Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis J. M. Anderson, 1 D. W. Hampton, 1 R. Patani, 1 G. Pryce, 2 R. A. Crowther, 3 R. Reynolds, 4 R. J. M. Franklin, 1,5 G. Giovannoni, 2 D. A. S. Compston, 1 D. Baker, 2 M. G. Spillantini 1 and S. Chandran 1 1 Department of Clinical Neurosciences, Cambridge Centre for Brain Repair, University of Cambridge, Forvie Site, Robinson Way, Cambridge, 2 Neuroimmunology Unit, Neuroscience Centre, Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, 3 Medical Research Council, Laboratory of Molecular Biology, Hills Road, Cambridge, 4 Department of Cellular and Molecular Neuroscience, Imperial College Faculty of Medicine, Charing Cross Hospital Campus, Fulham Palace Road, London and 5 Department of Veterinary Medicine, University of Cambridge, Madingley Road, Cambridge, UK Correspondence to: Siddharthan Chandran, Department of Clinical Neurosciences, Cambridge Centre for Brain Repair, University of Cambridge, Forvie Site, Robinson Way, Cambridge, UK E-mail: [email protected] The pathological correlate of clinical disability and progression in multiple sclerosis is neuronal and axonal loss; however, the underlying mechanisms are unknown. Abnormal phosphorylation of tau is a common feature of some neurodegenerative disorders, such as Alzheimer’s disease.We investigated the presence of tau hyperphos- phorylation and its relationship with neuronal and axonal loss in chronic experimental autoimmune encephalo- myelitis (CEAE) and in brain samples from patients with secondary progressive multiple sclerosis.We report the novel finding of abnormal tau phosphorylation in CEAE. We further show that accumulation of insoluble tau is associated with both neuronal and axonal loss that correlates with progression from relapsing^remitting to chronic stages of EAE. Significantly, analysis of secondary progressive multiple sclerosis brain tissue also revealed abnormally phosphorylated tau and the formation of insoluble tau. Together, these observations pro- vide the first evidence implicating abnormal tau in the neurodegenerative phase of tissue injury in experimental and human demyelinating disease. Keywords: tau; secondary progressive multiple sclerosis; experimental autoimmune encephalomyelitis; axonopathy; neuronal loss Abbreviations: CEAE = chronic experimental autoimmune encephalomyelitis; CREAE = chronic relapsing experimental autoimmune encephalomyelitis; LFB = Luxol fast blue; mAb = monoclonal antibody; NGS = normal goat serum; PB = phosphate buffer; PBS = phosphate buffered saline; RM2EAE = second remission experimental autoimmune encephalomyelitis; TX-PBS = Triton-phosphate buffered saline Received February 5, 2008. Revised April 29, 2008. Accepted May 12, 2008. Advance Access publication June 21, 2008 Introduction Multiple sclerosis is the commonest cause of acquired neurological disability in young adults. Although the cause is unknown, it is well-established that an interplay of genetic and environmental factors results in a multi-focal and multi- phasic disease defined histologically by inflammatory demye- lination, axonal injury, astrocytosis and varying degrees of remyelination (Compston et al., 2006). The most common form of the disease has two distinct clinical phases, reflecting inter-related pathological processes: inflammation drives activity during the relapse–remitting stage, whereas neuro- nal and axonal degeneration represents the principal substrate for progressive disability seen in the secondary progressive stage (Davie et al., 1995; De Stefano et al., 1998; Wegner et al., 2006). The cause of progressive axonal loss is unknown, although accumulating disability is known to occur in the absence of significant inflammatory activity (Coles et al., 1999). Axonal swellings and accumulation of amyloid precursor protein have been observed in MS lesions suggesting that disruption of axonal transport may doi:10.1093/brain/awn119 Brain (2008), 131 , 1736 ^1748 ß The Author (2008). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: [email protected] by guest on May 16, 2016 http://brain.oxfordjournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Abnormally phosphorylated tau is associated withneuronal and axonal loss in experimental autoimmuneencephalomyelitis and multiple sclerosisJ. M. Anderson,1D.W. Hampton,1 R. Patani,1 G. Pryce,2 R. A.Crowther,3 R. Reynolds,4 R. J. M. Franklin,1,5
G.Giovannoni,2 D. A. S. Compston,1D. Baker,2 M.G. Spillantini1 and S. Chandran1
1Department of Clinical Neurosciences, Cambridge Centre for Brain Repair, University of Cambridge, Forvie Site, RobinsonWay, Cambridge, 2Neuroimmunology Unit, Neuroscience Centre, Institute of Cell and Molecular Science, Barts and theLondon School of Medicine and Dentistry, Queen Mary University of London, London, 3Medical Research Council,Laboratory of Molecular Biology, Hills Road, Cambridge, 4Department of Cellular and Molecular Neuroscience, ImperialCollege Faculty of Medicine, Charing Cross Hospital Campus, Fulham Palace Road, London and 5Department of VeterinaryMedicine, University of Cambridge, Madingley Road, Cambridge, UK
Correspondence to: Siddharthan Chandran, Department of Clinical Neurosciences, Cambridge Centre for Brain Repair,University of Cambridge, Forvie Site, RobinsonWay, Cambridge, UKE-mail: [email protected]
The pathological correlate of clinical disability and progression in multiple sclerosis is neuronal and axonal loss;however, the underlying mechanisms are unknown. Abnormal phosphorylation of tau is a common feature ofsome neurodegenerative disorders, such as Alzheimer’s disease.We investigated the presence of tau hyperphos-phorylation and its relationship with neuronal and axonal loss in chronic experimental autoimmune encephalo-myelitis (CEAE) and in brain samples frompatients with secondary progressive multiple sclerosis.We report thenovel finding of abnormal tau phosphorylation in CEAE.We further show that accumulation of insoluble tau isassociated with both neuronal and axonal loss that correlates with progression from relapsing^remitting tochronic stages of EAE. Significantly, analysis of secondary progressive multiple sclerosis brain tissue alsorevealed abnormally phosphorylated tau and the formation of insoluble tau.Together, these observations pro-vide the first evidence implicating abnormal tau in the neurodegenerative phase of tissue injury in experimentaland human demyelinating disease.
Keywords: tau; secondary progressive multiple sclerosis; experimental autoimmune encephalomyelitis; axonopathy;neuronal loss
Abbreviations: CEAE=chronic experimental autoimmune encephalomyelitis; CREAE=chronic relapsing experimentalautoimmune encephalomyelitis; LFB=Luxol fast blue; mAb=monoclonal antibody; NGS=normal goat serum;PB=phosphate buffer; PBS=phosphate buffered saline; RM2EAE=second remission experimental autoimmuneencephalomyelitis; TX-PBS=Triton-phosphate buffered saline
Received February 5, 2008. Revised April 29, 2008. Accepted May12, 2008. Advance Access publication June 21, 2008
IntroductionMultiple sclerosis is the commonest cause of acquiredneurological disability in young adults. Although the cause isunknown, it is well-established that an interplay of geneticand environmental factors results in a multi-focal and multi-phasic disease defined histologically by inflammatory demye-lination, axonal injury, astrocytosis and varying degrees ofremyelination (Compston et al., 2006). The most commonform of the disease has two distinct clinical phases, reflectinginter-related pathological processes: inflammation drives
activity during the relapse–remitting stage, whereas neuro-nal and axonal degeneration represents the principalsubstrate for progressive disability seen in the secondaryprogressive stage (Davie et al., 1995; De Stefano et al., 1998;Wegner et al., 2006). The cause of progressive axonal loss isunknown, although accumulating disability is known tooccur in the absence of significant inflammatory activity(Coles et al., 1999). Axonal swellings and accumulation ofamyloid precursor protein have been observed in MSlesions suggesting that disruption of axonal transport may
doi:10.1093/brain/awn119 Brain (2008), 131, 1736^1748
� The Author (2008). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: [email protected]
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

contribute to axonal pathology (Ferguson et al., 1997;Trapp et al., 1998; Shriver and Dittel, 2006). Altered axonaltransport is known to play a role in a number of neuro-degenerative conditions, such as Alzheimer’s disease andhereditary fronto-temporal dementias, characterized byhyperphosphorylation and aggregation of tau (Mandelkowet al., 2003; Stokin et al., 2005; Goedert and Spillantini,2006). Tau is a microtubule-associated protein involved inthe assembly and stabilization of microtubules that arenecessary for axonal transport. Abnormal phosphorylationof tau leads to decreased microtubule stability and canresult in the formation of potentially neurotoxic aggregates(Allen et al., 2002; Johnson and Stoothoff, 2004;Bandyopadhyay et al., 2007).Recent studies highlight the potential value of chronic-
relapsing experimental autoimmune encephalomyelitis(CREAE) as a quantifiable histological and behaviouralmodel that mirrors some aspects of progressive multiplesclerosis (Kornek et al., 2000; Pryce et al., 2005;Papadopoulos et al., 2006). In Biozzi ABH mice, repeatedneurological insults occur in CREAE with a relativelypredictable time course, starting with a relapsing–remittingphase (RL/RM) and later progressing to a chronic, typicallynon-relapsing, stage (CEAE) that exhibits progressivedisability (Baker et al., 1990; Petzold et al., 2003; Pryceet al., 2005). In addition to paralysis, CEAE animals accu-mulate further clinical signs, including tremor and spas-ticity, which are comparable with patients with secondaryprogressive multiple sclerosis (Pryce et al., 2005).In this study, we show that tau is hyperphosphorylated in
progressive CEAE and multiple sclerosis. Furthermore, neu-ronal and axonal loss is associated with abnormally phos-phorylated and insoluble tau in post-inflammatory CEAEmice that exhibit progressive disability, Importantly, we alsodemonstrate abnormal tau phosphorylation and insolubletau formation in cerebral tissue from post-mortem cases ofsecondary progressive multiple sclerosis.
Material and MethodsAnimals and surgeryAll procedures were performed in compliance with national andinstitutional guidelines [UK Animals (Scientific Procedures) Act1986 and the University of Cambridge and London Animal CareCommittees].
Induction of chronic relapsing EAE (CREAE)Biozzi ABH mice were inoculated as previously described (Bakeret al., 1990). Briefly, spinal cord homogenate, emulsified incomplete Freund’s adjuvant, was injected subcutaneously in bothhind-flanks and repeated 7 days later. Mice were monitored dailyand scored according to motor disability and weight loss (typicalat the onset of a relapse), as previously documented (Baker et al.,1990; Pryce et al., 2005). Mice were sacrificed during therelapsing–remitting phase at day 40–50 post-inoculation whilstin remission (typically after two attacks, referred to as RM2EAE)
or during the chronic progressive phase at least 65 days post-inoculation (typically after four attacks, referred to as CEAE).
Immunohistochemistry and histologyAnimals for immunohistochemistry were perfused with 4%paraformaldehyde/PB at the stated time-points, with age-matchedcontrol ABH mice (at least four animals per group). The brainand individual cervical spinal cord segments were dissected, post-fixed for 4 h and cryoprotected. Sagitttal cerebral and parasagittalspinal cord sections (16 mm) were processed by immunofluore-sence as previously described (Hampton et al., 2007). Briefly,slides were washed in PBS and blocked for 1 h using 3% normalgoat serum (NGS) in 0.2% Triton-X100/0.1M phosphate bufferedsaline (TX-PBS). Primary antibodies applied overnight in TX-PBScontaining 1% NGS, were: mouse monoclonal antibody (mAb) anti-glial fibrillary acid protein (GFAP)-Cy3 conjugate (1 : 500; Sigma,Poole, UK), biotinylated mouse mAb specific for Neuronal Nuclei(NeuN) antigen (1 : 100; Chemicon, Chandlers Ford, UK), rat mAbspecific for the macrophage/microglial F4/80 antigen (1 : 200; AbDSerotec, Kidlington, UK), rat polyclonal antibody specific for myelinbasic protein (MBP) (1 : 100; AbD Serotec, UK), mouse mAb anti-tau phospho-dependent AT8 (1 : 1000; Ser202/Thr205) and rabbitpolyclonal antibody specific for neurofilament 200 (NF200) antigen(1: 200; Sigma). Primary species antibody was detected withappropriate secondary antibodies (conjugated Alexa Ab:MolecularProbes/Invitrogen, Paisley, UK. 1 : 500) diluted in TX-PBS contain-ing 1% NGS and Bis-benzamide (Sigma, 1 : 4000).Sections for Luxol fast blue (LFB) staining were dehydrated
before incubation in 50 : 50 chloroform–ethanol for 6 h. Sectionswere washed in ethanol and incubated in LFB solution [LFB-MBS10% w/v, 95% ethanol (v/v), 87mM glacial acetic acid] overnightat 40–55�C. Following water washes sections were differentiated in6.8mM lithium carbonate (Sigma) solution. The sections werecounterstained with cresyl violet, dehydrated and placed undercoverslips using DPX (RA Lamb, Eastbourne, UK).Sections for modified Bielschowsky staining, which labels
neuronal components, were washed in water and then incubatedin pre-warmed 10% silver nitrate for 25min at 40�C. Sectionswere then removed to permit addition of ammonium hydroxideto just clear the silver precipitate, after which they were returnedto the silver ammoniacal solution and incubated for a further45min at 40�C. Sections were then placed in developing solutionfor 40–50 s until the stain was visualized. Staining was subse-quently stopped by incubation in 1% ammonium hydroxidesolution for 1min. After washing, the sections were incubated in5% sodium thiosulphate solution for 5min, washed, dehydratedand placed under coverslips with DPX.
Semithin section processingAnimals for resin embedding (five per group) were perfused using4% glutaraldehyde. The spinal cord was dissected and segmentswere post-fixed in 4% glutaraldehyde, before fixation with 2%osmium tetraoxide (Oxkem Limited, Reading, UK) overnight at4�C. Tissue was dehydrated and embedded in resin (TAAB Labs,Aldermaston, UK). One micro metre semithin sections were cuton a Leica RM2065 ultra microtome and dried onto Polysineslides (VWR international, Lutterworth, UK). Sections were thenstained with toluidine blue (5% in a Borax solution) and placedunder coverslips with DPX.
Abnormal tau in demyelinating disease Brain (2008), 131, 1736^1748 1737
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

Western blotsAnimals were sacrificed and the spinal cord of control, RM2EAEand CEAE ABH mice were dissected. Tissue was immediately snapfrozen. The cervical cord was used for biochemical analysis unlessotherwise specified. Tissue was homogenized in a Tris-based salineextraction buffer containing: Mini Complete Protease Inhibitor(Roche, Burgess Hill, Sussex, UK) Phosphatase Inhibitor Cocktail I& II (Sigma), 100mM sodium fluoride, 10mM b-glycerophosphateand 2.5mM sodium pyrophosphate. After centrifugation, thesupernatant was harvested and the remaining pellet underwentfurther extraction using the above extraction buffer plusdetergents (0.1% SDS, 1%NP40, 0.25% sodium deoxycholate).Protein concentration of the lysates was estimated using theBicinchoninic acid (BCA) assay.The presence of insoluble tau can be biochemically demon-
strated by immunoblotting of sarcosyl insoluble protein extracts.Sarcosyl-insoluble tau preparation was performed as previouslydescribed (Goedert et al., 1992). Due to the volume of tissuerequired for an efficient extract, cervical cords of two animals werepooled for a single preparation (n=3, i.e. six animals). Anteriorforebrain was processed as individual animal extracts (n= 4, i.e.four CEAE animals). Tissue was homogenized in a 10� volume ofhigh salt buffer of 10mM Tris pH 7.4, 0.8M sodium chloride,1mM EGTA, 10% sucrose (w/v). The extract was centrifuged at45 000g for 30min at 4�C, after which the supernatant wascollected (S1), and the pellet exposed to a further extraction stepwith a 5� volume of the same buffer. The subsequent extract wascentrifuged and the two supernatants pooled (S1 + S2). Thesupernatant fraction was incubated with 1% sarcosyl for 1 h andthen centrifuged at 260 000g for 1 h at 4�C. The subsequent pelletrepresented the sarcosyl-insoluble tau preparation, potentiallyenriched for tau filaments. This was re-suspended in 50mMTris–HCl (pH 7.4), 0.8 ml/mg of starting tissue and stored at 4�C.Samples were resolved by 10% sodium dodecyl sulphate (SDS)
polyacrylamide gel electrophoresis (PAGE) and transferred topolyvinylidene difluoride (PVDF) membrane (Millipore, Watford,UK). Membranes were blocked in 5% bovine serum albumin(BSA), fraction IV (Sigma) and probed with either the phospho-independent mouse mAb Tau5 (1 : 1500, Calbiochem,Nottingham, UK;) or the following anti-tau phospho-dependentmouse mAbs: AT8 (1 : 1000; Ser202/Thr205), AT180 (1 : 1000;Thr 231/Ser 235), AT270 (1 : 1500; Thr 181), AT100 (dilution1 : 1000; Ser 212/Thr 214)—all from Autogen Bioclear, Wiltshire,UK—and PHF-1 (1 : 2000; Ser396/Ser404; a kind gift fromDr P Davies, Albert Einstein College of Medicine, Bronx, NY).Membrane-bound primary antibody was probed with horse radishperoxidase (HRP)-conjugated, rabbit polyclonal anti-mouseimmunoglobulin antibody (Dako, Ely, UK) and visualizedwith Amersham’s basic or enhanced chemi-luminescence kits(Amersham, Buckinghamshire, UK).Controls with secondary antibody alone or no antibody were
performed for all blots and non-specific binding was negligible.In addition to the BCA, membranes were probed with mousemonoclonal b-actin antibody (1 : 10 000, Sigma), to control for theamount of protein loaded. All western blots were replicated threetimes.
Electron microscopyImmunogold labelling of sarcosyl-insoluble material with AT8,MT1 (antiserum MT1 recognizes the three adult mouse tau
isoform but fails to recognize human tau isoforms; a kind gift
from Dr M. Goedert), BR134 (anti-serum BR134 recognizes the
C terminus of both human and mouse tau), was performed
as described previously (Goedert et al., 1992). Micrographs
were recorded on a Philips EM208S Microscope at 40 000�
magnification.
Human brain tissueSamples of fronto-parietal cerebral tissue (4% paraformaldehyde
fixed and snap frozen) were supplied by the UK Multiple Sclerosis
Tissue Bank. This study was approved by the Multi Centre
Research Ethics Committee (MREC). Human post-mortem
material from seven pathologically confirmed cases of multiple
sclerosis (four females and three males, aged 34–57 years, mean 43
years: disease duration 8–17 years, mean 12.9 years), known to
have entered the secondary progressive phase in life, were studied.
The cause of death in all cases was non-CNS sepsis. We also
studied a rare acute relapsing–remitting female case, aged 32 years
(2 years total disease duration). Fronto-parietal cerebral tissue
from three female controls (aged 60–69 years, mean 64 years),
with no known neuro-pathological disease, were also used. In
addition, occipital cortex tissue from a patient with Alzheimer’s
disease was used as a positive control for abnormally phosphory-
lated insoluble tau. In the individuals with multiple sclerosis, the
average time to post-mortem from death was 15.6 h, but 25.5 h in
the controls.Tissue used in this study was either 4% paraformaldehyde fixed
or snap frozen, unfixed blocks. Snap-frozen sections (12 mm) were
fixed in 90% ethanol or 4% paraformaldehyde for 10min before
use. Serial sections were immunohistochemically labelled with
LN3mAb [reactive with non-polymorphic human leucocyte
antigen (HLA)-DR antigens on B cells, monocytes, macrophages
and some activated T cells] and counterstained with LFB or
modified Bielschowsky silver impregnation, in order to assess
inflammation in association with demyelination and axonal
pathology, respectively.Sections requiring antigen retrieval (not used for LN3) were
microwaved in 10mM citrate buffer (pH6) (2� 5min at 600W)
after rehydration. Endogenous peroxidase activity was blocked
using 3% hydrogen peroxide in 10% methanol and non-specific
antibody binding blocked with 5% NGS. Primary antibodies used
were: mouse mAb anti-human B lymphocyte (Clone LN3 : 1: 50;
MP Biomedicals, Cambridge, UK), mouse mAb anti-human GFAP
(1: 500; Covance, Harrogate, UK), mouse mAb anti-amyloid pre-
cursor protein (APP)-A4, (Clone 22C11; 1 : 100; Chemicon),
mouse mAb anti-non-phopsphorylated neurofilament (Clone
SMI32; 1 : 1000; Covance), aforementioned anti-tau phospho-
dependent antibodies (AT8 and AT180 1 : 1000, PHF-1 1 : 1500)
and NF200. Primary antibody binding was revealed using a
biotinylated donkey anti-mouse IgG (1 : 200; Jackson Immuno
Research Europe Ltd, Newmarket, UK) and a Vector Laboratories
ABC Elite kit (Peterborough, UK). Labelling was visualized using a
3030-diaminobenzidine fast kit (Sigma). To confirm tau immuno-
reactivity specificity a further control was performed: the primary
antibody mix was adsorbed against a small aliquot of insoluble tau
extract prior to application (data not shown).Snap-frozen tissue from the cases and controls was used for
immunoblotting. Several white matter lesions were identified by
systematic review of stained sections. This allowed a ‘lesion map’
1738 Brain (2008), 131, 1736^1748 J. M. Anderson et al.
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

to be created on the relevant tissue block enabling discrete lesionsto be sampled.
AnalysisImages were captured using Lucia or Leica software (Nikon,Kingston upon Thames, UK) via a digital camera. SigmaScan Pro(SPSS, Chicago, IL, USA) was used for GFAP and brain NeuNquantitative measurements. Four animals were used for eachgroup. Immunopositive cell density measurements were made onoverlays of transverse spinal cord segment images. This analysisused an automated thresholding procedure (Hampton et al., 2007)that measures the density of immunopositive objects independentof their individual intensities. For GFAP densitometric measure-ments, representative areas through the dorsal spinal tracts weresampled using a mask overlay of six 25� 50 mm boxes. For NeuNanalysis, irrespective of the density reading, it was possible todefine how much of the area measured contained a positive pixel.In the brain, motor cortex regions M1 and M2 were analysed witha parallel 25 mm strip mask overlay, running through the laminalayers, 1, 1.5 and 2mm lateral from the midline. Results werenormalized by applying thresholds to the raw images prior toanalysis.Dorsal horn NeuN quantification was performed by formal cell
counts on NeuN immunohistochemistry of C5 dorsal horns.Multiple counts (2–4 according to size of lamina) were performedat 60� magnification under a defined grid (160 mm� 16 mm) ineach of the lamina, with the observer blinded to the animal group.Both dorsal horns were analysed using a minimum of threesections per animal. Four animals were analysed in the controland RM2EAE group and five in the CEAE group. Data waspresented as number of NeuN-positive cells per 1mm2 of tissue.Axonal counts on toluidine blue semithin sections were performedblinded. All healthy myelinated axons that dissected a grid lineacross dorsal spinal tracts (from surface to a depth of 480 mm)were counted.Immunoblots were scanned and NIH image J software used to
measure pixel intensity. The same CEAE cervical spinal cord lysatewas used as a loading reference between independent immunoblots.Statistical analysis was performed using SigmaStat 3.0 software.
Unpaired two tailed t-tests were used to assess significance(P50.01) and linear regression analysis performed to measureassociation. Values expressed are the mean� SEM, from aminimum of four animals, unless otherwise stated.
ResultsDemyelination, gliosis, neuronal and axonalloss in CEAE Biozzi-ABH miceAll studies were undertaken in post relapse-remission,chronic stage ABH mice (CEAE) that displayed progressivedisability (Fig. 1A; day 70–90 post inoculation; Baker et al.,1990) unless specified otherwise. Analysis was restricted tothe spinal cord, since earlier sampling showed that pathologywas largely confined to this region of the nervous system(Baker et al., 1990). LFB staining of myelin was used todetermine demyelination in RM2EAE and CEAE (Fig. 1Dand E). Loss of myelin was most evident in CEAE throughoutthe white matter tracts of the spinal cord, includingdorsal, lateral and anterior tracts. However, these multifocal
lesions were variable except in the dorsal funiculus of thecervical spinal cord and so all further analysis was restrictedto the dorsal funiculus. Reactive astrocytes were identifiedby positive GFAP staining (Fig. 1F–H). A significantincrease in glial reactivity was present in CEAE comparedto control sections and this was quantified by measuring
4.0Relapsing-Remitting EAE (RREAE) Chronic EAE (CEAE)
AP RL 1
RM2
RM1
RM3
RL 2 RL 33.5
Mea
n D
isea
se S
core
± S
EM
3.0
2.5
2.0
1.5
1.0
0.5
0.010 15 20 25 30 35 40 45
Time Post-Inoculation (Days)50 55 60 65 70 75 80 85
Fig. 1 Biozzi ABH mouse CREAE is clinically characterized by aninitial relapsing^remitting phase with subsequent progression toirreversible progressive disability with histopathological evidenceof demyelination and gliosis. (A) Schema showing disease progres-sion in CREAE with motor disability score of animals post induc-tion of EAE (see ‘Material and Methods’ section; 0=no deficit;1= tail paresis; 2= impaired righting reflex; 3=hind-limb paresis;4=hind-limb paralysis; 5=moribund). Following the acute phase(AP) and relapses (RL), where disease episodes were associatedwith worsening in clinical score and associated weight loss, animalswent into remission (RM) and were sacrificed during the post-relapse remission phase (RM2, day 40^50 post-inoculation) orthe late chronic progressive disease stage (CEAE, after 65 dayspost-inoculation). Results represent the mean� SEM for11animals.Histology and immunohistochemistry of spinal cord of CEAE andcontrols. (B and C) Myelin staining was identified using LFB innormal biozzi ABH mice.The boxed area in B highlights the dorsalfuniculus and this is the region that the subsequent images focuson. (D and E) This white matter tract shows a lack of myelinstaining (LFB), which is modest in RM2EAE (D) but marked inCEAE (E) C5 spinal cord. (F^H) GFAP immunohistochemistry ofcontrol, RM2EAE and CEAE C5 spinal cord shows increasedimmunoreactivity throughout the dorsal funiculus being mostmarked in CEAE (H) compared to control (F). (I^K) F4/80immunohistochemistry of C5 spinal cord, showing marked specificimmunoreactivity in RM2EAE (J) compared to both CEAE (K) andcontrol (I). Scale bars: (B)=500mm; (C^K)=25mm.
Abnormal tau in demyelinating disease Brain (2008), 131, 1736^1748 1739
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from
the proportional density of GFAP immunoreactivitythroughout the dorsal spinal tracts (98.67� 3.41% versus52.56� 15.54%; P50.001; Fig. 1F and H). Microglia wereimmunolabelled with F4/80 antibody; an increased numberwere observed in the dorsal spinal tracts, particularly inRM2EAE compared with control (Fig. 1I–K).In order to evaluate neuronal and axonal loss, we
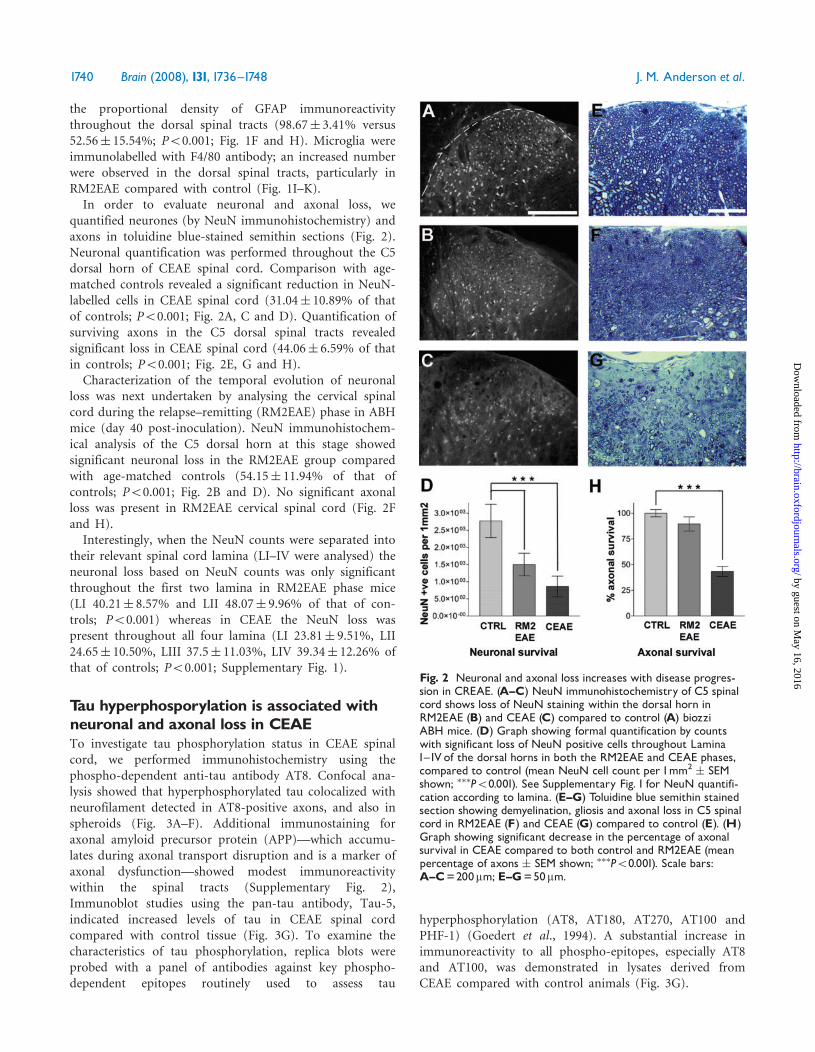
quantified neurones (by NeuN immunohistochemistry) andaxons in toluidine blue-stained semithin sections (Fig. 2).Neuronal quantification was performed throughout the C5dorsal horn of CEAE spinal cord. Comparison with age-matched controls revealed a significant reduction in NeuN-labelled cells in CEAE spinal cord (31.04� 10.89% of thatof controls; P50.001; Fig. 2A, C and D). Quantification ofsurviving axons in the C5 dorsal spinal tracts revealedsignificant loss in CEAE spinal cord (44.06� 6.59% of thatin controls; P50.001; Fig. 2E, G and H).Characterization of the temporal evolution of neuronal
loss was next undertaken by analysing the cervical spinalcord during the relapse–remitting (RM2EAE) phase in ABHmice (day 40 post-inoculation). NeuN immunohistochem-ical analysis of the C5 dorsal horn at this stage showedsignificant neuronal loss in the RM2EAE group comparedwith age-matched controls (54.15� 11.94% of that ofcontrols; P50.001; Fig. 2B and D). No significant axonalloss was present in RM2EAE cervical spinal cord (Fig. 2Fand H).Interestingly, when the NeuN counts were separated into
their relevant spinal cord lamina (LI–IV were analysed) theneuronal loss based on NeuN counts was only significantthroughout the first two lamina in RM2EAE phase mice(LI 40.21� 8.57% and LII 48.07� 9.96% of that of con-trols; P50.001) whereas in CEAE the NeuN loss waspresent throughout all four lamina (LI 23.81� 9.51%, LII24.65� 10.50%, LIII 37.5� 11.03%, LIV 39.34� 12.26% ofthat of controls; P50.001; Supplementary Fig. 1).
Tau hyperphosporylation is associated withneuronal and axonal loss in CEAETo investigate tau phosphorylation status in CEAE spinalcord, we performed immunohistochemistry using thephospho-dependent anti-tau antibody AT8. Confocal ana-lysis showed that hyperphosphorylated tau colocalized withneurofilament detected in AT8-positive axons, and also inspheroids (Fig. 3A–F). Additional immunostaining foraxonal amyloid precursor protein (APP)—which accumu-lates during axonal transport disruption and is a marker ofaxonal dysfunction—showed modest immunoreactivitywithin the spinal tracts (Supplementary Fig. 2),Immunoblot studies using the pan-tau antibody, Tau-5,indicated increased levels of tau in CEAE spinal cordcompared with control tissue (Fig. 3G). To examine thecharacteristics of tau phosphorylation, replica blots wereprobed with a panel of antibodies against key phospho-dependent epitopes routinely used to assess tau
hyperphosphorylation (AT8, AT180, AT270, AT100 andPHF-1) (Goedert et al., 1994). A substantial increase inimmunoreactivity to all phospho-epitopes, especially AT8and AT100, was demonstrated in lysates derived fromCEAE compared with control animals (Fig. 3G).
Fig. 2 Neuronal and axonal loss increases with disease progres-sion in CREAE. (A^C) NeuN immunohistochemistry of C5 spinalcord shows loss of NeuN staining within the dorsal horn inRM2EAE (B) and CEAE (C) compared to control (A) biozziABH mice. (D) Graph showing formal quantification by countswith significant loss of NeuN positive cells throughout LaminaI^IV of the dorsal horns in both the RM2EAE and CEAE phases,compared to control (mean NeuN cell count per 1mm2
� SEMshown; ���P50.001). See Supplementary Fig. 1 for NeuN quantifi-cation according to lamina. (E^G) Toluidine blue semithin stainedsection showing demyelination, gliosis and axonal loss in C5 spinalcord in RM2EAE (F) and CEAE (G) compared to control (E). (H)Graph showing significant decrease in the percentage of axonalsurvival in CEAE compared to both control and RM2EAE (meanpercentage of axons � SEM shown; ���P50.001). Scale bars:A^C=200mm; E^G=50mm.
1740 Brain (2008), 131, 1736^1748 J. M. Anderson et al.
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

Tau phosphorylation can result in disruption of micro-tubule stability and formation of insoluble tau aggregates.Analysis of RM2EAE and CEAE cervical spinal cordinsoluble sarcosyl extract with AT8 antibody, revealed astrongly reactive band of 60 kDa and a weaker band of64 kDa (Fig. 4A). A significant increase in insolublehyperphosphorylated tau in CEAE was evident comparedto RM2EAE (Fig. 4A: P50.005). This pattern of reactivitywas uniform throughout the CEAE spinal cord (Supple-mentary Fig. 3). To confirm that insoluble tau is not afeature of all CNS regions in EAE, we next examinedforebrain tissue from CEAE, since this is known to becomparatively spared in CEAE, consistent with our ownfinding of negligible neuronal loss (Supplementary Fig. 4)(Baker et al., 1990). Immunoblot analysis of insolublesarcosyl extracts from CEAE forebrain revealed significantlyless tau phosphorylation (Fig. 4B: 11.82-fold reductioncompared to CEAE—cervical cord; P50.0001).To determine whether there was a relationship between
the presence of insoluble tau and neuronal or axonal loss, wenext compared levels of insoluble tau (semi-quantitativewestern blot analysis) with both neuronal and axonal quan-tification data from control, RM2EAE and CEAE cervicalspinal cord. Samples were selected at specific time points.
Linear regression analysis showed a clear association betwe-en accumulation of insoluble tau and the neuronal loss thataccompanies progression of EAE (Fig 4C: r2 = 0.8875).In addition, there was an increase in insoluble tau betweenRM2EAE and CEAE which mirrored the axonal loss.
Tau phosphorylation in secondaryprogressive multiple sclerosisWe investigated whether chronic white matter lesions fromcases with secondary progressive multiple sclerosis alsodisplay abnormal tau phosphorylation and formation ofinsoluble tau. Initial studies using post-mortem samples offronto-parietal cerebral tissue confirmed the key patholog-ical features of progressive multiple sclerosis. Active,chronic active or inactive lesions, plus shadow plaqueswere identified based on inflammatory cell infiltration(HLA-DR immunohistochemistry; LN3) and the extent ofdemyelination (LFB) (Fig. 5A–H). The majority of thelesions identified within the cohort and subsequentlybiochemically studied were chronically active. Gliosis, asrevealed by GFAP-immunoreactive astrocytes, was promi-nent (Fig. 5P–R). Widespread axonal injury was confirmedby dystrophic axons and loss of neuronal elements on
Fig. 3 Hyperphosphorylated tau is present in axons and in a soluble fraction in CEAE. (A and B) AT8 (red) immunohistochemistry inlongitudinal sections of CEAE thoracic spinal cord demonstrating focal areas of strong immunoreactivity (boxed area) with spheroidformation (arrow) in the dorsal white matter tract. Spheroid is distinct from cell bodies as revealed by DAPI (blue) counterstain.(C^F) Importantly, AT8 immunoreactivity colocalizes with NF200 immunopositive axonal compartment (C^E; single channel images;F, merge with accompanying z plane images). Scale bars=200mm (A and B), 20mm (C^F). (G) Biochemical analysis of control and CEAEsamples for soluble tau by western blot using phospho-dependent anti-tau antibodies AT8, AT180, AT270, AT100, PHF-1 and the phospho-independent pan anti-tau antibodyTau-5. b-actin confirms equal protein loading. Marked hyperphosphorylation of soluble tau in CEAE isobserved at all phospho-epitopes.
Abnormal tau in demyelinating disease Brain (2008), 131, 1736^1748 1741
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

Bielschowsky staining (Fig. 5I and J). Numerous axonslabelled with dephosphorylated neurofilament (SMI-32)showed axonal end-bulb accumulations or ‘axonal ovoids’representing the terminal ends of transected axons(Fig. 5K–M; Trapp et al., 1998). Some partially demyelin-ated axons were noted to end in ovoids immunopositive fordephosphorylated neurofilament in their demyelinatedsections (Fig. 5M). In addition, swellings and spheroidsimmunopositive for axonal amyloid precursor protein(APP) were abundant in active lesions and at the borderof chronic active lesions (Fig. 5I–M).
We next examined for the presence of hyperphosphory-lated tau using phosphorylation-dependent anti-tau anti-bodies combined with immunohistochemical and westernblot analysis. Immunohistochemistry of fronto-parietalcerebral tissue revealed labelling with PHF-1, AT180 andAT8 antibodies predominantly in diseased white matterwith a predilection for the border zone of chronic activelesions (Fig. 6A–C, Supplementary Fig. 5). No positivelabelling with these antibodies was present in control tissue(e.g. AT180, Fig. 6D; other data not shown). Furthermore,using confocal analysis, co-localization of phosphorylated
Fig. 4 Insoluble tau is regionally and temporally graded in CREAE. (A) Western blot analysis of sarcosyl-insoluble tau preparations fromcervical spinal cord of control, RM2EAE and CEAE mice. AT8 immunoreactive insoluble tau is detected in CEAE and RM2EAE, but not incontrol samples. Positive control material derived from an Alzheimer’s disease case corroborates strong immunoreactivity of the 60kDaband and weaker reactivity of the 64kDa band. Densitometric analysis revealed a significant increase of insoluble tau in CEAE samplescompared to RM2EAE (P50.005). (B) Immunoblot analysis of sarcosyl-insoluble tau preparations from anterior forebrain (FB) shows asignificant decrease in insoluble tau compared to cervical spinal cord (CSp) in CEAE (P50.0001). Densitometric analysis (A and B)represents mean � SEM from three independent samples (six animals per group). (C) Graph showing a positive association betweeninsoluble tau deposition and neuronal loss in cervical cord of RM2EAE and CEAE, with a linear regression analysis value of r2=0.8875.
1742 Brain (2008), 131, 1736^1748 J. M. Anderson et al.
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

tau (AT180 or PHF-1) with neurofilament (NF200)-positivespheroids confirmed that phosphorylated tau is present inneurons (Fig. 6F–H).Tissue protein extracts from 19 of 24 lesions in fronto-
parietal white matter, obtained from six cases, showedstrong immunoreactive bands at 50–60 kDa of solublehyperphosphorylated tau using the following anti-tauantibodies; AT8, AT180, AT270, AT100 and PHF-1(representative immunoblots from three cases studied:
Fig. 6E). Phosphorylation at these different phospho-epitopes was variable between lesions. Negligible phosphor-ylation was seen in control samples. Immunoblot using thepan anti-tau antibody Tau 5 demonstrated that, despiteequal actin levels, there was either a marked reduction oran apparent increment in tau levels in lesion lysatescompared with controls (Fig. 6E). Immunoblotting ofinsoluble sarcosyl protein extracts from cerebral whitematter lesions was next performed to determine whether
Fig. 5 Immunohistochemical and histological characterization of multiple sclerosis tissue. (A^H) Characterization of secondaryprogressive multiple sclerosis lesions using LFB/LN3 immunohistochemistry and a modified Bielschowsky axonal stain.The four lesion typesare demonstrated: (A and B) activeçcharacterized by increased infiltration of inflammatory cells (HLA-DR/LN3) and a loss of myelinstaining (LFB), associated with a decrease in axonal staining; (C and D) chronic-activeçshowed an expanding border of HLA-DR+veinflammatory cells and a demyelinated core with significant reduction in axon staining; (E and F) chronic inactiveçdemyelinatedthroughout with fewer HLA-DR +ve inflammatory cells and marked loss of axons; (G andH) shadow plaques were also observed showingmyelin pallor along with evidence of axonal disruption. Further characterization of chronic active lesions shows widespread axonal injury.(I and J) Axonal pathology occurs in the border, with evidence of significant loss at the centre of the plaque.Modified Bielschowsky staining(I and J) combined with SMI-32 (dephosphorylated neurofilament) (K^M) and APP (N andO) immunohistochemistry reveals disruption ofaxons and the presence of axonal swellings and spheroids.White arrows indicate axonal swellings (I,M and O); pink arrows indicatespheroids (I,K, L and N). SMI-32 labelled axons (green) also show regions denuded of myelin (MBP: red) that terminate in end-bulbs (K).(P^R) Gliosis is also an established feature within chronic lesions and GFAP immunohistochemistry (counterstained with LFB) revealsreactive enlarged multi-polar astrocytes (Q) at the borders of the lesion with dense fibrillary immunoreactivity within the lesion itself (R).Enlarged images (L^M and Q^R) are high-power images of boxed regions shown in K and P, respectively. Scale bars=500mm (A^H) and25mm (I^L).
Abnormal tau in demyelinating disease Brain (2008), 131, 1736^1748 1743
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

phosphorylation is associated with the formation ofinsoluble tau. Strong immunoreactive bands were presentin insoluble extracts derived from six separate cases, but notfrom control samples (representative immunoblots fromthree cases studied: Fig. 6I). Whereas soluble extracts wereimmunoreactive to a broad spectrum of phospho-dependentantibodies (Fig. 6E), insoluble extracts had more lim-ited immunoreactivity, preferentially to the PHF-1 andAT100 antibodies.Sarcosyl insoluble preparations both from CEAE and
multiple sclerosis tissue samples containing insoluble tau
were next studied by electron microscopy. In neitherinstance were tau filaments observed although occasionalaccumulations of material with unclear specificity andstructure did show immunogold labelling.
DiscussionTau is a microtubule stabilizing protein that is essential forefficient axonal transport (Johnson and Stoothoff, 2004).Abnormal tau phosphorylation and the formation ofinsoluble tau are defining features of common
Fig. 6 Abnormal phosphorylated tau is present in secondary progressive multiple sclerosis. (A^D) Immunohistochemistryfor phosphorylated tau using antibodies AT8/AT180/PHF-1 in fronto-parietal tissue from cases with multiple sclerosis revealsspheroid-like intensities not seen in age-matched controls [(D) control with AT180]. (F^H) AT180 (red) is also shown to colocalizewith NF200 (green) in spheroids which compartmentalized to axons using confocal microscopy: [(H) merged images with accompanyingz plane images]. Scale bars=100mm (A^D); 20mm (F^H). (E) Western blot analysis was performed on white matter tissue lysates,from controls (Ctrl) and from several fronto-parietal white matter lesions (lanes A^G) from three representative cases withsecondary progressive multiple sclerosis. Several phospho-dependent epitope antibodies, including AT8, AT100, AT180, AT270and PHF-1, were used to show site-specific phosphorylation of tau in the lesions of multiple sclerosis. b-Actin confirmed equalprotein loading. Significant phosphorylation is evident in two lesions (lanes C and F), with AT180- and PHF-1-directed epitopesbeing the most immunoreactive. Tau5 demonstrated the variability of total tau concentration within multiple sclerosis lesions.(I) Western blot of sarcosyl-insoluble extracts showing immunoreactivity against PHF-1, AT100 and Tau5. All six casesof secondary progressive multiple sclerosis studied were immunoblot positive. Representative samples from three separate casesare shown.
1744 Brain (2008), 131, 1736^1748 J. M. Anderson et al.
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

neurodegenerative diseases such as Alzheimer’s and fronto-temporal dementia. The pathological correlate of clinicaldisability and progression in multiple sclerosis is neuronaland axonal loss. Therefore, we hypothesized that taupathology may also be important in the neurodegenerativephase of multiple sclerosis. We report the novel finding ofsoluble and specifically insoluble hyperphosphorylated tauboth in chronic EAE and secondary progressive multiplesclerosis. We also show a clear temporal associationbetween the accumulation of insoluble tau and theevolution of both neuronal and axonal loss in theprogression of EAE.Although reports of neuronal and axonal injury in
multiple sclerosis date from the earliest pathologicaldescriptions of the disease, it is only with the comparativelyrecent emergence of improved imaging techniques andmore detailed histological analyses that axonal loss has beenidentified as the main pathological substrate for progressivedisability (Davie et al., 1995; De Stefano et al., 1998; Kornekand Lassmann, 1999). Despite early documentation ofcortical lesions (Brownell and Hughes, 1962), neuronalpathology has been comparatively understudied. An emerg-ing body of evidence suggests that grey matter lesions arefrequent and contribute to brain atrophy (Kidd et al., 1999;Peterson et al., 2001; Vercellino et al., 2005; Wegner et al.,2006). The relationship between neuronal and axonaldegeneration is not clearly defined. However, indirect evi-dence supporting an association is suggested in a recentstudy of the anterior optic pathway in multiple sclerosiscases (Evangelou et al., 2001).Improved understanding of the mechanisms underlying
chronic neuronal and axonal loss in multiple sclerosis hasbeen hampered by the relative absence of a suitableexperimental model of secondary progression. Most experi-mental models of multiple sclerosis tend to mirror acuteinflammation or primary progression (Gold et al., 2006). Incontrast, ABH mouse CEAE models key aspects of sec-ondary progression. The natural history is consistent withrelapse–remission followed by a secondary phase associatedwith progressive clinical disability (Baker et al., 1990;Petzold et al., 2003; Pryce et al., 2005). Our findings ofdemyelination, gliosis, axonal and neuronal loss in ABHCEAE mice extend earlier studies and indicate that thismodel reproduces many of the pathological features of sec-ondary progressive multiple sclerosis (Petzold et al., 2003).In addition, we show significant axonal loss in the dorsalspinal tracts including the ascending sensory fibres runningin the dorsal funiculus in the chronic progressive phase ofEAE. Recent reports of axonal loss in multiple sclerosis alsoidentify significant axonal loss in the equivalent tracts of thecervical spinal cord (DeLuca et al., 2004). Neuronal loss isalso evident in the cervical spinal cord and increases withdisease progression from the relapse–remitting phase tochronic secondary progression (post relapse–remissionphase). Whilst these observations document significantneuronal loss occurring earlier in the disease course than
the axonal loss, this is likely to be a sampling effect ratherthan a reflection on the natural history. The axonal densityanalysis was restricted to the dorsal spinal columns, andtherefore did not take into account any pathology in otheraxonal tracts which may have been differentially affected atRM2EAE. Further sub-analysis of neuronal loss accordingto spinal cord lamina demonstrated that NeuN cell loss inRM2EAE occurred predominantly in Lamina I and II of thedorsal horn whilst NeuN cell loss in CEAE was evidentthroughout all four lamina (Supplementary Fig. 1). Neu-rones in Lamina I and II are known to cross the mid-lineand project to the upper cervical cord in the spinothalamic,spinoreticular and spinomesencephalic tracts (Willis andCoggeshall, 2004). These tracts run through the lateralspinal cord, not through the dorsal column–medial lem-niscus pathway (DC–ML) and so the relevant axonal pro-jections of these Lamina I–II neurones would not beidentified in our analysis. However, the post-synaptic dorsalcolumn pathway (which is comprised of tract cells residentin the dorsal horns of the spinal cord through laminaIII–VI) does project to the dorsal column nuclei, throughthe DC–ML tracts. Therefore, at the later time-point of theCEAE mice, as the neuronal loss progresses so does theaxonal loss that is now represented as loss observed throughthe dorsal funiculus. In contrast, negligible neuronal loss isobserved in the anterior forebrain in CEAE.
Axonal injury in the context of acute inflammation ispresent early in the course of multiple sclerosis (Fergusonet al., 1997; Trapp et al., 1998; Bitsch et al., 2000). However,significant axonal loss in normal appearing white matterand ongoing loss in inflammatory quiescent chronic lesionssuggest that additional mechanisms may contribute tochronic axonal pathology in multiple sclerosis (Evangelouet al., 2000; Kornek et al., 2000; Lovas et al., 2000). This isreinforced by the clinical observation of progressive dis-ability in the absence of ongoing inflammation in humans(Coles et al., 1999) and animal models (Pryce et al., 2005)following immunomodulation strategies. The presence ofaxonal injury, including disruption of axonal transport, iswell established from studies of active multiple sclerosislesions and models of acute inflammatory demyelination(Ferguson et al., 1997; O’Neill et al., 1998; Trapp et al.,1998; Aboul-Enein et al., 2006; Shriver and Dittel, 2006). Inaddition, one study of hyper-acute EAE modelling acuteinflammation, showed abnormal phosphorylation of solubletau (Schneider et al., 2004). We focused on chronic lesionsthat do not display active inflammation and are dominatedby gliosis, demyelination and neuronal injury. The directcontribution of demyelination to neuronal and axonal lossis uncertain, although increasing evidence suggests a rolefor oligodendrocyte-derived signals in maintaining neuronaland axonal health including modulation of axonal transport(Kornek et al., 2000; Lappe-Siefke et al., 2003; Wilkinset al., 2003; Yin et al., 2006).
Against this background, we first examined whetherprogressive neuronal degeneration in CEAE is associated
Abnormal tau in demyelinating disease Brain (2008), 131, 1736^1748 1745
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

with accumulation of soluble and insoluble hyperphos-phorylated tau. It is well established that abnormalphosphorylation of tau can lead to neuronal injury throughreduced microtubule stability and/or formation of neuro-toxic protein aggregates (Gustke et al., 1992; Alonso et al.,1997). We demonstrated hyperphosphorylated tau localizedwithin axons in CEAE. Soluble tau was found to be heavilyphosphorylated at multiple epitopes using a panel ofantibodies that detect proline-directed kinase phosphoryla-tion sites (Goedert et al., 1994; Gustke et al., 1992). Thesesame epitopes are known to be phosphorylated in degen-erative tauopathies, characterized by a toxic gain of func-tion and the formation of insoluble filamentous aggregates(Goedert et al., 1992; Forman et al., 2002; Zhukareva et al.,2002a, b; Goedert, 2004; Mott et al., 2005). Hyperphos-phorylation of the soluble form causes tau to dissociatefrom the microtubule and increases the propensity for theformation of insoluble tau (Gustke et al., 1992; Alonsoet al., 1997; Johnson and Stoothoff, 2004). In support ofthis, we report the presence of insoluble tau in the spinalcord of the ABH CEAE mouse. The graded accumulation ofinsoluble hyperphosphorylated tau in CEAE correlatedpositively with significant neurodegeneration. Importantly,this temporal trend of accumulation of insoluble tauparallels the development of progressive disability inCREAE (Baker et al., 1990; Pryce et al., 2005). Electronmicroscopy of preparations containing insoluble tau didnot show tau filaments. Interestingly, occasional smallamorphous aggregates of uncertain significance wereobserved. Insoluble, non-filamentous tau has been reportedto cause axonopathy and muscle weakness in the singleisoform human 4R tau transgenic mouse (Probst et al.,2000). Furthermore, mouse tau protein is not consistentlypresent in filaments (Allen et al., 2002).Previous studies investigating tau in multiple sclerosis
have been restricted to cerebrospinal fluid analysis of totaltau as a potential biomarker of neuronal injury (Kapakiet al., 2000; Teunissen et al., 2005). Our study is the first toimplicate abnormal phosphorylation of tau in the progres-sion of multiple sclerosis; fronto-parietal white matterplaques from six cases with established secondary progres-sion all showed abnormal soluble tau phosphorylation. Allcases were aged557 years (mean age 51 years) making thepresence of changes due to Alzheimer’s disease highlyimprobable. The complex pattern of the axonal tau‘phosphoprint’ in lesions, with variable phosphorylation atdifferent epitopes, is perhaps not surprising given theheterogeneity of pathology observed in MS (Lassmannet al., 2001). Importantly, phosphorylated tau colocalizedwith neurofilament immunopositive spheroids in axons ofchronic multiple sclerosis lesions.Significantly, all six cases of secondary progressive mul-
tiple sclerosis displayed evidence of an insoluble fraction oftau within chronic lesions. One postulated mechanismof tau-mediated neurodegeneration involves formation offilamentous aggregates. Ultrastructural analysis revealed
occasional accumulations of immunogold-labelled materialbut no filaments. It is unclear why the insoluble aggregatesobserved were not filamentous. However, our findings showthat in the multiple sclerosis cases not all epitopes werephosphorylated—as they are in normal tauopathies—andthis could contribute to different conformations. Emergingevidence from Drosophila melanogaster tauopathy modelsadditionally suggests that abnormally phosphorylated taumay be neurotoxic in the absence of filaments (Wittmannet al., 2001; Jackson et al., 2002). Studies in the P301S tautransgenic mouse have also shown evidence for neuronaldysfunction, with synaptic loss and behavioural deficits thatprecede the emergence of insoluble filamentous taupathology (Yoshiyama et al., 2007). Furthermore, a recentin vitro study has shown that aggregation of hyperphos-phorylated tau reduces its ability to inhibit microtubuleassembly, promoting sequestration of normal tau, andproposes that soluble hyperphosphorylated tau or tauoligomers are the more toxic species (Alonso et al., 2006).
Our finding of insoluble tau is unlikely to represent aninvariant feature of neurodegeneration, given the absence ofabnormal tau phosphorylation in other neurodegenerativeconditions such as idiopathic Parkinson’s disease(Trojanowski et al., 1998). Furthermore, the absence ofinsoluble tau in a rare acute active case of relapsing–remitting multiple sclerosis (data not shown) providessome support for the involvement of insoluble tau in theaetiopathogenesis of progression in multiple sclerosis. Aninterpretation of our observation is that there is an evo-lution of tau pathology from a soluble hyperphosphorylatedstate to an insoluble phosphorylated fraction that occurswith disease progression.
This study demonstrates a positive association betweeninsoluble tau and both neuronal and axonal loss in theABH mouse CREAE model of multiple sclerosis, andreports accumulation of insoluble tau in secondary pro-gressive multiple sclerosis. Together, we report the firstevidence implicating tau dysfunction in both experimentaland human demyelinating disease. Our findings provide aplatform for further evaluating the contribution of taupathology to progression in multiple sclerosis.
Supplementary materialSupplementary material is available at Brain online.
AcknowledgementsThese studies were supported by the Webb Trust Fund,Sir David Walker Trust Fund, Husky Foundation, MedicalResearch Council and MS Society of Great Britain andNorthern Ireland and the National Multiple SclerosisSociety, USA. Tissue samples were supplied by the UKMS Tissue Bank, funded by the Multiple Sclerosis Society ofGreat Britain and Northern Ireland, registered charity207495.
1746 Brain (2008), 131, 1736^1748 J. M. Anderson et al.
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

ReferencesAboul-Enein F, Weiser P, Hoftberger R, Lassmann H, Bradl M. Transient
axonal injury in the absence of demyelination: a correlate of clinical
disease in acute experimental autoimmune encephalomyelitis. Acta
Neuropathol 2006; 111: 539–47.
Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, et al. Abundant
tau filaments and nonapoptotic neurodegeneration in transgenic mice
expressing human P301S tau protein. J Neurosci 2002; 22: 9340–51.
Alonso A del C, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal
phosphorylation of tau and the mechanism of Alzheimer neurofibrillary
degeneration: sequestration of microtubule-associated proteins 1 and 2
and the disassembly of microtubules by the abnormal tau. Proc Natl
Acad Sci USA 1997; 94: 298–303.
Alonso A del C, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of
hyperphosphorylated tau into filaments eliminates its inhibitory activity.
Proc Natl Acad Sci USA 2006; 103: 8864–9.
Baker D, O’Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL.
Induction of chronic relapsing experimental allergic encephalomyelitis in
Biozzi mice. J Neuroimmunol 1990; 28: 261–70.
Bandyopadhyay B, Li G, Yin H, Kuret J. Tau aggregation and toxicity in a
cell culture model of tauopathy. J Biol Chem 2007; 282: 16454–64.
Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal
injury in multiple sclerosis. Correlation with demyelination and
inflammation. Brain 2000; 123: 1174–83.
Brownell B, Hughes JT. The distribution of plaques in the cerebrum in
multiple sclerosis. J Neurol Neurosurg Psychiatry 1962; 25: 315–20.
Coles AJ, Wing MG, Molyneux P, Paolillo A, Davie CM, Hale G, et al.
Monoclonal antibody treatment exposes three mechanisms underlying
the clinical course of multiple sclerosis. Ann Neurol 1999; 46: 296–304.
Compston A, Confavreux C, Lassmann H, McDonald IR, Miller D,
Noseworthy J, et al. McAlpine’s multiple scerosis. London: Churchill
Livingstone; 2006.
Davie CA, Barker GJ, Webb S, Tofts PS, Thompson AJ, Harding AE, et al.
Persistent functional deficit in multiple sclerosis and autosomal
dominant cerebellar ataxia is associated with axon loss. Brain 1995;
118: 1583–92.
De Stefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS,
et al. Axonal damage correlates with disability in patients with relapsing-
remitting multiple sclerosis. Results of a longitudinal magnetic
resonance spectroscopy study. Brain 1998; 121: 1469–77.
DeLuca GC, Ebers GC, Esiri MM. Axonal loss in multiple sclerosis: a
pathological survey of the corticospinal and sensory tracts. Brain 2004;
127: 1009–18.
Evangelou N, Esiri MM, Smith S, Palace J, Matthews PM. Quantitative
pathological evidence for axonal loss in normal appearing white matter
in multiple sclerosis. Ann Neurol 2000; 47: 391–5.
Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM. Size-
selective neuronal changes in the anterior optic pathways suggest a
differential susceptibility to injury in multiple sclerosis. Brain 2001; 124:
1813–20.
Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute
multiple sclerosis lesions. Brain 1997; 120: 393–9.
Forman MS, Zhukareva V, Bergeron C, Chin SS, Grossman M, Clark C,
et al. Signature tau neuropathology in gray and white matter of
corticobasal degeneration. Am J Pathol 2002; 160: 2045–53.
Goedert M. Tau protein and neurodegeneration. Semin Cell Dev Biol
2004; 15: 45–9.
Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E,
Vandermeeren M, et al. Epitope mapping of monoclonal antibodies to
the paired helical filaments of Alzheimer’s disease: identification of
phosphorylation sites in tau protein. Biochem J 1994; 301: 871–7.
Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science 2006;
314: 777–81.
Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of
Alzheimer paired helical filaments: abnormal phosphorylation of all six
brain isoforms. Neuron 1992; 8: 159–68.
Gold R, Linington C, Lassmann H. Understanding pathogenesis and
therapy of multiple sclerosis via animal models: 70 years of merits and
culprits in experimental autoimmune encephalomyelitis research. Brain
2006; 129: 1953–71.
Gustke N, Steiner B, Mandelkow EM, Biernat J, Meyer HE, Goedert M,
et al. The Alzheimer-like phosphorylation of tau protein reduces
microtubule binding and involves Ser-Pro and Thr-Pro motifs. FEBS
Lett 1992; 307: 199–205.
Hampton DW, Steeves JD, Fawcett JW, Ramer MS. Spinally upregulated
noggin suppresses axonal and dendritic plasticity following dorsal
rhizotomy. Exp Neurol 2007; 204: 366–79.
Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S,
et al. Human wild-type tau interacts with wingless pathway components
and produces neurofibrillary pathology in Drosophila. Neuron 2002; 34:
509–19.
Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell
function and dysfunction. J Cell Sci 2004; 117: 5721–9.
Kapaki E, Paraskevas GP, Michalopoulou M, Kilidireas K. Increased
cerebrospinal fluid tau protein in multiple sclerosis. Eur Neurol 2000;
43: 228–32.
Kidd D, Barkhof F, McConnell R, Algra PR, Allen IV, Revesz T. Cortical
lesions in multiple sclerosis. Brain 1999; 122: 17–26.
Kornek B, Lassmann H. Axonal pathology in multiple sclerosis. A
historical note. Brain Pathol 1999; 9: 651–6.
Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T,
et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a
comparative quantitative study of axonal injury in active, inactive, and
remyelinated lesions. Am J Pathol 2000; 157: 267–76.
Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, et al.
Disruption of Cnp1 uncouples oligodendroglial functions in axonal
support and myelination. Nat Genet 2003; 33: 366–74.
Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiple sclerosis
pathogenesis: implications for diagnosis and therapy. Trends Mol Med
2001; 7: 115–21.
Lovas G, Szilagyi N, Majtenyi K, Palkovits M, Komoly S. Axonal changes
in chronic demyelinated cervical spinal cord plaques. Brain 2000; 123:
308–17.
Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of
axons by tau, inhibition of axonal traffic and starvation of synapses.
Neurobiol Aging 2003; 24: 1079–85.
Mott RT, Dickson DW, Trojanowski JQ, Zhukareva V, Lee VM,
Forman M, et al. Neuropathologic, biochemical, and molecular
characterization of the frontotemporal dementias. J Neuropathol Exp
Neurol 2005; 64: 420–8.
O’Neill JK, Baker D, Morris MM, Gschmeissner SE, Jenkins HG, Butt AM,
et al. Optic neuritis in chronic relapsing experimental allergic
encephalomyelitis in Biozzi ABH mice: demyelination and fast axonal
transport changes in disease. J Neuroimmunol 1998; 82: 210–8.
Papadopoulos D, Pham-Dinh D, Reynolds R. Axon loss is responsible for
chronic neurological deficit following inflammatory demyelination in
the rat. Exp Neurol 2006; 197: 373–85.
Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites,
apoptotic neurons, and reduced inflammation in cortical multiple
sclerosis lesions. Ann Neurol 2001; 50: 389–400.
Petzold A, Baker D, Pryce G, Keir G, Thompson EJ, Giovannoni G.
Quantification of neurodegeneration by measurement of brain-specific
proteins. J Neuroimmunol 2003; 138: 45–8.
Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, et al.
Axonopathy and amyotrophy in mice transgenic for human four-repeat
tau protein. Acta Neuropathol 2000; 99: 469–81.
Pryce G, O’Neill JK, Croxford JL, Amor S, Hankey DJ, East E, et al.
Autoimmune tolerance eliminates relapses but fails to halt progression
in a model of multiple sclerosis. J Neuroimmunol 2005; 165: 41–52.
Schneider A, Araujo GW, Trajkovic K, Herrmann MM, Merkler D,
Mandelkow EM, et al. Hyperphosphorylation and aggregation of tau in
experimental autoimmune encephalomyelitis. J Biol Chem 2004; 279:
55833–9.
Abnormal tau in demyelinating disease Brain (2008), 131, 1736^1748 1747
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from

Shriver LP, Dittel BN. T-cell-mediated disruption of the neuronal
microtubule network: correlation with early reversible axonal dysfunc-
tion in acute experimental autoimmune encephalomyelitis. Am J Pathol
2006; 169: 999–1011.
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL,
et al. Axonopathy and transport deficits early in the pathogenesis of
Alzheimer’s disease. Science 2005; 307: 1282–8.
Teunissen CE, Dijkstra C, Polman C. Biological markers in CSF and
blood for axonal degeneration in multiple sclerosis. Lancet Neurol 2005; 4:
32–41.
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal
transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338:
278–85.
Trojanowski JQ, Goedert M, Iwatsubo T, Lee VM. Fatal attractions:
abnormal protein aggregation and neuron death in Parkinson’s disease
and Lewy body dementia. Cell Death Differ 1998; 5: 832–7.
Vercellino M, Plano F, Votta B, Mutani R, Giordana MT, Cavalla P. Grey
matter pathology in multiple sclerosis. J Neuropathol Exp Neurol 2005;
64: 1101–7.
Wegner C, Esiri MM, Chance SA, Palace J, Matthews PM. Neocortical
neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 2006;
67: 960–7.
Wilkins A, Majed H, Layfield R, Compston A, Chandran S. Oligoden-
drocytes promote neuronal survival and axonal length by distinct
intracellular mechanisms: a novel role for oligodendrocyte-derived
glial cell line-derived neurotrophic factor. J Neurosci 2003; 23: 4967–74.
Willis WD, Coggeshall R.E. Sensory Mechanisms of the Spinal Cord.
New York: Kluwer Academic/Plenum, 2004.
Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J,
Hutton M, et al. Tauopathy in Drosophila: neurodegeneration without
neurofibrillary tangles. Science 2001; 293: 711–4.
Yin X, Baek RC, Kirschner DA, Peterson A, Fujii Y, Nave KA, et al.
Evolution of a neuroprotective function of central nervous system
myelin. J Cell Biol 2006; 172: 469–78.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al.
Synapse loss and microglial activation precede tangles in a P301S
tauopathy mouse model. Neuron 2007; 53: 337–51.
Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, et al.
Sporadic Pick’s disease: a tauopathy characterized by a spectrum of patho-
logical tau isoforms in gray and white matter. AnnNeurol 2002a; 51: 730–9.
Zhukareva V, Shah K, Uryu K, Braak H, Del Tredici K, Sundarraj S, et al.
Biochemical analysis of tau proteins in argyrophilic grain disease,
Alzheimer’s disease, and Pick’s disease: a comparative study. Am J
Pathol 2002b; 161: 1135–41.
1748 Brain (2008), 131, 1736^1748 J. M. Anderson et al.
by guest on May 16, 2016
http://brain.oxfordjournals.org/D
ownloaded from
Related Documents











