neurons. In the striatum there was an abundance of TH
staining and GAD and SSR were rarely found separately. This concentration of the
pathway for GHB production in striatal neurons indicates that GHB plays a role in
dopaminergic regulation

33
Aim of Dissertation The aim of this dissertation is to investigate the effects of GBL administration on
locomotor activity, and monoamine neurotransmitter metabolism in mice. Since GBL is
rapidly metabolized in the body to GHB, these studies have relevance to GHB drug abuse
and its other potential uses as outlined in this chapter. A clear understanding of the role
that GHB plays in the CNS remains to be defined, specifically any long-term effects from
chronic use. Currently there are no known antidote treatments for GHB toxicity or
overdose, even though its use as a recreational and date rape drug continues to increase.
The specific aims of this dissertation are designed to further our understanding of GHB
and its effect in the CNS as follows:
1) To characterize the acute effects of GBL administration on locomotor function,
and to correlate behavioral and monoamine metabolite (DA & 5-HT) changes in
mice.
2) To determine the effects of dopamine elevating drugs or dopamine agonists on
GBL induced loss of locomotor function.
3) To determine the effects of long term / chronic treatment with GBL on
locomotor function, and brain monoamine neurotransmitter metabolism.

34
CHAPTER TWO
Materials and Methods
Chemicals and Solvents
Chemicals
The following chemicals were used in assays, either as reagents, or as standards.
The chemicals were purchased from Sigma (St. Louis, MO): 3,4-Dihydroxy-L-
Phenylalanine (L-DOPA), 3,4-Dihydroxyphenylacetic Acid (DOPAC), 3-
Methoxytyramine (3-MT), 3-O-Methyldopa (3-OMD), 4-Hydroxy-3-
Methoxyphenylacetic Acid (HVA), 5-Hydroxyindole-3-Acetic Acid (5-HIAA), 5-
Hydroxytryptamine (5-HT) Creatine Sulfate Complex, Ethylenediaminetetraacetic acid
(EDTA), Apomorphine Hydrochloride, Bicuculline Methiodide, Dopamine (DA),
Gamma Butyrolactone (GBL), Gamma Hydroxybutyric Acid (GHB), NCS-382,
Nomifensine Maleate, Norepinephrine (NE), Pargyline Hydrochloride, Pergolide
Mesylate, Potassium Di-Hydrogen Phosphate, Sodium Octyl Sulfate (OSS), Taurine
(TAU) and Tolcapone.
Solvents
HPLC grade methanol, 85% phosphoric acid, and 70% perchloric acid (PCA)
were purchased from Mallinckrodt Chemicals (Phillipsburg, NJ). Reagent ethanol was
purchased from Allegiance (McGraw, IL).

35
Animals
Mice
The male mice (6-8 weeks old) used were of the C57 strain (Harlan Laboratories,
Indianapolis, IN). The mice were individually housed in cages in the animal facility.
The conditions of the facility were temperature regulated at 21± 2°C, a light/dark cycle of
12 hours, and standard mouse diet and water provided ad libitum. All mice were moved
and transported by the tail. Baylor University Medical Center’s Institute of Animal Care
Committee approved all procedures utilizing animals.
Drug Injections
All compounds to be injected were prepared fresh daily. The compounds were
dissolved in phosphate buffered saline and adjusted with 85% phosphoric acid to a
physiologic pH of 7.2-7.5. Injection volumes were 500 µl unless poor solubility of the
compound required a larger volume to accommodate the appropriate dose. No injections
required greater than 800 µl for the proper dose. All injections were made using
tuberculin syringes (1mL; 25G; 5/8 in.) and were given intraperitoneally (i.p.).
HPLC Methods
Monoamine Analysis
Monoamine neurotransmitters (DA and 5-HT) and their metabolites (DOPAC,
HVA, 3-MT and 5-HIAA) were analyzed using HPLC equipped with electrochemical
detection. Compounds were eluted on a reverse-phase Gemini 5μ C18 250x3.0 mm
column (Phenomenex, Fullerton, CA, USA). The column temperature was maintained at
35°C with a flow rate of 0.35 ml/min. The mobile phase consisted of 50mM potassium
dihydrogen phosphate, 1mM octyl sodium sulfate, 54μM EDTA, and 14% methanol.

36
Deionized water was passed through a Sep-Pak C18 cartridge to remove impurities. All
other components were added and then the pH was adjusted to 2.47 with 85% phosphoric
acid. Monoamine stock standards were prepared with water and ascorbic acid (1mg/ml)
and kept at -80°C until needed. They were then diluted with 0.1M PCA to a final
concentration of 1μM. Standards and tissue extracts were injected into the HPLC system
(10μl injection volume) by means of a refrigerated autosampler kept at 4°C. Detection of
the monoamines and metabolites was performed using the Microdialysis cell 5014B and
ESA Guard cell 5020. The cell potentials were E1= +50mV, E2 = +450 mV, and the
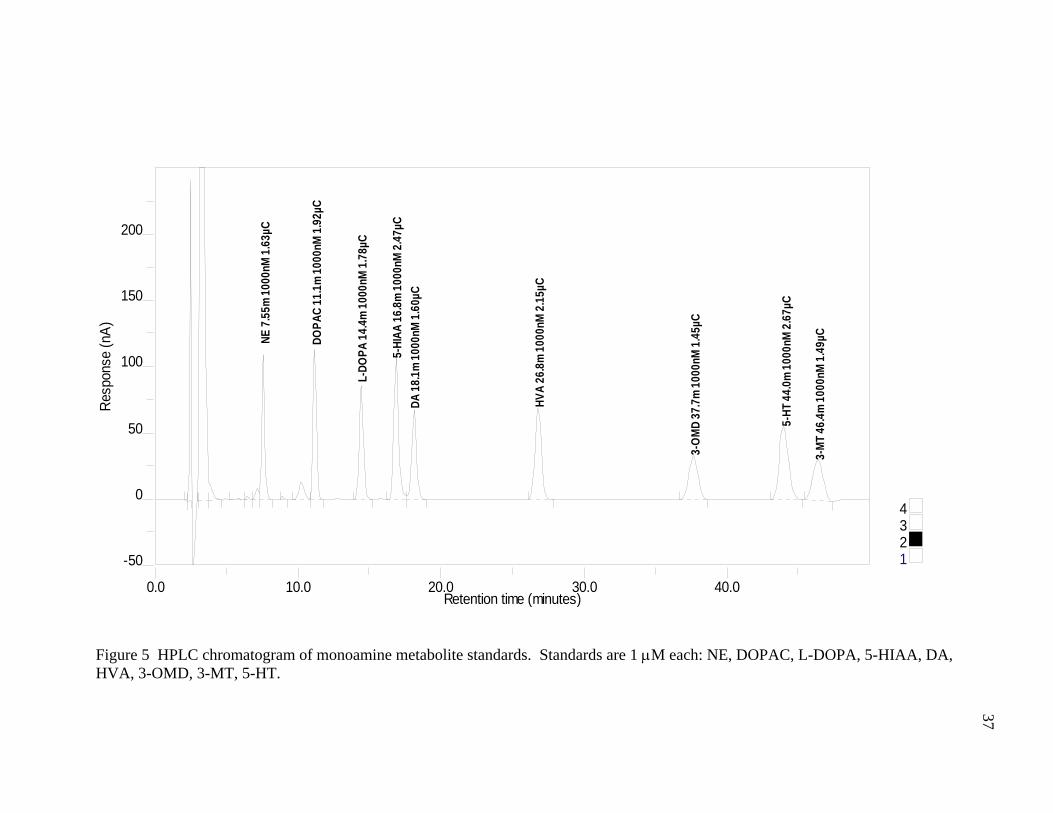
guard cell = +600mV. HPLC chromatograms of a 1μM standard and a deproteinized
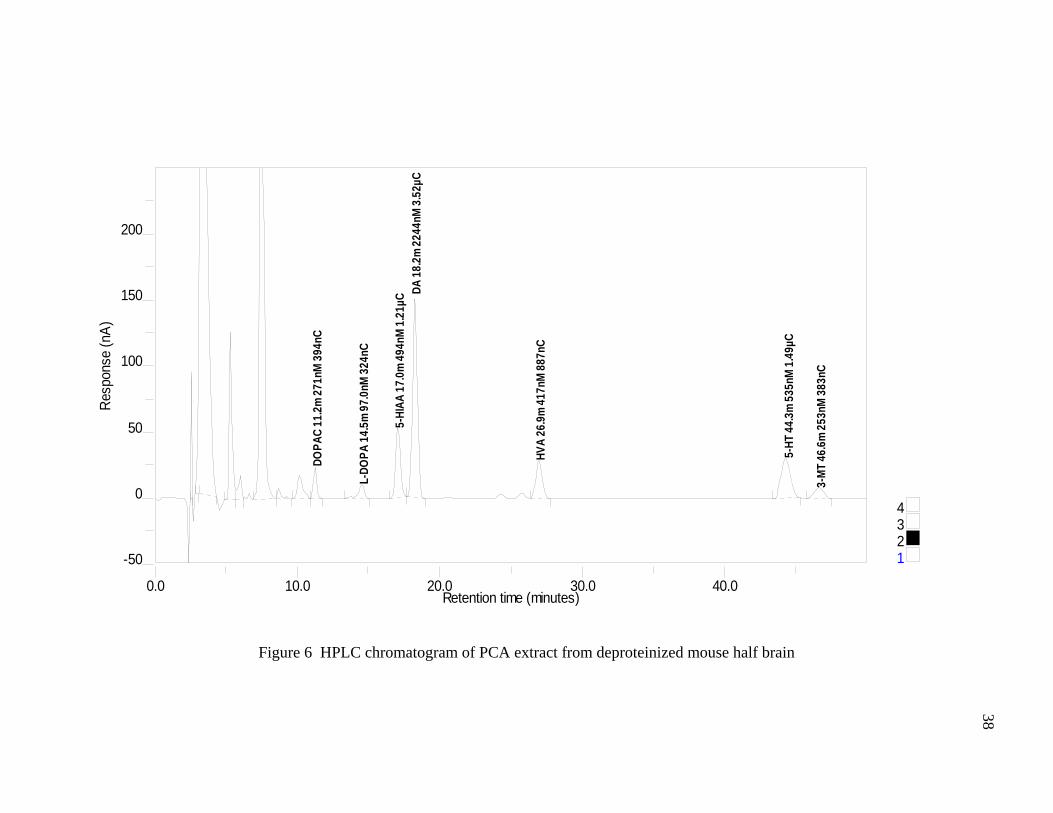
mouse half brain are shown in Figures 5 and 6 respectively.
Brain Tissue Preparation
Mice were asphyxiated by means of CO2 and sacrificed via cervical dislocation.
Brain tissue was immediately dissected and separated from the cerebellum. Brain halves
were rinsed in ice cold phosphate buffered saline (PBS), placed in eppendorf tubes, and
stored at -80°C until time of analysis. Half brains were deproteinized 1:4
(weight/volume) in ice cold 0.1M PCA using a motorized teflon pestle in an eppendorf
tube. Homogenized tissue was then centrifuged at 11,000 rpm for 15 minutes at 4°C.
Clear PCA supernatants were immediately transferred to an autosampler vial and injected
into the HPLC system. Remaining tissue extract was stored at -80°C.
0.0 10.0 20.0 30.0 40.0
-50
0
50
100
150
200
Retention time (minutes)
Resp
onse
(nA)
4321
NE 7
.55m
100
0nM
1.6
3µC
DOPA
C 11
.1m
100
0nM
1.9
2µC
L-DO
PA 1
4.4m
100
0nM
1.7
8µC
5-HI
AA 1
6.8m
100
0nM
2.4
7µC
DA 1
8.1m
100
0nM
1.6
0µC
HVA
26.8
m 1
000n
M 2
.15µ
C
3-O
MD
37.7
m 1
000n
M 1
.45µ
C
5-HT
44.
0m 1
000n
M 2
.67µ
C
3-M
T 46
.4m
100
0nM
1.4
9µC
Figure 5 HPLC chromatogram of monoamine metabolite standards. Standards are 1 μM each: NE, DOPAC, L-DOPA, 5-HIAA, DA, HVA, 3-OMD, 3-MT, 5-HT.
37
0.0 10.0 20.0 30.0 40.0
-50
0
50
100
150
200
Retention time (minutes)
Resp
onse
(nA)
4321
DOPA
C 11
.2m
271
nM 3
94nC
L-DO
PA 1
4.5m
97.
0nM
324
nC
5-HI
AA 1
7.0m
494
nM 1
.21µ
C DA 1
8.2m
224
4nM
3.5
2µC
HVA
26.9
m 4
17nM
887
nC
5-HT
44.
3m 5
35nM
1.4
9µC
3-M
T 46
.6m
253
nM 3
83nC
Figure 6 HPLC chromatogram of PCA extract from deproteinized mouse half brain 38

39
Behavioral Methods
Behavioral experiments were performed using two different testing systems. The
TruScan® and rotarod systems (Coulbourn Instruments, Allentown, PA) were used for
investigating open-field behavior and fine motor coordination respectively.
TruScan®
The TruScan® system is designed to monitor open-field behavior in rodents. The
system used for studies presented in this dissertation was configured for mice. The
TruScan® system is comprised of a clear Plexiglas cage that is 25.40 cm wide, 25.40 cm
deep, and 40.64 cm tall. There are two sensor rings placed around the cage (Figure 7).
These rings monitor the movements of the mice via infrared beams arranged in a grid
pattern. The beams are 1.52 cm apart giving a 0.76 cm resolution. One sensor ring is
placed at the base of the cage and monitors movements in the XY plane by means of
coordinate changes. These coordinate changes are a result of breaking an intersection of
two beams. The other ring is 7.62 cm from the base of the cage and monitors movements
or breaks in the vertical plane. In order to make the cage appear uniform to the test
subjects as well as blocking outside stimuli, the walls of the Plexiglas cage were
completely covered with blue fabric. The only exception to this covering was the space
for the sensor rings. There are 60 different observations that can be measured by the
floor plane and 20 observations measured in the vertical plane. Three floor plane
observations and one vertical plane observation were recorded as data for each trial. The
floor plane parameters were: Total Movement Episodes, Total Movement Time and Total
Movement Distance. The vertical plane parameter was: Vertical Plane Entries (VPE).

40
Figure 7 Picture of the TruScan®system Total movement episodes measured the total number of coordinate changes in each trial.
Total movement time is the sum of the time spent in all movements in the floor plane.
Total movement distance measured the sum of the distances of all coordinate changes in
the floor plane. Vertical plane entries are the sum of all occurrences when any part of the
mouse’s body broke the vertical plane. This action is also known as rearing.
The TruScan® was implemented in three different protocols. All protocols
involved injecting each mouse two times. In all three protocols, the mice were injected
twice with varying compounds and then their behavior was monitored for a specified
period of time during the course of each trial. These experiments explored different
compounds with the aim of antagonizing the locomotor effects of GBL by either pre-
treating the mice or giving treatment after GBL had been administered. Therefore, all
animals received two injections to either simulate pre or post-treatment conditions. The
first injection was given before the behavioral monitoring began, at time zero. Once the

41
first injection had been administered, the mouse was placed in the TruScan® chamber
and the program started. After 10 minutes, the program paused to allow for the second
injection. The mice were removed from the chamber, injected with the second
compound, and placed back into the chamber and the program restarted. Initial studies
monitored behavior for a total of 130 minutes. The 10 minutes between injections
provided an opportunity for the first compound to be absorbed and begin taking effect.
The 120 minutes monitored after the second injection measured the affects of both
compounds together. Once a working dose of GBL was determined, the studies were
shortened to 40 minutes. Again, there were 10 minutes between injections and 30
minutes monitoring both compounds together. The third protocol was the same as the
two previously described except that the total time was 70 minutes. Upon completion of
the behavioral monitoring, the mice were asphyxiated by means of CO2 and then
sacrificed via cervical dislocation. The brain tissue was then harvested as half brains
without the cerebellum and stored at -80°C until analyzed.
Rotarod
The rotarod test is specifically designed to quantitatively assess coordination and
locomotor activity in rodents. Various models or brands are commercially available but
all have the same basic design principle of a rotating drum or rod. The system used in
studies presented in this dissertation includes a rotating drum in a cage designed with a
shock floor. The cage is 17.78 cm wide, 17.78 cm long, and 30.48 cm tall with a metal
shock floor and a rotating drum that is 8.2 cm long and has a diameter of 5 cm. The
mouse cage is connected to a Habitest Linc System (Coulbourn Instruments, Allentown,
PA) that controls all variables of the cage, rotating drum, and shock floor while

42
interacting with the Graphic State 3.0 (Coulbourn Instruments, Allentown, PA) computer
software. This software collects data while the experiment is taking place.
All mice were allowed to adjust to ambient light and temperature in the testing
room for one hour prior to the trials. To begin each trial, mice were placed on the drum,
which was already rotating at 5 rpm. The speed of the drum was increased by 10
rpm/minute until reaching a top speed of 35 rpm. At 35 rpm the drum continued for an
additional two minutes making the total run time five minutes. If the mouse fell to the
floor at any point in the experiment, the shock floor administered a 0.2 mV shock for
three seconds. The level of shock (voltage) was determined previously by Ho and others
(Ho and others 1997). After three seconds, the drum stopped rotating and the trial was
finished. The three second shock time differs from that of Ho and others (Ho and others
1997) in order to allow the mouse to rotate completely while hanging onto the drum. If
the mouse was able to get back onto the drum within the three second shock, the floor
ceased shocking and the trial continued. After a total time of five minutes had elapsed,
the drum stopped and the mouse was removed from the testing chamber immediately.
All mice were given a training regimen of five trials per day for four consecutive days
with all individual trials being at least 30 minutes apart. Data collected using this
protocol was the total time of each trial and the speed of the drum when each trial
finished.
Data and Statistics
Data was collected and stored in Microsoft Excel. Statistical analysis was
performed using Graph Pad Prism 4.0 (Graph Pad Software, San Diego, CA). Numerical
data was presented as mean ± standard deviation and graphed data was presented as mean

43
± standard error of the mean unless otherwise indicated. Behavioral results were
analyzed using a Two-way ANOVA with the Bonferroni posttests. Monoamine
neurotransmitter metabolite results were analyzed using a One-way ANOVA with
Bartlett's test for equal variances and Dunnett's Multiple Comparison Test.

CHAPTER THREE
Acute Effects of GBL on Locomotor Activity in Mice
Introduction
In animals GHB induces sedation, seizures, loss of rearing ability and loss of
locomotor activity (Davies 1978; Dudek and Fanelli 1980; Kaufman and others 1990;
Snead 1990; Cook and others 2002). The loss of locomotor activity and loss of rearing
ability have been previously studied in both rats and mice (Davies 1978; Gianutsos and
Moore 1978). Despite the similarities between rats and mice, the effects of GHB and
GBL have been shown to differ between the groups. The doses that lead to sedation and
loss of locomotor activity have a tendency to be higher in rats than in mice (Cook and
others 2002; Carter and others 2005). Additionally, the doses of GHB were higher than
the doses of GBL required to produce the same type of behavioral results or loss of acute
locomotor function seen in rodents.
The aims of the experiments presented in this chapter are to study:
1. Behavioral variables of GBL administration over 120 minute time period
(Acute Study 1),
2. To characterize dose response effects of GBL in mice (Acute Study 1),
3. To obtain behavioral data that will be used for comparative purposes in future
studies with other drug treatments (Acute Study 2).
44

45
The time course of studies included varying doses of GBL in order to establish the
optimal doses for further testing. We predicted a low dose would show little to no effect
in the mice and a high dose would show complete sedation as well as awakening from the
sedation within the allotted time. The findings in Acute Study 1 (120 minutes post GBL)
assisted in the design of Acute Study 2 (30 minutes post GBL). Acute Study 2 produced
control data composed of mice given a placebo (saline), the low dose, and the high dose
of GBL. This control data then served as the benchmark in future studies of
pharmacological intervention detailed in succeeding chapters.
Mice were chosen as the rodent species for studies presented in this dissertation,
for two reasons. The first is the increased sensitivity mice have to the effects of GHB and
GBL. The second centered upon economics. Drug treatments are costly, and on a mg/kg
basis smaller amounts can be used in mice compared to rats.
Materials & Methods
In Acute Study 1, four groups of mice were monitored in the TruScan® cage for
130 minutes. The first injection was administered at time zero and the second injection
was administered at 10 minutes. That resulted in 120 minutes of exposure to GBL. The
treatments in these four groups were control (no GBL, saline placebo), 50 mg/kg, 150
mg/kg, and 200 mg/kg GBL. All solutions were prepared fresh daily and administered
via i.p. injection. The behavioral parameters monitored in the TruScan® were movement
episodes, time moved, distance moved, and vertical plane entries (VPE). After each
experiment, all animals were immediately asphyxiated by means of CO2 and sacrificed
via cervical dislocation. The brains were harvested for further monoamine
neurotransmitter analysis. Brains were collected as half brains without the cerebellum.

46
Acute Study 2 examined three groups of mice in the TruScan® cage for 40
minutes. The first injection was administered at time zero and the second injection was
administered at 10 minutes. That resulted in 30 minutes of exposure to GBL. The
treatment groups were a control group (saline), low dose (50 mg/kg GBL), and a high
dose (150 mg/kg GBL). The behavioral parameters monitored and brain tissue collection
procedures were the same as described for Acute Study 1.
Behavioral results were analyzed using a Two-way ANOVA with the Bonferroni
posttests.
Results
Acute Study 1- 120 Minutes Post GBL
The results for Acute Study 1 (120 minutes post GBL) are shown in Figures
8,9,10, and 11.
Movement episodes. The control group and the 50 mg/kg group’s movements
remained relatively constant until the 70 minute mark (See Figure 8). At that point they
both began to show a slight decrease in movement that was not significant. At 90
minutes, the 50 mg/kg group had a significant increase (p<0.01) in the number of
movements when compared to controls, but then returned to mirror the control group for
the duration of the experiment. The groups of mice that received GBL doses of 150 and
200 mg/kg GBL showed dramatic, statistically significant decreases (p<0.01, p<0.05) in
movement episodes at 20 and 15 minutes respectively when compared to controls. The
150 mg/kg group had significantly fewer (p<0.01) movement episodes between 20 and
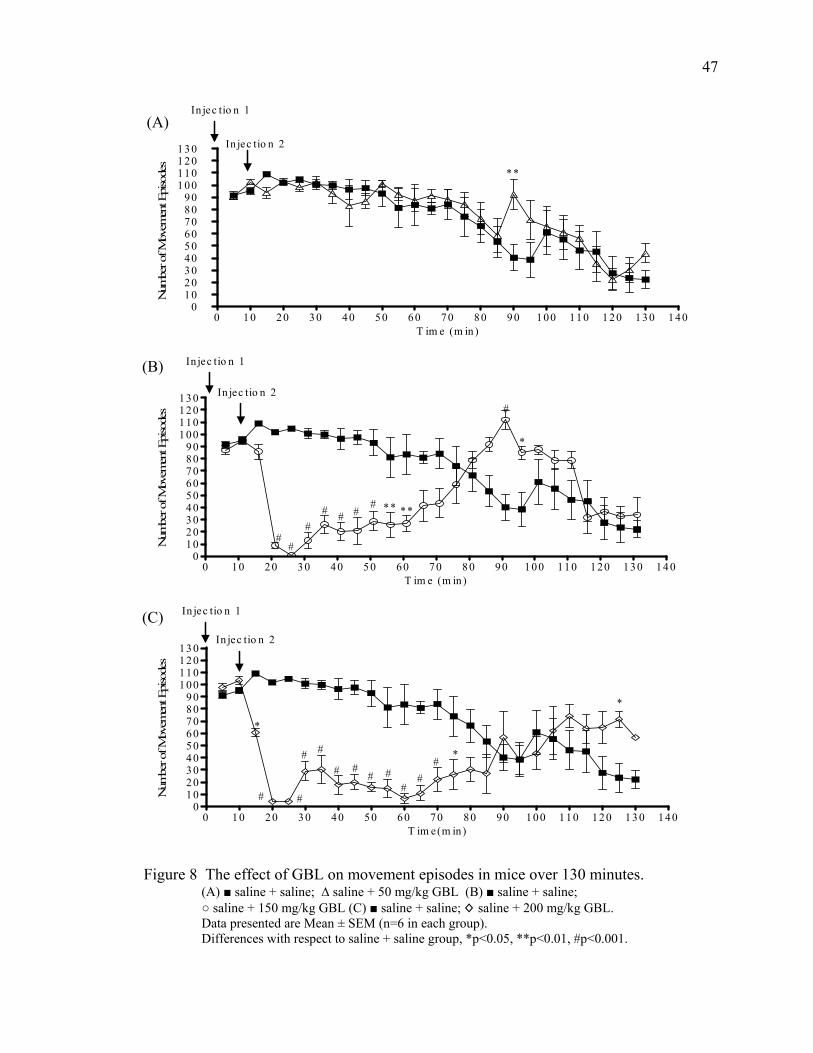
47
0 10 20 30 40 50 60 70 80 90 100 110 120 130 1400
102030405060708090
100110120130
**
In jec tio n 1
In jec tio n 2
T im e (m in )
Num
ber o
f Mov
emen
t Epi
sode
s
(A)
0 10 20 30 40 50 60 70 80 90 100 110 120 130 1400
102030405060708090
100110120130
****
*
#
### #
#
##
In jec tio n 1
In jec tio n 2
T im e (m in)
Num
ber o
f Mov
emen
t Epi
sode
s
(B)
0 10 20 30 40 50 60 70 80 90 100 110 120 130 1400
102030405060708090
100110120130
*
*
*
#
#
##
#
## # #
#
#
In jec tio n 1
In jec tio n 2
T im e(m in)
Num
ber o
f Mov
emen
t Epi
sode
s
(C)
Figure 8 The effect of GBL on movement episodes in mice over 130 minutes.
(A) ■ saline + saline; ∆ saline + 50 mg/kg GBL (B) ■ saline + saline; ○ saline + 150 mg/kg GBL (C) ■ saline + saline; ◊ saline + 200 mg/kg GBL. Data presented are Mean ± SEM (n=6 in each group). Differences with respect to saline + saline group, *p<0.05, **p<0.01, #p<0.001.

48
60 minutes when compared to controls and at 90 and 95 minutes had significantly more
(p<0.05) movement episodes than the control mice. The 200 mg/kg group, however,
moved more (p<0.05) than controls at 125 minutes. Otherwise that group had
significantly fewer (p<0.05) movements between 15 and 75 minutes when compared to
controls.
Time moved. The graphs showing the time spent moving are similar to the graphs
of movement episodes (See Figure 9). The 50 mg/kg group differed from controls only at
90 minutes when on average, they moved a greater (p<0.05) amount of time. Both the
150 and 200 mg/kg groups showed rapid decreases (p<0.001) in the amount of time
moved between 15 and 25 minutes. That decrease in the amount of time spent moving
continued until 60 and 75 minutes respectively. Between 85 and 95 minutes, the 150
mg/kg group spent a greater (p<0.05) amount of time moving than the control group.
The 200 mg/kg group did not show the same increase in activity during that time. Again,
at 90 minutes, both the 50 and 150 mg/kg groups moved more (p<0.05) than the control
group.
Distance moved. The 50 mg/kg group did not show a difference from the control
group at any point during the experiment (See Figure 10). At 20 minutes, the 150 and
200 mg/kg groups showed decreases (p<0.01) in distance moved that continued until 60
and 70 minutes respectively, with the exception of 65 minutes for the 200 mg/kg group.
At 90 minutes, the 150 mg/kg group moved a greater (p<0.01) distance than the control
group. None of the three treatment groups demonstrated any statistically reliable
differences in distance moved when compared to controls after 90 minutes.
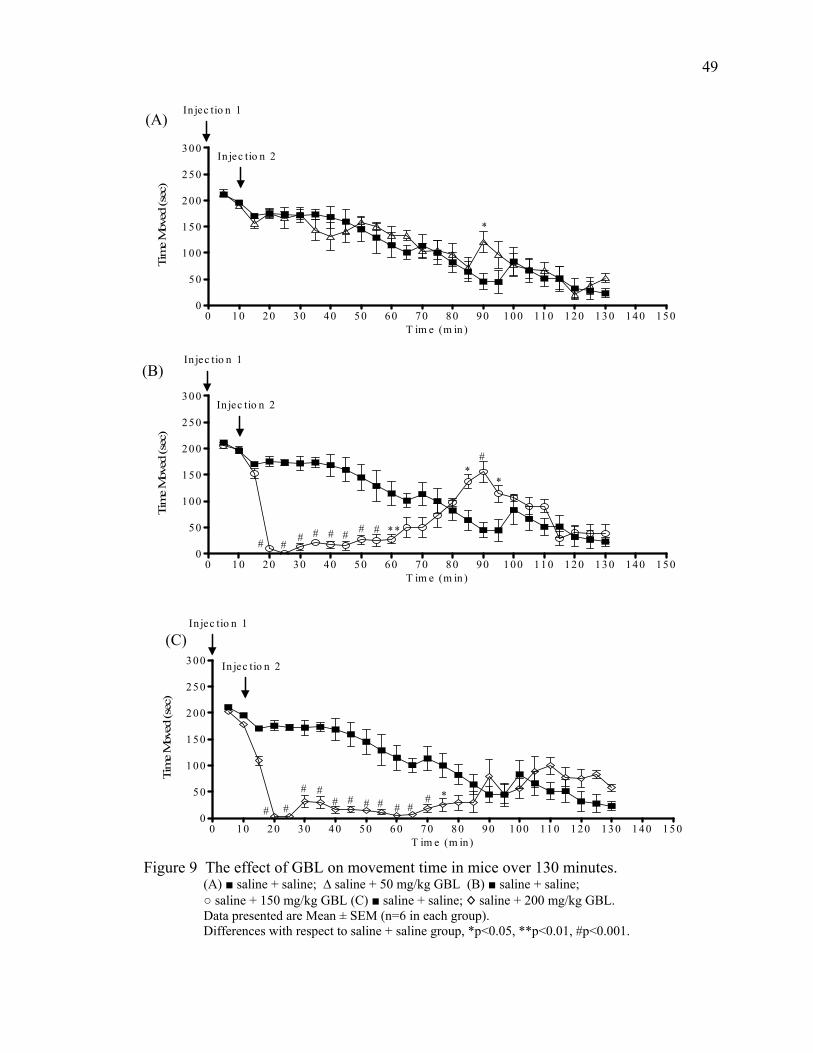
49
0 1 0 20 30 40 50 60 70 80 90 10 0 110 1 20 130 140 1500
50
100
150
200
250
300
*
In jec tio n 1
In jec tio n 2
T im e (m in )
Tim
e M
oved
(sec
)
(A)
0 1 0 20 30 40 50 60 70 80 90 100 110 12 0 130 140 1500
50
100
150
200
250
300
**
**
#
########
In jec tio n 1
In jec tio n 2
T im e (m in )
Tim
e M
oved
(sec
)
(B)
(C)
Figure 9 The effect of GBL on movement time in mice over 130 minutes. (A) ■ saline + saline; ∆ saline + 50 mg/kg GBL (B) ■ saline + saline; ○ saline + 150 mg/kg GBL (C) ■ saline + saline; ◊ saline + 200 mg/kg GBL. Data presented are Mean ± SEM (n=6 in each group). Differences with respect to saline + saline group, *p<0.05, **p<0.01, #p<0.001.
0 10 2 0 30 4 0 50 6 0 70 8 0 90 1 00 11 0 1 20 13 0 1 40 15 00
5 0
1 00
1 50
2 00
2 50
3 00
# *
In jec tio n 1
In jec tio n 2
# #
## # # # # #
#
T im e (m in)
Tim
e M
oved
(sec
)
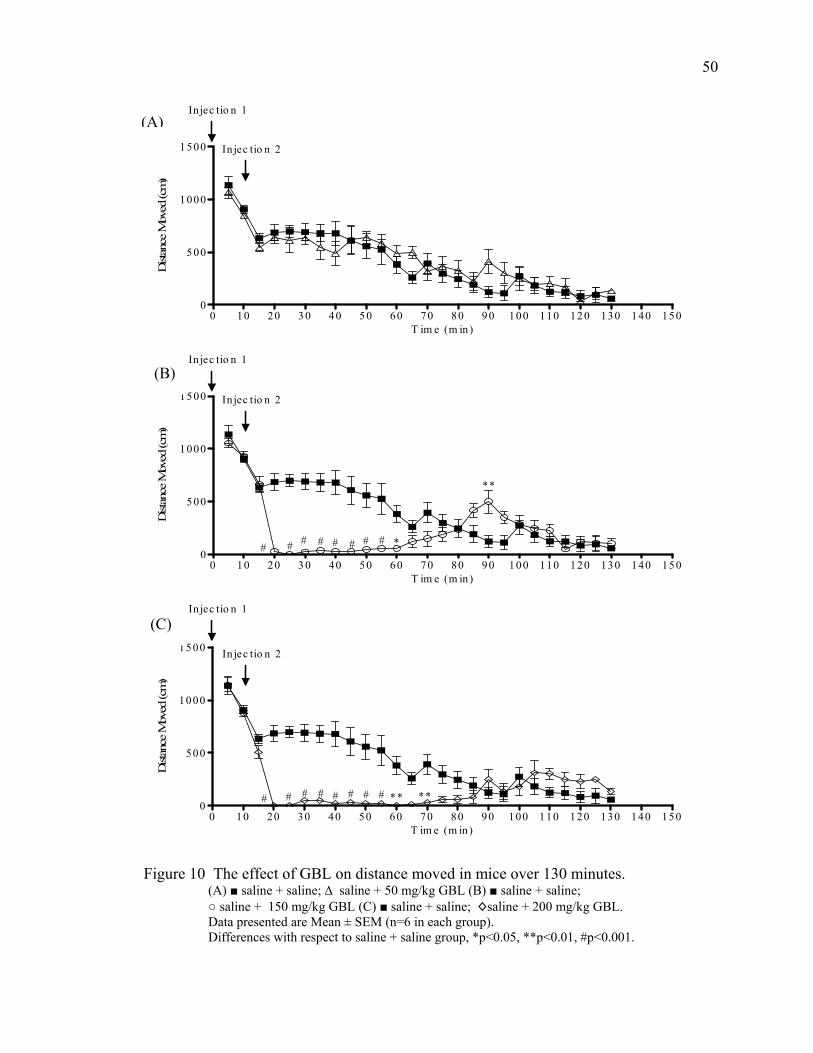
50
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 1500
500
1000
1500
In jec tio n 1
In jec tio n 2
T im e (m in )
Dist
ance
Mov
ed (c
m)
(A)
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 1500
500
1000
1500
*
**
# ## ## ###
In jec tio n 1
In jec tio n 2
T im e (m in )
Dist
ance
Mov
ed (c
m)
(B)
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 1500
500
1000
1500
** **########
In jec tio n 1
In jec tio n 2
T im e (m in)
Dist
ance
Mov
ed (c
m)
(C)
Figure 10 The effect of GBL on distance moved in mice over 130 minutes.
(A) ■ saline + saline; ∆ saline + 50 mg/kg GBL (B) ■ saline + saline; ○ saline + 150 mg/kg GBL (C) ■ saline + saline; ◊saline + 200 mg/kg GBL.
Data presented are Mean ± SEM (n=6 in each group). Differences with respect to saline + saline group, *p<0.05, **p<0.01, #p<0.001.

51
Vertical plane entries (VPE). The graphs of vertical plane entries are dissimilar to
the other graphs in this experiment (See Figure 11). All of the groups had their highest
number of VPE at 10 minutes. The 150 and 200 mg/kg groups showed no vertical plane
entries between 20 and 55 minutes, a difference which was reliably different (p<0.001)
than the control group. Due to a spike in activity by the control group, both 150 and 200
mg/kg groups had fewer VPE at 70 minutes (p<0.01 and p<0.001 respectively). This is
the only behavioral parameter that did not show the 150 mg/kg group with a reliable
difference in activity at 90 minutes compared to the control group.
Summary of Behavioral Changes.
Three of the four parameters measured show remarkably similar results. The
movement episodes, time moved, and distance moved graphs all showed the control and
50 mg/kg group with very similar time dependent profiles. Generally, activity showed a
decreasing trend throughout the experiment with both control and 50 mg/kg groups
ending with nearly the same measurements. The 150 and 200 mg/kg groups appeared to
follow the same trends in those three parameters until 40-50 minutes, at which point the
150 mg/kg group showed a trend of increasing activity, although it was not statistically
reliable. The 150 and 200 mg/kg groups did not show an increase in the number of VPE
between 30 and 40 minutes, but began their recovery at 60 minutes. The dramatic spike
in activity seen at 90 minutes in the other three parameters did not occur with the vertical
plane entries parameter.
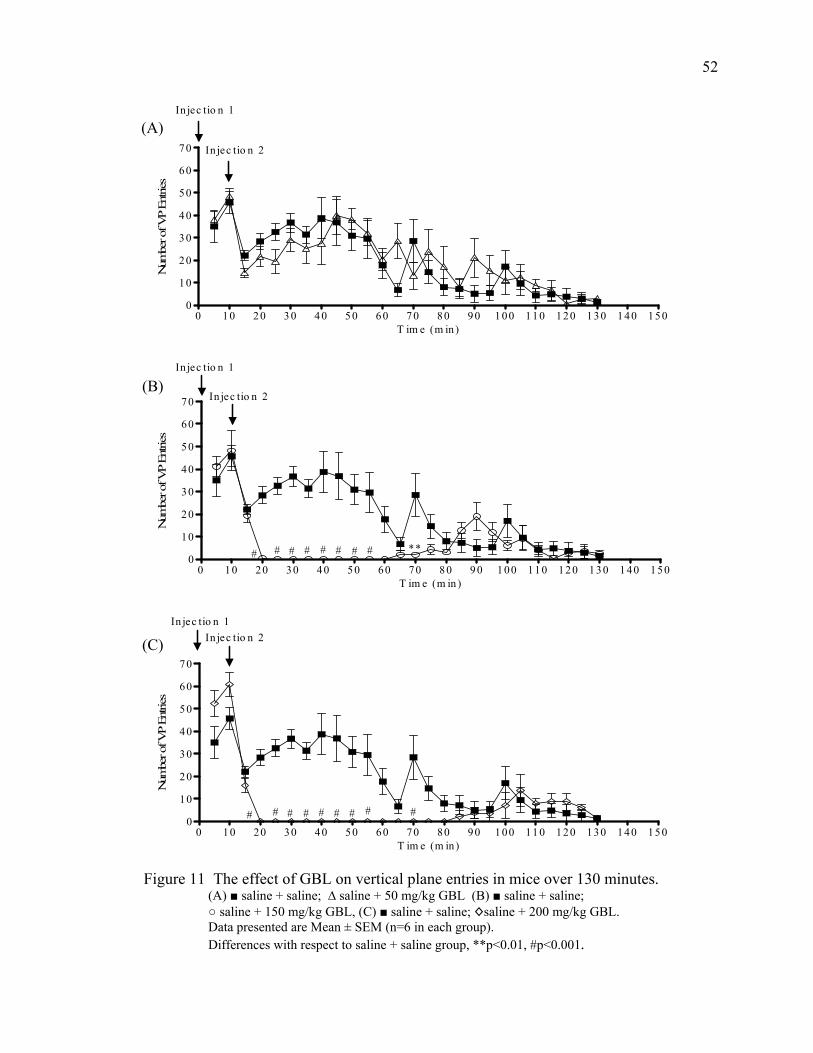
52
0 1 0 2 0 3 0 4 0 5 0 6 0 7 0 8 0 9 0 1 00 1 10 1 20 1 30 1 40 1 500
10
20
30
40
50
60
70
*
In jec tio n 1
In jec tio n 2
T im e (m in )
Num
ber o
f VP
Entri
es
(A)
0 10 20 30 40 50 60 70 8 0 9 0 1 00 1 10 1 20 1 30 1 40 1 500
10
20
30
40
50
60
70
**########
In jec tio n 1
In jec tio n 2
T im e (m in )
Num
ber o
f VP
Entri
es
(B)
0 10 20 30 40 50 60 70 80 90 1 00 11 0 1 20 130 1 40 1500
10
20
30
40
50
60
70
#########
In jec tio n 1In jec tio n 2
T im e (m in)
Num
ber o
f VP
Entri
es
(C)
Figure 11 The effect of GBL on vertical plane entries in mice over 130 minutes. (A) ■ saline + saline; ∆ saline + 50 mg/kg GBL (B) ■ saline + saline; ○ saline + 150 mg/kg GBL, (C) ■ saline + saline; ◊saline + 200 mg/kg GBL. Data presented are Mean ± SEM (n=6 in each group).
Differences with respect to saline + saline group, **p<0.01, #p<0.001.

53
Acute Study 2 – 30 Minutes Post GBL
The results for Acute Study 2 (30 minutes post GBL) are shown in Figure 12 A-
D. In this experiment, the total time was reduced to 40 minutes and included a control
group, a low dose of GBL (50 mg/kg) and a high dose of GBL (150 mg/kg).
Movement episodes. As can be seen in the movement episodes graph (See Figure
12A), the control and 50 mg/kg groups showed very similar numbers of movements. The
150 mg/kg group had fewer movements (p<0.001) than the control group beginning at 15
minutes and continuing for the duration of the experiment. Track plots of this data are
shown in Figure 13.
Time moved. The time moved graph was similar to the movement episodes graph
(See Figure 12B). The 50 mg/kg group never differed from the saline control group. The
150 mg/kg group moved a shorter (p<0.001) amount of time between 15 and 40 minutes
when compared to controls.
Distance moved. When measuring the distance moved, all three groups moved
more than 1000 cm during the first 5 minutes (See Figure 12C). Between 15 and 20
minutes, the 150 mg/kg group decreased to almost no distance moved when compared to
controls, which was a reliable difference (p<0.001). That decrease continued for the
remainder of the experiment.
Vertical plane entries (VPE). The VPE graph showed the 150 mg/kg group
beginning with more VPE than either of the other two groups (See Figure 12D). All
three treatment groups had nearly the same number of vertical plane entries at 10 minutes
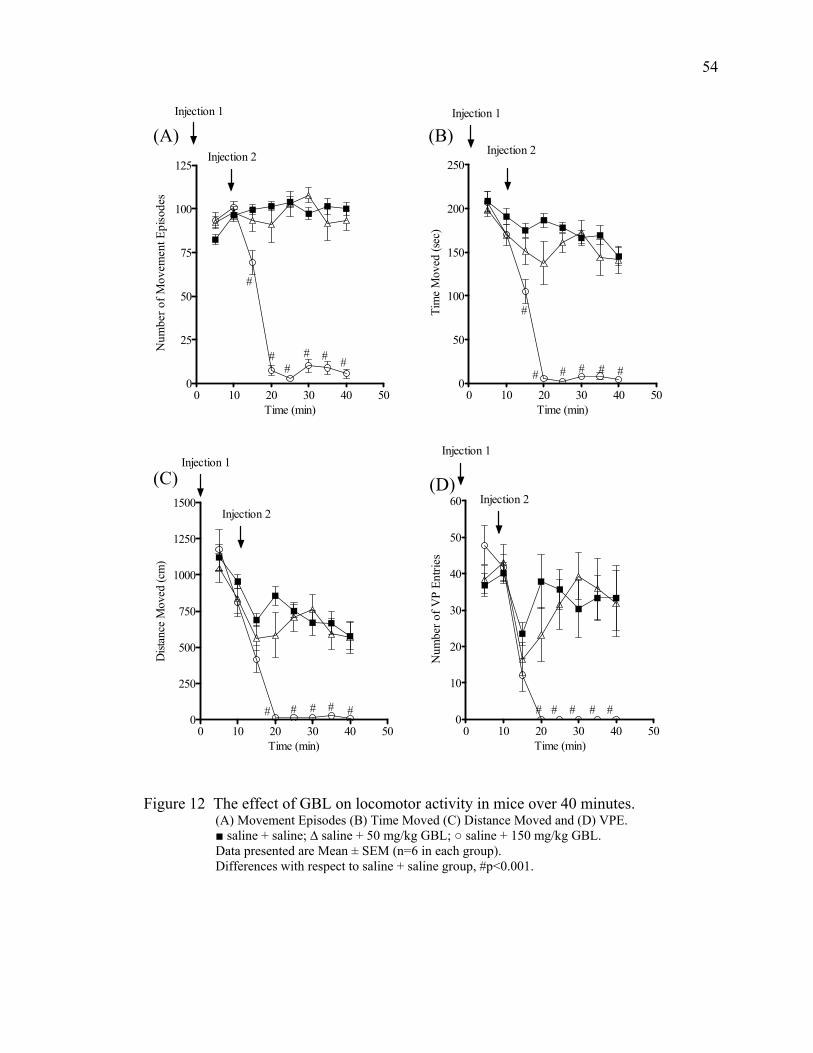
54
0 10 20 30 40 500
25
50
75
100
125
#
##
# # #
Injection 2
Injection 1
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
0 10 20 30 40 500
50
100
150
200
250
#
# # # # #
Injection 2
Injection 1
Time (min)
Tim
e M
oved
(sec
)
(A) (B)
0 10 20 30 40 500
250
500
750
1000
1250
1500
# # # # #
Injection 1
Injection 2
Time (min)
Dist
ance
Mov
ed (c
m)
0 10 20 30 40 500
10
20
30
40
50
60
# # # # #
Injection 1
Injection 2
Time (min)
Num
ber o
f VP
Entri
es
(C) (D)
Figure 12 The effect of GBL on locomotor activity in mice over 40 minutes. (A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; ∆ saline + 50 mg/kg GBL; ○ saline + 150 mg/kg GBL. Data presented are Mean ± SEM (n=6 in each group). Differences with respect to saline + saline group, #p<0.001.
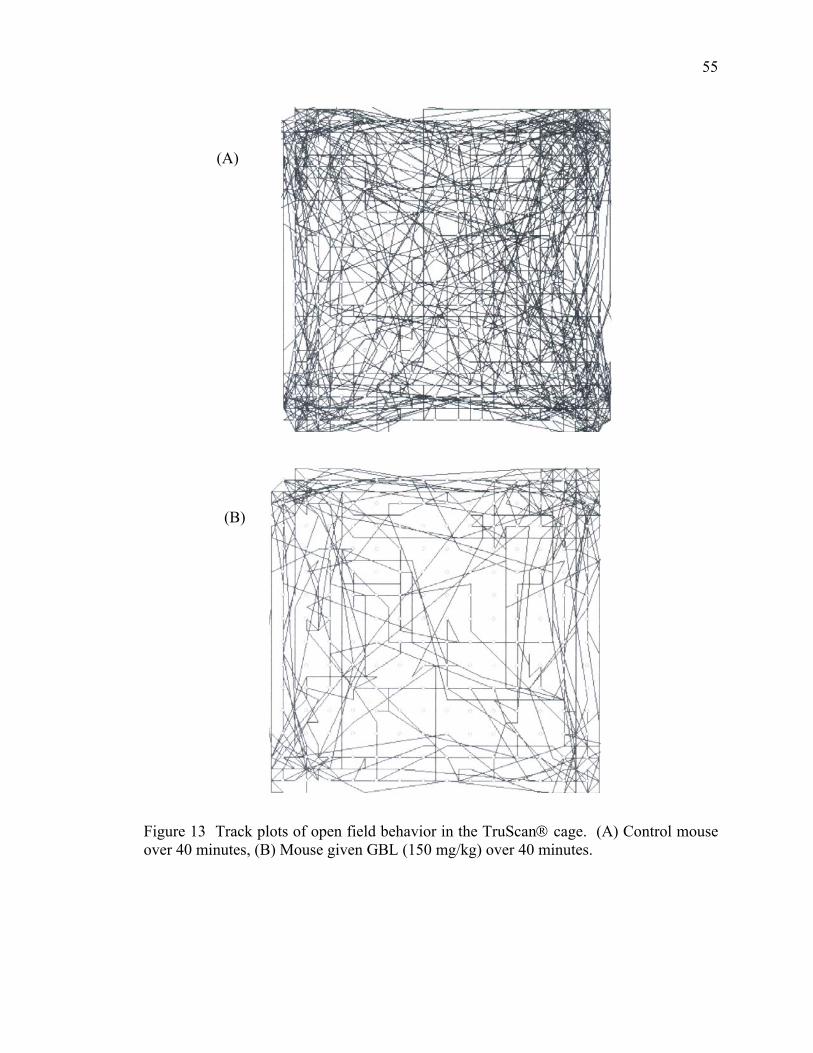
55
(A)
(B)
Figure 13 Track plots of open field behavior in the TruScan® cage. (A) Control mouse over 40 minutes, (B) Mouse given GBL (150 mg/kg) over 40 minutes.

56
and all showed a marked decrease (not statistically significant) over the following five
minutes. The 150 mg/kg group recorded no vertical plane entries after 20 minutes, which
was reliably different (p<0.001) than the control group.
Summary of Behavioral Changes.
The control group and the group of mice given 50 mg/kg GBL showed very
similar behavior in all four parameters measured. The 150 mg/kg group exhibited very
low activity after 15 minutes in all parameters and had measurements of zero or very
close to zero in the time moved, distance moved, and VPE graphs beginning at 20
minutes and lasting the duration of the experiment.
Discussion
The aims of the experiments presented in this chapter were to study:
1. Behavioral variables of GBL administration over 120 minute time period
(Acute Study 1),
2. To characterize dose response effects of GBL in mice (Acute Study 1),
3. To obtain behavioral data that will be used for comparative purposes in future
studies with other drug treatments (Acute Study 2).
Acute Study 1 (120 minutes post GBL) focused on the first two aims. The
observed changes in three of the four categories monitored by the TruScan® were very
similar (See Figures 8, 9, and 10). The control mice exhibited the expected exploratory
behavior that is typical of mice that have never been exposed to the TruScan® chamber.
During the first 15-20 minutes the mice were constantly moving around the TruScan®
and standing up on their hind legs (rearing) in order to familiarize themselves with the

57
chamber. This is an expected pattern of open-field behavior that has been demonstrated
by other investigators (Crawley 2000). It has also been noted that as the size of the open
field chamber decreases, the rate of habituation increases (Crawley 2000). One
interesting observation was that of the vertical plane entries. Between 5 and 10 minutes,
a trend of increasing VPE contrasted with a trend of decreasing activity in the time
moved and distance moved parameters. However, at 10 minutes the second injection was
given which lead to a marked, statistically non-significant decrease in VPE for all of the
groups. Generally, all four categories measured by the TruScan® showed decreases over
time as habituation occurred. The 50 mg/kg group showed very similar behavior to the
control group as demonstrated by the similarities between the graphs, indicating that this
dose of GBL had little effect on open-field behavior in mice. However, between 30 and
40 minutes, the 50 mg/kg group showed a slight but statistically non-significant decrease
in all four parameters. This trend was less pronounced in the VPE than in the other
parameters, possibly indicating that the ability of rearing was not affected to the same
degree as total activity or movement. Although there were no sedative effects observed,
it is likely that the decrease in activity between 30 and 40 minutes is due in part to the
GBL. Perhaps it is at this point that enough of the GBL had been metabolized to cause a
small locomotor effect. In all parameters, after the decrease between 30 and 40 minutes,
the mice showed an increasing trend in activity for the following 10 minutes. This could
be the extent of the effects caused by such a low dose of GBL.
Between 10 and 20 minutes, both the 150 and 200 mg/kg groups showed
dramatic, statistically significant decreases in activity that lead to measurements of
almost no movement at all. The second injection was given at 10 minutes and when

58
taken into account, indicated that the GBL took approximately 10 minutes to exert its
effects. As the mice recovered, the effects of the GBL wore off and the 150 and 200
mg/kg groups increased their movements in the TruScan® arena. All categories, except
vertical plane entries, showed this trend of increasing activity.
Vertical plane entries did not show any evidence of recovery from the effects of
GBL until 60 minutes. This indicates that the rearing ability was either affected more
than the basic ability to move in the XY plane or that this ability was slowest to recover
from the effects of GBL over time. The 150 mg/kg group, in all categories, except VPE,
showed a statistically significant spike in activity at 90 minutes, when compared to
controls, suggesting that a full recovery of locomotor function had not been reached.
The doses selected for the remaining studies were a low dose of 50 mg/kg and a
high dose of 150 mg/kg because the 150 and 200 mg/kg groups behaved in a very similar
fashion all four behavioral parameters. After 25 minutes both groups appeared to exhibit
trends of increasing activity that implied recovery from the sedation and loss of
locomotor activity. However, the 200 mg/kg group experienced another trend of
decreasing activity between 40 and 60 minutes. The continued sedation of the 200 mg/kg
group lead us to select a GBL dose of 150 mg/kg for the remaining experiments. Both
150 and 200 mg/kg groups showed sedation and loss of locomotor activity. However,
because we intended to test the ability of other compounds to antagonize the effects of
GBL, it would be advantageous to choose a dose that does not result in extended
sedation. Therefore, the 150 mg/kg group, which did not experience a second bout of
sedation, was used as the high dose.

59
The total time the remaining studies was also determined from the results of the
120 minute post GBL experiment. The first injection was a saline placebo. In the
remaining studies, the first injection was a compound or drug thought to antagonize the
effects of GBL. Therefore, the compounds in the first injection will have had 10 minutes
to act before the GBL was injected. The second injection, which was GBL, needed only
10 minutes to exert its effects on locomotor activity. It was anticipated that the recovery
from sedation and loss of locomotor activity would be present within the 10 to 20 minute
range after the second injection if a pre-treatment drug is effective. The mice injected
with 150 mg/kg GBL appeared to recover from sedation between 25 and 30 minutes and
continued to show trends of increasing locomotor activity. By 45 minutes, the mice
seemed to no longer be effected by the GBL. Thus, any antagonism of the effects of
GBL should be observed within 10 to 20 minutes and any after effects would be observed
before the mice return to pre-treatment activity levels between 30 and 40 minutes. It was
determined to be undesirable to include the peak of activity seen at 90 minutes, as the
cause could not be attributed to one factor alone. Therefore, the total time the remaining
studies was 40 minutes.
As expected, the results from Acute Study 2 (30 minutes post GBL) were very
similar to the results of the mice observed for 120 minutes post GBL. This set of
experiments served as both behavioral and monoamine metabolite controls to which
remaining pharmacological interventions were compared. The control mice in this study
had about the same number of movements in each five minute block and actually
appeared to move more in the last five minutes than they did during the first five minutes.
In the graphs of time moved and distance moved, the control group showed a general

60
trend of decreasing activity in both parameters throughout the experiment. As previously
mentioned, this was most likely due to habituation to the test chamber over time and
therefore a progressive disinterest in further exploration. Between 10 and 15 minutes, the
control group showed a steep drop (not statistically significant) in the number of vertical
plane entries. This was seen in the 120 minute post GBL trials at the same time,
immediately after the second injection was given. It is possible that after being removed
from the chamber to receive the second injection and then being returned (this was only
accomplished by lifting the mice out of the top of the chamber), the mice experienced
some fear about being captured again and remained as low in the chamber as possible.
After five minutes, the number of vertical plane entries returned to levels just below those
seen previously.
The 50 mg/kg mice again showed behavior similar to that of the control mice.
The distance moved and number of vertical plane entries both decreased in a statistically
reliable fashion between 10 and 20 minutes, immediately after the second injection was
given. It is not likely that this decrease in activity was due to GBL, but more likely a
fearful response to being removed from the chamber and then returned to it. Overall, the
50 mg/kg group showed habituation to the testing chamber, decreased movement over
time, and few side effects of the GBL. Between 20 and 30 minutes, the 50 mg/kg mice
did show trends of increasing activity in all four parameters.
The 150 mg/kg mice initially showed exploratory behavior much the same as the
other two groups. After the second injection at 10 minutes, there was a statistically
significant decrease in all four categories. All of the graphs showed little or no
movement between 20 and 40 minutes. This indicates that the GBL was responsible for

61
the sedative effects and loss of locomotor activity. An observation of note was the VPE
parameter. Beginning at 20 minutes, there were no vertical plane entries for the duration
of the experiment. This again leads to speculation about whether GBL affected the
ability of rearing more than other aspects of movement or if that ability took more time to
recover than the other categories. As this particular parameter of open field behavior did
not show recovery in the 40 minute time limit of the experiment, VPE had the potential to
be a valuable marker of GBL antagonism in the remaining studies.
The data presented in this chapter is consistent with results seen in other studies.
A study comparing the effects of GBL and 1,4-BD on locomotor activity in Swiss-
Webster mice showed that GBL doses of 100 and 150 mg/kg lead to statistically
significant decreases in movement episodes between 10 and 40 minutes and 10 and 50
minutes respectively (de Fiebre and others 2004). Those time courses of decreased
activity were consistent with the statistically reliable decreases in activity induced by 150
and 200 mg/kg GBL reported in this chapter. An experiment by Davies (Davies 1978)
showed that GBL doses of 100 and 200 mg/kg induced remarkable decreases in
locomotor activity. However, in that particular study there was a period of hyperactivity
reported after the period of decreased activity. The studies described here showed similar
decreases in locomotor activity, but no period of hyperactivity was observed. Cook and
others (Cook and others 2002) tested the effects of GHB in male Swiss-Webster mice and
male Long Evans rats in the Functional Observational Battery (FOB). The mice showed
statistically reliable increases in the time required to complete the inverted screen test, a
the distance observed in the hind limb splay test, and a statistically significant decrease in
the forelimb grip strength test (Cook and others 2002). However, there was a dose

62
dependent, significant decrease in the number of rears in both the home cage and open
field when GHB was administered. This is in agreement with the significant decreases
observed in VPE in this study.
Previous work in this lab has also shown that GHB causes marked decreases in
locomotor activity after a single injection of GHB (500 mg/kg) (Bottiglieri and others
2001; Anderson and others 2002). A study by Schmidt-Mutter (Schmidt-Mutter and
others 1998) assessed spontaneous locomotor activity in male Long-Evans rats after
administration of GHB. Doses of GHB that were 150 mg/kg or less showed no decreases
in locomotor activity when compared to animals that were not given GHB. Doses of 300
and 600 mg/kg did show decreases in activity leading to the conclusion that sedation had
also occurred. Zerbib and others (Zerbib and others 1992) showed that the locomotor
activity of mice was markedly decreased 30 minutes after administration when GHB (250
mg/kg) was administered as either an acute or chronic injection.
There are several studies demonstrating a faster onset of action and more potent
effects of GBL when compared to GHB at equal doses (mg/kg). Studies by Carter and
others (Carter and others 2005) showed the order of effectiveness in inhibiting locomotor
activity in mice to be GBL > 1,4-BD > GHB. In that study, reliable decreases in
locomotor activity were observed when doses of GBL were greater than 100 mg/kg. That
corresponds to the inhibition of locomotor activity seen in this study for GBL doses of
150 and 200 mg/kg. Few studies have examined the discriminative stimulus effects
between GHB and GBL. Male Sasco Sprague Dawley rats were trained to discriminate
GHB (300 mg/kg IG 30 min) and GBL (150 mg/kg i.p. 15 min) (Baker and others 2005).
Responses on lever pressing were used to indicate successful discrimination. The

63
animals trained to discriminate GBL met the criteria with fewer sessions than those
trained to discriminate GHB. This supports the theory that GBL is more potent than
GHB and acts more quickly.
In these experiments, we did not use GHB in comparison with GBL.
Unfortunately, when these experiments were conducted, GHB was unavailable. Future
studies should include both GHB and GBL in order to more clearly understand the
molecular mechanisms taking place when each drug is administered. However, our use
of GBL strengthened the data base regarding the respective doses and time course over
which GBL exerts its effects.
It is becoming clear that GHB and GBL differ in these properties. Studies to be
presented in future chapters will show differences in the abilities of certain compounds to
antagonize the effects of GBL. That aspect of GBL has not been investigated as
thoroughly as GHB and therefore it deserves attention.

CHAPTER FOUR
Acute Effects of GBL on Brain Neurotransmitter Metabolism in Mice
Introduction
The role of GHB in brain neurotransmitter metabolism is still not completely
understood. Previous studies have shown variable monoamine neurotransmitter
responses to GHB or precursors such as GBL (Feigenbaum and Howard 1996a). Most
investigators have directed their attention to the effect of GHB on dopamine synthesis
and release, with studies showing conflicting results (Feigenbaum and Howard 1996a).
Numerous studies have investigated GHB or GBL’s role in dopamine release. Many
studies have reported a decrease of impulse flow in mesolimbic and nigrostriatal
dopaminergic neurons for 60 minutes following GHB administration (Aghajanian and
Roth 1970; Roth and Suhr 1970; Roth and others 1973; Stock and others 1973; Walters
and Roth 1974; Roth 1976; Hechler and others 1991). Studies of GHB in rats have
shown a decrease in extracellular 3-MT levels, which indicates inhibition of dopamine
release (Bottiglieri and others 2001; Anderson and others 2002). In in vivo microdialysis
experiments, Hechler and others (Hechler and others 1991) showed a biphasic affect of
GHB on the release of dopamine. In that study, GHB decreased dopamine release in the
striatum when low doses were administered (300 mg/kg or less), and increased dopamine
release when high doses were administered (>300 mg/kg). The increase of dopamine
release in many microdialysis experiments was shown by Feigenbaum and Howard
(Feigenbaum and Howard 1996a) to be the result of high concentrations of calcium in the
64

65
perfusion fluid or the use of an anesthetic that alters the release or regulation of
dopamine.
The effects of GHB or GBL on serotonin have also been investigated, with reports
of conflicting results. Microdialysis experiments performed in rats have shown a
statistically significant decrease in extracellular 5-HT and 5-HIAA in the cortex and
striatum following GHB (4 mmol/kg i.p.) administration (Gobaille and others 2002).
Further studies of the same GHB dose in tissue slices showed an increase in 5-HIAA in
frontal cortex (39%), striatum (57%), hippocampus (53%), and Raphe nuclei (60%).
Serotonin levels were unaffected in those same brain regions. The authors suggested that
the increase in 5-HIAA and stable 5-HT levels indicate increased 5-HT turnover induced
by GHB. In a separate study, Speciale and Friedman (Speciale and Friedman 1975)
reported increases of 5-HT in the midbrain and brainstem of rats after GBL
administration (350 mg/kg i.p.). To further complicate matters, some studies have shown
increases in serotonin synthesis and degradation induced by GHB, while the specific
levels of 5-HT and 5-HIAA were unchanged (Spano and Przegalinsky 1973; Hedner and
Lundborg 1983). Serotonin synthesis in those studies was determined by accumulation
of its precursor 5-HTP after decarboxylase inhibition by NSD 1015. The results of
studies reported in this chapter will help to clarify the role of GHB or GBL in the
dopaminergic and serotonergic systems, by defining the effect of GBL on brain
monoamine neurotransmitter metabolism. Data presented was obtained from a drug
administration paradigm as described in Chapter Three.

66
Materials & Methods
There were two sets of brain samples analyzed in this study. The first set was
comprised of 24 half brains obtained from mice monitored in Acute Study 1 (120 minutes
post GBL). The second set was 18 half brains from the mice monitored in Acute Study 2
(30 minutes post GBL). A single half brain (without cerebellum) for each mouse was
analyzed for monoamine neurotransmitters DA and 5-HT and metabolites DOPAC,
HVA, 3-MT, and 5-HIAA. The protocol for monoamine analysis was described
previously in Chapter Two. Monoamine metabolite results were analyzed using a One-
way ANOVA with Bartlett's test for equal variances and Dunnett's Multiple Comparison
Test.
Results
Acute Study 1 - 120 minutes post GBL
Dopamine metabolites. The dopamine metabolites and turnover are shown in
Table 3. There were no statistically reliable changes seen in DA, DOPAC, or 3-MT
levels for any of the three treatment groups when compared to control mice. Among the
metabolites analyzed, only HVA showed elevated levels in this study. Both the 150 and
200 mg/kg groups had more (p<0.05 and p<0.01 respectively) HVA than the control
group while the 50 mg/kg group showed no change. Dopamine turnover was assessed by
using the following calculation: DOPAC + HVA / DA. None of the three GBL groups
had different rates of dopamine turnover.
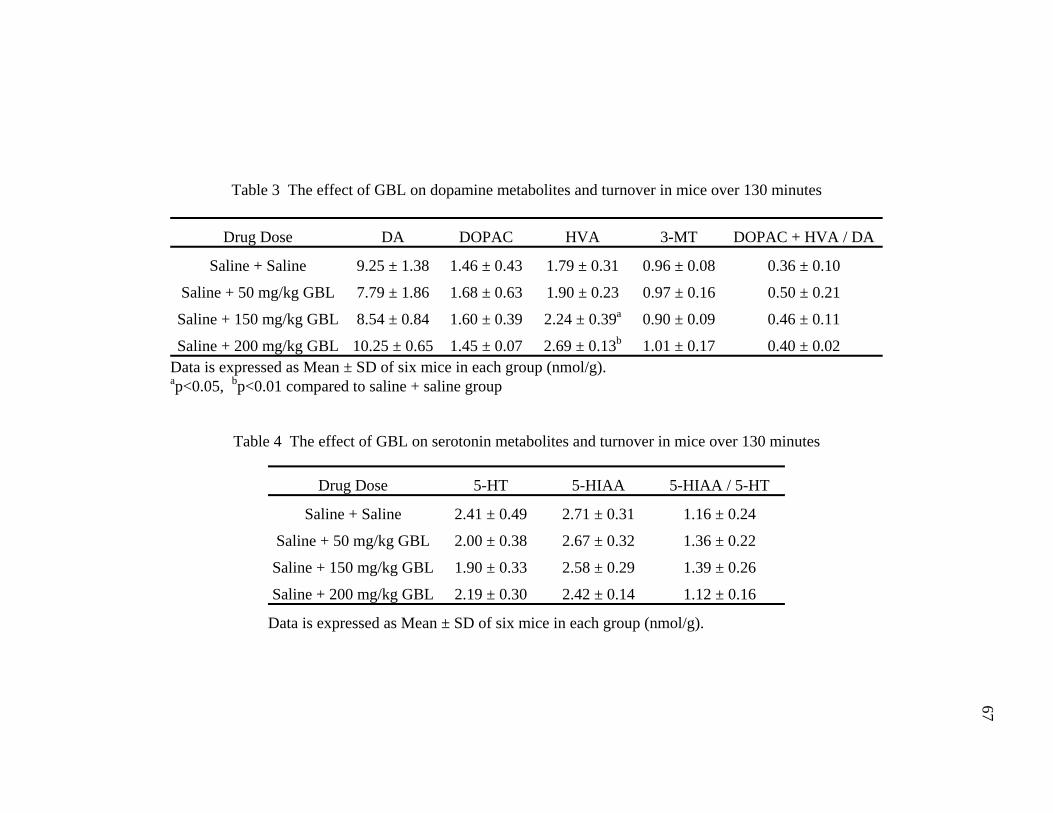
67
Table 3 The effect of GBL on dopamine metabolites and turnover in mice over 130 minutes
Drug Dose DA DOPAC HVA 3-MT DOPAC + HVA / DA
Saline + Saline 9.25 ± 1.38 1.46 ± 0.43 1.79 ± 0.31 0.96 ± 0.08 0.36 ± 0.10
Saline + 50 mg/kg GBL 7.79 ± 1.86 1.68 ± 0.63 1.90 ± 0.23 0.97 ± 0.16 0.50 ± 0.21
Saline + 150 mg/kg GBL 8.54 ± 0.84 1.60 ± 0.39 2.24 ± 0.39a 0.90 ± 0.09 0.46 ± 0.11
Saline + 200 mg/kg GBL 10.25 ± 0.65 1.45 ± 0.07 2.69 ± 0.13b 1.01 ± 0.17 0.40 ± 0.02 Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.05, bp<0.01 compared to saline + saline group
Table 4 The effect of GBL on serotonin metabolites and turnover in mice over 130 minutes
Drug Dose 5-HT 5-HIAA 5-HIAA / 5-HT
Saline + Saline 2.41 ± 0.49 2.71 ± 0.31 1.16 ± 0.24
Saline + 50 mg/kg GBL 2.00 ± 0.38 2.67 ± 0.32 1.36 ± 0.22
Saline + 150 mg/kg GBL 1.90 ± 0.33 2.58 ± 0.29 1.39 ± 0.26
Saline + 200 mg/kg GBL 2.19 ± 0.30 2.42 ± 0.14 1.12 ± 0.16
Data is expressed as Mean ± SD of six mice in each group (nmol/g).

68
Serotonin metabolites. The serotonin metabolites and turnover are shown in
Table 4. None of the three GBL treatment groups had statistically different levels of
either 5-HT or 5-HIAA. The rate of serotonin turnover was calculated using the formula
5-HIAA / 5-HT. There were no statistically reliable changes in serotonin turnover.
Summary of Monoamine Metabolite Changes.
The monoamine neurotransmitter metabolites showed fluctuating trends between
all three GBL treatment groups in this study. Of the metabolites examined, only the
HVA levels changed reliably. The 150 and 200 mg/kg groups both had more HVA
(p<0.05 and p<0.01 respectively )than the control group. Neither the dopamine or
serotonin turnover rates changed significantly for any treatment group.
Acute Study 2 - 30 minutes post GBL
Dopamine metabolites. The dopamine metabolites and turnover are shown in
Table 5. The group of mice given 50 mg/kg GBL showed no reliable changes in
dopamine metabolites or turnover. The group of mice given 150 mg/kg GBL had greater
(p<0.05) levels of dopamine and less HVA (p<0.01), 3-MT (p<0.01) and a lower rate of
dopamine turnover (p<0.01) when compared to control mice. DOPAC levels did not
show any reliable changes from controls.
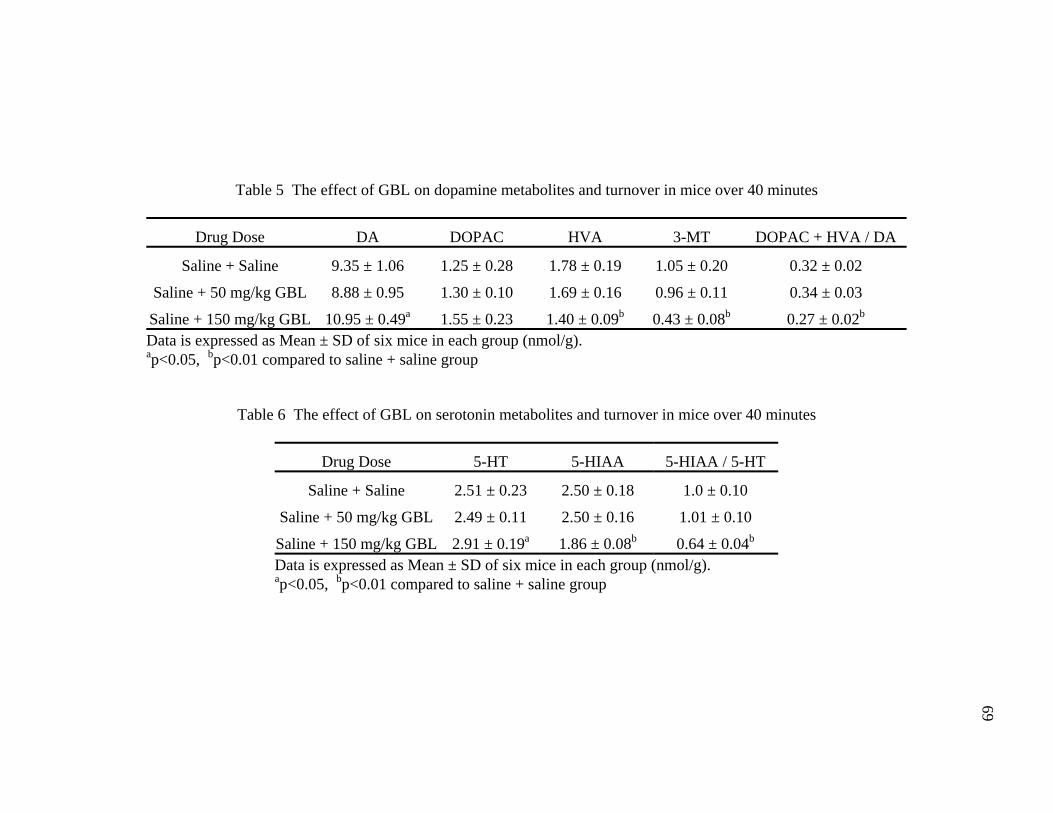
69
Table 5 The effect of GBL on dopamine metabolites and turnover in mice over 40 minutes
Drug Dose DA DOPAC HVA 3-MT DOPAC + HVA / DA
Saline + Saline 9.35 ± 1.06 1.25 ± 0.28 1.78 ± 0.19 1.05 ± 0.20 0.32 ± 0.02
Saline + 50 mg/kg GBL 8.88 ± 0.95 1.30 ± 0.10 1.69 ± 0.16 0.96 ± 0.11 0.34 ± 0.03
Saline + 150 mg/kg GBL 10.95 ± 0.49a 1.55 ± 0.23 1.40 ± 0.09b 0.43 ± 0.08b 0.27 ± 0.02b
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.05, bp<0.01 compared to saline + saline group
Table 6 The effect of GBL on serotonin metabolites and turnover in mice over 40 minutes
Drug Dose 5-HT 5-HIAA 5-HIAA / 5-HT
Saline + Saline 2.51 ± 0.23 2.50 ± 0.18 1.0 ± 0.10
Saline + 50 mg/kg GBL 2.49 ± 0.11 2.50 ± 0.16 1.01 ± 0.10
Saline + 150 mg/kg GBL 2.91 ± 0.19a 1.86 ± 0.08b 0.64 ± 0.04b
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.05, bp<0.01 compared to saline + saline group

70
Serotonin metabolites. The serotonin metabolites and turnover are shown in
Table 6. The 150 mg/kg group showed an increase in 5-HT (p<0.05) and a decrease in 5-
HIAA (p<0.01) and serotonin turnover (p<0.01) when compared to control mice.
Summary of Monoamine Metabolite Changes.
In Acute Study 2 (30 minutes post GBL) there were no reliable changes in
metabolite levels for the group of mice given 50 mg/kg GBL when compared to controls.
However, with the exception of DOPAC, all metabolite levels from the group of mice
given 150 mg/kg GBL were different (p<0.05) from control levels.
Discussion
In the two studies described here, the effects of GBL on monoamine
neurotransmitters were monitored over two different time courses of 120 and 30 minutes.
The mice given 50 mg/kg GBL had no statistically significant monoamine metabolite
changes in either the 120 or 30 minute post GBL studies. This suggested that a low dose
of GBL did not significantly alter the metabolism of either dopamine or serotonin when
administered in a single dose. When paired with the behavioral observations from
Chapter Three it was clear that a single dose of 50 mg/kg GBL did not affect behavior or
monoamine neurotransmitter metabolism.
The only reliable changes observed for the GBL doses of 150 and 200 mg/kg in
the 120 minute post GBL study were increased levels of HVA. This suggested the end of
a dopaminergic rebound or “catch up” process. As the inhibition of dopamine release
ended, it was followed by a rapid release of the intracellular dopamine stores (Cheramy
1977). Changes in all metabolite levels may have occurred because of that burst release
of dopamine and then returned to normal levels over time. HVA is one of the last

71
metabolites to be formed and thus would be the last to return to basal levels. That return
to basal levels had not yet occurred as evidenced by the increase in HVA levels at 120
minutes post GBL.
In contrast to the dose of 50 mg/kg, the dose of 150 mg/kg GBL did affect the
metabolism of both dopamine and serotonin over the course of 30 minutes. The
increased dopamine (Handforth and Sourkes 1975; Booth and others 1994), decreased 3-
MT (Bottiglieri and others 2001; Anderson and others 2002), and decreased dopamine
turnover rates observed for the 30 minute post GBL study were indicative of a decrease
in dopamine release and subsequent intraneuronal accumulation (Gessa and others 1968).
Decreased synaptic dopamine lead to decreased dopamine metabolism and metabolites.
That was supported by the significant decrease of HVA levels in the mice given 150
mg/kg GBL in the 30 minute post GBL study. As a metabolite of dopamine, HVA
cannot be formed if there is no dopamine in the synaptic cleft. Thus, a reduction of
postsynaptic dopamine would result in decreased levels of HVA as well. In theory,
DOPAC levels should have decreased for the same reason HVA levels decreased.
However, DOPAC is formed intraneuronally. Inhibition of dopamine release leads to
intraneuronal dopamine accumulation, which can be metabolized into DOPAC. Perhaps
the speed at which DOPAC is converted into HVA is slow, or this metabolic activity has
been inhibited as a secondary effect of decreased dopamine release. The decreased
turnover rate is explained by the increase in dopamine and the decrease in HVA.
The decrease in dopamine release induced by GBL explains the loss of locomotor
activity described in Chapter Three. This is supported by earlier work showing that
administration of GHB inhibits locomotor activity in rats (Bottiglieri and others 2001;

72
Anderson and others 2002). In both studies, the mice given 150 mg/kg GBL experienced
sedation and significant loss of locomotor function for 30 minutes post GBL
administration. Recovery occurred within the following 30 minutes. Evidence of this
recovery was shown in Chapter Three (see Figure 3). No recovery was observed in the
30 minute post GBL experiment. Thus, recovery from the sedative and behavioral effects
of GBL occurred between 30 and 60 minutes. The monoamine metabolite changes seen
30 minutes post GBL must also have recovered along with the behavioral deficits in the
following 30 minutes. That recovery lead to the lack of metabolite changes seen in the
120 minute post GBL experiment. The inhibition of dopamine release caused by GBL
must also have stopped between 30 and 120 minutes. As shown in this chapter (See
Table 3), only the HVA levels for the mice given 150 mg/kg GBL in the 120 minutes
post GBL study were significantly greater than controls. Over the 90 minutes that
separated the two studies, the release in dopamine returned to normal levels, leading to
the recovery of locomotor activity. After 120 minutes had passed, GBL’s affects on the
monoamine metabolites returned to normal levels. The increased dopamine turnover rate
in all three GBL treatment groups after 120 minutes could have indicated the end of a
dopaminergic “catch up” process due to an earlier inhibition of release. A possible
explanation for the significant increase in HVA levels was a rebound effect. As the GBL
was metabolized, dopamine release returned to normal levels and the dopamine that
accumulated intraneuronally was released very quickly as a sort of rebound response. A
rapid release of dopamine after inhibited release has been suggested previously (Chearmy
1977; Hechler and others 1991). The metabolism of dopamine then had to “catch up” as
well, leading to an increase in metabolite levels. The results described here indicate that

73
HVA was still present at high levels as a result of this recovery process. It seems that 3-
MT levels should also have been elevated as a result of this process. Dopamine
metabolism has two pathways, both of which end with HVA. However, 3-MT is formed
in only one of those pathways. When dopamine is metabolized directly by COMT, 3-MT
is formed. 3-MT can then be further metabolized into 3-methoxyphenylacetaldehyde,
which is then converted into HVA. If MAO first metabolizes dopamine, 3,4-
dihydroxyphenylacetaldehyde is formed and then metabolized further into DOPAC, and
finally HVA. Both of these pathways result in HVA while 3-MT is only one step in one
of the pathways. Therefore, any increase in 3-MT levels would have been observed
earlier in the 120 minute post GBL study and after 120 minutes would have been
metabolized into HVA.
The effects of GBL on the serotonergic system are not completely clear. In the
120 minute post GBL study, there were no reliable changes in 5-HT, 5-HIAA, or
serotonin turnover. There were, however, changes in the 30 minute post GBL study. The
group of mice given 150 mg/kg GBL had higher levels of 5-HT, lower levels of 5-HIAA,
and a lower rate of serotonin turnover. Those effects are similar to those seen in the
dopamine metabolites in that they were acute changes that recovered between 30 and 120
minutes. In contrast to the present findings, decreases in serotonin levels due to GHB or
GBL have been observed previously in microdialysis studies (Gobaille and others 2002).
The same studies did show increased 5-HIAA levels after GHB administration, which is
in agreement with the results described in this chapter. The authors also observed an
increase in 5-HIAA levels, but not serotonin levels, in tissue slices of frontal cortex,
striatum, hippocampus, and Raphe nuclei. Those results indicated an increase in

74
serotonin turnover, which is contrary to our findings of decreased serotonin turnover.
Other studies have shown increases in 5-HT and 5-HIAA in the striatum after GHB
administration (Waldmeier and Fehr 1978; Hedner and Kundborg 1983).
It should be mentioned that the monoamine metabolite results presented here were
obtained using half brains. In contrast, much of the previous work regarding the effects
of GHB or GBL on brain monoamines has been performed in microdialysis experiments
or utilized regional dissection (Cheramy and others 1977; Hechler and others 1985,
1991). The primary reason for choosing half brains was to eliminate any variation in
dissection errors when dealing with the small size of 6-8 week old mouse brains.
Preliminary studies in which the striatum was dissected from other regions produced a
large variation in DA and metabolites, either because the striatum was not completely
removed or additional adjacent tissue was also dissected along with it
The inhibition of dopamine release could have neurochemical effects other than
those discussed here. Decreasing the amount of dopamine release could lead to changes
in various receptor systems. Presynaptic receptors might down regulate in order to
inhibit reuptake and maximize the dopamine in the synaptic cleft. At the same time,
postsynaptic receptors might increase in sensitivity in order to maximize the amount of
dopamine that is used postsynaptically. This could also lead to more postsynaptic
receptors present in the synapse as a response to not enough dopamine being
metabolized. These changes in the regulation of dopamine receptors are unlikely to occur
in the time courses of our experiments thus far. Previous work has shown that GBL is
converted to GHB very quickly after administration via blood born lactonases (Roth and
Giarman 1965; Fishbein and Bessman 1966; Roth and others 1966; Lettieri and Fung

75
1978) and that the conversion’s half life has been estimated at less than one minute (Roth
and Giarman 1966; Roth and others 1966). Therefore, it is unlikely that significant
changes in receptor systems would occur after a one-time administration of GBL.
Chronic administration could produce such changes and should be investigated as GHB
has recently been approved as a treatment for cataplexy associated with narcolepsy
(Anonymous 2002a). Considering the decreases in dopamine and serotonin turnover, it is
also likely that chronic GBL administration would lead to changes in the regulation of
both systems. Such changes could include decreased sensitivity to elevated levels of
dopamine and serotonin. There could also be decreases in dopamine and serotonin
receptors as a response to higher levels of the monoamine neurotransmitters. Chronic
administration of GBL would make it possible to monitor such changes and should be
performed to gain a further understanding of the effects of long term exposure to GBL.
In response to the results seen in Chapters Three and Four, it was important to
look for a way to antagonize the behavioral effects of GBL. Based on previous reports of
GHB’s interactions with GABA, dopamine, serotonin, and other systems, the ability of a
compound to antagonize the effects of GBL was most likely dependent on its ability to
increase central dopaminergic activity. Such compounds include dopamine agonists,
dopamine reuptake inhibitors, GABA receptor antagonists, COMT inhibitors, and MAO
inhibitors. The results of testing those compounds will be presented in the next chapter.

CHAPTER FIVE
Pre-Treatment Effect of GABAA and GHB Receptor Antagonists and Compounds Affecting Dopamine Metabolism on GBL Induced Loss of Locomotor Activity in Mice
Introduction
As demonstrated in Chapter Three, GBL (150 mg/kg) causes a complete loss of
locomotor activity in mice, with maximal sedation occurring 10 minutes after
administration and recovery occurring 30 minutes later. In addition to the loss of
locomotor activity, there were several significant changes related to monoamine
neurotransmitter metabolites and turnover rates of both dopamine and serotonin. Several
investigators have attempted to antagonize these effects of GHB or GBL using
pharmacological agents as pre-treatments. These compounds were often chosen based on
their ability to antagonize GABAB receptors (Matthiessen and Wright 1869; Maitre and
others 1990; Schmidt and others 1991; Bottiglieri and others 2001; Blandini and others
2003) or act as dopamine agonists (Matthiessen and Wright 1869; Anderson and others
2002). Based on the findings presented in Chapters Three and Four, we chose to focus on
compounds that would increase central dopaminergic activity such as: receptor agonists,
re-uptake inhibitors, MAO inhibitors, and COMT inhibitors. While central dopaminergic
activity was the main focus of these studies, the role of GHB acting at GABA receptors
was also considered in the investigation. Studies have shown that GHB is not only
derived from GABA, but also can bind both to GABAA and GABAB receptors (Hosli and
others 1983; Bourguignon and others 1988; Zvosec and others 2001). Work in this
laboratory has previously studied the effect of GABA receptor antagonists in protecting B
76

77
against GHB toxicity (Bottiglieri and others 2001). Therefore, GABA antagonists were
also tested for their ability to antagonize both the behavioral and monoamine metabolism
effects of GBL. All of the compounds that were tested are shown in Table 7.
Table 7 Classification of compounds tested for GBL antagonism
Drug Mechanism of Action
Pergolide D1/D2 Dopamine Receptor Agonist NCS-382 GHB Receptor Antagonist Pargyline Monoamine Oxidase Inhibitor Nomifensine Monoamine Reuptake Inhibitor
Catechol-O-Methyl Transferase Inhibitor Tolcapone Apomorphine D1/D2 Dopamine Receptor Agonist Taurine Amino Acid Bicuculline GABA-A Receptor Antagonist
Pergolide
There have been conflicting reports regarding pergolide’s affinity for the various
dopamine receptors. Traditionally, pergolide is known as a D /D1 2 dopamine receptor
agonist (Fuller and Clemens 1991), but has also shown affinity for the D3 receptor
(Sokoloff and others 1992). Pergolide typically exerts greater effects at the pre and
postsynaptic D receptors as opposed to D2 1 receptors (Fuller and others 1979; Clemens
and Phebus 1988; Clow and others 1992). Levadopa (L-dopa) is the primary treatment
for patients with Parkinson’s disease (PD). After a few years of levadopa use, the
frequency of dyskinesias and other motor related effects in patients tends to increase
(Fahn 1992). Pergolide’s primary pharmacological use has been as an adjunct treatment
for Parkinson’s disease (Clow and others 1992). This allows physicians to decrease the
dose of L-dopa, which helps decrease the dyskinesias and motor fluctuations (Tanner and

78
others 1982; Ilson and others 1983; Klawans and others 1983; Gonce and Delwaide
1985). The Weaver Mutant Mouse is characterized by ataxia, fine tremor, cerebellum
atrophy, and up to a 50% decrease in dopamine concentrations. When given pergolide
(1mg/kg), the locomotor activity of the Weaver Mouse increased considerably compared
to control mice (Schmidt and others 1982). These examples lead us to believe that
pergolide would be successful in antagonizing the loss of locomotor activity induced by
GBL.
NCS-382
6,7,8,9-tetrahydro-5-[H]benzocyclo-hepten-5-ol-4-ylideneacetic acid (NCS-382)
is a structural analogue of GHB and GHB receptor antagonist (Waszkielewicz and
Bojarski 2004) that has been shown to compete in vitro with both the high and low
affinity GHB binding sites (Maitre and others 1990). NCS-382 has been reported to
antagonize many neuropharmacological effects of GHB induced seizures (Maitre and
others 1990). When tested in vivo, NCS-382 antagonized petit mal seizures and sedative
effects induced by GHB (Schmidt and others 1991). In behavioral studies, NCS-382 (30
mg/kg) did not block inhibitory effects of GHB on rearing in the functional observational
battery (FOB), but did antagonize the decrease in total distance traveled in the open field
(Cook and others 2002).
Pargyline
Pargyline is a monoamine oxidase inhibitor (MAOI). While there are two types
of monoamine oxidase enzymes, MAOA and MAOB, pargyline is a non-specific MAOI
(Cumming and others 1992; Tuomainen and others 1996). Dopamine is catabolized by
two pathways: either MAO or COMT, giving rise to DOPAC and 3-MT. In rat striatum,

79
approximately 70-90% of intraneuronal DA turnover is through MAO and 10-30% of
extracellular DA turnover is through COMT (Westerink 1984; Wood and others 1987).
In our lab, rats were pretreated with pargyline, which prevented the GHB induced loss of
locomotor activity and the decrease in 3-MT levels (Anderson and others 2002).
Nomifensine
Nomifensine is a monoamine reuptake inhibitor (Hansard and others 2002) and
has caused some improvement when used in the treatment of motor symptoms in
Parkinson’s disease patients (Teychenne and others 1976; Bedard and others 1977; Park
and others 1977, 1981; Delwaide and others 1983; Goetz and others 1984). In one study,
nomifensine (7 mg/kg i.p.) was administered as a pre-treatment to electrical stimulation
in the brain. The effects on dopamine transients were then measured via microdialysis.
The result was a 2.5 fold increase of [DA]max in the nucleus accumbens and the olfactory
tubercle (Robinson and Wightman 2004). In separate studies, adult male marmosets were
treated with N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) to induce PD like
motor deficits (Hansard and others 2002). When treated with nomifensine (25 mg/kg),
there was a significant increase in locomotor activity as well as improvement in head
checking movements, alertness, balance, coordination, and posture. Capillary
electrophoresis combined with laser induced florescence detection (CE-LIFD) was used
to study the effects of nomifensine and apomorphine on extracellular dopamine,
glutamate and aspartate in the striatum (Bert and others 2002). Administration of 20 μM
nomifensine resulted in a 3100% increase of striatal dopamine (Bert and others 2002).

80
Tolcapone
Tolcapone is an extracellular catechol-O-methyl transferase (COMT) inhibitor. It
is able to cross the blood brain barrier, so may act centrally as well as a peripherally
(Blandini and others 2003). Tolcapone is able to increase extracellular dopamine levels
by preventing the conversion to 3-MT via COMT (Mannisto and others 1992). By
inhibiting both central and peripheral COMT, tolcapone causes L-dopa to accumulate,
which in turn would increase dopamine synthesis and possibly release as well (Zurcher
and others 1990a, 1990b, 1993). Tunbridge and others tested tolcapone on male Sprague
Dawley rats using extra-dimensional (ED) set shifting, a component of the Wisconsin
card sorting task (WCST). Various studies cited by the authors reference the prefrontal
cortex (PFC) as the driving source of ED shifting in primates, humans, and rats. It is also
known that ED shifting is mediated by catecholamines (Downes and others 1989; Roberts
and others 1994; Middleton and others 1999; Rogers and others 1999; Crofts and others
2001; Ragozzino 2002). The rats in that study showed significant improvement in the
ED shifting after being given tolcapone (30 mg/kg). In a retrospective study of tolcapone
in patients with PD, Balndini and others (Blandini and others 2003) showed that after two
weeks and again after three months, daily administration of tolcapone (300 mg) plus
standard L-dopa treatment increased the amount of L-dopa and dopamine in both plasma
and platelets.
Apomorphine
/D Apomorphine is a D1 2 dopamine receptor agonist, was derived from morphine
over 135 years ago (Matthiessen and Wright 1869, and was suggested as a treatment for
PD in 1884(Chen and Obering 2005). It was approved by the FDA for subcutaneous

81
administration in 2004 (Chen and Obering 2005) making it the first injectable dopamine
agonist in the U.S. Apomorphine has been shown to prevent protein oxidation in the
brain as well as in the mitochondria (Sokoloff and others 1990). It is also able to inhibit
both MAOA and MAOB (Youdim and others 1999). In vivo it protects against
neurodegeneration in the striatum caused by MPTP, an agent commonly used to induce
PD like motor effects. In a study using male Wistar rats, apomorphine (0.5 mg/kg)
induced an increase in a conditioned learning response (CAR) test (Reis and others
2004). Apomorphine also has been shown to cause disruptions in sensorimotor gating
and significant disruption of prepulse inhibition (PPI) in male Swiss mice (Malone and
others 2004).
Taurine
Taurine is an amino acid found in plasma at 50 μmol/l and in millimolar
concentrations in heart, liver, kidney, retina, and neural tissue of animals (Nandhini and
others 2003). Taurine is involved in several functions such as membrane stabilization,
regulation of intracellular calcium concentrations, cytoprotection, neuromodulation, and
osmoregulation (Huxtable 1992; Sturman 1993; Timbrell and others 1995). It has also
been shown to interact with GABAA and GABAB receptors in varying sections of the
brain (Saransaari and Oja 2000). SSADH deficiency knock out mice die of fatal seizures
between 16 and 22 days of life. This corresponds to the weaning period, thus implying a
possible role of mother’s milk (Hogema and others 2001). Taurine is found in extremely
high concentrations in mother’s milk (Sarwar and others 1998), so might offer a
protective role from high levels of GHB. SSADH knock out mice given daily doses of
250 mg/kg had a survival rate of 55.6%. Among the four doses tested, 250 mg/kg had the

82
best results. Doses higher than 1000 mg/kg/day led to lower survival rates than controls
indicating a toxic effect (Gupta and others 2002).
Bicuculline
Bicuculline is a GABAA antagonist. It is often used to induce seizures in rodents
when testing anti-seizure medication (Rejdak and others 2000; Snead and others 2000).
In a study involving brain slices obtained from female Wistar rats, the addition of
bicuculline to the superfusion fluid increased dopamine outflow to 120% of basal levels
(Li and others 2005).
Materials & Methods
In these drug treatment studies, mice were monitored in the TruScan® cage for 40
minutes. The treatments in these groups were the compounds pergolide, NCS-382,
pargyline, nomifensine, tolcapone, apomorphine, taurine, and bicuculline. The first
injections were the compounds being tested and they were administered at time zero.
The second injections were either saline or GBL (150 mg/kg) and were administered at
10 minutes. That resulted in 10 minutes of exposure to the compounds being tested and
30 minutes of exposure to GBL. All solutions were prepared fresh daily and were
administered via i.p. injection. The behavioral parameters monitored in the TruScan®
were movement episodes, time moved, distance moved, and vertical plane entries. After
each experiment, all animals were immediately asphyxiated by means of CO2 and
sacrificed via cervical dislocation. The brains were harvested for further monoamine
neurotransmitter analysis. Brains were collected as half brains without the cerebellum.

83
The monoamine metabolite analysis was identical to that previously described in
Chapter Four. A single half brain (without cerebellum) for each mouse was analyzed for
the monoamine neurotransmitters DA and 5-HT and metabolites DOPAC, HVA, 3-MT,
and 5-HIAA.
Behavioral results were analyzed using a Two-way ANOVA with the Bonferroni
posttests. Monoamine metabolite results were analyzed using a One-way ANOVA with
Bartlett's test for equal variances and Dunnett's Multiple Comparison Test.
Behavioral Results
Pergolide Treatment
The behavioral data for the pergolide pre-treatment is shown in Figure 14 A-D.
Movement episodes. Beginning at 15 minutes and continuing for the remainder of
the experiment, the mice that were given pergolide (10 mg/kg) before GBL (150 mg/kg)
showed an increase (p<0.01) in movement episodes when compared to the mice that were
given only GBL (150 mg/kg) (See Figure 14A). That same group of mice also had
movement episodes that were equal to the number of movement episodes observed in the
saline + saline control group by the end of the study.
Time moved. The graph of time moved (See Figure 14B), shows that the mice
that were given pergolide (10 mg/kg) before GBL (150 mg/kg) spent less (p<0.05) time
moving during the first five minutes than the mice given saline + GBL (150 mg/kg).
Between 20 and 40 minutes, the group of mice given pergolide (10 mg/kg) + GBL (150
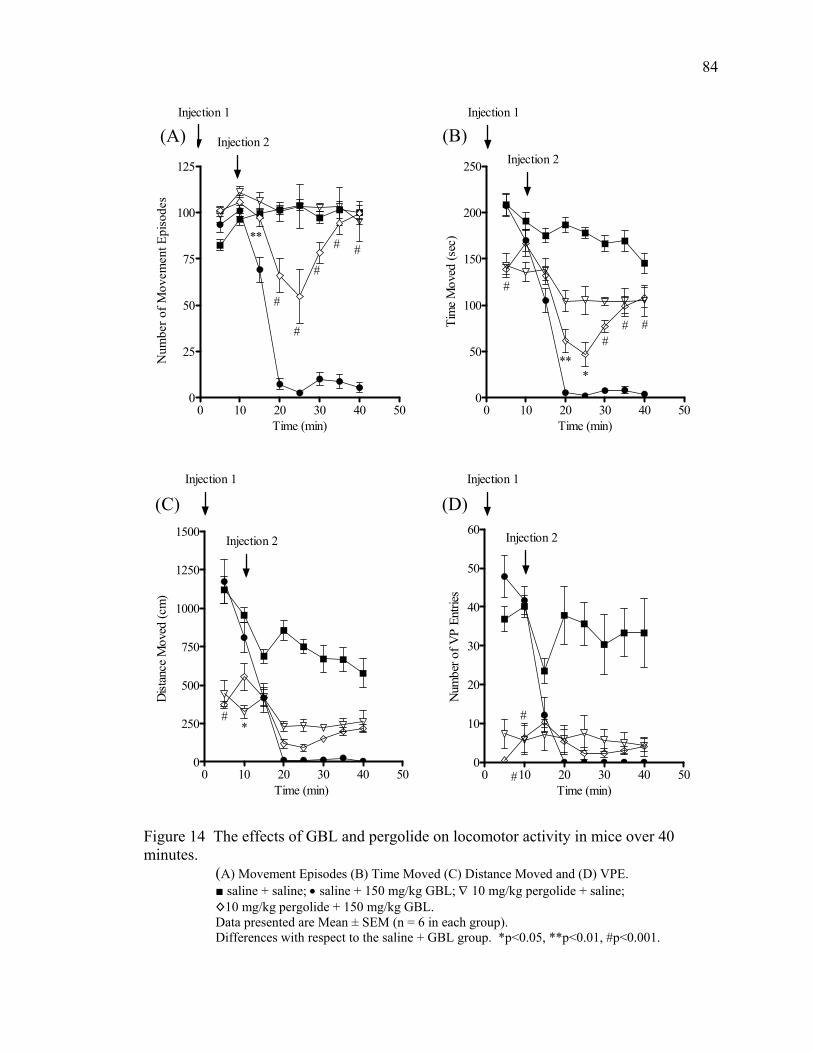
84
0 10 20 30 40 500
50
100
150
200
250
#
###
***
Injection 1
Injection 2
Time (min)
Tim
e M
oved
(sec
)
0 10 20 30 40 500
25
50
75
100
125
#
**
#
#
# #
Injection 1
Injection 2
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
(B) (A)
0 10 20 30 40 500
10
20
30
40
50
60
#
#
Injection 2
Injection 1
Time (min)
Num
ber o
f VP
Entri
es
0 10 20 30 40 500
250
500
750
1000
1250
1500
#*
Injection 2
Injection 1
Time (min)
Dist
ance
Mov
ed (c
m)
(C) (D)
Figure 14 The effects of GBL and pergolide on locomotor activity in mice over 40 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; • saline + 150 mg/kg GBL; ∇ 10 mg/kg pergolide + saline; ◊10 mg/kg pergolide + 150 mg/kg GBL. Data presented are Mean ± SEM (n = 6 in each group). Differences with respect to the saline + GBL group. *p<0.05, **p<0.01, #p<0.001.

85
mg/kg) spent more (p<0.05) time moving than the mice given saline + GBL (150 mg/kg).
Both groups of mice given pergolide (10 mg/kg) moved less time (p<0.05) than the mice
given saline + saline (data not shown in graph) for the entire 40 minutes.
Distance moved. The mice given pergolide (10 mg/kg) + GBL (150 mg/kg) did
not move farther than the mice given saline + GBL (150 mg/kg) at any time during the
experiment (See Figure 14C). Both groups of mice that received pergolide (10 mg/kg)
moved a shorter distance than the mice given saline + saline for the first 10 minutes
(p<0.001) and between 20 and 40 minutes (p<0.05) (data not shown in graph).
Vertical plane entries (VPE). The group of mice given pergolide (10 mg/kg) +
GBL (150 mg/kg) had fewer (p<0.01) VPE between 5 and 10 minutes than the group of
mice given saline + GBL (150 mg/kg), but never differed reliably for the remainder of the
experiment (See Figure 14D). Both groups of mice that received pergolide (10 mg/kg)
had fewer VPE (p<0.05) for the entire 40 minutes than the mice that received saline +
saline (data not shown in graph).
NCS-382 Treatment
The behavioral data for the NCS-382 pre-treatment is shown in Figure 15 A-D.
NCS-382 showed little to no effect in any of the four parameters studied. The movement
episodes, time moved, and distance moved graphs showed the group of mice given NCS-
382 (150 mg/kg) + GBL (150 mg/kg) with almost identical activity to the mice given
saline + GBL (150 mg/kg).
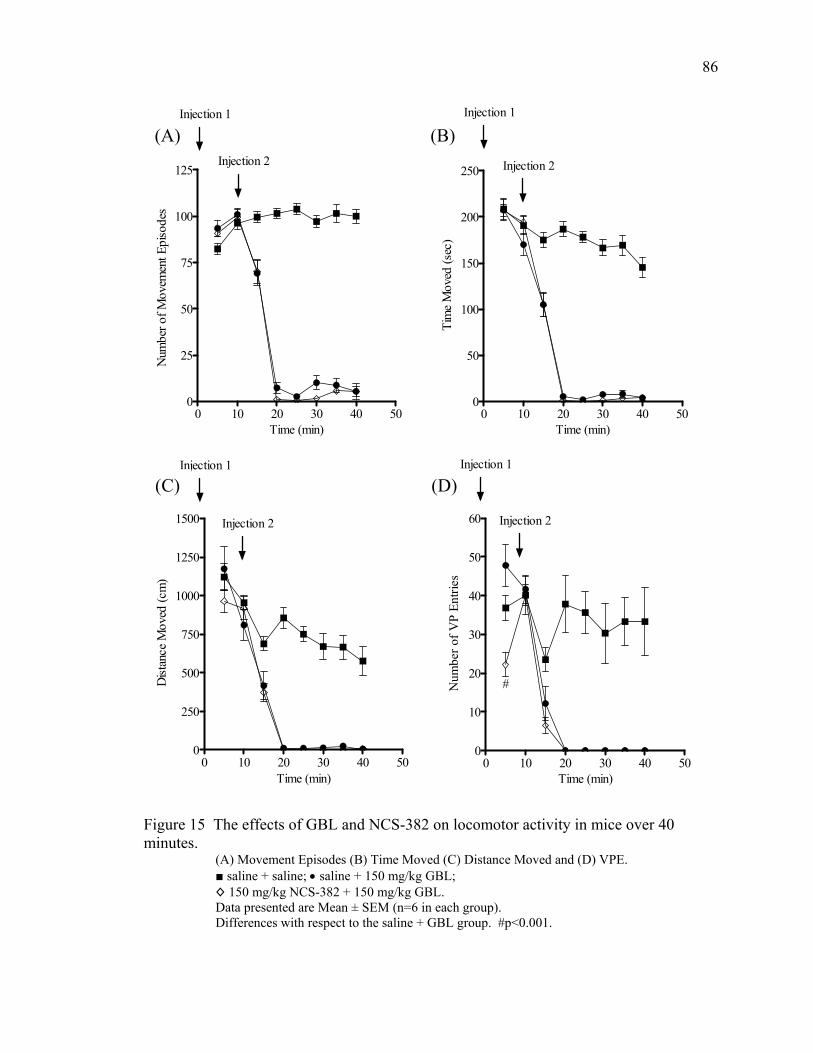
86
0 10 20 30 40 500
50
100
150
200
250
Injection 1
Injection 2
Time (min)Ti
me
Mov
ed (s
ec)
0 10 20 30 40 500
25
50
75
100
125
Injection 1
Injection 2
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
(A) (B)
0 10 20 30 40 500
10
20
30
40
50
60
#
Injection 1
Injection 2
Time (min)
Num
ber o
f VP
Entri
es
0 10 20 30 40 500
250
500
750
1000
1250
1500
Injection 1
Injection 2
Time (min)
Dist
ance
Mov
ed (c
m)
(C) (D)
Figure 15 The effects of GBL and NCS-382 on locomotor activity in mice over 40 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; • saline + 150 mg/kg GBL; ◊ 150 mg/kg NCS-382 + 150 mg/kg GBL. Data presented are Mean ± SEM (n=6 in each group). Differences with respect to the saline + GBL group. #p<0.001.

87
Between 5 and 10 minutes of the experiment, the mice given NCS-382 (150
mg/kg) + GBL (150 mg/kg) had fewer (p<0.001) VPE (See Figure 15D) than the mice
given saline + GBL (150 mg/kg). This statistically significant difference occurred before
GBL had been administered.
Pargyline Treatment
The behavioral data for the pargyline pre-treatment is shown in Figure 16 A-D.
In all four parameters, the mice given pargyline (100 mg/kg) prior to GBL (150 mg/kg)
showed a trend of decreasing locomotor activity over time that lead to little or no activity
after 20 minutes. In the parameters of movement episodes and time moved (See Figures
16A & B respectively), the mice given pargyline (100 mg/kg) + GBL (150 mg/kg)
showed less activity (p<0.001) between 10 and 15 minutes than the group of mice given
saline + GBL (150 mg/kg). The group of mice given pargyline (100 mg/kg) + GBL (150
mg/kg) also showed less (p<0.001) distance moved (See Figure 16C) at 5 minutes and
fewer (p<0.01) VPE (See Figure 16D) for the first 10 minutes when compared to mice
given saline + GBL (150 mg/kg). The mice pretreated with pargyline did not show an
increase in any of the four parameters when compared with mice given GBL (150
mg/kg).
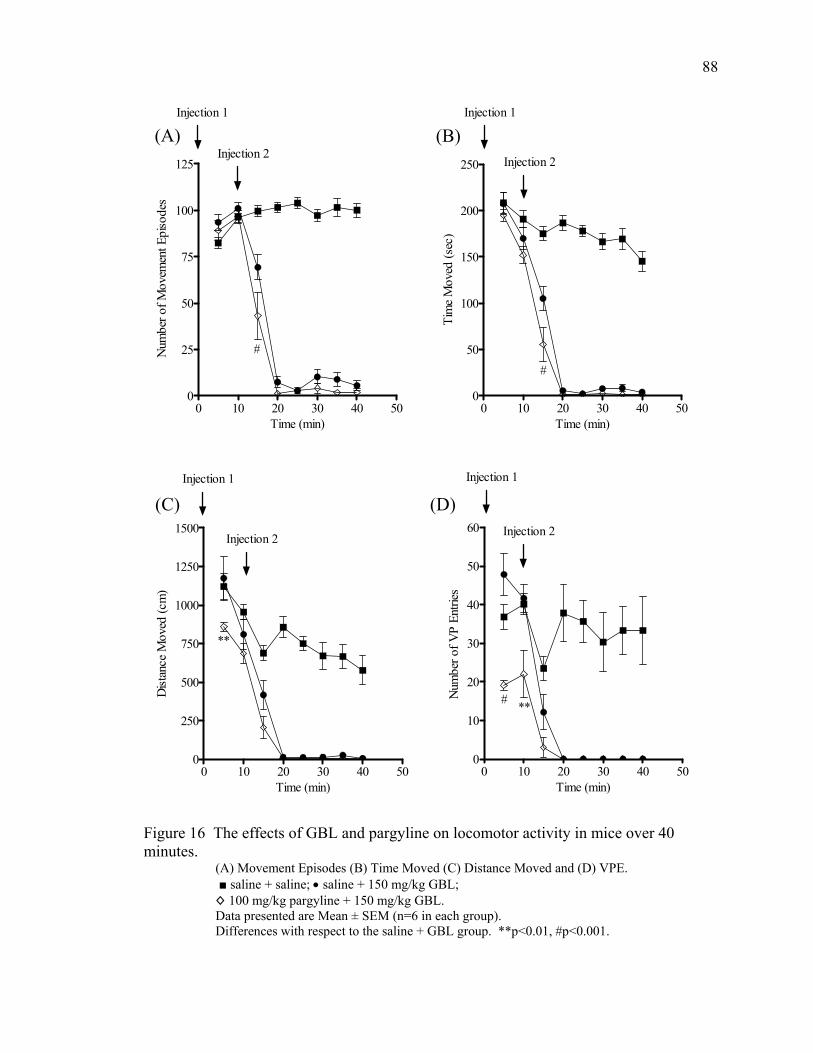
88
0 10 20 30 40 500
50
100
150
200
250
#
Injection 2
Injection 1
Time (min)
Tim
e M
oved
(sec
)
0 10 20 30 40 500
25
50
75
100
125
#
Injection 1
Injection 2
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
(A) (B)
0 10 20 30 40 500
10
20
30
40
50
60
# **
Injection 1
Injection 2
Time (min)
Num
ber o
f VP
Entri
es
0 10 20 30 40 500
250
500
750
1000
1250
1500
**
Injection 1
Injection 2
Time (min)
Dist
ance
Mov
ed (c
m)
(C) (D)
Figure 16 The effects of GBL and pargyline on locomotor activity in mice over 40 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; • saline + 150 mg/kg GBL; ◊ 100 mg/kg pargyline + 150 mg/kg GBL. Data presented are Mean ± SEM (n=6 in each group). Differences with respect to the saline + GBL group. **p<0.01, #p<0.001.

89
Nomifensine Treatment
The behavioral data for the nomifensine pre-treatment mice is shown in Figure 17
A-D.
Movement episodes. Between 5 and 10 minutes, the group of mice that received
nomifensine (20 mg/kg) + GBL (150 mg/kg) had fewer (p<0.001) movement episodes
than the group that received saline + GBL (150 mg/kg) (See Figure 17A). From 20 to 40
minutes, the group of mice that was pre-treated with nomifensine (20 mg/kg) followed by
GBL (150 mg/kg) showed more (p<0.01) movements than the mice given saline + GBL
(150 mg/kg). At 10, 20, and 25 minutes both groups of mice that were given nomifensine
(20 mg/kg) had fewer movement episodes (p<0.05) than the group of mice given saline +
saline (data not shown in graph).
Time moved. The mice that received nomifensine (20 mg/kg) + GBL (150 mg/kg)
showed a decrease of more than 50% in the amount of time moved between 10 and 20
minutes. Between 10 and 40 minutes, the mice that were pretreated with nomifensine (20
mg/kg) followed by GBL (150 mg/kg) moved for a greater (p<0.01) amount of time than
the mice pretreated with saline and followed with GBL (150 mg/kg) (See Figure 17B).
Between 15 and 40 minutes, the mice given nomifensine (20 mg/kg) + saline moved for
more time (p<0.05) than the mice given saline + saline (data not shown in graph). That
same group of mice given nomifensine (20 mg/kg) + saline also moved for more time
(p<0.001) than the mice given saline + saline between 20 and 40 minutes (data not shown
in graph).
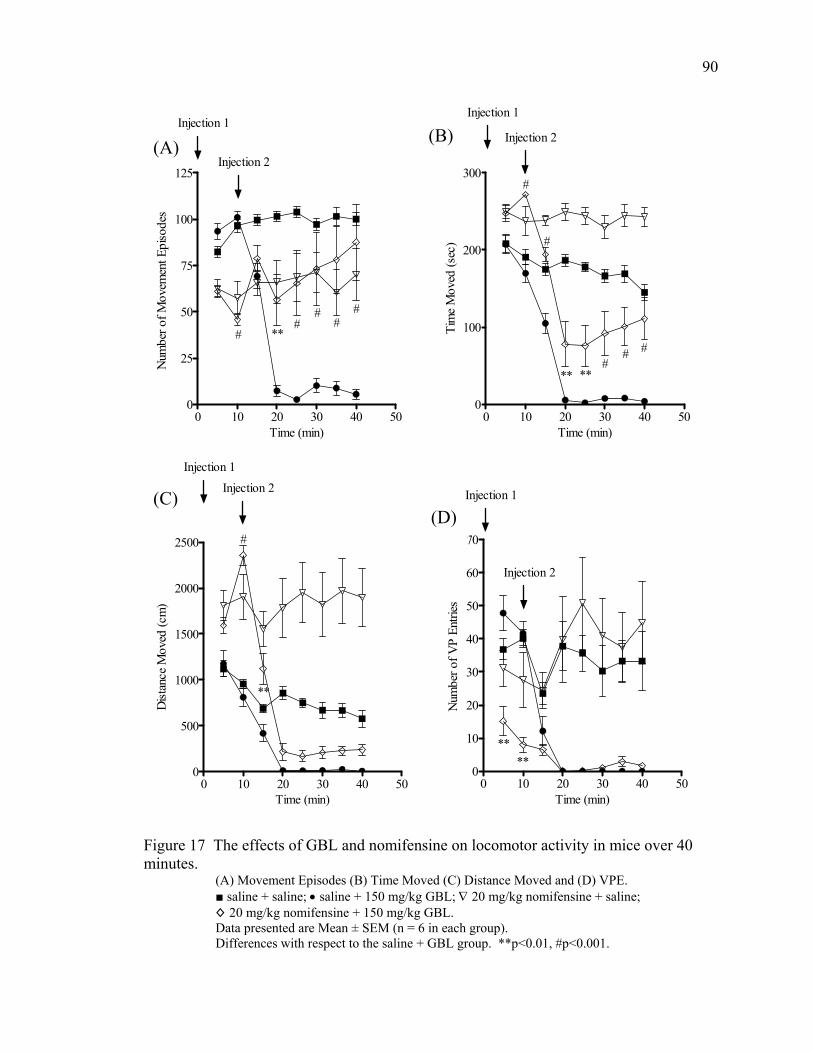
90
0 10 20 30 40 500
100
200
300
** **
#
#
###
Injection 2
Injection 1
#
Time (min)Ti
me
Mov
ed (s
ec)
0 10 20 30 40 500
25
50
75
100
125
#
**#
##
#
Injection 1
Injection 2
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
(B) (A)
0 10 20 30 40 500
500
1000
1500
2000
2500
**
#
Injection 1
Injection 2
Time (min)
Dist
ance
Mov
ed (c
m)
(C)
0 10 20 30 40 500
10
20
30
40
50
60
70
****
Injection 1
Injection 2
Time (min)
Num
ber o
f VP
Entri
es
(D)
Figure 17 The effects of GBL and nomifensine on locomotor activity in mice over 40 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; • saline + 150 mg/kg GBL; ∇ 20 mg/kg nomifensine + saline; ◊ 20 mg/kg nomifensine + 150 mg/kg GBL. Data presented are Mean ± SEM (n = 6 in each group). Differences with respect to the saline + GBL group. **p<0.01, #p<0.001.

91
Distance moved. Between 5 and 15 minutes, the mice given nomifensine (20
mg/kg) + GBL (150 mg/kg) moved a greater (p<0.01) distance than the mice given saline
+ GBL (150 mg/kg) (See Figure 17C). After 15 minutes, there were no statistically
significant differences between the mice pre-treated with nomifensine followed by GBL
and the mice pre-treated with saline followed by GBL (150 mg/kg). For the entire study,
the mice that received nomifensine (20 mg/kg) + saline moved a greater distance
(p<0.05) than the mice that received saline + saline (data not shown in graph). The mice
given nomifensine (20 mg/kg) + saline also moved a greater distance (p<0.001) than the
mice given nomifensine (20 mg/kg) + GBL (150 mg/kg) between 20 and 40 minutes
(data not shown in graph).
Vertical plane entries (VPE). The mice given nomifensine (20 mg/kg) + GBL
(150 mg/kg) had fewer (p<0.01) VPE than the mice given saline + GBL (150 mg/kg)
during the first 10 minutes of the experiment (See Figure 17D). After 10 minutes, the
group of mice given nomifensine (20 mg/kg) + GBL (150 mg/kg) did not differ from the
mice given saline + GBL (150 mg/kg). Between 20 and 40 minutes, the mice given
nomifensine (20 mg/kg) + saline had more VPE (p<0.01) than the group of mice given
nomifensine (20 mg/kg) + GBL (150 mg/kg) (data not shown in graph).

92
Tolcapone Treatment
The behavioral data from the tolcapone pre-treatment is shown in Figure 18 A-D.
Only the VPE graph showed any statistically significant changes (See Figure 18D).
During the first five minutes, the group given tolcapone (30 mg/kg) before GBL (150
mg/kg) had fewer (p<0.01) VPE than the group given saline + GBL (150 mg/kg). There
were no other statistically significant changes resulting from pre-treatment with
tolcapone.
Apomorphine Treatment
The behavioral data for the apomorphine pre-treatment is shown in Figure 19
A-D. All statistically significant differences in this experiment were negative in regard to
locomotor activity with the exception of movement episodes between 10 and 15 minutes.
In that five minute block, the mice given apomorphine (3 mg/kg) + GBL (150 mg/kg)
had more movement episodes (p<0.05) than the mice given saline + GBL (150 mg/kg)
(See Figure 19A). The group of mice given apomorphine (3 mg/kg) before GBL (150
mg/kg) moved for less time (p<0.01) during the first five minutes (See Figure 19B) than
the group of mice given saline before GBL (150 mg/kg). The same GBL treatment group
also moved a shorter distance (p<0.01) in the first 10 minutes than that of the mice given
saline + GBL (150 mg/kg) and had fewer (p<0.001) VPE in the first 10 minutes than the
group of mice that were given saline before GBL (150 mg/kg).
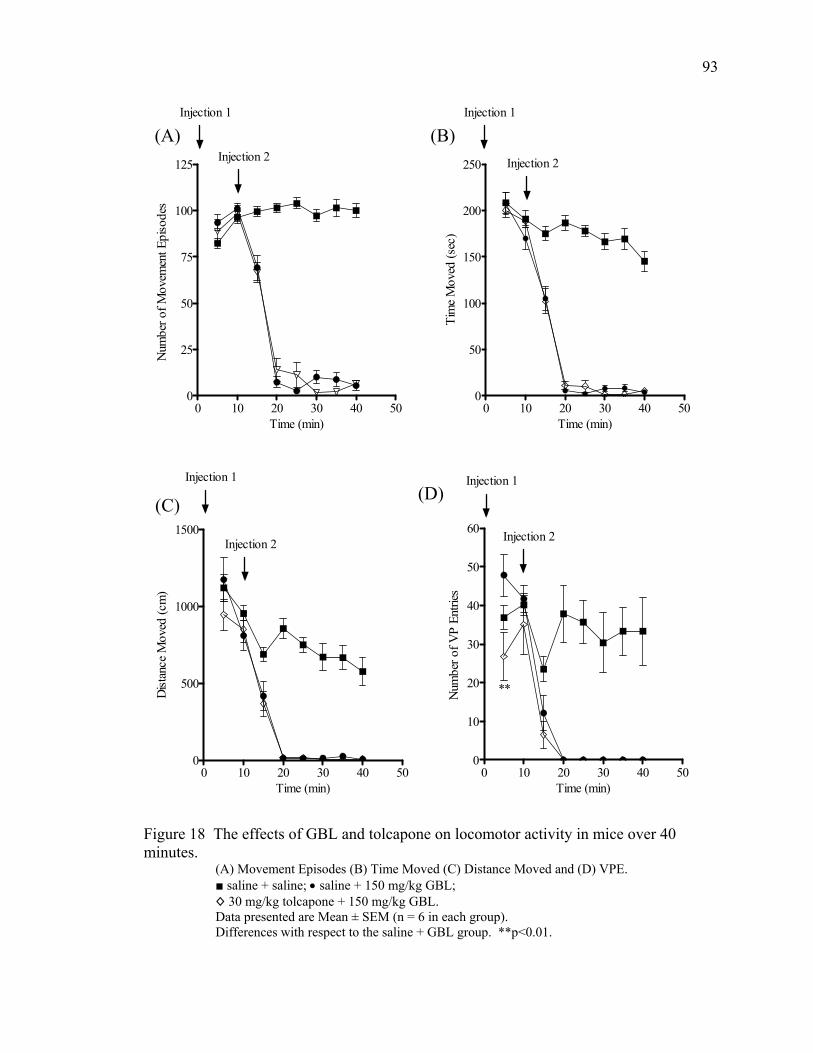
93
0 10 20 30 40 500
25
50
75
100
125 Injection 2
Injection 1
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
0 10 20 30 40 500
50
100
150
200
250 Injection 2
Injection 1
Time (min)
Tim
e M
oved
(sec
)
(A) (B)
0 10 20 30 40 500
500
1000
1500Injection 2
Injection 1
Time (min)
Dist
ance
Mov
ed (c
m)
0 10 20 30 40 500
10
20
30
40
50
60
**
Injection 2
Injection 1
Time (min)
Num
ber o
f VP
Entri
es
(D) (C)
Figure 18 The effects of GBL and tolcapone on locomotor activity in mice over 40 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; • saline + 150 mg/kg GBL; ◊ 30 mg/kg tolcapone + 150 mg/kg GBL. Data presented are Mean ± SEM (n = 6 in each group). Differences with respect to the saline + GBL group. **p<0.01.
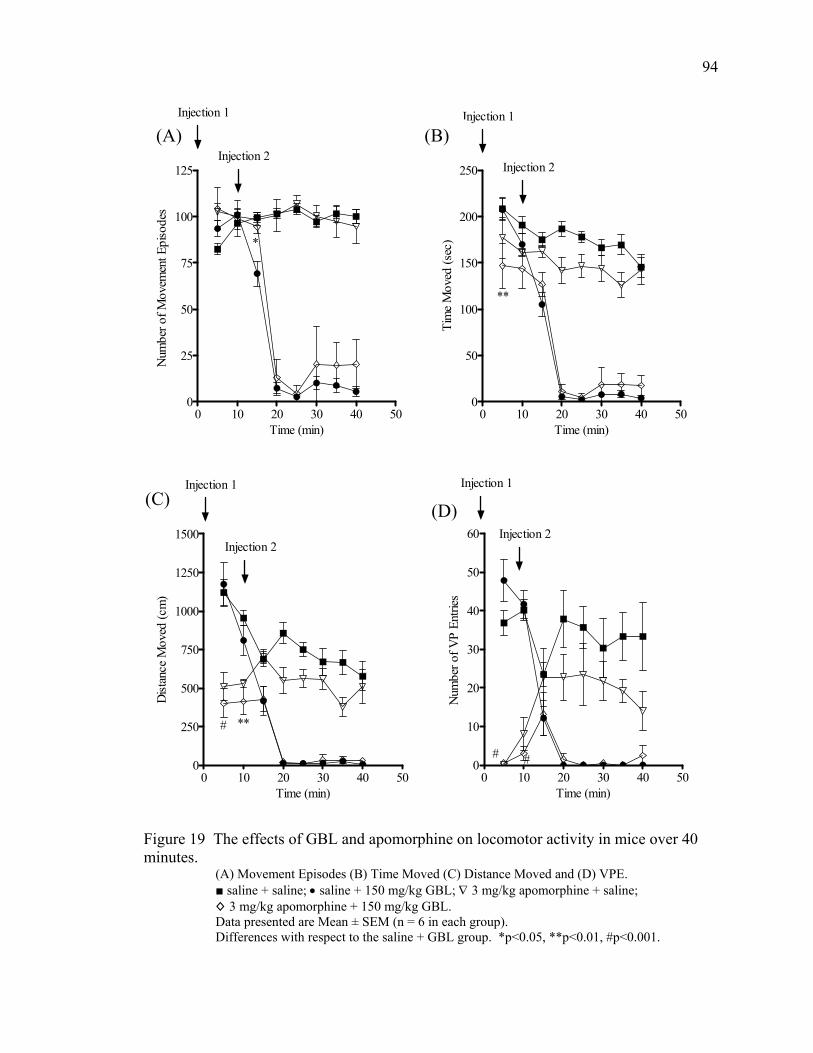
94
0 10 20 30 40 500
25
50
75
100
125
*
Injection 2
Injection 1
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
0 10 20 30 40 500
50
100
150
200
250
**
Injection 2
Injection 1
Time (min)Ti
me
Mov
ed (s
ec)
(A) (B)
0 10 20 30 40 500
10
20
30
40
50
60
# #
Injection 1
Injection 2
Time (min)
Num
ber o
f VP
Entri
es
0 10 20 30 40 500
250
500
750
1000
1250
1500
# **
Injection 2
Injection 1
Time (min)
Dist
ance
Mov
ed (c
m)
(C) (D)
Figure 19 The effects of GBL and apomorphine on locomotor activity in mice over 40 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; • saline + 150 mg/kg GBL; ∇ 3 mg/kg apomorphine + saline; ◊ 3 mg/kg apomorphine + 150 mg/kg GBL. Data presented are Mean ± SEM (n = 6 in each group). Differences with respect to the saline + GBL group. *p<0.05, **p<0.01, #p<0.001.

95
Taurine Treatment
The behavioral data for the taurine pre-treatment is shown in Figure 20 A-D. The
only statistically significant increase in this experiment was seen between 10 and 15
minutes (See Figure 20B). During that five minute time period, the mice given taurine
(350 mg/kg) spent more (p<0.05) time moving than the mice that were given saline +
GBL (150 mg/kg). Otherwise all differences occurred between 0 and 10 minutes. Those
differences were all decreases in locomotor activity before GBL was administered.
Bicuculline Treatment
The behavioral data for the bicuculline pre-treatment is shown in Figure 21 A-D.
The group of mice that received bicuculline (8 mg/kg) + GBL (150 mg/kg) showed an
increase (p<0.01) in movement episodes (See Figure 21A) between 20 and 25 minutes
when compared to mice given saline + GBL (150 mg/kg). That was the only statistically
significant difference between the mice pretreated with bicuculline and the mice
pretreated with saline.
Summary of Behavioral Changes
Only two compounds showed statistically significant increases in any parameters
of locomotor activity when compared to mice that were affected by GBL. Pergolide and
nomifensine both showed increases in movement episodes and time moved. The distance
moved rarely increased reliably for any of the compounds tested. Pergolide and
nomifensine showed no improvement in distance moved except for nomifensine between
10 and 15 minutes (p<0.001). Neither of these two compounds increased the number of
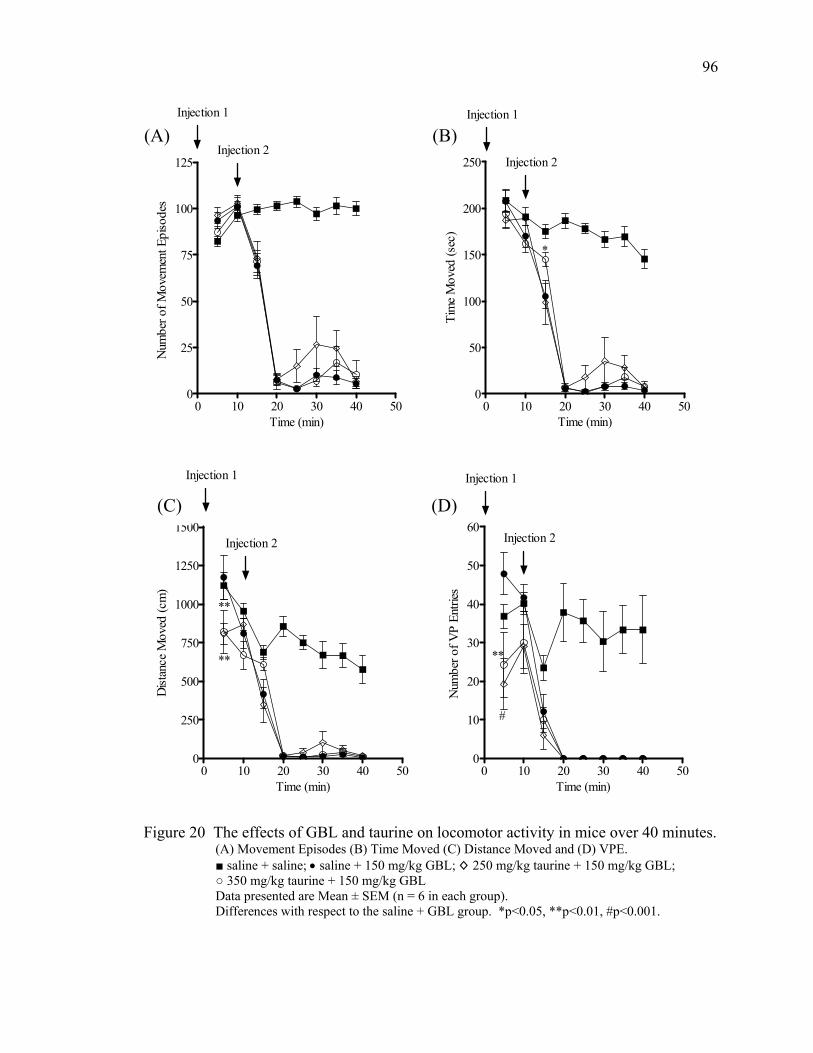
96
0 10 20 30 40 500
25
50
75
100
125Injection 2
Injection 1
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
0 10 20 30 40 500
50
100
150
200
250
*
Injection 2
Injection 1
Time (min)
Tim
e M
oved
(sec
)
(A) (B)
0 10 20 30 40 500
250
500
750
1000
1250
1500
**
**
Injection 2
Injection 1
Time (min)
Dist
ance
Mov
ed (c
m)
0 10 20 30 40 500
10
20
30
40
50
60
#
**
Injection 2
Injection 1
Time (min)
Num
ber o
f VP
Entri
es
(C) (D)
Figure 20 The effects of GBL and taurine on locomotor activity in mice over 40 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; • saline + 150 mg/kg GBL; ◊ 250 mg/kg taurine + 150 mg/kg GBL; ○ 350 mg/kg taurine + 150 mg/kg GBL Data presented are Mean ± SEM (n = 6 in each group). Differences with respect to the saline + GBL group. *p<0.05, **p<0.01, #p<0.001.
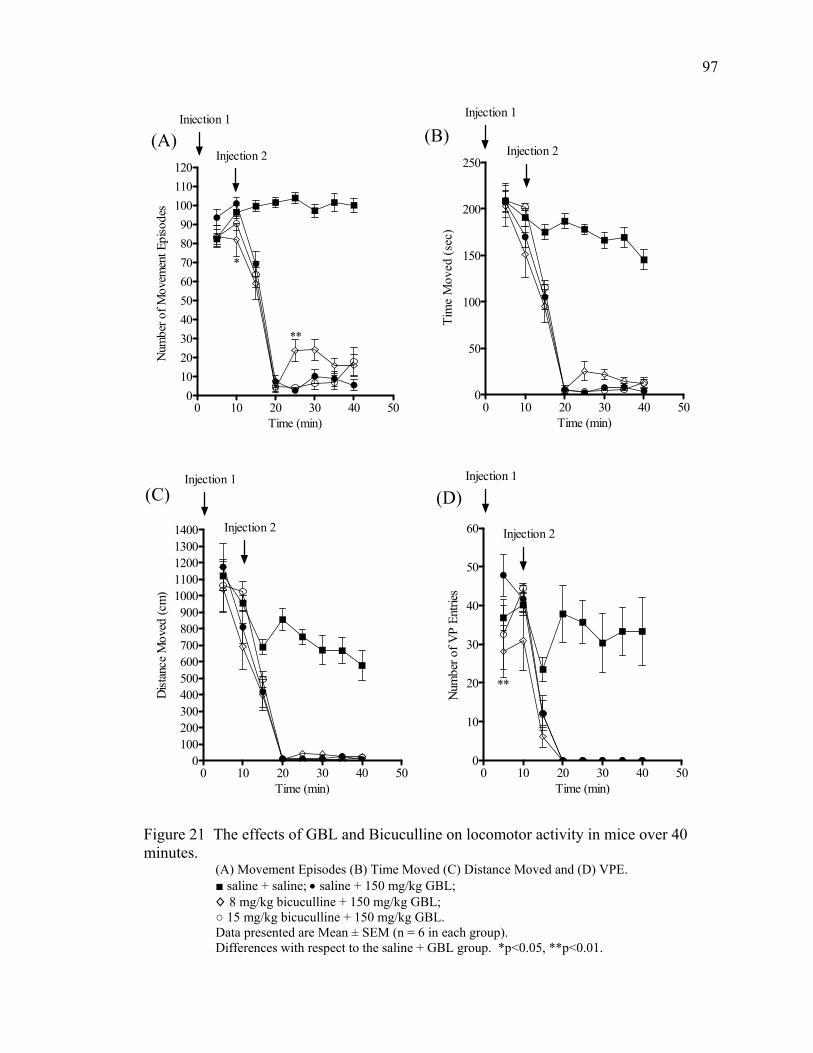
97
0 10 20 30 40 500
50
100
150
200
250Injection 2
Injection 1
Time (min)
Tim
e M
oved
(sec
)
0 10 20 30 40 500
102030405060708090
100110120
*
**
Injection 2
Injection 1
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
(B) (A)
0 10 20 30 40 500
10
20
30
40
50
60
**
Injection 2
Injection 1
Time (min)
Num
ber o
f VP
Entri
es
0 10 20 30 40 500
100200300400500600700800900
10001100120013001400 Injection 2
Injection 1
Time (min)
Dist
ance
Mov
ed (c
m)
(C) (D)
Figure 21 The effects of GBL and Bicuculline on locomotor activity in mice over 40 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; • saline + 150 mg/kg GBL; ◊ 8 mg/kg bicuculline + 150 mg/kg GBL; ○ 15 mg/kg bicuculline + 150 mg/kg GBL. Data presented are Mean ± SEM (n = 6 in each group). Differences with respect to the saline + GBL group. *p<0.05, **p<0.01.

98
VPE when compared with mice given saline + GBL (150 mg/kg). Pergolide and
nomifensine were the only compounds that successfully antagonized the behavioral
effects of GBL; thus they were the only compounds whose effects on the monoamine
neurotransmitter metabolites were investigated. Therefore, the mice that were treated
with pergolide and nomifensine were sacrificed and their brains used for monoamine
metabolite analysis.
Monoamine Metabolite Results
Pergolide Treatment
Dopamine metabolites. The dopamine metabolites and turnover for the pergolide
pre-treatment are shown in Table 8. Mice given pergolide (10 mg/kg) + saline had more
(p<0.01) dopamine and less HVA (p<0.01), 3-MT (p<0.05), and a lower dopamine
turnover rate (p<0.01) than mice given saline + saline. When compared to the mice that
were given saline + GBL (150 mg/kg), the mice that were given pergolide (10 mg/kg) +
GBL (150 mg/kg) had higher (p<0.01) levels of dopamine. The same group of mice pre-
treated with pergolide had less DOPAC (p<0.01), HVA (p<0.01), and a lower dopamine
turnover rate (p<0.01) than the saline + GBL (150 mg/kg) group.
Serotonin metabolites. The serotonin metabolites and turnover for the pergolide
pre-treatment are shown in Table 9. The group of mice given pergolide (10 mg/kg) +
saline had more (p<0.01) 5-HT, less (p<0.01) 5-HIAA, and a lower (p<0.01) serotonin
turnover rate than the group of mice given saline + saline. The group of mice treated
with pergolide (10 mg/kg) + GBL (150 mg/kg) had more (p<0.01)
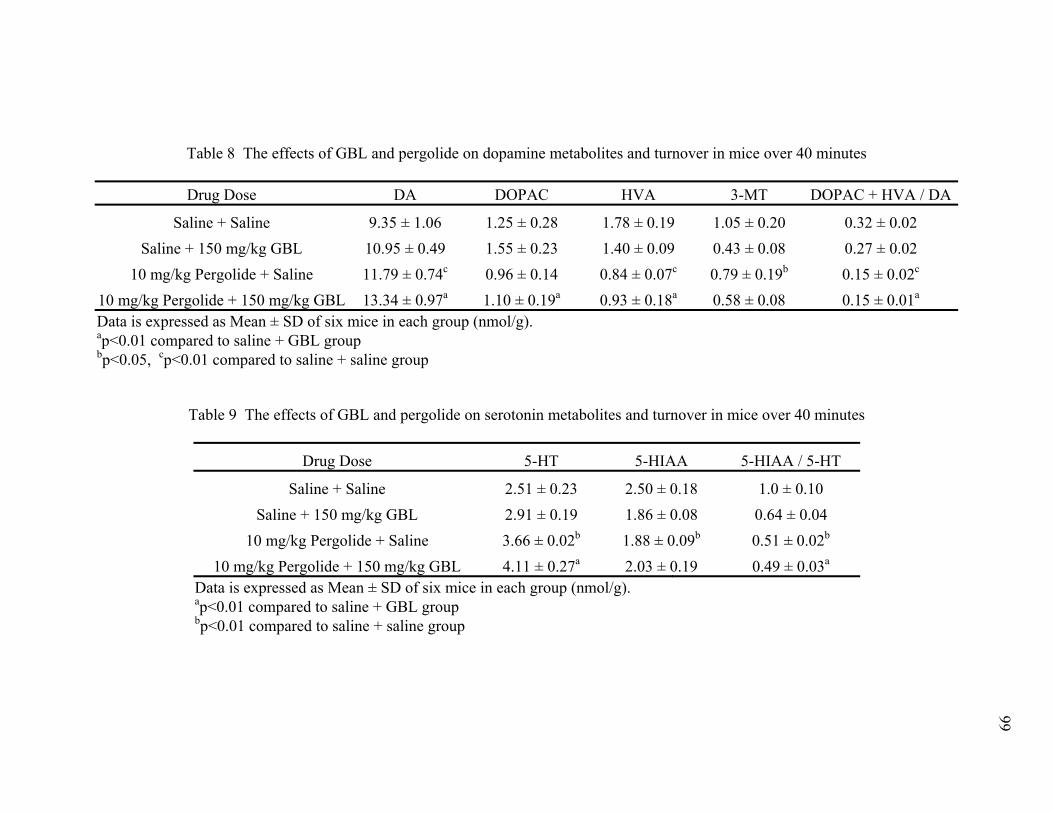
99
Table 8 The effects of GBL and pergolide on dopamine metabolites and turnover in mice over 40 minutes
Drug Dose DA DOPAC HVA 3-MT DOPAC + HVA / DA
Saline + Saline 9.35 ± 1.06 1.25 ± 0.28 1.78 ± 0.19 1.05 ± 0.20 0.32 ± 0.02 Saline + 150 mg/kg GBL 10.95 ± 0.49 1.55 ± 0.23 1.40 ± 0.09 0.43 ± 0.08 0.27 ± 0.02
10 mg/kg Pergolide + Saline 11.79 ± 0.74c 0.96 ± 0.14 0.84 ± 0.07c 0.79 ± 0.19b 0.15 ± 0.02c
0.15 ± 0.0110 mg/kg Pergolide + 150 mg/kg GBL 13.34 ± 0.97a 1.10 ± 0.19a 0.93 ± 0.18a 0.58 ± 0.08 a
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.01 compared to saline + GBL group bp<0.05, cp<0.01 compared to saline + saline group
Table 9 The effects of GBL and pergolide on serotonin metabolites and turnover in mice over 40 minutes
Drug Dose 5-HT 5-HIAA 5-HIAA / 5-HT
Saline + Saline 2.51 ± 0.23 2.50 ± 0.18 1.0 ± 0.10 Saline + 150 mg/kg GBL 2.91 ± 0.19 1.86 ± 0.08 0.64 ± 0.04
10 mg/kg Pergolide + Saline 3.66 ± 0.02b 1.88 ± 0.09b 0.51 ± 0.02b
0.49 ± 0.0310 mg/kg Pergolide + 150 mg/kg GBL 4.11 ± 0.27a 2.03 ± 0.19 a
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.01 compared to saline + GBL group p<0.01 compared to saline + saline group b

100
5-HT and a lower (p<0.01) serotonin turnover rate than the saline + GBL (150 mg/kg)
treated mice. There were no statistically significant changes in 5-HIAA levels for that
particular group.
Nomifensine Treatment
Dopamine metabolites. The dopamine metabolites and turnover for the
nomifensine pre-treatment are shown Table 10. Mice given nomifensine (20 mg/kg) +
saline had less (p<0.01) dopamine and DOPAC (p<0.01) than mice given saline + saline.
The mice treated with nomifensine (20 mg/kg) + GBL (150 mg/kg) had decreases in
dopamine (p<0.01), DOPAC (p<0.01), and HVA (p<0.01) levels when compared to mice
treated with saline + GBL (150 mg/kg). The group of mice that received nomifensine (20
mg/kg) + GBL (150 mg/kg) did not have different levels of 3-MT when compared with
mice treated with only GBL. The rate of dopamine turnover did not change reliably for
any of the groups of mice that received nomifensine.
Serotonin metabolites. The serotonin metabolites and turnover for the
nomifensine pre-treatment are shown in Table 11. The mice given nomifensine (20
mg/kg) + saline had more (p<0.01, p<0.05) 5-HT and 5-HIAA than the mice given saline
+ saline. Mice treated with nomifensine (20 mg/kg) followed by GBL (150 mg/kg) did
not show changes in 5-HT levels. However, that group did have less (p<0.05) 5-HIAA
and a lower (p<0.05) serotonin turnover rate when compared to mice treated only with
GBL (150 mg/kg).
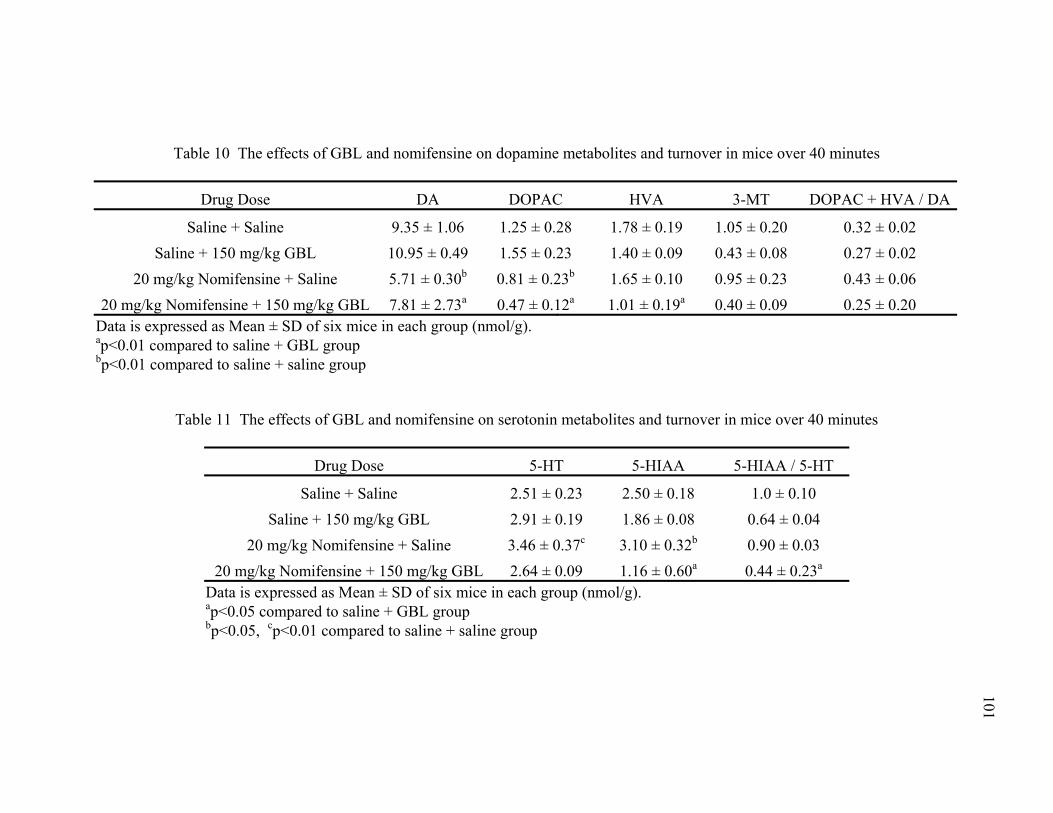
101
Table 10 The effects of GBL and nomifensine on dopamine metabolites and turnover in mice over 40 minutes
Drug Dose DA DOPAC HVA 3-MT DOPAC + HVA / DA
Saline + Saline 9.35 ± 1.06 1.25 ± 0.28 1.78 ± 0.19 1.05 ± 0.20 0.32 ± 0.02 Saline + 150 mg/kg GBL 10.95 ± 0.49 1.55 ± 0.23 1.40 ± 0.09 0.43 ± 0.08 0.27 ± 0.02
20 mg/kg Nomifensine + Saline 5.71 ± 0.30b 0.81 ± 0.23b 1.65 ± 0.10 0.95 ± 0.23 0.43 ± 0.06 0.25 ± 0.20 20 mg/kg Nomifensine + 150 mg/kg GBL 7.81 ± 2.73a 0.47 ± 0.12a 1.01 ± 0.19a 0.40 ± 0.09
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.01 compared to saline + GBL group bp<0.01 compared to saline + saline group
Table 11 The effects of GBL and nomifensine on serotonin metabolites and turnover in mice over 40 minutes
Drug Dose 5-HT 5-HIAA 5-HIAA / 5-HT
Saline + Saline 2.51 ± 0.23 2.50 ± 0.18 1.0 ± 0.10 Saline + 150 mg/kg GBL 2.91 ± 0.19 1.86 ± 0.08 0.64 ± 0.04
20 mg/kg Nomifensine + Saline 3.46 ± 0.37c 3.10 ± 0.32b 0.90 ± 0.03 0.44 ± 0.2320 mg/kg Nomifensine + 150 mg/kg GBL 2.64 ± 0.09 1.16 ± 0.60a a
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.05 compared to saline + GBL group bp<0.05, cp<0.01 compared to saline + saline group

102
Summary of Monoamine Metabolite Changes
In the pergolide treatment study, the monoamine metabolites of the mice treated
with pergolide (10 mg/kg) and GBL (150 mg/kg) were investigated. Pre-treating mice
with pergolide before administering GBL resulted in several statistically significant
changes in dopamine and serotonin metabolites. Dopamine and 5-HT levels increased
(p<0.01). In contrast, DOPAC and HVA levels as well as dopamine and serotonin
turnover rates all decreased (p<0.01) in mice given pergolide (10 mg/kg) + GBL (150
mg/kg) when compared to mice treated with saline + GBL (150 mg/kg). The group of
mice that were treated with nomifensine (20 mg/kg) followed by GBL (150 mg/kg) had
more (p<0.01) dopamine than the group of mice treated only with GBL (150 mg/kg).
That same group of mice had less DOPAC (p<0.01), HVA (p<0.01), 5-HIAA (p<0.05),
and serotonin turnover (p<0.05) than mice treated only with GBL (150 mg/kg).
Discussion
The overall aim of the studies presented in this chapter was to investigate compounds or
pharmacological agents that may antagonize the sedation and loss of locomotor activity
induced by GBL. Eight compounds were tested of which only two showed the ability to
antagonize the loss of locomotor function induced by GBL in mice. The success of each
compound is summarized in Table 12.
NCS-382 did not prevent sedation and at times appeared to make the loss of
locomotor activity worse than GBL alone. There were no increases in locomotor activity
in any of the four parameters. While NCS-382 is a GHB receptor antagonist (Maitre and
others 1990), the results in this study are consistent with others (Castelli and others 2004)
who have shown NCS-382 to be incapable of antagonizing the loss of locomotor activity

103
Table 12 Compounds tested for antagonism of GBL
Compound Antagonism of GBLPergolide + NCS-382 - Pargyline -
Nomifensine + Tolcapone -
Apomorphine - Taurine -
Bicuculline - due to GHB. It was expected that NCS-382 would prevent GBL derived GHB from
binding to its receptors and prevent the loss of locomotor activity. However, the lack of
any effect indicates that GHB receptors are not likely to be involved in the control of
locomotor function. These findings may also indicate that GHB may bind to receptors
other than GHB specific receptors. Numerous studies have shown the potential of GHB
to bind to both GABAA/B receptor subtypes (Bourguignon and others 1988). This
suggests that GHB may concurrently bind to both GHB specific and GABA receptors.
Furthermore, GHB specific receptors might not directly influence dopamine receptors or
metabolism. If that were the case, antagonizing GHB specific receptors would not
prevent the sedative or behavioral effects of GHB or GBL.
Pargyline showed approximately the same effects as NCS-382 and did not
antagonize the loss of locomotor activity at any point during the 40 minute study. The 10
minutes before GBL was administered showed less distance moved than was normally
seen in the mice treated with saline + GBL. The lack of antagonism by pargyline is in
contrast to previous findings in this lab, which indicated that pre-treatment with pargyline
antagonized the sedation and loss of locomotor activity induced by GHB in rats

104
(Anderson and others 2002). Possible reasons for the discrepancy between the two
studies include differences in absorption and metabolism of GHB and GBL between
rodent species (rats versus mice) and differences in absorption and metabolism of
pargyline between rodent species. Another possibility is the greater speed with which
GBL exerts its effects compared to GHB, which has been mentioned previously. Perhaps
pargyline was successful against GHB because it had more time to act before GHB
exerted its effects.
Tolcapone caused no improvement in any of the four parameters when compared
to mice given only GBL. As a COMT inhibitor, we expected tolcapone to increase
extracellular levels of dopamine by inhibiting the conversion of dopamine to 3-MT.
However, if dopamine release is inhibited by GBL (as has been indicated in earlier
chapters), perhaps extracellular levels of dopamine are so low that COMT inhibition is
ineffective in increasing synaptic dopamine. While some improvement in locomotor
activity was expected, the results indicate that the inhibition of dopamine release is more
severe than previously thought or that inhibiting postsynaptic COMT is not adequate to
antagonize the effects of GBL.
As a dopamine receptor agonist, apomorphine’s inability to antagonize GBL was
unexpected. Compounds aimed at increasing dopaminergic activity are the basis of the
attempts to antagonize GBL as described in this dissertation. The hypothesis is that
increasing central dopaminergic activity should prevent the GBL induced loss of
locomotor activity. However, there were no improvements in any of the four behavioral
parameters studied. The dose of apomorphine was small at 3 mg/kg and it appeared that
the effect size for movement episodes was close to statistical significance as the

105
experiment continued. It is possible that a higher dose of apomorphine might lead to
significant increases in locomotor activity and should be further explored in the future.
The experimental study with taurine doses of either 250 or 350 mg/kg did not
successfully antagonize the sedation or loss of locomotor activity induced by GBL in
mice. Previous studies performed in this laboratory have shown that taurine (250 mg/kg)
was able to antagonize the sedation and loss of locomotor activity induced by GHB in
rats (Anderson and Bottiglieri unpublished data). It is therefore surprising that taurine
was not effective in mice when administered after GBL. The discrepancy between the
two studies include species differences for metabolism of taurine and the use of GBL
(150 mg/kg), which is absorbed and metabolized differently and may result in higher
concentrations of GHB in tissue. Taurine has also been used successfully in extending
the life of the SSADH knock out mouse (Gupta and others 2002). In that study, the mice
used were SSADH knock out mice which had high levels of GHB from birth, but also
had a constant supply of taurine in mother’s milk. When the diets were supplemented
with taurine, that constant supply continued. It is likely that the acute injection in our
study was the major difference in the success of antagonizing GBL when compared to the
constant supply of taurine given to the SSADH knock out mice. As previously
mentioned, it is not known if GBL derived GHB has the same properties as endogenous
GHB. This is one possible reason the current results differ from the previous
experiments. Another is the species difference between mice and rats. This species
difference has already been implicated as a confounding factor in the experiments
regarding pargyline and may also be a determining factor in the results presented in this
chapter. The metabolism of GBL may also be different between rodent species. Finally,

106
GBL is rapidly absorbed and converted to GHB, and may exert its effects more rapidly
than GHB that is injected directly.
Bicuculline showed a similar effect to taurine in that the lower dose showed a
greater response than the higher dose. There was an increase in movement episodes in
the mice given bicuculline (8 mg/kg) + GBL (150 mg/kg) compared to mice given saline
+ GBL (150 mg/kg). As a GABA antagonist, we expected better results from this
compound. However, GHB and its precursors have been antagonized more readily with
GABA antagonists than with GABAB A antagonists. As a GABAA antagonist, bicuculline
lends support to a stronger affinity between GHB and GABA receptors. B
Pergolide was one of two successful compounds tested. Initially, three doses of
pergolide were tested: 5 mg/kg (data not shown), 10 mg/kg, and 30 mg/kg (data not
shown). The results from 5 mg/kg showed some positive trends towards antagonizing the
locomotor effects of GBL, but that dose was discontinued when results for 10 mg/kg
were better. After observing the effects of 10 mg/kg, a higher dose of 30 mg/kg was
attempted. Some increases in activity were observed, but they were not statistically
significant (data not shown) and were worse than the activity seen with 10 mg/kg. The
results from 10 mg/kg are shown in the graphs in Figure 14. When mice were pre-treated
with pergolide (10 mg/kg) before receiving GBL (150 mg/kg), the sedation and loss of
locomotor activity were antagonized. Beginning at 15 minutes and continuing for the
duration of the study, the number of movement episodes and amount of time moved were
higher than in the mice treated only with GBL. These increases in locomotor activity
were paralleled by changes in the dopamine metabolites. DA levels increased while

107
DOPAC, HVA, and the dopamine turnover rate all decreased in mice treated with
pergolide (10 mg/kg) + GBL (150 mg/kg).
Pergolide is a D /D1 2 dopamine receptor agonist. By administering pergolide to
the mice before giving GBL, the aim was to stimulate postsynaptic dopamine receptors,
which would in turn help maintain consciousness and locomotor capability. GBL has
been shown to inhibit dopamine release in Chapter Four of this dissertation as evidenced
by the decrease in 3-MT levels. Inhibition of dopamine release has also been shown in
other studies (Bustos and Roth 1972; Hechler and others 1991; Bottiglieri and others
2001; Anderson and others 2002). This inhibition activates a feedback mechanism,
which causes TH activity to increase, and more dopamine to be produced intracellularly
(Gessa and others 1968; Cheramy and others 1977; Roth and others 1980; Hechler and
others 1991; Diana and others 1993). Thus, neuronally, the mice functioned as if
dopamine were still being released. That explains the increase in dopamine levels, and
the decrease in DOPAC and HVA levels. With dopamine release inhibited, downstream
metabolism into DOPAC and HVA could not occur. The changes in 3-MT reveal a
different effect than observed thus far. When mice were given pergolide (10 mg/kg) +
saline, the 3-MT levels decreased when compared to mice given saline + saline. When
pergolide (10 mg/kg) was followed by GBL (150 mg/kg), there was no change when
compared to mice given saline + GBL (150 mg/kg). It appears that pergolide alone can
cause a decrease in 3-MT levels by stimulating the D /D1 2 receptors. The decrease in
dopamine turnover rate is a result of dopamine release being inhibited, which lowers
levels of the respective metabolites.

108
The serotonin metabolite changes are less easily explained. Changes in serotonin
have not been monitored in cases of GHB to the extent that changes in dopamine have.
The analysis in Chapter Four indicated that GHB might have a role in regulating
serotonin metabolites and turnover. In the pergolide study, the levels of serotonin in mice
treated with pergolide (10 mg/kg) + GBL (150 mg/kg) increased when compared to mice
treated with only GBL. The levels of 5-HIAA however, did not change reliably. This
indicates that the decrease in 5-HIAA levels induced by GBL over 40 minutes was
antagonized by pergolide. This leads to speculation regarding the D2 receptor and its role
in the metabolism of 5-HT into 5-HIAA. Due to the increase in 5-HT in mice treated
with pergolide (10 mg/kg) + GBL (150 mg/kg) the rate of serotonin turnover decreased
from the rate seen in mice treated with only GBL. This is similar to the trend seen in
Acute Study 2 where despite increased 5-HT there was a decrease in serotonin turnover.
This further implies that GBL might have a role in serotonergic activity or the regulation
of MAO.
The group of mice given pergolide (10 mg/kg) + saline were used to determine
the effects of pergolide when not being tested against GBL. The increased dopamine
levels indicate an inhibition of release and subsequent intraneuronal accumulation of
dopamine. The inhibition of DA release is due to the postsynaptic stimulation of D2
receptors by pergolide. The decreased levels of HVA, 3-MT, and decrease in dopamine
turnover are verification that dopamine release was inhibited. Furthermore, the decrease
in 3-MT levels has previously been shown to be an indicator of the inhibition of
dopamine release induced by GHB in rats (Bottiglieri and others 2001; Anderson and
others 2002).

109
The group of mice given pergolide (10 mg/kg) + saline also showed increased 5-
HT levels, decreased 5-HIAA levels, and a decreased rate of serotonin turnover.
Increased 5-HT levels indicate an increase in either synthesis or accumulation of 5-HT.
The decreases in 5-HIAA levels explain the reduced rate of serotonin turnover.
Newman-Tancredi and others (Newman-Tancredi and others 2002) have shown that
pergolide acts as an agonist at h5-HT1A, h5-HT1B, h5-HT1D, h5-HT2A, h5-HT2B, and h5-
HT2C serotonin receptors. 5-HT1A (Barnes and Sharp 1999; Millan and others 2000), 5-
HT1B (Sarhan and others 2000), and 5-HT2A (Ng and others 1999; De Deurwaerdere and
Spampinato 2001,) receptors have all shown an interaction with either locomotor function
or dopamine release. These interactions could explain the changes observed in the
serotonergic system. The serotonin metabolite data presented here is contrary to previous
studies by Gobaille and others (Gobaille and others 2002). That study showed decreased
5-HT and increased 5-HIAA levels after GHB or GBL administration. This discrepancy
between the two studies may be due to the methodology used. This study used half brain
analysis and the study by Gobaille and others used microdialysis.
Nomifensine was successful in antagonizing the sedation and loss of locomotor
activity. Both 10 mg/kg (data not shown) and 20 mg/kg doses were tested on mice prior
to GBL administration. Both doses showed improvement, but the 20 mg/kg dose proved
better at antagonizing the effects of GBL. As can be seen in the graphs of movement
episodes (See Figure 17A) and time moved (See Figure 17B), the mice treated with
nomifensine (20 mg/kg) before administration of GBL (150 mg/kg) were more active
between 15 and 40 minutes than the mice treated with only GBL.

110
Nomifensine is a monoamine reuptake inhibitor. Administration prior to GBL
would prevent any dopamine that is in the synaptic cleft from being reabsorbed
presynaptically. That would lead to more dopamine in the synaptic cleft and therefore,
more dopamine to act pre and postsynaptically. As previously stated, studies have shown
GBL to inhibit dopamine release, but the degree to which that occurs is a subject of
debate. Some dopamine may still be released. It is also possible that intraneuronally
accumulating dopamine leaks out of the neuron. If either of these processes occurred,
nomifensine would keep that dopamine in the synaptic cleft until it was metabolized by
COMT or used postsynaptically, and the effects of GBL might then be partially
antagonized.
When mice were pre-treated with nomifensine (20 mg/kg) followed by GBL (150
mg/kg) administration, the DA, DOPAC, and HVA levels all decreased. Interestingly, in
mice that received nomifensine (20 mg/kg) + saline the levels of dopamine decreased by
an even greater amount when compared to their saline + saline controls. This is likely
due the effects of GBL. When GBL was administered, it increased TH activity, and thus
increased DA synthesis. The mice that received nomifensine (20 mg/kg) + saline did not
have the increase in TH activity. If dopamine release were inhibited, that would also
cause less dopamine to be present, which in turn would lead to lower levels of the
respective metabolites. In this experiment, DOPAC and HVA were lower than in mice
given only GBL. The 3-MT levels were relatively the same with no difference between
mice that received saline + GBL (150 mg/kg) and mice that received nomifensine (20
mg/kg) + GBL (150 mg/kg). If nomifensine did prevent dopamine reuptake, it would

111
have been metabolized postsynaptically into 3-MT. The dopamine turnover rate was
relatively unchanged in this experiment.
The changes in serotonin metabolites in this study were less pronounced than in
the pergolide experiment. The decrease in 5-HT levels in the mice treated with
nomifensine (20 mg/kg) followed by GBL (150 mg/kg) was not statistically significant,
but the group of mice given nomifensine (20 mg/kg) + saline had more 5-HT than the
mice treated only with GBL. The same observation is present in 5-HIAA levels except
that the decrease seen in the group of mice given nomifensine prior to GBL was
statistically significant. As a result of these metabolite levels, the serotonin turnover rate
was lower in mice pre-treated with nomifensine followed by GBL than it was in mice
given only GBL. As previously mentioned, little is known about the role GHB plays in
serotonergic regulation or MAO regulation. However, the results from this experiment
were more straightforward. When both 5-HT and 5-HIAA increased, the serotonin
turnover rate was increased.
The group of mice given nomifensine (20 mg/kg) + saline was used to investigate
the effects of nomifensine by itself. The decreases in DA and DOPAC indicate that after
reuptake was inhibited, all of the dopamine in the synaptic cleft was metabolized. The
rate of metabolism was such that DA and DOPAC were metabolized quickly, but the rate
of metabolism for HVA and 3-MT had not yet “caught up”.
The rate of serotonin turnover in the mice given nomifensine (20 mg/kg) + saline
was not significantly different from mice given saline + saline despite the increases in 5-
HT and 5-HIAA. Thus the metabolism in the serotonergic system is unaffected, but the

112
synthesis or accumulation of both 5-HT and 5-HIAA has increased due to nomifensine’s
effects on the dopaminergic system.
There was a drawback to the results seen in both the pergolide and nomifensine
studies. Neither compound caused an improvement in the distance moved or VPE
parameters. There are several possible explanations for this. A single movement episode
started when a mouse began to move in the cage. As long as the mouse continued to
move without stopping for longer than 0.5 seconds, that activity was considered one
movement episode. The time moved began when a mouse first started moving and
stopped when the mouse stopped moving for longer than 0.5 seconds. Thus it was
possible to have one movement episode that lasted 60 seconds or 12 movement episodes
lasting five seconds each. The number of movement episodes would be different, but the
total time moved would be the same. The distance moved was defined as the total
distance moved in each five minute block of time. This value could be very large if the
mouse explored a large area of the cage. The value could also be very small if the mouse
only moved far enough to break another set of coordinates. Breaking the intersection of
two perpendicular beams in the cage is what defined a movement. With many of the
drugs tested, it appeared that the distance the mice moved was very low compared to the
number of movements and time spent moving. This was the case for pergolide and
nomifensine. It is reasonable to think that while the two compounds produced significant
increases in movement episodes and movement time, the mice did not move very far
when they did move. Rather than exploring the total area of the cage, it appears that
when the mice received pre-treatment with pergolide or nomifensine, they moved very
short distances, but took longer in doing so. The reason for shorter movements was

113
unclear. It could have been based on a depletion of neural dopamine or part of the
muscular control effects of GBL induced loss of locomotor activity. The other parameter
that did not show any improvement was vertical plane entries. That has been shown in
earlier studies of GHB (Cook and others 2002). None of the compounds seemed to have
any effect on the ability of the mice to regain the ability to rear. Rearing requires a good
degree of coordination and muscle strength, which is markedly affected by GHB and
GBL. It is clear that while pergolide and nomifensine treatments were effective in
preventing sedation induced by GBL, and increasing movement episodes, they were not
effective at fully restoring rearing.
Currently, no treatment exists for an overdose of GHB or its precursors GBL and
1,4-BD. Since pergolide and nomifensine successfully antagonized the effects of GBL,
they were investigated further. These two compounds were tested as a form of post-
treatment therapy rather than a pre-treatment therapy with the thought of clinical
application taken into account.

CHAPTER SIX
Post-Treatment Effect of Dopamine Agonists on GBL Induced Loss of Locomotor Activity in Mice
Introduction
The previous chapter demonstrated the ability of both the D1/D2 receptor agonist
pergolide, and the monoamine reuptake inhibitor nomifensine, to antagonize the sedation
and loss of locomotor activity induced by GBL. Various studies have shown the ability
to antagonize the loss of locomotor function induced by GHB or GBL by pre-treatment
using the GABAB antagonists, CGP 35348 (Bottiglieri and others 2001; Carter and others
2005) and CGP 36742 (Bottiglieri and others 2001). The GABAB antagonists SCH
50911 and CGP 46381 have also both been administered as a pre-treatment to GHB with
some success (Carai and others 2001). Other agents tested include NCS-382 and
naloxone. NCS-382 is often cited as having the ability to antagonize the effects of GHB
when used as a pre-treatment (Nissbrandt and Engberg 1996). Naloxone, when given as
a pre-treatment, has been shown to antagonize the decrease in striatal dopamine release
caused by GHB (Feigenbaum and Howard 1997). While pre-treating the mice with
compounds can be successful at antagonizing the effects of GBL, this pharmacological
approach may have limited success following an overdose with GHB or GBL, or in
benefiting patients with SSADH deficiency. Antagonizing the behavioral effects of GHB
or GBL via post-treatment has rarely been reported. In fact, only two studies have shown
success using a post-treatment approach. Carai and others (Carai and others 2001) have
shown that SCH 59011, when administered after GHB, reduced the time needed to regain
the righting reflex in mice in a dose dependent fashion. Pargyline has been shown to
114

115
antagonize the effects of GHB in rats when given either before or after GHB (Anderson
and others 2002).
The illicit use of GHB and GBL is of increasing concern. GBL is still widely
advertised on the internet as an industrial cleaner, and instructions on making GHB from
GBL are readily available. Currently there is no antidote treatment for overdosing with
GHB or its analogues GBL or 1,4-BD (Shannon and Quang 2000). Cases of GHB
overdose seen in the ER increased so dramatically that the FDA banned its sale in 1990
(Okun and others 2000). Survival from an overdose of GHB is dependent on admission
to an emergency room, where the individual is intubated and placed on a respirator until
they have stabilized and regained consciousness.
Based on the results from the experiments in Chapter Five, the effect of both
pergolide and nomifensine as a post-treatment to GBL administration have been
investigated. The results presented in this chapter indicate that both drugs tested have the
potential to effectively antagonize the effects of GBL, suggesting that they may be useful
as a rescue or antidote therapy.
Materials & Methods
In these experiments, mice were monitored in the TruScan® cage for 60 minutes.
The first injection was administered at time zero before any testing and consisted of
either saline (control experiments for pergolide/nomifensine) or GBL (testing vs.
pergolide/nomifensine). The second injection was administered at 10 minutes of testing
and consisted of either saline (control experiments for GBL) or pergolide or nomifensine.
All solutions were prepared fresh daily and were administered via i.p. injection.

116
The behavioral parameters monitored in the TruScan® were movement episodes,
time moved, distance moved, and vertical plane entries. After each experiment, all
animals were immediately asphyxiated by means of CO2 and sacrificed via cervical
dislocation. The brains were harvested for further monoamines and metabolite analysis.
Brains were collected as half brains without the cerebellum.
The monoamine and metabolite neurotransmitter analysis is identical to that
previously described in Chapter Four. A single half brain (without cerebellum) for each
mouse was analyzed for the monoamines DA and 5-HT and metabolites DOPAC, HVA,
3-MT, and 5-HIAA.
Behavioral results were analyzed using a Two-way ANOVA with the Bonferroni
posttests. Monoamine metabolite results were analyzed using a One-way ANOVA with
Bartlett's test for equal variances and Dunnett's Multiple Comparison Test.
Behavioral Results
GBL Treatment
The behavioral data for the GBL treatment is shown in Figures 22 A-D.
Movement episodes. The first set of experiments consisted of control mice (saline
+ saline) and mice given GBL (150 mg/kg) followed by saline. These two groups of
mice served as controls for the studies utilizing pergolide and nomifensine as post-
treatment therapies. The group of mice given GBL (150 mg/kg) showed that the loss
(p<0.001) of locomotor function (movement episodes) occurred within 10 minutes and
continued for the duration of the experiment (See Figure 22A). There were less than 10
moves per five minute time block between 10 and 45 minutes.
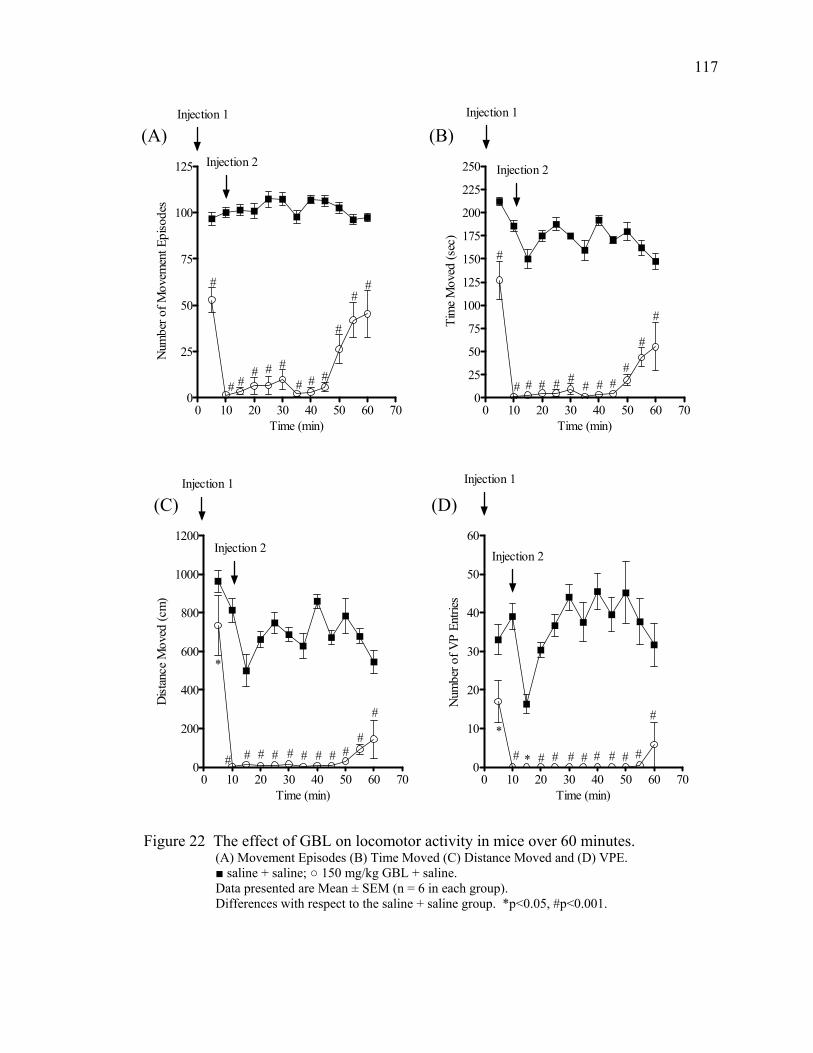
117
0 10 20 30 40 50 60 700
25
50
75
100
125
150
175
200
225
250 Injection 2
Injection 1
# # # # # #
#
##
#
#
#
Time (min)
Tim
e M
oved
(sec
)
0 10 20 30 40 50 60 700
25
50
75
100
125
#
Injection 2
Injection 1
#
# ## # #
# #
#
##
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
(A) (B)
0 10 20 30 40 50 60 700
10
20
30
40
50
60
*
*
Injection 2
Injection 1
#
#########
Time (min)
Num
ber o
f VP
Entri
es
0 10 20 30 40 50 60 700
200
400
600
800
1000
1200
*
Injection 2
Injection 1
#
##########
Time (min)
Dist
ance
Mov
ed (c
m)
(C) (D)
Figure 22 The effect of GBL on locomotor activity in mice over 60 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; ○ 150 mg/kg GBL + saline. Data presented are Mean ± SEM (n = 6 in each group). Differences with respect to the saline + saline group. *p<0.05, #p<0.001.

118
Between 50 and 60 minutes there was a slow, steady increase in the number of movement
episodes with a value of 45 movement episodes at 60 minutes. That final point at 60
minutes was still reliably lower than the control mice (p<0.001).
Time moved. The mice that received GBL moved for less time (p<0.001) than
the control mice beginning at five minutes and lasting until 60 minutes (See Figure 22B).
Distance moved. The group of mice given GBL (150 mg/kg) showed less than
100 cm of movement in every five minute segment between 5 and 50 minutes, which was
less (p<0.01) than the control group (See Figure 22C).
Vertical plane entries (VPE). The mice given GBL (150 mg/kg) showed VPE in
only two five minute time blocks (See Figure 22D). Those blocks were between 5 and 10
minutes and between 55 and 60 minutes. Otherwise, the mice showed no VPE in the
experiment. Between 5 and 60 minutes, the mice given saline + GBL (150 mg/kg) had
fewer (p<0.001) VPE than the control mice.
Pergolide Treatment
The behavioral data for the pergolide post-treatment is shown in Figures 23 A-D.
Movement episodes. The group of mice given pergolide (10 mg/kg) after GBL
(150 mg/kg) showed an increase (p<0.05) in movement episodes that began at 15 minutes
and continued for the duration of the experiment (See Figure 23A). From the beginning
of the study, the mice given saline + pergolide (10 mg/kg) had more movement episodes
(p<0.001) than the mice given GBL (150 mg/kg) + pergolide (10 mg/kg) (data not shown
in graph).
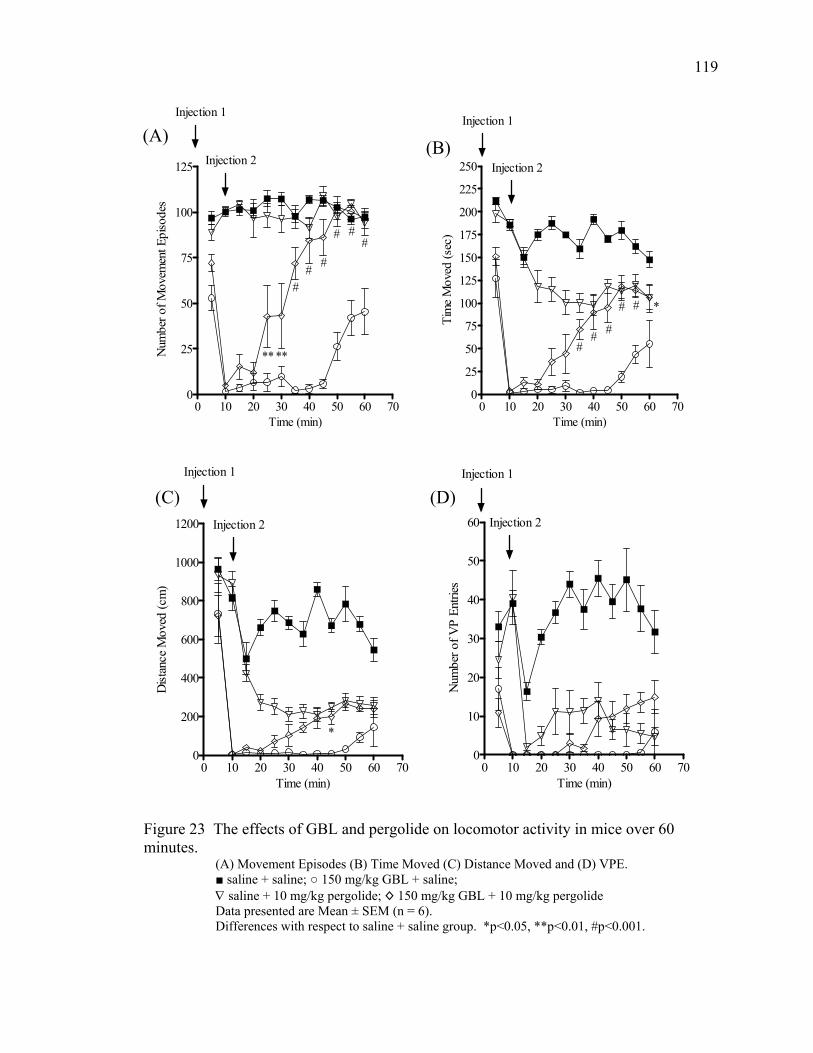
119
0 10 20 30 40 50 60 700
25
50
75
100
125
****
###
###
Injection 2
Injection 1
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
0 10 20 30 40 50 60 700
25
50
75
100
125
150
175
200
225
250
*##
###
Injection 2
Injection 1
Time (min)
Tim
e M
oved
(sec
)
(A) (B)
0 10 20 30 40 50 60 700
200
400
600
800
1000
1200
*
Injection 2
Injection 1
Time (min)
Dist
ance
Mov
ed (c
m)
0 10 20 30 40 50 60 700
10
20
30
40
50
60 Injection 2
Injection 1
Time (min)
Num
ber o
f VP
Entri
es
(C) (D)
Figure 23 The effects of GBL and pergolide on locomotor activity in mice over 60 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; ○ 150 mg/kg GBL + saline; ∇ saline + 10 mg/kg pergolide; ◊ 150 mg/kg GBL + 10 mg/kg pergolide Data presented are Mean ± SEM (n = 6). Differences with respect to saline + saline group. *p<0.05, **p<0.01, #p<0.001.

120
However, the mice given GBL (150 mg/kg) + pergolide (10 mg/kg) did show movement
episodes equal to those of the mice given saline + saline and the mice given saline +
pergolide (10 mg/kg) by the end of the study.
Time moved. The group of mice given GBL (150 mg/kg) + pergolide (10 mg/kg)
showed a response to the pergolide between 30 and 35 minutes (See Figure 23B). There
was then an increase (p<0.001) in the amount of time moved between 35 and 60 minutes
when compared to mice given GBL (150 mg/kg) + saline. The mice given GBL (150
mg/kg) + pergolide (10 mg/kg) moved for less time (p<0.01) than the mice given saline +
saline for the first 55 minutes in the study (data not shown in graph). After 35 minutes,
there was no reliable difference in the amount of time spent moving between the group of
mice given saline + pergolide (10 mg/kg) and the group of mice given GBL (150 mg/kg)
+ pergolide (10 mg/kg) (data not shown in graph).
Distance moved. The mice given pergolide (10 mg/kg) after GBL (150 mg/kg)
showed a gradual increase in the distance moved between 20 and 60 minutes (See Figure
23C). However, only the distance moved between 40 and 45 minutes was reliably greater
(p<0.05) than the mice given saline + GBL (150 mg/kg). The mice given GBL (150
mg/kg) + pergolide (10 mg/kg) moved less distance (p<0.05) than the mice given saline +
saline for the entire study (data not shown in graph). The group of mice that received
saline + pergolide (10 mg/kg) moved less and distance (p<0.001) than the mice that
received saline + saline between 20 and 60 minutes (data not shown in graph). After 20
minutes there was no statistically significant difference between the distances moved by
the two groups of mice that received pergolide (data not shown in graph).

121
Vertical plane entries (VPE). There mice given pergolide (10 mg/kg) as a post-
treatment experienced no statistically significant increase in VPE when compared with
mice given GBL (150 mg/kg) + saline (See Figure 23D). When compared to the mice
given saline + saline, the mice given saline + pergolide (10 mg/kg) had fewer VPE
(p<0.001) beginning at 20 minutes and continuing for the duration of the study (data not
shown in graph). The group of mice given GBL (150 mg/kg) + pergolide (10 mg/kg) had
fewer VPE (p<0.05) than the group of mice that received saline + saline (data not shown
in graph). That decrease in VPE was observed for the entire 60 minutes in the study.
Nomifensine Treatment
The behavioral data for the nomifensine post-treatment is shown in Figures 24 A-
D.
Movement episodes. The mice given GBL (150 mg/kg) + nomifensine (20
mg/kg) showed a rapid, but statistically non-significant spike in movement episodes
between 10 and 25 minutes (See Figure 24A). Between 45 and 60 minutes, the mice
given GBL (150 mg/kg) + nomifensine (10 mg/kg) showed an increase in activity that
was greater (p<0.001, p<0.01) than mice given only GBL. During that same period of
time, the mice given GBL (150 mg/kg) + nomifensine (10 mg/kg) showed movement
episodes that were not statistically different from the mice given saline + saline (data not
shown in graph). Between 20 and 30 minutes and 45 and 60 minutes, the two groups of
mice that received nomifensine (10 mg/kg) did not have statistically significant
differences in the number of movement episodes (data not shown in graph).
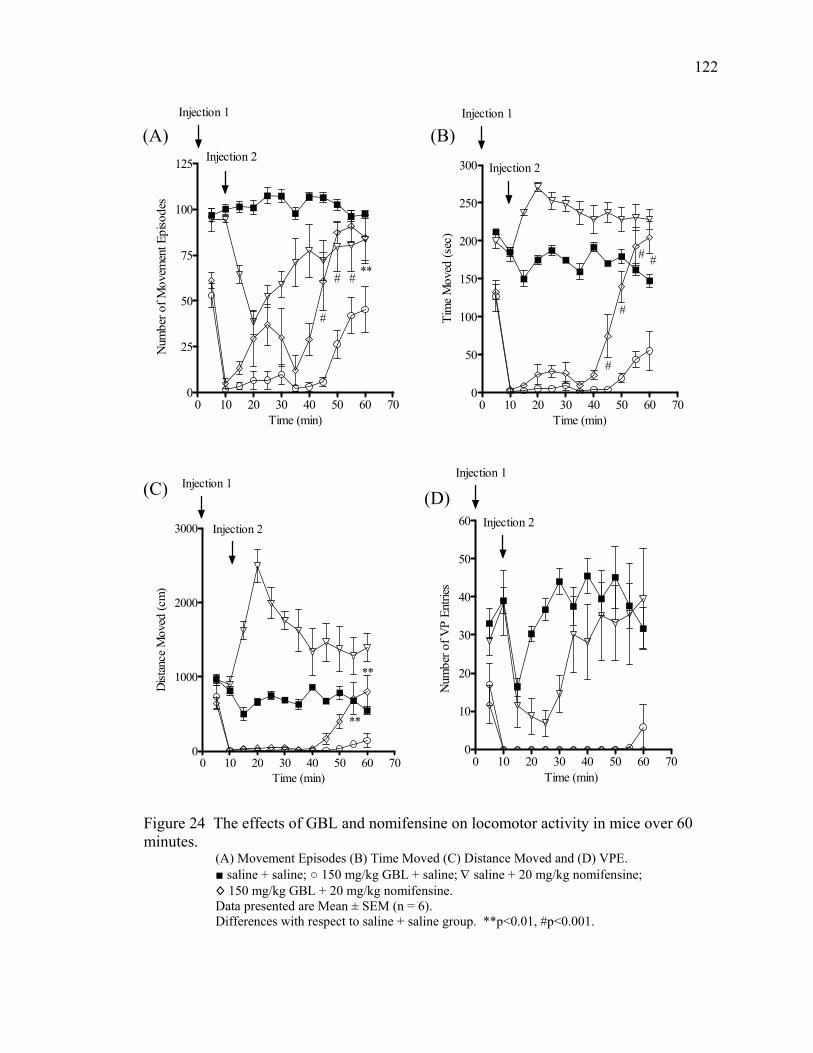
122
0 10 20 30 40 50 60 700
25
50
75
100
125
#
# # **
Injection 2
Injection 1
Time (min)
Num
ber o
f Mov
emen
t Epi
sode
s
0 10 20 30 40 50 60 700
50
100
150
200
250
300
#
Injection 2
Injection 1
#
# #
Time (min)
Tim
e M
oved
(sec
)
(A) (B)
0 10 20 30 40 50 60 700
10
20
30
40
50
60 Injection 2
Injection 1
Time (min)
Num
ber o
f VP
Entri
es
0 10 20 30 40 50 60 700
1000
2000
3000
**
**
Injection 1
Injection 2
Time (min)
Dist
ance
Mov
ed (c
m)
(C) (D)
Figure 24 The effects of GBL and nomifensine on locomotor activity in mice over 60 minutes.
(A) Movement Episodes (B) Time Moved (C) Distance Moved and (D) VPE. ■ saline + saline; ○ 150 mg/kg GBL + saline; ∇ saline + 20 mg/kg nomifensine; ◊ 150 mg/kg GBL + 20 mg/kg nomifensine. Data presented are Mean ± SEM (n = 6). Differences with respect to saline + saline group. **p<0.01, #p<0.001.

123
Time moved. The group of mice given GBL (150 mg/kg) + nomifensine (20
mg/kg) showed very little activity until 45 minutes (See Figure 24B). After 45 minutes
that group of mice began a steady and statistically significant increase (p<0.001) in the
amount of time moved that lasted the duration of the experiment when compared with the
group of mice given GBL (150 mg/kg) + saline. The mice that were given saline +
nomifensine (10 mg/kg) moved for more time between 15 and 30 minutes (p<0.01), at 45
minutes (p<0.01), and between 55 and 60 minutes (p<0.001) when compared to mice that
were given saline + saline (data not shown in graph). Between 55 and 60 minutes, the
two groups of mice that received nomifensine (10 mg/kg) no longer moved for reliably
different amounts of time (data not shown in graph).
Distance moved. The group of mice given GBL (150 mg/kg) + nomifensine (20
mg/kg) showed very little evidence of any distance moved from 10 to 40 minutes (See
Figure 24C). Within the final 20 minutes, those mice showed an increase in the distance
moved that became statistically reliable (p<0.01) for the final 10 minutes of the
experiment when compared to the group of mice given GBL (150 mg/kg) + saline. The
mice given saline + nomifensine (10 mg/kg) moved a greater distance between 15 and 35
minutes (p<0.001) and between 45 and 60 minutes (p<0.05) than the mice given saline +
saline (data not shown in graph).
Vertical plane entries (VPE). The mice given nomifensine (20 mg/kg) as a post-
treatment to GBL (150 mg/kg) did not increase their number of VPE when compared to
mice that received GBL (150 mg/kg) + saline (See Figure 24D). Between 20 and 30
minutes, the group of mice given saline + nomifensine (10 mg/kg) had fewer VPE
(p<0.05) than the group of mice given saline + saline (data not shown in graph).

Serotonin metabolites. The serotonin metabolites and turnover for the control
experiment are shown in Table 14. The mice that were given GBL (150 mg/kg) + saline
had decreases in 5-HIAA (p<0.05) and the serotonin turnover rate (p<0.01) when
compared to mice given saline + saline.
showed increases in DA (p<0.01), DOPAC (p<0.01), and a decrease in 3-MT (p<0.01)
when compared to mice that received saline + saline.
124
Summary of Behavioral Changes
In these experiments, both pergolide and nomifensine showed some degree of
efficacy at antagonizing the behavioral effects of GBL when given as a post-treatment.
The greatest antagonism was observed in the movement episodes parameter. Both
pergolide and nomifensine caused increases (p<0.01) in movement episodes beginning at
25 and 45 minutes respectively. At 35 and 45 minutes, both pergolide and nomifensine
also showed increases (p<0.05 and p<0.001 respectively) in the amount of time moved.
Significant increases in the distance moved were never observed before 45 minutes in
either experiment. There was no improvement seen in the number of VPE in these
experiments.
Monoamine Metabolite Results
GBL Treatment
Dopamine metabolites. The dopamine metabolites and turnover for the control
experiment comparing mice given saline + saline to mice given GBL (150 mg/kg) +
saline are shown in Table 13. The group of mice that received GBL followed by saline
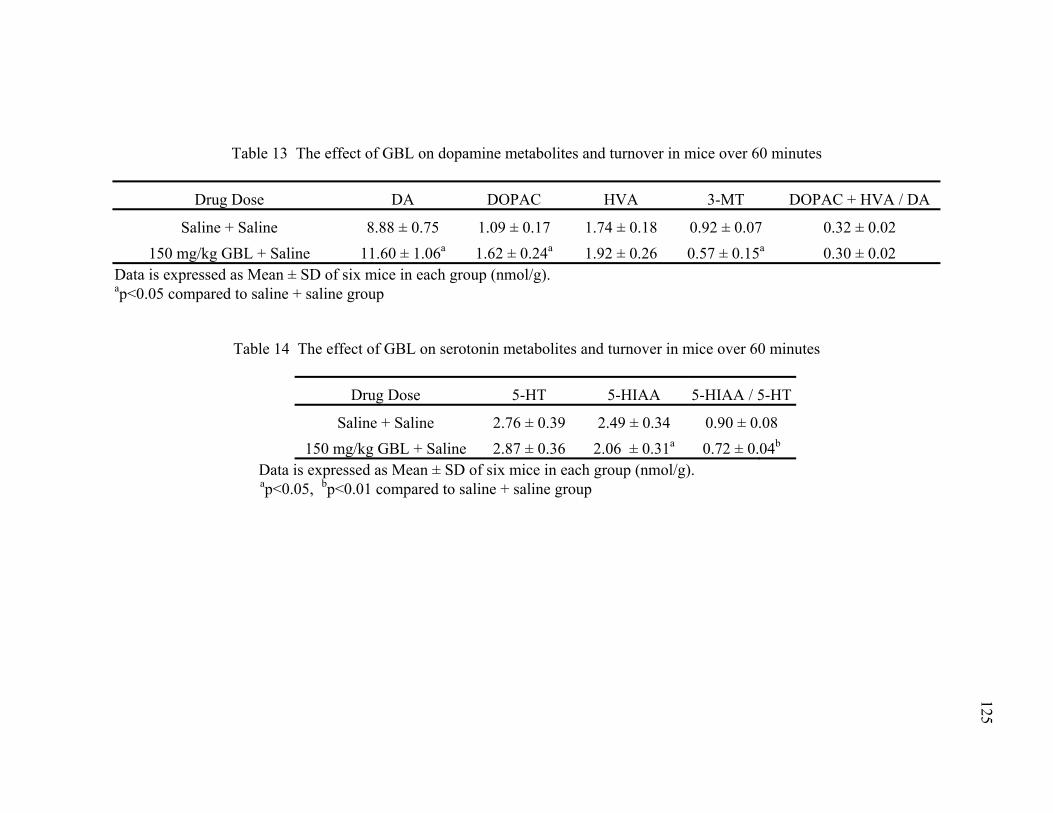
125
Table 13 The effect of GBL on dopamine metabolites and turnover in mice over 60 minutes
Drug Dose DA DOPAC HVA 3-MT DOPAC + HVA / DA
Saline + Saline 8.88 ± 0.75 1.09 ± 0.17 1.74 ± 0.18 0.92 ± 0.07 0.32 ± 0.02 0.30 ± 0.02 150 mg/kg GBL + Saline 11.60 ± 1.06a 1.62 ± 0.24a 1.92 ± 0.26 0.57 ± 0.15a
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.05 compared to saline + saline group
Table 14 The effect of GBL on serotonin metabolites and turnover in mice over 60 minutes
Drug Dose 5-HT 5-HIAA 5-HIAA / 5-HT
Saline + Saline 2.76 ± 0.39 2.49 ± 0.34 0.90 ± 0.08 0.72 ± 0.04150 mg/kg GBL + Saline 2.87 ± 0.36 2.06 ± 0.31a b
Data is expressed as Mean ± SD of six mice in each group (nmol/g). p<0.01 compared to saline + saline group bap<0.05,

126
Pergolide Treatment
Dopamine metabolites. The dopamine metabolites and turnover from the
pergolide post-treatment study are shown in Table 15. The mice given saline + pergolide
(10 mg/kg) had lower DOPAC (p<0.01), HVA (p<0.01), 3-MT (p<0.01) levels and a
lower dopamine turnover rate (p<0.01) when compared to mice given saline + saline.
When comparing mice given GBL (150 mg/kg) + pergolide (10 mg/kg) to mice given
GBL (150 mg/kg) + saline, there were no statistically significant changes in dopamine or
3-MT levels. However, there were decreases in DOPAC (p<0.01), HVA (p<0.01) and
dopamine turnover (p<0.01).
Serotonin metabolites. The serotonin metabolites and turnover for the pergolide
post-treatment study are shown in Table 16. The mice that received saline + pergolide
(10 mg/kg) had increases in 5-HT (p<0.05), and decreases in 5-HIAA (p<0.01), and
serotonin turnover (p<0.01) when compared to mice that received saline + saline. When
compared to mice that received only GBL (150 mg/kg), the group of mice that received
GBL (150 mg/kg) + pergolide (10 mg/kg) had an increase (p<0.01) in 5-HT levels and a
decrease in 5-HIAA levels (p<0.01) and serotonin turnover (p<0.01).
Nomifensine Treatment
Dopamine metabolites. The dopamine metabolites from the nomifensine post-
treatment study are shown in Table 17. The mice given saline + nomifensine (20 mg/kg)
had a lower (p<0.01) dopamine turnover rate than mice given saline + saline. There were
no statistically significant changes seen in DA, HVA, DOPAC, or 3-MT levels in mice
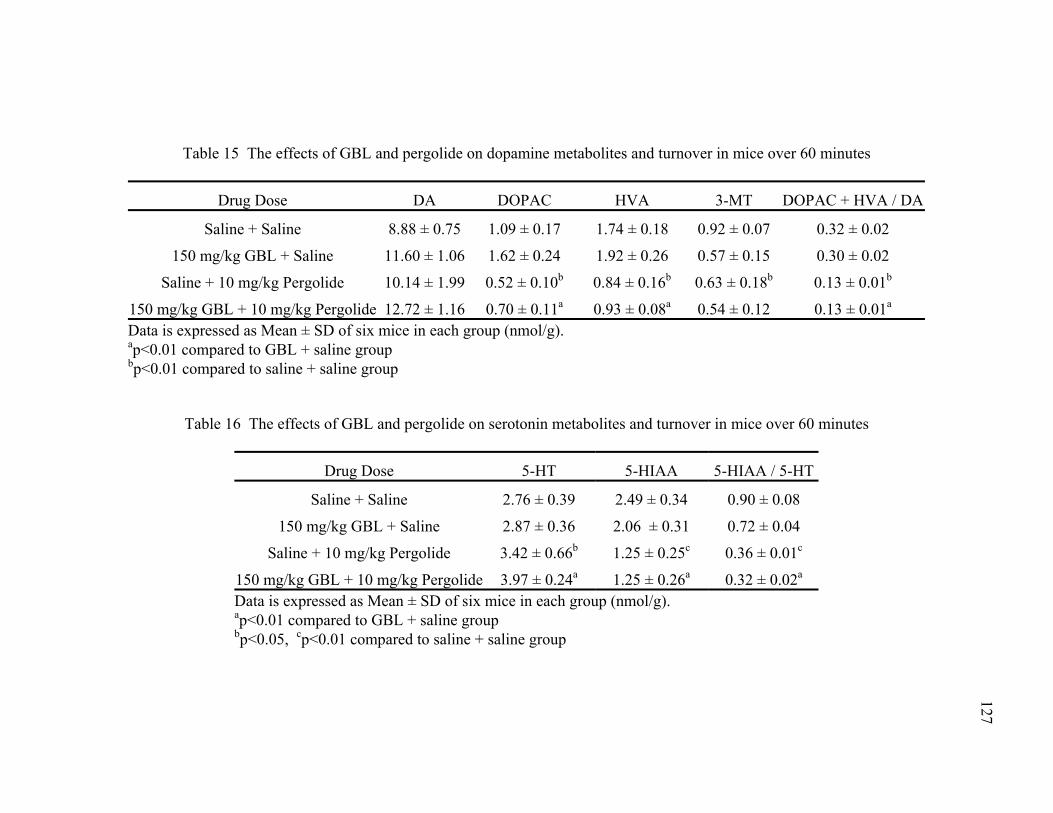
Table 15 The effects of GBL and pergolide on dopamine metabolites and turnover in mice over 60 minutes
Drug Dose DA DOPAC HVA 3-MT DOPAC + HVA / DA
Saline + Saline 8.88 ± 0.75 1.09 ± 0.17 1.74 ± 0.18 0.92 ± 0.07 0.32 ± 0.02
150 mg/kg GBL + Saline 11.60 ± 1.06 1.62 ± 0.24 1.92 ± 0.26 0.57 ± 0.15 0.30 ± 0.02
Saline + 10 mg/kg Pergolide 10.14 ± 1.99 0.52 ± 0.10b 0.84 ± 0.16b 0.63 ± 0.18b 0.13 ± 0.01b
150 mg/kg GBL + 10 mg/kg Pergolide 12.72 ± 1.16 0.70 ± 0.11a 0.93 ± 0.08a 0.54 ± 0.12 0.13 ± 0.01a
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.01 compared to GBL + saline group bp<0.01 compared to saline + saline group
Table 16 The effects of GBL and pergolide on serotonin metabolites and turnover in mice over 60 minutes
Drug Dose 5-HT 5-HIAA 5-HIAA / 5-HT
Saline + Saline 2.76 ± 0.39 2.49 ± 0.34 0.90 ± 0.08
150 mg/kg GBL + Saline 2.87 ± 0.36 2.06 ± 0.31 0.72 ± 0.04
Saline + 10 mg/kg Pergolide 3.42 ± 0.66b 1.25 ± 0.25c 0.36 ± 0.01c
150 mg/kg GBL + 10 mg/kg Pergolide 3.97 ± 0.24a 1.25 ± 0.26a 0.32 ± 0.02a
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.01 compared to GBL + saline group bp<0.05, cp<0.01 compared to saline + saline group
127
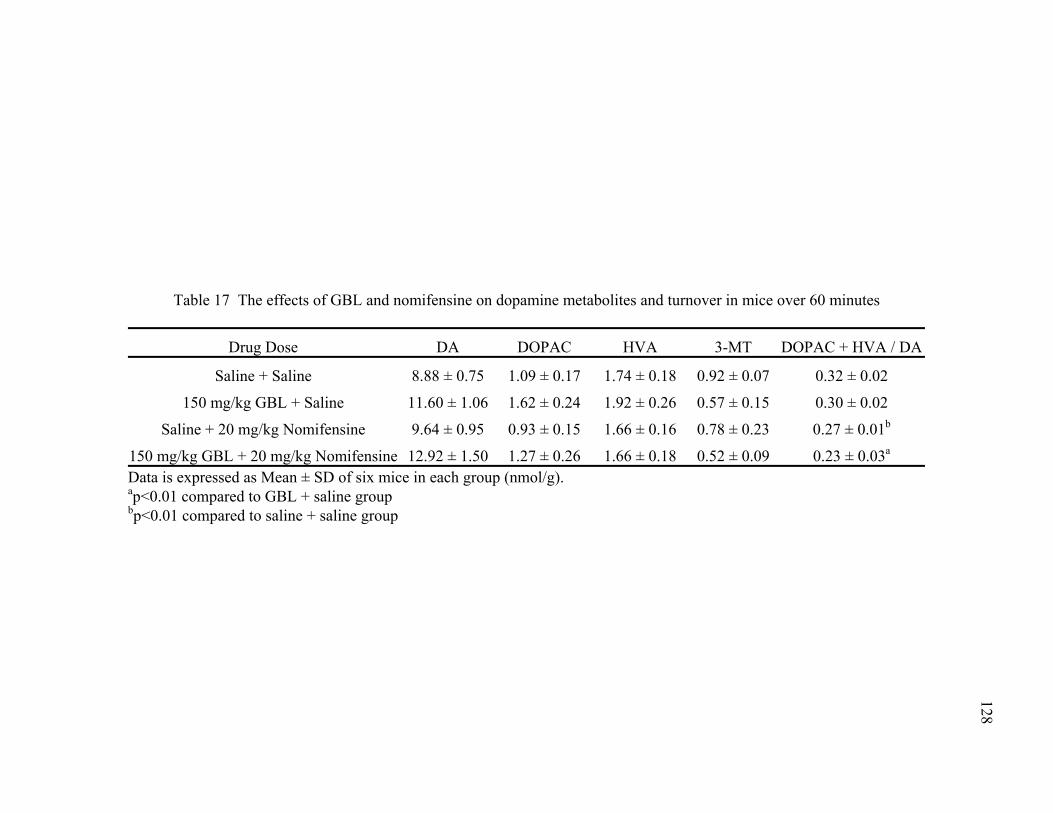
128
Table 17 The effects of GBL and nomifensine on dopamine metabolites and turnover in mice over 60 minutes
Drug Dose DA DOPAC HVA 3-MT DOPAC + HVA / DA
Saline + Saline 8.88 ± 0.75 1.09 ± 0.17 1.74 ± 0.18 0.92 ± 0.07 0.32 ± 0.02
150 mg/kg GBL + Saline 11.60 ± 1.06 1.62 ± 0.24 1.92 ± 0.26 0.57 ± 0.15 0.30 ± 0.02
Saline + 20 mg/kg Nomifensine 9.64 ± 0.95 0.93 ± 0.15 1.66 ± 0.16 0.78 ± 0.23 0.27 ± 0.01b
0.23 ± 0.03150 mg/kg GBL + 20 mg/kg Nomifensine 12.92 ± 1.50 1.27 ± 0.26 1.66 ± 0.18 0.52 ± 0.09 a
Data is expressed as Mean ± SD of six mice in each group (nmol/g). ap<0.01 compared to GBL + saline group bp<0.01 compared to saline + saline group

129
given GBL (150 mg/kg) followed by nomifensine (20 mg/kg). The group of mice that
received GBL (150 mg/kg) + nomifensine (20 mg/kg) had a lower rate of dopamine
turnover (p<0.01) when compared with the group of mice that received GBL (150 mg/kg)
+ saline.
Serotonin metabolites. The serotonin metabolites and turnover rates for the
nomifensine post-treatment study are shown in Table 18. The group of mice that
received saline + nomifensine (20 mg/kg) had lower 5-HIAA (p<0.01) levels and
serotonin turnover rate (p<0.01) than the group of mice that received saline + saline. The
mice given GBL (150 mg/kg) followed by nomifensine (20 mg/kg) had more 5-HT
(p<0.01) and a lower serotonin turnover rate (p<0.01) than the mice given only GBL (150
mg/kg).
Summary of Monoamine Metabolite Changes
DOPAC levels and the dopamine and serotonin turnover rates were affected more
by pergolide and nomifensine than the remainder of the metabolites. When compared to
mice that received only GBL, all groups of mice that received pergolide as a post-
treatment to GBL, had lower (p<0.01) levels of DOPAC. Both drugs decreased (p<0.01)
the rate of dopamine turnover as well as the rate of serotonin turnover (p<0.01).
Additionally, 5-HT levels in mice given either GBL (150 mg/kg) + pergolide (10 mg/kg)
or GBL (150 mg/kg) + nomifensine (20 mg/kg) were higher (p<0.01) than the levels seen
in mice given only GBL (150 mg/kg).
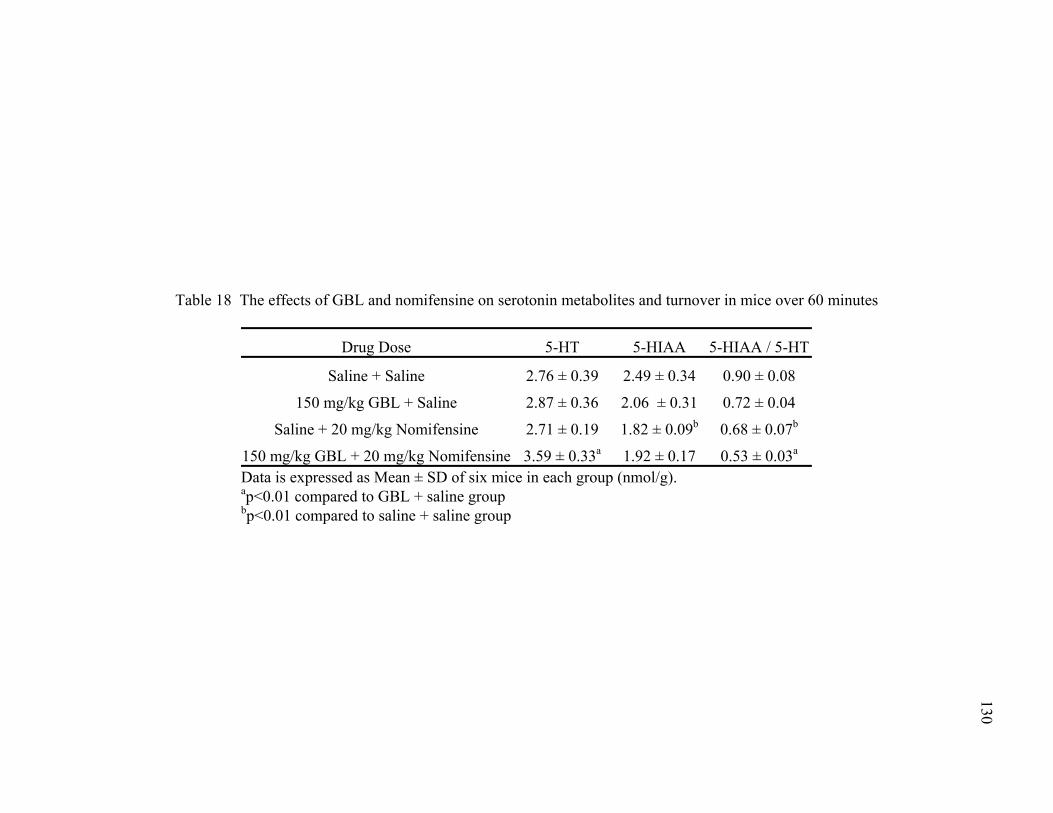
130
Table 18 The effects of GBL and nomifensine on serotonin metabolites and turnover in mice over 60 minutes
Drug Dose 5-HT 5-HIAA 5-HIAA / 5-HT
Saline + Saline 2.76 ± 0.39 2.49 ± 0.34 0.90 ± 0.08
150 mg/kg GBL + Saline 2.87 ± 0.36 2.06 ± 0.31 0.72 ± 0.04
Saline + 20 mg/kg Nomifensine 2.71 ± 0.19 1.82 ± 0.09b 0.68 ± 0.07b
0.53 ± 0.03150 mg/kg GBL + 20 mg/kg Nomifensine 3.59 ± 0.33a 1.92 ± 0.17 a
Data is expressed as Mean ± SD of six mice in each group (nmol/g).
ap<0.01 compared to GBL + saline group p<0.01 compared to saline + saline group b

131
Discussion
The experiments described in this chapter evaluated the ability of pergolide and
nomifensine to antagonize the behavioral effects of GBL when administered as post-
treatments. The design of these experiments more closely resembled an acute overdose
condition or state than the experiments described in Chapter Five.
When pergolide was given as a post-treatment for GBL, it caused an increase in
movement episodes and time moved, beginning 15 and 25 minutes respectively, after
administration and continuing for the duration of the experiment. The other two
parameters did not show any statistically significant improvement other than the distance
moved between 45 and 50 minutes. These results show that pergolide partially
antagonized the locomotor effects of GBL when administered as a post-treatment.
As a D /D1 2 dopamine receptor agonist, pergolide caused stimulation of dopamine
receptors. In Chapters Three and Four, GBL was shown to inhibit dopamine release
leading to loss of locomotor function. By administering pergolide, the dopaminergic
system was stimulated, even though dopamine release was inhibited by GBL, as indicated
by decreased 3-MT levels and decreased dopamine turnover rate. It appeared, however,
that stimulating the dopamine receptors (D /D1 2) with an agonist only partially
antagonized the locomotor effects of GBL. Partial antagonism was unable to improve the
distance moved and VPE. The results regarding rearing (VPE) have been observed
previously to some degree. In a study testing NCS-382 as a GHB antagonist, it was able
to antagonize the decrease in forelimb strength and distance traveled caused by GHB
(Cook and others 2002). However, NCS-382 was unable to antagonize the dose
dependent decrease in the number of rears induced by GHB.

132
The mice given saline + pergolide (10 mg/kg) verified the effects that pergolide
had on the dopaminergic system before combining with GBL. When pergolide
stimulated D /D1 2 dopamine receptors, a feedback mechanism decreased the presynaptic
release of dopamine. This was confirmed by the decreases in DOPAC, HVA, 3-MT, and
turnover. Those same mice had higher levels of 5-HT, 5-HIAA, and serotonin turnover.
That result indicates that pergolide affects the serotonin system in addition to stimulating
D /D1 2 dopamine receptors. It is possible that the increase in serotonin metabolites is a
secondary effect of decreased dopamine release. Pergolide has been shown to be an
agonist at h5-HT1A, h5-HT1B, h5-HT1D, h5-HT2A, h5-HT2B, and h5-HT2C serotonin
receptors subtypes (Newman-Tancredi and others 2002). 5-HT1A receptors are localized
in areas such as the striatum, nucleus accumbens, and frontal cortex, all areas involved
with motor function (Barnes and Sharp 1999; Millan and others 2000). Pergolide could
be stimulating 5-HT1B receptors, which can inhibit DA release in the striatum (Sarhan
and others 2000) and therefore account for a role in the decreased dopamine metabolites.
5-HT2A receptors are upregulated when nigrostriatal dopaminergic input is inhibited
(Numan and others 1995; Barnes and Sharp 1999; Gresch and Walker 1999). When 5-
HT2A receptors are activated, they enhance DA release in the striatum (Ng and others
1999; De Deurwaerdere and Spampinato 2001). These receptors could account for some
of the increases observed in both dopamine and serotonin metabolites.
The partial antagonism of GBL was accompanied by decreases in DOPAC, HVA,
and the dopamine turnover rate. Over 60 minutes, the inhibition of dopamine release
causes intracellular dopamine to accumulate, but also leads to depletion of dopamine
metabolites as seen by the significant in DOPAC and HVA levels. The serotonin

133
metabolite analysis showed decreased 5-HIAA and serotonin turnover and an increase in
5-HT. The increase in 5-HT levels has been shown previously (Gobaille and others
2002) although such increases are often accompanied by increases in 5-HIAA
(Waldmeier and Fehr 1978; Hedner and Kundborg 1983), which is in contrast to the
results reported in this chapter. In the present study, pergolide, which stimulates
dopamine receptors and restores dopaminergic activity, did not affect the decrease in 5-
HIAA and the serotonin turnover rate induced by GBL as is indicated by the levels
presented in Table 16. However, the 5-HT levels increased more, when compared to
saline control mice, when both GBL and pergolide were administered together than when
pergolide was administered independently. However, the greatest increase in 5-HT levels
occurred when pergolide was the only compound administered to the mice. That result
suggests that pergolide had a more profound effect on 5-HT levels than did the
combination of pergolide and GBL. This indicates that the effects of GBL on the
serotonergic system are not solely dependent on D /D dopamine receptor activation. 1 2
Nomifensine also partially antagonized the locomotor effects of GBL.
Statistically significant increases in movement episodes and time moved were present in
both parameters beginning at 45 minutes, and continued until the end of the experiment.
The distance moved showed a reliable change at 55 and 60 minutes. The mice
experienced no increase in VPE when compared to mice given only GBL.
Nomifensine, a monoamine reuptake inhibitor, functioned as a post-treatment for
GBL toxicity by allowing dopamine to remain in the synaptic cleft, prolonging synaptic
stimulation. This method of increasing central dopaminergic activity appeared to take
some time as the locomotor effects in the mice did not improve until 35 minutes after

134
nomifensine was given. There were other factors that contributed to the time in which
nomifensine is able to exert its effects. The uptake and absorption of nomifensine
throughout the body could take a long time, as could adequate concentration at central
target sites. Additionally, the physiological response of nomifensine could take longer
than other compounds. Manipulating the dopaminergic system by altering reuptake was
not sufficient to completely antagonize all locomotor deficits induced by GBL.
Mice given saline + nomifensine (20 mg/kg) provided clarification of the effects
that nomifensine had on dopamine and serotonin levels before it was given as a post-
treatment for GHB. When compared with mice given saline + saline, the mice given
saline + nomifensine (20 mg/kg) had lower dopamine turnover, 5-HIAA, and serotonin
turnover. While the dopaminergic metabolites did not change significantly, as a reuptake
inhibitor, nomifensine kept dopamine in the synaptic cleft until metabolized. This lead to
a slow down for the formation of all metabolites and thus a lower total dopamine
turnover. The decreased 5-HT turnover implied that the serotonergic system was down
regulated due to nomifensine. This was most likely a secondary effect of dopamine
reuptake inhibition.
Nomifensine’s partial antagonism of the effects of GBL leads to few monoamine
neurotransmitter changes. None of the dopamine metabolites were statistically different.
The only change was a decrease in dopamine turnover. The inhibition of dopamine
release likely left little dopamine reuptake to be prevented. However, DOPAC and HVA
levels did not change significantly, indicating some source of dopamine. After 60
minutes, perhaps the post inhibition release of dopamine had occurred and the
metabolites were replenished, which lead to the levels shown here. The reduced turnover

135
rate suggests that if dopamine release did occur, it was late in the time period. Despite
the decrease in turnover, nomifensine did partially antagonize the effects of GBL. The
serotonin levels in this study were increased and the serotonin turnover rate was
decreased. Serotonin was relatively unaffected until both GBL and nomifensine were
administered together. In combination those compounds lead to increased 5-HT levels,
but decreased serotonin turnover. This suggests that increasing synaptic dopamine levels
may have a role in either 5-HT synthesis or accumulation.
Both pergolide and nomifensine showed the ability to partially antagonize the
effects of GBL when administered as post-treatments. This is only the third successful
attempt at antagonizing the effects of GHB or GBL with a post-treatment. One
successful antagonism used pargyline as a pre-treatment to GHB in rats (Anderson and
others 2002). A second success was observed by Carai and others (Carai and others
2001) when SCH 59011 reduced the time needed for mice to regain the righting reflex
when administered after GHB. A case report from Berkeley University described an
attempt to revive an individual in a GHB induced coma by administering crystal
methamphetamine intranasally (Kohrs and others 2004). This “treatment” induced
movement, but did not reverse the cognitive effects of GHB. While methamphetamines
antagonized the GHB to a certain extent, it is difficult to definitively label this as a
successful antagonism of GHB. This report does, however, suggest that amphetamines
and methamphetamines should be utilized as another way to clarify the mechanism of
action GHB has in the CNS. Therefore, to our knowledge, the data presented in this
chapter is the first successful antagonism of the effects of GBL using a post-treatment.
Pergolide appeared to exert its effects more rapidly after administration than nomifensine,

136
but each showed a return to approximately the same number of movement episodes as the
other. However, nomifensine showed a greater amount of time moved per five minutes
by the end of the study than pergolide did. Both drugs also showed evidence that GBL’s
effects on the serotonergic system are dependent on dopamine. Further investigation
using regional dissection of brain regions that are heavily innervated with serotonin
neurons might provide a better insight at how GBL and dopamine interact in those
neurons or systems.
While pergolide and nomifensine were successful in this study, it is unlikely they
would be used in clinical situations involving GHB or GBL overdoses. GHB or GBL
overdoses are often associated with the use of other drugs of abuse and almost always by
alcohol (Catalano and others 2001). It is also difficult to ascertain how much GHB has
been consumed as patients often refer to the number of capfuls (GHB or GBL in liquid
form) they have consumed (Okun and others 2001). Toxicology screens can be negative
(Catalano and others 2001) and few hospitals have the equipment or assay for testing
GHB or GBL levels. When examining clinical situations, SSADH patients should be
considered, as their diagnosis is clear. Pergolide and nomifensine could be useful in
treating the ataxia that is seen in SSADH patients. However, increasing central
dopaminergic activity does not combat the characteristically high levels of GABA and
GHB seen in those patients.
Both of these drugs antagonized the effects of GBL although not completely in all
four parameters. The results suggests that stimulation of central dopaminergic activity is
one pharmacological strategy that can protect against GHB toxicity. Other compounds
that have shown sporadic results such as NCS-382, pargyline, and taurine should not be

137
abandoned, but should be considered as augmentation to compounds that have shown
success. Pergolide and NCS-382 should complement each other, as they would affect
different systems shown to have a role in the metabolism of GHB or GBL. Raevskii and
others (Raevskii and others 2002) described an example of this type of combination
therapy. GBR 12909, a dopamine reuptake inhibitor, increased extracellular striatal
dopamine concentration s by 220-280%. When tolcapone was administered in
conjunction with GBR 12909, dopamine levels increased by 500-600%. A single drug or
compound may not fully antagonize the behavioral effects of GBL. Thus, multiple
compounds should be considered in future studies. In addition to antagonizing the
locomotor effects of GBL, the mechanism of action might also be more fully understood.

CHAPTER SEVEN
Chronic Effect of GBL on Locomotor Activity and Brain Monoamine Neurotransmitter Metabolism in Mice
Introduction
Very few studies have focused on the behavioral effects that develop from long
term use of GHB or GBL. This is surprising, as the abuse of GHB is a rising health care
concern. In 1990, GHB received a Schedule I rating by the FDA, due to the increase in
overdoses and ER cases seen across the country. GHB was, however, approved for use in
treating cataplexy associated with narcolepsy in 2002 (Anonymous 2002a). Furthermore,
SSADH patients have high levels of GHB associated a variety of locomotor and muscular
control symptoms (Jakobs and others 1993; Gibson and others 1997). Knock out mouse
models of SSADH deficiency all experience high levels of GHB from birth and
subsequently die within 22 days of life (Gupta and others 2003). Despite these examples
of long term exposure to GHB in a variety of arenas, the consequences of long term use
are poorly understood.
Chronic use of GHB has most often resulted in tolerance to sedative effects and
locomotor activity deficits. Furthermore, in humans when chronic GHB use is abruptly
discontinued, withdrawal symptoms appear very quickly and resemble symptoms of
ethanol withdrawal. Two studies in baboons have investigated the behavioral effects and
physical dependence of chronic GHB administration (Goodwin and others 2005; Weerts
and others 2005). The first study showed decreased performance on a fine motor task
between 10 and 15 days in three of four baboons. That was then followed by gradual
138

139
improvement between days 20 and 25 (Weerts and others 2005). Ataxia, muscle
relaxation (lipdroop) and decreases in food maintained behavior were also present in the
baboons. Within six hours of discontinued GHB administration, withdrawal symptoms
consisting of disruption on a fine motor task, body tremors, aggression, and limb tremors
occurred (Weerts and others 2005). In the second study, four baboons exhibited a dose
dependent decrease in the number of food pellets earned in a lever pressing task during
the chronic treatment schedule (Goodwin and others 2005). The same study showed no
deficits in fine motor task capability one hour after GHB administration, but significant
deficits were observed two hours after GHB administration. Those deficits were present
in all baboons given the two highest doses of GHB (320 & 420 mg/kg). In addition to the
difficulties on the fine motor task observed at high doses, tremors, ataxia, jerks, and
gastrointestinal discomfort were observed for all doses of GHB. Those symptoms of
chronic GHB administration seen in baboons have also been observed in humans
(Galloway and others 1997; Nicholson and Balster 2001).
A study by Colombo and others (Colombo and others 1995) tested the behavioral
tolerance to GHB (1g/kg) administration in rats over a nine day period of administration.
Rats were tested on a rotarod immediately before GHB administration and 60 minutes
post GHB administration. By the end of the nine days, the mice demonstrated less
impairment on the rotarod, 60 minutes post GBL, than on the first six days of the study.
Separate studies of GHB in rats found doses between 0.25 and 2 g/kg given every three
hours for six days resulted in tolerance to the intoxicating effects of GHB (Bania and
others 2003). Itzhak and others (Itzhak and Ali 2002) showed tolerance to

140
hypolocomotion after six days of GHB administration (200 mg/kg/day). After 14 days
increased levels of striatal dopamine were observed.
Long term use of GHB has also shown to lead to physical dependence as is
evidenced by withdrawal upon discontinuation. Multiple studies have documented cases
of GHB withdrawal from a clinical perspective. The most common symptoms associated
with GHB withdrawal were tremors, insomnia, anxiety, muscle cramps, severe agitation,
disorientation, paranoia, auditory and visual hallucinations, tachycardia, elevated blood
pressure, and diaphoresis (Craig and others 2000). In most cases, the insomnia lasted
only three days (Nicholson and Balster) and the remaining signs of withdrawal subsided
after nine days (Craig and others 2000). A common approach to treating GHB
withdrawal has been the use of benzodiazepines, specifically lorazepam for agitation
(Craig and others 2000). In a review of the literature, Catalano and others (Catalano and
others 2001) described six patients who had a median duration of 24 months of GHB
abuse followed by withdrawal. In contrast, Mamelak and others (Mamelak and others
1986) reported no tolerance to GHB in 48 patients over the course of nine years in a
clinical trial for GHB as a treatment for narcolepsy.
Case reports of GBL withdrawal are rarer than those of GHB withdrawal.
Catalano and others (Catalano and others 2001) reported the first three cases of GBL
withdrawal in 2001. GBL withdrawal symptoms included insomnia, chills, shaking,
vivid nightmares, paranoia, agitation, and auditory, visual, and tactile hallucinations. The
agitation was treated with lorazepam (Catalano and others 2001) as it had proved useful
in earlier GHB withdrawal cases and is often used for ethanol withdrawal (Hernandez
and others 1998; Addolorato and others 1999; Craig and others 2000).

141
The experiments in this chapter examined the effects of low and high dose GBL
administration in mice for 14 consecutive days. The behavioral effects induced by GBL
were analyzed using a rotarod system, which measured fine motor coordination and
locomotor activity. It was anticipated that chronic administration of GBL and the
inhibition of dopamine release would result in a decrease in fine motor coordination over
the course of the experiment. Dopamine, serotonin and related metabolites were also
determined after chronic treatment with GBL.
Materials & Methods
The mice were divided into four groups of six. The four treatment groups were
saline, 50 mg/kg, 150 mg/kg, and 300 mg/kg. GBL injections were prepared fresh daily
and administered via i.p. injection.
The locomotor activity and coordination of the mice were monitored in the
rotarod system. In earlier studies, mice with motor deficits have had difficulty remaining
on a rotarod for a total of five minutes with a top speed of 40 rpm (Crawley 2000). All
mice in this study were allowed to adjust to ambient light and temperature in the testing
room for one hour prior to the experiments. To begin each trial, mice were placed on the
drum, which was already rotating at 5 rpm. The speed of the drum increased by 10
rpm/minute until reaching a top speed of 35 rpm. The drum then continued for an
additional two minutes making the entire run time five minutes. If the mouse fell to the
cage floor at any point in the experiment, a 0.2 mV shock was administered for three
seconds. After three seconds, the drum stopped rotating and the trial was finished. If the
mouse was able to get back onto the drum within the three second shock, the shocking
ceased and the experiment continued. After a total time of five minutes had elapsed, the

142
drum stopped and the mouse was removed from the testing chamber immediately. This
training schedule was adapted from Ho and others (Ho and others 1997). All mice were
given a training regimen of five trials per day for four consecutive days with all
individual trials being at least 30 minutes apart. After the training days, each mouse was
tested on days 0, 1, 7, 8, 14, and 15. Days 0, 8, and 15 tested each mouse in five trials as
described. Day 8 was after seven injections had been given and day 15 was after all 14
injections had been given. On those particular days, the GBL injections were
administered after the five trials had been completed. Days 1, 7, and 14 used a different
testing protocol. The mice received their injections as scheduled and were then tested on
the rotarod one, two, and four hours post injection. The parameters monitored in these
experiments were the amount of time each mouse remained on the drum and the speed of
the drum when each mouse fell off. The aim of the testing on days 0, 8, and 15 was to
assess the coordination of the mice prior to any injections, after day seven, and after day
14 of the chronic treatment schedule. The aim of testing on days 1, 7, and 14 was to
evaluate the time course of the effects of GBL immediately after the injections.
After each experiment, all animals were immediately asphyxiated by means of
CO2 and sacrificed via cervical dislocation. The brains were harvested for further
monoamine analysis. Brains were collected as half brains without the cerebellum.
The monoamine and metabolite analysis is identical to that previously described
in Chapter Four. A single half brain (without cerebellum) for each mouse was analyzed
for the monoamine neurotransmitters DA and 5-HT and metabolites DOPAC, 5-HIAA,
HVA, 3-MT, and 5-HIAA.

143
Behavioral results were analyzed using a Two-way ANOVA with the Bonferroni
posttests. Monoamine metabolite results were analyzed using a One-way ANOVA with
Bartlett's test for equal variances and Dunnett's Multiple Comparison Test.
Behavioral Results
The results from the rotarod experiments are shown in Figures 25-28. The first
two graphs show data for days 0, 8, and 15 (See Figures 25 A & B). The data displayed
is the total time spent on the rotating drum (See Figure 25A) and the speed of the drum
when the mice fell off (See Figure 25B). All four treatment groups began with times
between 150 and 200 seconds on day zero (the day before the GBL injections began) of
the study. That range of times corresponded to speeds between 25 and 30 rpm. Days
eight and 15 (24 hours after the last injection) both showed times between 100 and 250
seconds spent on the drum. Those times corresponded to speeds between 20 and 35 rpm.
On these three days of testing, none of the three GBL treatment groups showed any
significant differences in time spent on the rotating drum or the speed of the drum when
compared to saline controls.
Figures 26-28 show the results of the rotarod trials that were one, two, and four
hours post injection. There were several important changes observed over the course of
14 days. On day one, the saline group and the 50 mg/kg group spent between 100-150
and 200-250 seconds respectively on the rotarod for all three testing times (See Figure
26A). This translated to top speeds of between 20-25 and 30-35 rpm respectively (See
Figure 26B). The 150 mg/kg group had a behavioral change two hours after the GBL
injections on day one. That group had a top speed that was higher (p<0.05) than the top
speed of the control group two hours after the injections (See Figure 26B).
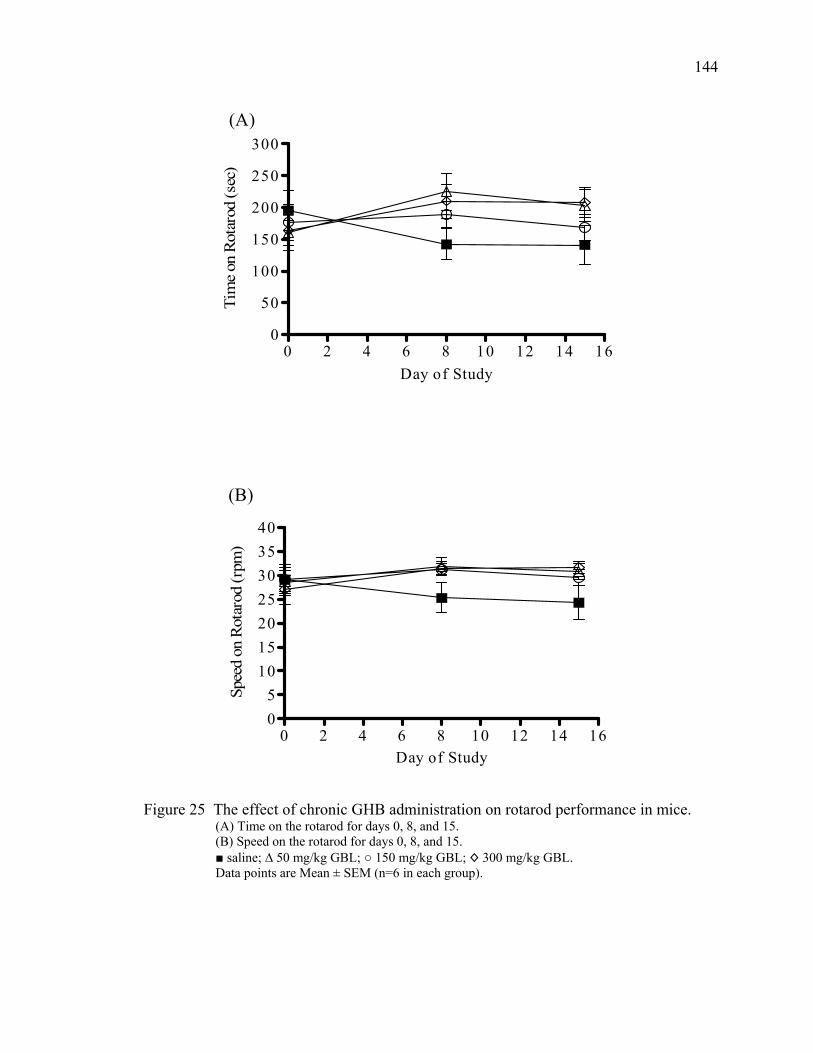
144
0 2 4 6 8 10 12 14 160
50
100
150
200
250
300(A)
Day of Study
Tim
e on
Rot
arod
(sec
)
0 2 4 6 8 10 12 14 1605
10152025303540
(B)
Day of Study
Spee
d on
Rot
arod
(rpm
)
Figure 25 The effect of chronic GHB administration on rotarod performance in mice.
(A) Time on the rotarod for days 0, 8, and 15. (B) Speed on the rotarod for days 0, 8, and 15. ■ saline; ∆ 50 mg/kg GBL; ○ 150 mg/kg GBL; ◊ 300 mg/kg GBL. Data points are Mean ± SEM (n=6 in each group).
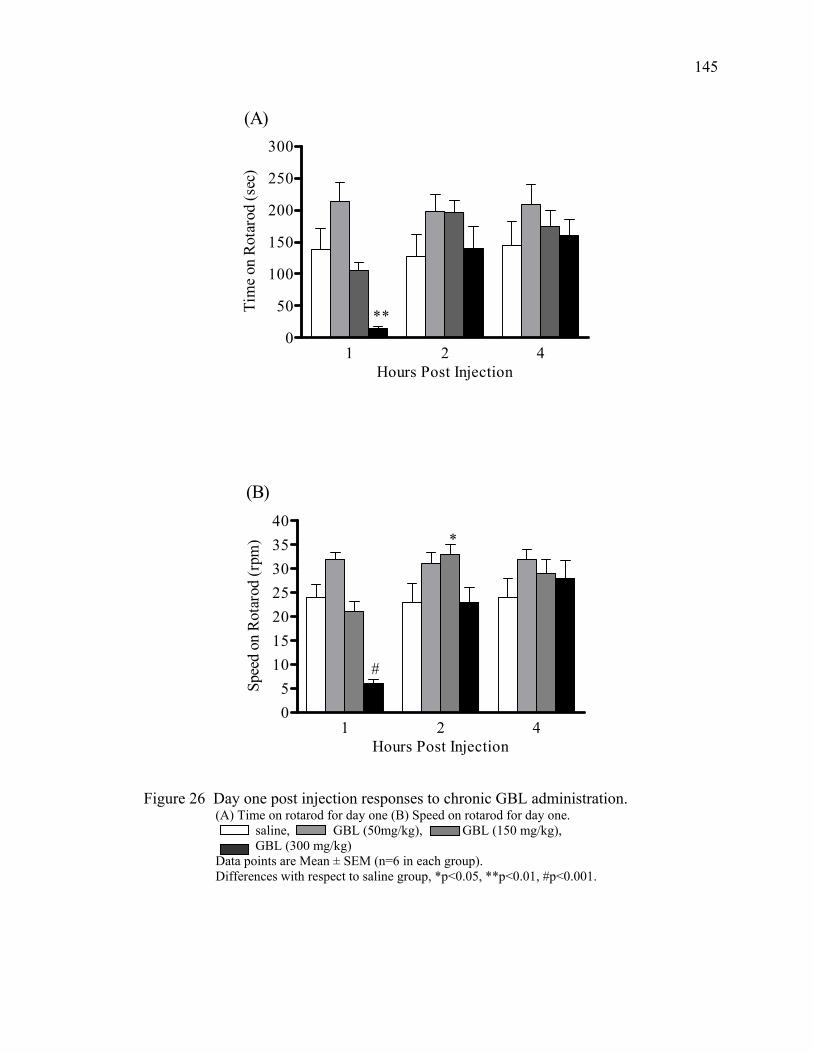
145
1 2 40
50
100
150
200
250
300
**
(A)
Hours Post Injection
Tim
e on
Rot
arod
(sec
)
1 2 405
10152025303540
*
#
(B)
Hours Post Injection
Spee
d on
Rot
arod
(rpm
)
Figure 26 Day one post injection responses to chronic GBL administration.
(A) Time on rotarod for day one (B) Speed on rotarod for day one. saline, GBL (50mg/kg), GBL (150 mg/kg), GBL (300 mg/kg) Data points are Mean ± SEM (n=6 in each group). Differences with respect to saline group, *p<0.05, **p<0.01, #p<0.001.
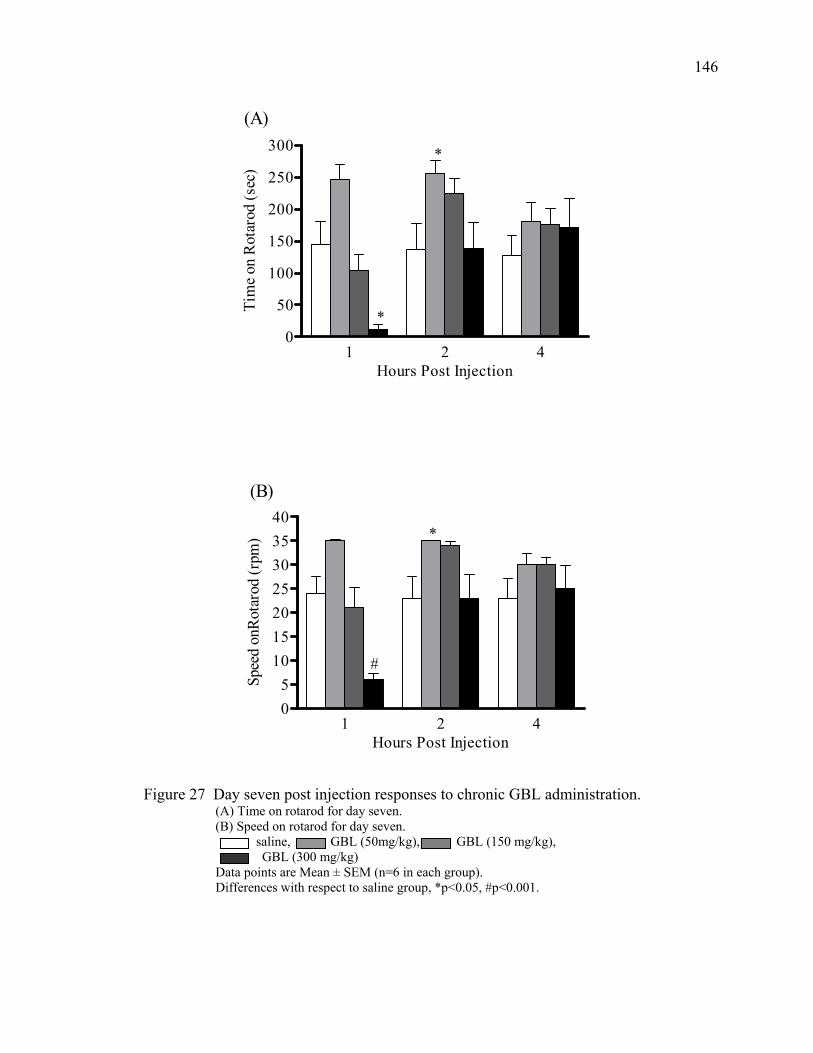
146
1 2 40
50
100
150
200
250
300 *
*
(A)
Hours Post Injection
Tim
e on
Rot
arod
(sec
)
1 2 405
10152025303540
*
#
(B)
Hours Post Injection
Spee
d on
Rot
arod
(rpm
)
Figure 27 Day seven post injection responses to chronic GBL administration.
(A) Time on rotarod for day seven. (B) Speed on rotarod for day seven. saline, GBL (50mg/kg), GBL (150 mg/kg), GBL (300 mg/kg) Data points are Mean ± SEM (n=6 in each group). Differences with respect to saline group, *p<0.05, #p<0.001.
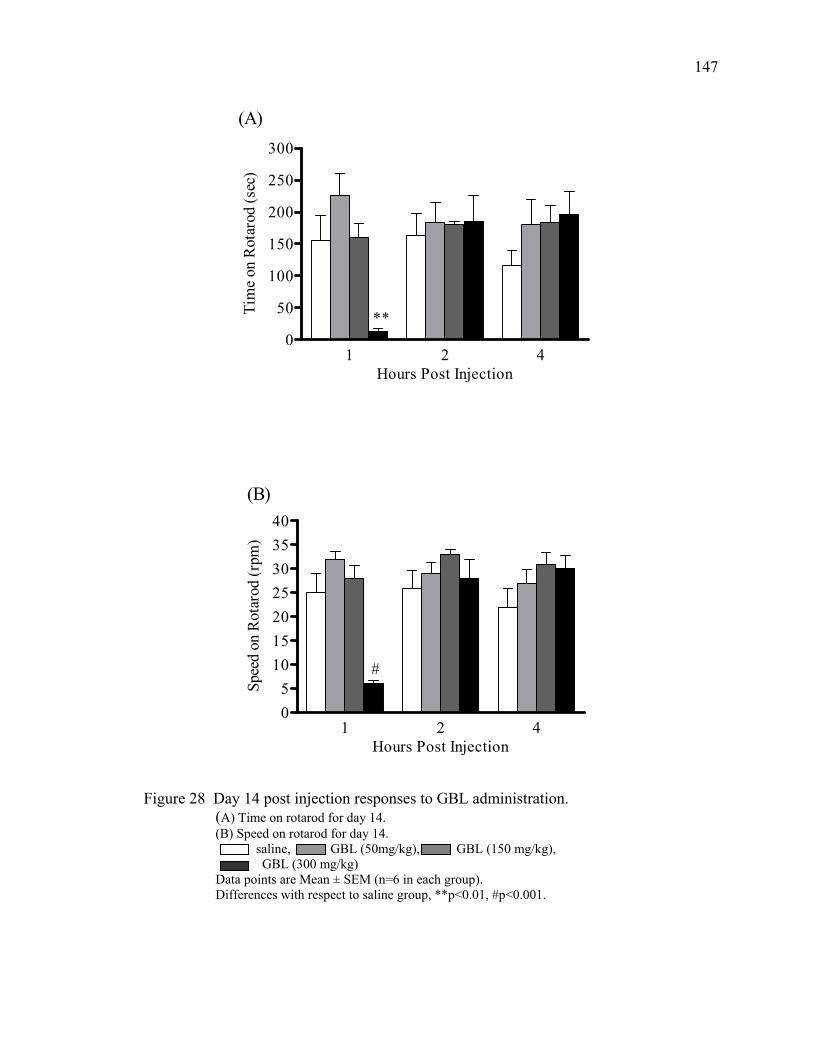
147
1 2 40
50
100
150
200
250
300
**
(A)
Hours Post Injection
Tim
e on
Rot
arod
(sec
)
1 2 405
10152025303540
#
(B)
Hours Post Injection
Spee
d on
Rot
arod
(rpm
)
Figure 28 Day 14 post injection responses to GBL administration.
(A) Time on rotarod for day 14. (B) Speed on rotarod for day 14. saline, GBL (50mg/kg), GBL (150 mg/kg), GBL (300 mg/kg) Data points are Mean ± SEM (n=6 in each group). Differences with respect to saline group, **p<0.01, #p<0.001.

148
Despite this difference in top speed, the 150 mg/kg group did not differ from controls in
the amount of time spent on the drum two hours after injection (See Figure 26A). That
difference was the only one observed two hours after the injections on day one and there
were no reliable changes observed four hours after the injections on day one. The 300
mg/kg group remained on the rotarod for less time (p<0.01) and had a top speed that was
lower (p<0.001) than controls one hour after the GBL injections on day one. Those
statistically significant deficits were observed again on days 7 and 14 (See Figures 26, 27,
& 28).
The results for day seven were somewhat similar to the results for day one. On
day seven the 50 mg/kg group had an increase in activity. The 50mg/kg group spent
more (p<0.05) time on the rotarod and had a higher (p<0.05) top speed than the control
group (See Figures 27 A & B). The 150 mg/kg group had no statistically significant
changes from controls on day seven. Again, the 300 mg/kg group spent less time
(p<0.05) on the rotatrod and had a lower (p<0.001) top speed than controls one hour after
injections (See Figures 27 A & B). There were no statistically significant changes
observed four hours after the injections.
Day 14 showed the fewest changes of the three post injection study days. The
300 mg/kg group spent less time (p<0.01) on the rotarod and had a top speed that was
lower (p<0.001) than controls one hour after the GBL injections (See Figures 28 A & B).
There were no other changes observed on day 14. Overall, the four treatment groups had
their highest top speeds on the rotarod on day 14 despite receiving daily injections of
GBL over the previous 14 days.

149
Monoamine Metabolite Results
Dopamine metabolites. The dopamine metabolites and turnover rate are shown in
Table 19. In this study, there were no statistically significant changes in any of the
dopamine metabolites or the turnover rate.
Serotonin metabolites. The serotonin metabolites and turnover rate are shown in
Table 20. In this study, there were no statistically significant changes in any of the
serotonin metabolites or the turnover rate.
Discussion
Overall, chronic treatment with GBL did not result in marked deficits in
locomotor activity. In Chapter Three, acute administration of GHB lead to decreases in
locomotor activity and sedation. It was expected that chronic administration would have
the same results compounded over time. However, no tolerance effect was observed in
mice tested on the rotarod. This is in contrast to other studies that have shown tolerance
to the inhibition of locomotor activity induced by GHB. The two studies of chronic GHB
administration in baboons (Goodwin and others 2005; Weerts and others 2005) showed
difficulty with a fine motor task until almost two weeks had passed. Only then did
tolerance to the behavioral effects of GHB develop. The study described here was only
15 days long and was expected to show decreased performance on the rotarod task
without any tolerance. However, there were rarely statistically significant decreases in
locomotor activity on the rotarod. Colombo and others (Colombo and others 1995)
showed 100% failure of a rotarod task beginning the first day of chronic GHB (1 g/kg)
administration.
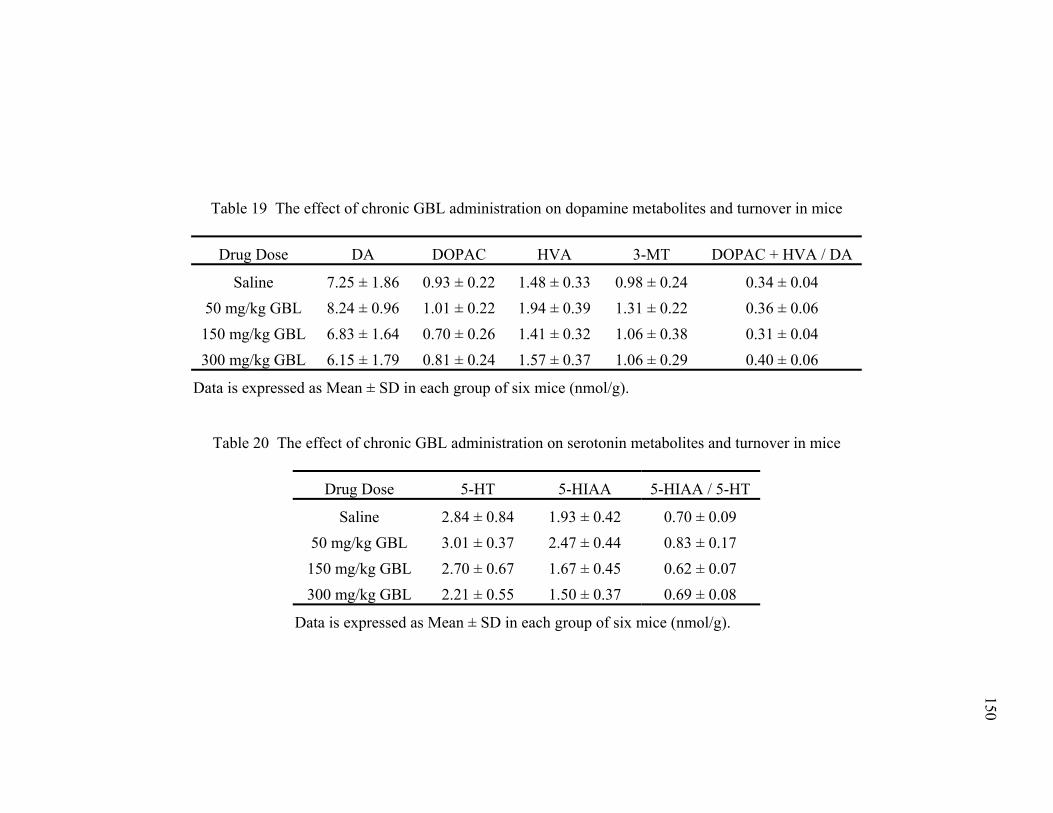
150
Table 19 The effect of chronic GBL administration on dopamine metabolites and turnover in mice
Drug Dose DA DOPAC HVA 3-MT DOPAC + HVA / DA
Saline 7.25 ± 1.86 0.93 ± 0.22 1.48 ± 0.33 0.98 ± 0.24 0.34 ± 0.04 50 mg/kg GBL 8.24 ± 0.96 1.01 ± 0.22 1.94 ± 0.39 1.31 ± 0.22 0.36 ± 0.06 150 mg/kg GBL 6.83 ± 1.64 0.70 ± 0.26 1.41 ± 0.32 1.06 ± 0.38 0.31 ± 0.04 300 mg/kg GBL 6.15 ± 1.79 0.81 ± 0.24 1.57 ± 0.37 1.06 ± 0.29 0.40 ± 0.06
Data is expressed as Mean ± SD in each group of six mice (nmol/g).
Table 20 The effect of chronic GBL administration on serotonin metabolites and turnover in mice
Drug Dose 5-HT 5-HIAA 5-HIAA / 5-HT
Saline 2.84 ± 0.84 1.93 ± 0.42 0.70 ± 0.09 50 mg/kg GBL 3.01 ± 0.37 2.47 ± 0.44 0.83 ± 0.17 150 mg/kg GBL 2.70 ± 0.67 1.67 ± 0.45 0.62 ± 0.07 300 mg/kg GBL 2.21 ± 0.55 1.50 ± 0.37 0.69 ± 0.08
Data is expressed as Mean ± SD in each group of six mice (nmol/g).

151
It was between six and nine days that those mice developed any tolerance to the
behavioral effects. The experiments described in this chapter showed no significant
decrease in locomotor activity or coordination on the rotarod with the exception of the
highest dose of GBL at one hour post injection. The 300 mg/kg group always spent less
time on the drum and had a top speed that was lower than controls. Those changes were
not entirely unexpected. It has previously been demonstrated that a GBL dose of 150
mg/kg was a high enough dose to cause sedation and loss of locomotor activity over the
course of 40 – 50 minutes. This was supported by the experimental results presented in
Chapter Three of this dissertation. Thus, it was not surprising when a 300 mg/kg dose
caused sedation and loss of locomotor activity that lasted longer than the 150 mg/kg dose.
During most of the one hour post injection trials, the mice given 300 mg/kg were still
sedated to some degree when they were placed on the drum. When placed onto the drum,
the mice made very few attempts to walk or move and most often fell off as the drum
rotated. This is similar in nature to observations of baboons presented with a fine motor
task after high doses of GHB were administered (Weerts and others 2005). After several
days of GHB administration two of the baboons were noted as non-responsive and did
not participate in the task at all for 48 hours. The 300 mg/kg group of mice recovered
very quickly in the second hour and spent similar amounts of time on the drum four hours
post injection when compared to controls.
The recovery of locomotor activity within two hours of GBL administration was
shown in Chapter Three in Acute Study 1 (120 minutes post GBL). Mice given GBL
doses of 150 or 200 mg/kg recovered from the behavioral effects within 120 minutes of
an acute injection. That explains the observations on day one, but the same results were

152
not expected to continue as the study progressed. The rapid recovery of activity was
most likely due to the accumulated dopamine being released into the synapse after the
GBL was completely metabolized. Four hours after the injections, the level of locomotor
activity returned to normal, indicating the dopamine levels had returned to normal as
well. The metabolite levels for the 300 mg/kg group showed no statistically significant
changes.
We expected to see some long term effects on the dopaminergic system as a result
of daily injections of 300 mg/kg GBL. However, the lack of such changes must have
been due to the rapid metabolism of GBL and an equally rapid action on dopamine
metabolism. As stated previously, the half-life of GBL before conversion to GHB via
blood born lactonases has been estimated at less than a minute (Roth and Giarman 1966).
The behavioral data shows that activity levels recovered by two hours and normalized by
four hours. Thus, the metabolite levels did the same. Therefore, daily injections of 300
mg/kg GBL did not have any long term negative effect on behavior or monoamine
neurotransmitters. Interestingly, on day 15, the 300 mg/kg group ended with the highest
amount of time spent on the rotarod and the highest top speed. It should be noted that
those values were not statistically significant.
The other statistically significant changes occurred on day seven and were
observed in the 50 mg/kg group. That group spent more time on the rotarod and had a
higher top speed when compared to controls two hours after injection. This observation
was unexpected based on the results of previous acute studies in Chapter Three. In that
chapter 50 mg/kg was used as a testing dose and showed no effect on open field behavior.

153
It would appear that low doses of GBL may have a stimulatory effect on locomotor
activity as assessed by the rotarod test in the experiments described here.
Previously, microdialysis studies have shown an increase in dopamine release
caused by GHB (Maitre and others 1990; Hechler and others 1991). However, those
studies are thought to have falsely induced dopamine release via dialysate calcium
concentrations greater than 3.4 mM (Feigenbaum and Howard 1996a). Other studies
involving GHB induced increases in dopamine release were performed in awake animals
and used anesthetics such as halothane and chloral hydrate (Roth and others 1973; Mereu
and others 1984; Nissbrandt and others 1994). Both anesthetics have been shown to alter
dopamine release (Nieoullon and others 1977; Savaki and others 1986; Spampinato and
others 1986; Lane and Blaha 1987; Collin and others 1988; Zhang and others 1989;
Osborne and others 1990; Stahle and others 1990; Hamilton and others 1992). Based on
those reports, an increase in dopamine release induced by GHB would not be responsible
for the increased activity observed in this study. However, there is little evidence to
suggest that the increased activity was not a result of an increase in dopamine release.
This study investigated the long term effects of chronic exposure to GBL using
the rotarod, which is specifically designed to test for coordination and locomotor activity.
Other tests of locomotor function or coordination such as the grip strength, vertical pole,
and hanging wire tests were not performed. It is possible that these other tests would
reveal more subtle locomotor and coordination deficits than the rotarod and should be
considered for further studies. Testing the chronic effects of GHB or GBL on memory
function would also be of interest. A common side effect of GHB or GBL abuse is
amnesia, which is caused by a hyperpolarization of neurons in the hippocampus, the brain

154
structure responsible for memory (Waszkielewicz and Bojarski 2004). Memory testing
should utilize nose poke and water maze protocols.
In Figure 25A the 50 and 150 mg/kg groups showed a decreasing trend in the
amount of time spent on the rotarod from day 8 to day 15. From first hand observation, it
was noted that on occasion, mice attempted to stand up while the drum was rotating. If
standing up was possible, the mice attempted to escape the cage by jumping towards one
of the walls of the cage. This would result in the mouse landing on the floor, receiving a
shock, and ending the trial before 300 seconds had elapsed. This by itself does not
explain the decreasing trends in time spent on the rod, but may have been a contributing
factor.
Another difference between this study and others is the route of administration for
GBL. The study described here used intraperitoneal (i.p.) injections. The studies in
baboons used intragastric (IG) catheters and pumps to administer GHB either
continuously (Weerts and others 2005) throughout the day or in single infusions
(Goodwin and others 2005). A constant, steady infusion of GHB would stabilize blood
levels and have markedly different effects over time than single injections. Other options
for drug delivery include adding GHB or GBL to drinking water or administration by oral
gavage. These methods of administration certainly affect the uptake and metabolism of
GHB and may possibly explain some of the differing results.
One area of concern is the amount of exposure each mouse was given to the
rotarod protocol. There were four training days and then six testing days all in the span
of 19 total days. Perhaps the mice showed very few deficits because they had so many
opportunities to practice the procedure after GBL administration. The mice could have

155
learned to perform the rotarod task while slightly impaired by the doses of GBL. This
same idea has been suggested previously (Nicholson and Balster 2001) in response to the
tolerance seen in the study by Colombo and others (Colombo and others 1995). In
retrospect, the two facets of this study should have been performed on different groups of
mice to get a more accurate picture of the effects of GBL both long term and immediately
after injections.
Unexpectedly, there were few behavioral or monoamine neurotransmitter
metabolite changes in mice treated with GBL for 14 days. This is in contrast to studies in
baboons, which showed decreased performance in a fine motor task in the first two weeks
of chronic GHB administration (Weerts and others 2005). We expected the daily
exposure of GBL to have some effect on the dopaminergic system due to the repeated
inhibition of dopamine release followed by the rapid release of dopamine. However, the
lack of any statistically significant changes is most likely due to the rapid metabolism
time of GBL. Future studies should concentrate on higher doses as well as a longer
period of time. Chronic studies in baboons have administered GHB for as many as 3
days (Weerts and others 2005). Longer studies may show behavioral or molecular
changes that take longer to develop.

CHAPTER EIGHT
General Conclusions
The illicit use of GHB and its prodrugs GBL and 1,4-BD is a growing healthcare
concern in the United States. Reports of GHB overdoses continue to increase in
emergency rooms across the country. In addition, GHB was approved by the FDA as a
treatment for cataplexy associated with narcolepsy (Anonymous 2002a). Furthermore,
SSADH deficiency, an inherited metabolic disorder, leads to a marked elevation of GHB
in the body, which is associated with neurological deficits. Despite these examples of
increased use or accumulation of GHB, its long term effects and mechanism of action in
the CNS are not completely understood.
The studies presented in this dissertation used mice as a model to study the effects
of GBL on locomotor function. Initial studies in this dissertation characterized the dose
response and time course of GBL in open field behavior. A low dose of GBL (50 mg/kg)
had no statistically significant effects on locomotor function. However, a high dose (150
mg/kg) resulted in sedation and loss of locomotor activity. The difference in behavioral
responses for the two doses examined shows a steep dose effect for GBL in mice. This
loss of locomotor activity induced by GBL is similar to the loss of locomotor activity
induced by GHB that has been demonstrated by other investigators (Bottiglieri and others
2001; Anderson and others 2002; Itzhak and Ali 2002; de Fiebre and others 2004). These
initial studies investigated only one aspect of locomotor activity. Other methods that
could be used to test fine motor function in future studies may include grip strength,
vertical pole, and the hanging wire screen test. Memory and water maze testing were not
156

157
used, but should be considered for cognitive studies, since GHB has been shown to affect
memory and cause amnesia in humans (Waszkielewicz and Bojarski 2004).
Monoamine neurotransmitter metabolite analysis showed decreased 3-MT levels
after GBL administration. Previous studies in this lab have shown decreases in striatal 3-
MT levels in rats after GHB administration (Bottiglieri and others 2001; Anderson and
others 2002). Decreased 3-MT levels are indicative of an inhibition of dopamine release.
To our knowledge, no other laboratory has measured decreased 3-MT levels as an
indicator of inhibited dopamine release in studies involving GHB or GBL. Furthermore,
decreased 3-MT levels have been observed in both rats and mice as well as after GHB
and GBL administration. These results have proven to be a reproducible method by
which to test for the inhibition of dopamine release induced by GHB or GBL. Earlier
studies have reported conflicting results when using microdialysis to study the effects of
GHB on dopamine release. Those studies that have shown an increase in dopamine
release as a result of GHB often used perfusion fluids with high calcium concentrations
or anesthetics that are known to alter dopamine release. Monoamine metabolite analysis
also showed an increase in dopamine levels in half brains. This is consistent with
previous work showing intraneuronal accumulation of DA when release is inhibited
(Gessa and others 1966; Gessa and others 1968; Aghajanian and Roth 1970; Spano and
others 1971; Pericic and Walters 1976; Lundborg and others 1980; Dyck and Kazakoff
1982). The neurochemical findings in this dissertation are limited to monoamine
neurotransmitters (DA & 5-HT) and their metabolites. Other neurotransmitter systems
may be affected by GHB or GBL, such as NE, GABA, and acetylcholine. Future studies
should focus on these pathways as potential targets for GHB toxicity.

158
Experimental studies in this dissertation were extended to investigate compounds
for their ability to antagonize the loss of locomotor function induced by GBL. The eight
compounds tested were chosen on their ability to act as GHB receptor antagonists,
GABAA antagonists or increase central dopaminergic activity (Chapter 5, Table 13).
Both NCS-382 (GHB receptor antagonist) and bicuculline (GABAA antagonist)
failed to block the effects of GBL on locomotor activity. Tolcapone, apomorphine and
pargyline, which targeted the dopamine pathway, also failed to antagonize the effect of
GBL on locomotor activity. The lack of success with pargyline was unexpected, as
pargyline has previously been shown to successfully antagonize the effects of GHB in
rats in this lab (Anderson and others 2001). The discrepancies in the two studies may be
dependent on the species of rodent used and the use of GHB vs. GBL.
It was also unexpected that taurine was unable to antagonize the loss of locomotor
activity induced by GBL. It was anticipated that it would be effective against the effects
of GBL since previous studies in this laboratory have shown that taurine administered
prior to GHB protected against the loss of locomotor function in rats (Anderson and
Bottiglieri unpublished data). Other studies have shown that taurine can increase survival
of the SSADH knock out mouse (Gupta and others 2002). Taurine is present in a high
concentration in mother’s milk, and is thought to protect the SSADH knock out mouse
from birth until the weaning period. Mice supplemented with taurine after weaning had
an increase in survival rate compared to mice that were fed a normal diet. This may
suggest that a constant supply of taurine is required to protect against the effects of GHB.
Two of the eight compounds tested were successful at partially antagonizing the
locomotor effects of GBL: pergolide and nomifensine. Both drugs antagonized the loss

159
of locomotor activity induced by GBL and caused increases in the movement episodes
and time moved parameters, but no improvement in the distance moved or VPE
parameters. To our knowledge, no other studies have shown antagonism of the
locomotor effects of GBL using pergolide or nomifensine. Future studies should
consider combining drugs in an effort to increase dopaminergic activity as well as
antagonize the inhibitory functions of GABA and GHB receptor stimulation. A previous
study combining GBR 12909 and tolcapone showed beneficial effects that were not seen
when they were tested separately (Raevskii and others 2002).
Based on the antagonism of GBL by pergolide and nomifensine, both drugs were
investigated further as post-treatments of GBL. Pergolide and nomifensine both showed
antagonism of the loss of locomotor activity caused by GBL. Again, the movement
episodes and time moved were improved while the distance moved and VPE parameters
were not. This is the first time antagonism of GBL has been achieved by post-treatments.
Only two studies have shown antagonism of GHB with a post-treatment. One study used
pargyline as a post-treatment in rats (Anderson and others 2001). The second study
showed a dose dependent reduction in the time needed to regain the righting reflex in
mice when SCH 59011 was administered after GHB (Carai and others 2001). Using
these drugs as post-treatments has application for a GHB or GBL overdose condition or
possible treatment for SSADH patients. There are currently no effective treatments for
GHB overdose or symptoms associated with SSADH deficiency.
Despite the experimental success of these compounds, they may prove difficult to
test clinically in cases of GHB or GBL overdoses. Typical ER admissions include
teenagers that frequent clubs and abuse a variety of narcotics including GHB,

160
amphetamines, barbiturates, and alcohol. Concomitant substance abuse makes a clear
diagnosis and pharmacological treatment of GHB toxicity difficult.
However, pergolide and nomifensine may be useful as an antidote in cases of
GHB overdose in narcoleptic patients that misuse Xyrem medication. The current
information regarding Xyrem overdose is limited. If alcohol or other drugs are suspected
in addition to Xyrem overdose, gastric decontamination is recommended (Jazz
Pharmaceuticals 2006). Intubation is also suggested to combat any difficulty in
breathing. Bradycardia accompanying Xyrem overdose has shown to be responsive to
atropine administration, but naloxone and flumazenil have not been successful in
reversing CNS depressive effects. As indicated, there is little pharmacological
intervention recommended in cases of Xyrem overdose. Based on the results presented in
this dissertation, pergolide and nomifensine should be investigated for potential
application in such cases.
Pergolide and nomifensine may also be of benefit to SSADH patients. In a
review of 60 SSADH patients, ataxia was a symptom in nearly 50% of the group (Pearl
and others 2003). Both pergolide and nomifensine have been shown to increase
locomotor activity when administered after GHB (Chapter Six). Therefore, these two
compounds could possibly provide symptomatic relief from the ataxia that is present in
many SSADH patients. While decreasing ataxia in these individuals does not address the
elevated levels of GABA or GHB, it has the potential to dramatically increase the quality
of life for those affected. Perhaps combining one of these drugs with drugs aimed at
reducing GABA and GHB levels would prove even more beneficial.

161
Chronic effects of GHB have been monitored in baboons (Goodwin and others
2005; Weerts and others 2005), but rarely in rats or mice. We sought to investigate the
effects of chronic GBL administration over the course of 14 days. These studies used a
rotarod, which is used for testing fine motor function. There were relatively few negative
effects observed after 14 days of chronic GBL administration. This is in contrast to
tolerance studies by Colombo and others (Colombo and others 1995). Those studies
showed decreased motor performance over the first six days of chronic GHB
administration, after which tolerance developed and performance improved. The present
study showed relatively few negative effects of chronic GBL administration. However,
there were minimal improvements due to GBL. Mice given a low dose GBL (50 mg/kg)
showed improvement in time spent on the rotarod two hours after injection on day seven.
In contrast, mice given a high dose of GBL (300 mg/kg) spent less time on the rotarod
and had a lower top speed one hour post GBL administration on days 1, 7, and 14. Those
results support a biphasic effect of GHB or GBL that has been previously observed in the
Swiss-Webster mouse (de Fiebre and others 2004). In that study, both GBL and 1,4-BD
had increases in the number of open field movements when given low doses (50 mg/kg).
High doses (150 mg/kg) lead to decreases in open field movements.
Over the course of 14 days of chronic GBL administration, fine motor
coordination was investigated by a rotarod system. Other measures of motor function
should be used in future studies for a complete analysis of the effects of chronic GBL
administration. In addition to behavioral testing, the neurochemical analysis should
expand beyond monoamine neurotransmitters. GABA and glutamate are important in the
synthesis of endogenous GHB and need to be explored further. Neuropathological

162
changes were not studied in this dissertation and may be another potential area of
investigation. The effects of GHB or GBL on cell signaling changes and cell apoptotic
mechanisms should also be investigated in order to determine detrimental neurological
effects from chronic GBH or GBL use. Another aspect of chronic substance abuse that
deserves attention is physical dependence and withdrawal. Studies have shown physical
dependence in animals (Goodwin and others 2005; Weerts and others 2005) after chronic
GHB use. Case reports of humans have shown withdrawal symptoms after discontinuing
GHB or GBL (Craig and others 2000; Catalano and others 2001). Due to the nature of
the experimental design in the studies presented here, withdrawal was not examined.
Further studies should incorporate the monitoring of withdrawal symptoms into their
design.
The studies presented in this dissertation are novel and show that the loss of
locomotor activity induced by GBL can be antagonized by both pergolide and
nomifensine regardless of whether those drugs are administered as pre or post-treatments.
It was also shown that 14 days of chronic GBL administration showed few negative
effects on fine motor skills. Further examination of the effects of GBL should include
more in depth neurochemical analysis and additional measures of locomotor and
behavioral activity.

163
WORKS CITED
Addolorato G, Balducci G, Capristo E, Atilla ML, Taggi F, Gasbarrini G, Ceccanti M. 1999. Gamma-hydroxybutyric acid (GHB) in the treatment of alcohol withdrawal syndrome: a randomized comparative study versus benzodiazepine. Alcohol Clin Exp Res 23(10):1596-1604.
Addolorato G, Caputo F, Capristo E, Bernardi M, Stefanini F, Gasbarrini G. 1999. A
case of gamma-hydroxybutyric acid withdrawal syndrome during alcohol addiction treatment: utility of diazepam administration. Clin Neuropharmacol 22:60-62.
Addolorado G, Castelli E, Stefanini G, Casella G. 1996. An open multicenter study
evaluating 4-hydroxybutyric acid sodium salt in the medium term treatment of 179 alcohol dependent subjects. Alcohol 31(4):341-345.
Aghajanian GK, Roth RH. 1970. γ-hydroxybutyrate-induced increase in dopamine:
localization by fluorescence microscopy. J Pharmacol Exp Ther 175:131-138. Alter CA, O’Neil S, Marshall JF. 1984. Sensorimotor impairment and elevated levels of
dopamine metabolites in the neostriatum occur rapidly after intranigral injection of 6-hydroxy-dopamine or gamma-hydroxybutyrate in awake rats. Neuropharmacology 23:309-318.
Anden NE, Magnussen T, Stock G. 1973. Effect of drugs influencing monoamine
mechanisms on the increase in brain dopamine produced by axotomy or treatment with gamma hydroxybutyric acid. Arch Pharmacol 278:363-372.
Anderson D, Bottiglieri T, Gibson KM. 2002. Pargyline antagonizes the effect of
gamma-hydroxybutyrate on locomotor activity in the rat. 749.15. Abstract Viewer/Itinerary Planner. Washington DC. Soc Neurosci:Online.
Andriamampandry C, Siffert JC, Schmitt M, Garnier JM, Muller C, Gobaille S, Mark J,
Maitre M. 1988. Cloning of a rat brain succinic semialdehyde reductase involved in the synthesis of the neuromodulator γ-hydroxybutyrate. Biochem 334:43-50.
[Anonymous]. 1997. Gamma-hydroxybutyrate use – New York and Texas, 1995-1996.
Morb Mortal Wkly Rep 46(13):281-283. [Anonymous]. 2002a. FDA approves XYREM for cataplexy attacks in patients with
narcolepsy. Washington: FDA Talk Paper. Available via http://www.fda.gov/bbs/topics/ANSWERS/2002/ANS01157.html. Accessed 2006 Feb 10.

164
[Anonymous]. 2002b. Xyrem (sodium oxybate) Questions and Answers. USFDA: Center for Drug Evaluation and Research. Available via http://www.fda.gov/cder/drug/infopage/xyrem/xyrem_qa.htm. Accessed 2006 Feb 12.
[Anonymous]. 2006. Club drugs include alcohol, LSD (Acid), MDMA (Ecstasy), GHB,
GBL, ketamine (Special-K), fentanyl, rohypnol, amphetamines and methamphetamine. Mountainside Treatment Center Website. Available via http://www.drug-addiction.net/clubdrugs.htm. Accessed 2006 Feb 12.
Bania TC, Ashar T, Press G, Carey PM. 2003. Gamma-hydroxybutyric acid tolerance
and withdrawal in a rat model. Acad Emerg Med 10(7):697-704. Baker LE, Van Tilburg TJ, Brandt AE, Poling A. 2005. Discriminative stimulus effects
of gamma-hydroxybutyrate (GHB) and its metabolic precursor, gamma-butyrolactone (GBL) in rats. Psychopharmacology 181(3):458-466.
Barker SA, Snead OC, Poldrugo F, Liu CC, Fish FP, Settine RL. 1985. Identification
and quantification of 1,4-butanediol in mammalian tissues: An alternative biosynthetic pathway for gamma-hydroxybutyric acid. Biochem Pharmacol 34:1849-1852.
Barnes NM, Sharp T. 1999. A review of central 5-HT receptors and their function.
Neuropharmacology 38:1083-1152. Bedard P, Parkes JD, Marsden CD. 1977. Nomifensine in Parkinson’s disease. Br J
Clin Pharmacol 4:(Suppl) S187-S190. Behar KL, Rothman DL, Peterson KF, Hooten M, Delaney R, Petroff OA, Shulman GI,
Navarro V, Petrakis IL, Charney DS and others. 1999. Preliminary evidence of low cortical GABA levels in localized 1H-MR spectra of alcohol-dependent and hepatic encephalopathy patients. Am J Psychiatry 156:952-954.
Benavides J, Rumigny JF, Bourguignon JJ, Cash C, Wermuth CG, Mandel P, Vincendon
G, Maitre M. 1982a. High affinity binding site for γ-hydroxybutyric acid in rat brain. Life Sci 30:953-961.
Benavides J, Rumigny JF, Bourguignon J, Wermuth CG, Mandel P, Maitre M. 1982. A
high affinity, Na+-dependent uptake system for γ-hydroxybutyrate in membrane vesicles prepared from rat brain. J Neurochem 38:1570-1575.
Berge F, Carpanini MT. 2000. Safety and tolerability of gamma-hydroxybutyric acid in
the treatment of alcohol-dependent patients. Alcohol 20:223-225.

165
Bernasconi R, Lauber J, Marescaux C, Vergnes M, Martin P, Rubio V, Leonhardt T, Reymann N, Bittiger H. 1992. Experimental absence seizures: potential role of gamma-hydroxybutyric acid and GABAB receptors. J Neural Transm 35:155-177.
Bernasconi R, Mathivet P, Bischoff S, Marecaux C. 1999. Gamma-hydroxybutyric acid:
an endogenous neuromodulator with abuse potential? Trends Pharmacol Sci 20: 135-141.
Bert L, Parrot S, Robert F, Desvignes C, Denoroy L, Suaud-Chagny MF, Renaud B.
2002. In vivo temporal sequence of rat striatal glutamate, aspartate and dopamine efflux during apomorphine, nomifensine, NMDA and PDC in situ administration. Neuropharmacology 43:825-835.
Blandini F, Nappi G, Fancellu R, Mangiagalli A, Samuele A, Riboldazzi G, Calandrella
D, Pacchetti C, Bono G, Martignoni E. 2003. Modifications of plasma and platelet levels of L-DOPA and its direct metabolites during treatment with tolcapone or entacapone in patients with Parkinson’s disease. J Neural Transm 110:911-922.
Booth RG, Baldessarini RJ, Marsh E, Owens CE. 1994. Actions of (+/-)-7-hydroxy-
N,N-dipropylaminotetralin (7-OH-DPAT) on dopamine synthesis in limbic and extrapyramidal regions of rat brain. Brain Res 662:283-288.
Bottiglieri T, Anderson D, Gibson KM, Froestl W, Diaz-Arrastia R. 2001. Effect of
gamma-hydroxybutyrate on locomotor activity and brain dopamine metabolism in the rat. Soc Neurosci Abstr 27:971.1.
Bourguignon JJ, Schoenfelder A, Schmitt M, Wermuth CG, Hechler V, Charlier B,
Maitre M. 1988. Analogues of gamma-hydroxybutyric acid. Synthesis and binding studies. J Med Chem 31(5):893-897.
Brown GK, Cromby CH, Manning NJ, Pollitt RJ. 1987. Urinary organic acids in
succinic semialdehyde dehydrogenase deficiency: evidence of alpha-oxidation of 4-hydroxybutyric acid, interaction of succinic semialdehyde with pyruvate dehydrogenase and possible secondary inhibition of mitochondrial beta-oxidation. J Inherit Metab Dis 10:367-375.
Broxterman HJ, Noach EL, Van Valkenburg CFM, Wijling A. 1981. Cross-tolerance of
dopamine metabolism to baclofen, γ-butyrolactone and HA-966 in the striatum and olfactory tubercle of the rat. Life Sci 28:973-981.
Bustos G, Roth RH. 1972. Effect of γ-hydroxybutyrate on the release of monoamines
from the rat striatum. Br J Pharmacol 44:817-820.

166
Carai MAM, Colombo G, Brunetti G, Melis A, Serra S, Vacca G, Mastinu S, Pistuddi AM, Solinas C, Cignarella G, and others. 2001. Role of GABAB receptors in the sedative/hypnotic effect of γ-hydroxybutyric acid. Eur J Pharmacol 428:315-321.
Carai MA, Columbo G, Reali R, Serra S, Mocci I, Castelli MP, Cignarella G, Gessa GL.
2002. Central effects of 1,4-butanediol are mediated by GABA(B) receptors via its conversion into gamma-hydroxybutyric acid. Eur J Pharmacol 441:157-163.
Carter LP, Wu H, Chen W, Matthews MM, Mehta AK, Hernandez RJ, Thomson JA,
Ticku MK, Coop A, Koek W, France CP. 2005. Novel γ-Hydroxybutyric Acid (GHB) Analogs Share Some, but Not All, of the Behavioral Effects of GHB and GABAB Receptor Agonists. J Pharmacol Exp Ther 313:1314-1323.
Cash C. 1994. Gammahydroxybutyrate: An overview of the pros and cons for it being
a neurotransmitter and/or a useful therapeutic agent. Neurosci Biobehav Rev 18(2):291-304.
Castelli MP, Mocci I, Langlois X, Gommerendagger W, Luyten WH, Leysen JE, Gessa
GL. 2000. Quantatative autoradiographic distribution of gamma-hydroxybutyric acid binding sites in human and monkey brain. Brain Res Mol Brain Res 78:91-99.
Castelli MP, Pibiri F, Carboni G, Piras AP. 2004. A review of pharmacology of NCS-
382, a putative antagonist of gamma-hydroxybutyric acid (GHB) receptor. CNS Drug Rev 10(3):243-260.
Catalano MC, Glass JM, Catalano G, Burrows SL, Lynn WA, Weitzner BS. 2001. Gamma Butyrolactone (GBL) withdrawal syndromes. Psychosomatics 42:83-88. Chambliss KL, Zhang Y-A, Rossier E, Vollmer B, Gibson KM. 1995. Enzymatic and
immunologic identification of succinic semialdehyde dehydrogenase in rat and human neural and non-neural tissues. J Neurochem 65:851-855.
Chan-Palay V, Wu JY, Palay SL. 1979. Immunocytochemical localization of
γ-aminobutyric acid transaminase at cellular and ultrastructural levels. Proc Natl Acad Sci USA 76:2067-2071.
Chen JJ, Obering C. 2005. A review of intermittent subcutaneous apomorphine
injections for the rescue management of motor fluctuations associated with advanced parkinson’s disease. Clin Ther 27(11):1710-1724.
Cheramy A, Nieoullon A, Glowinski J. 1977. Stimulating effects of γ-
hydroxybutyrate on dopamine release from the caudate nucleus and the substantia nigra of the cat. J Pharmacol Expl Ther 202:283-293.

167
Clemens JA, Phebus LA. 1988. Dopamine depletion protects striatal neurons from ischemia-induced cell death. Life Sci 42:707-713.
Clow A, Hussain T, Glover V, Sandler M, Walker M, Dexter D. 1992. Pergolide can
induce soluble superoxide dismutase in rat striate. J of Neural Trans 90(1):27-31. Colombo G, Agabio R, Lobina C, Reali R, Fadda F, Gessa GL. 1995. Cross- tolerance
to ethanol and gamma-hydroxybutyric acid. Eur J Pharmacol 95(273):235-238. Colombo G, et al. 2002. Behavioral pharmacology of γ-hydroxybutyrate. In Gamma
Hydroxybutyrate (Tunnicliff G and Cash CD, eds), pp 150-168, Taylor & Francis. Collin AK, Stahle L, Ungerstedt U. 1988. Effects of halothane anaesthesia on
extracellular levels of DA, DOPAC, HVA, and 5-HIAA in rat striatum. Acta Physiologies Scandanovean 132:30-39.
Cook CD, Aceto MD, Coop A, Beardsley PM. 2002. Effects of the putative antagonist
NCS382 on the behavioral pharmacological actions of gammahydroxybutyrate in mice. Psychopharmacology (Berl) 160:99-106.
Couper F, Logan B. 2000. Determination of gamma-hydroxybutyrate (GHB) in
biological specimens by gas chromatography – mass spectrometry. J Anal Toxicol 24:1-7.
Craig K, Gomez HF, McManus JL, Bania TC. 2000. Severe gamma-hydroxybutyrate
withdrawal: A case report and literature review. Journal of Emerg Med 18:65-70. Crawley JN. 2000. What’s Wrong With My Mouse? New York: Wiley-Liss 329 p. Crofts HS, Dalley JW, Collins P, VanDenderen JCM, Everitt BJ, Robbins TW, Roberts
AC. 2001. Differential effect of 6-OHDA lesions of the frontal cortex and caudate nucleus on the ability to acquire an attentional set. Cereb Cortex 11:1015-1026.
Cumming P, Brown E, Damsma G, Fibiger H. 1992. Formation and clearance of
interstitial metabolites of dopamine and serotonin in the rat striatum: an in vivo microdialysis study. J Neurochem 59:1905-1914.
DaPrada M, Keller HH. 1976. Baclofen and γ-hydroxybutyrate: similar effects on
cerebral dopamine neurons. Life Sci 19:1253-1264. Davies JA. 1978. The effect of gamma-butyrolactone on locomotor activity in the rat.
Psychopharmacology (Berl) 78(60):67-72.

168
DeBoer P, Damsma G, Fibiger HC, Timmerman W, de Vries JB, Westerink BHC. 1990. Dopaminergic-cholinergic interactions in the striatum: the critical significance of calcium concentrations in brain microdialysis. Naunyn Schmiedebergs Arch Pharmacol 342:528-534.
De Deurwaerdere P, Spampinato U. 2001. The nigrostriatal dopamine system: a
neglected target for 5-HT2C receptors. Trends Pharmacol Sci 22:502-503. de Fiebre C, de Fiebre N, Coleman S, Forster M. 2004. Comparison of the actions of
γ-butyrolactone and 1,4-butanediol in Swiss-Webster mice. Pharmacol Biochem Behav 77:705-710.
Delwaide PJ, Martinelli P, Schoenen J. 1983. Mazindol in the treatment of Parkinson’s
disease. Arch Neurol 40:788-790. Diana M, Pistis M, Muntoni A, Gessa G. 1993. Heterogeneous responses of substantia
nigra pars reticulata neurons to γ-hydroxybutyric acid administration. Eur J Pharmacol 230:363-365.
Doherty JD, Hattox SE, Snead OC, Roth RH. 1978. Identification of endogenous
γ-hydroxybutyrate in human and bovine brain and its regional distribution in human, guinea pig, and rhesus monkey brain. J Pharmacol Exp Ther 207:130-139.
Doherty JD, Snead OC, Roth RH. 1975a. A sensitive method for quantitation of
γ-hydroxybutyric acid and γ-butyrolactone in brain by electron capture gas chromatography. Anal Biochem 69:268-277.
Downes JJ, Roberts AC, Sahakian BJ, Evenden JL, Morris RG, Robbins TW. 1989.
Impaired extra-dimensional shift performance in medicated and unmedicated Parkinson’s disease: evidence for a specific attentional dysfunction. Neuropsychologia 27:1329-1343.
Dudek BC, Fanelli RJ. 1980. Effects of gamma-butyrolactone, amphetamine, and
haloperidol in mice differing in sensitivity to alcohol. Psychopharmacology (Berl) 68:89-97.
Dyck LE, Kazakoff CW. 1982. Acceleration of the biosynthesis of rat striatal dopamine
by incubation and by administration of γ-butyrolactone. J Neurosci Res 8:57-65. Eli M, Cattabeni F. 1983. Endogenous γ-hydroxybutyrate in rat brain areas: postmortem
changes and effects of drugs interfering with γ-aminobutyric acid metabolism. J Neurochem 41:524-530.
Ellinwood EH, Gonzalez AE, Dougherty GG. 1983. γ-butyrolactone effects on behavior
induced by dopamine agonists. Biol Psychiatry 18:1023-1032.

169
Emri Z, Antal K, Crunelli V. 1996. Gamma-hydroxybutyric acid decreases thalamic sensory excitatory postsynaptic potentials by an action on presynaptic GABAB receptors. Neurosci Lett 216:121-124.
Enberg G, Nissbrandt H. 1993. Gamma-hydroxybutyric acid (GHBA) induces
pacemaker activity and inhibition of substantia nigra dopamine neurons by activating GABAB receptors. Naunyn-Schmiedebergs Arch Pharmacol 348:491-497.
Entholzner E, Mielke L, Pichmeier R, Weber F, Schneck H. 1995. EEG changes during
sedation with gamma-hydroxybutyric acid. Anaesthesist 44:345-350. Fahn S. 1992. Adverse effects of levadopa. In: Olanow, CW, Lieberman AN, editors.
The Scientific Basis for the Treatment of Parkinson’s disease. Park Ridge: The Parthenon Publishing Group Ltd. p 89-112.
Fattore L, Cossu G, Martellotta MC, Deiana S, Fratta W. 2001. Baclofen antagonizes
intravenous self-administration of gamma-hydroxybutyric acid in mice. Neuroreport 12:2243-2246.
Feigenbaum JJ, Howard SG. 1996a. Does gamma-hydroxybutyrate inhibit or stimulate
central dopamine release? Int J Neurosci 88(1-2):53-69. Feigenbaum JJ, Howard SG. 1996b. Gamma-hydroxybutyrate is not a GABA agonist.
Prog Neurobiol 50(1):1-7. Feigenbaum JJ, Howard SG. 1996c. Naloxone reverses the inhibitory effect of gamma-
hydroxybutyrate on central DA release in vivo in awake animals: a microdialysis study. Neurosci Lett 218:5-8.
Feigenbaum JJ, Howard SG. 1997. Naloxone reverses the inhibitory effect of γ-
hydroxybutyrate on central DA release in vivo in awake animals: a microdialysis study. Neurosci Lett 224:71-74.
Ferrara S, Tedeschi L, Frison G. 1993. Therapeutic gamma-hydroxybutyric acid
monitoring in plasma and urine by gas chromatography – mass spectrometry. J Pharm Biomed Anal 11:483-487.
Fishbein WN, Bessman SP. 1966. Purification and properties of an enzyme in human
blood and rat liver microsomes catalizing the formation and hydrolysis of γ-lactones. II. Metal ion effects, kinetics, and equilibria. J Biol Chem 241:4842-4847.
Freese TE, Miotto K, Reback CJ. 2002. The effects and consequences of selected club
drugs. J Subst Abuse Treat 23:151-156.

170
Fuller RW, Clemens JA. 1991. Pergolide: A dopamine agonist at both D1 and D2 receptors. Life Sci 49:925-930.
Fuller RW, Clemens JA, Kornfeld EC, Snoddy HD, Smalstig EB, Bach N. 1979. Effect
of 8(B)-8-[(methylthio) methyl]-6-propylergoline on dopaminergic function and brain dopamine turnover in rats. Life Sci 24(4):375-382.
Gallimberti L, Canton G, Gentile N, Ferri M. 1989. Gamma-hydroxybutyric acid for the
treatment of alcohol withdrawal syndrome. Lancet 30,2(8666):787-789. Galloway GP, Frederick SL, Staggers FE, Gonzales M, Stalcup SA, Smith DE. 1997.
Gamma-hydroxybutyrate: an emerging drug of abuse that causes physical dependence. Addiction 92(1):89-96.
Gessa GL, Crabai F, Vargiu L, Spano PF. 1968. Selective increase of brain dopamine
induced by γ-hydroxybutyrate: study of the mechanism of action J Neurochem 15:377-381.
Gessa GL, Vargiu L, Crabai F, Boero GC, Caboni F, Camba R. 1966. Selective increase
of brain dopamine induced by gamma-hydroxybutyrate. Life Sci 5:1921-1930. Gianutsos G, Moore KE. 1978. Tolerance to the effects of baclofen and gamma-
butyrolactone on locomotor activity and dopaminergic neurons in the mouse. J Pharmacol Exp Ther 207:859-869.
Gianutsos G, Suzdak PD. 1984. Evidence for downregulation of GABA receptors
following long-term gamma-butyrolactone. Naunyn-Schiederbergs Arch Pharmacol 328:62-68.
Gibson KM. 2005. Gamma-hydroxybutyric aciduria: A biochemist’s education from a
heritable disorder of GABA metabolism. J Inher Metab Dis 28:247-265. Gibson KM, Christensen E, Jakobs C, Fowler B, Clarke MA, Hammersen G, Raab K,
Kobori J, Moosa A, Vollmer B and others. 1997. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria): case reports of 23 new patients. Pediatrics 99:567-574.
Gibson KM, DeVivoDC, Jakobs C. 1989. Vigabatrin therapy in succinic semialdehyde
dehydrogenase deficiency. Lancet 2(8671):1105-1106. Gibson KM, Gupta M, Callan M, and others. 2002. Intraperitoneal adenoviral rescue
from lethal seizures in mice deficient for succinate semialdehyde dehydrogenase (SSADH :Aldh5a1). Program no. 619.2. Abstract viewer/itinerary planner. Washington, DC: Society for Neuroscience, 2002c. online

171
Gibson KM, Jakobs C, Ogier H, Hagenfeldt L, Eeg-Olofsson KE, Eeg-Olofsson O, Aksu F, Weber HP, Rossier E, Vollmer B, and others. 1995. Vigabatrin therapy in six patients with succinic semialdehyde dehydrogenase deficiency. J Inher Metab Dis 18:143-146.
Gibson KM, Schor DSM, Gupta M, Guerand WS, Senephansiri H, Burlingame TG,
Bartels H, Hogema BM, Bottiglieri T, Froestl W and others. 2002. Focal neurometabolic alterations in mice dificient for succinate semialdehyde dehydrogenase. J Neurochem 81:71-79.
Gobaille S, Schleef C, Hechler V, Viry S, Aunis D, Maitre M. 2002. Gamma-
hydroxybutyrate increases tryptophan availability and potentiates serotonin turnover in rat brain. Life Sci 70:2101-2112.
Godin Y, Mark J, Mandel P. 1968. The effects of 4-hydroxybutyric acid on the
biosynthesis of amino acids in the central nervous system. J Neurochem 15:1085-1091.
Goetz CG, Tanner CM, Klawans HL. 1984. Bupropion in Parkinson’s disease.
Neurology 34:1092-1094. Gold BI, Roth RH. 1977. Kinetics of in vivo conversion of γ-[3H]aminobutyric acid to
γ-[3H]hydroxybutyric acid by rat brain. J Neurochem 28:1069-1073. Gonce M, Delwaide PJ. 1985. Clinical study of pergolide in Parkinson’s disease.
Presse Med 14:1409-1411. Goodwin AK, Froestl W, Weerts EM. 2005. Involvement of gamma-hydroxybutyrate
GHB) and GABA-B receptors in the acute behavioral effects of GHB in baboons. Psychopharmacology 180:342-351.
Gresch PJ, Walker PD. 1999. Serotonin2 receptor stimulation normalizes striatal
preprotachykinin messenger RNA in an animal model of Parkinson’s disease. Neuroscience 93:831-841.
Guidotti A, Ballotti PL. 1970. Relationship between pharmacological effects and blood
and brain levels of gamma-butyrolactone and gamma-hydroxybutyrate. Biochem Pharmacol 19:883-894.
Gupta M, Greven R, Jansen EEW, Jakobs C, Hogema BM, Froestl W, Snead OC, Bartels
H, Grompe M, Gibson KM. 2002. Therapeutic intervention in mice deficient for succinate semialdehyde dehydrogenase. Pharmacol Exp Ther 302:180-187.

172
Gupta M, Hogema BM, Grompe M, Botiglieri TG, Concas A, Biggio G, Sogliano C, Rigamonti AE, Pearl PL, Snead III OC, Jakobs C, Gibson KM. 2003. Murine succinate semialdehyde dehydrogenase deficiency. Ann Neurol 54(6 Suppl):S81-S90.
Haller C, Mende M, Schuier F, Schuh R, Schrock H, Kuschinsky W. 1990. Effect of
γ-hydroxybutyrate on local and globalglucose metabolism in the anesthetized cat brain. J Cereb Blood Flow Metab 10:493-498.
Hamilton ME, Mele A, Pert A. 1992. Striatal extracellular dopamine in conscious vs.
anaesthetized rats: effects of chloral hydrate anaesthetic on responses to drugs of different classes. Brain Res 597:1-7.
Handforth A, Sourkes TL. 1975. Inhibition by dopamine agonists of dopamine
accumulation following hydroxybutyrate treatment. Eur J Pharmacol 34:311-319.
Hansard MJ, Smith LA, Jackson MJ, Cheetham SC, Jenner P. 2002. Dopamine, but not
norepinephrine or serotonin, reuptake inhibition reverses motor deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates. J Exp Pharmacol Ther 303:952-958.
Hechler V, Bourguignon JJ, Wermuth CG, Mandel P, Maitre M. 1985.
γ-Hydroxybutyrate uptake by rat brain striatal slices. Neurochem Res 10:387-396. Hechler V, Gobaille S, Bourguignon JJ, Maitre M. 1991. Extracellular events induced
by gamma-hydroxybutyrate in striatum : a microdialysis study. J Neurochem 56:938-944.
Hechler V, Mersel M, Dreyfus H, Maitre M. 1990a. Effects of phospholipases,
proteases, and neuraminidase on gamma-hydroxybutyrate binding sites. Mol Cell Biochem 93:87-94.
Hechler V, Peter P, Gorbaille S, Bourguignon JJ, Schmitt M, Ehrhardt JD, Mark J, Maitre
M. 1993. γ-Hydroxybutyrate ligands possess antidopaminergic and neuroleptic- like activities. J Pharmacol Exp Ther 264:1406-1414.
Hechler V, Schmitt M, Bourguignon JJ, Maitre M. 1990b. Trans-gamma-
hydroxycrotonic acid binding sites in brain: evidence for a subpopulation of gamma-hydroxybutyrate sites. Neurosci Lett 110:204-209.
Hedner T, Lundborg P. 1983. Effect of gammahydroxybutyric acid on serotonin
synthesis, concentration and metabolism in the developing rat brain. J Neural Transm 57(1-2):39-48.

173
Hedou G, Chasserot-Golaz S, Kemmel V, Gobaille S, Roussel G, Artault JC, Andriamampandry C, Aunis D, Maitre M. 2000. Immunohistochemical studies of the localization of neurons containing the enzyme that synthesizes dopamine, GABA, or gamma-hydroxybutyrate in the rat substantia nigra and striatum. J Comp Neurol 426(4):549-560.
Hernandez M, McDaniel Ch, Costanza CD, Hernandez J. 1998. GHB-induced delirium:
a case report and review of the literature on gamma hydroxybutyric acid. Am J Drug Alcohol Abuse 24:179-183.
Ho CS, Grange RW, Joho RH. 1997. Pleiotropic effects of a disrupted K1 channel gene:
Reduced body weight, impaired motor skill and muscle contraction, but no seizures. Proc Natl Acad Sci 94:1533-1538.
Hogeman BM, Gupta M, Senephansiri H, Burlingame TG, Taylor M, Jakobs C,
Schutgens RB, Froestl W, Snead OC, Diaz-Arristia R, Bottiglieri T, Grompe M, Gibson KM. 2001. Pharmacological rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet 29:212-216.
Hornfeldt C, Lothridge K, Upshaw JC. 2002. Forensic science update: gamma-
hydroxybutyrate (GHB). Forensic Sci Commun 4(1). Hosli E, Hosli L. 1983. Binding sites for [3H]γ-hydroxybutyrate on cultured neurones of
rat cerebellum and spinal cord. Neurosci Lett 42:145-148. Hosli L, Hosli E, Lehmann R, Schneider J, Borner M. 1983. Action of gamma-
hydroxybutyrate and GABA on neurons of cultured rat central nervous system. Neurosci Lett 31(3):257-260.
Hutchins DA, Rayevsky KS, Sharman DF. 1972. The effect of sodium
γ-hydroxybutyrate on the metabolism of dopamine in the brain. Br J Pharmacol 46:409-415.
Huxtable RJ. 1992. Physiological actions of taurine. Physiol Rev 72:101-163. Ilson J, Fahn S, Mayeux R, Cote LJ, Snider SR. 1983. Pergolide treatment in
parkinsonism. Adv Neurol 37:85-94. Ishige K, Aizawa M, Ito Y, Fukuda H. 1995. γ-Butyrolactone-induced absence-like
seizures increase nuclear CRE and AP-1 DNA binding activities in mouse brain. Neuropharmacol 35:45-55.
Ito Y, Ishige K, Zaitsu E, Anzai K, Fukuda H. 1995. γ-Hydroxybutyric acid increases
intracellular Ca2+ concentration and nuclear cyclic AMP-responsive element and activator protein 1 DNA binding activities through GABAB receptor in cultured cerebellar granule cells. J Neurochem 65:75-83.

174
Itzhak Y, Ali SF. 2002. Repeated administration of gamma-hydroxybutyric acid (GHB) to mice: assessment of the sedative and rewarding effects of GHB. Ann New York Acad Sci 965:451-460.
Jakobs C, Bojasch M, Monch E, Rating D, Siemes H, Hanfeld F. 1981. Urinary
excretion of gamma-hydroxybutyric acid in a patient with neuroligical abnormalities: The probability of a new inborn error of metabolism. Clin Chim Acta 111:169-178.
Jakobs C, Jaeken J, Gibson KM. 1993. Inherited disorders of GABA metabolism.
J Inher Metab Dis 16:704-715. Jazz Pharmaceuticals. 2006. Xyrem. U.S. Prescribing Information. Palo Alto, CA. 5 p.
Available from http://xyrem.info/usprescribe.htm. Accessed 2006 February 20. Kaufman EE, Nelson T. 1987. Evidence for the participation of a cytosolic NADP-
dependent oxidoreductase in the catabolism of γ-hydroxybutyrate in vivo. J Neurochem 48:1953-1941.
Kaufman EE, Porrino LJ, Nelson T. 1990. Pyretic action of low doses of gamma-
hydroxybutyrate in rats. Biochem Pharmacol 40:2637-2640. Kaufman EE, Relkin N, Nelson T. 1983. Regulation and properties of an NADP+
oxioreductase which functions as a γ-hydroxybutyrate dehydrogenase. J Neurochem 40:1639-1646.
Kehr W, Carlsson A, Lindqvist M, Magnusson T, Atack C. 1972. Evidence for a
receptor mediated feedback control of striatal tyrosine hydroxylase activity. J Pharm Pharmacol 24:744-747.
Kelly PH, Moore KE. 1978. Dopamine concentrations in the rat brain following
injections into the substantia nigra of baclofen, γ-aminobutyric acid, γ-hydroxybutyric acid, apomorphine and amphetamine. Neuropharmacology 17:169-174.
Kemmel V, Taleb O, Andriamampandry C, Aunis D, Maitre M. 2003.
γ-Hydroxybutyrate receptor function determined by stimulation of rubidium and calcium movements from NCB-20 neurons. Neuroscience 116:1021-1031.
Kemmel V, Taleb O, Perard A, Andriamampandry C, Siffert JC, Mark J, Maitre M.
1998. Neurochemical and electrophysiological evidence for the existence of a functional gamma-hydroxybutyrate system in NCB-20 neurons. Neuroscience 86:989-1000.
Klawans HL, Tanner CM, Glatt S, Goetz CG. 1983. A 6-month trial of pergolide
mesylate in the treatment of idiopathic Parkinson’s disease. Adv Neurol 37:75-83.

175
Kohrs FP, Mann C, Greenberg R. 2004. The use of amphetamine in gamma- hydroxybutyrate overdose: a case report. J Psychoactive Drugs 36(3):401-402.
Kolin A, Brezina A, Mamelak M. 1991. Cardioprotective effects of sodium
gamma-hydroxybutyrate (GHB) on brain induced myocardial injury. In Vivo 5:429-432.
Kozhechkin SX. 1980. Microiontophoretic study of the mechanism of action of gamma-
hydroxybutyric acid. Bull Exp Biol Med 88:1293-1296. Laborit H. 1964. Sodium 4-hydroxybutyrate. Int J Neuropharmacol 3:433-452. Laborit H. 1973. Gamma-hydroxybutyrate, succinic semialdehyde and sleep.
Prog Neurobiol 1:257-274. Lane RF, Blaha CD. 1978. Acute thioridazine stimulates mesolimbic but not
nigrostriataldopamine release: demonstration by in vivo electrochemistry. Brain Res 408:317-320.
Lapierre O, Montplaisir J, Lamarre M, Bedard MA. 1990. The effect of gamma-
hydroxybutyrate on nocturnal and diurnal sleep of normal subjects: further considerations on REM sleep-triggering mechanisms. Sleep 13:24-30.
Lavyne MH, Hairiri RJ, Tankosic T, Babiac T. 1983. Effect of low dose γ-butyrolactone
therapy on forebrain neuronal ischemia in the unrestrained, awake rat. Neurosurgery 12:430-434.
Leinwand D. 2001 July 25. Club drugs sending more youths to hospitals. USA
Today.com. Available via http://www.usatoday.com/news/health/2001-07-25-drugs-usat.htm. Accessed 2005 Nov 9.
Lettieri J, Fung HL. 1978. Improved pharmacological activity via pro-drug
modification: Comparative pharmacokinetics of sodium γ-hydroxybutyrate and γ-butyrolactone. Res Commun Chem Pathol Pharmacol 22:107-118.
Li T, Thumen A, Moser A. 2005. Modulation of a neuronal network by electrical high
frequency stimulation in striatal slices of the rat in vitro. Neurochem Int 48(2):83-86.
Lingenhoel K, Brom R, Heid J, Beck P, Froestl W, Kaupmann K, Bettler B, Mosbacher J.
1999. Gamma-hydroxybutyrate is a weak agonist at recombinant GABA(B) receptors. Neuropharmacology 38:1667-1673.

176
Lloyd J. 2002. Gamma Hydroxybutyrate (GHB). Washington: White House Office of National Drug Control Policy. Available from http://www.whitehousedrugpolicy.gov/ publications/factsht/gamma. Accessed 2006 Feb 10.
Louagie H, Verstraete A, De Soete C. 1997. A sudden awakening from a near coma
after combined intake of gamma-hydroxybutyric acid (GHB) and ethanol. J Toxicol Clin Toxicol 35:591-594.
Lundborg P, Hedner T, Engel J. 1980. Catecholamine concentration in the developing
rat brain after gamma-hydroxybutyric acid. J Neurochem 35:425-429. MacMillan V. 1980a. Effects of gamma-hydroxybutyrate and gamma-butyrolactone
on cerebral energy metabolism during exposure and recovery from hypoxia-oligemia. Stroke 11:271-277.
MacMillan V. 1980b. Sequential alterations of cerebral carbohydrate metabolism
associated with gamma-hydroxybutyrate. Brain Res 183:123-134. Madden TE, Johnson SW. 1998. Gamma-hydroxybutyrate is a GABA B receptor
agonist that increases the potassium conductance in rat ventral tegmental dopamine neurons. J Pharmacol Exp Ther 287(1):261-265.
Maitre M. 1997. The γ-hydroxybutyrate signalling system in brain: Organization and
functional implications. Prog Neurobiol 51:337-361. Maitre M, Andriamampandry C, Kemmel V, Schmidt C, Hode Y, Hechler V, Gobaille S.
2000. Gamma-hydroxybutyric acid as a signaling molecule in brain. Alcohol 20:277-283.
Maitre M, Cash CD, Weissmann-Nanopoulos D, Mandel P. 1983a. Depolarization-
evoked release of γ-hydroxybutyrate from rat brain slices. J Neurochem 41:287-290.
Maitre M, Hechler V, Vayer P, Gobaille S, Cash CD, Schmitt M, Bourguignon JJ. 1990.
A specific gamma-hydroxybutyrate receptor ligand possesses both antagonistic and anticonvulsant properties. J Pharmacol Exp Ther 255:657-663.
Maitre M, Rumigny JF, Cash C, Mandel P. 1983c. Subcellular distribution of
γ-hydroxybutyrate binding sites in rat brain. Principal localization in the synaptomsomal fraction. Biochem Biophys Res Commun 110:262-265.
Malone DT, Long LE, Taylor DA. 2004. The effect of SR 141716 and apomorphine on sensorimotor gating in Swiss mice. Pharmacol Biochem Behavior 77:839-845.

177
Mamelak M. 1989. Gamma-hydroxybutyrate: An endogenous regulator of energy metabolism. Neurosci Biobehav Rev 13(4):187-198.
Mamelak M. 1997. Neurodegeneration, sleep, and cerebral energy metabolism: A
testable hypothesis. J Geriatr Psychiatry Neurol 10(1):29-32. Mamelak M, Escriu JM, Stokan O. 1977. The effects of γ-hydroxybutyrate on sleep.
Biol Psychiat 12:273-278. Mamelak M, Scharf MB, Woods M. 1986. Treatment of narcolepsy with gamma-
hydroxybutyrate. A review of clinical and sleep laboratory findings. Sleep 9:285-289.
Mannisto PT, Tuomainen P, Tuominen RK. 1992. Different in vivo properties of three
new inhibitors of catechol-O-methyltransferase in the rat. Br J Pharmacol 105:569-574.
Marcu RJ, Winters WD, Mori K, Spooner CE. 1967. EEG And behavioral comparison
of the effects of gamma-hydroxybutyrate, gamma-butyrolactone and short chain fatty acids in the rat. Int J Neuropharmacol 6:175-185.
Marcus R, Winters W, Hultin E. 1976. Neuropharmacological effects induced by
butanol, 4-hydroxybutyrate, 4-mercaptobutyric acid thiolactone, tetrahydrofuran, pyrolidine, 2-deoxy-D-glucose and related substances in the rat. Neuropharmacology 15:29-38.
Martellota MC, Fattore L, Cossu G, Fratta W. 1997. Rewarding properties of gamma-
hydroxybutyric acid: an evaluation through place preference paradigm. Psychopharmacology (Berl) 132:1-5.
Matern D, Lehnert W, Gibson KM, Korinthenberg R. 1996. Seizures in a boy with
succinic semialdehyde dehydrogenase deficiency treated with vigabatrin (γ-vinyl-GABA). J Inherit Metab Dis 19:313-318.
Mathivet P, Bernasconi R. 1997. Binding characteristics of gamma-hydroxybutyric acid
as a weak but selective GABA B receptor agonist. Eur J Pharmacol 321(1):67-75.
Mathivet P, Bernasconi R, De Barry J, Marescaux C, Bittiger H. 1997. Binding
characteristics of gamma-hydroxybutyric acid as a weak but selective GABAB receptor agonist. Eur J Pharmacol 321:67-75.
Matsuda M, Hoshino M. 1977. Natural occurrence of succinic semialdehyde in mouse
brain. Jikeikai Med J 24:33-36.

178
Matthiessen A, Wright CR. 1869. Research into the chemical constitution of the opium bases. Part I: On the action of hydrochloric acid on morphia. Proc R Soc Lond B Biol Sci 17:455-460.
Maxwell R, Roth RH. 1972. Conversion of 1,4-butanediol to γ-hydroxybutyric acid in
rat brain and in peripheral tissue. Biochem Pharmacol 21:1521-1533. McCabe ER, Layne EC, Sayler DF, Slusher N, Bessman, SP. 1970. Synergy of ethanol
and a natural soporific-gammahydroxybutyrate. Science 171:404-406. Mereu G, Fadda F, Gessa GL. 1984. Ethanol stimulates the firing rate of nigral
dopaminergic neurons in unanesthetized rats. Brain Res 292(1):63-69. Middleton HC, Sharma A, Agouzoul D, Sahakian BJ, Robbins TW. 1999. Idazoxan
potentiates rather than antagonizes some of the cognitive effects of clonidine. Psychopharmacology (Berl) 145:401-411.
Millan MJ, Lejeune F, Gobert A. 2000. Reciprocal autoreceptor and heteroreceptor
control of serotonergic, dopaminergic, and noradrenergic transmission in the frontal cortex: relevance to the actions of antidepressant agents. Psychopharmacology 14:114-138.
Miotto K, Darakjian J, Basch J, Murray S, Zogg J, Rawson R. 2001. Gamma-
hydroxybutyric acid: patterns of use, effects and withdrawal. Am J Addict 10:232-241.
Moghaddam B, Bunney BS. 1989. Ionic composition of microdialysis perfusing
solution alters the pharmacological responsiveness and basal outflow of striatal dopamine. J Neurochem 53:652-654.
Muller C, Viry S, Miehe M, Andriamampandry C, Aunis D, Maitre M. 2002. Evidence
for a γ-hydroxybutyrate (GHB) uptake by rat brain synaptic vesicles. J Neurochem 80:899-904.
Nagatsu T, Levitt M, Udenfriend S. 1964. Tyrosine hydroxylase : The initial step in
norepinephrine biosynthesis. J of Biol Chem 239(9):2910-2917. Nandihini TA, Anuradha CV. 2003. Inhibition of lipid peroxidation, protein glycation
and elevation of membrane ion pump activity by taurine in RBC exposed to high glucose. Clinica Chimica Acta 336:129-135.
Nelson T, Kaufman E, Kline J, Sokoloff L. 1981. The extraneural distribution of
γ-hydroxybutyrate. J Neurochem 37:1345-1349.

179
Newman-Tancredi A, Cussac D, Quentric Y, Touzard M, Verrielle L, Carpentier N, Millan MJ. 2002. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. III. Agonist and antagonist properties at serotonin, 5HT1 and 5HT2 receptor subtypes. J Pharmacol Exp Ther 303(2):815-822.
Ng NK, Lee HS, Wong PTH. 1999. Regulation of striatal dopamine release through 5-
HT1 and 5-HT2 receptors. J Neeurosci Res 55:600-607. Nicholson KL, Balster RL. 2001. GHB: a new and novel drug of abuse. Drug Alcohol
Depend 63:1-22. Nieoullan A, Cheramy A, Glowinski J. 1977. An adaptation of the push-pull cannula
method to study the in vivo release of (3H) tyrosine in the cat caudate nucleus: effects of various physical and pharmacological treatments. J Neurochem 28:819-828.
[NIH/NINDS]. 2006. Narcolepsy Fact Sheet. Bethesda, MD. Available from
http://www.ninds.nih.gov/disorders/narcolepsy/detail_narcolepsy.htm. Accessed 2006 Feb 12.
Nissbrandt H, Elverfors A, Engberg G. 1994. Pharmacologically induced cessation of
burst activity in nigral dopamine neurons: significance for the terminal dopamine efflux. Synapse 17:217-224.
Nissbrandt H, Engberg G. 1996. The GABA-receptor antagonist, CGP 35348,
antagonizes gamma-hydroxybutyrate and baclofen induced alterations in locomotor activity and forebrain dopamine levels in mice. J Neural Transm 103:1255-1263.
Nissbrandt H, Elverfors A, Engberg G. 1994. Pharmacologically induced cessation of
burst activity in nigral dopamine neurons: Significance for the terminal dopamine efflux. Synapse 17:217-224.
Numan S, Lundgren KH, Wright DE, Herman JP, Seroogy KB. 1995. Increased
expression of 5-HT2 receptor mRNA in rat striatum following 6-OHDA lesions of the adult nigrostriatal pathway. Mol Brain Res 29:391-396. O'Connell T, Kaye L, and Plosay. 2000. Am Fam Physician 1;62(11):2478-83. Okun M, Bartfield RB, Doering PL. 2000. GHB Toxicity: What You Need to Know.
Emerg Med 10-23. Okun M, Boothby L, Bartfield R, Doering P. 2001. GHB: An important pharmacologic
and clinical update. J Pham Pharmaceut Sci 4(2):167-175.

180
Olpe HR, Karlsson G, Pozza MF, Brugger F, Steinmann M, Van Riezen H, Fagg G, Hall RG, Froestl W, Bittiger H. 1990. CGP 35348: a centrally active blocker of GABAB receptors. Eur J Pharmacol 187:27-38.
Osborne PG, O’Connor WT, Drew KL, Ungerstedt U. 1990. An in vivo characterization
of extracellular dopamine and GABA in the dorsolateral striatum of awake freely moving, and halothane anaesthetized rats. J Neurosci Methods 34:99-105.
Park DM, Findley LJ, Teychenne PF. 1977. Nomifensine in Parkinsonism.
J Clin Pharmacol 4:(Suppl) S185-S186. Park DM, Findley LJ, Teychenne PF. 1981. Nomifensine: effect in Parkinsonian
patients not receiving levodopa. J Neurol Neurosurg Psychiatry 44:352-354. Pattarelli PP, Nyhan WL, Gibson KM. 1988. Oxidation of (U-14C) succinic
semialdehyde in cultured human lymphoblasts: Measurement of residual succinic semialdehyde dehydrogenase activity in 11 patients with 4-hydroxybutyric aciduria. Pediat Res 24:455-460.
Pearl PL, Novotny EJ, Acosta MT, Jakobs C, Gibson KM. 2003. Succinic semialdehyde
dehydrogenase deficiency in children and dults. Ann Neurol 54(6 Suppl):S73-S80.
Pericic D, Walters JR. 1976. Dopamine in substantia nigra and cortex after
γ-butyrolactone treatment. J Pharm Pharmacol 28:527-530. Poldrugo F, Snead OC. 1984. 1,4 Butanediol, gamma-hydroxybutyric acid and ethanol:
Relationships and interactions. Neuropharmacology. 84(23):109-113. Poldrugo F, Snead OC. 1986. 1,4 Butanediol and ethanol compete for degradation in rat
brain and liver. Alcohol 3:367-370. Quang LS, Desai, MC, Kraner JC, Shannon MW, Woolf AD, Maher TJ. 2002a. Enzyme
and receptor antagonists for preventing toxicity from the gamma-hydroxybutyric acid precursor 1,4-butanediol in CD-1 mice. Ann NY Acad Sci 965:461-472.
Quang LS, Shannon MW, Woolf AD, Desai MC, Maher TJ. 2002b. Pretreatment of
CD-1 mice with 4-methylpyrazole blocks toxicity from the gamma-hydroxybutyrate precursor, 1,4-butanediol. Life Sci 71:771-778.
Raess BU, Tunnicliff G. 2002. Abuse potential and toxicology of γ-hydroxybutyrate.
In Gamma-Hydroxybutyrate (Tunnicliff G and Cash CD, eds), pp188-196, Taylor & Francis.

181
Raevskii KS, Gainetdinov RR, Budygin EA, Mannisto P, Wightman M. 2002. Dopaminergic transmission in the rat striatum in vivo in conditions of pharmacological modulation. Neurosci Behav Physiol 32(2):183-188.
Ragozzino ME. 2002. The effects of dopamine D1 receptor blockade in the prelimbic-
infralimbic areas on behavioural flexibility. Learn Mem 9:18-28. Rating D, Hanefeld F, Siemes H, Kneer J, Jacobs C, Hermier M, Divry P. 1984.
4-Hydroxybutyric aciduria: A new inborn error of metabolism. J Inher Met Dis 7:90-96.
Ratomponirina C, Hode Y, Hechler V, Maitre M. 1995. γ-Hydroxybutyrate receptor binding in rat brain is inhibited by guanyl nucleotides and pertusis toxin. Neurosci Lett 189:51-53.
Reis FLV, Masson S, de Oliveira AR, Brandao ML. 2004. Dopaminergic mechanisms
in the conditioned and unconditioned fear as assessed by the two-way avoidance and light switch-off tests. Pharmacol Biochem Behav 79:359-365.
Rejdak K, Rejdak R, Czuczwar SJ, Kleinrok Z, Sieklucka-Dziuba M. 2000. The
influence of CGP-40116 on bicuculline evoked seizures in mice exposed to transient episode of brain oligemia. J Neural Transm 107:1263-1272.
Roberts AC, De Salvia MA, Wilkinson LS, Collins P, Muir JL, Everitt BJ, Robbins TW.
1994. 6-Hydroxydopamine lesions of the prefrontal cortex in monkeys enhance performance on an analog of the Wisconsin card sort test: possible interaction with subcortical dopamine. J Neurosci 4:2531-2544.
Robinson DL, Wightman RM. 2004. Nomifensine amplifies subsecond dopamine
signals in the ventral striatum of freely-moving rats. J Neurochem 90:894-903. Roffler-Tarlov S, Sharman DF, Tegerdine P. 1971. 3,4-dihydroxyphenylacetic acid and
4-hydroxy-3-methoxy-phenylacetic acid in the mouse striatum: a reflection of intra-and extra-neuronal metabolism of dopamine? Br J Pharmacol 42:335-343.
Rogers RD, Blackshaw AJ, Middleton HC, Matthews K, Hawtin K, Crowley C,
Hopwood A, Wallace C, Deakin JF, Sahakian BJ, Robbins TW. 1999. Tryptophan depletion impairs stimulus-reward learning while methylphenidate disrupts attentional control in healthy young adults: implications for the monoaminergic basis of impulsive behaviour. Psychopharmacology (Berl) 146:482-491.
Roth RH. 1971. Effect of anaesthetic doses of gamma-hydroxybutyrate on subcortical
concentrations of homovanillic acid. Eur J Pharmacol 15:52-59. Roth RH. 1976. Striatal dopamine and gamma-hydroxybutyrate. Pharmacol Ther
2:71-88.

182
Roth RH, Delgado JM, Giarman NJ. 1966. Gamma-butyrolactone and gamma- hydroxybutyric acid: II. The pharmacologically active form. Int J Neuropharmacol 5:421-428.
Roth RH, Doherty JD, Walters JR. 1980. Gamma-hydroxybutyrate: A role in the
regulation of dopaminergic neurons? Brain Res 189:556-560. Roth RH, Giarman NJ. 1965. Preliminary report on the metabolism of γ-butyrolactone
and γ-hydroxybutyric acid. Biochem Pharmacol 14:177-178. Roth RH, Giarman NJ. 1966. Gamma-butyrolactone and gamma-hydroxybutyric acid:
I. Distribution and metabolism. Biochem Pharmacol 15:1333-1348. Roth RH, Giarman J. 1970. Natural occurrence of gamma-hydroxybutyrate in
mammalian brain. Biochem Pharmacol 19:1087-1093. Roth RH, Suhr Y. 1970. Mechanism of γ-hydroxybutyrate-induced increase in brain
dopamine and its relation to “sleep.” Biochem Pharmacol 19:3001-3012. Roth RH, Walters, JR, Aghajanian GK. 1973. Effect of impulse flow on the release and
synthesis of dopamine in the rat striatum. In: Usdin E, Snyder S, editors. Frontiers in catecholamine research. New York: Pergammon Press. p 567-574.
Rumigny JF, Cash CD, Mandel P, Maitre M. 1981. Evidence that a specific succinic
semialdehyde reductase is responsible for γ-hydroxybutyrate synthesis in brain slices. FEBS Lett 134:96-98.
Rumigny JF, Maitre M, Cash CD, Mandel P. 1980. Specific and nonspecific succinic
semialdehyde reductases from rat brain. Isolation and properties. FEBS Lett 117:111-116.
Rumigny JF, Maitre M, Cash C, Mandel P. 1981. Regional and subcellular localization
in rat brain of the enzymes that can synthesize γ-hydroxybutyric acid. J Neurochem 36:1433-1438.
Santaniello E, Manzocchi A, Tosi L. 1978. Evaluation of gamma-hydroxybutyrate
formed from L-glutamate by mouse brain homogenate. J Neurochem 34:1117-1118.
Saransaari P, Oja SS. 2000. Taurine and neural cell damage. Amino Acids 19:509-526. Sarhan H, Grimaldi B, Hen R, Fillion G. 2000. 5-HT1B Receptors modulate
release of [3H]dopamine from rat striatal synaptosomes further evidence using 5-HT moduline, polyclonal 5-HT1B receptor antibodies and 5-HT1B receptor knock-out mice. Naunyn-Schmiedebergs Arch Pharmacol 361:12-18.

183
Sarwar G, Botting HG, Davis TA, Darling P, Pencharz PB. 1998. Free amino acids in milks of human subjects, other primates and non-primates. Br J Nutr 79:129-131.
Schmidt C, Gobaille S, Hechler V, Schmitt M, Bourguignon JJ, Maitre M. 1991. Anti-
sedative and anti-cataleptic properties of NCS-382. A γ-hydroxybutyrate receptor antagonist. Eur J Pharmacol 203:393-397.
Schmidt MJ, Sawyer BD, Perry KW, Fuller RW, Foreman MM, and Ghetti B. 1982.
Dopamine Deficiency in the Weaver Mutant Mouse. J Neurosci 2(3):376-380. Schmidt-Mutter C, Muller C, Zwiller J Gobaille S, Maitre M. 1999. Gamma-
hydroxybutyrate and cocaine administration increases mRNA expression in dopamine D1 and D2 receptors in rat brain. Neuropsychopharmacology 21:662-669.
Schmidt-Mutter C, Pain L, Sandner G, Gobaille S, and Maitre M. 1998. The anxioloytic
effect of γ-hydroxybutyrate in the elevated plus maze is reversed by the benzodiazepine receptor anatgonist, flumazenil. Eur J Pharmacol 342:21-27.
Schneidereit T, Burkhart K, Donovan JW. 2000. Butanediol toxicity delayed by
preingestion of ethanol. Int J Med Toxicol 3:1. Schoerken U, Sprenger GA. 1998. Thiamin-dependent enzymes as catalysts in
chemoenzymatic syntheses. Biochem Biophys Acta 1385:229-243. Schousboe A, Saito K, and Wu JY. 1980. Characterization and cellular and subcellular
localization of GABA-transaminase. Brain Res Bull 5:71-76. Scotti De Carolis A, Massotti M. 1978. Electroencephalographic and behavioral
investigations on “gabaergic” drugs: muscimol, baclofen, and sodium γ-hydroxybutyrate. Implications on human epileptic studies. Prog Neuro-Psychopharmacol 2:431-432.
Scrima L, Hartman PG, Johnson FH, Thomas EE, Hiller FC. 1990. The effects of
gamma-hydroxybutyrate on the sleep of narcolepsy patients: A double-blind study. Sleep 13(6):479-490.
Serra M, Sanna E, Foddi C, Concas A, Biggio G. 1991. Failure of gamma-
hydroxybutyrate to alter the function of the GABAA receptor complex in the rat cerebral cortex. Psychopharmacol 104:351-355.
Shannon M, Quang LS. 2000. Gamma-hydroxybutyrate, gamma-butyrolactone, and 1,4-butanediol : a case report and review of the literature. Pediatr Emerg Care 16:435-440.

184
Shaw LMJ, Westerfeld WW. 1968. A study of the enzymatic reactions involved in the formation of 5-hydroxy-4-ketohexanoic acid and its isomer, 5-keto-4-hydroxyhexanoic acid. Biochemistry 7:1333-1338.
Savaki HE, Girault JA, Spampinato U, Truong NA, Glowinski J, Besson MJ. 1986.
Release of newly synthesized 3H-dopamine in the striatum: An adaptation of the push-pull cannula method to awake restrained and anesthetized rats. Brain Res Bull 16:149-154.
Smith KM, Larive LL, Romanelli F. 2002. Club drugs:
methylenedioxymethamphetamine, flunitrazepam, ketamine hydrochloride, and gamma-hydroxybutyrate. Am J Health Syst Pharm 59:1067-1076.
Snead OC. 1977. Minireview: gamma hydroxybutyrate. Life Sci 20:1935-1944. Snead OC. 1978. Gamma-hydroxybutyrate in the monkey, electroencephalographic,
behavioral, and pharmacokinetic studies. Neurology 28(7):636-642. Snead OC. 1982. An investigation of the relationship between the dopaminergic and
electroencephalographic effects of gamma-butyrolactone. Neuropharmacology 21:539-543.
Snead OC. 1987. γ-Hydroxybutyric acid in subcellular fractions of rat brain.
J Neurochem 48:196-201. Snead OC. 1990. Gamma-hydroxybutyric acid-induced seizures bear no relation to core
temperature. Epilepsia 31:253-258. Snead OC. 1992. GABAB receptor mediated mechanisms in experimental absence
seizures in rat. Pharmacol Commun 2:63-69. Snead OC. 1994. The ontogeny of [3H]-γ-hydroxybutyrate and [3H] GABAB
binding sites: relation to the development of experimental absence seizures. Brain Res 659:147-156.
Snead OC. 1996. Relation of the [3H] gamma-hydroxybutyric acid (GHB) binding site
to the gamma-aminobutyric acid B (GABAB) receptor in rat brain. Biochem Pharmacol 52:1235-1243.
Snead OC. 2000. Evidence for a G-protein-coupled gamma-hydroxybutyric acid
receptor. J Neurochem 75:1986-1996. Snead OC. 2002. γ-Hydroxybutyrate and absence seizure activity. In Gamma-
Hydroxybutyrate (Tunnicliff G and Cash CD, eds.) pp 120-131, Taylor & Francis.

185
Snead III OC, Banerjee PK, Burnham M, Hampson D. 2000. Modulation of absence seizures by the GABAA receptor: A critical role for metabotropic glutamate receptor 4 (mGluR4). J Neurosci 20(16):6218-6224.
Snead OC, Bearden LJ, Pegram P. 1980. Effect of acute and chronic anticonvulsant
administration on endogenous γ-hydroxybutyrate in rat brain. Neuropharmacology 19:47-52.
Snead III OC, Furner R, Liu CC. 1989. In vivo conversion of gamma-amino-butyric
acid and 1,4-butanediol to gamma-hydroxybutyric acid in rat brain. Studies using stable isotopes. Biochem Pharmacol 89(38):4375-4380.
Snead OC, Liu CC. 1984. Gamma-hydroxybutyric acid binding sites in rat and human
brain synaptosomal membranes. Biochem Pharmacol 33:2587-2590. Snead OC, Liu CC. 1993. GABAA receptor function in the γ-hydroxybutyrate model of
generalized absence seizures. Neuropharmacol 32:401-409. Snead OC, Liu CC, Bearden LJ. 1982. Studies on the relation of gamma-hydroxybutyric
acid (GHB) to gamma-aminobutyric acid (GABA). Evidence that GABA is not the sole source for GHB in rat brain. Biochem Pharmacol 32:3917-3923.
Snead OC, Nichols AC. 1987. Gamma-hydroxybutyric acid binding sites: evidence for
coupling to a chloride anion channel. Neuropharmacol 26:1519-1523. Snead OC, Nichols AC, Liu CC. 1992. γ-Hydroxybutyric acid binding sites: interaction
with the GABA-benzobiazepine-picrotoxin receptor complex. Neurochem Res 17:201-204.
Sokoloff P, Andrieux M, Besancon R, Pilon C, Martes MP, Giros B, Schwartz JC. 1992.
Pharmacology of human dopamine D3 receptor expressed in mammalian cell line: Comparison with D2 receptor. Eur J Pharmacol 225:331-337.
Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. 1990. Molecular cloning
and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347:146-151.
Sonnewald U, McKenna M. 2002. Metabolic compartmentation in cortical
synaptosomes: influence of glucose and preferential incorporation of endogenous glutamate into GABA. Neurochem Res 27:43-50.
Spampinato U, Girault JA, Danguir J, Savaki HE, Glowinski J, Besson MJ. 1986.
Apomorphine and haloperidol effects on striatal [3H] dopamine release in anaesthetized, awake, restrained and freely moving animals. Brain Res Bull 16:1161-1166.

186
Spano PF, Przegalinsky E. 1973. Stimulation of serotonin synthesis by anesthetic and non-anesthetic doses of gamma-hydroxybutyrate. Pharmacol Res Comm 5:55-69.
Spano PF, Tagliamonte A, Tagliamonte P, Gessa GL. 1971. Stimulation of brain
dopamine synthesis by gamma-hydroxybutyrate. J Neurochem 18:1831-1836. Speciale SG, Friedman AH. 1975. Gamma-butyrolactone Sleep: A 24-hour rhythm
paralleling normal sleep in the rat and CNS amine changes. Pharmacol Biochem Behav 3:761-764.
Stahle L, Collin AK, Ungerstedt U. 1990. Effect of halothane anaesthesia on
extracellular levels of dopamine , dihydroxyphenylacetic acid, homovanillic acid, and 5-hydroxy-indoleacetic acid in rat striatum: a microdialysis study. Naunyn Schmiedeberg Arch Pharmacol 342:136-140.
Stock G, Magnusson T, Anden NE. 1973. Increase in brain dopamine after axotomy or
treatment with gammahydroxybutyric acid due to elimination of the nerve impulse flow. Naunyn Schmiedebergs Arch Pharmacol 278:347-361.
Sturman JA. 1993. Taurine in development. Physiol Rev 73:119-147. Tanner CM, Goetz CG, Glantz RH, Glatt SL, Klawans HL. 1982. Pergolide mesylate
and idiopathic Parkinson disease. Neurology 32:1175-1179. Teter CJ, Guthrie SK. 2001. A comprehensive review of MDMA and GHB: two
common club drugs. Pharmacotherapy 21:1486-1513. Teychenne PF, Park DM, Findley LJ, Rose CF, Calne DB. 1976. Nomifensine in
parkinsonism. J Neurol Neurosurg Psychiatry 39:1219-1221. Timbrell JA, Seabra V, Waterfield CJ. 1995. The in vivo and in vitro protective
properties of taurine. Gen Pharmacol 26:453-462. Timmerman W, Westerink BHC. 1991. Importance of the calcium content infused
during microdialysis for the effects induced by D-2 agonists on the release of dopamine in the striatum of the rat. Neuroscience Letters 131:93-96.
Tunbridge EM, Bannerman DM, Sharp T, Harrison PJ. 2004. Catechol-O-
Methyltransferase improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J Neurosci 24(23):5331-5335.
Tuomainen P, Tornwall M, Mannisto PT. 1996. Minor effect of tolcapone, a catechol-
O-methyltransferase inhibitor, on extracellular dopamine levels modified by amphetamine or pargyline: a microdialysis study in anesthetized rats. Pharmacol Toxicol 78:392-396.

187
Van Cauter E, Plat L, Scharf M, Leproult R. 1997. Simultaneous stimulation of slow wave sleep and growth hormone secretion by gamma-hydroxybutyrate in normal young men. J Clin Invest 100(3):745-753.
Vayer P, Ehrhardt JD, Gobaille S, Mandel P, Maitre M. 1988. Gamma hydroxybutyrate
distribution and turnover rates in discrete brain regions of the rat. Neurochem Intl 12:53-59.
Vickers MD. 1969. Gamma hydroxybutyric acid. Int Anesthesiol Clin 7:75-79. Waldmeier PC. 1991. The GABA-B antagonist, CGP 35348, antagonizes the effects of
baclofen, γ-butyrolactone and HA 966 on rat striatal dopamine synthesis. Naunyn-Schmiedbergs Arch Pharmacol 343:173-178.
Waldmeier PC, Fehr B. 1978. Effects of baclofen and γ-hydroxybutyrate on rat striatal
and mesolimbic 5-HT metabolism. Eur J Pharmacol 49:177-184. Walters JR, Roth RH. 1972. Effect of gamma-hydroxybutyrate on dopamine and
dopamine metabolites in the rat striatum. Biochem Pharmacol 31:2111-2121. Walters JR, Roth RH. 1974. Dopaminergic neurons: Drug induced antagonisms of the
increase in tyrosine hydroxylase activity produced by a cessation of impulse flow. J Pharmacol Exp Ther 191:82-91.
Waszkielewicz A, Bojarski J. 2004. γ-Hydroxybutyric Acid (GHB) and its chemical
modifications: A review of the GHBergic system. Pol J of Pharmacol 56:43-49. Weerts EM, Goodwin AK, Griffiths RR, Brown PR, Froestl W, Jakobs C, Gibson KM.
2005. Spontaneous and precipitated withdrawal after chronic intragastric administration of gamma-hydroxybutyrate (GHB) in baboons. Psychopharmacology 179:678-687.
Westerink BH. 1984. Determination of normetanephrine, 3,4-
dihydroxyphenylethyleneglycol (free and total), and 3-methoxy-4-hydroxyphenylethyleneglycol (free and total) in rat brain by high performance liquid chromatography with electrical detection and effects of drugs on regional concentrations. J Neurochem 42:934-942.
Westerink BHC, Hofsteede HM, Damsma G, de Vries JB. 1988. The significance of
extracellular calcium for the release of dopamine, acetylcholine and amino acids in conscious rats, evaluated by microdialysis. Naunyn Schmiedebergs Arch Pharmacol 337:373-378.
Whittle SR, Turner AJ. 1978. Effects of the anticonvulsant sodium valproate on
γ-aminobutyrate and aldehyde metabolism in ox brain. J Neurochem 31: 1453-1459.

188
Williams SR, Turner JP, Crunelli V. 1995. Gamma-hydroxybutyrate promotes oscillatory activity of rat and cat thalamocortical neurons by a tonic GABAB receptor-mediated hyperpolarization. Neuroscience 66:133-141.
Winters WD, Spooner CE. 1965. A neurophysiological comparison of gamma-
hydroxybutyrate with pentobarbital in cats. Electroencephalog Clin Neurophysiol 18:287-296.
Wolfson LI, Sakurada O, Sokoloff L. 1977. Effects of γ-butyrolactone on local cerebral
glucose utilization in rat. Neurochem 29:777-783. Wood PL, Kim HS, Marien MR. 1987. Intracerebral dialysis: direct evidence for the
utility of 3-MT measurements as an index of dopamine release. Life Sci 41:1-5. Wong C, Bottiglieri T, Snead O. 2003. GABA, γ-hydroxybutyric acid, and
neurological disease. Ann Neurol 54(6 Suppl):S3-S12. Wong C, Gibson K, Snead O. 2004. From the street to the brain: neurobiology of the
recreational drug γ-hydroxybutyric acid. Trends Pharmacol Sci 25(1):29-34. Xie X, Smart TG. 1992. Gamma-hydroxybutyrate depresses monosynaptic excitatory
and inhibitory postsynaptic potentials in rat hippocampal slices. Eur J Pharmacol 223:193-196.
Xyrem Intrnational Study Group. 2005. Further evidence supporting the use of sodium
oxybate for the treatment of cataplexy: a double-blind, placebo-controlled study in 228 patients. Sleep Med 6:415-421.
Youdim MB, Grunblatt E, Mandel S. 1999. The pivotal role of iron in NF-kappa B
activation and nigrostriatal dopaminergic neurodegeneration. Prospects for neuroprotection in Parkinson’s disease with iron chelators. Ann NY Acad Sci 890:7-25.
Zerbib R, Pierrefiche G, Ferran C, Laborit H. 1992. Potential antidepressant activity of
gamma-hydroxybutyrate in the mouse ‘behavioral despair’ test : Correlation with the central dopaminergic system. Res Commun Psychol Psychiatr Behav 17:109-122.
Zhang WQ, Tilson HA, Stachowiak MK, Hongs JS. 1989. Repeated haloperidol administration changes basal release of striatal dopamine and subsequent response to haloperidol challenge. Brain Res 484:389-392.
Zurcher G, Colzi A, Da Prada M. 1990a. Ro 40-7592: inhibition of COMT in rat brain
and extracerebral tissues. J Neural Transm 32:(Suppl) S375-S380.

189
Zurcher G, Keller HH, Kettler R, Borgulya J, Bonnetti EP, Eigenmann, Da Prada M. 1990b. Ro 40-7592, a novel, very potent and orally active inhibitor of catechol-O-methyltransferase: a pharmacological study in rats. Adv. Neurol 53:497-503.
Zurcher G, Dingemanse J, Da Prada M. 1993. Potent COMT inhibition by Ro 40-7592
in the periphery and in the brain. Preclinical and clinical findings. Adv. Neurol 60:641-647.
Zvosec DL, Smith SW, McCutcheon BS, Spillane J, Hall BJ, Peacock EA. 2001.
Adverse events, including death associated with the use of 1,4-butanediol. New Eng J Med 344(2):87-94.
Related Documents

![DETERMINATION OF GAMMA-HYDROXYBUTYRATE (GHB) IN … · 0.1 m g/m l in plasm a and about 2.5 mg/m l in urine – m a - te ri als col lected from liv ing per sons [28], oth ers pub](https://static.cupdf.com/doc/110x72/6146d391f4263007b1356d6a/determination-of-gamma-hydroxybutyrate-ghb-in-01-m-gm-l-in-plasm-a-and-about.jpg)


![[Open Drugs] GHB Manual](https://static.cupdf.com/doc/110x72/5449ce0baf79598c188b460e/open-drugs-ghb-manual.jpg)






